
HIGH PERFORMANCE ELECTRONIC STRUCTURE
THEORY
Mark S. Gordon, Klaus Ruedenberg
Ames Laboratory
Iowa State University
BBG

OUTLINE
• Methods and Strategies– Correlated electronic structure methods– Distributed Data Interface (DDI)– Approaches to efficient HPC in chemistry– Scalability with examples

CORRELATED ELECTRONIC STRUCTURE METHODS
• Well Correlated Methods Needed for– Accurate relative energies, dynamics– Treatment of excited states, photochemistry– Structures of diradicals, complex species
• Computationally demanding: Scalability important• HF Often Reasonable Starting Point for Ground
States, Small Diradical Character– Single reference perturbation theory
• MP2/MBPT2 Scales ~N5
• Size consistent• Higher order MBPT methods often perform worse

SINGLE REFERENCE COUPLED CLUSTER METHODS
– Cluster expansion is more robust• Can sum all terms in expansion• Size-consistent
– State-of-the-art single reference method• CCSD, CCSDT, CCSDTQ, …• CCSD(T), CR-CCSD(T): efficient compromise
– Scales ~N7
• Methods often fail for bond-breaking: consider N2
– Breaking 3 bonds: – Minimal active space = (6,6)
Internuclear Separation (A)1.01.52.02.53.03.54.0Relative Energy (kJ/mol)01002003004005006007008009001000
MBPT(2)CCSD(T) CASPT2MRCIMCSCF (10,8)

MCSCF METHODS
• Single configuration methods can fail for– Species with significant diradical character– Bond breaking processes– Often for excited electronic states– Unsaturated transition metal complexes
• Then MCSCF-based method is necessary
• Most common approach is– Complete active space SCF (CASSCF/FORS)
• Active space = orbitals+electrons involved in process• Full CI within active space: optimize orbitals & CI coeffs• Size-consistent

MULTI-REFERENCE METHODS
• Multi reference methods, based on MCSCF– Second order perturbation theory (MRPT2)
• Relatively computationally efficient• Size consistency depends on implementation
– Multi reference configuration interaction (MRCI)• Very accurate, very time-consuming• Highly resource demanding• Most common is MR(SD)CI• Generally limited to (14,14) active space• Not size-consistent
– How to improve efficiency?

Internuclear Separation (A)1.01.52.02.53.03.54.0Relative Energy (kJ/mol)01002003004005006007008009001000
MBPT(2)CCSD(T) CASPT2MRCIMCSCF (10,8)

DISTRIBUTED PARALLEL COMPUTING
• Distribute large arrays among available processors
• Distributed Data Interface (DDI) in GAMESS– Developed by G. Fletcher, M. Schmidt, R. Olson– Based on one-sided message passing– Implemented on T3E using SHMEM– Implemented on clusters using sockets or MPI,
and paired CPU/data server

The virtual shared-memory model. Each large box (grey) represents the memory available to a given CPU. The inner boxes represent the memory used by the parallel processes (rank in lower right). The gold region depicts the memory reserved for the storage of distributed data. The arrows indicate memory access (through any means) for the distributed operations: get, put and accumulate.

FULL shared-memory model:
All DDI processes within a node attach to all the shared-memory segments.
The accumulate operation shown can now be completed directly through memory.

CURRENTLY DDI ENABLED
• Currently implemented– Closed shell MP2 energies & gradients
• Most efficient closed shell correlated method when appropriate (single determinant)
• Geometry optimizations• Reaction path following• On-the-fly “direct dynamics
– Unrestricted open shell MP2 energies & gradients• Simplest correlated method for open shells
– Restricted open shell (ZAPT2) energies & grad• Most efficient open shell correlated method• No spin contamination through second order

CURRENTLY DDI ENABLED
– CASSCF Hessians• Necessary for vibrational frequencies, transition state
searches, building potential energy surfaces
– MRMP2 energies• Most efficient correlated multi-reference method
– Singles CI energies & gradients• Simplest qualitative method for excited electronic states
– Full CI energies• Exact wavefunction for a given atomic basis
– Effective fragment potentials• Sophisticated model for intermolecular interactions

COMING TO DDI
• In progress– Vibronic (derivative) coupling (Tim Dudley)
• Conical intersections, photochemistry
– GVVPT2 energies&gradients: Mark Hoffmann– ORMAS energies, gradients
• Joe Ivanic, Andrey Adsatchev• Subdivides CASSCF active space into subspaces
– Coupled cluster methods • Ryan Olson, Ian Pimienta, Alistair Rendell• Collaboration w/ Piotr Piecuch, Ricky Kendall
• Key Point:– Must grow problem size to maximize scalability

FULL CI: ZHENGTING GAN
– Full CI = exact wavefunction for given atomic basis– Extremely computationally demanding
• Scales ~ eN
• Can generally only be applied to atoms & small molecules
• Very important because all other approximate methods can be benchmarked against Full CI
• Can expand the size of applicable molecules by making the method highly scalable/parallel
• CI part of FORS/CASSCF

0
4
8
12
16
20
24
28
32
0 4 8 12 16 20 24 28 32 36NProcs
Speedup
FCI(14,14)*FCI(14,15)**
– Parallel performance for FCI on IBM P3 cluster• * singlet state of H3COH:
– 14 electrons in 14 orbitals– 11,778,624 determinants
• ** singlet state of H2O2
– 14 electrons in 15 orbitals– 41,409,225 determinants
JCP, 119, 47 (2003)

0
32
64
96
128
160
192
224
256
0 32 64 96 128 160 192 224 256MSPs
SpeedUp SpeedUp
– Parallel performance for FCI on Cray X1 (ORNL)• O-
– Aug-cc-pVTZ atomic basis, O 1s orbitals frozen– 7 valence electrons in 79 orbitals– 14,851,999,576 determinants: ~ 8-10 Gflops/12.5 theoretical
– Latest results:aug-cc-pVTZ C2, 8 electrons in 68 orbitals
– 64,931,348,928 determinants, < 4 hours wall time!
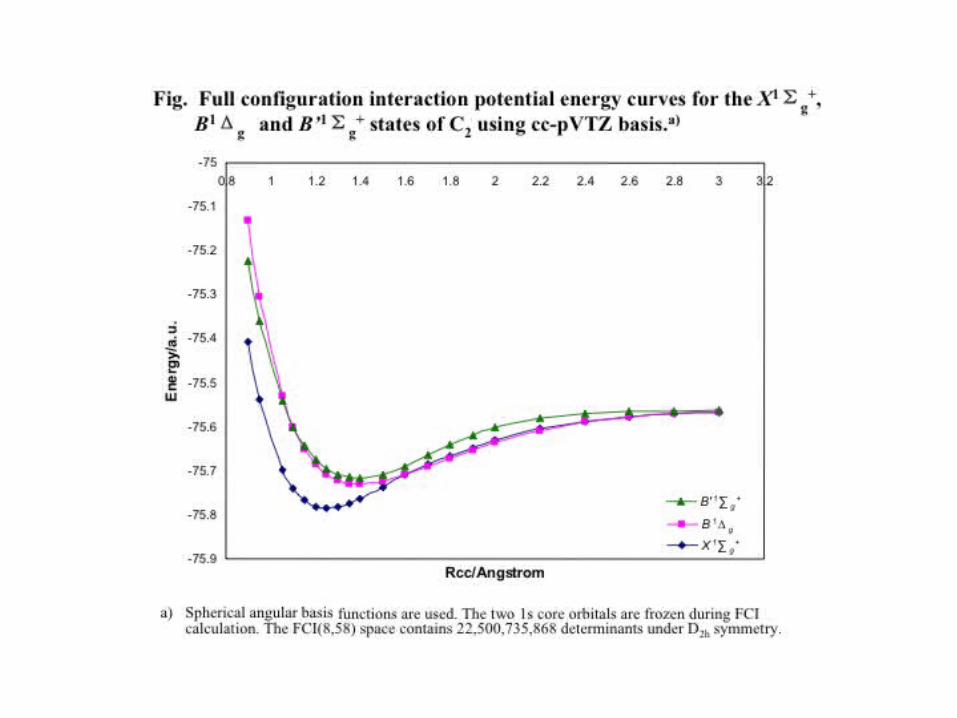
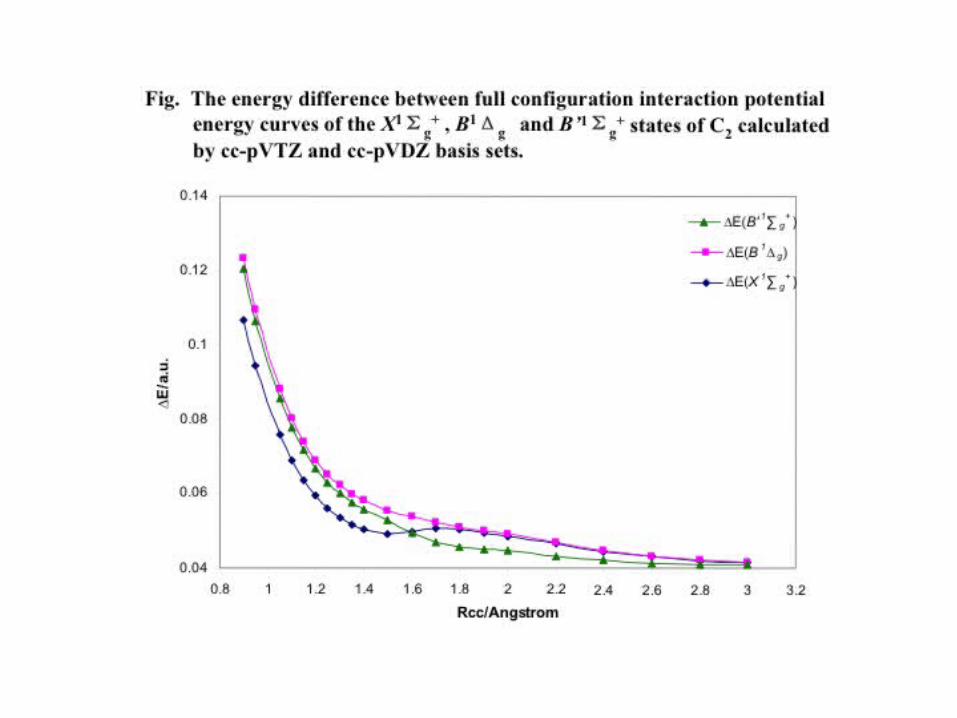

C2 Vertical excitation energies (eV):
cc-pVTZ cc-pVQZ a-cc-pVTZ a-cc-pVQZ
1Δg(1Ag):E -OM CCSD 4.68 4.76 4.67 4.76CR-EOM-CCSD(T) .48 .48 .56 .57FCI .18
1Πu(1Bu):E -OM CCSD 1.33 1.30 1.3 1.30CR-EOM-CCSD(T) 1.31 1.30 1.30 1.30FCI 1.8
1Σu+(1B1u):
E -OM CCSD 5.6 5.56 5.58 5.55CR-EOM-CCSD(T) 5.8 5.51 5.5 5.51FCI 5.47
1Πg(1Bg):E -OM CCSD 6.49 6.53 6.45 6.51CR-EOM-CCSD(T) 4.45 4.4 4.50 4.50FCI 4.38
– Comparison with Coupled Cluster

Correlation Energy Extrapolation by Intrinsic Scaling: CEEISAn Alternative Approach to Full CI
Rigorous variational energyDetermined as complete basis set (CBS) limit of FCI calculations in termsof systematically consistent basis sets.
Extrapolation to the CBS limitExtrapolation formulas for Dunning DZ, TZ, ... XZ AO bases
FCI for a given orbital basisRequires solution of the eigenvalue problem for Ψ=su mo f A LL Slaterdeterminantsgeneratedb y th eorbitalbasis.Th is is impossiblebecaus etheexpansion s a remuc h too long.
However:The ycontainove r 99%deadwood.
Question:Howt o selectaprioriall liv e woo d thatisrequiredforachievinganaccuracyof ≤1m /hmolecul e≈0.6kcal/molei nth e energy?
J .Chem.Phys.11,1085(004)J.Chem.Phys.11,10905(004)J .Chem.Phys.11,10919(004) J .Chem.Phys.1,154110(005)

Full CI for a given orbital basis
Natural orbital ordering for a wavefunction ΨOccupation s>0.1: Principa l NOsOccupation s<0.1: SecondaryNOs,“dynamicallycorrelating”
Correspondingl y ordere ddeterminantexpansiono f C I wavefunctionΨ=Ψ0+Ψcorr Ψcorr=correlatin gpa rt ofΨ
Ψ0:Zeroth-orde r wavefunctio ncontain sonl y principal NOsSCF(on e determinant) orMCSCF(man y determinants)
Dynamiccorrelatio n termΨcorr=∑xΨx=Ψ1+Ψ+Ψ3+...Ψn+....
Ψx=∑kcxkΨxk
{Ψxk}=all -x tupl e excitation s wi threspec t toΨo,i.e.al l determinantscontaining x seconda ryorbitals
WhileΨcorr=∑xΨxconverge sfas (t 6-8termsfo r mhaccuracy),th e determinant al expansion sΨx=∑kcxkΨxkconverg e veryslowly.But,f orn>3,theycontai n over99%deadwoo .d

Correlation energy as a sum of incremental contributions fromsuccessive excitation levels
Preliminary calculationsΨob y SC F o (r sma )ll MCSCFFullSD-C I → SD-C -I NOs
A ll excitation s generatedfro m thes e natura l orbitals
Resolutioni n termso f NO-basedexcitatio n contributionsExcitatio nincrementsum: ETotal−E0=ΔEcorr=∑xΔE(x)
ΔE(x)= (E x)–E( -x 1)=incrementa l contributionfro m -x tupleexcitationswhe re E( )x =total energ yu p toan dincludin gx-tupl e excitations
Incrementa l contribution sΔE(x) asorbita l limitsΔE(x)=limi tΔE(x|m) f or m→ M, where
ΔE(x|m)=analogou s toΔE(x)above,exce pt thatonl y th e fir stmcorrelatin gnatura l orbital s ar e used,an d M=totalnumbero f correlatin g orbitals


New relations between contributions from different excitation levels x
Considering ΔE(x|m)a safunctio n of mfo r fixed ,x w einfer:T hevaluesforx≥4a re relatedt o thos e fo r lowe r x by
ΔE(x|m)=axΔE(x−|m)+cx
whencefo >r x4:
ΔE(x; -x 1|m)=[E(x|m)–E(x-|m)]=AxΔE(|m)+BxΔE(3|m)+Cx
an dalso:
E(al l excitation |s )m– (E 3|m)]= A ΔE(|m)+ B ΔE(3|m)+C
Controll edenerg y extrapolatio n byintrinsi c scalin g (CEEIS)(i)Obtaincoefficient sA,B,Cb yLM S fittingt o lo wvalueso f mwit h a modera te numbe r ofdeterminants.(i )i T hedesire d value so f ΔE(x)=ΔE(x|M)fo r x≥4ar e thenobtainedfro mth e valu es fo r x=,3and =m M.


Accurate binding energies of C2, N2, O2, F2
(i) Full CI energies, including all valence correlations are determined using theCEEIS extrapolation for the cc-pV2Z, cc-pV3Z, cc-pV4Z basis sets
ii) The FCI energies are extrapolated to the complete basis set (CBS) limitSCF energy: EX(SCF) = ECBS(SCF) + c exp(−γ )X
Correlatio n energy: EX(Corr)=ECBS(Corr)+aX−3
Yieldsthenon-relativistic,valence-only-correlate d energieso f th e fourmoleculesand the correspondin gatoms.
)iii Experimentallyknow nar eth etotal atomi cenergiesassu m of ionizationpotentialsan dth etotalmolecul ardissociatio nenergies.
iv)T orelat ethes etheoreticalan dexperimentalquantities,onemus t accountforth efollowin geffect :s•Scalarrelativist iceffectsinatomsan dmolecules,•Spin-orbitcouplin g in the atom s C, ,O F,•Zero-poin t vibrationalan dlo w rotational energiesinmolecules,• In-corean dcore-valen ceelectro ncorrelations.

Comparison of CEEIS-FCI-CBS and experimental energies for C2, N2, O2, F2
Energy (mh) C C2 2C → C
Experimentall y Measured −3785.0 −75935.6 −31.8±0.8Vibrati on-Rotation 0.0 4. 4.Scala r Relativistic −6.65 −13.0 0.3S pinOrbi tCoupling −0.15 0.0 0.3C oreCorrelations −55.0 −11.4 −.4
Nonrelativist icValen ceTotal −37790.0 −75814.1 −34.1
CEEI -S FC I -CBS −37790.6 −75813.5 -3.3
NNN→ N
Experimentall y Measured −54610.0 −109578.5 −358.5±0.04Vibrati on-Rotation 0.0 5.4 5.4Scala r Relativistic −0.7 −41. 0.S pinOrbi tCoupling 0.0 0.0 0.0C oreCorrelations −58.8 −119.0 −1.4
Nonrelativist icValen ceTotal −54530.5 −10943.7 −36.7
CEEI -S FC I -CBS −54531. −10945.1 −36.7

Comparison of experimental and CEEIS-FCI-CBS energies for C2, N2, O2, F2
Energy (mh) O O2 2O → O
Experimentall y Measured −75106.45 −150400.9 −188.0±0.00Vibrati on-Rotation 0.0 3.6 3.6Scala r Relativistic −38.35 −76.4 0.3S pinOrbi tCoupling −0.35 0.0 0.7C oreCorrelations −6.1 −14.9 −0.7
Nonrelativist icValen ceTotal −75005.3 −1500.5 −191.9
CEEI -S FC I -CBS −75006.4 −15004.0 −191.
FFF→ F
Experimentall y Measured −99785.3 −1999.5 -58.9±0.Vibrati –on Rotation 0.0 .1 .1Scala r Relativistic −70.9 −141.8 0.0S pinOrbi tCoupling −0.6 0.0 1.C oreCorrelations −65.4 −130.8 0.0
Nonrelativist icValen ceTotal −99668.7 −199399.6 −6.CEEI -S FC I -CBS −99669.5 −199399.3 −60.3

CEEIS-FCI vs. Complete FCI Determinants Requiredfor C2, N2, O2, F2 (cc-pVQZ Basis)
C2 N2 O2 F2
CEEIS 6.4x107 3.2x107 2.0x108 1.1x108
FCI 3.6x1012 1.6x1015 1.7x1017 3.7x1019
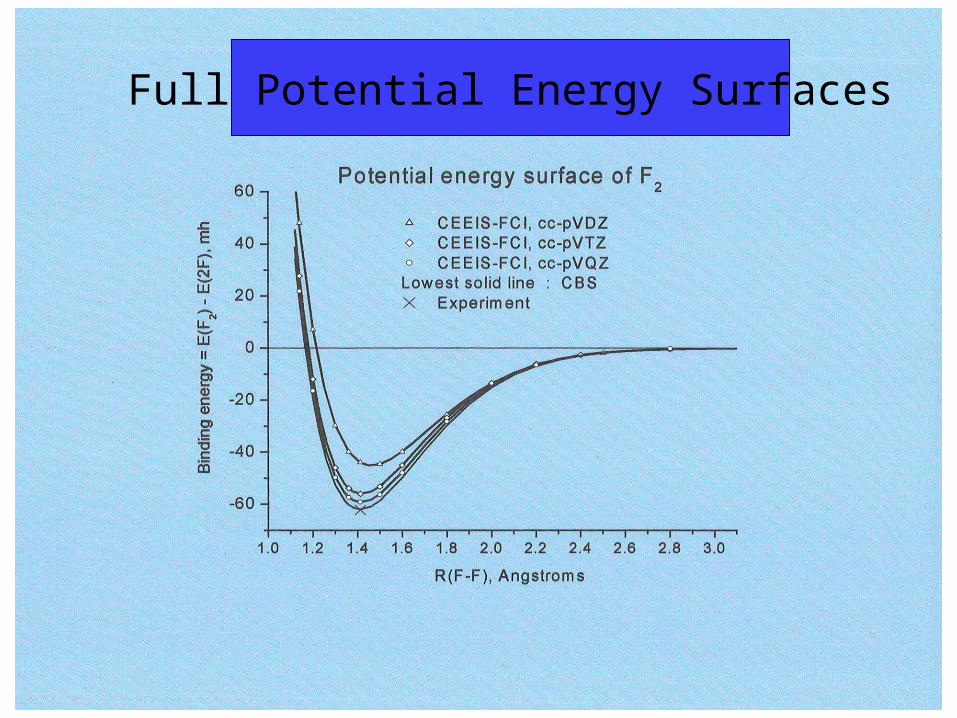
Full Potential Energy Surfaces
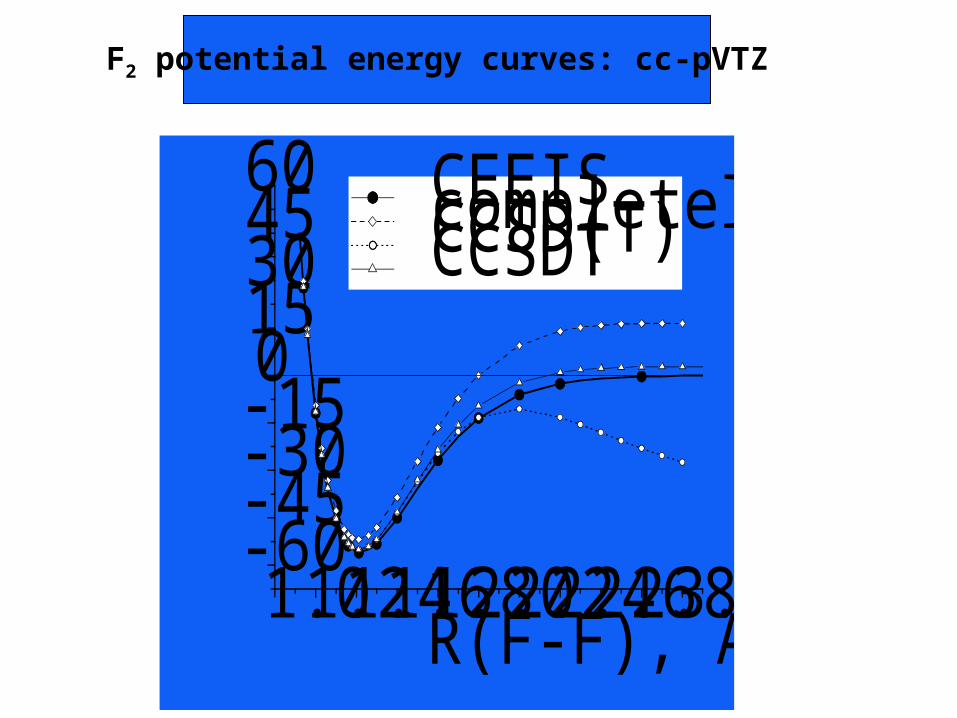
1.01.21.41.61.82.02.22.42.62.83.0-60-45-30-15015304560
Binding energy = E(F
2) - E(2F), mh
R(F-F), Angstroms
CEEIS completely renormalized CCSD(T) CCSD(T) CCSDT
F2 potential energy curves: cc-pVTZ

Summary
CEEIS deduces the correlation contributions of quadruple andhigher excitation levels from those of single, double and tripleexcitations.
The FCI energy, generated from a given atomic basis, can beobtained from energy values calculated in only a very small partof the full configuration space, e.g. 107 vs. 1019 determinants forF2 with a QZ basis.
Combining these CEEIS full CI energies with extrapolation tothe CBS limit, the complete full CI energy is approached tochemical accuracy.
The binding energies obtained agree with experimental valueswithin the chemical accuracy criterion of 1 kcal/mol.

MCSCF HESSIANS: TIM DUDLEY
– Analytic Hessians generally superior to numerical or semi-numerical
– Finite displacements frequently cause artificial symmetry breaking or root flipping
– Necessary step for derivative coupling– Computationally demanding: Parallel efficiency
desirable– DDI-based MCSCF Hessians– IBM clusters, 64-bit Linux

Speedup of CAS(2|3) Hessian Calculation of Cyclopentadienyl Complex of Zirconium
y = 0.95x + 0.11
R2 = 0.9997
1
2
3
4
5
6
7
8
9
1 2 3 4 5 6 7 8 9
# Processors
Speedup
Zr
304 basis fxns, small active spaceDominated by calc of derivative integrals

Speedup of CAS(2|3) Hessian Calculation of Zirconium Cyclopentadienyl Complex
0
4
8
12
16
20
24
28
32
0 16 32 48 64 80 96 112 128
# CPUs
Speedup
Derivative Integrals CPMCSCF Total Freq. Calc.
Zr

Speedup of CAS(16|12) Hessian Calculation of Silicon Dioxide
y = 0.97x + 0.03
R2 = 1
1
2
3
4
5
6
7
8
9
1 2 3 4 5 6 7 8 9
# Processors
Speedup
SiO O
Large active space, small AO basisDominated by calc of CI blocks of H

Speedup of CAS(16|12)/6-31G* Hessian Calculation of Silicon Dioxide
0
4
8
12
16
20
24
28
32
0 16 32 48 64 80 96 112 128
# CPUs
Speedup
Total Freq. Calc.
SiO O

Speedup of CAS(10|9) Hessian Calculation of 7-Azaindole
y = 0.96x + 0.09
R2 = 0.9995
1
2
3
4
5
6
7
8
9
1 2 3 4 5 6 7 8 9
# Processors
Speedup
N NH
216 basis fxns, full active spaceCalc is mix of all bottlenecks

Speedup of CAS(10|9)/TZV Hessian Calculation of 7-azaindole
0
4
8
12
16
20
24
28
32
0 16 32 48 64 80 96 112 128
# CPUs
Speedup
Derivative Integrals CPMCSCF Total Freq. Calc.
N NH

ZAPT2 BENCHMARKS
• IBM p640 nodes connected by dual Gigabit Ethernet– 4 Power3-II processors at 375 MHz– 16 GB memory
• Tested – Au3H4
– Au3O4
– Au5H4
– Ti2Cl2Cp4
– Fe-porphyrin: imidazole

Au3H4
• Basis set– aug-cc-pVTZ on H– uncontracted SBKJC with 3f2g polarization
functions and one diffuse sp function on Au– 380 spherical harmonic basis functions
• 31 DOCC, 1 SOCC• 9.5 MWords replicated• 170 MWords distributed
Au
HAu
HAu
H H

Au3O4
• Basis set– aug-cc-pVTZ on O– uncontracted SBKJC with 3f2g polarization
functions and one diffuse sp function on Au– 472 spherical harmonic basis functions
• 44 DOCC, 1 SOCC• 20.7 MWords replicated• 562 MWords distributed
O
OAu Au
O
O
Au

Au5H4
• Basis set– aug-cc-pVTZ on H– uncontracted SBKJC with 3f2g polarization
functions and one diffuse sp function on Au– 572 spherical harmonic basis
functions• 49 DOCC, 1 SOCC• 30.1 MWords replicated• 1011 MWords distributed
H
HAuAu
H
H
Au
AuAu

Ti2Cl2Cp4
• Basis set– TZV– 486 basis functions (N = 486)
• 108 DOCC, 2 SOCC• 30.5 MWords replicated• 2470 MWords distributed
Fe-porphyrin: imidazole
• Two basis sets– MIDI with d polarization functions (N = 493)– TZV with d,p polarization functions (N = 728)
• 110 DOCC, 2 SOCC• N = 493
– 32.1 MWords replicated– 2635 MWords distributed
• N = 728– 52.1 MWords replicated– 5536 MWords distributed

Speedup Curve
0.0
10.0
20.0
30.0
40.0
50.0
60.0
70.0
80.0
0 10 20 30 40 50 60 70
Number of processors
Speedup
Au3H4 (380)
Au3O4 (472)
Au5H4 (570)
Ti2Cl2Cp4 (486)
Fe-porphyrin (493)
Fe-porphyrin (728)
Linear

Load Balancing
• Au3H4 on 64 processors– Total CPU time ranged from 1124 to 1178 sec.– Master spent 1165 sec.– average: 1147 sec.– standard deviation: 13.5 sec.
• Large Fe-porphyrin on 64 processors– Total CPU time ranged from 50679 to 51448 sec.– Master spent 50818 sec.– average: 51024 sec.– standard deviation: 162 sec.

THANKS!
• GAMESS Gang
• DOE SciDAC program
• IBM SUR grants






