early diagnosis of melanoma: what do we know?
TRANSCRIPT

Early diagnosis of melanoma: what do we know?
Malignant melanoma is a potentially lethal disease with anexcellent prognosis if detected early. In this paper, we review thepathophysiology, clinical presentation, epidemiology, and riskfactors for melanoma. We also examine clinical models for pre-dicting risk and survival, and critically appraise data on theeffectiveness of screening for melanoma.
KEY WORDS: Melanoma, diagnosis - Melanoma, pathology - Skinneoplasms.
Pathophysiology/clinical features
Melanoma orginates from melanocytes, pigmentcells that are mostly from neural crest cells or
neuroectoderm cells (the latter form retinal pigmentepithelium). Melanocyte precursors differentiate andmigrate from the neural crest to skin and other tissuesduring early gestation. More than 95% of melanomasare cutaneous,1 and while they may be located any-where on the skin surface, they are often on the trunk(especially the back) in men and legs in women.2Primary extracutaneous sites include eyes, gastroin-testinal tract, genitourinary tract, leptomeninges, andlymph nodes (unknown melanoma primary).1 TheClark model describes a stepwise tumor transfor-mation starting with a proliferation of normalmelanocytes nesting to form nevi, followed by hyper-plasia/aberrant differentiation, atypia and dysplasia,
radial growth (intraepidermal proliferation), verti-cal growth (dermal invasion), and metastasis.3 Whileit is thought that most dysplastic melanocytic nevi areterminal lesions that do not progress to melanoma, thefrequency of nevi progression to malignancy or neviregression is not known.3, 4 Biologically, melanomasmay be quite heterogeneous, with variable rates ofprogression.5 However, there are no known biolog-ic indicators of indolent melanomas, and the molec-ular differences between these and clinically impor-tant early melanomas are not clear.6
Epidemiology
Cutaneous melanoma is a public health problemamong fair-skinned populations, and the number ofcases worldwide is increasing rapidly.7 Melanomahas ranked as the fourth most common cancer in
1Department of MedicineNew Mexico Veterans Affairs Health Care System
University of New Mexico, Albuquerque, NM , USA 2Department of Medicine
New Mexico Veterans Affairs Health Care SystemUniversity of New Mexico
Cancer Research Treatment Center, Albuquerque, NM, USA3Division of Epidemiology
University of New Mexico Cancer Research Treatment CenterPopulation Science Program, Albuquerque, NM, USA
Address reprint requests to: E. F. T. Yee, MD, 1501 San Pedro Drive SE(Mail Code 111-GIM) Albuquerque, NM 87110, USA.E-mail: [email protected]
E. F. T. YEE 1, R. M. HOFFMAN 2, M. BERWICK 3
Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 55
REVIEWSG ITAL DERMATOL VENEREOL 2007;142:55-70
CUTANEOUS MALIGNANCY UPDATE:MELANOMA IN 2007

YEE EARLY DIAGNOSIS OF MELANOMA
56 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
Australia and New Zealand, the seventh most com-mon in the United States and Canada, the tenth mostcommon in Scandinavia, and the eighteenth mostcommon in England, Wales, and Scotland.8 Mela-noma incidence is closely associated with skin col-or and geography.9 Between the early 1960s and thelate 1980s, annual incidence rates increased 3-7%in populations of mainly European origins,10 withan estimated doubling of rates every 10-20 years.11
The highest incidence rates (standardized to the worldpopulation) are seen in Australia with 40.5 cases/100000 in males and 31.8 cases/100 000 in females, andNew Zealand with 36.7 cases/100 000 in males and34.9 cases/100 000 in females. By comparison, thestandardized rates in the United States are 13.3/100 000 in males and 9.4/100 00 in females, whilestandardized rates in Italy are 4.6/100 000 in malesand 5.5/100 000 in females.7, 12, 13
Annual mortality rates vary with the highest rates(standardized to the world population) in males seenin New Zealand: 5.3/100 000, Australia: 4.8/100 000,and Norway 3.3/100 000. The highest rates in femalesare seen in New Zealand: 3.2/100 000, Norway:2.7/100 000, and Australia 2.5/100 000.13 Mortalityrates increased in most populations of European ori-gin throughout most of the 20th century, but stabi-lized or even declined in some populations.14 In Swe-den, age-standardized mortality rates leveled offsince the mid-1980s and declined from 1987 to 1996in women.15 In Australia, age-standardized mortali-ty rates appear to have peaked in 1985 and thenplateaued.16 Mortality rate increases are consistent-ly lower than the increase seen in incidence rates.14
Five-year survival rates have increased from approx-imately 40% in the 1940s to over 90%.7, 17 Mostmelanomas are localized (e.g., confined to primarysite); US SEER data from 1988 to 2002 show that82% of cutaneous melanomas were localized at diag-nosis, 10% were regional, and 3% had distant metas-tases.18 Older persons have a higher burden of mor-bidity and mortality from melanoma,19 especiallyolder men who account for nearly two thirds of allmelanoma deaths in Australia and the United States.20
In the United States in 2002-2003, the median age atdiagnosis for melanoma was 58 years of age and themajority of cases (77%) were diagnosed in personsaged 45 and older. The median age at death frommelanoma was 67 years of age.17
The difference between the trends in incidence
and mortality rates may be partly due to the increaseddiagnosis of thin melanomas. A study of seven pop-ulations in the 1980s showed that relative and absoluteincidence increased most for the thinnest melanomasand least for the thickest lesions.10 In Central Europe,the median tumor thickness decreased from 1.2 mmin 1986 to 0.8 mm in 1996 (P<0.001), while the per-centage of melanomas ≤0.75 mm increased from29.8% to 46.4% (P<0.001).11 Data from the popula-tion-based Queensland Cancer Registry show anincreasing age-standardized incidence, with a shift toproportionately more in situ lesions, and stable mor-tality rates from 1991 to 2002.21
Questions have been raised regarding whether theincreased incidence in melanoma reflects a trueincrease in the disease or is a result of more intensivesurveillance and overdiagnosis.22, 23 More intensivemelanoma surveillance activity is associated withincreased diagnoses of thin melanomas, but not thatof advanced tumors. The large increase in melanomadiagnosis without a corresponding increase inadvanced tumors and mortality do not fit with a pre-sumed malignant behavior of thin melanomas.Melanoma incidence may be artificially increasedby increased melanoma surveillance intensity if his-tologically “malignant” but biologically benignlesions are excised. There is little available evidenceto suggest an actual increase in the frequency of bio-logically malignant tumors.24
A population based ecological study usingMedicare claims data examined changes in skin biop-sy rates in persons aged 65 and older to determine therelation to changes in the incidence of melanoma.During 1986 to 2001, the average biopsy rateincreased 2.5-fold among people aged 65 and older(2 847 to 7 222 per 100 000 population) while theaverage incidence of melanoma increased 2.4-fold (45to 108 per 100 000 population). Most of the increasewas due to early stage (in situ and local) rather thanlate stage (regional and nodal) disease, and mortali-ty due to melanoma did not change appreciably dur-ing this period. The authors concluded that melanomaincidence is associated with biopsy rates, and theincreased incidence could be attributed to increaseddiagnostic scrutiny (skin lesions being biopsied thatwould not have been in the past) rather than anincrease in the disease incidence.25 Although this isa plausible hypothesis, the authors were unable toseparate biopsies for suspicious melanoma versus

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 57
EARLY DIAGNOSIS OF MELANOMA YEE
non-melanoma skin cancer. However, this type ofmisclassification would bias their results to the nullrather than inflate the estimates.
Mortality rates and the diagnosis rate for thicktumors (>1.5 mm thick) have not gone up along withthe increased rate of melanoma diagnosis. It is notclear whether the increase in diagnosed melanomacases reflects an actual increase in real disease.Aggressive surveillance might result in the identifi-cation of pigmented skin tumors of limited or nonex-istent potential for malignant behavior. The improve-ment in melanoma prognosis may be a consequenceof removing biologically benign pigmented tumorsthat are inadvertently classified as malignant.26
Diagnosis of melanoma depends on the examina-tion of a biopsy specimen and the interpretation of apathologist. Pathology reports lack lexicon and for-mat standardization. Though well-established crite-ria for diagnosis and staging exist, subjective inter-pretation can result in discordant diagnoses.27
Histopathology is qualitative and subjective, and sen-sitivity in diagnosis melanoma is important due to therisks of missing a cancer diagnosis. Setting a lowerdiagnostic threshold to increasing sensitivity canresult in lower specificity and more false positivediagnoses.22 Four histopathologists evaluating 140slides and classifying lesions as “melanoma” or “oth-er pigmented lesions” agreed on 74% of slides (kap-pa = 0.61).28 Eight expert pathologists reviewing 37slides and classifying specimens as benign, malignant,or indeterminate had unanimous agreement or onlyone discordant on 62% of slides (kappa = 0.50).29 Aretrospective study of 5 136 specimens reviewed byone pathologist found that 11% had a significantdiagnosis change from the referral diagnosis. Criti-cal revisions included 1.2% being changed frommalignant to benign, and 1.1% from benign to malig-nant. The rest, 8.5%, had an upgrade or downgradein severity sufficient enough to change treatment,prognosis, and research.30 In a density ratio model, asmany as 40% of lesions diagnosed as melanoma in1988 would not have been so diagnosed in 1978.31
Risk factors for melanoma
A recent meta-analysis assessed major risk fac-tors for melanoma, including number of commonnevi on the whole body and the arms, number ofatypical nevi on the whole body, sun exposure, fam-
TABLE I.—Pooled estimates for risk of melanoma.
Risk factor RR (95% CI)
# Common neviWhole body
0-15 1.0016-40 1.47 (1.36-1.59)41-60 2.24 (1.90-2.64)61-80 3.26 (2.55-4.15)81-100 4.74 (3.44-6.53)101-120 6.89 (4.63-10.25)
Arms0 1.00 1-5 1.44 (1.29-1.60)5-10 2.48 (1.90-3.23)11-15 4.82 (3.05-7.62)
Atypical nevi-all body # of nevi
0 1.001 1.60 (1.38-1.85)2 2.56 (1.91-3.43)3 4.10 (2.64-6.35)4 6.55 (3.65-11.75)5 10.49 (5.05-21.76)
Sun exposure*Total sun exposure 1.34 (1.02-1.77)Intermittent sun exposure — All data 1.61 (1.31-1.99)— Aus, US, Can, UK 1.14 (0.90-1.44)— Other countries 2.08 (1.55-2.78)Chronic sun exposure 0.95 (0.87-1.04)Sunburns 2.03 (1.73-2.37)
Family history, actinic damage, and phenotypic factorsFamily History 1.74 (1.41-2.14)Actinic damage:— Premalignant/skin CA 4.28 (2.80-6.55)— Other actinic indiactors 2.02 (1.24-3.29)High density of freckles 2.10 (1.80-2.45)Phototype— I vs IV 2.09 (1.67-2.58)— II vs IV 1.84 (1.43-2.36)— III vs IV 1.77 (1.23-2.56)Eye color— Blue vs Dark 1.47 (1.28-1.69)— Green vs Dark 1.61 (1.06-2.45)— Hazel vs Dark 1.52 (1.26-1.83)Hair color— Red vs Dark 3.64 (2.56-5.37)— Blond vs Dark 1.96 (1.41-2.74)— Light brown vs Dark 1.62 (1.11-2.34)Skin color Light vs Dark 2.06 (1.68-2.52)
*Sun exposure was defined as: total sun exposure (amount of sun exposure of allkinds); intermittent sun exposure (amount of intermittent pattern of sun exposure);chronic sun exposure (amount of more continuous pattern of sun exposure); sunburns(number of episodes of sunburns). Adapted from Gandini et al.32-34
ily history, actinic damage, and phenotypic factorssuch as eye color, hair color, and skin photo type(type I-always burn, never tan to type IV-never burn,tan easily). Pooled relative risks (RR) (Table I)demonstrate approximately 6-fold relative risks for

YEE EARLY DIAGNOSIS OF MELANOMA
58 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
developing melanoma with a large number of neviand 5 or more atypical nevi. In addition, a large num-ber of nevi on the arms, actinic damage, and red hairincreased risk approximately 4-fold while otherimportant risk factors, such as freckling, light skintype, light skin color, a history of severe sunburnsand a family history of melanoma increase risk moremodestly, approximately 2-fold. Intermittent sunexposure increased risk while chronic sun exposureappeared to be inversely related to melanoma.32-34
Constant sun exposures is postulated to lead toincreased tanning and skin thickening conferring aprotective effect. Intermittent exposure may stimulatefrequent melanocyte activity and lead to more cellularchanges resulting in cell transformation than wouldoccur with constant exposure.14
Other proposed risk factors include exposure totanning beds and sunlamps, obesity, industrial occu-pation, personal history of skin cancer, higher socioe-conomic status, brown hair, male sex, and endogenoushormones have been proposed. However, the evi-dence for an association between any of these risk fac-tors and melanoma is weak.1
Risk models for developing melanoma
Investigators have evaluated risk for melanomausing either number of risk factors or risk algorithmsto form risk prediction models. A Western Australiastudy 35 examined the cross-sectional associationbetween the number of skin cancer/melanoma riskfactors and the diagnosis of a malignant melanoma.Data were analyzed from 5 950 subjects evaluatedbetween 1996 and 2003 in skin cancer screening clin-ics targeting high risk individuals. A questionnairewas used to collect information on 9 risk factors: per-sonal history of malignant melanoma, family historyof malignant melanoma, 5 or more moles on the fore-arm, previous removal of non-cancerous moles, pre-vious skin cancer, a mole/freckle that has changedsize, shape, or color, fair skin that always burns, pre-vious blistering sunburn, and non-healing or inflamedskin sores. No relationship was found with logisticregression analyses between the number of risk fac-tors, adjusted for age and gender, and a histopatho-logically-confirmed melanoma. This study was lim-ited by the small number (18) of confirmedmelanomas and the reliance on self-reporting of thenumber of risk factors which is subject to recall biasand over- or under-statement of participant risk.
MacKie et al. 36 used information from a case-control study of all patients with cutaneous malignantmelanoma first diagnosed in Scotland in 1987 toderive a personal risk-factor chart. This was intend-ed for use by medical professionals and the generalpublic. The authors created 4 risk groups for bothmen and women based on the presence or absence ofthe 4 strongest risk factors identified by conditionallogistic regression: having 20 or more benign pig-mented nevi above 2 mm diameter; freckling ten-dency; having some clinically atypical nevi (over 5mm diameter and having an irregular edge, irregularpigmentation, or inflammation); and having a his-tory of severe sunburn at any time in life. Patientsin the lowest risk group had a relative risk of beingdiagnosed with melanoma ranging from 1.5, with95% Confidence Interval (95% CI 0.96-2.2) to 15.6(95% CI 1.9-130) (compared to patients with none ofthe 4 risk factors), while patients in the highest riskgroup had relative risks ranging from 18.2 (95% CI16.1-54.0) to 587 (95% CI 33.7-10 200).
A prediction model to estimate the 5-year absoluterisk of melanoma was developed using case-controldata from 718 non-Hispanic white patient cases withinvasive cutaneous melanoma from melanoma clin-ics in Philadelphia, PA and San Francisco, CA, and945 matched controls. The intent was to help identi-fy high-risk individuals for potential interventions.Sex-specific relative risk models were combinedwith incidence and mortality rates by United Statesgeographic areas to develop estimates of the absoluterisk of developing melanoma within 5 years. Theareas under the receiver operating characteristiccurves (AUROC) estimated by the models rangedfrom 0.70 for women aged 50 years and older to 0.80for men 20 to 49 years of age. Individual absolute riskcan be estimated using a program available onlineat http://dceg.cancer.gov/melanomarisktool. The riskassessment tool factors in geographic residence(northern, central or southern United States), gen-der, race, age, number of small and large moles onback, complexion (light, medium, or dark), tanning(deeply, moderately, lightly, or not at all) after severe,prolonged exposure to sunlight, the extent of freck-ling (by comparison to standard photographs), andsun exposure (blistering sunburn, severe solar dam-age on the shoulders). These projections are notintended to identify current melanoma cases, can-not be used to predict risk in races other than non-His-

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 59
EARLY DIAGNOSIS OF MELANOMA YEE
panic whites, and are applicable only to those livingin the United States.37
Predictors of melanoma survival
A model predicting 10-year survival for patientswith primary cutaneous melanoma and no apparentmetastatic disease found 4 independent predictors:thickness, site, age, and gender. Adjusted odds ratioswere: for lesions <0.76 mm thick, OR=50.8, (95% CI18.5-140), lesions 0.76-1.69 mm, OR=9.5 (95% CI4.0-22.5), lesions 1.70-3.60mm, OR=2.9, (95% CI1.29-6.5), primary lesion on an extremity, OR=4.4(95% CI 2.3-8.4), age ≤60 years at diagnosis, OR=3.0(95% CI 1.7-5.3), and female sex, OR=2.0 (95% CI1.2-3.6). The four-variable model was significantlymore accurate than a one-variable model using tumorthickness alone.38 However, a study validating thisprognostic model found no significant differencesfor accuracy between the data sets for the four- andone-variable models.39
An analysis of a multicenter database from 30 450melanoma patients, found that survival was related tothickness of the melanoma (Breslow depth, mea-sured in millimeters) and regional node status. Inlocalized disease (stage I and II), the strongest prog-nostic factors were thickness of the tumor and pres-ence of ulceration. In regional metastatic disease(stage III, including regional nodal involvement, intransit or satellite metastases), node status (number,clinical versus occult), and tumor ulceration wereimportant prognostic factors. With distant metas-tases (stage IV), anatomic site was the most signifi-cant factor.40 The 10-year survival rates for stage Itumors ≤1.0 mm are 88% without ulceration and83% with ulceration or Clark level IV, V invasion. The10-year survival rates for other stages range from64%-32% for stage II and 63%-15% for stage III.With stage IV, rates decrease to 16% for skin andsubcutaneous lesions, 6% for other visceral lesions,and 2% for lung lesions.41
Other prognostic factors include serum LDH, anindependent predictor of disseminated metastases,41
and tumor mitotic rate (TMR), expressed asmitoses/mm2. TMR has been shown to be an impor-tant prognostic factor in a number of multivariateanalyses.42, 43 In a study examining data from 3 661patients in the Sydney Melanoma Unit database,patients with a TMR of 0 mitoses/mm2 had signifi-cantly better survival compared to those with 1 mito-
sis/mm2 (P<0.0001). Two different TMR groupingswere used for a Cox regression analysis- method A:0, 1-4, 5-10, and ≥11 mitosis/mm2 and method B:0-1, 2-4, and ≥5 mitosis/mm2. TMR was a highlysignificant independent prognostic factor with bothgroupings. With method A, TMR was second only totumor thickness in predicting survival (P<0.0001),and was more powerful than age and ulceration. Withmethod B, thickness was the most powerful predic-tor, followed by age, ulceration, and TMR.42 In apopulation-based series of 650 consecutive invasivecutaneous malignant melanoma cases ascertainedfrom the Connecticut Tumor Registry, TMR was asignificant predictor, but tumor ulceration was nolonger an independent prognostic factor after adjust-ing for TMR.43
Detecting melanoma
The skin examination
ABCDE AND THE UGLY DUCKLING: OPPORTUNISTIC
MELANOMA DETECTION
A visual skin exam is the most commonly usedscreening method to detect skin cancers.44 Clinicalrecognition and differentiation of cutaneous melanomafrom clinically atypical moles can be difficult.45 Twoclinical indicators for visual recognition of melanomainclude the ABCDE mnemonic and the Ugly Duck-ling sign. The ABCD (Asymmetry, Border irregular-ity, Color variegation, Diameter greater than 6 mm)melanoma screening mnemonic was developed in1985 to assist lay persons and primary care providersin the early detection of melanomas.46 Evolvinglesions (E) was later added to expand the criteria toABCDE.47 However, a melanoma may be missed if alesion does not have the typical ABCDE features.44
Another clinical indicator used to recognizemelanomas is the “ugly duckling sign.” In a givenindividual, nevi generally share characteristics. Anevus with different features from other nevi is con-sidered to be an ugly duckling and should raise theconsideration of a possible melanoma.48
TOTAL BODY SKIN EXAM: SYSTEMATIC MELANOMA
DETECTION
Using the total-body skin exam for screening ratherthan a partial exam has been advocated because 80%

YEE EARLY DIAGNOSIS OF MELANOMA
60 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
of melanomas appear in areas of the body normallycovered by clothing.49 In a study of 2 239 persons seenat the Manhattan Melanoma/Skin Cancer DetectionScreening Program, patients with complete skinexaminations were 6.4 times more likely to have amelanoma detected compared to those with partialexaminations (P=0.025). Thirteen out of fourteenmelanomas found were on anatomic sites normallycovered by clothing.50 Important incidental lesionssuch as melanoma, lentigo maligna, dysplastic nevi,and congenital nevi are found in a significantly high-er proportion of patients receiving total body skinexam compared to those receiving a partial exami-nation.51
PHYSICIAN-DETECTED MELANOMAS ARE USUALLY THIN-NER THAN PATIENT-DETECTED LESIONS
Multiple studies have found that primary melanomais most often detected by the patient, followed by adoctor, spouse or partner, or other source.49, 52-57
Physician-detected lesions are usually thinner thanpatient or spouse-detected lesions, and dermatolo-gist-detected lesions are usually thinner than otherphysician-detected lesions. Whether that means thatphysician-detection will be more effective is notproven by these data due to the lack of informationabout efficacy of screening or early detection in pre-venting mortality. A population-based study of 3 772Queensland residents with a histologically confirmedmelanoma diagnosed between 2000 and 2003 foundthat patients detected 44% of lesions while physi-cians detected 25%. Melanomas detected by doctorswere more likely to be thin (< 0.75 mm) than those
detected by the patient or other layperson. Overall,81% of physician-detected lesions were thin com-pared to 62% of layperson-detected lesions.Melanomas detected during a deliberate skin exam-ination were thinner than those detected incidental-ly.52 A study of 102 patients with newly detected pri-mary cutaneous melanoma seen at the John HopkinsMelanoma Center found that patients detected 55%of lesions compared to 24% detected by physicians.Physician detection was associated with thinnerlesions compared to detection by self or others (medi-an thickness 0.23 mm vs 0.9 mm, P<0.001).53 A studyof 1 515 consecutive patients with melanoma seen atthe University of Michigan,54 a review of 471 new-ly diagnosed melanoma patients at Sloan Kettering,55
and a retrospective cohort analysis of 218 patientswith melanoma at the Yale Pigmented Lesion Clin-ic all found that physician-detected initial lesionswere more likely to be thinner than self/non-physi-cian detected lesions. Thin lesions were also associ-ated with detection by a dermatologist compared toother physicians,49 a finding also noted in a study of813 patients consecutively diagnosed with melanomain 11 Italian Clinical Centers. Detection by a der-matologist and skin self-examination were signifi-cantly associated with thinner lesions compared tothose found by patients or other physicians.56 Aprospective survey of 590 consecutive melanomapatients in France found that tumor thickness waslower (median 0.94 vs 1.50 mm, mean 1.88 vs 2.82mm respectively, P<0.001) when first seen by a der-matologist as compared to another physician.57
Accuracy of the skin examination
ACCURACY BY LESION: ABCDE AND UGLY DUCKLING
A review of studies examining the diagnostic accu-racy of the ABCDE criteria found that sensitivityand specificity varied depending on whether theywere used alone or in combination (Table II).47, 58 Inone study, using all five ABCDE criteria had thehighest specificity (100%) but the lowest sensitivity(43%). In other studies, one or more of the ABCDEshad a sensitivity of 92% (95%CI 82-96),59 and a 3-variable model (the BCD criteria used together) hada sensitivity of 100% (95% CI, 54-00) and a speci-ficity of 98% (95% CI, 95-99).60
A prospective French study described themelanoma recognition process by immediately sur-
TABLE II.—Sensitivities, specificities, and likelihood ratios associa-ted with ABCDE criteria.
Criteria Sensitivity Specificity Likelihood(%) (%) Ratio
A 57 72 2.0B 57 71 2.0C 65 59 1.6D 90 63 2.4E 84 90 8.4≥1 Criterion 97 36 1.5≥2 Criteria 89 65 2.5≥3 Criteria 66 81 3.5≥4 Criteria 54 94 9.0≥5 Criteria 43 100 ∝
Adapted from Abbasi et al.47 and Thomas et al.58

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 61
EARLY DIAGNOSIS OF MELANOMA YEE
veying 135 dermatologists after they clinically eval-uated pigmented lesions (n=4 036) that were subse-quently biopsied. Features most predictive of accu-rately diagnosing melanoma were the impression ofoverall irregularity of the lesion, the perception thatthe lesion is an “ugly duckling” compared to the oth-er nevi, and knowledge that the lesion had changedor evolved via patient report. A, C, and D criteriahad lower predictive values.61
CORRELATION BETWEEN EXAMINERS REGARDING SKIN
EXAMINATION AND RISK FACTORS
A study to determine feasibility of targeted ear-ly detection for melanoma found limited agreementbetween patient and dermatologist assessments ofrisk factors. The kappa score was best for hair col-or (0.67), while the kappa scores were 0.22 forprevalence of ≥10 moles on head and neck, 0.36for reporting of skin type I or II, and 0.13 for report-ing moderate or many freckles. Correlation mayhave been low as patients tended to under-reporttheir risk level. While a high nevi count is an impor-tant risk factor for melanoma, patients appearedreluctant to categorize themselves in the mostextreme group having a lot of moles.62 In a study ofclinician agreement on clinical characteristics recog-nition of cutaneous melanoma (number and typeof nevi), variation was seen for the index of agree-ment (calculated as the intra-class correlation coef-ficient). Agreement was low for freckling on theright forearm (60%) and on the shoulders (69%), andpalpable nevi of the arms (36%). Agreement washigher (88%) for total arm nevi (both palpable andnon-palpable), as well as total number of atypicalnevi on the body (87%), and total number of alltypes of nevi (92%).63
ACCURACY OF THE TOTAL-BODY SKIN EXAMINATION:ONE OR MORE LESIONS
We define a low positive predictive value (PPV) asnumber of biopsy-proven melanomas divided bynumber of screens with presumed melanoma diag-nosis. A high PPV is defined as the number of biop-sy-proven lesions divided by the number of presumedmelanoma with biopsy data.
The reported sensitivity of a total-body skin exam-ination in a community-based screening programwas 70 % (95% CI 51.3-84), specificity was 98%
(95% CI 97.2-97.9), low (PPV) was 11%, and neg-ative predictive value (NPV) was 99.9% formelanomas diagnosed within one year of a screen.The low PPV was 10% if the body site was takeninto account. All negative screens were followedthrough a population-based cancer registry to cal-culate sensitivity of the screening diagnosis. Screenswere conducted by volunteer plastic surgeons anddermatologists.64 In another community-based screen-ing program, primary care physicians had a reportedspecificity of 86% (95% CI 86-87), and low PPV of15% using a denominator of number of screens withpresumed melanoma diagnosis, or high PPV of 20%using a denominator of number of presumedmelanoma with biopsy data. However, this study didnot follow up on negative screens, and the specifici-ty calculations were based on expected number offalse-negative cases.65
In other studies, the low PPV of a total body skinexam ranged from 7-17%, and high PPV from 12-20% 64, 66-70 (Table III). One of the largest studies toevaluate screening reviewed 242 374 skin cancerscreenings on more than 206 000 Americans whoattended the American Academy of DermatologyNational Skin Cancer Screening Programs duringthe period 1992-1994. Investigators contacted 96%of 3 476 screenees with a presumptive diagnosis ofmelanoma or possible melanoma. Follow-up recordswere obtained for 73% of screenees, and 363 hadhistologically proven melanoma. Middle-aged andolder men (age ≥50 years) comprised only 25% ofscreenees but comprised 44% of those with a con-firmed diagnosis of melanoma. Men age ≥50 yearswith a changing mole had a low PPV=22%, and lowPPV=28% if they had skin types I and II.68
When sensitivities and predicted values are report-ed in the studies, it appears that only one study,Fritschi et al.,64 attempted to account for body site ofthe melanoma to improve validity. A reported suspi-cious lesion may not be the lesion that was biopsied.If a melanoma was found at a different site than whatwas considered suspicious, then the sensitivity andpositive predictive value will be overestimated. Anoth-er caution when interpreting these data is that scree-nees are self-selected, and results might not be applic-able to the general population. Sensitivity and preva-lence of the preclinical disease influence the PPV ofa screening test. The PPV from studies in a commu-nity or screenee group with a high prevalence of

YEE EARLY DIAGNOSIS OF MELANOMA
62 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
melanoma will be higher than in those with a lowerprevalence of melanoma.
ACCURACY OF SKIN EXAM: DERMATOLOGY VS PRIMARY
CARE
Studies comparing the accuracy of dermatologistsand primary care physicians (PCPs) in identifyingpigmented lesions suggestive of melanoma and mak-ing the appropriate management decisions regard-ing performing a biopsy or referring the patient havefound mixed results. A systematic review of 32 stud-ies published between January 1966 and October1999 found that the sensitivity for diagnosingmelanoma was 81% to 100% for dermatologists and42% to 100% for PCPs. None of the studies report-ed specificity for dermatologists and only one report-ed a specificity for PCPs of 98%. The biopsy or refer-ral accuracy sensitivity ranged from 82 to 100% fordermatologists and 70 to 88% for PCPs. The speci-ficity ranged from 70 to 89% for dermatologists and70 to 87% for PCPs. However, authors were unableto detect any significant differences in the areas underthe receiver operating characteristic curves for biop-sy and referral ability between dermatologists andPCPs.71
A subsequent study assessed differences betweendermatologists and PCPs in accurately diagnosingmelanoma and appropriately managing (based ondecisions to refer/biopsy) using photographs of sus-picious pigmented lesions. Dermatologists had bet-ter diagnostic accuracy and ability to manage pig-mented lesions than PCPs. Investigators used a sur-vey of a random sample of 30 photographs of pig-mented lesions with known pathology administeredto 101 dermatologists and 115 PCPs. Likelihoodsthat a photographed lesion was melanoma and that thelesion should be biopsied/referred were scored on a1 to 10 scale. Accuracy of melanoma diagnosis andappropriateness of pigmented lesion managementwere compared between dermatologists and PCPsby using the areas under the receiver operating char-acteristic curves (AUROC). Dermatologists weresuperior to PCPs in diagnosing melanomas (AUROC0.89 vs 0.80, P<0.001) and appropriately managingpigmented lesions (AUROC 0.84 vs 0.76, P<0.001).72
Suggestions have been made for additional educationand training of PCPs due to concerns regarding ashortage of dermatologists. PCPs have the opportu-nity to manage pigmented lesions in patients pre-senting to them. However it is unclear that this would
TABLE III.—Results of screening program studies, positive predictive values for melanoma.
Confirmed Presumed Biopsies of Low Highmelanomas melanoma presumed PPV PPVSubjects and setting Patients Screening Examiner
(N) at screen melanoma at (%) (%)(N) screen (N)
Aitken Attendees of skin screening 16 383 TSE PCP 33 222 161 14.9 20.52006 66 clinics Australia
Fritschi Attendees of Lion Cancer Clinics, 7 436 TSE Derm or 23 (1 yr) 201 11.4 (1 yr)2006 64 Australia targeted to patients plastic *21 (1 yr)* 211 *10.0 (1 yr)*
with skin cancer risks surgeon (site specific)*
Swetter Veterans attending skin cancer 374 TSE Derm 1 14 8 7.1 12.52003 67 screening clinics, targeted
to patient with skin cancer risksUnited States
Geller Attendees of American Academy 242 374 Partial, Derm 363 3476 10.42002 68 of Dermatology TSE, or site
Screening Program United States specific
Engelberg Attendees of skin cancer 520 Sun exposed Derm 1 6 6 16.7 16.71999 69 screening clinics British Columbia or site specific
Jonna Attendees of skin cancer screening 464 TSE Derm 2 35 13 5.7 15.41998 70 San Diego, targeted to patient
with skin cancer risks
Low PPV: number of confirmed melanomas/number with a presumed or suspected melanoma diagnosis at screen; High PPV: number of confirmed melanomas/num-ber of presumed or suspected melanomas at screen with biopsy results; TSE: total skin examination; Derm: dermatologist; PCP: primary care physician. *: Fritschi’sstudy=accounting for body-site.

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 63
EARLY DIAGNOSIS OF MELANOMA YEE
be feasible or useful given the lack of data that ear-ly detection of melanoma leads to a decrease in latestage melanoma.6, 72, 73
EARLY DIAGNOSIS PUBLIC EDUCATION CAMPAIGNS:PROGAMS TO ENCOURAGE SKIN SURVEILLANCE
Public education programs to promote early detec-tion have been carried out in Scotland, the UnitedKingdom (UK), and Australia. In Scotland, the num-ber of referrals to a melanoma clinic and number ofmelanomas diagnosed increased following an edu-cation campaign. Thin tumor percentage increased.The incidence of thick tumors decreased in women,and a decrease in melanoma mortality was seen inwomen.74 Subsequent studies of a similar educationcampaign conducted during 1987-1989 in 6 areasof England and one in Scotland showed an increasein referrals to pigmented lesion clinics and anincrease in melanoma rates following the campaign.There was an apparent rise in thin melanomas, butthe incidence of thick tumors did not appear todecrease.75-79 An analysis of melanoma mortalityrates during 1981 to 1996 (adjusted or pre-inter-vention rates) showed no decrease in cumulativemortality in intervention areas compared to nonintervention areas. As these educational campaignswere not trials, there were no control groups, andthe actually effectiveness of the intervention is dif-ficult to measure. Information regarding whoreceived and acted on the health education couldnot be ascertained, and it is possible that the healtheducation could have reached non-intervention areasand decreased the ability to detect a difference inmortality.80 As these education campaigns were notpopulation screening trials, the results should notbe applied to screening.2, 22
Rationale for screening
Early stage melanoma is associated with excel-lent survival and the goal of screening for melanomais to decrease morbidity and mortality. A case forscreening is predicated on the assumptions thatmelanoma has a long latency period during which itcan be treated with a high rate of success, and that ear-ly detection prevents progression of early-stage can-cers to advanced, lethal stages.81 Melanoma is usu-ally visible on the skin at a curable stage.82 Skinexams are quite feasible and relatively non-invasive,
and primary care physicians and the public can beeducated to perform them.
YIELD OF SCREENING PROGRAMS
Skin cancer screening programs have been con-ducted worldwide. Helfand et al. reviewed data from24 screening programs published in papers between1994 and June 1999. They found that rates for sus-pected melanoma ranged from 0 to 9 per 100 scree-nees, referral for follow-up of suspicious lesionsranged from 2 to 34 per 100 screenees, and biopsyranged from 4 to 31 per 100 screenees. Rates of con-firmed melanoma and melanoma-in-situ ranged from1 to 4 per 1 000 screenees,19 except for one study inSweden with no melanomas found in 152 suspectedcancers in 1 654 people screened,83 and one studyin Australia that had 8 confirmed melanomas per100 screenees.84
A retrospective study of melanoma screening clin-ics in the United Kingdom examined data from 15970 physician-referred patients screened from 1997to the end of 2004. There were 403 primary invasivemelanomas detected, and 190 in situ melanomas(37/1 000 screenees).85 The Lions Cancer InstituteSkin Cancer Screening Program in Australia hasoffered screening by dermatologists and plastic sur-geons since 1990. Advertisements for attendees tar-get individuals with self-assessed skin cancer riskfactors. During 1994 and 2002 there were 7 436attendees, most had 3 or more risk factors. Thirty-three melanomas were diagnosed within one yearof a screen (4/1 000 screenees), and 16 more werediagnosed between 1-2 years after a screen (total7/1 000 screenees).64 In 1998, the Queensland Aus-tralia Cancer Fund started the first phase of a ran-domized trial of population screening to increaseskin screening rates. During the 3-year interventionperiod, 16 383 total body skin exams by primarycare physicians resulted in 2 302 referrals (14%) for4 129 suspicious lesions. Subsequently, 1 822 indi-viduals visited a doctor with their referral, 1 417lesions were biopsied, and 1 343 lesions were diag-nosed histopa-thologically. Of these, 33 weremelanomas (2/1 000 screenees).66In Northern Italy,a regional program to evaluate the efficacy of earlydetection of melanoma by general practitioners dur-ing 1997-1998 yielded 11 040 examinations. Sub-sequently, 820 patients (7%) were referred to a der-matologist, and 38 melanomas were detected (3/1000

YEE EARLY DIAGNOSIS OF MELANOMA
64 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
screenees), most of which (18) were ≤1.5 mm thick.86
A study of 374 veterans in northern California attend-ing free skin cancer screening clinics targeted at ahigh risk population during 1997-2000 found thatdermatologists 203 referred patients (54%) for sus-picious lesions, including 14 for possible melanomaversus dysplastic nevi. Of the 101 patients that inves-tigators were able to follow-up, only 1 out of 8 pos-sible melanomas versus dysplastic nevi was con-firmed by biopsy (3/1 000 screenees, if data extrap-olated to 1 000 screenees). The high referral ratemay reflect the highly selected screening popula-tion that was mostly male, caucasian, and older(mean age 63).67 When the database of the morethan 242 374 screenings from the American Acad-emy of Dermatology National Skin Cancer Screen-ing Programs during 1992-1994 was analyzed, 3476 screenees had a presumptive diagnosis ofmelanoma. The number of confirmed diagnoses ofmelanomas was 363, giving an overall yield of 1.5melanomas per 1 000 screenings. In men aged ≥50years, the yield was 3/1 000 screenees, 4/1 000 ifthey had skin types I and II, and 5/1 000 if they hada changing mole.68
EVIDENCE FOR BENEFIT WITH SCREENING
The gold standard for evidence-based benefits ofscreening are large-scale, prospective randomized tri-als demonstrating decreases in cancer-specific mor-tality in screened versus non-screened populations.There are no such completed randomized controlledprospective screening trials for melanoma.14
Melanoma is relatively rare in the general populationand a randomized clinical trial to test efficacy ofscreening would be very costly. The QueenslandAustralia project was the largest randomized trialof population screening for melanoma. In it, 18 com-munities were randomized to either intervention(community and professional education, and skinscreening by a doctor) or control.65 This project wasnot funded for follow up, and currently there are nodata that provide direct evidence that screening leadsto reduced morbidity and mortality. In the absenceof such data, we examine observational studies. Caremust be taken when interpreting data from obser-vational studies because of inherent biases, and thesebiases are discussed later. A population-based, case-control study to investigate whether early detectionthrough skin self-examination (SSE), (defined as
“conducting a careful, deliberate, and purposefulexamination of the skin”) is associated with adecreased risk of lethal melanoma was conducted in1 199 Caucasian residents of the state of Connecti-cut. The study included 650 individuals with newlydiagnosed cutaneous melanoma, and 549 age- andsex-frequency matched control subjects from thegeneral population. In 5 years of follow-up, therewere 110 “lethal” melanomas. SSE was practicedby only 15% of all subjects, and was associated witha reduced risk of the development of melanoma(OR=0.66, 95% CI=0.44-0.99). The results furtherindicated that SSE may reduce the risk of advanceddisease among melanoma patients (unadjusted riskratio = 0.58; 95% CI=0.31-1.11) and that when com-bining the incidence reduction with the preventionof mortality, SSE may reduce mortality frommelanoma by 63% (adjusted OR=0.37; 95%CI=0.16-0.84.87 These results are provocative butneed to be confirmed with longer follow-up andstudies in other populations.
SCREENING SELECTED HIGH RISK POPULATIONS
Screening certain high-risk populations may resultin detection of thinner lesions. In a screening studyof 555 members of hereditary melanoma kindredswith dysplastic nevi, the average thickness of 28 sur-veillance-detected melanomas was 0.52 mm, com-pared to 0.55 mm for 64 non-surveillance incidentmelanomas (diagnosed before entry into the sur-veillance program), and 1.44 mm for 48 index lesions(P<0.001). The proportion of cases with level I or IIinvasion was 61% in the surveillance group, 58% inthe non-surveillance group, and 36% in the indexgroup (P=0.002).88
COST EFFECTIVENESS OF SCREENING
Studies examining the cost effectiveness of screen-ing for melanoma have found that melanoma screen-ing could be cost effective. Losina et al. used a com-puter simulation Markov model to evaluate the cost-effectiveness of four types of screening strategiesbeginning at 50 years of age: background screeningonly, and screening once, every two years, and annu-ally. Expected outcomes included life expectancy,quality-adjusted life expectancy, and lifetime costs.Cost-effectiveness ratios were calculated in dollars perquality-adjusted life year (US dollars/QALY) gained.

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 65
EARLY DIAGNOSIS OF MELANOMA YEE
In the general population, screening 1 time, every 2years, and annually saved 1.6, 4.4, and 5.2 QALYs per1 000 persons screened respectively. Cost-effective-ness ratios were $ 10 100/QALY, $ 80 700/QALY, and$ 586 800/QALY, respectively. In siblings of patientswith melanoma with a relative risk of 2.24 comparedwith the general population, 1-time, every-2-years,and annual screenings saved 3.6, 9.8, and 11.4QALYs per 1 000 persons screened. Cost-effective-ness ratios were $ 4 000/QALY, $ 35 500/QALY,and $ 257 800/QALY, respectively. In higher risksiblings of patients with melanoma with a relativerisk of 5.56, 1-time, every-2-years, and annual screen-ings cost-effectiveness ratios were $ 900/QALY, $14 7000/QALY, and $ 99 800/QALY, respectively.89
Cost-effectiveness thresholds cited in literature foracceptable interventions range from $ 50 000-$100 000/QALY gained,90 Using these thresholds,acceptable strategies include a one-time melanomascreening of the general population older than 50years, and screening every 2 years in siblings ofpatients with melanoma. Limitations in this studyincluded possible lead time and length time bias thatcould influence the survival benefit of earlier detec-tion of melanoma. In addition, the actual rate ofmelanoma progression is unknown and could impacton results of screening every two years. Non-melanoma skin cancers, particularly basal cell car-cinomas with little potential for progression, may bedetected by screening and add to costs.89
Other analyses have also found that screeningcould be cost effective. A computer model simulat-ed a melanoma screening program over a 20-yearperiod, using cohorts of Australians 50 years of ageat the start of the program. The sensitivity of thescreening test (visual inspection of the skin) was setat 60%. Cost effectiveness ranged from $ 6 853 (Aus-tralian, AUS) per life year saved for men if screeningwas undertaken every five years to $ 12 137 if screen-ing was every two years. For women, it ranged fromAUS $ 11 102 for screening every five years to AUS$ 20 877 for screening every two years.91 A decisionanalysis determined the effectiveness and costs of aone time visual screen by a dermatologist to diagnosemalignant melanoma in high-risk persons ages 20and older. Using a cost of $ 30 per screen, a cost-effectiveness ratio of $ 29 170 per year of life saved(YLS) with screening was found. The cost effec-tiveness ratio for screening remained below $
50 000/YLS if the prevalence of melanoma in thescreened population was at least 0.0009 (9/1 000)and the probability that a melanoma detected inscreening was localized was at least 94.8%, or the costof each screen was below $ 57.92 The prevalence of9/10 000 is highly unlikely at this time.
BIASES IN OBSERVATIONAL STUDIES
Most of the data regarding melanoma screeningare derived from observational studies. These designsare subject to a number of potential biases includinglead-time, length, and selection/referral. Effectivescreening for melanoma should diagnose the diseaseearlier than it would be without screening but wouldalso prevent progression of the disease. Biases couldmake screening appear beneficial when in actualityit is less or not effective.
Lead time is the period between the detection ofmelanoma by screening and when it otherwise wouldbe diagnosed once symptoms appeared. Lead timebias occurs when early diagnosis appears to prolongsurvival because disease is picked up at an earlierstage. Even when early treatment does not increasesurvival, the apparent survival time following ascreening diagnosis will exceed that following a clin-ical diagnosis. In the only case-control study ofscreening with skin self exam (SSE), SSE was shownto be associated with a decreased risk of melanomamortality (adjusted OR=0.37,95% CI 0.16-0.84).Lead-time bias could potentially influence the sec-ondary preventive impact of SSE as case subjectswho practiced screening discover their cancers ear-lier in the course of the disease. If SSE results onlyin earlier diagnosis, but treatment is not effective,then the apparent improvement in survival could bedue to lead-time bias. The survival curves showedthat the curve for SSE case patients approached aplateau after approximately 2.5 years of follow up,whereas failures (lethality) continue to occur in thosethose who did not practice SSE, lending suggestiveevidence that the observed benefit is due to actualsurvival rather than lead-time bias.19, 87
Length bias arises when screening preferentiallydetects less aggressive disease which may have alonger asymptomatic period and better prognosis.Aggressive disease, which has short asymptomaticperiod and worse prognosis, is less likely to be detect-ed by screening. Some melanomas may be moreindolent than others and survival time is related to

YEE EARLY DIAGNOSIS OF MELANOMA
66 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
lesion thickness at the time of resection. Length biasis suspected when observational studies of screeningreport detecting higher proportions of thinner mel-onomas.24
Clinical studies are very likely biased by the factthat patients are often referred and may not repre-sent the general population. Referral bias is likelyto occur in specialty clinics that evaluate pigment-ed lesions and attract high risk individuals. A highprevalence of melanomas will be seen in this highrisk group.93 The variation in positive predictivevalues may be related to variation in the prevalenceof disease, which in turn could be related to thetype of patients recruited (high risk or not targeted).Attendees of screening programs were all self-selected, and they may not represent the generalpopulation.
Potential risks of screening
Potential adverse effects of screening include beinglabeled with melanoma which may impact qualityof life or an individual’s finances. Mislabeling andmisdiagnosis can have similar consequences. Patientsmay receive an ambiguous result or false positiveresult and undergo more testing or be treated unnec-essarily.19 The diagnosis of melanoma is based onhistopathology, and is subjective. Some pathologistswill call an abnormality melanoma while others willnot.28-30 The concern that pathologists may diagnoseborderline lesions as melanoma because of liabilityissue has been raised.26 Screening can also increasebiopsies of benign conditions and can be costly if itleads to more procedures, and treatment that maynot be benign.19
There is potential embarrassment or discomfortdue to screening. A study of 201 female veteransfound that 15% expressed embarrassment about afull body skin exam (FBSE), and 16% would refusethe exam if the primary care provider were of theopposite sex.94 Another downside to screening isthat it may distract physicians from issues the patientconsiders more important. During a clinic visit,limited time may cause physicians to spend lesstime addressing patient concerns in order to com-plete screening.95 Finally, while screening mayresult in an earlier diagnosis of melanoma, a con-sequent risk is the possibility of increased anxietyas the patient must live with knowledge of the dis-ease for longer.
Consequences of not screening: delayed diagnosis
CONSEQUENCES OF DELAYED DIAGNOSIS
Studies examining the consequences of delayeddiagnosis have found inconsistent results.19, 96 In thelargest population-based analysis of the role of lengthof time to diagnosis, Baade et al. examined the rela-tionship between melanoma thickness and reportedtime from first recognition and from first physiciancontact to the diagnosis of invasive melanoma in3 772 participants in Queensland, Australia. Theyfound no significant association between melanomathickness and reported time to diagnosis for allmelanomas combined, superficial spreading melano-mas, or nodular melanomas. The exception was apositive association between melanoma thicknessand post-presentation delay of physician-detectednodular melanomas. The authors concluded that thismay be due to varying biological characteristics ofmelanomas, as well as the methodological limita-tions of retrospective studies when trying to mea-sure this association. They investigators did not con-trol for patient knowledge of melanoma symptomswhich could have influenced association.97 Sevenother studies have reported a null association andtwo reported an inverse association between delay inseeking diagnosis and lesion thickness.96
Campaigns to reduce delay in diagnosis by a com-bination of professional and public education havebeen reported from several centers around the world.The effects of these campaigns in reducing the depthdistribution of cutaneous malignant melanoma havebeen mixed. In the absence of clear evidence ofdecreased mortality from melanoma due to earlydetection, opportunistic promotion of early detec-tion may not be cost effective and will fail to reach allof the at-risk population.2
Conclusions
There is no direct evidence that screening formelanoma reduces mortality and studies to date do notsupport population-based screening. While periodictotal skin examination appears to increase the detec-tion of thin melanoma, it is not clear if this detec-tion leads to a reduction in mortality from melanoma.A number of organizations have made recommen-dations regarding skin cancer screening. The US Pre-ventive Services Task Force (USPSTF) 81 concluded

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 67
EARLY DIAGNOSIS OF MELANOMA YEE
that “the evidence is insufficient to recommend for oragainst routine screening for skin cancer using atotal body examination for the early detection ofcutaneous melanoma, basal cell cancer, or squa-mous cell skin cancer.” Clinical considerationsinclude “being aware of groups at high risk formelanoma including fair-skinned men and womenaged >65, patients with atypical moles, and thosewith >50 moles”. In addition, “clinicians shouldremain alert for skin lesions with malignant featuresnoted in the context of physical examinations per-formed for other purposes.” Recommendations fromvarious organizations 81, 98-101 are summarized in TableIV, and are noted to lack uniformity and consistency.
Implications of screening recommendations
Recommendations of medical organizations havesignificant implications for the medical and lay com-munities including medicolegal ramifications througha direct or implied standard of care. Recommend-ing population-based screening for melanoma means
that total skin examinations should be performed onall patients in a specific age group. However, there isno direct evidence that such screening would decreasemorbidity or mortality from melanoma and suchscreening may result in a systematic overdiagnosis ofmelanoma.22 Selective screening of high risk popu-lations may be the most promising strategy,44 butthere is no consensus on which risk factors to use todefine a population that would benefit most fromscreening. More research is needed to understandhow to define high risk individuals, to identify themolecular biology of melanoma tumor progression,and to determine the optimal strategies to improveearly detection of melanoma.
Riassunto
Diagnosi precoce di melanoma: cosa sappiamo?
Il melanoma maligno è una patologia potenzialmenteletale che tuttavia, se diagnosticato precocemente, ha unaprognosi eccellente. In questo lavoro ricediamo gli aspettifisiopatologici, la presentazione clinica, l’epidemiologia e i
TABLE IV.—Skin cancer screening recommendations.
Organization Recommendation
US Preventive Services Task Force 81
American Cancer Society 98
Canadian Task Force on Preventive Health Care 99
American College of Preventive Medicine 100
National Institutes of Health 101
The Cancer Council Australia 102
National Cancer Institute 103
Institute of Medicine 104
Evidence is insufficient to recommend for or against routine screening for skin cancer usinga total body examination for the early detection of cutaneous melanoma, basal cell cancer,or squamous cell skin cancer. Clinical considerations include being aware of groups athigh risk for melanoma including fair-skinned men and women aged >65, patients with aty-pical moles, and those with >50 moles. In addition, clinicians should remain alert for skinlesions with malignant features noted in the context of physical examinations performed forother purposes
On the occasion of a periodic health exam, cancer-related check up should include examina-tion for skin cancer for those aged ≥20 years
Poor evidence to include or exclude total body skin examination from the periodic healthexamination of the general population; there is fair evidence for the inclusion of total bodyskin examination for a very select sub-group of individuals. Insufficient evidence to recom-mend for or against counseling patients to perform periodic self-examination of the skin
Periodic total cutaneous examinations, targeting populations at high risk for malignant mela-noma (family or personal history of skin cancer, predisposing phenotypic characteristics,increased occupational or recreational exposure to sunlight, or clinical evidence of pre-cursor lesions)
Screen for melanoma as part of routine primary care
Evidence is insufficient to recommend screening for melanoma of the general asymptomaticpopulation
In asymptomatic populations, the effect of visual skin examination on mortality from non-melanomatous skin cancers is unknown. Evidence is inadequate to determine whethervisual examination of the skin in asymptomatic individuals would lead to a reduction in mor-tality from melanomatous skin cancer
Insufficient evidence to recommend skin cancer screening; physicians and patients should remainalert to common signs of skin malignancies, with special attention to older white men

YEE EARLY DIAGNOSIS OF MELANOMA
68 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
fattori di rischio per melanoma. Esamineremo anche i mod-elli clinici per predire il rischio e la sopravvivenza e val-uteremo criticamente i dati sull’efficacia dello screening peril melanoma.
PAROLE CHIAVE: Melanoma, diangosi - Melanoma, anatomiapatologica - Cute neoplasie.
References
1. Markovic SN, Erickson LA, Rao RD, Weenig RH, Pockaj BA, Bar-dia A, et al. Malignant melanoma in the 21st century, part 1: Epi-demiology, risk factors, screening, prevention, and diagnosis. MayoClin Proc 2007;82:364-80.
2. Austoker J. Melanoma: Prevention and early diagnosis. BMJ1994;308:1682-6.
3. Clark WH Jr, Elder DE, Guerry D 4th, Epstein MN, Greene MH, VanHorn M. A study of tumor progression: The precursor lesions ofsuperficial spreading and nodular melanoma. Hum Pathol 1984;15:1147-65.
4. Miller AJ, Mihm MC Jr. Melanoma. N Engl J Med 2006; 355:51-65.5. Burton RC, Armstrong BK. Current melanoma epidemic: A non-
metastasizing form of melanoma? World J Surg 1995;19:330-3.6. Halpern AC, Lieb JA. Early melanoma diagnosis: A success story that
leaves room for improvement. Curr Opin Oncol 2007;19:109-15.7. Lens MB, Dawes M. Global perspectives of contemporary epidemi-
ological trends of cutaneous malignant melanoma. Br J Dermatol2004;150:179-85.
8. Marks R. Epidemiology of melanoma. Clin Exp Dermatol 2000;25:459-63.
9. Diepgen TL, Mahler V. The epidemiology of skin cancer. Br J Dermatol2002; 146 Suppl 61:1-6.
10. Armstrong BK, Kricker A. Cutaneous melanoma. Cancer Surv 1994;19-20:219-40.
11. Garbe C, McLeod GR, Buettner PG. Time trends of cutaneousmelanoma in queensland, australia and central europe. Cancer 2000;89:1269-78.
12. World Health Organization. GLOBOCAN: Cancer Incidence, Mor-tality, and Prevalence Worldwide, IARC Cancerbase No.5, Version 1.0.Lyon:IARC Press, 2001.
13. Berwick M, Weinstock MA. Epidemiology Current Trends. In: BalchCM, Houghton AN, Sober AJ, Soon SJ, editors. Cutaneous Melanoma.4th edition. Canada: Quality Medical Publishing; 2003.p.15-23.
14. Elwood M, Aitken JF, English DR. Prevention and screening. In:Balch CM, Houghton AN, Sober AJ, Soong SJ (eds). CutaneousMelanoma. 4th edition. Canada: Quality Medical Publishing, Inc.;2003.p.93-120.
15. Cohn-Cedermark G, Mansson-Brahme E, Rutqvist LE, Larsson O,Johansson H, Ringborg U. Trends in mortality from malignantmelanoma in sweden, 1970-1996. Cancer 2000;89:348-55.
16. Giles GG, Armstrong BK, Burton RC, Staples MP, Thursfield VJ.Has mortality from melanoma stopped rising in australia? analysis oftrends between 1931 and 1994. BMJ 1996;312:1121-5.
17. Ries LAG, Harkins D, Krapcho M, Mariotto A, Miller BA, Feuer EJet al. SEER Cancer Statistics Review, 1975-2003, National CancerInstitute. Bethesda, MD, http://seer.cancer.gov/csr/1975_2003/, basedon November 2005 SEER data submission, posted to the SEER website 2006. http://www.seer.cancer.gov/statfacts/html/melan.html?stat-facts_page=melan.html&x=10&y=18. Accessed 3-1-07.
18. Surveillance, Epidemiology, and End Results (SEER) Program(www.seer.cancer.gov). SEER*Stat Database:Incidence-SEER 17Regs Public-use, Nov 2005 Sub (1973-2003 Varyingi), National Can-cer Institute, DCCPS, Surveillance Research Program, Cancer StatisticsBranch, released April 2006, based on the November 2005 submission.
19. Helfand M, Mahon SM, Eden KB, Frame PS, Orleans CT. Screeningfor skin cancer. Am J Prev Med 2001; 20:47-58.
20. Koh HK. Melanoma screening: Focusing the public health journey.Arch Dermatol 2007;143:101-3.
21. Coory M, Baade P, Aitken J, Smithers M, McLeod GR, Ring I. Trendsfor in situ and invasive melanoma in queensland, australia, 1982-2002. Cancer Causes Control 2006;17:21-7.
22. Edman RL, Klaus SN. Is routine screening for melanoma a benignpractice? JAMA 2000;284:883-6.
23. Lamberg L. “Epidemic” of malignant melanoma: True increase orbetter detection? JAMA 2002;287:2201.
24. Swerlick RA, Chen S. The melanoma epidemic: More apparent thanreal? Mayo Clin Proc 1997;72:559-64.
25. Welch HG, Woloshin S, Schwartz LM. Skin biopsy rates and incidenceof melanoma: Population based ecological study. BMJ 2005;331:481.
26. Swerlick RA, Chen S. The melanoma epidemic. is increased surveil-lance the solution or the problem? Arch Dermatol 1996;132:881-4.
27. Farmer ER. The fundamental issue of diagnosis. Arch Dermatol2002;138:684-5.
28. Corona R, Mele A, Amini M, De Rosa G, Coppola G, Piccardi P et al.Interobserver variability on the histopathologic diagnosis of cuta-neous melanoma and other pigmented skin lesions. J Clin Oncol1996;14:1218-23.
29. Farmer ER, Gonin R, Hanna MP. Discordance in the histopatholog-ic diagnosis of melanoma and melanocytic nevi between expert pathol-ogists. Hum Pathol 1996; 27:528-31.
30. McGinnis KS, Lessin SR, Elder DE, Guerry D 4th, Schuchter L, MingM et al. Pathology review of cases presenting to a multidisciplinarypigmented lesion clinic. Arch Dermatol 2002;138:617-21.
31. Qin J, Berwick M, Ashbolt R, Dwyer T. Quantifying the change ofmelanoma incidence by breslow thickness. Biometrics 2002;58:665-70.
32. Gandini S, Sera F, Cattaruzza MS, Pasquini P, Zanetti R, Masini C etal. Meta-analysis of risk factors for cutaneous melanoma: III. fami-ly history, actinic damage and phenotypic factors. Eur J Cancer 2005;41:2040-59.
33. Gandini S, Sera F, Cattaruzza MS, Pasquini P, Picconi O, Boyle P etal. Meta-analysis of risk factors for cutaneous melanoma: II. sunexposure. Eur J Cancer 2005;41:45-60.
34. Gandini S, Sera F, Cattaruzza MS, Pasquini P, Abeni D, Boyle P et al.Meta-analysis of risk factors for cutaneous melanoma: I. commonand atypical naevi. Eur J Cancer 2005;41:28-44.
35. Williams HA, Fritschi L, Beauchamp C, Katris P. Evaluating the use-fulness of self-reported risk factors in a skin cancer screening program.Melanoma Res 2006;16:341-5.
36. MacKie RM, Freudenberger T, Aitchison TC. Personal risk-factorchart for cutaneous melanoma. Lancet 1989; 2:487-90.
37. Fears TR, Guerry D,4th, Pfeiffer RM, Sagebiel RW, Elder DE, HalpernA et al. Identifying individuals at high risk of melanoma: A practicalpredictor of absolute risk. J Clin Oncol 2006;24:3590-6.
38. Schuchter L, Schultz DJ, Synnestvedt M, Trock BJ, Guerry D, ElderDE et al. A prognostic model for predicting 10-year survival in patientswith primary melanoma. the pigmented lesion group. Ann Intern Med1996;125:369-75.
39. Margolis DJ, Halpern AC, Rebbeck T, Schuchter L, Barnhill RL, FineJ et al. Validation of a melanoma prognostic model. Arch Dermatol1998;134:1597-601.
40. Balch CM, Soong SJ, Gershenwald JE, Thompson JF, Reintgen DS,Cascinelli N et al. Prognostic factors analysis of 17,600 melanomapatients: Validation of the american joint committee on cancermelanoma staging system. J Clin Oncol 2001;19:3622-34.
41. Balch CM, Buzaid AC, Soong SJ, Atkins MB, Cascinelli N, Coit DGet al. Final version of the american joint committee on cancer stagingsystem for cutaneous melanoma. J Clin Oncol 2001;19:3635-48.
42. Azzola MF, Shaw HM, Thompson JF, Soong SJ, Scolyer RA, WatsonGF et al. Tumor mitotic rate is a more powerful prognostic indicatorthan ulceration in patients with primary cutaneous melanoma: An

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 69
EARLY DIAGNOSIS OF MELANOMA YEE
analysis of 3661 patients from a single center. Cancer 2003;97:1488-98.
43. Barnhill RL, Katzen J, Spatz A, Fine J, Berwick M. The importanceof mitotic rate as a prognostic factor for localized cutaneous melanoma.J Cutan Pathol 2005;32:268-73.
44. Lange JR, Balch CM. Screening for cutaneous melanoma. Surg OncolClin N Am 2005;14:799-811.
45. Tsao H, Atkins MB, Sober AJ. Management of cutaneous melanoma.N Engl J Med 2004;351:998-1012.
46. Friedman RJ, Rigel DS, Kopf AW. Early detection of malignantmelanoma: The role of physician examination and self-examinationof the skin. CA Cancer J Clin 1985;35:130-51.
47. Abbasi NR, Shaw HM, Rigel DS, Friedman RJ, McCarthy WH,Osman I, et al. Early diagnosis of cutaneous melanoma: Revisiting theABCD criteria. JAMA 2004;292:2771-6.
48. Grob JJ, Bonerandi JJ. The “ugly duckling” sign: Identification ofthe common characteristics of nevi in an individual as a basis formelanoma screening. Arch Dermatol 1998;134:103-4.
49. Fisher NM, Schaffer JV, Berwick M, Bolognia JL. Breslow depth ofcutaneous melanoma: Impact of factors related to surveillance of theskin, including prior skin biopsies and family history of melanoma. JAm Acad Dermatol 2005;53:393-406.
50. Rigel DS, Friedman RJ, Kopf AW, Weltman R, Prioleau PG, Safai Bet al. Importance of complete cutaneous examination for the detectionof malignant melanoma. J Am Acad Dermatol 1986;14:857-60.
51. Lee G, Massa MC, Welykyj S, Choo J, Greaney V. Yield from total skinexamination and effectiveness of skin cancer awareness program.findings in 874 new dermatology patients. Cancer 1991;67:202-5.
52. McPherson M, Elwood M, English DR, Baade PD, Youl PH, AitkenJF. Presentation and detection of invasive melanoma in a high-risk pop-ulation. J Am Acad Dermatol 2006;54:783-92.
53. Epstein DS, Lange JR, Gruber SB, Mofid M, Koch SE. Is physiciandetection associated with thinner melanomas? JAMA 1999;281:640-3.
54. Schwartz JL, Wang TS, Hamilton TA, Lowe L, Sondak VK, JohnsonTM. Thin primary cutaneous melanomas: Associated detection pat-terns, lesion characteristics, and patient characteristics. Cancer2002;95:1562-8.
55. Brady MS, Oliveria SA, Christos PJ, Berwick M, Coit DG, Katz J etal. Patterns of detection in patients with cutaneous melanoma. Can-cer 2000; 89:342-7.
56. Carli P, De Giorgi V, Palli D, Maurichi A, Mulas P, Orlandi C et al. Der-matologist detection and skin self-examination are associated withthinner melanomas: Results from a survey of the italian multidisci-plinary group on melanoma. Arch Dermatol 2003;139:607-12.
57. Richard MA, Grob JJ, Avril MF, Delaunay M, Gouvernet J, Wolken-stein P et al. Delays in diagnosis and melanoma prognosis (II): Therole of doctors. Int J Cancer 2000;89:280-5.
58. Thomas L, Tranchand P, Berard F, Secchi T, Colin C, Moulin G.Semiological value of ABCDE criteria in the diagnosis of cutaneouspigmented tumors. Dermatology 1998;197:11-7.
59. Healsmith MF, Bourke JF, Osborne JE, Graham-Brown RA. An eval-uation of the revised seven-point checklist for the early diagnosis ofcutaneous malignant melanoma. Br J Dermatol 1994;130:48-50.
60. McGovern TW, Litaker MS. Clinical predictors of malignant pig-mented lesions. A comparison of the glasgow seven-point checklist andthe american cancer society’s ABCDs of pigmented lesions. J DermatolSurg Oncol 1992;18:22-6.
61. Gachon J, Beaulieu P, Sei JF, Gouvernet J, Claudel JP, Lemaitre M etal. First prospective study of the recognition process of melanomain dermatological practice. Arch Dermatol 2005;141:434-8.
62. Melia J, Harland C, Moss S, Eiser JR, Pendry L. Feasibility of targetedearly detection for melanoma: A population-based screening study. BrJ Cancer 2000; 82:1605-9.
63. Roush GC, Barnhill RL, Ernstoff MS, Kirkwood JM. Inter-clinicianagreement on the recognition of clinical pigmentary characteristics ofpatients with cutaneous malignant melanoma. studies of melanocyt-ic nevi, VI. Br J Cancer 1991; 64:373-6.
64. Fritschi L, Dye SA, Katris P. Validity of melanoma diagnosis in acommunity-based screening program. Am J Epidemiol 2006;164:385-90.
65. Aitken JF, Elwood JM, Lowe JB, Firman DW, Balanda KP, Ring IT.A randomised trial of population screening for melanoma. J MedScreen 2002;9:33-7.
66. Aitken JF, Janda M, Elwood M,Youl PH, Ring IT, Lowe JB. Clinicaloutcomes from skin screening clinics within a community-basedmelanoma screening program. J Am Acad Dermatol 2006;54:105-14.
67. Swetter SM, Waddell BL, Vazquez MD, Khosravi VS. Increased effec-tiveness of targeted skin cancer screening in the veterans affairs pop-ulation of northern california. Prev Med 2003;36:164-71.
68. Geller AC, Sober AJ, Zhang Z, Brooks DR, Miller DR, Halpern A etal. Strategies for improving melanoma education and screening for menage >or= 50 years: Findings from the american academy of derma-tological national skin cancer sreening program. Cancer 2002;95:1554-61.
69. Engelberg D, Gallagher RP, Rivers JK. Follow-up and evaluation ofskin cancer screening in british columbia. J Am Acad Dermatol1999;41:37-42.
70. Jonna BP, Delfino RJ, Newman WG, Tope WD. Positive predictive val-ue for presumptive diagnoses of skin cancer and compliance withfollow-up among patients attending a community screening program.Prev Med 1998;27:611-6.
71. Chen SC, Bravata DM, Weil E, Olkin I. A comparison of dermatolo-gists’and primary care physicians’accuracy in diagnosing melanoma:A systematic review. Arch Dermatol 2001;137:1627-34.
72. Chen SC, Pennie ML, Kolm P, Warshaw EM, Weisberg EL, Brown KMet al. Diagnosing and managing cutaneous pigmented lesions: Primarycare physicians versus dermatologists. J Gen Intern Med 2006;21:678-82.
73. Berwick M. Melanoma: Is more diagnosis better? J Gen Intern Med2006; 21:800.
74. MacKie RM, Hole D. Audit of public education campaign to encour-age earlier detection of malignant melanoma. BMJ 1992;304:1012-5.
75. Melia J. Early detection of cutaneous malignant melanoma in britain.Int J Epidemiol 1995;24 Suppl 1:S39-44.
76. Melia J, Cooper EJ, Frost T, Graham-Brown R, Hunter J, Marsden Aet al. Cancer research campaign health education programme to pro-mote the early detection of cutaneous malignant melanoma. II. char-acteristics and incidence of melanoma. Br J Dermatol 1995;132:414-21.
77. Whitehead SM, Wroughton MA, Elwood JM, Davison J, Stewart M.Effects of a health education campaign for the earlier diagnosis ofmelanoma. Br J Cancer 1989;60:421-5.
78. Graham-Brown RA, Osborne JE, London SP, Fletcher A, Shaw D,Williams B et al. The initial effects on workload and outcome of apublic education campaign on early diagnosis and treatment ofmalignant melanoma in leicestershire. Br J Dermatol 1990;122:53-9.
79. Herd RM, Cooper EJ, Hunter JA, McLaren K, Chetty U, Watson ACet al. Cutaneous malignant melanoma. publicity, screening clinicsand survival—the edinburgh experience 1982-90. Br J Dermatol1995;132:563-70.
80. Melia J, Moss S, Coleman D, Frost T, Graham-Brown R, Hunter JAet al. The relation between mortality from malignant melanoma andearly detection in the cancer research campaign mole watcher study.Br J Cancer 2001;85:803-7.
81. US Preventive Services Task Force. Screening for skin cancer: Rec-ommendations and rationale. Am J Prev Med 2001;20:44-6.
82. Weinstock MA. Early detection of melanoma. JAMA 2000;284:886-9.
83. Tornberg S, Mansson-Brahme E, Linden D, Ringborg U, Krakau I,Karnell R et al. Screening for cutaneous malignant melanoma: A fea-sibility study. J Med Screen 1996;3:211-5.

YEE EARLY DIAGNOSIS OF MELANOMA
70 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
84. Katris P, Crock JG, Gray BN. Research note: The lions cancer insti-tute and the western australian society of plastic surgeons skin cancerscreening programme. Aust N Z J Surg 1996;66:101-4.
85. Weatherhead SC, Lawrence CM. Melanoma screening clinics: Are wedetecting more melanomas or reassuring the worried well? Br J Der-matol 2006;154:539-41.
86. Veronesi A, Pizzichetta MA, De Giacomi C, Gatti A, Trevisan G,Regional Committee for the Early Diagnosis of Cutaneous Melanoma.A two-year regional program for the early detection of cutaneousmelanoma. Tumori 2003;89:1-5.
87. Berwick M, Begg CB, Fine JA, Roush GC, Barnhill RL. Screening forcutaneous melanoma by skin self-examination. J Natl Cancer Inst1996;88:17-23.
88. Masri GD, Clark WH,Jr, Guerry D,4th, Halpern A, Thompson CJ,Elder DE. Screening and surveillance of patients at high risk formalignant melanoma result in detection of earlier disease. J Am AcadDermatol 1990;22:1042-8.
89. Losina E, Walensky RP, Geller A, Beddingfield FC 3rd, Wolf LL,Gilchrest BA et al. Visual screening for malignant melanoma: A cost-effectiveness analysis. Arch Dermatol 2007;143:21-8.
90. Ubel PA, Hirth RA, Chernew ME, Fendrick AM. What is the price oflife and why doesn’t it increase at the rate of inflation? Arch Intern Med2003;163:1637-41.
91. Girgis A, Clarke P, Burton RC, Sanson-Fisher RW. Screening formelanoma by primary health care physicians: A cost-effectivenessanalysis. J Med Screen 1996;3:47-53.
92. Freedberg KA, Geller AC, Miller DR, Lew RA, Koh HK. Screeningfor malignant melanoma:A cost-effectiveness analysis. J Am Acad Der-matol 1999;41:738-45.
93. Halpern AC, Lieb JA. Early melanoma diagnosis: A success storythat leaves room for improvement. Curr Opin Oncol 2007;19:109-15.
94. Federman DG, Kravetz JD, Haskell SG, Ma F, Kirsner RS. Full-body skin examinations and the female veteran: Prevalence and per-spective. Arch Dermatol 2006; 142:312-6.
95. Welch HG. Informed choice in cancer screening. JAMA 2001;285:2776-8.
96. Berwick M. Evaluating early detection in the diagnosis of melanoma.Arch Dermatol 2006;142:1485-6.
97. Baade PD, English DR,Youl PH, McPherson M, Elwood JM, AitkenJF. The relationship between melanoma thickness and time to diagnosisin a large population-based study. Arch Dermatol 2006;142:1422-7.
98. Smith RA, Cokkinides V, Eyre HJ. American cancer society guide-lines for the early detection of cancer, 2006. CA Cancer J Clin2006;56:11-25; quiz 49-50.
99. Feightner JW. Prevention of skin cancer. Ottawa: Health Canada,1994; In: Canadian Task Force on the Periodic Health Examination.Canadian Guide to Clinical Preventive Health Care. Ottawa: HealthCanada; 1994.p.850-9.
100. Ferrini RL, Perlman M, Hill L. American college of preventive med-icine policy statement: Screening for skin cancer. Am J Prev Med1998;14:80-2.
101. NIH consensus conference. diagnosis and treatment of earlymelanoma. JAMA 1992;268:1314-9.
102. Cancer Council Australia The Cancer Council of Australia 2004.National Cancer Prevention Policy 2004-06. NSW: The CancerCouncil Australia. Available at: http://www.cancer.org.au/docu-ments/ NCPP_full_contents_links.pdf. Accessed March 30, 2007.
103. National Cancer Institute. Skin Cancer (PDQ) Screening: HealthProfessional Version http://www.cancer.gov/cancertopics/pdq/screen-ing/skin/HealthProfessional. Accessed March 30, 2007.
104. Field MJ, Lawrence RS, Zwanziger L, Eds., for the Institute of Med-icine. Extending Medicare Coverage for preventive and other services.Washington D.C.: National Academy Press; 2000.

G ITAL DERMATOL VENEREOL 2007;142:71-82
The molecular mechanisms of melanoma tumorigenesis:an update
Therapeutic resistance and proclivity for metastasis cast thedismal prognosis of cutaneous melanoma. Although recentdiscoveries in genetic, epidemiological and genomic studiesbring some hope of improved understanding of melanomapathogenesis, questions regarding underlying molecular hete-rogeneity of melanoma genesis remain. Mining these com-plexities will hopefully reveal clues essential to understandingdisease pathogenesis, clinical behavior and response to the-rapy. Current advances in high-resolution genome-wide tech-nologies, as well as gene-specific mutational analysis, in conjunc-tion with genetic and phenotypic analyses, improved animalmodels, may ultimately help to better define the seminal mole-cular events contributing to disease pathogenesis and ultima-tely identify more effective therapeutic targets.
KEY WORDS: Melanoma - MITF protein, human - Genes, p53 -AKT - PI-3K.
Cutaneous melanoma derives from the malignanttransformation of melanocytes, the pigment-pro-
ducing cells that reside in the basal layer of the epi-dermis in human skin. The incidence of cutaneousmelanoma has increased dramatically at a rate greaterthan that of any other cancer type worldwide, despiteincreased levels of awareness, and targeted health edu-cation. Although cutaneous melanoma accounts foronly 4% of all skin cancers, it is among a handful ofcancers whose dimensions are reported in millime-ters, however, it can demonstrate aggressive clinical
behavior and a high mortality rate with metastatic dis-ease. At early stage, melanoma can essentially be curedby surgical excision with wide local margins. Oncemetastasis develops, however, it is resistant to mostcurrent therapies. A median survival of 6-9 month isreported for AJCC Stage IV metastatic disease withonly a 14% 5 year survival rate.1
The etiology of melanoma is complex. As in manycancers, gene-environment interactions are felt to con-tribute to disease pathogenesis.2 In fact, epidemio-logical evidence suggests that melanoma is linked tointermittent sun exposure and a history of sunburns.3This association has led to enhanced patient educa-tion geared towards ultraviolet (UV) protection, recog-nition of early signs of skin cancer and consequently,diagnosis of disease at an earlier stage. These effortsmay be responsible for the slight improvement in sur-vival rate despite the increased incidence of disease (i.e.diagnosis of melanoma at an earlier stage is more like-ly to be cured by surgical excision). It has been demon-strated that UV may induce signature mutations (CC–> TT), however, they are not significantly found inmelanoma tumors. In fact, despite the epidemiologicevidence as sited above, the precise molecular mech-
1Division of DermatologyDepartment of Internal Medicine, Washington University
School of Medicine, St. Louis, MO, USA2Department of Pathology and Immunology, Washington
University School of Medicine, St. Louis, MO, USA
Address reprint requests to: L. Gao, Division of Dermatology, Depart-ment of Internal Medicine, Washington University School of Medicine, St.Louis, MO, USA. E-mail: [email protected]
L. GAO 1, H. ZHAO 2, L. A. CORNELIUS 1
Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 71

GAO THE MOLECULAR MECHANISMS OF MELANOMA TUMORIGENESIS
72 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
anisms of the contribution of UV to melanoma patho-genesis has not been clearly defined. In this papar, wewill attempt to review the most recent advances thathave been demonstrated with respect to the molecularevents contributing to melanomagenesis.
CDKN2A and INK4a/RB-ARF/p53 pathwaysStrong risk factors for melanoma development
include a family history of melanoma, multiple benignnevi (>50) or atypical nevi (any number), and a pre-vious history of primary melanoma. Other risk fac-tors include specific phenotypic characteristics, suchas fair skin, red or blond hair, blue eyes and a tenden-cy to freckling.4 Although familial melanoma is notcommon (~10% of melanoma cases), genetic studiesin melanoma-prone families have identified specificmolecular events and pathways that are key to thetransformation of melanocytes that contribute to bothfamilial and sporadic melanoma development. Tradi-tional linkage studies, combined with cytogenetic andloss of heterozygosity (LOH) analysis, have identi-fied several susceptibility loci predisposing tomelanoma. Further studies using positional cloningand sequence analysis have idntified CDKN2A (p16)at the 9p21 locus and CDK4 (12q13) as high pene-
trance melanoma genes and melanocortin 1 receptorgene (MC1R) as a low penetrance melanma gene at16q24.
The first cytogenetic analysis was performed near-ly 20 years ago and demonstrated loss or transloca-tion of 9p21 in a cohort of familial melanomaspatients.5 Homozygous deletions of this region inmelanoma suggested the existence of a tumor sup-pressor gene,6 that was further supported by germlinedeletion of this region in a patient with multiple primarymelanomas.7 Subsequent genetic studies in largemelanoma-prone families ultimately led to the cloningand identification, from the 9p21 region, of the firsthigh pentrance melanoma susceptibility gene, cyclin-dependent kinase inhibitor (CDKN) 2A tumor sup-pressor gene.8-11
CDKN2A is the major high-risk melanoma suscep-tible gene recognized to date. It is composed of 4exons: exon 1a, 1b, 2 and 3. It is unique in thatCDKN2A encodes 2 distinct tumor suppressor pro-teins, p16INK4A and p14ARF (p19ARF for mouse), viaalternatively using the fist exons (1a for p16INK4A and1b for p14ARF). The shared exon 2 and 3 of the twotranscripts are translated in different reading frames,thus encoding two proteins with no amino acid homol-ogy.12, 13 Inherited CDKN2A missense mutations inexons 1a and 2 are the most common genetic defects,although deletions and mutations in noncoding regionsand in the introns were also found in a small portion ofmelanoma families.10, 14-19 Both p16INK4A and p14ARF
are potent inhibitors of cell cycle progression and actthrough the retinoblastoma (RB) and p53 pathways, thetwo critical pathways important in cell cycle control.As illustrated in Figure 1, p16INK4A sequesters CDK4/6,abrogates the binding of these kinases to cyclin D andthereby prevents CDK4/6-mediated phosphorylationand subsequent inactivation of RB. Active RB repress-es E2F mediated gene transcription and results in aG1 to S cell cycle arrest.20 Loss of this repression,therefore, leads to unregulated cell cycle progression.On the other hand, p14ARF binds to and inactivates theMDM2 protein, which acts as an E3 ubiquitin ligase totarget p53 for proteasomal degradation. Thus, expres-sion of p14ARF stabilizes p53 which induces growtharrest, senescence or apoptosis via the specific down-stream effectors.21-23 Furthermore, loss of theINK4A/ARF locus has also been associated withreduced repair of UV-induced DNA damage and, there-fore increased gene mutations.24 Unlike several other
CDKNA2A
CHD5
p16INK4A p14ARF
HDM2
Cyclin D
CDK4/6
pRB
E2F
G1 to S transition Apoptosis
p21 Bax
p53
Figure 1.—CDKN2A signaling pathway.

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 73
THE MOLECULAR MECHANISMS OF MELANOMA TUMORIGENESIS GAO
tumors, including squamous cell carcinoma of theskin, mutations in p53 are uncommon in melanoma,although dysregualtion of the p53 pathway may occurdue to mutations in its upstream regulator, p14ARF asdescribed above.
The functional importance of CDKN2A locus in themelanoma development has been elucidated exten-sively in mice during the past few years. Mice har-boring deletions in exons 2 and/or 3 of murineCDKN2A, which specifically lack either CDKN2AINK4A (INK4A -/-) or ARF (ARF-/-) both have been gen-erated.25-29 Although these mice have increased pre-disposition to tumor development, predominantly lym-phomas and sarcomas, very few spontaneousmelanomas were observed. However, when a consti-tutively active mutant of HRAS (Tyr-RAS+), anupstream activator of MAPK signaling (see below),was coexpressed in cells of melanocytic lineage oneither null background (INK4A -/- or ARF -/-),enhanced melanoma development was demonstrat-ed.29 In addition, INK4A and ARF double null mice arehighly prone to cutaneous melanoma, and tumors hada shorter latency and higher penetrance compared withtheir single gene knockout counterpart mice.30 Thesestudies support the role of p16INK4A and p14ARF asbona fide tumor suppressors in melanoma tumorige-nesis although either of these mutations alone doesnot appear to be sufficient for melanoma development.
As previously stated, approximately 10% ofmelanomas occur in patients with a family history ofdisease. Of these, in familial melanoma cohorts stud-ied to date, 50% demonstrate a genetic linkage to the9p21 locus, and only 20-40% of these have been iden-tified to have germline alterations in CDKN2A. Thehigh linkage to the 9p21 region together with the rel-atively low detection of CDKN2A mutations supportsthe existence of additional melanoma susceptibilitygenes in this region. Analysis of somatic deletions intumors indicated that at least 2 additional tumor sup-pressor loci centromeric and telomeric to the CDKN2Aare linked to the development of melanoma.31-33 Iden-tification of these genes and characterization of theirfunction will hopefully provide additional insightsinto the molecular genesis of melanoma.
A second melanoma susceptibility gene is CDK4located on chromosome 12q13. However, CDK4 muta-tions have only been documented in a small number ofmelanoma families to date.34-36 The CDK4 mutationidentified so far located in codon 24 of exon 2 and
changes a critical arginine residue, crucial for bindingto INK4A. As anticipated, this CDK4 mutation is func-tionally identical to INK4A loss, and renders the CDK4kinase constitutively active, inactivating RB and leadsto unchecked cell cycle progression. Therefore, phe-notypic characteristics of the families carrying CDK4germline mutations are indistinguishable to CDKN2Aaffected families. Mice that carry the human CDK4mutation have been shown to be susceptible tomelanoma development following a second exoge-nous carcinogen exposure.37 Despite these findings,a low detection rate of CDK4 mutations have beenfound in melanoma patients, possibly due to targetedscreening performed to date (exon 2), the patientcohorts examined, or an actual low incidence of thismutation in humans. Other investigations have puta-tively identified other candidate melanoma gene(s) onchromosomes 1p22, 1p36, 11p15, and 4, although thesignificance of these findings remains unclear.32, 38, 39
A striking recent advance in our understanding ofINK4A/RB and ARF/p53 pathways in human cancerdevelopment is the identification of CHD5 (chro-modomain-helicase-DNA binding-5) as a tumor sup-pressor on chromosome 1p36.40, 41 Although its rele-vance to melanomagenesis is unclear at this time, dele-tions of 1p36 are widely reported in human cancers,including melanoma.42 However, identification of the1p36 tumor suppressor has remained elusive to date.By using chromosomal engineering, Bagchi et al. gen-erated mouse strains with a gain and loss of a genom-ic interval corresponding to human 1p36. Using thispowerful model, these investigators identified CHD5as a tumor suppressor at this locus using a retrovirus-based shRNA strategy. Most interestingly, all CHD5-regulated cellular processes, such as proliferation andapoptosis, were p53 dependent. Furthermore, CHD5has been shown to function upstream of p53 and mostlikely RB through regulation of INK4A and ARFexpression. Although the data shows that CHD5 mayfunction as a novel tumor suppressor gene, specificmechanisms addressing its affect on p53 and RB path-ways remain to be elucidated. Again, whether CHD5is altered or acts as a tumor suppressor in melanomahas not yet been determined.
A low penetrance melanoma susceptibility gene isthe MC1R, located on chromosome 16q24 and hasbeen shown to act as a genetic modifier of melanomarisk in CDKN2A germline mutation carriers.43, 44 MC1Rencodes the G-protein coupled receptor (GPCR) for a-

GAO THE MOLECULAR MECHANISMS OF MELANOMA TUMORIGENESIS
74 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
melanocyte stimulating hormone (a-MSH) and playsa central role in regulating melanin synthesis. Ligandbinding induces tyrosinase transcription and melaninproduction. The MC1R gene is highly polymorphicin the Caucasian population, and more than 30 allelicvariants have been described. Some of these variantscause loss of gene function (LOF) resulting in the pro-duction of phaeomelanin, as opposed to the normaleumelanin, pigment. Specific LOF variants may con-fer an increased overall risk of melanoma develop-ment, even in patients without red hair and blue eyes.
RAS/BRAF/MAPK/ERK pathway
As in many other human cancers, aberrant cellgrowth/proliferation plays a critical role in melanomatumorigenesis and progression. The mitogen-activat-ed protein kinase (MAPK)/extracellular signal-regu-lated kinase (ERK) pathway has emerged as the cen-tral growth-stimulating pathway in melanoma tumori-genesis.45 RAS and RAF kinases are components of theMAPK/ERK cascade. Activation of this pathway inmelanoma can not only occur through RAF/RAS muta-tions, but can also occur following exposure toautocrine growth factors, such as basic fibroblastgrowth factor, hepatocyte grow factor (HGF).46, 47
Growth factors such as these, certain cytokines andhormones activate RAS through their engagement oftheir cognate receptors, adaptor proteins or het-erotrimeric G-proteins. Once activated, Ras proteinsbind to and activate their downstream effectors, suchas RAF proteins which are serine/threonine proteinkinases. RAF proteins then phosphorylate and acti-vate the MEK protein kinases, which in turn phos-phorylate and activate the ERK kinases. ERK is alsoa serine/threonine kinase that phosphorylate numeroussubstrates to regulate a wide range of cellular func-tions, such as apoptosis, migration and cell prolifera-tion.48
Although RAS and RAF germline mutations inmelanoma have not been found, somatic mutations ofNRAS have been demonstrated in approximately 15%of melanoma tumors and this activating mutation dri-ves activation of ERK. With regards to RAF, threemammalian isoforms exist – ARAF, BRAF and CRAF(RAF-1). ARAF and RAF-1 are rarely mutated inmelanomas, however BRAF mutations have been deter-mined to have a seminal role n melanoma develop-ment. BRAF is mutated in up to 70% melanoma tumors
and cell lines.49 Reported mutations to date have beenfound in the BRAF kinase domain, encoded by exons11 and 15. Over 90% of those reported affect one crit-ical amino acid, resulting in a valine to glutamic acidsubstitution at residue 600 (BRAF V600E), leading toconstitutive kinase activation of BRAF, and conse-quently downstream ERK activation. In support of itsrole in melanoma formation, studies employing smallinterfering RNA against mutated BRAF but not CRAFinhibit the transformation of melanocytes harboring thisactivating BRAF mutation.
Interestingly, NRAS and BRAF mutations are mutu-ally exclusive in melanoma – they have an epistaticrelationship. It has been postulated that this mutualexclusivity is due to the ability of each activating muta-tion to activate the Ras pathway, and consequentlydownstream ERK, independently, similarly deregu-lating this common effector pathway. Although theinduction of these somatic mutations is not well under-stood, current evidence suggests that BRAF and NRASmutations may be instigated by distinct mechanisms.In support of this, genome-wide CGH profiling and tar-geted re-sequencing of primary melanoma tumorshave demonstrated that distinct patterns of genomicalterations, including BRAF and NRAS mutation fre-quencies, can be identified in melanomas arising atdifferent anatomic sites, different subtypes and withvarying UV exposure history (acral melanoma, mucos-al melanoma, and histologic evidence of chronic sundamage).50 As previously noted, this study confirmedthat these mutations are not signature UV-induced thy-madine dimer mutations. In addition, a recent microar-ray analysis in melanoma indicates that BRAF andNRAS mutations have overlapping but distinct tran-scriptional profiles.51
Following the determination of the high mutationrate of BRAF in melanoma, several groups investigat-ed BRAF mutational status in benign nevi, in an attemptto determine whether BRAF activating mutations werean early, or late, event, in melanocyte transformation.Although unexpected, it has been determined thatBRAF mutation occurs at a similar frequency in benignnevi as in primary and metastatic melanomas, impli-cating BRAF mutation as an early event in the devel-opment of melanocytic neoplasia. In addition, thesefindings demonstrate that BRAF dysfunction per se isnot sufficient to drive melanoma development and thatcells of melanocyte lineage in nevi must acquire addi-tional molecular “hits” for transformation to melanoma.

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 75
THE MOLECULAR MECHANISMS OF MELANOMA TUMORIGENESIS GAO
In support of this multi-hit hypothesis, melanocyte-specific expression of BRAFV600E, but not wild-typeBRAF, led to increased nevi formation in a zebra-fishmelanoma model.52 When combined with p53 defi-ciency, however, the BRAFV600E induced melanocytelesions rapidly progressed to invasive melanoma.
Further understanding of the molecular mechanismsunderlying the contribution of BRAF activating muta-tions to the pathogenesis of benign nevi and melanomamay be garnered from studies that have evaluated theeffect of the BRAFV600E on cell cycling in melanocytes.Interestingly, sustained BRAFV600E expression inhuman melanocytes (and consequently prolonged ERKactivation) has been found to induce cell senescence byincreasing the expression of both p16INK4A and thewell recognized senescence-associated marker acidicb-galactosidase activity. Along these lines, it has beenpreviously demonstrated that although rapid and tran-sient ERK activation stimulates cell proliferation, pro-longed ERK activation induces cell senescence in avariety of cell types, including melanocytes andmelanoma cell lines. In recent studies, BRAF mutationhas been found to predict sensitivity to MEK inhibition,findings that may have implications for melanomatreatment strategies. In this study, small moleculeinhibitors of MEK were used in tumors and cells thatwere either WT for BRAF and NRAS or those harbor-ing gain of function mutations. Compared to WT orcells having a NRAS mutation, V600E activating muta-tion of BRAF was found to enhance cell sensitivity toMEK inhibition, completely abrogating tumor growthin mutant xenografts.53 In vivo, BRAF mutation statusmay affect cell response to MEK or ERK specificphosphotases, endogenous and physiologic inhibitorsof these kinases.
Another interesting finding is that BRAF mutationalters the stability of BRAF protein.54 Unlike the oth-er RAF isoforms ARAF and CRAF, WT BRAF doesnot require Hsp90 chaperone for protein stabilization.In contrast, mutated, activated BRAF does bind to anHsp90-cdc37 chaperone complex, required for its sta-bility and function. Exposure of melanoma cells andtumors harboring this mutation to Hsp90 inhibitorsresults in the degradation of mutant BRAF and sub-sequent inhibition of ERK activation, cell proliferationand tumor progression, again, findings that may haveimplications in melanoma treatment.
Although RAS and RAF proteins are well charac-terized upstream activators of ERK pathway in
melanoma as described above, a recent report hasshown enhanced ERK activity despite a low frequen-cy of BRAF and NRAS mutations in one melanomasubtype, acral melanoma. These findings suggest thatfactors other than BRAF and NRAS gain of functionmutations may contribute to constitutive activation ofERK pathway in certain tumors.55 Based upon work inour laboratory, one potential candidate is RAS-asso-ciated protein-1 (Rap1), a member of the RAS smallGTPase superfamily.56 Like RAS, Rap1 functions asa molecular switch, cycling between an inactive, GDP-bound and an active, GTP-bound form. Rap1 guaninenucleotide exchange factors (GEFs) activate Rap1 byfacilitating the replacement of GDP with GTP, where-as Rap1 GTPase-activating proteins (GAPs) inhibitRap1 activity by enhancing the intrinsic GTPase activ-ity of Rap1 to hydrolyze the bound GTP to GDP. Theeffect of Rap1 on ERK activation is complex and high-ly cell type specific. In several cell types, Rap1 atten-uates growth factor-induced, RAS-mediated ERK acti-vation by sequestration of CRAF and other RAS effec-tors in an inactive complex. On the other hand, Rap1has been shown to stimulate ERK through the directbinding and activation of BRAF in certain, if not all,BRAF expressing cells. Finally, Rap1 has been report-ed fail to interfere with RAS signaling towards ERKin certain cell types. In support of the role of Rap1 inmelanoma development, we have recently shown thatRap1 is activated in both human metastatic melanomatissues and in several melanoma cell lines. Our in vit-ro experiments demonstrated that Rap1 positively reg-ulates ERK activation induced by HGF, a growth fac-tor that has been implicated in melanomagenesis andby 8CPT-2Me-cAMP, an activator of Epac, a Rap1GEF downstream of cAMP (by increasing levels ofactivated Rap1). Furthermore, Rap1 has demonstrat-ed the ability to bind to BRAF in vitro. Taken togeth-er, these data support the model postulated in Figure2, in which Rap1 couples GPCR and RTK signalingpathways to ERK activation in melanoma cells. Inaddition to modulating ERK activity, we have shownthat Rap1 also induces avb3 integrin activation andconsequently increases melanoma cell adhesion andmigration, processes previously implicated in tumorprogression.57
Accumulating evidence suggests that dysregulationof Rap1 activity, either by Rap1 over-expression orchanges in levels of Rap1 activators, such as GEFs, andinhibitors, such as tuberin and Rap1 GAPs, has been

GAO THE MOLECULAR MECHANISMS OF MELANOMA TUMORIGENESIS
76 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
associated with human malignancies.58-64 Mice defi-cient in SPA-1, a principal Rap1 GAP in hematopoi-etic cells, develop a spectrum of myeloid disordersthat resemble human chronic myelogenous leukemia.65
Rap1-GAP, which is expressed in melanoma cells,maps to chromosomal region 1p36, which is deleted ina wide range of human cancers.10, 39, 66-68 As describedabove, CHD5 has been recently identified as a tumorsuppressor in 1p36 and functions upstream of p53/RBpathways. Since alterations of the p53 and RB path-ways alone do not induce melanoma in several in vivomodels, distinct tumor suppressor genes in other path-ways must contribute tumor development. We are cur-rently investigating whether mutations of Rap1-GAP,may be implicated in this process.
PTEN/PI-3K/AKT pathway
A third critical molecular pathway involved inmelanoma pathogenesis is the phophatidylinositol 3-kinases (PI-3Ks)/AKT (PKB) signaling module.69 Acti-vated AKT phosphorylates and regulates the functionof many downstream molecules which control sur-vival/apoptosis, proliferation/cell growth, metabolismand neo-vascularization. AKT exerts its antiapoptoticactivity by several distinct mechanisms. First, AKT
phosphorylates and inactivates the proapoptotic fac-tors procaspase-9 and BAD, which antagonizes bcl-2and triggers the release of cytochrome c from the mito-chondria, a key step in stress-induced apoptosis. Sec-ond, AKT phosphorylates and activates IKK-a, whichphosphorylates IkB leading to the release and activationof NF-kB which results in the expression of antiapop-totic genes. Third, AKT phosphorylates the FOXOtranscription factors, which results in the nuclear exclu-sion of FOXO proteins and a decrease of the expressionof genes critical for apoptosis, such as the death recep-tor Fas ligand and proapoptotic Bim. With respect toproliferation and cell growth, active AKT promotescell cycle progression by phosphorylation and inacti-vation of GSK-3b to avert cyclin D1 degradation. AKTalso directly phosphorylates the cell cycle inhibitorsp21WAF1 and p27Kip1, leading to the sequestration ofthese inhibitors in the cytoplasm, and been shown to ter-minate FOXO-mediated inhibition of the expressionof p21WAF1, p27Kip1, cyclin D1 and D2. Finally, AKTincreases MDM2 phosphorylation and the translocationof MDM2 into the nucleus, where it downregulatesp53 and antagonizes p53-mediated cell cycle arrest. Amajor cellular metabolic pathway regulated by AKT liesin mTOR (target of Rapamycin). In this context, AKTphosphorylates TSC2 and prevents its dimerizationwith TSC1. The TSC1-TSC2 complex functions as aGAP for RAS-like small GTPase, RHEB, which pro-motes the activity of the kinase mTOR. Therefore,AKT activation inhibits the GAP activity of TSC1-TSC2 towards to RHEB and consequently activatesmTOR. Activated mTOR phosphorylates a number ofsubstrates and induces expression of cyclin D1, c-myc,and vascular endothelial growth factor (VEGF).
As illustrated in Figure 3, extracellular stimuli trans-duced through RTK or GPCR activate PI-3Ks, whichphosphorylate phosphatidylinositol and its phospho-rylated derivatives, mainly phosphatidylinositol 4, 5-bisphosphate (PIP2), generating the second messengerphosphatidylinositol 3, 4, 5-triphosphate (PIP3). Thehigh level of PIP3 induces the translocation of the ser-ine-threonine kinase AKT from the cytoplasm to theplasma membrane. Recruitment of AKT by PIP3 resultsin the activation of AKT. PIP3 can be dephosphory-lated to PIP2 by phosphatase and tensin homolog delet-ed in chromosome ten (PTEN). Thus, PTEN functionsas a negative regulator of PI-3K/AKT signaling and,as such, a tumor suppressor.
As outlined above, hyperactivation of AKT is asso-
GPCR
GPs
cAMP
Epac
Rap1
Integrins
Motilitymetastasis
RTK
RAS
B-Raf
MEK
ERK
GEF?
Proliferation
Figure 2.—RAS/BRAF/ERK pathway.

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 77
THE MOLECULAR MECHANISMS OF MELANOMA TUMORIGENESIS GAO
ciated with resistance to apoptosis and increased cellproliferation/growth, events that favor tumorigenesis.It is not surprising that a PI-3K/AKT pathway dereg-ulation occurs in many human cancers, includingmelanoma.70-74 Similar to other tumors, several mech-anisms potentially contribute to AKT hyperactivationin melanoma. First, inactivating mutations or deletionsof PTEN may lead to AKT activation. LOH at chro-mosome 10q, which harbors the PTEN locus, has beenreported in up to 50% of melanomas.75 Secondly, ampli-fication and overexpression of PI-3K subunits isobserved in certain types of human cancers. Morerecently, mutations in PIK3CA, the gene that encodesthe catalytic subunit p110a, and in the regulatory sub-unit, p85 of PI-3K have been identified in human tumorcell lines and tissues,76, 77 although not in melanoma to
date. Interestingly, activating RAS mutations can acti-vate AKT through direct binding to the p110 catalyticsubunit of PI-3K, which may have implications inmelanoma in that 15% of melanomas harbor NRASmutations (as described above), and the previouslyreported finding that NRAS mutations and PTEN alter-ations are epistatic in melanoma (see below). Finally,amplification of AKT3 has been identified in melanomaand may be responsible for the high frequency of AKTactivation in some tumors. A role for AKT in melanomaprogression is demonstrated by the finding by onegroup that overexpression of AKT in radial growthphase melanoma converts it to an invasive, moreadvanced vertical growth tumor.78 Upregulation ofVEGF expression and increased production of super-oxide ROS were implicated in tumor progression, how-
PIP2
PIP3
PDK1
AKT
BAD
Angiogenesis
Protein synthesis
RAS
RTK
PI-3K PTEN
VEGF
HIF1α
S6K
TSC1/2
RHEB
mTOR
4IF4E
4EBP
Apoptosis Cell cycle progression
IKK-α FOXO
BlmFASLNF-κBcas-9
HMDM2 GSK-3β FOXO
p53 p21/p27
Cyclin D
Figure 3.—PTEN/PI-3K/AKT pathway.

GAO THE MOLECULAR MECHANISMS OF MELANOMA TUMORIGENESIS
78 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
ever, the precise AKT downstream effector pathwaysinvolved remain to be elucidated. As previouslydescribed, many AKT downstream effectors, such asFOXO and TSC2, function as tumor suppressors inother malignancies, although their contribution tomelanoma tumorigenesis has not been demonstrated.
A recent study has revealed high level of phospho-AKT in both primary and metastatic melanomas (49%and 77%, respectively) 74. Studies by Tsao et al. havedemonstrated PTEN alterations in approximately 20%of uncultured tumors,75 suggesting that factors otherthan PTEN alterations contribute to constitutively acti-vation of the PI3K signaling pathway. Interestingly, fur-ther characterization by Tsao et al. demonstrated thatmelanomas segregate into one of three mutational pro-files: a mutation in NRAS alone, concurrent PTEN andBRAF mutations, or BRAF mutations alone. Fromthese findings, and the recognition that benign nevihave a low NRAS mutation frequency and a relativelyhigh BRAF mutation rate, these investigators proposedthat NRAS acts to stimulate both downstream BRAFand PI3K, and consequently MAPK and AKT. Isolat-
ed BRAF activation may require the cooperation ofPTEN alterations, possibly a later event in melanomadevelopment, to activate AKT.
Oncogenic effects of MITF and NEDD9 onmelanoma genesis
Melanocytes are derived from neural crest. Microph-thalmia-associated transcription factor (MITF) is a lin-eage-specific melanocyte gene and a master regulatorof melanocyte development and function. MITF belongsto the MiT family of helix-loop-helix/leucine-zippertranscription factors.79 The binding of α-MSH to MC1Rinduces cAMP, which increases MITF expression ina positive regulatory loop. MITF in turn stimulates thetranscription of genes governing melanin synthesis.Both activated ERK induced by RTK/RAS/BRAF sig-naling pathway and β-catenin in the WNT signalingpathway can function to activate MITF (Figure 4). Inaddition to its role in melanocyte pigmentation, MITFis also essential for melanocyte survival. In fact, micelacking functional MITF lack melanocytes and arephenotypically albino.80 In humans, MITF mutationsresult in pigmentary disorders such as Waardenberg’sType II and Tietz syndrome.
MITF regulates melanocyte survival by transcrip-tionally activating the Bcl-2 gene, a key antiapoptot-ic factor that is characteristically expressed at highlevels in melanocytes and melanoma. In normalmelanocytes and cells of melanocyte lineage, MITF isrequired for the induction of cell cycle exit and acti-vation of the differentiation programme via activationof p16INK4A and p21Cip1 genes.81, 82 Another recentfinding supports the role of MITF-induced cell cyclearrest in melanocytes where high levels of MITFexpression were found to counteract BRAF-stimulat-ed melanocyte proliferation.83
In contrast to the role of MITF in normalmelanocytes of differentiation cell cycle exit, a recent,seemingly contradictory, finding in melanoma hasidentified MITF as a lineage-dependent oncogeneamplified in a subset of these tumors.84 Using an inte-grated analysis of high-resolution single nucleotidepolymorphism maps and gene expression databasesthe authors found that the chromosome region (3p13-3p14) containing the MITF gene was amplified inmost melanoma cell lines. MITF gene was also ampli-fied (from 5 to 119 copies) in about 10% of primarymelanomas and up to 20% of metastatic melanomas in
MITF
TYR, TYRP1DCT
Dia1, Tbx2, CDK2,p21, p16INK4A
C-MetHIF1α
Bcl-2HIF1α
DifferentiationPigmentation
Cell cycle progression MotilityMetastasis
Apoptosis
ERK
MEK
BRaf
Ras b-catenin
RTK Wnt
GFs Frizzled
CREB
PKA
cAMP
AC
GPCR
α-MSH
Figure 4.—MITF signaling pathway.

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 79
THE MOLECULAR MECHANISMS OF MELANOMA TUMORIGENESIS GAO
human tissue samples. The MITF amplification is sig-nificantly correlated to a decreased five-year survivalin patients with advanced melanoma. Moreover, invitro functional studies showed that MITF togetherwith mutated BRAF transformed immortalized humanmelanocytes, thereby confirming MITF’s function asan oncogene in melanoma. These findings in melanomamay support a role for MITF as a potential target fortreatment. A number of mechanisms may, in fact, sup-port this proposed oncogenic effect of MITF onmelanoma cells. First, MITF promotes cell cycle pro-gression and invasiveness of melanoma cells by thesuppression of CDK inhibitor p27kip1 via regulatingDia1 and actin polymerization.85 Second, one of theMITF target genes, Tbx2 (T-Box transcription factor2), has been recently implicated in proliferation andsuppression of senescence in melanoma through itsability to repress ARF and p21 promoter expression.86,
87 Third, MITF has been shown to regulate cell cycleprogression by modulation of CDK2, which is essen-tial for melanoma clonogenic growth 88. Fourth, MITFbinds to the HIF1a promoter and strongly stimulatesits transcriptional activities, including the inductionof VEGF expression in response to hypoxia.89 Last, c-Met, the receptor for HGF has been identified as adirect transcriptional target of MITF. HGF/c-Met sig-naling has been shown to be a key pathway in bothmelanocyte development and melanoma progression.90
Through these mechanisms, MITF may exemplify oneof a newly recognized group of lineage survival genesthat potentially contribute to tumorigenesis.
Employing genome-wide high-resolution array-CGH of nonmetastatic parental and metastatic vari-ants of murine melanoma cells, a recent study by Kimet al. identified a recurrent focal gene amplificationassociated with the metastatic cells.91 The amplifiedregion identified in mouse is syntenic to 6p24-25(NEDD9) in humans, which, is amplified in 36% ofmetastatic as opposed to of primary melanomas 92, 93
and may function as a novel melanoma metastasisgene. It has been proposed that NEDD9 interacts withfocal adhesion kinase in mediating cell invasion, andas such, tumor progression.
Concluding remarks
In one model of melanomagenesis, Clark porposesthe histologic changes accompany the progressionfrom normal melanocytes to melanoma. Five distinct
stages have been proposed: common acquired andcongenital nevi without dysplastic changes; dysplas-tic nevi with structural and architectural atypia; radi-al growth phase melanoma; vertical growth melanomaand metastatic melanoma. This model suggests thatcertain key molecular events occur at each stage, dri-ving the progression from melanocyte transformationto tumor formation and metastasis. However, withincreasing understanding of the molecular pathogen-esis of melanoma, tumor heterogeneity is becomingincreasingly evident, and with several distinct, andsometimes synergistic molecular mechanisms at play.As has long been recognized, melanomas arising ondifferent body sites exhibit markedly distinct biolog-ical and clinical behaviors. Lentigo maligna melanomasare indolent tumors that develop over decades on chron-ically sun exposed area such as the face. In contrast,acral lentigenous melanomas, which develop on sun-protected regions, are often diagnosed at a later stage,and some propose that they tend to exhibit more aggres-sive behavior. Breslow thickness clearly has strongprognostic value, however, there exists a subset of thinmelanomas that metastasize.
The ‘hallmarks of cancer’ model offers a frame-work describing the genetic complexity of human can-cers. According to this model, detailed dissection oftumorigenic pathways and identification of the ‘mol-ecular signature’ of melanoma at each stage and sub-type may aid in our attempt to prevent tumor devel-opment and provide insight into treatment strategies.From this review, however, it should be evident that thecurrently recognized chromosomal alterations andmutational signatures of key melanoma genes do notprovide a complete understanding of melanomagene-sis. As in most cancers, processes such as epigeneticmodification, transcriptional profiling of both codingand noncoding RNAs, and gene-environment inter-face most certainly contribute to tumor development,and these mechanisms have not yet been clearly definedin melanoma.
Riassunto
Meccanismi molecolari della tumoregenesi del melanoma:un aggiornamento
La resistenza alla terapia e la tendenza a metastatizzarespiegano la pessima prognosi del melanoma cutaneo. Seb-bene recenti scoperte nel settore della genetica, e studi epi-demiologici e gnomici che hanno alimentato una qualche

GAO THE MOLECULAR MECHANISMS OF MELANOMA TUMORIGENESIS
80 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
speranza di migliorare la comprensione della patogenesi delmelanoma, restano senza risposta le domande relative all’e-terogeneità molecolare sottostante alla genesi del melano-ma. La comprensione di queste complessità si spera chesarà di aiuto per identificare gli aspetti essenziali dellapatogenesi del tumore e per capirne il comportamento cli-nico e la risposta alla terapia. Gli attuali progressi nelletecnologie ad alta risoluzione per ampi tratti di genoma,così come l’analisi mutazionale gene-specifica, assieme aquella genetica e fenotipica e ai migliorati modelli anima-li, possono in ultima analisi aiutare a meglio definire glieventi molecolari fondamentali che contribuiscono allapatogenesi della malattia e ad identificare gli obiettivi tera-peutici più efficaci.PAROLE CHIAVE: Melanoma - Linfonodo sentinella - Biopsialinfonodale - Metastasi linfatiche - Linfoangiogenesi.
References
1. Miller AJ, Mihm MC, Jr. Melanoma. N Engl J Med 2006;355:51-65. 2. Merlino G, Noonan FP. Modeling gene-environment interactions in
malignant melanoma. Trends Mol Med 2003;9:102-8. 3. Thompson JF, Scolyer RA, Kefford RF. Cutaneous melanoma. Lan-
cet 2005;365:687-701. 4. Chudnovsky Y, Khavari PA, Adams AE. Melanoma genetics and the
development of rational therapeutics. J Clin Invest 2005;115:813-24. 5. Cowan JM, Halaban R, Francke U. Cytogenetic analysis of mela-
nocytes from premalignant nevi and melanomas. J Natl Cancer Inst1988;80:1159-64.
6. Fountain JW, Karayiorgou M, Ernstoff MS, Kirkwood JM, VlockDR, Titus-Ernstoff L et al. Homozygous deletions within humanchromosome band 9p21 in melanoma. Proc Natl Acad Sci U S A1992;89:10557-61.
7. Petty EM, Bolognia JL, Bale AE,Yang-Feng T. Cutaneous malignantmelanoma and atypical moles associated with a constitutional rear-rangement of chromosomes 5 and 9. Am J Med Genet 1993;45:77-80.
8. Nobori T, Miura K, Wu DJ, Lois A, Takabayashi K, Carson DA. Dele-tions of the cyclin-dependent kinase-4 inhibitor gene in multiplehuman cancers. Nature 1994;368:753-6.
9. Kamb A, Shattuck-Eidens D, Eeles R, Liu Q, Gruis NA, Ding W et al.Analysis of the p16 gene (CDKN2) as a candidate for the chromoso-me 9p melanoma susceptibility locus. Nat Genet 1994;8:23-6.
10. Hussussian CJ, Struewing JP, Goldstein AM, Higgins PA, Ally DS,Sheahan MD et al. Germline p16 mutations in familial melanoma. NatGenet 1994;8:15-21.
11. Kamb A, Gruis NA, Weaver-Feldhaus J, Liu Q, Harshman K, Tavti-gian SV et al. A cell cycle regulator potentially involved in genesis ofmany tumor types. Science 1994;264:436-40.
12. Chin L, Garraway LA, Fisher DE. Malignant melanoma: geneticsand therapeutics in the genomic era. Genes Dev 2006;20:2149-82.
13. de Snoo FA, Hayward NK. Cutaneous melanoma susceptibility andprogression genes. Cancer Lett 2005;230:153-86.
14. Liu L, Dilworth D, Gao L, Monzon J, Summers A, Lassam N et al.Mutation of the CDKN2A 5’UTR creates an aberrant initiation codonand predisposes to melanoma. Nat Genet 1999;21:128-32.
15. Orlow I, Begg CB, Cotignola J, Roy P, Hummer AJ, Clas BA et al.CDKN2A Germline Mutations in Individuals with Cutaneous Mali-gnant Melanoma. J Invest Dermatol 2007 in press.
16. MacKie RM, Andrew N, Lanyon WG, Connor JM. CDKN2A germ-line mutations in U.K. patients with familial melanoma and multipleprimary melanomas. J Invest Dermatol 1998;111:269-72.
17. Harland M, Holland EA, Ghiorzo P, Mantelli M, Bianchi-Scarra G,
Goldstein AM et al. Mutation screening of the CDKN2A promoter inmelanoma families. Genes Chromosomes Cancer 2000;28:45-57.
18. Petronzelli F, Sollima D, Coppola G, Martini-Neri ME, Neri G,Genuardi M. CDKN2A germline splicing mutation affecting bothp16(ink4) and p14(arf) RNA processing in a melanoma/neurofibro-ma kindred. Genes Chromosomes Cancer 2001;31:398-401.
19. Loo JC, Liu L, Hao A, Gao L, Agatep R, Shennan M et al. Germlinesplicing mutations of CDKN2A predispose to melanoma. Oncogene2003;22:6387-94.
20. Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature1993;366:704-7.
21. Kamijo T, Weber JD, Zambetti G, Zindy F, Roussel MF, Sherr CJ.Functional and physical interactions of the ARF tumor suppressorwith p53 and Mdm2. Proc Natl Acad Sci U S A 1998;95:8292-7.
22. Pomerantz J, Schreiber-Agus N, Liegeois NJ, Silverman A, Alland L,Chin L et al. The Ink4a tumor suppressor gene product, p19Arf, inte-racts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell1998;92:713-23.
23. Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degrada-tion and stabilizes p53: ARF-INK4a locus deletion impairs both theRb and p53 tumor suppression pathways. Cell 1998;92:725-34.
24. Sarkar-Agrawal P, Vergilis I, Sharpless NE, DePinho RA, RungerTM. Impaired processing of DNA photoproducts and ultraviolethypermutability with loss of p16INK4a or p19ARF. J Natl CancerInst 2004;96:1790-3.
25. Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA.Role of the INK4a locus in tumor suppression and cell mortality. Cell1996;85:27-37.
26. Krimpenfort P, Quon KC, Mooi WJ, Loonstra A, Berns A. Loss ofp16Ink4a confers susceptibility to metastatic melanoma in mice.Nature 2001;413:83-6.
27. Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH,Aguirre AJ et al. Loss of p16Ink4a with retention of p19Arf predisposesmice to tumorigenesis. Nature 2001;413:86-91.
28. Kamijo T, Bodner S, van de Kamp E, Randle DH, Sherr CJ. Tumorspectrum in ARF-deficient mice. Cancer Res 1999;59:2217-22.
29. Sharpless NE, Ramsey MR, Balasubramanian P, Castrillon DH,DePinho RA. The differential impact of p16(INK4a) or p19(ARF)deficiency on cell growth and tumorigenesis. Oncogene 2004;23:379-85.
30. Chin L, Pomerantz J, Polsky D, Jacobson M, Cohen C, Cordon-Car-do C et al. Cooperative effects of INK4a and ras in melanoma suscep-tibility in vivo. Genes Dev 1997;11:2822-34.
31. Pollock PM, Welch J, Hayward NK. Evidence for three tumor sup-pressor loci on chromosome 9p involved in melanoma development.Cancer Res 2001;61:1154-61.
32. Geffrotin C, Crechet F, Le Roy P, Le Chalony C, Leplat JJ, IannuccelliN et al. Identification of five chromosomal regions involved in pre-disposition to melanoma by genome-wide scan in the MeLiM swinemodel. Int J Cancer 2004;110:39-50.
33. van der Velden PA, Sandkuijl LA, Bergman W, Hille ET, Frants RR,Gruis NA. A locus linked to p16 modifies melanoma risk in Dutchfamilial atypical multiple mole melanoma (FAMMM) syndrome fami-lies. Genome Res 1999;9:575-80.
34. Zuo L, Weger J,Yang Q, Goldstein AM, Tucker MA, Walker GJ et al.Germline mutations in the p16INK4a binding domain of CDK4 infamilial melanoma. Nat Genet 1996;12:97-9.
35. Soufir N, Avril MF, Chompret A, Demenais F, Bombled J, Spatz A etal. Prevalence of p16 and CDK4 germline mutations in 48 melanoma-prone families in France. The French Familial Melanoma StudyGroup. Hum Mol Genet 1998;7:209-16.
36. Goldstein AM, Struewing JP, Chidambaram A, Fraser MC, TuckerMA. Genotype-phenotype relationships in U.S. melanoma-pronefamilies with CDKN2A and CDK4 mutations. J Natl Cancer Inst2000;92:1006-10.
37. Sotillo R, Garcia JF, Ortega S, Martin J, Dubus P, Barbacid M et al.

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 81
THE MOLECULAR MECHANISMS OF MELANOMA TUMORIGENESIS GAO
Invasive melanoma in Cdk4-targeted mice. Proc Natl Acad Sci U S A2001;98:13312-7.
38. Gillanders E, Juo SH, Holland EA, Jones M, Nancarrow D, Freas-LutzD et al. Localization of a novel melanoma susceptibility locus to1p22. Am J Hum Genet 2003;73:301-13.
39. Bale SJ, Dracopoli NC, Tucker MA, Clark WH, Jr., Fraser MC, Stan-ger BZ et al. Mapping the gene for hereditary cutaneous malignantmelanoma-dysplastic nevus to chromosome 1p. N Engl J Med1989;320:1367-72.
40. Schuster EF, Stoger R. CHD5 defines a new subfamily of chromo-domain-SWI2/SNF2-like helicases. Mamm Genome 2002;13:117-9.
41. Bagchi A, Papazoglu C, Wu Y, Capurso D, Brodt M, Francis D et al.CHD5 is a tumor suppressor at human 1p36. Cell 2007;128:459-75.
42. Poetsch M, Dittberner T, Woenckhaus C. Microsatellite analysis at1p36.3 in malignant melanoma of the skin: fine mapping in search ofa possible tumour suppressor gene region. Melanoma Res 2003;13:29-33.
43. van der Velden PA, Sandkuijl LA, Bergman W, Pavel S, van MourikL, Frants RR et al. Melanocortin-1 receptor variant R151C modifiesmelanoma risk in Dutch families with melanoma. Am J Hum Genet2001;69:774-9.
44. Box NF, Duffy DL, Chen W, Stark M, Martin NG, Sturm RA et al.MC1R genotype modifies risk of melanoma in families segregatingCDKN2A mutations. Am J Hum Genet 2001;69:765-73.
45. Tsao H, Atkins MB, Sober AJ. Management of cutaneous melanoma.N Engl J Med 2004;351:998-1012.
46. Satyamoorthy K, Li G, Gerrero MR, Brose MS, Volpe P, Weber BLet al. Constitutive mitogen-activated protein kinase activation in mela-noma is mediated by both BRAF mutations and autocrine growthfactor stimulation. Cancer Res 2003;63:756-9.
47. Li G, Schaider H, Satyamoorthy K, Hanakawa Y, Hashimoto K, HerlynM. Downregulation of E-cadherin and Desmoglein 1 by autocrinehepatocyte growth factor during melanoma development. Oncogene2001;20:8125-35.
48. Gray-Schopfer VC, da Rocha Dias S, Marais R. The role of B-RAFin melanoma. Cancer Metastasis Rev 2005;24:165-83.
49. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S et al.Mutations of the BRAF gene in human cancer. Nature 2002;417:949-54.
50. Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner Het al. Distinct sets of genetic alterations in melanoma. N Engl J Med2005;353(20):2135-47.
51. Pavey S, Johansson P, Packer L, Taylor J, Stark M, Pollock PM, et al.Microarray expression profiling in melanoma reveals a BRAF muta-tion signature. Oncogene 2004;23:4060-7.
52. Patton EE, Widlund HR, Kutok JL, Kopani KR,Amatruda JF, MurpheyRD et al. BRAF mutations are sufficient to promote nevi formation andcooperate with p53 in the genesis of melanoma. Curr Biol 2005;15:249-54.
53. Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A et al.BRAF mutation predicts sensitivity to MEK inhibition. Nature2006;439:358-62.
54. Grbovic OM, Basso AD, Sawai A, Ye Q, Friedlander P, Solit D et al.V600E B-Raf requires the Hsp90 chaperone for stability and is degra-ded in response to Hsp90 inhibitors. Proc Natl Acad Sci U S A2006;103:57-62.
55. Takata M, Goto Y, Ichii N,Yamaura M, Murata H, Koga H et al. Con-stitutive activation of the mitogen-activated protein kinase signalingpathway in acral melanomas. J Invest Dermatol 2005;125:318-22.
56. Hattori M, Minato N. Rap1 GTPase: functions, regulation, and mali-gnancy. J Biochem (Tokyo) 2003;134:479-84.
57. Gao L, Feng Y, Bowers R, Becker-Hapak M, Gardner J, Council L etal. Ras-associated protein-1 regulates extracellular signal-regulatedkinase activation and migration in melanoma cells: two processesimportant to melanoma tumorigenesis and metastasis. Cancer Res2006;66:7880-8.
58. Apicelli AJ, Uhlmann EJ, Baldwin RL, Ding H, Nagy A, Guha A etal. Role of the Rap1 GTPase in astrocyte growth regulation. Glia2003;42:225-34.
59. Lau N, Uhlmann EJ, Von Lintig FC, Nagy A, Boss GR, Gutmann DHet al. Rap1 activity is elevated in malignant astrocytomas independentof tuberous sclerosis complex-2 gene expression. Int J Oncol2003;22:195-200.
60. Gutmann DH, Saporito-Irwin S, DeClue JE, Wienecke R, Guha A.Alterations in the rap1 signaling pathway are common in human glio-mas. Oncogene 1997;15:1611-6.
61. Altschuler DL, Ribeiro-Neto F. Mitogenic and oncogenic propertiesof the small G protein Rap1b. Proc Natl Acad Sci U S A 1998;95:7475-9.
62. Zhang Z, Mitra RS, Henson BS, Datta NS, McCauley LK, Kumar Pet al. Rap1GAP inhibits tumor growth in oropharyngeal squamous cellcarcinoma. Am J Pathol 2006;168:585-96.
63. Fischer U, Meltzer P, Meese E. Twelve amplified and expressed geneslocalized in a single domain in glioma. Hum Genet 1996;98:625-8.
64. Wienecke R, Konig A, DeClue JE. Identification of tuberin, the tube-rous sclerosis-2 product. Tuberin possesses specific Rap1GAP acti-vity. J Biol Chem 1995;270:16409-14.
65. Ishida D, Kometani K,Yang H, Kakugawa K, Masuda K, Iwai K et al.Myeloproliferative stem cell disorders by deregulated Rap1 activationin SPA-1-deficient mice. Cancer Cell 2003;4:55-65.
66. Goldstein AM, Goldin LR, Dracopoli NC, Clark WH, Jr., TuckerMA. Two-locus linkage analysis of cutaneous malignant melano-ma/dysplastic nevi. Am J Hum Genet 1996;58:1050-6.
67. Dracopoli NC, Harnett P, Bale SJ, Stanger BZ, Tucker MA, Hou-sman DE et al. Loss of alleles from the distal short arm of chromosome1 occurs late in melanoma tumor progression. Proc Natl Acad Sci US A 1989;86:4614-8.
68. Tsao H, Zhang X, Majewski P, Haluska FG. Mutational and expres-sion analysis of the p73 gene in melanoma cell lines. Cancer Res1999;59:172-4.
69. Wu H, Goel V, Haluska FG. PTEN signaling pathways in melano-ma. Oncogene 2003;22:3113-22.
70. Isshiki K, Elder DE, Guerry D, Linnenbach AJ. Chromosome 10 alle-lic loss in malignant melanoma. Genes Chromosomes Cancer1993;8:178-84.
71. Guldberg P, thor Straten P, Birck A, Ahrenkiel V, Kirkin AF, ZeuthenJ. Disruption of the MMAC1/PTEN gene by deletion or mutation isa frequent event in malignant melanoma. Cancer Res 1997;57:3660-3.
72. Tsao H, Zhang X, Benoit E, Haluska FG. Identification ofPTEN/MMAC1 alterations in uncultured melanomas and melanomacell lines. Oncogene 1998;16:3397-402.
73. Stahl JM, Sharma A, Cheung M, Zimmerman M, Cheng JQ, Bosen-berg MW et al. Deregulated Akt3 activity promotes development ofmalignant melanoma. Cancer Res 2004;64:7002-10.
74. Dai DL, Martinka M, Li G. Prognostic significance of activated Aktexpression in melanoma: a clinicopathologic study of 292 cases. JClin Oncol 2005;23:1473-82.
75. Haluska FG, Tsao H, Wu H, Haluska FS, Lazar A, Goel V. Genetic alte-rations in signaling pathways in melanoma. Clin Cancer Res 2006;12(7Pt 2):2301s-07s.
76. Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: thePI3K pathway as an integrator of multiple inputs during tumorigenesis.Nat Rev Cancer 2006;6:184-92.
77. Bader AG, Kang S, Zhao L, Vogt PK. Oncogenic PI3K deregulatestranscription and translation. Nat Rev Cancer 2005;5:921-9.
78. Govindarajan B, Sligh JE, Vincent BJ, Li M, Canter JA, Nickoloff BJet al. Overexpression of Akt converts radial growth melanoma to ver-tical growth melanoma. J Clin Invest 2007;117:719-29.
79. Levy C, Khaled M, Fisher DE. MITF: master regulator of melanocytedevelopment and melanoma oncogene. Trends Mol Med 2006;12:406-14.
80. Steingrimsson E, Moore KJ, Lamoreux ML, Ferre-D’Amare AR,

GAO THE MOLECULAR MECHANISMS OF MELANOMA TUMORIGENESIS
82 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
Burley SK, Zimring DC et al. Molecular basis of mouse microphthal-mia (mi) mutations helps explain their developmental and phenotypicconsequences. Nat Genet 1994;8:256-63.
81. Loercher AE, Tank EM, Delston RB, Harbour JW. MITF links diffe-rentiation with cell cycle arrest in melanocytes by transcriptional acti-vation of INK4A. J Cell Biol 2005;168:35-40.
82. Carreira S, Goodall J, Aksan I, La Rocca SA, Galibert MD, Denat Let al. Mitf cooperates with Rb1 and activates p21Cip1 expression toregulate cell cycle progression. Nature 2005;433:764-9.
83. Wellbrock C, Marais R. Elevated expression of MITF counteracts B-RAF-stimulated melanocyte and melanoma cell proliferation. J CellBiol 2005;170:703-8.
84. Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Rama-swamy S et al. Integrative genomic analyses identify MITF as a linea-ge survival oncogene amplified in malignant melanoma. Nature2005;436:117-22.
85. Carreira S, Goodall J, Denat L, Rodriguez M, Nuciforo P, Hoek KSet al. Mitf regulation of Dia1 controls melanoma proliferation andinvasiveness. Genes Dev 2006;20:3426-39.
86. Carreira S, Liu B, Goding CR. The gene encoding the T-box factorTbx2 is a target for the microphthalmia-associated transcription fac-tor in melanocytes. J Biol Chem 2000;275:21920-7.
87. Vance KW, Carreira S, Brosch G, Goding CR. Tbx2 is overex-pressed and plays an important role in maintaining proliferation
and suppression of senescence in melanomas. Cancer Res2005;65:2260-8.
88. Du J, Widlund HR, Horstmann MA, Ramaswamy S, Ross K, HuberWE et al. Critical role of CDK2 for melanoma growth linked to itsmelanocyte-specific transcriptional regulation by MITF. Cancer Cell2004;6:565-76.
89. Busca R, Berra E, Gaggioli C, Khaled M, Bille K, Marchetti B et al.Hypoxia-inducible factor 1{alpha} is a new target of microphthal-mia-associated transcription factor (MITF) in melanoma cells. J CellBiol 2005;170:49-59.
90. McGill GG, Haq R, Nishimura EK, Fisher DE. c-Met expression isregulated by Mitf in the melanocyte lineage. J Biol Chem2006;281:10365-73.
91. Kim M, Gans JD, Nogueira C, Wang A, Paik JH, Feng B et al. Com-parative oncogenomics identifies NEDD9 as a melanoma metastasisgene. Cell 2006;125:1269-81.
92. Bastian BC, LeBoit PE, Hamm H, Brocker EB, Pinkel D. Chromo-somal gains and losses in primary cutaneous melanomas detected bycomparative genomic hybridization. Cancer Res 1998;58:2170-5.
93. Namiki T,Yanagawa S, Izumo T, Ishikawa M, Tachibana M, Kawaka-mi Y et al. Genomic alterations in primary cutaneous melanomasdetected by metaphase comparative genomic hybridization with lasercapture or manual microdissection: 6p gains may predict poor outcome.Cancer Genet Cytogenet 2005;157:1-11.

G ITAL DERMATOL VENEREOL 2007;142:83-97
Current perspectives on the pathologic diagnosis and reportingof melanocytic tumors
One of the major goals in the pathologic evaluation of mela-nocytic tumors is to distinguish benign lesions (nevi) from mali-gnant ones (melanomas). Although this may appear to be asimple task, the assessment of melanocytic tumors can be oneof the most difficult of all areas of pathologic diagnosis. Histo-logic features favoring a benign diagnosis are symmetry, cir-cumscription, a V-shaped silhouette, dermal maturation andabsence of mitoses. Poor circumscription, lateral or peripheralpagetoid epidermal spread, expansile dermal growth, high cel-lularity, necrosis, nuclear pleomorphism and frequent or abnor-mal mitoses raise the possibility of malignancy. Although mosttumors are readily diagnosable with the application of standardcriteria, there are unusual variants of both nevi and melanomasthat require awareness of pathologic diagnostic pitfalls andcareful clinicopathologic correlation to avoid misdiagnosis.The performance of a complete excision biopsy, wherever pos-sible, is crucial for the optimal pathologic assessment of chal-lenging lesions. For melanomas, the pathology report shouldinclude all pathologic factors that are important in determiningthe patient’s prognosis and management. The use of a format-ted synoptic pathology report can facilitate this and presentthe required information to the clinician clearly. The use ofthe sentinel lymph node biopsy is now well established in themanagement of patients with melanoma, as is the usefulness offine needle biopsy in the assessment of clinically-suspectedmetastatic melanoma. Determining the prognosis of patientswith melanoma requires integration of all pertinent clinico-
pathologic features and this may be aided by the use of com-puter programs in the future. Molecular diagnostic techniquesmay further refine the diagnosis of challenging lesions andprovide more accurate prognostic estimates. KEY WORDS: Nevus, pathology - Nevus, diagnosis - Dermoscopy -Biopsy, fine-needle - Melanoma.
Melanocytic tumors are amongst the most commontumors that occur in humans. Corresponding-
ly, excised skin specimens bearing melanocytic lesionsare commonly encountered in the surgical pathologylaboratory. They are usually removed either for cos-metic reasons, or for the purpose of making or exclud-ing a diagnosis of melanoma. In the vast majority ofinstances, the pathologic diagnosis of these lesions isstraightforward, but in some cases, diagnosis can beextremely challenging for several reasons. For exam-
1Department of Anatomical PathologyRoyal Prince Alfred Hospital, Sydney, Australia
2Department of Pathology, Yong Loo Lin School of Medicine,National University of Singapore
National University Hospital, Singapore3Sydney Melanoma Unit, Sydney Cancer CentreRoyal Prince Alfred Hospital, Sydney, Australia 4Melanoma and Skin Cancer Research Institute
Sydney, Australia 5Discipline of Pathology, Faculty of Medicine
The University of Sydney, Sydney, Australia6Discipline of Surgery, Faculty of MedicineThe University of Sydney, Sydney, Australia
Acknowledgements.—Dr R. Murali and Dr C. Arnold are supported bythe Cancer Institute NSW Clinical Fellowship Program. Dr K-B Tan is sup-ported by the China Medical Board Fellowship, the National University ofSingapore and the National University Hospital, Singapore.
Address reprint requests to: Prof. R. Scolyer, Department of Anatomi-cal Pathology, Royal Prince Alfred Hospital, Missenden Road, Camper-down, N.S.W. 2050 Australia. E-mail: [email protected]
K.-B. TAN 1, 2, R. MURALI 1, 3, 4, 5, J. F. THOMPSON 3, 4, 6,C. J. ARNOLD 1,3, 4, S. W. MCCARTHY 1, 3, 4, R. A. SCOLYER 1, 3, 4, 5
Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 83
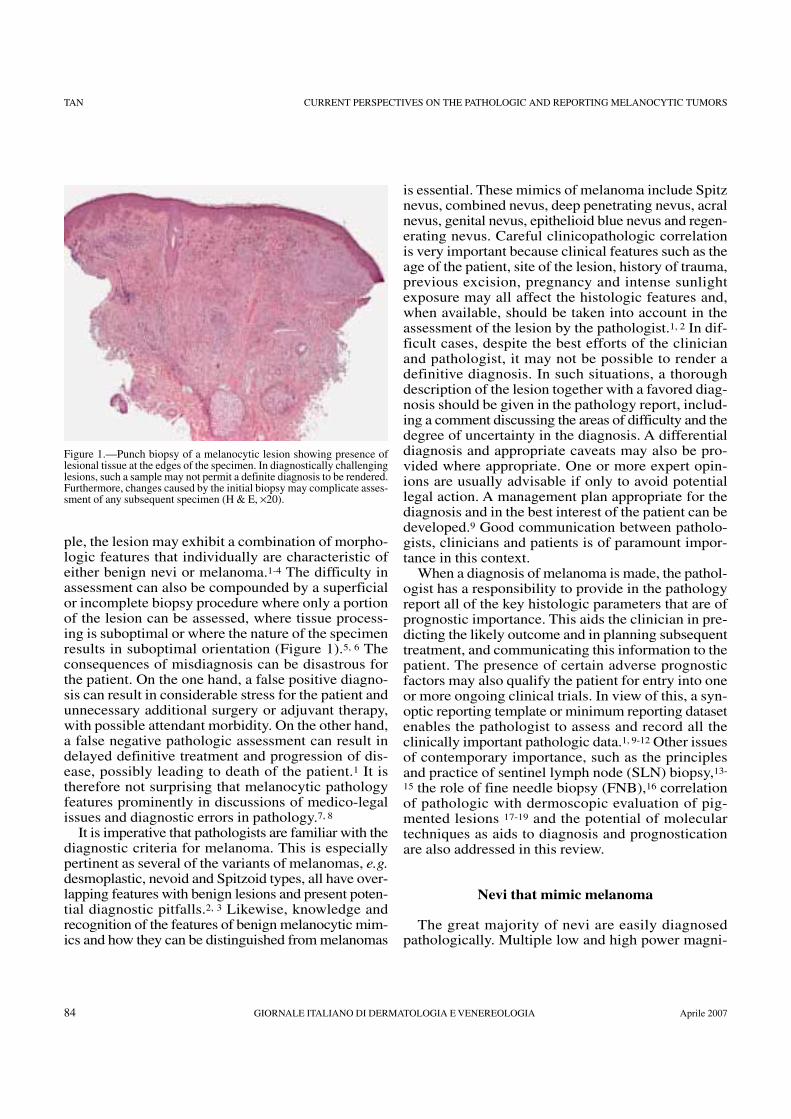
TAN CURRENT PERSPECTIVES ON THE PATHOLOGIC AND REPORTING MELANOCYTIC TUMORS
84 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
ple, the lesion may exhibit a combination of morpho-logic features that individually are characteristic ofeither benign nevi or melanoma.1-4 The difficulty inassessment can also be compounded by a superficialor incomplete biopsy procedure where only a portionof the lesion can be assessed, where tissue process-ing is suboptimal or where the nature of the specimenresults in suboptimal orientation (Figure 1).5, 6 Theconsequences of misdiagnosis can be disastrous forthe patient. On the one hand, a false positive diagno-sis can result in considerable stress for the patient andunnecessary additional surgery or adjuvant therapy,with possible attendant morbidity. On the other hand,a false negative pathologic assessment can result indelayed definitive treatment and progression of dis-ease, possibly leading to death of the patient.1 It istherefore not surprising that melanocytic pathologyfeatures prominently in discussions of medico-legalissues and diagnostic errors in pathology.7, 8
It is imperative that pathologists are familiar with thediagnostic criteria for melanoma. This is especiallypertinent as several of the variants of melanomas, e.g.desmoplastic, nevoid and Spitzoid types, all have over-lapping features with benign lesions and present poten-tial diagnostic pitfalls.2, 3 Likewise, knowledge andrecognition of the features of benign melanocytic mim-ics and how they can be distinguished from melanomas
is essential. These mimics of melanoma include Spitznevus, combined nevus, deep penetrating nevus, acralnevus, genital nevus, epithelioid blue nevus and regen-erating nevus. Careful clinicopathologic correlationis very important because clinical features such as theage of the patient, site of the lesion, history of trauma,previous excision, pregnancy and intense sunlightexposure may all affect the histologic features and,when available, should be taken into account in theassessment of the lesion by the pathologist.1, 2 In dif-ficult cases, despite the best efforts of the clinicianand pathologist, it may not be possible to render adefinitive diagnosis. In such situations, a thoroughdescription of the lesion together with a favored diag-nosis should be given in the pathology report, includ-ing a comment discussing the areas of difficulty and thedegree of uncertainty in the diagnosis. A differentialdiagnosis and appropriate caveats may also be pro-vided where appropriate. One or more expert opin-ions are usually advisable if only to avoid potentiallegal action. A management plan appropriate for thediagnosis and in the best interest of the patient can bedeveloped.9 Good communication between patholo-gists, clinicians and patients is of paramount impor-tance in this context.
When a diagnosis of melanoma is made, the pathol-ogist has a responsibility to provide in the pathologyreport all of the key histologic parameters that are ofprognostic importance. This aids the clinician in pre-dicting the likely outcome and in planning subsequenttreatment, and communicating this information to thepatient. The presence of certain adverse prognosticfactors may also qualify the patient for entry into oneor more ongoing clinical trials. In view of this, a syn-optic reporting template or minimum reporting datasetenables the pathologist to assess and record all theclinically important pathologic data.1, 9-12 Other issuesof contemporary importance, such as the principlesand practice of sentinel lymph node (SLN) biopsy,13-
15 the role of fine needle biopsy (FNB),16 correlationof pathologic with dermoscopic evaluation of pig-mented lesions 17-19 and the potential of moleculartechniques as aids to diagnosis and prognosticationare also addressed in this review.
Nevi that mimic melanoma
The great majority of nevi are easily diagnosedpathologically. Multiple low and high power magni-
Figure 1.—Punch biopsy of a melanocytic lesion showing presence oflesional tissue at the edges of the specimen. In diagnostically challenginglesions, such a sample may not permit a definite diagnosis to be rendered.Furthermore, changes caused by the initial biopsy may complicate asses-sment of any subsequent specimen (H & E, ×20).

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 85
CURRENT PERSPECTIVES ON THE PATHOLOGIC AND REPORTING MELANOCYTIC TUMORS TAN
fication microscopic features are assessed, enablinga diagnosis to be made in most cases.2 Features favor-ing benignity are symmetry, circumscription, pres-ence of maturation, uniform small nuclei, absence ofdermal mitoses and absence of pagetoid intraepidermalinvasion. Features that raise the possibility of malig-nancy include asymmetry and poor circumscription,poor maturation, frequent, abnormal or deep dermalmitoses, lateral pagetoid intraepidermal spread, nuclearpleomorphism, expansile or sheet-like cellular growthpattern in the dermis, tumor necrosis and vascularinvasion.2 None of these morphologic features in iso-lation is diagnostic of a benign or malignant lesion.Definitive pathologic assessment requires a consider-ation of all the microscopic features in a particularcase and a comparison of these with standard diag-nostic criteria. The importance of clinicopathologiccorrelation cannot be overemphasized. For partialbiopsies, including punch and some shave biopsies,if the clinical and pathologic features are not compat-ible, consideration should be given to performing afurther biopsy.5
Certain characteristic nevi are known to present adiagnostic pitfall to both the clinician and the pathol-ogist as they may exhibit features typical of benignlesions but also possess worrying features seen inmalignant lesions. It is important to regard all the fea-tures observed in a particular case within the appro-priate context of these challenging melanocytic lesions.In such cases a credible diagnosis can only be estab-lished following assessment of all architectural andcytologic features, and features of the host response andcorrelated with clinical data including site of the lesionand age of the patient. It requires a sound knowledgeof diagnostic criteria, a keen awareness of potentialpitfalls, and finally, the ability to make a logical andwell reasoned judgement on the basis of all the infor-mation available.
Dysplastic nevus
The dysplastic nevus takes up an intermediate posi-tion between the common nevus and melanoma inboth morphology and underlying genetic changes (Fig-ure 2).3, 20 The presence of a dysplastic nevus is also amarker for an increased risk of developing a melanoma,especially in the context of the dysplastic nevus syn-drome.11 Clinically, there is some degree of asymme-try, mild border irregularity and color variegation. His-tologically, the dysplastic nevus is characterized by 2major and 4 minor criteria. The major criteria are: 1)
a proliferation of atypical and primarily basally-locat-ed epidermal melanocytes, and 2) the melanocyticproliferation takes the form of either a lentiginousgrowth pattern or an epithelioid and nested growthpattern. The presence of papillary dermal lamellarfibroplasia, increased upper dermal vascularity, a lym-phocytic infiltrate and fusion of adjacent junctionalmelanocytic nests are minor criteria. It is usually nec-essary for the lesion to include a total of at least 2major and 2 minor criteria for the pathologic diagno-sis of a dysplastic nevus.3, 20
Melanoma in situ (MIS) is pathologically distin-guished from dysplastic nevus by the presence of moremarked cytologic atypia, more florid intraepidermalpagetoid spread of single atypical melanocytes (oftenreaching the granular layer) and greater architecturaldisorder.
Pigmented spindle cell nevus/pigmented epithelioidcell nevus
This heavily pigmented nevus is centered in thejunctional zone and is another notable mimic ofmelanoma both clinically and pathologically.21, 22 Theremay occasionally be a superficial dermal componentas well. Features that are often present and that maycause some concern include the presence of pagetoidintraepidermal spread, mild-to-moderate cellular atyp-ia and heavy pigmentation. The lesion is, however,typified by symmetry, circumscription, absence ofmarked cytologic atypia, rapid maturation with depthof the dermal component and the absence of frequentor abnormal mitoses.
Figure 2.—Compound dysplastic nevus. There is fusion of adjacent nevo-melanocytic nests and lamellar dermal fibrosis (H & E, ×100).

TAN CURRENT PERSPECTIVES ON THE PATHOLOGIC AND REPORTING MELANOCYTIC TUMORS
86 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
“Irritated” nevus
The histologic features of a common nevus can bemarkedly modified by concurrent skin pathology.2Nevi that have been subjected to trauma, strong sun-light, or surface irritant agents may show features suchas pagetoid intraepidermal spread, mild cellular atyp-ia and occasional mitotic figures. In such cases, cor-relation with clinical information is often critical inarriving at a correct (benign) diagnosis. Histological-ly, the presence of parakeratosis, hypergranulosis andepidermal acanthosis provide hints that there was pres-ence of a superimposed irritant etiology. Nevi that arepresent on skin afflicted by eczema (Meyerson nevus)may also display similar features. Again, clinical cor-relation is important, however, the diagnosis wouldbe supported by the histologic presence of epidermalspongiosis and an inflammatory infiltrate composed oflymphocytes and some eosinophils.
Regenerating nevus
A regenerating nevus is one that recurs after phys-ical damage in particular following previous incompleteexcision.5, 6 It may have alarming features and hasbeen termed ‘pseudomelanoma’.23-25 Such featuresinclude pagetoid intraepidermal spread, some degreeof cellular atypia and an occasional mitotic figure. Animportant clue to the diagnosis is the presence of a
dermal scar in the vicinity of the regenerating nevuscells. On closer examination, the reactive-appearingregenerating nevus cells can sometimes be seen tointerface and merge with the pre-existing banal nevuscells. This is in contrast to melanoma occurring in thecontext of a preexisting nevus where the malignantcells stand out as a distinct population featuring highcellularity, nuclear pleomorphism, prominent nucleoliand frequent mitoses.
Acral nevus
Nevi occurring on the palms, sole, fingers and toesoften display features that would favor a melanomawhen present in a melanocytic tumor occurring atanother site. The most notable of these is pagetoidintraepidermal spread (Figure 3).26-29 In our experi-ence, this feature can be present in up to 71% of acralnevi.30 Intraepidermal spread is also seen in acrallentiginous melanoma in situ (MIS), but the pagetoidspread in the latter is usually denser and more hap-hazard (Figure 4). The pagetoid cells in MIS are usu-ally markedly atypical while those in acral nevi arebland or pyknotic. Other features that favor acral neviover acral melanomas include greater likelihood toform junctional hests of melanocytes and a paucity ofjunctional and subjunctional lumphocytes.30
Many cases of acral nevi also show poor circum-scription and asymmetry. The latter two features, how-
Figure 3.—Acral nevus. There is presence of pagetoid intraepidermalspread of melanocytes. Note that the lesional cells show only show mildatypia (H & E, ×100).
Figure 4.—Acral lentiginous melanoma in situ. There is a lentiginousgrowth pattern of moderately to severely atypical melanocytes and focalpagetoid intraepidermal spread (H & E, ×100).

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 87
CURRENT PERSPECTIVES ON THE PATHOLOGIC AND REPORTING MELANOCYTIC TUMORS TAN
ever, may be related to the direction/plane the sectionsof such skin lesions have been taken for pathologicexamination. In a study on acral nevi by Signoretti etal., such features were less frequently encounteredwhen sections were taken perpendicular to the skindermatoglyphics in contrast to when sections weretaken parallel to the markings.29 The former thereforeis the recommended way to process acral skin speci-mens bearing pigmented lesions.
Spitz nevus
This nevus is one of the most notable of melanomaimposters (Figures 5 A, B).31, 32 Clinically, it presentsas a dome-shaped, pale, red or pigmented skin nodule,usually in young patients. Histologically, the tumoris usually a circumscribed junctional or compoundmelanocytic proliferation of more or less uniform largecells which can be epithelioid or spindle, the latteroften orientated in a plane perpendicular to the over-lying epidermis, giving rise to a ‘raining-down’appear-ance. The overlying epidermis typically shows acan-thosis and variable hypergranulosis. Many cases exhib-it some nuclear pleomorphism, dermal mitoses andpagetoid epidermal spread, features which raise con-cern for melanoma. In contrast to melanoma exhibit-ing Spitz nevus-like morphology (so-called ‘Spitzoidmelanoma’), Spitz nevi lack atypical mitotic figures,contain fewer than 2 mitoses per mm2 and lack mitosesnear the deep edge of the lesion. Pagetoid spread may
be seen in the central part of otherwise typical Spitznevi, but is a cause for concern when occurring at theperipheral edge of the lesion.33 Other features thatsupport a diagnosis of Spitz nevus are the presence ofdermal maturation, clustered and sizable Kamino bod-ies, terminal junctional nests, cell uniformity in a hor-izontal plane across the lesion HMB-45 negativity anda low Ki67 proliferative index (<5% of melanocyticcells) in the dermal component of the lesion.34
Spitz nevus-like lesions with atypical features (so-called ‘atypical Spitzoid tumors’) show overlappingfeatures between Spitz nevus and melanoma.2 It iswell documented that it is not possible to predict withcertainty the biologic behavior of such lesions fromtheir pathologic features. These difficulties were high-lighted in a disturbing study involving the assessmentof 30 Spitzoid melanocytic tumors by 10 internation-al authorities; 17 cases yielded no clear consensusdiagnosis, in only one case did 6 or more of the pathol-ogists agree on the diagnosis, and some lesions that ulti-mately proved fatal were categorized as benign bymost of the pathologists.35 In a recent study from theSydney Melanoma Unit, distinction between atypicalSpitz tumors exhibiting and lacking sentinel lymphnode metastases was not possible on the basis of his-tologic features (unpublished data). This study foundthat SLN biopsy offers a means of assessing themetastatic potential of atypical Spitzoid tumors andaids in the management of these patients by selectingpatients who may benefit from a regional node field dis-
Figure 5.—A) Spitz nevus: at low magnification power, the lesion appearssymmetrical, is well circumscribed and shows epidermal hyperplasia anddermal maturation. (H & E, ×20). B) Spitz nevus. At higher magnificationpower, the cells have enlarged but uniform nuclei and show the typical‘raining-down’ appearance. (H & E, ×100).

TAN CURRENT PERSPECTIVES ON THE PATHOLOGIC AND REPORTING MELANOCYTIC TUMORS
88 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
section and those in whom the use of adjuvant thera-pies could be considered. The recommended clinicalmanagement for atypical Spitzoid tumors is for com-plete excision and careful follow up. At the SydneyMelanoma Unit, patients with such tumors that are >1mm in thickness are offered sentinel lymph node biop-sy as a form of prognostication.
Blue nevus
Dendritic blue nevus and deep penetrating nevususually presents clinically as a heavily pigmentedsmooth-surfaced nodule. In the cellular variant of bluenevus, the dermal proliferation of plump ovoid andspindled nevus cells are typically admixed with heav-ily pigmented melanophages and sometimes brancheddendritic melanocytes, and forms nodular masses,often with a dumb-bell shaped configuration.35 It maypossess worrying features including high cellularity,nuclear hyperchromasia and the presence of occa-sional dermal mitotic figures. Given its dermal location,the differential diagnosis includes metastatic melanomawhich on rare occasions may be difficult to distin-guish.37 In contrast to cellular blue nevus, melanoma(whether primary or metastatic) can usually be iden-tified by the presence of marked nuclear pleomor-phism, frequent mitoses, necrosis, a lymphocytic infil-trate, and with clinicopathologic correlation.
The epithelioid blue nevus is a variant associatedwith Carney myxoma syndrome.38 Histologically, there
are pigmented epithelioid melanocytic cells with raremitoses admixed with branched dendritic cells. Thelesional cells are often HMB-45 positive. The absenceof marked nuclear pleomorphism and the lack of fre-quent or abnormal mitoses argue against a diagnosis ofmelanoma. Even so, there is some overlap of histo-logic features between this lesion and two other enti-ties: animal-type melanoma and pigmented epithe-lioid melanocytoma (vide infra).1, 2
Deep penetrating nevus
This lesion is thought to represent a distinct entitybut with some overlapping features with blue nevus andSpitz nevus.39-41 Deep penetrating nevus is character-ized by a wedge-shaped histologic profile with thebase towards the epidermis and the apex directeddeeply, and composed of epithelioid and spindle-shaped lesional cells with prominent admixed pig-ment-containing melanophages (Figure 6 A, B). Thenuclei are often enlarged and may contain character-istic nuclear pseudoinclusions. The lesion has a ten-dency to surround skin adnexal structures and neu-rovascular bundles and may feature a rare mitosis.Immunohistochemically, the lesional cells are typi-cally HMB-45 positive. Awareness of this entityenables it to be differentiated from melanoma whichcommonly exhibits asymmetry, expansile dermalgrowth, nuclear pleomorphism and frequent mitoses.
Figure 6.—A) Deep penetrating nevus: at low magnification power, the lesion shows a wedge-shaped profile (H & E, ×20). B) Deep penetratingnevus. The lesional cells display moderate degree of nuclear pleomorphism and occasional intranuclear inclusions. Admixed melanin-containingmacrophages are present (H & E, ×100).

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 89
CURRENT PERSPECTIVES ON THE PATHOLOGIC AND REPORTING MELANOCYTIC TUMORS TAN
Combined nevus
The heterogeneity of the cellular components ofthis nevus may give rise to a variegated and worryingclinical appearance, prompting its biopsy.42, 43 Histo-logically, the lesion contains at least 2 nevomelanocyticcomponents (Figure 7). It typically partners a com-mon nevus with a deep penetrating nevus (62% of cas-es in one study 43), blue nevus or Spitz nevus. As aresult of the admixture of different populations ofnevomelanocytic cells and the asymmetry that oftenresults, this lesion has been a source of histologic mis-diagnosis. This is compounded by the increased cel-lularity and sometimes atypical nuclear features ofthe secondary component and the presence of an occa-sional dermal mitotic figure. Knowledge of this enti-ty and the assessment of the lesion using standard cri-teria should enable it to be accurately classified.
Cellular nodule in congenital nevus
A congenital nevus may occasionally include a local-ized expansile proliferation of a nodule of tumor cells,featuring enlarged cells with atypia and the presenceof mitotic figures.44, 45 Not unexpectedly, such nod-ules may be misinterpreted as melanoma superven-ing in a nevus. Genetic studies of such cellular noduleshave shown results supporting the benignity of thelesion.46, 47 Pathologic features favoring a malignant
lesion are the presence of marked nuclear pleomor-phism, prominent nucleoli, poor circumscription,abrupt transition between the cells in the nodule and theadjacent banal nevus cells (rather than merging of thetwo components) frequent and atypical mitoses andthe presence of necrosis.2 Particularly difficult caseswith ambiguous morphologic features may be desig-nated as an atypical nodular proliferation within a con-genital nevus with a recommendation for completeexcision and close follow-up.
Pigmented epithelioid melanocytoma
This is probably a heterogeneous group ofmelanocytic lesions first characterized by Zembowiczet al.48 In that series, the entity encompassed lesionsoriginally reported as epithelioid blue nevus and ani-mal-type melanoma. Histologically, the tumor is situ-ated in the dermis and composed of heavily-pigment-ed epithelioid and spindle cells (Figure 8). Althoughmost cases behaved in an indolent manner, 46% ofcases showed metastatic deposits in regional lymphnodes. On histology, it was not possible to segregate themetastasizing and non-metastasizing tumors. A reviewof similar cases from the Sydney Melanoma Unit alsoshowed overlapping features with epithelioid bluenevus and animal-type melanoma, corroborating theobservations of Zembowicz et al., although the fre-
Figure 7.—Combined nevus. Admixture of a deep penetrating nevus com-ponent (left side) and a common nevus component (right side) noted (H &E, ×400).
Figure 8.—Pigmented epithelioid melanocytoma. Low power magnifica-tion view showing a symmetrical dermally-located heavily pigmentedtumor with overlying epidermal hyperplasia (H & E, ×20).

TAN CURRENT PERSPECTIVES ON THE PATHOLOGIC AND REPORTING MELANOCYTIC TUMORS
90 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
quency of lymph node metastases appeared to be low-er.49 The behavior and clinical outcome of these lesionsare yet to be resolved and further studies with long termfollow-up are needed to further characterize them, themalignant potential of which is at present, considered tobe uncertain.
Potentially problematic variants of melanoma
Although most cases of melanoma show overt malig-nant features, there are a few notable variants whichmimic benign melanocytic and non-melanocytictumors. Awareness of these entities and their respec-tive features will help in their recognition.
Nevoid melanoma
Nevoid melanoma mimics the common nevus byappearing circumscribed and having a variable, oftenscant, intraepidermal component.50, 51 Although dermalmaturation appears to be present, closer examinationreveals pleomorphic cells in the deep aspect of thetumor with nuclear hyperchromasia and often promi-nent nucleoli. Mitoses are almost always present andare often frequent and with abnormal forms. There isoften expansion of the nests of tumor cells in the der-mis. The overlying epidermis may be attenuated withrete ridges that may be elongated or lost.
Desmoplastic melanoma
Desmoplastic melanoma usually presents clinical-ly as a firm skin nodule or plaque in the sun-exposedskin of elderly patients.52-54 Histologically, the tumorshows a variably cellular dermal proliferation of oftenelongated spindle cells with hyperchromatic nucleiand sometimes prominent nucleoli. The spindle cellsinfiltrate between dermal collagen bundles and in thisregard, the tumor closely mimics scar tissue (Figure 9).Closer inspection reveals rare, sparse or frequentmitoses and the characteristic presence of lympho-cytic aggregates and perineural invasion. Althoughnot present in all cases, the presence of an atypicaljunctional melanocytic component, as well as the char-acteristic lymphoid aggregates are valuable clues to thediagnosis.
As the differential diagnosis of desmoplasticmelanoma includes scar tissue, in re-excision speci-mens for cutaneous melanoma, it is important that thepathologist is made aware if the original tumor was adesmoplastic melanoma. Close examination for theabove-mentioned characteristics and use of S-100 pro-tein immunohistochemistry should enable detectionof any residual tumor and assessment of the status ofthe excision margins. However, it is important to notethat this variant of melanoma is usually negative for theother traditional melanoma markers such as HMB-45and Melan-A (unless epithelioid cells are present) and,furthermore, that the fibroblasts in scar tissue may bepositive for S100.55, 56
Regressing/regressed melanoma
A regressing melanoma may show a paucity oflesional cells, which are often concealed or obscuredby a lymphocytic infiltrate and fibroblastic tissue.2 Ithas to be distinguished from other regressing cuta-neous tumors, e.g. basal cell carcinoma and Merkelcell carcinoma. The presence of an atypical junction-al melanocytic component may provide a clue to aregressed melanoma. In this regard, examination offurther blocks or levels of tissue may be warranted. Ifa small residual cellular tumor is present, S-100, HMB-45 and Melan-A immunohistochemistry may helpdelineate melanoma. Basal cell carcinoma is positivefor Ber-EP4 while dot-like positivity for cytokeratin 20characterizes Merkel cell carcinoma. For tumors thatare completely regressed where only fibroblastic tis-sue and melanin containing macrophages (melano-
Figure 9.—Desmoplastic melanoma. There are infiltrative atypical spind-le cells with an associated lymphocytic infiltrate (left side). Part of a hairfollicle is present on the right side of the photomicrograph (H & E, ×200).

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 91
CURRENT PERSPECTIVES ON THE PATHOLOGIC AND REPORTING MELANOCYTIC TUMORS TAN
phages) remain, it may be impossible to determinethe nature of the regressed tumor with certainty sinceregressed pigmented basal cell carcinomas, melanomasand some other tumors may result in a similar mor-phologic appearance.
A diagnostic approach
The histologic evaluation of a melanocytic lesionmust take into account a constellation of pathologic fea-tures including a range of architectural and cytologicfeatures and features of the host response. There existsan effective strategy for the pathologist to first assessthe histology of a melanocytic lesion without priorknowledge of the clinical history. All the pertinent his-tologic features are sought and on balance, a provi-sional diagnosis and relevant differential diagnosesare arrived at. The clinical features are then noted orsought for, correlated with the morphologic appear-ances, and the diagnosis refined with re-examinationof the histology if necessary. This strategy of initial‘blinded’ histologic assessment without the bias ofclinical foreknowledge has been effectively appliedin other areas of histopathology, e.g. liver core biopsyinterpretation.57 As there are quite a number of histo-logic features that need to be examined and taken intoaccount for the overall diagnostic impression of amelanocytic lesion, there is a strong case for an initialobjective and comprehensive diagnostic assessment.Final clinicopathologic appraisal then enables a sounddiagnosis to be made.
Handling difficult melanocytic lesions
Despite the faithful use of established criteria in thediagnostic work-up of melanocytic tumors, there area small number of cases that will prove difficult evenfor the experienced pathologist. A practical way ofcategorizing such difficult melanocytic lesions intotwo main groups has been proposed by Elder.58, 59 Thefirst group comprises lesions that are superficiallylocated in the epidermis, junctional zone and superfi-cial papillary dermis. These include dysplastic nevi,pigmented spindle cell nevi, junctional Spitz nevi,nevi of special sites (e.g. acral and genital) and someregenerating nevi. The malignant counterpart of suchsuperficial lesions is the radial growth phase melanoma,including MIS. When standard histologic criteria cou-
pled with clinicopathologic correlation have been uti-lized and yet a lesion in this category defies definiteclassification, a label of superficial melanocytic pro-liferation of uncertain significance may be appropri-ate. The most adverse outcome of such lesions is thatof local recurrence and, thus, complete excision withclear margins and clinical follow-up are recommend-ed.
The second tumor group is dermally-situated andcomprise atypical Spitz tumors and some variants ofblue nevus, including the cellular and epithelioidtypes, atypical deep penetrating nevus and pigment-ed epithelioid melanocytoma. The malignant coun-terpart of this group is the vertical growth phasemelanoma, a tumor that has significant metastaticcapability. In tackling an ambiguous or difficult lesionin this category, Elder proposes the term: melanocyt-ic tumor of uncertain malignant potential. In such asituation, following optimal clinician-pathologist andclinician-patient communication, there is a case for thepatient to be managed as per the guidelines for amalignant lesion. The pathologist’s report (vide infra)should address all of the parameters normally report-ed in a synoptic report for a melanoma. Excision witha margin of normal tissue is usually recommendedwith additional consideration for a sentinel lymphnode biopsy for lesions greater than 1mm in thick-ness.
The melanoma pathology report
The melanoma pathology report must contain allof the pathologic information that is deemed to benecessary to determine the optimal clinical manage-ment of the patient. Together with clinical staging, awell documented record of the pathologic features andparameters of the excised tumor provides the bestavailable information for the prediction of prognosis.1This will help the clinician to counsel the patient appro-priately on the need and extent of any further surgeryor adjuvant therapy that may be required.
To help in the documentation of all the requiredprognostic factors, there are a number of minimumdata sets or synoptic reporting templates that are avail-able.1, 10 By providing a checklist, it standardizes thereport format and minimizes the frequency with whichimportant information is omitted from the pathologyreport (unpublished data). In the busy clinics andwards, a synoptic reporting template will also facilitate

TAN CURRENT PERSPECTIVES ON THE PATHOLOGIC AND REPORTING MELANOCYTIC TUMORS
92 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
the expedient extraction of crucial prognostically-important information from the pathology report by theclinician. An example of a synoptic reporting tem-plate is provided (Table I).
Important issues in melanoma reporting
Tumor thickness
The Breslow thickness and Clark level are two of thetime-honored and easily assessed prognostic featuresof primary melanoma. The former is of greater prog-nostic value and is the primary determinant of thetumor (T) score in the AJCC staging system formelanoma.60, 61 Additionally, the Clark level has beenshown to stratify prognosis in patients with thinmelanomas (less than 1mm in thickness).62 In a studyinvestigating patients with melanoma 0.6 mm to 1.1mm thick, Clark level IV tumors had a distinctly poor-er prognosis.63
Ulceration
Ulceration is an adverse prognostic feature thatreflects the aggressiveness of melanoma.64 It has been
shown to be an important independent prognostic fac-tor and has been incorporated into the latest AJCCmelanoma staging system.61 Histologically, the ulcerhas to be in direct association with the tumor and mustbe distinguished from secondary ulceration due totrauma or other exogenous factors; such distinction isoften difficult, and correlation with the clinical histo-ry (e.g. history of trauma or prior topical treatment) mayhelp. The width of the ulcer is also held to be impor-tant, with a width of greater than 6mm being associatedwith a higher incidence of nodal metastasis.65
Dermal mitotic rate
Tumor mitotic rate has been shown to be an impor-tant predictor of survival in several studies.66-69 In astudy of 3 661 patients from the Sydney MelanomaUnit, the tumor mitotic rate was an independent pre-dictor of survival and emerged second to Breslowthickness and ahead of the presence of ulceration, in itsprognostic power.69
Mitotic rate is a conveniently measured light micro-scopic parameter. However, as for some other solidtumors, there may be significant intra- and inter-observ-er variability.70 With a standardized approach, thisvariability can be minimized. The ‘hot-spot’method isrecommended for assessment of the mitotic rate. Thehistologic section of the melanoma is first surveyed atlow magnification to find area(s) where mitoses arecommon. Such areas are examined at high magnifi-cation, and mitoses are counted and expressed as afigure per mm2.1 Using this technique, a recent studyfrom the Sydney Melanoma Unit showed that pathol-ogists with differing backgrounds and varying levelsof experience were able to attain excellent interob-server correlation for assessing tumor mitotic rate(intraclass correlation coefficient [ICC] 0.76).71 It isenvisaged that with more widespread use of stan-dardized assessment of mitotic rate, this parametermay be considered for incorporation into future revi-sions of the AJCC staging system.
Regression
Tumor regression has been held to be an adverseprognostic factor in melanoma, although not all stud-ies have supported this.72-74 Histologically, regressionis recognized by an absence of melanoma cells andreplacement by a lymphohistiocytic infiltrate and/orfibrosis. Regression can be graded into early, inter-
Pathologic feature
SexSiteDiagnosisHistologic subtypeVertical growth phaseBreslow thicknessUlceration (diameter in mm)Dermal mitotic rate (per mm2)Clark levelVascular or lymphatic invasionNeurotropismDesmoplasia (% of dermal invasive tumor)SatellitesFeatures of regression:
— early (TILs)
— intermediate (angiofibroplasia +/- TILs)— late (fibrosis and loss of rete ridges)
Predominant cell typeAssociated nevusNearest lateral margin to insitu componentNearest lateral margin to dermal invasive
componentDistance from tumor to deep marginSolar elastosis
TABLE I.—An example of a synoptic pathology report for a primarycutaneous melanoma.
Example
MaleLeft forearmMelanomaSuperficial spreadingPresent2.7 mmPresent (3.1 mm)10IVAbsentAbsentAbsentAbsent
Mild and focal (non-brisk)
AbsentAbsentEpithelioidNil10.9 mm4.1 mm
7.1 mmMild (1+)

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 93
CURRENT PERSPECTIVES ON THE PATHOLOGIC AND REPORTING MELANOCYTIC TUMORS TAN
mediate and late.75, 76 Early changes are essentiallythe presence of tumor-infiltrating lymphocytes, inter-mediate changes are characterized by angiofibroplasiaand late changes incorporate dermal fibrosis and lossof rete ridges. The reason for regression being anadverse prognostic factor is not entirely clear. Onepossible reason is that when regression is present in thedeep dermal portion of a tumor, it may cause an under-estimation of the original Breslow thickness of thelesion and therefore its metastatic risk is underesti-mated. From a pathogenic point of view, it has beenproposed that although a robust immune response to thetumor causes many cells to undergo apoptosis, there isa paradoxical pressure on tumor cells to undergo pro-gressive genetic aberrations, leading to the ultimategeneration of more aggressive clones of tumor cells.72
Sentinel lymph node biopsy
Sentinel lymph node (SLN) biopsy has emerged asone of the most powerful tools to assess prognosis inmelanoma.13-15, 77 From a treatment point of view, on theone hand, a negative SLN has been established to behighly predictive of negative lymph nodes in the restof the draining region, thus sparing such patients a com-pletion lymph node dissection and its associated com-plications and morbidity. On the other hand, completionregional lymph node dissection (RLND) followingdetection of metastatic melanoma in the SLN biopsyhas been shown to confer a survival advantage whencompared with patients who undergo RLND after theoccurrence of clinically evident nodal disease.14 SLNbiopsy is currently recommended for patients withmelanoma of intermediate thickness (1 to 4 mm).78 Intumors less than 1mm thick, SLN biopsy is consideredif there is presence of tumor ulceration or regression orif the tumor is in vertical growth phase.78 SLN biopsyhas also been suggested for young patients and thosewith a high tumor mitotic rate, as these two factors areassociated with a higher incidence of SLN positivity.79
SLN are most efficiently identified by lym-phoscintigraphy combined with injection of methylene-blue dye. Intraoperatively, a hand-held gamma probeis often used to aid the identification of the SLN. Inhigh volume centers, there is a reported failure rate ofabout 2%, which may either be due to the inability toidentify and remove the true SLN or the inability of thehistopathologic process to detect metastatic diseasewithin the SLN.80, 81 In our department, each SLN is
initially sliced along the longitudinal axis through thecentral meridian at 3 mm intervals and histologic exam-ination of four sequential 4 µm thick sections carriedout.82 Sections 1 and 4 are stained with hematoxylin-eosin and sections 2 and 3 are stained immunohisto-chemically for S-100 protein and HMB-45 respec-tively. Intraoperative frozen section evaluation of SLNsfrom melanoma patients is not recommended becauseit may reduce the accuracy of pathologic assessment.83
The complication rate has been reported to be about5% with hematoma, seroma, wound infection and lym-phedema being the more commonly reported mor-bidities.84, 85 There is no evidence for an increased riskof in-transit metastases arising from the performanceof SLN biopsy.14
Although the current standard of care is for patientswith a positive SLN biopsy to undergo completionRLND, only 8-30% of such patients have furthertumor-positive lymph nodes in the RLND.1, 86, 87
Recently, there have been attempts to identify specif-ic features, including those in a tumor-harboring SLN,that strongly predict tumor involvement of other region-al lymph nodes. Potentially, completion RLND couldthen be performed only on high risk patients thus spar-ing the rest of the patients the need for RLND and itsattendant morbidity. Not unexpectedly, several studieshave shown that it is the tumor burden within the SLNthat most accurately predicts the risk of tumor metas-tases in the rest of the draining lymph node basin. Ina recent study from the Sydney Melanoma Unit, itwas found that the tumor penetrative depth (distancebetween the nodal capsule and the deepest aspect of thetumor deposit) of >2 mm, area of tumor deposit >10mm2, effacement of nodal architecture and presence oftumor cells in the perinodal lymphatic spaces were allsignificantly correlated with increased risk of furthernodal disease.88 A predictive algorithmic model basedon a multitude of features (relative tumor area in theSLN, Breslow thickness and density of dendritic cellsin the nodal paracortex) has been developed byCochran et al and appears to be of use in predictingwhich patients are likely to have non-SLN metastasesbut is reliant on the use of specialized equipment, lim-iting its practicability for widespread use.89
Role of fine needle biopsy
Fine needle biopsy (FNB) is a rapid, convenient andaccurate means for the assessment of suspected recur-

TAN CURRENT PERSPECTIVES ON THE PATHOLOGIC AND REPORTING MELANOCYTIC TUMORS
94 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
rent or metastatic melanoma. Smears of FNBs ofmelanoma are characterized by a dispersed popula-tion of epithelioid or spindle shaped cells with pleo-morphic nuclei, prominent nucleoli and variable pres-ence of melanin pigment. In challenging cases, a cellblock specimen can be prepared and the diagnosis ofmelanoma can be confirmed with the use of immuno-chemistry. In a recent study of 2 204 cases at the Syd-ney Melanoma Unit, the role of FNB in the evalua-tion of suspected metastatic melanoma sites was reaf-firmed by a high sensitivity of 96% and specificity of98%.13 FNB should therefore be regarded as the first-line investigation for the pathologic confirmation ofclinically- and/or radiologically-suspected recur-rent/metastatic melanoma.16
Correlation of dermoscopic features withpathology
Dermoscopy has emerged as a useful adjunct for theclinical diagnosis of melanocytic lesions.17, 19 Its use hashelped to improve the clinical diagnostic accuracy ofsuch lesions and has also been shown to reduce the exci-sion biopsy rates of nevi. The introduction of digital der-moscopic monitoring further allows the detection of sub-tle changes of a melanocytic lesion over time, thusenhancing the diagnosis of early melanomas. Morerecently, digital dermoscopic analysis offers automatedevaluation of clinically challenging lesions in an attemptto distinguish atypical nevi from early melanomas.18 Thedevelopment of these dermoscopic techniques hasrequired close correlation of the clinical, imaging andpathologic features, as histopathologic evaluation isregarded as the ‘gold standard’. Nevertheless, there havebeen instances where suspicious areas detected on der-moscopy were not sampled during pathologic process-ing. In this regard, close clinician-pathologist commu-nication is required with the clinician guiding the pathol-ogist to more intensely sample specific areas for histologicevaluation. Such clinician-pathologist cooperation wouldhelp to further refine dermoscopic diagnostic criteria.1
The future trends of refinements to diagnosis andprognostication
Recent advances in molecular biology have enhancedthe understanding of the pathology of melanocyticlesions. Using the technique of comparative genomic
hybridization, Spitz nevi were shown to have a distinctgenetic signature compared with melanomas.90-92 Mostcases of melanoma have aberrations of chromosomes 9,10, 7, 6 while most Spitz nevi do not show these aber-rations. However, some Spitz nevi show an isolatedgain of the short arm of chromosome 11. Gene expres-sion studies have also delineated desmoplasticmelanomas, which demonstrate increased expressionof neurotropic and extracellular matrix production fac-tors; this at least partially explains the characteristichistologic features of neurotropism and desmoplasia(vide supra).93 A recent analysis of single atypicalmelanocytes beyond the histopathologically apparent insitu component of acral melanomas by comparativegenomic hybridization showed that these cells oftenharbor similar genetic changes to those of the melanoma.It is uncertain whether these cells represent the periph-eral part of the melanoma or are reflective of a “fieldeffect”, raising the issue whether such tumors mayrequire the consideration of wider excision margins.94
For assessing prognosis, decreased mRNA expressionof the melastatin, a melanocyte-specific gene, has beenshown to be an adverse prognostic factor for patientswith melanoma.95 In the effort to increase the sensitiv-ity of SLN biopsies, reverse transcriptase-polymerasechain reactions (RT-PCR) have been used although falsepositive results remain an issue.1 The use of multimarkerRT-PCR in combination with conventional histologyand immunohistochemistry appears promising.96 It islikely that molecular methods will play an increasing rolein the diagnosis and prognostication of patients withmelanoma.96
The integration of all the relevant clinical, patho-logic and molecular features points the way forwardtowards more effective prognostication of patientswith melanoma. There has been development of com-puter programs that use a selection of salient clinico-pathologic parameters to give a recurrence, sentinelnode metastasis or survival estimate for patients withmelanoma.97 It is likely that additional pathologic andmolecular parameters that are proved to be predictiveof outcome will be incorporated into such programs tobetter refine the prognostication of individual patients.98
Conclusions
For a patient with a challenging melanocytic lesion,a chain of events is essential for a correct diagnosis tobe reached and appropriate management to be insti-

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 95
CURRENT PERSPECTIVES ON THE PATHOLOGIC AND REPORTING MELANOCYTIC TUMORS TAN
tuted. The clinician makes a provisional clinical diag-nosis on the basis of the clinical history and examina-tion, often with the aid of dermoscopy. If the circum-stances allow, an excision biopsy with narrow mar-gins provides the best opportunity for the optimalpathologic assessment of the melanocytic lesion. Patho-logic assessment is based on assessment of a constel-lation of microscopic features in corroboration with allthe relevant clinical factors. Effective clinician-pathol-ogist and clinician-patient communication is crucial forthe management of the patient with a difficultmelanocytic lesion. For patients with melanoma, asynoptic pathology report is an effective means of pre-senting all the relevant pathologic parameters for accu-rate prognostication and management. Molecular para-meters are not yet in contemporary use in the diagno-sis of melanocytic tumors but hold promise for refin-ing the diagnostic and prognostication process.
Riassunto
Prospettive attuali sulla diagnosi istologica e sulla referta-zione dei tumori melanocitari
Uno degli obiettivi fondamentali della valutazione isto-logica dei tumori melanocitari è rappresentato dalla distin-zione tra lesioni benigne (nevi) e quelle maligne (melanomi).Sebbene possa sembrare semplice, la valutazione dei tumo-ri melanocititari è tra le più difficili nell’ambito delle diagnosiistologiche. Gli aspetti istologici a favore della benignitàsono rappresentati dalla simmetria, dalla delimitazione del-la lesione, dall’aspetto a V, dalla maturazione dermica e dal-l’assenza di mitosi. Al contrario, una scarsa delimitazione del-la lesione, la disseminazione epidermica laterale o periferi-ca pagetoide, l’accrescimento del derma, l’elevata cellularità,la presenza di necrosi, il pleiomorfismo nucleare e la presenzadi mitosi frequenti o anormali aumentano il sospetto di mali-gnità. Sebbene la maggior parte dei tumori siano pronta-mente diagnosticabili con l’applicazione dei criteri standard,vi sono varianti inusuali sia per i nevi che per i melanomi cherichiedono molta attenzione per evitare errate diagnosi isto-logiche e un’attenta correlazione citopatologica per evitarela mancata diagnosi. L’esecuzione di una biopsia escissionalecompleta, laddove possibile, è cruciale per poter fare unavalutazione istologica ottimale delle lesioni sospette. Per imelanomi, il referto istologico dovrebbe comprendere tuttigli aspetti istologici che sono importanti per determinare laprognosi e la gestione del paziente. L’utilizzo di un modulosinottico standard può facilitare questo aspetto e consentiredi presentare le informazioni al medico in modo chiaro.Attualmente il ricorso alla biopsia del linfonodo sentinella èben stabilito nella gestione dei pazienti con melanoma, cosìcome è utile l’agoaspirato in caso di sospetto clinico di mela-
noma metastatico. La valutazione prognostica dei pazienti conmelanoma dipende dall’integrazione di tutti gli aspetti cli-nicoistologici pertinenti e questa può essere favorita in futu-ro dall’utilizzo di programmi informatici. Le tecniche didiagnostica molecolare possono ulteriormente affinare ladiagnosi delle lesioni sospette e fornire stime prognostichepiù accurate.PAROLE CHIAVE: Parole chiave: Nevi, analtomia patologica -Nevi, diagnosi - Dermoscopia - Agobiopsia - Melanoma.
References
1. Scolyer RA, Thompson JF, Stretch JR, Sharma R, McCarthy SW.Pathology of melanocytic lesions: new, controversial, and clinicallyimportant issues. J Surg Oncol 2004;86:200-11.
2. McCarthy SW, Scolyer RA. Melanocytic lesions of the face: diag-nostic pitfalls. Ann Acad Med Singapore 2004;33(4 Suppl):3-14.
3. Liu V, Mihm MC. Pathology of malignant melanoma. Surg Clin NorthAm 2003;83:31-60.
4. Cerroni L, Kerl H. Tutorial on melanocytic lesions. Am J Der-matopathol 2001;23:237-41.
5. Scolyer RA, Thompson JF, McCarthy SW, Strutton GM, Elder DE.Incomplete biopsy of melanocytic lesions can impair the accuracyof pathological diagnosis. Australas J Dermatol 2006;47:71-3.
6. Scolyer RA, McCarthy SW, Elder DE. Frontiers in melanocytic pathol-ogy. Pathology 2004;36:385-6.
7. Troxel DB. Pitfalls in the diagnosis of malignant melanoma: find-ings of a risk management panel study. Am J Surg Pathol2003;27:1278-83.
8. Troxel DB. Diagnostic errors in surgical pathology uncovered by areview of malpractice claims. Part IV. Melanoma. Int J Surg Pathol2001;9:61-3.
9. Thompson JF, Scolyer RA. Cooperation between surgical oncolo-gists and pathologists: a key element of multidisciplinary care forpatients with cancer. Pathology 2004; 36: 496-503.
10. Cochran AJ, Bailly C, Cook M, Crotty K, McCarthy S, Mihm M et al.Recommendations for the reporting of tissues removed as part of thesurgical treatment of cutaneous melanoma. The Association of Direc-tors of Anatomic and Surgical Pathology. Am J Clin Pathol1998;110:719-22.
11. Thompson JF, Scolyer RA, Kefford RF. Cutaneous melanoma. Lancet2005;365:687-701.
12. Scolyer RA, Thompson JF, Stretch JR, McCarthy SW. Collaborationbetween clinicians and pathologists: a necessity for the optimal mana-gement of melanoma patients. Cancer Forum 2005; 29: 76-81.
13. Gershenwald JE, Mansfield PF, Lee JE, Ross MI. Role for lymphaticmapping and sentinel lymph node biopsy in patients with thick (> or= 4 mm) primary melanoma. Ann Surg Oncol 2000;7:160-5.
14. Morton DL, Thompson JF, Cochran AJ, Mozzillo N, Elashoff R,Essner R et al. Sentinel-node biopsy or nodal observation in melano-ma. N Engl J Med 2006;355:1307-17.
15. Balch CM, Cascinelli N. Sentinel-node biopsy in melanoma. N EnglJ Med 2006;355:1370-1.
16. Murali R, Doubrovsky A, Watson GF, McKenzie PR, Lee CS, McLeodDJ et al. Diagnosis of metastatic melanoma by fine-needle biopsy:analysis of 2,204 cases. Am J Clin Pathol 2007;127:385-97.
17. Menzies SW, Zalaudek I. Why perform dermoscopy? The evidence forits role in the routine management of pigmented skin lesions. Arch Der-matol 2006;142:1211-2.
18. Rubegni P, Burroni M, Andreassi A, Fimiani M. The role of der-moscopy and digital dermoscopy analysis in the diagnosis of pig-mented skin lesions. Arch Dermatol 2005;141:1444-6.
19. Crotty KA, Menzies SW. Dermoscopy and its role in diagnosing

TAN CURRENT PERSPECTIVES ON THE PATHOLOGIC AND REPORTING MELANOCYTIC TUMORS
96 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
melanocytic lesions: a guide for pathologists. Pathology 2004; 36:470-7.
20. Clemente C, Cochran AJ, Elder DE Levene A, MacKie RM, Mihm MCet al. Histopathologic diagnosis of dysplastic nevi: concordance amongpathologists convened by the World Health Organization MelanomaProgramme. Hum Pathol 1991;22:313-9.
21. Weedon D, Little JH. Spindle and epithelioid cell nevi in childrenand adults. A review of 211 cases of the Spitz nevus. Cancer1977;40:217-25.
22. Sagebiel RW, Chinn EK, Egbert BM. Pigmented spindle cell nevus.Clinical and histologic review of 90 cases. Am J Surg Pathol1984;8:645-53.
23. Dymock RB, Menz J. Recurrent melanocytic nevi following partialremoval (pseudomelanoma). Australas J Dermatol 1986;27:67-9.
24. Kornberg R, Ackerman AB. Pseudomelanoma: recurrent melanocyt-ic nevus following partial surgical removal. Arch Dermatol1975;111:1588-90.
25. Suster S. Pseudomelanoma. A pathologist’s perspective. Int J Der-matol 1986;25:506-7.
26. Haupt HM, Stern JB. Pagetoid melanocytosis. Histologic features inbenign and malignant lesions. Am J Surg Pathol 1995;19:792-7.
27. Clemente C, Zurrida S, Bartoli C, Bono A, Collini P, Rilke F. Acral-lentiginous nevus of plantar skin. Histopathology 1995;27:549-55.
28. Cook MG. Benign melanocytic lesions mimicking melanomas. Pathol-ogy 2004;36:414-8.
29. Signoretti S, Annessi G, Puddu P, Faraggiana T. Melanocytic nevi ofpalms and soles: a histological study according to the plane of section.Am J Surg Pathol 1999;23:283-7.
30. Tan KB, Moncrieff M, Thomson JF. Subungual melanoma: a study of124 cases highlighting features of early lesions, potential pitfalls indiagnosis and guidelines for histologic reporting. Am J Surg Pathol.In press.
31. Dahlstrom JE, Scolyer RA, Thompson JF, Jain S. Spitz nevus: diag-nostic problems and their management implications. Pathology2004;36:452-7.
32. Crotty KA, Scolyer RA, Li L, Palmer AA, Wang L, McCarthy SW.Spitz nevus versus Spitzoid melanoma: when and how can they be dis-tinguished? Pathology 2002;34:6-12.
33. Scolyer RA, Crotty KA, Palmer AA, McCarthy SW. Reply to SomersG (letter). Pagetoid melanocytosis in Spitz naevi. Pathology 2002;34: 592.
34. Li L-X L, Crotty KA, Scolyer RA, Thompson JF, Kril JJ, Palmer AA,McCarthy SW. Use of multiple cytometric markers improves discri-mination between benign and malignant melanocytic lesions: a studyof DNA microdensitometry, karyometry, argyrophilic staining ofnucleolar organizer regions (AgNORs) and MIB1-Ki67 immuno-reactivity. Melanoma Res 2003;13:581-6.
35. Barnhill RL, Argenyi ZB, From L, Glass LF, Maize JC et al. AtypicalSpitz nevi/tumors: lack of consensus for diagnosis, discrimination frommelanoma, and prediction of outcome. Hum Pathol 1999; 30:513-20.
36. Calonje E, Blessing K, Glusac E, Strutton G. Blue naevi. In: LeBoitPE, Burg G, Weedon D, Sarasin A eds. World Health OrganizationClassification of Tumors. Pathology and Genetics: Skin Tumors.Lyon: IARCPress; 2006.p.95-9.
37. Busam KJ. Metastatic melanoma to the skin simulating blue nevus. AmJ Surg Pathol 1999;23:276-82.
38. Carney JA, Ferreiro JA. The epithelioid blue nevus. A multicentricfamilial tumor with important associations, including cardiac myxo-ma and psammomatous melanotic schwannoma. Am J Surg Pathol1996;20:259-72.
39. Seab JA Jr, Graham JH, Helwig EB. Deep penetrating nevus. Am J SurgPathol 1989;13:39-44.
40. Mehregan DA, Mehregan AH. Deep penetrating nevus. Arch Der-matol 1993;129:328-31.
41. Scolyer RA, McCarthy SW. Case 12: Deep penetrating naevus-likemelanoma. In: Scolyer RA, McCarthy SW, Elder DE eds. Melanocyt-
ic Skin Lesions: Difficult Problems, Important Issues and Contro-versies. Brisbane: Knowledge Books & Software; 2004.p.73-90.
42. Pulitzer DR, Martin PC, Cohen AP, Reed RJ. Histologic classificationof the combined nevus. Analysis of the variable expression ofmelanocytic nevi. Am J Surg Pathol 1991;15:1111-22.
43. Scolyer RA, Zhuang L, Palmer AA, Thompson JF, McCarthy SW.Combined nevus: a benign lesion frequently misdiagnosed both clin-ically and pathologically as melanoma Pathology 2004;36:419-27.
44. Hendrickson MR, Ross JC. Neoplasms arising in congenital giantnevi: morphologic study of seven cases and a review of the literature.Am J Surg Pathol 1981;5:109-35.
45. Lowes MA, Norris D, Whitfeld M. Benign melanocytic proliferativenodule within a congenital nevus. Australas J Dermatol 2000;41:109-11.
46. Bastian BC, Xiong J, Frieden IJ, Williams ML, Chou P, Busam K etal. Genetic changes in neoplasms arising in congenital melanocyticnevi: differences between nodular proliferations and melanomas. AmJ Pathol 2002;161:1163-9.
47. Bastian BC. Understanding the progression of melanocytic neoplasiausing genomic analysis: from fields to cancer. Oncogene 2003;22:3081-6.
48. Zembowicz A, Carney JA, Mihm MC. Pigmented epithelioidmelanocytoma: a low-grade melanocytic tumor with metastatic poten-tial indistinguishable from animal-type melanoma and epithelioidblue nevus. Am J Surg Pathol 2004;28:31-40.
49. Scolyer RA, Thompson JF, Warnke K, McCarthy SW. Pigmentedepithelioid melanocytoma. Am J Surg Pathol 2004;28:1114-5.
50. McNutt NS, Urmacher C, Hakimian J, Hoss DM, Lugo J. Nevoidmalignant melanoma: morphologic patterns and immunohistoche-mical reactivity. J Cutan Pathol 1995;22:502-17.
51. Zembowicz A, McCusker M, Chiarelli C, Dei Tos AP, Granter SR,Calonje E et al. Morphological analysis of nevoid melanoma: a studyof 20 cases with a review of the literature. Am J Dermatopathol2001;23:167-75.
52. McCarthy SW, Scolyer RA, Palmer AA. Desmoplastic melanoma: adiagnostic trap for the unwary. Pathology 2004;36:445-51.
53. McCarthy SW, Crotty KA, Scolyer RA. Desmoplastic Melanomaand Desmoplastic Neurotropic Melanoma. In: LeBoit PE, Burg G,Weedon D, Sarasian A eds. WHO Classification of Tumours. Pathol-ogy and Genetics of Skin Tumours. IARC Press: Lyon 2006.p.76-8.
54. Scolyer RA, Thompson JF. Desmoplastic melanoma: a heterogeneousentity in which subclassification as “pure” or “mixed” may haveimportant prognostic significance. Ann Surg Oncol 2005;12:197-9.
55. Chorny JA, Barr RJ. S100-positive spindle cells in scars: a diagnos-tic pitfall in the re-excision of desmoplastic melanoma. Am J Der-matopathol 2002;24:309-12.
56. Robson A, Allen P, Hollowood K. S100 expression in cutaneous scars:a potential diagnostic pitfall in the diagnosis of desmoplastic melanoma.Histopathology 2001;38:135-40.
57. Wee A. Hepatitis—a diagnostic approach to histopathologic inter-pretation of liver biopsy specimens. Ann Acad Med Singapore1989;18:416-23.
58. Elder DE. Pathology of melanoma. Clin Cancer Res 2006;12(7 Pt2):2308s-2311s.
59. Elder DE, Xu X. The approach to the patient with a difficult melano-cytic lesion. Pathology 2004;36:428-34.
60. Prade M, Bognel C, Charpentier P, Gadenne C, Duvillard P, Sancho-Garnier H et al. Malignant melanoma of the skin: prognostic factorsderived from a multifactorial analysis of 239 cases. Am J Der-matopathol 1982;4:411-2.
61. Balch CM. Cutaneous Melanoma. In: Greene FL, Page DL, Flem-ing ID, Fritz AG, Balch CM, Haller DG et al. eds. American Joint Com-mittee on Cancer. Cancer Staging Manual. 6th ed. New York: Springer;2002.p.209-17.
62. Buttner P, Garbe C, Bertz J, Burg G, d’Hoedt B, Drepper H et al. Pri-mary cutaneous melanoma. Optimized cutoff points of tumor thick-

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 97
CURRENT PERSPECTIVES ON THE PATHOLOGIC AND REPORTING MELANOCYTIC TUMORS TAN
ness and importance of Clark’s level for prognostic classification.Cancer 1995;75:2499-506.
63. Kelly JW, Sagebiel RW, Clyman S, Blois MS. Thin level IV malignantmelanoma. A subset in which level is the major prognostic indicator.Ann Surg 1985;202:98-103.
64. Balch CM, Wilkerson JA, Murad TM Soong SJ, Ingalls AL, Mad-dox WA. The prognostic significance of ulceration of cutaneousmelanoma. Cancer 1980 15;45:3012-7.
65. Day CL Jr, Lew RA, Harrist TJ. Malignant melanoma prognostic fac-tors 4: ulceration width. J Dermatol Surg Oncol 1984;10:23-4.
66. Cochran AJ. Histology and prognosis in malignant melanoma. J Pathol1969;97:459-68.
67. Scolyer RA, Thompson JF, Shaw HM, McCarthy SW. The impor-tance of mitotic rate as a prognostic factor for localized primary cuta-neous melanoma. J Cutan Pathol 2006;33:395-6.
68. Francken AB, Shaw HM, Thompson JF, Soong SJ, Accortt NA, Azzo-la MF et al. The prognostic importance of tumor mitotic rate confir-med in 1317 patients with primary cutaneous melanoma and longfollow-up. Ann Surg Oncol 2004;11:426-33.
69. Azzola MF, Shaw HM, Thompson JF, Soong SJ, Scolyer RA, WatsonGF et al. Tumor mitotic rate is a more powerful prognostic indicatorthan ulceration in patients with primary cutaneous melanoma: ananalysis of 3661 patients from a single center. Cancer 2003 ;97:1488-98.
70. Thunnissen FB, Ambergen AW, Koss M, Travis WD, O’Leary TJ,Ellis IO. Mitotic counting in surgical pathology: sampling bias, het-erogeneity and statistical uncertainty. Histopathology 2001;39:1-8.
71. Scolyer RA, Shaw HM, Thompson JF, Li L-XL, Colman MH, LoSK et al. Interobserver reproducibility of histopathologic prognosticvariables in primary cutaneous melanomas. Am J Surg Pathol2003;27:1571-6.
72. Ronan SG, Eng AM, Briele HA, Shioura NN, Das Gupta TK. Thinmalignant melanomas with regression and metastases. Arch Derma-tol 1987;123:1326-30.
73. Prehn RT. The paradoxical association of regression with a poor prog-nosis in melanoma contrasted with a good prognosis in keratoacan-thoma. Cancer Res 1996;56:937-940.
74. Kelly JW, Sagebiel RW, Blois MS. Regression in malignant melanoma.A histologic feature without independent prognostic significance.Cancer 1985;56:2287-91.
75. Barr RJ. The many faces of completely regressed malignant melanoma.Pathology (Phila) 1994;2:359-70.
76. Tefany FJ, Barnetson RS, Halliday GM, McCarthy SW, McCarthy WH.Immunocytochemical analysis of the cellular infiltrate in primaryregressing and non-regressing malignant melanoma. J Invest Der-matol 1991;97:197-202.
77. Yee VS, Thompson JF, McKinnon JG, Scolyer RA, Li LX, McCarthyWH et al. Outcome in 846 cutaneous melanoma patients from a sin-gle center after a negative sentinel node biopsy. Ann Surg Oncol2005;12:429-39.
78. Crowson AN, Magro CM, Mihm MC. Prognosticators of melanoma,the melanoma report, and the sentinel lymph node. Mod Pathol 2006;19Suppl 2:S71-87.
79. Sondak VK, Taylor JM, Sabel MS, Wang Y, Lowe L, Grover AC et al.Mitotic rate and younger age are predictors of sentinel lymph node posi-tivity: lessons learned from the generation of a probabilistic model. AnnSurg Oncol 2004;11:247-58.
80. Thompson JF, McCarthy WH, Bosch CM, O’Brien CJ, Quinn MJ,Paramaesvaran S et al. Sentinel lymph node status as an indicator ofthe presence of metastatic melanoma in regional lymph nodes.Melanoma Res 1995;5:255-60.
81. Scolyer RA, Thompson JF, Li L-XL, Beavis A, Dawson M, Doble Pet al. Failure to remove “true” sentinel nodes can cause failure of thesentinel node biopsy technique. Evidence from antimony quantitation
of false negative sentinel nodes from melanoma patients. Ann SurgOncol 2004 11: 174S-178S.
82. Li LX, Scolyer RA, Ka VS, McKinnon JG, Shaw HM, McCarthySW et al. Pathologic review of negative sentinel lymph nodes inmelanoma patients with regional recurrence: a clinicopathologic studyof 1152 patients undergoing sentinel lymph node biopsy. Am J SurgPathol 2003;27:1197-202.
83. Scolyer RA, Thompson JF, Gershenwald, JE, Ross, MI, McCarthy SW,Cochran AJ. Intraoperative frozen section evaluation may reduce theaccuracy of pathologic assessment of sentinel nodes in melanomapatients. J Am Coll Surg 2005;201:821-3.
84. McMasters KM, Noyes RD, Reintgen DS, Goydos JS, Beitsch PD,Davidson BS et al. Lessons learned from the Sunbelt Melanoma Trial.J Surg Oncol 2004;86:212-23.
85. Morton DL, Cochran AJ, Thompson JF, Elashoff R, Essner R, GlassEC et al. Sentinel node biopsy for early-stage melanoma: accuracy andmorbidity in MSLT-1, an international multicenter trial. Ann Surg2005;242:302-13.
86. Joseph E, Brobeil A, Glass F, Glass J, Messina J, DeConti R et al.Results of complete lymph node dissection in 83 melanoma patientswith positive sentinel nodes. Ann Surg Oncol 1998;5:119-25.
87. Starz H, Balda BR, Kramer KU, Buchels H, Wang H. A micromor-phometry-based concept for routine classification of sentinel lymphnode metastases and its clinical relevance for patients with melanoma.Cancer 2001;91:2110-21.
88. Scolyer RA, Li LX, McCarthy SW, Shaw HM, Stretch JR, Sharma Ret al. Micromorphometric features of positive sentinel lymph nodespredict involvement of nonsentinel nodes in patients with melanoma.Am J Clin Pathol 2004;122:532-9.
89. Cochran AJ, Wen DR, Huang RR, Wang HJ, Elashoff R, Morton DL.Prediction of metastatic melanoma in nonsentinel nodes and clinicaloutcome based on the primary melanoma and the sentinel node. ModPathol 2004;17:747-55.
90. Bastian BC. Molecular cytogenetics as a diagnostic tool for typingmelanocytic tumors. Recent Results Cancer Res 2002;160:92-9.
91. Harvell JD, Kohler S, Zhu S, Hernandez-Boussard T, Pollack JR, vande Rijn M. High-resolution array-based comparative genomichybridization for distinguishing paraffin-embedded Spitz nevi andmelanomas. Diagn Mol Pathol 2004;13:22-5.
92. Harvell JD, Bastian BC, LeBoit PE. Persistent (recurrent) Spitz nevi:a histopathologic, immunohistochemical, and molecular pathologicstudy of 22 cases. Am J Surg Pathol 2002;26:654-61.
93. Busam KJ, Zhao H, Coit DG, Kucukgol D, Jungbluth AA, NobregaJ et al. Distinction of desmoplastic melanoma from non-desmoplasticmelanoma by gene expression profiling. J Invest Dermatol2005;124:412-8.
94. Bastian BC, Kashani-Sabet M, Hamm H, Godfrey T, Moore DH 2nd,Brocker EB et al. Gene amplifications characterize acral melanomaand permit the detection of occult tumor cells in the surrounding skin.Cancer Res 2000;60:1968-73.
95. Duncan LM, Deeds J, Cronin FE, Donovan M, Sober AJ, KauffmanM et al. Melastatin expression and prognosis in cutaneous malignantmelanoma. J Clin Oncol 2001;19:568-76.
96. Morton DL, Hoon DS, Cochran AJ, Turner RR, Essner R, TakeuchiH et al. Lymphatic mapping and sentinel lymphadenectomy for ear-ly-stage melanoma: therapeutic utility and implications of nodalmicroanatomy and molecular staging for improving the accuracy ofdetection of nodal micrometastases. Ann Surg 2003;238:538-49.
97. Wong SL, Katten MW, McMasters KM, Coit DG. A nomogram thatpredicts the presence of sentinel node metastasis in melanoma with bet-ter discrimination than the American Joint Committee on Cancerstaging system. Ann Surg Oncol 2005;12:282-8.
98. Thompson JF, Shaw HM, Hersey P, Scolyer RA. The history andfuture of melanoma staging. J Surg Oncol 2004;86:224-35.

G ITAL DERMATOL VENEREOL 2007;142:99-108
Dermoscopic patterns of melanomaThe Good, The Bad, and The Gray
Dermoscopy is an effective tool for differentiating melanomafrom benign melanocytic nevi. A number of score-based andgestalt-based algorithms are available to help accomplish the taskof correctly identifying melanoma while at the same time cor-rectly classifying most clinically atypical nevi as benign lesions,which need not be biopsied. We describe a simple and easyapproach to learn pattern analysis that can be utilized to helpclinicians differentiate melanoma from benign nevi. Most mela-nocytic nevi manifest one of nine well defined benign patterns.These benign patterns are symmetric, uniform and organizedand include the: (1) diffuse reticular network, (2) patchy reticularnetwork, (3) peripheral reticular network with central hypo-pigmentation, (4) peripheral reticular network with centralhyperpigmentation, (5) peripheral reticular network with cen-tral globules, (6) globular, (7) peripheral globules with centralreticular network or starburst, (8) homogeneous, and the (9)symmetric multi-component pattern. Melanoma on the otherhand tend to reveal a pattern that deviates from the above men-tioned benign patterns by manifesting asymmetry and an archi-tecture that is disordered. Most melanomas will also containat least one of the following eight local features: atypical network,streaks, atypical dots or globules, negative pigment network,off center blotch, blue-white veil, and atypical vascular struc-tures. Thus, by simply knowing a set of nine benign nevus pat-tern and eight local melanoma specific features one can startimplementing dermoscopy into the daily routine practice.KEY WORDS: Melanoma, pathology - Dermoscopy - Skin.
Early diagnosis of cutaneous melanoma remainsthe best course for ensuring a good prognosis for
patients with this malignancy. The added benefit ofdermoscopy, above and beyond simple unaided visu-al inspection, has been firmly established. In the handsof experienced users, dermoscopy boosts diagnosticaccuracy by more than 40%.1 Additionally, it improvesthe clinician’s confidence level in the diagnosis beingrendered.2 The ultimate sequale of an improved diag-nostic accuracy and confidence level is a decrease inthe total number of unnecessary biopsies while at thesame time insuring that malignancies are not missed– a fact that translates into an improved benign tomalignant biopsy ratio.3
The learning curve for dermoscopy, however issteep. The observer needs to recognize a multitude ofdermoscopic structures and colors, many of which canat times be subtle. In addition, they need to recognizethe patterns created by the placement and distributionof these colors and structures. These patterns can great-ly facilitate the clinician in correctly diagnosing a par-ticular cutaneous lesion. A number of diagnostic algo-rithms and approaches have been described with theaim of teaching novices to correctly interpret dermo-scopic primary morphology.4-8 In general, the algo-rithms can be classified as gestalt-based (e.g., patternanalysis 7, 9) or score based (e.g., ABCD rule of der-moscopy,6 Menzie’s method or 7-point check list 4).
Dermatology ServiceMemorial Sloan-Kettering Cancer Center, New York, NY USA
Address reprint requests to: A. A. Marghoob, MD, Dermatology Service,Memorial Sloan-Kettering Cancer Center, 160 East 53rd Street, New York,NY 10022, USA. E-mail: [email protected]
S. Q. WANG, A. SCOPE, A. A. MARGHOOB
Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 99

WANG DERMOSCOPIC PATTERNS OF MELANOMA
100 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
Various studies 4-8 have demonstrated that there is nosingle ideal way to diagnose melanoma dermosco-pally, and most likely, something can be learned fromall these methods. However, pattern analysis is themost frequently used approach by experts.1, 8 Experi-ence plays a major role in this approach. An expertsees the features and patterns in a lesion, and almostsimultaneously he or she compares this lesion with amental database of dermoscopic images. Usually, in aspan of few seconds, the expert reaches a conclusionand renders a diagnosis. The major advantage of pat-tern analysis over the score-based approach is thespeed by which one can reach a diagnosis for mostpigmented skin lesions. Since this process requiresadequate knowledge and experience, it is perhaps themost difficult method to teach novices. Despite thischallenge, we will attempt to describe dermoscopicpatterns of melanoma through a three-phase approach.We will first describe the classic patterns of the benignmelanocytic nevi (the good), then the typical patternsof frank melanomas (the bad), and finally the patternsencountered in some early melanomas and some atyp-ical nevi (the gray).
The Good: classic patternsof benign melanocytic nevus
Learning the normal patterns of benign nevi is thefirst step. In medical school, students first learn the nor-mal physiology before attempting to understand thepathophysiology of different disease states. Likewise, tounderstand the dermoscopic patterns of melanoma,novices first need to replete a mental database with nor-mal melanocytic lesions. Fortunately, benign nevi tendto manifest one of a finite number of well recognized pat-terns (Table I).10 If a lesion adheres to one of these benigndermoscopic nevus patterns while at the same time notmanifesting any of the local features attributed tomelanoma (Table II), then it is highly improbable that thelesion will be a melanoma. Conversely, if a lesion devi-ates from one of the classic dermoscopic patterns ofbenign nevi, then the index of suspicion for melanomashould be raised, especially in the presence of one ormore of the local features attributed to melanoma (TableII). Hence, by knowing a set of classic dermoscopic pat-terns encountered in benign nevi and a few local fea-tures that occur in melanoma one can easily implementdermoscopy in daily routine practice.
Diffuse reticular network
Patchy reticular network
Peripheral reticular network with central hypopigmentation
Peripheral reticular network with central hyperpigmentation
Peripheral reticular network with central globules
Globular
Homogeneous pattern
Peripheral globular with central reticular network or centrallyhomogeneous area (this pattern also includes the starburst pat-tern)
Symmetric multi-component
TABLE I.—Description of nine benign patterns of melanocytic nevi.
Benign patterns Description
— Uniform network distributed diffusely and throughout the lesion. The linesof network represents the presence of melanocytes or pigmented kerati-nocytes along the epidermal rete ridges. The holes in the network representthe dermal papillae
— Patches of uniform network scattered throughout the lesion. There are struc-tureless areas interspersed among the patches of network.
— Uniform network is located at the periphery and surrounding a central struc-tureless or light brown homogeneous area
— Uniform network is located at the periphery and surrounding a central darklypigmented and homogeneous area. There may also be a small number ofdots present in the center or overlying the network lines.
— Uniform network is located at the periphery and surrounding a cluster ofglobules in the center of the lesion
— Uniform globules are the predominant feature. The globules correspond tonests of melanocytes. Depending on the depth of these nests, the globulescan be either light brown, dark brown or black in color. Close proximity andjuxtaposition of these globules can form a “cobble-stoned” appearingpattern
— Brown homogeneous pigment throughout the entire lesion. On occasion onemay see a few globules and/or remnants of a network
— A rim of uniformly distributed globules surrounding an area of network.This pattern is frequently seen in growing nevi. If instead of seeing globulesone sees pseudopods around the periphery of the lesion (starburst pattern) thenthe most likely diagnosis is a Spitz nevus
— Combination of three or more structures distributed symmetrically. Multi-com-ponent lesions should always be viewed with caution. Only if the structuresare completely symmetrically distributed should one consider this a benignpattern

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 101
DERMOSCOPIC PATTERNS OF MELANOMA WANG
The benign nevus patterns share some commonmotifs. In general, benign nevi have fewer (<3) colorsand fewer numbers of dermoscopic structures. Mostimportantly, benign lesions tend to be uniform, reveal-ing symmetry in the distribution of dermoscopic struc-tures and colors and they tend to be architecturallyorganized.11 Most benign lesions have at least one axisof symmetry. Uniformity is a more elusive concept. Itapplies to the size and shape of the dermoscopic struc-tures that are present. For example, uniform networkis composed of a mesh network with similar sizes ofholes and width of lines, and uniform globules arecomposed of globules of similar shape, size and col-or. Architectural organization is the most difficult con-cept to convey in description, though most people havean intuitive understanding of this concept. It is one ofthe key concepts described in the CASH algorithm.11
Perhaps the best way to illustrate this idea is by takinga page from the architectural layout of two major cities:
New York and Rome. New York streets are laid out ina grid fashion where most of the streets and avenues arestraight, aligned either in north-south or east-westdirections. In contrast, the streets in Rome are hap-hazardly, although charmingly, laid out. As we dis-cuss each of the classic benign patterns of nevi, hope-fully the concept of architectural order will becomeclearer.
Specifically, the nine classic dermoscopic patternsof benign melanocytic nevi include: 1) diffuse retic-ular network; 2) patchy reticular network; 3) periph-eral reticular network with central hypopigmentation;4) peripheral reticular network with central hyper-pigmentation; 5) peripheral reticular network withcentral globules; 6) globular; 7) peripheral globuleswith central reticular network or starburst; 8) homo-geneous and 9) multi-component patterns.12 A wordof caution is warranted here that although a com-pletely symmetric multi-component lesion is usually
Atypical network (including branched streaks)
Streaks (includes pseudopods and radial streaming)
Atypical dots and globules
Negative or reverse pigment network
Off center blotch
Blue white veil overlying flat (macular) areas and/or the presen-ce of blue-gray granules (peppering)
Blue white veil overlying raised areas
Vascular structures
TABLE II.—Dermoscopic structures commonly seen in melanomas.
Structures Descriptions
— Black, brown or gray network with focal irregular mesh with thick lines anddifferent sized holes. The lines represent the melanocytes and melanin alongthe rete ridges, and the holes represent the dermal papillae. Branched streaksrepresent remnants of pigmented rete ridges resulting from bridging of nestsof melanocytes at the dermal-epidermal junction
— Linear pigmented projections at the periphery of the lesion. When theselinear structures terminate with a bulbous projection they are called pseu-dopods. Histologically they represent confluent junctional nests of mela-nocytes
— Black, brown or gray dots or globules, varied in size and distributed hapha-zardly within the lesions. They frequently occur at the periphery of thelesion and are not associated with the network. Histologically, black dotsrepresent melanin in the stratum cornium, gray dots represents melanin freein dermis or within melanophages, and brown dots represent small nevo-melanocytic nests at the tips of the rete ridges. Brown globules representslarger nevomelanocytic nests along the dermo-epidermal junction or in thedermis
— The lines of the network appear lighter (almost white) in color compared tothe holes which are darker in color. Histologically this probably representsnarrow and hypopigmented rete ridges accompanied by the presence of lar-ge melanocytic nests within a widened papillary dermis
— A blotch is a darkly pigmented area in which one cannot discern any struc-tures. Histologically it represents large concentrations of melanin in the epi-dermis and/or dermis
— Irregular, confluent, gray-blue to whitish blue pigmentation overlying flatareas within the lesion. Histologically this represents regression. One can alsosee peppering which represents melanin or melanophages in the papillary der-mis. If the regression is complete then one will see a white scar like areainstead
— Irregular, confluent, blue-white hazy pigmentation overlying raised areaswithin the lesion. Histological it represents melanocytes in the dermis togetherwith compact orthokeratosis
— Presence of different shaped blood vessels can be seen in melanoma. The mostcommon are dotted, linear irregular and polymorphous vessels

WANG DERMOSCOPIC PATTERNS OF MELANOMA
102 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
benign, one should always be cautious whenever eval-uating such lesions since melanomas frequently pos-sesses a multi-component pattern. With that beingsaid, these aforementioned nine benign nevus pat-terns are derived from three fundamental dermoscopicfeatures, namely reticular network, globules andhomogeneous areas. Benign nevi may reveal just oneof these three features or may reveal a combination ofthese three features 10 (Figure 1). The detailed descrip-tion and schematic illustration of each pattern can befound in Table I and Figure 2, respectively. Dermo-scopic images representing each pattern are shownin Figures 3-11. In general, lesions with any of theabove nine patterns are uniform and architecturallyordered. Lesions manifesting these benign patternswhile at the same time not exhibiting any of themelanoma specific structures mentioned in Table II arehighly unlikely to represent melanoma.
The Bad: classic patterns of frank melanoma
To a certain degree, learning the dermoscopic pat-terns of melanoma is easy when one knows the motifsand patterns of benign nevi. Dermoscopic patterns of
melanoma should share little to no similarity withthose principles and patterns described for benignnevi. Unlike nevi, the motifs in melanomas usuallyreveal asymmetry, multiple colors (>3) and many der-moscopic structures. The classic dermoscopic patternsof melanoma display asymmetry in at least one axis,and most have two axis asymmetry. They usually revealnon-uniformity in shape and size of the dermoscopicstructures. More importantly, the structures arearranged in a haphazard distribution, and there is archi-tectural disorder. On a gestalt level, the dermscopicpatterns of melanoma are defined by chaos and disor-ganization, and they create a level of unease in the eyeof the examiners.
Unlike the nine dermoscopic patterns encounteredin benign nevi, there is no finite set of dermoscopicpatterns that are characteristic of melanomas. Thus, itis not possible to categorize all the infinite variationsof different dermoscopic patterns of melanomas. Fornovices without extensive training the following guide-line may be helpful. melanomas are lesions that 1)do not fit into any of the aforementioned nine benigndermoscopic patterns associated with nevi; 2) haveasymmetry, non-uniformity, and architectural disor-der and 3) have any of the local features attributed tomelanoma (Table II). Figures 12-16 illustrate theexamples of dermoscopic patterns of frank mela-nomas.
This contrast and comparison between the “good”and the “bad” dermoscopic patterns has also beendescribed as “the beauty and the beast” sign. “Beau-ty” connotes the dermoscopic patterns of benign nevirevealing symmetry and engendering a sense of easein the observer. “Beast” refers to the features and pat-terns of melanomas, most of which reveal asymmetryand engender a sense of unease in the viewer. Inessence, the dermoscopic diagnosis of melanomarelies on the understanding of the benign patternsseen in melanocytic nevi. This should not be a surprise,because in nearly all branches of medicine the recog-nition of pathology requires the understanding of thenormal.
The Gray: the dermoscopic patterns ofindeterminate melanocytic lesions
Biologic systems are complex and often cannot beorganized within simple dichotomies. It is almostimpossible to neatly classify all lesions as either good
Reticular(Network)
Homogeneous(Structureless) Globular
R-H R-G
R-G-H
G-H
Figure 1.—Dermoscopic patterns encountered in melanocytic lesions arecreated by the presence of one or more of the following three structures:reticular network (R), homogeneous areas (H), and globules (G).

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 103
DERMOSCOPIC PATTERNS OF MELANOMA WANG
Figure 2.—Schematic illustration of the nine benign melanocytic nevus dermoscopic patterns includes starbust patterns.
Figure 3.—Diffuse network pattern. There are a few brown dots presentwithin the lesion. However, dots overlying the network lines is common inbenign nevi and they can generally be ignored. Figure 4.—This lesion has a classic patchy network pattern.

WANG DERMOSCOPIC PATTERNS OF MELANOMA
104 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
or bad, black or white, beautiful or ugly. In addition,our current understanding of tumor progression and thesensitivity of diagnostic instruments are not adequateto allow us to accurately classify all the melanocyticlesions as “good” or “bad”. For these reasons, there are
Figure 5.—This benign melanocytic nevus reveals a peripheral network witha central hypopigmented area.
Figure 6.—This benign melanocytic nevus reveals a peripheral network witha central hyperpigmented area.
Figure 7.—This lesion has a benign homogeneous pattern.
Figure 8.—This lesion has a peripheral network with centrally placed glo-bules, another benign pattern.
Figure 9.—This lesion has a peripheral rim of globules. A peripheral rimof globules is seen in benign nevi that are in the process of enlarging.Lesions revealing a peripheral rim of pseudopods (streaks) instead of glo-bules usually represent Spitz nevi.
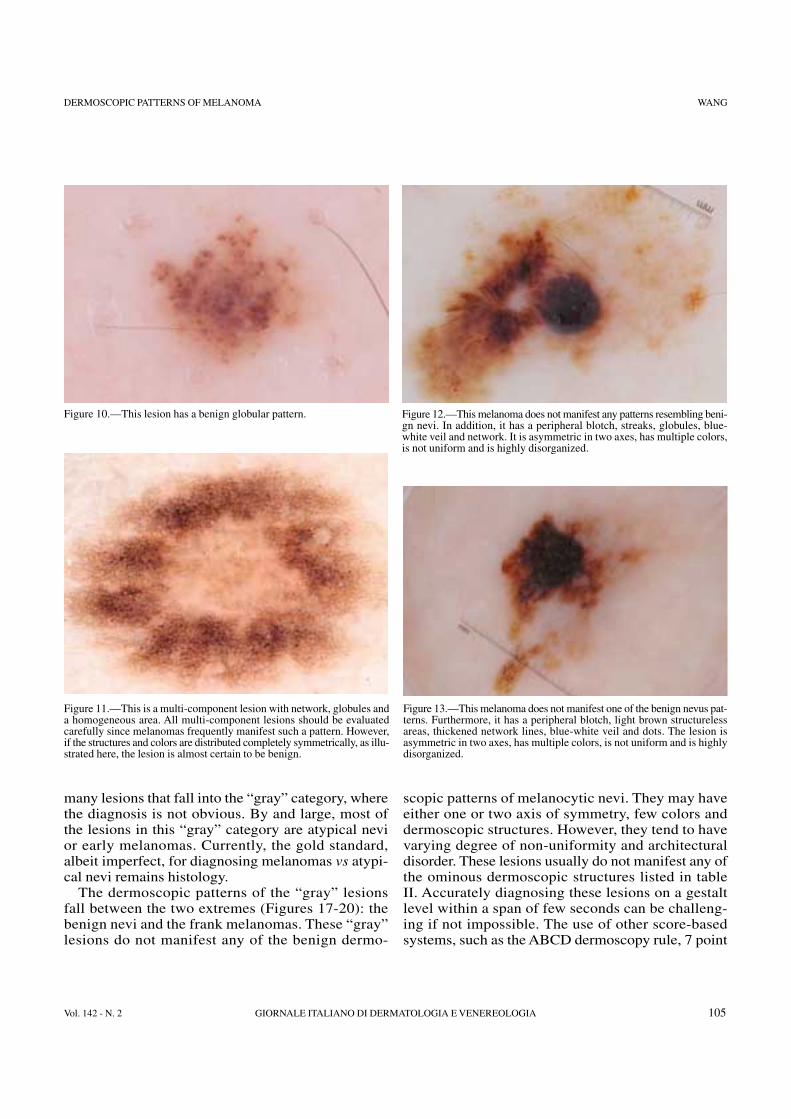
Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 105
DERMOSCOPIC PATTERNS OF MELANOMA WANG
many lesions that fall into the “gray” category, wherethe diagnosis is not obvious. By and large, most ofthe lesions in this “gray” category are atypical nevior early melanomas. Currently, the gold standard,albeit imperfect, for diagnosing melanomas vs atypi-cal nevi remains histology.
The dermoscopic patterns of the “gray” lesionsfall between the two extremes (Figures 17-20): thebenign nevi and the frank melanomas. These “gray”lesions do not manifest any of the benign dermo-
scopic patterns of melanocytic nevi. They may haveeither one or two axis of symmetry, few colors anddermoscopic structures. However, they tend to havevarying degree of non-uniformity and architecturaldisorder. These lesions usually do not manifest any ofthe ominous dermoscopic structures listed in tableII. Accurately diagnosing these lesions on a gestaltlevel within a span of few seconds can be challeng-ing if not impossible. The use of other score-basedsystems, such as the ABCD dermoscopy rule, 7 point
Figure 10.—This lesion has a benign globular pattern.
Figure 11.—This is a multi-component lesion with network, globules anda homogeneous area. All multi-component lesions should be evaluatedcarefully since melanomas frequently manifest such a pattern. However,if the structures and colors are distributed completely symmetrically, as illu-strated here, the lesion is almost certain to be benign.
Figure 12.—This melanoma does not manifest any patterns resembling beni-gn nevi. In addition, it has a peripheral blotch, streaks, globules, blue-white veil and network. It is asymmetric in two axes, has multiple colors,is not uniform and is highly disorganized.
Figure 13.—This melanoma does not manifest one of the benign nevus pat-terns. Furthermore, it has a peripheral blotch, light brown structurelessareas, thickened network lines, blue-white veil and dots. The lesion isasymmetric in two axes, has multiple colors, is not uniform and is highlydisorganized.

WANG DERMOSCOPIC PATTERNS OF MELANOMA
106 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
check list, and Menzies method may be helpful forsuch lesions, but even the score-based systems willusually fail us when it comes to these “gray” lesions.Perhaps the most helpful method, short of perform-ing a biopsy, for correctly classifying these “gray”lesions is via short term mole monitoring.13, 14 Shortterm mole monitoring is based on the premise thatbiologically relevant melanomas will reveal dermo-scopic changes when monitored over a three monthperiod and benign nevi will generally not show anychange.
Management of patients with these “gray” lesionsdepends on a host of factors. The patient’s own histo-ry and observation are important elements in the diag-nostic consideration. Lesions with symptoms of pain,bleeding, itching or change should be biopsied or mon-itored closely. Comparison of the lesion with neigh-boring lesions is another sound practice principle.Lesion that appears different (either via clinical ordermoscopic examination) relative to its neighbors,the “ugly duckling sign”, should be biopsied or mon-itored closely.
Figure 15.—This melanoma does not manifest one of the benign nevus pat-terns. It also has dotted vessels, pink veil (indicating increased vasculature),blue-white veil and light brown structureless areas.
Figure 14.—This melanoma reveals network, streaks, light brown struc-tureless areas and a subtle blue-white veil. The lesion is asymmetric in oneaxis, has multiple colors and is somewhat disorganized. It clearly doesnot conform to one of the benign nevus patterns depicted in figure 2.
Figure 16.—This melanoma has regression structures, globules and networkdistributed asymmetrically. The colors and structures and not uniform andare disorganized. This lesion does not conform to one of are the benignnevus pattern.
Figure 17.—This lesion is composed of mostly light brown globules, andit is symmetric in two axes. However, it contains area suggestive of areverse network. The histologic diagnosis was dysplastic nevus.

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 107
DERMOSCOPIC PATTERNS OF MELANOMA WANG
In summary, dermoscopy is a valuable tool fordifferentiating most melanomas from benignmelanocytic nevi. For dermoscopists, learning torecognize melanoma requires knowing the patternsand structures encountered in benign nevi. Physi-cians should consider melanoma in the differentialdiagnosis anytime they encounter a lesion that devi-ates from one of the nine benign nevus patterns(Table I, Figure 2), especially if the lesion also revealsone or more of the ominous dermoscopic structureslisted in Table II.
Riassunto
Modelli dermoscopici del melanoma. Il Buono, Il Cattivo, IlGrigio
La dermoscopia rappresenta uno strumento efficace per dif-ferenziare il melanoma dal nevo melanocitico benigno. Sonodisponibili diversi algoritmi basati sul punteggio e sulla for-ma per identificare il melanoma correttamente e contempo-raneamente classificare correttamente i nevi clinicamenteatipici come lesioni benigne, per cui non è necessario effet-tuare una biopsia. Descriviamo il modello di approccio ana-litico per imparare le interpretazioni dermoscopiche deimelanomi e delle lesioni melanocitiche benigne. Per le lesio-ni benigne vi sono nove modelli classici: 1) aspetto reticolarediffuso, 2) aspetto reticolare a chiazze, 3) aspetto reticolareperiferico con ipopigmetazione centrale, 4) aspetto reticolareperiferico con iperpigmentazione centrale, 5) aspetto reti-colare periferico con globuli centrali, 6) aspetto globulare, 7)globuli periferici con aspetto reticolare centrale o starburst,8) aspetto omogeneo e, 9) aspetto caratterizzato da compo-nenti multiple. Per i melanomi maligni l’aspetto classiconon ricorda nessuno dei nove precedenti. Il melanoma mali-gno presenta anche asimmetria, architettura disordinata emolteplicità di colori. Infine, il melanoma ha anche struttu-re locali inquietanti, quali velature bluastre, striature atipichee vasi sanguigni irregolari.
PAROLE CHIAVE: Melanoma, anatomia patologica - Dermo-scopia - Cute.
References
1. Kittler H, Pehamberger H, Wolff K, Binder M. Diagnostic accuracyof dermoscopy. Lancet Oncol 2002;3:159-65.
Figure 18.—The lesion has few colors, atypical network, structurelessarea and branched streaks. The histologic diagnosis was melanoma insitu.
Figure 19.—The lesion has peripheral dark and light brown globules andfocal network. At the inferior portion of the lesion, there is a hypopig-mented structureless area and blue white veil. The histologic diagnosiswas dysplastic nevus.
Figure 20.—The lesion is composed of brown network with relative unifor-mity. It also has focal atypical network and focal peripheral streaks. Thehistologic diagnosis was melanoma in situ.

WANG DERMOSCOPIC PATTERNS OF MELANOMA
108 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
2. Benvenuto-Andrade C, Dusza SW, Hay JL, Agero AL, Halpern AC,Kopf AW et al. Level of confidence in diagnosis: clinical examinationversus dermoscopy examination. Dermatol Surg 2006;32:738-44.
3. Carli P, De Giorgi V, Crocetti E, Mannone F, Massi D, Chiarugi A etal. Improvement of malignant/benign ratio in excised melanocyticlesions in the ‘dermoscopy era’: a retrospective study 1997-2001. BrJ Dermatol 2004;150:687-92.
4. Argenziano G, Fabbrocini G, Carli P, De Giorgi V, Sammarco E, Del-fino M. Epiluminescence microscopy for the diagnosis of doubtfulmelanocytic skin lesions. Comparison of the ABCD rule of derma-toscopy and a new 7-point checklist based on pattern analysis. ArchDermatol 1998;134:1563-70.
5. Menzies SW, Ingvar C, Crotty KA, McCarthy WH. Frequency andmorphologic characteristics of invasive melanomas lacking specificsurface microscopic features. Arch Dermatol 1996;132:1178-82.
6. Nachbar F, Stolz W, Merkle T, Cognetta AB, Vogt T, Landthaler M etal. The ABCD rule of dermatoscopy. High prospective value in the dia-gnosis of doubtful melanocytic skin lesions. J Am Acad Dermatol1994;30:551-9.
7. Pehamberger H, Steiner A, Wolff K. In vivo epiluminescence micro-scopy of pigmented skin lesions. I. Pattern analysis of pigmentedskin lesions. J Am Acad Dermatol 1987;17:571-83.
8. Argenziano G, Soyer HP, Chimenti S, Talamini R, Corona R, Sera Fet al. Dermoscopy of pigmented skin lesions: results of a consensusmeeting via the Internet. J Am Acad Dermatol 2003;48:679-93.
9. Pehamberger H, Binder M, Steiner A, Wolff K. In vivo epiluminescencemicroscopy: improvement of early diagnosis of melanoma. J InvestDermatol 1993;100:356S-62S.
10. Hofmann-Wellenhof R, Blum A, Wolf IH, Piccolo D, Kerl H, GarbeC et al. Dermoscopic classification of atypical melanocytic nevi (Clarknevi). Arch Dermatol 2001;137:1575-80.
11. Henning JS, Dusza SW, Wang SQ, Marghoob AA, Rabinovitz HS, Pol-sky D et al. The CASH (color, architecture, symmetry, and homoge-neity) algorithm for dermoscopy. J Am Acad Dermatol 2007;56:45-52.
12. Malveyh J, Braun SPP, Marghoob AA, Kopf AW, eds. Hand book ofdermoscopy. London & New York: Taylor & Francis; 2006.
13. Kittler H, Pehamberger H, Wolff K, Binder M. Follow-up of mela-nocytic skin lesions with digital epiluminescence microscopy: patternsof modifications observed in early melanoma, atypical nevi, and com-mon nevi. J Am Acad Dermatol 2000;43:467-76.
14. Menzies SW, Gutenev A, Avramidis M, Batrac A, McCarthy WH.Short-term digital surface microscopic monitoring of atypical or chan-ging melanocytic lesions. Arch Dermatol 2001;137:1583-9.

G ITAL DERMATOL VENEREOL 2007;142:109-21
New molecular biomarkers for malignant melanoma
Melanoma is the most lethal of human skin cancer and its inci-dence is increasing worldwide. Melanomas often metastasizeearly during the course of the disease and are then almost intrac-table by current therapeutic regimens. Consequently, under-standing the factors that maintain melanocyte homeostasis andprevent their neoplastic transformation into melanoma is of out-standing interest. Targeted molecular therapeutics are tailoredto genetic abnormalities that are associated with tumor pro-gression. In this review, we report about our quest for new dia-gnostic biomarkers and therapeutic targets, respectively, usingdifferent molecular techniques and approaches, including cDNAmicroarrays, RT-PCR, conventional immunohistochemistry andtissue-microarrays from in vivo and in vitro samples. We willdescribe our recent findings on the newly discovered markers indetail, among them phosphorylated pRb Ser795, the retinobla-stoma protein binding protein 2-homolog 1 (RBP2-H1) and thedipeptidyl peptidase IV (DPPIV), and link them to alreadyknown mechanisms of melanoma pathogenesis. Since the repor-ted markers may be causally linked to malignant transformation,their molecular signaling mechanisms deserve to be studied inmore detail in future investigations. KEY WORDS: Melanoma, pathology - Biological markers - Skinneoplasms.
Since the 1960s, melanoma incidence has risen by3% to 8% per year in the Caucasian population.1
Although early primary melanomas are curable by
surgery, treatment of advanced metastatic diseaseremains futile and the strategies employed in the last30 years have not significantly improved survival rates,which are 3-16% (10-years survival rate) in stage IVmelanoma.2 Thus, advances in understanding the genet-ic and functional alterations in melanocytic tumorshave sustained hopes for a breakthrough in the thera-py of disseminated melanoma.3 A current paradigmis that genetic and epigenetic alterations play an impor-tant role in the etiology and pathogenesis of cancer.Apparently, mammalian cells have numerous safe-guards that protect against neoplastic transformation,and only when lesions occur in multiple genes, inva-sive cancer can develop.4
As a consequence of this multifactorial pathogene-sis of melanoma, the application of one single bio-marker for targeted therapy or diagnosis frequentlyfails. For example, the recent identification of acti-vating mutations in BRAF in over 60% of cases ofmelanoma has caused much excitement in themelanoma community and may actually offer the firstopportunity for a rational treatment program.2 However,since more than 80% of common benign nevi showthe same mutation,5 BRAF can not be applied as a bio-marker for differentiation between melanoma and nevi.Particularly, the discrimination of borderline lesionssuch as dysplastic nevi 6 or deep penetrating nevi 7
Department of DermatologyRegensburg University Medical Center, Regensburg, Germany
The presented data were partly funded by the German Research Foun-dation, DFG-Grant: KR 2135/1-1 and the Dr. H. Maurer-Foundation.
Address reprint requests to: A. Roesch, MD, Department of Dermato-logy, Regensburg University Medical Center, Franz-Josef Strauss-Allee 11,D-93053 Regensburg, Germany. E-mail: [email protected]
A. ROESCH, T. VOGT
Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 109

ROESCH NEW MOLECULAR BIOMARKERS FOR MALIGNAT MELANOMA
110 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
still remains critical at all levels of the diagnosticprocess, i.e. clinical appearance, dermatohistopathol-ogy and possible molecular work-up.6, 8, 9 Conse-quently, further molecular markers are highly neededthat may also be suitable as therapeutic co-targets.
In this review, we report about our quest for newmelanoma biomarkers. We are targeting on both 1)the detection of new markers for discriminationbetween benign and malignant tumor growth and 2) thetherapeutic disclosure of these markers in melanomadevelopment considering functional linkages to alreadyidentified melanoma signaling pathways, such as theCDKN2A/CDK/pRb pathway. Different approachesare conceivable for detection of new markers for malig-nancy. In our group, we usually start from a biologickey process that is involved in melanoma develop-ment, as e.g. malignant invasion or disruption of cellcycle control, and either analyze empirically chosenmarkers in a functional setting or systematically screenfor markers. In particular, we will focus on the mostpromising examples of markers with therapeutic impli-cations, such as phosphorylated pRb Ser795,retinoblastoma protein-binding protein 2-homolog 1and dipeptidyl peptidase IV, respectively.
Detection of a gene expression profiledetermining the invasive potential of malignant
melanoma
Most melanomas begin to grow within the epider-modermal junction and progress through a period ofradial expansion to an invasive phenotype that acquiresmetastatic capacity possibly in a stepwise fashion.10, 11
Currently, the genes that orchestrate this transitionhave not been fully established.12 In recent years, somePCR- or immunohistochemistry-based approachescould detect a potential association of the invasivecharacter of melanoma cells and the expression of cer-tain singular genes, e.g. metalloproteinase-2 13 andannexin VII.14 But, a systematic analysis of geneexpression patterns of the invasive margin was lacking,although such an analysis could provide clues for theunderstanding of this pivotal step in progression.Recent technical advances, the combined applicationof laser pressure catapulting microdissection withgenome wide expression profiling using cDNAmicroarrays, have opened the gateway to this kind ofinvestigations.15
cDNA arrays allow an effective investigation of dif-
ferential gene expression patterns using a systematicand comprehensive approach.16 Analysing primarypatient tissues, Golub et al.17 showed that a reliablediscrimination between acute lymphoblastic leukemiaand acute myeloid leukaemia could not be made basedon single genes. In contrast, predictions based on theexpression levels of a multitude of genes were highlyaccurate. For malignant melanoma, it was also demon-strated that a classification based upon differentialgene expression might be possible.18 But, in contrastto Golub’s study, genetic profiling of melanoma wasmainly founded on the investigation of cultured celllines 18 or mouse models.19 Consequently, predictionsabout the in vivo situation of human melanomas werestill rather limited, because in vitro strategies cannotreproduce the cutaneous environment, which repre-sents a unique determinant for the invading cells. Theapplication of laser microdissection of tissue sectionscould overcome this problem, because it allows thedetection of changes in the transcriptome of in situcells also reflecting intercellular cross-talk.15, 20 Incontrast to former microdissection approaches (e.g.needle microdissection), laser-assisted microdissec-tion allows the isolation of a small number of tumorcells avoiding contamination.21
In a previous study,22 we combined laser microdis-section and cDNA array analysis to detect differencesin the gene expression profile between the invasivemargins and the cores of nine nodular malignantmelanomas. For data analysis, we suggested a novelstrategy for gene sorting according to the signal-to-noise statistic.17, 23 In addition, for each of the 9 genesrelative expression factors (invasive rim/tumor cen-tre) were calculated for all cases. Using such algo-rithms, a profile of 9 selected markers was establishedto distinguish between samples derived from invasivetumor regions and samples from non-invasive regionsin a hierarchical cluster analysis. Taken together, 15 of18 tumor regions (83.3%) were classified correctly.
Regarding the differential expression between tumormargins and cores of all melanomas, phospho-enolpyruvate carboxykinase 1 (PEPCK) and twoanonymous ESTs (R52541 and N73761) were typi-cally upregulated in the invading edge. PEPCK is amain switch for regulation of gluconeogenesis in orderto adjust glucose production to physiologic require-ments. In terms of melanoma expansion, the role ofPEPCK remains unclear. The upregulation of PEPCKin the invasive margin could reflect the higher degree

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 111
NEW MOLECULAR BIOMARKERS FOR MALIGNAT MELANOMA ROESCH
of glucose metabolism in those cells, which may needmore energy for migration and invasion. New datagenerated in hepatoma cell lines also showed an upreg-ulation of PEPCK in cells cultured at high densitysuggesting a possible role of PEPCK in the metabolismof cells competing with each other.24
In contrast to that, Homo sapiens similar to S. cervisi-ae SSM4 (TEB4) had higher relative expression ratiosin the melanoma centres. In S. cerevisiae, mutations inthe SSM4 locus, suppress the cell growth phenotypeas well as mRNA instability.25, 26 Therefore, the rela-tive absence of TEB4 activation in the invading edgemay increase growth, possibly by an alteration ofnuclear mRNA stability. We further measured anincreased central expression value of the human homo-logue of A. nidulans SudD suppressor of bimD6 homo-logue across 8 of nine melanoma experiments. In A.nidulans, BimD6 mutation is thought to be associatedwith a failure of chromosomes to correctly attach to thespindle microtubules, resulting in an increase in chro-mosome loss. SudD is identified to be a suppressor ofthe bimD6 mutation. Its product is a highly conservedprotein that is found in a variety of eukaryotes from fun-gi to man.27 Accordingly, in humans SudD could rep-resent an important regulator of chromosomal home-ostasis, but its exact function in the pathogenesis ofmelanoma remains to be elucidated. Ribosomal pro-tein L19 was shown to be induced in the tumor centre.In human breast cancer with overexpression of erbB-2, mRNA levels of L19 were also found to be moreabundant than in cells with low erbB-2 expressionsuggesting a potential role in carcinogenesis.28 How-ever, for cutaneous neoplasms no relationship has beenfound so far. The a subunit of the IL-3 receptor, a veryimportant player in the transmission of growth regu-latory signals, also showed a higher expression in cellsmicrodissected from melanoma centres. In the lastyears, the impact of the IL-3 receptor subunits a and bon cell cycle regulation and apoptotic pathways havebeen controversially discussed. For instance inhematopoetic cells, both subunits may be involved intumor proliferation as well as in growth inhibition.But, in contrast to the b chain, the less characterised asubunit is supposed to be much more ligand specific.29
Hence, an association of IL-3 seems to be conceiv-able. Inositol 1,4,5-triphosphate 3-kinase isoenzyme,was also found to be upregulated in melanoma cores.Being part of Ca2+-mediated intracellular signalling,this kinase phosphorylates the second messenger mol-
ecule inositol 1,4,5-triphosphate (IP3) to inositol1,3,4,5-tetrakisphosphate (IP4). In contrast to IP3, IP4mediates slower and more prolonged responses in thecell, but both messengers have important functions inthe regulation of cell proliferation.30 Conclusively, anenhanced conversion of IP3 to IP4 as well as describedalterations of TEB4, PEPCK, SudD, L19 and Il-3receptor a subunit might influence cell proliferation andinfluence melanoma growth, and its infiltrative char-acter, respectively.
Taken together, with this straightforward approach,we could specify some novel differences between cellsfrom the invasive margin and the centre of melanomasthat emerge from transcriptome-wide screening. Thesefindings could both foster our understanding of tumorbiology and spark new functional investigations. Inaddition, genes exhibiting an enhanced activation in theinvasive margin could potentially serve as prognosticmarkers.
Dipeptidyl peptidase IV as a new marker fordiscrimination of malignant melanomas from
deep penetrating nevi
In 1989, Seab et al.7 reported a series of highly inva-sive, but non-metastasizing pigmented melanocytictumors and invented the term deep penetrating nevus(DPN). DPN mostly appear as darkly pigmentedpapules or nodules with no or mild epidermal changesand are most commonly found in the face, on the uppertrunk or proximal extremities of patients at the age of10 to 30 years.7, 31 Histopathologic growth patternsare often worrisome showing a wedge-shaped inva-sive growth extending from the upper dermis into thesubcutaneous fat tissue not rarely following preformedstructures, e.g. hair follicles or sweat glands.7, 32 Thus,DPN are often mistaken for vertical growth phasenodular malignant melanomas (NMM), both clinical-ly and histologically.7, 32-37 Estimates exist that, depend-ing on the criteria used for classification, misdiag-noses as melanoma occur in 29% to 40% of the cases.7,
31, 32 Beyond the clinical demand for precise diagnosis,the DPN may also represent a valuable natural modelfor melanocytic invasion without metastatic potentialand deserves further elucidation.
In previous studies, several immunohistochemicalattempts with standard melanoma markers, such as S-100, failed to differentiate DPN from NMM.7, 38 Beforewe initiated our experiments, only one study existed

ROESCH NEW MOLECULAR BIOMARKERS FOR MALIGNAT MELANOMA
112 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
suggesting PCNA (proliferating cell nuclear antigen)as a possible discriminating marker.39 PCNA repre-sents an accessory protein of DNA δ-polymerase whichis increased during the late G1 growth phase and peaksin the S phase of the cellular cycle. However, PCNAnever entered routine diagnostics, probably due to alack of studies with higher numbers of cases. So, weexpanded on the search for new discriminating mark-ers analyzing an empirical selection of common can-didate markers determining cell proliferation controlor melanocytic invasion. However, against our expec-tations, semi-quantitative assessment of bothimmunolocalization and immunoreactivity of the pro-liferation markers MIB-1/Ki-67, retinoblastoma pro-tein (pRb) and its activity-reduced form phospho-retinoblastoma protein Ser795 (p-pRbSer795) revealedno consistent differences between DPN and matched
cases of NMM. Moreover, also the invasion-relatedmarkers matrix metalloproteinase-1, matrix metallo-proteinase-2, membrane-type matrix metalloproteinase-1 and integrin β 3 showed no significant differences inexpression.9 According to the highly invasive charac-ter of DPN, matrix metalloproteinase-1 and matrixmetalloproteinase-2 immunostaining of some DPNeven exceeded that of NMM, thereby, confirming theirrole in tumor infiltration, but, at the same time, ques-tioning their importance for metastasizing.
In addition, we stained for a new marker that waspreviously shown to affect both invasion and prolif-eration, the dipeptidyl peptidase IV (DPPIV).40-42 Inter-estingly, in our immunostainings, the mean expres-sion scores for DPN clearly exceeded that of NMMwith P<0.001. All DPN stained highly positive, where-as most melanoma samples showed low or no expres-
DPN
NMM
A B
C D
Controls
E F
Figure 1.—DPPIV represents a potential discriminating markerbetween DPN and NMM (modified from Roesch et al.,9 with kindpermission by the Nature Publishing Group). A) DPPIV expres-sion in a DPN with its typical diffuse staining pattern (100x). B) 400xmagnification. C) Example of a NMM showing only single positivecells (100x), D) 400x magnification. E) Negative control (100xoverview). F) Positive control (sebaceous gland, 400x magnification).G) Immunoreactivity was semi-quantitatively assessed regardingstaining quantity and subcellular staining intensity in 11 DPN and16 NMM. Staining quantity and intensity were summarized to asingle expression score (ES) as recently published. The ES valuesof each sample were grouped into one of 4 ES categories (ES 0,ES 0-40, ES 40-80 and ES >80) and displayed as percentage of allsamples of one entity. Differences were statistically significant withhigher scores in DPNs (U-test P<0.001).
Perc
enta
ge o
f tu
mor
sam
ple
100
80
60
40
20
0DPN (n=11) NMM (n=6)
ES >80 ES 40-80 ES 0-40 ES 0
G
P <0.001

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 113
NEW MOLECULAR BIOMARKERS FOR MALIGNAT MELANOMA ROESCH
sion (Figure 1). The intratumoral immunolocalizationof DPPIV in both entities showed a diffuse expres-sion pattern with focal accumulation close to cuta-neous adnexes or at the tumor periphery. Thus, onlyDPPIV kept its promise to be a protein that possiblyseparates infiltrative tumor growth from truemelanocytic malignancy.
DPPIV (CD26) is a 110-kD, trans-membrane, cellsurface peptidase expressed by normal melanocytesand common nevi, but primary and advanced malignantmelanomas almost invariably lose or alter their DPPIVexpression.40-42 DPPIV has numerous functions includ-ing involvement in T-cell activation, cell adhesion,digestion of proline containing peptides in the kidneyand intestines, HIV infection and apoptosis, and reg-ulation of tumorigenicity in certain melanoma cells.43
Functionally, DPPIV re-expression leads to re-differ-entiation and an acquired dependence on exogenousgrowth factors, but it also favors loss of tumorigenic-ity and anchorage-independent growth.42 Thus, highDPPIV expression was found correlated with lessmetastatic potential in vivo.44 The effect of DPPIVappears to be mediated through several mechanisms:1) by up-regulation of other factors such as E-cad-herin and tissue inhibitors of matrix metallopro-teinases;45 2) by its ability to bind components of theextracellular matrix such as collagen or fibronectin;43,
44, 46 and, most interesting for melanoma biology, 3) byits inhibition of mitogen-activated protein kinase(MAPK)-extracellular signal-regulated kinase(ERK)1/2 activation.47 Since loss of dipeptidyl pepti-dase IV may be causally linked to the transition ofinvasive to metastatic phenotypes, the molecular mech-anisms downstream of dipeptidyl peptidase IV deserveto be studied in more detail in future investigations.
Taken together, in the case of DPPIV, a strategy thatsingles out the most promising candidates based onprevious evidence led to the establishment of a novelmarker also with therapeutic implications.
Hyperphosphorylated retinoblastoma proteinrepresents a possible prognostic marker in the
progression of malignant melanoma
The main principle of cell cycle homeostasis is theintegrated phosphorylation-dependent activation anddeactivation of regulatory proteins upstream of theretinoblastoma protein (pRb), which represents thecentral cycle-controlling element. At the G1/S transi-
tion, the antiproliferative function of active, hypophos-phorylated pRb is mediated by its binding capacity topro-proliferative transcription factors such as E2F orc-Myc.48 Surprisingly, with the exception of retinoblas-toma, osteosarcoma and small cell lung carcinoma,in the vast majority of human cancers the overall rateof pRb-mutations is either extremely low or not exis-tent.49 Also in melanoma, the loss of cell cycle controlis thought to be due to a lack of pRb-activity and notto lack of expression or mutation.50, 51 Currently, theconcept is arguably accepted that persistent inactiva-tion and hyperphosphorylation of wild-type pRb inmelanoma is caused by a sustained cyclin dependentkinase activity (CDK2, CDK4, CDK6).52, 53 Substan-tial experimental evidence shows that this persistentkinase activity can be due to 1) genetic alterationseliminating the activity of cyclin dependent kinaseinhibitors (CKIs) such as p16/INK4a; 2) a downregu-lation of CKIs, e.g. p27/KIP1; 3) the persistent over-production or mutation of several cyclins (e.g. D1, E,A); or 4) a combination of those factors.54
Due to a lack of studies on pRb phosphorylationstatus in in vivo samples of cutaneous melanocytictumors, we analyzed total and phospho-pRb expressionin tumor biopsies of progressive and non-progressivemelanocytic lesions including common and dysplas-tic nevi, superficial spreading (SSM) and nodularmalignant melanomas (NMM), melanoma metastasesand, as control, further neuroectodermal tumors suchas glioblastomas, schwannomas and neurofibromas.8Relative measurements of Rb mRNA expression inmicrodissected melanocytic tumors biopsies revealeda significant almost 3-fold overexpression of Rb mRNAin melanoma and melanoma metastases versus benignmelanocytic nevi. In contrast, expression levels of var-ious non-neuroectodermal malignant control tissues(lung carcinoma, breast carcinoma, colon carcinoma)showed uniformly low expression levels. In accor-dance with the RT-PCR data, tissue microarray (TMA)-based immunohistochemistry confirmed gross con-stitutive differences in pRb-expression (Figure 2).Interestingly at phospho-pRb Ser795, we found analmost complete phosphorylation of the progressive-ly overexpressed total pRb, whereas in common nevionly a fraction of total pRb was phosphorylated. Phos-phorylation at Ser807/811 generally occurred to a less-er extent, but significantly higher in metastases com-pared to nevi. In case of Ser780, no significant differ-ential phosphorylation was detectable. To analyse pos-

ROESCH NEW MOLECULAR BIOMARKERS FOR MALIGNAT MELANOMA
114 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
sible intratumoral heterogeneity that may be missed byTMAs due to limited punch size, we also analyzedsections with preserved full tumor architecture (Figure3). Expression scoring for total pRb in complete tumorsections was in support of our RT-PCR results, i.e. aprogressive increase of pRb in malignant melanocyt-ic tumors. Comparing expression scores from com-plete sections with TMA-based results, the progressiveincrease in phosphorylation could be confirmed forall three phospho sites analyzed. Furthermore, thecomplete tissue sections did reveal considerable intra-tumoral heterogeneity of total pRb expression andpRb phosphorylation. In SSM, subepidermal vs dermalexpression scores were significantly different with themost intense staining result in the subepidermal-lateralportions (P<0.005). In NMM, the invasive part vstumor centre (P<0.001) and in melanoma metastases
outer margins vs centers (P<0.003) also showed sig-nificant differences. Other benign neuroectodermaltumors that served as independent controls such asschwannomas and neurofibromas showed a compa-rably low “melanocytic nevus-like” pRb-expressionwith no significant intratumoral heterogeneity. Inmetastases, an accumulation of phospho-pRb stain-ing was mainly detected in the outer margins com-pared to the tumor centre. In contrast, in benign anddysplastic nevi as well as schwannomas/neurofibromaspRb phosphorylation was almost negligible.
Our data suggest that particularly Ser795 is a goodchoice for estimating both the presence of pRb andits degree of inactivation. Specifically Ser795 is amongthose phosphorylation-sites that undergo hyperphos-phorylation by CDK2-E in late G1. This results in therelease of E2F that finally leads to an enhanced tran-
Bad quality spots Negative Positive
p-pRb Ser780
p-pRb Ser795 p-pRb Ser807/811
Total pRb
A B
%
100
80
60
40
20
0MN MMM
15
48.33
36.67
16.67
21.67
61.67
%
100
80
60
40
20
0MN MMM
15
51.67
33.33
63.33
15
21.67
C D
%
100
80
60
40
20
0MN MMM
15
63.33
21.67
20
16.67
63.33
%100
80
60
40
20
0MN MMM
13.33
85
1.07
58.33
23.33
18.33
Figure 2.—Tissue microarray-based evaluation of total pRb- and phosphorylated pRb Ser795/Ser780/Ser807/811-expression in 60melanocytic nevi MN versus 60 melanoma metastases MMM (from Roesch et al.,8 with kind permission by the Nature PublishingGroup). Depicted are percentages of positively and negatively stained samples as well as bad quality spots.

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 115
NEW MOLECULAR BIOMARKERS FOR MALIGNAT MELANOMA ROESCH
Figure 3.—Staining patterns of pRb and phosphorylated pRb Ser795 in melanocytic tumors (from Roesch et al.,8 with kind permissionby the Nature Publishing Group). First row (200x magnification): negative control with omitted primary antibody (epidermis, A), posi-tive controls (epidermis, B and sebaceous gland, C). Second row (200x magnification): increasing total pRb expression in malignantmelanocytic tumors: benign melanocytic nevus (MN, D), nodular melanoma (NMM, E) and melanoma metastasis (MMM, F). Third row:phospho-pRb-staining of a superficial spreading melanoma (SSM). Overview (100x magnification) showing a high regional variance ofphospho-pRb staining with a maximum in subepidermal-lateral nests versus dermal melanoma nests (G). Details: 400x magnification ofa highly phosphorylated subepidermal melanoma nest (H) in comparison to dermal nests (I). Forth row: phospho-pRb-stained nodularmelanoma (NMM). Overview (100x magnification) showing an increase of pRb-phosphorylation in the invasive basal front (J). Details:400x magnification of the invasive front (K) and almost complete absence of phospho-pRb in central tumor areas (L).

ROESCH NEW MOLECULAR BIOMARKERS FOR MALIGNAT MELANOMA
116 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
scription of S-phase-genes such as DNA polymerasea and dihydrofolate reductase.48, 55-58 In contrast toSer795, Ser780 and Ser807/811 are exclusively phos-phorylated by CDK4-D.48, 56, 57, 59 InterestinglyEzhevsky et al. reported that CDK4-D-mediated phos-phorylation of pRb is a prerequisite for the transitionof unphosphorylated inactive pRb into hypophospho-rylated active pRb.60 This suggests that the increasein pRb phosphorylation at Ser780 detected in benignnevi represents active cell cycling suppressing pRb.Therefore it could be worthwhile to further unravelthe details of the phospho status of all 16 pRb phosphosites in melanoma. However, currently the usefulnessand practical value of a diversified analysis of all 16phospho residues of pRb remains elusive due to lim-ited availability of suitable antibodies.
To disclose a potential association of the histologi-cally detectable degree of pRb phosphorylation andthe prognosis of melanoma patients, we additionallyanalyzed a panel of long-term survivors with thick,high-risk primary melanoma (TD >3.5 mm). We founda clear trend to higher phospho-pRb expression inshort-time survivors versus exceptional long-time sur-vivors in our cohort of matched survivors and non-survivors. Low phospho-pRb expression was signifi-cantly correlated with a prolonged survival, whereashigh phospho-pRb expression was correlated with ear-ly death (log-rank-rest P=0.03). This suggests a pos-sible Breslow-(tumor-thickness)-independent prog-nostic impact of pRb-phosphorylation.
In conclusion, we could confirm that 1) in cuta-neous malignant melanocytic tumors total pRb-expres-sion is progressively upregulated and that 2) simulta-neously, augmented total pRb is probably functional-ly inactive due to protein hyperphosphorylation, par-ticularly at Ser795. Interestingly, a high degree of pRb-phosphorylation can be observed in tumor-regionswith expansive growth activity, i.e., the deep invasivefront of the vertical growth phase nodular melanomasand the lateral-subepidermal parts of superficial spread-ing melanomas. Accordingly, the benign counterpartsanalyzed (melanocytic nevi, schwannomas, neurofi-bromas) show a moderate, mostly homogeneousexpression of pRb and pRb-phosphorylation is dif-fuse and negligibly weak. Furthermore, Kaplan-Meieranalysis of advanced melanomas with long-time fol-low-up suggested a significant negative impact of pRb-phosphorylation on survival independent of tumorthickness.
The retinoblastoma binding protein 2-homolog 1:a new tumor suppressive marker downregulated
in malignant melanomas
As described above, the loss of cell cycle controlin malignant melanomas is thought to be due to a lackof retinoblastoma protein (pRb)-activity and not to alack of its expression or mutation.50, 51, 61 To find newpRb-binding proteins (RBPs) functioning as poten-tial pRb-modulating factors, Defeo-Jones et al.screened a human expression cDNA library with arecombinant Rb probe and identified two novel proteinstermed RBP-1 and RBP-2 (alternatively “RBBP1,RBP1; RBBP2, RBP2”). Both proteins contain high-ly conserved pRb-binding motifs, which show a strik-ing homology with viral oncogenic proteins such as E7,large T and E1A.62
In 1999, we have described a novel homolog ofRBP-2, termed RBP2-homolog 1 (RBP2-H1, NCBIgenebank Acc. No. AF087481).63 The correspondingtranscript was detected due to its UV-B responsive-ness in normal non-transformed human melanocytesusing RNA fingerprinting. It encodes a protein with a54% amino acid identity with RBP-2. Further com-puterized sequence analyses revealed highly conservedmotifs with possible functional implications. Amongthose, two DNA-binding zinc finger (leukemia-asso-ciated protein, LAP) motifs, a rhombotin-2 (RBTN2,LMO2) binding domain and a domain possibly medi-ating a direct binding and interaction with pRb (non-T/E1A-pRb binding domain) were most remarkable.Since the sequence analysis also found the ancienthomeodomain dri (dead ringer protein), it was recent-ly proposed to group RBP-2-homologue proteins intothe superfamily of ARID (AT rich interactive domain)DNA binding proteins.64 Two more RBP2-H1-homologtranscripts have been described so far that are differ-entially regulated in breast carcinomas, RBBP2H1aand PLU-1.65, 66 RBBP2H1a, PLU-1 and RBP2-H1represent three alternative splicing variants of the PLU-1 gene.67 RBP2-H1 is distinguished by one extra exonof 108 bp. The RBBP2H1a transcript has a muchlonger 5’-UTR and an ATG upstream of that used forRBP2-H1 and PLU-1. On cDNA level, RBP2-H1shows a 98.5% identity to PLU-1 and a 98.0% identi-ty to RBBP2H1a. The comparison of RBP2-H1 withRBP-2 reveals a cDNA homology of 37.8%. On pro-tein level, 54.0% of the amino acids of RBP2-H1 andRBP-2 are identical. Sequence analysis of the threesplicing variants revealed further highly conserved

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 117
NEW MOLECULAR BIOMARKERS FOR MALIGNAT MELANOMA ROESCH
functional motifs. Applying the new HUGO nomen-clature,64 PLU-1 gene derivates would now be termedas JARID1B variants, e.g. JARID1B_v1 for PLU-1,JARID1B_v2 for RBP2-H1 and JARID1B_v3 forRBBP2-H1a according to the respective dates of pub-lication. The corresponding protein isoforms should bedenoted as JARID1B_i1, JARID1B_i2 andJARID1B_i3.
Since previous work has gained preliminary evi-dence that RBP2-H1 mRNA can be downregulated inUV-irradiated melanocytes and possibly in malignantmelanomas,63 we subsequently investigated mRNAexpression of RBP2-H1 by real-time RT-PCR.68 Com-pared to melanocytic nevi, RBP2-H1 mRNA was notsignificantly decreased in melanomas, but significantlydecreased in melanoma metastases. The splicing vari-ant RBBP2H1a also showed the most prominentexpression in benign melanocytic tumors. There wasa similar, but not significant trend to a deficient expres-sion in melanomas and metastases. Interestingly, sim-ilar to melanomas and melanoma metastases, glioblas-tomas, used as external controls, also showed a relativeloss of RPB2-H1 compared to fetal brain. In contrast,epithelial cancers, in particular breast cancer, showeda comparably high RBP2-H1 expression and only mar-ginal RBBP2H1a expression. The progressive defi-ciency of RBP2-H1 mRNA from common nevi tomelanoma and melanoma metastasis could be alsoconfirmed on protein level. TMAs were examined toevaluate gross constitutive differences in RBP2-H1-expression between benign melanocytic lesions andmalignant melanocytic tumors (Figure 4). In accor-dance to our RT-PCR data, TMA-based immunohis-tochemistry confirmed a progressive deficiency ofRBP2-H1 expression from nevi (TMA No. 1, n=52) tomelanoma (TMA No. 2, n=60) and melanoma metas-tases (TMA No. 3, n=60). Almost 70% of the spottednevus samples were classified as “RBP2-H1-expres-sors”. Strikingly, in melanomas, RBP2-H1 stainingwas only detected in primary radial growth phasemelanomas with a cut-off tumor thickness of ≤1.6mm. In this group 31.6% were positive which addsup to only 10% of all melanomas spotted on TMANo. 2. In thicker more advanced melanomas (>1.6mm) no staining signals could be found. However, inmelanoma metastases, 30% of the samples did expressRBP2-H1. With regard to the whole tumor architecture,no region-specific distribution (e.g. tumor front ortumor core) of RBP2-H1 could be detected in completetissue sections. However, certain heterogeneity of
staining results (number, intensity) across the full sec-tions could be seen. In accordance with our mRNAmeasurements and the TMA analyses, the highestRBP2-H1 expression scores were found in nevi witha significant decrease in both melanomas and metas-tases, respectively (Figure 5). Other benign neuroec-todermal tumors that served as independent controlssuch as schwannomas and neurofibromas showed acomparably high “melanocytic nevus-like” RBP2-H1-expression.
Since RBP-2 was suggested to be involved in pRb-phosphorylation control by direct protein-protein inter-action,62 we addressed the question whether RBP2-H1, the closest homolog of RBP-2, could also bind topRb. While RBP2-H1 lacks the typical Leu-X-Cys-X-Glu-motif of some pRb-interacting proteins, it bearsa homolog-domain of the non-T/E1A-pRb bindingdomain of RBP-2.63 The latter could theoretically con-fer a direct interaction with pRb and make RBP2-H1a true pRb-binding protein. Thus, co-immunoprecip-itation studies were performed with cells that endoge-nously express RBP2-H1 at sufficient levels, such asMCF-7 breast carcinoma cells. By this, we coulddemonstrate that wild type RBP2-H1 can truly bind topRb suggesting a possible functional interaction ofboth proteins.68
In a following study,69 we re-established the pRbmodulating function of RBP2-H1 in highly metastat-ic A375-SM melanoma cells by re-expressing its C-
Perc
enta
ge o
f sta
ined
sam
ple
spot
s 100
80
60
40
20
0Nevus
TMA No. 1MelanomaTMA No. 2
MetastasisTMA No. 3
Bad quality spots Negative Positive
11.67 18.33
78.3351.67
30
10
69.23
21.15
9.62
Figure 4.—Tissue microarray-based evaluation of RBP2-H1-expres-sion (from Roesch et al.,68 with kind permission by the Nature Pub-lishing Group). TMA No. 1 comprised 52 melanocytic nevi, TMANo. 2 60 malignant melanomas and TMA No. 3 60 melanomametastases. Depicted are percentages of positively and negativelystained samples as well as bad quality spots.

ROESCH NEW MOLECULAR BIOMARKERS FOR MALIGNAT MELANOMA
118 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
term (cRBP2-H1) encoding the non-T/E1A-pRb bind-ing domain. Our results demonstrated that theexpressed cRBP2-H1 binds to pRb and, moreover,exerts a continuous hypophosphorylation at pRbSer795 during the whole G1 phase of the cell cycle,whereas in untreated A375-SM and control virus infect-ed A375-SM cells, phosphorylation at Ser795 slight-ly increases in late G1. Ser795 has been described tobe a phosphoacceptor site which is implicitly requiredfor activation of pRb cell cycle arrest function.70 As asubstrate of cdk4/cyclin D and cdk2/cyclin E,70 Ser795is supposed to be among those phospho-sites thatundergo progressive hyperphosphorylation from ear-ly to late G1 phase and, therefore, might be directlyinvolved in G1/S transition. The observation that inuntreated A375-SM cells, Ser795 is partly phospho-rylated already in early G1, perhaps due to an addi-tionally uncontrolled cdk4/cyclin D activity general-ly discussed in melanomas,71 further reinforces therole of cRBP2-H1 as a stabilisator of hypophospho-rylated pRb, since in cRBP2-H1-transduced cells,hypophosphorylated Ser795 was detected already inearly G1, too. The exclusively cdk4-targeted phos-phoacceptor sites Ser780 and Ser807/811 lacked toshow significant differences in phosphorylation in ear-ly as well as late G1 in cRBP2-H1-transfected cells.Therefore, we suggested that RBP2-H1 does not act asa cdk-specific inhibitor, but as a pRb phosphoaccep-tor site-specific stabilisator, although we have not yetinvestigated the remaining 12 pRb phospho-sites ofpRb. A further argument in favor of this hypothesis isthat a direct interaction of retinoblastoma binding pro-teins with cdks has never been detected so far.
As demonstrated by cDNA microarrays and con-
firmed by quantitative RT-PCR, we found a differen-tial expression of various melanoma-associated geneswhich are, only in part, directly linked to the pRb/E2Fpathway; e.g. BMP2, FST and TGFA.72 TGFA, down-regulated in cRBP2-H1-transduced cells, was shownto be involved in autocrine growth of melanoma cells.73
FST, also downregulated, inhibits activin-mediatedgrowth reduction and apoptosis in melanoma cells.74
Finally, BMP-2, upregulated in transduced cells, wasshown to reduce the secretion of HGF,75 which repre-sents a potent pro-proliferative cytokine eliciting mito-genic, motogenic and morphogenic responses inmelanoma cells.76 Consistent with BMP-2 upregula-tion in cRBP2-H1-transduced cells, HGF was found tobe strongly suppressed. Thus, for all four candidates,FST, TGFA, BMP-2 and HGF, we found transcrip-tional responses typical for a more benign melanocyt-ic phenotype. Beside to E2F downstream target genes,further target genes of other pRb-inhibited transcrip-tion factors, such as SPI1 and PP-1alpha2 (notshown),58 were identified by IngenuityTM.
Since pRb itself was not differentially expressedafter transduction, the transcriptional regulation ofdownstream genes might be caused by a functionalmodulation of pRb and pRb-inhibited factors on theprotein level, e.g. due to an altered E2F release afterRBP2-H1/pRb-interaction. However, common E2F-transactivated S phase genes, such as DHFR, TK1,DNA-polymerase alpha were not found differentiallyexpressed. Therefore, further, yet unknown mecha-nisms, independent from pRb, might be involved, too.One example of cRBP2-H1-activity that is probablyindependent of pRb is the upregulation of TCF4 whichrepresents another highly melanoma-relevant path-
Figure 5.—Staining patterns of RBP2-H1 in melanocytic lesions (from Roesch et al.,68 with kind permission by the Nature Publishing Group).Immunohistochemical staining of whole tissue sections (200x magnification) reflects the deficiency of RBP2-H1 in malignant melanomaand melanoma metastases. Common melanocytic nevus (A), advanced nodular melanoma (B), and melanoma metastasis (C).

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 119
NEW MOLECULAR BIOMARKERS FOR MALIGNAT MELANOMA ROESCH
way. Transcription factors of the TCF family are down-stream effectors of the Wnt/‚-catenin signaling path-way which is implicated in developmental processesand progression of some tumors, especially colon car-cinoma and melanoma.77, 78 Depending on the inter-action with co-regulative factors like ‚-catenin or Grou-cho (both not regulated), TCF can act as transcrip-tional activator or repressor. In absence of ‚-catenin,TCF represses the transcription of several target genes,including DCT and TYRP1.79, 80 Next to tyrosinase,both DCT and TYRP1 represent major pigmentationenzymes of melanocytes.81 Recently, Widlund and co-workers suggested that also MITF is a target gene ofWnt/‚-catenin/TCF signaling pathway.81 MITF, strong-ly downregulated upon re-expression of RBP2-H1, isdiscussed as the master gene for melanocytic survivalas well as the key transcription factor for regulation ofmelanocytic protein expression.77, 81 Therefore, thestriking downregulation of MITF in our experimentsalso indicated a more benign gene expression pheno-type of cells re-expressing cRBP2-H1 and assignsRBP2-H1 to the genes with tumor-suppressive action.
As a possible result of the observed changes in pRbinteraction and gene expression, a block in G1/S tran-sition was detected in cell cycle profile analyses ofRBP2-H1-transduced cells. This observation and arecorded decrease in cellular proliferation also sup-port the hypothesis that RBP2-H1 exerts a tumor-sup-pressive function in melanocytes. In conclusion, ourdata suggest that the progressive loss of RBP2-H1might not only serve as a diagnostic and prognosticmarker for melanoma, but re-establishing parts of itsfunction in metastatic melanomas may open newavenues of experimental strategies for cure of metasta-tic malignant melanoma.
Concluding remarks
In the last years, we have detected several new can-didate markers of melanocytic tumorigenesis affectingcell cycle control, stroma infiltration and gene tran-scription regulation. We linked their functions to knowntumor pathways and found, especially for RBP2-H1and DPPIV, a potential involvement in malignant trans-formation and melanoma progress. We propose thatRBP2-H1 and DPPIV deserve further investigations infuture projects targeting on both the establishment asdiagnostic parameters and the exploration in a possi-ble therapeutic context. However, since it was repeat-
edly demonstrated that melanomagenesis is not causedby failure of one cellular check point, but by a concertof disrupted regulatory factors, we suppose that mark-er-targeted diagnosis and therapy of malignantmelanoma can only be significantly improved using amultifactorial approach.
Riassunto
Nuovi biomarcatori molecolari per il melanoma Il melanoma rappresenta il carcinom cutaneo più letale
per il genere umano e la sua incidenza è in aumento in tuttoil mondo. Spesso i melanomi metastatizzano precocementenel corso della malattia e divengono quasi intrattabili con gliattuali regimi terapeutici. Di conseguenza, la comprensionedei fattori che mantengono l’omeostasi dei melanociti e laprevenzione della loro trasformazione neoplastica nel mela-noma rappresentano degli aspetti di fondamentale interesse.Le terapie molecolari mirate sono dirette nei confronti delleanormalità genetiche che sono associate alla progressionedel tumore. In questa review gli autori prendono in conside-razione la ricerca di nuovi biomarcatori diagnostici e di nuo-vi obiettivi terapeutici, utilizzando diverse tecniche ed approc-ci molecolari, quali i cDNA microarray, la RT-PCR, l’im-munoistochimica convenzionale e i microarray tissutali sucampioni in vivo e in vitro. Gli autori inoltre descrivono in det-taglio i loro dati relativi ai marcatori più recenti, quali il pRbSer795 fosforilato, l’omologo 1 della proteina 2 legante la pro-teina del retinoblastoma (retinoblastoma protein binding pro-tein 2-homolog 1: RBP2-H1) e la dipeptidil peptidasi IV(DPPIV) e li correleremo ai meccanismi già noti della pato-genesi del melanoma. Dal momento che è possibile che imarcatori descritti possano correlare casualmente con la tra-sformazione maligna, i loro meccanismi di segnale moleco-lare devono essere studiati più in dettaglio in futuro.PAROLE CHIAVE: Melanoma, anatomia patologica - Markerbiologici - Cute, neoplasie.
References
1. Thompson JF, Scolyer RA, Kefford RF. Cutaneous melanoma. Lancet2005;365:687-701.
2. Smalley KS, Herlyn M. Targeting intracellular signaling pathwaysas a novel strategy in melanoma therapeutics. Ann N Y Acad Sci2005;1059:16-25.
3. Chudnovsky Y, Khavari PA, Adams AE. Melanoma genetics and thedevelopment of rational therapeutics. J Clin Invest 2005;115:813-24.
4. Vogelstein B, Kinzler KW. Cancer genes and the pathways they con-trol. Nat Med 2004;10:789-99.
5. Pollock PM, Harper UL, Hansen KS, Yudt LM, Stark M, RobbinsCM et al. High frequency of BRAF mutations in nevi. Nat Genet2003;33:19-20.
6. Roesch A, Burgdorf W, Stolz W, Landthaler M, Vogt T. Dermatosco-py of “dysplastic nevi”: a beacon in diagnostic darkness. Eur J Der-matol 2006;16:479-93.

ROESCH NEW MOLECULAR BIOMARKERS FOR MALIGNAT MELANOMA
120 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
7. Seab JA Jr., Graham JH, Helwig EB. Deep penetrating nevus. Am JSurg Pathol 1989;13:39-44.
8. Roesch A, Becker B, Meyer S, Hafner C, Wild PJ, Landthaler M et al.Overexpression and hyperphosphorylation of retinoblastoma protein inthe progression of malignant melanoma. Mod Pathol 2005;18:565-72.
9. Roesch A, Wittschier S, Becker B, Landthaler M, Vogt T. Loss ofdipeptidyl peptidase IV immunostaining discriminates malignantmelanomas from deep penetrating nevi. Mod Pathol 2006;19:1378-85.
10. Clark WH, Jr., Elder DE, Van Horn M. The biologic forms of malig-nant melanoma. Hum Pathol 1986;17:443-50.
11. Gutgemann A, Golob M, Muller S, Buettner R, Bosserhoff AK. Iso-lation of invasion-associated cDNAs in melanoma. Arch DermatolRes 2001;293:283-90.
12. Albino AP, Reed, JA, McNutt, NS. Molecular biology of cutaneousmalignant melanoma. In: DeVita V, Hellman S, Rosenberg SA eds.Cancer: Principles and practice of oncology. 5th edition. Philadel-phia: Lippincott-Raven Publishers; 1997.p.1935-46.
13. Hofmann UB, Westphal JR, Van Kraats AA, Ruiter DJ, Van MuijenGN. Expression of integrin alpha(v)beta(3) correlates with activa-tion of membrane-type matrix metalloproteinase-1 (MT1-MMP) andmatrix metalloproteinase-2 (MMP-2) in human melanoma cells invitro and in vivo. Int J Cancer 2000;87:12-9.
14. Kataoka TR, Ito A, Asada H, Watabe K, Nishiyama K, Nakamoto Ket al. Annexin VII as a novel marker for invasive phenotype of malig-nant melanoma. Jpn J Cancer Res 2000;91:75-83.
15. Fink L, Kohlhoff S, Stein MM, Hanze J, Weissmann N, Rose F et al.cDNA array hybridization after laser-assisted microdissection fromnonneoplastic tissue. Am J Pathol 2002;160:81-90.
16. Brown PO, Botstein D. Exploring the new world of the genome withDNA microarrays. Nat Genet 1999;21:33-7.
17. Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, MesirovJP et al. Molecular classification of cancer: class discovery and classprediction by gene expression monitoring. Science 1999;286:531-7.
18. Bittner M, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M et al.Molecular classification of cutaneous malignant melanoma by geneexpression profiling. Nature 2000;406:536-40.
19. Rumpler G BB, Hafner C, McClelland M, , Stolz W LM, Schmitt R,Bosserhoff AK, Vogt T. Identification of differentially expressed genesin models of melanoma progression by cDNA array analysis: SPARC,MIF and a novel cathepsin protease characterize aggressive phenotypes.Exp Derm 2002:12:761-71.
20. Mariani L, McDonough WS, Hoelzinger DB, Beaudry C, KaczmarekE, Coons SW et al. Identification and validation of P311 as a glioblas-toma invasion gene using laser capture microdissection. Cancer Res2001;61:4190-6.
21. Emmert-Buck MR, Bonner RF, Smith PD, Chuaqui RF, Zhuang Z,Goldstein SR et al. Laser capture microdissection. Science1996;274:998-1001.
22. Roesch A, Vogt T, Stolz W, Dugas M, Landthaler M, Becker B. Dis-crimination between gene expression patterns in the invasive marginand the tumour core of malignant melanomas. Melanoma Res2003;13:503-9.
23. Pomeroy SL, Tamayo P, Gaasenbeek M, Sturla LM, Angelo M,McLaughlin ME et al. Prediction of central nervous system embry-onal tumour outcome based on gene expression. Nature 2002;415:436-42.
24. Zeitouni N, Eubank DW, Lee AQ, Oxford MG, Freeman TL, MailliardME et al. Phosphoenolpyruvate carboxykinase is induced in growth-arrested hepatoma cells. Biochem Biophys Res Commun2002;290:1513-20.
25. Mandart E, Dufour ME, Lacroute F. Inactivation of SSM4, a newSaccharomyces cerevisiae gene, suppresses mRNA instability due torna14 mutations. Mol Gen Genet 1994;245:323-33.
26. Rouillard JM, Dufour ME, Theunissen B, Mandart E, Dujardin G,Lacroute F. SLS1, a new Saccharomyces cerevisiae gene involved inmitochondrial metabolism, isolated as a syntheticlethal in associati-on with an SSM4 deletion. Mol Gen Genet 1996;252:700-8.
27. Anaya P, Evans SC, Dai C, Lozano G, May GS. Isolation of theAspergillus nidulans sudD gene and its human homologue. Gene1998;211:323-9.
28. Henry JL, Coggin DL, King CR. High-level expression of the ribo-somal protein L19 in human breast tumors that overexpress erbB-2.Cancer Res 1993;53:1403-8.
29. Blalock WL, Weinstein-Oppenheimer C, Chang F, Hoyle PE, WangXY, Algate PA et al. Signal transduction, cell cycle regulatory, and anti-apoptotic pathways regulated by IL-3 in hematopoietic cells: possiblesites for intervention with anti-neoplastic drugs. Leukemia1999;13:1109-66.
30. Mengubas K, Jabbar SA, Nye KE, Wilkes S, Hoffbrand AV, Wickre-masinghe RG. Inactivation of calcium ion-regulating inositol polyphos-phate second messengers is impaired in subpopulations of humanleukemia cells. Leukemia 1994;8:1718-25.
31. Robson A, Morley-Quante M, Hempel H, McKee PH, Calonje E.Deep penetrating naevus: clinicopathological study of 31 cases withfurther delineation of histological features allowing distinction fromother pigmented benign melanocytic lesions and melanoma. Histo-pathology 2003;43:529-37.
32. Mehregan DA, Mehregan AH. Deep penetrating nevus. Arch Der-matol 1993;129:328-31.
33. Barnhill RL, Barnhill MA, Berwick M, Mihm MC, Jr. The histolog-ic spectrum of pigmented spindle cell nevus: a review of 120 cases withemphasis on atypical variants. Hum Pathol 1991;22:52-8.
34. Barnhill RL, Mihm MC, Jr., Magro CM. Plexiform spindle cell nae-vus: a distinctive variant of plexiform melanocytic naevus. Histopa-thology 1991;18:243-7.
35. Hoss DM, Grant-Kels JM. Significant melanocytic lesions in infan-cy, childhood, and adolescence. Dermatol Clin 1986;4:29-44.
36. Cooper PH. Deep penetrating (plexiform spindle cell) nevus. A fre-quent participant in combined nevus. J Cutan Pathol 1992;19:172-80.
37. Pulitzer DR, Martin PC, Cohen AP, Reed RJ. Histologic classificationof the combined nevus. Analysis of the variable expression ofmelanocytic nevi. Am J Surg Pathol 1991;15:1111-22.
38. Skelton HG, 3rd, Smith KJ, Barrett TL, Lupton GP, Graham JH. HMB-45 staining in benign and malignant melanocytic lesions. A reflectionof cellular activation. Am J Dermatopathol 1991;13:543-50.
39. Mehregan DR, Mehregan DA, Mehregan AH. Proliferating cell nuclearantigen staining in deep-penetrating nevi. J Am Acad Dermatol1995;33:685-7.
40. Houghton AN,Albino AP, Cordon-Cardo C, Davis LJ, Eisinger M. Cellsurface antigens of human melanocytes and melanoma. Expression ofadenosine deaminase binding protein is extinguished with melanocytetransformation. J Exp Med 1988;167:197-212.
41. Morrison ME, Vijayasaradhi S, Engelstein D, Albino AP, HoughtonAN. A marker for neoplastic progression of human melanocytes is acell surface ectopeptidase. J Exp Med 1993;177:1135-43.
42. Wesley UV, Albino AP, Tiwari S, Houghton AN. A role for dipep-tidyl peptidase IV in suppressing the malignant phenotype ofmelanocytic cells. J Exp Med 1999;190:311-22.
43. Pethiyagoda CL, Welch DR, Fleming TP. Dipeptidyl peptidase IV(DPPIV) inhibits cellular invasion of melanoma cells. Clin Exp Meta-stasis 2000;18:391-400.
44. Pro B, Dang NH. CD26/dipeptidyl peptidase IV and its role in cancer.Histol Histopathol 2004;19:1345-51.
45. Kajiyama H, Kikkawa F, Khin E, Shibata K, Ino K, Mizutani S. Dipep-tidyl peptidase IV overexpression induces up-regulation of E-cad-herin and tissue inhibitors of matrix metalloproteinases, resulting indecreased invasive potential in ovarian carcinoma cells. Cancer Res2003;63:2278-83.
46. Abdel-Ghany M, Cheng H, Levine RA, Pauli BU. Truncated dipep-tidyl peptidase IV is a potent anti-adhesion and anti-metastasis pep-tide for rat breast cancer cells. Invasion Metastasis 1998;18:35-43.
47. Wesley UV, McGroarty M, Homoyouni A. Dipeptidyl peptidaseinhibits malignant phenotype of prostate cancer cells by blocking

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 121
NEW MOLECULAR BIOMARKERS FOR MALIGNAT MELANOMA ROESCH
basic fibroblast growth factor signaling pathway. Cancer Res2005;65:1325-34.
48. Kaelin WG, Jr. Recent insights into the functions of the retinoblastomasusceptibility gene product. Cancer Invest 1997;15:243-54.
49. Ho A, Dowdy SF. Regulation of G(1) cell-cycle progression by onco-genes and tumor suppressor genes. Curr Opin Genet Dev 2002;12:47-52.
50. Bartkova J, Lukas J, Guldberg P, Alsner J, Kirkin AF, Zeuthen J et al.The p16-cyclin D/Cdk4-pRb pathway as a functional unit frequentlyaltered in melanoma pathogenesis. Cancer Res 1996;56:5475-83.
51. Halaban R, Miglarese MR, Smicun Y, Puig S. Melanomas, from thecell cycle point of view. Int J Mol Med 1998;1:419-25.
52. Czajkowski R, Drewa T, Wozniak A, Krzyzynska-Malinowska E.Cell cycle in sporadic melanoma. Int J Dermatol 2002;41:550-6.
53. Halaban R, Cheng E, Smicun Y, Germino J. Deregulated E2F tran-scriptional activity in autonomously growing melanoma cells. J ExpMed 2000;191:1005-16.
54. Halaban R. Melanoma cell autonomous growth: the Rb/E2F path-way. Cancer Metastasis Rev 1999;18:333-43.
55. Gius DR, Ezhevsky SA, Becker-Hapak M, Nagahara H, Wei MC,Dowdy SF. Transduced p16INK4a peptides inhibit hypophos-phorylation of the retinoblastoma protein and cell cycle progressionprior to activation of Cdk2 complexes in late G1. Cancer Res1999;59:2577-80.
56. Knudsen ES, Wang JY. Differential regulation of retinoblastoma pro-tein function by specific Cdk phosphorylation sites. J Biol Chem1996;271:8313-20.
57. Zarkowska T, Mittnacht S. Differential phosphorylation of theretinoblastoma protein by G1/S cyclin-dependent kinases. J BiolChem 1997;272:12738-46.
58. Taya Y. RB kinases and RB-binding proteins: new points of view.Trends Biochem Sci 1997;22:14-7.
59. Kitagawa M, Higashi H, Jung HK, Suzuki-Takahashi I, Ikeda M,Tamai K et al. The consensus motif for phosphorylation by cyclinD1-Cdk4 is different from that for phosphorylation by cyclin A/E-Cdk2. Embo J 1996;15:7060-9.
60. Ezhevsky SA, Nagahara H, Vocero-Akbani AM, Gius DR, Wei MC,Dowdy SF. Hypo-phosphorylation of the retinoblastoma protein (pRb)by cyclin D:Cdk4/6 complexes results in active pRb. Proc Natl AcadSci U S A 1997;94:10699-704.
61. Becker B, Roesch A, Hafner C, Stolz W, Dugas M, Landthaler M etal. Discrimination of melanocytic tumors by cDNA array hybridiza-tion of tissues prepared by laser pressure catapulting. J Invest Dermatol2004;122:361-8.
62. Defeo-Jones D, Huang PS, Jones RE, Haskell KM, Vuocolo GA,Hanobik MG et al. Cloning of cDNAs for cellular proteins that bindto the retinoblastoma gene product. Nature 1991;352:251-4.
63. Vogt T, Kroiss M, McClelland M, Gruss C, Becker B, Bosserhoff AKet al. Deficiency of a novel retinoblastoma binding protein 2-homologis a consistent feature of sporadic human melanoma skin cancer. LabInvest 1999;79:1615-27.
64. Wilsker D, Probst L, Wain HM, Maltais L, Tucker PW, Moran E.Nomenclature of the ARID family of DNA-binding proteins. Genomics2005;86:242-51.
65. Kashuba V, Protopopov A, Podowski R, Gizatullin R, Li J, Klein G et
al. Isolation and chromosomal localization of a new human retinob-lastoma binding protein 2 homologue 1a (RBBP2H1A). Eur J HumGenet 2000;8:407-13.
66. Lu PJ, Sundquist K, Baeckstrom D, Poulsom R, Hanby A, Meier-Ewert S et al. A novel gene (PLU-1) containing highly conservedputative DNA/chromatin binding motifs is specifically up-regulatedin breast cancer. J Biol Chem 1999;274:15633-45.
67. Barrett A, Madsen B, Copier J, Lu PJ, Cooper L, Scibetta AG et al.PLU-1 nuclear protein, which is upregulated in breast cancer, showsrestricted expression in normal human adult tissues: a new cancer/testisantigen? Int J Cancer 2002;101:581-8.
68. Roesch A, Becker B, Meyer S, Wild P, Hafner C, Landthaler M et al.Retinoblastoma-binding protein 2-homolog 1: a retinoblastoma-bind-ing protein downregulated in malignant melanomas. Mod Pathol2005;18:1249-57.
69. Roesch A, Becker B, Schneider-Brachert W, Hagen I, Landthaler M,Vogt T. Re-expression of the retinoblastoma-binding protein 2-homo-log 1 reveals tumor-suppressive functions in highly metastatic mela-noma cells. J Invest Dermatol 2006;126:1850-9.
70. Connell-Crowley L, Harper JW, Goodrich DW. Cyclin D1/Cdk4 reg-ulates retinoblastoma protein-mediated cell cycle arrest by site-spe-cific phosphorylation. Mol Biol Cell 1997;8:287-301.
71. Bachmann IM, Straume O, Akslen LA. Altered expression of cellcycle regulators Cyclin D1, p14, p16, CDK4 and Rb in nodular mela-nomas. Int J Oncol 2004;25:1559-65.
72. Bracken AP, Ciro M, Cocito A, Helin K. E2F target genes: unravelingthe biology. Trends Biochem Sci 2004;29:409-17.
73. Gordon-Thomson C, Mason RS, Moore GP. Regulation of epider-mal growth factor receptor expression in human melanocytes. ExpDermatol 2001;10:321-8.
74. Stove C, Vanrobaeys F, Devreese B, Van Beeumen J, Mareel M,Bracke M. Melanoma cells secrete follistatin, an antagonist of activin-mediated growth inhibition. Oncogene 2004;23:5330-9.
75. Chattopadhyay N, J TFH, Godbole MM, Brown EM. Transforminggrowth factor beta receptor family ligands inhibit hepatocyte growthfactor synthesis and secretion from astrocytoma cells. Brain Res MolBrain Res 2004;121:146-50.
76. Noonan FP, Dudek J, Merlino G, De Fabo EC. Animal models ofmelanoma: an HGF/SF transgenic mouse model may facilitate experi-mental access to UV initiating events. Pigment Cell Res 2003;16:16-25.
77. Poser I, Bosserhoff AK. Transcription factors involved in develop-ment and progression of malignant melanoma. Histol Histopathol2004;19:173-88.
78. Goding CR. Melanocyte development and malignant melanoma.Forum (Genova) 2000;10:176-87.
79. Furumura M, Potterf SB, Toyofuku K, Matsunaga J, Muller J, Hear-ing VJ. Involvement of ITF2 in the transcriptional regulation ofmelanogenic genes. J Biol Chem 2001;276:28147-54.
80. Brantjes H, Barker N, van Es J, Clevers H. TCF: Lady Justice castingthe final verdict on the outcome of Wnt signalling. Biol Chem2002;383:255-61.
81. Widlund HR, Horstmann MA, Price ER, Cui J, Lessnick SL, Wu Met al. Beta-catenin-induced melanoma growth requires the down-stream target Microphthalmia-associated transcription factor. J CellBiol 2002;158:1079-87.

G ITAL DERMATOL VENEREOL 2007;142:123-9
Prognostic factors in localized cutaneous melanomawith particular reference to thin primary lesions
Sentinel lymph node biopsy is a highly accurate method forthe staging of patients with localized cutaneous melanoma.The status of the sentinel node is the single most importantpredictor of recurrence and survival in patients with clinical-ly node-negative melanoma. While this procedure has foundwidespread acceptance in treatment of patients with melanoma>1 mm in thickness, its use in patients with thinner lesions ismore controversial. Overall, approximately 5% of all melanomapatients with lesions ≤1 mm are reported to harbor identifi-able metastases in the sentinel nodes at the time of diagnosis.There is clearly an unmet clinical need for improved predictivebiomarkers of regional metastasis in clinically localized pri-mary melanomas, especially in patients with thin melanomas.This review summarizes the present knowledge regarding therelationship between clinical and histologic prognostic factorsrelated to sentinel node involvement in melanoma, with par-ticular reference to this important group of thin lesions, andintroduces the concept of considering host polymorphisms aswell as tumor-derived factors as potential biomarkers for refin-ing our understanding of melanoma metastasis.
KEY WORDS: Melanoma - Sentinel node biopsy - Lymphatic metas-tasis - Lymphangiogenesis.
As the incidence of melanoma continues to riseworldwide, so does the proportion of patients
diagnosed with early-stage (Stage I/II) disease. In a
recent evaluation, 78% of cutaneous melanomasreported to the US National Cancer Institute’s Sur-veillance, Epidemiology, and End Results (SEER)cancer registry were American Joint Committee onCancer/International Union Against Cancer (AJCC/UICC) Stage I disease, with a 5-year survival rate of97.7%.1 Yet even though the majority (81%) of thesepatients have thin tumors (Breslow thickness <1.00mm),2 in the SEER database 15% of all melanomadeaths are in these “low risk” patients.1 Given thelarge numbers of early stage patients and the world-wide efforts at early detection, this percentage ofdeaths attributable to thin melanomas is likely to riseconsiderably in the future. Thus, identification of riskfactors for disease progression are perhaps mostimportant in this group of patients.
1Department of Pathology University of South Florida College of Medicine
Tampa, Florida, USA2Department of Interdisciplinary Oncology
Cutaneous Oncology ProgramH. Lee Moffitt Cancer Center & Research Institute
University of South Florida College of MedicineTampa, Florida, USA
3Department of Surgery University of South Florida College of Medicine
Tampa, Florida, USA
The authors have received no financial support for the publication ofthis manuscript.
Address reprint requests to: J. L. Messina, M.D., H. Lee Moffitt Can-cer Center & Research Institute, 12902 Magnolia Drive, Tampa, FL, USA.E-mail: [email protected]
J. L. MESSINA 1, 2, V. K. SONDAK 2, 3
Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 123

MESSINA PROGNOSTIC FACTORS IN LOCALIZED CUTANEOUS MELANOMA
124 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
Sentinel lymph node biopsy is a highly accuratemethod for the staging of patients with localized cuta-neous melanoma. The status of the sentinel node isthe single most important predictor of recurrence andsurvival in patients with clinically node-negativemelanoma.3, 4 While this procedure has found wide-spread acceptance in treatment of patients withmelanoma >1 mm in thickness, its use in patients withthinner lesions is more controversial.5 Overall, approx-imately 5% of all melanoma patients with lesions ≤1mm are reported to harbor identifiable metastases in thesentinel nodes at the time of diagnosis.6-7 The inci-dence of sentinel node positivity in thin melanomapatients has ranged from as low as 0% to as high as9.7% in most reported series.8, 9 This variability appearsto be due, at least in part, to the differing criteria forselection of patients with melanomas <1 mm for thisprocedure, as well as to the relatively low numbers ofpatients (especially node-positive patients) in moststudies. There is clearly an unmet clinical need forimproved predictive biomarkers of regional metasta-sis in clinically localized primary melanomas, espe-cially in patients with thin melanomas. The follow-ing summarizes the present knowledge regarding therelationship between clinical and histologic prognos-tic factors related to both sentinel node involvement andclinical outcome in melanoma, with particular referenceto this important group of thin lesions.
Patient age
Older patients are more likely to present with thick,ulcerated melanomas.10 Melanoma survival decreas-es with advancing age, as was demonstrated in a largeretrospective study of 17 600 melanoma patients:patients younger than 30 years have a 5-year sur-vival rate of 87% compared to 78%, 71%, and 60%respectively for those in their 60’s, 70’s, and 80’s.11
The influence of age on nodal status, however, is theinverse of this phenomenon. Sondak et al. reviewedthe University of Michigan experience with over 400consecutively treated melanoma patients undergo-ing sentinel node biopsy, with almost all melanomasbeing at least 1.0 mm in thickness.12 Their findingsrevealed that younger age was a significant predictorof an increased likelihood of nodal involvement inpatients. In this study, the rate of nodal progressiondecreased steadily as patient age increased, a findingthat has now been confirmed in an extension of this
study involving 1130 patients,13 as well as in otherseries.14-16
Less is known about the influence of age on thelikelihood of nodal progression in thin melanomapatients. In 2003, a retrospective review by Bleicher etal. showed that younger age (defined in their study as<44 years of age) was associated with nodal progres-sion in patients with melanomas ≤1.50 mm in depth.6A number of recent series have examined the impactof a variety of prognostic factors on sentinel node pos-itivity in thin melanoma patients, but all of these havebeen negative or inconclusive with respect to the influ-ence of age,7, 8, 17, 18 except for a recent series fromUniversity of Cincinnati. In this series of 64 thinmelanoma patients undergoing sentinel node biopsy,patients with sentinel nodes found to be either histo-logically or PCR positive were significantly younger(mean age 48.3 years) than those with negative sentinelnodes (50.6 years).19 Several other single-institutionseries of sentinel node biopsy for melanomas 1.00mm or thinner have found that patients with nodalprogression were confined to those 60 years of age oryounger, without finding a direct correlation for age asa continuous variable.7, 18 However, data presented byLeiter et al. showed age was significant only in oldermen with melanomas <0.75 mm in depth.20 Thus, atpresent, age alone cannot be considered to be a vali-dated criterion for recommending or avoiding sentinelnode biopsy in thin melanoma patients.
Gender
Many studies suggest that, overall, men are morelikely to die of melanoma than women. While mostrecent series have not found a significant correlationof gender with sentinel node involvement in thinlesions,7, 8, 18-20 Gimotty et al. found male gender to beprognostically significant for increased risk of pro-gression in thin melanomas.21 Based on the availableevidence, gender alone cannot be considered to be avalidated criterion for recommending or avoiding sen-tinel node biopsy in thin melanoma patients.
Location
Several studies have demonstrated a correlationbetween tumor location and prognosis. In one study ofmelanomas of all thicknesses, lesions on the extrem-

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 125
PROGNOSTIC FACTORS IN LOCALIZED CUTANEOUS MELANOMA MESSINA
ity had a significantly improved 10-year overall survival(90%) compared to 70% for those on the trunk, heador neck.22 In a more recent analysis of 1 130 patientsundergoing sentinel node biopsy, trunk or head andneck locations were more predictive of sentinel nodeinvolvement than upper and lower extremity locationsin both univariate and multivariate models.13 Althoughanalyses have been limited, tumor location has notbeen shown to have a significant impact on sentinelnode involvement in thin melanomas to date. How-ever, sentinel node biopsy for head and neck primariesis more difficult, riskier and associated with higherrates of false negative findings.23, 24 This should betaken into account when advising a patient with a thinmelanoma regarding sentinel node biopsy.
Breslow thickness
Breslow thickness, the tumor depth in millimetersfrom the granular layer of the epidermis to the deep-est point of tumor invasion, is universally consideredto be one of the most useful prognostic factors inpatients with early stage melanoma.10, 15 Many studieshave demonstrated that aside from sentinel node sta-tus, Breslow depth is the most powerful predictor ofclinical outcome.3, 4, 25 In the most recent version of theAJCC/IUCC staging system, the threshold for T2melanoma (marking the beginning of “intermediatethickness”) was changed from 0.75 to 1.01 mm. Somemelanoma centers, including ourselves, continue touse 0.75 mm as the threshold for selecting patientsfor sentinel node biopsy. But few centers routinelyperform sentinel node biopsy for melanomas thinnerthan 0.75 mm in the absence of some other factor indi-cating an increased risk for progression. While nodalmetastasis has been described in melanomas thinnerthan 0.75 mm, as noted by Stitzenberg et al.,7 it appearsto be exceedingly rare. Kesmodel et al. found onlyone patient with a positive sentinel node among 91vertical growth phase melanomas <0.76 mm in thick-ness.16 In a series of 118 patients with melanomas≤0.75 mm, Bleicher et al. found only 2 positive sentinelnodes (1.7%).6 Our own experience is consistent withthe two reports just cited: less than 1% of those patientswith melanomas <0.75 mm selected for sentinel nodebiopsy were found to have a positive node (Messina JL,unpublished data). Routine use of sentinel node biop-sy for melanomas below 0.75 mm does not appear tobe justified based on the available data.
Clark level
In the current AJCC staging scheme, nonulceratedinvasive melanomas ≤1.00 mm confined to the pap-illary dermis (Clark level II or III) are classified asT1a, while Clark IV (into the reticular dermis) andulcerated tumors are classified as T1b. This has ledmany surgeons to restrict the use of sentinel nodebiopsy for thin melanomas to patients with Clark lev-el IV or ulcerated Clark level II and III lesions. Theevidence to support this approach, however, is lack-ing. In our recent series at Moffitt Cancer Center of409 patients with melanomas 0.75 to 1.00 mm inthickness, we found an overall sentinel node positiv-ity rate of 4.9%. In this series, Clark level was not asignificant predictor of nodal positivity: 5.7% of ClarkIV tumors had at least one positive sentinel node,compared to 4.4% of patients with level II and IIIlesions, a difference that was not statistically signif-icant and likely reflected the slight preponderance oflesions 0.9 mm to 1.00 mm among Clark IV cases.26
Greater numbers of thin melanoma patients with Clarklevel II and III melanomas, in total more node-posi-tive cases were found in the level II and III group thanamong the Clark level IV cases. In fact, nearly allreviewed reports reveal that Clark level does not cor-relate with nodal progression in thin melanomas.7, 16
The available evidence strongly suggests that if Clarklevel and ulceration are used as the primary selectioncriteria for recommending sentinel node biopsy inpatients with melanomas of 1.00 mm or thinner, thena substantial number of node-positive cases wouldbe missed. Less well studied, but equally concern-ing, it appears that many patients with Clark level IVlesions ≤0.75 mm are being subjected to sentinel nodebiopsy with very little likelihood of finding a posi-tive node.
Mitotic rate
Mitotic rate is generally measured as the numberof mitoses per square mm, the area of six 40× high-power microscopic fields. In considering melanomasof all thickness, a cutoff of >6 mitoses/mm2 was foundto be indicative of a significantly worse prognosis.27
Investigators at the Sydney Melanoma Unit in Aus-tralia evaluated tumor mitotic rate as a prognostic indi-cator for disease progression.15, 27 Their findings in aseries of 3 661 patients showed that those with a mitot-

MESSINA PROGNOSTIC FACTORS IN LOCALIZED CUTANEOUS MELANOMA
126 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
ic rate of 0 had a significantly better survival rate thanpatients with 1 or more mitoses per mm2. In their data,mitotic rate was a more significant prognostic factorthan ulceration when both were taken into account.Sondak et al. also found mitotic rate to be a significantpredictor of nodal progression, and in this series ulcer-ation did not retain statistical significance as a prog-nostic factor in multivariate analysis once mitotic ratewas taken into account.12, 13 In the University of Penn-sylvania experience with patients with vertical growthphase melanomas 1.00 mm or thinner, Kesmodel etal.16 and Gimotty et al.21 found that a mitotic rate >0(so-called “mitogenicity”) correlated with both nodalprogression and decreased overall survival. The Indi-ana University group found that mitoses >6/mm2 weresignificant predictors of sentinel node positivity in astudy of 184 thin melanoma patients.17 However,reviews of small numbers of sentinel node-positivethin melanoma patients from the University of NorthCarolina 7 and from the Roswell Park Cancer Insti-tute 18 did not suggest that nodal progression was mostcommonly from higher mitotic rate tumors. Thus,while we believe strongly that tumor mitotic rate is asignificant predictor of sentinel node positivity, theoptimal cutoffs to use this biomarker to select patientsfor sentinel node biopsy remains to be defined.
Ulceration
Ulceration is an uncommon pathologic finding inthin melanomas, and when present may be secondaryto trauma. While ulceration correlated with increasedrates of disease progression in patients with thinmelanomas in the large AJCC data set (in which mitot-ic rate was not evaluated),11 it has not correlated withnodal progression in most series of sentinel node biop-sy for the same patients. In a number of series, includ-ing the previously described analysis of the MoffittCancer Center data 25 and reports from the Universi-ty of Pennsylvania 16 and the University of North Car-olina at Chapel Hill 7 no patients with ulcerated thin pri-mary melanomas were found to have evidence of nodalmetastases.
Regression
Regression is a pathologic finding suggesting that amelanoma had at one time penetrated to a greater
degree than was evident at the time of biopsy andcould theoretically identify thin lesions with a partic-ularly high risk of nodal progression. To date, however,no study has found regression to be a statistically sig-nificant independent predictor of nodal metastasiseither for all patients with melanoma or only forpatients with thin melanoma. In fact, it appears thatregression may be negatively correlated with nodalmetastases: patients with regression were actually lesslikely to spread to the sentinel node than othermelanoma, adjusting for thickness and mitotic rate.13
Based on the available evidence, the presence of regres-sion should not be used to select patients with thinmelanomas for sentinel node biopsy.
Tumor infiltrating lymphocytes
Clark et al. are credited with first evaluating and cat-egorizing tumor infiltrating lymphocytes (TILs) as aprognostic biomarker in primary melanoma.25 Clarkdescribed “brisk” tumor infiltration by lymphocytesas a favorable factor, while “nonbrisk” infiltration wasintermediate and “absent or slight” infiltration wasunfavorable. A subsequent study by Clemente et al. ina different cohort of patients also established the pres-ence of brisk TILs to be an independent positive pre-dictive factor.28 A prospective randomized clinical tri-al of 259 patients with clinically localized melanomaconducted by the Southwest Oncology Group foundthe presence of brisk TILs (defined as lymphocytescompletely surrounding the nodule in a band-like man-ner, with no defect in the band of greater than 0.30mm) to be strongly correlated with a favorable out-come.29 Whether the patient had “nonbrisk” or “absentor slight” TILs was not associated with any differencein outcome. The 10-year survival rate for the 30 patientswith brisk TILs was 93%, compared with 58% for non-brisk and 55% for absent or slight TILs. The relative riskof recurrence in this study for nonbrisk or absent TILssuperseded both ulceration and mitotic rate in multi-variate analysis. The study stresses the importance ofincluding all known potential prognostic factors in anymultivariate model of outcome in melanoma. Muchless is known about the implications of TILs formelanoma progression as assessed by regional nodalmetastasis, however, as most recent studies have notincluded this factor. The group at the University ofPennsylvania recently described TILs as an independentprognostic factor for sentinel node metastasis in patients

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 127
PROGNOSTIC FACTORS IN LOCALIZED CUTANEOUS MELANOMA MESSINA
with stage I and II melanoma, with patients with absentTILs having a three-fold increased relative risk of nodalmetastasis.30 They found that the presence or absenceof TILs was second only to tumor thickness in a regres-sion tree analysis for predicting risk of sentinel nodemetastasis in melanomas greater than 1 mm. Thesefindings, which have yet to be validated in an inde-pendent data set, support a potential but as yet incom-pletely defined role of TILs as a biomarker formelanoma regional progression.
Growth phase
The first step in melanoma progression is referred toas radial growth phase, which can be found either innon-invasive (in situ) or early invasive melanomas. Asoriginally described by Clark et al. in radial growthphase lesions, melanocytes in the dermis are present asnests smaller than those nests in the epidermis, withoutmitotic activity.25 The onset of vertical growth phase,which heralds increased potential for metastatic behav-ior, is characterized by either mitotic activity (“mito-genic vertical growth phase”) or dermal tumor nestslarger than those in the epidermis (“tumorigenic verti-cal growth phase”). Several groups have demonstratedthat growth phase is an independent prognostic indicatorof both sentinel node positivity and outcome.31-34
Histologic type
Generally speaking, histologic type does not cor-relate directly with the likelihood of finding a posi-tive sentinel lymph node.13 It has been suggested thatpatients with “pure” desmoplastic melanomas have avery low incidence of nodal metastasis and sentinelnode biopsy may be unnecessary.35, 36 Our experiencehas been that desmoplastic melanomas have a lowerrate of nodal metastasis adjusted for tumor thickness,but that sentinel node biopsy is still indicated fordesmoplastic lesions ≥1.00 mm in thickness.37 Therole of sentinel node biopsy for the very rare thindesmoplastic melanoma is undefined.
The relationship of tumor and host factors
Although this discussion has focused primarily onthose clinicopathologic prognostic factors found in the
primary melanoma which predict risk of metastasis, itis highly likely that melanoma development and metas-tasis is influenced by both “seed” and “soil.” That is,host factors undoubtedly play a key role as well astumor factors. For example, the development ofmelanoma is related to either acute or chronic ultravi-olet light exposure, or both, as well as the host’s abil-ity or inability to respond to that exposure in a protec-tive manner. This has newly appreciated clinical rele-vance as well: melanomas that arise in the setting ofsolar elastosis (a “host” response to chronic ultravioletexposure) appear to have a better overall outcome,38
as well as distinct genetic abnormalities compared toother melanomas.39 The tumor and host molecular fac-tors underlying lymphangiogenesis in melanoma are atpresent almost totally unknown, but there is increasingpreclinical and clinical evidence that nodal metastasisis associated with lymphangiogenesis.40-42 Lymphan-giogenesis can now be visualized and quantified byobserving definable lymphatic endothelial cells in theregion of the tumor, and it appears to correlate withmelanoma nodal metastasis.43, 44
Over the next several years, we believe that improvedbiomarkers of melanoma nodal metastasis can be foundthrough an improved understanding of the biologicprocesses involved in tumor growth and the hostresponse that growth incites. For example, our currentresearch has focused on STAT3 as a potential new bio-marker. STAT3, phosphorylated to an activated form,leads to the transcription of a number of proteins impor-tant to cancer cell survival,45, 46 and its activation iscommon in melanoma.47 When some of the potentialconsequences of STAT3 activation – increased tumorproliferation (increased mitotic rate), decreased hostimmune response (diminished or absent tumor infil-trating lymphocytes), and increased angiogenesis/lym-phangiogenesis – are considered in the context of knownpredictors of melanoma metastasis, it becomes appar-ent that STAT3 activation could serve as a unifyingbiomarker explaining several observations. Incorpo-rating an understanding of the host responsiveness to theeffects of STAT3 activation potentially could allow thispredictive model to be refined even further. To cite twoconcrete examples, it is now known that STAT3 acti-vation leads to the production of VEGF (an angiogen-esis/lymphangiogenesis factor) and IL-10 (an immuno-suppressive cytokine).48, 49 Polymorphisms in theseproteins and/or their receptors are known to occur, andto influence the degree of host response to malignan-

MESSINA PROGNOSTIC FACTORS IN LOCALIZED CUTANEOUS MELANOMA
128 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
cy their expression will generate.50, 51 Thus, one couldenvision a model of tumor progression where primarymelanomas with low levels of STAT3 activation havea low risk of metastasis and manifest concordant patho-logic predictors: low mitotic rate, brisk tumor infiltra-tion by lymphocytes and limited or no lymphangio-genesis, but primary melanomas with high levels ofSTAT3 activation have a variable risk of metastasis –and potentially manifest discordant pathologic pre-dictors – based on host polymorphisms affecting sen-sitivity to the tumor-derived factors produced as a con-sequence of that activation. This important concept oftumor and host factors both contributing to biomarkerutility deserves to be carefully evaluated in a large-scale, well characterized cohort of patients to deter-mine if it has potential clinical relevance, independentof the factors currently known to be associated withsentinel node metastasis.
Riassunto
Fattori prognostici nel melanoma cutaneo localizzato
La biopsia del linfonodo sentinella rappresenta un metodo alta-mente accurato per la stadiazione dei pazienti con melanomacutaneo localizzato. Lo stato del linfonodo sentinella rappre-senta il predittore singolo più importante per quanto riguarda larecidiva del tumore e la sopravvivenza dei pazienti con melanomaclinicamente linfonodo-negativo. Mentre questa procedura haincontrato un ampio consenso nel trattamento dei pazienti conmelanoma >1 mm di spessore, il suo utilizzo nei pazienti conlesioni più sottili è più controverso. In generale, in circa il 5% ditutti i melanomi con lesioni ≤1 mm è possibile identificare meta-stasi nei linfonodi sentinella al momento della diagnosi. Esisteun chiaro e non ancora soddisfatto bisogno clinico di avere adisposizione dei biomarcatori predittivi migliori per le meta-stasi regionali nei melanomi primitivi clinicamente localizzati,specialmente nei pazienti con melanomi sottili. Questa reviewriassume le attuali conoscenze circa la correlazione esistentetra i fattori prognostici clinici e istologici e il coinvolgimento dellinfonodo sentinella nel melanoma, con particolare attenzione aquesto gruppo di lesioni sottili e introduce il concetto dei poli-morfismi dell’ospite così come dei fattori tumore-derivati qua-li biomarcatori potenziali per il miglioramento della nostra com-prensione delle metastasi del melanoma.
Parole chiave: Melanoma - Linfonodo sentinella - Biopsialinfonodale - Metastasi linfatiche - Linfoangiogenesi.
References
1. Gimotty PA, Elder DE, Fraker DL, Botbyl J, Sellers K, Elenitsas R etal. Identification of high-risk patients among those diagnosed with thincutaneous melanomas. J Clin Oncol 2007;25:1129-34.
2. Gimotty PA, Botbyl J, Soong S, Guerry D. A population-based vali-dation of the AJCC melanoma staging system. J Clin Oncol 2005;23:8065-75.
3. Gershenwald JE, Thompson W, Mansfield PF, Lee JE, Colome MI,Tseng CH et al. Multi-institutional melanoma lymphatic mappingexperience: the prognostic value of sentinel lymph node status in 612stage I or II melanoma patients. J Clin Oncol 1999;17:976-83.
4. Morton DL, Thompson JF, Cochran AJ, Mozzillo N, Elashoff R, Ess-ner R et al. MSLT Group. Sentinel-node biopsy or nodal observationin melanoma. N Engl J Med 2006;355:1307-17.
5. Johnson TM, Sondak VK, Bichakjian CK, Sabel MS. The role of sen-tinel lymph node biopsy for melanoma: evidence assessment. J AmAcad Dermatol 2006:54:19-27.
6. Bleicher RJ, Essner R, Foshag LJ, Wanek LA, Morton DL. Role of sen-tinel lymphadenectomy in thin invasive cutaneous melanomas. J ClinOncol 2003;21:1326-31.
7. Stitzenberg KB, Groben PA, Stern SL, Thomas NE, Hensing TA,Sansbury LB et al. Indications for lymphatic mapping and sentinel lym-phadenectomy in patients with thin melanoma (Breslow thickness≤1.0 mm). Ann Surg Onc 2004:11:900-6.
8. Wong SL, Brady MS, Busam KJ, Coit DG. Results of sentinel lymphnode biopsy in patients with thin melanoma. Ann Surg Oncol2006;13:302-9.
9. Thompson JF, Shaw HM. Is sentinel lymph node biopsy appropriatein patients with thin melanomas: too early to tell? Ann Surg Oncol2006;13:279-81.
10. Demierre MF, Chung C, Miller DR, Geller AC. Early detection ofthick melanomas in the United States: beware of the nodular sub-type. Arch Dermatol 2005;141:745-50.
11. Balch CM, Soong SJ, Gershenwald JE, Thompson JF, Reintgen DS,Cascinelli N et al. Prognostic factors analysis of 17,600 melanomapatients: validation of the American Joint Committee on Cancermelanoma staging system. J Clin Oncol 2001;19:3622-34.
12. Sondak VK, Taylor JM, Sabel MS, Wang Y, Lowe L, Grover AC et al.Mitotic rate and younger age are predictors of sentinel lymph node pos-itivity: lessons learned from the generation of a probabilistic model.Ann Surg Oncol 2004;11:247-58.
13. Paek SC, Griffith KA, Johnson TM, Sondak VK, Wong SL, Chang AEet al. The impact of factors beyond Breslow depth on predicting sen-tinel lymph node positivity in melanoma. Cancer 2007:109:100-8.
14. Chao C, Martin RC 2nd, Ross MI, Reintgen DS, Edwards MJ, NoyesRD et al. Correlation between prognostic factors and increasing agein melanoma. Ann Surg Oncol 2004;11:259-64.
15. Thompson JH, Shaw HM. Should tumor mitotic rate and patient age,as well as tumor thickness, be used to select melanoma patients for sen-tinel node biopsy? Ann Surg Oncol 2004;11:233-5.
16. Kesmodel SB, Karakousis GC, Botbyl JD, Canter RJ, Lewis RT, WahlPM et al. Mitotic rate as a predictor of sentinel lymph node positivi-ty in patients with thin melanomas. Ann Surg Oncol 2005;12:1-10.
17. Ranieri JM, Wagner JD, Wenck S, Johnson CS, Coleman JJ 3rd. Theprognostic importance of sentinel lymph node biopsy in thin melanoma.Ann Surg Oncol 2006;13:927-32.
18. Vaquerano J, Kraybill WG, Driscoll DL, Cheney R, Kane JM 3rd.American Joint Committee on Cancer clinical stage selection criteri-on for sentinel lymph node biopsy melanoma. Ann Surg Oncol2006;13:198-204.
19. Hershko DD, Robb BW, Lowy AM, Ahmad SA, Ramadas GH, Sol-dano DA et al. Sentinel lymph node biopsy in thin melanoma patients.J Surg Oncol 2006;93:279-85.
20. Leiter U, Buettner PG, Eigentler TK, Garbe C. Prognostic factors ofthin cutaneous melanoma: an analysis of the central malignantmelanoma registry of the German Dermatological Society. J ClinOncol 2004;22:3660-7.
21. Gimotty PA, Guerry D, Ming ME, Elenitsas R, Xu X, Czerniecki Bet al. Thin primary cutaneous malignant melanoma: a prognostic treefor 10-year metastasis is more accurate than American Joint Com-mittee on Cancer staging. J Clin Oncol 2004;22:3668-76.

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 129
PROGNOSTIC FACTORS IN LOCALIZED CUTANEOUS MELANOMA MESSINA
22. Schuchter L, Schultz DJ, Synnestvedt M, Trock BJ, Guerry D, ElderDE et al. A prognostic model for predicting 10-year survival in patientswith primary melanoma. The Pigmented Lesion Group. Ann InternMed 1996;125:368-75.
23. Schmalbach CE, Nussenbaum B, Rees RS, Schwartz J, Johnson TM,Bradford CR. Reliability of sentinel lymph node mapping with biop-sy for head and neck cutaneous melanoma. Arch Otolaryngol HeadNeck Surg 2003;129:61-5.
24. Wagner JD, Ranieri J, Evdokimow DZ, Logan T, Chuang TY, John-son CS et al. Patterns of initial recurrence and prognosis after sentinellymph node biopsy and selective lymphadenectomy for melanoma.Plast Reconstr Surg 2003;112:486-97.
25. Clark WH Jr, Elder DE, Guerry D 4th, Braitman LE, Trock BJ, SchultzD et al. Model predicting survival in stage I melanoma based ontumor progression. J Natl Cancer Inst 1989;81:1893-904.
26. Puleo CA, Messina JL, Riker AI, Glass LF, Nelson C, Cruse CW etal. Sentinel node biopsy for thin melanomas: which patients shouldbe considered? Cancer Control 2005;12:230-5.
27. Azzola MF, Shaw HM, Thompson JF, Soong SJ, Scolyer RA, WatsonGF et al. Tumor mitotic rate is a more powerful prognostic indicatorthan ulceration in patients with primary cutaneous melanoma: ananalysis of 3661 patients from a single center. Cancer 2003;97:1488-98.
28. Clemente CG, Mihm MC Jr, Bufalino R, Zurrida S, Collini P, Cascinel-li N. Prognostic value of tumor infiltrating lymphocytes in the verti-cal growth phase of primary cutaneous melanoma. Cancer1996;77:1303-10.
29. Tuthill RJ, Unger JM, Liu PY, Flaherty LE, Sondak VK; SouthwestOncology Group. Risk assessment in localized primary cutaneousmelanoma: Southwest Oncology Group study evaluating nine fac-tors and a test of the Clark logistic regression prediction model. AmJ Clin Pathol 2002;118:504-11.
30. Kruper LL, Spitz FR, Czerniecki BJ, Fraker DL, Blackwood-ChirchirA, Ming ME et al. Predicting sentinel node status in AJCC stage I/IIprimary cutaneous melanoma. Cancer 2006;107:2436-45.
31. Bedrosian I, Faries MB, Guerry D 4th, Elenitsas R, Schuchter L, MickR, Spitz FR et al. Incidence of sentinel node metastasis in patients withthin primary melanoma (≤1 mm) with vertical growth phase. AnnSurg Oncol 2000;7:262-7.
32. Cook MG, Spatz A, Brocker E, Ruiter DJ. Identification of histolog-ical features associated with metastatic potential of thin (<1.0 mm) cuta-neous melanoma with metastasis: a study on behalf of the EORTCMelanoma Group. J Pathol 2002;197:188-93.
33. Lefevre M, Vergier B, Balme B, Thiebault R, Delaunay M, ThomasL et al. Relevance of vertical growth pattern in thin level II cutaneoussuperficial spreading melanomas. Am J Surg Pathol 2003;27:717-24.
34. Oliveira Filho RS, Ferreira LM, Biasi LJ. Vertical growth phase andpositive sentinel node in thin melanoma. Braz J Med Biol Res2003;36:347-50.
35. Gyorki DE, Busam K, Panageas K. Sentinel lymph node biopsy for
patients with cutaneous desmoplastic melanoma. Ann Surg Oncol2003;10:403-7.
36. Pawlik TM, Ross MI, Prieto VG, Ballo MT, Johnson MM, MansfieldPF et al. Assessment of the role of sentinel lymph node biopsy for pri-mary cutaneous desmoplastic melanoma. Cancer 2006;106;900-6.
37. Su LD, Fullen DR, Lowe L, Wang TS, Schwartz JL, Cimmino VM etal. Desmoplastic and neurotropic melanoma. Analysis of 33 patientswith lymphatic mapping and sentinel lymph node biopsy. Cancer2004;100:598-604.
38. Berwick M, Armstrong BK, Ben-Porat L, Fine J, Kricker A, Eberle Cet al. Sun exposure and mortality from melanoma. J Natl Cancer Inst2005;97:195-9.
39. Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner Het al. Distinct sets of genetic alterations in melanoma. N Engl J Med2005;353:2135-47.
40. Gershenwald JE, Fidler IJ. Targeting lymphatic metastasis. Science2003;296:1811-2.
41. Dadras SS, Lange-Asschenfeldt B, Velasco P, Nguyen L, Vora A,Muzikansky A et al. Tumor lymphangiogenesis predicts melanomametastasis to sentinel lymph nodes. Mod Pathol 2005;18:1232-42.
42. Tobler NE, Detmar M. Tumor and lymph node lymphangiogenesis –impact on cancer metastasis. J Leukoc Biol 2006;80:691-6.
43. Niakosari F, Kahn HJ, Marks A, From L. Detection of lymphaticinvasion in primary melanoma with monoclonal antibody D2-40. Anew selective immunohistochemical marker of lymphatic endotheli-um. Arch Dermatol 2005;141:440-4.
44. Van der Auwera I, Cao Y, Tille JC, Pepper MS, Jackson DG, Fox SBet al. First international consensus on the methodology of lymphan-giogenesis quantification in solid human tumors. Br J Cancer2006;95:1611-25.
45. Yu H, Jove R. The STATs of cancer – new molecular targets come ofage. Nature Rev Cancer 2004;4:97-105.
46. Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer andimmune cells: role of STAT3 in the tumour microenvironment. NatureRev Immunol 2007;7:41-51.
47. Niu G, Bowman T, Huang M, et al. Roles of activated Src and Stat3signaling in melanoma tumor cell growth. Oncogene 2002;21:7001-7010.
48. Jung JE, Lee HG, Cho IH, Chung DH, Yoon SH, Yang YM et al.STAT3 is a potential modulator of HIF-1–mediated VEGF expres-sion in human renal carcinoma cells. FASEB J 2005;19:1296-8.
49. Staples KJ, Smallie T, Williams LM, et al. IL-10 induces IL-10 inprimary human monocyte-derived macrophages via the transcriptionfactor Stat3. J Immunol 2007:178:4779-4785.
50. McCarron SL, Edwards S, Evans PR, et al. Influence of cytokinegene polymorphisms on the development of prostate cancer. CancerRes 2002;62:3369-3372.
51. Kesmodel SB, Wang Y, Vo A. IL-10 gene polymorphisms predict pri-mary tumor ulceration and risk of recurrence in melanoma patients[abstract]. Ann Surg Oncol 2007;14 [February supplement]:3.

G ITAL DERMATOL VENEREOL 2007;142:131-5
Risk of melanoma in congenital melanocytic nevi
It is an undoubted fact that melanoma may arise in melanocyticnevi, either of aquired or of congenital type. However, themagnitude of this risk is debated controversially. The decisiontowards a prophylactic excision, especially in the case of largercongenital melanocytic nevi (CMN), importantly depends on theassessment of the melanoma risk. In this article, scientific datafrom the literature are summarized to determine the exact roleof CMN as melanoma precursors. First, clinical studies provi-ding a systematic follow-up of patients with CMN are analysed.It is shown that those mostly smaller - studies reporting a highincidence of melanoma probably underly selection bias.Furthermore, the melanoma risk strongly depends on the sizeof CMN and is highest in very large, so-called “garment“ nevi.Second, the significance of histological studies focussing onremnants of “congenital“ nevi in melanoma specimens is cri-tically evaluated.KEY WORDS: Melanoma, diagnosis - Melanoma, pathology - Nevus,pigmented.
CMN represent neural crest-derived hamartomasthat are visible as pigmented lesions of the skin at
or shortly after birth.1 The diameter of these lesionsvaries between a few millimeters and large parts ofthe body surface. The most common classificationdiscriminates small (<1.5 cm), medium (1.5-20 cm),and large CMN (>20 cm), according to their largestdiameter.2 CMN are found in 0.2% to 2% of newborninfants, mostly being very small. These small CMN lat-er may hardly be distinguishable from melanocytic
nevi aquired during childhood or adolescence. Theincidence of larger CMN is much lower. Until now,only one study provided data on a cohort large enoughto estimate this incidence. Castilla et al. identifiednevi ≥4 cm in 15:100 000 and nevi ≥10 cm in 5:100 000South American newborns.3 In another population-based study, Berg and Lindelöf gave incidences of“small“ and “large“ CMN without properly definingthese terms.4
In 1963, Pers from Denmark published the first ret-rospective series of 110 patients with “giant“ CMN, inwhich three cases of melanoma were found during 23years of follow-up.5 Since then, numerous epidemio-logical studies, either prospective or retrospective,have been performed, reporting a melanoma risk ofup to 42% in CMN.6 Moreover, several authors triedto assess this risk by determining the frequency ofremnants of “congenital“ melanocytic nevi in histo-logical melanoma specimens. The goal of this articleis to critically review the published data in order tobetter define the risk of melanoma in CMN.
Epidemiological studies
The plenitude of scientific articles dealing with themelanoma risk of CMN makes it necessary to identi-fy those articles bearing epidemiologically relevant
Dermatological Practice, Lübeck, Germany
Address reprint requests to: S. Krengel, Dermatological Practice, Moi-slinger Allee 160, 23558 Lübeck, Germany. E-mail:[email protected]
S. KRENGEL
Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 131

KRENGEL RISK OF MELANOMA IN CONGENITAL MELANOCYTIC NEVI
132 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
data. It is often difficult to define whether an article rep-resents a selection of individual cases or a systematiccase collection in the sense of a retrospective orprospective study. In a systematic analysis of the lit-erature, we therefore decided that studies with lessthan 20 patients or with a mean follow-up of less than3 years should be regarded as epidemiologically lesssignificant.7 As the result of a literature search in Med-line (1966-October 2005), 14 articles meeting these cri-teria were chosen for further analysis.4, 8-20
The studies varied significantly with respect to studydesign (source of cases; retrospective vs prospectiveanalysis), age of patients, follow-up time, and nevussize. The proportion of melanomas in the 14 studiesranged from 0.05% to 10.7% of the CMN cases. Tak-en together, 49 melanomas were reported in 46 out of6 571 patients (0.7%). The mean follow-up rangedbetween 3.4 and 23.7 years. The mean age of thepatients at entry into the study ranged between 0 and19 years. The sample size of the studies ranged between39 and 3 922 patients. Importantly, smaller studiesreported higher incidences of malignant transformation(P<0.0001). This finding strongly indicates selectionbias. Cases from referral centres and retrospective casecollections are likely to result in highly selected cohortsof patients that are at an increased risk for melanoma.
Melanoma of childhood is a very rare disease. It haslong been reported that, in patients with CMN, a sig-nificant percentage of melanomas arises in early child-hood.21, 22 In the 46 melanoma patients of our literatureanalysis, the mean age at diagnosis was 15.5 years(median: 7 years). This seems to confirm a maximumrisk for melanoma in childhood and adolescence. How-ever, most studies only included information betweenchildhood and early adulthood; therefore, younger cas-es might have been relatively over-reported. By com-parison with age-matched data from the Surveillance,Epidemiology, and End Results database (SEER) wecalculated that patients with CMN carry an approxi-mately 465-fold increased risk of developing melanomaduring childhood and adolescence.
However, these general data are of limited value forthe counselling of parents or patients regarding theirindividual risk. Assuming that the melanoma risk isprobably influenced by the size or localisation of thenevus, it appears necessary to analyse subgroups ofCMN. Some studies demonstrated that the vast major-ity of melanomas in their samples arose in very largeCMN,8, 23 so-called “garment“ nevi. Therefore, we
analysed the data from the 14 selected studies withregard to the CMN sizes. For this subgroup analysis,we counted the numbers of large CMN (>20 cm diam-eter; LCMN) and garment nevi (GN) which we definedas a nevus situated on the trunk which measures >40cm in largest diameter or is expected to reach this sizein adulthood. In those studies that gave sufficient infor-mation on the CMN size (9/14 studies), 39 melanomaswere reported in 1 539 LCMN (2.5%). The number ofGN was stated in only 3/14 studies.8, 9, 18 In these threestudies, the proportion of melanomas was 3.1%(20/636). Compared to the overall incidence of 0.7%in all 14 studies, this obviously shows a higher inci-dence of melanoma in LCMN and GN. Additionally,we analyzed the size of the underlying CMN in themelanoma cases. Sufficient clinical information wasavailable in 41/46 melanoma cases. In 30/41 cases(73.2%), the patients had a GN; in only 5/41 cases(12.2%), the patients had a non-garment LCMN. Thesefigures underscore that the risk of malignancy ismarkedly elevated in GN in comparison to non-garmentLCMN.
Although the above-mentioned data suggest a cor-relation between nevus size and risk magnitude, the riskof small CMN remains to be systematically evaluated.Additionally, it remains to be investigated whetherthis risk exceeds that of acquired nevi of the same size.Earlier reports that suggest relatively high incidencesof melanoma in small CMN 24, 25 certainly requirerevision because their data are based on histologicalrather than epidemiological findings.
Given that melanoma in CMN patients may alsodevelop in deep structures or at extracutaneous sites,removal of the CMN is no guarantee to protect thepatients against melanoma. Moreover, larger CMNmay be too large to be completely excised. Therefore,the impact of prophylactic surgical or other therapeu-tic measures, e.g., dermabrasion. on the risk ofmelanoma is difficult to assess. Each technique thatremoves significant numbers of melanocytic cells cer-tainly reduces this risk. However, a quantitative analy-sis is hampered by the heterogeneity of the studiesconcerning mode and extent of prophylactic therapy.
Histological studies
CMN are defined as melanocytic nevi clinicallydetectable either at birth or, at the latest, during the firstweeks of postnatal life. Histological criteria for the

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 133
RISK OF MELANOMA IN CONGENITAL MELANOCYTIC NEVI KRENGEL
diagnosis of CMN irrespective of their size have beenoriginally suggested by Mark et al.26 These include: 1)presence of nevus cells in the lower two thirds of thereticular dermis or in the subcutis; 2) disposition ofthese deep nevus cells between collagen bundles singlyor in Indian files; 3) involvement of skin appendages,nerves and vessels (i.e. presence of nevus cells inside theepithelium, perineurium or vessel wall). In their study,the first two of these criteria were encountered in 59out of 60 anamnestically congenital lesions and onlyin 2 out of 60 acquired nevi from the control group.Similar findings were presented by Walsh et al.27 Wal-ton et al. in 1976, were the first to demonstrate thatsmaller CMN often lack the above-mentioned histo-logical characteristics.28 Accordingly, Barnhill and Fleis-chli 29 demonstrated a direct correlation between thesize of the lesion and the depth of nevus cell infiltration.Other groups confirmed that: 1) acquired nevi oftenexhibit congenital histological features;30-33 2) congen-ital nevi may present with predominantly junctionalhistological changes.32-39 In contrast, when only thesubgroup of larger CMN is regarded, histological find-ings are much more sensitive and specific, and providesome evidence that these nevi are based on a distinctpathogenic background. Recently, Saida proposed thatthe pathogenesis may be essentially the same in smallcongenital and acquired melanocytic nevi, because bothdevelop from melanocytes situated at the dermo-epi-dermal junction, whereas larger CMN are derived frommigrating neural-crest cells, so-called melanoblasts.40 Acommon molecular finding in small congenital andacquired melanocytic nevi is the presence of BRAF-mutations, which are much less frequent in largerCMN.41 Similarly, Zalaudek et al., based on dermo-scopic findings (globular/cobblestone pattern), haveproposed that small CMN and aquired melanocytic nevidetectable during childhood should be regarded as apathogenic entity.42
Taken together, despite certain typical histologicalpatterns in (mostly larger) congenital nevi, the diagnosisof congenitalness is not possible solely on histologi-cal grounds. Regarding the assessment of melanomarisk, a histological rather than clinical definition ofCMN has produced significant confusion. Most impor-tantly, several authors counted congenital features ofnevus remnants in specimens of malignant melanomaand concluded that congenital nevi, irrespective oftheir size, carry an increased risk for the developmentof melanoma.24, 25 It is an undoubted fact that a
melanoma may arise in congenital (and aquired) neviof any size.43 However, as shown above, a statistical-ly increased risk of malignancy has only been provenfor larger CMN. The risk of malignant degeneration insmall and medium-sized congenital nevi is definitelynot estimable by histological studies of malignantmelanoma showing remnants of congenital nevi.Unlike other authors,44 we believe that the mere fact ofthe congenital presence of a small or medium-sizedmelanocytic nevus does not justify treatment deci-sions.
Conclusions
Epidemiologically, the rarity of (large) CMN andthe heterogeneity of studies only allow a rough assess-ment of the risk of melanoma. However, the overall riskseems to be lower than it has long been suspected.The higher incidence of melanomas in smaller studiesindicates selection bias. The risk strongly depends onthe size of the CMN and is highest in those nevi tra-ditionally designated as “garment“ nevi. In these nevi,preliminary data point to a maximum risk in child-hood and adolescence. Regarding the relatively shortfollow-up in most of these studies, conclusions on thelifetime risk of melanoma should always be drawnwith caution.
The fact that nearly 3 of 4 melanomas in our sys-tematic analysis appeared in very large, garment CMN,leads us to propose that larger CMN should be subdi-vided into distinct groups for clinical handling andfor further studies. One big advantage of the Kopfclassification is that it is very easy to assess the largestdiameter (although the measurement of the nevus areawould be more accurate to describe the size). There-fore, we agree with Ruiz-Maldonado,45 who recentlyrecommended to classify CMN according to theirlargest diameter as follows: small <1.5 cm; medium1.5-10 cm; large 11-20 cm; giant G>20 cm; G1 21-30cm; G2 31-40 cm; G3 >40 cm (patients with giantnevi and with more than 50 small or medium-sizesatellite nevi should be classified one group abovetheir corresponding size classification).
It is not possible to deduce the risk of melanoma inCMN from histological studies on melanoma speci-mens. Eventually, similar studies will get feasiblewhen better molecular markers for CMN and CMNsubgroups, respectively, have been defined. Until then,the term “congenital nevus“ comprises a heteroge-

KRENGEL RISK OF MELANOMA IN CONGENITAL MELANOCYTIC NEVI
134 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
nous group of skin lesions in terms of pathogenesis, his-tology, clinical appearance, and prognosis. This shouldbe borne in mind when clinical consequences aredrawn.
Riassunto
Rischio di melanoma nel caso di nevi melanocitici congeni-ti
È un fatto assodato che il melanoma può derivare dai nevimelanocitici, sia di tipo acquisito che congenito. Tuttavia, ladimensione di questo rischio è ancora oggetto di controver-sie. La decisione di ricorrere ad una escissione profilattica,specie nei casi con nevi melanocitici congeniti di grandidimensioni (congenital melanocytic nevi: CMN), dipendefondamentalmente dalla valutazione del rischio di compar-sa di melanoma. In questo articolo vengono riassunti i datipresenti nella letteratura scientifica, per determinare l’esat-to ruolo dei CMN quali precursori del melanoma. Anzitut-to sono stati analizzati gli studi clinici che hanno avuto un fol-low-up sistematico dei pazienti con CMN. Si è dimostrato chegli studi, spesso con scarsa numerosità, che hanno evidenziatoun’elevata incidenza di melanoma, presentavano probabil-mente degli errori di selezione. Inoltre, il rischio di melanomadipende strettamente dalla dimensione del CMN ed esso èmaggiore per i nevi molto grandi, i cosiddetti nevi “gar-ment”. In secondo luogo è stato valutato criticamente il signi-ficato degli studi istologici focalizzati sui resti dei nevi “con-geniti” nei campioni bioptico di melanoma.PAROLE CHIAVE: Melanoma, diagnosi - Melanoma, anatomiapatologica - Nevi pigmentati.
References
1. Krengel S. Nevogenesis. New thoughts regarding a classical prob-lem. Am J Dermatopathol 2005;27:456-65.
2. Kopf AW, Bart RS, Hennessey P. Congenital nevocytic nevi and malig-nant melanomas. J Am Acad Dermatol 1979;1:123-30.
3. Castilla EE, da Graca Dutra M, Orioli-Parreiras IM. Epidemiology ofcongenital pigmented naevi. I. Incidence rates and relative frequen-cies. Br J Dermatol 1981;104:307-15.
4. Berg P, Lindelöf B. Congenital melanocytic naevi and cutaneousmelanoma. Melanoma Res 2003;13:441-5.
5. Pers M. Naevus pigmentosus giganticus. Ugeskr Laeger 1963;125:613-9.
6. Watt AJ, Kotsis SV, Chung KC. Risk of melanoma arising in large con-genital melanocytic nevi: a systematic review. Plast Reconstr Surg2004;113:1968-74.
7. Krengel S, Hauschild A, Schäfer T. Melanoma risk in congenitalmelanocytic naevi ñ a systematic review. Br J Dermatol 2006;155:1-8.
8. Greeley PW, Middleton GA, Curtin JW. Incidence of malignancy ingiant pigmented nevi. Plast Reconstr Surg 1965;36:26-37.
9. Lorentzen M, Pers M, Bretteville-Jensen G. The incidence of malig-nant transformation in giant pigmented nevi. Scand J Plast ReconstrSurg 1977;11:163-7.
10. Arons MS, Hurwitz S. Congenital nevocellular nevus: a review ofthe treatment controversy and a report of 46 cases. Plast ReconstrSurg 1983;72:355-65.
11. Quaba AA, Wallace AF. The incidence of malignant melanoma (0 to15 years of age) arising in “large” congenital nevocellular nevi. PlastReconstr Surg 1986;78:174-81.
12. Ruiz-Maldonado R, Tamayo L, Laterza AM, Duran C. Giant pig-mented nevi: clinical, histopathologic, and therapeutic considera-tions. J Pediatr 1992;120:906-11.
13. Swerdlow AJ, English JS, Qiao Z. The risk of melanoma in patientswith congenital nevi: a cohort study. J Am Acad Dermatol 1995;32:595-9.
14. Dawson HA, Atherton DJ, Mayou B. A prospective study of con-genital melanocytic naevi: progress report and evaluation after 6 years.Br J Dermatol 1996;134:617-23.
15. Sahin S, Levin L, Kopf AW, Rao BK, Triola M, Koenig K et al. Riskof melanoma in medium-sized congenital melanocytic nevi: a fol-low-up study. J Am Acad Dermatol 1998;39:428-33.
16. Egan CL, Oliveria SA, Elenitsas R, Hanson J, Halpem AC. Cuta-neous melanoma risk and phenotypic changes in large congenitalnevi: a follow-up study of 46 patients. J Am Acad Dermatol1998;39:923-32.
17. Foster RD, Williams ML, Barkovich AJ, Hoffman WY, Mathes SJ,Frieden IJ. Giant congenital melanocytic nevi: the significance ofneurocutaneous melanosis in neurologically asymptomatic children.Plast Reconstr Surg 2001;107:933-41.
18. Bett BJ. Large or multiple congenital melanocytic nevi: occurrence ofcutaneous melanoma in 1008 persons. J Am Acad Dermatol2005;52:793-7.
19. Ka VS, Dusza SW, Halpern AC, Marghoob AA. The associationbetween large congenital melanocytic naevi and cutaneous melanoma:preliminary findings from an Internet-based registry of 379 patients.Melanoma Res 2005 ;15:61-7.
20. Hale EK, Stein J, Ben-Porat L, Panageas KS, Eichenbaum MS,Marghoob AA et al. Association of melanoma and neurocutaneousmelanocytosis with large congenital melanocytic naevi—results fromthe NYU-LCMN registry. Br J Dermatol 2005;152:512-7.
21. Kaplan EN. The risk of malignancy in large congenital nevi. PlastReconstr Surg 1974;53:421-8.
22. DeDavid M, Orlow SJ, Provost N, Marghoob AA, Rao BK, HuanfCL et al. A study of large congenital melanocytic nevi and associ-ated malignant melanomas: review of cases in the New York Uni-versity Registry and the world literature. J Am Acad Dermatol1997;36:409-16.
23. Bittencourt FV, Marghoob AA, Kopf AW, Koenig KL, Bart RS. Largecongenital melanocytic nevi and the risk for development of malignantmelanoma and neurocutaneous melanocytosis. Pediatrics2000;106:736-41.
24. Rhodes AR, Melski JW. Small congenital nevocellular nevi and the riskof cutaneous melanoma. J Pediatr 1982;100:219-24.
25. Massi D, Carli P, Franchi A, Santucci M. Nevus-associated melano-mas: cause or chance? Melanoma Res 1999;9:85-91.
26. Mark G, Mihm M, Liteplo M, Reed R, Clark W. Congenital melanocyt-ic nevi of the small and garment type. Hum Pathol 1973;4:395-418.
27. Walsh MY, MacKie RM. Histological features of value in differenti-ating small congenital melanocytic naevi from acquired naevi.Histopathology 1988;12:145-54.
28. Walton R, Jacobs A, Cox A. Pigmented lesions in newborn infants. BrJ Dermatol 1976;95:389-96.
29. Barnhill R, Fleischli M. Histologic features of congenital melanocyt-ic nevi in infants 1 year of age or younger. J Am Acad Dermatol1995;33:780-5.
30. Clemmensen O, Kroon S. The histology of “congenital features“ in ear-ly acquired melanocytic nevi. J Am Acad Dermatol 1988;19:742-6.
31. Brandenburg K, Paul E. Quantitative Untersuchungen an kongenita-len und erworbenen Nävuszellnävi. Hautarzt 1992;43:775-80.
32. Rhodes AR, Silverman RA, Harrist TJ, Melski JW. A histologic com-

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 135
RISK OF MELANOMA IN CONGENITAL MELANOCYTIC NEVI KRENGEL
parison of congenital and acquired nevomelanocytic nevi. Arch Der-matol 1985;121:1266-73.
33. Cribier BJ, Santinelli F, Grosshans E. Lack of clinical-pathological cor-relation in the diagnosis of congenital naevi. Br J Dermatol1999;141:1004-9.
34. Kühnl-Petzoldt C, Kunze J, Mueller R, Volk B, Petres J. Histology ofcongenital nevi during the first year of life. Am J Dermatopathol1984;6(Suppl 1):81-8.
35. Stenn KS, Arons M, Hurwitz S. Patterns of congenital nevocellularnevi. A histologic study of thirty-eight cases. J Am Acad Dermatol1983;9:388-93.
36. Zitelli JA, Grant MG, Abell E, Boyd JB. Histologic patterns of con-genital nevocytic nevi and implications for treatment. J Am AcadDermatol 1984;11:402-9.
37. Silvers DN, Helwig EB. Melanocytic nevi in neonates. J Am Acad Der-matol 1981;4:166-75.
38. Nickoloff BJ, Walton R, Pregerson-Rodan K, Jacobs AH, Cox AJ.Immunohistologic patterns of congenital nevocellular nevi. Arch Der-matol 1986;122:1263-8.
39. Everett MA. Histopathology of congenital pigmented nevi. Am J Der-matopathol 1989;11:11-2.
40. Saida T Histogenesis of congenital and acquired melanocytic nevi: aunifying concept. Am J Dermatopathol 2006;28:377-9.
41. Ichii-Nakato N, Takata M, Takayanagi S, Takashima S, Lin J, MurataH et al. High frequency of BRAFV600E mutation in acquired nevi andsmall congeital nevi, but low frequency in medium-sized congenitalnevi. J Invest Dermatol 2006;126:2111-8.
42. Zalaudek I, Hofmann-Wellenhof R, Soyer HP, Ferrara G, ArgenzianoG. Naevogenesis: new thoughts based on dermoscopy. Br J Dermatol2006;154:793-4.
43. Illig L, Weidner F, Hundeiker M, Gartmann H, Biess B, Leyh F etal. Congenital nevi < 10 cm as precursors to melanoma. Arch Dermatol1985;121:1274-81.
44. Zaal L, Mooi W, Sillevis Smitt J, van der Horst CM. Classification ofcongenital melanocytic naevi and malignant transformation: a reviewof the literature. Br J Plast Surg 2004;57:707-19.
45. Ruiz-Maldonado R. Measuring congenital melanocytic nevi. Pedi-atr Dermatol 2004;21:178-9.

G ITAL DERMATOL VENEREOL 2007;142:137-47
Melanoma chemoprevention
The incidence and mortality of melanoma is steadily increasingand therapies for advanced melanoma lack efficacy. Melano-ma has been proposed to originate via a series of stepwisegenotypic and phenotypic changes– these stepwise changescreate targets for chemoprevention. Investigation of multiplenovel agents that block UV radiation, prevent activation ofoncogenes and oxidative stress, exploit apoptosis, and boostthe immune system suggest promising strategies for melanomachemoprevention. A better understanding of cancer biomarkersof toxicity and drug efficacy, improved chemoprevention trialdesign, and heightened public awareness of the benefits of can-cer prevention will facilitate efficient identification of usefulchemopreventive agents. Because no agent yet emerges as aclear choice for effective melanoma chemoprevention, advi-sing patients to avoid excessive sun exposure remains the main-stay of melanoma prevention for persons at high risk. Thisreview summarizes recent evidence regarding potential mela-noma chemopreventive agents and discusses current barriersto providing chemoprevention to patients at high risk of deve-loping melanoma.
KEY WORDS: Melanoma - Chemoprevention - Photoprotection -Hydroxymethylglutaryl-CoA reductase inhibitors - Diet - Immunologicfactors - Retinoids - Vaccines - Neoplasms, prevention and control.
Pigment producing cells, melanocytes, migrate fromthe neural crest to the skin, hair follicles, eye
(choroid, ciliary body and iris), the leptomeninges andthe inner ear (cochlea) during embryogenesis. Complexligand-receptor interactions localize melanocytes tothe basal layer of the epidermis where melanocytesreside. Melanin is produced within a unique intracel-lular organelle known as the melanosome and by trans-ferring melanin to neighboring keratinocytes, themelanocyte controls cutaneous pigmentation and pro-tects the skin form ultraviolet radiation.1 Errors inprocesses regulating melanocyte proliferation can leadto malignant transformation and melanoma.
Melanoma is the most deadly form of skin cancer,causing nearly 80% of skin cancer related deaths, andmelanoma incidence is increasing faster than any oth-er cancer worldwide.2-9 Fortunately, melanoma mor-tality rates, while increasing, have not paralleled inci-dence rates. The annual increase in incidence withinfair-skinned Caucasian populations has been 3-7%per year, producing a doubling of melanoma incidenceevery 10-20 years.2 More than 62 000 patients wereexpected to develop melanoma and 7 910 patients todie from their disease in 2006 in the United States,
1Department of DermatologyUniversity of Colorado Health Sciences Center, Aurora
Colorado, USA2Department of Veterans Affairs Medical Center, Denver
Colorado, USA
Funding.—This review was supported by the DermatoepidemiologyResearch Unit of the Department of Dermatology at the University ofColorado, by the University of Colorado Cancer Center, by grant K-07CA92550 (R.P. Dellavalle) from the National Cancer Institute, and bygrant T32 AR07411 (S. Freeman) from the National Institutes of Health,Bethesda, MD.
Conflicts of interest.—Dr. Dellavalle owns 100 shares of Merck com-mon stock in a retirement account. Merck makes the statin drug lovastatin.
Address reprint requests to: R. P. Dellavalle, MD, VA Medical Center-Department of Dermatology , 1055 Clermont Street, Mail Stop 165, Den-ver, CO 80220, USA. E-mail: [email protected]
S. R. FREEMAN 1, R. P. DELLAVALLE 1, 2
Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 137

FREEMAN MELANOMA CHEMOPREVENTION
138 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
with men over 50 counting for around two thirds ofthese deaths.10 The incidence rate for melanoma inthe United States has more than doubled from 5.7% to14.3% in the past 30 years.10
Risk factors strongly associated with developmentof melanoma are personal history of melanoma ormultiple benign or atypical nevi, presence of associatedgenetic abnormalities and family history of melanoma.1,
11 Other risk factors include skin phototypes I and II,intermittent UV light exposure, blistering burns dur-ing childhood, large congenital nevi over greater than5% of the body surface area, immune suppression,older age, male gender, higher socioeconomic status,equatorial latitudes, and inheritance of rare DNA repairdefects (Xeroderma Pigmentosum).12, 13 Epidemio-logic observations suggest that chronic or low-gradeexposure to UV light induces protection against DNAdamage, whereas intense, intermittent exposure caus-es genetic damage.11
Patients with thin melanomas who receive earlytreatment experience excellent outcomes, with 10-year metastasis-free survival rates between 91% and100%.14 Treatment modalities for advanced melanoma
are largely ineffective; they provide modest survivalbenefit and can be associated with serious side effects.15
The results of multivariate analyses using a databasederivedfrom records of 17 600 patients with melanomafrom 13 cancer centers and cooperative groups fromNorth America, Europe, and Australia identified lowsurvival rates for patients with advanced stages ofmelanoma.16, 17 Melanoma patients with nodal metas-tases had five-year survival rates ranging from 69%for patients with nonulcerated melanomas who had asingle clinically occult nodal metastasis to 13% forpatients with ulcerated melanomas who had four ormore clinically apparent nodal metastases.16, 17 Sur-vival rates for patients with metastatic disease, unfor-tunately, is often measured in months; few survivebeyond one year.16, 17
Educational and melanoma screening programs havebeen proposed to improve melanoma outcomes. Whilethese systems are appealing, worldwide health initia-tives including physician directed and patient self-assessment screening programs have so far failed toprovide conclusive data on effectiveness.18 The steadyincrease in melanoma incidence and mortality cou-
TABLE I.—Possible melanoma chemoprevention agents and proposed mechanisms of action.
Agent Photoprotection Anti- Anti- ProAnti-
Anti- Immunomodulatory Oncogeneoxidant inflammatory apoptosis
proliferativeangiogenesis inhibition(Cell cycle
arrest)
Sunscreen √Forskolin √HMG-CoA reductase inhibitors √ √ √ √ √ √PPAR-alpha inhibitors √ √Sorafenib √Cox-2 Inhibitors √ √ √IRM * √ √Retinoids √ √Perillyl alcohol √Vaccines √EGCG° √ √ √ √ √Vitamin E √ √ √ √Vitamin D √ √ √Beta-carotene √ √Lycopene √ √Flavonoids √ √ √ √ √Resveratrol √ √ √ √Selenium √Apomine √Curcumin √ √DFMO ** √Betulinic acid √Genistein √ √T4NV °° √
*Immune response modifiers; Imiquimod and Resiquimod; °Epigallocetechin 3-gallate; **Difluoromethylornithine; °°T4 endonuclease V.

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 139
MELANOMA CHEMOPREVENTION FREEMAN
pled with the meager efficacies of current interven-tions for advanced disease has spawned great interestin prevention of this disease.
Chemoprevention
The Clark model of melanoma details the cellularchanges that occur as normal melanocytes progressin a stepwise manner toward malignancy.11 The pro-liferation of melanocytes resulting in the formationof nevi, and the subsequent development of dyspla-sia, hyperplasia, invasion, and metastasis observed inthe growth phases of melanoma is hypothesized tocorrespond to the accumulation of genetic mutations.11,
17 The presence of such stepwise histologic changes arelinked to specific biologic and molecular changes with-in the transforming melanocytes that have created newtargets for therapeutic intervention. Melanoma chemo-prevention is being explored as one such interventionaimed at targeting these molecular events.
Chemoprevention is a novel approach to cancermanagement and can be defined as the use of naturalor synthetic agents to reverse, suppress, or preventmolecular or histologic premalignant lesions from
progressing to invasive cancer.19, 20 The first approvedmedication for the prevention of breast cancer inwomen at high risk was Tamoxifen, though otherchemopreventive agents have followed including Cele-coxib for Familial Adenomatous Polyposis syndrome,Diclofenac for actinic keratoses, retinoids for headand neck cancers and Imiquimod for actinic keratosesand superficial basal cell carcinoma. Gardesil, a vac-cine proven 89% effective in guarding against 4 com-mon strains of human papillomavirus, was recentlyapproved for use in the United States. This vaccinecould prevent many of the 300 000 cervical cancerrelated deaths that occur worldwide each year.21
Recent discoveries in melanoma biology have illu-minated many of the biochemical pathways this can-cer depends upon for malignant transformation, localinvasion and metastasis and have identified a myriadof potential targets of interest from a chemopreventionstandpoint.11, 17 A variety of natural and syntheticagents that block ultraviolet radiation, prevent activa-tion of oncogenes and oxidative stress, exploit apop-tosis, and boost the immune system have been sug-gested as promising strategies that may delay, reverse,suppress, or prevent pre-malignant lesions from pro-gressing to cancer.17, 22
TABLE II.—Skin cancer summary data for promising chemopreventive agents.
Agents Population Studied Results
Sunscreen 23 Human Conflicting resultsCelecoxib 24, 25 Murine Protective against nonmelanoma skin cancer
Human Studies ongoing for treatment of actinic keratoses and nonmelanoma skin cancer
Retinoids* 24, 26-29 Human Protective against nonmelanoma skin cancerEpigallocetechin 3-Gallate (EGCG) 24, 30, 31 Murine Protective against nonmelanoma skin cancerLycopene 24, 32 Murine Protective against nonmelanoma skin cancer
Human Antineoplastic in lung, colon and breast
Curcumin 24, 33-36 Murine Protective against nonmelanoma skin cancerHuman Induces apoptosis in human basal cell and melanoma cell lines
Difluoromethylornithine (DFMO) 24, 37, 38 Murine Protective against nonmelanoma skin cancerHuman Ongoing Phase I trials
Genestein 24, 39 Murine Protective against nonmelanoma skin cancerHuman Decreased UVB induced erythema
T4NV 24, 40, 41 Murine Protective against nonmelanoma skin cancerHuman Protective against actinic keratoses and basal cell carcinoma
HMG-CoA Reductase Inhibitors (statins) 42 Murine Inhibits in melanoma cell linesForskolin 43 Murine Protective against tumor formationImmune Response Modifiers 44 Human Effective in treating Bowen’s, actinic keratoses and lentigo maligna
*Isotretinoin, Acitretin, Etretinate.

FREEMAN MELANOMA CHEMOPREVENTION
140 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
This review summarizes recent evidence regardingpotential melanoma chemopreventive agents (TablesI, II)23-44 and discusses current barriers to providingchemoprevention to a patients at high risk of developingmelanoma.
Melanoma chemoprevention agents and mechanism of action
Sunscreen
Sunscreen is frequently recommended as a UVprotective compound with presumed skin cancer pro-tective properties, though its association withmelanoma is unclear. Meta-analyses of observation-al studies have failed to show an inverse relationshipbetween sunscreen use and melanoma.23, 45 Resultsfrom individual studies included in the meta-analy-ses are inconsistent, with some studies reporting pos-itive, negative or no association between sunscreenuse and melanoma risk.17, 45-47 The lack of conclu-sive quantitative evidence is not surprising given theretrospective nature and large methodological dis-crepancies of many of the included studies. Vari-ability in sunscreen application practices, sun expo-sure duration and frequency, and inferior UV pro-tection provided by the sunscreens studied likelyimpacted the results of these studies.23
In spite of this controversy, photoprotection is cur-rently a mainstay of melanoma protection, with appro-priate sunscreen use and peak hour sun avoidancebeing ideal photoprotective measures. Emphasis shouldalso be placed on comprehensive sun protection withUV protective clothing, sunglasses and use of shadestructures while outdoors. It is currently unclearwhether UVA, UVB, or both forms of radiation aresignificant in melanoma, though a recent mouse mod-el suggests that UVB may be most important.48 Sun-screens protecting against both UVA and UVB arestrongly recommended and agents including Parsol1789 and titanium dioxide are effective in providingthis coverage.17 Both animal and human studies havedemonstrated that sunscreens reduce UV-inducedimmunosuppression, and animal studies have beenshown to protect mice from p53 mutations.49
The association between sunscreen use and thedevelopment of nevi has not been confirmed and resultsof several studies have reported conflicting data.50-58 Itis reassuring that a recent randomized controlled trial
found that children wearing broad-spectrum SPF30sunscreen developed fewer nevi compared to controlgroup children, particularly since nevi are a surrogatefor melanoma risk.23, 50
Forskolin
Decreased ability to tan in response to UV light isassociated with an increased susceptibility tomelanoma. Melanin synthesis and distribution con-trols pigmentation and alpha-melanocyte stimulatinghormone (α-MSH) is the main physiologic regulatorof skin pigmentation.1 Binding of α-MSH to themelanocortin 1 receptor (MC1R) leads to increasedintracellular signal transduction through the cyclic-AMP pathway (c-AMP), causing increased expres-sion of enzymes responsible for generation of melanin.1Alterations in melanin synthesis that result in defectiveUV-induced tanning response have been document-ed in many persons with fair skin.17, 43 Althoughmelanin synthesis can be disrupted at any step, func-tional changes in the MC1R have been shown to havea strong association with fair skin and weak tanningresponses to UV light exposure.17, 43
Forskolin, a labdane diterpene produced by the Indi-an Coleus plant Plectranthus barbatus can affectmelanin synthesis and is being studied as a potentialchemopreventive agent. Forskolin resensitizesmelanocortin-1 receptors by activating the enzymeadenylyl cyclase which increases the intracellular lev-els of cAMP and ultimately leads to increased melaninsynthesis.17 A recent study showed that mice withinactivating mutations in MC1R that were unable to uti-lize UV to form pigment became pigmented after top-ical application of forskolin.43 Further investigationwith mice carrying xeroderma-pigmentosum-com-plementation-group-C-deficient (Xpc -/-) revealed thatmice treated with forskolin had a decreased incidenceof tumorigenesis. Forskolin holds chemopreventivepromise should it induce melanogenesis in human skinwithout promoting melanoma.
Lipid lowering medications
The oncogenes N-RAS and BRAF are strongly asso-ciated with melanoma development and somatic muta-tions of these genes can cause abnormal activation ofthe mitogen-activated protein kinase (MAPK) path-way and stimulate tumorigenesis through the consti-tutive activation of serine-threonine kinases.11, 59, 60

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 141
MELANOMA CHEMOPREVENTION FREEMAN
This signaling pathway represents an important tar-get for melanoma chemoprevention.
Medications known as statins inhibit the rate-limit-ing enzyme Hydroxymethylglutaryl-coenzyme AReductase (HMG-CoA) in the cholesterol synthesispathway, thereby lowering cholesterol. These med-ications are used to treat hypercholesterolemia andprevent cardiovascular diseases. Surprisingly, ran-domized controlled trials studying cardiovascular dis-ease reported a statistically significant decrease inmelanoma and other cancers in patients taking statins.17
Subsequently a multitude of studies have reported pro-apoptotic, anti-inflammatory, immunomodulatory andanti-angiogenic effects of statins on tumor cells.17, 60,
61 For example, statins affect the RAS signaling path-way by preventing the activation of RAS proteins. Theoncogenic properties of RAS proteins depend on iso-prenylation—they must have a farnesyl and geranylgroups attached to accelerate cell growth and cell cycleprogression.17 The cholesterol synthesis pathway isintegral to the isoprenylation of RAS proteins and byinhibiting HMG-CoA Reductase, statins prevent theattachment of key pro-tumor control groups.17
No consensus exists on the effectiveness of statinsas antineoplastic agents, either as a therapeutic orchemopreventive. Recently statins have been proposedby a limited case control study as possibly exacerbat-ing bladder cancer.62 In the case of melanoma, a ran-domized control trial of lovastatin found a decreasedincidence of melanoma in patients exposed to thedrug,63 while a case-control study reported no associ-ation.64 Additionally, meta-analyses of randomizedcontrolled trials of melanoma found no associationbetween melanoma and standard low doses of statins.65-
67 In vitro and animal studies of statins provide strongevidence of the melanoma killing potential of statinsand further investigation is needed to reveal their rolein melanoma prevention.17
Large randomized control trials of lipid loweringmedications have also found decreased incidence ofmelanoma in patients exposed to fibrate medications.68
These medications inhibit peroxisome proliferator-activator receptors (PPAR) but the mechanisms bywhich they may perhaps retard or prevent the devel-opment of melanoma are largely unknown. Meta-analyses of studies of these medications have failed toidentify an association between fibrates andmelanoma.66, 67
Other agents that exhibit chemoprentive properties
in melanoma by acting on similar pathways includeperillyl alcohol and apomine. Perillyl alcohol, whichis found in fruits and vegetables, has been shown toinhibit the farnesylation of RAS in vitro.69 Apomine isa member of the family of cholesterol synthesisinhibitors known as 1,1-bisphosphonate esters. Thiscompound has been shown to increase the rate ofHMG-CoA Reductase degradation, decrease prolif-eration and induce apoptosis in tumor cells at lowerconcentration than statins.70
Sorafenib
BRAF mutations lead to constitutive activation ofserine-threonine kinases in the MAPK pathway andare detected in up to 66% of melanomas.11, 19, 71 Thehigh prevalence of BRAF mutations highlights theircentral role in melanoma development and makes allassociated pathways appealing targets for chemopre-vention.17, 19, 71 Sorafenib is an oral multikinase inhibitorthat inhibits Raf kinase and receptor tyrosine kinases,thereby specifically targeting the Raf/extracellular sig-nal-regulated kinase (ERK) kinase (MEK)/ERK sig-naling pathway.72 Inhibiting Raf/MEK/ERK signalingleads to apoptosis in melanoma cells and clinical dataon sorafenib demonstrates significant activity againstneovascularization and tumor progression.73, 74
Epigallocatechin-3-gallate
Epigallocatechin-3-gallate (EGCG) is a polyphe-nolic compound present in green and black teas andgrape seeds and possesses both antioxidant and sun-screen activity.24 EGCG has been shown to decreasecell proliferation, increase apoptosis, and inhibit thecolony forming ability of multiple melanoma cell lines,and has little effect on healthy melanocytes.17 EGCGexerts its effects through modulations of cyclin kinaseinhibitor-cyclin-cyclin dependent kinase machineryand B-cell leukemia/lymphoma 2 (Bcl2) proteins.11, 75
Multiple murine models report strong photoprotec-tive properties, especially against UVB radiation, andhave shown that EGCG reduces the incidence of skincancers in mice exposed to UVB radiation.24, 76, 77
COX-2 inhibitors
Non-steroidal anti-inflammatory drugs (NSAIDs)inhibit cyclo-oxygenase (COX) enzymes (COX-1 andCOX-2), thereby preventing the formation of

FREEMAN MELANOMA CHEMOPREVENTION
142 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
prostaglandins and other pro-inflammatory mole-cules.17, 78 UVB radiation has been shown to increaseCOX-2 expression and subsequent prostaglandin pro-duction like prostaglandin E2 (PGE-2) in the epider-mis and studies have demonstrated a strong correlationwith increased PGE-2 levels and the development andmetastasis of cancer.17, 79-82 The COX pathway mayprove to be key component in melanoma tumor devel-opment, thus making NSAIDS potential candidatesfor melanoma chemoprevention.17, 78, 83 Mouse mod-els have shown the selective COX-2 inhibitor cele-coxib to be effective both at increasing skin cancerlatency and limiting tumor multiplicity.24 Case-controland retrospective studies in humans demonstratedecreased incidence, recurrence rates, and metastasisof melanoma in patients prescribed a COX-inhibitor.84
Retinoids
Retinoid is a term used to describe vitamin A(retinol) and all of its derivatives.24 Retinoids bind toall-trans retinoic acid receptors (RAR) and rexinoidreceptors (RXR) in nuclei, and control epidermal dif-ferentiation, proliferation, and apoptosis.85
Clinical trials of oral retinoids in patients at moderateto high risk for nonmelanoma skin cancer have demon-strated significantly lower risk of developing a firstnew squamous cell carcinoma (SCC) when comparedto placebo group.24, 79 Due to melanoma’s relativeresistance to apoptosis, retinoids have been proposedas possible chemoprevention agents.
Difluoromethylornithine
Difluoromethylornithine (DFMO) is an irreversibleinhibitor of ornithine decarboxylase (ODC).24 ODCis the rate-limiting enzyme in the synthesis ofpolyamines, a process thought to be critical to the for-mation and survival of several cancers.24, 86 DFMOstudies in mice have shown this agent effective in pre-venting epidermal tumors, and Phase I studies inhumans are underway.24, 87
Resveratrol
Resveratrol is a plant polyphenol found in grapes, redwine, berries, and peanuts inhibits the growth of var-ious tumor cell lines, including melanoma and squa-mous cell carcinoma.88 The mechanisms through whichresveratrol acts are thought to be linked to cell cycle
arrest and angiogenesis suppression.88, 89 Besideschemopreventive effects, resveratrol also appears toexhibit therapeutic effects against melanoma cells invitro.90 Resveratrol has been shown safe in humansand analogues of this drug are being studied withemphasis on improved bioavailability.17, 88
Vaccines
Tumor-associated antigens (TAAs) include non-mutated, overexpressed, or inappropriately expressedtissue differentiation antigens and are increasinglybeing targeted in laboratory and clinical cancerresearch.17 Melanoma cells are immunogenic andTAAs have been identified allowing for the specific tar-geting of malignant cells.17, 91 The aims of current vac-cine systems in melanoma include enhancing cellu-lar and humoral responses, improving melanoma anti-gen presentation to antigen presenting cells like den-dritic cells and combat local immunosuppressive effectsthat melanoma has on tissues.92 Melanoma vaccinesbeing studied are diverse, including polyvalent agentscreated from autologous and allogeneic tumors, anti-gen specific vaccines and DNA based vaccines.92
Challenges facing melanoma vaccines includeimmunologic recognition escape common to malig-nancies and development of effective in vivo vaccinedelivery systems.92, 93 Multiple phase I, II and III clin-ical trials have been performed utilizing various vac-cination approaches.92, 94 Despite encouraging suc-cess in phase I and II trials, the outcomes from phaseIII trials have largely been disappointing, though addi-tional studies are ongoing.17
Immune response modifiers
Another approach to preventing and treating cuta-neous malignancy is the use of Immune ResponseModifiers (IRMs). IRMs include imiquimod andresiquimod and these agents are able to stimulate boththe innate and acquired immune responses leading tothe local production of interleukins (ILs), interferons(IFNs) and tumor necrosis factor alpha (TNF-α).44
Subsequently, monocytes, macrophages, toll-like recep-tor-7 bearing dendritic cells, and cytotoxic T-lym-phocytes are activated and this activity is thought toincrease the skin’s ability to fight cancer cells.44, 95
Imiquimod is a member of the imidazoquinoline fam-ily and the 5% cream preparation is currently approvedfor the treatment of actinic keratoses, squamous cell

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 143
MELANOMA CHEMOPREVENTION FREEMAN
carcinoma in situ, superficial basal cell carcinoma andgenital warts.96, 97 Imiquimod has been successful in 2small studies at treating lentigo maligna (LM), a pre-cursor to melanoma.98, 99 The largest of these studieswas an open-label case series that included 30 patientswith histologically proven LM. After daily applica-tion of 5% imiquimod for 3 months, 93% of subjectsexperienced complete clinical and histological clear-ance.44, 99 A smaller study (6 patients) looked at theeffectiveness of topical imiquimod 5% at treating LMwhen used once daily for 13 weeks. There was a 100%response rate in all 6 subjects and no recurrences at 18-month follow- up.98
Resiquimod is an analogue of imiquimod that isbeing investigated for treatment of actinic keratoses.44
Like imiquimod, this medication stimulates the pro-duction of ILs, IFNs and TNF-α, but unlike imiquimod,resiquimod has not been shown to induce apoptosisin basal cell carcinoma and melanoma cells in vitro.44
T4 endonuclease V
The accumulation of UVB-induced pyrimidinedimers in DNA is carcinogenic and can lead to themalignant transformation of epidermal cells.24 T4endonuclease V (T4NV) is a bacterial DNA repairenzyme proven to accelerate the removal of harmfulphotoinduced DNA adducts and is currently beingstudied in humans as a potential chemopreventiveagent. Topical T4NV effectively decreases skin tumorsin UVB exposed mice, and a study in patients withxeroderma pigmentosum found decreased incidencesof both actinic keratoses and basal cell carcinomas inpatients exposed to this medication topically.24, 40, 41 Anongoing phase II clinical trial is underway to measurethe chemopreventive efficacy of topical T4NV in renaltransplant patients.24
Other agents
Many agents exist that have limited evidence inmelanoma chemoprevention. These include vitaminE, beta-carotene, selenium, lycopene, curcumin, genis-tein, vitamin D, flavonoids (found in fruits and veg-etables), silymarin (found in milk thistle) and betulin-ic acid (found in bark and roots of certain flora). Ofthese compounds, vitamin E, beta-carotene and sele-nium have been studied specifically for their roles inphotodamage and photocarcinogenesis and found inef-fective.24 Lycopene, which gives some fruits and veg-
etables a red color, is an antioxidant that exhibits anti-tumor properties in mice exposed to UVB.24 Curcumin(diferuloylmethane) is found in many foods and isderived from the root of the Curcuma longa plant.While studies in mice suggest that this compound hasantitumor properties, its mechanism of action is large-ly unknown and studies in humans are lacking.24 Genis-tein is a phytochemical antioxidant found in high con-centration in soybeans. This compound is chemopre-ventive in mice exposed to UVB and at least one smallstudy suggests that it is photoprotective in humans.24
The role of vitamin D in cancer prevention is a con-troversial issue. Studies demonstrating decreased riskof non-Hodgkin’s lymphoma and improved survival inpatients with non-metastatic melanoma in which chron-ic sun damage was present have led many to cogitatethat vitamin D is responsible.100-102 Data from an Aus-tralian study suggests that sun exposure may have apositive effect on melanoma outcome, but linking thisto vitamin D is premature.17, 103
Discussion
Melanoma incidence and mortality is increasingand current therapies for advanced disease are large-ly ineffective.15 Molecular studies have described thestep-wise genotypic and phenotypic transformationsthat takes place in UV-induced melanoma and haveidentified potential molecular targets for chemopre-vention.11 Effective prevention strategies could savecountless lives, though no agents currently exist to fillthe need. While some of the agents targeting thesepathways were reviewed in this article, most are stillin the preclinical stages of investigation. Studies ofmelanoma agents in humans have been, for the mostpart, limited to retrospective studies which are sub-ject to recall bias and other confounding factors.17
While policy makers remain open to chemopren-tive strategies and understand the potential benefitsthese agents may confer to populations at risk formelanoma, they require more conclusive evidence.18
Efficient generation of convincing evidence for anagent’s chemopreventive effect will require carefulattention to the hurdles preventative medicine faces.Current research environments focus mainly on ther-apeutic interventions for established diseases, not dis-ease prevention, and the public has very little under-standing of the importance and potential of cancerpreventive science.21 The lack of public knowledge

FREEMAN MELANOMA CHEMOPREVENTION
144 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
and the relatively small investment in chemopreventiveresearch at industry and government levels furtherimpede cancer preventive research because the stud-ies needed to prove benefit are complex, expensive,long and require many subjects.21, 104 Campaigns aimedat improving the public knowledge of cancer preven-tion science should be initiated in order to ensureresearch study recruitment and aid in disseminationof important trial findings.105
Clear designs for clinical research models shouldbe established.17, 19 Of great importance is safety. Giv-en that cancer prevention trials by nature will includehealthy individuals followed for considerable lengthsof time, the tolerance for adverse events associatedwith trial drugs is very low. Effort must be made toestablish safe, effective dosing regimens based oninformation collected from transgenic murine mod-els.92, 105 Ideal chemoprevention trials should utilizemolecular safety biomarkers that allow close surveil-lance of drug associated toxicity and disease specificbiomarkers that allow for identification of those at riskand are linked to disease occurrence or progression.17,
105 Such biomarkers need to be identified and validat-ed.19, 106-108 Better understanding and identification ofthose at risk for developing melanoma should allow forsmaller studies that still possess the power to generateaccurate assessment of drug benefit.
Chemoprevention of cancer, including melanoma, isin its infancy, but significant progress is being made.Evidence from in vitro, animal, and epidemiologicstudies continue to identify key melanoma pathwaysand raise the possibility that a chemopreventive agentis feasible.17 New insights into the normal physiolog-ic tanning response to UV radiation may help identi-fy a new class of chemopreventive agents, but until amelanoma chemoprevention agent is found, advisingpatients to avoid excessive sun exposure remains themost reasonable tool available to prevent melanoma.17,
109 Currently, there is no evidence proving that provideror patient skin examinations prevent melanoma butrecent studies show these interventions are cost-effec-tive, logistically possible and effective at targeting at-risk populations.18, 110
Riassunto
Chemioprevenzione del melanomaL’incidenza e la mortalità del melanoma sono costante-
mente in aumento e le terapie per il melanoma avanzato per-
dono efficacia. È stato proposto che il melanoma sia il risul-tato di una serie di modificazioni successive di tipo genoti-pico e fenotipico; queste modificazioni successive possonodiventare dei bersagli per la chemioprevenzione. La ricercasu nuovi e diversi agenti che bloccano la radiazione UV, cheprevengono l’attivazione degli oncogeni e lo stress ossidativo,che sfruttano l’apoptosi e che amplificano la risposta immu-nitaria, ha suggerito strategie promettenti per la chemiopre-venzione del melanoma. Una migliore comprensione deibiomarcatori cancerosi di tossicità e di efficacia farmacolo-gica, il miglioramento del disegno degli studi clinici sulla che-mioprevenzione e l’attenzione pubblica sempre più alta cir-ca i benefici della prevenzione tumorale faciliteranno l’i-dentificazione degli agenti chemiopreventivi più efficaci.Dal momento che attualmente non è ancora stato identifica-to un agente chiaramente efficace per la chemioprevenzionedel melanoma, l’avvisare i pazienti di evitare di esporsieccessivamente alla luce solare resta la forma principale diprevenzione del melanoma per le persone ad alto rischio.Questa review riassume le evidenze relative ai potenzialiagenti chemiopreventivi del melanoma e discute le attuali dif-ficoltà nel mettere in atto la chemioprevenzione in pazientiad alto rischio di sviluppare melanoma.PAROLE CHIAVE: Melanoma - Chemoprevenzione - Fotopro-tezione - Statine - Dieta - Fattori immunologici - Retinoidi -Vac-cini - Neoplasie, prevenzione e controllo.
References
1. Bolognia J, Jorrizo JL, Rapinin RP. Melanocyte biology. In: Derma-tology Online. Ohiladelphia:Mosby; 2007. p. 935-45.
2. Lens MB, Dawes M. Global perspectives of contemporary epidemi-ological trends of cutaneous malignant melanoma. Br J Dermatol2004;150:179-85.
3. McCarthy WH. The Australian experience in sun protection andscreening for melanoma. J Surg Oncol 2004;86:236-45.
4. Burton RC. Malignant melanoma in the year 2000. CA Cancer J Clin2000;50:209-13.
5. Elwood JM. Developing areas in cancer in New Zealand. Jpn J ClinOncol 2002;32(Suppl 1):S43-51.
6. de Vries E, Coebergh JW, van der Rhee H. Trends, causes, approachand consequences related to the skin-cancer epidemic in the Nether-lands and Europe. Ned Tijdschr Geneeskd 2006;150:1108-15.
7. Aviles JA, Lazaro P, Lecona M. Epidemiology and survival of cuta-neous melanoma in Spain: a report of 552 cases (1994-2003). Rev ClinEsp 2006;206:319-25.
8. Bulliard JL, Panizzon RG, Levi F. [Melanoma prevention in Switzer-land: where do we stand?] Rev Med Suisse 2006;2:1122-5. French.
9. Holme SA, Malinovsky K, Roberts DL. Malignant melanoma inSouth Wales: changing trends in presentation (1986–98). Clin Exp Der-matol 2001;26:484-9.
10. American Cancer Society. Cancer facts and figures 2005. Atlanta,GA: American Cancer Society; 2005. http://www.cancer.org/doc-root/home/index.asp
11. Miller AJ, Mihm MC. Melanoma. N Engl J Med 2006;355:51-65.12. Gandini S, Sera F, Cattaruzza MS, Pasquini P, Abeni D, Boyle P et al.
Meta-analysis of risk factors for cutaneous melanoma: I. Common andatypical nevi. Eur J Cancer 2005;41:28-44.
13. Gandini S, Sera F, Cattaruzza MS, Pasquini P, Zanetti R, Masini C etal. Meta-analysis of risk factors for cutaneous melanoma: III. Fami-

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 145
MELANOMA CHEMOPREVENTION FREEMAN
ly history, actinic damage and phenotypic factors. Eur J Cancer2005;41:2040-59.
14. Fears TR, Guerry D, Pfeiffer RM, Sagebiel RW, Elder DE, HalpernA et al. Identifying individuals at high risk of melanoma: a practicalpredictor of absolute risk. J Clin Oncol 2006;24:3590-6.
15. Sasse AD, Sasse EC, Clark LGO, Ulloa L, Clark OAC. Chemoim-munotherapy versus chemotherapy for metastatic malignant melanoma.Cochrane Database Syst Rev 2007;1:CD005413.
16. Balch CM, Soong SJ, Atkins MB, Buzaid AC, Cascinelli N. Coit DGet al. An evidence-based staging system for cutaneous melanoma.CA Cancer J Clin 2004;54:131-49.
17. Collier, AP, Francis SO, Mahlberg MJ, McLaughlin JA, Dellavalle RP.Chemoprevention: a role in melanoma? Expert Rev Dermatol2007;2:51-8.
18. Koh HK. Melanoma screening: focusing the public health journey. ArchDermatol 2007;143:101-3.
19. Demierre MF, Sondak VK. Cutaneous melanoma: pathogenesis andrationale for chemoprevention. Crit Rev Oncol Hematol 2005;53:225-39.
20. Sporn MB, Dunlop NM, Newton DL, Smith JM. Prevention of chem-ical carcinogenesis by vitamin A and its synthetic analogs (retinoids).Fed Proc 1976;35:1332-8.
21. Herberman RB, Pearce HL, Lippman SM, Pyenson BS, Alberts DS.Cancer chemoprevention and cancer preventive vaccines. A call toaction: leaders of diverse stakeholder groups present strategies forovercoming multiple barriers to meet an urgent need. Cancer Res2006;66:11540-9.
22. Demierre MF, Nathanson L. Chemoprevention of melanoma: an unex-plored strategy. J Clin Oncol 2003;21:158-65.
23. Diffey BL. Sunscreens and melanoma: the future looks bright. Br J Der-matol 2005;153:378-81.
24. Wright TI, Spencer JM, Flowers FP. Chemoprevention of non-melanoma skin cancer. J Am Acad Dermatol 2006;54:933-46; quiz947-50.
25. Mittal A, Elmets CA, Katiyar SK. Dietary feeding of proanthocyani-dins from grape seeds prevents photocarcinogenesis in SKH-1 hair-less mice: relationship to decreased fat and lipid peroxidation. Car-cinogenesis 2003;24:1379-88.
26. Moon TE, Levine N, Cartmel B, Banger JL, Rodney S, Dong Q et al.Effect of retinol in preventing squamous cell skin cancer in moderate-risk subjects: a randomized, double-blind, controlled trial. SouthwestSkin Cancer Prevention Study Group. Cancer Epidemiol BiomarkersPrev 1997;6:949-56.
27. Kraemer KH, DiGiovanna JJ, Peck GL. Chemoprevention of skincancer in xeroderma pigmentosum. J Dermatol 1992;19:715-8.
28. Tangrea JA, Edwards BK, Taylor PR, Hartman AM, Peck GL, SalascheSJ et al. Long-term therapy with low-dose isotretinoin for preven-tion of basal cell carcinoma: a multicenter clinical trial. Isotretinoin-Basal Cell Carcinoma Study Group. J Natl Cancer Inst 1992;84:328-32.
29. Levine N, Moon TE, Cartmel B, Bangert JL, Rodney S, Dong Q et al.Trial of retinol and isotretinoin in skin cancer prevention: a random-ized, double-blind, controlled trial. Southwest Skin Cancer PreventionStudy Group. Cancer Epidemiol Biomarkers Prev 1997;6:957-61.
30. Valcic S, Timmermann BN, Alberts DS, Wachter GA, Krutzsch M,Wyner J et al. Inhibitory effect of six green tea catechins and caf-feine on the growth of four selected human tumor cell lines. Anti-cancer Drugs 1996;7:461-8.
31. Zhao J, Wang J, Chen Y, Agarwal R. Anti-tumor-promoting activity ofa polyphenolic fraction isolated from grape seeds in the mouse skintwo-stage initiation-promotion protocol and identification of pro-cyanidin B5-3’-gallate as the most effective antioxidant constituent.Carcinogenesis 1999;20:1737-45.
32. Ahmad N, Gilliam AC, Katiyar SK, O’Brien TG, Mukhtar H. A defin-itive role of ornithine decarboxylase in photocarcinogenesis. Am JPathol 2001;159:885-92.
33. Limtrakul P, Lipigorngoson S, Namwong O,Apisariyakul A, Dunn FW.
Inhibitory effect of dietary curcumin on skin carcinogenesis in mice.Cancer Lett 1997;116:197-203.
34. Conney AH, Lysz T, Ferraro T, Abidi TF, Manchand PS, Laskin JD etal. Inhibitory effect of curcumin and some related dietary compoundson tumor promotion and arachidonic acid metabolism in mouse skin.Adv Enzyme Regul 1991;31:385-96.
35. Jee SH, Shen SC, Tseng CR, Chiu HC, Kuo ML. Curcumin inducesa p53-dependent apoptosis in human basal cell carcinoma cells. JInvest Dermatol 1998;111:656-61.
36. Bush JA, Cheung KJ Jr, Li G. Curcumin induces apoptosis in humanmelanoma cells through a Fas receptor/caspase-8 pathway indepen-dent of p53. Exp Cell Res 2001;271:305-14.
37. Carbone PP, Pirsch JD, Thomas JP, Douglas JA, Vrma AK, Larson POet al. Phase I chemoprevention study of difluoromethylornithine in sub-jects with organ transplants. Cancer Epidemiol Biomarkers Prev2001;10:657-61.
38. Carbone PP, Douglas JA, Larson PO, Verma AK, Blair IA, PomplunM et al. Phase I chemoprevention study of piroxicam and alpha-diflu-oromethylornithine. Cancer Epidemiol Biomarkers Prev 1998;7:907-12.
39. Wei H, Saladi R, Lu Y, Wang Y, Palep SR, Moore J et al. Isoflavonegenistein: photoprotection and clinical implications in dermatology.J Nutr 2003;133(11 Suppl 1):3811S-3819S.
40. Yarosh D, Alas LG,Yee V, Oberyszyn A, Kibitel JT, Mitchell D et al.Pyrimidine dimer removal enhanced by DNA repair liposomes reducesthe incidence of UV skin cancer in mice. Cancer Res 1992;52:4227-31.
41. Yarosh D, Klein J, O’Connor A, Hawk J, Rafal E, Wolf P. Effect of top-ically applied T4 endonuclease V in liposomes on skin cancer in xero-derma pigmentosum: a randomised study. Xeroderma PigmentosumStudy Group. Lancet 2001;357:926-9.
42. Feleszko W, Balkowiec EZ, Sieberth E, Marczak M, Dabrowska A,Giernasz A et al. Lovastatin and tumor necrosis factor-alpha exhibitpotentiated antitumor effects against Ha-ras-transformed murinetumor via inhibition of tumor-induced angiogenesis. Int J Cancer1999;81:560-7.
43. D’Orazio JA, Nobuhisa T, Cui R, Arya M, Spry M, Wakamatsu K etal. Topical drug rescue strategy and skin protection based on the roleof Mc1r in UV-induced tanning. Nature 2006;443:340-44.
44. Woodmansee C, Pillow J, Skinner RB. The role of topical immuneresponse modifiers in skin cancer. Drugs 2006;66:1657-64.
45. Huncharek? M, Kupelnick B. Use of topical sunscreens and the riskof malignant melanoma: a meta-analysis of 9067 patients from 11case-control studies. Am Eur J Public Health 2002; 92:1173-7.
46. Holly? EA, Aston DA, Cress RD, Ahn DK, Kristiansen JJ. Cutaneousmelanoma in women. I. Exposure to sunlight, ability to tan, and otherrisk factors related to ultraviolet light. Am J Epidemiol1995;41:923-33.
47. Autier? P, Dore JF, Schifflers E, Cesarini JP, Bollaerts A, KoelmelKF et al. Melanoma and use of sunscreens: an EORTC case-controlstudy in Germany, Belgium and France. The EORTC Melanoma Co-operative Group.Int J Cancer 1995;61:749-55.
48. De Fabo EC, Noonan FP, Rears T, Merlino G. Ultraviolet B but notultraviolet A radiation initiates melanoma. Cancer Res 2004;64):6372-6.
49. IARC Handbooks on Cancer Prevention. Vol 5. Sunscreens. Gene-va:IARC;2001.
50. Lee TK, Rivers JK, Gallagher RP. Site-specific protective effect of bro-ad-spectrum sunscreen on nevus development among white school-children in a randomized trial. J Am Acad Dermatol 2005;52:786-92.
51. Luther H, Altmeyer P, Garbe C, Ellwanger U, Jahn S, Hoffmann K etal. Increase of melanocytic nevus counts in children during 5 years offollow-up and analysis of associated factors. Arch Dermatol1996;132:1473-8.
52. Darlington S, Siskind V, Green L, Green A. Longitudinal study of mela-nocytic nevi in adolescents. J Am Acad Dermatol 2002;46:715-22.

FREEMAN MELANOMA CHEMOPREVENTION
146 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
53. Dulon M, Weichenthal M, Blettner M, Breibart M, Hetzer M, Grein-ert R et al. Sun exposure and number of nevi in 5- to 6-year-old Euro-pean children. Clin Epidemiol 2002;55:1075-81.
54. Wachsmuth RC, Turner F, Barrett JH, Gaut R, Randerson-Moor JA,Bishop DT et al. The effect of sun exposure in determining nevusdensity in UK adolescent twins. J Invest Dermatol 2005;124:56-62.
55. Bauer J, Buttner P, Wiecker TS, Luther H, Garbe C. Effect of sunscreenand clothing on the number of melanocytic nevi in 1812 German chil-dren attending day care. Am J Epidemiol 2005;161:620-7.
56. Autier P, Dore JF, Cattaruzza MS, Renard F, Renard F, Luther H,Gentiloni-Silveri F et al. Sunscreen use, wearing clothes, and numberof nevi in 6- to 7-year-old European children. European Organizationfor Research and Treatment of Cancer Melanoma Cooperative Group.J Natl Cancer Inst 1998;90:1873-80.
57. Gallagher RP, Rivers JK, Lee TK, Bajdik CD, McLean DI, ColdmanAJ. Broad-spectrum sunscreen use and the development of new neviin white children: a randomized controlled trial. JAMA 2000;283:2955-60.
58. Bauer J, Buttner P, Wiecker TS, Luther H, Garbe C. Interventional studyin 1232 young German children to prevent the development ofmelanocytic nevi failed to change sun exposure and sun protectivebehavior. Int J Cancer 2005;116:755-61.
59. Levy C, Khaled M, Fisher DE. MITF: master regulator of melanocytedevelopment and melanoma oncogene. Trends Mol Med 2006;12:406-14.
60. Demierre MF. What about chemoprevention for melanoma? CurrOpin Oncol 2006;18:180-4.
61. Depasquale1 I, Wheatley DN. Action of Lovastatin (Mevinolin) on anin vitro model of angiogenesis and its co-culture with malignantmelanoma cell lines. Cancer Cell Int 2006;6:9.
62. Hoffmann P, Roumeguère T, Schulman C, van Velthoven R. Use ofstatins and outcome of BCG treatment for bladder cancer. N Engl J Med2006;355:2705-7.
63. Downs JR, Clearfield M, Weis S, Whitney E, Shapiro DR, Beere PAet al. Primary prevention of acute coronary events with lovastatin inmen and women with average cholesterol levels: results ofAFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Pre-vention Study. JAMA 1998;279:1615-22.
64. Kaye JA, Jick H. Statin use and cancer risk in the general practiceresearch database. Br J Cancer 2004;90:635-7.
65. Dale KM, Coleman CI, Henyan NN, Kluger J, White CM. Statinsand cancer risk: a meta-analysis. JAMA 2006;295:74-80.
66. Dellavalle RP, Drake A, Graber M, Heilig LF, Hester EJ, JohnsonKR et al. Statins and fibrates for preventing melanoma.CochraneDatabase Syst Rev 2005;4:CD003697.
67. Freeman SR, Drake AL, Heilig LF, Graber M, McNealy K, SchillingLM et al. Statins, fibrates, and melanoma risk: a systematic review andmeta-analysis. J National Cancer Inst 2006;98:1538-46.
68. Rubins HB, Robins SJ, Collins D, Fye CL, Anderson JW, Elan MB etal. Gemfibrozil for the secondary prevention of coronary heart diseasein men with low levels of high-density lipoprotein cholesterol. VeteransAffairs High-Density Lipoprotein Cholesterol Intervention Trial StudyGroup. N Engl J Med 1999;341:410-8.
69. Fischer Jde S, Carvalho PC, Gattass CR, Paschoal EM, da Motae Sil-va MS, Carvalho Mda G. Effects of perillyl alcohol and heat shock tre-atment in gene expression of human lung adenocarcinoma cell lineA549. J Exp Ther Oncol 2006;5:301-7.
70. Flach J, Antoni I, Villemin P, Bentzen CL, Niesor EJ. The meval-onate/isoprenoid pathway inhibitor apomine (SR-45023A) is antipro-liferative and induces apoptosis similar to farnesol. Biochem Bio-phys Res Commun 2000;270:240-6.
71. Haluska FG, Ibrahim N. Therapeutic targets in melanoma: map kinasepathway. Curr Oncol Rep 2006;8:400-5.
72. Escudier B, Lassau N, Angevin E. Phase I trial of Sorafenib in com-bination with IFN {alpha}-2a in patients with unresectable and/ormetastatic renal cell carcinoma or malignant melanoma. Clin CancerRes 2007;13:1801-9.
73. Strumberg D. Preclinical and clinical development of the oral multi-kinase inhibitor sorafenib in cancer treatment. Drugs Today2005;41:773-84.
74. Smalley KS, Herlyn M. Targeting intracellular signaling pathwaysas a novel strategy in melanoma therapeutics. Ann N Y Acad Sci2005;1059:16-25.
75. Nihal M, Ahmad N, Mukhtar H, Woods GS. Anti-proliferative andproapoptotic effects of (-)epigallocatechin-3-gallate on humanmelanoma: possible implications for the chemoprevention ofmelanoma. Int J Cancer 2005;114:513-521.
76. Katiyar SK, Agarwal R, Wang ZY, Bhatia AK, Mukhtar H. (-)Epi-gallocatechin-3-gallate in Camellia sinensis leaves from Himalayanregion of Sikkim: inhibitory effects against biochemical events andtumor initiation in Sencar mouse skin. Nutr Cancer 1992;18:73-83.
77. Wang ZY, Agarwal R, Bickers DR, Mukhtar J. Protection againstultraviolet B radiation-induced photocarcinogenesis in hairless miceby green tea polyphenols. Carcinogenesis 1991;12:1527-30.
78. Harris RE, Beebe-Donk J, Doss H, Burr Doss D. Aspirin, ibuprofen,and other non-steroidal anti-inflammatory drugs in cancer preven-tion: a critical review of non-selective COX-2 blockade. Oncol Rep2005;13:559-83.
79. Wright TI, Spencer JM, Flowers FP. Chemoprevention of non-melanoma skin cancer. J Am Acad Dermatol 2006;54:933-946.
80. Harris RE, Beebe-Donk J, Namboodiri KK. Inverse association ofnon-steroidal anti-inflammatory drugs and malignant melanomaamong women. Oncol Rep 2001;8:655-7.
81. Vogt T, McClelland M, Jung B, Popova S, Bogenrieder T, Becker Bet al. Progression and NSAID-induced apoptosis in malignantmelanomas are independent of cyclooxygenase II. Melanoma Res2001;11:587-99.
82. Cahlin C, Gelin J, Delbro D, Lonnroth C, Doi C, Lundholm K. Effectof cyclooxygenase and nitric oxide synthase inhibitors on tumor gro-wth in mouse tumor models with and without cancer cachexia relatedto prostanoids. Cancer Res 2000;60:1742-9.
83. Denkert C, Kobel M, Berger S, Siegert A, Leclere A, Treefzer U et al.Expression of cyclooxygenase 2 in human malignant melanoma. Can-cer Res 2001;61:303-8.
84. Ramirez CC, Ma F, Federman DG, Kirsner RS. Use of cyclooxyge-nase inhibitors and risk of melanoma in high-risk patients. DermatolSurg 2005;31(7 Pt 1):748-52.
85. Hansen LA, Sigman EC, Andreola F, Ross SA, Kellof GJ, De LucaLM. Retinoids in chemoprevention and differentiation therapy. Car-cinogenesis 2000;21:1271-9.
86. Weiss RL, Calhoun KH, Ahmal AE, Stanley D. Ornithine decar-boxylase activity in tumor and normal tissue of head and neck cancerpatients. Laryngoscope 1992;102:855-7.
87. Rebel H, van Steeg H, Beems RB, Schouten R, de Gruijl FR, TerlethC. Suppression of UV carcinogenesis by difluoromethylornithine innucleotide excision repair-deficient Xpa knockout mice. Cancer Res2002;62:1338-42.
88. Aggarwal BB, Bhardwaj A, Aggarwal RS, Seeram NP, Shishodia S,Takada Y. Role of resveratrol in prevention and therapy of cancer:preclinical and clinical studies. Anticancer Res 2004;24:2783-840.
89. Fuggetta MP, D’Atri S, Lanzilli G, Tricarico M, Cannavo E, ZambrunoG et al. In vitro antitumour activity of resveratrol in human melanomacells sensitive or resistant to temozolomide. Melanoma Res2004;14:189-96.
90. Niles RM, McFarland M, Weimer MB, Redkar A, Fu YM, MeadowsGG. Resveratrol is a potent inducer of apoptosis in human melanomacells. Cancer Lett 2003;190:157-63.
91. van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E,Van den Eynde B et al. A gene encoding an antigen recognized bycytolytic T lymphocytes on a human melanoma. Science1991;254:1643-7.
92. Demierre MF, Allten S, Brown R. New treatments for melanoma.Dermatol Nurs 2005;17:287-95.

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 147
MELANOMA CHEMOPREVENTION FREEMAN
93. Adamina M, Schumacher R, Zajac P, Weber WP, Rosenthal R,Groeper C et al. Advanced liposomal vectors as cancer vaccines inmelanoma immunotherapy. J Liposome Res 2006;16:195-204.
94. Pilla L, Valenti R, Marrari A, Patuzzo R, Santinami M, Parmiani Get al. Vaccination: role in metastatic melanoma. Expert Rev Anti-cancer Ther 2006;6:1305-18.
95. Berman B, Perez OA, Zell D. Immunological strategies to fight skincancer. Skin Therapy Lett 2006;11:1-7.
96. Peris K, Micantonio T, Fargnoli MC, Lozzi GP, Chimenti S.Imiquimod 5% cream in the treatment of Bowen’s disease and inva-sive squamous cell carcinoma. J Am Acad Dermatol 2006;55:324-7.
97. Peris K, Campione E, Micantonio T, Marulli GC, Fargnoli MC, Chi-menti S. Imiquimod treatment of superficial and nodular basal cellcarcinoma: 12-week open-label trial. Dermatol Surg 2005;31:318-23.
98. Wolf IH, Cerroni L, Kodama K, Kerl H. Treatment of lentigo maligna(melanoma in situ) with the immune response modifier imiquimod.Arch Dermatol 2005;141:510-4.
99. Naylor MF, Crowson N, Kuwahara R, Teague K, Garcia C, Macin-nis C et al. Treatment of lentigo maligna with topical imiquimod. BrJ Dermatol 2003;149 (Suppl 66):66-70.
100. Egan KM, Sosman JA, Blot WJ. Sunlight and reduced risk of cancer:is the real story vitamin D? J Natl Cancer Inst 2005;97:161-3.
101. Smedby KE, Hjalgrim H, Melbye M, Torrang A, Rostgaard K, Munks-gaard L et al. Ultraviolet radiation exposure and risk of malignant lym-phomas. J Natl Cancer Inst 2005;97:199-209.
102. Berwick M, Armstrong BK, Ben-Porat L, Fine J, Kricker A, Eberle
C et al. Sun exposure and mortality from melanoma. J Natl CancerInst 2005;97:195-9.
103. Boniol M, Armstrong BK, Dore JF. Variation in incidence and fatal-ity of melanoma by season of diagnosis on New South Wales, Aus-tralia. Cancer Epidemiol Biomarkers Prev 2006;15:524-6.
104. Holick MF. Vitamin D: importance in the prevention of cancers, typeI diabetes, heart disease, and osteoporosis. Am J Clin Nutr2004;79:362-71.
105. Kellof GJ, Lippman SM, Dannenberg AJ, Sigman CC, Pearce HL,Reid BJ et al. Progress in chemoprevention drug development: the pro-mise of molecular biomarkers for prevention of intraepithelial neo-plasia and cancer—a plan to move forward. Clin Cancer Res2006;12:3661-97.
106. Hampton T. Clinical trials point to complexities of chemopreven-tion of cancer. JAMA 2005;294:29-31.
107. SchatzkinA, Gail M. The promise and peril of surrogate end pointsin cancer research. Nat Rev Cancer 2002;2:19-27.
108. Meyskens FL, Szabo E. How should we move the field of chemo-preventive agent development forward in a productive manner?Recent Results Cancer Res 2005;166:113-24.
109. Cui R, Widlund HR, Feige E, Lin JY, Wilensky DL, Igras VE et al.Central role of p53 in the suntan response and pathologic hyperpig-mentation. Cell 2007;128:853-64.
110. Losina E, Walensky RP, Geller A, Beddingfield FC, Wolf LL, GilchrestBA et al. Visual screening for malignant melanoma: a cost-effec-tiveness analysis. Arch Dermatol 2007;143:21-8.

G ITAL DERMATOL VENEREOL 2007;142:149-60
The clinical experience of cancer vaccinesfor malignant melanoma
Much effort has been made to improve systemic treatment ofadvanced melanoma. However, clinically meaningful prolon-gation of survival in patients with metastatic melanoma remainselusive. Anecdotal reports of spontaneous cancer regressions aswell as durable remissions following immune modulating ther-apies (e.g. interleukin-2, interferon alpha, anti-CTLA4) continueto support the effort and hope that effective immune therapyof melanoma is an achievable goal. One approach in this efforthas been the attempt to improve immune destruction ofmelanoma through active immunotherapy via vaccinationagainst melanoma antigens. A broad range of strategies havebeen employed, from single peptide vaccinations to injectionsof modified dendritic cells; many of these vaccines have beentested in clinical trials in melanoma patients. Herein, we reviewthe breadth of clinical trials conducted to date using vaccinebased therapy of melanoma.KEY WORDS: Melanoma - Skin - Vaccines.
The incidence of malignant melanoma is increasingat an alarming pace, faster than any other form of
cancer.1 When diagnosed early, melanoma is curableby simple surgical resection. However, when diag-nosed after skin lesions have invaded more than 1 mmbeyond the granular layer of the epidermis (T2 andgreater), melanoma has a high propensity to metasta-size to regional lymph nodes and subsequently to vis-ceral organs. When metastatic, melanoma is amongthe most difficult of cancers to treat, as it is generallyresistant to all current forms of chemotherapy.2 This
lack of effective forms of chemotherapy has spurnedinvestigators to turn to novel strategies in melanomatreatment.
While metastatic melanoma is nearly impossible tocure with chemotherapy, primary tumor sites of cuta-neous melanoma frequently demonstrate histologicand clinical evidence of spontaneous partial regres-sion. Complete regressions are rare, but documentedfull regressions of both primary cutaneous melanomaand metastatic melanoma have been reported. A com-mon histologic feature of clinically regressing cuta-neous melanoma is the presence of cytotoxic T lym-phocytes in the regressing lesions.3 Likewise, autoim-mune phenomena such as vitiligo frequently accom-pany spontaneous regression of melanoma. This sug-gests that patients’ immune systems are capable ofdestroying malignant melanocytes. These data in addi-tion to a voluminous preclinical literature demon-strating the responsiveness of murine melanoma tumormodels to immunomodulatory/vaccine therapies serveas the basis for the ongoing clinical trial efforts in
1Department of Internal MedicineMayo Clinic College of Medicine, Rochester, MN, USA
2Division of HematologyDepartment of Internal Medicine
Mayo Clinic College of Medicine, Rochester, MN, USA
Address reprint requests to: S. Markovic, Department of Internal Medi-cine, Mayo Clinic College of Medicine, Rochester, MN, USA.E-mail: [email protected]
M. S. BLOCK 1, S. N. MARKOVIC 2
Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 149

BLOCK THE CLINICAL EXPERIENCE OF CANCER VACCINES FOR MALIGNANT MELANOMA
150 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
developing effective immune/vaccine based therapyfor melanoma in humans.
Several factors must be considered in the develop-ment of an effective melanoma vaccine. First, the vac-cine must provide tumor-associated antigens (TAA)that the patient’s immune system can recognize. TAAused in therapeutic vaccines may be highly definedpeptide or glycolipid moieties, or may be a host ofproteins found in cell lysates or irradiated cells. Pep-tides generally stimulate cellular (T cell-mediated)immunity, while glycolipids (commonly gangliosides)stimulate humoral (antibody-mediated) immunity;protein antigens may generate either cellular and/orhumoral immunity. Second, the TAA must be pre-sented in a manner leading to generation of a potentimmune response. This may be accomplished by theuse of immune stimulants (adjuvants), coadministeredcytokines, or by providing antigen presenting cells(APC) such as dendritic cells (DC) as part of the vac-cine treatment. Finally, an effective vaccine depends onthe ability of the patient’s immune system to mount aneffective immune response, as well as on the tumorbeing susceptible to immune-mediated destruction.Patients who have lost immune responsiveness willhave a poor clinical response to vaccination even whengiven an optimal vaccine. Thus, for a vaccine againstmelanoma to be clinically effective, appropriate TAAmust be presented in an adequate immune-stimulato-ry context to a patient capable of mounting an effec-tive immune response.
Vaccines that use endogenousantigen presentation
Tumor lysates
Among the first antigen sources used in clinical tri-als of melanoma vaccines were lysed melanoma tumorcells. Tumor lysate-based vaccines provide multipleTAA from killed melanoma cells. Early trials of wholelysates 4 or cell membranes that were lysed and formedinto liposomal preparations,5 showed little efficacy,with few clinical responses. Thus, tumor lysates alonehave not generally been regarded as effective vaccines.
Adjuvants have been used in an attempt to enhancethe immunogenicity of tumor cell lysates. One prepa-ration known as Melacine® combines lysates fromtwo melanoma cell lines with a modified Freund’sadjuvant.6 Phase III trials in stage IV patients com-
paring Melacine® to standard chemotherapy withBCNU plus tamoxifen 7 or to interferon α-2b mono-therapy 8 failed to demonstrate a survival benefit forpatients receiving vaccine therapy. When Melacine®
was compared to observation in the adjuvant setting,no significant difference in progression-free survivalwas found, although retrospective analysis showedbenefit in patients with certain HLA alleles.9 Unfor-tunately, discouraged by the overall results ofMelacine® in clinical testing, further developmenthas been abandoned.
Because viral infections generate significant cellularimmune responses, several trials have been conductedin which melanoma cell lines were incubated withviruses prior to lysis. In phase III trials in stage II 10 andstage IIB and III 11 melanoma patients, adjuvant immu-nization with lysates from allogeneic melanoma celllines incubated with vaccinia virus was compared toplacebo. No significant difference in progression-freesurvival or overall survival was seen, although bothstudies demonstrated a trend towards increased over-all survival in those receiving the cell lysate vaccine.Lysates from cell lines incubated with Norwalk diseasevirus (NDV) have also been studied. Survival ofpatients treated with NDV lysates was prolonged com-pared to historical controls,12 and patients who relapsedafter treatment had improved survival if they werereimmunized.13 However, a small randomized study ofan NDV lysate vaccine in stage III patients failed todemonstrate a benefit in relapse-free survival forpatients immunized with the NDV lysate vaccine.14
An additional strategy used to enhance lysate-basedvaccine efficacy involves affixing lysed membranesto silica microbeads. A phase I trial conducted in stageIV patients showed little toxicity from the microbead-membrane preparation; however, only 2 of 19 patientsstudied showed mixed or partial treatment respons-es.15 While tumor lysates, either alone or with addi-tional immune stimulants, would seem to have greatpotential as melanoma vaccines, they have been large-ly unsuccessful in clinical trials.
Irradiated tumor cells
Vaccines prepared from melanoma cells that havebeen irradiated are among the most studied vaccinetypes in melanoma clinical trials. Broadly speakingthere are two types of irradiated tumor cell vaccines:autologous and allogeneic. An autologous vaccine isgenerated by culturing tumor cells from a patient and

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 151
THE CLINICAL EXPERIENCE OF CANCER VACCINES FOR MALIGNANT MELANOMA BLOCK
then vaccinating the patient with the cultured cells,often after they have been modified to improveimmunogenicity, while allogeneic vaccines use oneor more “off the shelf” cultured human melanoma celllines. Autologous vaccines have the advantage of beingidentical in HLA type and TAA to the patient’s tumor,while allogeneic vaccines are far easier to prepare andmodify. One major means by which both autologousand allogeneic vaccines have been modified to enhancetheir immunogenicity has been through the use ofgenetic engineering.
The earliest irradiated tumor cell vaccines were com-prised of a mixture of several different allogeneic celllines.12, 16, 17 After irradiation, cells were injected withor without cyclophosphamide, used as a vaccine adju-vant. In phase II trials, clinical responses to vaccinewere seen in a minority of patients. One study showedcomplete responses in 3 of 40 and partial responses in6 of 40 patients.12 Subsequent allogeneic vaccines haveused other vaccine adjuvants such as bacillus Calmette-Guérin (BCG),13, 18 cyclophosphamide and otherdrugs,19, 20 or recombinant granulocyte-macrophagecolony-stimulating factor (GM-CSF). 21 Measurableimmune responses were demonstrated by delayed typehypersensitivity (DTH) testing in many cases, and sev-eral phase II studies have shown a modest improve-ment in 5 year survival relative to single-institutional his-toric controls.19, 20
To improve immunogenicity of allogeneic irradiat-ed melanoma cell vaccines, several investigators havetransfected the vaccine cells with genes encodingimmune-stimulating molecules. The earliest such tri-al, a phase I trial in advanced melanoma patients,involved allogeneic cells transfected with IL-4.22 Only1 of 12 patients mounted a measurable immuneresponse to autologous tumor, and no patient had sig-nificant tumor regression. Both IL-4 and IL-2 havebeen tried in different allogeneic tumor cell vaccines,with occasional clinical responses seen in early phaseclinical trials.23, 24 However, when in vitro immuneresponses were measured, one trial demonstratedimmune responses only to allogeneic and not to autol-ogous melanoma cells.
Because autologous vaccines require patientmelanoma cells, excised tumor is required for theirproduction. As a result, the majority of clinical trialsof irradiated autologous tumor cell vaccines have beendone in patients with stage IV disease. As with allo-geneic tumor cell vaccines, the vast majority of trials
have been nonrandomized trials with small numbers ofpatients. Adjuvant strategies used in autologous vac-cines have included: 1) modifying the tumor cells withthe hapten dinitrophenyl (DNP); 25-27 2) using BCG*173, 151; 3) modified Freund’s adjuvant; 28 4) co-administering cytokines such as interferons,29, 30 and/orGM-CSF.29-31 Many of the trials yielded responses ina minority (often 10-20%) of patients, with a few com-plete responses and long-term survivals reported; how-ever, no randomized trials have been conducted to ver-ify a beneficial clinical effect.
Autologous tumor cells have also been transfectedwith numerous cytokines and other immune proteinsin an attempt to enhance antitumor immune respons-es. The majority of trials conducted thus far have beensmall phase I trials. Transfected genes have includedIL-2,32-34 interferon gamma (IFNγ),35 IL-12,36 IL-7,37, 38
GM-CSF,38-45 a Th1/NK cell stimulating moleculeknown as d-SLIM, and an innate immune proteincalled tag7/PGRP-S.46 The majority of these trialsreport immune stimulation as measured by in vitroassays in a significant proportion of patients (over halfin some cases). However, clinical responses, whenseen, were present in a minority of patients.
Peptides
Peptide-based vaccines provide a highly definedantigen to which the patient immune response can eas-ily assessed. Furthermore, peptide vaccines are rela-tively easy to produce compared to vaccines requiringpatient tumor or immune cell harvesting. For thesereasons, peptide vaccines are among the most stud-ied types of melanoma vaccines. One of the limita-tions of peptide-based vaccines, however, is that theyrequire a particular HLA allele for peptide binding.Because of this, many trials of peptide vaccines areperformed only in HLA-A2-positive patients. Clinicaltrials to date have all been phase I or II trials; these havefocused on types of peptides, the use of HLA class II-binding peptides, and the use of adjuvants and otherimmune stimulants.
Melanocytes use multiple unique proteins not pro-duced by other cell types. As such, many of the pep-tides used in melanoma vaccines are specific tomelanocytes, and immune responses to peptide vac-cines would be expected to be restricted to tumor cellsand melanocytes. Among the earliest peptides used inclinical trials were derived from the gp100 protein.47-50
Peptides from MART-1/Melan A 51-53 and from the

BLOCK THE CLINICAL EXPERIENCE OF CANCER VACCINES FOR MALIGNANT MELANOMA
152 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
enzyme tyrosinase 54, 55 have also been widely used.MART-1 and gp100 peptides have generally beenfound to be more immunogenic than tyrosinase-derivedpeptides.56 Many vaccines utilize combinations of pep-tides to enhance and broaden the immune response.57-59
Additional peptides that have been used in trials includeNY-ESO-1,60 MAGE-A12,61 and multiple nonspecifiedpeptides complexed with heat shock proteins.62, 63 Fewimmune responses have been seen with peptides oth-er those derived from MART-1, gp100, MAGE-3, andtyrosinase.
While initial trials focused exclusively on HLA classI-binding peptides that stimulate cytotoxic T cells, theimportance of T helper cells in immune responses hasprompted trials involving the use of class II-bindingpeptides. Some trials have utilized class II-bindingpeptides from melanoma-associated proteins, such asgp100,64 MART-1,65 and NY-ESO-1; 60 others haveused unrelated peptides such as tetanus vaccine-derivedpeptides.48, 66 While one trial reported frequent immuneresponses to a class II-binding peptide,65 one reportedno effect on class I-restricted immune responses,60
and one actually reported worsening of the class I-restricted response with concomitant use of a class II-binding peptide.64 It is difficult to draw conclusionsfrom these limited data. However, it appears that theaddition of class II-binding peptides do not seem to pro-vide significant improvement to peptide-based vac-cines.
As peptide vaccines provide a defined antigen andthus an easily measurable immune response, trials ofpeptide vaccines frequently test novel adjuvants andimmune stimulants. One early trial used the cytokinesIL-2, GM-CSF, or IL-12, and found that each sup-ported a clinical tumor response in a significant minor-ity of patients.67 However, a later trial saw fewerimmune responses with concurrent versus delayed useof IL-2.68 Stimulants of the innate immune systemsuch as Toll-like receptor seven ligand (TLR7L) 69 andCpG oligonucleotides 53 appear to enhance immuneresponses in early trials; however, improvement inclinical responses has not been clearly shown. Final-ly, blocking antibodies to the immune inhibitor CTLA-4 have been added to a multi-epitope peptide vaccine,with a significant minority of patients (11%) experi-encing objective clinical responses.70 Albeit not over-ly successful in phase II clinical testing, some pep-tide vaccines have advanced to randomized phase IIIclinical trials. Most notable of these studies is E4697
where patients with surgically resected advanced stageIII and IV melanoma were treated with a mixture ofthree HLA class-I binding peptides (MART-1, gp100,tyrosinase) with/without adjuvant GM-CSF adminis-tered at a dose of 125 ?g/m2 for 14 days of a 28 daytreatment cycle. The vaccine was compared to GM-CSF alone and placebo. We eagerly await the resultsof the study.
Proteins
Whole protein vaccines have seen little use inmelanoma therapeutic trials, despite their widespreaduse in other applications. Compared to peptide vac-cines, they provide the theoretical advantages of beingapplicable to patients with different HLA types andof simultaneously providing HLA class I and class II-binding antigens capable of stimulating both cytotox-ic T cells and helper T cells. The first protein vaccinetrial for melanoma used recombinant MAGE-3 as theprotein antigen given in adjuvant as a series of intra-muscular injections.71 Humoral responses to MAGE-3 were elicited, but clinical responses were limited topartial and mixed responses in a minority of patients.A second trial studied the effects of immunizationwith recombinant NY-ESO-1, a cancer-testes antigenpresent in melanoma and sarcoma patients.72 Immu-nization with NY-ESO-1 produced highly potentimmune responses. The trial was not designed to assessclinical outcomes, but showed a favorable trend towardincreased relapse-free survival in patients receivingNY-ESO-1 plus adjuvant. Protein vaccines, althoughthey are little studied in melanoma, represent a viablemeans of immunization against TAA, and further studyto determine their clinical efficacy is warranted.
Gangliosides
Gangliosides are a class of glycosphingolipids thatcontain sialic acid. They are found on the plasma mem-brane of cells. Gangliosides are involved in many cel-lular functions including cell adhesion and signalingresponses to receptor-ligand interactions. Certain gan-gliosides that are overexpressed in a number of dif-ferent malignancies, including melanoma, have beenimplicated in enabling tumors to grow and metasta-size.73 As such, they are attractive therapeutic targets,since elimination of cells with altered gangliosideexpression would theoretically limit a tumor’s abilityto grow and metastasize. Several groups have attempt-

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 153
THE CLINICAL EXPERIENCE OF CANCER VACCINES FOR MALIGNANT MELANOMA BLOCK
ed to target gangliosides as antigens in cancer vac-cines. In an early phase III clinical trial comparingimmunization of stage III melanoma patients with theganglioside GM2 plus BCG versus BCG alone, nodifference was seen in progression-free survival oroverall survival; however, a post hoc analysis revealeddifferences in both progression-free survival and over-all survival in those patients without prior immunity toGM2.74 Various vaccine preparations have been tried,and GM2 is most immunogenic when complexed to thecarrier protein keyhole limpet hemocyanin (KLH).75
However, a phase III clinical trial comparing GM2-KLH to high-dose IL-2 in stage IIB/III patients demon-strated superiority of IL-2 over ganglioside vaccinein overall and progression-free survival.76 Other gan-glioside vaccines have been tried including GD3,77, 78
GD2,79 and GM3; 80 however, none of these havedemonstrated significant success in generating clini-cal responses. Thus, while ganglioside vaccines appearto be immunogenic and can generate clinical respons-es, they are clinically inferior to conventionalimmunotherapy thus far.
Anti-idiotype antibodies
One potential barrier to vaccination against melanomaand other cancers is that TAA are inherently “self”antigens. As such, TAA often promote immune toler-ance, limiting the immune system’s ability to effec-tively eliminate TAA-bearing cells. Furthermore, somepotential TAA such as gangliosides are difficult to pre-pare in vaccine form, as they are difficult to generatesynthetically. For these reasons, investigators have triedanti-idiotype vaccine strategies.81 Briefly, in order togenerate an anti-idiotype vaccine, a TAA is used toimmunize a host animal. A monoclonal antibody againstthe TAA is generated and is in turn used to immunizea second host animal. A new monoclonal antibody,specific for the antigen binding portion of the first anti-body (anti-idiotype) is formed. The antigen bindingsite of this second antibody closely resembles the orig-inal TAA. The anti-idiotype antibody, itself a potentimmunogen, is used as a therapeutic vaccine. Patientimmune responses to the anti-idiotype antibody arecross-reactive with the TAA and can promote tumorimmunity. Phase I studies have confirmed the safety ofanti-idiotype vaccines against tumor associated pro-teoglycans 82 and gangliosides. 83-85 However, ran-domized trials demonstrating the efficacy of anti-idio-type vaccines have yet to be reported.
Nucleic acid vectors
Vectors that contain genes coding for TAA haveseveral attractive features that have prompted interestin their use as melanoma vaccines. First, proteins pro-duced in eukaryotic cells frequently undergo post-translational modification, including glycosylation.Synthetic peptides and prokaryote-derived recombinantproteins lack these modifications, but vector-driveneukaryotic (patient cell) expression generates proteinsin a manner allowing for normal post-translationalmodification. Second, many vectors such as virusesinduce potent immune responses and thus serve astheir own adjuvants. Because of these qualities, inves-tigators have attempted to use several different typesof vectors to deliver TAA and other genes to patients.The first attempt to use gene delivery to immunizepatients against melanoma antigens was with an ade-novirus-based construct with genes encoding MART-1 or gp100.86 Although the vaccine was administeredsafely, high pre-existing levels of neutralizing anti-bodies to adenovirus precluded the generation of aneffective immune response to the TAA. An early plas-mid-based vaccine also failed to generate immuneresponses.87 Subsequent trials have employed differ-ent poxviruses including vaccinia,88-90 canarypox,91, 92 andfowlpox.90, 93 In addition to delivery of genes encod-ing TAA, viral vectors have been used to generateexpression of costimulatory molecules, includingCD80 and CD86 (B7-1 and B7-2).88 One strategy hasinvolved priming patients with canarypox-TAA con-structs, then boosting with free peptides.91, 92 In gen-eral, the use of poxvirus vectors has promoted the gen-eration of immune responses to most TAA, althoughthe tyrosinase antigen has been poorly immunogenicin this setting.89, 90 Despite this, clinical responses tovaccination with vectors encoding TAA have thus farbeen modest at best.
Vaccines that provide antigen presentation
The above vaccine strategies all involve injectingpatients with TAA in a manner designed to promoteeffective antigen presentation by the patients’ APC.These modes of vaccination depend on patients’ abil-ity to absorb antigen and present it in a manner sup-portive of a protective immune response. Alternativestrategies have been developed in which the vaccineincludes both TAA and APC. This circumvents the

BLOCK THE CLINICAL EXPERIENCE OF CANCER VACCINES FOR MALIGNANT MELANOMA
154 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
need for endogenous antigen processing and presen-tation. Most vaccines that provide APC employ DC.DC are specialized APC that are especially proficientin stimulating naïve T cells. Immature DC are capableof phagocytosing large amounts of antigen, whilemature DC can present antigens in the context of ahost of costimulatory molecules. Two groups of vac-cines employing APC have been used: 1) those inwhich the APC are loaded with a limited number ofdefined antigens; and 2) those in which APC are loadedwith a multitude of antigens that are not defined.
Antigen presenting cells with defined antigens
Most APC-based vaccines employ DC because oftheir ability to prime immune responses with naïve Tcells. However, DC are not present at significant num-bers in the circulation, and, therefore, must be mobi-lized prior to harvest or generated in vitro. There arenumerous techniques for culturing DC from peripheralblood mononuclear cells (PBMC). Most commonly,GM-CSF and IL-4 are used to generate immature DCfrom cultured PBMC.94-98 Other investigators haveused type 1 interferons to generate DC,98-100 but DCgenerated by type 1 interferons have not producedimmune responses as efficiently in patients as thosegenerated by GM-CSF and IL-4. After they are gen-erated, DC are often incubated with additionalcytokines to make them “mature” (upregulate expres-sion of costimulatory molecules). Commonly usedmaturation cytokines include IL-3,95, 100 IL-6,95, 98
tumor necrosis factor alpha (TNFα),95, 98, 101 and CD40ligand.102 An alternative method to culturing DC invitro from PBMC is to mobilize DC in vivo by inject-ing the patient with the kinase Flt3, then directly har-vesting DC from blood.102 Finally, PBMC have beenused as APC without in vitro maturation into DC, andhave been shown to generate anti-tumor immuneresponses when injected along with IL-12.103, 104
Investigators using peptide-pulsed APC in clinicaltrials have used a diverse array of peptide antigens.Some trials employ DC pulsed with a single peptideantigen, such as MAGE-3,105, 106 MelanA/MART-1,104,
107-109 or NA17.A2.100 Others use an array of tumor-associated peptides, making tumor immune escapemore difficult.94, 110 In addition to TAA, some investi-gators have used unrelated peptides/proteins to assesspatient immune competency and to compare antitu-mor responses with those against other foreign anti-gens. These include KLH,94, 101 tetanus toxoid,105 and
influenza matrix protein (Flu-MP).106, 110 Immuneresponses to each of the above tumor-associated pep-tides have been demonstrated, albeit with differentimmunization efficacy.
At this time, most clinical trials of peptide-pulsedAPC vaccines have been conducted in patients withmetastatic melanoma (stage IV). Despite this, many tri-als demonstrate frequent robust immune responses toTAA, as measured by DTH and in vitro assays.102, 110
Clinical responses are seen at a lower frequency inpatients with advanced disease. A number of phase Iclinical trials have recorded clinical responses, andcomplete responses have been documented.104 Fur-ther evidence that peptide-pulsed APC vaccines gen-erate an effective antitumor response is seen whenresidual tumor after a partial response is examined.Several investigators have noted that remaining tumorfollowing a partial or mixed response to vaccinationshows evidence of antigen loss.103, 105 To date, there ispublished data from only one phase III clinical trialusing peptide-pulsed DC vaccine in patients withmetastatic melanoma.98 Patients were randomized toreceive either DC pulsed with a cocktail of peptidesbound by various HLA alleles or dacarbazine. No dif-ference was seen in overall survival, progression-freesurvival, or objective response. A few trials have beendone in the adjuvant setting in patients with surgical-ly resected melanoma. One such trial in stage II patientsdemonstrated frequent durable immune responses butdid not assess clinical outcomes.109 In conclusion, pep-tide-pulsed APC vaccines show promise in that theyfrequently induce measurable immune responses toTAA in advanced melanoma patients; however, theyhave not yet been shown to be clinically superior to“conventional chemotherapy” in the setting ofadvanced disease.
Antigen presenting cells with undefined antigens
Peptide-pulsed APC vaccines share the main limi-tations of peptide vaccines in that any given peptiderequires a particular HLA allele for its proper presen-tation, and that individual tumors may lose expres-sion of a peptide antigen either prior to or in responseto therapy (antigen escape). Investigators have usedseveral vaccine strategies that exploit the T cell prim-ing capability of DC in a manner that provides a broadrange of patient-specific antigens. Early trials usedDC that had been pulsed with lysed tumor cells. 94, 111
Subsequent trials have used autologous DC with both

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 155
THE CLINICAL EXPERIENCE OF CANCER VACCINES FOR MALIGNANT MELANOMA BLOCK
autologous 97, 112 and allogeneic 113-115 melanoma celllysates. Other investigators have used DC pulsed withirradiated tumor cells as an antigen source.116-118 Yetanother strategy has been to fuse autologous tumorcells with allogeneic DC with the goal that immuneresponses to allogeneic HLA molecules in the fusioncells would stimulate or augment TAA-directedimmune responses. Finally, investigators have usedgene transfer strategies to transfect DC with mRNAfrom autologous tumor tissue, causing the vaccine DCto produce and present TAA.123, 124 As with peptide-pulsed APC vaccines, the various multiantigen vac-cine strategies have shown evidence of TAA-directedimmune responses in a majority of patients, but clin-ical responses are seen in a significantly lower per-centage.
In one early clinical trial of DC-based vaccines,investigators used either multiple peptides or tumorcell lysates as a source of TAA.94 Immune responseswere monitored by measuring DTH skin responsesto intradermal injections of DC pulsed with controlantigen (KLH) or a cocktail of tumor-associated pep-tides. No exogenous maturation cytokines were giv-en. Both tumor antigen-specific immune responsesand clinical responses were seen in subsets of patientstreated with either peptide-pulsed or lysate-pulsedDC. This early study was not designed to determinewhether peptides or lysates are more effective as anti-gens in DC-based immunotherapy. A second clinicaltrial compared immunization with DC loaded with acocktail of HLA class I-binding peptides versusmelanoma cell lysates.97 Here, TAA-directed immuneresponses as measured by DTH were seen in 5 of 15patients treated with lysate-pulsed DC and in approx-imately half the patients treated with peptide-pulsedDC. However, no clinical response was seen in thegroup treated with peptide-pulsed DC, while 3 partialresponses and one mixed response were seen in thegroup treated with lysate-pulsed DC. Although patientswere not randomized, these data argue for increasedefficacy of lysates over peptides as a source of TAAin DC-based vaccines.
Conclusions
A vast range of therapeutic vaccine strategies againstmelanoma have been developed and tested in clinicaltrials. Tested antigen sources include autologous andallogeneic melanoma cell lysates, irradiated tumor
cells, synthetic peptides and proteins, gangliosidesand anti-idiotype antibodies, and nucleic acids encod-ing protein and peptide antigens. Methods to increasethe immunogenicity of vaccines consist of chemicaland biological adjuvants, cytokines, viral vectors, APCincluding DC, and blocking antibodies to regulatorymolecules. Trials have taken place in patients fromstage II through stage IV melanoma, with the major-ity of trials being in patients with metastatic disease.Numerous clinical trials have produced promisingresults in terms of objective evidence of tumor antigen-specific immune responses, at times in the majorityof patients immunized. Clinical responses are seenless frequently. Despite this promising data, no phaseIII trial to date has demonstrated superiority ofmelanoma vaccines over standard cytokine therapy orchemotherapy.
Clinically useful vaccine treatment of melanomarequires the use of effective TAA presented in appro-priate immune-stimulatory context to a host capableof generating a robust response. Failure of effectivevaccine antigen selection is seen in immune escape,which has been documented in several clinical trials.Immune escape occurs when the immune responseselects out variant tumor cells that lack the TAA usedin the vaccine. Strategies to overcome immune escapeinclude the use of more than one TAA in the vaccineregimen; however, tumors that loose expression ofHLA class I molecules may evade even multiantigenvaccines. When a vaccine fails to generate an immune-stimulatory context, a clonal expansion of TAA-spe-cific T cells may be produced, but the cells are inca-pable of immune destruction of the tumor, either dueto anergy (tolerance) or activation of inappropriatefunctions. Based on measures of in vitro functional-ity of T cells (cytotoxicity and cytokine secretionassays), many strategies appear to generate appro-priate immune responses. The disparity between robustimmune functionality and inconsistent clinicalresponses may be due to local or systemic effects ofthe tumor on immune cell effector function in vivothat are not detectable in vitro (immunosuppressivetumor micro environment). Finally, some patients failto generate immune or clinical responses to vaccinesthat have appropriate antigens presented with appro-priate immune stimulation. This includes patientswho fail to mount a response to control antigens suchas Flu-MP or KLH. In these patients, the melanomaitself and/or chemotherapy may be causing global

BLOCK THE CLINICAL EXPERIENCE OF CANCER VACCINES FOR MALIGNANT MELANOMA
156 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
immune dysregulation such that effective immuneresponses cannot be generated.
Clinical trials of melanoma vaccines thus far haveseen sporadic success, but have not demonstrated aconsistent patient benefit. Overcoming this will requireinvestigators to understand tumor antigen escape mech-anisms as well as mechanisms of tumor immunosup-pression both local in the tumor micro-environment(IL10, TGFb overproduction at the tumor site) andsystemic (regulatory T cell mediated immune sup-pression).
Riassunto
L’esperienza clinica dei vaccini per il melanoma maligno
Sono stati fatti molti sforzi per migliorare il trattamentosistemico del melanoma avanzato. Tuttavia, l’allungamentoclinicamente significativo della sopravvivenza nei pazienticon melanoma metastatico resta deludente. Le comunica-zioni aneddotiche relative a regressioni spontanee del car-cinoma, così come a remissioni durature dopo terapie immu-nomodulanti (ad esempio interleuchina-2, alfa-interferone,anti-CTLA4) continuano a supportare lo sforzo e la speran-za che una terapia immunomodulante efficace per il mela-noma sia un obiettivo raggiungibile. In questo senso, unapproccio è stato il tentativo di migliorare la distruzioneimmunitaria del melanoma tramite un’immunoterapia atti-va mediante una vaccinazione diretta contro gli antigeni delmelanoma. È stato impiegato un ampio spettro di strategie,dalle vaccinazioni con un singolo peptide a quelle con cel-lule dendritiche modificate; molti di questi vaccini sono sta-ti testati negli studi clinici eseguiti su pazienti con melano-ma. In questo lavoro vengono sintetizzati gli ampi studi cli-nici condotti sinora sulla terapia per il melanoma basata suivaccini.
Parole chiave: Malanoma - Cute - Vaccini.
References
1. Markovic SN, Erickson LA, Rao RD, Weenig RH, Pockaj BA, Bar-dia A et al. Malignant melanoma in the 21st century, Part 1: epi-demiology, risk factors, screening, prevention, and diagnosis. MayoClin Proc 2007;82:364-80.
2. Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology andnew targeted therapy. Nature 2007;445:851-7.
3. King M, Spooner D, Rowlands DC. Spontaneous regression of metasta-tic malignant melanoma of the parotid gland and neck lymph nodes:a case report and a review of the literature. Clin Oncol (R Coll Radi-ol) 2001;13:466-9.
4. Elliott GT, McLeod RA, Perez J, Von Eschen KB. Interim results ofa phase II multicenter clinical trial evaluating the activity of a thera-peutic allogeneic melanoma vaccine (theraccine) in the treatment ofdisseminated malignant melanoma. Semin Surg Oncol 1993;9:264-72.
5. Adler A, Schachter J, Barenholz Y, Bar LK, Klein T, Korytnaya R et
al. Allogeneic human liposomal melanoma vaccine with or without IL-2 in metastatic melanoma patients: clinical and immunobiologicaleffects. Cancer Biother 1995;10:293-306.
6. Sondak VK, Sosman JA. Results of clinical trials with an allogenicmelanoma tumor cell lysate vaccine: melacine. Semin Cancer Biol2003;13:409-15.
7. Mitchell MS, Eschen KBv. Phase III trial of melacine melanoma ther-accine versus combination chemotherapy in the treatment of stageIV melanoma. Proc Am Soc Clin Oncol 1997;16:494a.
8. Mitchell MS. Immunotherapy as part of combinations for the treatmentof cancer. Int Immunopharmacol 2003;3:1051-9.
9. Sosman JA, Unger JM, Liu PY, Flaherty LE, Park MS, Kempf RA etal. Adjuvant immunotherapy of resected, intermediate-thickness,node-negative melanoma with an allogeneic tumor vaccine: impact ofHLA class I antigen expression on outcome. J Clin Oncol2002;20:2067-75.
10. Wallack MK, Sivanandham M, Ditaranto K, Shaw P, Balch CM, UristMM et al. Increased survival of patients treated with a vacciniamelanoma oncolysate vaccine: second interim analysis of data froma phase III, multi-institutional trial. Ann Surg 1997;226:198-206.
11. Hersey P, Coates AS, McCarthy WH, Thompson JF, Sillar RW,McLeod R et al. Adjuvant immunotherapy of patients with high-riskmelanoma using vaccinia viral lysates of melanoma: results of a ran-domized trial. J Clin Oncol 2002;20:4181-90.
12. Morton DL, Foshag LJ, Hoon DS, Nizze JA, Famatiga E, Wanek LAet al. Prolongation of survival in metastatic melanoma after activespecific immunotherapy with a new polyvalent melanoma vaccine. AnnSurg 1992;216:463-82.
13. Hsueh EC, Essner R, Foshag LJ, Ye W, Morton DL. Activeimmunotherapy by reinduction with a polyvalent allogeneic cell vac-cine correlates with improved survival in recurrent metastaticmelanoma. Ann Surg Oncol 2002;9:486-92.
14. Voit C, Kron M, Schwurzer-Voit M, Sterry W. Intradermal injectionof Newcastle disease virus-modified autologous melanoma cell lysateand interleukin-2 for adjuvant treatment of melanoma patients withresectable stage III disease. J Dtsch Dermatol Ges 2003;1:120-5.
15. Mitchell MS, Kan-Mitchell J, Morrow PR, Darrah D, Jones VE,Mescher MF. Phase I trial of large multivalent immunogen derived frommelanoma lysates in patients with disseminated melanoma. Clin Can-cer Res 2004;10:76-83.
16. Oratz R, Dugan M, Roses DF, Harris MN, Speyer JL, Hochster H etal. Lack of effect of cyclophosphamide on the immunogenicity of amelanoma antigen vaccine. Cancer Res 1991;51:3643-7.
17. Hoon DS, Foshag LJ, Nizze AS, Bohman R, Morton DL. Suppressorcell activity in a randomized trial of patients receiving active specif-ic immunotherapy with melanoma cell vaccine and low dosages ofcyclophosphamide. Cancer Res 1990;50:5358-64.
18. Mordoh J, Kairiyama C, Bover L, Solarolo E. Allogeneic cells vaccineincreases disease-free survival in stage III melanoma patients. A nonrandomized phase II study. Medicina 1997;57:421-7.
19. Morton DL, Hsueh EC, Essner R, Foshag LJ, O’Day SJ, Bilchik A etal. Prolonged survival of patients receiving active immunotherapywith Canvaxin therapeutic polyvalent vaccine after complete resectionof melanoma metastatic to regional lymph nodes. Ann Surg2002;236:438-48; discussion 48-9.
20. Hsueh EC, Essner R, Foshag LJ, Ollila DW, Gammon G, O’Day SJet al. Prolonged survival after complete resection of disseminatedmelanoma and active immunotherapy with a therapeutic cancer vac-cine. J Clin Oncol 2002;20:4549-54.
21. Barrio MM, de Motta PT, Kaplan J, von Euw EM, Bravo AI, ChaconRD et al. A phase I study of an allogeneic cell vaccine (VACCIMEL)with GM-CSF in melanoma patients. J Immunother 2006;29:444-54.
22. Arienti F, Belli F, Napolitano F, Sule-Suso J, Mazzocchi A, Gallino GFet al. Vaccination of melanoma patients with interleukin 4 gene-trans-duced allogeneic melanoma cells. Hum Gene Ther 1999;10:2907-16.
23. Osanto S, Schiphorst PP, Weijl NI, Dijkstra N, Van Wees A, Brouwen-

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 157
THE CLINICAL EXPERIENCE OF CANCER VACCINES FOR MALIGNANT MELANOMA BLOCK
stein N et al. Vaccination of melanoma patients with an allogeneic,genetically modified interleukin 2-producing melanoma cell line.Hum Gene Ther 2000;11:739-50.
24. Maio M, Fonsatti E, Lamaj E, Altomonte M, Cattarossi I, Santanto-nio C et al. Vaccination of stage IV patients with allogeneic IL-4- orIL-2-gene-transduced melanoma cells generates functional antibod-ies against vaccinating and autologous melanoma cells. CancerImmunol Immunother 2002;51:9-14.
25. Berd D, Maguire HC Jr, Schuchter LM, Hamilton R, Hauck WW,Sato T et al. Autologous hapten-modified melanoma vaccine as post-surgical adjuvant treatment after resection of nodal metastases. J ClinOncol 1997;15:2359-70.
26. Lotem M, Peretz T, Drize O, Gimmon Z, Ad El D, Weitzen R et al.Autologous cell vaccine as a post operative adjuvant treatment forhigh-risk melanoma patients (AJCC stages III and IV). The newAmerican Joint Committee on Cancer. Br J Cancer 2002;86:1534-9.
27. Lotem M, Shiloni E, Pappo I, Drize O, Hamburger T, Weitzen R et al.Interleukin-2 improves tumour response to DNP-modified autolo-gous vaccine for the treatment of metastatic malignant melanoma.Br J Cancer 2004;90:773-80.
28. Eton O, Kharkevitch DD, Gianan MA, Ross MI, Itoh K, Pride MW etal. Active immunotherapy with ultraviolet B-irradiated autologouswhole melanoma cells plus DETOX in patients with metastaticmelanoma. Clin Cancer Res 1998;4:619-27.
29. Dillman RO, Nayak SK, Barth NM, DeLeon C, Schwartzberg LS,Spitler LE et al. Clinical experience with autologous tumor cell linesfor patient-specific vaccine therapy in metastatic melanoma. CancerBiother Radiopharm 1998;13:165-76.
30. Dillman RO, Wiemann M, Nayak SK, DeLeon C, Hood K, DePriestC. Interferon-gamma or granulocyte-macrophage colony-stimulat-ing factor administered as adjuvants with a vaccine of irradiated autol-ogous tumor cells from short-term cell line cultures: a randomizedphase 2 trial of the cancer biotherapy research group. J Immunother2003;26:367-73.
31. Leong SP, Enders-Zohr P, Zhou YM, Stuntebeck S, Habib FA, AllenRE Jr et al. Recombinant human granulocyte macrophage-colonystimulating factor (rhGM-CSF) and autologous melanoma vaccinemediate tumor regression in patients with metastatic melanoma. JImmunother 1999;22:166-74.
32. Mackensen A, Veelken H, Lahn M, Wittnebel S, Becker D, Kohler Get al. Induction of tumor-specific cytotoxic T lymphocytes by immu-nization with autologous tumor cells and interleukin-2 gene trans-fected fibroblasts. J Mol Med 1997;75:290-6.
33. Schreiber S, Kampgen E, Wagner E, Pirkhammer D, Trcka J, KorschanH et al. Immunotherapy of metastatic malignant melanoma by a vac-cine consisting of autologous interleukin 2-transfected cancer cells:outcome of a phase I study. Hum Gene Ther 1999;10:983-93.
34. Palmer K, Moore J, Everard M, Harris JD, Rodgers S, Rees RC et al.Gene therapy with autologous, interleukin 2-secreting tumor cells inpatients with malignant melanoma. Hum Gene Ther 1999;10:1261-8.
35. Abdel-Wahab Z, Weltz C, Hester D, Pickett N, Vervaert C, BarberJR et al.A Phase I clinical trial of immunotherapy with interferon-gam-ma gene-modified autologous melanoma cells: monitoring the humoralimmune response. Cancer 1997;80:401-12.
36. Sun Y, Jurgovsky K, Moller P, Alijagic S, Dorbic T, Georgieva J et al.Vaccination with IL-12 gene-modified autologous melanoma cells: pre-clinical results and a first clinical phase I study. Gene Ther 1998;5:481-90.
37. Moller P, Sun Y, Dorbic T, Alijagic S, Makki A, Jurgovsky K et al. Vac-cination with IL-7 gene-modified autologous melanoma cells canenhance the anti-melanoma lytic activity in peripheral blood of patientswith a good clinical performance status: a clinical phase I study. Br JCancer 1998;77:1907-16.
38. Wittig B, Marten A, Dorbic T, Weineck S, Min H, Niemitz S et al. Ther-apeutic vaccination against metastatic carcinoma by expression-mod-
ulated and immunomodified autologous tumor cells: a first clinicalphase I/II trial. Hum Gene Ther 2001;12:267-78.
39. Soiffer R, Lynch T, Mihm M, Jung K, Rhuda C, Schmollinger JC etal. Vaccination with irradiated autologous melanoma cells engineeredto secrete human granulocyte-macrophage colony-stimulating fac-tor generates potent antitumor immunity in patients with metastaticmelanoma. Proc Natl Acad Sci U S A 1998;95:13141-6.
40. Chang AE, Li Q, Bishop DK, Normolle DP, Redman BD, NickoloffBJ. Immunogenetic therapy of human melanoma utilizing autolo-gous tumor cells transduced to secrete granulocyte-macrophagecolony-stimulating factor. Hum Gene Ther 2000;11:839-50.
41. Kusumoto M, Umeda S, Ikubo A, Aoki Y, Tawfik O, Oben R et al.Phase 1 clinical trial of irradiated autologous melanoma cells aden-ovirally transduced with human GM-CSF gene. Cancer ImmunolImmunother 2001;50:373-81.
42. Mahvi DM, Shi FS, Yang NS, Weber S, Hank J, Albertini M et al.Immunization by particle-mediated transfer of the granulocyte-macrophage colony-stimulating factor gene into autologous tumorcells in melanoma or sarcoma patients: report of a phase I/IB study.Hum Gene Ther 2002;13:1711-21.
43. Soiffer R, Hodi FS, Haluska F, Jung K, Gillessen S, Singer S et al. Vac-cination with irradiated, autologous melanoma cells engineered tosecrete granulocyte-macrophage colony-stimulating factor by aden-oviral-mediated gene transfer augments antitumor immunity in patientswith metastatic melanoma. J Clin Oncol 2003;21:3343-50.
44. Luiten RM, Kueter EW, Mooi W, Gallee MP, Rankin EM, GerritsenWR et al. Immunogenicity, including vitiligo, and feasibility of vac-cination with autologous GM-CSF-transduced tumor cells in metasta-tic melanoma patients. J Clin Oncol 2005;23:8978-91.
45. Parney IF, Chang LJ, Farr-Jones MA, Hao C, Smylie M, Petruk KC.Technical hurdles in a pilot clinical trial of combined B7-2 and GM-CSF immunogene therapy for glioblastomas and melanomas. J Neu-rooncol 2006;78:71-80.
46. Moiseyenko VM, Danilov AO, Baldueva IA, Danilova AB, Tyukav-ina NV, Larin SS et al. Phase I/II trial of gene therapy with autologoustumor cells modified with tag7/PGRP-S gene in patients with dis-seminated solid tumors: miscellaneous tumors. Ann Oncol2005;16:162-8.
47. Rosenberg SA, Yang JC, Schwartzentruber DJ, Hwu P, MarincolaFM, Topalian SL et al. Immunologic and therapeutic evaluation of asynthetic peptide vaccine for the treatment of patients with metasta-tic melanoma. Nat Med 1998;4:321-7.
48. Slingluff CL Jr, Yamshchikov G, Neese P, Galavotti H, Eastham S,Engelhard VH et al. Phase I trial of a melanoma vaccine withgp100(280-288) peptide and tetanus helper peptide in adjuvant:immunologic and clinical outcomes. Clin Cancer Res 2001;7:3012-24.
49. Smith JW, 2nd, Walker EB, Fox BA, Haley D, Wisner KP, Doran T etal. Adjuvant immunization of HLA-A2-positive melanoma patientswith a modified gp100 peptide induces peptide-specific CD8+ T-cellresponses. J Clin Oncol 2003;21:1562-73.
50. Chianese-Bullock KA, Woodson EM, Tao H, Boerner SA, Smolkin M,Grosh WW et al. Autoimmune toxicities associated with the admin-istration of antitumor vaccines and low-dose interleukin-2. JImmunother 2005;28:412-9.
51. Wang F, Bade E, Kuniyoshi C, Spears L, Jeffery G, Marty V et al. PhaseI trial of a MART-1 peptide vaccine with incomplete Freund’s adju-vant for resected high-risk melanoma. Clin Cancer Res 1999;5:2756-65.
52. Cebon J, Jager E, Shackleton MJ, Gibbs P, Davis ID, Hopkins W etal. Two phase I studies of low dose recombinant human IL-12 withMelan-A and influenza peptides in subjects with advanced malig-nant melanoma. Cancer Immun 2003;3:7.
53. Speiser DE, Lienard D, Rufer N, Rubio-Godoy V, Rimoldi D, Leje-une F et al. Rapid and strong human CD8+ T cell responses to vac-cination with peptide, IFA, and CpG oligodeoxynucleotide 7909. J ClinInvest 2005;115:739-46.

BLOCK THE CLINICAL EXPERIENCE OF CANCER VACCINES FOR MALIGNANT MELANOMA
158 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
54. Scheibenbogen C, Schmittel A, Keilholz U, Allgauer T, Hofmann U,Max R et al. Phase 2 trial of vaccination with tyrosinase peptides andgranulocyte-macrophage colony-stimulating factor in patients withmetastatic melanoma. J Immunother 2000;23:275-81.
55. Scheibenbogen C, Schadendorf D, Bechrakis NE, Nagorsen D, Hof-mann U, Servetopoulou F et al. Effects of granulocyte-macrophagecolony-stimulating factor and foreign helper protein as immunolog-ic adjuvants on the T-cell response to vaccination with tyrosinasepeptides. Int J Cancer 2003;104:188-94.
56. Lee P, Wang F, Kuniyoshi J, Rubio V, Stuges T, Groshen S et al.Effects of interleukin-12 on the immune response to a multipeptide vac-cine for resected metastatic melanoma. J Clin Oncol 2001;19:3836-47.
57. Pullarkat V, Lee PP, Scotland R, Rubio V, Groshen S, Gee C et al. Aphase I trial of SD-9427 (progenipoietin) with a multipeptide vac-cine for resected metastatic melanoma. Clin Cancer Res 2003;9:1301-12.
58. Di Pucchio T, Pilla L, Capone I, Ferrantini M, Montefiore E, UrbaniF et al. Immunization of stage IV melanoma patients with Melan-A/MART-1 and gp100 peptides plus IFN-alpha results in the activa-tion of specific CD8(+) T cells and monocyte/dendritic cell precursors.Cancer Res 2006;66:4943-51.
59. Markovic SN, Suman VJ, Ingle JN, Kaur JS, Pitot HC, Loprinzi CLet al. Peptide vaccination of patients with metastatic melanoma:improved clinical outcome in patients demonstrating effective immu-nization. Am J Clin Oncol 2006;29:352-60.
60. Khong HT, Yang JC, Topalian SL, Sherry RM, Mavroukakis SA,White DE et al. Immunization of HLA-A*0201 and/or HLA-DPbe-ta1*04 patients with metastatic melanoma using epitopes from theNY-ESO-1 antigen. J Immunother 2004;27:472-7.
61. Bettinotti MP, Panelli MC, Ruppe E, Mocellin S, Phan GQ, White DEet al. Clinical and immunological evaluation of patients with metasta-tic melanoma undergoing immunization with the HLA-Cw*0702-associated epitope MAGE-A12:170-178. Int J Cancer 2003;105:210-6.
62. Pilla L, Patuzzo R, Rivoltini L, Maio M, Pennacchioli E, Lamaj E etal. A phase II trial of vaccination with autologous, tumor-derivedheat-shock protein peptide complexes Gp96, in combination withGM-CSF and interferon-alpha in metastatic melanoma patients. Can-cer Immunol Immunother 2006;55:958-68.
63. Belli F, Testori A, Rivoltini L, Maio M, Andreola G, Sertoli MR et al.Vaccination of metastatic melanoma patients with autologous tumor-derived heat shock protein gp96-peptide complexes: clinical andimmunologic findings. J Clin Oncol 2002;20:4169-80.
64. Phan GQ, Touloukian CE, Yang JC, Restifo NP, Sherry RM, Hwu Pet al. Immunization of patients with metastatic melanoma using bothclass I- and class II-restricted peptides from melanoma-associatedantigens. J Immunother 2003;26:349-56.
65. Wong R, Lau R, Chang J, Kuus-Reichel T, Brichard V, Bruck C etal. Immune responses to a class II helper peptide epitope in patientswith stage III/IV resected melanoma. Clin Cancer Res 2004;10:5004-13.
66. Woodson EM, Chianese-Bullock KA, Wiernasz CJ, Bissonette EA,Grosh WW, Neese PY et al. Assessment of the toxicities of systemiclow-dose interleukin-2 administered in conjunction with a melanomapeptide vaccine. J Immunother 2004;27:380-8.
67. Rosenberg SA, Yang JC, Schwartzentruber DJ, Hwu P, MarincolaFM, Topalian SL et al. Impact of cytokine administration on the gen-eration of antitumor reactivity in patients with metastatic melanomareceiving a peptide vaccine. J Immunol 1999;163:1690-5.
68. Slingluff CL Jr, Petroni GR, Yamshchikov GV, Hibbitts S, GroshWW, Chianese-Bullock KA et al. Immunologic and clinical outcomesof vaccination with a multiepitope melanoma peptide vaccine pluslow-dose interleukin-2 administered either concurrently or on a delayedschedule. J Clin Oncol 2004;22:4474-85.
69. Shackleton M, Davis ID, Hopkins W, Jackson H, Dimopoulos N, TaiT et al. The impact of imiquimod, a Toll-like receptor-7 ligand
(TLR7L), on the immunogenicity of melanoma peptide vaccinationwith adjuvant Flt3 ligand. Cancer Immun 2004;4:9.
70. Attia P, Phan GQ, Maker AV, Robinson MR, Quezado MM,Yang JCet al. Autoimmunity correlates with tumor regression in patients withmetastatic melanoma treated with anti-cytotoxic T-lymphocyte anti-gen-4. J Clin Oncol 2005;23:6043-53.
71. Marchand M, Punt CJ, Aamdal S, Escudier B, Kruit WH, Keilholz Uet al. Immunisation of metastatic cancer patients with MAGE-3 pro-tein combined with adjuvant SBAS-2: a clinical report. Eur J Cancer2003;39:70-7.
72. Davis ID, Chen W, Jackson H, Parente P, Shackleton M, Hopkins Wet al. Recombinant NY-ESO-1 protein with ISCOMATRIX adjuvantinduces broad integrated antibody and CD4(+) and CD8(+) T cellresponses in humans. Proc Natl Acad Sci U S A 2004;101:10697-702.
73. Fredman P, Hedberg K, Brezicka T. Gangliosides as therapeutic tar-gets for cancer. Biodrugs 2003;17:155-67.
74. Livingston PO, Wong GY, Adluri S, Tao Y, Padavan M, Parente R etal. Improved survival in stage III melanoma patients with GM2 anti-bodies: a randomized trial of adjuvant vaccination with GM2 gan-glioside. J Clin Oncol 1994;12:1036-44.
75. Helling F, Zhang S, Shang A, Adluri S, Calves M, Koganty R et al.GM2-KLH conjugate vaccine: increased immunogenicity in melanomapatients after administration with immunological adjuvant QS-21.Cancer Res 1995;55:2783-8.
76. Kirkwood JM, Ibrahim JG, Sosman JA, Sondak VK, Agarwala SS,Ernstoff MS et al. High-dose interferon alfa-2b significantly prolongsrelapse-free and overall survival compared with the GM2-KLH/QS-21vaccine in patients with resected stage IIB-III melanoma: results of inter-group trial E1694/S9512/C509801. J Clin Oncol 2001;19:2370-80.
77. Ragupathi G, Meyers M, Adluri S, Howard L, Musselli C, LivingstonPO. Induction of antibodies against GD3 ganglioside in melanomapatients by vaccination with GD3-lactone-KLH conjugate plusimmunological adjuvant QS-21. Int J Cancer 2000;85:659-66.
78. Chapman PB, Wu D, Ragupathi G, Lu S, Williams L, Hwu WJ et al.Sequential immunization of melanoma patients with GD3 gangliosidevaccine and anti-idiotypic monoclonal antibody that mimics GD3ganglioside. Clin Cancer Res 2004;10:4717-23.
79. Chapman PB, Morrisey D, Panageas KS, Williams L, Lewis JJ, IsraelRJ et al. Vaccination with a bivalent G(M2) and G(D2) ganglioside con-jugate vaccine: a trial comparing doses of G(D2)-keyhole limpethemocyanin. Clin Cancer Res 2000;6:4658-62.
80. Guthmann MD, Bitton RJ, Carnero AJ, Gabri MR, Cinat G, KolirenL et al. Active specific immunotherapy of melanoma with a GM3ganglioside-based vaccine: a report on safety and immunogenicity. JImmunother 2004;27:442-51.
81. Bhattachary-Chatterjee M, Nath Baral R, Chatterjee SK, Das R, ZeytinH, Chakraborty M et al. Counterpoint. Cancer vaccines: single-epi-tope anti-idiotype vaccine versus multiple-epitope antigen vaccine.Cancer Immunol Immunother 2000;49:133-41.
82. Quan WD, Jr., Dean GE, Spears L, Spears CP, Groshen S, Merritt JAet al. Active specific immunotherapy of metastatic melanoma with anantiidiotype vaccine: a phase I/II trial of I-Mel-2 plus SAF-m. J ClinOncol 1997;15:2103-10.
83. Foon KA, Sen G, Hutchins L, Kashala OL, Baral R, Banerjee M et al.Antibody responses in melanoma patients immunized with an anti-idio-type antibody mimicking disialoganglioside GD2. Clin Cancer Res1998;4:1117-24.
84. Yao TJ, Meyers M, Livingston PO, Houghton AN, Chapman PB.Immunization of melanoma patients with BEC2-keyhole limpet hemo-cyanin plus BCG intradermally followed by intravenous boosterimmunizations with BEC2 to induce anti-GD3 ganglioside antibod-ies. Clin Cancer Res 1999;5:77-81.
85. Alfonso M, Diaz A, Hernandez AM, Perez A, Rodriguez E, Bitton Ret al. An anti-idiotype vaccine elicits a specific response to N-glycolylsialic acid residues of glycoconjugates in melanoma patients. JImmunol 2002;168:2523-9.

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 159
THE CLINICAL EXPERIENCE OF CANCER VACCINES FOR MALIGNANT MELANOMA BLOCK
86. Rosenberg SA, Zhai Y,Yang JC, Schwartzentruber DJ, Hwu P, Mar-incola FM et al. Immunizing patients with metastatic melanomausing recombinant adenoviruses encoding MART-1 or gp100melanoma antigens. J Natl Cancer Inst 1998;90:1894-900.
87. Rosenberg SA, Yang JC, Sherry RM, Hwu P, Topalian SL,Schwartzentruber DJ et al. Inability to immunize patients withmetastatic melanoma using plasmid DNA encoding the gp100melanoma-melanocyte antigen. Hum Gene Ther 2003;14:709-14.
88. Zajac P, Oertli D, Marti W, Adamina M, Bolli M, Guller U et al.Phase I/II clinical trial of a nonreplicative vaccinia virus express-ing multiple HLA-A0201-restricted tumor-associated epitopes andcostimulatory molecules in metastatic melanoma patients. HumGene Ther 2003;14:1497-510.
89. Meyer RG, Britten CM, Siepmann U, Petzold B, Sagban TA, LehrHA et al. A phase I vaccination study with tyrosinase in patientswith stage II melanoma using recombinant modified vaccinia virusAnkara (MVA-hTyr). Cancer Immunol Immunother 2005;54:453-67.
90. Lindsey KR, Gritz L, Sherry R, Abati A, Fetsch PA, Goldfeder LCet al. Evaluation of prime/boost regimens using recombinantpoxvirus/tyrosinase vaccines for the treatment of patients withmetastatic melanoma. Clin Cancer Res 2006;12:2526-37.
91. van Baren N, Bonnet MC, Dreno B, Khammari A, Dorval T, Piper-no-Neumann S et al. Tumoral and immunologic response after vac-cination of melanoma patients with an ALVAC virus encoding MAGEantigens recognized by T cells. J Clin Oncol 2005;23:9008-21.
92. Spaner DE, Astsaturov I, Vogel T, Petrella T, Elias I, Burdett-RadouxS et al. Enhanced viral and tumor immunity with intranodal injectionof canary pox viruses expressing the melanoma antigen, gp100.Cancer 2006;106:890-9.
93. Rosenberg SA,Yang JC, Schwartzentruber DJ, Hwu P, Topalian SL,Sherry RM et al. Recombinant fowlpox viruses encoding the anchor-modified gp100 melanoma antigen can generate antitumor immuneresponses in patients with metastatic melanoma. Clin Cancer Res2003;9:2973-80.
94. Nestle FO, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R et al.Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med 1998;4:328-32.
95. Mackensen A, Herbst B, Chen JL, Kohler G, Noppen C, Herr W etal. Phase I study in melanoma patients of a vaccine with peptide-pulsed dendritic cells generated in vitro from CD34(+) hematopoi-etic progenitor cells. Int J Cancer 2000;86:385-92.
96. Panelli MC, Wunderlich J, Jeffries J, Wang E, Mixon A, RosenbergSA et al. Phase 1 study in patients with metastatic melanoma ofimmunization with dendritic cells presenting epitopes derived fromthe melanoma-associated antigens MART-1 and gp100. J Immunother2000;23:487-98.
97. Hersey P, Menzies SW, Halliday GM, Nguyen T, Farrelly ML, DeSil-va C et al. Phase I/II study of treatment with dendritic cell vaccinesin patients with disseminated melanoma. Cancer ImmunolImmunother 2004;53:125-34.
98. Schadendorf D, Ugurel S, Schuler-Thurner B, Nestle FO, Enk A,Brocker EB et al. Dacarbazine (DTIC) versus vaccination withautologous peptide-pulsed dendritic cells (DC) in first-line treatmentof patients with metastatic melanoma: a randomized phase III tri-al of the DC study group of the DeCOG. Ann Oncol 2006;17:563-70.
99. Banchereau J, Ueno H, Dhodapkar M, Connolly J, Finholt JP,Klechevsky E et al. Immune and clinical outcomes in patients withstage IV melanoma vaccinated with peptide-pulsed dendritic cellsderived from CD34+ progenitors and activated with type I interfer-on. J Immunother 2005;28:505-16.
100. Trakatelli M, Toungouz M, Blocklet D, Dodoo Y, Gordower L,Laporte M et al. A new dendritic cell vaccine generated with inter-leukin-3 and interferon-beta induces CD8+ T cell responses againstNA17-A2 tumor peptide in melanoma patients. Cancer ImmunolImmunother 2006;55:469-74.
101. Fay JW, Palucka AK, Paczesny S, Dhodapkar M, Johnston DA,Burkeholder S et al. Long-term outcomes in patients with metasta-
tic melanoma vaccinated with melanoma peptide-pulsed CD34(+)progenitor-derived dendritic cells. Cancer Immunol Immunother2006;55:1209-18.
102. Davis ID, Chen Q, Morris L, Quirk J, Stanley M, Tavarnesi ML et al.Blood dendritic cells generated with Flt3 ligand and CD40 ligandprime CD8+ T cells efficiently in cancer patients. J Immunother2006;29:499-511.
103. Gajewski TF, Fallarino F, Ashikari A, Sherman M. Immunizationof HLA-A2+ melanoma patients with MAGE-3 or MelanA peptide-pulsed autologous peripheral blood mononuclear cells plus recom-binant human interleukin 12. Clin Cancer Res 2001;7:895s-901s.
104. Peterson AC, Harlin H, Gajewski TF. Immunization with Melan-Apeptide-pulsed peripheral blood mononuclear cells plus recombi-nant human interleukin-12 induces clinical activity and T-cell respons-es in advanced melanoma. J Clin Oncol 2003;21:2342-8.
105. Thurner B, Haendle I, Roder C, Dieckmann D, Keikavoussi P, JonuleitH et al. Vaccination with mage-3A1 peptide-pulsed mature, mono-cyte-derived dendritic cells expands specific cytotoxic T cells andinduces regression of some metastases in advanced stage IVmelanoma. J Exp Med 1999;190:1669-78.
106. Schuler-Thurner B, Dieckmann D, Keikavoussi P, Bender A, MaczekC, Jonuleit H et al. Mage-3 and influenza-matrix peptide-specificcytotoxic T cells are inducible in terminal stage HLA-A2.1+melanoma patients by mature monocyte-derived dendritic cells. JImmunol 2000;165:3492-6.
107. Butterfield LH, Ribas A, Dissette VB,Amarnani SN, Vu HT, OsegueraD et al. Determinant spreading associated with clinical response indendritic cell-based immunotherapy for malignant melanoma. ClinCancer Res 2003;9:998-1008.
108. Ribas A, Glaspy JA, Lee Y, Dissette VB, Seja E, Vu HT et al. Roleof dendritic cell phenotype, determinant spreading, and negativecostimulatory blockade in dendritic cell-based melanomaimmunotherapy. J Immunother 2004;27:354-67.
109. Tuettenberg A, Becker C, Huter E, Knop J, Enk AH, Jonuleit H.Induction of strong and persistent MelanA/MART-1-specific immuneresponses by adjuvant dendritic cell-based vaccination of stage IImelanoma patients. Int J Cancer 2006;118:2617-27.
110. Banchereau J, Palucka AK, Dhodapkar M, Burkeholder S, Taquet N,Rolland A et al. Immune and clinical responses in patients withmetastatic melanoma to CD34(+) progenitor-derived dendritic cellvaccine. Cancer Res 2001;61:6451-8.
111. Chakraborty NG, Sporn JR, Tortora AF, Kurtzman SH, Yamase H,Ergin MT et al. Immunization with a tumor-cell-lysate-loaded autol-ogous-antigen-presenting-cell-based vaccine in melanoma. CancerImmunol Immunother 1998;47:58-64.
112. Nagayama H, Sato K, Morishita M, Uchimaru K, Oyaizu N, Inaza-wa T et al. Results of a phase I clinical study using autologoustumour lysate-pulsed monocyte-derived mature dendritic cell vac-cinations for stage IV malignant melanoma patients combined withlow dose interleukin-2. Melanoma Res 2003;13:521-30.
113. Vilella R, Benitez D, Mila J, Lozano M, Vilana R, Pomes J et al. Pilotstudy of treatment of biochemotherapy-refractory stage IV melanomapatients with autologous dendritic cells pulsed with a heterologousmelanoma cell line lysate. Cancer Immunol Immunother 2004;53:651-8.
114. Escobar A, Lopez M, Serrano A, Ramirez M, Perez C, Aguirre A etal. Dendritic cell immunizations alone or combined with low dosesof interleukin-2 induce specific immune responses in melanomapatients. Clin Exp Immunol 2005;142:555-68.
115. Salcedo M, Bercovici N, Taylor R, Vereecken P, Massicard S, Duri-au D et al. Vaccination of melanoma patients using dendritic cellsloaded with an allogeneic tumor cell lysate. Cancer ImmunolImmunother 2006;55:819-29.
116. Krause SW, Neumann C, Soruri A, Mayer S, Peters JH, AndreesenR. The treatment of patients with disseminated malignant melanomaby vaccination with autologous cell hybrids of tumor cells and den-dritic cells. J Immunother 2002;25:421-8.

BLOCK THE CLINICAL EXPERIENCE OF CANCER VACCINES FOR MALIGNANT MELANOMA
160 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
117. O’Rourke MG, Johnson M, Lanagan C, See J,Yang J, Bell JR et al.Durable complete clinical responses in a phase I/II trial using anautologous melanoma cell/dendritic cell vaccine. Cancer ImmunolImmunother 2003;52:387-95.
118. Dillman R, Selvan S, Schiltz P, Peterson C, Allen K, Depriest C et al.Phase I/II trial of melanoma patient-specific vaccine of proliferatingautologous tumor cells, dendritic cells, and GM-CSF: planned inter-im analysis. Cancer Biother Radiopharm 2004;19:658-65.
119. Haenssle HA, Krause SW, Emmert S, Zutt M, Kretschmer L, Schmid-berger H et al. Hybrid cell vaccination in metastatic melanoma: clin-ical and immunologic results of a phase I/II study. J Immunother2004;27:147-55.
120. Trefzer U, Herberth G, Wohlan K, Milling A, Thiemann M, SherevT et al. Vaccination with hybrids of tumor and dendritic cells inducestumor-specific T-cell and clinical responses in melanoma stage III andIV patients. Int J Cancer 2004;110:730-40.
121. Trefzer U, Herberth G, Wohlan K, Milling A, Thiemann M, SharavT et al. Tumour-dendritic hybrid cell vaccination for the treatment ofpatients with malignant melanoma: immunological effects and clin-ical results. Vaccine 2005;23:2367-73.
122. Wei Y, Sticca RP, Holmes LM, Burgin KE, Li J, Williamson J et al.Dendritoma vaccination combined with low dose interleukin-2 inmetastatic melanoma patients induced immunological and clinicalresponses. Int J Oncol 2006;28:585-93.
123. Kyte JA, Mu L, Aamdal S, Kvalheim G, Dueland S, Hauser M et al.Phase I/II trial of melanoma therapy with dendritic cells transfectedwith autologous tumor-mRNA. Cancer Gene Ther 2006;13:905-18.
124. Markovic SN, Dietz AB, Greiner CW, Maas ML, Butler GW, PadleyDJ et al. Preparing clinical-grade myeloid dendritic cells by elec-troporation-mediated transfection of in vitro amplified tumor-derivedmRNA and safety testing in stage IV malignant melanoma. J TranslMed 2006;4:35.w

G ITAL DERMATOL VENEREOL 2007;142:161-70
Molecular targeted therapy of melanoma
The increasing understanding of the molecular pathology ofmalignancies will have impact on future therapeutic approaches.Effective, non-toxic intervention in a broad range of cancersincluding melanoma may potentially be provided by targetedtherapeutics which are tailored towards the molecular abnor-malities that cause tumour progression. a) Constitutive acti-vation of the Ras/MAPK- and PI3K/Akt-signal transductionpathways, b) epigenetic deregulation of gene expression dueto drastic changes in DNA methylation and histone modifica-tion and c) loss of stability of tumour suppressor proteins havebeen demonstrated to be involved in melanomagenesis. Smallmolecular weight inhibitors interfering with these deregula-ted events, as well as the results of first clinical trials applyingthese substances are covered in this review.
KEY WORDS: Melanoma, diagnosis - Melanoma, drug therapy -Skin.
The current standard therapy of inoperable metas-tasised melanoma is largely based on the use of
cytotoxic and cytostatic substances.1 These substances,which unselectively target all proliferating cells in thebody, have improved significantly cure and survivalrates for several malignancies. However, for the treat-ment of malignant melanoma, the outcome is poor.2 Incontrast to this cessation concerning therapies, tremen-dous progress has been made in understanding themechanisms of melanomagenesis by applying modernbiochemical and molecular techniques.3 Now, the
development of drugs targeting the molecular abnor-malities that cause melanoma has started, hopefullyleading to effective and specific therapeutics. Clini-cal research however, is still at the beginning and timewill tell whether targeted therapy can fulfil these hopes.
Activation of the MAPK pathway in melanoma
Raf kinases have been shown to be components ofan evolutionarily conserved signalling cascade thatlinks receptor activation at the cell membrane to themodification of cytoplasmic or nuclear targets whichare required for the execution of developmental pro-grams as well as for cell survival and proliferation.4 Animportant linker coupling receptor activation with theinduction of Raf kinase activity is the small G proteinRas that can switch between an inactive GDP boundand an active GTP bound form. GTP-Ras recruits Rafto the plasma membrane initiating a complex activa-tion process.5 Active Raf (MAP kinase kinase kinase)phosphorylates and activates MEK (MAP kinasekinase), and MEK phosphorylates and activates ERK1/2 (p44/p42 MAP kinases). While Raf and MEKappear largely restricted to only one class of substrates,ERK targets more than 70 substrates including mem-
Department of Dermatology, Venereology and AllergologyJulius-Maximilians University, Würzburg, Germany
Address reprint requests to: J. C. Becker, Department of Dermatology,Universitätsklinik Würzburg, Josef-Schneider-Str. 2, 97080 Würzburg,Germany. E-mail: [email protected]
R. HOUBEN, S. ORTMANN, D. SCHRAMA, E.-B. BRÖCKER, J. C. BECKER
Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 161

HOUBEN MOLECULAR TARGETED THERAPY OF MELANOMA
162 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
brane, cytoskeletal, cytoplasmic, nuclear and evenmitochondrial proteins.6
The Raf family of serin/threonine kinases consistsof three isoforms: A-, B- and C-Raf. In 2003, the can-cer genome project revealed B-Raf to be affected byactivating mutations in several cancers most frequentlyin melanoma.7 The incidence of B-Raf mutations inmelanoma varies between different subtypes but amean frequency of 46% in 2 338 melanoma samplesanalysed (Catalogue of somatic mutations in cancer,[COSMIC], http://www.sanger.ac.uk/genetics/CGP/cosmic/), demonstrates the exceptional importance ofthis alteration. Additional activators of the MAP kinasecascade which are found to be mutated in melanomaare N-Ras (mutated in 19% of melanoma according tothe COSMIC database) and the stem cell factor recep-tor Kit.8
For B-Raf the exchange of a single amino acid thevaline at position 600 to a charged residue (glutamicacid, aspartic acid, lysine or arginine) accounts formore than 90% of the B-Raf mutations found inmelanoma. These exchanges render B-Raf constitu-tively active 9 and it is believed that this is due to mim-icry of activating phosphorylations at nearby positionsin this part of the protein which is termed the kinaseactivation segment. In B-Raf, the threonine at posi-tion 599 and the serin at position 602 both requirephosphorylation to achieve maximal kinase activityand both are indeed phosphorylated upon recruitmentof B-Raf to the plasma membrane by activated Ras.10
For N-Ras the activating mutations are paradoxicallyinactivating the enzymatic activity of N-Ras as aGTPase. Thereby, N-Ras is constitutively in the GTPbound form ready to initiate the activation of Raf andfurther effectors among which the Phosphoinositide 3-kinase (PI3K) may be the most important. However, themutual exclusivity of B-Raf and N-Ras mutations inmelanoma 11 suggests that the major effector pathwayfor mutated N-Ras is the Raf-MAPK signal transduc-tion pathway. Activation of the MAP kinase pathwaycan also occur by upstream signalling proteins likereceptor tyrosine kinases and receptor associated tyro-sine kinases which are also frequently activated incancer by mutation or increased ligand availability.In melanoma activating mutations in the stem cell fac-tor receptor tyrosine kinase Kit have been described tobe present in certain subsets of melanoma which arecharacterised by a more rare appearance of B-Raf andN-Ras mutations.8
Inhibitors of the Ras-MAPK signal-transductionpathway
A relatively specific inhibition of the different intra-cellular signal transduction cascades was enabled bythe development of a set of low molecular weightkinase inhibitors. Among those are Raf inhibitors suchas sorafenib (Bay43-9006) and Raf 265 (CHIR-265),MEK inhibitors such as PD184352, CI-1040 andU0126 and tyrosine kinase inhibitors including Gefi-tinib (IressaTM), Erlotinib (TarcevaTM) and Imatinib(GlivecTM). The kinase inhibitor molecules work inmany cases through competitive binding to the ATPpocket displacing ATP which is necessary to providethe phosphate for the kinase reaction. This commonmechanism explains why the specificity of many kinaseinhibitors is limited. Sorafenib developed as C-Rafinhibitor acts on B-Raf isoforms as well but is alsoinhibitory to more distantly related kinases includingvascular-endothelial growth factor receptors (VEG-FR-2 and VEGFR-3), platelet-derived growth factorreceptor (PDGFR), Flt-3 and Kit.
Only one year following the publication in “Nature”reporting activating B-Raf mutations in melanoma,first results of a phase-I/II study demonstrating thera-peutic activity of sorafenib in combination with car-boplatin/placitaxel were presented by Flaherty et alat the VIII Perspectives in Melanoma Meeting in Berlinin September 2004. 28 of 35 patients showed eitherregression or stable disease.12, 13 Currently there are 8ongoing clinical trials evaluating sorafenib in combi-nation with different chemotherapeutics for the treat-ment of advanced melanoma (http://www.clinicaltri-als.gov) and we can expect a lot of information fromthese studies. However, the results reported so far ,are not confirming the initial impact on disease pro-gression (http://www.medicalnewstoday.com/med-icalnews.php?newsid=58112).
Sorafenib was also evaluated as mono-therapy foradvanced melanoma. However, since no benefit forthe treated patients could be demonstrated 13 sorafenibas a single agent is not further under investigation.Interestingly, sorafenib failed also as a therapeutic insome animal models while application of the MEKinhibitors U0126 or CI-1040 was successful. In a C-Raflung adenoma as well as in a B-RafV600E melanomametastasis mouse model inhibition of MEK led totumour reduction and inhibition of metastasis, respec-tively, while sorafenib did not show any effect.14, 15
Therefore, although there is still hope that next gen-

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 163
MOLECULAR TARGETED THERAPY OF MELANOMA HOUBEN
eration Raf inhibitors like Raf-265, which is in vitro 50-to 100-fold more potent than sorafenib,11 will improveRaf targeted therapy, it may arise that MEK is the bet-ter target for treating melanoma.
The MEK inhibitors UO126 and CI-1040, howev-er, due to bad pharmacokinetics and lack of anti-tumouractivity observed in patients with several advancedmalignancies are considered insufficient to warrantfurther development.16, 17
In contrast, PD0325901, a derivate of CI-1040 thatblocks MEK at much higher potency, still holdspromise for the use of the compound as a therapeuticagent. In a phase I/II study enrolling 41 patients (27melanoma) possible anticancer activity has been eval-uated. Two partial responses were observed inmelanoma patients, while 8 patients (5 melanoma, 2non small cell lung cancer [NSCLC] and 1 colon can-cer) achieved stable disease lasting 3-7 months.
ARRY-142886 (AZD6244), another novel and high-ly-selective oral MEK inhibitor, is also under studyin clinical trials currently. A study including melanomapatients as well as patients with other advanced can-cers has completed enrollment and first results of thisstudy were reported at the EORTC-NCI-AACR Sym-posium on “Molecular Targets and Cancer Therapeu-tics” in November 2006. Sixteen of the 20 patientswith melanoma completed at least one cycle of treat-ment. Twelve had stable disease after completion ofcycle two, with stable disease persisting for at leastfive months in six patients (range, 5-13 months; medi-an, 6.5 months) compared to 3 of 19 patients with oth-er malignancies.17
In both above mentioned studies the therapeuticimpact of the MEK inhibitors seems to be more pro-nounced for melanomas than for other cancers. B-Rafmutant melanoma cell lines have been demonstrated tobe particularly sensitive to MEK inhibition comparedto cells with wt B-Raf (including cells with N-Rasmutation).18 The genotype of the treated tumours wasnot reported yet. It will be interesting to see whetherin patients there is also a prevalence of MEK inhibitionbeing more effective in B-Raf mutant tumours.
The lack of specificity of protein kinase inhibitorsmay sometimes be of advantage. Sorafenib developedas a Raf inhibitor has now been approved by the USFood and Drug Administration for the treatment ofpatients with advanced renal cell carcinoma or kid-ney cancer. However, since in several models sorafenibinhibited tumour growth without affecting MAPK sig-nalling in the tumour cells, it is now believed that
sorafenib acts by exerting potent antivascular effectsvia inhibition of VEGF receptors.19
PIP3 signalling
A second downstream signalling pathway of Rasand many growth factor receptors which is thought tobe one of the major survival pathways, involves class1 phosphoinositide-3 kinases (PI3Ks) and the Aktkinase. PI3Ks constitute a large family of lipid kinas-es and class 1 PI3Ks phosphorylate phosphatidyli-nositol 4,5- bisphosphate (PIP2) to produce phos-phatidylinositol 3,4,5-trisphosphate (PIP3), a key lipidsecond messenger that controls a wide range of cellu-lar responses via downstream effectors such as adap-tor proteins, protein kinases (most important the phos-phoinositide-dependent kinase 1 (PDK-1) andAkt/PKB) and nucleotide-exchange factors or GTPase-activating proteins (GAPs) for GTPases of the Rho, Rasand Arf families.20 Growth factor receptors drive acti-vation of PI3Ks and thereby PIP3 formation eitherthrough direct binding and phosphorylation or viaassociated tyrosine kinases, heterotrimeric G proteinsor Ras.20 The antagonist of the PI3Ks down regulatingPIP3 levels is the phosphatase and tensin homologuedeleted on chromosome 10 (PTEN). PTEN is a phos-phatidylinositol phosphate phosphatase which is fre-quently inactivated in human cancers includingmelanoma.11
Akt contains an NH2-terminal pleckstrin homologydomain, which interacts with PIP3 synthesized at theplasma membrane. Akt recruitment to the plasma mem-
Figure 1.—Inhibitors of the MAP kinase pathway.

HOUBEN MOLECULAR TARGETED THERAPY OF MELANOMA
164 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
brane results in a conformational change enablingphosphorylation by PDK-1, which also requires PIP3for activation and by a second kinase whose identity isnot yet clear. Established antiapoptotic modes of actionof Akt are phosphorylation and inactivation of theproapoptotic protein Bad, phosphorylation of pro-cas-pase-9 rendering it resistant to processing and activa-tion, phosphorylation of proapoptotic Forkhead tran-scription factors and activation of the NF-κB pathwaypossibly through phosphorylation of I-κB.21 Akt hasalso been implicated to be involved in driving the cellcycle via a couple of direct and indirect targets includ-ing Myc, Mdm2/p53, the Retinoblastoma protein,cyclin D and cyclin dependent kinase inhibitors (seetumour suppressor section). Moreover, Akt plays arole in angiogenesis, cell migration and metastasisformation.21 This multitude of cancer related func-tions explains why inhibiting PI3K or Akt are regard-ed as some of the most promising molecular targets forfuture therapies.
Targeting PI3K dependent signalling
Unfortunately, most of the inhibitors developed sofar display only restricted specificity.22 Nevertheless,preclinical studies applying the PI3K inhibitorLy294002 in xenotransplantation or transgenic mousetumour models demonstrated high antitumoural activ-ity following topical application.23,24 The Akt inhibitortriciribine has been tested in phase I/II clinical trials for
the treatment of advanced breast cancer. The outcomeof this study was a lack of activity and toxicity at high-er doses.25 However, at that time triciribine was test-ed due to its ability to inhibit DNA and protein syn-thesis without the knowledge of triciribine being an Aktinhibitor. Our days patients with metastatic cancerwhose tumours must be shown to be phospho-Aktpositive are recruited for a triciribine study (http://www.clinicaltrials.gov).
Further downstream the PI3K/Akt pathway the pro-tein kinase mTOR (mammalian target of rapamycin)gets activated. mTOR is a member of the recently iden-tified family of PIKKs (PI3K and related kinases) whichare involved in cell cycle regulation.26 Activation ofmTOR can be induced by hypoxia, which results in twoindependent cellular responses. On the one hand pro-liferation is accelerated, on the other hand via the acti-vation of hypoxia inducible factor (HIF)-1· genes getactivated which adopt the tissue to oxygen deficiency.Both mTOR induced responses are essential for hypox-ia induced neoangiogenesis and enable thereby the socalled angiogenic switch of solid tumours.27 Interest-ingly, according to the data base of the American OrganProcurement and Transplant Networt/United Networkfor Organ Sharing (OPTN/ UNOS) the risk to developcancer post transplantation for patients treated by mTORinhibitors is markedly reduced compared to patientstreated with traditional calcineurin-inhibitors. Indeed,immunosuppression by e.g. cyclosporine or tacrolimuscan be associated with high cancer rates and can be thecause for the subsequent death of the transplant recip-ient.28 Analogs of rapamycin, like CCI-779 (tem-sirolimus),AP23573 and RAD001 (everolimus) are rel-atively specific inhibitors of mTOR which block G1/Stransition and activation of HIF-1·.26 In several phase IIclinical trials (4 melanoma) the effectivity of CCI-779as monotherapy and in combination with other agents(combination with sorafenib in 2 melanoma studies) iscurrently evaluated. In a study including 109 patientswith advanced breast cancer CCI-779 demonstrated amodest but clear cut activity with one third of the patientshaving partial remission or stable disease lasting formore than two months.29 It should be noted that thenumber of patients bearing tumours with PTEN-muta-tion or Her2/neu overexpression – which both shouldlead to activated PI3K/Akt signalling – was increasedamong the responders. The conclusion however, of astudy with 31 patients with advanced melanoma treat-ed with CCI-779 as a single agent was that CCI-779 is
Figure 2.—Inhibitors of the PI3K/Akt pathway.

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 165
MOLECULAR TARGETED THERAPY OF MELANOMA HOUBEN
not sufficiently active in melanoma to warrant furthertesting as a single agent.30 So far CCI-779 was tested inclinical trials for its direct effect on tumour cells. Asdescribed above inhibition of mTOR should also atten-uate neoangiogenesis and thereby indirectly tumourgrowth. Therefore, currently conducted studies com-bining CCI-779 with other potentially antiangiogenicsubstances may provide interesting results.
Tumour suppressor genes
Tumour suppressor genes code for proteins forwhich loss of function, frequently achieved byrepressed expression, contributes to carcinogenesis.The first tumour suppressor to be identified is the
product of the Retinoblastoma gene (Rb) which dis-covered in retinoblastoma is also inactivated in manyother cancers. Rb is a transcriptional repressor inhibit-ing entrance into the cell cycle. It can be inactivatedby phosphorylation through cyclin D/cyclin depen-dent kinase (cdk) complexes.31 A cellular defencemechanism against oncogene driven deregulated Rbphosphorylation is expression of the cdk inhibitorp16ink4a.32 In melanoma the Rb pathway is frequent-ly affected by loss of expression of p16ink4a and thisseems to be mediated mainly by so called epigenet-ic mechanism.33, 34
PTEN the above mentioned antagonist of PI3Ks isa further example of a tumour suppressor whoseexpression is frequently down regulated in melanoma.This is associated with PTEN promoter methylation,
Figure 3.—Inhibiting DNA methyl transferases and histone deacetylases.

HOUBEN MOLECULAR TARGETED THERAPY OF MELANOMA
166 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
and demethylation using the demethylation agent 5-aza-2’-deoxycytidine leads to PTEN reexpression.35
Epigenetic silencing
Cancer is viewed today as a genetic as well as an epi-genetic disease. DNA methylation and histone modifi-cations are two of the most important epigenetic mech-anism through which hereditary variants in gene expres-sion without alteration of the DNA sequence occur.36
Histones are proteins that, tightly associated with theDNA, build up the nucleosomes. Three dimensionalarrangements of the nucleosomes form the so calledchromatin and the structure of chromatin plays a crit-ical role in gene expression. Post translational modi-fications (acetylation, methylation and phosphoryla-tion) of histone tails lead to architectural changes ofnucleosomes and chromatin remodelling. Chromatinremodelling may finally enhance or repress gene tran-scription.37 Histone acetylation takes place at the N-ter-mini of the histones that have been highly conservedthroughout evolution and contain a number of lysineresidues which are the targets for acetylation. Twoopposite enzymatic activities, histone acetyl trans-ferase (HAT) and histone deacetylase (HDAC), regu-late the level of histone acetylation. Imbalancesbetween the activities of HAT and HDAC, leading tohistone hypo-acetylation and repression of gene expres-sion are regarded as a necessary step in human car-cinogenesis.38
DNA methylation occurs at cytosines within CpG-rich DNA regions and is catalysed by DNA methyl-transferases.39 Methylation of DNA in promoter regionscan prevent binding of the basal transcriptional machin-ery and ubiquitous transcription factors to their cognateDNA sequences.40 Therefore, DNA hypermethylationis a hallmark of gene silencing and was described inmelanoma and other cancers for the promoters of manyrelevant genes.41, 42
Derepression of silenced genes
Enzymes that regulate DNA methylation and histonemodifications have been successfully targeted by avariety of small molecule drugs including the DNAmethyltransferase inhibitor decitabine (5-aza-2’-deoxy-cytidine) and the HDAC inhibitors suberoylanilidehydroxamic acid (SAHA), valproic acid and the beza-mide-derivative MS275.43, 44 These drugs can poten-tially revert the epigenetic modifications and reactivatetumour suppressor genes or genes that increase thesensitivity of target tissues to anticancer drugs. Fol-lowing a hierarchical model of cancer development,cancer stem cells represent the main therapeutic target,as only they have the ability to initiate and maintaintumour growth.45 Current therapies, however, are oftenlargely aimed at reducing the tumour mass, primarilyaffecting cells undergoing cell division. Slowly-pro-liferating or dormant cells, which presumably includecancer stem cells, are spared. Modulation of epige-netic imprinting offers the possibility of also targetingthe cancer stem cell population.
The methyltransferase inhibitor decitabine has beenevaluated in a phase I clinical trial in combination withhigh doses of the immune stimulator IL-2 for the treat-ment of melanoma and renal cell carcinoma. The treat-ment was well tolerated and 5 of 16 melanoma patientsresponded with partial regression suggesting thatdecitabine may enhance the activity of IL-2 inmelanoma (response rate 15%).46 Two phase I studiesevaluating decitabine as mono therapy for melanomatreatment have not yet been reported. Most clinicaltrials on HDAC inhibitors in humans are also still inphase I/II. The HDAC inhibitor SAHA has shown sig-nificant anticancer activity against both hematologicand solid tumours at doses well tolerated by patients.47
A drug application was approved by the US Food andDrug Administration for SAHA for the treatment ofcutaneous T-cell lymphoma.48 For the treatment of
Figure 4.—Effectors of proteasome inhibitors.

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 167
MOLECULAR TARGETED THERAPY OF MELANOMA HOUBEN
melanoma evaluation of SAHA has just started; onestudy applying SAHA is recruiting patients. Valproicacid, which has been used for decades in the treat-ment of epilepsy, has also been recognized as HDACinhibitor.49 The discovery of its inhibitory effect cameafter thorough evaluation of its main side effect ofcausing birth defects such as defective closure of thevertebral canal and disproportionate facial bone for-mation. The embryotoxic effect was found to be causedby inhibition of HDAC. Other side effects, and unfor-tunately also antitumour effects of this substance class,are relatively moderate, and it can be presumed that suf-ficient therapeutic effectiveness can only be realized bysuitable combination therapies, e.g., with methyl-transferase inhibitors, cytostatics, differentiation induc-ers or proteasome inhibitors (see following section). Forexample a recently reported phase I/II clinical trialcombining the DNA methylation inhibitor decitabinewith valproic acid for the treatment of patients withadvanced leukaemia demonstrated that the combina-tion of epigenetic therapy in leukaemia was safe andactive (12 objective responders (10 complete remis-sions) within a group of 54 patients), and was associ-ated with transient reversal of aberrant epigeneticmarkers.50
Proteasome structure and function
A proteasome is a highly selective, multicatalyticproteinase complex that degrades and thereby inacti-vates intracellular proteins.51 Proteasomes consist oftwo complex components, a cylindrical 20S core com-plex (or 20S proteasome) formed by 28 subunits and
the 19S caps, docked on the ends of the core. Theentire complex, referred to as 39S proteasome, isviewed as the biologically active unit within the cell.For proteasomal degradation proteins are marked byconjugation with several ubiquitin molecules, which aresmall ubiquitinously expressed 76 amino acid pro-teins. Ubiquitin is among the evolutionary most high-ly conserved proteins – yeast and human ubiquitindiffer only by three amino acids. Ubiquitin is initial-ly activated in an ATP dependent manner, prior to itsbinding to one of hundreds of ubiquitin ligases, whichspecifically recognize the proteins to be degraded. Theprocess is repeated several times to form an ubiquitinchain of at least four residues. The longer the chain, themore rapid is the degradation of the ubiquitinated pro-tein. Marked proteins are bound by the 19S complex,unfolded energy dependently, and drawn into the lumenof the 20S proteasome cylinder where they are cutinto peptides of variable size (3–22 amino acids).52
The proteasome as a therapeutic target
Protein degradation by proteasomes is an impor-tant physiological process ensuring exact regulation ofthe breakdown of metabolic enzymes, transcriptionfactors, and proteins regulating cell cycle and celldeath, such as cyclins, CDK inhibitors or p53. Pro-teasomes are targeted in the treatment of malignanttumours, since the inhibition of their activity reducesthe degradation rate of proteins with anti-tumour activ-ity, leading to their accumulation. The main targetsfor proteasome inhibition are thought to be primarilythe NF-κB pathway and to a lesser extend p53 degra-
TABLE I.—Examples of the described therapeutic agents.
Active principle Agent Substance Class
MAPK pathway inhibitors Sorafenib (Bay 43-9006) Raf-, VEGF-InhibitorRAF-265 Raf-InhibitorCI-1040 MEK inhibitorU0126 MEK inhibitor
ARRY-142886 MEK inhibitorPI3K/Akt pathway inhibitors Ly294002 PI3K inhibitor
Triciribine Akt inhibitorCCI-779 (Temsirolimus) mTOR inhibitorRAD001 (Everolimus) mTOR inhibitor
Derepression of tumour SAHA HDAC inhibitorsuppressor genes Valproic acid HDAC inhibitor
FK228 HDAC inhibitorDecitabine Methyltransferase inhibitor
Protein stabilization Bortezomib Proteasome inihibitor

HOUBEN MOLECULAR TARGETED THERAPY OF MELANOMA
168 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
dation and some other proteins like heat shock pro-teins, Jnk and caspases.51, 53 The tumour suppressorp53 is responsible for the transcriptional activation ofa series of proteins involved in cell cycle control, apop-tosis and senescence. Activation of p53 occurs main-ly through protein stabilization, i.e. decreasedubitquitinylation by its specific ubiquitin ligase mdm2and decreased proteasomal degradation.54 NF-κB isa transcription factor that activates the expression of anumber of genes whose products mediate cell prolif-eration and the prevention of apoptosis. NF-κB is acti-vated by proteasomal degradation of its cytoplasmicbinding partner I-κB which prevents NF-κB nucleartranslocation. Increased levels of I-κB in the cyto-plasm as a result of proteasome inhibition reducesproliferation of cancer cells, as well as resistance to celldeath programs, and moreover increases the proapop-totic effect of conventional chemotherapy regimes andradiation therapy.31
Proteasome inhibitors
Bortezomib (VelcadeTM), a dipeptidyl boronic acid,is a synthetically manufactured highly specific pro-teasome inhibitor. It inhibits growth in vitro in a num-ber of cancer cell lines and has also demonstratedgrowth-inhibitory effects on xenotransplanted humantumours in mice; in some models it has been associatedwith a notably longer survival rate of laboratory ani-mals.55 In in vitro studies, hypoxia enhanced theproapoptotic effect of bortezomib. In addition, in vivoactivity was partly due to the pronounced anti-angio-genetic effect of the proteasome inhibitor. A numberof phase I clinical trials have shown activity of borte-zomib against advanced-stage melanoma, and sever-al studies are currently evaluating its efficacy as amonotherapy or in combination with cytotoxic sub-stances (http://www.clinicaltrials.gov). One borte-zomib/melanoma study however, was already closeddue to early evidence of insufficient clinical efficacyfollowing an interim analysis after treating 27patients.56 In contrast, for the treatment of multiplemyeloma bortezomib has entered clinical practice asthird- and second-line treatment and is currently underevaluation as first-line treatment in patients with new-ly diagnosed multiple myeloma in combination withmelphalan/prednisone in comparison to standard mel-phalan/prednisone therapy.53
Perspectives
Therapy of stage IV melanoma has not been fun-damentally improved over the last years. Therefore,there is a pressing need for new therapeutic approach-es. Although the first attempts of targeted therapy ofadvanced melanoma did not fulfil the great expecta-tions, the success of some targeted therapeutics in thetreatment of other malignancies maintains the hopethat a critical molecular target and the optimal agent tointerfere with will be discovered in the near future.Moreover, targeting multiple cancer pathways by thecombination of inhibitors or combination of targetedwith traditional therapeutics may provide the futurebreakthrough. However, since the enormous multi-tude of possible combinations of agents can not beevaluated in patients meaningful preclinical test sys-tem are warranted.
Riassunto
Terapia target molecolare per il melanomaLa crescente comprensione della patologia molecolare
dei tumori maligni avrà un impatto sui futuri approcci tera-peutici. L’intervento efficace, privo di tossicità, per un ampiagamma di neoplasie, compreso il melanoma, può poten-zialmente derivare dai farmaci mirati nei confronti delleanormalità molecolari che provocano la progressione deltumore: 1) L’attivazione costitutiva della cascata legata allatraduzione del segnale Ras/MAPK- e PI3K/Akt; 2) la dere-golazione epigenetica dell’espressione genica dovuta a muta-zioni radicali della mutilazione del DNA e a modificazionedell’istone; e 3) la perdita della stabilità delle proteine sop-primenti il tumore sono eventi per i quali è stato dimostratoil coinvolgimento nella melanomagenesi. In questa reviewvengono presi in considerazione gli inibitori a basso pesomolecolare che interferiscono con questi eventi deregolati,così come i risultati dei primi studi clinici relativi a questesostanze.PAROLE CHIAVE: Melanoma, diagnosi - Melanoma, terapiafarmacologica - Cute.
References
1. Eigentler T K, Caroli UM, Radny P, Garbe C. Palliative therapy of dis-seminated malignant melanoma: a systematic review of 41 randomisedclinical trials. Lancet Oncol 2003;4:748-59.
2. Ross J S, Schenkein DP, Pietrusko R, Rolfe M, Linette GP, Stec J,etal. Targeted therapies for cancer 2004. Am J Clin Pathol 2004;122:598-609.
3. Thompson JF, Scolyer RA, Kefford RF. Cutaneous melanoma. Lancet2005;365:687-701.

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 169
MOLECULAR TARGETED THERAPY OF MELANOMA HOUBEN
4. Schreck R, Rapp UR. Raf kinases: oncogenesis and drug discovery.Int J Cancer 2006;119:2261-71.
5. Mercer KE, Pritchard CA. Raf proteins and cancer: B-Raf is identi-fied as a mutational target. Biochim.Biophys Acta 2003;1653:25-40.
6. Chen Z, Gibson TB, Robinson F, Silvestro L, Pearson G, Xu B et al.MAP kinases. Chem Rev 2001;101:2449-76.
7. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S et al.Mutations of the B-RAF gene in human cancer. Nature 2002;417:949-54.
8. Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation ofKIT in distinct subtypes of melanoma. J Clin Oncol 2006;24:4340-6.
9. Houben R, Becker JC, Kappel A, Terheyden P, Brocker EB, Goetz Ret al. Constitutive activation of the Ras-Raf signaling pathway inmetastatic melanoma is associated with poor prognosis. J Carcinog2004;3:6.
10. Zhang BH, Guan KL. Activation of B-Raf kinase requires phospho-rylation of the conserved residues Thr598 and Ser601. EMBO J2000;19:5429-39.
11. Chin L, Garraway LA, Fisher DE. Malignant melanoma: geneticsand therapeutics in the genomic era. Genes Dev 2006;20:2149-82.
12. Strumberg D, Richly H, Hilger RA, Schleucher N, Korfee S, TewesM et al. Phase I clinical and pharmacokinetic study of the Novel Rafkinase and vascular endothelial growth factor receptor inhibitor BAY43-9006 in patients with advanced refractory solid tumours. J ClinOncol 2005;23:965-72.
13. Eisen T, Ahmad T, Flaherty KT, Gore M, Kaye S, Marais R et al.Sorafenib in advanced melanoma: a Phase II randomised discontin-uation trial analysis. Br J Cancer 2006;95:581-6.
14. Kramer BW, Gotz R, Rapp UR. Use of mitogenic cascade blockers fortreatment of C-Raf induced lung adenoma in vivo: CI-1040 stronglyreduces growth and improves lung structure. BMC Cancer 2004;4:24.
15. Sharma A, Tran MA, Liang S, Sharma AK, Amin S, Smith CD et al.Targeting mitogen-activated protein kinase/extracellular signal-reg-ulated kinase kinase in the mutant (V600E) B-Raf signaling cascadeeffectively inhibits melanoma lung metastases. Cancer Res2006;66:8200-9.
16. Rinehart J, Adjei AA, Lorusso PM, Waterhouse D, HechtJR, NataleRB et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040,in patients with advanced non-small-cell lung, breast, colon, and pan-creatic cancer. J Clin Oncol 2004;22:4456-62.
17. Wang D, Boerner SA, Winkler JD, Lorusso PM. Clinical experienceof MEK inhibitors in cancer therapy. Biochim BiophysActa
18. Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A et al.B-RAF mutation predicts sensitivity to MEK inhibition. Nature2006;439:358-62.
19. Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA etal. Discovery and development of sorafenib: a multikinase inhibitorfor treating cancer. Nat Rev Drug Discov 2006;5:835-44.
20. Vanhaesebroeck B, Ali K, Bilancio A, Geering B, Foukas LC. Sig-nalling by PI3K isoforms: insights from gene-targeted mice. TrendsBiochem Sci 2005;30:194-204.
21. Kim D, Chung J. Akt: versatile mediator of cell survival and beyond.J Biochem Mol Biol 2002;35:106-15.22. Barnett SF, Bilodeau MT, Lindsley CW. The Akt/PKB family ofprotein kinases: a review of small molecule inhibitors and progresstowards target validation. Curr Top Med Chem 2005;5:109-25.
23. Bedogni B, O’Neill MS, Welford SM, Bouley DM, Giaccia AJ, DenkoNC et al. Topical treatment with inhibitors of the phosphatidylinosi-tol 3’-kinase/Akt and Raf/mitogen-activated protein kinasekinase/extracellular signal-regulated kinase pathways reducesmelanoma development in severe combined immunodeficient mice.Cancer Res 2004;64:2552-60.
24. Bedogni B, Welford SM, Kwan AC, Ranger-Moore J, Saboda K,Powell MB. Inhibition of phosphatidylinositol-3-kinase and mito-gen-activated protein kinase kinase 1/2 prevents melanoma develop-ment and promotes melanoma regression in the transgenic TPRasmouse model. Mol Cancer Ther 2006;5:3071-7.
25. Hoffman K, Holmes FA, Fraschini G, Esparza L, Frye D, Raber MNet al. Phase I-II study: triciribine (tricyclic nucleoside phosphate) formetastatic breast cancer. Cancer Chemother Pharmacol 1996;37:254-8.
26. Mita MM, Mita M, Rowinsky EK. The molecular target of rapamycin(mTOR) as a therapeutic target against cancer. Cancer Biol Ther2003;2:S169-S177.
27. Kerbel R, Folkman J. Clinical translation of angiogenesis inhibitors.Nat Rev Cancer 2002;2:727-39.
28. Rowinsky EK. Targeting the molecular target of rapamycin (mTOR).Curr Opin Oncol 2004;16:564-75.
29. Chan S. Targeting the mammalian target of rapamycin (mTOR): anew approach to treating cancer. Br J Cancer 2004;91:1420-4.
30. Margolin K, Longmate J, Baratta T, Synold T, Christensen S, WeberJ et al. CCI-779 in metastatic melanoma: a phase II trial of the Cali-fornia Cancer Consortium. Cancer 2005;104:1045-8.
31. Vogelstein B, Kinzler KW. Cancer genes and the pathways they con-trol. Nat Med 2004;10:789-99.
32. Ohtani N, Yamakoshi K, Takahashi A, Hara E. The p16INK4a-RBpathway: molecular link between cellular senescence and tumoursuppression. J Med Invest 2004;51:146-53.
33. Marini A, Mirmohammadsadegh A, Nambiar S, Gustrau A, RuzickaT, Hengge UR. Epigenetic inactivation of tumour suppressor genes inserum of patients with cutaneous melanoma. J Invest Dermatol2006;126:422-31.
34. van der Velden PA, Metzelaar-Blok JA, Bergman W, Monique H,Hurks H, Frants RR et al. Promoter hypermethylation: a commoncause of reduced p16(INK4a) expression in uveal melanoma. CancerRes 2001;61:5303-6.
35. Mirmohammadsadegh A, Marini A, Nambiar S, Hassan M, TannapfelA, Ruzicka T et al. Epigenetic silencing of the PTEN gene inmelanoma. Cancer Res 2006;66:6546-52.
36. Lu Q, Qiu X, Hu N, Wen H, Su Y, Richardson BC. Epigenetics, dis-ease, and therapeutic interventions. Ageing Res Rev 2006;5:449-67.
37. Moggs JG, Goodman JI, Trosko JE, Roberts RA. Epigenetics andcancer: implications for drug discovery and safety assessment. Toxi-col Appl Pharmacol 2004;196:422-30.
38. Liu T, Kuljaca S, Tee A, Marshall GM. Histone deacetylase inhibitors:multifunctional anticancer agents. Cancer Treat Rev 2006;32:157-65.
39. Chen T, Li E. Establishment and maintenance of DNA methylation pat-terns in mammals. Curr Top Microbiol Immunol 2006;301:179-201.
40. Wade PA. Methyl CpG binding proteins: coupling chromatin archi-tecture to gene regulation. Oncogene 2001;20:3166-73.
41. Ducasse M, Brown MAS. Epigenetic aberrations and cancer. MolCancer 2006;5:60.
42. Muthusamy V, Duraisamy S, Bradbury CM, Hobbs C, Curley DP,Nelson B et al. Epigenetic silencing of novel tumour suppressors inmalignant melanoma. Cancer Res 2006;66:11187-93.
43. Brown R, Strathdee G. Epigenomics and epigenetic therapy of can-cer. Trends Mol Med 2002;8:S43-S48.
44. Piekarz R, Bates S.A review of depsipeptide and other histone dea-cetylase inhibitors in clinical trials. Curr Pharm Des 2004;10:2289-98.
45. Al Hajj M, Becker MW, Wicha M, Weissman I, Clarke MF. Therapeuticimplications of cancer stem cells. Curr Opin Genet Dev 2004;14:43-7.
46. Gollob JA, Sciambi CJ, Peterson BL, Richmond T, Thoreson M,Moran K et al. Phase I trial of sequential low-dose 5-Aza-2’-deoxy-cytidine plus high-dose intravenous bolus interleukin-2 in patientswith melanoma or renal cell carcinoma. Clin Cancer Res2006;12:4619-27.
47. Duvic M, Zhang C. Clinical and laboratory experience of vorinostat(suberoylanilide hydroxamic acid) in the treatment of cutaneous T-celllymphoma. Br J Cancer 2006;95 (Suppl 1):S13-S19.
48. Richon VM. Cancer biology: mechanism of antitumour action ofvorinostat (suberoylanilide hydroxamic acid), a novel histone deacety-lase inhibitor. Br J Cancer 2006;95(Suppl 1):S2-S6.

HOUBEN MOLECULAR TARGETED THERAPY OF MELANOMA
170 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
49. Gottlicher M. Valproic acid: an old drug newly discovered as inhibitorof histone deacetylases. Ann Hematol 2004;83 (Suppl 1):S91-S92.
50. Garcia-Manero G, Kantarjian HM, Sanchez-Gonzalez B, Yang H,Rosner G, Verstovsek S et al. Phase 1/2 study of the combination of5-aza-2’-deoxycytidine with valproic acid in patients with leukemia.Blood 2006;108:3271-9.
51. Adams J. The proteasome: a suitable antineoplastic target. Nat RevCancer 2004;4:349-60.
52. Smith DM, Benaroudj N, Goldberg A. Proteasomes and their associat-ed ATPases: a destructive combination. J Struct Biol 2006;156:72-83.
53. Kropff M, Bisping G, Wenning D, Berdel WE, Kienast J. Proteasomeinhibition in multiple myeloma. Eur J Cancer 2006;42:1623-39.
54. Lavin MF, Gueven N.The complexity of p53 stabilization and acti-vation. Cell Death Differ 2006;13:941-50.
55. Roccaro AM, Hideshima T, Richardson PG, Russo D, Ribatti D, Vac-ca D et al. Bortezomib as an antitumour agent. Curr Pharm Biotech-nol 2006;7:441-8.
56. Markovic SN, Geyer SM, Dawkins F, Sharfman W, Albertini M,Maples W et al. A phase II study of bortezomib in the treatment ofmetastatic malignant melanoma. Cancer 2005;103:2584-9.

G ITAL DERMATOL VENEREOL 2007;142:171-95
The surgical management of cutaneous melanoma
Melanoma is an enigmatic cancer that can be deadly in anyand all of its forms if left unchecked or undiscovered. The inci-dence of melanoma is rapidly increasing over the last decade,the reasoning being multi-factorial combined with severalknown risk factors within the environment as well as per-sonal behaviors. Although often overlooked in terms of impor-tance, it cannot be stressed enough that we will have the mostimpact upon overall survival if we re-focus our efforts onthe early detection and prevention of melanoma. Oncemelanoma has spread to the lymphatic system or hematoge-nously, the overall survival of all patients dramaticallydecreases in comparison to the outstanding survival of thosepatients who undergo the appropriate margins of excisionfor melanoma in situ. Indeed, the surgical management ofmelanoma has changed dramatically within the last 20 years.We have been guided by the results of well-designed prospec-tive, randomized trials addressing the optimal surgical mar-gins of excision of the primary melanoma, able to modifyand refine the way that we surgically manage such patients.Further studies have definitively addressed the efficacy ofthe elective lymph node dissection, with recent studies show-ing the central role of selective lymphadenectomy in the man-agement of the draining lymph node basin(s). We will discussall of these issues and more, providing evidence-based datato support (or refute) current surgical thought and decisionmaking. Some issues still remain controversial due to a pauci-ty of evidence-based data to support their use. Other on-going studies may further guide us in the surgical manage-ment of melanoma. Lastly, we will revisit the past surgicaladvances for melanoma and how we can learn from them. In
turn, this will guide us as to how we will proceed and advancethis field in the future. KEY WORDS: Melanoma - Metastases - Sentinel lymph node biop-sy - Lymphoscintigraphy - Lymph node dissection - Multidisci-plinary cancer care - Surgery - Cytoreductive surgery.
The history of surgical treatment for melanoma
There are but a few diseases that are capable ofeliciting a true sense of fear in individuals, with
melanoma renowned for being such a deadly disease.Some of this fear is real, while other long-held beliefsare but the result of folklore and tales passed downfrom one generation to the next. The surgical man-agement of melanoma can be generally divided into 3eras: the 1st period defined as the John Hunter era,with the surgical philosophy of “small cancer, largeoperation”. The 2nd period, often described during thelate 1800’s, is defined by the work of Dr. H.E. Snow,who published his observations advocating extensivesurgery for melanoma, consisting of a large excisionof the primary melanoma with concomitant excisionof the draining lymph nodes.1 The 3rd period began in
1Mitchell Cancer InstituteCutaneous Oncology Program
University of South Alabama, USA 2Division of Dermatology, University of South Florida, USA
3Division of Plastic Surgery, USA
Address reprint requests to: A. I. Riker, MD, F.A.C.S., Chief, SurgicalOncology, Associate Professor of Surgery, University of South Alabama,Mitchell Cancer Institute, 307 North University Blvd., MSB 2015, Mobi-le, Alabama 36688.E-mail: [email protected]
A. I. RIKER 1, M. J. D’ALESSIO 1, R. M. HAGMAIER 1, L. F. GLASS 2, C. WAYNE CRUSE 3
Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 171

RIKER THE SURGICAL MANAGEMENT OF CUTANEOUS MELANOMA
172 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
the early 1960’s and continues through today, definedby the design and conduction of experimental clinicaltrials based upon hypothesis driven ideas as to theoptimal surgical management of melanoma.
Our history reveals that cutaneous melanoma hasafflicted humans as far back as paleopathologists areable to examine, first noting rounded, melanotic mass-es on the skin of pre-Columbian Incan mummies, nowpresent day Peru. These mummies were estimated to be2 400 years old by radiocarbon dating, with the calve-ria and long bones revealing metastatic tumor deposits.Melanoma was also described in the writings of Hip-pocrates in the 5th century B.C. and by the Greek physi-cian Rufus of Ephesus, circa 60-120 A.D.2 Over thecenturies, there have been only a few well-describedcases of pigmented malignant lesions, with the firstpublished account discussing the surgical treatment ofmelanoma by John Hunter in 1787.3 Although he nev-er described the excised tumor in any great detail, theactual specimen, the lower mandible of a male withmelanoma, remains well preserved as Hunter’s speci-men # 219 in the Hunterian Museum of the Royal Col-lege of Surgeons in England. Hunter described hisspecimen as “…a cancerous fungous excres-cence….part white and part spongy, soft and black”.3
In 1806, it was Renee Laennec, more famous forhis invention of the stethoscope, who first describedmelanoma as “la melanose” to the Faculté de Medicinein Paris, France.4 He later coined the term “melanosis”(derived from the Greek word meaning “black”) in1812.5 In 1820, William Norris described the first caseof melanoma in the English literature, later publishingthe first comprehensive study of melanoma entitled,“Eight cases of melanosis with pathological and ther-apeutic remarks”.6, 7 This landmark article was thefirst observational analysis of a group of patients withmelanoma, accurately describing many of the epi-demiological, clinical and pathological features ofpatients with melanoma, of which many of his obser-vations remain true today (Table I).
In 1834, David Williams may have been the first tocomment on the vertical and horizontal growth phas-es of a superficial spreading melanoma. He describesa spilus (tumor), that “increase(ed) in size and …con-tinued to spread gradually and after the spilus hadattained the circumference of a shilling, an excres-cence, similar in color to itself, began to rise in itscenter”.8 Oliver Pemberton published his observationsof 60 cases of cutaneous and ocular melanoma in 1858,
by far the largest series of patients with melanoma forthe time period, noting the post-mortem findings in33 cases.9 One of his reported cases is that of a 29year old male from Madagascar, the first describedcase of cutaneous melanoma in a black person. Hereported the site as along the side of the man’s foot, anuncommon site for caucasians. Pemberton was alsoone of the first surgeons to note the futility of manytreatments for advanced disease and was a strong advo-cate of surgical management of melanoma with wideexcision of the primary and extensive resection andremoval of the draining lymph node basins. However,such radical concepts of surgical management werenot uniformly accepted, shunned by many in favor oftraditional local therapies with salves and other med-icinal treatments passed down from previous genera-tions.
Over time, the excision of the primary lesion withwide margins was slowly gaining favor with a smallgroup of surgeons. In 1903, Frederick Eve describeda case series of 45 patients with melanoma treated atthe London Hospital, commenting that ~80% of themelanoma cases had originated from pigmented moleson the skin.10 He strongly expressed his views on thesurgical management of melanoma, stating in his lec-ture, “The treatment of melanoma can be given in a fewwords, free excision or amputation, in accordancewith the position and extent of disease…The removalof the nearest chain of lymphatic glands, whether pal-pably involved or not, should never be omitted; for itmay be taken as a matter of certainty that in the greatmajority of cases they are infected.”
TABLE I.—Observations on melanoma (Dr. William Norris, 1857).7
— There is a correlation between moles and melanoma— The majority of patients have light-colored hair and fair complexions— Family history of melanoma in some cases and probably a hereditary
predisposition to the disease/cancer may run in families— Although often black in color, the degree of pigmentation varied, with
some amelanotic— Tumor were often nodular and pedunculated/satellite tumors can deve-
lop around the primary growth/Subcutaneous deposits could developanywhere
— Widespread dissemination could involve the lungs, liver, bone, heart— They were more often men and in heavy smokers— They usually remained in good health until a very late stage of the
disease— Fever was not a feature, in contrast to tuberculosis— Local lesions can recur with minimal excisions— Wide excision is essential for long-term survival (reported an 8-year
survivor with this treatment)— Neither medical or surgical treatment was effective when the disease
was widely disseminated

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 173
THE SURGICAL MANAGEMENT OF CUTANEOUS MELANOMA RIKER
During his research fellowship at Middlesex Hospi-tal in London 1907, William Sampson Handley studiedthe lymphatic spread of a melanoma in individuals. Hesummarized his results in a Hunterian Lecture entitled“Melanotic growths in relation to their operative treat-ment,” in which he strongly supports the views of Fred-erick Eve, advocating wide excision of the primarymelanoma in combination with elective regional lymphnode dissection or possibly amputation in selected cas-es.11 In this manuscript, he accurately notes that the“permeation of the lymphatics is the principle agent inthis local centrifugal spread” of melanoma, recom-mending a circular incision of about one inch from theedge of the tumor, and another two inches into the sub-cutaneous fat. Handley’s work, published in The Lancetin 1907, is of great historical significance as his rec-ommendations became the basis for the surgical man-agement of melanoma for the next 75 years (Figure 1).
Until the 1960s, invasive melanoma was considereda high-risk disease that required an extensive localexcision for all tumors. Wallace H. Clark, Jr., a pathol-ogist at the Massachusetts General Hospital, had apassion for the morphology and biology of neoplasticpigment cells, becoming renowned for his descrip-tions of melanocytes and melanosomes seen with elec-tron microscopy.12 In 1969, he used the microscopicmorphology of melanoma cells as a basis for classi-fying tumors into various stages of development. Hisstudies revealed the progression of melanomas from aradial growth phase to that of a nodular, or verticalgrowth phase. Clark also recognized variant patternsof the radial growth phase, further developing a clas-sification system (“Clark’s level”). He classified pri-mary cutaneous melanoma samples based upon theextent of tumor invasion related to the anatomic lay-ers of the skin, correlating the level of invasion to thatof overall survival.13 This system constituted the firstwidely used prognostic model for melanoma, followedshortly thereafter by Alexander Breslow who added asecond method of measurement, based upon the truevertical thickness of the tumor, measured in millime-ters.14 This system provided a more accurate and repro-ducible method of measurement than previous meth-ods, providing an excellent correlation with overall5-year survival. A comparison of the two systemsalong with other histologic parameters reveal that thetumor thickness, measured in millimeters, was a bet-ter predictor of metastasis and overall survival com-pared to the Clark’s level of tumor invasion.15
In the early 1970s, Donald L. Morton observed that
the body developed serum antibodies against tumorcell surface antigens in melanoma patients. His pio-neering discoveries led to a major program in vaccinedevelopment against melanoma antigens. Additional-ly, he added to our understanding of the lymphaticsystem and how it relates to the drainage of melanomacells from the primary lesion, developing the sentinellymph node concept in 1990.16 He conclusively showedthat sentinel lymph node mapping was able to accu-rately identify the first draining lymph node that wasconnected to the primary tumor site, further reveal-ing that extensive microscopic analysis of a singlelymph node was possible and predictive of the statusof the remaining nodal basin.17, 18 The utility of sentinellymph node mapping has resulted in a true paradigmshift in our surgical approach to staging patients withmelanoma.
Steven Rosenberg continues to spend most of hiscareer, which has lasted more than 3 decades to date,focused upon the immunotherapy of cancer, in par-ticular melanoma. He was the first to utilize inter-leukin-2 to treat patients with metastatic melanoma,currently the only FDA approved agent to treat suchpatients. His group within the surgery branch of theNational Cancer Institute continues to develop ground-breaking treatments for patients with advancedmelanoma. They have recently developed a novelapproach, termed adoptive cell transfer (ACT) thera-py, which involves the isolation, and in vitro growth oflarge numbers of highly active, melanoma-specificautologous T cells. This is followed by infusion ofthese reactive T-cells back into the same patient, usu-ally given in combination with high dose IL-2 as well.This approach represents a promising therapy for thetreatment of advanced melanoma.19 The initial studiesusing cloned melanoma-antigen specific T cells, withor without IL-2, seemed to be relatively ineffective atinducing an objective anti-tumor response.20, 21 Others
Figure 1.—A depiction of Handley’s original diagram advocating widelocal excision for primary melanoma. The dotted lines (arrowheads) repre-sent his recommended margin of excision.

RIKER THE SURGICAL MANAGEMENT OF CUTANEOUS MELANOMA
174 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
approachs utilized tumor-infiltrating lymphocytes(TIL), which has hypothetical advantages over otherapproaches due to the diverse effector population com-prised of both CD4+ and CD8+ T cells. In clinicalstudies where TIL were used in conjunction with highdose IL-2, objective tumor regressions were seen in33% of the patients treated, unfortunately, these clin-ical responses were of short duration with the persis-tence of the transferred TIL only transient.22, 23
To enhance the efficacy of ACT utilizing TIL, alymphodepleting, non-myeloablative pre-condition-ing chemotherapy regimen consisting of cyclophos-phamide and fludarabine was given before the admin-istration of highly reactive melanoma-antigen spe-cific TIL combined with high dose IL-2.24 The ini-tial study involved 13 patients with progressivemelanoma despite multiple previous treatments,including high dose IL-2, 6/13 patients achieving anobjective clinical responses and 4/13 exhibiting amixed response with regression of some lesions butwith progression in other lesions.24 In a recent fol-low-up study, cancer regression in patients with refrac-tory metastatic melanoma with large, vascularizedtumors was found in a striking 18/35 patients (51%response rate), including 4 patients with a completeregression of all metastatic disease.25 To date, thisimmunotherapeutic approach is the highest reportedresponse rate ever reported for the treatment of patientswith stage IV melanoma.
Recently, the Rosenberg group examined the utili-ty of autologous peripheral blood lymphocytes trans-duced ex vivo with a retrovirus encoding a T-cell recep-tor for the MART-1 antigen.26 This experimental reg-imen was administered to a total of 15 patients in con-junction with high dose IL-2 in a lymphodepleting,
non-myeloablative setting. A total of 2/15 patientsachieved objective clinical responses that are ongo-ing after a year, demonstrating for the very first timethe efficacy of gene therapy combined with a multi-modal approach (IL-2, non-myeloablative chemother-apy) to treating stage IV melanoma patients.
It is important to recognize the important contribu-tions of past physicians and surgeons, learning fromtheir experiences in the clinical management of patientswith melanoma. Their exceedingly insightful obser-vations have paved the way for improvements in ourpresent day surgical management. Other such obser-vations and tenets have since been dismissed basedupon the results of several well designed multi-insti-tutional and cooperative prospective, randomized clin-ical trials. It is clear that we must continue down thepathways of our predecessors and strive to improvethe quality of surgical care for all melanoma patients.The future holds great promise for the development ofnovel treatment strategies for those patients withadvanced disease, many of which do not require thescalpel. However, the basic tenets for the surgical man-agement of primary melanoma and regional nodeshave been forged from previous trials, while questionsstill remain as to the optimal management of patientswith late stage, advanced disease.
Early detection and prevention of melanoma
It is imperative that the diagnosis of cutaneousmelanoma be made as early as possible, as this direct-ly correlates with long-term outcome. For decades,physicians have utilized the clinical examination ofthe skin as the primary screening modality for detect-
Figure 2.—The ABCDE’s of melanoma diagnosis. A: asymmetry. B: border irregularity. C: color change. D: diameter increase. E: elevation.

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 175
THE SURGICAL MANAGEMENT OF CUTANEOUS MELANOMA RIKER
ing melanoma. Yet, the ability of the clinician to accu-rately identify those lesions that are melanoma ishighly variable in most cases, making the correctdiagnosis in only about 65% of all cases.27-32 Cur-rent methods of diagnosis depend heavily upon thephysician’s ability to identify the classic signs of acutaneous melanoma, primarily the A-B-C technique(Figure 2). Although many of newly diagnosed lesionsmay have one or more of these signs, some may not,such as the amelanotic melanoma that is void of anypigmentary change (Figure 3). It is very common tohave the patient diagnose their own melanoma, oftencoming in to see the physician after noticing a recentchange in the lesion or an episode of bleeding. Some-times, it is the spouse that first notices a distinctchange in a skin lesion. Regardless, we currently rec-ommend that everyone perform full body, naked skinexams on a monthly basis at home, in combinationwith a yearly full body skin examination by theirphysician. All lesions that are deemed suspicious onphysical examination should undergo a biopsy, withthe continued observation of such suspicious lesionsdiscouraged.
The accuracy rate of detection can be improved by10-20% with the addition of other imaging tools, suchas epiluminescence microscopy and sequential fullbody photography.33, 34 However, no matter what obser-vational threshold is being followed, many lesions thatare deemed suspicious for melanoma will ultimatelyundergo a biopsy to obtain a definitive diagnosis.Obtaining a tissue sample by means of whatevermethod of biopsy, followed by histological examina-tion of the tissue is still considered the “gold stan-dard” for accurately making the diagnosis of primarycutaneous melanoma.
Biopsy techniques
The majority of clinical management guidelinesrecommend that a pigmented lesion or mole that isdeemed suspicious undergo an excisional biopsy asthe preferred method of biopsy, obtaining a margin ofnormal skin of 1-2 mm.27-29 The depth of the biopsyshould encompass the subcutaneous fat, with com-plete removal of the lesion in order to provide an unen-cumbered histologic examination that is inclusive of theentire lesion. This will provide an accurate Breslow’sdepth of invasion in addition to other important prog-nostic determinates.35-37 The definitive surgical pro-cedure of the primary melanoma should be deferreduntil the final histologic diagnosis has been made,even for suspected thin melanomas such as melanomain-situ.38-40 It is imperative for the clinician to be cog-nizant of cosmetically sensitive areas when perform-ing a biopsy as this will dictate the type of biopsy per-formed and the necessity for possibly specialty surgi-cal consultation. Definitive excision of such areas mustbe deferred until the final diagnosis has been com-
Figure 3.—Patient with an amelanotic melanoma of the forehead (arrow).
Figure 4.—Techniques for biopsy of suspicious skin lesions. A) Punchbiopsy is the preferred technique for pigmented lesions suspicious formelanoma, as it provides an accurate full thickness measurement of thelesion. B) Shave biopsy technique used for sampling of raised lesions.Shave biopsies should be avoided in any lesion suspicious for melanomaas they fail to provide an accurate Breslow thickness staging.

RIKER THE SURGICAL MANAGEMENT OF CUTANEOUS MELANOMA
176 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
pleted, as often the pathological diagnosis yields abenign result that does not require any further surgicalintervention.41, 42
If a punch biopsy is performed, one should obtainthe sample from the thickest portion of the lesion, avoid-ing areas of crusting, ulceration or necrosis that maygrossly underestimate the overall thickness of the tumor(Figure 4). It is not recommended to perform a punchbiopsy at the edge of the lesion, as this may markedlyunderestimate the true thickness of the lesion and maynot be representative of the sample as a whole. If the sur-rounding architecture is to be included, then a deepshave (scallop) biopsy of the entire lesion should thenbe performed (Figure 4). Although the preferred methodof biopsy is the excisional biopsy, many physicians willperform a deep shave, or saucerization, of a lesion sus-pected of being melanoma. This is usually done witheither a scalpel or a single-edged razor blade held in asemi-curved position.27 A saucerization is essentiallya modified shave biopsy that adequately samples thedeeper dermis, and is achieved by pinching the skinaround the lesion while curving the razor blade.43
One potential drawback of either method is thatthere remains the possibility of transecting the baseof the lesion, thus resulting in a deep margin that isinvolved with melanoma. This is problematic in that thetrue Breslow’s thickness is not known, creating a diag-nostic dilemma for the surgeon in terms of the decision-making for the appropriate surgical margins andwhether the draining lymph node basin needs to beevaluated. A second consideration as to the type ofbiopsy performed is the cosmetic outcome. A shavebiopsy site will heal by secondary intention, often tak-ing several weeks before completely filled in with scar
tissue. This type of healing process is inferior in termsof cosmetic outcome compared to other techniques,such as a punch biopsy with primary closure. However,the biggest advantage of performing a punch biopsy isthat the specimen can be accurately measured for truedepth of invasion. The Breslow’s depth of melanomainvasion is what will ultimately dictate the appropriatecourse of surgical management for the majority ofpatients. The resulting defect after a punch biopsy iseasily closed primarily with 1 or 2 interrupted sutures,resulting in a superior cosmetic outcome comparedto a shave biopsy that heals by secondary intention. Thelargest available punch biopsy is 6 mm in greatestdiameter, thus for larger primary lesions, this may notbe able to completely remove such lesions. This maylimit the amount of adjacent normal skin collectedand thus limit the interpretation of the histologic archi-tecture of the entire specimen, which in combinationwith other cytological features, is of particular impor-tance when diagnosing melanoma.44
Additionally, there are several other important featuresthat require special attention in order to obtain an accu-rate diagnosis of melanoma, such as the presence ofasymmetry, the lack of circumscription and the pres-ence (or absence) of scattered atypical melanocytesthroughout the epidermis and adnexal epithelium. Suchfeatures may not be present if a small punch biopsyonly is performed and the type of biopsy must be tak-en into account by the dermatopathologist.43 In cases ofinadequate sampling, it may be necessary to complete-ly remove the lesion with an excisional biopsy in orderto confirm the diagnosis of a suspected melanoma.
Excision of the primary melanoma and surgicalmargins
The surgical management of cutaneous melanomamust always begin with the proper identification andtreatment of the primary lesion. When melanoma isdiagnosed in its earliest stages, over 90% will be essen-tially cured by surgical excision alone.45, 46 It is impor-tant to understand that removal of the appropriate sur-gical margins with the excision of a primary melanomais instrumental in minimizing the most importantpotential complication associated with surgical exci-sion, that of local tumor recurrence. Local recurrenceof disease may be due to the lack of excising adequatesurgical margins, with the possibility of tumor cells pre-sent outside of the margins of excision (Figure 5). It is
Figure 5.—Patient with a local recurrence of melanoma after inadequateexcision.

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 177
THE SURGICAL MANAGEMENT OF CUTANEOUS MELANOMA RIKER
often impossible to differentiate between local per-sistence and metastasis. The development of a localrecurrence, defined as tumor recurring within 2 cm ofthe surgical scar, has been associated in many studieswith a dismal prognosis, with 40-60% of all patientswith a local recurrence of melanoma dying of theirdisease (Table II).
The majority of cutaneous melanomas can be effec-tively and adequately excised utilizing standard exci-sions that are elliptically placed with the long axis ofthe ellipse directed towards the regional nodal basin.The length of the long axis of the ellipse can be esti-mated to ensure an adequate closure without undo ten-sion or cosmetically displeasing “dog-ears” at theends. In terms of the wide local excision, efforts shouldbe made to decrease the amount of wound tensionalong the entire length of the incision. This can beaccomplished through careful dissection and under-mining of the surrounding skin edges. Transverse inci-sions that cut across and disrupt the flow of lymphat-ics may contribute to lymphedema, especially in thelower extremity.
In general, attempts should be made to adequatelyexcise the primary lesion while minimizing scar for-mation and the need for skin grafts. In some anatom-ic locations, however, such as the distal extremities,joint lines, face, head and neck, scalp, hands and feet,a split thickness skin graft and/or flap closure is the pre-ferred closure, as primary closure is often not possibleor increases the risk of undo tension along the incision.Partial or full thickness skin grafts are associated witha higher surgical morbidity, cosmetic disfigurementand overall cost compared to primary closure. Thedonor site should be examined and chosen pre-opera-tively and the possibility of skin graft necrosis andpoor healing of the graft should be fully explained tothe patient prior to surgery. In the era of sentinel lymphnode biopsy, we have increasingly turned to full-thick-
ness skin grafts harvested from the node biopsy site toclose wide excision wounds that would otherwiserequire split-thickness skin grafting from a third sur-gical site (Figure 6).47
The standard operative approach in the past usual-ly included a 3-5 cm margin of normal skin measuredfrom the outer edge of the melanoma in all directions,with most patients requiring a split-thickness skin graftto cover the resulting defect. This extensive surgicalprocedure resulted in a prolonged hospital stay andassociated perioperative complications such as woundinfection and skin graft necrosis. Fortunately, the extentof surgical resection and margins began to be ques-tioned, allowing for the completion of several prospec-tive, randomized trials that have addressed this issue.The first trial addressed the efficacy of 2 versus 4 cmmargins for primary melanomas between 1 and 4 mmin Breslow’s thickness.48 The Intergroup MelanomaCommittee found a local recurrence rate of 0.8% forpatients who had 2 cm margins compared to 1.7% forthose who had 4 cm margins taken at the time ofsurgery. The differences in local recurrence were notstatistically significant. There was, however, a signif-icant difference in the number of skin grafts needed,with 46% of the 4 cm group requiring a skin graft butonly 11% of the 2 cm group. This same trial was recent-ly updated to provide 10-years of follow-up, againrevealing no significant differences in the local recur-rence rate, disease-free or overall survival.49 This tri-al clearly demonstrated that a 2 cm margin is safe andeffective compared to a 4 cm margin for primarymelanomas between 1 and 4 mm, with a markeddecrease in the need for skin grafting. There have beentwo other trials that have examined 2 cm vs 5 cm mar-gins for intermediate thickness primary melanomas<2 mm in Breslow’s thickness, with both studies show-ing no difference in local recurrence rates or overall sur-vival.50, 51
Several randomized trials have firmly establishedthat the overall thickness of the primary melanomadramatically influences the likelihood of a local recur-rence.52 The first trial to address this issue was theWorld Health Organization (WHO) Melanoma Groupstudy by Veronesi et al., which prospectively ran-domized patients with primary melanomas ≤2.00 mmin Breslow’s thickness to 1 cm versus 3 cm surgicalmargins.53 There were no local recurrences at all amongpatients with primary melanomas of <1 mm, regard-less of whether a 1 cm or 3 cm margin was taken. Inthose patients with primary melanomas between 1 and
TABLE II.—Local recurrence and associated mortality.
Primary tumor Recurrent 5-year 10-year
thickness rate survival survivalrate rate
Ng et al.57 <1 to >4 mm 7% 61% —Dong et al.58 0.76 to 4.0 mm — 51% 35%Soong et al.59 <1 to >4 mm 5% 41% 37%Urist et al.60 0.76 to 4 mm 5% 50% (3-yr) 20%Reintgen et al.61 0.76 to 4 mm — 55% —Kalady et al.62 <1 mm 15% 60% 45%

RIKER THE SURGICAL MANAGEMENT OF CUTANEOUS MELANOMA
178 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
2 mm, there were 4 local recurrences, all occurringwithin the group that had received 1 cm margins. How-ever, there were no differences noted in either group interms of disease-free or overall survival. This trial hasbeen recently updated with 15-year follow-up, andthere were still no difference noted in disease-free oroverall survival.54 This important study provides aclear demonstration that a surgical excision margin of1 cm is safe and provides excellent local control formelanomas <1.00 mm in Breslow’s thickness.
For primary melanomas with a tumor thicknessbetween 1-2 mm, current NCCN guidelines suggestthat the margin of excision can be between 1 and 2cm, depending upon the anatomic circumstances. Inmost cases, it will be possible to perform a 2-cm mar-gin of excision with a smaller 1-cm margin accept-able if placement of a skin graft or an excessively highamount of skin tension will result from taking a larg-er 2-cm margin. Recently, Thomas et al. prospective-ly examined the excision margins in a defined “high-risk group” of patients with primary melanoma, con-
sidered >2 mm in Breslow’s thickness in this study.55
All patients were randomized to either 1 cm or 3 cmmargins of excision, finding that a 1 cm margin ofexcision for melanomas of at least 2 mm in Breslow’sthickness was associated with a significantly greaterrisk of combined (local and regional) recurrence whencompared to a 3 cm margin. It is important to notethat this high risk group included all primary lesions>2 mm in thickness (median tumor thickness was 3mm), and therefore the results and conclusions of thistrial cannot be directly applied to those patients withonly thick (>4 mm) primary lesions. Regardless, thisis an important trial because it is the first time that a ran-domized trial examining surgical margins of excisionhas demonstrated a significant increase in combinedlocoregional recurrence with a narrower 1-cm mar-gin. However, there was no statistically significantdifference noted in the death rate from melanoma asso-ciated with a narrow (1 cm or less) margin of exci-sion for thicker melanomas.
The data addressing the recommended surgical mar-
Figure 6.—Technique for full thickness skin grafting after wide excision of melanoma using sentinel lymph node biopsy wound as donor site. A) Pri-mary melanoma of the dorsal wrist requiring wide excision. This site is not readily amenable to primary or local flap closure. B) Axillary sentinel lymphnode biopsy site marked for full thickness skin graft donation. C) Full thickness skin graft reconstruction after wide excision. D) Axillary full thick-ness donor site wound used for access to perform sentinel node biopsy. E) Late postoperative appearance of the reconstruction showing excellent cosme-tic and functional result. F) Postoperative appearance of the axillary donor site demonstrating excellent cosmesis and full range of motion.

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 179
THE SURGICAL MANAGEMENT OF CUTANEOUS MELANOMA RIKER
gins for thick melanomas (>4 mm) have been addressedwith both retrospective and prospective analyses. Amulti-institutional retrospective review of surgicalmargins and prognostic factors in patients with thick(>4 mm) primary melanomas was performed on 278patients, showing no difference in local recurrencerate, disease-free survival or overall survival if marginslarger than 2 cm were taken.56 Indeed, this is addressedby Ng et al., who analyzed 1 155 patients who hadtheir primary melanoma excised, finding 84 (7%)patients overall with local recurrences, and 33 of those84 (39%) patients dying of their disease.57 The 5-yearsurvival for all patients with local recurrence was 59%,compared to 86% for those without a local recurrence(P<0.0001). Other studies examining local tumor recur-rence and its impact upon long term survival haveyielded similar results.58-61
Even patients with thin melanomas (≤1 mm in thick-ness) deserve an appropriate surgical margin, as recur-rence does occur in this group and is a true harbingerof very poor prognosis and outcome.62 Although it isrecognized that a 2-cm margin of excision is adequatefor melanomas >1 mm, there may not be overwhelm-ing support for a trial that wishes to address the valueof a smaller margin, such as a 1 cm margin comparedto a 2 cm margin of excision for primary melanoma.This would certainly be interesting to conduct such atrial for cosmetically sensitive areas such as the face andhead and neck, however, this would require a tremen-dous amount of resources in order to answer this ques-tion. Rather, our efforts may be better directed towardsresearch into developing better prognostic markers ofoutcome, whereby we can apply such markers to spe-cific subgroups of melanoma patients. This may allowus to select those patients that will benefit the most(and exclude those that will not) from a given proce-dure, such as SLNB for thin or thick melanoma. Thecurrent recommendations for excision margins for pri-
mary cutaneous melanoma are outlined in Table III. Interms of the need to remove (or not remove) the deepmuscular fascia, there does not appear to be any clearadvantage (or disadvantage) to removing the deepmuscular fascia as part of the definitive excision ofthe primary melanoma. Several studies have addressedthis issue and it does not appear that there is any sig-nificant difference in recurrence rates, locally or dis-tant, when the fascia was either left in place or removedas part of the definitive surgery.63, 64
Mohs micrographic surgery
The primary goal of the surgical management ofprimary cutaneous melanoma is to completely removethe entire tumor in order to minimize the chance of alocal recurrence. However, removal of the optimalsurgical margins may be compromised when dealingwith melanomas in cosmetically sensitive areas, suchas the head and neck. The utility of Mohs micrographicsurgery (MMS) in treating melanoma has developedover the years to become a recognized form of surgi-cal therapy in select patients with melanoma. Forexample, MMS may provide some advantages in treat-ing head and neck melanoma, such as with the removalof the lentigo maligna subtype. The utility of MMSin this situation has been demonstrated to have a lowlocal recurrence rate (0.2% for MMS vs 9% for con-ventional surgery) and smaller tissue defect due to theminimization of the skin margin removed.65 Addi-tionally, MMS for primary cutaneous head and neckmelanomas may contribute to favorable outcomeswhere extensive sub-clinical spread can be found inmany cases.
However, it should be recognized that when MMSis applied to melanoma lesions, it is important to exam-ine 100% of the surgical margin for each tumor, accom-plished by either frozen section or standard histolog-ical analysis. If frozen section analysis is to be per-formed at the time of MMS, it is essential that theMohs surgeon be adequately trained to interpret suchresults accurately and that a trained and experiencedhistotechnician be available for the proper prepara-tion of thin marginal analysis without undo processingartifact.66 Although there may be some advantages toperforming MMS on primary melanomas of the headand neck due to the minimization of removing excessskin, its role in truncal and most extremity melanomasis limited. In general, there is only a small difference
TABLE III.—Recommendations for excision margins of primary cuta-neous melanoma.
Location Tumor Thickness Margins
Trunk/proximal extremity Melanoma in-situ 0.5 cm≤1 mm 1 cm1-2 mm 1-2 cm
>2-4 mm 2 cm> 4 mm 2 cm
Head/neck, distal extremity(or cosmetically sensitive area) ≤1 mm 1 cm
>1 mm at least 1 cm

RIKER THE SURGICAL MANAGEMENT OF CUTANEOUS MELANOMA
180 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
in excess tissue removed with MMS compared to thatof standard surgical excision based upon Breslow’stumor thickness. Regardless, MMS requires bothmeticulous surgical and pathological analysis in orderto optimize patient outcomes and minimize local recur-rence rates.66
Truncal and extremity melanoma
The surgical management of truncal and extremitymelanoma is fairly straight-forward, with the basictenets of surgical therapy to remove the primarymelanoma with the appropriate surgical margins. How-ever, certain situations and anatomic locations mayalter the surgeon’s approach to management, such asmelanomas located along the forearm, leg and digits.In particular, a melanoma >2 mm in Breslow’s thick-ness on the forearm will require a 2-cm circumferen-tial excisional margin with a resultant defect of at least
4 x 4 cm. Due to the anatomic limitations of skinmobility in such areas, it is often necessary to utilizea split-thickness skin graft (STSG) for adequate cov-erage, often taken from the anterolateral aspect of thethigh. Other possible donor sites may include a fullthickness skin graft from the lower quadrant of theabdomen with primary closure of this defect, therebysparing the patient the increased pain and discomfortassociated with a STSG from the thigh.
The majority of primary melanomas located on theback can be treated with the appropriate excision mar-gin followed by skin edge approximation and prima-ry closure (Figure 7). The skin on the back is generallythicker and has more laxity compared to other areas ofthe body, with the resulting defect successfully closedprimarily without the need of skin grafting. In order tominimize the amount of tension along the mid-por-tion of the defect, attention should be given to the opti-mal orientation of the surgical excision related to theoptimal lines of skin tension in order to minimize theneed for extensive undermining of the surroundingskin edges. Occasionally, the surgeon may encounteran undo amount of skin tension and this situation is besttreated with the placement of a STSG or possibly oneof several plastic reconstructive options such as a rota-tional, advancement or rhomboid skin flap.
The surgical management of headand neck melanoma
Special attention should be paid to the patient witha primary melanoma of the head and neck due to theadded anatomic complexity posed within this region.Although the established guidelines are generally fol-lowed whenever possible, melanomas arising withinaesthetic areas of the face often require a compromisein such margins. Every attempt should be made toobtain the appropriate surgical margins and concomi-tantly achieving the best cosmetic outcome with thelowest possible chance of local recurrence. It is imper-ative that a thorough discussion of the planned excisionbe made with the patient, outlining the operative planand any associated reconstruction being performed.The risks, benefits and expected cosmetic outcomesshould be carefully discussed with the patient, dis-cussing any unrealistic expectations of any surgicalprocedure.
The surgical treatment of the primary tumor of thehead and neck includes planning the complete excision
Figure 7.—Technique for wide excision and primary closure of a thick mela-noma of the back. A) The lesion is injected with vital blue dye prior to exci-sion for intraoperative lymphatic mapping. B) The primary lesion is exci-sed with a 2 cm margin circumferentially. An elliptical shaped excision isused, oriented with the lines of tension to facilitate primary closure. C) Thedefect has been closed primarily with mobility of the wound edges obtai-ned by simple skin undermining. Note the nylon stay sutures placed atthe center of the closure to alleviate tension.

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 181
THE SURGICAL MANAGEMENT OF CUTANEOUS MELANOMA RIKER
of the primary melanoma as well as the reconstruc-tive procedures simultaneously.67 The surgeon shouldbe well-versed in the anatomy of the face, consideringthe relaxed skin tension lines and functional aesthet-ic units of the entire face. Special consideration shouldbe given to primary melanoma excisions that involveoverlying lymph node-bearing areas, such as the parotidgland and neck. A pre-auricular vertical incision fol-lowed by the development of an anterior cervico-facialflap is able to adequately expose the parotid gland orperi-auricular and upper neck lymph nodes. In theneck, an upper-neck transverse incision or a mid-neckposterior vertical incision provide optimal exposureto the appropriate cervical lymph node basin.
The method of reconstruction of the primarymelanoma excision site depends upon several factorssuch as the location and size of the defect, the func-tional and aesthetic requirements and the overall med-ical condition of the patient. There are numerous pos-sible reconstructive options such as the utility of aSTSG, local vascularized and regional tissue flaps aswell as myocutaneous flaps (Figure 8). The most com-
mon surgical excision of a primary scalp melanomainvolves the removal of the appropriate skin marginsand underlying subcutaneous fat down to the galea.The underlying periosteum is well-vascularized andprovides a good base for the proper healing of a STSG.For smaller excisions, local random rotational skinflaps may be suitable in lieu of skin grafting. In rare cas-es, extensive surgical excision of the primary melanomawith a large residual defect may require a free flap foradequate wound closure, possibly obtained from sev-eral sites such as the rectus, latissimus or radial fore-arm muscle.
Small excisions of the cheek can usually be closedwithin the exaggerated “smile lines” on the face. Forlarger defects involving the medial portion of the cheek,an inferiorly-based cervico-facial rotation advance-ment flap may provide the optimal aesthetic result.For upper lip defects that are lateral to and above thevermillion border, we utilize a cheek advancementflap for good cosmetic results. Defects along the medi-al and central upper lip are best suited for a lower lip-cross lip flap. For lower lip defects, local rotation flaps
Figure 8.—Intraoperative photograph of an excsion and sentinel lymph node biopsy of a intermediate thickness melanoma of the cheek. A) The exci-sion margins and planned flap have been marked. The the sentinel node is located with the gamma probe. B) The lesion has been widely excised andthe rotational flap has been raised. C) The skin flap has been retracted and the sentinel node located and excised. D) The completed rotational flap recon-struction.

RIKER THE SURGICAL MANAGEMENT OF CUTANEOUS MELANOMA
182 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
are often utilized, bearing in mind that if the defect isa result of a complete excision of the lip, muscle, andmucosa, then one of several lip advancement tech-niques can be employed. The orbicularis oris myocu-taneous flap can be an excellent reconstructive choicefor lip excisions that have removed between 25% and75% of the lower lip. If the entire lower lip must beexcised, utilization of a radial forearm free flap may benecessary as part of the reconstructive process.
The role of elective lymph node dissection
The evaluation and management of the draininglymph node basins for patients with clinically localizedmelanomas >1 mm in Breslow’s thickness has evolvedover the last 20 years. In the past, we relied upon thestaging information gained from performing an elec-tive lymph node dissection (ELND). Although suchinformation on staging was obtained, several prospec-tive, almost all of the randomized trials examining thetherapeutic role of ELND have failed to show an over-all survival advantage for this technique. However,there is one trial of special note, the IntergroupMelanoma Surgical Trial, reporting 10-year survivalresults for those patients undergoing ELND.68 Aprospective, stratified subgroup of patients were foundto have a significant decrease in their overall mortal-ity, namely those with non-ulcerated melanomas (30%reduction), tumor thickness between 1-2 mm in Bres-low’s thickness (30% reduction), age <60 years old(27% reduction) and location of the melanoma on anextremity (27% reduction).68 Regardless, an electivelymph node dissection provides durable local controland accurate staging for most patients with occultlymph node metastases. A therapeutic lymph nodedissection, performed for patients with clinically evi-dent regional lymphadenopathy, provides locoregionalcontrol of disease and a chance of cure, with 5-year sur-vival rates of 20-40%.69-71 Meyer et al. performed 144therapeutic lymph node dissections in 140 melanomapatients (14 cervical, 49 axillary, 73 groin) and founda 5-year survival for all patients of 30%.72
Secondly, there is some evidence of a potential sur-vival benefit for early evaluation and management ofthe draining lymph nodes from the WHO trial thatcompared ELND to observation.73 This trial random-ized those patients with a primary melanoma >1.5 mminto either observation versus an immediate ELND.Although there was no evidence of an overall survival
benefit, the group who underwent an immediate ELNDand found to have positive lymph nodes had a 5-yearsurvival of 48% compared to the observation arm anddelayed CLND with an overall 5-year survival of only27% (P=0.04). A second trial by Morton et al. com-pared the survival of matched groups of patients under-going immediate (after a positive SLN) versus delayed(after nodal recurrence) CLND.74 They found a sig-nificantly higher survival rate (at 5, 10 and 15 years)in the group that underwent immediate CLND fol-lowing a positive SLN compared to the latter group thatunderwent a delayed procedure.
The role of sentinel lymphadenectomy for thesurgical staging of melanoma
Intraoperative lymphatic mapping and selective lym-phadenectomy was originally developed to provide aminimally invasive staging procedure that reduced theassociated morbidity and expense of an ELND. Theminimally invasive technique of sentinel lymph nodebiopsy (SLNB) for appropriately selected patientswith melanoma has become part of the standard dis-cussion of operative intervention, with the availabledata showing that SLNB is the best available diag-nostic test to date in detecting nodal metastases. Obvi-ous advantages of SLNB over ELND include a lessinvasive procedure, smaller skin incisions, reducedperi- and post-operative complications, less extensivedissection of the nodal basin, and improved staginginformation for both the physician and patient.
The concept of SLNB is based upon the hypothesisthat cancer cells from the primary lesion gain entranceinto the lymphatic channels and travel to the nearbydraining nodal basin. The SLNB technique is furthersupported by a large amount of data that clearly indi-cates that the identification of a tumor-free SLN willtranslate into a remaining draining nodal basin that iswithout evidence of metastatic cancer cells.75 The eval-uation of the draining lymph node basins for patientswith clinically localized melanoma has resulted in atrue paradigm shift in our surgical management.
It is vitally important that we continue to learn frompast trials in order to improve upon the way that wetreat patients with melanoma presently and in thefuture. There are four major reasons to perform SLNB,all driven to provide the most accurate informationavailable.76 The first reason is to improve the accura-cy of staging. The second is to facilitate early thera-

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 183
THE SURGICAL MANAGEMENT OF CUTANEOUS MELANOMA RIKER
peutic lymph node dissection for those patients withnodal metastases. Thirdly, SLNB identifies patientswho are candidates for adjuvant treatment with inter-feron-α 2b. Fourth, SLNB identifies homogeneouspatient populations for entry into experimental clini-cal trials. The latter reason is critically important, rec-ognizing the importance of enrolling similarly stagedpatients to strengthen results and develop more effec-tive treatment strategies for such patients.
It should be clearly stated that the conceptual devel-opment and subsequent utility of the SLNB procedurewas never designed as, or meant to be used as, a ther-apeutic procedure. The utility of SLNB is that of adiagnostic test that provides a significant improve-ment in the overall sensitivity, specificity and accura-cy of identifying metastatic disease within the SLNof the draining nodal basin. Few would argue thatSLNB provides the most accurate staging informa-tion available for melanoma patients. However, thereare those that will continue the discussion as to theutility of SLNB, insisting that it must also provide atherapeutic advantage before being accepted into stan-dard surgical practice. It is akin to the argument for thedevelopment and utility of the PET scanner, a similarlynew technology developed as a staging tool. McMas-ters et al. eloquently describes this parallel developmentand reinforces the notion that we never should have toraise the bar any higher for a diagnostic procedurethat was never meant to show or give an overall survivalbenefit to patients.75 Thus, although SLNB is not 100%accurate, it is a far better test than clinical staging ofregional lymph nodes, and it would seem illogical torevert back to a delayed nodal dissection for thosepatients that present with palpable regional disease.77
Others have strongly argued that since this proceduredoes not provide a survival advantage, “that it shouldbe abandoned immediately, without question”.78, 79
Additionally, those opposed to SLNB argue that theinvolvement of the lymph nodes (sentinel or not) withmelanoma is a “harbinger of distant spread of diseaseand associated poor outcome in all cases, with theonly uncertainty being when the metastatic diseasewill manifest itself”.78,79 The current available evi-dence would strongly refute this notion, faced withseveral lines of evidence showing that surgical thera-py alone can result in long-term cures of patients withstage III melanoma. Young et al. recently evaluatedthe role of definitive surgical management for patientswith stage III disease.80 At a maximum follow-up of
386 months (32 years) for the total population of 1422patients in the trial, the 15-, 20- and 25-year melanoma-specific survival rates were 36%, 35% and 35%, respec-tively. Additionally, they found that survival rates weresignificantly lower if the regional nodes were palpable.Although some risk factors decreased the likelihood oflong-term survival, the high overall survival rates in allgroups clearly supports the utility of surgery as theprimary treatment option for regional metastaticmelanoma.
The Multicenter Sentinel Lymphadenectomy Trial(MSLT-I) is a prospective, randomized, multination-al trial specifically designed to address the possibletherapeutic utility of SLNB. In this study, a total of1269 patients with intermediate thickness melanomas(1.2 to 3.5 mm) were randomized to either wide exci-sion only followed by observation (no SLNB) or towide excision and SLNB. In the observation onlygroup, complete lymphadenectomy was performedonly when there was clinical evidence of nodal recur-rence (delayed), while the SLNB group underwent acomplete (immediate) lymphadenectomy if nodalmicrometastases were detected within any of theSLN’s. The results from this trial were first reported inDecember 2004, updated in may 2005 and most recent-ly published in September 2006 based upon the 3rd
interim analysis.81, 82 They found that the mean esti-mated 5-year disease-free survival rate was signifi-cantly higher in the SLNB group compared to theobservation only group (78.3% vs 73.1%, respective-ly, P=0.009). Although 5-year melanoma-specific sur-vival rates were similar in the two groups, the presenceof metastatic disease within the SLN was found to bethe single most important prognostic factor predictiveof overall survival. The 5-year survival rate was 72.3%in those patients with tumor-positive SLN’s and 90.2%in those with tumor-negative SLN’s.
Interestingly, the incidence of SLN micrometas-tases was 16% as compared to a very similar rate ofnodal relapse of 15.6% in the observation only group,strongly suggesting that occult micrometastases in thesentinel node usually progress to aggressive regionalor distant disease, rather than remaining clinicallyinsignificant within the SLN [82]. Furthermore, themean number of clinically detectable tumor-positivelymph nodes in the delayed CLND group was 3.3,compared to 1.4 lymph nodes within the immediateCLND group. This would indicate that disease pro-gression is indeed occurring during the observational

RIKER THE SURGICAL MANAGEMENT OF CUTANEOUS MELANOMA
184 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
period, with a significant difference noted betweenthe two groups in both disease-free survival and the rateof nodal relapse (4.0% in patients with tumor-negativeSLN’s and 15.6% in the observation only group). Itwould appear that such results support the notion thatobservation of metastatic lymph nodes allows enlarge-ment and adjacent spread to other nodes, therebyincreasing the chances of the patient developing dis-tant metastatic disease and a poorer long-term sur-vival.82 This is most evident when a direct comparisonis made between the immediate vs. delayed CLNDgroups, revealing a melanoma-specific survival of72.3% for the immediate CLND compared to 52.4%in the delayed CLND group. Thus, this recent data isquite compelling in support of performing a SLNB inthose patients with an intermediate-thicknessmelanoma as compared to observation only (noSLNB). Immediate CLND after the finding of SLN-positive disease prolongs survival compared to a
delayed CLND performed only upon the discovery ofclinically evident disease.81, 82
We believe that it is in the patient’s best interest tobe fully informed as to the possible diagnostic andtherapeutic tests available to them. A thorough dis-cussion as to the risks and benefits of SLNB should beincluded for those patients deemed appropriate can-didates. It has been our experience that the vast major-ity of melanoma patients who are candidates for SLNBwill chose to undergo this procedure in order to gainuseful information about their case. The discussion isnot focused on the argument of whether we are pro-viding a survival benefit, but rather centered on if weare providing a diagnostic procedure capable ofanswering the question of a primary melanoma cellmetastasizing to the draining lymph node basin(s).Thus, the available data supports a role for SLNB,able to accurately identify the first draining lymphnodes in a basin and to predict the outcome and stag-ing for melanoma patients. We currently utilize a dualtechnique, combining the pre-operative radiolym-phoscintigraphy with intra-operative injection of vitalblue dye (Lymphazurin, 1% isosulfan blue dye), result-ing in a high accuracy rate for the proper identificationof the SLN. (Figure 9).
Sentinel lymph node mapping for thin melanoma(<1 mm)
The risk of regional nodal involvement for thinmelanomas (<1 mm in Breslow’s thickness) is in therange of 0-9.7%, with an average risk of about 5%.85
For those primary melanomas with other higher riskfactors or between 0.75 mm and 1 mm, the risk ofnodal involvement is slighter higher to about 5-8%.It is well recognized that patients within this groupalso are heterogeneous in nature, with some patients ata much higher risk as compared to others that mayhave minimal to no risk. There are several factorswhich may be considered as higher risk when dis-cussing the risks and benefits of SLNB in the patientwith a thin melanoma. The presence of a verticalgrowth phase, Clark’s level of invasion IV or V, ulcer-ation, regression, high mitotic rate and a younger ageare all considered high risk features for those with thinmelanoma.46, 86-90 There is strong evidence that indi-cates that tumor mitotic rate and a younger age aremore powerful predictors than most, if not all, otherprognostic factors available today.88-90 The focus of
Figure 9.—Techniques of preoperative and intraoperative lymphatic map-ping. A) Preoperative Tc-99 lymphoscintigraphy of a chest wall melano-ma demonstrating lymphatic uptake in both axillae. B) Preoperativelymphoscintigraphy of a trunk melanoma demonstrating lymphatic uptakein the ipsilateral axilla and contralateral groin. C) Technique for injectionof a primary melanoma lesion with vital blue dye for intraoperative lympha-tic mapping. Note the uptake of dye within the afferent dermal lymphatics(arrow). D) Intraoperative appearance of a blue sentinel node.

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 185
THE SURGICAL MANAGEMENT OF CUTANEOUS MELANOMA RIKER
our discussion for patients with thin melanomas iscentered upon issues of whether the incidence of nodalprogression of ~5% warrants the added morbidity andcost of SLNB and if there are specific prognostic/pre-dictive risk factors that could be utilized to determinethe need for SLNB among thin melanoma patients.91
There are many patients and physicians who considera 5% likelihood of nodal metastasis an adequate riskin order to justify the relatively low morbidity of SLNB.However, one cannot forget the intrinsic false-negativerate of about 3% with the procedure itself, with thecaveat that such statistics are only meaningful for thosepatients who indeed have positive lymph nodes, pro-viding a minimal true benefit to most of the overallpatient population with a thin melanoma. Thus, a thor-ough discussion is necessary describing the risks andbenefits of SLNB for patients with thin melanoma.Clearly, there are certain recommendations that canbe made for melanomas that are very close to 1 mm inBreslow’s thickness, with a strong argument made forperforming a SLNB for those melanomas within a fewtenths of a millimeter of 1 mm. Although we all havea few anecdotal cases, it is extremely rare to have a tru-ly thin melanoma <0.75 mm metastasize via the lym-phatic channels or hematogenously. Such patients areunlikely to benefit from a SLNB and it is fairly safe torecommend to the vast majority of patients (if not all)with melanomas <0.75 mm that they may be safelyand adequately treated with a 1 cm margin of exci-sion without a SLNB.92 A recent analysis of 223patients with thin melanomas who underwent a SLNBreveals that there was a 3.6 % rate of nodal metastasis,all in patients with a primary melanoma >0.75 mm inBreslow’s thickness.85 Furthermore, Thompson et al.reported on 187 similar patients with thin melanomasfrom the Sydney Melanoma Unit, finding a positiveSLN rate of 5%, with all patients having a primarymelanoma >0.9 mm in Breslow’s thickness.92
It is important to take an evidence-based approachwhen considering the role of SLNB for thin melanomapatients due to the increasingly large number of patientswithin this subset. Many have tried to define a specif-ic population of patients that will benefit the greatestfrom this staging procedure. The biopsy specimens ofthose patients within the so-called “grey zone” (primarymelanoma between 0.75 and 1 mm) must be exam-ined very carefully histologically for all of the previ-ously described pathological features which may swaythe surgeon to proceed with a SLNB. We approach
such patients by first re-examining all of the patho-logical slides from the original biopsy or excision, uti-lizing the expertise and experience of a der-matopathologist. The presence or absence of all patho-logic features should then be carefully examined anda full and detailed report issued.
It has been our practice to fully explain the risksand benefits involved of the SLNB and weigh themagainst the small risk of nodal metastasis of about 5%.In particular, if a patient is found to have any one (ormore) of the previously considered poor prognosticfeatures, we will thoroughly discuss the role of SLNBwith each patient. For those patients with no evidenceof such prognostic features, we will still have an in-depth discussion of performing the SLNB with thefull understanding of the risks and benefits of the pro-cedure. It has been our experience that the over-whelming majority of patients within this subset willchoose to undergo the SLNB after carefully examin-ing their options of either excision of the primary(alone) versus combining this with SLNB.
Role of sentinel lymph node mapping for thickmelanoma (>4 mm)
The patient with a thick primary melanoma (>4mm) has an overall poor prognosis, with an average 5-and 10-year survival of 45% and 39%, respectively.45,
46 For this reason, many question the utility of per-forming a SLNB in this group of patients, since it is pre-sumed that the patient will already have evidence ofeither microscopic or macroscopic systemic disease.However, the role of SLNB in this group of patientsremains a valuable tool for the most accurate stagingand associated survival data based upon clinical andpathological stage. Although there is a clear consensusthat such patients have a much poorer long term sur-vival, it is of benefit to perform the SLNB in this set-ting in order to gain the most accurate surgical stagingand to achieve early locoregional control of disease.The best opportunity to provide locoregional diseasecontrol is at the earliest possible time of diagnosis,which often represents an earlier time in the progres-sion of the disease process. It is fairly well recognizedamong surgical oncologists that locoregional controlof microscopic disease is associated with a markedlydecreased morbidity with a much simpler surgical pro-cedure as compared to surgical excision of bulkyregional lymphadenopathy, which is fraught with intra-

RIKER THE SURGICAL MANAGEMENT OF CUTANEOUS MELANOMA
186 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
operative and postoperative complications rangingfrom nerve dysfunction, chronic pain, decreased func-tional ability and life-long lymphedema.75
The final decision to undergo a SLNB for a prima-ry cutaneous melanoma must be made by the patient.We believe it is our obligation as physicians and sur-geons to have an objective, data-driven and under-standable discussion with our melanoma patientsaddressing the risks of nodal involvement and whatthis actually means to the patient. This is equallyweighed against the morbidity and possible adverseoutcomes of the SLNB procedure, to include a dis-cussion about the costs, false-negative rates and thecurrent paucity of data in regards to any proven survivalbenefit from this staging procedure.
Role of sentinel lymph node mapping for headand neck melanoma
In general, primary cutaneous melanoma of the headand neck has an overall worse prognosis compared tothose located in other locations, such as the trunk andextremities, and therefore it is of particular impor-
tance to obtain the most accurate staging informationavailable which will then dictate the adjuvant courseof management. The lymphatic drainage of the headand neck is variable and involves a higher number oflymph nodes within the five levels of the cervicalregion. There are also added concerns in regards toaesthetic outcome that can often be overlooked forhead and neck melanoma in comparison to other loca-tions such as the trunk and extremities.
Due to the relatively small anatomic region of thecervical triangles within the head and neck, sentinellymph nodes may be located very close to, and occa-sionally directly beneath, the primary site. This maypose some difficulties in visualization as to the exactlocation of the sentinel node(s) as they may be oblit-erated by the “shine through” effect of the Te-99 injec-tion around the primary site as seen on preoperativeradiolymphoscintigraphy. It is important to performa thorough exploration of the area directly beneaththe primary tumor site for evidence of SLN’s, espe-cially in the area of the pre- and post-auricular faceand neck. An additional tool that may be helpful isthe utilization of a small amount of vital blue dye
Figure 10.—Intraoperative photograph of an excision and sentinel node biopsy of a head and neck melanoma. A) The primary lesion has been injec-ted with vital blue dye and the excision margins have been marked. The sentinel node is located using the hand-held gamma probe. B) The sentinelnode is identified by its uptake of blue dye. C) The completed rotational flap reconstruction.

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 187
THE SURGICAL MANAGEMENT OF CUTANEOUS MELANOMA RIKER
injected around the primary melanoma site. It is onlynecessary to utilize a small amount of dye, usually of0.5 ml. or less, as larger amounts can diffuse into thesurrounding skin of the face and neck, possibly result-ing in the permanent discoloration of the area outsideof the margins of excision. The excision of the pri-mary tumor site should be done simultaneously withthe lymphatic mapping and sentinel node removal,thereby eliminating the background radiation fromthe primary tumor site (Figure 10).
Once the primary melanoma has been completelyremoved, sub-platysmal skin flaps are raised for iden-tification and localization of the SLN utilizing thehand-held gamma probe to pinpoint the exact location.Careful blunt and sharp dissection is critical within allanatomic nodal levels of the cervical region as manyvital structures are at risk for injury and transection.Utilizing the combined techniques of pre-operativeradiolymphoscintigraphy and intra-operative vitalblue dye injection is the most accurate method ofSLN identification, with an accuracy rate of >90%.75
One caveat of performing SLNB in the head and neckis the essential inclusion of “side-view” three-dimen-sional pre-operative images that note whether the sen-tinel nodes are within the posterior versus anteriortriangle or deeper portions of the cervical region (Fig-ure 11).
There are several structures within the neck thatdemand particular attention. The greater auricularnerve, external jugular vein, spinal accessory nerve,and branches of the facial nerve should be sparedfrom division during the sentinel lymph node exci-sion. The great auricular nerve to the ear should beidentified and can be retracted anteriorly and poste-riorly to remove sentinel lymph nodes in the jugu-lodigastric area. The spinal accessory nerve is oftenidentified deep in the jugulodigastric area along theposterior border of the sternocleidomastoid muscleduring sentinel lymphadenectomy. The marginalmandibular branch of the facial nerve must be care-fully protected and is particularly prone to a retrac-tion injury against the hardened surface of themandible. Blunt dissection around the submandibu-lar gland spares any sharp division of the facial nerveat this site.
Lymphatic mapping of the scalp will usually iden-tify sentinel lymph nodes within the occipital and post-auricular area, or along the surface or within the super-ficial portion of the parotid gland. The parotid gland isexposed through a pre-auricular, vertical skin incision
followed by retraction and raising of a facial flap. Theparotid gland can be fully visualized and careful bluntdissection is utilized to identify and remove the SLNthat may be within the parenchyma of the superficiallobe of the parotid gland. The hand-held gamma probeis very helpful in guiding the surgeon towards the iden-tification of a radioactive SLN that has a hint of bluestaining within it. The SLN in this area can often befound between the parotid gland fascia and the auric-ular cartilage, while others may be identified alongthe posterior surface of the facial vein that lies withinthe tail of the parotid gland.
If micrometastatic melanoma is found within theSLN of the head and neck, a completion neck dissec-tion should be performed. This will usually involve amodified radical neck dissection in order to remove allof the lymph node-bearing tissue within the various cer-vical regions. If micrometastatic disease is found with-in a SLN removed from the parotid gland, a superficialparotidectomy with preservation of the facial nerve isalso performed in combination with a modified radi-cal neck dissection.
Complete and selective lymph node dissection
Patients with melanoma may often present withmore advanced regional disease. The surgical man-agement of bulky lymphadenopathy of the axilla orgroin is best managed with a thorough and completeoperative removal of levels I and II within the axilla,with level III lymph nodes carefully palpated and
Figure 11.—Biplanar imaging for head and neck lymphoscintigraphy. A)AP projection of a Tc-99 lymphoscintigraphy of a neck melanoma, demon-strating uptake within the contralateral neck. B) MLO projection of the samepatient showing uptake within multiple nodes in the ipsilateral neck, whi-ch were obscured by the injection site in the AP projection.

RIKER THE SURGICAL MANAGEMENT OF CUTANEOUS MELANOMA
188 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
examined in order to determine if further dissectionneeds to be performed. If there is evidence of palpableadenopathy within level III, they should be removed,as it is very possible that this represents extension ofdisease into this region. If necessary, transection ofthe pectoralis muscle is performed in order to improvethe overall exposure of this area. Operative removal canbe problematic as matted adenopathy of the axilla canbe associated with involvement and possibly encase-ment of adjacent structures such as the thoracodorsalneurovascular bundle, long thoracic and intercostal-brachial nerve, axillary vein and even the brachialplexus. Dissection may result in injury of these struc-tures and attention to detail and anatomic localizationof the thoracodorsal and long thoracic nerves is essen-tial; sacrifice of either of these nerves should bereserved for cases when the tumor is intimately involv-ing these nerves. Within the groin, complete surgicalresection of disease can be equally challenging in thepresence of bulky lymphadenopathy, with the overallcomplication rate with regional lymphadenectomyrelatively high.93, 94
Observations about Cloquet’s node
For palpable inguinal lymphadenopathy, the main-stay of surgical therapy will usually include a super-ficial inguinal lymphadenectomy with inclusion ofthe deep pelvic region for either clinical evidence ofdisease (palpable pelvic lymph nodes), radiographic orintra-operative evidence of obvious lymph nodeinvolvement. Intra-operative pathologic analysis ofclinically suspicious lymph nodes may be necessary inorder to determine the presence of metastatic diseaseand possibly more extensive nodal dissection. Hugh-es et al. describes 132 patients presenting with clini-cally palpable inguinal lymphadenopathy, with theextent of dissection determined by pre-operative radi-ographic studies, clinical suspicion of pelvic nodes,patient morbidity and performance status and evalua-tion of Cloquet’s node.95 They found that those patientsthat underwent a superficial and deep nodal dissec-tion (N=72) had a lower regional recurrence rate com-pared to those whom underwent a superficial dissec-tion only (N=60), with no statistical difference in over-all survival for either group.
Cloquet’s node is defined as the highest lymph nodewithin the superficial inguinal node basin, often locat-ed at the entrance of the femoral triangle at the inguinalligament, sometimes adjacent and medial to the femoralvein or within the femoral canal (Figure 12). Thislymph node is considered by many to be the last (high-est) lymph node within the superficial inguinal regionprior to entrance into the deep iliac and obturator nodalsystem. Thus, it has been hypothesized that the statusof Cloquet’s node can be predictive of metastaticinvolvement of the deep nodal basin, thereby selec-tively performing a deep pelvic lymphadenectomyonly in those patients with a positive Cloquet’s node.The sensitivity and predictive value of Cloquet’s nodeis variable, ranging from 44-90%.96-98
Recently, Essner et al. have updated their previousexperience with Cloquet’s node and have reported apositive predictive value of 66% (negative predictivevalue of 97%) when basing the decision to perform adeep groin dissection upon the status of Cloquet’snode being positive or negative.99 They found that thepathologic status of Cloquet’s node was superior tothe clinical status of the superficial groin nodes forpredicting occult iliac node metastases. However,although most surgeons will agree as to the utility ofperforming a superficial inguinal lymphadenectomy formetastatic disease within this nodal basin, it remains
Figure 12.—Diagram of the inguinal and pelvic lymphatics. S: superficialinguinal nodes; D: deep inguinal node; E: external iliac nodes; O: obturatornode; L: inguinal ligament. Cloquet’s node (curved arrow) is the highestsuperficial inguinal node.

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 189
THE SURGICAL MANAGEMENT OF CUTANEOUS MELANOMA RIKER
controversial as to the utility and benefits of perform-ing a deep pelvic lymphadenectomy, regardless of thestatus of Cloquet’s node.100, 101 In summary, surgeonsshould use all possible available data in order to deter-mine whether a patient should undergo further dis-section and removal of deep pelvic lymph nodes.
Isolated limb perfusion/infusion for melanoma
The technique of isolated limb perfusion/infusion(ILP) was originally developed by Creech and Kre-mentz at Tulane University in 1958, as a way to deliv-er higher concentrations of regional chemotherapycompared to systemic administration, but without thesystemic side effects.102 In about 10% of extremitymelanoma cases, the recurrence of melanoma is con-fined to the same extremity where the previous exci-sion was performed, referred to as in-transit disease orsatellitosis. Although surgical resection should be con-sidered as the first line of treatment in all cases if pos-sible, there are those patients where resection is not aviable option, either due to an overwhelming tumorburden within the extremity or possibly the disease isspread out over a large area and thus precluding a sur-gical option. In such situations, ILP may be a favorabletherapeutic option as a limb-sparing regional treat-ment modality. The surgical technique of ILP is a com-plex and highly invasive procedure that is associatedwith a number of possible intra- and post-operativecomplications. However, although complex and requir-ing a considerable amount of experience from the sur-geon, ILP has an established role for patients with in-transit and satellite metastases of the extremity. Theaddition of hyperthermia (mild hyperthermia, 39-40 °C)to the technique of ILP has further enhanced the over-all response rates, while limiting the toxicity associatedwith higher temperatures (40-43 oC) (Figure 13).
The reported complete response (CR) rates for mel-phalan-alone, mild hyperthermic ILP range from 40-82%, with a median response rate of 54% in a large ret-rospective meta-analysis.103 The addition of tumornecrosis factor-· (TNF) to melphalan was previouslythought to enhance the complete response rate to 60-85%, with suspected increased therapeutic value inpatients with large, bulky lesions or recurrent diseaseafter a previous ILP.103 However, a recent random-ized, multi-center trial conducted through ACOSOGhas compared hyperthermic ILP with melphalan aloneto melphalan + TNF.102 The results revealed that in
locally advanced extremity melanoma treated withILP, the addition of TNF to melphalan did not demon-strate a significant enhancement of short-term responserates (3 month follow-up) compared to melphalanalone. Furthermore, the addition of TNF to melphalanwas found to significantly increase the overall com-plication rate. Other excellent reviews have describedthe clinical experience with melphalan alone, or incombination with several other agents, such as TNF,interferon-· (IFN) and chemotherapeutic agents, con-cluding that ILP has earned a permanent place in thetreatment of patients with unresectable or recurrentmelanoma of the extremity.104-107
Despite the clear evidence for the effectiveness ofILP, it is a technique that is only utilized in a select fewhospitals around the world. Although the technicalconcept is simple and elegant, it continues to chal-lenge surgeons as a technically complex, labor-inten-sive and time-demanding procedure that remains cost-ly.106 For these reasons, Thompson et al. developedthe isolated limb infusion (ILI) technique in 1994 as asimpler and less invasive technique compared to ILP.107
Similar in concept to ILP, ILI was designed as a methodwith broader applicability, such as in the elderly orthose patients with co-morbid conditions that wouldmake an ILP prohibitive.108 The overall response ratesof ILI with melphalan and actinomycin D for treat-ment of recurrent or advanced melanoma of the extrem-
DrugArterial tempVenous tempArterial pressure
Heatexchanger
Oxygenator
Rollerpump
Limbtemps
Figure 13.—Diagram of a circuit for hyperthermic isolated limb perfu-sion.

RIKER THE SURGICAL MANAGEMENT OF CUTANEOUS MELANOMA
190 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
ity are comparable to that achieved with convention-al ILP with melphalan alone.107 It appears that ILIprovides an equivalent treatment option compared toILP, possibly providing an alternative technique that istechnically easier to perform with less post-operativecomplications versus ILP.
Reappraising the role of surgical intervention foradvanced melanoma
It is well established that the overall prognosis forpatients with advanced stage III and IV melanoma ispoor.45, 46 Most patients who experience recurrencewith regional nodal disease and subsequently under-go a complete lymphadenectomy will have a 5-yearsurvival of about 25%, while those that recur at any oth-er distant site carry a much poorer prognosis of about5%.110 The surgical options for patients with metasta-tic melanoma will generally fall into three categories:curative, palliative or immunotherapeutic. It is veryimportant that detailed discussions are undertaken
with the patient in order to address the ultimate intentof surgical intervention with a special focus upon therealistic view of expected complications, outcomesand goals.
Curative attempts with surgery for metastaticmelanoma should carefully weigh the risks of thesurgery against the potential benefits. Recent data onthe surgical management of metastatic melanoma notethat certain factors are associated with an improvedoverall survival: 1) ability to achieve a complete sur-gical resection with negative margins; 2) location of theinitial site of metastasis; 3) the extent of metastaticdisease (single or multiple sites); 4) disease-free inter-val after surgical removal of the primary melanomaand 5) stage of initial disease.111 Favorable sites ofresection include the skin, subcutaneous tissue, lymphnodes, lung, and gastrointestinal tract, with unfavorablesites associated with metastasis to the brain, adrenal,and liver.112 Skin and subcutaneous sites had the bestlong-term outcome with a 20-30 % 5-year survivaland a median survival of 48 months. Among thepatients with metastases to unfavorable sites, there
Figure 14.—Intraoperative photograph of a palliative resection for bulky metastatic melanoma. A) Initial presentation with bulky primary lesion in pla-ce and massive bilateral inguinal adenopathy. This patient had difficulty wearing pants. B) Intraoperative photograph demonstrating a bulky inguinallymph node. C) Postoperative appearance 6 weeks after resection. The patient ultimately succumbed to diffuse visceral metastatic disease 1 yearlater.

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 191
THE SURGICAL MANAGEMENT OF CUTANEOUS MELANOMA RIKER
were no 5-year survivors, with a median survival ofonly 18.2 months. In reality, the overall survival andpossible long-term outcomes for most patients withstage IV disease seems to ultimately depend upon theintrinsic biologic activity and nature of the melanomaitself.
Often, surgical intervention for metastatic melanomais performed for palliation, with the primary goal torelieve identifiable symptoms directly caused by grow-ing and metastatic tumor deposits. For instance, patientswill often present with bulky, recurrent lym-phadenopathy of the axilla or groin that cause severepain in addition to motor and sensory limitations of theinvolved extremity (Figure 14). Other sites for suc-cessful surgical palliation may include a single metasta-tic deposit within the gastrointestinal tract that is caus-ing either bleeding or a high-grade obstruction. Singlemetastatic deposits within the brain can also be resect-ed for palliative intent, as waiting may result in sur-rounding brain edema that may be fatal if left untreat-ed. However, the outcome for such patients has beenuniformly dismal, with a median survival of <1 year.112
Careful consideration should be given to any palliativesurgical intervention, weighing the benefits of surgeryin alleviating the symptoms against the true morbidi-ty and complexity of the surgical procedure.
The last type of surgical intervention to be per-formed for patients with stage IV melanoma can bestbe described as cytoreductive immunotherapeuticsurgery. An immunotherapeutic surgical approach hasbeen described, whereby complete cytoreductivesurgery is hypothesized to play a central role in elim-inating the tumor burden. This, in turn, allows for animproved overall function of the host anti-tumorimmune response.113 It is thought that removing thetumor burden, or “tumor cell factory” as described byMorton et al, through complete cytoreductive surgicalresection may allow the host immune system to over-come tumor-induced immunosuppression.113 Indeed,Morton has recently described his results from thepremature closure of the onamelatucel-L (Canvaxin)trial, designed to assess the efficacy of this vaccine inboth stage III and IV melanoma patients.
Although both trials were closed to further accrualearly due to an interim analysis that revealed no prob-able efficacy over placebo, some very interesting resultswere nonetheless found in stage IV patients. The designof the Canvaxin trial for stage IV patients requiredthat all patients receive definitive surgical removal of
all metastatic disease prior to entry into the trial.Although their was no advantage to receiving the Can-vaxin vaccine over placebo, they found that a remark-ably high 40% of all patients (in both arms) were aliveat 5 years, suggesting that prolonged survival may bedue not to the vaccine, but instead to complete surgi-cal resection of metastatic disease.114 It is clear that ourunderstanding of the tumor biology of melanomaremains limited, however, there is a suggestion thatcomplete surgical resection of metastatic disease mayplay an important role in long-term survival in select-ed patients with stage IV disease.
Multidisciplinary care of the melanoma patient
The multidisciplinary care of the melanoma patientbegins with the initial diagnosis. Once the definitivebiopsy has been performed, usually in the office, everyattempt should be made for a subsequent follow-upvisit to thoroughly discuss the pathology results and toexamine the biopsy site for signs of infection and theoverall healing process. We believe it is in poor formto provide such results over the telephone, often del-egated to a member of the office staff that may be ill-prepared for questions and or emotional outbursts asa result of a patient receiving the diagnosis of cancerover the telephone. Every attempt should be made bythe treating physician to sit down with the patient (andfamily members) in a quiet and comfortable settingand thoroughly review the entire pathology report indetail, allowing the patient to comprehend the resultsand possibly ask pertinent questions about their situ-ation.
The care of the patient with melanoma can be com-plex, often requiring the opinions of peers from mul-tiple specialties. The management of a thin primarymelanoma that simply requires a 5 mm or 1 cm exci-sion is fairly straightforward. However, a consider-able higher level of expertise and complexity are addedto the patient with an intermediate or thick primarymelanoma. The discussion must be understandable tothe patient, no matter what their educational back-ground, and encompass a global perspective on whatlies ahead for them. This, of course, should include athorough discussion as to the surgical treatment optionsas well as the possible pathological outcomes and itsimplications in regards to further surgical or medicaltherapy. In regards to the planned surgical intervention,all options should be discussed, focusing on the cur-

RIKER THE SURGICAL MANAGEMENT OF CUTANEOUS MELANOMA
192 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
rent data and literature that support the procedure. Adetailed explanation of SLNB and other such stagingprocedures should be given, including all of the pos-sible risks and benefits of the procedure.
We encourage the participation of all family mem-bers in these discussions, often providing an extra “setof ears” and to ask intuitive questions that the patientmay not ask. Ample time should be given for the patientand family to ask questions about the surgery and alsoabout the “bigger picture” of their disease. Suchdetailed conversations are very important to alleviatethe fears and to dispel many of the common mythsassociated with cancer surgery. It is important toremember that the patient with melanoma, or any can-cer for that matter, can be emotionally distraught oversuch news and may not be focused on the discussionat hand. Receiving the diagnosis of cancer is a lifechanging event and it is imperative that the surgeon becognizant of the emotional issues at hand, providing fora compassionate and supportive environment for thepatient and family members.
The patient’s case will often be presented in detailat a weekly multidisciplinary cutaneous oncology con-ference, attended by the radiologist (radiographic inter-pretation of studies), dermatopathologist (review ofall biopsy slides), medical oncologists (adjuvant optionsfor appropriate patients), surgical oncologists (expertsurgical opinions), radiation oncology (expert opin-ion on adjuvant radiation therapy) and dermatologist(whom may have done the original biopsy). Otherimportant members of the multidisciplinary teamshould include the clinical and research nurses, socialworkers, physician extenders and a recorder for eachmeeting that will provide written documentation ofeach weekly proceeding.
Many hospitals and institutions will not have theluxury of such coordinated care of the melanomapatient. Indeed, there are only a handful of places in theUnited States able to provide such comprehensive carewithin a single institution, while the vast majority ofmelanoma patients will be seen outside of such a set-ting. It is our recommendation that patients that arediagnosed with intermediate and thick primarymelanomas be referred to an institution with an estab-lished record of multidisciplinary care of the melanomapatient, such as an NCI-designated cancer center. If thegeographic location does not allow for this, referralto an academic medical center may benefit the patientas most institutes will have at least a few physicians
with a special interest and training for melanomapatients. Optimally, this would include a surgical oncol-ogist who is well versed in the latest surgical treat-ment options and who is up-to-date on the most recentliterature in regards to the surgical management ofmelanoma.
Conclusions
Receiving the diagnosis of melanoma is a life-chang-ing event for the patient as well for all of the familymembers involved. The spectrum of mental distressrelated to this diagnosis is highly variable, rangingfrom acceptance and rapid treatment to overt stages ofanger and depression for more advanced cases. Wemust never forget the melanoma patient in all of thisdiscussion, for it is impossible for those of us who are“cancer-free” to experience the emotional rollercoasterthat many, if not all, will experience. In regards to theutility of SLNB in treating patients with melanoma, itis clear that they do not accept a “wait and see”approach supported by other physicians. Patientsshould clearly understand that the primary role is oneof accurate staging, with most patients overwhelmingchoosing to undergo this procedure. Regardless ofsuch deep-seeded beliefs about SLNB, the diagnosticutility of SLNB is unmatchable, firmly established asa necessary part of the discussion that we have on a dai-ly basis with our patients. Lastly, the surgical man-agement of the melanoma patient continues to evolveas we improve our understanding of the complex inter-actions that are occurring within the tumor microen-vironment and the host immune system.
Riassunto
Gestione chirurgica del melanoma cutaneoIl melanoma è un carcinoma enigmatico che può essere
mortale in qualsiasi sua forma se non identificato o misco-nosciuto. Negli ultimi 10 anni l’incidenza del melanoma staaumentando rapidamente, essendoci una multifattorialitàcombinata con diversi fattori di rischio noti sia ambientali checomportamentali. Sebbene spesso sovrastimato in terminidi importanza, non si può dimenticare che il maggior impat-to che riusciremo ad avere sulla sopravvivenza globale saràil risultato di nuovi sforzi tesi all’identificazione precoce ealla prevenzione del melanoma. Dal momento che il mela-noma si diffonde per via linfatica o ematogena, la sopravvi-venza globale di tutti i pazienti diminuisce drammaticamente

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 193
THE SURGICAL MANAGEMENT OF CUTANEOUS MELANOMA RIKER
rispetto a quella notevole di quei pazienti che si sottopongonoall’asportazione del melanoma in situ con ampi margini diescissione. Infatti, la gestione chirurgica del melanoma èmutata radicalmente negli ultimi 20 anni. Siamo stati guidatidai risultati di studi clinici ben disegnati, prospettici, ran-domizzati, che hanno evidenziato l’importanza dell’esten-sione dell’asportazione chirurgica del melanoma primitivoche, se ottimale, è in grado di modificare e di raffinare ilmodo con il quale gestiamo chirurgicamente tali pazienti.Studi ulteriori hanno definitivamente sottolineato l’effica-cia dell’asportazione linfonodale elettiva e studi recenti stan-no dimostrando il ruolo centrale della linfoadenectomiaselettiva nella gestione del drenaggio linfonodale. Gli auto-ri discutono tutti questi aspetti ed altri, fornendo dati basatisull’evidenza a favore (o contro) l’attuale modo di pensaredei chirurghi e del processo decisionale. Alcuni aspetti resta-no ancora controversi a causa della scarsità di dati basatisull’evidenza a favore del loro utilizzo. Altri studi in corsopotranno guidarci ulteriormente nella gestione chirurgicadel melanoma. Infine, gli autori prendono in considerazio-ne i progressi chirurgici del passato e come possiamo impa-rare da essi. Tutto questo guiderà su come procedere e avan-zare in futuro nell’ambito di questo settore.PAROLE CHIAVE: Melanoma - Metastasi - Biopsia linfonodosentinella - Linfoscintigrafia - Dissezione linfonodale - Inte-ressamento multidisciplinare - Chirurgia citoriduttiva.
References
1. Snow H. Melanotic cancerous disease. Lancet 1892;2: 872.2. Urteaga OB, Pack GT. On the antiquity of melanoma. Cancer
1967;19:607-10.3. Home Sir Everard. Observations on Cancer. London: 1805.p.48.4. Laennec RTH. Sur les melanoses. Bulletin de la Faculte de Medici-
ne de Paris 1812;1:2.5. Carswell R. Melanosis. In Cyclopedia of Practical Medicine. London
3:92, 1834.6. Norris W. Case of fungoid disease. Edinburgh Med. Surg J
1820;16:562.7. Norris W. Eight cases of melanosis with pathological and therapeu-
tical remarks on that disease. Longman, Brown, Green, Longmanand Roberts, London, UK, 1857.
8. Silvers DN. On the subject of primary cutaneous melanoma: an histo-rical perspective. Progress in Surgical Pathology Volume IV 1982;277.
9. Pemberton O. Observations on the history, pathology and treatmentof cancerous diseases 8. London: Churchill; 1858.
10. Eve F. Lecture on melanoma. Practitioner 1903;70:165.11. Handley WS. The pathology of melanotic growths in relation to their
operative treatment (II). Lancet 1907;1:996-1001.12. Wallace H, Clark Jr MD. A Biography and Annotated Bibliography.
J Invest Dermatol 1993;100:246-50.13. Clark WH Jr, From L, Bernardino EA, Mihm MC Jr. The histogene-
sis and biologic behavior of primary human malignant melanomas ofthe skin. Cancer Res 1969;29:705-27.
14. Breslow A. Thickness, cross-sectional areas and depth of invasion inthe prognosis of cutaneous melanoma. Ann Surg 1970;172:902-8.
15. Breslow A, Cascinelli N, van der Esch EP, Morabito A. Stage I mela-noma of the limbs: assessment of prognosis by levels of invasion andmaximum thickness. Tumori 1978;64:273-8.
16. Morton D, Cagle L, Wong J, et al. Intraoperative lymphatic mapping
and selective lymphadenectomy: technical details of a new procedu-re for clinical stage I melanoma. Presented at the Annual Meeting ofthe Society of Surgical Oncology; Washington, DC; 1990.
17. Morton DL, Wen DR, Wong JH, Economou JS, Cagle LA, Storm FKet al. Technical details of intraoperative lymphatic mapping for earlystage melanoma. Arch Surg 1992;127:392-9.
18. Morton DL, Wanek L, Nizze JA, Elashoff RM, Wong JH. Improvedlong-term survival after lymphadenectomy of melanoma metastatic toregional nodes: Analysis of prognostic factors in 1134 patients fromthe John Wayne Cancer Clinic. Ann Surg 1991;214:491-9.
19. Dudley ME, Rosenberg SA. Adoptive -cell-transfer therapy for thetreatment of patients with cancer. Nat Rev Cancer 2003;3:666-75.
20. Dudley ME, Wunderlich J, Nishimura MI,Yu D,Yang JC, Topalian SLet al. Adoptive transfer of cloned melanoma-reactive T lymphocytesfor the treatment of patients with metastatic melanoma. J Immunother2001;24:363-73.
21. Dudley ME, Wunderlich JR, Yang JC, Hwu P, Schwartzentruber DJ,Topalian SL et al. A phase I study of non-myeloablative chemotherapyand adoptive transfer of autologous tumor antigen-specific T lymphocy-tes in patients with metastatic melanoma. J. Immunother 2002;25:243-51.
22. Rosenberg SA,Yannelli JR,Yang JC, Topalian SL, SchwartzentruberDJ, Weber JS et al. Treatment of patients with metastatic melanomawith autologous tumor-infiltrating lymphocytes and interleukin 2. J NatlCancer Inst 1994;86:1159-66.
23. Rosenberg SA, Aebersold P, Cornetta K, Kasid, A, Morgan RA, MoenR et al. Gene transfer into humans: immunotherapy of patients withadvanced melanoma, using tumor-infiltrating lymphocytes modifiedby retroviral gene transduction. N Engl J Med 1990;323:570-8.
24. Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwart-zentruber DJ et al. Cancer regression and autoimmunity in patients afterclonal repopulation with antitumor lymphocytes. Science 2002;298:850-4.
25. Rosenberg SA, Dudley ME. Cancer regression in patients with meta-static melanoma after the transfer of autologous antitumor lymphocy-tes. Proc Natl Acad Sci 2004;101:14639-45.
26. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC,Sherry RM et al. Cancer Regression in Patients after Transfer ofGenetically Engineered Lymphocytes. Science 2006;314:126-9.
27. Salopek TG, Slade J, Marghoob AA, Rigel DS, Kopf AW, Bart RS etal. Management of cutaneous malignant melanoma by dermatologi-sts of the American Academy of Dermatology. I. Survey of biopsy prac-tices of pigmented lesions suspected as melanoma. J Am Acad Der-matol 1995;33:441-50.
28. Kanzler MH, Mraz-Gernhard S. Primary cutaneous malignant mela-noma and its precursor lesions: diagnostic and therapeutic overview.J Am Acad Dermatol 2001;54:260-76.
29. Thomas L, Tranchard P, Berard F, Secchi T, Colin C, Moulin G.Semiological value of ABCD criteria in the diagnosis of cutaneous pig-mented tumors. Dermatology 1998;197:11-7.
30. Weinstock MA. Early detection of melanoma. J Am Med Assoc2000;284: 886-9.
31. Nachbar F, Soltz W, Merkle T, Cognetta AB, Vogt T, Landthaler M etal. The ABCD rule of dermatoscopy. High prospective value in the dia-gnosis of doubtful melanocytic skin lesions. J AmAcad Dermatol1994;30:551-9.
32. McGovern TW, Litaker MS. Clinical predictors of malignant pig-mented lesions. A comparison of the Glasgow seven-point checklistand the American Cancer Society's ABCD of pigmented lesions. JDermatol Surg Oncol 1992;18:22-6.
33. Argenziano G, Soyer H. Dermoscopy of pigmented skin lesions - avaluable toll for early diagnosis of melanoma. Lancet Oncol2001;2:443-9.
34. Argenziano G, Fabbrocini G, Carli P, DeGiogi V, Sammarco E, Del-fino M. Epiluminescence microscopy for the diagnosis of doubtfulmelanocytic skin lesions. Comparison of the ABCD rule of derma-toscopy and a new 7-point checklist based on pattern analysis. ArchDermatol 1998;134:1563-70.

RIKER THE SURGICAL MANAGEMENT OF CUTANEOUS MELANOMA
194 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
35. Breslow A. Prognostic factors in the treatment of cutaneous melano-ma. J Cut Pathol 1979;6:208-12.
36. Clark WH Jr, Elder DE, Guerry D, Braitman LE, Trock BJ, SchultzE et al. Model predicting survival in stage I melanoma based on tumorprogression. J Natl Cancer Inst 1989;81:1893-904.
37. Farmer ER. Why a skin biopsy? Arch Dermatol 2000;136: 779-80.38. Herlyn M, Ferrone S, Ronai Z, Finerty J, Pelroy R, Mohla S. Mela-
noma biology and progression. Cancer Res 2001;61:4642-3.39. Johnson TM, Sondak VK. Melanoma margins: the importance and need
for more evidence-based trials. N Engl J Med 2004;350:757-66.40. Landthaler M, Braun-Falco O, Leitl A, Konz B, Holzel D. Excisional
biopsy as the first therapeutic procedure versus primary wide local exci-sion of malignant melanoma. Cancer 1989;64:1612-6.
41. Levi F, Randimbison L, LaVecchia C, Te VC, Frandeschi S. Progno-stic factors for cutaneous malignant melanoma in Vaud, Switzerland.Int J Cancer 1998;78:315-9.
42. Macy-Roberts E, Ackerman AB. A critique of techniques for biopsyof clinically suspected malignant melanomas. Am J Dermatopathol1982;4:391-8.
43. Riker AI, Glass F, Perez I, Cruse CW, Messina J, Sondak VK. Cuta-neous melanoma: methods of biopsy and definitive surgical excision.Dermatol Ther 2005;18:387-93.
44. Roses D. Biopsy technique for suspected melanoma. Pigment Cell1985;7:8-17.
45. Balch CM, Buzaid AC, Soong SJ, Atkins MB, Cascinelli N, Coit DGet al. Final version of the American Joint Committee on Cancer sta-ging system for cutaneous melanoma. Cancer 2001;88:3635-48.
46. Balch CM, Soong SJ, Gershenwald JE, Thompson JF, Reintgen DS,Cascinelli N et al. Prognostic factors analysis of 17,600 melanomapatients. Validation of the American Joint Committee on Cancer mela-noma staging system. J Clin Oncol 2001;19:3622-34.
47. Dresel A, Kuhn JA, McCarty TM. Sentinel node biopsy site used asa full thickness skin graft donor for cutaneous melanoma. Am J Surg2002;184:176-8.
48. Balch CM, Urist MM, Karakousis CP, Smith TJ, Temple WJ,Drzewiecki K et al. Efficacy of 2-cm surgical margins for intermediatethickness melanomas (1-4 mm). Results of a multi-institutional ran-domized surgical trial. Ann Surg 1993;218:262-7.
49. Balch CM, Soong SJ, Smith T, Ross MI, Urist MM, Karakousis CPet al. Long term results of a prospective trial comparing 2 cm vs. 4 cmexcision margins for 740 patients with 1-4 mm melanomas. Ann SurgOncol 2001;8:101-8.
50. Khayat D, Rixe O, Martin G, Soubrane C, Banzet M, Bazex JA et al.Surgical margins in cutaneous melanoma (2 cm versus 5 cm for lesionsmeasuring less than 2.1 mm thick). Cancer 2003;97:1941-6.
51. Cohn-Cedermark G, Rutqvist LE, Andersson R, Breivald M, IngvarC, Johansson H et al. Long term results of a randomized study bythe Swedish Melanoma Study Group on 2 cm versus 5 cm resectionmargins for patients with cutaneous melanoma with a thickness of0.8 to 2.0 mm. Cancer 2000;89:1495-501.
52. Tanabe KK, Reintgen DS, Balch CM. Local recurrences and theirmanagement. In: Balch CM, Houghton AN, Sober AJ, Soong SJ eds.Cutaneous Melanoma. 4th Edition. St. Louis, Missouri: Quality Medi-cal Publishing; 2003.p.263-73.
53. Veronesi U, Cascinelli N, Adamus J, Balch C, Bandiera D, BarchukA et al. Thin stage I primary cutaneous malignant melanoma: com-parison of excision with margins of 1 or 3 cm. N Engl J Med1988;322:1159-62.
54. Santinarmi M, Maurichi A, Patuzzo R, Pennacchioli E, Cascinelli Net al. Impact of clinical trials on the treatment of melanoma. SurgOncol Clin N Am 2001;10:935-47.
55. Thomas JM, Newton-Bishop J, A'Hern R, Coombes G, Timmons M,Evans J et al. Excision margins in high-risk malignant melanoma. NEngl J Med 2004;350:757-66.
56. Heaton KM, Sussman JJ, Gershenwald JE, Lee JE, Reintgen DS, Man-sfield PF et al. Surgical margins and prognostic factors in patients withthick (>4 mm) primary melanoma. Ann Surg Oncol 1998;5:322-8.
57. Ng AK, Jones WO, Shaw JH. Analysis of local recurrence and opti-mizing excision margins for cutaneous melanoma. Br J Surg2001;88:137-42.
58. Dong XD, Tyler D, Johnson JL, DeMatos P, Seigler HF. Analysis ofprognosis and disease progression after local recurrence of melano-ma. Cancer 2000;88:1063-71.
59. Soong S, Harrison RA, McCarthy WH, Urist MM, Balch CM. Factorsaffecting survival following local, regional, or distant recurrence fromlocalized melanoma. J Surg Oncol 1998;67:228-33.
60. Urist MM, Balch CM, Soong SJ, Shaw HM, Milton GW, MaddoxWA. The influence of surgical margins and prognostic factors pre-dicting the risk of local recurrence in 3445 patients with primary cuta-neous melanoma. Cancer 1985;55:1398-402.
61. Reintgen DS, Cox C, Slingluff CL, Seigler HF. Recurrent malignantmelanoma: the identification of prognostic factors to predict survival.Ann Plast Surg 1992;28:45-9.
62. Kalady MF, White RR, Johnson JL, Tyler DS, Seigler HF. Thin mela-nomas: Predictive lethal characteristics from a 30-year clinical expe-rience. Ann Surg 2003;238: 528-37.
63. Kenady DE, Brown BW, McBride CM. Excision of underlying fasciawith a primary malignant melanoma: effect on recurrence and survi-val rates. Surgery 1982;92:615-8.
64. Sondergaard K, Schou G. Therapeutic and clinico-pathological fac-tors in the survival of 1,469 patients with primary cutaneous malignantmelanoma in clinical stage I. A multivariate regression analysis. Vir-chows Arch Am Pathol Anat Histopathol 1985;408:249-58.
65. Temple CLF, Arlette JP. Mohs micrographic surgery in the treatmentof lentigo maligna and melanoma. J Surg Oncol 2006;94:287-92.
66. Bricca GM, Brodland DG, Ren D, Zitelli JA. Cutaneous head andneck melanoma treated with Mohs micrographic surgery. J Am AcadDermatol 2005;52:92-100.
67. Cruse CW, Wells KE, Reintgen DS Treatment of the primary in mali-gnant melanoma of the skin. Ann Plast Surg 1992;28:22-5.
68. Balch CM, Soong SJ, Ross MI, Urist MM, Karakousis CP, Temple WJet al. Long-term results of a multi-institutional randomized trial com-paring prognostic factors and surgical results for intermediate thick-ness melanomas (1-4mm): Intergroup Melanoma Surgical Trial. AnnSurg Oncol 2000;7:87-97.
69. Morton DL, Wanek L, Nizze JA, Elashoff RM, Wong JH. Improvedlong-term survival after lymphadenectomy of melanoma metastatic toregional nodes. Ann Surg 1991;214:491-501.
70. Warso MA, Das Gupta TK. Melanoma recurrence in a previouslydissected lymph node basin. Arch Surg 1994;129:252-5.
71. Karakousis CP. Therapeutic lymph node dissection in malignant mela-noma. Ann Surg Oncol 1998;5:473-8.
72. Meyer T, Merkel S, Gohl J, Hohenberger W. Lymph node dissectionfor clinically evident lymph node metastases of malignant melanoma.Eur J Surg Oncol 2002;28:424-30.
73. Cascinelli N, Morabito A, Santinami M, Mackie RM, Belli F. Imme-diate or delayed dissection of regional nodes in patients with melanomaof the trunk: a randomized trial. Lancet 1998;351:793-6.
74. Morton DL, Hoon DS, Cochran AJ, Turner RR, Essner R, TakeuchiH et al. Lymphatic mapping and sentinel lymphadenectomy for earlystage melanoma: therapeutic utility and implications of nodal microa-natomy and molecular staging for improving the accuracy of detec-tion of nodal micrometastases. Ann Surg 2003;238:538-49.
75. Johnson TM, Sondak VK, Bichakjian CK, Sabel MS. The role of sen-tinel lymph node biopsy for melanoma: Evidence assessment. J AmAcad Dermatol 2005;54:19-27.
76. McMasters KM, Reintgen DS, Ross MI. Sentinel lymph node biopsyfor melanoma: Controversy despite widespread agreement. J ClinOncol 2001;19:2851-5.
77. McMasters KM. What good is sentinel lymph node biopsy for melanomaif it does not improve survival? Ann Surg Oncol 2004;11:810-2.
78. Medalie NS, Ackerman B. Sentinel lymph node biopsy has no bene-fit for patients with primary cutaneous melanoma metastatic to alymph node: An assertion based on comprehensive, critical analysis,Part I. Am J Dermatopathol 2003;25:399-417.

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 195
THE SURGICAL MANAGEMENT OF CUTANEOUS MELANOMA RIKER
79. Medalie NS, Ackerman B. Sentinel lymph node biopsy has no bene-fit for patients with primary cutaneous melanoma metastatic to alymph node: An assertion based on comprehensive, critical analysis,Part II. Am J Dermatopathol 2003;25:473-84.
80. Young SE, Martinez SR, Faries MB, Essner R, Wanek LA, Morton DL.Can surgical therapy alone achieve long-term cure of melanoma meta-static to the regional nodes. Cancer J 2006;12:207-11.
81. Morton DL. Interim analysis of the multicenter selective lymphade-nectomy trial (MSLT-I) in clinical stage I melanoma. Presented atthe American Society of Clinical Oncology, May 14th, 2005.
82. Morton DL, Thompson JF, Cochran AJ, Mozzillo N, Elashoff R,Essner R et al. Sentinel-node biopsy or nodal observation in melanoma.N Engl J Med 2006;355:1307-17.
83. Wick MR, Patterson JW. Sentinel node biopsies for cutaneous mela-noma. Am J Surg Pathol 2005;29:412-4.
84. Gershenwald JE, Thompson W, Mansfield PF, Lee JE, Colome MI,Tseng CH et al. Multi-institutional melanoma lymphatic mappingexperience: the prognostic value of sentinel lymph node status in 612stage I or II melanoma patients. J Clin Oncol 1999;17:976-83.
85. Wong SL, Brady MS, Busam KJ, Coit DG. Results of sentinel lymphnode biopsy in patients with thin melanoma. Ann Surg Oncol2006;13:302-9.
86. Kalady MF, White RR, Johnson JL, Tyler DS, Seigler HF. Thin mela-nomas: predictive lethal characteristics from a 30-year clinical expe-rience. Ann Surg 2003;238:528-35.
87. Stitzenberg KB, Groben PA, Stern SL. Indications for lymphatic map-ping and sentinel lymphadenectomy in patients with thin melanoma(Breslow's thickness < 1 mm). Ann Surg Oncol 2004;11:900-6.
88. Sondak VK, Taylor JMG, Sabel MS, Wang Y, Lowe L, Grover AC etal. Mitotic rate and younger age are predictors of sentinel lymph nodepositivity: Lessons learned from the generation of a probabilisticmodel. Ann Surg Oncol 2004;11:247-58.
89. Kesmodel SB, Karakousis Gc, Botbyl JD, Canter RJ, Lewis RT, WahlPM, et al. Mitotic rate as a predictor of sentinel lymph node positivityin patients with thin melanoma. Ann Surg Oncol 2005;12:449-58.
90. Azzola MF, Shaw HM, Thompson JF, Soong SJ, Scolyer RA, WatsonGF et al. Tumor mitotic rate is a more powerful prognostic indicator thanulceration in patients with primary cutaneous melanoma: an analysis of3661 patients from a single center. Cancer 2003;97:1488-98.
91. Puleo CA, Messina JL, Riker AI, Glass LF, Nelson C, Cruse CW etal. Sentinel node biopsy for thin melanoma: Which patients should beconsidered? Cancer Control 2005;12:230-5.
92. Thompson JF, Shaw HM. Is sentinel lymph node biopsy appropriatein patients with thin melanomas: Too early to tell? Ann Surg Oncol2006;13:279-81.
93. Urist MM, Maddox WA, Kennedy JE, Balch CM. Patient risk fac-tors and surgical morbidity after regional lymphadenectomy in 204melanoma patients. Cancer 1983;51:2152-6.
94. Tonouchi H, Ohmori Y, Kobayashi M, Konishi N, Tanaka K, Mohri Yet al. Operative morbidity associated with groin dissections. SurgToday 2004;34:413-8.
95. Hughes TM, A'Hern RP, Thomas JM. Prognosis and surgical mana-gement of patients with palpable inguinal lymph node metastasesfrom melanoma. Br J Surg 2000;87:892-901.
96. Shen P, Conforti AM, Essner R, Cochran AJ, Turner RR, MortonDL. Is the node of Cloquet the sentinel node for the iliac/obturatornode group? Cancer J 2000;6:93-7.
97. Coit DG, Brennan MF. Extent of lymph node dissection in melano-ma of the trunk and lower extremity. Arch Surg1989;124:162-6.
98. Strobbe LJ, Jonk A, Hart AA, Peterse JL, Wobbes T, Nieweg OE et al.The value of Cloquet's node in predicting melanoma nodal metastasesin the pelvic lymph node basin. Ann Surg Oncol 2006;8:209-14.
99. Essner R, Scheri R, Kavanagh M, Torisu-Itakura H, Wanek LA, Mor-ton DL. Surgical management of the groin lymph nodes in melano-ma in the era of sentinel lymph node dissection. Arch Surg2006;141:877-84.
100. Karakousis CP, Emrich LJ, Rao U. Groin dissection in malignantmelanoma. Am J Surg 1986;152:491-5.
101. Beitsch P, Balch C. Operative morbidity and risk factor assessmentin melanoma patients undergoing inguinal lymph node dissection. AmJ Surg 1992;164:462-6.
102. Creech O, Krementz ET, Ryan RF, Winblad JN. Chemotherapy of can-cer: regional perfusion utilizing an extracorporeal circuit. Ann Surg1958;148:616-32.
103. Grunhagen DJ, deWilt JHW, vanGeel AN, Eggermont AMM. Isola-ted limb perfusion for melanoma patients-a review of its indicationsand the role of tumor necrosis factor-alpha. EJSO 2006;32:371-80.
104. Cornett WR, McCall LM, Petersen RP, Ross MI, Briele HA, NoyesRD et al. Randomized multicenter trial of hyperthermic isolatedlimb perfusion with melphalan alone compared with melphalan plustumor necrosis factor: American College of Surgeons OncologyGroup Trial Z0020. J Clin Oncol 2006;24:4196-201.
105. Noorda EM, Vrouenraets BC, Nieweg OE, vanCoevorden F, KroonBBR. Isolated limb perfusion: What is the evidence for its use? AnnSurg Oncol 2004;11:837-45.
106. Kroon BBR, Noorda EM, Vrouenraets BC, Nieweg OE. Isolatedlimb perfusion for melanoma. J Surg Oncol 2002;79:252-5.
107. Thompson JF, deWilt JHW. Isolated limb perfusion in the manage-ment of patients with recurrent limb melanoma:An important but limi-ted role. Ann Surg Oncol 2001;8:564-5.
108. Thompson JF, Kam PC, Waugh RC, Harman CR. Isolated limb infu-sion with cytotoxic agents: A simple alternative to isolated limb per-fusion. Semin Surg Oncol 1998;14:238-47.
109. Thompson JF, Kam PC. Isolated limb infusion for melanoma: A sim-ple but effective alternative to isolated limb perfusion. J Surg Oncol2004;88:1-3.
110. Allen PJ, Coit DG. The surgical management of metastatic melano-ma. Ann Surg Oncol 2002;9:762-70.
111. Wong SL, Coit DG. Role of surgery in patients with stage IV mela-noma. Curr Opin Oncol 2004;16:155-60.
112. Essner R, Lee JH, Wanek LA, Itakura H, Morton DL. Contemporarysurgical treatment of advanced-stage melanoma. Arch Surg2004;139:961-7.
113. Morton DL, Ollila DW, Hsueh EC, Essner R, Gupta RK. Cytore-ductive surgery and adjuvant immunotherapy: A new managementparadigm for metastatic melanoma. Cancer J Clin 1999;49:101-6.
114. Morton DL. Surgery prolongs survival in stage IV melanoma. Sym-posium at the Society of Surgical Oncology, San Diego, 2006.

G ITAL DERMATOL VENEREOL 2007;142:197-206
Sentinel lymph node biopsy for melanoma:therapeutic procedure or diagnostic test?
Although sentinel lymph node (SLN) biopsy is considered stan-dard of care by many surgical oncologists and dermatologi-sts, it remains controversial among others. Clinical practitio-ners in both surgery and dermatology have used the same avai-lable evidence to both support and refute the sentinel nodehypothesis and the role SLN biopsy should play in the mana-gement of cutaneous melanoma. Much of the disagreementcenters on whether one views SLN biopsy as a therapeuticintervention meant to improve survival or a diagnostic testmeant to stratify risk and select patients for further therapy.This article will review the available data, including the mostrecent data from the Multicenter Selective LymphadenectomyTrial-I (MSLT-I), the first prospective randomized study ofSLN biopsy in melanoma.
KEY WORDS: Melanoma - Biopsy - Sentinel lymph node biopsy.
Although it is still the greatest source of debateamong physicians who treat melanoma, the
importance of the regional nodal basin in the man-agement of melanoma was recognized in the late1800’s.1 Today, the great majority of patients with pri-mary cutaneous melanoma present with clinically neg-ative (nonpalpable) regional lymph node basins, but1/5th of these patients harbor occult regional metas-tases. Prior to the introduction of lymphatic mappingand sentinel lymph node (SLN) biopsy by Morton etal.,2 this posed a clinical dilemma. Patients and theirphysicians were faced with two choices. With the
understanding that primary melanomas often spread toregional nodal basins before metastasizing widely,some surgeons advocated elective lymph node dis-section (ELND). This approach hinged on the ideathat early clearance of tumor deposits in the regionalnodal basin could prevent subsequent disseminationand improve survival, an idea supported by retro-spective studies.3, 4 Elective node dissection, though,exposed the 80% of patients who were node-negativeto the morbidity of a nodal dissection.
The alternative approach was that of limiting nodedissections to those patients with documented metas-tases, the therapeutic lymph node dissection (TLND).Clinically node negative patients underwent wide exci-sion alone. If they developed clinically palpable nodaldisease, but were without evidence of distant disease,they underwent TLND. This spared the node negativepatients the morbidity of a lymph node dissection, butrisked the possibility that in the time period betweenthe primary excision and when the nodal recurrencebecame evident, melanoma cells may have metasta-sized systemically from the node, losing the chance forcure.
1Department of General SurgeryUniversity of Michigan Hospital
Ann Arbor, MI, USA2Division of Surgical Oncology
University of Michigan Comprehensive Cancer CenterAnn Arbor, MI
Address reprint requests to: M. S. Sabel, 3304 Cancer Center, 1500East Medical, Center Drive, Ann Arbor, MI 48109, USA.E-mail: [email protected]
C. VEMURI 1, M. S. SABEL 2
Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 197

VERUMI SENTINEL LYMPH NODE BIOPSY FOR MELANOMA: THERAPEUTIC PROCEDURE OR DIAGNOSTIC TEST?
198 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
This dilemma ultimately led to several randomizedtrials, each of which showing no overall survival advan-tage to elective node dissection, but perhaps some ben-efit among certain subsets of patients.5-8 The Inter-group Melanoma Surgical Program randomized 740stage I and II melanoma patients to ELND or obser-vation.6 While there was no difference in survivalbetween the two groups overall, in a subgroup analy-sis, ELND was seen to confer a survival benefit inpatients with nonulcerated melanomas and in patientswith tumor thickness between 1 and 2 mm. This datasuggested, although by no means proved, that theredid exist a portion of patients for whom the earlyremoval of microscopic disease from the regionalnodes would improve survival. The results of thesetrials prompted many surgeons to abandon ELNDwhile others chose to use ELND selectively. As therewas no way to specifically identify patients with micro-scopic disease, surgeons based the decision to per-form ELND on clinical features such as patient age,gender, tumor location, tumor thickness and theabsence of ulceration.
This point became moot with the introduction oflymphatic mapping and SLN biopsy, a minimally inva-sive procedure capable of identifying that very subset;patients with melanoma harboring synchronous occultmicroscopic disease in the lymph nodes. With thisprocedure, the management of melanoma changedswiftly, allowing node-negative patients to avoid unnec-essary lymphadenectomies without sacrificing accu-rate staging. Today, SLN biopsy is considered stan-dard of care by most surgical oncologists for stagingthe regional lymph nodes of patients with primarycutaneous melanomas >1 mm thickness. Patients withthin melanomas (<1 mm) have a low incidence ofregional metastases, and so SLN biopsy is not rou-tinely recommended. In some cases, however, the pres-ence of other adverse features (ulceraton, mitotic rate,young age, or Clark’s level IV or V tumors) mayprompt SLN biopsy in patients with melanoma <1mm.9, 10
Despite the clinical acceptance of SLN biopsy, itsapplication is not without controversy and there isbroad disparity of opinions regarding SLN biopsyamong dermatologists.11-14 While some of this debatemay be fueled by the shift in who is responsible for thetreatment of melanoma (although this is only a minor-ity of melanoma patients as most do not fall withinthe current guidelines for SLN biopsy15), there are
many unanswered questions and legitimate concernsthat had not been addressed by current trial data. ShouldSLN biopsy be accepted as the standard of care in themanagement of melanoma? To best answer this ques-tion, one must address several issues surrounding theprocedure and ask not only whether SLN delivers whatis expected of it, but what precisely is expected of it.
The SLN technique is designed to identify the lymphnode or nodes that accurately represent the status of thedraining nodal basin. The concept is not a new one. Inthe mid-19th century, Virchow described the concept oflymphatic drainage from a given body site to a specificlymph node. Based on studies in cats and humans withvital dye, Braithwaite first described the “glands sen-tinel” as the lymph node which drains a particulararea. In 1960, Gould described a “sentinel node” thatdirectly drained the parotid gland and proposed that aradical neck dissection should be performed if thisnode contained micrometastatic disease. And, in 1976,Cabanas suggested that the sentinel node of the peniscould be used to determine the need for regional nodedissection for penile cancer. However, the use of intra-operative lymphatic mapping to identify the sentinelnode was truly brought forward by Donald Mortonfor the treatment of malignant melanoma.
The hypothesis underpinning SLN biopsy is thatwhile the mechanism by which melanoma cells metas-tasize to the lymph nodes is a complex and difficult topredict process, the manner in which they metasta-size is orderly and definable by mapping the lymphaticdrainage from the site of the melanoma. Typically,two methods are employed for identifying the sen-tinel node; a blue dye and a radiolabelled colloid solu-tion. The radiolabelled colloid is injected 1 to 4 h pre-operatively and the blue dye is injected intradermallyat the site of the primary tumor a few minutes beforethe sentinel node biopsy incision is made. The sur-geon then uses a hand-held gamma probe to identifythe “hot spot” marking the location of the sentinelnode, thereby minimizing the size of the skin incisionneeded. Once the incision is made, the surgeon iden-tifies the sentinel node by either following blue-stainedlymphatics or by finding the areas with the highestsignal.
The prevailing argument is that identification andremoval of the SLN will accurately stage the patient.This presumes two things. First, the lymph node(s)that take up the tracers are truly the nodes most like-ly to harbor micrometastases if present. In other words,

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 199
SENTINEL LYMPH NODE BIOPSY FOR MELANOMA: THERAPEUTIC PROCEDURE OR DIAGNOSTIC TEST? VERUMI
it is highly unlikely that if the SLN is negative (has noidentifiable micrometastases) there are melanoma cellsin other lymph nodes. The second presumption is thatthe identification of melanoma cells in the SLN por-tends a poor prognosis as compared with patients whoare SLN negative. In other words, is this a true stagingprocedure that stratifies patients by projected out-come? If these two presumptions are true, then thenext question is whether there are any interventionsavailable to improve outcome among SLN positivepatients. If not, then outside of accurate staging forresearch purposes, SLN biopsy provides minimal ben-efit to the patient and is not justified outside of a clin-ical trial. If so, the final question is whether the poten-tial benefit to the patient is worth the cost and morbidityof the procedure.
Does the sentinel lymph node accurately reflectthe status of the regional basin?
The SLN biopsy typically begins with the injectionof a radiolabeled colloid tracer in 4 quadrants aroundthe melanoma or biopsy scar. Lymphoscintigraphythen demonstrates the anatomic locations of the SLN(s)in the draining basin(s). The patient comes to the oper-ating room where a fat-soluble blue dye is injected ina similar manner. The hand-held gamma probe is usedto identify the vicinity of the SLN within each basin bymeans of elevated counts and a small incision is madein the skin. Any lymph node within the basin that isblue or has blue-stained lymphatics, or has high countson the gamma probe is excised and labeled a “sen-tinel node.” Any lymph node that is clinically suspi-cious on digital examination of the basin is also excisedfor pathologic examination. The procedure is com-pleted when all blue nodes have been removed andcounts have dropped to 10% of the highest node countex vivo. This approach will identify the SLN in over97% of cases.16-18 The incision is closed and the sen-tinel node(s) are sent to pathology. An equally impor-tant aspect of the SLN biopsy is the pathologic analy-sis of the specimens. Step sectioning of the harvestednodes increases detection of micrometastases. If stepsectioning and routine hematoxylin and eosin (H&E)staining is negative for metastasis, then immunohis-tochemical staining for melanoma markers such as S-100, Melan-A, and HMB-45 is performed.19-22
Does the SLN truly reflect the lymph node status?As one can imagine, there are several missteps that
can occur during the procedure that can lead to a false-negative finding- calling the patient node-negativewhen in reality spread to the regional nodes did occur(Table I). Initial studies of the false negative rate forSLN biopsy were pathologic in nature. Patients whounderwent lymphatic mapping and SLN biopsy had acomplete node dissection so that the status of the SLNcould be compared to the status of the nonsentinellymph nodes (NSLN). Multiple studies have demon-strated that, when the SLN is negative, the likelihoodof finding disease in any NSLN is quite low.16, 23, 24
This finding, however, assumes that the 99mTc-labeledcolloid sulfur and lymphoscintigraphy accurately iden-tified all draining basins. For example, if a flankmelanoma metastasized to the inguinal nodes, but thelymphoscintogram only showed drainage to the axil-lary basin, then the absence of disease in the axillarysentinel and nonsentinel nodes does not mean that thisis a true negative.
The second measure of the accuracy of the SLN inpredicting the nodal status is the regional recurrencerate among patients who are SLN negative. Severalsingle-institution series have demonstrated a relative-ly low false-negative rate in this situation, althoughfollow-up for some of these series has been limited.23,
25-27 The most recent data comes from the MulticenterSelective Lymphadenectomy Trial-I (MSLT-I); aprospective, randomized trial comparing wide localexcision alone to wide local excision and SLN biop-sy, with complete lymph node dissection for SLN pos-itive patients.28 In this trial, the false negative rate was3.4% at 5 years.
The regional recurrence rate, however, does not
TABLE I.—Possible mechanisms of false negative sentinel lymph node(SLN) biopsy.
— 99mTc-labeled colloid sulfur and lymphoscintigraphy fail to identify cor-rect basin(s).
— Tracer travels to correct basin but moves past SLN to second-tierlymph nodes (may be related to time between injection and procedu-re).
— Tracer travels to correct basin but collects in NSLN (serial versusparallel drainage).
— Surgeon fails to remove all true sentinel nodes (background countsdo not drop to <10% of highest node ex vivo).
— Tumor emboli within lymphatics block flow of tracer into the SLN.— Clinically involved nodes, without tracer uptake, missed by surgeon.— Lack of thorough pathologic evaluation of the SLN (no step sectioning,
lack of immunohistochemistry)— Crush or cautery artifacts preclude identification of micrometastes.— Microscopic disease in lymph node not identified despite appropria-
te methods.

VERUMI SENTINEL LYMPH NODE BIOPSY FOR MELANOMA: THERAPEUTIC PROCEDURE OR DIAGNOSTIC TEST?
200 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
absolutely define the accuracy of the SLN procedureeither. For patients who have micrometastases limitedto the SLN but missed by the pathologist on histolog-ic examination, they would be false negatives butwould not clinically recur because the SLN wasexcised. In one series, 4.1% of patients who devel-oped a nodal recurrence after a negative SLN had re-evaluation of the SLN and 80% had evidence of missedoccult metastasis.27 It is certainly feasible that a por-tion of patients who did not recur also had missedoccult metastases. It is also possible that some false-negative cases may have residual microscopic diseasein a NSLN that simply didn’t recur, possibly kept incheck by the immune system or capable of spread butincapable of growth and survival. On the other hand,patients who suffer a regional recurrence may havebeen truly node negative at the time of their SLN biop-sy, but in-transit disease in the lymphatic vessels mayhave reached a node subsequent to that procedure.
Therefore, the precise accuracy of SLN biopsy couldbe questioned for either method of assessing the falsenegative rate. However, when one looks at the compi-lation of data, combining both the low rate of identi-fying disease in the NSLN on completion dissectionafter a negative SLN with the low regional recurrencerate after a negative SLN biopsy not followed byCLND, the evidence to date strongly supports the SLNhypothesis.
Does finding a positive sentinel lymph node implya worse prognosis?
While the evidence strongly suggests the SLN isthe most likely to harbor micrometastatic cells if theyexist, this is meaningless if these cells are clinicallyinsignificant. If the SLN procedure finds melanomacells that are not indicative of the metastatic potentialof the cancer, then it is of little benefit. One exampleof this might be if a great number of the foci identifiedwithin the SLN were there secondary to mechanicaldislodgement, incapable of spread on their own. Anoth-er example would be if a high percentage of patientshad melanomas capable of true spread to the lymphnodes but incapable of further growth or hematogenousspread. In these cases, SLN positive patients wouldhave a prognosis not too dissimilar to SLN negativepatients.
As with the accuracy of the procedure, the prog-nostic worth of the procedure is also strongly sup-
ported by the literature. There is an overwhelmingpreponderance of evidence that SLN status is the mostsignificant factor for clinical outcome and is a criticalcomponent of melanoma staging.29-33 Despite the factthat SLN biopsy is often restricted to patients withtumors between 1 and 4 mm in depth, studies havevalidated the prognostic significance of the SLN biop-sy in both thin (<1 mm) and thick (>4 mm)melanoma.34-38 In the MSLT-I trial, the status of theSLN was the most important prognostic factor, with the5 year survival dropping from 90% among SLN neg-ative patients to 72% among SLN positive patients.28
While the literature supports that the SLN proce-dure is both accurate and provides the most importantprognostic information available, this still does not initself justify the procedure (outside of the context of aclinical trial or for research purposes). Prognostic signsoffer an accurate assessment of the likelihood of recur-rence and death so physicians and patients make choic-es as to the relative risks and benefits of either surgeryor adjuvant therapy. But what if there are no choices foradditional therapy? While one might argue that havingaccurate prognostic information is beneficial topatients, the true benefit of prognostic and predictivemarkers lies in our ability to intervene in those patientswith potentially poor outcomes. Simply knowingpatient A has a worse outcome than patient B is rela-tively meaningless unless we can offer patient A some-thing to improve that outcome.
When one talks of staging cancer patients, it is typ-ically with adjuvant therapy in mind. While theiroptions are limited, high-risk melanoma patients dohave one adjuvant therapy available to them; high-dose interferon (HDI) with interferon alpha-2b (Intron-A). Adjuvant HDI is a controversial topic. While threestudies have clearly demonstrated a benefit to disease-free survival for HDI, only two of these three studiesdemonstrated an overall survival benefit 39, 40 whileone found no survival advantage.41 Furthermore, anideal subset of high-risk melanoma patients who ben-efit the most from HDI has not been identified. A dis-cussion of the relative pros and cons of adjuvant IFNis beyond the scope of this paper.42-45 However, if afterreviewing the data one agrees that the evidence supportsat least offering patients adjuvant HDI, then the prog-nostic information provided by the SLN procedure iscrucial.
The other avenue by which SLN biopsy may providea survival benefit to patients is through additional

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 201
SENTINEL LYMPH NODE BIOPSY FOR MELANOMA: THERAPEUTIC PROCEDURE OR DIAGNOSTIC TEST? VERUMI
surgery, specifically a completion lymph node dis-section (CLND) in patients found to harbormicrometastases. If one excludes any potentialimprovements in survival obtained with HDI, then thetrue benefit of identifying node-positive patients is ifsurvival is improved by the early eradication of that dis-ease, as compared with delaying surgery until thosepatients would recur.
Is the sentinel lymph node biopsy a therapeuticprocedure that improves survival?
There are several arguments that one can make infavor of sentinel node biopsy irrespective of whetheroverall survival is improved. SLN biopsy will provideaccurate staging for appropriate counseling, decision-making and prognostication. In this way, surveillancepatterns may be adjusted accordingly, increasing sur-veillance of high risk individuals while the 80% ofpatients who are found to be node negative can bespared some of the anxiety (and the health care systemcan be spared some of the cost) of an intensive sur-veillance schedule. There is indirect evidence thatSLN biopsy and immediate CLND will provide betterregional control than delayed CLND.27, 46, 47 In addi-tion, the CLND for a positive SLN has decreased com-plications as compared with CLND for palpable dis-ease.48 As patients who recur after wide local exci-sion alone often have more advanced regional disease(multiple involved nodes, extranodal extension), theyoften require radiation therapy to optimize regionalcontrol.49 The use of SLN biopsy would decrease theneed for this, and the associated cost and morbidity.These relative advantages can be argued back andforth, however the most important question, and argu-ment for the routine application of SLN biopsy, iswhether identifying this disease at an early stage andremoving it before it is clinically apparent improvessurvival.
Another way to frame this question is whether, in theinterim of time between when the primary melanomais treated and regional metastases are identified, couldmelanoma cells have spread from the nodes to othersites and recur as distant metastases that would havebeen prevented had the nodes been excised at the timeof the primary wide excision? If the answer to thatquestion is yes, the next question is how large a sub-set of patients would this represent? Patients with clin-ically node negative melanoma essentially fall into 4
categories, only one of which would realize a survivalbenefit:
1. Patients with no metastases to the regional nodes.Obviously these patients would not experience anysurvival advantage to the SLN biopsy, and they rep-resent the overwhelming majority of patients (approx-imately 75% to 80%).
2. Patients with microscopic disease in the SLNwho have not yet metastasized but will in the time ittakes those regional mets to be clinically detectable.This is the group that does benefit from SLN biopsyand the subsequent CLND.
3. Patients with microscopic disease in the SLNwho still have no distant disease when they suffer theirregional recurrence and undergo a TLND.
4. Patients with microscopic disease in the SLNwho already have distant disease when their primarymelanoma is discovered. This is the biggest questionmark. If almost all patients with microscopic diseasein their nodes already harbor distant metastases, thengroups 2 and 3 would represent such a small fractionof the SLN positive patients that it is unlikely thatSLN biopsy and CLND for node positive patientswould impact survival in any more than a negligibleway. However, clinical evidence does not support thisnotion, as multiple prospective studies have demon-strated a significant percentage of long-term survivorswith stage III disease.
The relative distribution of patients into these fourcategories is dependent upon multiple factors. Firstand foremost is the biology of melanoma and thosefactors that may favor lymphatic versus hematoge-nous spread. These fractions will also change withearlier diagnosis of melanoma, the accuracy of SLNbiopsy and our ability to detect regional and distantmetastases on imaging. Therefore, the role that SLNbiopsy may play in the management of melanoma is ina constant state of flux, and could be impacted tomor-row by improvements in early diagnosis, imaging stud-ies or adjuvant therapies.
As for today, is SLN biopsy, with CLND for nodepositive patients, a therapeutic surgical procedure? Ifone considers SLN biopsy a therapeutic procedure,one must seek a survival advantage among all patientsto whom the procedure is applied. Using this thresh-old, which many have done in arguments against theuse of SLN biopsy, then the answer is clearly no. Pri-or to the onset of SLN biopsy, the overriding questionin melanoma surgery was whether ELND would

VERUMI SENTINEL LYMPH NODE BIOPSY FOR MELANOMA: THERAPEUTIC PROCEDURE OR DIAGNOSTIC TEST?
202 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
improve survival over delaying lymph node dissec-tion until there was clinical evidence of recurrence.Despite retrospective data supporting ELND,3, 4 fourprospective randomized trials failed to demonstrateany survival advantage to the ELND versus a watch andwait approach.5-8
Would we expect SLN biopsy to change that? Itseems unlikely. It is true that the SLN biopsy procedurewill identify aberrant lymphatic drainage pathwaysand intercalated nodes (also known as ectopic or inter-val nodes) that would have been missed otherwise.However, this would be a small benefit, offset by afalse negative rate resulting in node-positive patientsnot undergoing node dissection. The SLN biopsy pri-marily serves to limit the morbidity of the node dis-section to those patients harboring microscopic dis-ease. Its direct impact on survival is unlikely to be sig-nificantly different than ELND as 80% of the patientsare still going to be node negative and not realize anysurvival benefit from the procedure. The survival ben-efit obtained from early eradication of nodal micro-scopic metastases would have to be large to demon-strate that benefit in a randomized study (or the studywould have to accrue a number of patients beyondwhat is feasible to achieve statistical significance). Asexpected, there has to date been no survival advan-tage to SLN biopsy compared with observation alonein MSLT-I (with a 5-year melanoma-specific survivalrate of 86.6% in the observation group and 87.1% inthe biopsy group, P=0.58).28
Many who argue against SLN biopsy use that datato support their arguments that SLN biopsy shouldnever be done outside a clinical trial as it infers nosurvival benefit to the patient population as a whole.11,
12 However, is this the correct yardstick by which wemeasure SLN biopsy?
Is the sentinel lymph node biopsy a diagnosticprocedure that identifies patients who may
benefit from further surgery?
Let us say hypothetically that we had a serum testthat identified patients at a high likelihood of harbor-ing regional metastases. The test was inexpensive andhad minimal morbidity, save those complications asso-ciated with phlebotomy, and was accurate in about95% of cases (with both a high sensitivity and speci-ficity). If performing CLND on those patients whohad a positive serum test was shown to improve their
survival, would you order the test? Obviously theanswer is an overwhelming yes. We order similar testsfor a variety of malignancies every day. Upon diagnosismelanoma patients often undergo chest X-rays, a hostof blood tests, CT scans and PET scans even thoughnone of these have been shown to impact survival.50 Itis, therefore, hard to imagine there is any practitionerwho would not order our imaginary blood test if it tru-ly identified patients for whom survival might beimproved by further surgery.
Following this logic, the question is whether SLNbiopsy functions in the same manner, as a diagnostictest meant to identify a portion of patients who maybenefit from intervention. If the answer is yes, thenthe only remaining question is whether the costs andside effects of this surgical diagnostic test (obviouslymore substantial than a blood test or an X-ray) are jus-tified by the benefit to the patient.
To assess the performance of a diagnostic test, onemust ask whether it accurately identifies a patient pop-ulation who derives benefit from further intervention,in this case a survival benefit. As a diagnostic proce-dure, SLN biopsy is meant to identify node-positivepatients, so one must ask whether survival is improvedin this subset by CLND. This benefit must be furthertempered against the false negative rate of the proce-dure.
Prior to the MSLT-I trial, there was significant evi-dence that subsets of patients might benefit from ear-ly removal of nodal metastases. As discussed previ-ously, the Intergroup Melanoma Surgical Program,while demonstrating no overall survival differencebetween ELND and observation, did show in sub-group analysis a survival benefit to ELND in patientswith nonulcerated melanomas and in patients withtumor thickness between 1 and 2 mm.6 Further evi-dence comes from the World Health Organization(WHO) Melanoma Group Program 14 Trial, whichrandomized patients with truncal melanoma to wideexcision plus ELND or wide excision plus observation,with subsequent lymph node dissection if patientsrecurred.7 Again, there was no overall survival bene-fit, but when survival of patients with microscopic dis-ease on ELND were compared with those who hadregional recurrences, the survival was significantlyimproved in the ELND group (48.2% vs 26.6%,P=0.04). While this data is strongly suggestive, it cer-tainly does not prove that even among node positivepatients the node dissection provides that degree of

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 203
SENTINEL LYMPH NODE BIOPSY FOR MELANOMA: THERAPEUTIC PROCEDURE OR DIAGNOSTIC TEST? VERUMI
benefit, as there may be patients who were node pos-itive on ELND who would not have recurred had thenodes been left in place.
This all leads to the recently reported interim resultsof the Multicenter Selective Lymphadenectomy TrialI (MSLT-I) which is the first prospective randomizedtrial to specifically address the survival benefit of theSLN biopsy. As stated, to date there has been no sig-nificant difference in survival between the patientsrandomized to SLN biopsy versus those randomizedto excision alone. However, when one compares themelanoma specific survival among those patients whowere SLN positive to those patients who recurred afterexcision alone, there was a significant improvement insurvival in the SLN group. Among the node positivepatients, the 5-year survival rate with CLND for a pos-itive SLN was 72.3% versus 52.4% for patients whounderwent CLND for a recurrence (HR 0.51;95% CI,0.32 to 0.81; P=0.004). When one includes the falsenegative patients (those patients with a negative SLNbiopsy who had a regional recurrence) in the SLNbiopsy group, there is still a significant improvementin survival (66.2% vs 54.2%; HR 0.62; 95% CI 0.40 to0.95; P=0.02).28
Certainly, the same criticisms of the WHO Program14 data could be applied here; that there may havebeen a significant number of patients positive on SLNbiopsy who would not have recurred had they notundergone SLN biopsy. The initial report, in fact, didsuggest more patients who were SLN positive thanrecurred. However, with longer follow-up, there werenearly the same numbers of patients who were eitherSLN positive or recurred after a false negative SLN asthere were patients who had a regional recurrence afterwide excision alone. The cumulative incidence ofregional metastases in both the observation group andthe biopsy group were equal by 10 years of follow-up (about 20% in both groups).51 This suggests a verysmall number, if any, of patients who had nodal metas-tases that would not ultimately suffer a regional recur-rence.
This data, therefore, would strongly suggest thatSLN as a diagnostic procedure will accurately identi-fy patients who will experience a survival benefit fromfurther intervention, specifically completion node dis-section (and possibly further benefit from adjuvantHDI). It is hard to argue against its effectiveness as astaging procedure. Thus, the argument surroundingthe appropriateness of SLN biopsy shifts from whether
there is a benefit (there clearly is) to whether the costsand morbidity of a surgical staging procedure are jus-tified. Again, if this were a simple blood test or X-raymeant to identify patients who would experience asignificant decrease in mortality with further inter-vention, there would be no argument as to its worth.
The morbidity of the SLN procedure is low. Sever-al studies document postoperative complications afterSLN biopsy in the range of 5%.46, 52, 53 Complicationsare relatively minor, primarily consisting of woundinfection, seroma or hematoma. Allergic reaction to theblue dye is rare but potentially serious. Lymphedemaafter SLN biopsy is uncommon, and despite early con-cerns, SLN biopsy does not increase the likelihood ofin-transit recurrences.28, 54 The cost is another question.Much of the controversy surrounding the use of SLNbiopsy centers on the increased costs of a procedure thatbenefits a small subset of patients. For patients deemedappropriate candidates, surgical therapy shifts froman office-based procedure, which can be done forapproximately $1 000 to $1 750, to one where i.v.sedation or general anesthesia is utilized, nuclear med-icine is involved, and a time-consuming pathologicevaluation of the sentinel nodes is necessary. This rais-es the cost of treating melanoma to between $7 150 and$15 223.55, 56 Are these costs justified? In previouscost analyses of SLN biopsy, the answer was yes, butthis was only if one considered the survival benefitassociated with HDI or compared to ELND.57, 58 Thisfinal outstanding question; whether SLN biopsy isworth the cost, should be subjected to a cost-analysistaking into account the most recent information gainedfrom the MSLT-I study, and compared to other diag-nostic or therapeutic practices in oncology.
Conclusions
The SLN procedure for melanoma patients shouldbe thought of and evaluated as a staging procedure asopposed to a therapeutic procedure. Attempts to judgethe merits of the procedure as a therapeutic interven-tion will always come up lacking, as 80% of thesepatients are node negative and will derive no directsurvival benefit from the procedure. However, as adiagnostic procedure, SLN biopsy fulfills all of thecriteria. It is highly accurate in identifying a subset ofpatients who 1) have a significantly worse prognosisand 2) will benefit from further intervention. Certain-ly there are outstanding questions. Whether the cost and

VERUMI SENTINEL LYMPH NODE BIOPSY FOR MELANOMA: THERAPEUTIC PROCEDURE OR DIAGNOSTIC TEST?
204 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
morbidity of this staging procedure are justified can bedebated, but can only be truly addressed by a cost-benefit analysis that takes into account the most recentavailable data. It is also impossible to completely sep-arate the diagnostic aspect of the procedure from thetherapeutic, as to look at the SLN one must remove itand thus remove some (and possibly all) of the region-al disease (in essence an oncologic version of Heisen-berg’s uncertainty principle). In many patients, theSLN will be the only node(s) in which disease is iden-tified pathologically, although nonsentinel nodes arenot subjected to the same thorough pathologic exam-ination as the sentinel nodes. Unfortunately, predict-ing those patients that may not require CLND hasproven unreliable.59-61 The true benefit of the com-pletion node dissection is presently being prospec-tively addressed in the Multicenter Selective Lym-phadenectomy Trial II (MSLT-II), which randomizespatients with a positive SLN to CLND or observationwith serial ultrasounds of the regional basin.
Based on the available evidence, it is logical to con-sider SLN biopsy a standard diagnostic evaluation forpatients with cutaneous melanoma >1 mm and per-haps some patients with melanoma <1 mm.9, 10, 56, 62, 63
As with all interventions in oncology, there is no blackand white. Patients should be presented with avail-able data, with a balanced discussion of the risks andthe potential benefits. Models exist to help predict thelikelihood of finding a positive SLN that can help indi-vidualize the discussion.64, 65 It is reasonable thatpatients may weigh the pros and cons and opt to foregothe procedure and proceed with wide excision alone.However, given the present level of evidence; the prog-nostic information SLN biopsy will provide, the poten-tial benefit should they be SLN positive, and the futureoptions they may have (HDI, clinical trial participation)it does not seem reasonable to make this decision forthem.
Riassunto
Biopsia del linfonodo sentinella nel melanoma: procedura tera-peutica o test diagnostico?
Sebbene la biopsia del linfonodo sentinella venga considerata ilgold standard da parte di molti chirurghi oncologi e dermatologi, essacontinua ad essere oggetto di discussione da parte di altri. I clinici,sia chirurghi che dermatologi, hanno utilizzato la stessa evidenzadisponibile sia a favore che contro l’ipotesi del linfonodo sentinel-la e sul ruolo che la sua biopsia dovrebbe giocare nella gestionedel melanoma cutaneo. Molto del disaccordo nasce da un diversopunto di vista della biopsia del linfonodo sentinella, se considerar-
la un intervento terapeutico per migliorare la sopravvivenza o se con-siderarla un test diagnostico per stratificare il rischio e selezionarei pazienti per una terapia ulteriore. Questo articolo riassumerà idati disponibili, compresi quelli più recenti forniti da MulticenterSelective Lymphadenectomy Trial-I (MSLT-I), il primo studio pro-spettico randomizzato sulla biopsia del linfonodo sentinella nelmelanoma.PAROLE CHIAVE: Melanoma - Biopsia - Linfonodo sentinella.
References
1. Snow H. Melanotic cancerous disease. Lancet 1892;2:872.2. Morton DL, Wen DR, Wong JH, Economou JS, Cagle LA, Storm FK
et al. Technical details of intraoperative lymphatic mapping for ear-ly stage melanoma. Arch Surg 1992;127:392-9.
3. Balch CM, Cascinelli N, Milton GW. Elective node dissection: prosand cons. In: Balch CM, Milton GW editors. Cutaneous melanoma:clinical management and treatment results worldwide. Philadelphia:J.B. Lippincott; 1985. p. 131-57.
4. Balch CM. The role of elective lymph node dissection in melanoma:rationale, results and controversies. J Clin Oncol 1988;6:163-72.
5. Veronesi U, Adamus J, Bandiera DC, Brennhovd O, Caceres E,Cascinelli N et al. Delayed regional lymph node dissection in stage Imelanoma of the skin of the lower extremities. Cancer 1982;49:2420-30.
6. Balch CM, Soong SJ, Bartolucci AA, Urist MM, Karakousis CP,Smith TJ et al. Efficacy of an elective regional lymph node dissectionof 1 to 4 mm thick melanomas for patients 60 years of age and youn-ger. Ann Surg 1996;224:255-66.
7. Cascinelli N, Morabito A, Santinami M, MacKie RM, Belli F. Imme-diate or delayed dissection of regional nodes in patients with melanomaof the trunk: a randomized trial. Lancet 1998;351:793-6.
8. Sim FH, Taylor WF, Pritchard DJ, Soule EH. Lymphadenectomy inthe management of stage I malignant melanoma: a prospective ran-domized study. Mayo Clin Proc 1986;61:697-705.
9. Sondak VK, Taylor JM, Sabel MS, Wang Y, Lowe L, Grover AC et al.Mitotic rate and younger age are predictors of sentinel lymph node posi-tivity: lessons learned from the generation of a probabilistic model. AnnSurg Oncol 2004;11:247-58.
10. Bleicher RJ, Essner R, Foshag LJ, Wanek LA, Morton DL. Role of sen-tinel lymphadenectomy in thin invasive cutaneous melanomas. J ClinOncol 2003;21: 1326-31.
11. Medalie N,Ackerman AB. Sentinel node biopsy has no benefit for pati-ents whose primary cutaneous melanoma has metastasized to a lymphnode and therefore should be abandoned now. Br J Dermatol2004;151:298-307.
12. Pharis DB, Zitelli JA. The management of regional lymph nodes in can-cer. Br J Dermatol 2003;149:919-25.
13. Codiron BM. Sentinel node biopsy: who needs it? Int J Dermatol2000;39:807-11.
14. McMasters KM, Reintgen DS, Ross MI, Gershenwald JE, EdwardsMJ, Sober A et al. Sentinel lymph node biopsy for melanoma: con-troversy despite widespread agreement. J Clin Oncol 2001;19:2851-5.
15. Johnson TM, Bradford CR, Gruber SB, Sondak VK, Schwartz JL.Staging workup, sentinel node biopsy, and follow-up tests formelanoma: update of current concepts. Arch Dermatol 2004;140:107-13.
16. Morton DL, Thompson JF, Essner R, Elashoff R, Stern SL, NiewegOE et al. Validation of the accuracy of intraoperative lymphatic map-ping and sentinel lymphadenectomy for early-stage melanoma: amulticenter trial. Multicenter selective lymphadenectomy trial group.Ann Surg 1999;230:453-63.
17. Albertini JJ, Cruse CW, Rapaport D, Wells K, Ross M, DeConti R etal. Intraoperative radio-lymph-scintigraphy improves sentinel lymph

Vol. 142 - N. 2 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA 205
SENTINEL LYMPH NODE BIOPSY FOR MELANOMA: THERAPEUTIC PROCEDURE OR DIAGNOSTIC TEST? VERUMI
node identification for patients with melanoma. Ann Surg1996;223:217-24.
18. Harlow SP, Krag DN, Ashikaga T, Weaver DL, Meijer SJ, LoggieBW et al. Gamma probe guided biopsy of the sentinel node in malig-nant melanoma: a multi-centre study. Melanoma Res 2001;11:45-55.
19. Karimipour DJ, Lowe L, Su L, Hamilton T, Sondak V, Johnson TM etal. Standard immunostains for melanoma in sentinel lymph nodespecimens: which ones are most useful? J Am Acad Dermatol2003;50:759-64.
20. Cook MG, Green MA, Anderson B, Eggermont AM, Ruiter DJ, SpatzA et al. The development of optimal assessment of sentinel lymphnodes for melanoma. J Pathol 2003;200:314-9.
21. Spanknebel K, Coit DG, Bieligk SC, Gonen M, Rosai J, KlimstraDS. Characterization of micrometastatic disease in melanoma senti-nel lymph nodes by enhanced pathology: recommendations for stan-dardizing pathologic analysis. Am J Surg 2005;29:305-17.
22. Gietema HA, Vuylsteke RJ, de Jonge IA, van Leeuwen PA,Molenkamp BG, van der Sijp JR et al. Sentinel lymph node investi-gation in melanoma: detailed analysis of the yield from step section-ing and immunohistochemistry. J Clin Pathol 2004;57:618-20.
23. Vuylsteke RJ, van Leeuwen PA, Statius Muller MG, Gietema HA,Kragt DR, Meijer S. Clinical outcome of stage I/II melanoma patientsafter selective sentinel lymph node dissection: long-term follow-upresults. J Clin Oncol 2003;21:1057-65.
24. Thompson JF, McCarthy WH, Bosch CM, O’Brien CJ, Quinn MJ,Paramaesvaran S et al. Sentinel lymph node status as an indicator ofthe presence of metastatic melanoma in regional lymph nodes.Melanoma Res 1995;5:255-60.
25. Zogakis TG, Essner R, Wang HJ, Turner RR, Takasumi YT, GaffneyRL et al. Melanoma recurrence patterns after negative sentinel lym-phadenectomy. Arch Surg 2005;140:865-71.
26. Chao C, Wong SL, Ross MI, Reintgen DS, Noyes RD, Cerrito PB etal. Patterns of early recurrence after sentinel lymph node biopsy formelanoma. Am J Surg 2002;184:520-4.
27. Gershenwald JE, Colome MI, Lee JE, Mansfield PF, Tseng C, Lee JJet al. Patterns of recurrence following a negative sentinel lymph nodebiopsy in 243 patients with stage I or II melanoma. J Clin Oncol1998;16:2253-60.
28. Morton DL, Thompson JF, Cochran AJ, Mozzillo N, Elashoff R, Ess-ner R et al. Sentinel-node biopsy or nodal observation in melanoma.N Engl J Med 2006;355: 1307-71.
29. Kettlewell S, Moyes C, Bray C, Soutar D, MacKay A, Byrne D et al.Value of sentinel node status as a prognostic factor in melanoma:prospective observational study. BMJ 2006;332:1423-8.
30. Morton DL, Cochran AJ. The case for lymphatic mapping and sentinellymphadenectomy in the management of primary melanoma. Br JDermatol 2004;151:308-19.
31. Gershenwald JE, Thompson W, Mansfield PF, Lee JE, Colome MI,Tseng CH et al. Multi-institutional melanoma lymphatic mappingexperience: the prognostic value of sentinel lymph node status in 612stage I or II melanoma patients. J Clin Oncol 1999;17:976-83.
32. Balch CM, Soong SJ, Gershenwald JE, Thompson JF, Reintgen DS,Cascinelli N et al. Prognostic factors analysis of 17,600 melanomapatients: validation of the American Joint Committee on Cancer mela-noma staging system. J Clin Oncol 2001;19:3622-34.
33. Balch CM, Buzaid AC, Soong SJ, Atkins MB, Cascinelli N, Coit DGet al. Final version of the American Joint Committee on Cancer sta-ging system for cutaneous melanoma. J Clin Oncol 2001;19:3635-48.
34. Cascinelli N, Belli F, Santinami M, Fait V, Testori A, Ruka W et al. Sen-tinel lymph node biopsy in cutaneous melanoma: the WHO Melano-ma Program experience. Ann Surg Oncol 2000;7:469-74.
35. Cherpelis BS, Haddad F, Messina J, Cantor AB, Fitzmorris K, Reint-gen DS et al. Sentinel lymph node micrometastasis and other histo-logic factors that predict outcome in patients with thicker melanomas.J Am Acad Dermatol 2001;44:762-6.
36. Carlson GW, Murray DR, Hestley A, Staley CA, Lyles RH, Cohen C.
Sentinel lymph node mapping for thick (>=4mm) melanoma: shouldwe be doing it? Ann Surg Onc 2003;10:408-15.
37. Statius Muller MG, van Leeuwen PA, de Lange-De Klerk ES, vanDiest PJ, Pijpers R, Ferwerda CC, Vuylsteke RJ et al. The sentinellymph node status is an important factor for predicting clinical outcomein patients wtih stage I or II cutaneous melanoma. Cancer2001;91:2401-8.
38. Gershenwald JE, Mansfield PF, Lee JE, Ross MI. Role for lymphaticmapping and sentinel lymph node biopsy in patients with thick (>or= 4mm) primary melanoma. Ann Surg Oncol 2000;7:160-5.
39. Kirkwood JM, Strawderman MH, Ernstoff MS, Smith TJ, BordenEC, Blum RH. Interferon alfa-2b adjuvant therapy of high-risk resec-ted cutaneous melanoma: the Eastern Cooperative Oncology GroupTrial EST 1684. J Clin Oncol 1996;14:7-17.
40. Kirkwood JM, Ibrahim JG, Sosman JA, Sondak VK,Agarwala SS, Ern-stoff MS et al. High-dose interferon alfa-2b significantly prolongsrelapse-free and overall survival compared with the GM2-KLH/QS-21 vaccine in patients with resected stage IIB-III melanoma: resultsof intergroup trial E1694/S9512/C509801. J Clin Oncol 2001;19:2370-80.
41. Kirkwood JM, Ibrahim JG, Sondak VK, Richards J, Flaherty LE,Ernstoff MS et al. High- and low-dose interferon alfa-2b in high-riskmelanoma: first analysis of intergroup trial E1690/S9111/C9190. J ClinOncol 2000;18:2444-58.
42. Sabel MS, VK Sondak. Pros and cons of adjuvant interferon in the treat-ment of melanoma. Oncologist 2003;8:451-8.
43. Kirkwood JM, Manola J, Ibrahim J, Sondak V, Ernstoff MS, Rao U etal. A pooled analysis of Eastern Cooperative Oncology Group andIntergroup trials of adjuvant high-dose interferon for melanoma. ClinCancer Res 2004;10:1670-7.
44. Wheatley K, Ives N, Hancock B, Gore M, Eggermont A, Suciu S.Does adjuvant interferon alfa for high risk melanoma provide a worth-while benefit? A meta-analysis of the randomised trials. Cancer TreatRev 2003;29:241-52.
45. Lens MB, Dawes M. Interferon alfa therapy for malignant melanoma:a systematic review of randomized controlled trials. J Clin Oncol2002;20:1818-25.
46. Fife K, Thompson JF. Lymph-node metastases in patients withmelanoma: what is the optimum management? Lancet Oncol2001;2:614-21.
47. Wagner JD, Ranieri J, Evdokimow DZ, Logan T, Chuang TY, John-son CS et al. Patterns of initial recurrence and prognosis after sentinellymph node biopsy and selective lymphadenectomy for melanoma.Plast Reconstr Surg 2003;112:486-97.
48. Sabel MS, et al. Inguinal node dissection for melanoma in the era ofsentinel lymph node biopsy. Surgery 2007. In Press.
49. Ballo MT, Kian AK. Radiation therapy for malignant melanoma. SurgClin N Am 2003;83:323-42.
50. Johnson TM, Bradford CR, Gruber SB, Sondak VK, Schwartz JL.Staging workup, sentinel node biopsy, and follow-up tests formelanoma: update of current concepts. Arch Dermatol 2004;140:107-13.
51. Morton DL, Cochran AJ, Thompson JF. Sentinel-node biopsy inmelanoma. N Engl J Med 2007;356:418-21.
52. McMasters KM, Noyes RD, Reintgen DS, Goydos JS, Beitsch PD,Davidson BS et al. Lessons learned from the Sunbelt Melanoma Trial.J Surg Oncol 2004;86:212-23.
53. Blumenthal R, Banic A, Brand CU, Ris HB, Lardinois D. Morbidityand outcome after sentinel lymph node dissection in patients withearly stage malignant cutaneous melanoma. Swiss Surg 2002;8:209-14.
54. van Poll D, Thompson JF, Colman MH, McKinnon JG, Saw RP, Stret-ch JR et al. A sentinel node biopsy does not increase the incidence ofin-transit metastasis in patients with primary cutaneous melanoma. AnnSurg Oncol 2005;12:597-608.
55. Essner R, et al. Results of lymphatic mapping in melanoma. In: Nei-weg OE, Essner R, Reintgen DS, Thompson JF, editors. Lymphatic

VERUMI SENTINEL LYMPH NODE BIOPSY FOR MELANOMA: THERAPEUTIC PROCEDURE OR DIAGNOSTIC TEST?
206 GIORNALE ITALIANO DI DERMATOLOGIA E VENEREOLOGIA Aprile 2007
mapping and probe applications in oncology. New York: MarcelDekker; 2000. p. 101-24.
56. Agnese DM, Abdessalam SF, Burak WE Jr, Magro CM, PozderacRV, Walker MJ. Cost-effectiveness of sentinel lymph node biopsy inthin melanomas. Surgery 2003;134:542-8.
57. Wilson LS, Reyes CM, Lu C, Lu M,Yen C. Modelling the cost-effec-tiveness of sentinel lymph node mapping and adjuvant interferontreatment for stage II melanoma. Melanoma Res 2002;12:607-17.
58. Brobeil A, Cruse CW, Messina JL, Glass LF, Haddad FF, BermanCG et al. Cost analysis of sentinel lymph node biopsy as an alterna-tive to elective lymph node dissection in patients with malignantmelanoma. Surg Oncol Clin N Am 1999;8: 435-45.
59. Sabel MS, Griffith K, Sondak VK, Lowe L, Schwartz JL, Cimmino VMet al. Predictors of nonsentinel lymph node positivity in patients witha positive sentinel node for melanoma. J Am Coll Surg 2005;201:37-47.
60. Wagner JD, Gordon MS, Chuang TY, Coleman JJ 3rd, Hayes JT, JungSH et al. Predicting sentinel and residual lymph node basin disease
after sentinel lymph node biopsy for melanoma. Cancer 2000;89:453-62.
61. McMasters KM, Wong SL, Edwards MJ, Chao C, Ross MI, Noyes RDet al. Frequency of nonsentinel lymph node metastasis in melanoma.Ann Surg Oncol 2002;9:137-41.
62. Kesmodel SB, Karakousis GC, Botbyl JD, Canter RJ, Lewis RT, WahlPM et al. Mitotic rate as a predictor of sentinel lymph node positivi-ty in patients with thin melanomas. Ann Surg Onc 2005;12:449-58.
63. Puleo CA, Messina JL, Riker AI, Glass LF, Nelson C, Cruse CW etal. Sentinel node biopsy for thin melanomas: which patients shouldbe considered? Cancer Control 2005;12:230-5.
64. Paek S, Griffith KA, Johnson TM, Sondak VK, Wong SL, Chang AEet al. The impact of factors beyond Breslow depth on predicting sen-tinel lymph node positivity in melanoma. Cancer 2007;109:100-8.
65. Wong SL, Kattan MW, McMasters KM, Coit DG. A nomogram thatpredicts the presence of sentinel node metastasis in melanoma with bet-ter discrimination than the American Joint Committee on Cancer sta-ging system. Ann Surg Oncol 2005;12:282-8.