Download - Síndrome de Kallman

Dr. Mario Enrique Vega Carbó.
Endocrinólogo.
2015
Síndrome de Kallman.
Universidad de Ciencias Médicas de la Habana
Facultad de Ciencias Médicas “Comandante Manuel Fajardo”
Instituto Nacional de Endocrinología

Paciente: RLB Edad :22años
MasculinoHC: 955422
Egresado de sala 4D/3: 9julio 2012.
MI: “Baja estatura y no desarrollo genital”.HEA: Paciente masculino de 22 años que ingresa para estudio de Hipogonadismo hipogonadotropo de inicio prepuberal, baja talla e hiposmia. sin cefalea, ni trastornos visuales.APP: Agenesia renal derecha.APF: Hermano Hipogonadismo tratado.

INTERROGATORIO Sistema urogenital Deseo sexual y relaciones sexuales a 18 años, con erección, sin eyaculación. Sistema endocrinodesarrollo genital pobre, baja tallaAumento de peso 15 lbs en últimos mesesSistema nerviosoNo percibe olores de comidas, perfumes, aromas
fuertes como café ,vainilla.Estatura Madre:160cmPadre:172cm

Examen físico (+):General Ausencia de vello facial y corporal. Adrenales: Vello axilar ausente. Vello pubiano Tanner II.Panículo adiposo: aumentado en cintura pélvica
Regionaltiroides: grado 0mamas: No ginecomastia, ni galactorrea

Genitales externo:Teste I: V 2ml, en escroto, blando, no sensible,
superficie lisa.Teste D: V 1ml en canal inguinal que desciende al
agacharse retráctil, blando, no sensible, liso.Escrotos: poco desarrollo, hipopigmentados, poco
rugosos.Pene: Tanner I Largo: 3.5cm Circunsferencia: 5cm Glande: sin desarrolloSoma: Hipoatrofia muscular generalizada a predominio de
cintura escapular y miembros superiores.
S Nervioso Central: Hiposmia bilateral al café, vainilla, canela.

MesuracionesPeso:46.5kgTalla:1.53cm IMC:19.86Kg/m2
Brazada 165cm >12cm que la tallaVP:72cmPP:81cm PP > 9cm que VPDBA:32cmDBT:31.5C cintura:73cmC Cadera:87cm ICC =0.8

HPSocialPrenatales, Perinatales, Posnatales y DSM
Etapa escolar: Rendimiento intelectual medio. Comenzó a notar desde 8 años diferencias en su estatura con respecto a compañeros de escuela.
Adolescencia: Deprivación afectiva, aporte nutricional inadecuado, conflictos familiares.
Madre fallecida, padre internacionalista.

Bioquímica general EN PARÁMETROS NORMALES
Hormonales basales FSH=0,25UI/LLH =0,21UI/LPRL =298mU/LCORTISOL =400nmol/L17 OH PROGESTERONA =0,87ng/ml
Genéticos CARIOTIPO = 46XY 11 METAFASES

Prueba dinámica Test estimulación HCG Testosterona nmol/l y E2
basal 1.9924h 1.8748h 1.0272h 1.42

IMAGENRx selectivo de silla turca: Aumento del diámetro AP de
la silla turca.
RMN de cráneo: ausencia de surcos y giros rectos compatible con agenesia olfatoria.
Rx de manos izquierda y rodilla: edad ósea compatible con 13 años. Edad cronológica de 22 años.
Ultrasonido testicularTI y TD: hipoplasia de ambos bien vascularizados
localizados en escroto y canal inguinal respectivamente.
Síndrome de Kallmann
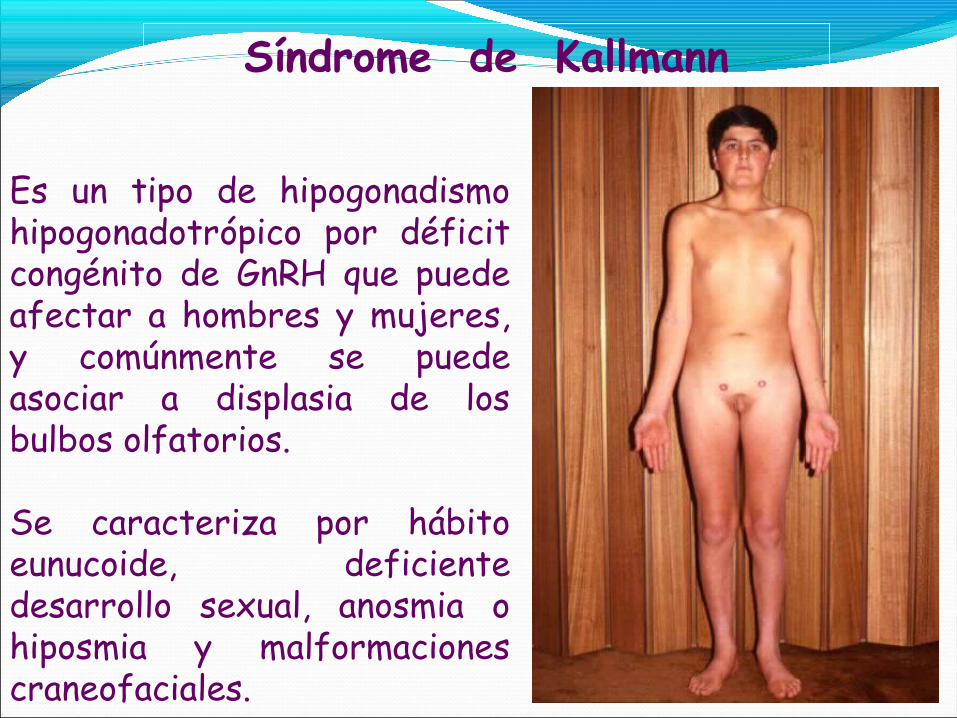
Es un tipo de hipogonadismo hipogonadotrópico por déficit congénito de GnRH que puede afectar a hombres y mujeres, y comúnmente se puede asociar a displasia de los bulbos olfatorios.
Se caracteriza por hábito eunucoide, deficiente desarrollo sexual, anosmia o hiposmia y malformaciones craneofaciales.

Síndrome de Kallmann
Formas clínicasEsporádica: 70%Familiar: 30%
Modelos familiares:Ligado al cromosoma X: 85%Autosómico dominante: 10%
Autosómico recesivo: 5%
Prevalencia1x10 mil en varones.
1x50 mil en hembras.

Patrón de herencia ligado al X
Gen KAL 1. Localizado en Xp 22.31
Codifica para la Anosmina-1.
Participa en la sinapsis bulbo-olfatoria.
Trasmiten mujeres portadoras a enfermos.
Síndrome de Kallmann

Patrón de herencia autosómico dominante
Gen KAL 2 FGFR1 8 p11.2-p12
Codifica para el receptor 1 del factor de crecimiento de fibroblastos, relacionados con la evaginación del bulbo olfatorio.
Síndrome de Kallmann

Patrón de herencia autosómico recesivo
Gen PROK 2 (KAL 3) Codifica para el receptor Procinecticina 2.
Gen PROK (KAL 4) Codifica para la Procinecticina 2.
Síndrome de Kallmann

Cuadro clínicoBebes: Micropene. Criptorquidea.Infancia: anosmia o hiposmia, no obligado
Adolescentes y adultos:Hipogonadismo. Desarrollo sexual incompleto.Volumen testicular: menor de 4 ml.Ausencia de caracteres sexuales secundarios.Aumento de masa muscular.Poca o ausencia de líbido.Disfunción eréctil.Infertilidad.Proporciones enucoides.

Otras manifestaciones clínicas:
Déficit olfatorio (anosmia o hiposmia).Agenesia renal unilateral (30%) es exclusivo del KAL 1.Perdida auditiva unilateral.Labio y/o paladar hendido.Agenesia de una o ambas piezas dentales.Sinequia de los dígitos.Braquidactilia o sindactiliaAgenesia del cuerpo calloso.Hipoplasia o aplasia del pabellón auricular.Pérdida del cartílago nasalSinquinesia bimanual o movimientos en espejo de miembros superiores.

Criterios diagnósticos del Síndrome de Kallmann
I. Clínicos:1.Hipogonadismo prepuberal2.Anosmia o hiposmia.3.Anomalías craneo-faciales.
II. Hormonales: Hipogonadismo hipogonadotrópico secundario.1.Testosterona total, FSH y LH baja.2.Se elevan FSH y LH con estimulación con GnRH.

III. Imagenológicos.1. Resonacia magnética de hipófisis con o sin gadolineo.Para descartar lesión hipotalamo-hipofisaria.Ausencia o hipoplasia del bulbo olfatorio.Ausencia del surco del giro recto.2. Ultrasonido de vías urinarias: Malformaciones.
IV. Examenes genéticos.1. Cariotipo normal.2. Prueba tipo FISH o por hibridación genómica comparativa. (Detecta delección de genes: KAL1-4)
Criterios diagnósticos del Síndrome de Kallmann

Tratamiento del Síndrome de Kallmann
Objetivos del tratamiento. Lograr desarrollo de caracteres sexuales secundarios y fertilidad.
Bases del tratamiento hormonal.1.Reemplazo hormonal con testosterona en prepuberes.2.Mejorar volumen testicular con HCG y HMG que pueda desarrollar espermatogénesis.3.Conseguir fertilidad con análogos de GnRH.
Psicoterapia de apoyo a pacientes y familiares.







