
CROWN ETHER COMPOUNDS: SYNTHESIS AND
ALKALI METAL CATION COMPLEXATION
by
MI-JA GOO, B.S., M.S.
A DISSERTATION
IN
CHEMISTRY
Submitted to the Graduate Faculty of Texas Tech University in
Partial Fulfillment of the Requirements for
the Degree of
DOCTOR OF PHILOSOPHY
Approved
Accepted
May, 1991

//^ LB
1991, The Graduate School, Texas Tech University.
11

TABLE OF CONTENTS
TABLE OF CONTENTS iii
LIST OF TABLES xii
LIST OF FIGURES xiv
I. INTRODUCTION 1
Discovery of Crown Ethers 1
Nature of Crown Ether Complexes with Cations 3
Assessment of Metal Ion Complexation Abilities of Crown
Ethers by Picrate Extraction 7
Proton-ionizable Crown Ethers 9
Solvent Extraction by Proton-ionizable Crown Ethers 1 1
Liquid Membrane Transport of Metal Ions by
Proton-ionizable Crown Ethers 1 8
Immobilization of Crown Ethers on Silica Gel 2 0
Statement of Research Goals 2 1
II . RESULTS AND DISCUSSION 2 6
Acyclic and Cyclic Polyether Derivatives of Salicylic Acid .... 2 6
Lipophilic Dibenzo-16-crown-5-oxyacetic Acids 3 2
Dibenzo-16-crown-5 Phosphonic Acid Monoalkyl Esters 3 4 Functionalized Crown Ethers for Attachment to Silica Gel 4 4
111

Picrate Extractions 5 1
Benzo-21-crown-7 and Dibenzo-21-crown-7 Compounds.. 5 1
Benzo-13-crown-4 Compounds 5 2
14-Crown-4 Compounds 5 6
Crown Ethers with Pendant Ferrocene Units 5 7
Crown Ethers with Pendant Pyridine Units 6 1
Molecular Receptors 6 5
Summary 7 0
I I I . EXPERIMENTAL PROCEDURES 7 2
Instrumentation and Reagents 7 2
General Procedure for Preparation of Tosylates of
Monobenzyl Glycols 62-64 7 3
Tosylate of Monobenzyl Ethylene Glycol (62) 7 3
Tosylate of Monobenzyl Diethylene Glycol (63) 7 4
Tosylate of Monobenzyl Triethylene Glycol (64) 7 4 General Procedure for Preparation of Carboxylic Acids
68-70 7 4
Methyl 2-[(l,4-Dioxa-5-phenyl)pentyl]benzoate (65) 7 5
2-[(l,4-Dioxa-5-phenyl)pentyl]benzoic Acid (68) 7 5
Methyl 2-[(l,4,7-Trixa-8-phenyl)octyl]benzoate (66) 7 5
2-[(l,4,7-Trioxa-8-phenyl)octyl]benzoic Acid (69) 7 6
IV

Methyl 2-[(l ,4,7,10-Tetraoxa-ll-phenyl)undecyl]benzoate (67) 7 6
2-[(l ,4,7,10-Tetraoxa-l l-phenyl)undecyl]benzoic Acid (70) 7 6
Preparation of (Benzyloxy)methyl-substituted Crown Ethers
71-73 7 6
(Benzyloxy)methyl-12-crown-4 (71) 7 6
(Benzyloxy)methyl-15-crown-4 (72) 7 7
(Benzyloxy)methyl-21-crown-4 (73) 7 8
General Procedure for Preparation of Hydroxy methyl Crown
Ethers 74-76 7 8
Hydroxymethyl-12-crown-4 (74) 7 9
Hydroxymethyl-15-crown-4 (75) 7 9
Hydroxymethyl-21-crown-4 (76) 7 9
General Procedure for Preparation of (Tosyloxy)methyl-
substituted Crown Ethers 77-82 7 9
(Tosyloxy)methyl-12-crown-4 (77) 8 0
3-[(Tosyloxy)methyl]-13-crown-4 (78) 8 0
(Tosyloxy)methyl-15-crown-5 (79) 8 0
(Tosyloxy)methyl-18-crown-6 (80) 8 0
(Tosyloxy)methyl-21-crown-7 (81) 8 0
(Tosyloxy)methyl-24-crown-8 (82) 8 1
l^fMNH

General Procedure for Preparation of Crown Ether
Carboxylic Acids 34, 35, and 38-41 8 1
Methyl 2-[(12-Crown-4)-methyloxy]benzoate (83) 8 2
2-[(12-Crown-4)-methyloxy]benzoic Acid (34) 82
Methyl 2-[3'-(13-Crown-4)-methyloxy]benzoate (84) .... 8 2
2-[3'-(13-Crown-4)-methyloxy]benzoic Acid (35) 8 2
Methyl 2-[(15-Crown-5)-methyloxy]benzoate (85) 8 3
2-f(15-Crown-5)-methyloxy]benzoic Acid (38) 8 3
Methyl 2-[(18-Crown-6)-methyloxy]benzoate (86) 8 3
2-[(18-Crown-6)-methyloxy]benzoic Acid (39) 8 3
Methyl 2-[(21-Crown-7)-methyloxy]benzoate (87) 8 4
2-[(21-Crown-7)-methyloxy]benzoic Acid (40) 8 4
Methyl 2-[(24-Crown-8)-methyloxy]benzoate (88) 8 4
2-[(24-Crown-8)-methyloxy]benzoic Acid (41) 8 4
sym-Ketodibenzo-16-crown-5 (90) 8 4
syni-(Methyl)(hydroxy)dibenzo-16-crown-5 (91) 8 5
General Procedure for Preparation of Crown Ether Alcohols 92 and 93 8 6
sym-(Hexyn(hydroxy)dibenzo-16-crown-5 (92) 8 7
sym-(Decyn(hydroxv)dibenzo-16-crown-5 (93) 8 7
General Procedure for Preparation of Crown Ether Carboxylic Acids 94-96 8 7
VI

syirL-(Methyl)dibenzo-16-crown-5-oxyacetic Acid (94) .. 8 8
sym-(Hexyl)dibenzo-16-crown-5-oxyacetic Acid (95) .... 8 8
iym.-(Decyl)dibenzo-16-crown-5-oxyacetic Acid (96) 8 8
Monoethyl sym-Dibenzo-16-crown-5-oxymethylphosphonic Acid (50) 8 9
General Procedure for Preparation of Crown Ethers Phosphonic Acid Monoethyl Esters 47-49 and 51-53 9 0
Diethyl iyi]i-Dibenzo-16-crown-5-oxyethylphosphonate (106) 9 0
Monoethyl sym-Dibenzo-16-crown-5-oxvethvlphosphonic Acid (51) 9 1
Diethyl sym-Dibenzo-16-crown-5-oxypropylphosphonate (107) 9 1
Monoethyl sym-Dibenzo-16-crown-5-ox v propyl phosphonic Acid (52) \ 9 1
Diethyl sym-Dibenzo-16-crown-5-oxvbutvlphosphonate (108) 9 1
Monoethyl sym-Dibenzo-16-crown-5-oxvbutvlphosphonic Acid (53) 9 2
Diethyl sym-(Decyl)dibenzo-16-crown-5-oxvethvl-phosphonate (103) 9 2
Monoethyl sym-(Decyl)dibenzo-16-crown-5-oxyethyl-phosphonic Acid (47) 9 2
Diethyl syni-(Decy l)dibenzo-16-crown-5-oxy propyl-phosphonate (104) 9 2
v i i

Monoethyl sym-(Decyndibenzo-16-crown-5-oxvpropvl-phosphonic Acid (48) 9 3
Diethyl sym.-(Decy l)dibenzo-16-crown-5-oxy butyl-phosphonate (105) 9 3
Monoethyl svm-(Decyndibenzo-16-crown-5-oxybutyl-phosphonic Acid (49) 9 3
Dimethyl ^yi]i-Dibenzo-16-crown-5-oxyethyl-phosphonate (109) 9 4
Monomethyl sym-Dibenzo-16-crown-5-oxvethyl-phosphonic Acid (110) 9 4
General Procedure for Preparation of Crown Ether Methanesulfonates 115-118 9 5
-(sym-Dibenzo-16-crown-5-oxy)-2-methanesulfonoxy)ethane (117) 9 5
- (sym-Dibenzo-16-crown-5-oxy)-3-methanesulfonoxy)propane (118) 9 5
-(sym-(Decyl)dibenzo-16-crown-5-oxv)-2 methanesulfonoxy)ethane (115) 9 6
- (sym-(Decyndibenzo-16-crown-5-oxy)-3-methanesulfonoxy)propane (116) 9 6
General Procedure for Preparation of Crown Ether Bromides
97 , 98 , 100 , and 101 9 6
1-(sym-Dibenzo-16-crown-5-oxy)-2-bromoethane (100) 9 7
l-(sxni-Dibenzo-16-crown-5-oxy)-3-bromopropane (101) 9 7
l-[(sxi] l-(Decyl)dibenzo-16-crown-5-oxy]-2-bromoethane (97) 9 7
V l l l

l-r(sym-(Decyndibenzo-l6-crown-5-oxy1-3-bromopropane (98) 9 7
General Procedure for Preparation of Crown Ether Bromides 99 and 102 9 8
l-(sxDl-Dibenzo-16-crown-5-oxy)-4-bromobutane (99)... 9 8
l-rsym-(Decyl)dibenzo-16-crown-5-oxy]-4-bromobutane (102) 9 8
General Procedure for Preparation of Crown Ether Esters
119-120 9 9
Ethyl (sxiIL-Dibenzo-16-crown-5-oxy)acetate (120) 9 9
Ethyl [(sxQi-(Decyl)dibenzo-16-crown-5-oxy)acetate (119) 9 9
General Procedure for Preparation of Crown Ether Alcohols 111 and 113 100
2-(sym-Dibenzo-16-crown-5-oxy)ethanol (113) 100
2-F(sym-(Decyl)dibenzo-16-crown-5-oxv1ethanol (111) .. 100
General Procedure for Preparation of Allyoxy Crown Ethers
121 and 122 1 0 1
3-(sym-Dibenzo-16-crown-5-oxy)-l-propene (122) 101
3-(sym-(Decyndibenzo- l6-crown-5-oxy1 1 -propene ( 1 2 1 ) 1 01
General Procedure for Preparation of Crown Ether Alcohols 112-114 102
3-(sym-Dibenzo-16-crown-5-oxy)propan-l-ol (114) 102
IX

3-[sym-(Decyl)dibenzo-16-crown-5-oxyJpropan-l-ol (112) 102
10-Methanesulfonoxy-l-decene (133) 103
10-Bromo-l-decene (134) 103
General Procedure for Preparation of Crown Ether Alcohols
124 and 125 104
sym-(9-Decenyn(hydroxy)dibenzo-14-crown-4 (124) 104
sxQi-(9-Decenyl)(hydroxy)dibenzo-16-crown-5 (125) 105
General Procedure for Preparation of Crown Ether Carboxylic Acids 135 and 136 105
syni-(9-Decenyl)dibenzo-14-crown-4-oxy acetic Acid (135) 105
sjTn-(9-Decenyl)dibenzo-16-crown-5-oxyacetic Acid (136) 106
General Procedure for Preparation of Crown Ether Esters 123 and 124 106
Ethyl sxni-(9-Decenyl)dibenzo-14-crown-4-oxyacetate (123) 106
Ethyl syni-(9-Decenyl)dibenzo-16-crown-5-oxyacetate (124) 107
General Procedure for Preparation of Crown Ether Esters 138 and 139 107
Ethyl syiTi-(10-Hydroxydecyl)dibenzo-14-crown-4-oxyacetate (138) 107
Ethyl sym-(10-Hydroxydecyl)dibenzo-16-crown-5-oxyacetate (139) 108
X
ni . I i i — . ^

11-Methanesulfonoxy-l-undecene (140) 108
2-Hydroxy-4-(10'-undecenoxy)benzoic Acid (141) 108
Methyl 2-Hydroxy-4-(10'-undecenoxy)benzoate (142) 109
General Procedure for Preparation of Methyl Esters 125-128 1 1 0
Methyl 2-[(12-Crown-4)methyloxy]-4-(lO'-undecenoxy)benzoate (125) 110
Methyl 2-[(15-Crown-5)methyloxy]-4-(lO'-undecenoxy)benzoate (126) 110
Methyl 2-[(18-Crown-6)methyloxy]-4-(lO'-undecenoxy)benzoate (127) I l l
Methyl 2-[(21-Crown-7)methyloxy]-4-
(10*-undecenoxy)benzoate (128) I l l
Preparation of Alkali Metal Picrates 1 1 1
Preparation of Alkylammonium Picrates 1 1 2
Preparation of N,N-Didecyl-7,16-diaza-18-crown-6
(185 ) 1 1 2
Decanoyl Chloride (183) 1 1 2
N,N-Didecanoyl-7,16-diaza-18-crown-6 (184) 1 12
N,N-Didecyl-7,16-diaza-18-crown-6 (185) 113
Procedure for Picrate Extractions 1 1 3
REFERENCES 1 1 5
XI

LIST OF TABLES
1. Comparison of Cation and Cavity Diameters 7
2. Lipophilicity of Salts of Crown Ethers 5-8 1 1
3. The Effect of Organic Solvent upon the Selectivity and Efficiency of Alkali Metal Solvent Extraction by Crown Ether Carboxylic Acid 18 1 8
4. Comparison of Bound and Analogous Unbound Crown Ether Interaction Constants with Metal Cations 2 3
5. Yields of Compounds 62-70 2 8
6. Yields of Compounds 34, 35, 38-41, and 77-88 3 2
7. Hydrolysis of Crown Ether Phosphonic Acid Dialkyl Esters 103-109 3 9
8. Picrate Extraction Data for 21-Crown-7 Compounds 5 3
9. Picrate Extraction Data for Benzo-13-crown-4 Compounds 5 5
10. Picrate Extraction Data for 14-Crown-4 Compounds 5 8
11. Picrate Extraction Data for Crown Ethers 171 and 172 6 0
12. Picrate Extraction Data for Crown Ethers 173-175 6 2
13. Picrate Extraction Data for Crown Ethers 176-178 6 3
14. Picrate Extraction Data for Molecular Receptors 179-181 66
15. Alkylammonium Picrate Data for Molecular Receptors 179-181 6 8
16. Alkylammonium Picrate Data for Molecular Receptor 182 6 9
Xl l
• ^ ^ N .

17. Alkylammonium Picrate Data for Crown Ether 185 7 0
Xl l l

LIST OF HGURES
1. Pedersen's Synthesis of Dibenzo-18-crown-6 1
2. A Dibenzo-18-crown-6 Complex with a Sodium Salt 2
3. The Template Effect in the Synthesis of 18-Crown-6 5
4. Reorganization of 18-Crown-6 upon Complexation 7
5. Complexation and Decomplexation of Proton-ionizable Crown Ethers with Metal Cations 9
6. Cram's Early Proton-ionizable Crown Ethers 1 0
7. Bartsch's Dibenzo Crown Ether Carboxylic Acids 1 2
8. Bartsch's Crown Ether Carboxylic Acids with Various
Ring Sizes 1 5
9. The Proposed Crown Ether Carboxylic Acids 1 5
10. Bartsch's Crown Ether Monoethyl Phosphonates 1 6
11 . The Proposed Crown Ether Monoethyl Phosphonates 17
12. Mechanism of Metal Cation Transport across a Liquid Membrane by a Proton-ionizable Crown Ether 1 9
13. Synthesis of Silica Gel-bound Crown Ethers by Bradshaw,
Izatt, and Coworkers 2 2
14. Functionalized Crown Ethers for Attachment to Silica Gel .... 4 5
15. Benzo-21-crown-7 and Dibenzo-21-crown-7 Compounds .... 5 2
16. Benzo-13-crown-4 Compounds 5 4
17. 14-Crown-4 Compounds 5 7
xiv

18. Crown Ethers with Pendant Ferrocene Units 5 9
19. Crown Ethers with Pendant Pyridine Units 6 1
20. The Proposed 2:2 Complex between Crown Ether 174 and Alkali Metal Cations 6 4
21 . The Proposed 1:1 Complex between Crown Ether 178 and Alkali Metal Cations 6 4
22. Molecular Receptors 6 5
XV

CHAPTER I
INTRODUCTION
Discovery of Crown Ethers
The earliest comprehensive report of macrocyclic polyethers
and their unique ability to solubilize metal salts appeared in 1967.ti]
Pedersen obtained a very small amount of a white, fibrous,
crystalline by-product in a preparation of bis[2-(o.-hydroxy-
phenoxy)ethyl] ether (3) by reacting bis(2-chloroethyl) ether (2)
with the sodium salt of 2-(o.-hydroxyphenoxy)tetrahydropyran (1)
which was contaminated with some catechol (Figure l).^^! The
unexpected compound was found to be insoluble in methanol, but
dissolved in methanol when sodium hydroxide was added. Pedersen
deduced the structure of the compound to be 2,3,11,12-dibenzo-
LO ^ ro, - a: :
OH CI CI "^^^=^0 O
4
Figure 1. Pedersen's Synthesis of Dibenzo-18-crown-6.

l,4,7,10,13,16-hexaoxacyclooctadeca-2,ll-diene (4) based on its
elementary composition, molecular weight, and nmr spectrum.
Compound 4 was formed by the reaction of two molecules of
bis(2-chloroethyl) ether with two molecules of the catechol
contaminant. In trying to explain the unusual solubility
characteristics of this compound in the presence of sodium salts,
Pedersen realized that the cyclic polyether ring formed a complex
with the sodium cation (Figure 2).[3]
O^ : O^^^is^ Anion % I ' 4. '
o' : ''o
Figure 2. A Dibenzo-18-crown-6 Complex with a Sodium Salt.
Since the systematic names of many of these polyethers were
too cumbersome for repeated use, abbreviated names have been
coined for their ready identification. Because of the appearance of its
molecular model and its ability to "crown" the cations, the cyclic
polyethers were designated "crown ethers" as a class and host-guest
chemistry was born. Pederson's nomenclature consisted of naming in
order: (1) the number and kind of substituents on the polyether ring;
(2) the total number of atoms in the polyether ring; (3) the class
name, crown; and (4) the number of oxygen atoms in the polyether
ring. Placement of the hydrocarbon rings and the oxygen atoms is

usually symmetrical and the exceptions are indicated by the prefix
"asym." Employing this nomenclature, macrocycle 4 is named
"Dibenzo-," "18-," "Crown-," and "-6," to give "Dibenzo-18-crown-6."
The five different methods for the preparation of the cyclic
polyethers reported by Pedersen^] are shown in Scheme 1, where R,
S, T, U, V, and W represent divalent organic groups which may or
may not be identical. Method 5 consisted of the hydrogenation of
benzo compounds to the corresponding cyclohexano derivatives using
ruthenium dioxide catalyst in p.-dioxane. Pedersen recognized that
method 2 was the most versatile for the preparation of compounds
containing two or more benzo groups. For example, method 2
produced dibenzo-14-crown-4 in a 27% yield, but method 3 gave no
recoverable amount of the desired product.
Pedersen found that the synthesis of crown ethers often
proceeded in a surprisingly smooth manner to give high yields of
cyclic products even without applying high dilution techniques.t^^l A
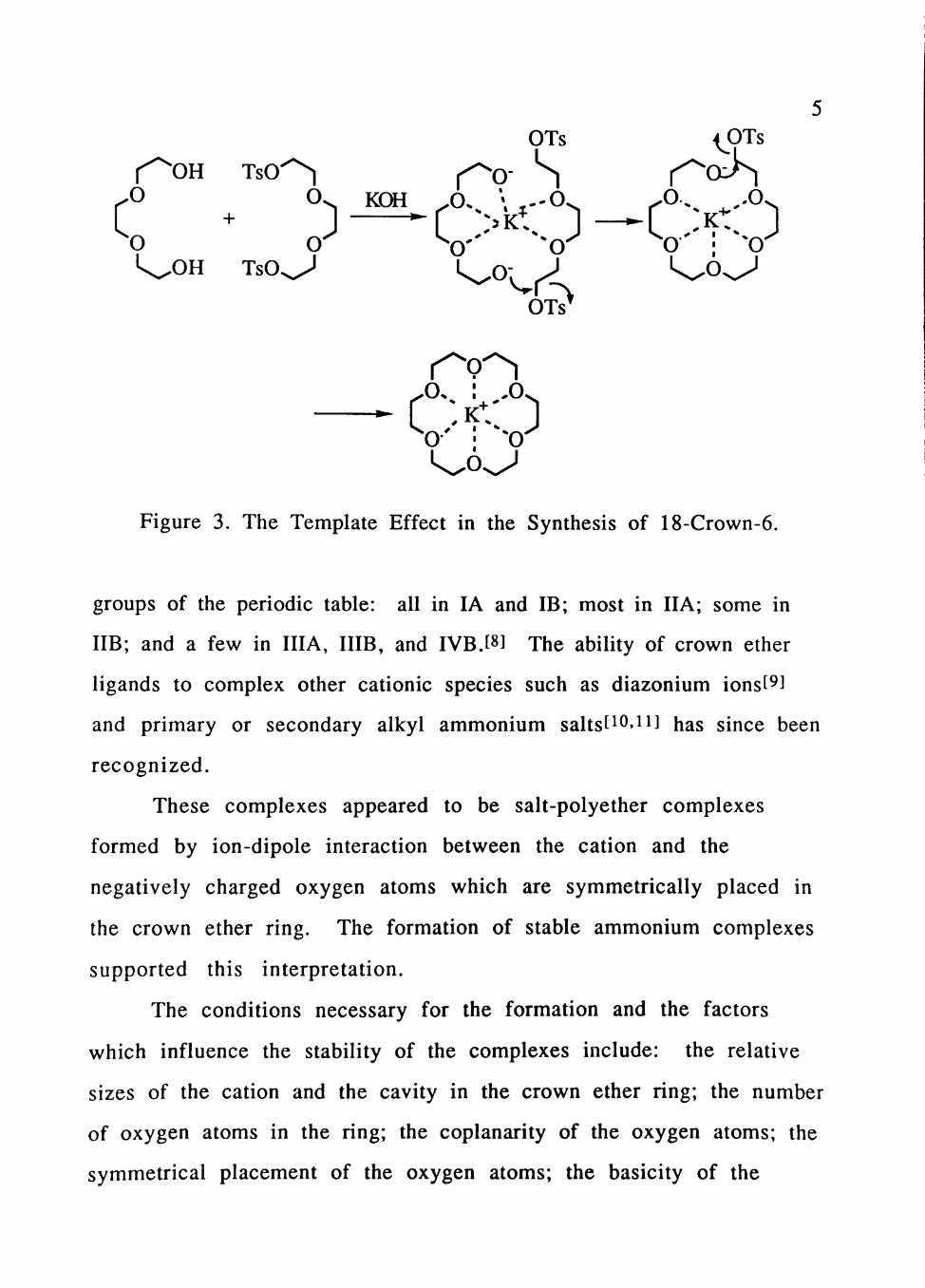
cation assisted cyclization, as depicted in Figure 3, is considered to be
responsible for the high yields. This kind of cyclization assistance is
called a template effect.t^^ The extent to which the template effect is
noticeable depends on the type of cation present.t^l In a few cases,
Cs"*" has proven to be very suitable.f^l
Nature of Crown Ether Complexes with Cations
Pedersen established that many crown ethers formed
complexes with the salts of the elements belonging to the following

Scheme 1
Method 1
,0H
^OH "^^55^0 + 2NaOH + Cl-R-Cl T X ^ ^
Method 2
' O ' ^ O - S - 0 ^ ^ ^ ^ ^ ^ " ^ C l - T - C l - ^ .<^^0-T-0
Method 5
O-S-0
Method 3
Method 4
^nr°" M-v<:,
4 NaOH + 2 Cl-U-Cl
+ o iSTnOH • ^^ i^awxx
- c
. if 1=
^ O - U - 0 .
^^o-u-o
s^O-V-O
= ^ 0 - V - 0
a::Do ^ H. -^^ Oil
5 OTs ^OTs
r oH Tso^ ^o- S r^ctK,
OTV
Figure 3. The Template Effect in the Synthesis of 18-Crown-6.
groups of the periodic table: all in lA and IB; most in IIA; some in
IIB; and a few in IIIA, IIIB, and IVB.tsi The ability of crown ether
ligands to complex other cationic species such as diazonium ionst^J
and primary or secondary alkyl ammonium saltsf^^.H] has since been
recognized.
These complexes appeared to be salt-polyether complexes
formed by ion-dipole interaction between the cation and the
negatively charged oxygen atoms which are symmetrically placed in
the crown ether ring. The formation of stable ammonium complexes
supported this interpretation.
The conditions necessary for the formation and the factors
which influence the stability of the complexes include: the relative
sizes of the cation and the cavity in the crown ether ring; the number
of oxygen atoms in the ring; the coplanarity of the oxygen atoms; the
symmetrical placement of the oxygen atoms; the basicity of the

oxygen atoms; steric hindrance in the polyether ring; the tendency of
the ion to associate with the solvent; and the electrical charge on the
ion.
A stable complex is not formed if the cation is too large to fit in
the cavity of the crown ether ring. The ionic diameters of the alkali
metal cations and crown ether cavities are listed in Table 1, which
shows that 12-crown-4 and Li+, 15-crown-5 and Na+, and 18-crown-
6 and K+ are well matched. The theoretically predicted spatial
relationships with inward-directed oxygen atoms are quite in
keeping with an ion-ball model of the 18-crown-6-K+ complex.^41
Measured complexation constants confirm the excellent fit of K+
within the 18-crown-6 ring.
Dunitz demonstrated from crystal structures of 18-crown-6
and those of its K+SCN- complex (Figure 4)n5,i6] that the host and its
complex have different conformational organizations. The potential
crown cavity of the host itself is filled with two inward-turned CH2
groups and the electron pairs of two oxygens face outward and away
from the center of the roughly rectangular structure. Thus, the free
host does not have a crown shape or a cavity. Only when the
oxygens become engaged with a guest such as K+ does a filled cavity
develop. The presence of a guest in the complex induces the electron
pairs to converge on the center of a crown-shaped object. In other
words, the guest conformationally reorganizes the host upon
complexation. Solvent molecules may play the same role as a
cationic guest.

Table 1. Comparision of Cation and Cavity Diameters.t^2,i3]
cation cation diameter[A]a crown ether cavity diameter[A]
U TJe 12-crown-4 1.2b . 1.5c
Na 1.90 15-crown-5 1.7 - 2.2
K 2.66 18-crown-6 2.6 - 3.2
Rb 2.98 19-crown-6 3.0 - 3.5
Cs 3.38 21-crown-7 3.4 - 4.3
a. The Shannon and Prewitt crystal radii are adopted because they come closer to representing reality than do the traditional values.
b. Estimated from Corey-Pauling-Koltun (CPK) models.
c. Estimated from Fisher-Hirschfelder-Taylor (FHT) models.
Figure 4. Reorganization of 18-Crown-6 upon Complexation.
Assessment of Metal Ion Complexation Abilities of Crown Ethers bv Picrate Extraction
The use of metal picrate salts as guest cations to provide a
spectrometric method for determining the stoichiometry of the
crown ether-cation complex was reported by Smid and co
workers. 7,18] The position of the absorption maximum of the

8
picrate anion in THE was noted to be sensitive to the ion pairing
nature of metal salt. For crown-cation complexes of 1:1
stoichiometry, the salt forms a tight ion pair which has an absorption
maximum between 350 and 362 nm, depending on the alkali metal
cation used. When the stoichiometry of the complex was two crown
ethers to one cation, the salt formed a "crown-separated" ion pair
with an absorption maximum between 375 and 390 nm.
Iwachido and co-workers later reported the distribution of
alkali metal picrate complexes between an aqueous phase and a
benzene solution of dibenzo-18-crown-6.[i9] xhe extraction constant
(Kex) was defined by the equilibrium between free metal picrate in
the aqueous phase ([M+]a and [A Ja), free crown in the organic phase
([Cr]o), and the complex ion pair in the organic phase ([MCrA]o)
according to the relationship shown in Equation 1:
[M+]a + [Cr]o + [A-]a ^=f=^ [MCrAJo. (1)
Kex = [MCrA]o/[M+]a[Cr]o[A-]a.
The extraction constant defined by Iwachido and co-workers
represents only the concentration equilibrium constant of the crown
ether with a particular metal picrate and does not include ionic
activity coefficients or any compensation for the increased
lipophilicity of the picrate salts as the ionic diameter increases. Thus,
the extraction constant is only a measurement of the activity of the
crown ether in single ion solvent extraction systems and has no
thermodynamic significance.

Proton-ionizable Crown Ethers
Crown ethers with ionizable pendant arms are known to form
stronger complexes with uni- and multivalent cations than their
neutral counterparts because the anion provides an internal
counterion for a complexed cation, as shown in Figure 5.
X-H
M"" ^ ( ]\^^ ) + H""
Figure 5. Complexation and Decomplexation of Proton-ionizable Crown Ethers with Metal Cations.
Cram and co-workers synthesized the first ionizable crown
ethers which are shown in Figure 6. ^ ^ Crown ethers 5-8 possess
carboxyl groups but its orientation into crown cavity can hinder ring
participation during complexation with the cation. Modification of
the pendant arm by insertion of spacer groups between the crown
ether ring and the carboxylic acid residue resulted in enhancement
of guest complex formation.[^2] Cram and co-workers prepared
crown ether carboxylic acids 9-12 which have ionizable pendant
arms of sufficient length to allow interaction with the guest by both
the crown ether ring oxygens and the ionizable group.[23]

10
n. 5 0 6 1 7 2 8 3
9, A=B=CH20CH2C02H, n=0 10, A=B=CH20CH2C02H, n=l 11, A=CH20CH2C02H, B=H, n=0 12, A=CH20CH2C02H, B=H, n=l
Figure 6. Cram's Early Proton-ionizable Crown Ethers.
Crown ethers 5-8 were tested for their abilities to lipophilize
Li+, Na+, K-"-, and Ca "*" by distributing their salts between
dichloromethane and water (Table 2). Maximum lipophilization of
each ion depends on the ring size of the host: for Li+, 18-crown-5;
for Na"*", 21-crown-6; for K+, 30-crown-8; for Ca^^, 18-crown-5.
The crystal structure of 6 showed that the potential cavity within the
molecule was filled with the carboxylic acid residue.[24] Therefore,
complexation of a guest species by 6 must be associated with a major
conformational reorganization of the host.
Bartsch and co-workers have synthesized many different types
of proton-ionizable crown ethers. Some of these are dibenzo crown
ether carboxylic acids and are shown in Figure 7. The two benzene
rings reduce the basicity of four ethereal oxygens through electron
delocalization, but provide ring rigidity which helps to preorganize
the ligand.

11
Table 2. Lipophilicity of Salts of Crown Ethers 5-8.
salt of
5
6
7
8
% of salt in die CH2CI2
Li+
1-4
7.2
6.1
3-4
Na+
1.5
7.9
8.7
5.2
K+
1-4
6.7
6.8
8.0
layer
Ca2+
1.1
4.8
1.8
2.9
Solvent Extraction bv Proton-ionizable Crown Ethers
Solvent extraction is a method of separation based on the
transfer of a solute from one immiscible solvent into another.[25] The
extraction efficiency of crown ethers has been markedly enhanced
by the introduction of crown ethers which bear a pendant carboxylic
acid group.[26] These proton-ionizable crown ethers possess a distinct
advantage over neutral crown ether compounds in the extraction of a
metal cation from an aqueous phase into an organic medium does not
require the concomitant transfer of the aqueous phase anion.[27]
Extraction efficiencies for a number of proton-ionizable crown ethers
are reported as Kex, the extraction constant. The extraction constant
is defined in Equation 2:
i C . = [MLlorg [H+]aq/[HL]org[M+]aq. ( 2 )

12
c 13 14 15 16
c
H^OCHjCOjH
';;o Y_
CH2CH2 CH2CH2CH2 CH2CH2OCH2CH2 CH2(CH20CH2)2CH:
^ 8 ^ 1 7
H^OCHCOoH
K : ^ ^ 0 ^
H^OCH.CO^H
0 ^ 0
1 7
2
CgHjy >^^OCH2C02H
o::::o ^ 0 ^
.)3
18 1 9
Figure 7. Bartsch's Dibenzo Crown Ether Carboxylic Acids.
Metal ion extraction efficiencies are influenced by many
factors. The lipophilicity of the molecule is an important
consideration in the design of extractant molecules. A proton-
ionizable crown ether extractant will be lost from the organic phase
upon deprotonation if it has insufficient lipophilicity, even if the
crown ether compound forms stable complexes with the metal ion
being extracted. Competitive alkali metal cation extraction from
aqueous solution into chloroform by the dibenzo crown ether

13
carboxylic acids 13-16 (see Figure 7) was examined.[27,28] it was
found that these proton-ionizable crown ethers were of insufficient
lipophilicity to remain completely in the organic phase during
extraction of alkali metal cations from alkaline aqueous phases. To
avoid such complications in extraction behavior, a lipophilic group
was attached either to each benzene ring, to the carboxylic acid
containing sidearm, or to the center carbon of the three-carbon
bridge of the dibenzo-16-crown-5 compound 15 to produce the
lipophilic dibenzo-16-crown-5-carboxylic acids 17, 18, and 19,
respectively (see Figure 7).[26-29] Compounds 17-19 were found to
be sufficiently lipophilic to remain completely in the chloroform
phases even when the contacting aqueous solutions of alkali metal
cations were highly alkaline.[26,28] por competitive solvent extraction
of alkali metal cations from aqueous solution into chloroform,
structural isomers 19 gave much higher Na+ selectivity than did 17
or 18. Hence the lipophilic group attachment site was shown to
influence the extraction selectivity.
A second factor which influences extraction selectivity is the
crown ether ring size. The series of lipophilic crown ether carboxylic
acids with varying ring sizes shown in Figure 8 was synthesized by
Bartsch and co-workers to study competitive solvent extraction of
alkali metal cations.[^0,31] Extraction selectivity for Li+ was observed
for crown ethers with 12-15-membered polyether rings containing
four oxygen atoms. For the 14-crown-4 carboxylic acids 23 and 24,
very high Li+/Na+ selectivity coefficients of 17-20 were observed

14
with no detectable extraction of K+, Rb+, or Cs+. The crown ether
carboxylic acids which contain 15-crown-5, 18-crown-6, and
21-crown-7 rings exhibited good selectivities for K+, Rb+, and Cs+
extraction, respectively. In contrast, poor extraction selectivity was
observed for crown ether carboxylic acids with 24-crown-8, 27-
crown-9, and 30-crown-lO rings. Thermodynamic data for alkali
metal cation complexation by this series of compound has not been
obtained because of their high lipophilicity which would not provide
solubility in the aqueous or aqueous alcoholic solvent in which such
measurements are usually performed. A new series of crown ether
carboxylic acids 34-41 which has no lipophilic group (Figure 9) is
needed for the determination of stability constants for interactions of
alkali metal cations by titration calorimetry.
Another potentially important structural parameter is the
length of the arm that connects the polyether ring to the ionizable
group. Unfortunately, systematic structural variation of the pendant
arm length for crown ether carboxylic acids presented certain
synthetic difficulties. (Surprisingly, there was no reaction of crown
ethers containing -0(CH2)nBr side arms with magnesium metal which
precluded the formation of Grignard reagents for which subsequent
reaction with carbon dioxide could conceivably produce carboxylic
acids. When crown ethers with these side arms were treated with
cyanide ion, substitution reactions to form the corresponding nitriles
did take place. However, no method has been found by which the
nitriles can be hydrolyzed to carboxylic acids.)[^2]

15 CioH2iv^!:^^OCH^E
'CO2H
2 0 2 1 2 2 2 3 2 4 2 5 2 6
CE 12-crown-4 13-crown-4(2) 13-crown-4(3) 14-crown-4(2) 14-crown-4(3) 15-crown-4(3) 15-crown-5
2 7 2 8 2 9 3 0 3 1 3 2 3 3
CE 16-crown 18-crown 19-crown 21-crown 24-crown 27-crown 30-crown
-5(3) -6 -6(2) -7 -8 -9 -10
[(2) or (3) designates attachment through a carbon of a two carbon bridge or the central carbon of the three-carbon bridge, respectively]
Figure 8. Bartsch's Crown Ether Carboxylic Acids with Various Ring Sizes.
aOCH^E
CO2H
CE CE 3 4 12-crown-4 3 8 15-crown-5 3 5 13-crown-4(3) 3 9 18-crown-6 3 6 14-crown-4(2) 4 0 21-crown-7 3 7 14-crown-4(3) 4 1 24-crown-8
Figure 9. The Proposed Crown Ether Carboxylic Acids.

16
A homologous series of crown ether phosphonic acid monoethyl
esters 42 -45 which have the same polyether and lipophilic
components as crown ether carboxylic acid 17 (Figure 10) was
accessible.[33] The observed selectivity orders were: Na'*'> Li+> K+>
Rb+, Cs+ for 42; Na-»-» K+> Li+> Cs+> Rb+ for 43; and Li+> Na+> K+> Rb+,
Cs+ for 44 and 45- It was proposed that the metal cation was
complexed within the crown ether rings of 42 and 4 3 , but
coordinated primarily with the monoethyl phosphonate center in 4 4
and 45. (Lipophilic phosphonic acid monoethyl esters which do not
have polyether units exhibited modest Li+ selectivity in competitive
solvent extraction of alkali metal cations into chloroform.)
O H^0(CH2i,P0H H r ^ C)Et 42 1
(CH3)3C-ft )Q-C(CH3)3 44 3 P O;-^-^ 45 4
^ o ^
Figure 10. Bartsch's Crown Ether Monoethyl Phosphonates.
The variation of the lipophilic group attachment site for crown
ether carboxylic acids 17-19 has been found to influence the
selectivity and efficiency of the solvent extraction process.[34.35] The
new series of lipophilic crown ether phosphonic acid monoethyl
esters 46-49 shown in Figure 11 would allow the influence of the
lipophilic group attachment site upon the sidearm length effect to be
compared with that reported for 42-45[331 in solvent extractions of

17
alkali metal cations. For the set of non-lipophilic crown ether
phosphonic acid monoethyl esters 50-53, titration calorimetry could
be used to assess the influence of side arm variation upon the
thermodynamics of alkali metal cation complexation.
O
f l OEt O O,
O O
^ 0 ^
46 47 48 49
R C10H21
C10H21
C10H21
C10H21
n. 1 2 3 4
5 0 5 1 5 2 5 3
R H H H H
n 1 2 3 4
Figure 11. The Proposed Crown Ether Monoethyl Phosphonates.
The effect of organic solvent variation by lipophilic crown ether
carboxylic acid, 2-r(sym-dibenzo-16-crown-5)oxyldecanoic acid (18),
was found in the examination of alkali metal solvent extraction in
chloroform, 1,1,1-trichloroethane, tetrahydronaphthalene, benzene,
toluene, and ^.-xylene as the organic solvents.[36] Table 3 shows the
organic phase loading data (assuming formation of 1:1 complexes) as
well as the Na+/Li+ and Na+/K+ concentration ratios for all six organic
solvents at an aqueous phase equilibrium pH of 8.7.
The organic phase loading is highest for chloroform and
decreases regularly in the order chloroform> l,l,l-trichloroethane>
tetrahydronaphthalene> benzene> toluene> ^.-xylene. This regular
ordering contrasts sharply with the observed extraction selectivities.
As would be predicted from the ratio of the cavity size of 18 and the
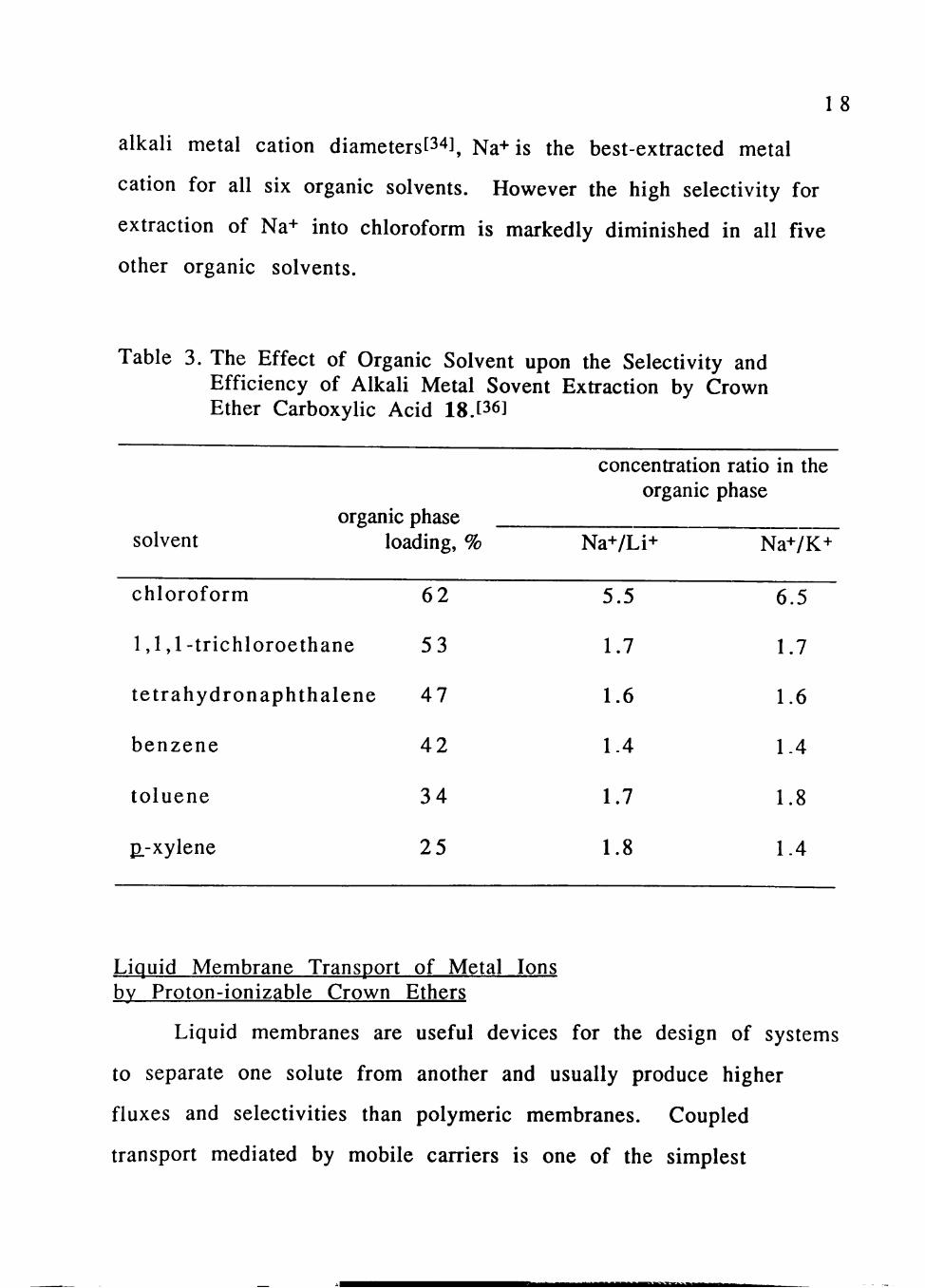
18
alkali metal cation diameters[34], Na+is the best-extracted metal
cation for all six organic solvents. However the high selectivity for
extraction of Na+ into chloroform is markedly diminished in all five
other organic solvents.
Table 3. The Effect of Organic Solvent upon the Selectivity and Efficiency of Alkali Metal Sovent Extraction by Crown Ether Carboxylic Acid 18.[36]
orga solvent
chloroform
1,1,1-trichloroethane
tetrahydronaphthalene
benzene
nic phase loading.
62
53
47
42
%
concentration ratio in the organic
Na+/Li+
5.5
1.7
1.6
1-4
phase
Na+/K+
6.5
1.7
1.6
1-4
toluene
p.-xylene
34
25
1.7
1.8
1.8
1-4
Liquid Membrane Transport of Metal Ions by Proton-ionizable Crown Ethers
Liquid membranes are useful devices for the design of systems
to separate one solute from another and usually produce higher
fluxes and selectivities than polymeric membranes. Coupled
transport mediated by mobile carriers is one of the simplest

19
mechanisms for the selective removal of a desired ion from a dilute
solution.[37] In such a system, the flux of one ion moving down its
concentration gradient may be used to drive the transport of the
desired cation up its concentration gradient. A pH gradient with
back-transport of protons is used most often to drive the transport of
another cationic species from a basic to an acid solution. The
mechanism of proton-coupled transport of a monovalent cation
across a liquid organic membrane is illustrated in Figure 12. The
carrier, which remains in the organic membrane, is deprotonated at
the organic phase-alkaline aqueous source phase interface and
Basic Aqueous Source Phase
H O ^ H-X
Organic Phase
H-X
M^
H^
Acidic Aqeous Receiving Phase
Step 1
- ^
Step 2
Step 3
Step 4
Overall
Figure 12. Mechanism of Metal Cation Transport across a Liquid Membrane by a Proton-ionizable Crown Ether.

20
complexes the metal cation (Step 1). The electroneutral complex
then diffuses across the organic membrane (Step 2). At the organic
phase-acidic aqueous receiving phase interface, the carrier is
protonated which releases the metal cation into the receiving phase
(Step 3). The carrier molecule then diffuses back across the organic
membrane (Step 4) to begin another cycle. Therefore, the net result
is transport of the metal ion from the source aqueous phase to the
receiving aqueous phase coupled with counter-transport of a proton.
Immobilization of Crown Ethers on Silica Gel
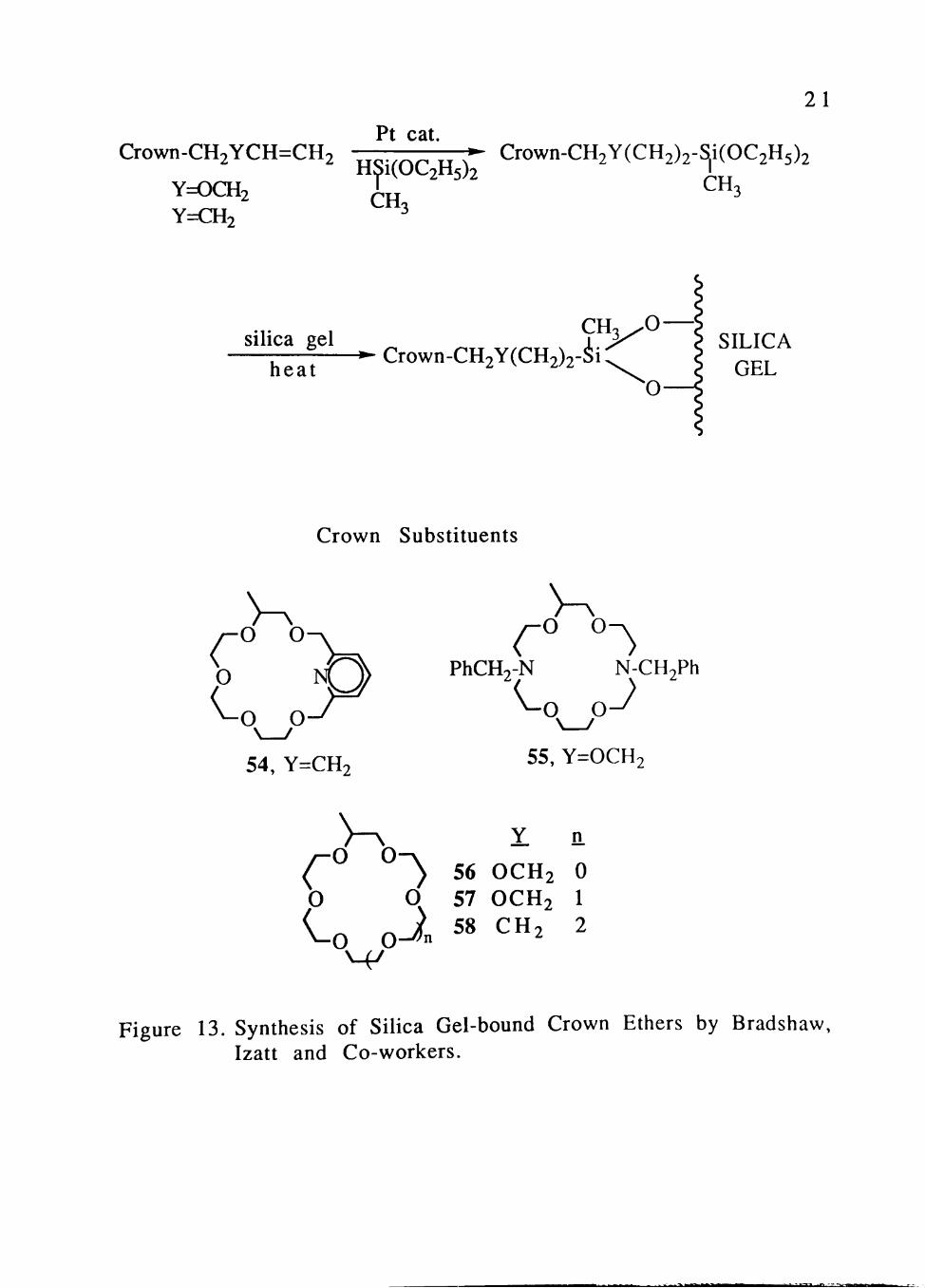
Crown ethers covalently attached to silica gel have been
prepared by Bradshaw, Izatt and co-workers (Figure 13)[38] to
circumvent the problem that the separation of metal ions using
crown ethers in extraction or liquid membrane systems may involve
the slow, but steady, loss of the expensive crown ether compounds
from the organic membrane or organic layer.
The log K values for the interaction of the silica gel-bound
crown ethers with various cations have been determined.[39] The
equilibrium expression for 1:1 cation-crown ether interaction is
given by Equation 3:
F(l-fKi[H+]+KiK2[H+]2) ^ - (l-f)[Mn+] • ^ ^
where f=the fraction of ligand sites containing bound cations, Ki and
K2 are the protonation constants applicable to 55 , and [M^+] and [H+]
are the equilibrium molar free cation and proton concentrations,
respectively. The quantities [Mn+] and [H+] are taken to be the
• ' ^ ^ — * ^
21
Crown-CH2YCH=CH2
Y=OCH2 Y=CH2
Pt cat. mKOCuA Crow„-CH,Y(CH,),-^i(OC3H5),
i CHo CH^ ^
silica gel
hea t Crown-CH2Y(CH2)2-^i SILICA
GEL
Crown Substituents
r o
o o
o o
54, Y=CH2
PhCH.-N N-CH.Ph 2
O
55, Y=0CH2
V^ Y n C^ "^ 56 OCH2 0 O O 57 OCH2 1
( ^ r^) 58 CH2 2 ^ O O - ^ "
Figure 13. Synthesis of Silica Gel-bound Crown Ethers by Bradshaw, Izatt and Co-workers.
- " • . - • - . - « . » . ^ r w ^ ^ - . > ^ ^ i J T I HI M I M i l l ^

22
effluent M"+ and H+ concentrations as determined by atomic
absorption (AA) spectroscopy and pH measurements when these
concentrations are equal to the input concentrations. The total
number of moles of ligand sites is known from the organic synthesis
and was checked by quantitatively loading every crown ether site
with a strongly interacting cation. After equilibrium was reached,
the column was stripped with pure water, a complexing agent or an
acidic solution. The resulting solution of known volume was
analyzed for cation concentration by AA spectroscopy. The fraction
of ligand sites containing the cation was calculated as moles of bound
cation/mole of ligand. The pKa values were determined by repeating
the above log K experiments for cations at several pH values and
curve fitting of the results according to Equation 3.
Table 4 shows complexation data for silica gel-bound and
analogous free crown ether interaction constants with metal ions.
The similarity of the log K values for the bound crown ethers to those
involving the unbound crown ethers suggests that metal separations
using silica gel-bound crown ether ligands should be possible.
Statement of Research Goals
The interest in macrocyclic polyethers as complexing agents for
metal cations, primary alkyl ammonium salts and neutral as well as
charged substrates has grown exponentially since Pedersen's original
publication.[^1 Methods for the design and synthesis of macrocyclic
polyethers have been developed to achieve high stability and high
selectivity for metal cations. Such development generally requires

23 Table 4. Comparison of Bound and Analogous Unbound Crown
Ether Interaction Constants with Metal Cations.[38]
crown ether
5 4
5 4
5 4
5 5
5 6
5 7
5 7
5 8
cation
H+
Ag+
Cu2+
Ag+
Ag+
Ag+
Ba2+
Ba2+
logK
bound
5.10
2.70
1.80
8.20
0.90
1.61
3.56
2.93
± 0 . 2 0
± 0.20
± 0.10
± 0.20
± 0.15
± 0.09
± 0.01
± 0.09
unbound
5.23a
5.50b
4.63b
7.80C
0.94
1.50
3.87t>
5-44
a. pKa value for pyridine in water.t oi The pKa value for unbound crown ether has not been reported.
b. Log K values measured are in methanol. These values have been shown to be 2-3 log K units higher than the log K values measured in water.
c. This value is for diaza-18-crown-6.

24
the use of functionalized macrocyclic polyethers containing structural
features that allow for further chemical modification. Indeed,
hundreds of original papers relating to various aspects of host-guest
chemistry have been published within the past decade and
considerable progress has been made.
A major portion of this dissertation deals with the synthesis of
new proton-ionizable crown ethers which will be utilized by others
to determine the effect of structural variation upon metal ion
complexation in solvent extraction and titration calorimetric studies.
Structural variations within the proton-ionizable crown ethers
include: (1) the identity of the proton-ionizable group; (2) the ring
size and rigidity of the crown ether ring; (3) the length of the "arm"
which connects the proton-ionizable group to the polyether
framework; and (4) the attachment site and nature of the lipophilic
group which is necessary to retain the ionized crown ether in the
organic phase during solvent extraction.[^H These compounds
include a series of acyclic and cyclic polyether derivatives of salicylic
acid, and series of non-lipophilic dibenzo-16-crown-5 phosphonic
acid monoalkyl esters and lipophilic dibenzo-16-crown-5 phosphonic
acid monoalkyl esters in which the sidearm length is systematically
varied.
For attachment to silica gel, functionalized crown ethers which
have dibenzo-14-crown-4 and dibenzo-16-crown-5 rings are to be
prepared. Also a set of functionalized crown ethers based on salicylic
acid are to be synthesized. These crown ether compounds have long

25
lipophilic tails with a terminal carbon-carbon double bond for
covalent attachment to silica gel.
The second portion of this dissertation will involve a
determination of the complexation efficiency of different neutral
crown ethers for alkali metal cations by the picrate extraction
method. Several series of novel crown ethers are to be investigated
in this manner.
- ^ ^ > > - • . ^ ^ . - w ^ ^ - w „

CHAPTER n
RESULTS AND DISCUSSION
Acyclic and Cyclic Polyether Derivatives of Salicylic Acid
Acyclic polyether derivatives of salicylic acid 68-70 were
synthesized for assessment of their alkali metal cation binding
properties by calorimetric titrations. Precursor tosylates 62-6 4
were prepared by reaction of g.-toluenesulfonyl chloride with the
corresponding alcohols 59-61, respectively, in pyridine (Scheme 2).
Scheme 2
i - A TsQ ir-\ HO 0)„CH2Ph pyridine ' TsO 0),CH2Ph
H 2-5 9 1 6 2 1 6 0 2 6 3 2 6 1 3 6 4 3
The synthetic route to 68-70 is summarized in Scheme 3.
Methyl salicylic acid was reacted with NaH (1.1 equivalents) in THE
and then tosylate 62 to give the methyl benzoate derivative 65 in
49% yield. The use of more NaH (2.0 or 4.0 equivalents) did not
increase the yield. Unreacted tosylate was still detected in the crude
reaction product mixture in all cases. Use of 1.1 equivalents of NaH
was found to be the best reaction stoichiometry for the coupling of
the tosylate 62 with the anion of methyl salicylate to give ester 65.
26

27
This stoichiometry was subsequently utilized for the preparation of
esters 66 and 67. Basic hydrolysis of the substituted methyl
benzoates 65-67 gave the benzoic acids 68-70. Table 5 shows the
yields of compounds 62-70.
Evaluation of the binding properties of acyclic polyether
carboxylic acids 68-70 for alkali metal cations in water by Dr. Moon
Hwan Cho['*2] using titration calorimetry was unsuccessful. The heat
change upon complexation was too small to allow for calculation of
log K values.[42]
Scheme 3
a" === ^C02Me
1) NaH, THE ^ .
2) TsO 0)nCH2Ph
n. 6 2 1 6 3 2 6 4 3
^ Y ^ 0),CH2Ph
^ ^ C 0 2 M e
n 6 5 1 6 6 2 6 7 3
NaOH ,^c^0^0)„CH2Ph
EtOH ' : ^C02H
n. 68 1 6 9 2 7 0 3

28
Table 5. Yields of Compounds 62-70.
percent yield of
(T^ r ^ ^ ^ O 0)„CH2Ph , ^ = ^ 0 0),CH2Ph TsO 0)„CH2Ph
C02Me ^^ CO2H n
95 49 89
79 79 88
67 75 94
Cyclic polyether derivatives of salicylic acid 34 ,35 , and 38-41
(see Figure 9) were prepared for determination of the stability
constants (log K values) for alkali metal cation complexation using
calorimetric titrations. For the preparation of cyclic polyether
derivatives of salicylic acid, hydroxymethyl-substituted crown ethers
were needed. Hydroxymethyl-13-crown-4(3), -18-crown-6, and -
24-crown-8 were available from other studies.[^3] The synthetic
routes to (benzyloxy)methyl crown ethers 71-73 are depicted in
Scheme 4. The (benzyloxy)methyl-12-crown-4 (71) was prepared in
62% yield by Okahara condensation[44] of 3-(benzyloxy)-l,2-
propanediol[45] with l,2-bis(2-chloroethoxy)ethane in a LiOtBu/LiBr/
t-BuOH reaction mixture. Cyclization of 3-(benzyloxy)-l,2-propane-
diol and tetraethylene glycol ditosylate with NaH in DMF-THF (4:1)
gave a 39% yield of (benzyloxy)methyl-15-crown-5 (72). The
' ^ - - * — — » "

29
(benzyloxy)methyl-21-crown-7 (73) was synthesized in 28% yield
by cyclization of 3,6-dioxo-5-(benzyloxy)methyl-l,8-diol and
tetraethylene glycol ditosylate in the presence of NaH in THE.
Quantitative hydrogenolysis of the protecting benzyl groups of 71 -
73 was achieved with palladium on carbon catalyst and a trace
amount of p.-toluenesulfonic acid in aqueous EtOH to yield
hydroxymethyl-substituted crown ethers 74-76 (Scheme 5).
Scheme 4
'-0CH2Ph
^O Cl H 0 , ^ 0 C H 2 P h LiOt-Bu, t-BuOH^ ^O O
L "*" J LiBr, H2O k^ ^J O Cl HO ^K P
7 1
0CH2Ph
h . HO^OCH,Ph NaH [ 3 ( un^ DMF-THF V ^ \ ^ O OTs " ^ (4-1) k . 0 ^
72
0CH2Ph
O + V " Y ^ NaH .0 O
/ THF IQ ^J
7 3
OTs
. . , , , - , . ^ - — . — . — , — ^ — — ^ • .

Scheme 5 30
0CH2Ph ^ O H
H. ^ ^O O.
^o p"^ Pd/C
7 1 1 7 4 1 7 2 2 7 5 2 7 3 4 7 6 4
The preparation of crown ether carboxylic acids 34, 35, and
38-41 is summarized in Scheme 6. Tosylates 77-82 were
synthesized from the corresponding hydroxymethyl crown ethers by
the reaction with p.-toluenesulfonyl chloride in pyridine. The
tosylates of hydroxymethyl-12-crown-4, -13-crown-4(3), -15-
crown-5, -18-crown-6, -21-crown-7, and -24-crown-8 were coupled
with methyl salicylate in the presence of NaH in THF.[30.3i] in the
initial coupling reaction of methyl salicylate anion with tosylate 79, a
very hygroscopic solid was eluted with difficulty when the crude
product was subjected to chromatography on alumina with CH2CI2-
MeOH (10:1) as eluent. This solid was identified as the crown ether
benzoic acid 38 after acidification with 5% HCl solution. Apparently
hydrolysis of the crown ether methyl benzoate ester 85 occurred on
the alumina column. Another coupling reaction was performed and
the crude product was readily purified by chromatography on silica
gel to give 85. Due to the success of this purification method.

31
chromatography on silica gel was used for crude crown ether esters
8 3 , 8 4 , 8 6 - 8 8 as well.
Scheme 6
a° " 1) NaH ^ C Y O C H Z C E
CO^Me 2)CECH20Ts M ^ C Q Me CE CE
7 7 12-crown-4 8 3 12-crown-4 7 8 13-crown-4(3) 8 4 13-crown-4(3) 7 9 15-crown-5 8 5 15-crown-5 8 0 18-crown-6 8 6 18-crown-6 8 1 21-crown-7 8 7 21-crown-7 8 2 24-crown-8 8 8 24-crown-8
NaOH ^ > ^ 2 Y ^ ^ " 2 C E
EtOH UL^^^^
CE 3 4 12-crown-4 3 5 13-crown-4(3) 3 8 15-crown-5 3 9 18-crown-6 4 0 21-crown-7 4 1 24-crown-8
Base-catalyzed hydrolysis of crown ether methyl benzoates
83-88 with sodium hydroxide in aqueous EtOH followed by
acidification gave the crown ether benzoic acids 34 ,35 , and 38-41
in high yields (Table 6).

32
Table 6. Yields of Compounds 34, 35, 38-41, and 77-88.
CE
12-crown-4
13-crown-4(3)
15-crown-5
18-crown-6
21 -c rown-7
24 -c rown-8
CECH2OTS
97
76
95
98
88
86
percent yield
^^>s-^0CH2CE
39
57
5 2
5 0
61
60
of
i ^-5^0CH2CE
^ C 0 2 H
88
97
93
92
84
88
The thermodynamics of alkali metal cation complexation by the
ionized forms of crown ether carboxylic acids 3 4 , 3 5 , and 38-41 in
90% methanol-10% water were determined by Drs. Moon Hwan Cho
and Visvanathan Ramesh[46] using titration calorimetry. A marked
influence of the crown ether ring size upon the stability constants for
alkali metal cation complexation was noted.[^61
Lipophilic Dibenzo-16-crown-5-oxyacetic Acids
Lipophilic crown ether carboxylic acids have been utilized for
solvent extraction of alkali and alkaline earth cations from aqueous

33
solutions as well as for the transport of these metal cations across
bulk liquid and liquid surfactant membranes.["^7.48] Recently Bartsch
and coworkers synthesized novel ion-exchange resins by
condensation polymerization of crown ether carboxylic acids with
formaldehyde in formic acid.[' 9] por this project, the crown ether
carboxylic acids 94-96 were prepared.
The synthetic route to lipophilic crown ether carboxylic acids
94-96 is summarized in Scheme 7. When subjected to Jones
oxidation,[50] crown ether alcohol 89 was converted into sym-
ketodibenzo-16-crown-5 (90)[5il in 58-78% yields. Reaction of
crown ether ketone 90 with Grignard reagents in THE provided 45%
and 62% yields of dibenzo crown ether alcohols 92 and 93,
respectively. However, the reaction of crown ether ketone 90 with
CH3MgI in THF did not give crown ether alcohol 91, because of the
limited solubility of the Grignard reagent in THF. When THF-dielhyl
ether (2:1) was used as the reaction solvent, crown ether alcohol 91
was produced in 41% yield. Crown ether alcohols 91-93 were
transformed into the corresponding lipophilic crown ether carboxylic
acids 94-96 in 72-80 % yields by reaction with NaH and then
bromoacetic acid in THF at room temperature. These alkylation
conditions produced considerably higher yields than when the
reaction was conducted at reflux or when a two-step reaction
sequence of alkylation with bromoacetate followed by hydrolysis
was utilized.[32]
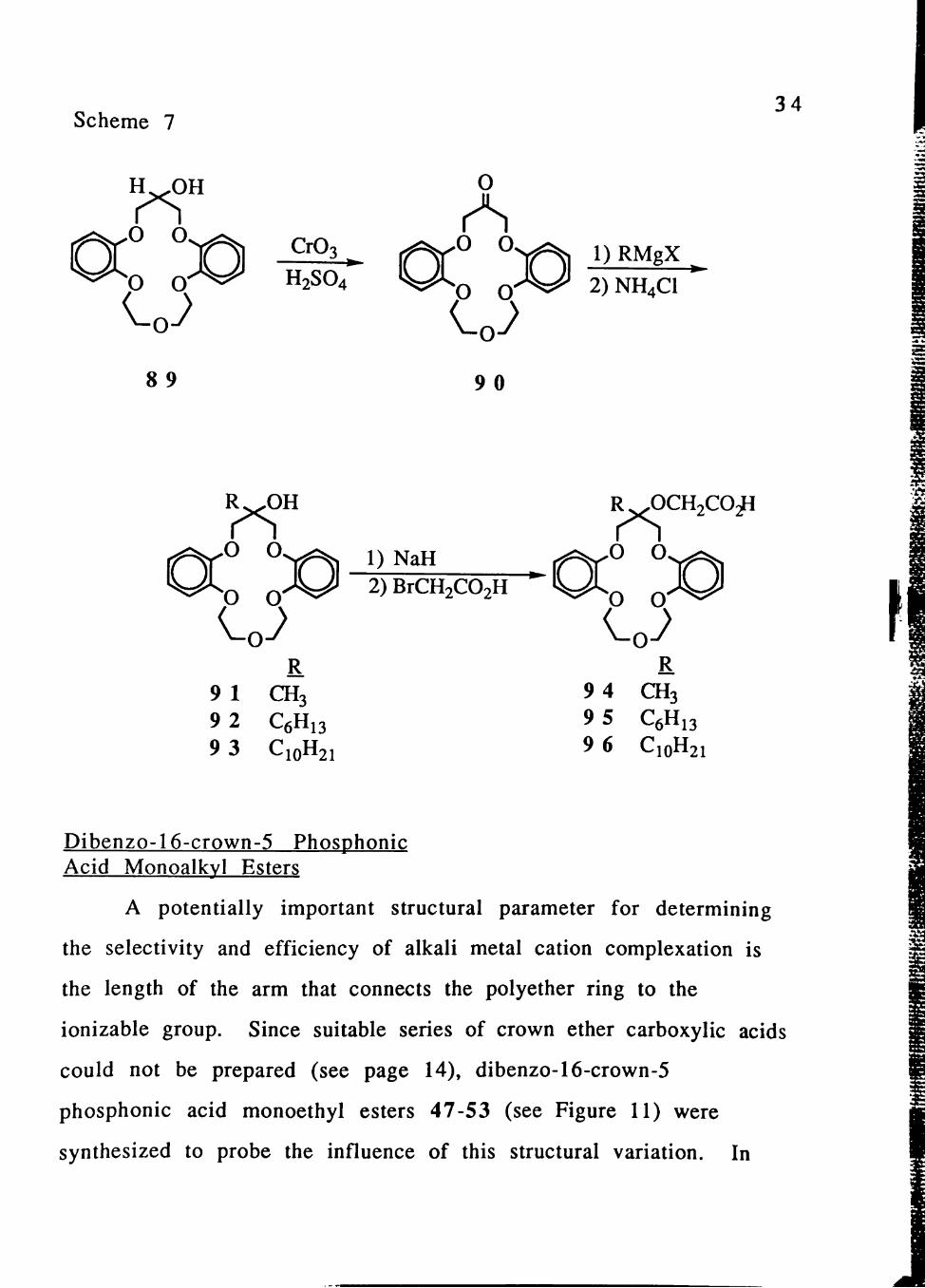
Scheme 7 34
H^OH
O O
p o
8 9
CrO. • ^ -
H2SO4
O
A o o
p o
90
1) RMgX 2) NH4CI
R^OH
c :3o • 15
K 9 1 CH3 9 2 C6H13 9 3 C10H21
1) NaH 2) BrCH2C02H
R ^ 0 C H 2 C 0 ^
-cc:::o IN.
9 4 CH3 9 5 C6H13 9 6 C,oH2,
\
Dibenzo-16-crown-5 Phosphonic Acid Monoalkyl Esters
A potentially important structural parameter for determining
the selectivity and efficiency of alkali metal cation complexation is
the length of the arm that connects the polyether ring to the
ionizable group. Since suitable series of crown ether carboxylic acids
could not be prepared (see page 14), dibenzo-16-crown-5
phosphonic acid monoethyl esters 47-53 (see Figure 11) were
synthesized to probe the influence of this structural variation. In

35
47-53 the number of methylene groups in the side arm is
systematically varied from one to four. Dibenzo-16-crown-5
phosphonic acid monoethyl esters 47-49 are designed for use in
solvent extraction and are sufficiently lipophilic to avoid loss of
complexing agents from an organic phase into a contacting aqueous
phase during the solvent extraction of alkali metal cations. The
dibenzo-16-crown-5 phosphonic acid monoethyl esters 50-53 which
do not possess lipophilic groups are designed for testing of their
alkali metal cation complexing abilities by titration calorimetry.
Scheme 8 shows the synthetic route to monoethyl sym-
dibenzo-16-crown-5-oxymethylphosphonic acid (50) which was
prepared in 33% yield by reaction of the alkoxide from crown ether
alcohol 89 with monoethyl iodomethylphosphonic acid. Monoethyl
iodomethyl phosphonic acid was prepared by the reaction of
diiodomethane with triethyl phosphite, according to the literature
method.[52]
I
Scheme 8
H ^ O H
o o
o o
8 9
l)2NaH
O
2)lCHoP0H I
OEt
O
ILX)CH2P0F
I I OEt O O ^^^
O O
5 0

36
The multistep syntheses of crown ether phosphonic acid
monoethyl esters 47-49 and 51-53 involved the initial preparation
of the crown ether substituted alkyl bromides 97-102 from crown
ether alcohols 89 and 93 by a different route for each bromide (vide
infra). Subsequently, bromides 97-102 were reacted with triethyl
phosphite to form crown ether phosphonic acid diethyl esters 103-
108 in 82-95% yields which produced monoethyl esters 47-49 and
51-53 upon basic hydrolysis (Scheme 9). Attempts to prepare the
crown ether phosphonic acid ester 106 by reaction of the mesylate
of 2-(^xQl"<^ib^r^zo-16-crown-5-oxy)ethanol with sodium diethyl
phosphonate[53] were unsuccessful.
When crown ether phosphonic acid diethyl esters 103 and 106
were subjected to basic hydrolysis by refluxing with NaOH in 95%
EtOH for 24 h, crown ether alcohols 89 and 93, respectively, were
recovered. Apparently a reverse Michael-type elimination was
taking place as shown in Scheme 10. When the basic hydrolyses of
103 and 106 were conducted at room temperature for 10 and 7
days, respectively, crown ether phosphonic acid monoethyl esters 4 7
and 51 were obtained, but in low to fair yields.
To improve the competition between the hydrolysis and
elimination, crown ether phosphonic acid dimethyl ester 109 was
prepared by the reaction of crown ether bromide 100 with trimethyl
phosphite in 75% yield (Scheme 11). Basic hydrolysis of 109 at room
temperature for 24 h gave a good yield of crown ether phosphonic
...^..^•ww——-•TMMfc"M'>~l'MI'>imfc^^ajifcM

Scheme 9 37
R^(CH2)„Br
O O,
p O (EtO)3P
-^^
O
R^(CH2)„ P(0Et)2
O O
p O
^ 0 ^
9 7 9 8 9 9 100 101 102
R ^10^21
^ 10 21
^10^21 H H H
n. 2 3 4 2 3 4
103 104 105 106 107 108
R
^10^21
^10^21
^10^21
H H H
n. 2 3 4 2 3 4
I
NaOH 95% EtOH
0
R^(CH2)nP0H
A A OEt
-oc :0 R n
4 7 C10H21 2 4 8 C10H21 3 4 9 C10H21 4 5 1 H 2 5 2 H 3 5 3 H 4

Scheme 10
R ^ - C H 2 - CH-P(OEt)2 ^JxP'
38
Scheme 11
H.^OCH2CH2Br
^ 0 ^
o IL/)CH2CH2l^(OMe)2
-a:::o ^ 0 ^
100 109
O
NaOH, EtOH - ^
H^CH2CH2POH l l OMe
O O,
RT,24h * ^ : ^ o O
1 1 0

39
acid monomethyl ester 110. Thus the problem of completing
elimination was greatly suppressed. Table 7 summarizes hydrolysis
conditions and yields of crown ether phosphonic acid dialkyl esters.
The synthetic route to the precursor crown ether alkyl
bromides 97, 98, 100 and 101 is presented in Scheme 12. Crown
ether mesylates 115-118 were prepared from the corresponding
crown ether alcohols 111-114 by reaction with methanesulfonyl
chloride in CH2CI2 in the presence of triethylamine. Subsequently,
the crown ether mesylates were reacted with sodium bromide in
acetone to form crown ether bromides 97, 98, 100, and 101 in
quantitative yields. Attempts to prepare crown ether bromides from
Table 7. Hydrolysis of Crown Ether Phosphonic Acid Dialkyl Esters 103-109.
compound
1 0 3
1 0 4
1 0 5
1 0 6
1 0 7
1 0 8
1 0 9
condition
RT, 10 d
reflux, 24 h
reflux, 24 h
RT, 7 d
reflux, 24 h
reflux, 24 h
RT, 24 h
yield (%)
29
80
80
40
53
55
54

40
crown ether alcohols by the reaction of Vilsmier reagent, phosphorus
tribromide in DMF, were unsuccessful.
Crown ether bromides 99 and 102 were synthesized in a
different way from that which is shown in Scheme 12. Scheme 13
presents the synthetic route to crown ether bromides 99 and 102.
A phase transfer catalyzed reaction of crown ether alcohol 89 and
Scheme 12
R^0(CH2)„0H
o o.
o o
R n 1 1 1 C10H21 2 1 1 2 C10H21 3 1 1 3 H 2 1 1 4 H 3
MsCl, Et3N
CH2a:
R^^O(CH2)nOMs
O O,
O O
R a 1 1 5 C10H21 2 1 1 6 C10H21 3 1 1 7 H 2 1 1 8 H 3
NaBr
acetone
R ^ ( C H 2 ) „ B r
O O,
O O
9 7 9 8
1 0 0 1 0 1
R n C10H21 2 C10H21 3 H 2 H 3

41
1,4-dibromobutane in a mixture of CH2Cl2and 50% aqueous NaOH in
the presence of tetrabutylammonium hydrogen sulfate gave crown
ether bromide 102 in 76% yield. However, when this method was
applied to the synthesis of crown ether bromide 99, the reaction
was found to be very sluggish and only a 34% yield of 99 was
obtained after 10 days. Presumably, the lipophilicity of crown ether
alcohol 93 was the causative factor. If the PTC reaction occurs at the
interface between the aqueous and organic phase, the lipophilic
group could hinder approach of the crown ether alcohol to the
interface.
Scheme 13
R ^ ) H
oc::]o ^ 0 ^
R 8 9 H 9 3 C10H21
Br(CH2)4Br
CH2Cl2-50% aq NaOH BU4NHSO4
PTC
R^(CH2)4Br
o::::o R
1 0 2 H 9 9 C10H21
The synthesis of crown ether alcohols 111 and 113,
from which the crown ether mesylates 115 and 117 were prepared,
is depicted in Scheme 14. Starting with crown ether alcohols 93 and
89, crown ether esters 119 and 120 were prepared by reaction with
ethyl bromoacetate in the presence of NaH in 66% and 76% yields.
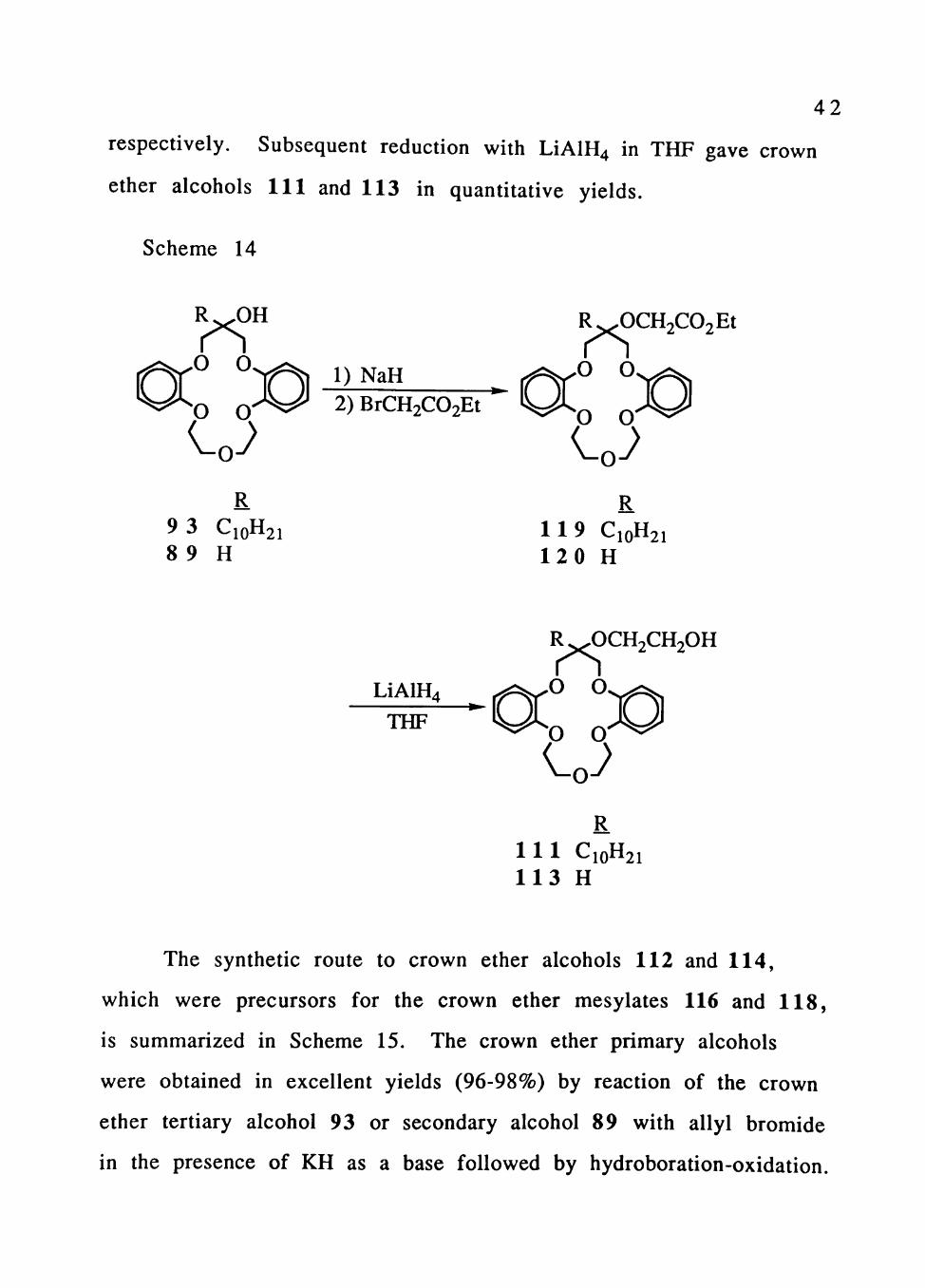
4 2
respectively. Subsequent reduction with LiAlH4 in THF gave crown
ether alcohols 111 and 113 in quantitative yields.
Scheme 14
RjK^H R^OCH2C02Et
^ 0-v^=^ D N a H ^ ^ - O O
O 0 ^ ^ = ^ 2)BrCH2C02Et %^^ ^
R R 9 3 C10H21 1 1 9 C10H21 8 9 H 1 2 0 H
R ^OCH2CH20H
LiAlH4 , ^ ^ ^ 0 O
O O THF
R 1 1 1 C10H21 1 1 3 H
The synthetic route to crown ether alcohols 112 and 114,
which were precursors for the crown ether mesylates 116 and 118 ,
is summarized in Scheme 15. The crown ether primary alcohols
were obtained in excellent yields (96-98%) by reaction of the crown
ether tertiary alcohol 93 or secondary alcohol 89 with allyl bromide
in the presence of KH as a base followed by hydroboration-oxidation.

43 Scheme 15
^ x O H R^OCH2CH=CH2
^ ^ V ^ 1)KH , ^ = ^ 0 O
O O " ^ ^ 2) BrCH2CH=CH2 " M ^ Q ^
R R ^ 3 C10H21 12 1 C10H21 8 9 H 1 2 2 H
R^OCHjCHjCH.OH
1) NaBH4, BF3.Et20 | f ^ ^ ^ ' 2) H2O2, NaOH ^ " W ^ o O
R 1 1 2 C10H21 1 1 4 H
The influence of the side arm length variation upon the
selectivity and efficiency of competitive alkali metal cation solvent
extraction into chloroform by the lipophilic monoethyl crown ether
phosphonates 47-49 was assessed by Dr. Wladyslaw Walkowiak.[54]
The side arm selectivity was found to be much higher with 47 than
for 48 or 49 which demonstrates a more appropriate sidearm length
in 47. The influence of sidearm length variation upon stability

4 4
constants for alkali metal cation complexation by monoethyl crown
ether phosphonates 50-53 in 90% methanol-10% water was
determined by Dr. Moon Hwan Cho by titration calorimetry.[^2]
Functionalized Crown Ethers for Attachment to Silica Gel
Crown ethers covalently attached to silica gel have two
advantages in solvent extraction and liquid membrane transport.
The first is to retain the crown ether species in the organic phase.
The second is to allow for facile recovery of the crown ether reagent.
Functionalized crown ethers 123-128 were synthesized for
attachment to silica gel (Figure 14). For binding of the molecules to
silica gel through the vinyl groups via the reactions shown in Figure
13, the carboxylic acid groups must be protected as esters.
Therefore, functionalized crown ethers 123-128 are esters rather
than carboxylic acids. Once bound to silica gel the esters can be
hydrolyzed to carboxylic acid functions.
Crown ether alcohols 130 and 131 were prepared (Scheme 16)
in 74% and 56% yields, respectively, by the reaction of crown ether
ketones 129 and 90 with the Grignard reagent obtained from
bromide 134. The preparation of bromide 134 is summarized in
Scheme 17. The 10-methanesulfonyl-l-decene (133) was prepared
from commercially available 9-decen-l-ol (132) in 89% yield by
reaction with methanesulfonyl chloride in CHoCh in the presence of
triethylamine. Subsequently, mesylate 133 was reacted with

OCH2C02Et 45
O O
u 1 2 3 CH2CH2CH2 1 2 4 CH2CH2OCH2CH2
"^7xs:> 1 2 5 1 2 6 1 2 7 1 2 8
n. 1 2 3 4
Figure 14. Functionalized Crown Ethers for Attachment to Silica Gel.
Scheme 16
O
A o o
u 1)
2) NH4CI
MgBr - ^ -
1 2 9 CH2CH2CH2 9 0 CH2CH2OCH2CH2
1 2 4 CH2CH2CH2 125 CH2CH2OCH2CH2

46
sodium bromide in acetone to give 10-bromo-l-decene (134) in 69%
yield.
Scheme 17
MsCl, Et3N
^ « CH2CI2 • " ^ ^ ^ 1 3 2 1 3 3
NaBr ^ .
acetone
The synthetic route to functionalized crown ethers 123 and
124 for attachment to silica gel is presented in Scheme 18. Crown
ether tertiary alcohols 130 and 131 were transformed into the
corresponding crown ether carboxylic acids 135 and 136 in 92-97%
yields by reaction with NaH and then bromoacetic acid in THF.
Subsequently, the crown ether carboxylic acids 135 and 136 were
esterified in EtOH with a catalytic amount of concentrated H2SO4 to
give crown ether esters 123 and 124 in 78% and 90% yields,
respectively. Attempts to directly prepare crown ether esters 123
and 124 from crown ether alcohols 130 and 131, respectively, by
the reaction with ethyl bromoacetate in the presence of NaH as a
base were unsuccessful (Scheme 19). Only the unreacted crown
ether alcohols were recovered.

Scheme 18 47
OH
O O
u 1) NaH 2) BrCH2C02H
OCH2CO2H
O O
u 1 3 0 CH2CH2CH2 1 3 1 CH2CH2OCH2CH2
1 3 5 CH2CH2CH2 136 CH2CH2OCH2CH2
H2SO4 EtOH
OCH2C02Et
Scheme 19
1 2 3 CH2CH2CH2 1 2 4 CH2CH2OCH2CH2
O O
u 1) NaH 2) BrCH2C02Et
• X - ^
OCH2C02Et
O O
u 1 3 0 CH2CH2CH2 1 3 1 CH2CH2OCH2CH2
1 2 3 CH2CH2CH2 1 2 4 CH2CH2OCH2CH2
4 8
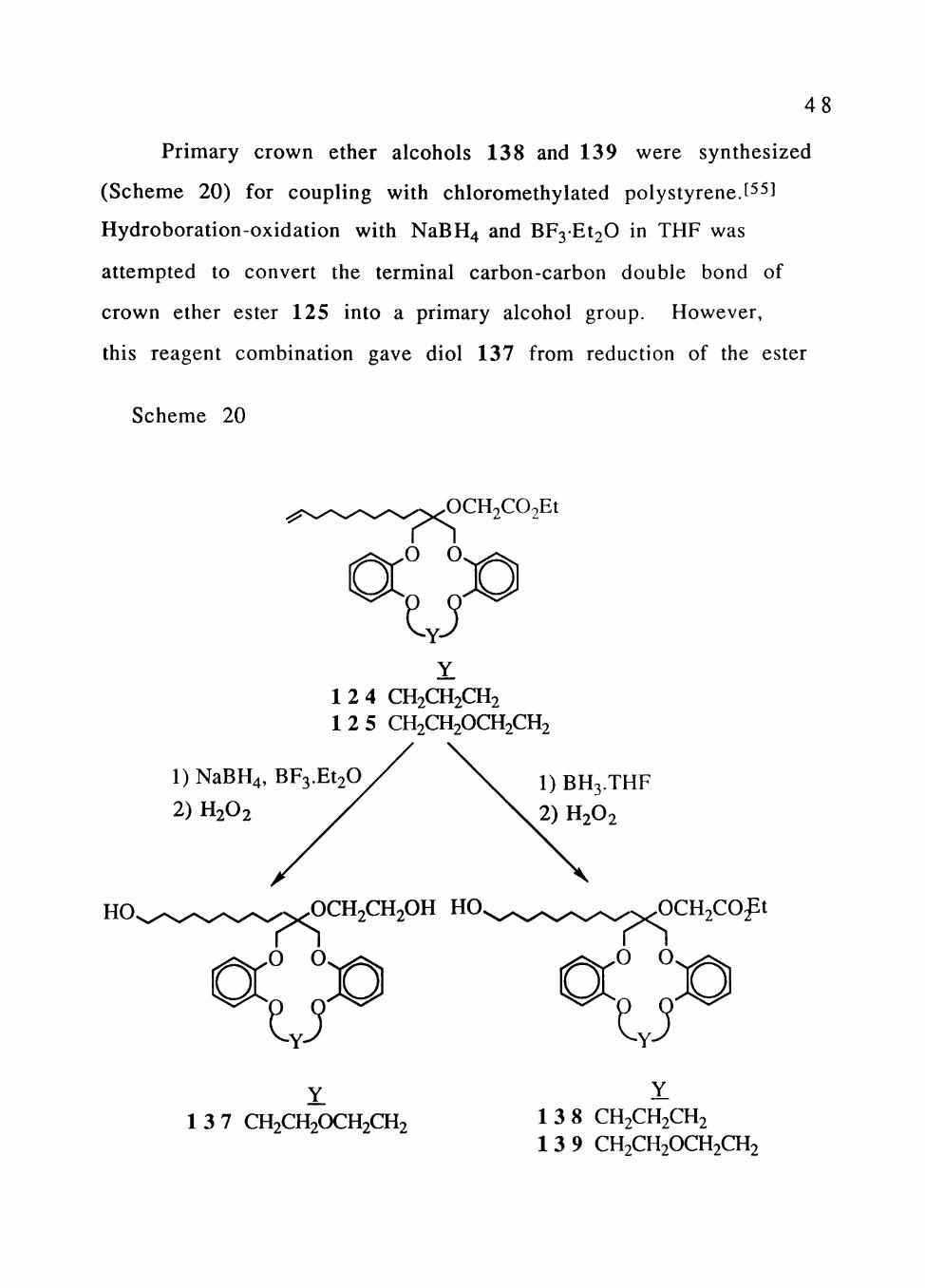
Primary crown ether alcohols 138 and 139 were synthesized
(Scheme 20) for coupling with chloromethylated polystyrene.[^^1
Hydroboration-oxidation with NaBH4 and BF3Et20 in THF was
attempted to convert the terminal carbon-carbon double bond of
crown ether ester 125 into a primary alcohol group. However,
this reagent combination gave diol 137 from reduction of the ester
Scheme 20
OCH2C02Et
O O
u 1 2 4 CH2CH2CH2 1 2 5 CH2CH2OCH2CH2
l)NaBH4, BF3.Et20
2) H2O2 1) BH3.THF
2) H2O2
HO. PS o o
u OCH2CH2OH HO,
PS o o
u 0CH2C0ft
1 3 7 CH2CH2OCH2CH2 1 3 8 CH2CH2CH2 1 3 9 CH2CH2OCH2CH2

49
function as well as conversion of the alkene to an alcohol group.
Hydroboration-oxidation of crown ether esters 124 and 125 with
BH3THF[56] gave desired products 134 and 135 which have a
hydroxyl group at the end of the long lipophilic tail.
A second group of functionalized crown ethers for attachment
to silica gel was also prepared. For 125-128 (see Figure 14), the
crown ether ring sizes are systematically varied from 12-crown-4 to
21-crown-7. The synthetic route to the substituted methyl salicylate
142 is presented in Scheme 21. Reaction of 2,4-dihydroxybenzoic
acid with potassium ethoxide and then mesylate 140 gave the
substituted salicylic acid 141 in 35% yield. Esterification of 141 in
Scheme 21
HO^^^^^OH .OMs -I-
CO2H
1 4 0
K' OEt' .^^.^^^^^^'^^^^0^.^:^^0n
ro2H
1 4 1
EtOH
H2SO4 ,jj;^-^^'^'x^^'v^0,,^^..^^v^0H MeOH
C02Me
142

50
MeOH with a catalytic amount of concentrated H2SO4 gave
substituted methyl salicylate 142 in 80% yield. Crown ether esters
125-128 were synthesized in 34-42% yields by the coupling
reaction of 142 with the corresponding tosylates of hydroxymethyl
crown ethers and NaH in THF (Scheme 22).
Scheme 22
' N^^=^^OH 1) NaH
r02Me u, 1 4 2
2) r^
^0 O Y C H 2 0 T S
^0 p
7 7 7 9 8 0 8 1
n. 1 2 3 4
•°^:x? n.
1 2 5 1 1 2 6 2 1 2 7 3 1 2 8 4
Attachment of functionalized crown ethers to silica gel
by the reactions shown in Figure 13 followed by hydrolysis of the

51
silica gel-bound crown ethers to carboxylic acids will be performed
by another member of the Bartsch Research Group. The behavior of
the silica gel-bound crown ether carboxylic acids in complexation of
alkali metal cation will be conducted by other members of the
Bartsch Research Group.
Picrate Extractions
The picrate extraction method, as discussed previously, is a
means for determining the complexation capacity and cation
selectivity of ionophores in a two-phased, single-ion solvent
extraction system. A solution of the ionophore to be investigated in
CDCI3 is shaken with an aqueous solution of the metal picrate salt.
The mixture is allowed to separate into two phases and the
equilibrium concentrations of metal picrate in the aqueous and
organic phases are determined spectrophotometrically. The degree
to which the metal picrate is transferred into the organic phase by
the ionophore is a measure of the ionophore's propensity to bind that
metal picrate. Extraction constants are calculated by Equation 1.
Benzo-21-crown-7 and Dibenzo-21-crown-7 Compounds
Benzo-21-crown-7 (143), 4-tert-butylbenzo-21 -crown-7
(144), dibenzo-21-crown-7 (145), and sym-dir4(5)-tert-butyl-
benzo]-21-crown-7 (146)[57] (Figure 15) were tested for their
selectivities in complexation with the alkali metal picrates. For this
ring size these crown ethers would be expected to favor extraction of
the larger alkali metal cations. Results of the extraction experiments

52
(Table 8) show that the complexation selectivity for crown ethers
143-146 is in the order Cs+, Rb+ > K+ » Na+ > Li+. The ratios of the
CsVNa"^ extraction selectivities for the dibenzo-21-crown-7
compounds are noted to be larger than those for the benzo-21-
crown-7 compounds. Selectivity for Cs" over Na" is important for the
removal of radioactive Cs" in the recycling of nuclear fuel rods.
i ^ ^ „ ^ n ^ r^ o- r o
R* A Q RfY^ o o
R R 1 4 3 H 1 4 5 H
R
1 4 4 C(CH3)3 14 6 C(CH3)3
Figure 15. Benzo-21-crown-7 and Dibenzo-21-crown-7 Compounds.
Benzo-13-crown-4 Compounds
As a part of a comprehensive study of the complexation of Li"*"
and Na" with small-ring crown ethers, a series of benzo-13-crown-4
compounds^^^^ was evaluated by the picrate extraction method for
their abilities to complex Li"*" and Na"* . The extraction of lithium
picrate from aqueous solution into CDCI3 was compared with the
extraction of sodium picrate by these crown ethers in the same
system. The benzo-13-crown-4 compounds tested are shown in
Figure 16. Table 9 shows that the log Kex values for lithium and

53
Table 8. Picrate Extraction Data for 21-Crown-7 Compounds.
compound M %Ex K ex selectivity'
1 4 3
1 4 4
1 4 5
1 4 6
;+ Li
Na
0.44
1.10
K+
Rb+
Cs+
Li+
Na+
K+
Rb-
Cs-
Li+
Na+
K+
Rb-
Cs+
Li+
Na-
17.3
38.0
39.1
0.29
1.00
19.3
35.5
36.6
0.17
0-41
10.9
17.6
22.1
0.11
0.39
(1.8
(4.6
(1.2
(6.3
(6.9
(1.2
(4.1
(1.4
(5.2
(5.5
(6.9
(1.7
(6.2
(1.3
(1.9
(2.8
(1.6
±
±
±
±
±
±
±
±
±
±
±
±
±
±
±
±
±
0.2)
0.4)
0.1)
0.7)
0.1)
0.1)
0.2)
0.2)
0.1)
0.1)
0.7)
0.2)
0.1)
0.1)
0.1)
0.2)
0.2)
X 10^
X 10^
10^
X 10 ^
102
X 102
X 10^
10^
X 10^
X 10^
X 10^
X 1()3
X 10"
10
X 10'
102
98
36
2.3
1.0
(1.0)
122
37
1.9
1.0
(1.0)
1 1 1
55
2.0
1.0
(1.0)
217
54

54
Table 8. (cont.)
K+
Rb+
Cs+
13.6
21.0
21.7
compound M" % Ex Kex selectivity*
1 4 6 K+ 13.6 (8.1 ± 0 . 1 ) X 103 1.6
(1.7 ± 0 . 3 ) X 10" 1.0
(1.8 ± 0 . 1 ) X 10- (1.0)
* Defined as the ratio of the percent extraction of Cs"*" to the percent extraction of the indicated cation.
sodium picrate extraction into CDCI3 are low in all cases. In general
the log Kex values for lithium picrate are somewhat smaller than
those for sodium picrate. Compound 148 which contains an exocyclic
methylene unit has the best selectivity for Na" .
O 0-^R^
o o - ^ R2
Rl R2 1 4 7 H H 1 4 8 =CH2 1 4 9 -CH2CCI2-1 5 0 Ph H 1 5 1 Bzl H 1 5 2 Bzl Bzl 1 5 3 BZIOCH2 Me 1 5 4 BZIOCH2 BZIOCH2 15 5 Et Et 1 5 6 PhC(0)NH Me
Figure 16. Benzo-13-crown-4 Compounds,

55
Table 9. Picrate Extraction Data for Benzo-13-crown-4 Compounds.
compound M"*" % Ex Kex
r?7
148
149
150
151
152
153
154
155
Li+
Na+
Li+
Na+
Li+
Na+
Li+
Na+
Li+
Na+
Li+
Na+
Li+
Na+
Li+
Na+
Li+
Na+
0.047
0.049
0.052
0.161
0.071
0.134
0.040
0.043
0.064
0.091
0.030
0.043
0.010
0.023
0.072
0.023
0.055
0.076
18 ± 2
20 ± 2
21 ± 2
64 ± 8
28 ± 5
52 ± 5
16 ± 6
17 ± 4
22 ± 4
34 ± 5
12 ± 3
17 ± 3
4 ± 2
9 ± 1
29 ± 2
9 ± 1
22 ± 5
30 ± 4

56 Table 9. (cont.)
compound M+ % Ex Kex
15 6 Li+ 0.040 16 ± 6
Na+ 0.053 2 1 + 4
14-Crown-4 Compounds
A variety of 14-crown-4 compounds' ^ ^ (Figure 17) were tested
for their abilities to extract lithium and sodium picrates from
aqueous solution into CDCI3 (Table 10). The percentages of lithium
and sodium picrates extracted into CDCI3 were low in all cases except
for compound 170. The efficiency of lithium extraction is found to
be higher than that of sodium extraction. Comparing extraction
abilities of the 14-crown-4 compounds with the benzo-13-crown-4
compounds it is noted that the 14-crown-4 compounds are more
selective for Li"*". Benzo-13-crown-4 compounds have one "odd side"
to the structure, while 14-crown-4 compounds have an equal
number of two and three carbon chains between donor atoms. The
opportunity for the reintroduction of molecular symmetry was
realized with 14-crown-4 compounds by keeping identical bridging
arms opposite each other in the ring system, thereby creating extra
planes and axes of symmetry with respect to the crown ring
structure. It has been found before that symmetry is an important
factor in the complexation of guests by the crown ether hosts. ^1

57
R i > y - o O A , R ; R 2 > - o o - ^ ^ ' '
157 158 159 160 1 6 1 1 6 2 1 6 3 164 165 166 167 168 169
Rl H
=CH2 Bzl
-CH2CCI2-Bzl Bzl Bzl Bzl Bzl(m-OMe) BZIOCH2 BZIOCH2 BZIOCH2 Et
170tf^"^ ^ C(0)N(Me)2
R2 H
Bzl
Bzl H Bzl Bzl H Me Me BZIOCH2 Et
Me
^1 H
=CH2 =CH2
-CH2CCI H Bzl Bzl Bzl Bzl(m-OMe) H BZIOCH2 BZIOCH2 Et
H
R4 H
2- (trans) H H H Bzl H H Me BZIOCH2 Et
H
Figure 17. 14-Crown-4 Compounds.
Crown Ethers with Pendant Ferrocene Units
Two crown ethers with pendant ferrocene units ^ l (Figure 18)
were tested by the picrate extraction method for their abilities to
extract alkali metal cations. Crown ethers 171 and 172 which have

58
Table 10. Picrate Extraction Data for 14-Crown-4 Compounds,
compound M+ % Ex Kex
158
159
160
161
162
163
164
165
Li+
Na+
Li+
Na+
Li+
Na+
Li+
Na+
Li+
Na+
Li+
Na+
Li+
Na+
Li+
Na+
Li+
Na+
1.35
0.079
1.14
0.028
0.081
0.053
0.85
0.60
0.26
0.052
0.36
0.026
0.15
0.026
0.16
0.030
0.43
0.13
560 ± 8
32 ± 5
489 ± 4 0
12 ± 3
31 ± 6
21 ± 4
354 ± 2 0
246 ± 2 2
106 ± 7
21 ± 3
144 ± 1 6
10 ± 2
59 ± 5
11 ± 2
64 ± 5
10 ± 2
170 ± 8
53 ± 5
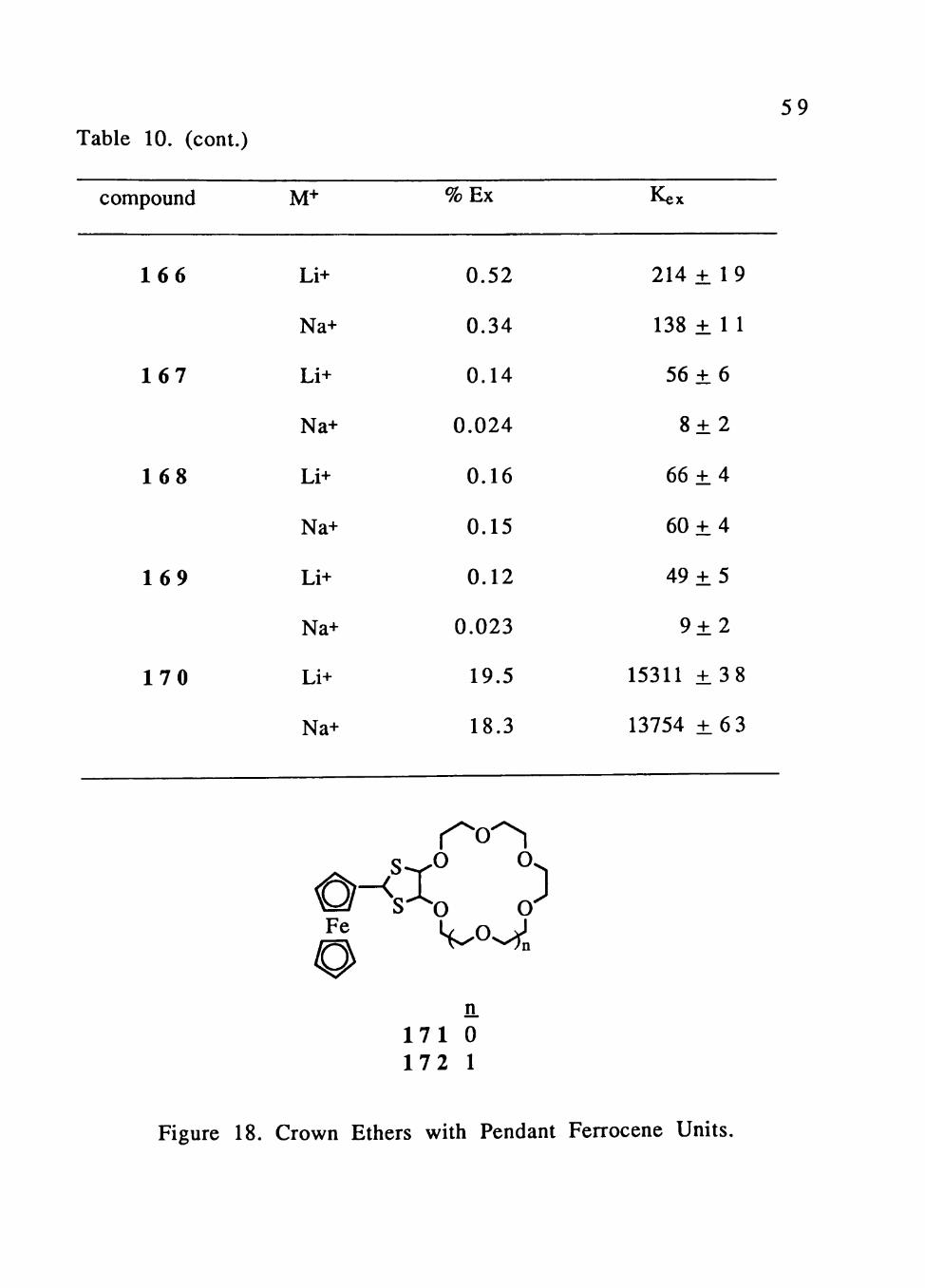
59 Table 10. (cont.)
compound
1 6 6
1 6 7
1 6 8
1 6 9
1 7 0
M+
Li+
Na+
Li+
Na+
Li+
Na+
Li+
Na+
Li+
Na+
%Ex
0.52
0.34
0.14
0.024
0.16
0.15
0.12
0.023
19.5
18.3
0-<sIo o Fe
n. 1 7 1 0 172 1
Kex
214 ± 19
138 ± 1 1
56 ± 6
8 ± 2
66 ± 4
60 ± 4
49 ± 5
9 ± 2
15311 ± 3 8
13754 ± 6 3
Figure 18. Crown Ethers with Pendant Ferrocene Units.

60
15-crown-5 and 18-crown-6 rings were found to preferentially
extract Na"*" and K"*" picrate, respectively. Since Na" should fit well
into a 15-crown-5 cavity and K"*" into a 18-crown-6 cavity the
observed selectivity is expected. For 171 the picrate selectivity is in
the order Na+ » K+, Li" > Rb" > Cs+ and for 172 are K" » Rb " > Cs"" »
Na-'>Li-^ (Table 11).
Table 11. Picrate Extraction Data for Crown Ethers 171 and 172.
compound M"*" % Ex Kex
T T l Li ^ TJl (6.3 ± 0.5) X 102
(4.9 ± 0.6) X 103
(7.2 ± 0.9) X 102
(5.0 ± 0.8) X 102
(3.0 ± 0.1) X 102
17 2 Li+ 0.91 (3.7 ±0.3) x 102
(1.3 ±0.8) X 103
(2.6 ± 0.1) X 105
(1.6 ±0.1) X 104
(7.1 ± 0.2) X 103
Li+
Na+
K+
Rb+
Cs+
Li+
Na+
K+
Rb+
Cs+
1.51
9.11
1.69
1.20
0.74
0.91
2.96
55.6
20.5
12.0

61
Crown Ethers with Pendant Pyridine Units
Non-lipophilic and lipophilic crown ethers with pendant
pyridine units[^9] (Figure 19) were tested by the picrate extraction
method for their selectivities in alkali metal cation complexation.
The extraction efficiencies for crown ethers 173-175 with R=H
(Table 12) were found to be very high and independent of the
number of atoms in the polyether ring and the identity of the alkali
metal cation. On the other hand the extraction efficiencies for crown
ethers 176-178 with R=alkyl group (Table 13) were very poor, but
somewhat selective for Na+. The X-ray crystal structures for 173-
175 [60] reveal that the pendant pyridine rings point away from the
crown ether cavities. Therefore the high efficiency but low
selectivity observed for compounds 173-175 may result from 2:2
R^0CH2
o o
U 1 7 3 1 7 4 1 7 5 1 7 6 1 7 7 1 7 8
R H H H C10H21 CH3
^10^21
Y CH2CH2 CH2CH2CH2 CH2CH2OCH2CH2
Cir{'^IrL'^Jji2 CH2CH2OCH2CH2 CH2CH2OCH2CH2
Figure 19. Crown Ethers with Pendant Pyridine Units.

Table 12. Picrate Extraction Data for Crown Ethers 173-175. 62
compound M+ %Ex Ke:
1 7 3
1 7 4
1 7 5
Li+
Na+
K+
Rb+
Cs+
Li+
Na+
K+
Rb+
Cs+
Li+
Na+
K+
Rb+
Cs+
67.0
67.5
65.3
69.9
61.4
62.2
59.8
60.0
61.0
56.1
63.9
61.8
57.2
60.0
57.2
(7.1
(7.1
(5.5
(8.9
(4.5
(4.5
(3.8
(3.6
(4.1
(2.7
(5.1
(4.3
(2.8
(3.6
(2.9
±0.2]
±0.3]
±0.6]
±0.1]
±0.5]
±0.4)
±0.1]
±0.6]
±0.5]
±0.2]
±0.2]
±0.1]
±o.i;
±o.i;
±o.i;
• x 105
• x 105
• X 105
I X 105
\ X 105
X 105
X 105
X 105
1 X 105
• X 105
1 X 105
1 X 105
1 X 105
1 X 105
) X 105

63 Table 13. Picrate Extraction Data for Crown Ethers 176-178.
compound M+ %Ex Kex
r76
177
178
Li+
Na+
K+
Rb+
Cs+
Li+
Na+
K+
Rb+
Cs+
Li+
Na+
K+
Rb+
Cs+
0.15
1.07
0.24
0.12
0.09
0.37
1.15
0.28
0.48
0.44
0.17
0-74
0.30
0.46
0.39
59.0 ± 8
440 ± 1 7
96.3 ± 1 3
50.0 ± 7
37.9 ± 8
155 ± 5
479 ± 2 9
112 ± 5
195 ± 9
179 ± 9
69.6 ± 4
306 ± 4
121 ± 4
186 ± 17
157 ± 9
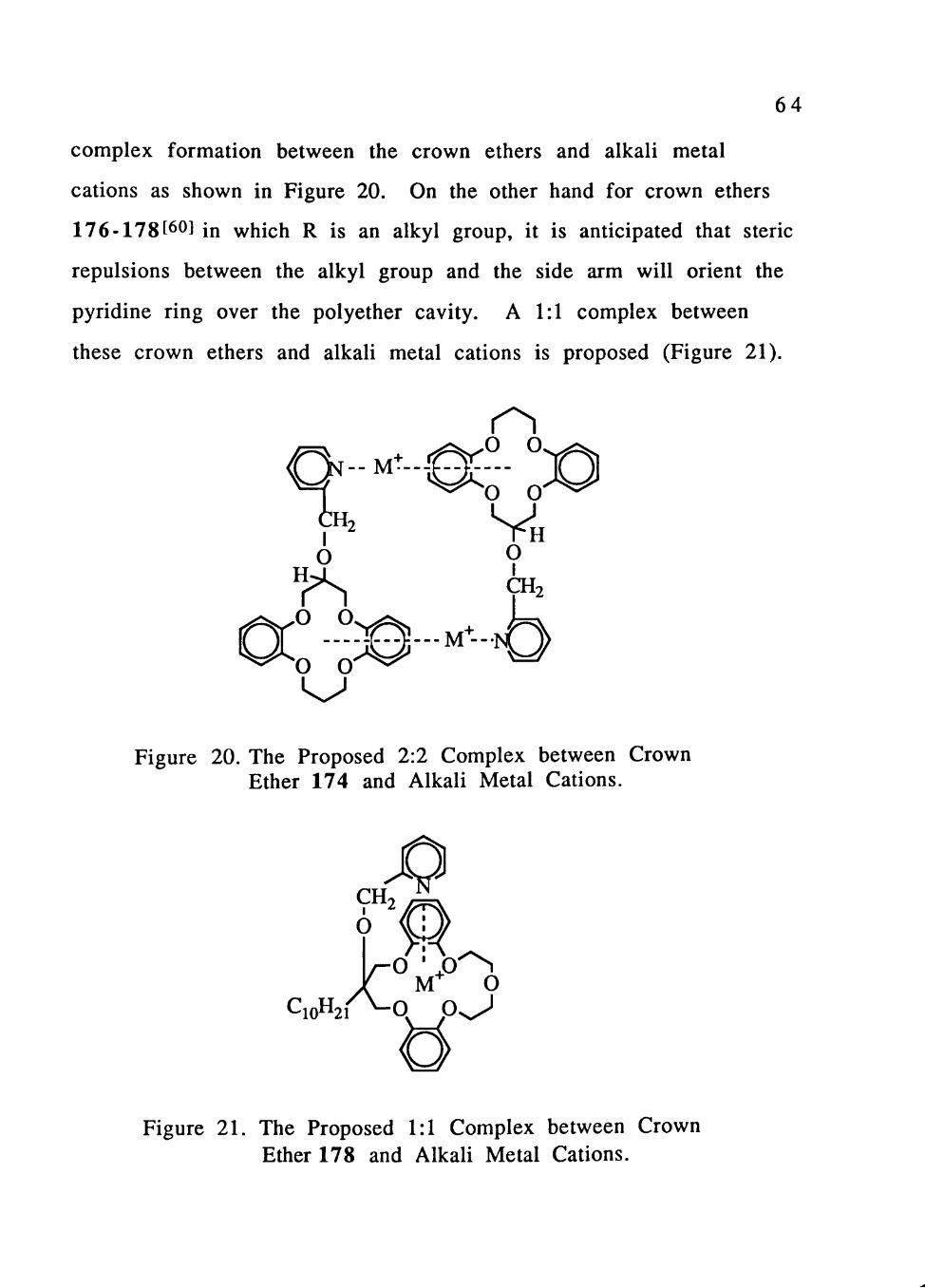
64
complex formation between the crown ethers and alkali metal
cations as shown in Figure 20. On the other hand for crown ethers
176-178[601 in which R is an alkyl group, it is anticipated that steric
repulsions between the alkyl group and the side arm will orient the
pyridine ring over the polyether cavity. A 1:1 complex between
these crown ethers and alkali metal cations is proposed (Figure 21).
Figure 20. The Proposed 2:2 Complex between Crown Ether 174 and Alkali Metal Cations.
CH2 O
^
r-O • fi'^
Figure 21. The Proposed 1:1 Complex between Crown Ether 178 and Alkali Metal Cations.

65
Molecular Receptors
The term "receptor" is preferable to define a collection of
ligating sites linked by covalent bonds because of the many
nonspecific uses of the word "ligand." The receptor-substrate
terminology[61] is preferred to the host-guest terminology[62]^ since
the latter covers all kinds of intermolecular associations including
inclusion compounds that exist only in the solid state, while the
former refers to physically characterizable species formed by well-
defined associations.
Molecular receptors 179-182[58] (Figure 22) which have both
hydrophobic and hydrophilic regions with in their cavities were
tested by the picrate extraction method to determine their abilities
to extract alkali metal cations. Molecular receptor 179 was found to
extract K+ the best (Table 14) and molecular receptors 180 and 181
O O N
O P ^ f n
1 7 9 1 8 0 1 8 1
m 1 1 2
n. 1 2 2
1 8 2
Figure 22. Molecular Receptors.

66
Table 14. Picrate Extraction Data for Molecular Receptors 179-181
compound M+ %Ex Ke:
1 7 9
1 8 0
1 8 1
Li+
Na+
K+
Rb+
Cs+
Li+
Na+
K+
Rb+
Cs+
Li+
Na+
K+
Rb+
Cs+
8.3
11.2
33.7
15.3
12.4
9.0
8.8
12.3
12.1
12.5
6.0
6.3
8.0
10.2
6.9
(4.3 ± 0.1
(6.3 ± 0.3
(4.6 ± 0.7
(1.0 ± 0.
(7.4 ± 0.
(4.8 ± 0.
(4.6 ± 0.
(7.3 ± 0.
(7.3 ± 0.3
(7.3 ± 0.
(2.9 ± 0.
(3.0 ± 0.
(4.0 ± 0.
(5.7 ± 0.6
(3.4 ± 0.1
X 103
X 103
X 104
X 104
X 103
X 103
X 103
X 103
X 103
X 103
X 103
X 103
X 103
X 103
X 103

67
to extract alkali metal cations with little selectivity because the host
cavities are so large.
Molecular receptors 179-181 [58] were also tested for their
abilities to extract alkylammonium ions from solutions of
alkylammonium picrate in water into CDCI3. Table 15 shows
extraction data for molecular receptors 179-181 and 182, which
have elongated cavities, with alkylammonium picrates. These
molecular receptors were found to extract propylammonium picrate
the best and the complexation selectivities are in order:
propylammonium > ethylammonium > butylammonium > t-butyl-
ammonium, methylammonium. The percent of propylammonium
picrate extracted into CDCI3 was the same for molecular receptors
179-182, but the extraction efficiencies for the other
alkylammonium picrates was highest with 182 (Table 16). The
greatest differences in alkylammonium picrate extraction abilities is
noted with receptor 181.
The model compound N,N-didecyl-7,16-diaza-18-crown-6
(185) was synthesized[62] (Scheme 23) to compare its complexation
selectivity for alkylammonium picrates with that for molecular
receptors 179-182. Decanoyl chloride (183) was prepared by the
reaction of decanoic acid with an excess of thionyl chloride in 96%
yield. Crown ether 185 was synthesized by the reaction of 7,16-
diaza-18-crown-6 and decanoyl chloride (183) followed by the
reduction of diamide 184 with BH3SMe2 in THF. Table 17 shows
that the complexation selectivities for 185 are the same as for the
molecular receptor 182. These results indicate that the

68 Table 15. Alkylammonium Picrate Data* for Molecular Receptors
1 7 9 - 1 8 1 .
compound picrate %Ex Ke;
17 9 methylammonium
ethylammonium
propylammonium
butylammonium
t-butylammonium
18 0 methylammonium
ethylammonium
propylammonium
butylammonium
t-butylammonium
18 1 methylammonium
ethylammonium
butylammonium
propylammonium
butylammonium
t-butylammonium
11.5
81.3
91.3
35.5
10.9
9.8
83.6
90.8
19.0
10.5
5.8
81.6
15.7
90.8
15-7
6.3
(6.7 ± 0.1
(3.6 ± 0.1
(2.6 ± 0.2
(5.2 ± 0.1
(6.1 ± 0.2
(5.3 ± 0.1
(5.2 ± 0.2
(1.6 ±0 .1
(1.4 ±0 .1
(5.9 ± 0.6
(2.8 ± 0.1
(4.6 ± 0.2
(1.1 ±0 .1
(1.7 ±0 .1
(1.1 ±0 .1
(3.1 ± 0.7
X 103
X 106
X 107
X 104
X 103
X 103
X 106
X 107
X 104
X 103
X 103
X 106
X 104
X 107
X 104
X 103
•Corrected from the values in the absence of receptor.

69 Table 16. Alkylammonium Picrate Data* for Molecular Receptor 182.
picrate % Ex Kex
methy lammonium 34.1 (4.6 ± 0.3) x 104
e thy lammonium 84.5 (6.1 ± 0.1) x 106
propy lammonium 93.0 (3.3 ± 0.1) x 107
bu ty lammonium 60.9 (3.7 ± 0.2) x 105
t -bu ty lammonium 32.3 (4.0 ± 0.1) x 104
•Corrected from the values in the absence of receptor.
Scheme 23
O O
C9Hi9^0H + SOCI2 ^ C9Hi9('!ci
1 8 3
d ^ ^ O ^ n r^ r-0 O < > II Et3N II \ / 11^
HN NH + C9H19CCI 1 • C9H19C-N N-CC9H19
^ O O - ^ 1 8 3 ^ O O-f
1 8 4
O O
^ " ^ • ^ ^ ^ ^ . C10H21-N N-C10H21
^ o o-^
1 8 5

70
Table 17. Alkylammonium Picrate Data* for Crown Ether 185.
picrate % Ex Kex
methylammonium 31.2 (3.8 ± 0.1) x 104
ethylammonium 84.8 (6.5 ± 0.1) x 106
propylammonium 92.6 (2.9 ± 0.1) x 107
butylammonium 60.3 (3.5 ± 0.2) x 105
t-butylammonium 30.8 (3.6 ± 0.1) x 104
*Corrected from the values in the absence of receptor.
complexation site for the molecular receptor 182 is the diaza crown
ether ring.
Summary
Several series of proton-ionizable crown ethers have been
synthesized to study the effect of structural variation upon metal ion
complexation. A series of cyclic polyether derivatives of salicylic
acid has been prepared to probe the ring size effect. Series of non-
lipophilic and lipophilic dibenzo-16-crown-5 phosphonic acid
monoalkyl esters have been prepared to investigate the effect of
sidearm length.
For attachment to silica gel, functionalized crown ethers which
have dibenzo-14-crown-4 and dibenzo-16-crown-5 rings have been
synthesized. Also a set of functionalized crown ethers based on
salicylic acid has been prepared.

71
Benzo-21-crown-7 and dibenzo-21-crown-7 compounds have
been investigated by the picrate extraction method for their abilities
to extract the alkali metal cations. The ratio of the Cs+/Na+ extraction
selectivities for the dibenzo-21-crown-7 compounds were noted to
be larger than those for the benzo-21-crown-7 compounds.
Series of benzo-13-crown-4 and 14-crown-4 compounds were
tested by the picrate extraction method for their abilities to extract
lithium and sodium cations. The effects of substituents attached to
the crown ether rings upon complexation selectivity and efficiency
were evaluated.
Crown ethers with pendant ferrocene units and pyridine units,
respectively, were also tested by the picrate extraction method for
alkali metal cation complexation selectivity. From the results, it is
postulated that crown ethers with pendent pyridine units can form
1:1 or 2:2 complexes with alkali metal cations depending upon the
crown ether structures.
Finally, molecular receptors were found to extract alkali metal
and alkylammonium cations selectively.

CHAPTER III
EXPERIMENTAL PROCEDURES
Instrumentation and Reagents
Melting points were determined on a Fisher-Johns melting
point apparatus and are uncorrected. H NMR spectra were obtained
with Varian EM-360, IBM AF-200, or IBM AF-300 spectrometers.
The chemical shifts are expressed in parts per million (ppm)
downfield from tetramethysilane. Splitting patterns are indicated as:
s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad
peak. Infrared spectra were obtained on either a Nicolet MS-X FT-IR
or a Perkin-Elmer 1600 Series FT-IR spectrometer on NaCl plates and
are given in wavenumbers (cm^O-
Unless specified otherwise, starting materials and solvents
were reagent grade and used as received. Dry solvents were
prepared as follows: pyridine and pentane were dried over KOH
pellets; N,N-dimethylformamide (DMF) was dried over 4A molecular
sieves, or K2CO3; acetone was distilled from NaHCOs; tetrahydrofuran
(THF) was distilled from LiAlILj; tert-butyl alcohol was distilled from
CaH2; MeOH was distilled from magnesium turnings with a crystal of
added iodine; and EtOH was dried by azeotropic distillation in the
presence of benzene.
Thin layer chromatography (TLC) was performed with either
Analtech Alumina GF or Silica GF prepared plates. The plates were
precoated with 250 mm silica gel or alumina. Column
72

73
chromatography was performed using either alumina (80-200 mesh)
or silica gel (60-200 mesh) from Fisher Scientific.
Elemental analyses were done by Desert Analytics of Tucson,
Arizona.
Hydroxymethyl-13-crown-4(3), hydroxymethyl-18-crown-6,
and hydroxymethyl-24-crown-8 were available from other
studies.[43]
General Procedure for Preparation of Tosylates of Monobenzyl Glycols 62-64
A solution of the benzyl glycol (15.3 mmol) in 10 mL of
pyridine was cooled to -10 °C and a solution of p-toluenesulfonyl
chloride (3.64 g, 19.1 mmol) in 10 mL of pyridine was added
dropwise. After the reaction mixture was stirred for 1 h at this
temperature, it was kept overnight at 4 °C and poured over ice. The
reaction mixture was acidified to pH 1 with cold 6N HCl and extracted
with CH2CI2 (2 X 30 mL). The combined extracts were washed with
water and dried over MgS04. Evaporation of the solvent gave the
pure tosylate.
Tosylate of Monobenzyl Ethylene Glycol (62)
A coloriess oil was obtained in 95% yield. IR (neat): 1357,
1190, 1177 (SO2); 1128, 1097 (C-0) cm-i. m NMR (CDCI3): 5 2.43 (s,
3H), 3.60-3.66 (m, 2H), 4.12-4.25 (m, 2H), 4.52 (s, 2H), 2.75-7.81 (m,
9H).

74 Tosylate of Monobenzyl Diethylene Glycol (63)
A colorless oil was obtained in 79% yield. IR (neat): 1357,
1190, 1177 (SO2); 1128, 1097 (C-0) cm-i. ^H NMR (CDCI3): 5 2.43 (s,
3H), 3.55-3.78 (m, 4H), 4.12-4.25 (m, 2H), 4.52 (s, 2H), 7.25-7.81 (m,
9H).
Tosylate of Monobenzyl Triethylene Glycol (64)
A colorless oil was obtained in 67% yield. IR (neat): 1357,
1190, 1177 (SO2); 1128, 1097 (C-0) cm-i. ^H NMR (CDCIs): 5 2.43 (s,
3H); 3.55-3.78 (m, 4H); 4.12-4.25 (m, 2H); 4.52 (s, 2H); 7.25-7.81 (m,
9H).
General Procedure for Preparation of Carboxylic Acids 68-7 0
Sodium hydride (0.50 g, 12.5 mmol, 60% dispersion in mineral
oil) was washed with pentane (2 X 20 mL) and suspended in 15 mL
of THF. With stirring under nitrogen, a solution of methyl salicylate
(1.53 g, 10 mmol) in 15 mL of THF was added dropwise during 45
min. After 1 h a solution of the tosylate of monobenzyl glycol (10
mmol) in 20 mL of THF was added dropwise during 30 min. The
reaction mixture was stirred at room temperature overnight and
then refluxed for 3 d. The solvent was evaporated in vacuo and
CH2CI2 was added. The resulting mixture was filtered and the filtrate
was evaporated in vacuo. The residue was chromatographed on
silica gel with CH2CI2 as eluent to give the carboxylic acid ester. The
ester (3.0 mmol) was dissolved in 20 mL of EtOH and a solution of

75
NaOH (1.0 g) in 5 mL of H2O was added. The mixture was refluxed
for 4 h and evaporated to dryness in vacuo. Water was added to the
residue. The mixture was acidified to pH 1 with 6N HCl and
extracted with CH2CI2 (3 X 20 mL). The combined extracts were
washed with water (30 mL), dried over MgS04 and evaporated in
vacuo to give the carboxylic acid.
Methyl 2-[( l ,4-Dioxa-5-phenyl)pentyl]-benzoate (65)
A colorless oil was obtained in 49% yield. IR (neat): 1730
(C=0); 1132, 1085 (C-0) cm-i. iH NMR (CDCI3): 5 3.86-4.25 (m, 7H),
4.67 (s, 2H), 6.95-7.81 (m, 9H).
2- [ ( l , 4 -Dioxa-5 -pheny l )penty l ]benzo ic Acid (68)
Basic hydrolysis of compound 65 gave a colorless oil in 89%
yield. IR (neat): 3266 (COOH); 1732 (C=0); 1125, 1105 (C-0) cm-i.
IH NMR (CDCI3): 8 3.66-4.40 (m, 4H), 4.62 (s, 2H), 7.00-8.20 (m,
9H), 11.09 (b, s, IH). Anal. Calcd. for C16H16O4: C, 70.57; H, 5.92.
Found: C, 70.35; H, 6.00.
Methyl 2-[(l,4,7-Trioxa-8-phenyl)octyI]-benzoate (66)
A colorless oil was obtained in 79% yield. IR (neat): 1729
(C=0); 1134, 1085 (C-0) cm-i. ^H NMR (CDCI3): 5 3.64-4.23 (m, IIH),
4.57 (s, 2H), 6.95-7.81(m, 9H).

76 2 - [ ( l , 4 , 7 - T r i o x a - 8 - p h e n y l ) o c t y l ] b e n z o i c Acid (69)
Basic hydrolysis of compound 66 gave a colorless oil in 88%
yield. IR (neat): 3269 (COOH); 1731 (C=0); 1127, 1101 (C-0) cm-J.
IH NMR (CDCI3): 5 3.66-4.40 (m, 8H), 4.57 (s, 2H), 7.00-8.20 (m, 9H),
11.09 (br s, IH). Anal. Calcd. for C18H20O5: C, 68.34; H, 6.37. Found:
C, 68.19; H, 6.46.
M e t h y l 2 . [ ( l , 4 , 7 , 1 0 - T e t r a o x a - l l - p h e n y l ) -undecyljbenzoate (67)
A colorless oil was obtained in 75% yield. IR (neat): 1730
(C=0); 1133, 1097 (C-0) cm-i. iH NMR (CDCI3): 5 3.62-4.22 (m, 15H),
4.56 (s, 2H), 6.95-7.81(m, 9H).
2 - ( l , 4 , 7 , 1 0 - T e t r a o x a - l l - p h e n y I ) -undecyljbenzoic Acid (70)
Basic hydrolysis of compound 67 gave a colorless oil in 94%
yield. IR (neat): 3274 (COOH); 1732 (C=0); 1126, 1101 (C-0) cm-i.
IH NMR (CDCI3): 6 3.62-4.36 (m, 12H), 4.55 (s, 2H), 7.00-8.20 (m, 9H),
11.09 (br s, IH). Anal. Calcl. for Anal. Calcd. for C20H24O6: C, 66.65; H,
6.71. Found: C, 66.54; H, 6.97.
Preparation of (Benzyloxy)methyl-substituted Crown Ethers 7 1 - 7 3
( B e n z y l o x y ) m e t h y l - 1 2 - c r o w n - 4 (71)
Under nitrogen, lithium metal (1.14 g, 0.16 mol) was added to
300 mL of tert-BuOH. After refluxing for 1 h, 3-(benzyloxy)-l,2-
propanediol[44,45| (lO.O g, 0.054 mol) was added dropwise. To the
cloudy, heterogeneous mixture, l,2-bis(2-chloroethoxy)ethane (10.28

77
g, 0.054 mol) was added followed by LiBr (4.69 g, 0.054 mol) and 10
mL of water. The reaction mixture was refluxed for 2 weeks. After
the solvent was removed in vacuo. 30 mL of water was added to the
residue and the mixture was neutralized with 6N HCl and extracted
with CH2Cl2(3 X 30 mL). The CH2CI2 solution was dried over MgS04
and evaporated in vacuo. The residue was purified by column
chromatography on alumina with petroleum ether/ethyl acetate (4:1)
as eluent to give 9.94 g (62% yield) of the product as a colorless oil.
IR (neat): 1249, 1126 (C-0) cm-i. iH NMR (CDCI3): 5 3.45-4.92 (m,
17H), 4.54 (s, 2H), 7.32 (s, 5H).
(Benzyloxy)methy-15-crown-5 (72)
Tetraethylene glycol ditosylate (2.74 g, 15 mmol) was diluted
to 12 mL with anhydrous DMF-THF (4:1) and taken up in a syringe.
3-(Benzyloxy)-l,2-propanediol (7.54 g, 15 mmol) was also diluted to
12 mL with DMF-THF (4:1) and taken up in a syringe. The two
solutions were simultaneously added with two syringe pumps during
38 h at room temperature to a mixture of NaH (0.96 g, 24 mmol, 60%
dispersion in mineral oil) and 20 mL of DMF-THF (4:1). After a total
time of 3 d, the reaction mixture was quenched with 20 mL of
saturated NaCl solution and the solution was extracted with CHCl3(3
X 40 mL). The combined CHCI3 extracts were dried over MgS04 and
evaporated in vacuo. The residual DMF was removed with high
vacuum. The residue was purified by column chromatography on
alumina with CH2CI2 and then ethyl acetate as eluents to give 1.90 g
(39% yield) of the product as a colorless oil. IR (neat): 1251, 1177

78
(C-O)cm-i. IH NMR (CDCI3): 5 3.50-3.86 (m, 21H), 4.55 (s, 2H), 7,32
(s, 5H).
(Benzyloxy) methyl-21-crown-7 (73)
Tetraethylene glycol ditosylate (7.54 g, 0.015 mol) was diluted
to 45 mL with THF and taken up in a syringe. 3,6-Dioxo-5-
(benzyloxy)methyl-l,8-diol (4.02 g, 0.015 mol) was also diluted to
45 mL with THF and taken up in a syringe. The two solutions were
simultaneously added with two syringe pumps during 10 h at room
temperature to a mixture of NaH (1.80 g, 0.045 mol, 60% dispersion
in mineral oil) and 150 mL of THF. After the reaction mixture was
refluxed for 24 h, 50 mL of saturated NaCl solution was added. The
reaction mixture was extracted with CH2CI2 (3 X 100 mL). The
combined CH2CI2 extracts were dried over MgS04 and evaporated i_n
vacuo. The residue was purified by column chromatography on
alumina with CH2CI2 and then ethyl acetate as eluents to give 1.80 g
(28% yield) of the product as a colorless oil. IR (neat): 1250, 1177
(C-O)cm-i. iH NMR (CDCI3): 5 3.50-3.86 (m, 29H), 4.54 (s, 2H), 7.33
(s, 5H).
General Procedure for Preparations of Hydroxymethyl Crown Ethers 74-76
To a solution of the (benzyloxy)methyl-substituted crown ether
(13-24 mmols) in 50 mL of EtOH was added 10% palladium on carbon
(100 mg/g of crown ether) and a catalytic amount of p.-toluene-
sulfonic acid monohydrate. The mixture was hydrogenated under

7 9
25 lbs pressure of hydrogen at room temperature for 48 h. The
reaction mixture was filtered and evaporated in vacuo. The residue
was taken up in CH2CI2. The CH2CI2 solution was dried over MgS04
and evaporated in vacuo to give the product.
H y d r o x y m e t h y l - 1 2 - c r o w n - 4 (74)
A colorless oil was obtained in 95% yield. IR (neat): 3237
(0-H); 1245, 1132 (C-0) cm-i. iH NMR (CDCI3): 6 2.34 (s, IH), 3.55-
3.82 (m, 17H).
H y d r o x y m e t h y l - 1 5 - c r o w n - 5 (75)
A colorless oil was obtained in 86% yield. IR (neat): 3312
(0-H); 1249, 1124 (C-0) cm-i. m NMR (CDCI3): 5 2.62 (br s, IH),
3.55-3.82 (m, 21H).
H y d r o x y m e t h y l - 2 1 - c r o w n - 7 (76)
A colorless oil was obtained in 91% yield. IR (neat): 3320
(0-H); 1245, 1130 (C-0) cm-i. ^H NMR (CDCI3): 5 2. 92 (br s, IH),
3.55-3.82 (m, 29H).
General Procedure for Preparation of (Tosyloxy)-methyl-substituted Crown Ethers 77 -82
To a solution of the hydroxymethyl crown ether (9-23 mmoles)
in 10 mL of pyridine at -10 °C under nitrogen was added dropwise
£-toluenesulfonyl chloride (1.25 equivalents ) in 10 mL of pyridine.
After keeping the mixture at 4 "C for 24 h, a cold solution of 6N HCl
and ice was added. The organic layer was separated and the aqueous

80
layer was extracted with CH2CI2 (3 X 10 mL). The combined extracts
were dried over MgS04 and evaporated in vacuo to give the tosylate.
( T o s y l o x y ) m e t h y l - 1 2 - c r o w n - 4 (77)
A colorless oil was prepared in 97% yield. IR (neat): 1358,
1190, 1171 (SO2); 1129, 1097 (C-0) cm-i. iH NMR (CDCI3): 6 2.45 (s,
3H), 3.50-4.20 (m, 17H), 7.55 (AB q, 4H).
3 - [ ( T o s y l o x y ) m e t h y l ) ] - 1 3 - c r o w n - 4 (78)
A colorless oil was prepared in 76% yield. IR (neat): 1358,
1190, 1171 (SO2); 1129, 1097 (C-0) cm-i. iH NMR (CDCI3): 5 2.21-
2.31 (m, IH), 2.45 (s, 3H), 3.30-3.70 (m, 16H), 4.08 (d, 2H), 7.55 (AB
q, 4H).
( T o s y l o x y ) m e t h y l - 1 5 - c r o w n - 5 (79)
A colorless oil was prepared in 95% yield. IR (neat): 1358,
1190, 1171 (SO2); 1129, 1097 (C-0) cm-i. iH NMR (CDCI3): 5 2.45 (s,
3H), 3.50-4.20 (m, 21H), 7.55 (AB q, 4H).
(Tosy loxy)m e t h y l - 1 8 - c r o w n - 6 (80)
A coloriess oil was prepared in 98% yield. IR (neat): 1358,
1190, 1171 (SO2); 1129, 1097 (C-0) cm-i. iH NMR (CDCI3): 5 2.45 (s,
3H), 3.50-4.20 (m, 25H), 7.55 (AB q, 4H).
( T o s y l o x y ) m e t h y l - 2 1 - c r o w n - 7 (81)
A coloriess oil was prepared in 88% yield. IR (neat): 1356,
1189, 1177 (SO2); 1129, 1097 (C-0) cm-i. ^H NMR (CDCI3): 5 2.45 (s,
3H), 3.50-4.20 (m, 29H), 7.55 (AB q, 4H).

81
( T o s y l o x y ) m e t h y l - 2 4 - c r o w n - 8 (82)
A colorless oil was prepared in 86% yield. IR (neat): 1356,
1189, 1177(S02); 1129, 1097 (C-0) cm-i. iH NMR (CDCI3): 5 2.45 (s,
3H), 3.50-4.20 (m, 33H), 7.55 (AB q, 4H).
General Procedure for Preparation of Crown Ether Carboxylic Acids 34. 35. and. 38-41
Under nitrogen, sodium hydride (0.26 g, 6.25 mmol, 60%
dispersion in mineral oil) was washed with pentane (2 X 20 mL) to
remove the protecting mineral oil and 10 mL of THF was added. To
the suspension, methyl salicylate (0.76 g, 5.0 mmol) in 15 mL of THF
was added slowly. After stirring at room temperature for 1 h, a
solution of the crown ether tosylate (5.0 mmol) in 10 mL of THF was
added and the mixture was refluxed for 3 d. The reaction mixture
was filtered and the filtrate was evaporated in vacuo. The residue
was chromatographed on silica gel with CH2CI2 and ethyl acetate as
eluents to give the crown ether carboxylic acid ester. The ester (3.0
mmol) was dissolved in 20 mL of EtOH and a solution of NaOH (1.0 g)
in 5 mL of water was added. The mixture was refluxed for 4 h and
evaporated to dryness in vacuo. Water (10 mL) was added to the
residue. The mixture was acidified to pH 1 with 6N HCl and
extracted with CH2Cl2(3 X 20 mL). The combined extracts were
washed with water (30 mL), dried over MgS04, and evaporated in
vacuo to give the crown ether carboxylic acid.

82 Methyl 2-[(12-Crown-4)-methyloxy] benzoate (83)
A colorless oil was obtained in 39% yield. IR (neat): 1730
(C=0); 1133, 1084 (C-0) cm-i. iH NMR (CDCI3): 6 3.60-4.20 (m, 20H),
6.96-7.85 (m, 4H).
2 - [ ( 1 2 - C r o w n - 4 ) - m e t h y l o x y ] b e n z o i c Acid (34)
Basic hydrolysis of compound 83 gave a colorless oil in 88%
yield. IR (neat): 3250 (COOH); 1729 (C=0); 1135, 1099 (C-0) cm-i.
IH NIVIR (CDCI3): 5 3.60-4.18 (m, 15H), 4.25-4.40 (m, 2H), 7.04-8.22
(m, 4H). Anal. Calcd. for C16H22O7: C, 58.88; H, 6.80. Found: C, 58.62;
H, 6.90.
Methyl 2 - [ 3 ' - ( 1 3 - C r o w n - 4 ) - m e t h y l o x y ] -benzoate (84)
A colorless oil was obtained in 57% yield. IR (neat): 1729
(C=0); 1133, 1085 (C-O) cm-i. IH NMR (CDCI3): 5 2.37-2.60 (m, IH),
3.50-3.78 (m, 16H), 3.86 (s, 3H), 4.15-4.25 (m, 2H), 6.96-7.85 (m,
4H).
2 - [ 3 ' - ( 1 3 - C r o w n - 4 ) - n i e t h y l o x y ] b e n z o i c Acid (35)
Basic hydrolysis of compound 84 gave a colorless oil in 97%
yield. IR (neat): 3277 (COOH); 1725 (C=0); 1133, 1107 (C-O) cnri .
iH NMR (CDCI3): 5 2.37-2.60 (m, IH), 3.50-3.78 (m, 16H), 4.30-4.40
(m, 2H), 7.09-8.22 (m, 4H). Anal. Calcd. for C17H24O7: C, 59.99; H,
7.11. Found: C, 59.80; H, 7.47.

83 Methyl 2-[(15-Crown-5)-methyIoxy]-benzoate (85)
A colorless oil was obtained in 52% yield. IR (neat): 1729
(C=0); 1131, 1049 (C-0) cm-i. ^H NMR (CDCI3): 5 3.60-4.20 (m, 24H),
6.96-7.85 (m, 4H).
2- [ (15-Crown-5) -methy loxy]benzo ic Acid (38)
Basic hydrolysis of compound 85 gave a colorless oil in 93%
yield. IR (neat): 3258 (COOH); 1729 (C-0); 1128, 1041 (C-0) cm-i.
IH NMR (CDCI3): 5 3.60-4.18 (m, 19H), 4.25-4.40 (m, 2H), 7.04-8.22
(m, 4H). Anal. Calcd. for C18H26O6: C, 58.37; H, 7.08. Found: C, 58.46;
H, 7.15.
Methyl 2-[(18-Crown-6)-methyIoxy]-benzoate (86)
A colorless oil was obtained in 50% yield. IR (neat): 1729
(C=0); 1124, 1049 (C-0). iH NMR (CDCI3): 6 3.60-4.20 (m, 28H), 6.96-
7.85 (m, 4H).
2- [ (18-Crown-6) -methy loxy]benzo ic Acid (39)
Basic hydrolysis of compound 86 gave a colorless oil in 92%
yield. IR (neat): 3260 (COOH); 1731 (C=0); 1119, 1041 (C-0) cm-i.
IH NMR (CDCI3): 5 3.60-4.00 (m, 22H), 4.01-4.15 (m, IH), 4.28-4.50
(m, 2H), 7.04-8.22 (m, 4H). Anal. Calcd. for C20H30O9: C, 57.96; H,
7.30. Found: C, 57.71; H, 7.44.

84 Methyl 2-[(21-Crown-7)-methyloxy] benzoate (87)
A colorless oil was obtained in 61% yield. IR (neat): 1732
(C=0); 1118, 1048 (C-0) cm-i. iH NMR (CDCI3): 5 3.60-4.20 (m, 32H).
6.96-7.85 (m, 4H).
2 - [ ( 2 1 - C r o w n - 7 ) - m e t h y l o x y ] b e n z o i c Acid (40)
Basic hydrolysis of compound 87 gave a colorless oil in 84%
yield. IR (neat): 3261 (COOH); 1732 (C=0); 1114, 1041 (C-0) cm-i.
IH NMR (CDCI3): 5 3.60-4.00 (m, 26H), 4.01-4.15 (m, IH), 4.30-4.50
(m, 2H), 7.04-8.22 (m, 4H). Anal. Calcd. for C22H34O10: C, 57.63; H,
7.47. Found: C, 57.49; H, 7.78.
Methyl 2-[ (24-Crown-8)-methyloxy]-benzoate (88)
A colorless oil was obtained in 60% yield. IR (neat): 1732
(C=0); 1124, 1048 (C-0) cm-i. IH NMR (CDCI3): 5 3.60-4.20 (m, 36H),
6.96-7.85 (m, 4H).
2 - [ ( 2 4 - C r o w n - 8 ) - m e t h y l o x y ] b e n z o i c Acid (41)
Basic hydrolysis of compound 88 gave a colorless oil in 88%
yield. IR (neat): 3260 (COOH); 1731 (C=0); 1114, 1041 (C-0) cm-i.
Anal. Calcd. for C24H38O11: C, 57.36; H, 7.62. Found: C, 57.04; H, 7.90.
<^yin-Ketodibenzo-16-crown-5[5i] (90)
With mechanical stirring, crown ether alcohol 89 (20.0 g, 57.8
mmol) was dissolved in 400 mL of acetone. To the cooled (ice bath)

85
and vigorously stirred solution was added 60 mL of Jones reagent[50]
(16.02 g of Cr03, 13.8 mL of concentrated H2SO4, and sufficient H2O to
make a 60 mL volume) over 2 h. After an additional 2 h of stirring,
the reaction mixture consisted of a tan solution and a green
precipitate. The tan solution was decanted, and the green precipitate
was washed with acetone (2 X 20 mL). The acetone washings were
combined with the tan solution. The solvent was evaporated partly
(to about 200 mL) in vacuo and 500 mL of water was added. After
the reaction mixture was kept in the refrigerator for 2 h, the
resulting solid was collected by filtration. The crude product was
dissolved in 400 mL of acetone, and decolorizing carbon was added
to remove yellow color. The mixture was refluxed for 30 min and
filtered while the mixture was still hot. The acetone was evaporated
partly in vacuo again. The resulting solid was filtered and allowed to
dry to give 90 in 58-78% yields with mp 141-143 «C (lit. mp 138-
139 °C). IR (deposit): 1737 (C=0) cm-i. m NMR (CDCI3): 3.80-4.50
(m, 12H), 4.93 (s, 4H), 6.85-7.05 (m, 8H).
fiXOL-(Me thy 1) ( h y d r o x y ) d i b e n z o -16-crown-5 (91)
To 0.84 g (34.5 mmol) of magnesium turnings in 150 mL of
anhydrous diethyl ether under nitrogen was added methyl iodide
(4.90 g, 34.5 mmol) dropwise. The reaction mixture was refluxed
undl the magnesium disappeared. Crown ether ketone 90 (4.0 g,
11.6 mmol) in 300 mL of THF was added to the Grignard reagent
solution dropwise, and refluxing was continued for 24 h. After
cooling the reaction mixture, 90 mL of 5% aqueous NH4CI solution

86
was added and the mixture was stirred for 2h. The solvent was
evaporated in vacuo and the residue was extracted with CH2CI2 (2 X
100 mL). The CH2CI2 solution was dried over MgS04 and evaporated
in vacuo. The crude product was recrystallized from pentane. The
white solid was purified by column chromatography on silica gel
with CH2Cl2as eluent to give 1.73 g (41%) of 91 as a white solid with
mp 110-111 °C. IR (deposit): 3460 (0-H); 1124, 1039 (C-0) cm-i. ^H
NMR (CDCI3): 6 0.88 (s, 3H), 3.45 (br s, IH), 3.90-4.30 (m, 12H), 6.84-
7.02 (m, 8H).
General Procedure for Preparation of Crown Ether Alcohols 92 and 93
To 0.56 g (23.0 mmol) of magnesium turnings under nitrogen
was added 40 mL of THF and 23.0 mmol of the 1-bromoalkane. The
mixture was refluxed until most of the magnesium had been
consumed. Then crown ether ketone 90 (3.96 g, 11.5 mmol) was
added and refluxing was continued for 5 h. After cooling the
reaction mixture to room temperature, 30 mL of 5% aqueous NH4CI
solution was added and the mixture was stirred for 10 h. The THF
was evaporated in vacuo and the residue was extracted with 50 mL
of CH2CI2. The CH2CI2 solution was dried over MgS04 and evaporated
in vacuo to afford a white solid which was stirred with 200 mL of
pentane for 1 h. The solid was filtered and dissolved in a small
amount of CH2CI2 and loaded onto a silica gel column. Elution with
CH2CI2 afforded the pure product as a white solid.

87 fiXm.-(Hexyl) ( h y d r o x y ) d i b e n z o -16-crown-5 (92)
A white solid with mp 121-123 ''C was obtained in 45% yield.
IR (deposit): 3395 (OH); 1122, 1039 (C-0) cm-i. iH NMR (CDCI3): 5
0.88 (t, 3H), 1.15-1.55 (m, 8H), 1.80-1.92 (m, 2H), 3.45 (br s, IH),
3.90-4.30 (m, 12H), 6.84-7.02 (m, 8H).
SXDL-(Decyl) ( h y d r o x y ) d i b e n z o -16-crown-5 (93)
A white solid with mp 90-92 °C was obtained in 62% yield. IR
(deposit): 3350 (0-H); 1125, 1039 (C-0) cm-i. ^H NMR (CDCI3): 6
0.88 (t, 3H), 1.15-1.55 (m, 16H), 1.80-1.92 (m, 2H), 3.45 (br s, IH),
3.90-4.30 (m, 12H), 6.84-7.02 (m, 8H).
General Procedure for Preparation of Crown Ether Carboxylic Acids 94-96
After removal of the protecting mineral oil from NaH (1.43 g,
35.8 mmol, 60% dispersion in mineral oil) by washing with pentane
(2 X 30 mL) under nitrogen, the crown ether alcohol 91-93 (6.0
mmol) in 200 mL of THF was added dropwise. The mixture was
stirred for 30 min at room temperature, and then bromoacetic acid
(1.78 g, 12.8 mmol) in 20 mL of THF was added dropwise. The
reaction mixture was stirred for 3 d at toom temperature. Careful
addition of ice (to decompose the excess NaH) and then 50 mL of
water was followed by evaporation of the THF in vacuo. To the oily
residue was added 20 mL of CH2CI2 and the mixture was acidified to
pH 1 with 6N HCl. The organic layer was separated, washed with 20

88
mL of water, dried over MgS04, and evaporated in vacuo to give a
white solid which was recrystallized from ethyl acetate.
s y m - ( M e t h y l ) d i b e n z o - 1 6 - c r o w n - 5 -oxyacetic Acid (94)
White crystals with mp 100-102 °C were prepared in 80%
yield. IR (deposit): 3400-3000 (weak) (COOH); 1766, 1736 (C=0);
1122, 1053 (C-0) cm-i. IH NMR (CDCI3): 5 1.50 (s, 3H), 3.80-4.15 (m,
lOH), 4.60 (d, 2H), 4.85 (s, 2H), 6.84-7.02 (m, 8H), 8.88 (br s, IH).
s y m - ( H e x y l ) d i b e n z o - 1 6 - c r o w n - 5 -oxyacetic Acid (95)
White crystals with mp 135-136 °C were prepared in 72%
yield. IR (deposit): 3400-3000 (weak) (COOH); 1735, 1699 (C=0);
1122, 1053 (C-0) cm-i. m NMR (CDCI3): 5 0.93 (t, 3H), 1.25-1.50 (m,
8H), 1.93-1.97 (m, 2H), 3.80-4.15 (m, lOH), 4.60 (d, 2H), 4.85 (s, 2H),
6.84-7.02 (m, 8H), 9.90 (br s, IH).
s y m - ( D e c v l ) d i b e n z o - 1 6 - c r o w n - 5 -oxyacetic Acid (96)
White crystals with mp 101-102 °C were prepared in 78%
yield. IR (deposit): 3400-3000 (weak) (COOH); 1766, 1745 (C=0);
1124, 1058 (C-O) cm-i. ^H NMR (CDCI3): 6 0.89 (t, 3H), 1.25-1.50 (m,
18H), 1.93-1.97 (m, 2H), 3.80-4.15 (m, lOH), 4.60 (d, 2H), 4.85 (s,
2H), 6.84-7.02 (m, 8H).

89 Monoethyl s v m - D i b e n z o - 1 6 - c r o w n - 5 oxymethylphosphonic Acid (50)
Under nitrogen NaH (0.32 g, 8.0 mmol, 60% dispersion in
mineral oil) was washed with pentane (2 X 20 mL) to remove the
mineral oil and was suspended in 50 mL of THF. A solution of sym-
hydroxydibenzo-16-crown-5 (1.36 g, 3.0 mmol) in 30 mL of THF was
added and the mixture was stirred for 1 h. A solution of monoethyl
iodomethylphosphonic acid (1.0 g, 4.0 mmol) in 40 mL of THF was
added during 30 min followed by stirring at room temperature for 5
h and refluxing for 24 h. The reaction mixture was evaporated in
vacuo and 10 mL of water was added to the cooled reaction mixture
followed by addition of 6N HCl to pH 1. The mixture was extracted
with CH2CI2 (3 X 50 mL), dried over MgS04, and evaporated in vacuo.
The residue was purified by column chromatography on silica gel
with CH2CI2 and CH2Cl2:MeOH (1:1) as eluents. Evaporation of eluent
gave the salt of 50. Water (lOmL) was added to the salt. The
solution was acidified to pH 1 with concentrated HCl, extracted with
CH2CI2 (3 X 20 mL), and evaporated in vacuo to give 0.61 g (33%
yield) of 50 as a white crystalline solid with mp 91-92 °C (lit. mp 48-
52 °C). IR (deposit): 3472, 2292, 1707 (PO-H); 1253 (P=0); 1041,
990, 935 (POEt) cm-i. iH NMR (CDCI3): 5 1.35 (t, 3H), 3.90-4.40 (m,
17H), 6.81-7.02 (m, 8H), 7,80 (br s, IH). Anal. Calcd. for
C22H29O9PO.25 CH2CI2: C, 54.57; H, 6.07. Found: C, 54,82; H, 5.93.

9 0 General Procedure for Preparation of Crown Ether Phosphonic Acid Monoethyl Esters 47-49 and •^1-i^3
Under nitrogen, the sxin-dibenzo-16-crown-5-oxyalkyl
bromide (1.1 mmol) and triethyl phosphite (0.42 g, 2.5 mmol) were
stirred at 140 °C for 24 h. Excess triethyl phosphite was removed by
vacuum distillation and the residue was purified by column
chromatography on silica gel with CH2CI2 and CH2Cl2-MeOH (10:1) as
eluents. The resultant diethyl phosphonate (1.0 mmol) was refluxed
for 24 h or stirred for 7 d at room temperature with 0.25 g of NaOH
in 50 mL of EtOH. The solution was cooled to 5 °C and evaporated in
vacuo. The residue was acidified to pH 1 with 6N HCl, extracted with
CH2CI2 (3 X 30 mL), dried over MgS04, and evaporated in vacuo to
give the crude product which was purified by column
chromatography on silica gel with CH2Cl2-MeOH (1:1) as eluent.
Evaporation of the eluent gave the salt of 47-49 and 51-53 which
was acidified to pH 1 with concentrated HCl. The acqueous solution
was extracted with CH2CI2 (3 X 20 mL) to give 47-49 and 51-53.
Diethyl s v m - D i b e n z o - 1 6 - c r o w n - 5 -oxye thy lphosphonate (106)
A colorless oil was obtained in 90% yield. IR (neat): 1259
(P=0); 1041, 950 (POEt) cm-i. ^H NMR (CDCI3): 6 1.41 (t, 6H), 2.00-
2.72 (m, 2H), 3.69-4.40 (m, 19H), 6,81-7.02 (m, 8H).

91 Monoethyl s v m - D i b e n z o - 1 6 - c r o w n - 5 -oxyethyl-phosphonic Acid (51)
Hydrolysis of 106 for 7d at room temperature gave a white
solid with mp 55-57 °C in 40% yield. IR (deposit): 3427, 2237, 1697
(PO-H); 1257 (P=0); 1055, 990, 931 (POEt) cm-i. iH NMR (CDCI3): 5
1.32 (t, 3H), 2.08-2.30 (m, 2H), 3.87-4.35 (m, 17H), 6,81-7.02 (m, 8H),
9.25 (br s, IH). Anal. Calcd. for C23H31O9PO.5 H2O: C, 56.21; H, 6.56.
Found: C, 56.35; H, 6.67.
Diethyl s y m - D i b e n z o - 1 6 - c r o w n - 5 -oxypropy l -phosphonate (107)
A colorless oil was obtained in 82% yield. IR (neat): 1259
(P=0); 1041, 950 (POEt) cm-i. iH NMR (CDCI3): 6 1.41 (t, 6H), 1.92-
2.20 (m, 4H), 3.69-4.40 (m, 19H), 6,81-7.02 (m, 8H).
Monoethyl s y m - D i b e n z o - 1 6 - c r o w n - 5 -oxypropyl-phosphonic Acid (52)
Hydrolysis of 107 for 24 h at reflux gave a white solid with mp
101-103 °C in 53% yield. IR (deposit): 3427, 2237, 1697 (PO-H);
1257 (P=0); 1055, 990, 931 (POEt) cm-i. iH NMR (CDCI3): 8 1.32 (t,
3H), 1.80-2.20 (m, 4H), 3.87-4.35 (m, 17H), 6,81-7.02 (m, 8H). Anal.
Calcd. for C24H3309P-2H20: C, 54.13; H, 6.25. Found: C, 54.78; H, 6.41.
Diethyl s y m - D i b e n z o - 1 6 - c r o w n - 5 -oxybuty l -phosphonate (108)
A colorless oil was obtained in 94% yield. IR (neat): 1253
(P=0); 1039, 962 (POEt) cm-i. iH NMR (CDCI3): 5 1.41 (t, 6H), 1.92-
2.20 (m, 4H), 3.69-4.40 (m, 19H), 6,81-7.02 (m, 8H).

if:
9 2
Monoethyl &xiIL-l>ibenzo-16-crown-5-oxybutyl-phosphonic Acid (53)
Hydrolysis of 108 for 24 h at reflux gave a colorless oil in 55%
yield. IR (neat): 3354,2276,1658 (PO-H); 1261 (P=0); 1045, 977
(POEt) cm-i. iH NMR (CDCI3): 6 1.32 (t, 3H), 1.80-2.20 (m, 6H), 3.87-
4.35 (m, 17H), 6,81-7.02 (m, 8H). Anal. Calcd. for C25H35O9P: C,
58.81; H, 6.91. Found: C, 58.60; H, 6.81.
Diethyl iXQL-(Decyl ) d i b e n z o - 1 6 - c r o w n - 5 -oxyethy l -phosphonate (103)
A colorless oil was obtained in 90% yield. IR (neat): 1257
(P=0); 1043, 958 (POEt) cm-i. iH NMR (CDCI3): 5 1.41 (t, 3H), 1.22-
1.38 (m, 22H), 1.80-2.20 (m, 2H), 2.04-2.23 (m, 2H), 3.91-4.36 (m,
18H), 6.81-7.02 (m, 8H). Anal. Calcd. for C35H55O9P: C, 64.59; H, 8.52.
Found: C, 64.77; H, 8.62.
Monoethyl s y m - ( D e c y n d i b e n z o - 1 6 -crown-5-oxyethylphosphonic Acid (47)
Hydrolysis of 103 for 10 d at room temperature gave a
colorless oil in 29% yield. IR (neat): 3400, 2358, 1682 (PO-H); 1257
(P=0); 1046, 960 (POEt) cm-i. m NMR (CDCI3): 5 0.88 (t, 3H), 1.22-
1.38 (19H), 1.80-2.20 (m, 2H), 2.04-2.23 (m, 2H). 3.91-4.36 (m, 16H),
6.81-7.02 (m, 8H). Anal. Calcd. for C33H5iO9P-0.5H2O: C, 62.74; H,
8.14. Found: C, 62.78; H, 8.35.
Diethyl s y m - ( D e c v l ) d i b e n z o - 1 6 - c r o w n -5 -oxypropy l -phosphonate (104)
A colorless oil was obtained in 85% yield. IR (neat): 1257
(P=0); 1056, 958 (POEt) cm-i. m NMR (CDCI3): 5 0.88 (t, 3H), 1.22-

93
1.38 (m, 22H), 1.80-2.20 (m, 6H), 3.91-4.36 (m, 16H), 6,85-5.94 (m,
8H). Anal. Calcd. for C36H57O9PITHF: C, 65.19; H, 8.89. Found: C,
65.56; H, 8.97.
Monoethyl &XQL-(Decyl) dibenzo-16-crown-5-oxypropylphosphonic Acid (48)
Hydrolysis of 104 for 24 h at refluxing temperature gave a
colorless oil in 80% yield. IR (neat): 3520, 2330, 1684 (PO-H); 1257
(P=0); 1046, 985 (POEt) cm-i. iH NMR (CDCI3): 5 0.88 (t, 3H), 1.22-
1.38 (m, 19H), 1.80-2.20 (m, 6H), 3.91-4.36 (m, 16H), 6.81-7.02 (m,
8H). Anal. Calcd. for C34H5309P-1THF: C, 65.19; H, 8.89. Found: C,
65.56; H, 8.89.
Diethyl SJJQL-(Decyl)dibenzo-16-crown-5-oxybutyl-phosphonate (105)
A colorless oil was obtained in 95% yield. IR (neat): 1257
(P=0); 1029, 959 (POEt) cm-i. iH NMR (CDCI3): 6 0.88 (t, 3H), 1.22-
1.38 (m, 22H), 1.80-2.20 (m, 8H), 3.91-4.36 (m, 18H), 6,81-7.02 (m,
8H). Anal. Calcd. for C37H59O9P: C, 65.46; H, 8.76. Found: C, 65.01; H,
8.94.
Monoethyl sym-(Decy l )d ibenzo-16-crown-5-oxybutyl-phosphonic Acid (49)
Hydrolysis of 105 for 24 h at refluxing temperature gave a
colorless oil in 80% yield. IR (neat): 3500, 2299, 1693 (PO-H); 1257
(P=0); 1046, 985 (POEt) cm-i. IH NMR (CDCI3): 5 0.88 (t, 3H), 1.22-
1.38 (m, 19H), 1.80-2.20 (m, 8H), 3.91-4.36 (m, 16H), 6.81-7.02 (m,

94
8H). Anal. Calcd. for C35H55O9PO.5H2O: C, 63.71; H, 8.40. Found: C,
63.97; H, 8.58.
Dimethyl s y m - D i b e n z o - 1 6 - c r o w n - 5 -oxye thy l -phophonate (109)
Under nitrogen, sym.-dibenzo-16-crown-5-oxyethyl bromide
(0.45 g, 1.1 mmol) and trimethyl phosphite (0.31 g, 2.5 mmol) were
stirred at 140 °C for 24 h. Excess trimethyl phosphite was removed
by vacuum distillation and the residue was purified by column
chromatography on silica gel with CH2CI2 and CH2Cl2-MeOH (10:1) as
eluents to give 0.40 g (75% yield) of the product as a colorless oil. IR
(neat): 1259 (P=0); 1041, 950 (POMe) cm-i. iH NMR (CDCI3): 5 2.11-
2.32 (m, 2H), 3.70-4.52 (m, 21H), 6.81-7.02 (m, 8H). Anal. Calcd. for
C23H31O9P: C, 57.26; H, 6.48. Found: C, 57.59; H, 6.32.
Monomethyl s y m - D i b e n z o - 1 6 - c r o w n - 5 -oxyethyphosphonic Acid (110)
A solution of the dimethyl crown ether phosphonate 109 (0.40
g, 0.83 mmol) and NaOH (0.17 g, 4.15 mmol) in 15 mL of 95% EtOH
was stirred for 24 h. The solvent was removed in vacuo, and 5 mL of
water was added to the residue. The aqueous solution was acidified
to pH 1 with 6N HCl and extracted with CH2CI2 (3 X 20 mL). The
CH2CI2 solution was dried over MgS04 and evaporated in vacuo. The
residue was purified by column chromatography on silica gel with
CH2Cl2-MeOH (1:1) as eluent. Evaporation of the eluent gave a salt of
110 which was acidified to pH 1 with concentrated HCl. The aqueous
solution was extracted with CH2CI2 (3 X 20 mL). The CH2CI2 solution

95
was dried over MgS04 and evaporated in vacuo to give 0.21 g (54%
yield) of the product as white crystals with mp 100-102 °C. IR
(deposit): 3427, 2237, 1697 (PO-H); 1257 (P=0); 1055, 990, 931
(POMe) cm-i. iH NMR (CDCI3): 5 2.61-2.80 (m, 2H), 3.75-4.42 (m,
18H), 6.81-7.02 (m, 8H). Anal. Calcd. for C22H29O9P: C, 56.41; H, 6.24.
Found: C, 56.47; H, 6.39.
General Procedure for Preparation of Crown Ether Methanesulfonates 115-118
To a solution of the sxni-dibenzo-16-crown-5-oxyalkyl alcohol
(10 mmol) and triethylamine (1.01 g, 10 mmol) in 100 mL of CH2CI2
which was cooled by an ice-salt bath, methanesulfonyl chloride (1.15
g, 10 mmol) was added dropwise. After the addition was completed,
the reaction mixture was stirred for 30 min at 0-10 ^C. The reaction
mixture was washed with water (2 X 100 mL), dried over MgS04, and
evaporated in vacuo without heating to give the mesylate.
l - ( s y m - D i b e n z o - 1 6 - c r o w n - 5 - o x y ) - 2 -(methanesu l fonoxy)e thane (117)
A colorless oil was obtained in 98% yield. IR (neat): 1350,
1174 (SO2); 1259, 1126 (C-0) cm-i. iH NMR (CDCI3): 5 3.08 (s, 3H),
3.85-4.27 (m, 17H), 6.81-6.98 (m, 8H). Anal. Calcd. for C22H28O9S: C,
56.40; H, 6.02. Found: C, 56.33; H, 6.05.
l - ( s v m - D i b e n z o - 1 6 - c r o w n - 5 - o x y ) - 3 -(methanesu l fonoxy)propane (118)
A coloriess oil was obtained in 95% yield. IR (neat): 1361,
1176 (SO2); 1251, 1140 (C-0) cm-i. ^H NMR (CDCI3): 1.85-2.15 (m.

96
2H), 2.95 (s, 3H), 3.81-4.52 (m, 17H), 6.81-6.98 (m, 8H). Anal. Calcd.
for C23H30O9S: C, 57.25; H, 6.27. Found: C, 57.04; H, 6.10.
1-[SXIlL-(Decyl)d ibenzo -16 -crown -5 -oxy ] -2-(methane-sulfonoxy)ethane (115)
A coloriess oil was obtained in 93% yield. IR (neat): 1354,
1174 (SO2); 1257, 1123 cm-i. iH NMR (CDCI3): 6 0.89 (t, 3H), 1.20-
1.35 (m, 16H), 1.80-1.94 (m, 2H), 3.06 (s, 3H), 3.66-4.47 (m, 16H),
6.81-6.96 (m, 8H). Anal. Calcd. for C32H48O9S: C, 63.13; H, 7.95.
Found: C, 63.33; H, 7.98.
1-[SXUL-(Decyl) d ibenzo -16 -crown-5 -oxy ] -3-(methane-sulfonoxy)propane (116)
A colorless oil was obtained in 82% yield. IR (neat): 1356,
1175 (SO2); 1257, 1123 cm-i. IH NMR (CDCI3): 5 0.88 (t, 3H), 1.20-
1.35 (m, 16H), 1.82-1.95 (m, 2H), 1.95-2.08 (m, 2H), 2.89 (s, 3H),
3.89-4.41 (m, 16H), 6.82-6.96 (m, 8H). Anal. Calcd. for
C33H5o09S-lCH2Cl2: C, 55.23; H, 7.12. Found: C, 55.65; H, 7.29.
General Procedure for Preparation of Crown Ether Bromides 97. 98. 100. and 101
A solution of the £XQl"di^^"zo-16-crown-5-oxy(methane-
sulfonoxy)alkane (5.0 mmol) in 50 mL of acetone was added to
sodium bromide (2.0 g) in 150 mL of acetone. The reaction mixture
was refluxed for 3 d. After the acetone was evaporated in vacuo.
20 mL of water and 20 mL of CH2CI2 were added to the residue. The
CH2CI2 layer was separated, washed with water (2 X 40 mL), dried

97
over MgS04, and evaporated in vacuo to give the sxni-dibenzo-16-
crown-5-oxyalkyl bromide.
l - (5XQL-Dibenzo-16-crown-5-oxy)-2-bromoethane (100)
A white solid with mp 100-102 °C was prepared in 65% yield.
IR (deposit): 1259, 1126 (C-0) cm-i. iH NMR (CDCI3): 6 3.32-4.50
(m, 17H), 6.81-6.98 (m, 8H). Anal. Calcd. for C2iH2506Br: C, 55.64; H,
5.56. Found: C, 55.52; H, 5.63.
1 - (SXQL-D i b e n z o -16 - c r o w n - 5 - o X y) - 3-bromopropane (101)
A white solid with mp 94-95 °C was prepared in 85% yield. IR
(deposit): 1253, 1125 (C-0) cm-i. iH NMR (CDCI3): 6 1.85-2.15 (m,
2H), 3.80-4.52 (m, 17H), 6.81-6.98 (m, 8H). Anal. Calcd. for
C22H26O6Br0.5THF: C, 57.26; H, 6.21. Found: C, 57.13; H, 5.83.
1-[SXDI-(Decyl) d ibenzo-16 -crown-5 -oxy ] -2-bromoethane (97)
A colorless oil was prepared in 90% yield. IR (neat): 1257,
1122 (C-0) cm-i. m NMR (CDCI3): 6 0.89 (t, 3H), 1.20-1.35 (m, 16H),
1.98-2.10 (m, 2H), 3.25 (t, 2H), 3.94-4.41 (m, 14H), 6.81-6.98 (m, 8H).
Anal. Calcd. for C3iH4506Br: C, 62.72; H, 7.64. Found: C, 62.89; H,
7.82.
1 - [iXHL-(D e cy 1) d i b e n z o -16 - c r o wn - 5 - oxy ] -3-bromo-propane (98)
A colorless oil was prepared in 92% yield. IR (neat): 1257,
1126 (C-0) cm-i. iH NMR (CDCI3): 5 0.88 (t, 3H), 1.20-1.35 (m, 16H),

98
1.81-1.92 (m, 2H), 2.05-2.16 (m, 2H), 3.57 (t, 3H), 3.86-4.33 (m, 14H),
6.81-6.98 (m, 8H). Anal. Calcd. for C32H4706Br-2THF: C,63.89; H, 8.40.
Found: C, 64.12; H, 8.20.
General Procedure for Preparation of Crown Ether Bromides 99 and 102
A solution of 1,4-dibromobutane (10.36 g, 48 mmol) and the
crown ether alcohol (16 mmol) in 36 mL of CH2CI2 was stirred
vigorously with tetrabutylammonium hydrogen sulfate (0.27 g, 0.8
mmol) in 50% aqueous NaOH (12 mL). After 24 h (for 99) or 10 d
(for 102), 40 mL of water and 200 mL of CH2CI2 were added. The
CH2CI2 layer was separated, washed with water (3 X 200 mL), dried
over MgS04, and evaporated in vacuo. The resultant oily residue was
purified by chromatography on silica gel with CH2CI2 and then ethyl
acetate as eluents to give the sym-dibenzo-16-crown-5-oxyalkyl
bromide.
1 - ( s v m - D i b e n z o - 1 6 - c r o w n - 5 - o x y ) - 4 -bromobutane (99)
A coloriess oil was obtained in 76% yield. IR (neat): 1251,
1114 (C-0) cm-i. m NMR (CDCI3): 1.81-2.25 (m, 4H), 3.42-4.50 (m,
17H), 6.81-6.98 (m, 8H). Anal. Calcd. for C23H29O6Br0.5THF: C,58.03;
H, 6.43. Found: C, 57.99; H, 6.12.
1 - [ S J J H - ( D e c y l ) - d i b e n z o - 1 6 - c r o w n - 5 -oxy] -4 -bromo-butane (102)
A coloriess oil was obtained in 34% yield. IR (neat): 1257,
1140 (C-0) cm-i. iH NMR (CDCI3): 6 0.88 (t, 3H), 1.20-1.31 (m, 16H),

99
1.69-1.76 (m, 2H), 1.83-2.10 (m, 6H), 3.47 (t, 2H), 3.77-4.35 (m, 14H),
6.81-6.98 (m, 8H). Anal. Calcd. for C33H4906Br: C, 63.76; H, 7.95.
Found: C, 63.54; H, 8.04.
General Procedure for Preparation of Crown Ether Esters 119 and 120
The protecting mineral oil from NaH (1.44 g, 36 mmol, 60%
dispersion in mineral oil) was removed by washing with pentane
(2 X 20 mL) under nitrogen, and the deprotected NaH was suspended
in 100 mL of THF. A solution of the crown ether alcohol (11.6 mmol)
in 150 mL of THF was added dropwise. After a solution of ethyl
bromoacetate (2.32 g, 13.9 mmol) in 50 mL of THF was added
dropwise during 1 h, the mixture was stirred for an additional 6 h
and refluxed for 30 min. The reaction mixture was filtered and the
filtrate was evaporated in vacuo. The crude product was
recrystallized from EtOH.
Ethyl ( s x Q L - D i b e n z o - 1 6 - c r o w n - 5 - o x y ) -acetate (120)
A white solid with mp 44-46 ®C was prepared in 76% yield. IR
(neat): 1755 (C=0); 1259, 1136 (C-0) cm-i. iH NMR (CDCI3): 6 1.29 (t,
3H), 3.88-4.45 (m, 15H), 4.58 (s, 2H), 6.81-7.03 (m, 8H).
Ethyl [ s x n i - ( D e c y l ) d i b e n z o - 1 6 - c r o w n - 5 -oxy]acetate (119)
A white solid with 80-81 °C was prepared in 66% yield. IR
(deposit): 1758 (C=0); 1258, 1122 (C-0) cm-i. ^H NMR (CDCI3): 6 0.88

4
100
(t, 3H), 1.23-1.55 (m, 19H), 1.90-2.07 (m, 2H), 3.84-4.50(m, 14H),
4.75 (s, 2H), 6.81-7.03 (m, 8H).
General Procedure for Preparation of Crown Ether Alcohols 111 and 113
Under nitrogen, the crown ether ester (11.56 mmol) in 50 mL
of THF was added dropwise to a mixture of LiAlH4 (1.75 g, 46.24
mmol) and 90 mL of THF at refluxing temperature with stirring
during 1 h. The mixture was refluxed for an additional 4 h and
cooled to room temperature. The unreacted LiAlH4 in the mixture
was decomposed carefully by adding ethyl acetate dropwise. The
mixture was poured into a cold solution of dilute sulfuric acid and
extracted with CH2CI2 (2 X 100 mL). The CH2CI2 solution washed with
water (2 X 100 mL), dried over MgS04 and evaporated in vacuo. The
crude product was recrystallized from CHCI3 to give white crystals.
2 - ( s y m - D i b e n z o - 1 6 - c r o w n - 5 - o x v ) e t h a n o l ( 1 1 3 )
A white solid with mp 127-129 °C was obtained in 90% yield.
IR (deposit): 3398 (0-H); 1263, 1132 (C-0) cm-i. m NMR (CDCI3): 6
2.90 (br s, IH), 3.80-4.35 (m, 17H), 6.83-7.02 (m, 8H). Anal. Calcd.
for C21H26O7: C, 64.60; H, 6.71. Found: C, 64.66; H, 6.74.
2-[s y m - ( D e c vl) d i b e n z o - 1 6 - c r o w n - 5 -oxy]ethanol (111)
A colorless oil was obtained in 98% yield. IR (neat): 3278 (O-
H); 1219, 1125 (C-0) cm-i. iH NMR (CDCI3): 5 0.88 (t, 3H), 1.10-1.48
(m, 16H), 1.90-2.07 (m, 2H), 2.55 (br s, IH), 3.68-4.57 (m, 16H), 6.83-

101
7.02 (m, 8H). Anal. Calcd. for C31H46O7: C, 70.16; H, 8.74. Found: C,
70.57; H, 8.86.
General Procedure for Preparation of Allyoxy Crown Ethers 121 and 122
The protecting mineral oil from KH (1.15 g, 10 mmol, 35%
dispersion in mineral oil) was removed by washing with pentane
(2 X 30 mL) under nitrogen. THF (20 mL) was added to the powdery
KH and a solution of the crown ether alcohol (5.0 mmol) in 20 mL of
THF was added dropwise. After the mixture was stirred for 1 h, a
solution of allyl bromide (1.73 g, 6.0 mmol) in 10 mL of THF was
added dropwise. The reaction mixture was stirred at room
temperature for 6 h and filtered. The filtrate was evaporated rn
vacuo., and the residue was purified by column chromatography on
silica gel with CH2CI2 as eluent to give the allyoxy crown ether.
3 - ( s v m - D i b e n z o - 1 6 - c r o w n - 5 - o x y ) - l -propene (122)
A white solid with mp 108-110 °C was obtained in 96% yield.
IR (deposit): 1290, 1129 (C-0) cm-i. iH NMR (CDCI3): 6 3.85-4.41 (m,
15H), 5.13-5.42 (m, 2H), 5.88-6.05 (m, IH), 6.82-7.06 (m, 8H).
3 - [ S X D L - ( D e c y l ) - d i b e n z o - 1 6 - c r o w n - 5 -o x y ] - l - p r o p e n e (121)
A coloriess oil was obtained in 98% yield. IR (neat): 1225,
1140 (C-0) cm-i. m NMR (CDCI3): 5 0.88 (t, 3H), 1.15-1.58 (m, 16H),
1.90-2.02 (m, 2H), 3.86-4.50 (m, 14H), 5.13-5.42 (m, 2H), 5.88-6.05

^
102
(m, IH), 6.82-7.06 (m, 8H). Anal. Calcd. for C32H46O6: C, 72.97; H,
8.80. Found: C, 72.99; H, 8.88.
General Procedure for Preparation of Crown Ether Alcohols 112 and 114
A solution of the allyloxy crown ether (13 mmol) in 120 mL of
THF was added dropwise to NaBH4 (0.50 g, 13 mmol) in 30 mL of THF
dropwise. Boron trifluoride etherate (2.25 mL) was added dropwise
at 0 °C, and the reaction mixture was stirred at this temperature for
2 h. Water (1.5 mL) was added to destroy the excess NaBH4. After
the addition of 7.5 mL of 2N aqueous NaOH, 9 mL of 30% H2O2 was
slowly added, and the reaction mixture was stirred overnight at
room temperature. The THF was evaporated in vacuo. The crude
product was extracted with CH2CI2 (2 X 50 mL) and dried over
MgS04. Evaporation of the CH2CI2 gave crown ether alcohol.
3 - ( s v m - D i b e n z o - 1 6 - c r o w n - 5 - o x y ) p r o p a n -l-ol (114)
A white solid with mp 82-83 °C was obtained in 26% yield. IR
(deposit): 3385 (OH); 1257, 1124 (C-0) cm-i. H NMR (CDCI3): 5
1.68-2.15 (m, 2H), 2.90 (br s, IH), 3.80-4.23 (m, 17H), 6.83-7.02 (m,
8H). Anal. Calcd. for C22H28O7: C, 65.33; H, 6.98. Found: C, 65.12; H,
6.74.
3 - [ iXiS-" (D e c y 1) d i b e n z o -16 - c r o w n - 5-oxy]propan-l-ol (112)
A colorless oil was obtained in 91% yield. IR (neat): 3314 (O-
H); 1256, 1129 (C-0) cm-^. iH NMR (CDCI3): 5 0.88 (t, 3H), 5 1.15-1.58

103
(m, 16H), 1.68-2.20 (m, 4H), 3.74-4.51 (m, 16H), 6.83-7.02 (m, 8H).
Anal. Calcd. for C32H48O7I.25H2O: C, 67.76; H, 8.53. Found: C, 67.80;
H, 8.68.
10-Methanesulfonoxy-l -decene (133)
Methanesulfonyl chloride (3.45 g, 0.03 mol) was added to a
solution of 9-decen-l-ol (5.0 g, 0.03 mol) and triethylamine (3.04 g,
0.03 mol) in 100 mL of CH2CI2 in an ice-salt bath. The reaction
mixture was stirred for an additional 1 h at room temperature and
washed with water (2 X 50 mL). The CH2CI2 solution was dried over
M g S 0 4 and evaporated in vacuo to give 6.64 g (89% yield) of the
mesylate. IR (neat): 1640 (C=C); 1356, 1176 (SO2) cm-i. ^H NMR
(CDCI3): 5 1.23-1.48 (m, lOH), 1.65-2.21 (m, 4H), 3.01 (s, 3H), 4.23 (t,
2H), 4.90-5.18 (m, 2H), 5.71-5.92 (m, IH).
10-Bromo-l-decene (134)
A solution of 10-methane-sulfonoxy-l-decene (6.50 g, 0.026
mol) and sodium bromide (6.69 g, 0.065 mol) in 20 mL of acetone
was refluxed for 5 d. After the mixture was cooled to room
temperature, the acetone was evaporated in vacuo. Then CH2CI2 (50
mL) and water (50 mL) were added to the residue. The CH2CI2 layer
was separated, washed with water (2 X 50 mL), dried over MgS04
and evaporated in vacuo to giye an oily residue which was purified
by column chromatography on silica gel with petroleum ether as
eluent. The product was obtained in 69% yield (3.92 g) as a colorless
oil. IR (neat): 1640 (C=C); 1254, 1126 (C-0) cm-i. iH NMR (CDCI3): 5

•Sf
104
1.23-1.48 (m, lOH), 1.65-2.21 (m, 4H), 3.41 (t, 2H), 4.90-5.18 (m, 2H),
5.71-5.92 (m, IH).
General Procedure for Preparation of Crown Ether Alcohols 124 and 125
To 0.26 g of magnesium turnings under nitrogen was added 30
mL of THF and 10-bromo-l-decene (2.30 g, 10.5 mmol). The mixture
was refluxed for 3 h, and the crown ether ketone (4.78 mmol) in 30
mL of THF was added dropwise at room temperature. The reaction
mixture was refluxed for 3 h. After 30 mL of 5% aqueous NH4CI was
added, the reaction mixture was stirred for 2 h. The THF was
evaporated in vacuo and the residue was extracted with CH2CI2. The
CH2CI2 solution was dried over MgS04 and evaporated in vacuo. The
resultant oil was purified by column chromatography on silica gel
with CH2Cl2and CH2Cl2-Et20 (1:1) as eluents to give the product.
s y m - ( 9 - D e c e n v l ) ( h y d r o x y ) d i b e n z o -14-crown-4 (124)
A white solid with mp 64-65 °C was prepared in 74% yield. IR
(deposit): 3375 (0-H); 1640 (C=C); 1254, 1120 (C-0) cm-i. ^H NMR
(CDCI3): 5 1.32-1.71 (m, 14H), 1.90-2.11 (m, 2H), 2.28-2.37 (m, 2H),
3.60 (br s, IH), 4.01-4.30 (m, 8H), 4.90-5.04 (m, 2H), 5.70-5.90 (m,
IH), 6.89-7.00 (m, 8H). Anal. Calcd. for C28H38O5I.25H2O: C, 73.25;
H, 8.45. Found: C, 73.40; H, 8.41.

105 &XQL-(9-Decenyl) ( h y d r o x y ) d i b e n z o -16-crown-5 (125)
A white solid with mp 82-83 °C was prepared in 56% yield. IR
(deposit): 3462 (0-H); 1640 (C-C); 1263, 1046 (C-0) cm-i. ^H NMR
(CDCI3): 5 1.32-2.05 (m, 16H), 3.26 (br s, IH), 3.90-4.26 (m, 12H),
4.90-5.04 (m, 2H), 5.70-5.90 (m, IH), 6.89-7.00 (m, 8H). Anal. Calcd.
for C29H40O6: C, 71.87; H, 8.32. Found: C, 71.88; H, 8.37.
General Procedure for Preparation of Crown Ether Carboxylic Acids 135 and 136
The protecting mineral oil from NaH (1.26 g, 31.5 mmol, 60%
dispersion in mineral oil) was removed by washing with pentane
(2 X 20 mL) and 20 mL of THF was added to the powdery NaH. The
crown ether alcohol (5.24 mmol) in 20 mL of THF was added
dropwise, and the mixture was stirred for 1 h. After a solution of
bromoacetic acid (1.46 g, 10.5 mmol) in 20 mL of THF was added
dropwise, the reaction mixture was stirred overnight at room
temperature and then refluxed for 6 h. After cooling, 40 mL of
water was added carefully to the reaction mixture to destroy the
excess NaH. The THF was evaporated in vacuo and the residue was
acidified to pH 1 with 6N HCl. The aqueous solution was extracted
with CH2CI2 (2 X 60 mL). The CH2CI2 solution was dried over MgS04
and evaporated in vacuo to give the product.
s y m - ( 9 - D e c e n y l ) d i b e n z o - 1 4 - c r o w n - 4 -oxyacetic Acid (135)
A colorless oil was prepared in 92% yield. IR (neat): 3450-
2600 (COOH); 1744 (C=0); 1251, 1120 (C-0) cm^. m NMR (CDCI3): 6

106
1.32-2.05 (m, 16H), 2.15-2.55 (m, 2H), 4.01-4.30 (m, 8H), 4.45 (s,
2H), 4.90-5.04 (m, 2H), 5.70-5.90 (m, IH), 6.89-7.00 (m, 8H). Anal.
Calcd. for C30H40O7: C, 70.29; H, 7.87. Found: C, 69.98; H, 8.01.
SXQL-(9 - D e c e n y 1) d i b e n z o -16 - c r o w n - 5-oxyacetic Acid (136)
A colorless oil was prepared in 97% yield. IR (deposit): 3357-
2600 (COOH); 1746 (C=0); 1256, 1154 (CO) cm-'. iH NMR (CDCI3): 6
1.32-2.05 (m, 16H), 3.90-4.26 (m, lOH), 4.56-4.62 (d, 2H), 4.85 (s.
2H), 4.90-5.04 (m,lH), 6.89-7.00 (m, 8H). Anal. Calcd. for C31H42O8:
C, 68.61; H, 7.80. Found: C, 68.51; H, 7.51.
General Procedure for Preparation of Crown Ether Esters 123 and 124
A solution of the crown ether carboxylic acid (5.53 mmol) and
2 drops of concentrated H2SO4 in 200 mL of absolute EtOH was
refluxed for 24 h. The water produced during the reaction was
removed by use of a Soxhlet thimble filled with anhydrous Na2S04.
After cooling, Na2C03 was added to neutralize the acid catalyst. EtOH
was evaporated in vacuo. The crude product was recrystallized from
EtOH.
Ethyl s y m - ( 9 - D e c e n y l ) d i b e n z o -1 4 - c r o w n - 4 - o x y a c e t a t e (123)
A colorless oil was obtained in 78% yield. IR (neat): 1757
(C=0); 1640 (C=C); 1256, 1119 (C-0) cm-i. iH NMR (CDCI3): 6 1.32-
2.05 (m, 19H), 2.15-2.55 (m, 2H), 4.01-4.30 (m, 8H), 4.49 (s, 2H),

107
4.90-5.04 (m, 2H), 5.70-5.90 (m, IH), 6.89-7.00 (m, 8H). Anal. Calcd.
forC32H4407: C, 71.08; H, 8.20. Found: C, 71.52; H, 8.11.
Ethyl sxiIL-(9-Decenyl)dibenzo-16-crown-5-oxyacetate (124)
A white solid with mp 85-86 °C was obtained in 90% yield. IR
(deposit): 1757 (C=0); 1640 (C=C); 1256, 1123 (C-0) cm-i. iH NMR
(CDCI3): 5 1.16-2.05 (m, 19H), 3.90-4.49 (m, 14H), 4.75 (s, 2H), 4.90-
5.04 (m, 2H), 5.70-5.90 (m, IH), 6.89-7.00 (m, 8H). Anal. Calcd. for
C33H46O8: C, 69.45; H, 8.13. Found: C, 69.50; H, 8.02.
General Procedure for Preparation of Crown Ether Esters 138 and 13 9
To the unsaturated crown ether ester (1.0 mmol) was added
0.5 M BH3.THF (0.70 mL, 0.35 mmol) at 0 °C. After the reaction
mixture had been stirred for 2 h at this temperature and 2 h at room
temperature, IN NaOH (0.21 mL) and 30% H2O2 (0.14 mL) were
added. The reaction mixture was extracted with CH2CI2. The CH2CI2
solution was dried over MgS04 and evaporated in vacuo to give the
product .
Ethyl s y m - ( 1 0 - H y d r o x y d e c y l ) d i b e n z o -14-crown-4-oxyace ta te (138)
A coloriess oil was obtained in 77% yield. IR (neat): 3412 (O-
H); 1754 (C=0); 1256, 1119 (C-0) cm-i. m NMR (CDCI3): 5 1.32-2.05
(m, 19H), 2.15-2.55 (m, 2H), 3.63 (t, 2H), 4.01-4.30 (m, lOH), 4.49 (s,
2H), 6.89-7.00 (m, 8H). Anal. Calcd. for C32H46O8-0.17CH2C12: C,
67.45; H, 8.16. Found: C, 67.63; H, 8.16.

108 Ethyl &xi lL- (10-Hydroxydecy l )d ibenzo-16-crown -5 -oxy a c e t a t e (139)
A colorless oil was obtained in 86% yield. IR (neat): 3412 (O-
H); 1754 (C=0); 1256, 1124 (C-0) cm-i. IH NMR (CDCI3): 6 1.16-2.05
(m, 21H), 3.64 (t, 2H), 4.01-4.49 (m, 14H), 4.75 (s, 2H), 6.89-7.00 (m,
8H). Anal. Calcd. for C32H46O8: C, 67.32; H, 8.23. Found: C, 67.54; H,
8.32.
1 1 - M e t h a n e s u l f o n o x y - l - u n d e c e n e (140)
A solution of methanesulfonyl chloride (11.31 g, 0.099 mol) in
75 mL of CH2CI2 was added to a cooled mixture of 10-undecen-l-ol
(13.50 g, 0.079 mol) and triethylamine (12.79 g, 0.13 mol) in 75 mL
of CH2CI2. The reaction mixture was stirred for 1 h at 0-10 "C and
washed with water (2 X 100 mL). The CH2CI2 solution was dried over
M g S 0 4 and evaporated in vacuo to give 19.50 g (99.5% yield) of the
product as a colorless oil. IR (neat): 1640 (C=C); 1356, 1177 (SO2)
cm-i. IH NMR (CDCI3): 5 1.20-1.48 (m, 12H), 1.68-1.85 (m, 2H), 1.48-
2.15 (m, 2H), 3.00 (s, 3H), 4.22 (t, 2H), 4.89-5.07 (m, 2H), 5.68-5.95
(m, IH).
2 - H y d r o x y - 4 - ( 1 0 * - u n d e c e n o x y ) b e n z o i c Acid (141)
Potassium metal (8.89 g, 0.227 mol) was added to 600 mL of
absolute EtOH, and the mixture was stirred until the potassium metal
disappeared. A solution of 2,4-dihydroxybenzoic acid (12.86 g, 0.083
mol) in 100 mL of EtOH was added, and the mixture was refluxed for
2 h. After a solution of mesylate 140 (18.80 g, 0.0758 mol) in 50 ml
of EtOH was added, the reaction mixture was refluxed for 6 d. The

109
solvent was evaporated in vacuo. The residue was dissolved in 50
mL of water, acidified to pH 1 with 6N HCl and extracted with CH2CI2
(2 X 50 mL). The combined extracts were washed with 50 mL of
water, dried over MgS04, and evaporated in vacuo. The crude
product was recrystallized from petroleum ether and a minimum
amount of CH2CI2 to give 8.15 g (35% yield) of the product as a solid
with mp 110-112 °C. IR (deposit): 3100-3050 (COOH); 1644, 1622
(C=0); 1242, 1176 (C-0) cm-i. iH NMR (CDCI3): 6 1.18-1.53 (m, 12H),
1.65-1.85 (m, 2H), 1.93-2.15 (m, 2H), 3.99 (t, 2H), 4.85-5.05 (m, 2H),
5.65-5.95 (m, IH), 6.35-6.50 (m, 2H), 7.79 (m, IH), 10.13 (br s, IH).
Methyl 2-Hydroxy-4-(10*-undecenoxy)-benzoate (142)
A solution of the benzoic acid 141 (5.61 g, 18.3 mmol) and 2 g
of concentrated sulfuric acid in 200 mL of CH3OH was refluxed for 48
h. The reaction mixture was cooled, and the solvent was removed in
vacuo. The residue was poured into ice-water (30 mL) and extracted
with diethyl ether (2 X 50 mL). The combined extracts were washed
with saturated aqueous NaHC03 (50 mL) and then with 50 mL of
water. The ether solution was dried over MgS04 and evaporated rn
vacuo to give 4.67 g (80% yield) of the product as a colorless oil. IR
(neat): 3077 (0-H); 1668, 1623 (C=0); 1224, 1191 (C-0) cm-i. iH
NMR (CDCI3): 5 1.18-1.53 (m, 12H), 1.65-1.85 (m, 2H), 1.93-2.15 (m,
2H), 3.82-4.04 (m, 5H), 4.85-5.05 (m, 2H), 5.65-5.95 (m, IH), 6.35-
6.50 (m, 2H), 7.65-7.78 (m, IH), 10.96 (s, IH).

I 10 General Procedure for Preparation of Methyl Esters 125-128
The protecting mineral oil from NaH (0.30 g, 7.56 mmol, 60%
dispersion in mineral oil) was removed by washing with pentane (2
X 20 mL) under nitrogen. THF (20 mL) was added to the powdery
NaH, and a solution of phenolic ester 142 (6.05 mmol) in 10 mL of
THF was added dropwise. After 1 h, a solution of the crown ether
tosylate (6.05 mmol) in 10 mL of THF was added dropwise. The
reaction mixture was refluxed for 3 d and filtered. The filtrate was
evaporated in vacuo, and the residue was purified by column
chromatography on silica gel with CH2CI2 and then ethyl acetate as
eluents to give the product.
Methyl 2-[(12-Crown-4)methyloxyJ-4-(lO'-undecenoxy)-benzoate (125)
A white solid with mp 52-53 °C was prepared in 36% yield. IR
(deposit): 1727, 1700 (C=0); 1640 (C=C); 1246, 1192 (C-0) cm-i. m
NMR (CDCI3): 5 1.18-1.53 (m, 12H), 1.65-1.85 (m, 2H), 1.93-2.15 (m,
2H), 3.56-4.18 (m, 22H), 4.85-5.05 (m, 2H), 5.65-5.95 (m, IH), 6.35-
6.50 (m, 2H), 7.80-7.89 (m, IH). Anal. Calcd. for C28H44O8: C, 66.11;
H, 8.72. Found: C, 66.16; H, 8.47.
Methyl 2-[(15-Crown-5)methyloxy]-4-(lO'-undecenoxy)benzoate (126)
A colorless oil was prepared in 34% yield. IR (neat):
1726,1699 (C=0); 1640 (C=C); 1251, 1193 (C-0) cm-i. ^H NMR (CDCI3):
5 1.18-1.53 (m, 12H), 1.65-1.85 (m, 2H), 1.93-2.15 (m, 2H), 3.56-4.18
(m, 26H), 4.85-5.05 (m, 2H), 5.65-5.95 (m, IH), 6.35-6.50 (m, 2H),

111
7.80-7.89 (m, IH). Anal. Calcd. for C30H48O9: C, 65.19; H, 8.75.
Found: C, 65.33; H, 8.58.
Methyl 2 - [ (18 -Crown-6 )methy loxy ] -4 -( lO' -undecenoxy)benzoate (127)
A colorless oil was prepared in 42% yield. IR (neat): 1726,
1700 (C=0); 1640 (C=C); 1249, 1194 (C-0) cm-i. iH NMR (CDCI3): 6
1.18-1.53 (m, 12H), 1.65-1.85 (m, 2H), 1.93-2.15 (m, 2H), 3.56-4.18
(m, 30H), 4.85-5.05 (m, 2H), 5.65-5.95 (m, IH), 6.35-6.50 (m, 2H),
7.80-7.89 (m, IH). Anal. Calcd. for C32H52O10O.2CH2CI2: C, 62.01; H,
8.61. Found: C, 63.19; H, 8.42.
Methyl 2 - [ (21 -Crown-7 )methy loxy] -4 -( lO' -undecenoxy)benzoate (128)
A colorless oil was prepared in 34% yield. IR (neat): 1725,
1700 (C=0); 1640 (C=C); 1251, 1193 (C-0) cm-i. 'H NMR (CDCI3): 6
1.18-1.53 (m, 12H), 1.65-1.85 (m, 2H), 1.93-2.15 (m, 2H), 3.56-4.18
(m, 34H), 4.85-5.05 (m, 2H), 5.65-5.95 (m, IH), 6.35-6.50 (m, 2H),
7.80-7.89 (m, IH). Anal. Calcd. for C34H56O11: C, 63.72; H, 8.81.
Found: C, 63.77; H, 9.01.
Preparation of Alkali Metal Picrates
Alkali metal picrates were prepared by dissolving picric acid in
a minimum amount of boiling distilled, deionized water and slowly
adding a stoichiometric amount of the alkali metal carbonate. After
allowing the solution to cool to room temperature, it was cooled in an
ice bath to promote crystallization. Crystals were collected, air-dried,
and recrystallized from distilled, deionized water. The recrystallized

1 12
alkali metal picrates were collected, air-dried, and dried in a vacuum
oven at 100 °C for 4 h. The dry alkali metal picrates were stored
under vacuum in the dark.
Preparation of Alkylammonium Picrates
The alkylamine was added to a saturated solution of picric acid
in distilled, deionized water. A precipitate was formed as the
alkylamine was added. The reaction mixture was filtered to give the
alkylammonium picrate as a yellow solid which was recrystallized
from distilled, deionized water.
Preparation of N.N-Didecyl-7.16-diaza-18-crown-6 (185)
Decanoyl Chloride (183)
Fresh distilled thionyl chloride (20 mL) was added to decanoic
acid (7.30 g, 42 mmol). The reaction mixture was heated slowly and
refluxed for 5 h. Excess thionyl chloride was evaporated in vacuo to
give 7.69 g of the product (96% yield) as a colorless oil. IR (neat):
1801 (C=0); cm-i. ^H NMR (CDCI3): 6 0.88 (t, 3H), 1.19-1.35 (m, 12H),
1.65-1.76 (m, 2H), 2.88 (t, 2H).
N , N - D i d e c a n o y l - 7 , 1 6 - d i a z a - 1 8 - c r o w n - 6 ( 1 8 4 )
A solution of decanoyl chloride (1.60 g, 8.38 mmol) in 15 mL of
THF was added dropwise to a stirred solution of 7,16-diaza-18-
crown-6 (1.00 g, 3.81 mmol) and triethylamine (0.93 g, 9.14 mmol)
in 30 mL of THF under nitrogen at 50 °C. After the reaction mixture
was stirred for 1 h at this temperature, it was filtered and

113
evaporated in vacuo. The crude product was recrystallized from
petroleum ether to give 1.82 g of the product (84% yield) as a white
solid with mp 64-65 °C. IR (deposit): 1639 (C=:0); 1250, 1120 (C-0)
cm-i. iH NMR (CDCI3): 6 0.88 (t, 6H), 1.19-1.35 (m, 2H), 1.63 (t, 4H),
2.33 (t, 4H), 3.49-3.75 (m, 24H). Anal. Calcd. for
C32H62O6N2-0.1CH2C12: C, 66.67; H, 10.84. Found: C, 66.88; H, 10.97.
N , N - D i d e c y l - 7 , 1 6 - d i a z a - 1 8 - c r o w n - 6 ( 1 8 5 )
To a solution of compound 184 (1.14 g, 2.0 mmol) in 10 mL of
THF was added 8.0 mL of 2M BH3-SMe2 complex. After the reaction
mixture was refluxed for 9 h, 5 mL of water was added slowly and
the solvent was evaporated in vacuo. The residue was treated with
6N HCl (10 mL) and water (10 mL). The resultant solution was
refluxed for 12 h, then aqueous ammonium hydroxide was added to
adjust the pH to 10. The aqueous solution was extracted with CH2CI2
(3 X 50 mL), dried over MgS04 and evaporated in vacuo. The crude
product was purified by column chromatography on alumina with
CH2Cl2-MeOH (1:1) as eluent to give 0.79 g of the product (73% yield)
as a colorless oil. IR (neat): 1250, 1178 (C-0) cm-i. 'H NMR (CDCI3):
5 0.88 (t, 6H), 1.12-1.38 (m, 32H), 2.48 (t, 4H), 2.78 (t, 8H), 3.51-3.72
(m, 16H). Anal. Calcd. for C32H66O4N2-0.13CH2C12: C, 69.67; H, 12.06.
Found: C, 69.85; H, 12.30.
Procedure for Picrate Extractions
Crown ether solutions (5.0 mM) were prepared in ethanol-free
deutriochloroform. Extraction experiments were conducted by

1 14
adding 0.50 mL of a 5.0 mM crown ether solution in
deuteriochloroform to 0.50 mL of 5.0 mM picrate solution in a
centrifuge tube and agitating the mixture with a vortex mixer for 1
min. Five identical samples were run concurrently. The mixture
were centrifuged for 10 min to assure complete separation of the
layers. Precisely measured aliquots were removed from each layer
with microsyringes and diluted in acetonitrile. Visible spectra of
these solutions were measured in the region 300-500 nm. The
absorbance at absorption maximum (375 nm) was measured and
compared with that for a known concentration of the alkali picrate.
From the absorbance values, the percent extraction was calculated.
From the percent extraction values, the log Kex value was calculated
using a computer program written by David A. Babb.[64]

REFERENCES
1. Pedersen, C. J. J. Am. Chem. Soc. 1967,89,7017.
2. Pedersen, C. J. Aldrichimica Acta 1967, 4, 1.
3. Pedersen, C. J. J. Am. Chem. Soc. 1967, 89, 2495.
4. Pedersen, C. J. Organic Synthesis 1972, 51, 66.
5. Mandolini, L.; Masci, B. J. Am. Chem. Soc. 1984. 106. 168.
6. Cook, F. L.; Caruson, T. C; Byrne, M. P.; Bowers, C. W.; Speck, D. H.; Liotta, C. L. Tetrahedron Lett. 1974, 4029.
7. Piepers, O.; Kellogg, R. M. J. Chem. Soc. Chem. Commun. 1978, 383.
8. Pedersen, C. J. J. Am. Chem. Soc. 1969, 91, 386.
9. Gokel, G. W.; Cram, D. J. J. Chem. Soc. Chem. Commun. 1973, 481.
10. Beckford, H. F.; King, R. F.; Stoddart, J. F.; Newton, R. F. Tetrahedron Lett. 1978, 171.
11. Larson, J. M.; Sousa, L. R. J. Am. Chem. Soc. 1978,100, 1943.
12. Shannon, R. D.; Prewitt, C. T. Acta Crvst. 1969, B15, 925.
13. Shannon, R. D. Acta Crvst. 1976, A31, 751.
14. Weber, E.; Vogtle, F. Host Guest Chemistry Macrocvcles: Springer-Verlag, 1985; p 18.
15. Dunitz, J. D.; Dobler, M.; Seller, P.; Phizackerley, R. P. Acta Cryst. 1974, B30. 2733.
16. Dunitz, J. D.; Seller, P. Acta Crvst. 1974, BM, 2739.
17. Wong, K. H.; Bourgoin, M.; Smid, J. .1. Chem. Soc, Chem. Commun. 1974, 715.
1 15

116
18. Bourgoin, M.; Wong, K. H.; Hui, J. Y.; Smid, J. J. Am. Chem. Soc. 1975, 9 1 , 3462.
19. Sadkane, A.; Iwachido, T.; Toei, K. Bull. Chem. Soc. Jpn. 1975, 48.
20. Nakamura, H.; Nishido,H.; Tadagi, M.; Ueno, K. Anal. Chim. Acta. 1982, 139., 219.
21. Newcomb, M.; Cram, D. J. J. Am. Chem. Soc. 1975, 97, 1257.
22. Wierenga, W.; Evans, B. R.; Woltersom, J. A. J. Am. Chem. Soc. 1979, M l , 1334.
23. Helgeson, R. C ; Timko, J. M.; Cram, D. J. J. Am. Chem. Soc. 1973, 95., 3023.
24. Goldberg, 1. Acta Crvst. 1975, B31. 2592.
25. Nakamura, H.; Takagi, M.; Ueno, K. Anal. Chem. 1980, 52, 1668.
26. Bartsch, R. A.; Heo, G. S.; Kang, S. 1.; Liu, Y.; Strzelbicki, J. J- Org. Chem. 1982, 47, 257.
27. Strzelbicki, J.; Bartsch, R. A. Anal. Chem. 1981, 51 , 1894.
28. Strzelbicki, J.; Heo, G. S.; Bartsch, R. A. Sep. Sci. Technol. 1982, LT., 635 .
29. Strzelbicki, J.; Bartsch, R. A. Anal. Chem. 1981, 53, 2251.
30. Czech, B.; Son, B.; Bartsch, R. A. Tetrahedron Lett. 1983, 24, 2923.
31. Czech, B.; Czech, A.; Son, B.; Lee, H. K.; Bartsch, R. A. L Heterocyclic Chem. 1986, 2 i , 465.
32. Bartsch, R. A. and co-workers, unpublished results, 1991.
33. Pugia, M. J.; Ndip. G.; Lee, H. K.; Yang, I. W.; Bartsch, R. A. Anal. Chem. 1986, 58., 2723.
34. Charewicz, W. A.; Bartsch, R. A. Anal. Chem. 1982, 52, 1668.

1 17
35. Charewicz, W. A.; Heo, G. S.; Bartsch, R. A. Anal. Chem. 1982, 54, 2094 .
36. Charewicz, W. A.; Walkowiak, W.; Bartsch, R. A. Anal. Chem. 1987, 59., 494.
37. Bartsch, R. A.; Charewicz, W. A.; Kang, S. 1.; Walkowiak, W. Liquid Membranes: Theory and Applications: ACS Symposium Series 347; American Chemical Society: Washington, D. C , 1987, p 86.
38. Bradshaw, J. S.; Bruening, R. L.; Krakowiak, K. E., Tarbet, B. J.; Bruening, M. L.; Izatt, R. M.; Christensen, J. J. J. Chem. Soc. Chem. Commun. 1988, 812.
39. Izatt, R. M.; Bradshaw, J. S.; Nielsen, S. A.; Lamb, J. D.; Christensen, 1 J. Pure Appl. Chem. 1985, 85, 271.
40. Bradshaw, J. S.; Nielsen, R. B.; Tse, R. K.; Arena, G.; Wilson, B. E.; Dalley, N. K.; Lamb, J. D. J. Heterocyclic Chem. 1986, 21, 361.
41. Bartsch, R. A. Sol. Extr. Ion Exch. 1989, 7, 829.
42 . Cho, M. H.; Bartsch, R. A., unpublished results,1991.
43. Provided by J. Krzykawski, T. W. Robinson, and J. L. Hallman, Texas Tech University.
44. Miyazaki, T.; Yanagida, S.; Itoh, A.; Okahara, M. Bull. Chem. Soc, Jpn. 1982, 55, 2005.
45. Golding, B. T.; loannou, P. V. Synthesis 1977, 423.
46. Cho, M. H.; Ramash, V.; Bartsch, R. A., unpublished results,1991.
47. Strzelbicki, J.; Bartsch, R. A. L Membr. Sci. 1982, 10, 35.
48. Strzelbicki, J.; Bartsch, R. A. T. Membr. Sci. 1983, 12, 323.
49. Hayashita, T.; Goo, M.-J.; Lee, J. C ; Kim, J. S.; Krzykawski, J.; Bartsch, R. A. Anal. Chem. 1990, 62, 2283.

118
50. Fieser, L. F.; Fieser, M. Reagents for Organic Synthesis: Wiley: New York, 1967; Vol. 1, p. 142.
51. Eastman, M. P.; Patterson, D. E.; Bartsch, R. A.; Liu, Y.; Filer, P. G. L Phvs. Chem. 1982, 86, 2052.
52. Ford-Moore, A. H.; Williams, J. H. J. Am. Chem. Soc. 1947, 69, 1465 .
53. Myers, T. C ; Preis, S.; Jensen, E. V. J. Am. Chem. Soc. 1954, 76. 4172 .
54. Walkowiak, W.; Bartsch, R.A., unpublished results, 1991.
55. Blasius, E.; Janzen, K.-P.; Adrian, W.; Klautke, G.; Lorscheider, R.; Maurer, P.-G.; Nguyen, V.P.; Nguyen, T. T.; Scholten, G.; Stockemer, J. Z. Anal. Chem. 1977, 284, 337.
56. Brown, H. C ; Keblys, K. A. J. Am. Chem. Soc. 1964, 86, 1795.
57. Provided by J. McDonough, Texas Tech University, 1990.
58. Provided by B. P. Czech, Technicon Instruments Corporation, 1990.
59. Provided by J. C. Lee, Texas Tech University, 1990.
60. Olsher, U.; Dalley, N. K.; Bartsch, R. A., unpublished results, 1991.
61. Lehn, J. M. Science 1985, 227, 849.
62. Cram, D. J.; Cram, J. M. Science 1974, H I , 803.
63. Cinquini, M. Synthesis 1976, 516.
64. Babb, D.A., Ph.D. Dissertation, Texas Tech University, 1985.



![Rigid-Strut-Containing Crown Ethers and [2]Catenanes for ...yaghi.berkeley.edu/pdfPublications/09rigidStrut.pdf · These crown ether based struts serve as ... Synthesis: In this section,](https://cdn.vdocument.in/doc/165x107/5a910bde7f8b9a4a268e7d00/rigid-strut-containing-crown-ethers-and-2catenanes-for-yaghi-crown-ether-based.jpg)




