Download - Green Solvents in Peptide Synthesis

Green Solvents in Peptide Synthesis
Lillian Worst
Submitted in Partial Fulfillment of the
Prerequisite for Honors in Biochemistry
under the advisement of Julia Miwa
May 2021
© 2021 Lillian Worst

1
COVID-19 impact statement
Due to the COVID-19 pandemic, the scope of this project had to be scaled back. Initially we planned on obtaining quantitative data on the use of propylene carbonate in peptide synthesis. However, due to my late start to this thesis (I originally started a thesis in a different lab, but had to switch due to the pandemic), a combined 3 quarantines between Professor Miwa and myself, and impacted supply chains for several of the required reagents, we were unable to begin collecting data until late March 2021. Though it is not the project we initially envisioned, it still lays a good groundwork for future research into propylene carbonate and its use as a green solvent in peptide synthesis.

2
Abstract Peptide synthesis is one of the most widely used and important sets of organic
reactions in biochemistry. With demand for peptides increasing, it is imperative that peptide synthesis becomes a greener process, better for both humans and the environment. One major means of “greening” peptide synthesis involves replacing the toxic solvent dimethylformamide (DMF) with a greener alternative. Previously, propylene carbonate, a very green solvent, has shown promise as a replacement for DMF in solution-phase peptide synthesis. This thesis focuses on the use of propylene carbonate as a solvent in the more prevalent solid-phase peptide synthesis, as well as the adaption of a substitution level assay to determine the quantitative success of solid-phase coupling in propylene carbonate. It was determined that solid-phase synthesis in propylene carbonate is possible, and that the substitution level assay can be adapted to work in propylene carbonate. Furthermore, all necessary reagents for peptide synthesis were shown to dissolve in propylene carbonate, making it a strong contender to replace DMF as the leading solvent for peptide synthesis. Future work will determine the quantitative comparison between propylene carbonate and DMF as solvents, and adapt the solid-phase peptide synthesis reactions for improved yields in propylene carbonate.

3
Table of Contents
Introduction 5 Peptides 5 Principles of Green Chemistry 6 Peptide Synthesis 9 Solvents in Peptide Synthesis 15 Coupling 17 Capping and Deprotection 23 Monitoring Peptide Synthesis 26 Specific Goals of the Thesis 27
Results and Discussion 28
Future Directions 36
Experimental Procedures 38 Coupling of amino acid to Chemmatrix Resin (general procedure) 38 Deprotection of Fmoc group from Fmoc-aminoacyl-chemmatrix resin: 39 Solubility Tests 39 NMR 40
References 41

4
Acknowledgements
I have many people I would like to thank for their help in this process. First, I would like to thank Professor Miwa for being an amazing teacher and mentor these last four years. Thank you for being a wonderful professor and agreeing to restart your lab to work on a thesis with me in these unprecedented times. I would also like to thank the members of my committee, Adam Matthews and Elizabeth Oakes, for meeting with us and helping us to get the project moving along. You are both wonderful professors and I am honored that you were willing to be a part of this project. Thank you also to Professors Chang and Radhakrishan for agreeing to attend my defense.
I need to thank my family and friends as well. Thank you to my parents and brother for supporting me over the phone and putting up with all of my nonsense for the last 22 years. I am sorry that I had to change projects just at the point where you were starting to understand what my first one was about, I hope you like this one too. Thank you to my friends, especially Ally, Gabe, and Landon, who have been amazing to block with through this pandemic and have been incredible sounding boards for some of my wilder ideas these past few months. I am looking forward to starting the next chapter of our lives together.

5
Introduction
Peptides
Peptides are short chains of amino acids, the building blocks for proteins. The
difference between peptides and proteins is fuzzy at best, but it is often determined by
length, manufacture, and use. Peptides are used commonly in many types of research
including helping to understand protein domains and epitopes (Tanabe, 2007), create
the next generation of antibiotics (Zhang, 2016), design new enzymes and binding sites
(Zhang et al., 2014), and activate immune responses (Tanabe, 2007) among other
things (PBRL). Peptides are likely to play an increasingly important role in medicine,
with 60 peptides currently on the market as therapeutics for a variety of conditions from
HIV to cancer, and over 600 more in development or clinical trials (Fosgerau et al.,
2015). Peptides make for great therapeutics due to their extreme specificity for one
target, which minimizes off-target interactions and side effects. Despite the promise of
peptides as medication, they are only just now beginning to hit the market due to the
difficulty of delivery to cells when taken orally. Enzymes in our stomachs are specifically
designed to break down peptides, meaning that strategies such as peptide stapling and
use of unnatural amino acids had to be developed before they could become viable as a
class of medicines (Cromm et al., 2015).
Both proteins (≥ 50 amino acids) and peptides (< 50 amino acids) are readily
prepared in the research laboratory. While proteins are most commonly produced by
cloning in bacterial cells, peptides are primarily produced via organic synthesis. While
peptide and protein production in cells is efficient and fast, peptide synthesis is much
more versatile, allowing for the addition of unnatural amino acids and modifications to

6
the peptide backbone. As processes such as peptide stapling rely on backbone
modifications and incorporation of unnatural amino acids, peptide synthesis is
increasingly important in research. Many academic and industrial labs complete peptide
synthesis on a large scale. This means that peptide synthesis is a common and
necessary set of organic reactions that is widely used. Any improvement to the process
of peptide synthesis has the potential to have a widespread impact.
Principles of Green Chemistry
Organic chemistry has been a distinct discipline of chemistry since the 1800s, but
only in the last 30 years has green chemistry become a concern to organic chemists
(Anastas and Beach, 2017). Green Chemistry was originally a response to the passage
of the Pollution Prevention Act of 1990, which aimed to improve the design of industrial
processes to decrease their toxicity and harm to human health and the environment.
This new effort contrasted with previous policies on pollution prevention that aimed to
mitigate the effects of harmful substances after the fact instead of designing the
processes themselves to be less harmful (Anastas and Beach, 2017). Throughout the
1990s, interest and funding for green chemistry grew, and in 1998, the 12 Principles of
Green Chemistry (Figure 1) were published as a guideline for future research. Overall,
the 12 principles provide criteria for designing new reactions and improving existing
reactions to decrease their environmental impact and potential harm to humans. There
are several different approaches to the goals that are laid out in the 12 principles. For
example, the first two principles, reducing waste and improving atom economy, both
aim to reduce the quantity of byproducts of a reaction. Waste prevention focuses more

7
on reducing the use of solvents and auxiliaries, while atom economy advocates for the
creation of reactions that do not produce unwanted byproducts, meaning that 100% of
the atoms present as reactants can be found in the products. Both of these principles
aid in creating reactions that are more efficient and generate less hazardous waste than
traditional syntheses. Efficient reactions also produce fewer byproducts. Byproducts and
reactants can be hard to separate from the product, requiring larger amounts of solvent
and other resources that become waste. Decreasing the number of byproducts
produced is therefore green in several ways. First, it reduces the waste of the
byproducts themselves, and second it decreases the solvent necessary in the reaction
and subsequent work-up, which would have otherwise become waste. Therefore, a

8
more efficient reaction that does a better job of converting the reactant into the desired
product will use fewer resources and produce less waste.
Other principles focus more on decreasing the toxicity of all organic species that
might be used during the reaction, from solvents, to waste, to byproducts, and even
products. One way to reduce the toxicities of these species is to find less toxic
alternatives to staples used in the reaction. Any alternative will need to have similar
properties to the original compound, but a lower toxicity. One example of this comes
from the field of solar cell development. Many of the compounds necessary for solar
cells tend to be both toxic and costly (defeating some of the purposes of solar energy).
Much chemical research in the last few decades has focused on the semiconductors
used in solar cells. Recently, Lokhande et al. have described replacing toxic CdTe and
CuInGaS2 (CIGS) quaternary chalcogenide compounds with the much safer Cu2ZnSnS4
compounds as semiconductors, therefore creating a much safer and greener production
process (Lokhande et al., 2016). This example also demonstrates another principle of
green chemistry: that if a new reaction is being designed from scratch, a good way to
decrease toxicity is simply to design reactions that do not require toxic materials. In the
case of Cu2ZnSnS4, it was chosen for use in solar cells partially due to the relative
abundance of its components, and relative ease of synthesis (Lokhande et al., 2016).
Unfortunately, designing reactions for a decrease in toxicity can lead to a
decrease in yield, which may increase waste. In other words, the principles of green
chemistry that call for a decrease in toxicity of products can directly conflict with the
principles that call for a decrease in waste, which is an important consideration
whenever trying to develop a greener reaction. However, using fewer toxic materials

9
ensures that even if more waste is produced, it will be less harmful to humans and the
environment, and usually easier to recycle or dispose of. Furthermore, once reactions
are initially developed for use of less toxic materials, yields can be incrementally
improved in the same way all other chemical reactions have been improved by
perfecting the reaction conditions over time.
Other principles of green chemistry consider having renewable feedstocks and
reducing the energy required to carry out a reaction by decreasing the heat or pressure
or using renewable sources of electricity. Overall, the principles of green chemistry
provide metrics for improving reactions to reduce their negative impacts on human
health and the environment. Greener chemical reactions are truly the future of
chemistry. It is vital that reactions developed before the emergence of green chemistry
be improved and made safer for humans and the environment. Since peptide synthesis
is such a widely used and important set of chemical reactions, it provides a great target
for improvement using green chemistry principles.
Peptide Synthesis
Both peptide synthesis and the biological production of proteins and peptides
require joining the carboxyl group of one amino acid to the amino group on another,
creating an amide (also referred to as a peptide) bond (Figure 2). Despite this
overarching similarity, the reactions carried out in peptide synthesis differ from those
carried out by cells in two key ways. First, chemical synthesis proceeds from the C-
terminus to the N-terminus while ribosomal translation occurs from the N-terminus to the
C-terminus. Second, because any chemical catalysts or coupling reagents used in

10
peptide synthesis cannot achieve the same amount of specificity as an enzyme, amino
acids for peptide synthesis must have protecting groups covering all reactive functional
groups except the one currently reacting to form the peptide bond. Specifically, the
carboxyl group on one amino acid, and the amino group on the other need to be
protected as well as any side chain functional groups that might react (such as the
carboxyl group on glutamic acid). These protecting groups ensure that only the correct
amino acids react with each other during the coupling process. Since peptides are
synthesized from C to N, the C-terminal protecting group must remain in place for the
entirety of the synthesis process (as do side chain protecting groups), while the N-
terminal protecting group must be removed after each step so that they next amino-
protected amino acid can be added. Thus, the synthesis requires orthogonal protection,
in which the “temporary” amino protecting group can be removed without removing the
“semi-permanent” protecting groups on the side chains and C-terminus. Therefore, even
though both biological synthesis of proteins and peptides essentially repeats the same
coupling step over and over, peptide synthesis also requires a deprotection step
between each coupling step.
Protecting groups affect the solubility of some amino acids. Since protecting
groups are often hydrophobic and they cover a hydrophilic amino group on each of the

11
incoming amino acids, protected amino acids are much less soluble than their
unprotected counterparts in a variety of solvents, including those commonly used in
peptide synthesis. This restricts the solvents that can be used in peptide synthesis,
especially when using the more common Fmoc-protected amino acids, which are
especially hard to dissolve in most solvents.
One important constraint of peptide synthesis is that it requires very high yield at
every step. This requirement is due to the sheer number of steps necessary to create
some peptides. While peptides are traditionally defined as under 50 amino acids, a
larger peptide could spell over 100 steps in a synthesis. A 50 amino acid peptide with a
95% yield of each coupling step could have an overall yield of less than 8%. In addition,
incomplete coupling steps lead to deletion impurities, meaning that the resulting peptide
will only be missing a single amino acid in the middle, which can be extremely difficult to
separate from the desired end product. In order to prevent the inclusion of these
deletion impurities, long synthesis processes often include a capping step after each
coupling step, wherein the unreacted free amine is capped with a third type of protecting
group. Capping changes deletion impurities to truncation impurities, making them much
easier to separate from the final product, but does not help maintain high yield. The
necessity of extremely high yield makes peptide synthesis a more difficult target for
green chemistry, as it is often the case that any change to a synthetic process will
initially decrease its yield. Since changes to any chemical process almost always initially
result in a decrease in the yield, all new techniques must be perfected in order to be
useful on an industrial scale.

12
Peptide synthesis was revolutionized in 1963 when R. B. Merrifield invented
solid-phase peptide synthesis (SPPS). Solid-phase peptide synthesis makes industrial
scale peptide synthesis possible by simplifying the purification processes and therefore
increasing the overall yield. Solid-phase synthesis involves anchoring the C-terminal
amino acid of the peptide to an insoluble resin bead. The first resin used for SPPS was
a functionalized polystyrene, and almost all SPPS resins in use today are derivatives of
polystyrene. The attachment of the peptide to the insoluble resin allows for easy
separation of the growing peptide chain from all solvents, unused reagents, and
byproducts. At each step, the solid resin with the peptides attached is simply filtered out
of the solution. Solid phase peptide synthesis can also increase yield by use of an
excess of reagents, forcing the reaction to completion through LeChatelier's principle.
Like other forms of peptide synthesis, SPPS involves repeated coupling and
deprotection steps (Figure 3).
Even though the chemistry of solid-phase peptide synthesis is essentially the
same as solution-phase peptide synthesis, solid-phase synthesis has several unique
concerns. First, the resin has to swell in the presence of the solvent, allowing for the
reagents to enter the interior of the resin matrix and react with the peptides anchored
there (as well as the ones on the surface) (AAAPTECH). The polystyrene resins used
in SPPS swell to large volumes in dichloromethane and dimethylformamide, solvents
commonly used in SPPS. In contrast, the resin shrinks to a small volume in methanol,
making it an unsuitable solvent for SPPS reactions.
Next, since SPPS relies on being able to separate the solid resin from the liquid
solution based on phase, it is imperative that no solid side products are produced during

13
any point in the synthesis (El-Faham and Albericio, 2011). Even with these constraints,
solid-phase peptide synthesis is the far superior form of peptide synthesis in most
cases, due to its increased yields and ease of application. Despite the fact that solution-
phase peptide synthesis was developed first, solid-phase peptide synthesis is the
industry (and academic) standard. Thus, applying green chemistry to solid-phase
peptide synthesis will have much more of an impact than applying it to solution-phase
peptide synthesis.

14
In 2017 Lawrenson et al. produced a paper focused on using cyclic carbonates, a
type of green solvent, in peptide synthesis (Lawrenson et al., 2017). Currently, the most
commonly used solvent for peptide synthesis is dimethylformamide (DMF) which is both
extremely toxic and hard to recycle. DMF is the leading standard solvent in peptide
synthesis because it easily dissolves Fmoc-protected amino acids (which are less
soluble in other organic solvents). Due to the large amounts of DMF used during the
peptide synthesis process, replacing it with a greener solvent could have a significant

15
impact on the overall greenness of the reaction. However, though Lawrenson et al. did
describe an environmentally friendly solvent that can replace DMF, they focused mainly
on solution-phase peptide synthesis. Though this was a good initial proof of concept, it
is imperative that cyclic carbonates and other green solvents be tested in solid-phase
peptide synthesis, since it is the predominant way peptides are manufactured in both
industry and academia.
Solvents in Peptide Synthesis
Replacing a solvent in an established reaction requires a knowledge of the
specific qualities of the original solvent. As previously mentioned, the most commonly
used solvent in peptide synthesis is currently dimethylformamide (DMF), though
dichloromethane is also used at certain points in the process. Both DMF and
dichloromethane are aprotic, polar solvents. This means that they have the ability to
dissolve a wide range of molecules, including both hydrophilic and hydrophobic
molecules, but they lack the ability to donate a proton to their solutes (Atherton).
Because of the protecting groups on the amino acids, this ability to dissolve a wide
range of hydrophobic and hydrophilic molecules is critical to the success of a peptide
synthesis reaction. Fmoc-protected amino acids especially are difficult to dissolve in
many polar solvents due to the hydrophobicity of the Fmoc group. On the other hand,
many coupling reagents are salts, meaning that they struggle to dissolve in nonpolar
solvents. DMF (and other polar aprotic solvents) tend to do a good job of spanning
these differences and dissolving both the hydrophobic protected amino acids, and the
hydrophilic coupling reagents. Thus, any solvent used in place of DMF must be similarly

16
polar and aprotic. It would also be beneficial to match other properties of the solvent,
such as the melting and boiling points. This is because the methods used for peptide
synthesis in DMF have been perfected for years, down to temperature, run time, and
concentrations. Deviation may at least temporarily decrease yield, which may
discourage the widespread adoption of greener solvents.
One family of candidates that has emerged as a green alternative to DMF in
peptide synthesis is cyclic carbonates (Figure 4). Cyclic carbonates are extremely green
solvents due to their relative safety, ease of recycling, and 100% atom economical
synthesis that sequesters CO2 (Figure 5). In fact, propylene carbonate was ranked as
the greenest solvent in 2014 (Prat et al., 2014). In 2017, Lawrenson et al. produced a
paper describing the use of propylene and ethylene carbonate as substitute solvents in
peptide synthesis (Lawrenson et al., 2017). Their initial findings suggest that propylene
carbonate holds great potential to replace DMF as a solvent in peptide synthesis, as
long as the process can be modified. Many of their qualitative results for synthesis in
propylene carbonate were comparable to synthesis in DMF (Lawrenson et al., 2017).
However, their work focused mostly on comparing propylene carbonate to another

17
cyclic carbonate, ethylene carbonate, in solution phase synthesis. They did gather some
data that shows solid-phase synthesis can be carried out in propylene carbonate but did
not quantitatively show its effects on the synthesis when compared to DMF or use
modern coupling reagents. Since solid-phase peptide synthesis is more commonly used
both industrially and in academic settings to make peptides, it is important to test this
solvent more quantitatively using common solid-phase reactions and coupling reagents.
Coupling
The key step in any peptide synthesis is the coupling of the two amino acids to
one another. In cells, this step is carried out by the ribosome, but in peptide synthesis, it
is facilitated by chemical catalysts known as coupling reagents. Coupling reagents work
by activating the carboxyl group of the incoming amino acid so that it can form the
peptide bond with the amino group of the growing peptide. On top of this, the coupling
step involves formation and breakage of bonds near the alpha carbon of the amino acid.
This means that for some amino acids such as histidine, there is a good chance that
side reactions can occur with the alpha carbon leading to racemization of the amino
acid (El-faham and Albericio, 2011). This can be a huge problem since peptides, like all
proteins, are stereospecific and derive their function from their three-dimensional

18
structure. Racemization of the amino acids during the coupling reaction could lead to a
significant decrease (up to 50%) in the yield for each step. Thus, coupling reagents that
have lower rates of racemization are preferred in modern peptide synthesis. There have
been many generations of coupling reagents, with the most important shown below
(Figure 6). The Lawrenson et al. paper from 2017 primarily relied on the use of ethyl-3-
(3-dimethylaminopropyl)carbodiimide (EDC) and dicyclohexylcarbodiimide (DCC) along
with various coupling additives (Figure 6). The carbodiimides DCC and EDC were two
of the original coupling reagents, with use in peptide synthesis dating back to the 1950s
(El-faham and Albericio, 2011). However, carbodiimides, and DCC in particular, are not
the best reagents available for solid-phase peptide synthesis. Their higher rates of
racemization can lead to a decrease in yield overall. Carbodiimide coupling occurs
through an addition of a carbonyl to the carboxyl oxygen of the amino acid and can
have unwanted side products that further decrease yield (Figure 7). While coupling
additives (Figure 6) can help decrease racemization, newer coupling reagents are less
complicated to use and often produce higher yields, making them preferable in most
cases. DCC is particularly unsuited to solid phase peptide synthesis, as one of the
natural side products of coupling (dicyclohexylurea) is soluble only in trifluoroacetic acid.
This means that in the absence of large amounts of trifluoroacetic acid (which is often
only used as a final step in synthesis to remove orthogonal protecting groups), it forms a

19
solid that cannot be separated from the resin by filtration (El-Faham and Albericio,
2011).
The use of these older coupling reagents may help to explain the lower yields
(between 75 and 80%) in the results from Lawrenson et al, and replacing the coupling
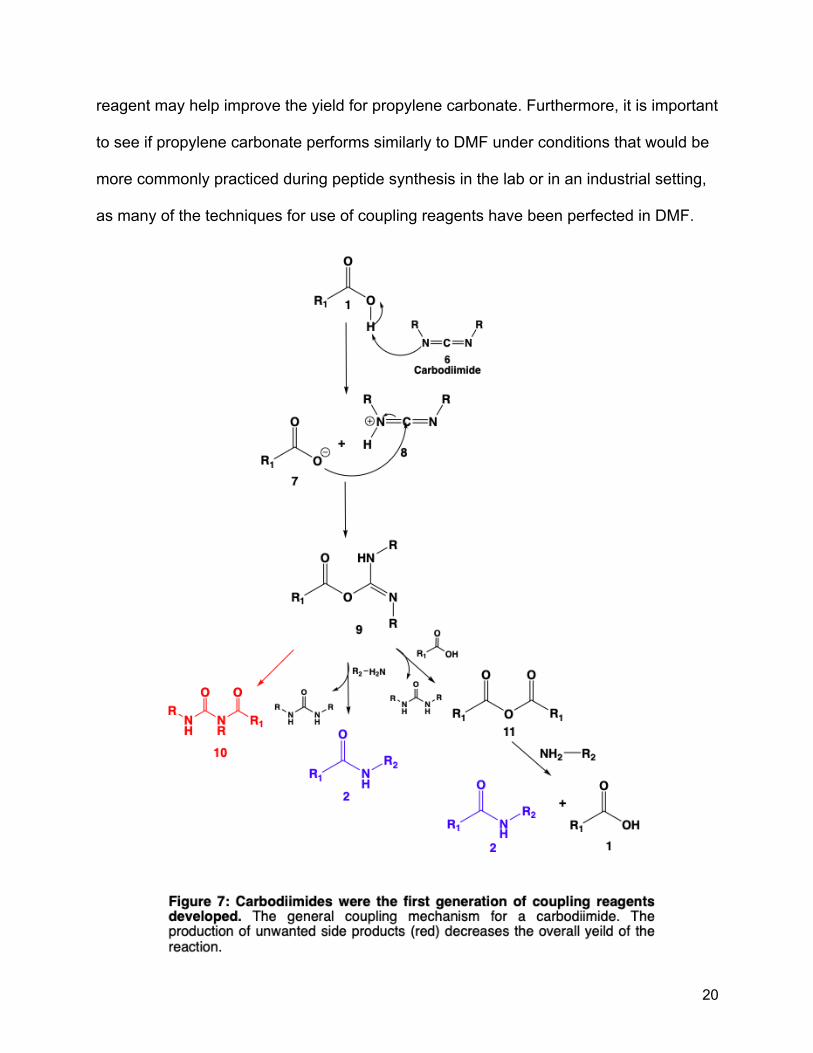
20
reagent may help improve the yield for propylene carbonate. Furthermore, it is important
to see if propylene carbonate performs similarly to DMF under conditions that would be
more commonly practiced during peptide synthesis in the lab or in an industrial setting,
as many of the techniques for use of coupling reagents have been perfected in DMF.

21
From a review of the literature, we determined that two of the most commonly used
coupling reagents in modern solid phase peptide synthesis are benzotriazol-1-yloxytris-
pyrrolidino-phosphonium hexafluorophosphate (PyBOP), a phosphonium salt, and
tetramethyluronium hexafluorophosphate (HBTU), a uronium salt (Figure 8A, Figure 6).
Both have similar mechanisms for activating the carboxyl group on the amino acid
(Figure 8B-C). The charged nitrogen on the coupling reagent facilitates the formation of
a temporary covalent bond with the carboxyl group on the amino acid. The carboxyl
group is then activated through the transfer of the nitrogen ring system to the carboxyl
oxygen. In both cases, the intermediate ester is also activated, and able to form a
peptide bond. However, due to the short lifespan of these intermediates, the peptide
bond formation is much more likely to occur with species 17 which is labeled as the
activated leaving group. Both HBTU and PyBOP have much lower rates of
racemization than carbodiimides due to the fact that they contain the racemization
suppressant HOBt (Figure 6) in their structure. The first coupling reagent to employ this
trick was BOP, the direct predecessor of PyBOP. Although BOP was a fantastic
coupling reagent, it produced a carcinogenic byproduct (hexamethylphosphoramide)
where PyBOP produces the relatively harmless species 19 (El-Faham and Albericio,
2011). Thus, PyBOP and several other similar phosphonium reagents were developed
to eliminate the production of toxic byproducts. This is an excellent example of ways in
which peptide synthesis has already been modified to make it a greener process. Unlike
carbodiimides, both PyBOP and HBTU have no side reactions that decrease the overall
yield of the reaction. Furthermore, they both have low racemization rates and are
relatively inexpensive as coupling reagents go, meaning that they are widely used for

22
peptide synthesis (Palasek et al., 2006 and Coste et al., 1990). Both PyBOP and HBTU
must prove effective in propylene carbonate in order for it to be a viable replacement for
DMF.
Another constraint of all coupling reagents is that not all coupling reactions are
created equally. Some pairs of amino acids are specifically more difficult to couple than
others due to the structures of their side chains, and their position in the growing

23
peptide. For example, it has been widely observed that using carbodiimide coupling
reagents leads to a difficulty in completing additions of amino acids 12-20 to the growing
peptide (Dunn et al., 1994). Furthermore, hydrogen bonding between unprotected side
chains such as those found on Gln and Asn can also lead to a decrease in the success
of an individual coupling step due to the formation of secondary structures (Dunn et al.,
1994). Many techniques are employed to help improve the success of each individual
coupling step, including using different coupling reagents for specific steps. Another
advantage of HBTU and PyBOP is their general fitness to most coupling steps. Though
some other coupling reagents may be better suited to a specific difficult coupling step,
HBTU and PyBOP (along with other phosphonium and uronium reagents) have the
ability to do a relatively good job coupling most steps (El-Fahan and Albericio, 2011).
This general promiscuity, combined with their low rates of racemization, relatively low
cost, and lack of toxicity (both themselves and in their by-products) have ensured that
they are widely used coupling reagents by most people doing peptide synthesis. Thus,
any green protocol for peptide synthesis must be compatible with both HBTU and
PyBOP.
Capping and Deprotection
The other major step in peptide synthesis is deprotection. Deprotection involves
removing the N-terminal protecting group from the growing peptide after each coupling
step, allowing for the next coupling to take place. There are two main types of protection
strategies commonly used in peptide synthesis: Boc/Benzyl and Fmoc/t-butyl. In both
cases, the first protecting group listed (Boc, Fmoc) refers to the temporary N-terminal

24
protecting group, and the second (benzyl, t-butyl) refers to the orthogonal semi-
permanent protecting groups on the side chains. In each case, the two types of
protecting groups are removed through different processes, allowing the N-terminal
protecting group to be removed before each coupling step while leaving the orthogonal
protection to the end (Albericio, 2000). Boc/benzyl protection had mostly fallen out of
favor for use in solid phase peptide synthesis due to the necessity of harsh conditions
required for deprotection of the amino group (trifluoroacetic acid), the necessity to
neutralize the acid before coupling can occur (Figure 9), and the requirement of a very
dangerous hydrofluoric acid procedure for the final removal of the side chain protecting
groups and cleavage of the peptide from the resin. Fmoc/t-butyl protection only requires
harsh conditions (trifluoroacetic acid) for the final cleavage from the resin and
deprotection of the orthogonal t-butyl groups. Since the Fmoc group is removed in
piperidine (a base), while the resin and side chain protecting groups are removed with
acid, the protection strategy is truly orthogonal. Furthermore, the basic conditions
necessary for coupling with reagents such as PyBOP are already present, and there is
no need to first neutralize the solution (Figure 9). Unfortunately, Fmoc protected amino
acids are harder to dissolve in most solvents when compared with Boc protected amino
acids. This led to the widespread adoption of DMF in peptide synthesis, as it is one of
the few solvents that dissolve Fmoc protected amino acids extremely well. Thus, it is
imperative that propylene carbonate demonstrate the ability to dissolve Fmoc amino
acids as well.

25
Another important step in the peptide synthesis process is capping. Capping
involves blocking the end of an unreacted peptide so that it cannot continue to react.
This prevents deletion impurities, which are much harder to separate from the final
product than the much shorter truncated impurities. Capping is usually accomplished by
addition of acetic anhydride, which reacts with the free amine to create an acetamide
group that cannot be removed by deprotection steps, thus truncating the peptide
(Verma et al., 2020). Capping is most important in the synthesis of long peptides where
deletion impurities would be much harder to separate from the desired product.

26
Ultimately, solid-phase peptide synthesis is a complex scheme of chemical
reactions that result in the ability to create any peptides regardless of sequence and
easily separate them from waste and byproducts. It is a powerful tool, and one that will
become increasingly important with the adoption of peptide medicines, but currently has
significant negative effects on people and the environment. It is imperative that greener
methods of peptide synthesis are developed and implemented.
Monitoring Peptide Synthesis
One of the most important parts of any complex reaction involves the ability to
monitor its progress and quantify its success after each stage. There are several
common methods for monitoring peptide synthesis reactions. The Kaiser test is a
commonly used method to monitor peptide synthesis. After each coupling step,
ninhydrin is added, which turns blue in the presence of free amine. By either looking at
the color of the solution or taking a spectrum, the general success of the coupling step
can be determined. Though the Kaiser test is very fast, and extremely useful
qualitatively, it does not provide quantitative results on the success of a coupling
reaction and can be inaccurate when used for certain amino acids (Fontenot et al.,
1997). On the other end of the spectrum, high performance liquid chromatography
(HPLC) can be used to get good quantitative data on the success of a coupling reaction
but is also very time and labor expensive. A happy medium can be struck by using a
substitution level analysis, which uses the fact that the byproduct of Fmoc deprotection,
dibenzofulvene, absorbs UV light. By using spectroscopy on an aliquot of deprotection
solution removed from the reaction vessel, it is possible to get some quantitative data

27
on the success of a peptide coupling reaction in much less time than it would take to
perform HPLC.
Specific Goals of the Thesis
This thesis will primarily focus on the methods needed to obtain quantitative data
from solid phase peptide synthesis in propylene carbonate. It will assess the solvation of
HBTU, PyBOP, Fmoc amino acids, and piperidine in propylene carbonate, as well as
address the use of ChemMatrix rink amide resin (which was selected due to its
compatibility with propylene carbonate). Furthermore, we will attempt to develop and
improve on a substitution level assay necessary to determine the success of a coupling
in propylene carbonate. Ultimately, we hope to prove that propylene carbonate can be a
viable replacement for DMF in modern solid phase peptide synthesis and set the stage
for a quantitative study directly comparing propylene carbonate to DMF.

28
Results and Discussion
The goal of this project was to adapt a solid-phase peptide synthesis protocol
using modern coupling reagents and the green solvent propylene carbonate. In order to
succeed in propylene carbonate, a solid phase synthesis reaction has several specific
requirements. First, a solid support resin must be able to demonstrate the appropriate
swelling in propylene carbonate. For this project, we used H-Rink Amide ChemMatrix
resin (same as Lawrenson et al).. ChemMatrix is a polyethylene glycol (PEG) based
resin rather than a polystyrene-based resin. It demonstrates excellent swelling in
propylene carbonate. Second, all reagents (protected amino acids and coupling
reagents) had to be able to dissolve in propylene carbonate at useful concentrations for
peptide synthesis. Third, the temporary N-terminal protecting groups (Fmoc in this case)
must be removable in propylene carbonate. Finally, there must be a reliable method to
measure the extent of coupling (the substitution level of protected amino acid on the
resin). The work done during this thesis project has focused primarily on the
development of the assay, assessment of solubility of a variety of protected amino
acids, and demonstration of successful deprotection and coupling on a sample synthetic
peptide.
Initially, we planned to only test the solubility of the reagents we planned to
actually use in the final coupling reaction. This included Fmoc-Val-OH, Fmoc-Phe,
PyBOP, HBTU, and tygon tubing (which we were hoping did not dissolve). Though the
results of this first round of solubility tests indicated coupling reagents and Fmoc-amino
acids can be dissolved in propylene carbonate, it also indicated that not all Fmoc amino
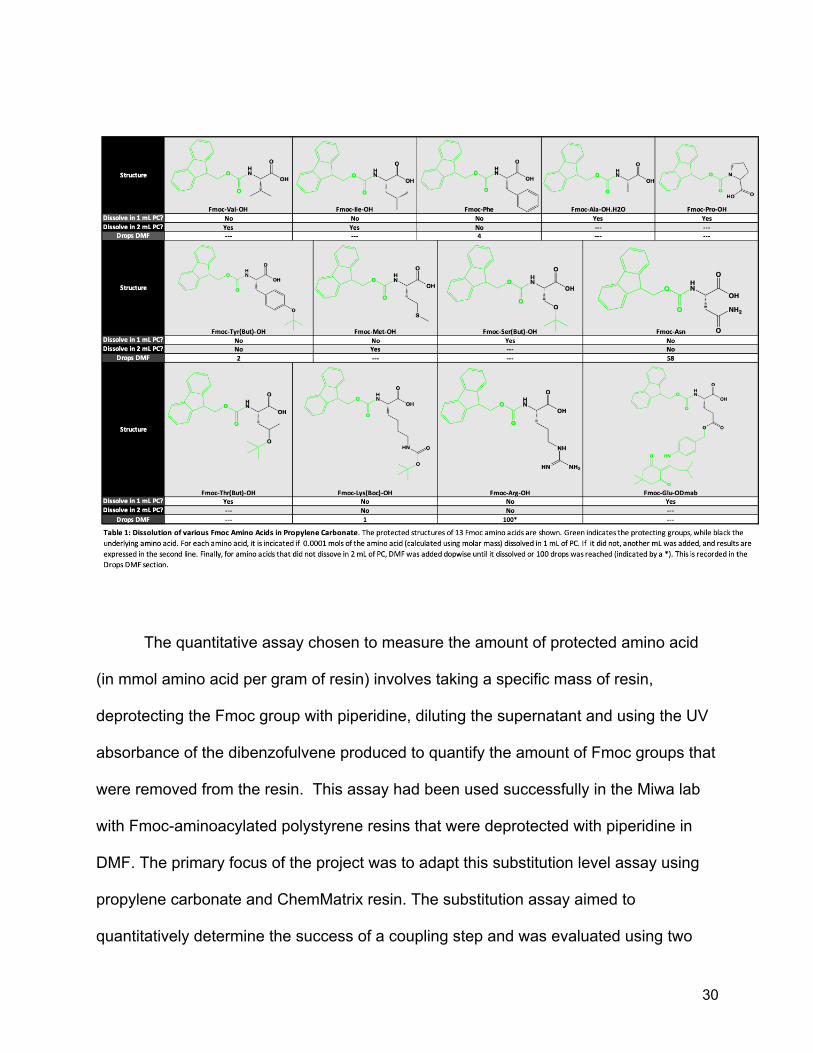
acids dissolve in propylene carbonate equally well. Specifically, in order to get the

29
Fmoc-Phe to dissolve, it was necessary to add a small amount of DMF. Thus, another
round of solubility tests was carried out, this time using all of the Fmoc amino acids on
hand (13 of the 20 naturally occurring amino acids were tested). To conserve solvent,
the concentration of amino acid typically present in a coupling reaction was calculated
(0.1M), and the amount to dissolve in 1 mL of propylene carbonate was determined
(0.0001 mol). The amino acid (0.0001 mol) was added to 1.0 mL of propylene
carbonate. Each sample was sonicated for one minute in an ultrasonic bath. Afterwards,
the samples that had dissolved were noted. For the samples that had not dissolved,
another 1 mL of propylene carbonate was added, and the samples were again placed in
the ultrasonic bath. Again, it was noted which amino acids had dissolved. For the five
amino acids that remained undissolved, DMF was added dropwise until either the amino
acid dissolved, or 100 drops was reached (Table 1). For arginine (Fmoc-Arg), it took
over 100 drops of DMF and would still not dissolve. An equivalent amount of the amino
acid was again weighed out and this time dissolved in 1mL of pure DMF to ensure that it
would dissolve in DMF. It did successfully dissolve in DMF, and subsequent dropwise
addition of 100 drops of propylene carbonate did not cause it to crash out of the
solution, suggesting that propylene carbonate could still be used to dilute the DMF, and
therefore make the reaction greener. It does seem that the propylene carbonate has
trouble dissolving any species with an unprotected nitrogen based functional group, as
the other amino acid that needed a significant amount of DMF to dissolve was
asparagine. These results are still very good however, as it is clear that PC can dissolve
the majority of the Fmoc-protected amino acids with minimal addition of DMF, and there
is a good chance that a greener cosolvent can be discovered in the future.
30
The quantitative assay chosen to measure the amount of protected amino acid
(in mmol amino acid per gram of resin) involves taking a specific mass of resin,
deprotecting the Fmoc group with piperidine, diluting the supernatant and using the UV
absorbance of the dibenzofulvene produced to quantify the amount of Fmoc groups that
were removed from the resin. This assay had been used successfully in the Miwa lab
with Fmoc-aminoacylated polystyrene resins that were deprotected with piperidine in
DMF. The primary focus of the project was to adapt this substitution level assay using
propylene carbonate and ChemMatrix resin. The substitution assay aimed to
quantitatively determine the success of a coupling step and was evaluated using two
31
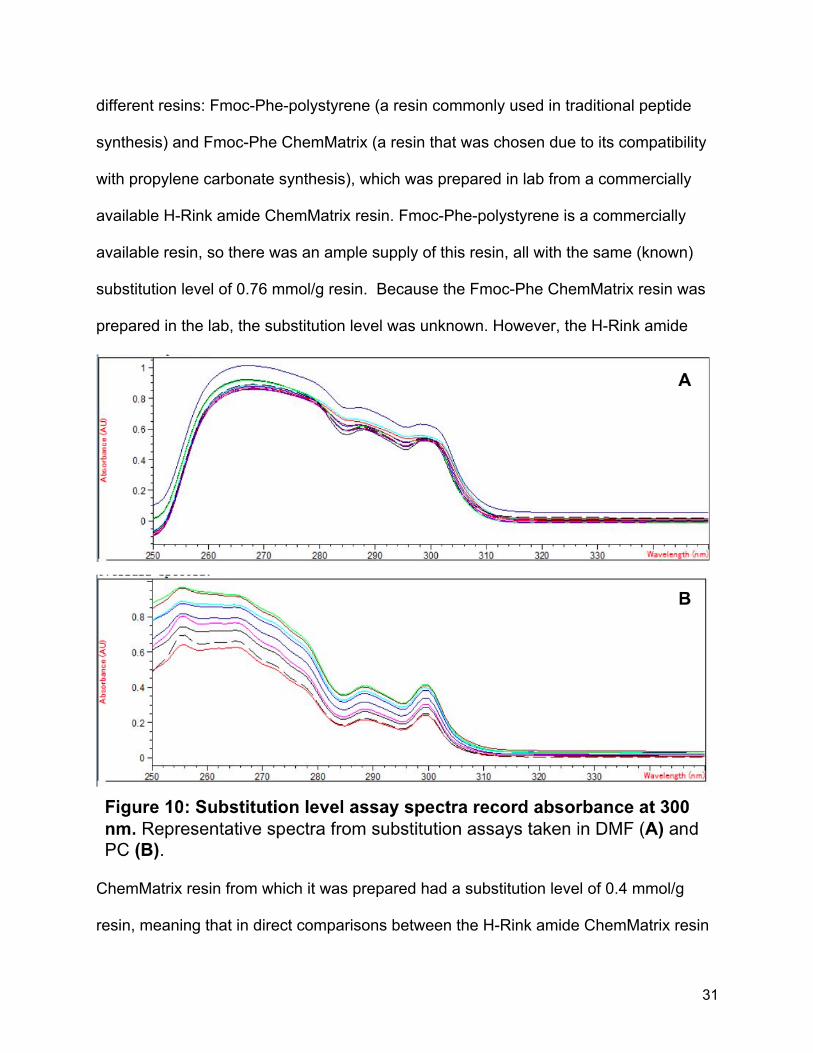
different resins: Fmoc-Phe-polystyrene (a resin commonly used in traditional peptide
synthesis) and Fmoc-Phe ChemMatrix (a resin that was chosen due to its compatibility
with propylene carbonate synthesis), which was prepared in lab from a commercially
available H-Rink amide ChemMatrix resin. Fmoc-Phe-polystyrene is a commercially
available resin, so there was an ample supply of this resin, all with the same (known)
substitution level of 0.76 mmol/g resin. Because the Fmoc-Phe ChemMatrix resin was
prepared in the lab, the substitution level was unknown. However, the H-Rink amide
ChemMatrix resin from which it was prepared had a substitution level of 0.4 mmol/g
resin, meaning that in direct comparisons between the H-Rink amide ChemMatrix resin

32
and the Fmoc-Phe-polystyrene resin, we would expect the ChemMatrix resin to have a
lower substitution level. While the two resins had very similar spectra, ultimately, the
Fmoc-Phe-Polystyrene in DMF gave more clustered data, while the ChemMatrix resin in
PC had more variation (Figure 10). This was at least partially due to the difference in
size of the resin particles. While the Fmoc-Phe-Polystyrene had a very small particle
size and tended to settle to the bottom in the DMF, the ChemMatrix resin had a much
larger particle size, especially when swelled, and remained suspended in the PC. This
led to difficulty in accurately pipetting aliquots of deprotection solution without plugging
the tip of the micropipette, or accidentally incorporating resin. An additional filtration step
after deprotection did decrease the variability of the spectra somewhat, though it was
still not as consistent as the data taken in DMF.
Initially, the substitution level assay had wide variation using Fmoc-Phe-
polystyrene in DMF. In order to improve the reproducibility and precision of the assay,
two major changes were made. The first goal was to improve the weighing, transfer,
and handling of the resin. Both the polystyrene resin and the ChemMatrix resin are
extremely likely to stick to the spatulas and the sides of the test tubes. This impacted
the accuracy of the measured masses of resin in each test tube, as resin on the side of
the test tube contributed to the overall mass of the resin in the tube but was unable to
be washed down into the reaction solution, and thus did not contribute to the amount of
dibenzofulvene in the end solution. In order to fix this problem, the following changes
were made. First, instead of diluting only one aliquot from each 1.00 mL sample, the
volume of deprotection solution was increased to 2.00 mL, and multiple (4-5) aliquots
were diluted and tested from each sample. This change ensured that any error resulting
33
from resin sticking to the test tube would be systematic rather than random. Second, the
original borosilicate test tubes were switched for microcentrifuge tubes with a much
smaller volume and therefore smaller surface area on the sides for the resin to stick.
The combination of these two changes helped decrease the noise in the data.
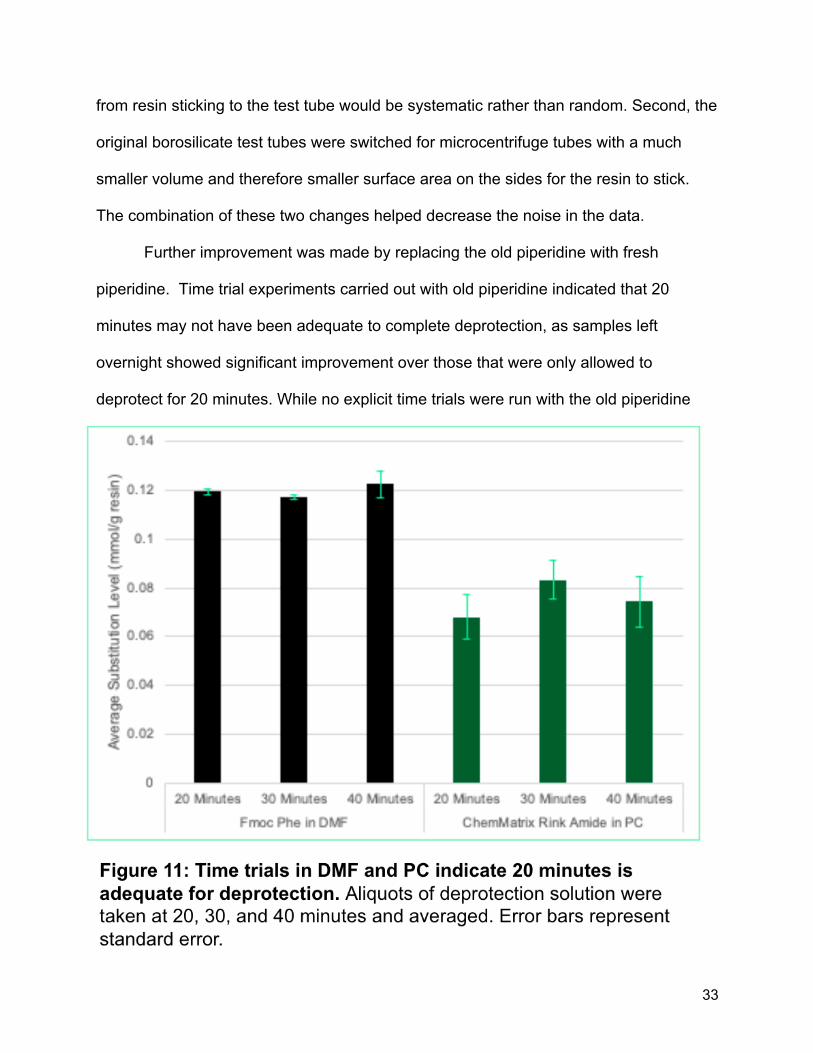
Further improvement was made by replacing the old piperidine with fresh
piperidine. Time trial experiments carried out with old piperidine indicated that 20
minutes may not have been adequate to complete deprotection, as samples left
overnight showed significant improvement over those that were only allowed to
deprotect for 20 minutes. While no explicit time trials were run with the old piperidine

34
(the new piperidine came in at the same time we were planning on running the time
trials), time trials with newly purchased piperidine showed no increase in deprotection
after 20 minutes, suggesting that 20 minutes was adequate time for deprotection
(Figure 11).
The other goal of this project was to prove that a solid-phase coupling reaction
using modern coupling reagents and Fmoc amino acids was possible in propylene
carbonate. To this end, a valine-phenylalanine dipeptide was synthesized on H-Rink
amide ChemMatrix resin using PyBOP as the coupling reagent and propylene
carbonate as the primary solvent. Though we were unable to get final data for the
success of this reaction, the substitution assay at each stage of synthesis (after Phe
addition, and after completion) indicates that the coupling was entirely successful
(Figure 12). Since the uncoupled resin does not have a protecting group in it it does not
show up at all in the substitution level assay, but the results of the assay for both the
phenylalanine-resin and the Val-Phe-resin are in the range we would expect from a
successful coupling reaction to the resin.
Overall, even though we were unable to get final characterization of a peptide
synthesized on solid phase in propylene carbonate, our results strongly suggest that it is
a viable option as a greener solvent in peptide synthesis. All of the required reagents
can dissolve in PC, the assay can be completed using PC, and it is possible to make a
dipeptide using PC as the primary solvent.

35

36
Future Directions
Now that it has been demonstrated that a modern solid phase coupling reaction
can take place in propylene carbonate, the next step is to get quantitative data
regarding the success of coupling in PC and compare it to the same reaction in DMF.
The assay has been improved to the point where it is able to give reasonably
reproducible data and could certainly be used for a comparison between the two
solvents. Once the direct comparison has been made, it is vital to adapt the synthesis to
propylene carbonate and attempt to improve yield. Furthermore, it is important to test
propylene carbonate as a solvent for more difficult coupling reactions and ensure that it
is a successful solvent in those as well. The Phe-Val coupling was initially chosen due
to its relative simplicity. By researching and testing more complex coupling reactions in
propylene carbonate, we would aim to prove that PC is as versatile a solvent for peptide
synthesis as DMF. This may also involve the use of coupling reagents besides HBTU
and PyBOP, as other reagents are more suited to specific difficult reactions.
One interesting result from this study was the correlation between an amino
acid’s inability to dissolve in PC, and the presence of an unprotected amine. Further
experimentation and research into why this might be may help to isolate alternative
cosolvents (other than DMF) that could be used to get these amino acids to dissolve.
Finally, it is important to determine whether the final step of peptide synthesis
(orthogonal deprotection and cleavage from the resin with TFA) can be performed in
propylene carbonate. Though this step does use less solvent in comparison to the
repetitive cycle of coupling and deprotection, its successful completion in PC would

37
mean that the entirety of the peptide synthesis process could be performed with PC as
the primary solvent.

38
Experimental Procedures
Substitution Assay for Aminoacylated Polystyrene Resin
A sample of the resin bound peptide was washed with either dichloromethane or
ethanol and dried under vacuum. A 10-20 mg sample of the dried resin was weighed
and mixed with 1.00 or 2.00 mL of 20% piperidine (vol/vol) in DMF. After 20 minutes,
0.200 mL of the solution was removed and diluted to a total volume of 4.00 mL using
either acetonitrile or ethanol. The absorbance at 300 nm was measured using a UV-Vis
Spectrometer (What type). Dibenzofulvene has an extinction coefficient (ε) of 7040 M-
1cm-1 at 300 nm. Thus, by Beer’s Law, for X g of resin, an absorbance of A, and Y mL of
initial deprotection solution, the substitution level of the resin sample is given by the
equation 𝑆𝐿 = (20 ∗ 𝑌 ∗ 𝐴)/(7040 ∗ 𝑋). It is extremely important to use fresh piperidine
in this assay. Initial trials were run in old piperidine, which was discolored yellow, and
caused much more variation in the end results.
Coupling of amino acid to ChemMatrix Resin (general procedure)
H-Rink amide ChemMatrix resin (0.1 to 0.5 g, 0.4 mmol/g) was added to a 10 mL
solid-phase synthesis reaction vessel. Fmoc-protected amino acid (1.1 equivalents) was
dissolved in 3 mL propylene carbonate. PyBOP (1.1 equivalents) was dissolved in 0.5
mL propylene carbonate. Propylene carbonate (2-5 mL) was added to the reaction
vessel to swell the resin. The mixture was shaken for 1 minute and the propylene
carbonate removed by filtration. This procedure was repeated three times. The Fmoc
amino acid solution, PyBOP solution and diisopropylethylamine (2.2 equivalents) were

39
added. The vessel was shaken for one hour. The solution was then removed by
filtration. The propylene carbonate washing procedure (add 2-5 mL of propylene
carbonate, shake for one minute, remove propylene carbonate by filtration) was
repeated four times. If necessary, an aliquot of resin was removed for testing at this
point following the procedure listed below.
Deprotection of Fmoc group from Fmoc-aminoacyl-ChemMatrix resin:
Fmoc-aminoacylated resin (0.5 - 1.0 g) was washed 3-4 times with propylene
carbonate. Then a 20% by volume solution of piperidine in propylene carbonate was
prepared and added to the reaction vessel until it covered the resin. The reaction vessel
was shaken for 20 to 30 minutes to allow the resin to deprotect, and then washed
thoroughly with propylene carbonate to remove the piperidine. This prepared the resin
for either a new coupling or the substitution assay described above.
Solubility Tests
The solubility of 13 Fmoc amino acids in propylene carbonate was determined.
Solubility tests were completed using 0.0001 mols of each amino acid and coupling
reagent. 1.00 mL of propylene carbonate was added to each test tube. Each sample
was given a minute in an ultrasonic bath. Afterwards, the samples that had dissolved
were noted. For the samples that had not dissolved, another 1.00 mL of propylene
carbonate was added, and the samples were again placed in the ultrasonic bath. It was
previously determined that a more dilute solution of the reagents was acceptable.
Again, it was noted which amino acids had dissolved. For the five amino acids that

40
remained undissolved, DMF was added dropwise until either the amino acid dissolved,
or 100 drops were reached. This data was recorded.
NMR
Many of our reagents were old and needed to be tested for purity. In order to do
this, NMR samples containing each reagent of interest dissolved in deuterated
chloroform with TMR were prepared and tested using a 500MHz NMR. The resulting
spectra were scrutinized for possible contaminants.

41
References
(1) Tanabe, S. Epitope Peptides and Immunotherapy. 10.
(2) Zhang, L. Antimicrobial Peptides. Current Biology 6.
(3) Zhang, C.; Xue, X.; Luo, Q.; Li, Y.; Yang, K.; Zhuang, X.; Jiang, Y.; Zhang, J.; Liu, J.; Zou, G.; Liang, X.-J. Self-Assembled Peptide Nanofibers Designed as Biological Enzymes for Catalyzing Ester Hydrolysis. 2014, 8 (11), 9.
(4) Fosgerau, K.; Hoffmann, T. Peptide Therapeutics: Current Status and Future Directions. Drug Discovery Today 2015, 20 (1), 122–128. https://doi.org/10.1016/j.drudis.2014.10.003.
(5) Cromm, P. M.; Spiegel, J.; Grossmann, T. N. Hydrocarbon Stapled Peptides as Modulators of Biological Function. ACS Chem. Biol. 2015, 10 (6), 1362–1375. https://doi.org/10.1021/cb501020r.
(6) Lokhande, A. C.; Gurav, K. V.; Jo, E.; Lokhande, C. D.; Kim, J. H. Chemical Synthesis of Cu 2 SnS 3 (CTS) Nanoparticles: A Status Review. Journal of Alloys and Compounds 2016, 656, 295–310. https://doi.org/10.1016/j.jallcom.2015.09.232.
(7) Fields, G. B. Introduction to Peptide Synthesis. Current Protocols in Protein Science 2001, 26 (1). https://doi.org/10.1002/0471140864.ps1801s26.
(8) Lawrenson, S. B.; Arav, R.; North, M. The Greening of Peptide Synthesis. Green Chem. 2017, 19 (7), 1685–1691. https://doi.org/10.1039/C7GC00247E.
(9) Atherton, E.; Sheppard, R. C. Solid Phase Peptide Synthesis: A Practical Approach; A Practical Approach.
(10) El-Faham, A.; Albericio, F. Peptide Coupling Reagents, More than a Letter Soup. Chem. Rev. 2011, 111 (11), 6557–6602. https://doi.org/10.1021/cr100048w.
(11) Kimmerlin, T.; Seebach, D. ‘100 Years of Peptide Synthesis’: Ligation Methods for Peptide and Protein Synthesis with Applications Toβ-Peptide Assemblies. Journal of

42
Peptide Research 2005, 65 (2), 229–260. https://doi.org/10.1111/j.1399-3011.2005.00214.x.
(12) Palasek, S. A.; Cox, Z. J.; Collins, J. M. Limiting Racemization and Aspartimide Formation in Microwave-Enhanced Fmoc Solid Phase Peptide Synthesis. Journal of Peptide Science 2007, 13 (3), 143–148. https://doi.org/10.1002/psc.804.
(13) Coste, J.; Le-Nguyen, D.; Castro, B. PyBOP: A New Peptide Coupling Reagent Devoid of Toxic By-Product. Tetrahedron Letters 1990, 31 (2), 205–208.
(14) Dunn, B.; Pennington, M.; Byrnes, M. Proceedures to Improve Difficult Couplings. In Peptide Synthesis Protocols; 1994; pp 1–16.
(15) Albericio, F. Orthogonal Protecting Groups for Nα-Amino and C-Terminal Carboxyl Functions in Solid-Phase Peptide Synthesis. Peptide Science 2000, 55 (2), 123–139. https://doi.org/10.1002/1097-0282(2000)55:2<123::AID-BIP30>3.0.CO;2-F.
(16) Al Musaimi, O.; Jad, Y. E.; Kumar, A.; El-Faham, A.; Collins, J. M.; Basso, A.; de la Torre, B. G.; Albericio, F. Greening the Solid-Phase Peptide Synthesis Process. 2-MeTHF for the Incorporation of the First Amino Acid and Precipitation of Peptides after Global Deprotection. Org. Process Res. Dev. 2018, 22 (12), 1809–1816. https://doi.org/10.1021/acs.oprd.8b00335.
(17) Deepshikha Verma; Pillai V N R; Giriraj Tailor. Role of Capping in Peptide Synthesis. ijrps 2020, 11 (4), 5225–5228. https://doi.org/10.26452/ijrps.v11i4.3134.
(18) Albericio, F. Encyclopedia of Reagents for Organic Synthesis; 2001.
(19) Anastas; Beach. History of Green Chemistry | Center for Green Chemistry & Green Engineering at Yale https://greenchemistry.yale.edu/about/history-green-chemistry (accessed Jan 23, 2021).
(20) Behrendt, R.; Huber, S.; Martí, R.; White, P. New t -Butyl Based Aspartate Protecting Groups Preventing Aspartimide Formation in Fmoc SPPS: NEW t -BUTYL BASED ASPARTATE PROTECTING GROUPS FOR FMOC SPPS. J. Pept. Sci. 2015, 21 (8), 680–687. https://doi.org/10.1002/psc.2790.
(21)

43
Chaudhari, M. I.; Muralidharan, A.; Pratt, L. R.; Rempe, S. B. Assessment of Simple Models for Molecular Simulation of Ethylene Carbonate and Propylene Carbonate as Solvents for Electrolyte Solutions. In Modeling Electrochemical Energy Storage at the Atomic Scale; Korth, M., Ed.; Topics in Current Chemistry Collections; Springer International Publishing: Cham, 2018; pp 53–77. https://doi.org/10.1007/978-3-030-00593-1_3.
(22) Cherkupally, P.; Ramesh, S.; de la Torre, B. G.; Govender, T.; Kruger, H. G.; Albericio, F. Immobilized Coupling Reagents: Synthesis of Amides/Peptides. ACS Comb. Sci. 2014, 16 (11), 579–601. https://doi.org/10.1021/co500126y.
(23) Declerck, V.; Nun, P.; Martinez, J.; Lamaty, F. Solvent-Free Synthesis of Peptides. Angewandte Chemie International Edition 2009, 48 (49), 9318–9321. https://doi.org/10.1002/anie.200903510.
(24) The Origins of Organic Chemistry https://chem.libretexts.org/Courses/Sacramento_City_College/SCC%3A_Chem_420_-_Organic_Chemistry_I/Text/01%3A_Introduction_and_Review/1.01%3A_The_Origins_of_Organic_Chemistry (accessed Jan 23, 2021).
(25) Frérot, E.; Coste, J.; Pantaloni, A.; Dufour, M.-N.; Jouin, P. PyBOP® and PyBroP: Two Reagents for the Difficult Coupling of the α,α-Dialkyl Amino Acid, Aib. Tetrahedron 1991, 47 (2), 259–270. https://doi.org/10.1016/S0040-4020(01)80922-4.
(26) Gescher, A. Metabolism of N,N-Dimethylformamide: Key to the Understanding of Its Toxicity. Chem. Res. Toxicol. 1993, 6 (3), 245–251. https://doi.org/10.1021/tx00033a001.
(27) Han, S.-Y.; Kim, Y.-A. Recent Development of Peptide Coupling Reagents in Organic Synthesis. Tetrahedron 2004, 60 (11), 20.
(28) Recent Development of Coupling Reagents.Pdf.
(29) Hu, L.; Xu, S.; Zhao, Z.; Yang, Y.; Peng, Z.; Yang, M.; Wang, C.; Zhao, J. Ynamides as Racemization-Free Coupling Reagents for Amide and Peptide Synthesis. J. Am. Chem. Soc. 2016, 138 (40), 13135–13138. https://doi.org/10.1021/jacs.6b07230.
(30)

44
Ingenito, R.; Bianchi, E.; Fattori, D.; Pessi, A. Solid Phase Synthesis of Peptide C-Terminal Thioesters by Fmoc/ t -Bu Chemistry. J. Am. Chem. Soc. 1999, 121 (49), 11369–11374. https://doi.org/10.1021/ja992668n.
(31) Kvsrg, P. APPLICATIONS OF PEPTIDE COUPLING REAGENTS – AN UPDATE. 8 (1), 12.
(32) Li, C.-J.; Trost, B. M. Green Chemistry for Chemical Synthesis. 6.
(33) Metcalfe, I. S.; North, M.; Pasquale, R.; Thursfield, A. An Integrated Approach to Energy and Chemicals Production. Energy Environ. Sci. 2010, 3 (2), 212–215. https://doi.org/10.1039/B918417A.
(34) Nehlig, E.; Motte, L.; Guénin, E. Nano-Organocatalysts Synthesis: Boc vs Fmoc Protection. Catalysis Today 2013, 208, 90–96. https://doi.org/10.1016/j.cattod.2012.10.027.
(35) Protein Biology Resource Library. Peptide Synthesis - US //www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/peptide-synthesis.html (accessed Feb 11, 2021).
(36) Riester, D.; Wiesmüller, K.-H.; Stoll, D.; Kuhn, R. Racemization of Amino Acids in Solid-Phase Peptide Synthesis Investigated by Capillary Electrophoresis. Anal. Chem. 1996, 68 (14), 2361–2365. https://doi.org/10.1021/ac9511511.
(37) Sheehan, J. C.; Hlavka, J. J. The Use of Water-Soluble and Basic Carbodiimides in Peptide Synthesis. J. Org. Chem. 1956, 21 (4), 439–441. https://doi.org/10.1021/jo01110a017.
(38) Sigma. Peptide Coupling Reagents Selection Guide https://www.sigmaaldrich.com/technical-documents/articles/chemistry/peptide-coupling-reagents-selection-guide.html (accessed Jan 29, 2021).
(39) Stawikowski, M.; Fields, G. B. Introduction to Peptide Synthesis. Curr Protoc Protein Sci 2002, CHAPTER, Unit-18.1. https://doi.org/10.1002/0471140864.ps1801s26.
(40) About Green Chemistry. Beyond Benign.

45
(41) Resins for Solid Phase Peptide Synthesis - Core Resins. AAPPTEC.
(42)
Fontenot JD, Ball JM, Miller MA, David CM, Montelaro RC. A survey of potential problems and quality control in peptide synthesis by the fluorenylmethoxycarbonyl procedure. Pept Res. 1991 Jan-Feb;4(1) 19-25. PMID: 1802234.
(43) Prat, Denis, John Hayler, and Andy Wells. "A survey of solvent selection guides." Green Chemistry 16, no. 10 (2014): 4546-4551.







