dysmoorphology
TRANSCRIPT

Genetics
1


Dysmorphology
Phenotypic features which when present in combination suggest the presence of chromosomal abnormalities ( numerical or structural ).

Head and CNS

Microcephaly
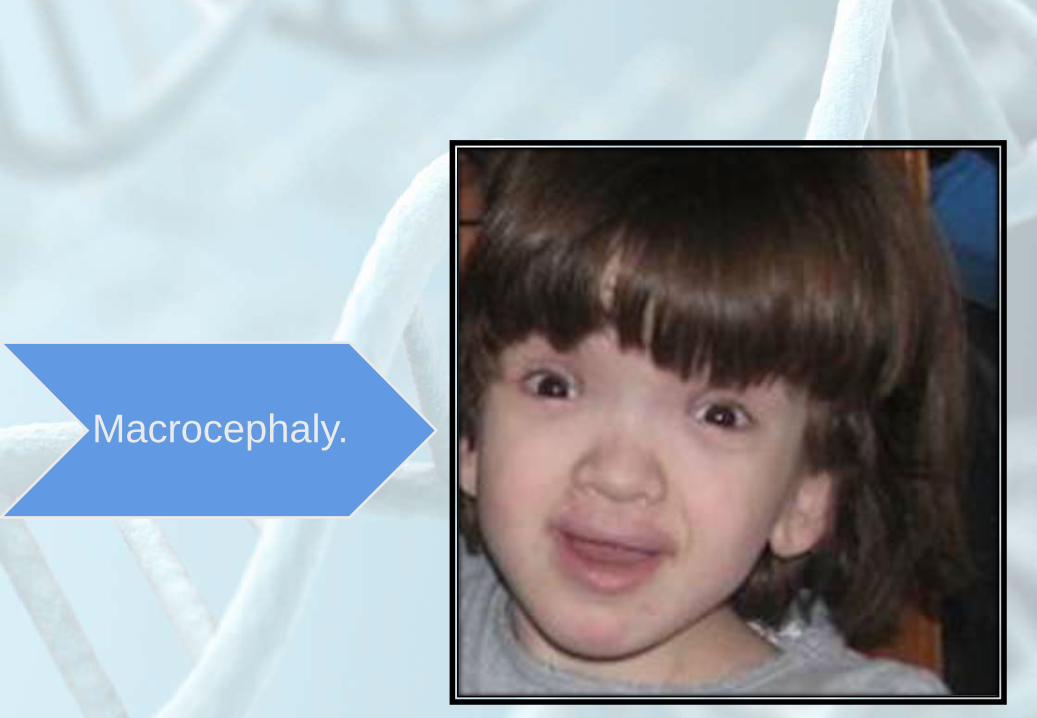
Macrocephaly.
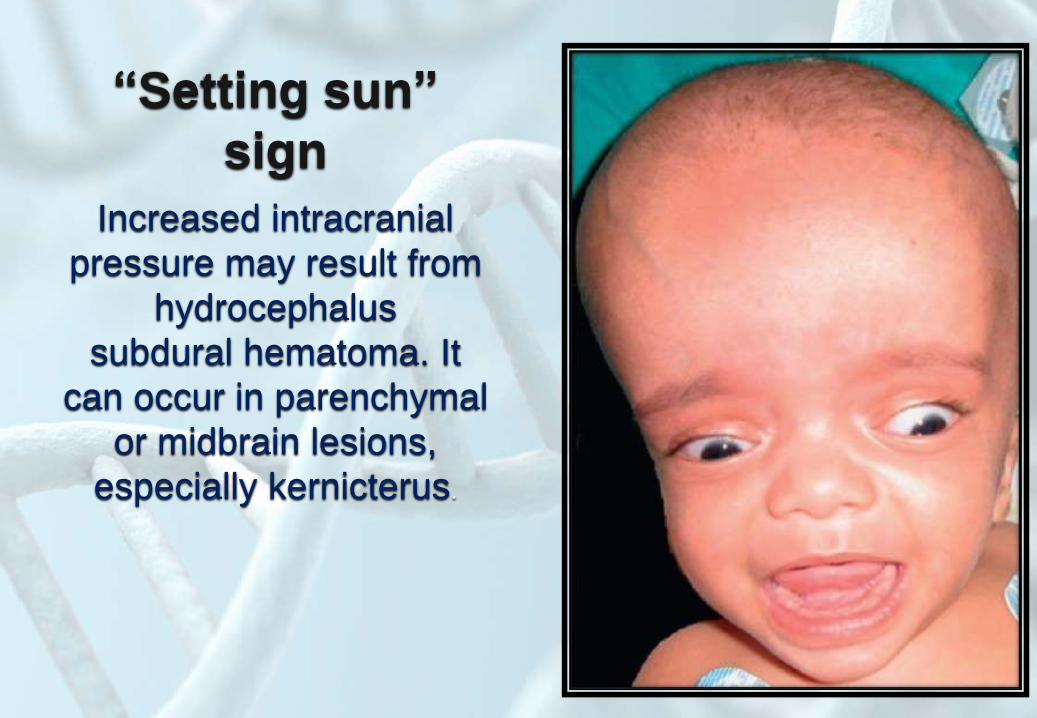
“Setting sun”
sign
Increased intracranial
pressure may result from
hydrocephalus
subdural hematoma. It
can occur in parenchymal
or midbrain lesions,
especially kernicterus.

Hydranencephalyfailure of the
development of
the cerebrum with
resulting gross dilatation
of the ventricles

Dandy-Walker
malformationa cyst-like dilation of the fourth
ventricle,
an abnormal cerebellar vermis,
elevation of the tentorium
cerebelli and lateral and
transverse sinuses and torcula
(torcular Herophilli),
lack of patency of the foramina
of Magendie and Luschka,
enlargement of the posterior
fossa,
hydrocephalus.

• lacunar skull
(lückenschädel):
defective calcification
of the skull bones
associated with neural
tube defects
(encephalocele and
meningocele).

Neural tube defects
• Anencephaly

Craniorachischisis

Encephaloceles
bony
defects of the skull with
protrusion
of the meninges which
may contain neural
tissue.
They are most common
in the
occipital area.


Floppy infant

Congenital parietal
foramina

Normal sutures

• Dolichocephaly :
sagittal suture remains
open.
Common in preterms.

• Scaphocephaly:
Premature fusion
of the sagittal
suture

Brachycephaly:
premature
fusion of the
coronal sutures

• Trigonocephalyis due to
premature fusion
of the metopic
suture and is
represented
clinically by a
triangular-shaped
head

Plagiocephaly (oblique-
shaped skull) occurs with
premature fusion of a
single suture (such as the
coronal or lambdoidal) or
with a congenital postural
deformity

Kleeblattschädel
(“cloverleaf ”skull) is
the result of premature
fusion of the sagittal and
coronal sutures. There is
a trilobed appearance of
the skull with indentations
in the center and in the
temporal regions.

Carpenter’s syndrome
(acrocephalopolysynd
actyly) high steep
protruding forehead,
turribrachycephaly due
to fused coronal
sutures, the flat midface,
the small pinched nose,
and the downward
slanting of the palpebral
fissures.


• Prominent occiput.

• Cutis aplasia
congenita:Associated with
embryologic
malformations such as
meningomyelocele
and spinal dysraphia,
omphalocele, and
gastroschisis.

Ears
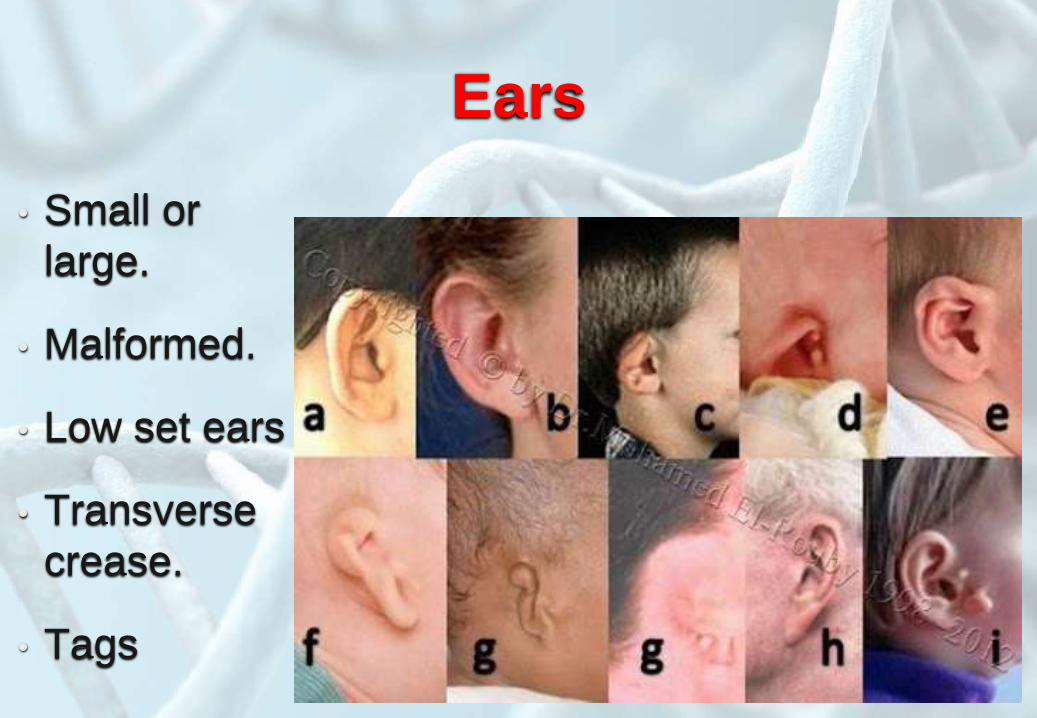
Ears
• Small or
large.
• Malformed.
• Low set ears
• Transverse
crease.
• Tags

MicrotiaHypoplasia of the pinna
associated with atresia of
the external auditory canal

Otocephaly
Bow- tie shaped due to branchial
arch embryopathy resulting in
agnathia, microstomia, and small
posteriorly positioned hypoplastic
tongue. The ears are very low set
and the lower lobes may be fused to
the neck which was short and thin.
Aural ascent does not occur due to
the lack of development of the jaw,
hence the low position of the ears.
These infants typically have
hypoplastic lungs.
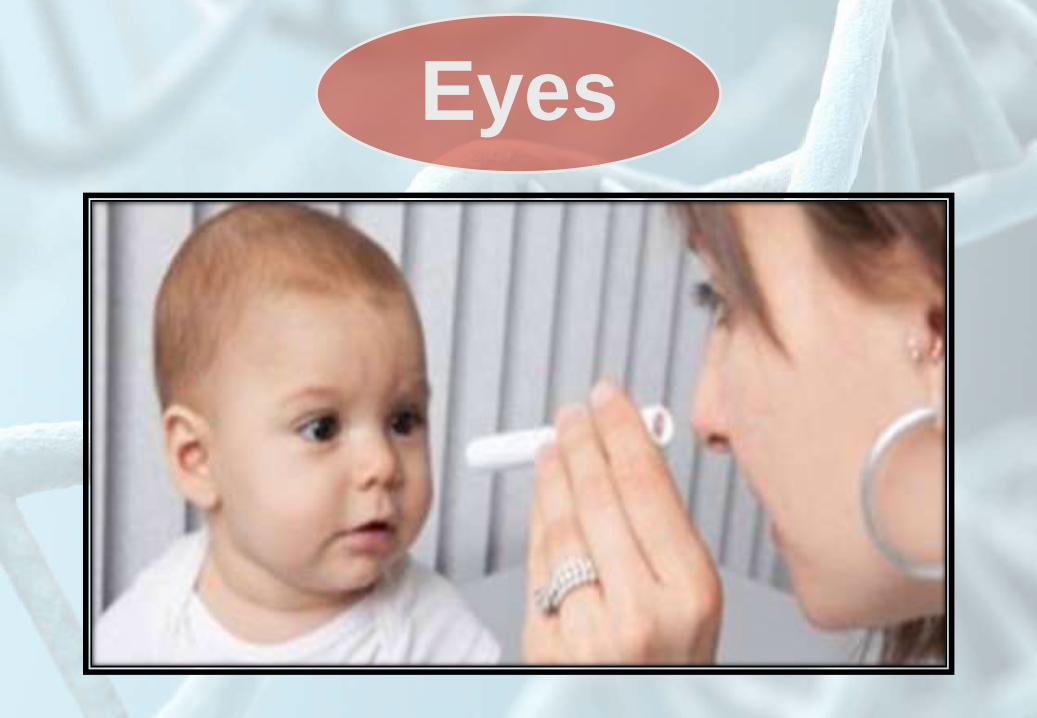
Eyes

• Upward or
downward slanting
of the palpebral
fissures .
• Hypertelorism
(Telecanthus ).
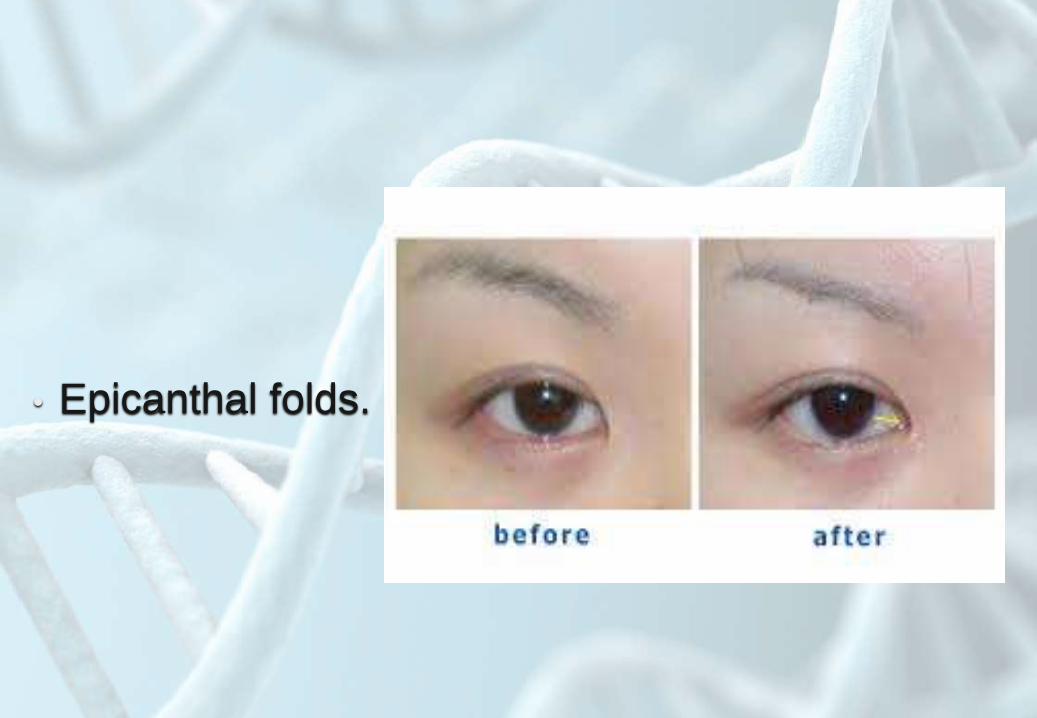
• Epicanthal folds.
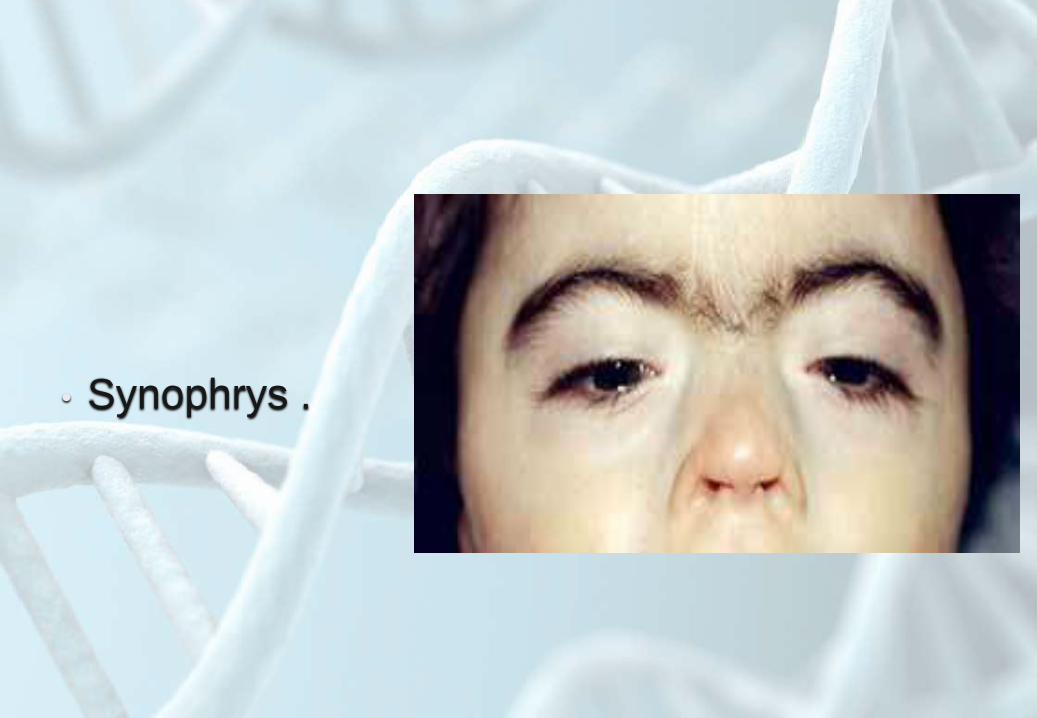
• Synophrys .

• Congenital glucoma.

• Congenital cataract.

Retinoblastoma

Brushfield eyes
brown irides
which are
pathognomonic
for Down
syndrome

Subconjunctival
hemorrhagesresolve
spontaneously
without any
consequences.

Dacryocystocele
may occur as an autosomal
dominant in families
The lacrimal sac is blocked
at both ends and a
sterile swelling appears as
a purplish swelling adjacent
to the base of the nose.
Simple lacrimal probing
allows
for a swift resolution of this
problem

Ankyloblepharon
filiforme adnatumfusion of the upper and
lower
eyelids by small filiform
attachments which can be
cut
with scissors.

Coloboma
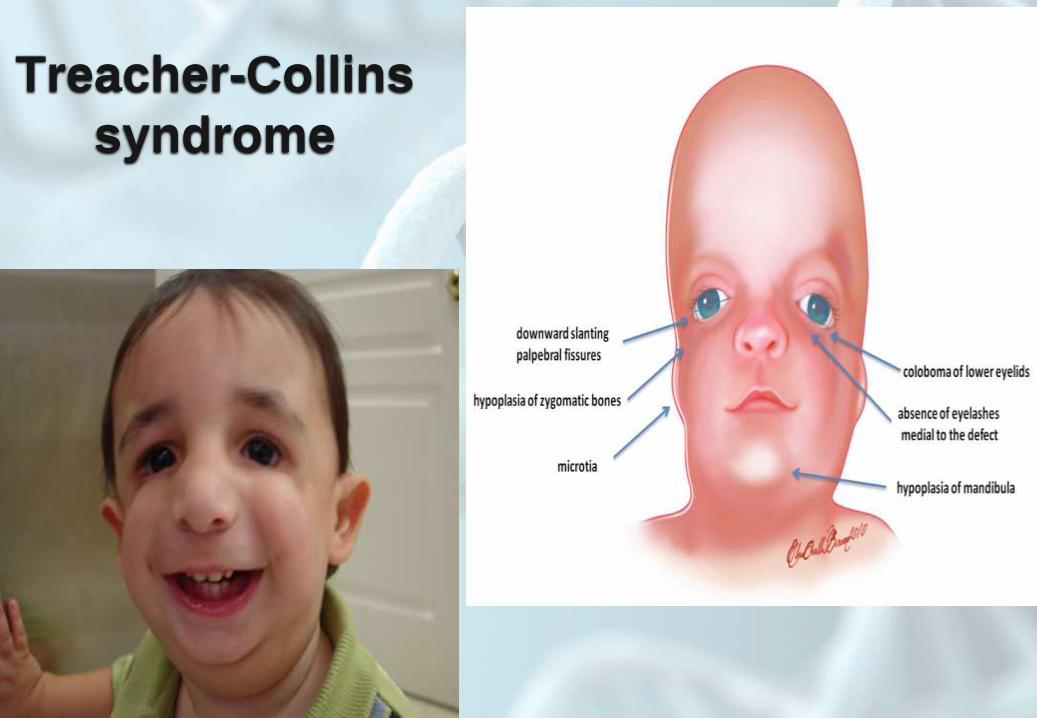
Treacher-Collins
syndrome
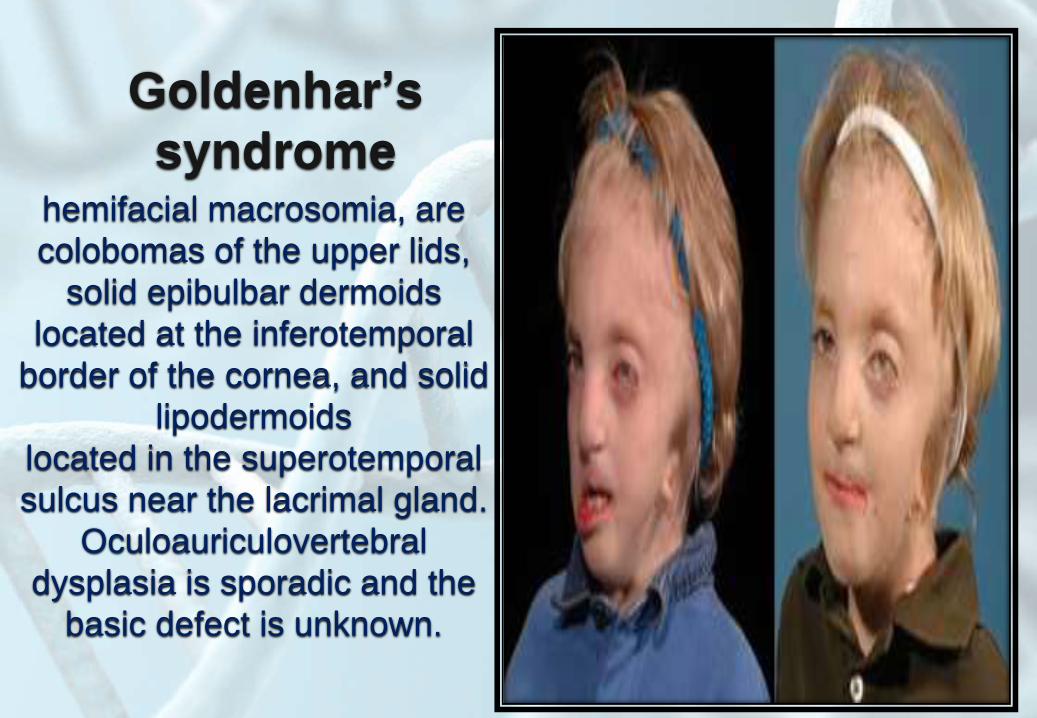
Goldenhar’s
syndromehemifacial macrosomia, are
colobomas of the upper lids,
solid epibulbar dermoids
located at the inferotemporal
border of the cornea, and solid
lipodermoids
located in the superotemporal
sulcus near the lacrimal gland.
Oculoauriculovertebral
dysplasia is sporadic and the
basic defect is unknown.
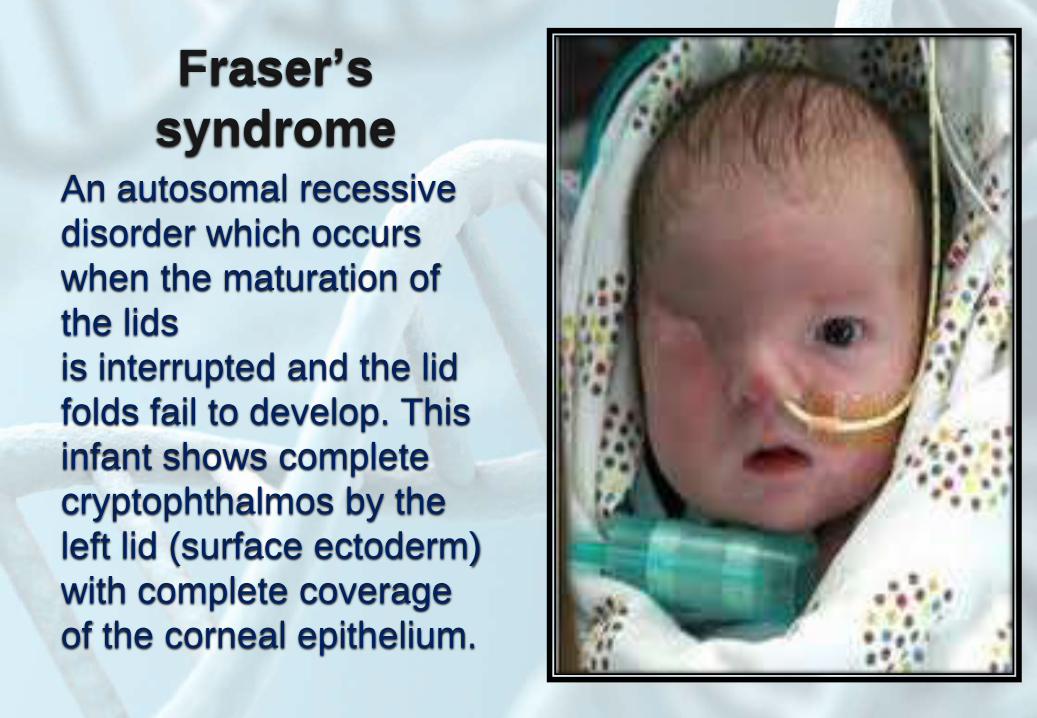
Fraser’s
syndromeAn autosomal recessive
disorder which occurs
when the maturation of
the lids
is interrupted and the lid
folds fail to develop. This
infant shows complete
cryptophthalmos by the
left lid (surface ectoderm)
with complete coverage
of the corneal epithelium.

Congenital
entropion
inturning of
the lower nasal lid such
that the lashes irritate the
cornea.
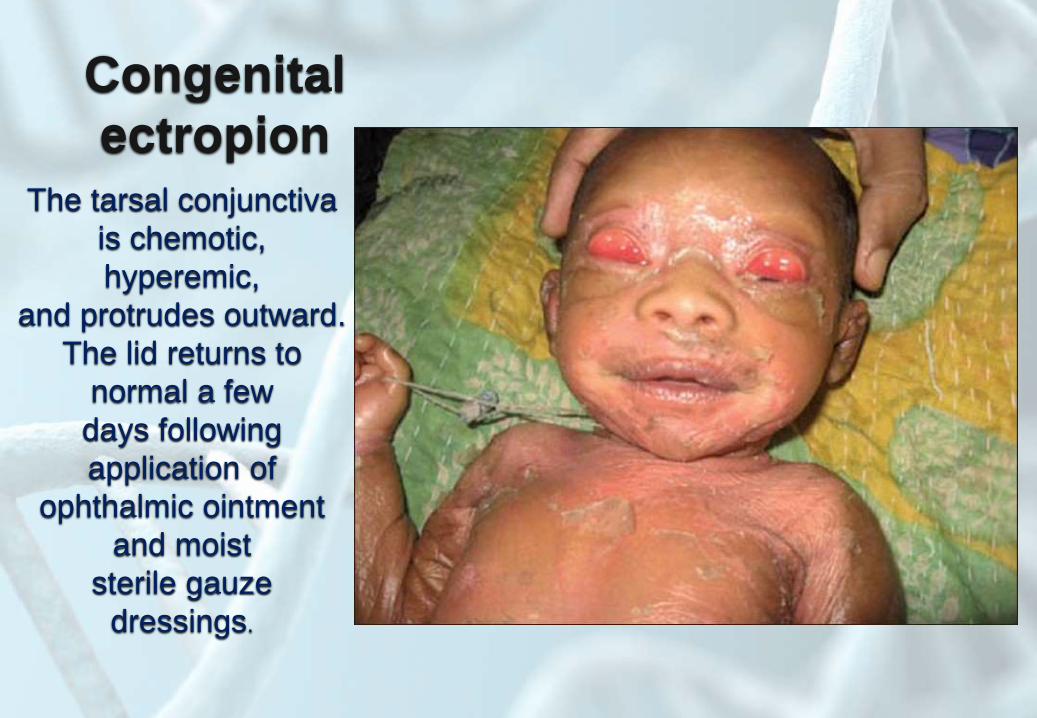
Congenital
ectropionThe tarsal conjunctiva
is chemotic,
hyperemic,
and protrudes outward.
The lid returns to
normal a few
days following
application of
ophthalmic ointment
and moist
sterile gauze
dressings.

Exophthalmos
Neonatal
Hyperthyroidism,
craniofacial anomalies
and shallow
Orbits e.g: Crouzon's
syndrome
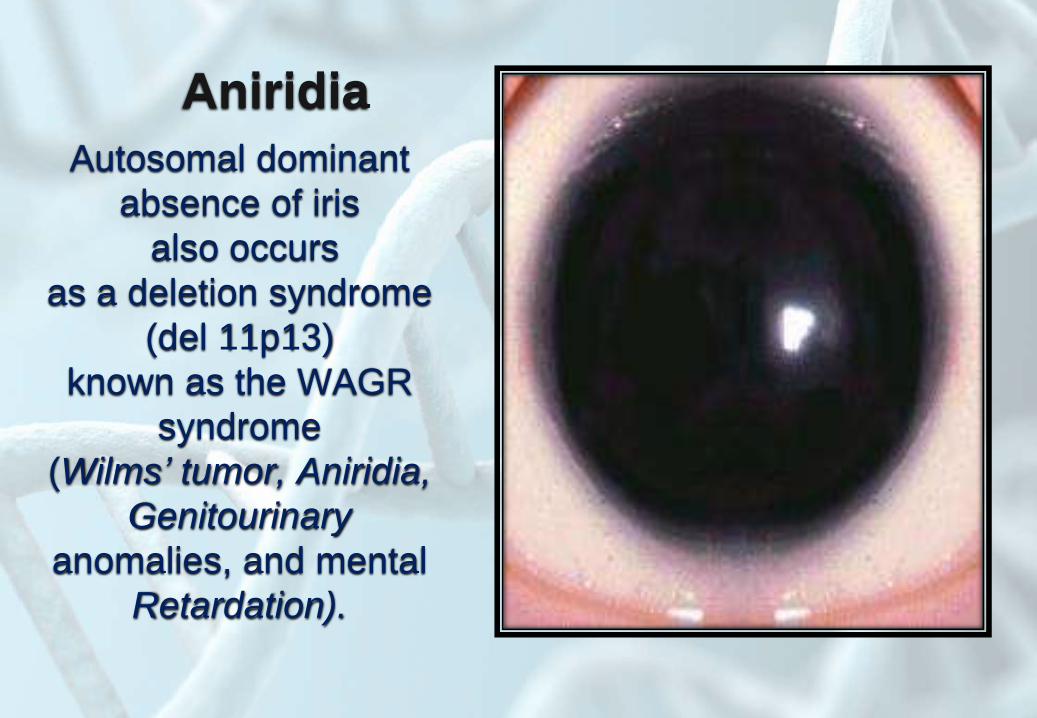
Aniridia
Autosomal dominant
absence of iris
also occurs
as a deletion syndrome
(del 11p13)
known as the WAGR
syndrome
(Wilms’ tumor, Aniridia,
Genitourinary
anomalies, and mental
Retardation).

Megalocornea
enlarged corneal
diameter present at
birth
intraocular pressure is
not
increased.

Heterochromia
Tuberous sclerosis,
waardenburg
syndrome
and normal individuals
Nose

• Depressed nasal
bridge.

Nasal cleft may occur
in otherwise
normal
infants.

• Peaked nose.

• Bulbous nose.
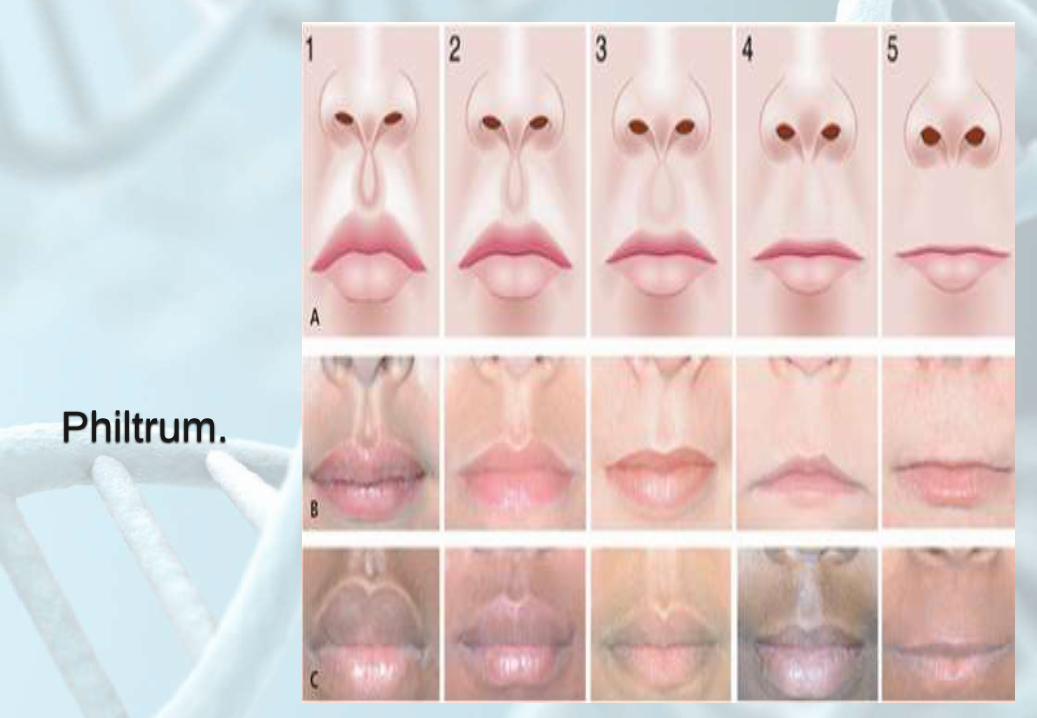
Philtrum.
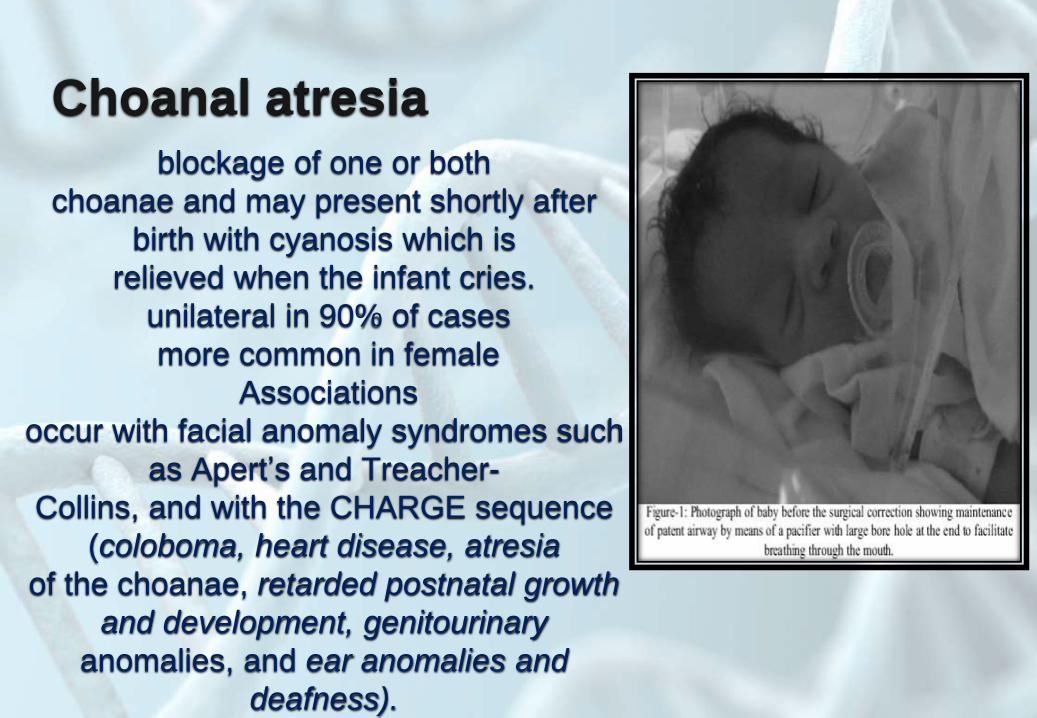
Choanal atresiablockage of one or both
choanae and may present shortly after
birth with cyanosis which is
relieved when the infant cries.
unilateral in 90% of cases
more common in female
Associations
occur with facial anomaly syndromes such
as Apert’s and Treacher-
Collins, and with the CHARGE sequence
(coloboma, heart disease, atresia
of the choanae, retarded postnatal growth
and development, genitourinary
anomalies, and ear anomalies and
deafness).
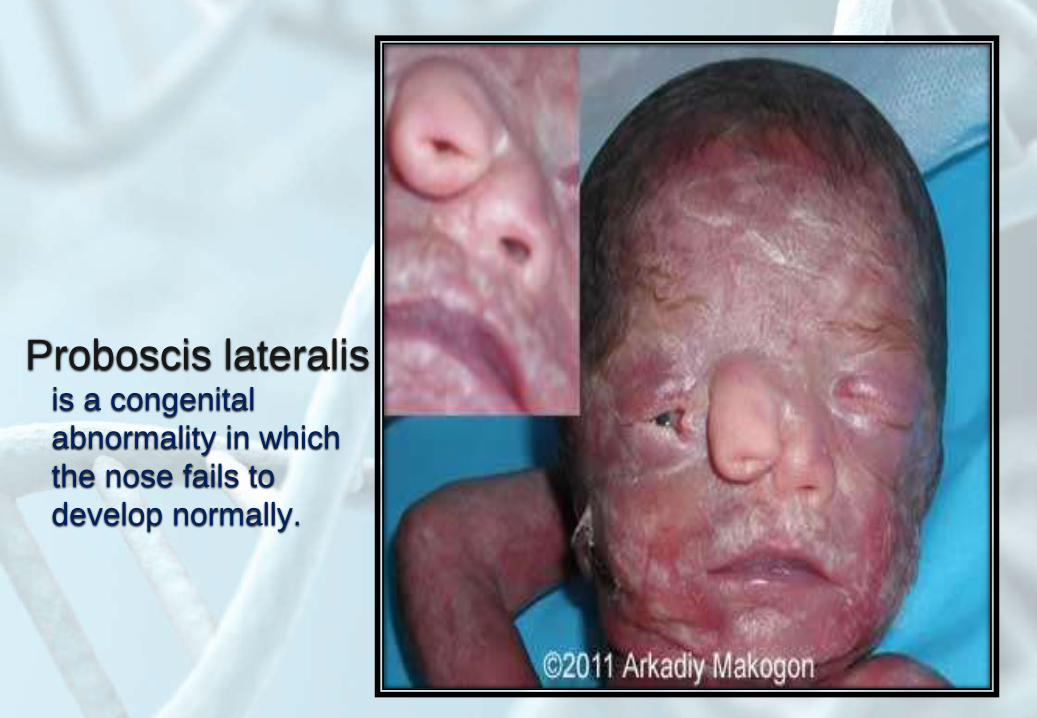
Proboscis lateralisis a congenital
abnormality in which
the nose fails to
develop normally.

Median cleft syndrome
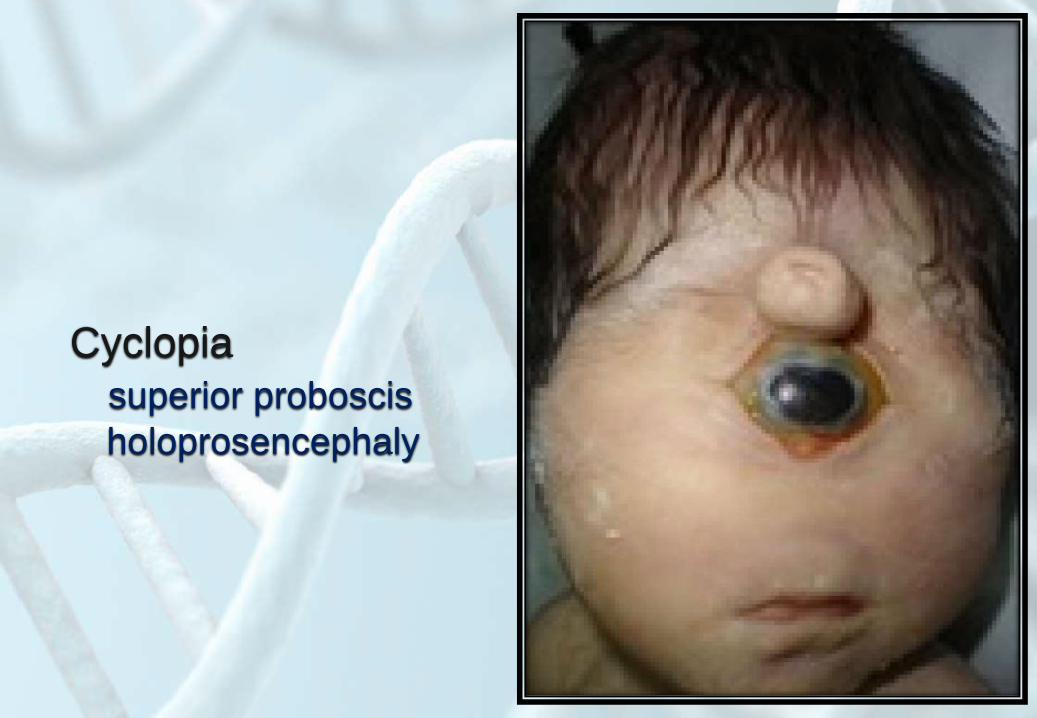
Cyclopia
superior proboscis
holoprosencephaly

Mouth

Lower jaw
• Retromicrognathia.

Mouth
• Cleft lip and palate.

Microstomia
may occur in normal
infants or is
associated with many
syndromes such as
Hallermann-Streiff
syndrome, Freeman-
Sheldon syndrome
and mosaic trisomy 8.

Macrostomia
may be unilateral or
bilateral and is associated
with deformities of the
outer ear, hypoplasia of
the mandible or maxilla,
and cleft palate.It is also
seen in Goldenhar’s
syndrome (unilateral
macrostomia,
antimongoloid slant to the
eyes, preauricular skin
tags, and deafness).

Eruption cysts in an
infant at birth
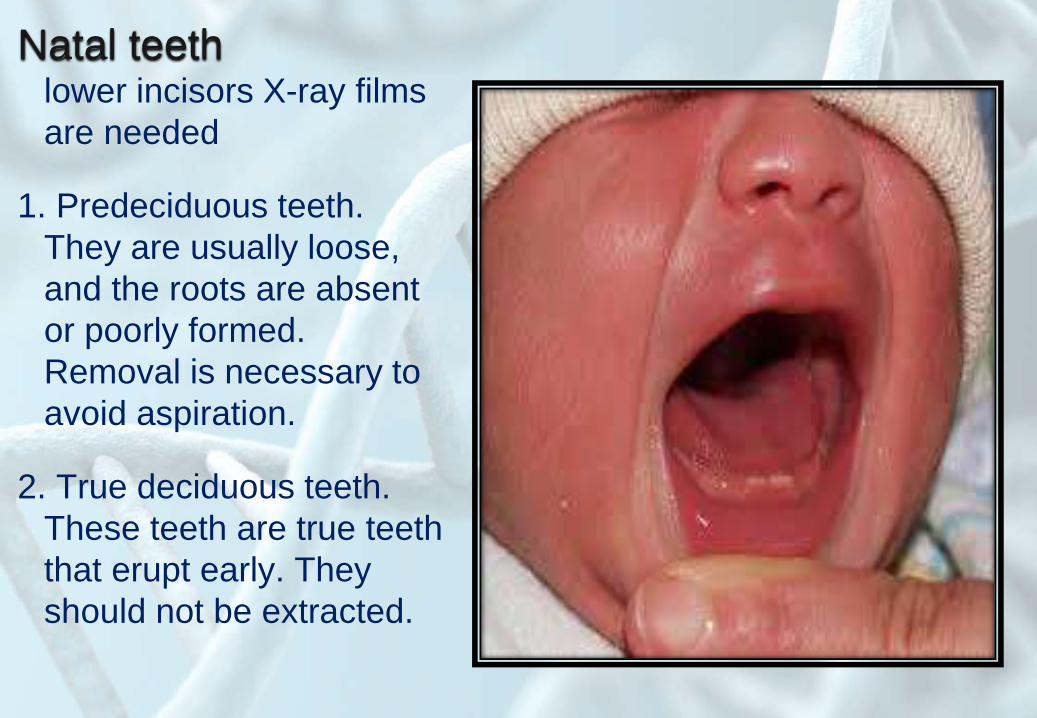
Natal teethlower incisors X-ray films
are needed
1. Predeciduous teeth.
They are usually loose,
and the roots are absent
or poorly formed.
Removal is necessary to
avoid aspiration.
2. True deciduous teeth.
These teeth are true teeth
that erupt early. They
should not be extracted.
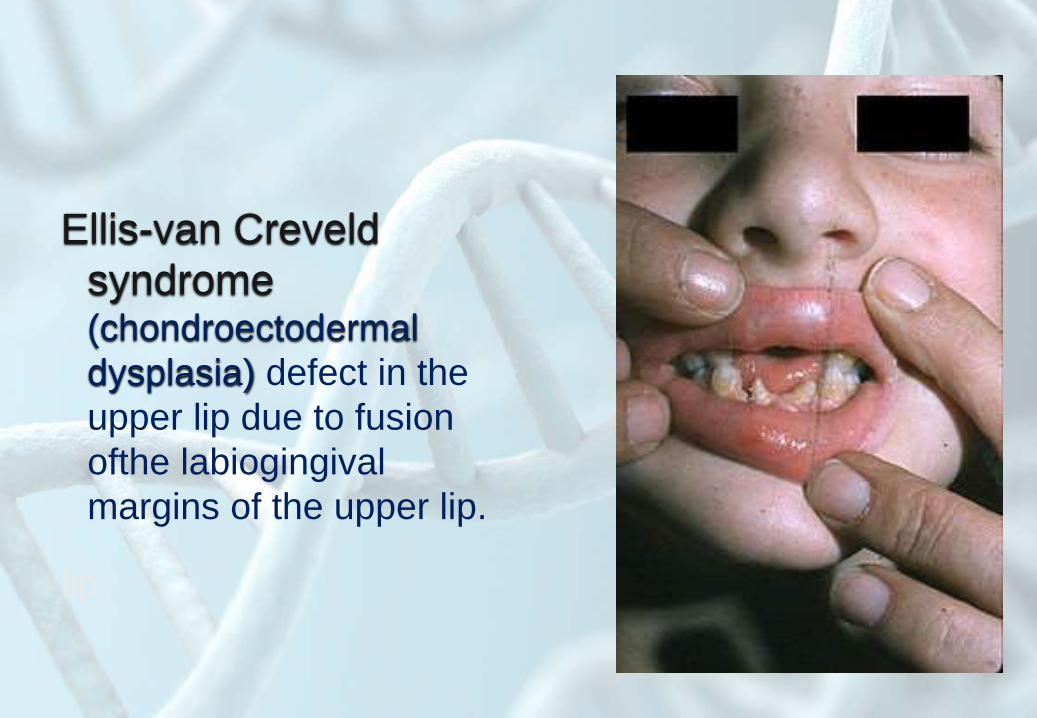
Ellis-van Creveld
syndrome (chondroectodermal
dysplasia) defect in the
upper lip due to fusion
ofthe labiogingival
margins of the upper lip.
lip

Gum hypertrophy due to a mother
treated with
phenytoin during
pregnancy.
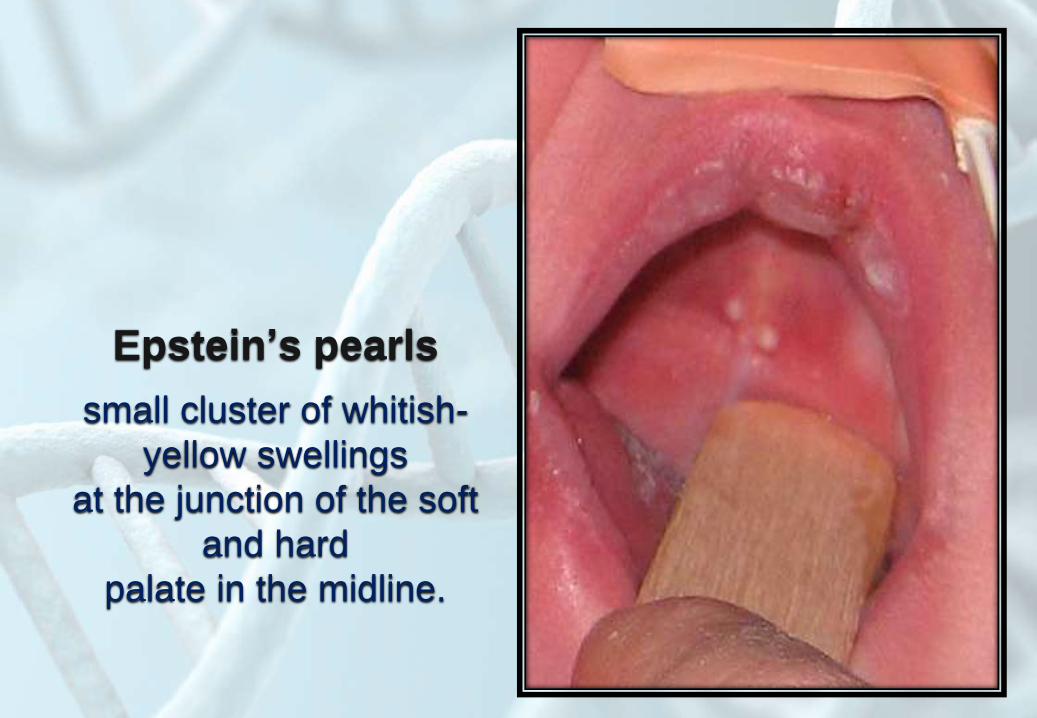
Epstein’s pearls
small cluster of whitish-
yellow swellings
at the junction of the soft
and hard
palate in the midline.

Bohn’s nodules
inclusion cysts on the
alveolar margin in this
infant.

Congenital epulis
a form of embryonal
hamartoma.
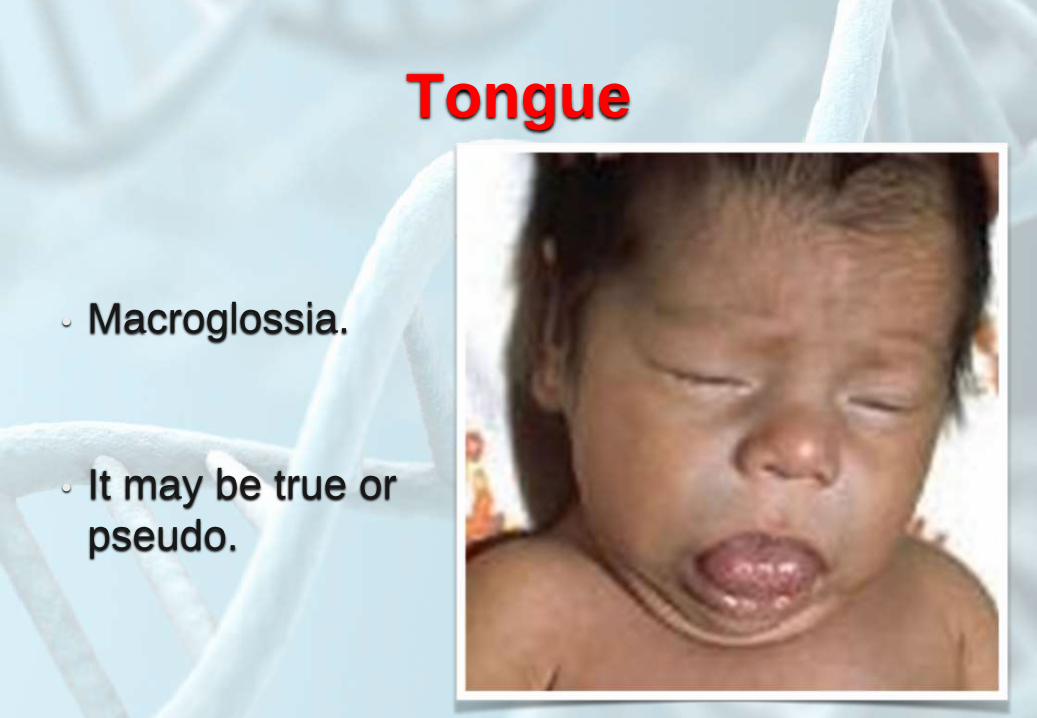
Tongue
• Macroglossia.
• It may be true or
pseudo.
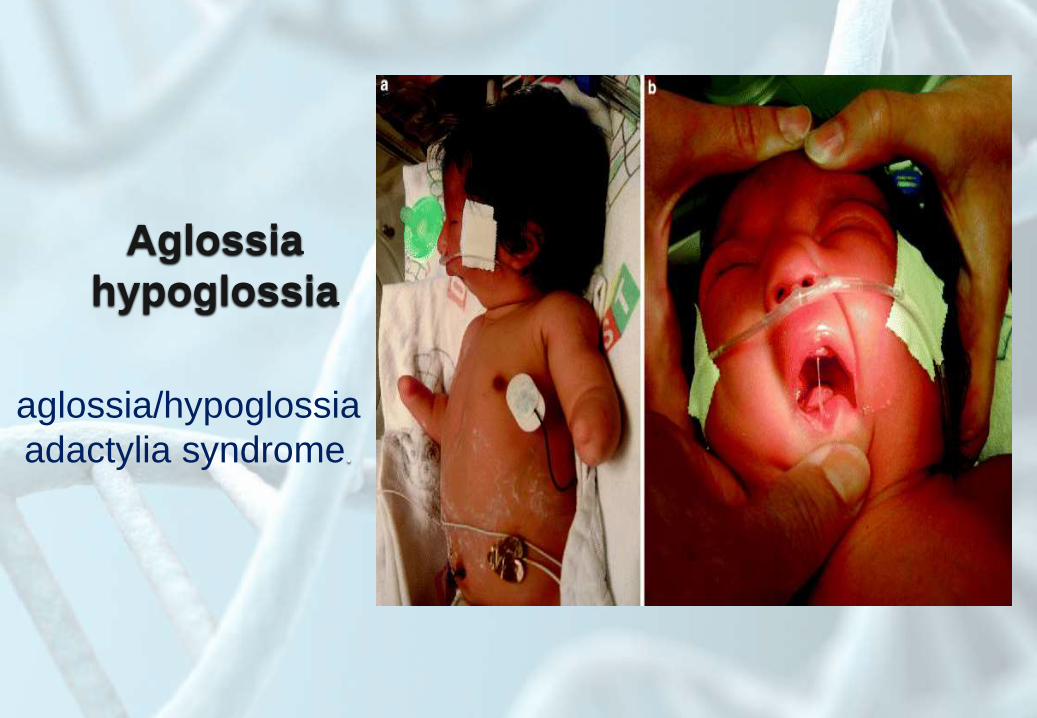
Aglossia
hypoglossia
aglossia/hypoglossia
adactylia syndrome.

Hamartomatous
masses on tongueOrofaciodigital
syndrome, Type I

Neck
• Short webbed
neck.

Congenital midline
cervical cleft
failure of
the branchial arches to
fuse
in the midline. It most
commonly
affects females

Sternomastoid
tumorWhen delivery has
involved
excessive rotation or
gross lateral rotation of
the
neck, a lump may appear
in the sternomastoid
muscle

Branchial clefts
result in remnants, fistulae or
cysts. Defects are usually
unilateral and the external
opening lies at the anterior edge
of the
sternocleidomastoid muscle,
usually at the lower third.
Secondary
bacterial infection and cyst
formation may occur. In this
infant
there is a branchial cleft remnant

Cystic hygroma
(lymphangioma)
soft fluctuating
mass. consist of
proliferation of lymph
vessels.
Although not malignant,
they may spread over
the
neck with extension into
the mouth.

Hand

Hand
• Polydactyl

• Syndactyly
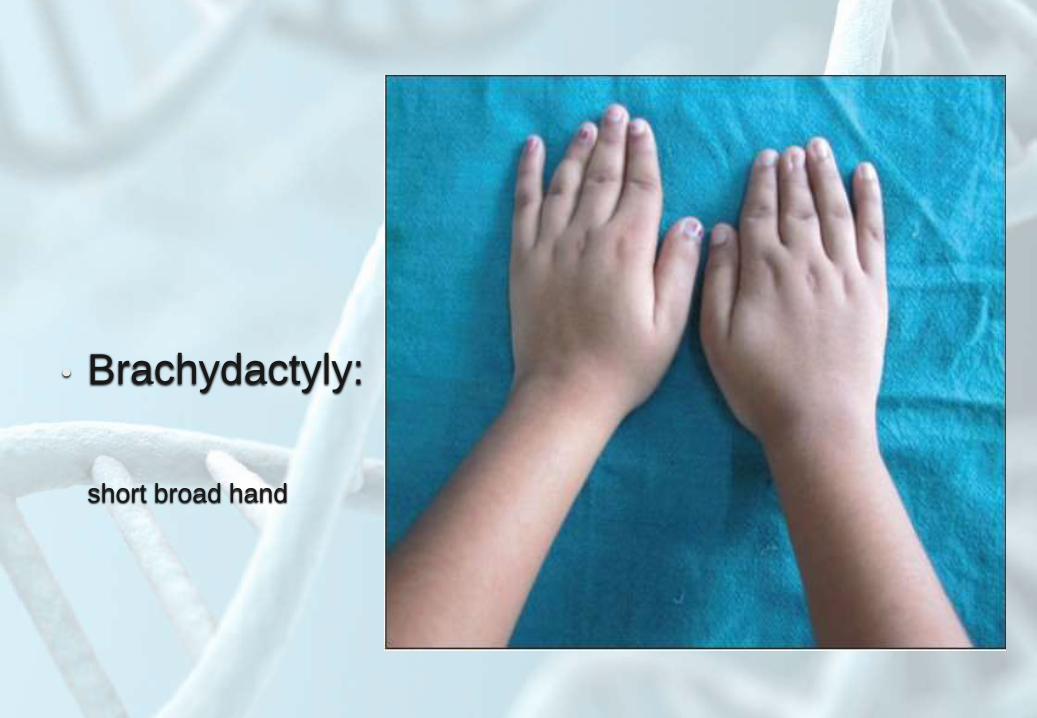
• Brachydactyly:
short broad hand

Camptodactylytrigger finger

• Clinodactyly
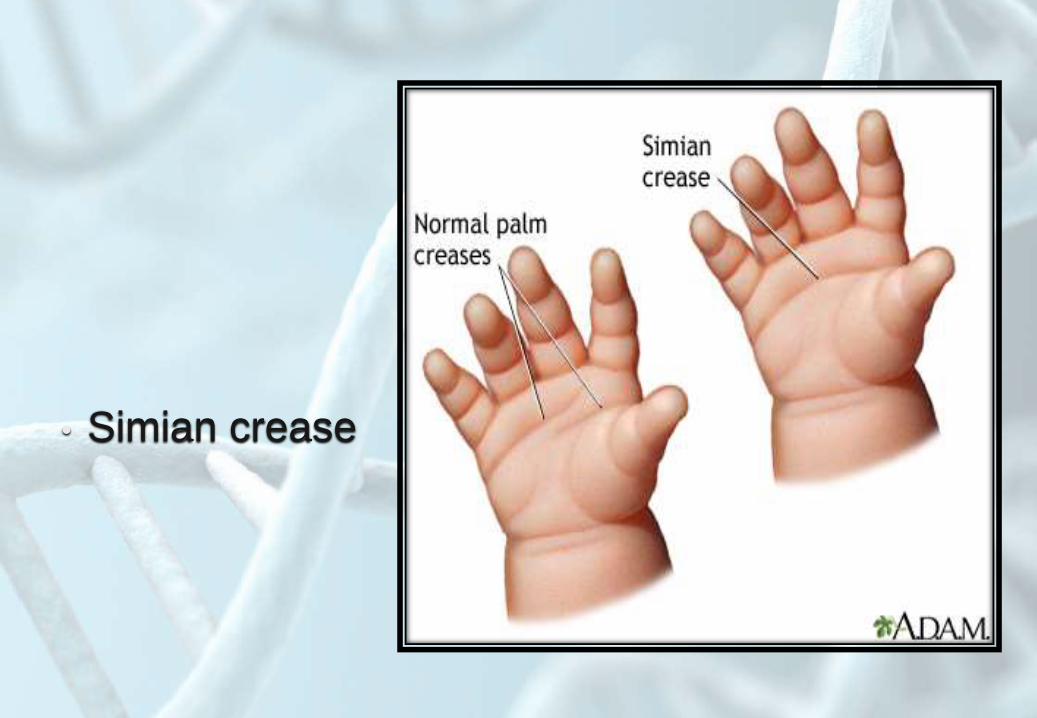
• Simian crease

• Clenched hand

Hypoplastic nails
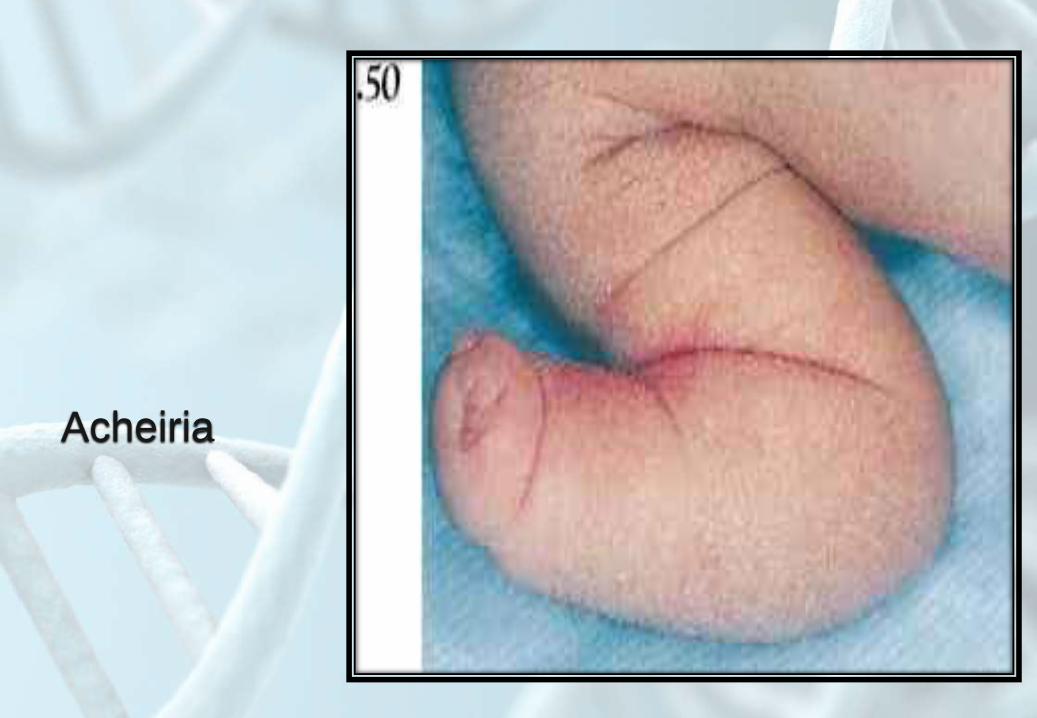
Acheiria

Floating thumb
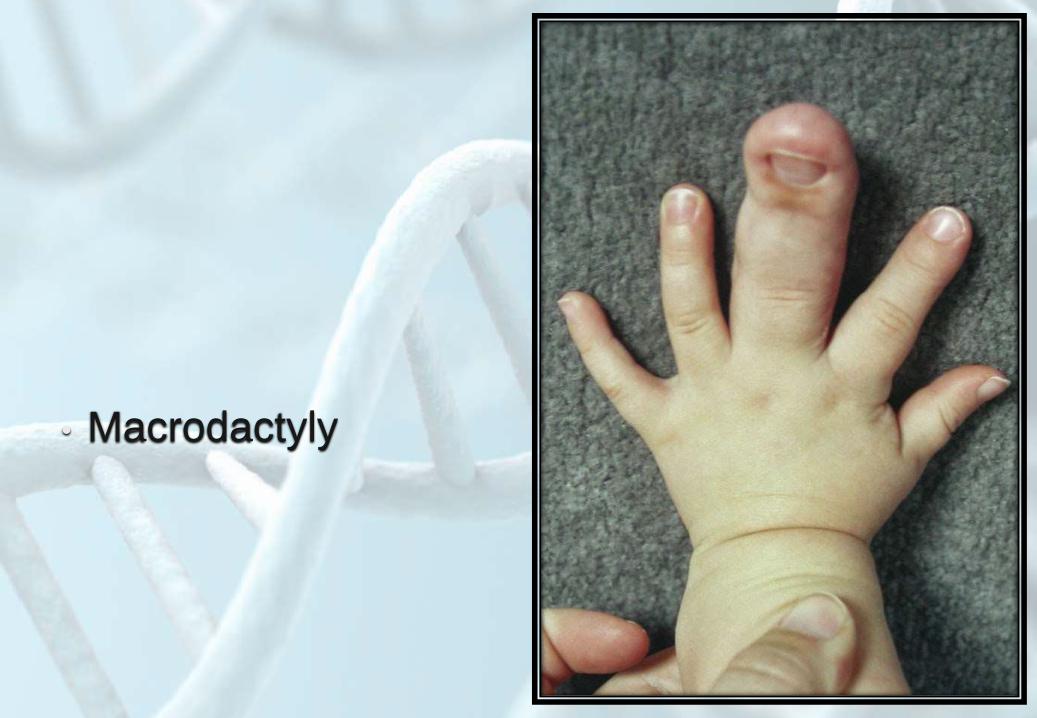
• Macrodactyly

Lobster-claw deformity (ectrodactyly, or split
hand/split foot
deformation).

Foot

Foot
• Talipus equinovarus

• Rocker bottom heel

• Polydactyly

Wide separation
between first and
second toes

limbs
Dislocation of the
hip:
common in
females

Congenital
absence of
patellae:in a normal infant. this
finding is also noted,
trisomy 8 and
Nievergelt syndrome.

limbs
• Amelia:
is absence of
the entire limb
structure

Phocomelia syndrome:more proximal portion of a
limb fails to develop
properly but distal
structures are relatively
intact.
Common with maternal
thalidomide ingestion

Sirenomelia

Caudal regression $

Absent radius:
TAR $
Fanconi
anemia
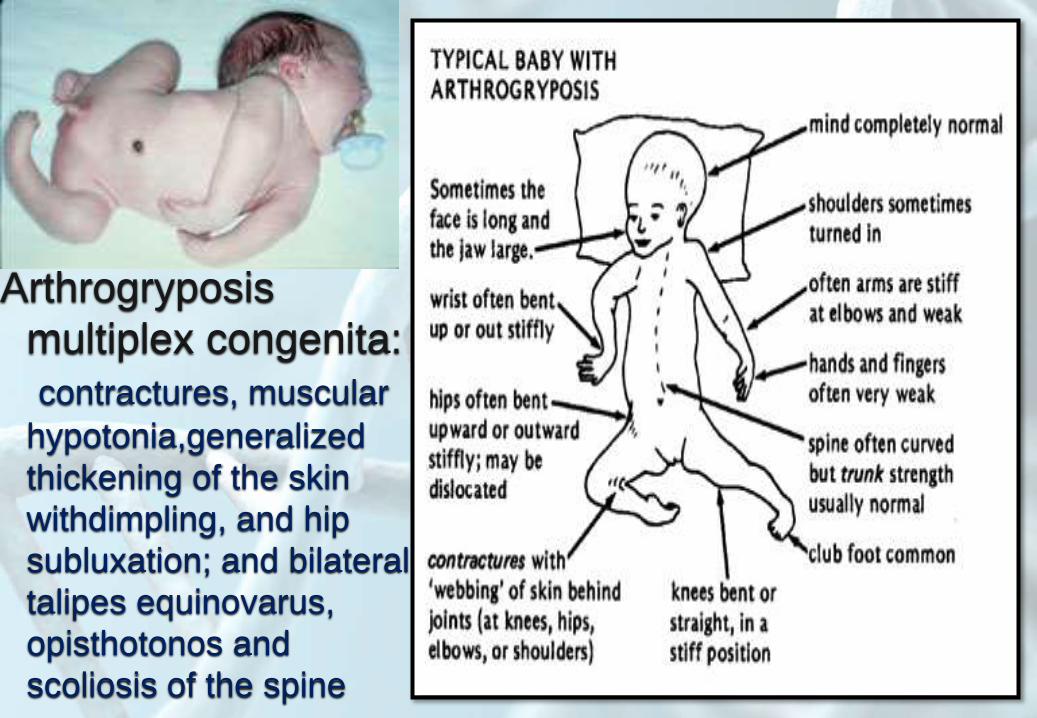
Arthrogryposis
multiplex congenita:
contractures, muscular
hypotonia,generalized
thickening of the skin
withdimpling, and hip
subluxation; and bilateral
talipes equinovarus,
opisthotonos and
scoliosis of the spine

Chest and abdomen

Asphyxiating
thoracic dystrophy
) Jeune's
syndrome(autosomal recessive
due to the short ribs results
in a small chest which limits
pulmonary expansion and
severely restricts respiration.
Because of the small thorax,
the whole liver lies in the
abdomen, producing the
rounded and enlarged
appearance.
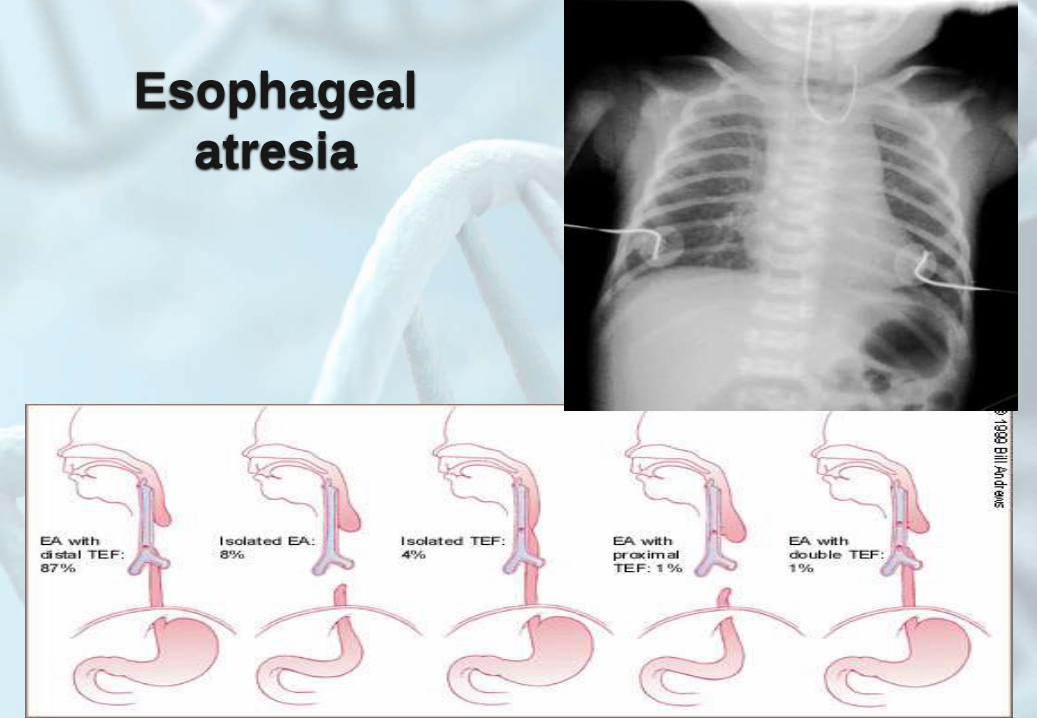
Esophageal
atresia
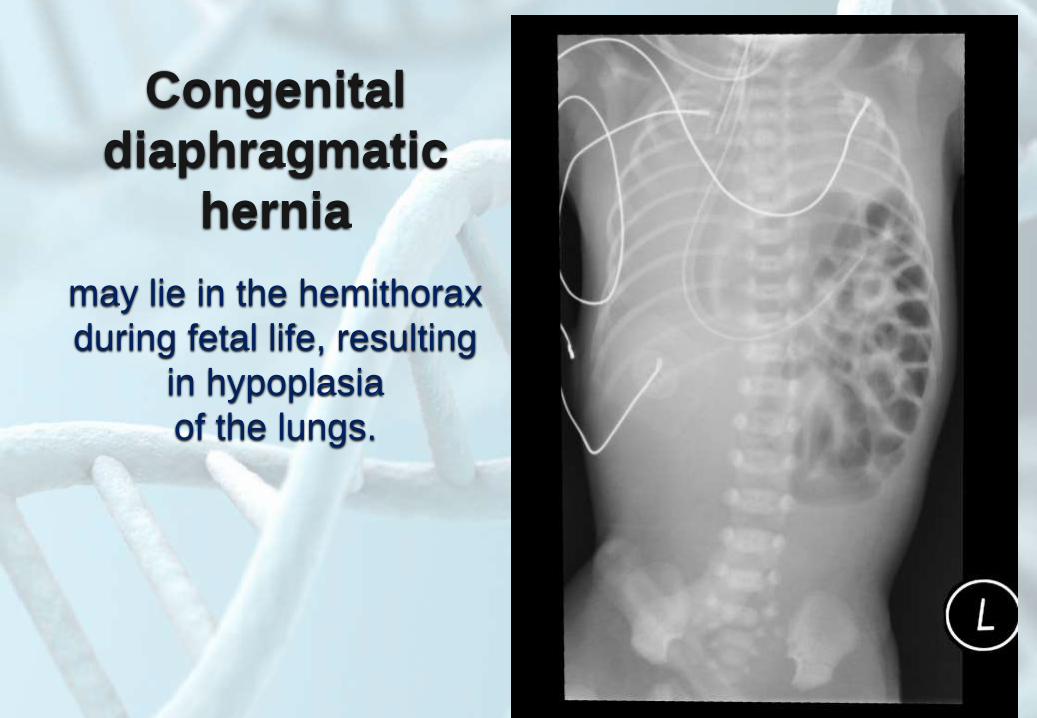
Congenital
diaphragmatic
hernia
may lie in the hemithorax
during fetal life, resulting
in hypoplasia
of the lungs.

Ectopia cordis
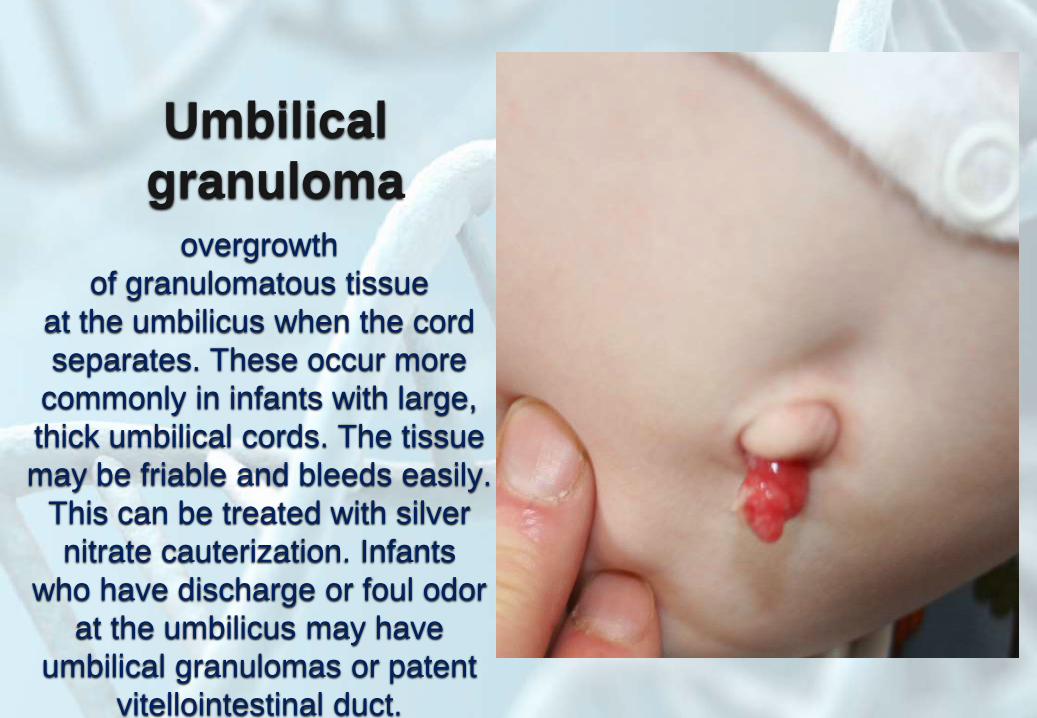
Umbilical
granulomaovergrowth
of granulomatous tissue
at the umbilicus when the cord
separates. These occur more
commonly in infants with large,
thick umbilical cords. The tissue
may be friable and bleeds easily.
This can be treated with silver
nitrate cauterization. Infants
who have discharge or foul odor
at the umbilicus may have
umbilical granulomas or patent
vitellointestinal duct.

Omphalocelefailure of the complete return of
intestines to the abdominal cavity in
early fetal life (10 weeks). Extra-
abdominal contents are positioned
midline. The umbilical cord is
incorporated and a sac is present.
Intestinal malrotation is a frequent
associated finding. Omphaloceles
may occur as isolated findings or
can be associated with other
congenital and chromosomal
abnormalities. It is frequently seen
in trisomy 13 and in
Beckwith-Wiedemann syndrome.

Gastroschisis
anterior abdominal
wall defect which is usually
paramedian to the right of
the umbilical cord insertion.
there is no covering
membrane.

Prune
belly syndromeabsence of the abdominal
musculature
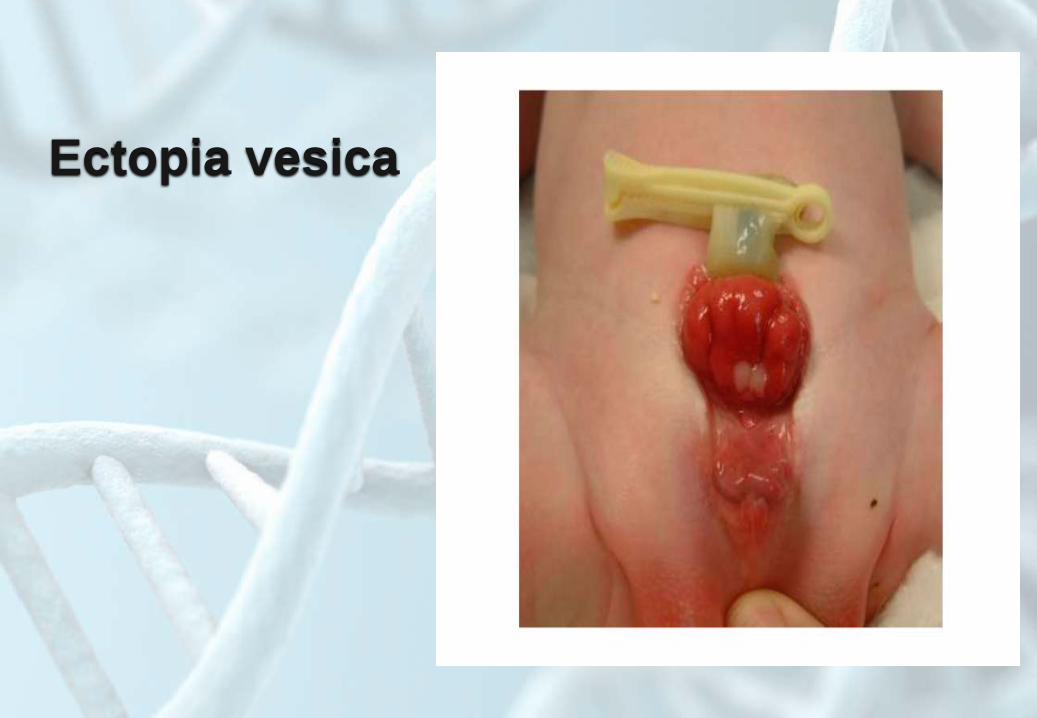
Ectopia vesica
119