
Heterogeneous lengths of copy number mutations in human coagulopathy revealed by genome-wide high-density SNP array
by Hee-Jin Kim, Duk-Kyung Kim, Ki-Young Yoo, Chur-Woo You, Jong-Ha Yoo, Ki-O Lee,In-Ae Park, Hae-Sun Choung, Hee-Jung Kim, Min-Jung Song, and Sun-Hee Kim
Haematologica 2011 [Epub ahead of print]
Citation: Kim HJ, Kim DK, Yoo KY, You CW, Yoo JH, Lee KO, Park IA, Choung HS,Kim HJ, Song MJ, and Kim SH. Heterogeneous lengths of copy number mutations inhuman coagulopathy revealed by genome-wide high-density SNP array. Haematologica. 2011; 96:xxx doi:10.3324/haematol.2011.052324
Publisher's Disclaimer. E-publishing ahead of print is increasingly important for the rapid dissemination of science.Haematologica is, therefore, E-publishing PDF files of an early version of manuscripts thathave completed a regular peer review and have been accepted for publication. E-publishingof this PDF file has been approved by the authors. After having E-published Ahead of Print,manuscripts will then undergo technical and English editing, typesetting, proof correction andbe presented for the authors' final approval; the final version of the manuscript will thenappear in print on a regular issue of the journal. All legal disclaimers that apply to the journal also pertain to this production process.
Haematologica (pISSN: 0390-6078, eISSN: 1592-8721, NLM ID: 0417435, www.haemato-logica.org) publishes peer-reviewed papers across all areas of experimental and clinicalhematology. The journal is owned by the Ferrata Storti Foundation, a non-profit organiza-tion, and serves the scientific community with strict adherence to the principles of openaccess publishing (www.doaj.org). In addition, the journal makes every paper publishedimmediately available in PubMed Central (PMC), the US National Institutes of Health (NIH)free digital archive of biomedical and life sciences journal literature.
Official Organ of the European Hematology AssociationPublished by the Ferrata Storti Foundation, Pavia, Italy
www.haematologica.org
Early Release Paper
Support Haematologica and Open Access Publishing by becoming a member of the EuropeHematology Association (EHA) and enjoying the benefits of this membership, which inc
participation in the online CME?program
Copyright 2011 Ferrata Storti Foundation.Published Ahead of Print on October 11, 2011, as doi:10.3324/haematol.2011.052324.

1
Heterogeneous lengths of copy number mutations in human
coagulopathy revealed by genome-wide high-density SNP array
Running head: Copy number mutations in human coagulopathy
Hee-Jin Kim,1,2 Duk-Kyung Kim,2,3 Ki-Young Yoo,4 Chur-Woo You,5 Jong-Ha Yoo,6
Ki-O Lee,7 In-Ae Park,7 Hae-Sun Choung,1 Hee-Jung Kim,1 Min-Jung Song,1
and Sun-Hee Kim1
1Department of Laboratory Medicine & Genetics, Samsung Medical Center, Sungkyunkwan
University School of Medicine, Seoul, Korea; 2Cardiac & Vascular Center, Samsung Medical Center,
Sungkyunkwan University School of Medicine, Seoul, Korea; 3Department of Medicine, Samsung
Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea 4Korea Hemophilia
Foundation, Seoul, Korea; 5Department of Pediatrics, Eulji University School of Medicine, Daejeon,
Korea; 6Department of Laboratory Medicine, National Health Insurance Corporation Ilsan Hospital,
Goyang, Korea, and 7Samsung Biomedical Research Institute, Samsung Medical Center, Seoul, Korea
Correspondence
Hee-Jin Kim, MD, PhD, Assistant Professor Department of Laboratory Medicine and
Genetics, Samsung Medical Center, Sungkyunkwan University School of Medicine, 50
Irwon-dong, Gangnam-gu, Seoul, Korea, 135-710. Phone: international +82.2.34102710.
Fax: international +82.2.34102719. E-mail: [email protected]
DOI: 10.3324/haematol.2011.052324

2
ABSTRACT
Background The recent advent of genome-wide molecular platforms has facilitated our
understanding of the human genome and disease, particularly copy number aberrations. The
authors performed genome-wide single nucleotide polymorphism-array in hereditary
coagulopathy to delineate the extent of copy number mutations and to assess its diagnostic
utility.
Design and Methods The study subjects were 17 patients with hereditary coagulopathy from
copy number mutations in coagulation genes detected by multiple ligation-dependent probe
amplification. Eleven had hemophilia (7 hemophilia A and 4 hemophilia B) and 6 had
thrombophilia (4 protein S deficiency and 2 antithrombin deficiency). Single nucleotide
polymorphism-array experiments were performed by using Affymetrix Genome-Wide
Human SNP arrays 6.0.
Results Copy number mutations were identified by single nucleotide polymorphism-array in
9 patients, which ranged in length from 51 Kb to 6,288 Kb harboring 2 to ~160 genes. Single
nucleotide polymorphism-array showed a neutral copy number status in 8 patients including 7
with either a single-exon copy number mutation or duplication mutations of PROS1.
Conclusions This study revealed unexpectedly heterogeneous lengths of copy number
mutations underlying human coagulopathy. Single nucleotide polymorphism-array had
limitations in detecting copy number mutations involving a single exon or those of genes with
homologous sequences such as a pseudogene. (Word count: 200)
Key words: coagulation, contiguous gene deletion, copy number, hemophilia, mutation,
single nucleotide polymorphism-array, thrombophilia
DOI: 10.3324/haematol.2011.052324

3
Introduction
The application of high-throughput genome-wide analysis of genetic variations has
facilitated our understanding of the human genome and the genotype-phenotype
correlations.1-4 The variability of the human genome is largely determined by 2 types
variations, single nucleotide variations and copy number (CN) variations. CN variations
represent genomic segments with a size range of ~100 bp to several Mbs demonstrating an
altered (or non-neutral) dosage status.5 They are typically introduced in the genome by
recombination-based or replication-based mechanisms. The detection of CN variations is
clinically important because they can be the genetic backgrounds of human diseases
including Mendelian disorders, contiguous gene deletion syndromes, and common complex
diseases.6-8 Two representative platforms to detect CN variations are array comparative
genomic hybridization (CGH) and single nucleotide polymorphisms (SNP)-array. SNP-array
produces genotype data from up to 1 million SNPs over the genome determined by the
hybridization signals. The SNP-array platform is proven to be a robust genome analysis tool
not only to obtain genotype data but also to detect CN variations, uniparental disomy, and
consanguinity. 9
Hereditary coagulation disorders encompass a variety of congenital deficiencies of proteins
involved in the physiological balance of the coagulation system. Depending on the molecule
involved, the clinical manifestation can be either hemophilic (pathologic bleeding) or
thrombophilic (pathologic clotting). Deficiencies of coagulation factor VIII (FVIII,
hemophilia A [HA]; MIM 306700) and factor IX (FIX, hemophilia B [HB]; MIM 306900)
from mutations in the F8 and F9 gene, respectively, are one of the representative human
Mendelian disorders, with X-linked recessive inheritance.10 On the other hand, deficiencies of
natural anticoagulants, protein C (PC def, MIM 176860), protein S (PS def, MIM 612336),
DOI: 10.3324/haematol.2011.052324

4
and antithrombin (AT def, MIM 613118), from mutations in PROC, PROS1, and SERPINC1,
cause hereditary thrombophilia. Traditionally, the molecular diagnosis of these hereditary
coagulation disorders has focused on the detection of point mutations by sequencing analyses,
except for the inv(22) mutation of F8 in HA. However, the recent introduction of technically
improved platforms for targeted CN mutation analysis, such as multiplex ligation-dependent
probe amplification (MLPA), have revealed that CN mutations involving one or more exons
to the whole gene are more common than has been recognized in hereditary
coagulopathy.11,12 Moreover, it was described that the deleted region involved unexpectedly
large genomic regions outside the coagulation gene of interest.13
In this regard, we performed high-throughput genome-wide SNP-array experiments in a
series of patients with hereditary coagulopathy to delineate the extent of causative CN
mutations and to assess its diagnostic utility.
Design and Methods
Study subjects
The study subjects were 17 patients with hereditary coagulopathy with CN mutations in the
causative coagulation genes. The characteristics of the patients are shown in Table 1. The
group of hemophilia (bleeders) consisted of 10 patients with hemophilia and 1 hemophilia
carrier (Table 1, H1-H11). Seven patients with HA (H1-H7) had severe FVIII deficiency
including 4 who were inhibitor-positive. Patient H7 was a 5-year-old girl with severe HA
who had been documented to have inherited inv(22) mutation of F8 from her father with
severe HA. Three patients with HB (H8-H10) had severe FIX deficiency including 1 who
was inhibitor-positive (H8). Individual H11 was a female obligatory carrier from a family
DOI: 10.3324/haematol.2011.052324

5
with HB and was tested for prenatal diagnosis. The group of thrombophilia (clotters)
consisted of 5 patients with venous thromboembolism (T1-T3, T5, and T6) and 1
asymptomatic individual (T4) who was screened for coagulopathy as a donor for liver
transplantation. Four had protein S deficiency based on functional coagulation tests (T1-T4),
and 2 had antithrombin deficiency (T5 and T6). The possibility of having CN mutations was
raised in these 17 patients or individuals based on the observation of negative point mutations
in the coding exons and flanking sequences of the target coagulation genes on direct
sequencing analyses. Particularly in male patients with hemophilia, PCR failure of one or
more exons of F8 and F9 genes on the X chromosome strongly indicated exon deletion
mutations and prompted to proceed with MLPA experiments to detect CN mutations.
MLPA analyses
Written informed consent was obtained from all patients or guardians for molecular genetic
diagnosis, under the approval of the Institutional Review Board. Genomic DNA was
extracted from peripheral blood leukocytes using the Wizard Genomic DNA Purification Kit
following the manufacturer's instructions (Promega, Madison, WI, U.S.A.). MLPA
experiments were performed by using commercial kits SALSA MLPA P178 F8, P207 F9,
P265 PROS1, and P227 SERPINC1 (MRC-Holland, Amsterdam, The Netherlands) as
described previously.14 The results of MLPA analyses in 5 of the study patients had been
reported previously (Table 1).
DOI: 10.3324/haematol.2011.052324

6
SNP-array analyses
For the SNP-array experiment, DNA concentration and purity were determined using the
NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies, Rockland, DE, USA). The
DNA samples were genotyped by using the Affymetrix Genome-Wide Human SNP Array
6.0 (Affymetrix, Santa Clara, CA, USA) according to the manufacturer’s instructions. Briefly,
each of 250 ng of DNA samples was digested with Nsp I and Sty I enzymes, followed by
adaptor-ligation and amplification. The amplified DNA was then fragmented, labeled, and
hybridized to the array. The arrays were then washed, scanned, and the image data were
analyzed with respect to the data from control DNA samples using the Hidden Markov Model
on the Genotyping Console 3.0.2 (Affymetrix). The UCSC Genome Browser Build
hg18/NCBI Map Viewer Build 36.3 was used to obtain information on the genomic segments
involved in CN mutations detected and the list of genes thereon.
Results
CN mutations in hemophilia genes
Eleven patients/carrier with hemophilia had CN mutations involving a single exon to the
whole gene identified by MLPA analyses (Table 1, Patients H1-H11). CN mutations of F8 in
7 patients with HA showed a heterogeneous range of segments involved, with only 1 patient
(H7) with a whole gene deletion (14%; 1/7). On the other hand, CN mutations of F9 in 4
patients/carrier of HB were mostly whole gene deletion (75%; 3/4). Both in HA and HB, CN
mutations were mostly deletions (deletion in 10 vs duplication in 1). SNP-array experiments
could successfully delineate the segments of CN mutations in 7 cases out of 10 with
successful results (70%). The SNP-array data were not successfully obtained in H4, possibly
DOI: 10.3324/haematol.2011.052324

7
from the poor quality of specimen (failure on quality control). The length of CN mutations in
hemophilia revealed by SNP-array ranged from ~51 Kb (H6) to ~6,288 Kb (H7) (Table 2 and
Figure 1). In all cases, the CN mutation involved other neighboring genes, from a single gene
(FUNDC2 in H6) to 161 genes (H7), compatible with contiguous gene deletion (Table 2,
Figure 1, and Supplementary Figure 1, A-E). The SNP-array experiment showed a neutral
CN status in 3 patients with hemophilia: Patient H3 with duplication of exon 24 of F8,
Patient H5 with deletion of exon 24 of F8, and Patient H8 with deletion of exon 1 of F9
(Supplementary Figure 2, A-C).
CN mutations in thrombophilia genes
Four patients with PS deficiency were shown to have CN mutations of PROS1 by MLPA, 2
with duplication of exons 5-10 (Table 1, T1 and T4) and one each with deletion of exon 1 (T1)
and exon 2 (T2). Two patients with AT deficiency had deletion of exon 1-6 (T5) and deletion
of exon 6 (T6), respectively. SNP-array experiments could successfully delineate the
segments of CN mutations in 2 cases out of 6 (33%), both of which involved other
neighboring genes (Table 2). In Patient T2 with deletion of exon 1 of PROS1 on MLPA,
SNP-array showed a deletion segment of ~67 Kb including the adjacent ARL13B gene
(Supplementary Figure 1F). In Patient T5 with deletion of exons 1 through 6 of SERPINC1
on MLPA, SNP-array showed a deletion segment of ~66 Kb including the adjacent RC3H1
gene (Supplementary Figure 1G). SNP-array showed a neutral CN status in 2 patents with
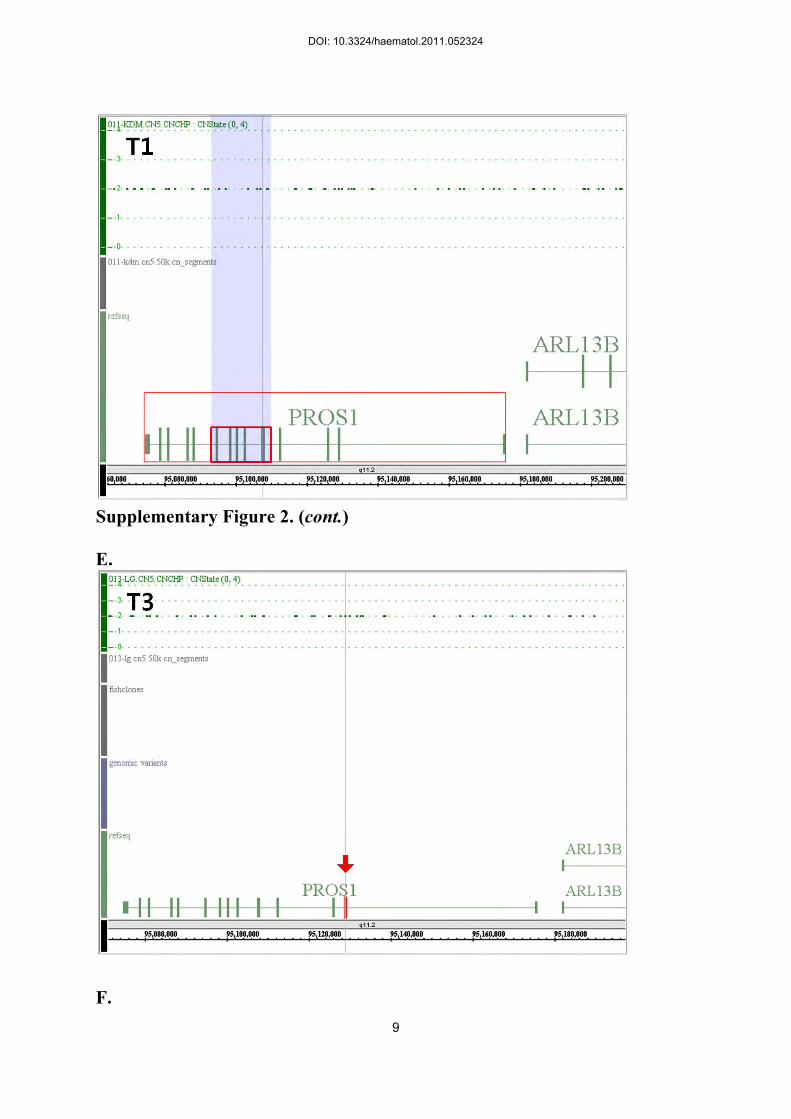
duplication of exons 5-10 of PROS1 (T1 and T4), and Patient T3 with exon 2 deletion of
PROS1, and Patient T6 with deletion of exon 6 of SERPINC1 (Supplementary Figure 2, D-G).
Discussion
DOI: 10.3324/haematol.2011.052324

8
In this study, SNP-array experiments demonstrated unexpectedly heterogeneous lengths of
segments of CN mutations underlying hereditary coagulopathies of Mendelian inheritance,
ranging from 51 Kb to 6.3 Mb. Notably, in all 9 patients with CN mutations detected by SNP-
array, the segment of CN mutation involved other neighboring genes, from 2 to 161 genes
(contiguous gene deletion) (Table 2 and Supplementary Table 1). In the group of hemophilia
(Table 1, Patients H1-H11), whole gene deletion was more frequent in HB (H8-H11, 75%)
than in HA (H1-H7, 14%). In comparison with in the group of hemophilia, the deletion
segments were relatively short in the thrombophilia group; half of patients had a single exon
aberration and only 2 patients had CN mutations involving one or more neighboring gene
(Table 2, T2 and T5). Particularly in thrombophilia, recent studies have shown that large CN
mutations involving one or more exons are particularly relevant to PS deficiency.12 In line
with this observation, PS deficiency from CN mutations of PROS1 accounted for the majority
of cases (67%), and AT deficiency from SERPINC1 mutations accounted for the rest (33%).
As for the genotype-phenotype correlations, we correlated the severity of disease and
inhibitor development in hemophilia cases with regard to the genetic defect identified. HA
and HB are categorized as severe, moderately severe, and mild, based on the residual factor
activity. All patients with HA/HB in our series had the severe phenotype (residual factor
activity <1%), which is in line with the previously acknowledged genotype-phenotype
correlation that large gene rearrangement mutations are associated with severe disease.
Approximately 20-30% of patients with severe HA and 3% of HB develop inhibitors to
defective coagulation factor, which is the most serious and challenging complication.15,16
Large, multi-domain CN mutations are known to be the single most significant risk factor to
inhibitor development with a frequency up to 80% in HA.17 Indeed, the frequency of inhibitor
in our series of large CN mutation was 71% in HA (5/7) and was 33% in HB (1/3) (Table 1).
DOI: 10.3324/haematol.2011.052324

9
Two patients with HA without inhibitor were a male patient with a single exon deletion (H3)
and a female patient with HA (H7). The patient with HB who had high-titer inhibitor (H8)
was previously reported to be the only patient with inhibitor among 33 HB patients.14 Two
patients with HB who were negative for inhibitor were a 2- and 3-year-old boy, respectively,
and thus careful management is mandatory in these young, high-risk patients.
Regarding the phenotypic contribution of the CN aberration in neighboring genes, no patient
among the 9 had definite phenotypic abnormalities other than coagulation defect (Table 2).
The nullisomic deletion of neighboring genes in hereditary coagulopathy was first reported in
2 unrelated patients with HB back in 1988.13 The patients were inhibitor-positive and had a
large deletion encompassing F9 and MCF2 genes on the X chromosome. Of note, they had
no clinical condition attributable to the loss of MCF2. In all 3 cases of HB with CN mutations
by SNP-array, the segments included MCF2 (Supplementary Table 1). Patient H7, a female
patient with HA, was compound heterozygous for inv(22) and the large CN mutation and
demonstrated completely skewed X chromosome inactivation status.18 The CN mutation
involved a genomic segment longer than 6 Mb harboring 161 genes (Figure 1B). Cytogenetic
analysis was not performed in the patient. A CN aberration involving a genomic segment
with a size 5-10 Mb can be detected on cytogenetic analyses. However, the CN mutation in
Patient H7 would not be readily visible because it is on a light band of the tip of X
chromosome (Xq28) on conventional G-bands. Family study revealed that the X chromosome
with the large CN mutation was inherited from her carrier mother, but there was no history of
HA of maternal side. We could not determine whether the large deletion was a de novo
occurrence in her mother or not, since no further family study was performed on the maternal
side. We speculated that a male individual with the large contiguous gene deletion might
have other clinical manifestations in addition to HA. In addition, a meticulous and targeted
DOI: 10.3324/haematol.2011.052324

10
phenotypic assessment could reveal relevant clinical and/or laboratory findings in at least
some of our series of patients with hereditary coagulopathy with contiguous gene deletion.
In 7 patients out of 16 (44%; not including Patient H4 with unsuccessful SNP-array results),
SNP-array showed neutral CN status in the region of interest (Supplementary Figure 2). The
CN mutations in these patients were either deletion/duplication of a single exon (Table 1, H3,
H5, H8, T3, and T6) or duplication mutation of PROS1 (T1 and T4).19 Total 7 patients in our
series (41%) had a CN mutation involving a single exon of the coagulation gene, exon 1
accounting for 43% (3/7). The segment of CN mutations in 2 of these 3 with exon 1
aberration involved neighboring genes. The failure of detection of CN mutation in small
segments involving a single exon, particularly when it was not exon 1, could be due to the
limited coverage of SNP markers in the region of interest. The single exon involved in CN
mutation was not exon 1 in all patients with neutral CN status by SNP-array, except in Patient
H8. On the other hand, the duplication mutation of exons 5-10 of PROS1 in Patients T1 and
T4 were not detected by SNP-array. The SNP-array used in this study has 5-9 SNP markers
in this region, which clearly revealed neutral CN status (Supplementary Figure 2, D and F).
The duplication mutation was previously proven by sequencing analysis.19 In these 2 cases
with PS deficiency, the presence of the pseudogene PROSP, with a high sequence homology
with PROS1 (exons 5-10 of PROS1 correspond to exons 1-6 of PROSP), could be a possible
mechanism underlying the discrepancy between MLPA and SNP-array results.
As demonstrated in the results of this study, MLPA analysis can be the first-line test to
detect CN mutation in single-gene disorders, since every single exon is specifically targeted
in a gene of interest. However, SNP-array can reveal the extent of CN mutation outside the
gene, and also can narrow down the rearrangement breakpoints. In addition, genomic
information other than local CN status can be obtained and utilized for other purposes, for
example, association studies for disease susceptibility. Lastly, the cost of SNP-array rapidly
DOI: 10.3324/haematol.2011.052324

11
decreases recently, which can also be taken into account for its diagnostic utility. The major
disadvantage of SNP-array is the limited coverage (sensitivity) and specificity. A particular
gene of interest might not be densely covered enough to detect or precisely identify the CN
mutation in the gene (as was in Patients H3, H5, H8, T3, and T6). In addition, results cannot
be reliably obtained in targets with high homologous sequences such as pseudogenes (as was
in Patients T1 and T4). To overcome these limitations, targeted SNP-arrays and exon-focused
or custom array-CGH have been developed more recently.
In conclusion, this study is the first investigation on the large CN mutations underlying
hereditary coagulation disorders in human by utilizing high-resolution SNP-array analyses.
SNP-array revealed deletion or duplication mutations involving a single exon of coagulation
genes to contiguous gene deletion. We believe the data from genome-wide SNP-array in
hereditary coagulation disorders would provide evidence to extend our knowledge on
genotype-phenotype correlations and susceptibility of large CN mutations by fine delineation
of boundaries of rearrangements.
Authorship and Disclosures
HK was the principal investigators who designed, performed the research, and wrote the
manuscript. DK, KY, CY, YC, and JY provided clinical data and blood samples of study
subjects, and discussed the content. KL and IP generated and analyzed experimental data, and
HC, HK, MS and SK analyzed the data and discussed the content.
The authors declare no financial conflict of interest to disclose.
DOI: 10.3324/haematol.2011.052324

12
Funding
This study was supported by a grant from the Korea Food & Drug Administration (KFDA).
References 1. Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, et al. Large-scale copy
number polymorphism in the human genome. Science. 2004;305(5683):525-8.
2. Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, et al. Global variation
in copy number in the human genome. Nature. 2006;444(7118):444-54.
3. Eichler EE, Nickerson DA, Altshuler D, Bowcock AM, Brooks LD, Carter NP, et al.
Completing the map of human genetic variation. Nature. 2007;447(7141):161-5.
4. Aitman TJ, Dong R, Vyse TJ, Norsworthy PJ, Johnson MD, Smith J, et al. Copy
number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans.
Nature. 2006;439(7078):851-5.
5. Zhang F, Gu W, Hurles ME, Lupski JR. Copy number variation in human health,
disease, and evolution. Annu Rev Genomics Hum Genet. 2009;10:451-81.
6. Ballif BC, Hornor SA, Jenkins E, Madan-Khetarpal S, Surti U, Jackson KE, et al.
Discovery of a previously unrecognized microdeletion syndrome of 16p11.2-p12.2. Nat
Genet. 2007;39(9):1071-3.
7. Balasubramaniam S, Rudduck C, Bennetts B, Peters G, Wilcken B, Ellaway C.
Contiguous gene deletion syndrome in a female with ornithine transcarbamylase
deficiency. Mol Genet Metab. 2010;99(1):34-41.
8. del Gaudio D, Yang Y, Boggs BA, Schmitt ES, Lee JA, Sahoo T, et al. Molecular
DOI: 10.3324/haematol.2011.052324

13
diagnosis of Duchenne/Becker muscular dystrophy: enhanced detection of dystrophin
gene rearrangements by oligonucleotide array-comparative genomic hybridization.
Hum Mutat. 2008;29(9):1100-7.
9. Schaaf CP, Wiszniewska J, Beaudet AL. Copy number and SNP arrays in clinical
diagnostics. Annu Rev Genomics Hum Genet. 2011;12:11.1-11.27.
10. Rogaev EI, Grigorenko AP, Faskhutdinova G, Kittler EL, Moliaka YK. Genotype
analysis identifies the cause of the "royal disease". Science. 2009;326(5954):817.
11. Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative
quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe
amplification. Nucleic Acids Res. 2002;30(12):e57.
12. Pintao MC, Garcia AA, Borgel D, Alhenc-Gelas M, Spek CA, de Visser MC, et al.
Gross deletions/duplications in PROS1 are relatively common in point mutation-
negative hereditary protein S deficiency. Hum Genet. 2009;126(3):449-56.
13. Anson DS, Blake DJ, Winship PR, Birnbaum D, Brownlee GG. Nullisomic deletion of
the mcf.2 transforming gene in two haemophilia B patients. EMBO J. 1988;7(9):2795-9.
14. Kwon MJ, Yoo KY, Kim HJ, Kim SH. Identification of mutations in the F9 gene
including exon deletion by multiplex ligation-dependent probe amplification in 33
unrelated Korean patients with haemophilia B. Haemophilia. 2008;14(5):1069-75.
15. Kreuz W, Becker S, Lenz E, Martinez-Saguer I, Escuriola-Ettingshausen C, Funk M, et
al. Factor VIII inhibitors in patients with hemophilia A: epidemiology of inhibitor
development and induction of immune tolerance for factor VIII. Semin Thromb Hemost.
1995;21(4):382-9.
16. Scharrer I, Neutzling O. Incidence of inhibitors in haemophiliacs. A review of the
literature. Blood Coagul Fibrinolysis. 1993;4(5):753-8.
17. Oldenburg J, Pavlova A. Genetic risk factors for inhibitors to factors VIII and IX.
DOI: 10.3324/haematol.2011.052324

14
Haemophilia. 2006;12 Suppl 6:15-22.
18 Song MJ, Kim HJ, Yoo KY, Park IA, Lee KO, Ki CS, Kim SH. Molecular
characterization of female hemophilia A by multiplex ligation-dependent probe
amplification analysis and X-chromosome inactivation study. Blood Coagul
Fibrinolysis. 2011;22(3):211-4.
19. Choung HS, Kim HJ, Gwak GY, Kim SH, Kim DK. Inherited protein S deficiency as a
result of a large duplication mutation of the PROS1 gene detected by multiplex
ligation-dependent probe amplification. J Thromb Haemost. 2008;6(8):1430-2.
20. Yoo JH, Kim HJ, Maeng HY, Kim YA, Sun YK, Song JW, et al. Hereditary protein S
deficiency from a novel large deletion mutation of the PROS1 gene detected by
multiplex ligation-dependent probe amplification (MLPA). Thromb Res.
2009;123(5):793-5.
21. You CW, Son HS, Kim HJ, Woo EJ, Kim SA, Baik HW. Mutation analysis of factor
VIII in Korean patients with severe hemophilia A. Int J Hematol. 2010;91(5):784-91.
22. Lee ST, Kim HJ, Kim DK, Schuit RJ, Kim SH. Detection of large deletion mutations in
the SERPINC1 gene causing hereditary antithrombin deficiency by multiplex ligation-
dependent probe amplification (MLPA). J Thromb Haemost. 2008;6(4):701-3.
DOI: 10.3324/haematol.2011.052324

15
Table 1. Seventeen study patients with hereditary coagulation disorders with copy number mutations identified by MLPA analyses.
Patient Age/sex Clinical Dx Coag Dx Gene MLPA Inhibitor
H1 5/M Bleeder HA, severe F8 Del, exon 7-26 21 (+)
H2 2/M Bleeder HA, severe F8 Del, exon 1-22 (+)
H3 24/M Bleeder HA, severe F8 Dup, exon 24 (‐)
H4 26/M Bleeder HA, severe F8 Del, exon 16-19 (+)
H5 43/M Bleeder HA, severe F8 Del, exon 24 (+)
H6 12/M Bleeder HA, severe F8 Del, exon 1 (+)
H7* 5/F Bleeder HA, severe F8 Del, WG 18 (‐)
H8 5/M Bleeder HB, severe F9 Del, exon 1 14 (+)
H9 3/M Bleeder HB, severe F9 Del, WG (‐)
H10 2/M Bleeder HB, severe F9 Del, WG (‐)
H11 60/F ASx HB, carrier F9 Del, WG N/A
T1 39/M PVT PS def. PROS1 Dup, exon 5-10 19 N/A
T2 44/F DVT & PE PS def. PROS1 Del, exon 1 20 N/A
T3 47/M VTE PS def. PROS1 Del, exon 2 N/A
T4 25/M ASx PS def. PROS1 Dup, exon 5-10 N/A
T5 27/M DVT & PE AT def. SERPINC1 Del, exon 1-6 22 N/A
T6 49/M DVT & SMVT AT def. SERPINC1 Del, exon 6 22 N/A
MLPA, multiplex ligation-dependent probe amplification; Dx, diagnosis; Coag, coagulation; HA, hemophilia A; HB, hemophilia B; ASx, asymptomatic; Del, deletion; Dup, duplication; WG, whole gene; N/A, not applicable; def, deficiency; PVT, portal vein thrombosis; DVT, deep vein thrombosis; PE, pulmonary embolism; SMVT, superior mesenteric vein thrombosis. *Also had inv(22) along with the whole gene deletion of F8.18
DOI: 10.3324/haematol.2011.052324

16
Table 2. Copy number mutations identified by SNP-array in 9 patients with hereditary coagulopathy.
Patient MLPA Loss/ gain
Chromosome band
Size (Kb)
Start marker
Start position
End marker
End position
No. of markers
No. of genes*
H1 F8_E7-26_del 21 Loss q28 ~157 CN_921539 153703648 CN_923648 153860284 68 5 (3**)
H2 F8_E1-22_del Loss q28 ~283 CN_923610 153755068 CN_923710 154038338 118 7 (6**)
H6 F8_E1_del Loss q28 ~51 CN_923660 153891415 CN_404047 153942523 15 2 (2**)
H7*** F8_WG_del Loss q28 ~6,288 CN_927985 148598983 SNP_A-8528843 154887040 3,307 161 (126**)
H9 F9_WG_del Loss q27.1 ~797 CN_932113 137863600 CN_940934 138660733 581 13 (7**)
H10 F9_WG_del Loss q26.3-q27.3 ~5,739 CN_932026 137633209 CN_930036 143372580 3,706 42 (28**)
H11 F9_WG_del Loss q26.3-q27.2 ~2,822 CN_932058 137702377 CN_912446 140524120 1,686 27 (16**)
T2 PROS1_E1_del 20 Loss 3q11.2 ~67 CN_1029570 95160010 CN_1031689 95226611 26 4 (3**)
T5 SERPINC1_E1-6_del 22 Loss 1q25.1 ~66 SNP_A-2092517 172143328 CN_009040 172209802 42 2 (2**)
SNP, single nucleotide polymorphism; Coag, coagulation; MLPA, multiplex ligation-dependent probe amplification; del, deletion; dup, duplication; WG, whole gene. *See Supplementary Table 1 for the list of genes. **Only including unique functional genes approved by the HUGO Gene Nomenclature Committee (HGNC). ***Also had inv(22) along with the whole gene deletion of F8.18
DOI: 10.3324/haematol.2011.052324

17
Legends for figures
Figure 1.
The screenshots of the Genotyping Console representing the SNP-array results that showed
copy number (CN) mutations causing hereditary coagulation disorders. The customized
screen consists of 4 parts: from top to bottom, 1) the CN status from target loci (0 to 4;
green dots or bars), 2) the genomic segment with CN aberrations detected (thick red bars;
arrowheads indicate CN loss), 3) refseq (reference sequence) genes in the corresponding
genome), and 4) the idiogram and location of the region. CN of 1 and 2 represent the
neutral status for X chromosome in male and female individuals, respectively. Also note
that transcript variants are shown in the refseq panel (for example, NM_000132 and
NM_019863 for F8 as indicated by blue boxes). A. Patient H6 with severe hemophilia A
with ~51 Kb deletion (CN 0; the genomic segment shaded in light red) involving the exon
1 of F8 (thick black arrow). Note that the adjacent FUNDC2 gene is also included in the
deletion segment. B. Female Patient H7 with severe hemophilia A showing ~6,299 Kb
deletion (CN 1; the genomic segment shaded in light red) involving the whole gene of F8
(small red box in the bottom indicated by a thick black arrow) and 161 neighboring genes.
She also had inv(22) mutation of paternal origin.
DOI: 10.3324/haematol.2011.052324

18
Figure 1A
DOI: 10.3324/haematol.2011.052324

19
Figure 1B
DOI: 10.3324/haematol.2011.052324

20
Supplementary Appendix
DOI: 10.3324/haematol.2011.052324

1
Supplementary Figure 1
The visual representation of copy number (CN) mutations causing hereditary coagulation
disorders revealed by SNP-array experiments obtained by using the Genotyping Console. A.
Patient H1 with severe hemophilia A (HA) with ~157 Kb deletion (CN 0) involving exons 7-
26 of F8. B. Patient H2 with severe HA with ~283 Kb deletion (CN 0) involving exons 1-22
of F8. Note that the adjacent genes, FUNDC2 and BRCC3, are also included in the deletion
segment. C. Patient H9 with severe hemophilia B (HB) with ~797 Kb deletion (CN 0)
involving the whole F9 gene (small red box in the bottom indicated by a red arrow), along
with adjacent genes, MCF2, ATP11C, and CXorf66. D. Patient H10 with severe HB with
~5,739 Kb deletion (CN 0) involving the whole F9 gene (small red box in the bottom
indicated by a red arrow), along with ~45 adjacent genes. E. Patient H11 with severe HB
with ~2,822 Kb deletion (CN 0) involving the whole F9 gene (small red box in the bottom
indicated by a red arrow), along with ~28 adjacent genes. F. Patient T2 with protein S
deficiency with ~67 Kb deletion (CN 1) involving the exon 1 of PROS1 (red arrow), along
with part of the adjacent ARL13B gene. G. Patient T5 with antithrombin deficiency with ~66
Kb deletion (CN 1) involving exons 1-6, along with part of the adjacent RC3H1 gene.
DOI: 10.3324/haematol.2011.052324

2
Supplementary Figure 1.
A.
B.
DOI: 10.3324/haematol.2011.052324

3
Supplementary Figure 1. (cont.)
C.
D.
DOI: 10.3324/haematol.2011.052324

4
Supplementary Figure 1. (cont.)
E.
F.
DOI: 10.3324/haematol.2011.052324

5
Supplementary Figure 1. (cont.)
G.
Supplementary Figure 2.
DOI: 10.3324/haematol.2011.052324

6
The visual representation of the SNP-array data showing neutral copy number (CN) status on
the genomic area of CN mutations causing hereditary coagulation disorders revealed by
multiplex ligation-dependent probe amplification. A. Patient H3 with severe hemophilia A
(HA) from a single exon (exon 24) duplication of F8 (red arrow). B. Patient H5 with severe
HA from a single exon (exon 24) deletion of F8 (red arrow). C. Patient H8 with severe
hemophilia B from a single exon (exon 1) deletion of F9 (red arrow). D. Patient T1 with
protein S (PS) deficiency from duplication of exons 5-10 PROS1 (small red box). Note that
the SNP markers covering this area represent neutral CN status (CN 2). E. Patient T3 with PS
deficiency from deletion of a single exon (exon 2) of PROS1 (red arrow). F. Patient T4 with
PS deficiency from duplication of exons 5-10 of PROS1 (small red box). Note that the SNP
markers covering this area represent neutral CN status (CN 2). G. Patient T6 with
antithrombin deficiency from deletion of a single exon (exon 6) of SERPINC1 (red arrow).
DOI: 10.3324/haematol.2011.052324

7
Supplementary Figure 2.
A.
B.
DOI: 10.3324/haematol.2011.052324

8
Supplementary Figure 2. (cont.)
C.
D.
DOI: 10.3324/haematol.2011.052324
9
Supplementary Figure 2. (cont.)
E.
F.
DOI: 10.3324/haematol.2011.052324

10
Supplementary Figure 2. (cont.)
G.
DOI: 10.3324/haematol.2011.052324
Patient H1
start stop Symbol O Cyto153704817 153717173 LOC100132963 ‐ Xq28153717260 153904192 F8 ‐ Xq28153766511 153767027 H2AFB1 + Xq28153767829 153769530 F8A1 + Xq28153790293 153800681 LOC553820 ‐ Xq28
*Only including unique functional genes approved by the HUGO Gene Nomenclature Committee (HGNC).
Patient H2
Start Stop Symbol O Cyto153717260 153904192 F8 ‐ Xq28153766511 153767027 H2AFB1 + Xq28153767829 153769530 F8A1 + Xq28153790293 153800681 LOC553820 ‐ Xq28153908258 153938385 FUNDC2 + Xq28153943080 153952830 MTCP1 ‐ Xq28153952904 154004543 BRCC3 + Xq28
*Only including unique functional genes approved by the HUGO Gene Nomenclature Committee (HGNC).
Patient H6
Start Stop Symbol O Cyto153717260 153904192 F8 ‐ Xq28153908258 153938385 FUNDC2 + Xq28
*Only including unique functional genes approved by the HUGO Gene Nomenclature Committee (HGNC).
Patient H7
Start Stop Symbol O Cyto148481974 148663581 LOC100130086 ‐ Xq28148575477 148604507 MAGEA11 ‐ Xq28148658294 148662686 LOC100132969 + Xq28148663576 148666336 HSFX1 + Xq28148671402 148677206 MAGEA9 + Xq28148697998 148698275 MAGEA7 + Xq28148770654 148775266 MAGEA8 + Xq28148845967 148847100 LOC642980 ‐ Xq28148847832 148850682 LOC100132460 + Xq28148851073 148857374 CXorf40B ‐ Xq28148864251 148865366 LOC100129660 + Xq28149033269 149035710 LOC643015 + Xq28149149697 149151799 LOC389901 ‐ Xq28149364378 149433104 MAMLD1 + Xq28149487727 149592272 MTM1 + Xq28149612527 149684233 MTMR1 + Xq28149660273 149660722 LOC100129373 + Xq28149685467 149817837 CD99L2 ‐ Xq28149902421 149909906 HMGB3 + Xq28149912632 149913222 LOC392557 + Xq28150094373 150099736 LOC100128688 ‐ Xq28
Region Displayed: 148,599K-154,887K bpNumber of Genes in Region: 161 (126*)
Number of Genes in Region: 5 (3*)Region Displayed: 153,704K-153,860K bp
Number of Genes in Region: 7 (6*)Region Displayed: 153,755K-154,038K bp
Number of Genes in Region: 2 (2*)Region Displayed: 153,891K-153,943K bp
DOI: 10.3324/haematol.2011.052324

150095717 150100588 GPR50 + Xq28150144832 150146845 LOC286456 ‐ Xq28150316363 150328494 LOC203547 + Xq28150428124 150435390 LOC100129236 + Xq28150482663 150595867 PASD1 + Xq28150617435 150620669 PRRG3 + Xq28150635164 150642322 FATE1 + Xq28150653874 150664692 CNGA2 + Xq27150823559 150834002 LOC100128125 ‐ Xq28150832017 150844298 MAGEA4 + Xq28150872252 150893807 GABRE ‐ Xq28150877705 150877785 MIRN224 ‐ Xq28150878755 150878839 MIRN452 ‐ Xq28151033182 151037100 MAGEA5 ‐ Xq28151053547 151057681 MAGEA10 ‐ Xq28151086290 151370487 GABRA3 ‐ Xq28151311346 151311426 MIRN105-1 ‐ Xq28151312548 151312656 MIRN767 ‐ Xq28151313539 151313619 MIRN105-2 ‐ Xq28151369753 151400152 KRT8P8 + Xq28151557293 151572481 GABRQ + Xq28151617901 151621470 MAGEA6 + Xq28151627399 151628403 CSAG2 ‐ Xq28151634487 151637752 MAGEA2B + Xq28151646936 151647478 LOC100130935 ‐ Xq28151649949 151653828 MAGEA12 ‐ Xq28151653884 151660174 CSAG1 + Xq28151669044 151673020 MAGEA2 ‐ Xq28151678543 151679391 CSAG3B + Xq28151685308 151688896 MAGEA3 ‐ Xq28151703603 151708516 psMAGEA ‐ Xq28151746527 151749957 CETN2 ‐ Xq28151750167 151788563 NSDHL + Xq28151833653 151892678 ZNF185 + Xq28151908024 151911417 PNMA5 ‐ Xq28151947786 151948925 LOC100128960 + Xq28151970965 151975594 LOC100129956 ‐ Xq28151975422 151979483 PNMA3 + Xq28151991521 151994057 PNMA6A + Xq28151994652 151996650 PNMA6B ‐ Xq28152041130 152046934 LOC728307 ‐ Xq28152134716 152139310 MAGEA1 ‐ Xq28152190272 152196454 LOC728317 + Xq28152237415 152253805 LOC649201 ‐ Xq28152262060 152270249 ZNF275 + Xq28152315381 152317501 LOC649238 ‐ Xq28152329775 152340280 ZFP92 + Xq28152363372 152365139 TREX2 ‐ Xq28152366318 152389283 UCHL5IP ‐ Xq28152392294 152405740 LOC389904 + Xq28152413605 152428198 BGN + Xq28152454774 152501581 ATP2B3 + Xq28152506577 152517775 FAM58A ‐ Xq28152521870 152523799 KRT18P48 ‐ Xq28152525166 152529360 LOC100131652 + Xq28152561182 152569971 DUSP9 + Xq28152580778 152581390 LOC347544 + Xq28
DOI: 10.3324/haematol.2011.052324

152588417 152592974 PNCK ‐ Xq28152606586 152615234 SLC6A8 + Xq28152619146 152643081 BCAP31 ‐ Xq28152643530 152663375 ABCD1 + Xq28152682905 152697989 PLXNB3 + Xq28152699704 152704381 SRPK3 + Xq28152704415 152713161 IDH3G ‐ Xq28152713288 152717148 SSR4 + Xq28152720817 152749197 PDZD4 ‐ Xq28152759900 152760217 CYCSP45 + Xq28152780581 152794505 L1CAM ‐ Xq28152799321 152807638 LCAP + Xq28152823564 152825834 AVPR2 + Xq28152826025 152844892 ARHGAP4 ‐ Xq28152848571 152853662 ARD1A ‐ Xq28152853917 152863426 RENBP ‐ Xq28152866202 152890013 HCFC1 ‐ Xq28152891185 152901840 TMEM187 + Xq28152929151 152938536 IRAK1 ‐ Xq28152940458 153016323 MECP2 ‐ Xq28153013619 153016356 LOC100128952 + Xq28153062939 153077701 OPN1LW + Xq28153077878 153098828 TEX28P2 ‐ Xq28153101361 153114725 OPN1MW + Xq28153114908 153136049 TEX28P1 ‐ Xq28153138461 153151953 OPN1MW2 + Xq28153152126 153176632 TEX28 ‐ Xq28153177345 153211894 TKTL1 + Xq28153230091 153252845 FLNA ‐ Xq28153254916 153255347 LOC100131857 ‐ Xq28153260981 153263075 EMD + Xq28153279912 153283874 RPL10 + Xq28153281816 153281950 SNORA70 + Xq28153282773 153293621 DNASE1L1 ‐ Xq28153293071 153303259 TAZ + Xq28153310172 153318056 ATP6AP1 + Xq28153318715 153325008 GDI1 + Xq28153325702 153332190 FAM50A + Xq28153339817 153355179 PLXNA3 + Xq28153358435 153360790 LAGE3 ‐ Xq28153365250 153368126 UBL4A ‐ Xq28153368842 153372189 SLC10A3 ‐ Xq28153387700 153397567 FAM3A ‐ Xq28153412800 153428981 G6PD ‐ Xq28153423653 153446455 IKBKG + Xq28153449493 153463455 LOC643894 + Xq28153452673 153453385 LOC100132967 + Xq28153462002 153463637 LOC340600 + Xq28153466610 153468269 CTAG1A + Xq28153499059 153500714 CTAG1B ‐ Xq28153513930 153514642 CXorf52 ‐ Xq28153520760 153529845 IKBKGP ‐ Xq28153533445 153535036 CTAG2 ‐ Xq28153540101 153541229 OR3B1P ‐ Xq28153556720 153632542 GAB3 ‐ Xq28153644344 153659154 DKC1 + Xq28153649997 153650128 SNORA36A + Xq28
DOI: 10.3324/haematol.2011.052324

153656467 153656595 SNORA56 + Xq28153660162 153686957 MPP1 ‐ Xq28153697668 153699090 LOC728776 ‐ Xq28153704817 153717173 LOC100132963 ‐ Xq28153717260 153904192 F8 ‐ Xq28153766511 153767027 H2AFB1 + Xq28153767829 153769530 F8A1 + Xq28153790293 153800681 LOC553820 ‐ Xq28153908258 153938385 FUNDC2 + Xq28153943080 153952830 MTCP1 ‐ Xq28153952904 154004543 BRCC3 + Xq28154097744 154121292 VBP1 + Xq28154140720 154147046 RAB39B ‐ Xq28154158694 154217180 CLIC2 ‐ Xq28154174358 154176025 PHF10P1 + Xq28154231158 154233743 LOC553939 + Xq28154263630 154263977 H2AFB2 + Xq28154264958 154266073 F8A2 + Xq28154340340 154341455 F8A3 ‐ Xq28154342274 154342790 H2AFB3 ‐ Xq28154372967 154495791 TMLHE ‐ Xq28154650645 154665311 SPRY3 + Xq28 and Yq12154710950 154712518 AMDP1 ‐ Xq22‐q28154764207 154826614 VAMP7 + Xq28 and Yq12154868104 154869205 TRPC6P ‐ Xq28;Yq12154880440 154893676 IL9R + Xq28 and Yq12
*Only including unique functional genes approved by the HUGO Gene Nomenclature Committee (HGNC).
Patient H9
Start Stop Symbol O Cyto138271765 138272307 CXorf19 + Xq27138356541 138358898 SRD5AP1 + Xq24‐qter138440561 138473283 F9 + Xq27.1‐q27.2138491596 138618047 MCF2 ‐ Xq27138630404 138630607 BCYRN1P1 + Xq27.1138636171 138742113 ATP11C ‐ Xq27.1138833972 138834055 MIRN505 ‐ Xq27.1138865550 138875345 LOC347487 ‐ Xq27.1138927087 138927864 LOC728660 + Xq27.1138942029 138943795 LOC643689 + Xq27.1139001492 139002736 LOC389895 + Xq27.1139125851 139126622 LOC729078 ‐ Xq27.1139306993 139307824 LOC266694 + Xq27.1
*Only including unique functional genes approved by the HUGO Gene Nomenclature Committee (HGNC).
Patient H10
Start Stop Symbol O Cyto137541401 137649181 FGF13 ‐ Xq26.3137716202 137716994 LOC100130620 + Xq26.3138271765 138272307 CXorf19 + Xq27138356541 138358898 SRD5AP1 + Xq24‐qter138440561 138473283 F9 + Xq27.1‐q27.2138491596 138618047 MCF2 ‐ Xq27138630404 138630607 BCYRN1P1 + Xq27.1
Number of Genes in Region: 13 (7*)Region Displayed: 137,864K-139,329K bp
Number of Genes in Region: 42 (28*)Region Displayed: 137,633K-143,373K bp
DOI: 10.3324/haematol.2011.052324

138636171 138742113 ATP11C ‐ Xq27.1138833972 138834055 MIRN505 ‐ Xq27.1138865550 138875345 LOC347487 ‐ Xq27.1138927087 138927864 LOC728660 + Xq27.1138942029 138943795 LOC643689 + Xq27.1139001492 139002736 LOC389895 + Xq27.1139125851 139126622 LOC729078 ‐ Xq27.1139306993 139307824 LOC266694 + Xq27.1139412818 139414891 SOX3 ‐ Xq27.1139421081 139624662 LOC286411 + Xq27.1139693091 139694389 CDR1 ‐ Xq27.1‐q27.2139912422 139913537 LOC100133171 + Xq27.1139924427 139925545 SPANXB1 + Xq27.1140060218 140061190 dJ507I15.1 + Xq27.1140097596 140098976 LDOC1 ‐ Xq27140163262 140164312 SPANXC ‐ Xq27.1140185124 140186001 LOC100132808 ‐ Xq27.2140302507 140303107 NDUFB3P5 + Xq26.3‐q27.3140418515 140565735 CXorf18 + Xq27.2140499472 140500502 SPANXA1 ‐ Xq27.1140505498 140506567 SPANXA2 + Xq27.1140541651 140554453 LOC645188 + Xq27.2140613234 140614264 SPANXD ‐ Xq27.1140753768 140813284 MAGEC3 + Xq27.2140819346 140824849 MAGEC1 + Xq26141063478 141091530 LOC392555 ‐ Xq27.2141117796 141120742 MAGEC2 ‐ Xq27141941370 141949732 SPANXN4 + Xq27.3142424230 142432973 SPANXN3 ‐ Xq27.3142510323 142511589 MYCL3 ‐ Xq27.2142543584 142550685 SLITRK4 ‐ Xq27.3142587891 142589106 LOC100129368 ‐ Xq27.3142622721 142632182 SPANXN2 ‐ Xq27.3142794839 142796028 UBE2NL + Xq27.3143219843 143220608 RRM2P4 + Xq27
*Only including unique functional genes approved by the HUGO Gene Nomenclature Committee (HGNC).
Patient H11
Start Stop Symbol O Cyto137716202 137716994 LOC100130620 + Xq26.3138271765 138272307 CXorf19 + Xq27138356541 138358898 SRD5AP1 + Xq24‐qter138440561 138473283 F9 + Xq27.1‐q27.2138491596 138618047 MCF2 ‐ Xq27138630404 138630607 BCYRN1P1 + Xq27.1138636171 138742113 ATP11C ‐ Xq27.1138833972 138834055 MIRN505 ‐ Xq27.1138865550 138875345 LOC347487 ‐ Xq27.1138927087 138927864 LOC728660 + Xq27.1138942029 138943795 LOC643689 + Xq27.1139001492 139002736 LOC389895 + Xq27.1139125851 139126622 LOC729078 ‐ Xq27.1139306993 139307824 LOC266694 + Xq27.1139412818 139414891 SOX3 ‐ Xq27.1139421081 139624662 LOC286411 + Xq27.1
Number of Genes in Region: 27 (16*)Region Displayed: 137,702K-140,524K bp
DOI: 10.3324/haematol.2011.052324
139693091 139694389 CDR1 ‐ Xq27.1‐q27.2139912422 139913537 LOC100133171 + Xq27.1139924427 139925545 SPANXB1 + Xq27.1140060218 140061190 dJ507I15.1 + Xq27.1140097596 140098976 LDOC1 ‐ Xq27140163262 140164312 SPANXC ‐ Xq27.1140185124 140186001 LOC100132808 ‐ Xq27.2140302507 140303107 NDUFB3P5 + Xq26.3‐q27.3140418515 140565735 CXorf18 + Xq27.2140499472 140500502 SPANXA1 ‐ Xq27.1140505498 140506567 SPANXA2 + Xq27.1
*Only including unique functional genes approved by the HUGO Gene Nomenclature Committee (HGNC).
Patient T2
Start Stop Symbol O Cyto95074647 95175395 PROS1 ‐ 3q11.295181672 95256813 ARL13B + 3q11.295190166 95190956 LOC100129555 ‐ 3q11.295215903 95230144 STX19 ‐ 3q11
*Only including unique functional genes approved by the HUGO Gene Nomenclature Committee (HGNC).
Patient T5
Start Stop Symbol O Cyto172139565 172153096 SERPINC1 ‐ 1q23‐q25.1172166975 172228833 RC3H1 ‐ 1q25.1
*Only including unique functional genes approved by the HUGO Gene Nomenclature Committee (HGNC).
**Above information was obtained form the UCSC Genome Browser Build hg18/NCBI Map Viewer Build 36.3.
Number of Genes in Region: 4 (3*)Region Displayed: 95,160K-95,227K bp
Number of Genes in Region: 2 (2*)Region Displayed: 172,143K-172,210K bp
DOI: 10.3324/haematol.2011.052324

Descriptionsimilar to hCG1808463coagulation factor VIII, procoagulant component (hemophilia A)
H2A histone family, member B1coagulation factor VIII‐associated (intronic transcript) 1eukaryotic translation elongation factor 1 alpha 1 pseudogene
*Only including unique functional genes approved by the HUGO Gene Nomenclature Committee (HGNC).
Descriptioncoagulation factor VIII, procoagulant component (hemophilia A)
H2A histone family, member B1coagulation factor VIII‐associated (intronic transcript) 1eukaryotic translation elongation factor 1 alpha 1 pseudogene
FUN14 domain containing 2mature T‐cell proliferation 1BRCA1/BRCA2‐containing complex, subunit 3
*Only including unique functional genes approved by the HUGO Gene Nomenclature Committee (HGNC).
Descriptioncoagulation factor VIII, procoagulant component (hemophilia A)
FUN14 domain containing 2*Only including unique functional genes approved by the HUGO Gene Nomenclature Committee (HGNC).
Descriptionhypothetical protein LOC100130086melanoma antigen family A, 11hypothetical protein LOC100132969heat shock transcription factor family, X linked 1melanoma antigen family A, 9melanoma antigen family A, 7, pseudogenemelanoma antigen family A, 8hypothetical LOC642980similar to LOC100126053 proteinchromosome X open reading frame 40Bsimilar to hCG2000002similar to nucleolar protein 11similar to ATP‐dependent DNA helicase 2 subunit 1 (ATP‐dependent DNA helicase II 70 kDa subunit) (Lupus Ku autoantigen protein p70) (Ku70) (70 kDa subunit of Ku antigen) (Thyroid‐lupus autoantigen) (TLAA) (CTC box‐binding factor 75 kDa subunit) (CT...
mastermind‐like domain containing 1myotubularin 1myotubularin related protein 1hypothetical protein LOC100129373CD99 molecule‐like 2high‐mobility group box 3similar to ribosomal protein L19hypothetical protein LOC100128688
DOI: 10.3324/haematol.2011.052324

G protein‐coupled receptor 50similar to NGFI‐A binding protein 1hypothetical protein LOC203547similar to hCG1653500PAS domain containing 1proline rich Gla (G‐carboxyglutamic acid) 3 (transmembrane)
fetal and adult testis expressed 1cyclic nucleotide gated channel alpha 2similar to hCG1653500melanoma antigen family A, 4gamma‐aminobutyric acid (GABA) A receptor, epsilonmicroRNA 224microRNA 452melanoma antigen family A, 5melanoma antigen family A, 10gamma‐aminobutyric acid (GABA) A receptor, alpha 3microRNA 105‐1microRNA 767microRNA 105‐2keratin 8 pseudogene 8gamma‐aminobutyric acid (GABA) receptor, thetamelanoma antigen family A, 6CSAG family, member 2melanoma antigen family A, 2Bsimilar to CSAG family, member 2melanoma antigen family A, 12chondrosarcoma associated gene 1melanoma antigen family A, 2CSAG family, member 3Bmelanoma antigen family A, 3melanoma antigen pseudogene, family Acentrin, EF‐hand protein, 2NAD(P) dependent steroid dehydrogenase‐likezinc finger protein 185 (LIM domain)paraneoplastic antigen like 5similar to hCG1645335hypothetical LOC100129956paraneoplastic antigen MA3paraneoplastic antigen like 6Aparaneoplastic antigen like 6Bsimilar to melanoma associated antigen (mutated) 1‐like 1melanoma antigen family A, 1 (directs expression of antigen MZ2‐E)
similar to melanoma associated antigen (mutated) 1‐like 1similar to paraneoplastic antigen MA3zinc finger protein 275similar to paraneoplastic antigen MA3zinc finger protein 92 homolog (mouse)three prime repair exonuclease 2UCHL5 interacting proteinsimilar to extracellular matrix protein 2biglycanATPase, Ca++ transporting, plasma membrane 3family with sequence similarity 58, member Akeratin 18 pseudogene 48hypothetical LOC100131652dual specificity phosphatase 9ribosomal protein L18a pseudogene
DOI: 10.3324/haematol.2011.052324

pregnancy upregulated non‐ubiquitously expressed CaM kinasesolute carrier family 6 (neurotransmitter transporter, creatine), member 8
B‐cell receptor‐associated protein 31ATP‐binding cassette, sub‐family D (ALD), member 1plexin B3SFRS protein kinase 3isocitrate dehydrogenase 3 (NAD+) gammasignal sequence receptor, delta (translocon‐associated protein delta)
PDZ domain containing 4cytochrome c, somatic pseudogene 45L1 cell adhesion moleculelung carcinoma‐associated proteinarginine vasopressin receptor 2 (nephrogenic diabetes insipidus)
Rho GTPase activating protein 4ARD1 homolog A, N‐acetyltransferase (S. cerevisiae)renin binding proteinhost cell factor C1 (VP16‐accessory protein)transmembrane protein 187interleukin‐1 receptor‐associated kinase 1methyl CpG binding protein 2 (Rett syndrome)hypothetical protein LOC100128952opsin 1 (cone pigments), long‐wave‐sensitive (color blindness, protan)
testis expressed 28 pseudogene 2opsin 1 (cone pigments), medium‐wave‐sensitive (color blindness, deutan)
testis expressed 28 pseudogene 1opsin 1 (cone pigments), medium‐wave‐sensitive 2testis expressed 28transketolase‐like 1filamin A, alpha (actin binding protein 280)hypothetical protein LOC100131857emerin (Emery‐Dreifuss muscular dystrophy)ribosomal protein L10small nucleolar RNA, H/ACA box 70deoxyribonuclease I‐like 1tafazzin (cardiomyopathy, dilated 3A (X‐linked); endocardial fibroelastosis 2; Barth syndrome)
ATPase, H+ transporting, lysosomal accessory protein 1GDP dissociation inhibitor 1family with sequence similarity 50, member Aplexin A3L antigen family, member 3ubiquitin‐like 4Asolute carrier family 10 (sodium/bile acid cotransporter family), member 3
family with sequence similarity 3, member Aglucose‐6‐phosphate dehydrogenaseinhibitor of kappa light polypeptide gene enhancer in B‐cells, kinase gamma
similar to Cyclic AMP‐dependent transcription factor ATF‐4 (Activating transcription factor 4) (DNA‐binding protein TAXREB67) (Cyclic AMP response element‐binding protein 2) (CREB2)
similar to hCG2042244cyclic‐AMP‐dependent transcription factor ATF‐4 pseudogene
cancer/testis antigen 1Acancer/testis antigen 1Bchromosome X open reading frame 52inhibitor of kappa light polypeptide gene enhancer in B‐cells, kinase gamma pseudogene
cancer/testis antigen 2olfactory receptor, family 3, subfamily B, member 1 pseudogene
GRB2‐associated binding protein 3dyskeratosis congenita 1, dyskerinsmall nucleolar RNA, H/ACA box 36A
DOI: 10.3324/haematol.2011.052324

small nucleolar RNA, H/ACA box 56membrane protein, palmitoylated 1, 55kDahigh‐mobility group nucleosome binding domain 1 pseudogene
similar to hCG1808463coagulation factor VIII, procoagulant component (hemophilia A)
H2A histone family, member B1coagulation factor VIII‐associated (intronic transcript) 1eukaryotic translation elongation factor 1 alpha 1 pseudogene
FUN14 domain containing 2mature T‐cell proliferation 1BRCA1/BRCA2‐containing complex, subunit 3von Hippel‐Lindau binding protein 1RAB39B, member RAS oncogene familychloride intracellular channel 2PHD finger protein 10 pseudogene 1trimethyllysine hydroxylase, epsilon pseudogeneH2A histone family, member B2coagulation factor VIII‐associated (intronic transcript) 2coagulation factor VIII‐associated (intronic transcript) 3H2A histone family, member B3trimethyllysine hydroxylase, epsilonsprouty homolog 3 (Drosophila)S‐adenosylmethionine decarboxylase pseudogene 1vesicle‐associated membrane protein 7transient receptor potential cation channel, subfamily C, member 6 pseudogene
interleukin 9 receptor*Only including unique functional genes approved by the HUGO Gene Nomenclature Committee (HGNC).
Descriptionchromosome X open reading frame 19steroid‐5‐alpha‐reductase, alpha polypeptide pseudogene 1 (3‐oxo‐5 alpha‐steroid delta 4‐dehydrogenase alpha pseudogene)
coagulation factor IX (plasma thromboplastic component, Christmas disease, hemophilia B)
MCF.2 cell line derived transforming sequencebrain cytoplasmic RNA 1, pseudogene 1ATPase, class VI, type 11CmicroRNA 505hypothetical LOC347487hypothetical protein LOC728660heterogeneous nuclear ribonucleoprotein A3 pseudogenesimilar to CG4768‐PAhypothetical protein LOC729078embryonic ectoderm development pseudogene
*Only including unique functional genes approved by the HUGO Gene Nomenclature Committee (HGNC).
Descriptionfibroblast growth factor 13hypothetical LOC100130620chromosome X open reading frame 19steroid‐5‐alpha‐reductase, alpha polypeptide pseudogene 1 (3‐oxo‐5 alpha‐steroid delta 4‐dehydrogenase alpha pseudogene)
coagulation factor IX (plasma thromboplastic component, Christmas disease, hemophilia B)
MCF.2 cell line derived transforming sequencebrain cytoplasmic RNA 1, pseudogene 1
DOI: 10.3324/haematol.2011.052324

ATPase, class VI, type 11CmicroRNA 505hypothetical LOC347487hypothetical protein LOC728660heterogeneous nuclear ribonucleoprotein A3 pseudogenesimilar to CG4768‐PAhypothetical protein LOC729078embryonic ectoderm development pseudogeneSRY (sex determining region Y)‐box 3hypothetical protein LOC286411cerebellar degeneration‐related protein 1, 34kDahypothetical protein LOC100133171SPANX family, member B1ribosomal protein L36a pseudogeneleucine zipper, down‐regulated in cancer 1SPANX family, member Chypothetical LOC100132808NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 3, 12kDa pseudogene 5
chromosome X open reading frame 18sperm protein associated with the nucleus, X‐linked, family member A1
SPANX family, member A2hypothetical LOC645188SPANX family, member Dmelanoma antigen family C, 3melanoma antigen family C, 1similar to Melanoma‐associated antigen C2 (MAGE‐C2 antigen) (MAGE‐E1 antigen) (Hepatocellular carcinoma‐associated antigen 587) (Cancer‐testis antigen 10) (CT10)
melanoma antigen family C, 2SPANX family, member N4SPANX family, member N3v‐myc myelocytomatosis viral oncogene homolog 3 (avian) (pseudogene)
SLIT and NTRK‐like family, member 4hypothetical LOC100129368SPANX family, member N2ubiquitin‐conjugating enzyme E2N‐likeribonucleotide reductase M2 polypeptide pseudogene 4
*Only including unique functional genes approved by the HUGO Gene Nomenclature Committee (HGNC).
Descriptionhypothetical LOC100130620chromosome X open reading frame 19steroid‐5‐alpha‐reductase, alpha polypeptide pseudogene 1 (3‐oxo‐5 alpha‐steroid delta 4‐dehydrogenase alpha pseudogene)
coagulation factor IX (plasma thromboplastic component, Christmas disease, hemophilia B)
MCF.2 cell line derived transforming sequencebrain cytoplasmic RNA 1, pseudogene 1ATPase, class VI, type 11CmicroRNA 505hypothetical LOC347487hypothetical protein LOC728660heterogeneous nuclear ribonucleoprotein A3 pseudogenesimilar to CG4768‐PAhypothetical protein LOC729078embryonic ectoderm development pseudogeneSRY (sex determining region Y)‐box 3hypothetical protein LOC286411
DOI: 10.3324/haematol.2011.052324

cerebellar degeneration‐related protein 1, 34kDahypothetical protein LOC100133171SPANX family, member B1ribosomal protein L36a pseudogeneleucine zipper, down‐regulated in cancer 1SPANX family, member Chypothetical LOC100132808NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 3, 12kDa pseudogene 5
chromosome X open reading frame 18sperm protein associated with the nucleus, X‐linked, family member A1
SPANX family, member A2*Only including unique functional genes approved by the HUGO Gene Nomenclature Committee (HGNC).
Descriptionprotein S (alpha)ADP‐ribosylation factor‐like 13Bsimilar to High‐mobility group nucleosome binding domain 1syntaxin 19
*Only including unique functional genes approved by the HUGO Gene Nomenclature Committee (HGNC).
Descriptionserpin peptidase inhibitor, clade C (antithrombin), member 1
ring finger and CCCH‐type zinc finger domains 1*Only including unique functional genes approved by the HUGO Gene Nomenclature Committee (HGNC).
**Above information was obtained form the UCSC Genome Browser Build hg18/NCBI Map Viewer Build 36.3.
DOI: 10.3324/haematol.2011.052324







