elucidating a role for cep290 in bardet-biedl syndrome and
TRANSCRIPT

University of IowaIowa Research Online
Theses and Dissertations
Summer 2013
Elucidating a Role for CEP290 in Bardet-BiedlSyndrome and other Cilia-related DisordersYan ZhangUniversity of Iowa
Copyright 2013 Yan Zhang
This dissertation is available at Iowa Research Online: https://ir.uiowa.edu/etd/4932
Follow this and additional works at: https://ir.uiowa.edu/etd
Part of the Genetics Commons
Recommended CitationZhang, Yan. "Elucidating a Role for CEP290 in Bardet-Biedl Syndrome and other Cilia-related Disorders." PhD (Doctor ofPhilosophy) thesis, University of Iowa, 2013.https://doi.org/10.17077/etd.6h1fycc9

1
ELUCIDATING A ROLE FOR CEP290 IN BARDET-BIEDL SYNDROME AND
OTHER CILIA-RELATED DISORDERS
by
Yan Zhang
A thesis submitted in partial fulfillment of the requirements for the Doctor of
Philosophy degree in Genetics in the Graduate College of
The University of Iowa
August 2013
Thesis Supervisor: Professor Val C. Sheffield

2
Copyright by
YAN ZHANG
2013
All Rights Reserved

Graduate College The University of Iowa
Iowa City, Iowa
CERTIFICATE OF APPROVAL
_______________________
PH.D. THESIS
_______________
This is to certify that the Ph.D. thesis of
Yan Zhang
has been approved by the Examining Committee for the thesis requirement for the Doctor of Philosophy degree in Genetics at the August 2013 graduation.
Thesis Committee: ___________________________________ Val C. Sheffield, Thesis Supervisor
___________________________________ Richard Smith
___________________________________ Robert Mullins
___________________________________ Anne Kwitek
___________________________________ Kamal Rahmouni

ii
2
To my grandparents, my parents and my friends. You are the wealth of my life.

iii
3
The scientist does not study nature because it is useful; he studies it because he delights in it, and he delights in it because it is beautiful. If nature were not beautiful, it would not be worth knowing, and if nature were not worth knowing, life would not be worth living.
Henri Poincaré
The value of science

iv
4
ACKNOWLEDGMENTS
First and foremost, I would like to give my deepest thanks to my mentor Dr. Val
Sheffield for his constant support throughout my graduate career. This thesis would have
been impossible without his guidance in my development as a scientist as well as a
person. His great personality lets me feel comfortable in working and living in a foreign
country. I would also like to thank Dr. Seongjin Seo for his support and patient guidance.
In addition, I thank my committee members: Dr. Richard Smith, Dr. Anne Kwitek, Dr.
Kamal Rahmouni and Dr. Robert Mullins for their support and insightful comments.
Additionally, I extend my gratitude to the past and current members of the
Sheffield lab. It has been a happy experience working around such a great number of
gifted, hardworking and nice people. A special thank you to Xitiz Chamling, Calvin
Carter and Xiaolei Lin. You have been not only colleagues but also friends.
Finally, I would like to acknowledge my family for endless support and for
keeping faith in my abilities, even when I had to leave you and travel halfway across the
world to realize my dream. Without you, I would not be the person I am today.

v
5
ABSTRACT
Ciliopathies are a group of heterogeneous diseases associated with ciliary
dysfunction. Diseases in this group display considerable phenotypic variation within
individual diseases as well as overlapping phenotypes among clinically distinct diseases.
In particular, mutations in CEP290 cause phenotypically diverse ciliopathies ranging
from isolated retinal degeneration, nephronophthisis (NPHP), and Bardet-Biedl
Syndrome (BBS) to the neonatal lethal Meckel-Gruber syndrome (MKS). However, the
underlying mechanisms of the variable expressivity in ciliopathies are not well
understood. This thesis focuses on evaluating the molecular and biological processes
behind the retinal degeneration and obesity observed in cilia disorders with respect to
CEP290 and other ciliopathy genes using the zebrafish and mouse model systems.
CEP290 is the most frequently mutated gene underlying the non-syndromic
blinding disorder, Leber's congenital amaurosis (LCA). We first aimed to characterize the
function of various CEP290 domains and to characterize a zebrafish model aimed at
progressing towards future therapy for patients with CEP290 LCA. To this end, an
antisense oligonucleotide [Morpholino(MO)] was used for gene knockdown. We showed
that cep290 MO-injected embryos have reduced Kupffer's vesicle size and delays in
melanosome transport, two phenotypes that are observed upon knockdown of BBS genes
in zebrafish. More importantly, the embryos had a statistically significant reduction in
visual function, and this vision impairment caused by the disruption of cep290 can be
rescued by expressing only the N-terminal region of the human CEP290 protein. These
data indicate a specific region of the CEP290 protein, which is necessary for visual
function.
We examined the contribution of BBS genes to the clinical variability of CEP290-
associated ciliopathies. We demonstrated that the BBSome binds to the N-terminal region
of CEP290 and co-localizes with CEP290 to the centriolar satellite in ciliated cells and to

vi
6
the connecting cilium of photoreceptor cells. We further showed that the BBSome is
required for proper localization of CEP290 in these structures. Genetic interactions were
tested using Cep290rd16, a Cep290 hypomorphic allele with an in-frame deletion of 299
residues, and Bbs4 null mutant mouse lines. Additional loss of Bbs4 alleles in
Cep290rd16/rd16 mutants results in increased body weight and accelerated photoreceptor
degeneration compared to mice without Bbs4 mutations. Furthermore, double
heterozygous mice (Cep290+/rd16; Bbs4+/-) have increased body weight compared to
single heterozygous animals. Our data indicated that genetic interactions between the
BBSome components and CEP290 underlie the variable expression and overlapping
phenotypes of ciliopathies caused by CEP290 mutations.
Finally, this work was extended to other cilia disorders through the
characterization of genetic interactions between CEP290 and other ciliopathy genes. We
found that different NPHP and MKS proteins interact with CEP290 via its different
regions, suggesting the central role of CEP290 in CEP290 biological/cellular functions.
To characterize the functional interaction between these proteins, we used in vitro
systems to double knockdown CEP290 with other NPHP and MKS genes and showed
that depletion of a certain combination set of these proteins disrupted the localization of
proteins into the cilia. The data indicated that the phenotypic variability of human
ciliopathies is associated with different degree of compromise of cilia function.

vii
7
TABLE OF CONTENTS
LIST OF TABLES………………………………………………………………………..ix
LIST OF FIGURES……………………………………………………………………….x
LIST OF ABBREVIATIONS……………………………………………………….….xiii
CHAPTER I INTRODUCTION ..........................................................................................1 Cilia and Intraflagellar Transport .....................................................................1CEP290 and CEP290-Related Cilia Disorders .................................................2
Basic Characteristics of CEP290 ...............................................................3CEP290 Mutations and Joubert syndrome ................................................4
CEP290 Mutations and Leber Congenital Amaurosis ...............................5CEP290 Mutations and Nephronophthisis ................................................6CEP290 Mutations and Meckel-Gruber Syndrome ..................................7
Proposed CEP290 Function in Cilia .................................................................7Vision in cep290 animal models .......................................................................8Clinical Features of Bardet-Biedl Syndrome ..................................................10Genetic Heterogeneity of Bardet-Biedl Syndrome .........................................12Structure and Function of BBS Proteins .........................................................14Animal Models ...............................................................................................16
Mus musculus ..........................................................................................16Danio rerio ..............................................................................................17
Specific Aims ..................................................................................................19
CHAPTER II FUNCTIONAL ANALYSIS OF CEP290 IN ZEBRAFISH ......................29 Introduction .....................................................................................................29Materials and Methods ...................................................................................30
Ethics Statement ......................................................................................30Reverse Transcriptase–polymerase Chain Reaction ...............................30MO Knockdown ......................................................................................31Analysis of KV Size and Melanosome Transport Time ..........................31Vision Startle Response Assay ................................................................31DNA Constructs ......................................................................................32Protein Localization and Rescue Experiments ........................................32Cell Culture and Immunofluorescence Microscopy ................................33
Results .............................................................................................................33cep290 is Expressed Throughout Development in Several Tissues ........33Knockdown of cep290 Results in Characteristic BBS Phenotypes .........34N-terminus of Human CEP290 Rescues the Vision Defect in cep290 Morphant Zebrafish ....................................................................35
Discussion .......................................................................................................37
CHAPTER III THE INTERACTION BETWEEN THE BBSOME AND CEP290 IS REQUIRED FOR MEDIATING CILIA FUNCTION .............................50 Introduction .....................................................................................................50Materials and Methods ...................................................................................51
Animal Studies ........................................................................................51

viii
8
Antibodies, Plasmids, and Reagents ........................................................52Cell culture, Transfection and Co-Immunoprecipitation Assay ..............53Quantitative Real-Time PCR ...................................................................54Immunoprecipitation ...............................................................................54Immunofluorescence Microscopy ...........................................................54Photoreceptor Outer Segment Isolation ...................................................55Histology and Immunohistochemistry ....................................................55Electroretinography (ERG) Recordings ..................................................56Leptin Resistance Study ..........................................................................57Analysis of Kupffer’s Vesicle .................................................................57Melanosome Transport Assay .................................................................57
Results .............................................................................................................58Physical Interaction Between the BBSome and CEP290 ........................58The Colocalization of the BBSome and CEP290 ....................................59Proper Localization of CEP290 to the Centriolar Satellite and Photoreceptor Connecting Cilium is BBSome-dependent ......................60Synergetic Interaction Between CEP290 and BBS Genes in vitro and in Zebrafish .......................................................................................61Increased Body Weight in Mice with Combined Loss of Cep290 and Bbs4 Genes .......................................................................................62Accelerated Retinal Degeneration in Mice with Combined Loss of Cep290 and Bbs4 alleles ..........................................................................64
Discussion .......................................................................................................65
CHAPTER IV INTERACTION BETWEEN CEP290 AND OTHER CILIOPATHY PROTEINS IS REQUIRED FOR THE CORRECT LOCALIZATION OF PROTEINS TO CILIA ...............................................97 Introduction .....................................................................................................97Materials and Methods ...................................................................................98
DNA Constructs, Reagents and Antibodies ............................................98Cell Culture, Transfection and Co-immunoprecipitation Assay .............99Quantitative Real-time PCR ..................................................................100Immunofluorescence Microscopy .........................................................100Sucrose Gradient Ultracentrifugation ....................................................101
Results ...........................................................................................................101Physical Interaction Between CEP290 and Other Ciliopathy Proteins ..................................................................................................101Requirement of CEP290 and Other NPHP and MKS Proteins for the Correct Localization of Ciliary Proteins ..........................................102
Discussion .....................................................................................................103
CHAPTER V SUMMARY, CONCLUSIONS AND FUTURE DIRECTIONS .............118 Summary .......................................................................................................118Conclusion ....................................................................................................120Future Directions ..........................................................................................122
REFERENCES ................................................................................................................126

ix
9
LIST OF TABLES
Table 1 Frequency of involvement in BBS and the function of the known BBS genes ..................................................................................................................21
Table 2 Percentages of embryos with body curvature defects based on MO dose .........42
Table 3 Compiled data set of KV, melanosome transport and vision startle response assay results for rescue experiments ..................................................46
Table 4 Body Weight of all the genotypes (male) from 1 month to 4 month .................88
Table 5 Body Weight of all the genotypes (female) from 1 month to 4 month ..............89
Table 6 Summarize of ERG data of 1-month old mice ..................................................95
Table 7 Summary of KO Phenotypes in NPHP Proteins and MKS1 Knockdown Cells .................................................................................................................117
Table 8 Summary of proteins interacting with CEP290 as well as the proteins for which CEP290 serves as a gatekeeper ............................................................125

x
10
LIST OF FIGURES
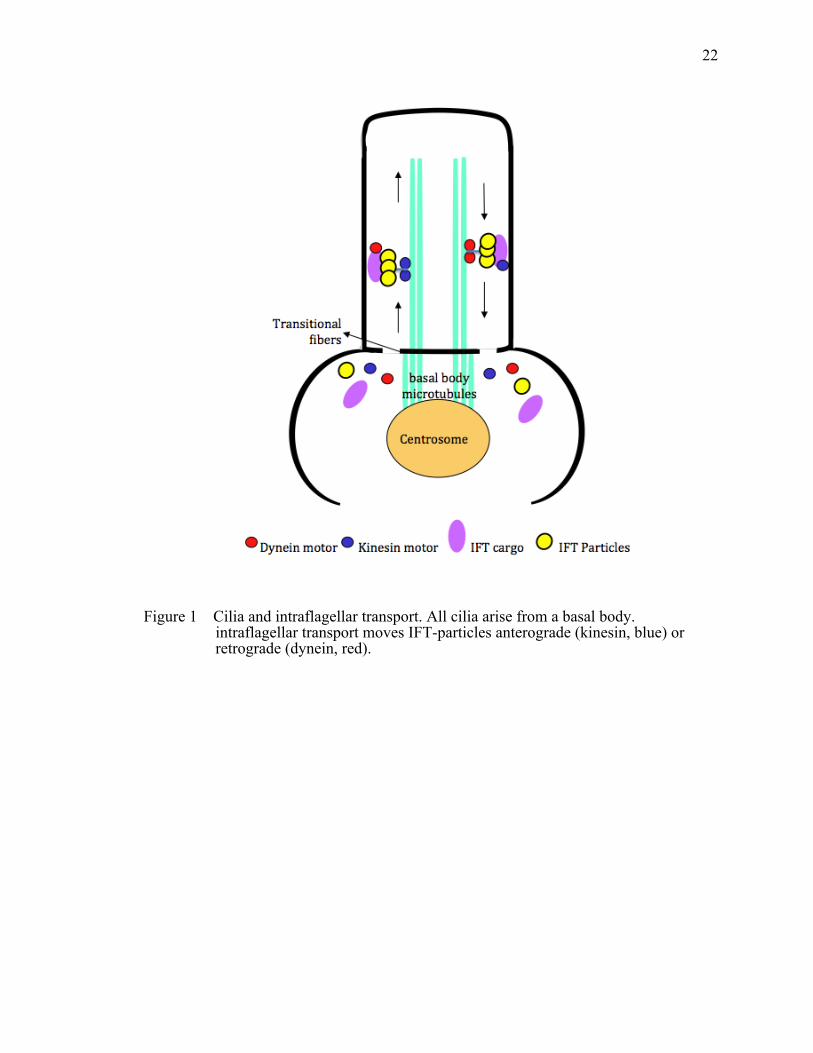
Figure 1 Cilia and intraflagellar transport. ....................................................................22
Figure 2 Schematic representation of the CEP290 gene and the CEP290 protein. ......23
Figure 3 Overview of all mutations in CEP290 identified in patients with cilia-related disorder. ..............................................................................................24
Figure 4 Overview of all mutations identified in JBTS patients. .................................25
Figure 5 Overview of all mutations identified in LCA patients. ..................................26
Figure 6 Overview of all mutations identified in NPHP patients with retinal degeneration. ..................................................................................................27
Figure 7 Overview of all mutations identified in MKS and MKS-like patients. ..........28
Figure 8 Expression of zebrafish cep290. .....................................................................39
Figure 9 cep290 gene targeting and knockdown efficacy. ............................................40
Figure 10 Gross cep290 morphant phenotypes. ..............................................................41
Figure 11 cep290 gene knockdown results in KV defects. .............................................43
Figure 12 cep290 gene knockdown results in melanosome transport delay. ..................44
Figure 13 Rescue of the cep290 morphant vision defect. ...............................................45
Figure 14 Both the N-and C-terminal constructs localize paracentrioler in undifferentiated ARPE-19 cells. .....................................................................47
Figure 15 N-terminal and C-terminal of CEP290 specifically localize to the mature centriole in ciliated ARPE-19 cells. ...................................................48
Figure 16 Cellular localization of the N-terminal and the C-terminal truncations of the human CEP290 protein. .......................................................................49
Figure 17 The BBSome interacts with CEP290. ............................................................70
Figure 18 Interaction of endogenous CEP290 and the BBSome in HEK293T cells and mouse retina. ............................................................................................71
Figure 19 Schematic representation of the CEP290 deletion mutants. ...........................72
Figure 20 PCM1-independent physical interaction between CEP290 and the BBSome. .........................................................................................................73
Figure 21 BBS4 interacts with CEP290..........................................................................74
Figure 22 Co-localization of CEP290 and the BBSome to centriolar satellites in cultured cells and to the connecting cilium of photoreceptor cells. ...............75

xi
11
Figure 23 Co-fractionation of the BBSome and Cep290 in the photoreceptor outer segment fraction. ............................................................................................76
Figure 24 Suppression of BBS gene expression by RNAi. ............................................77
Figure 25 CEP290 localization in the centriolar satellite is BBSome-dependent. .........78
Figure 26 CEP290 is not involved in BBS9 localization to cilia. ...................................79
Figure 27 Cep290 localization to the connecting cilium is BBSome-dependent. ..........80
Figure 28 Decreased ARL13B ciliary localization in BBS4 and CEP290 double knockdown cells. ............................................................................................81
Figure 29 Quantitation of ARL13B ciliary localization. ................................................82
Figure 30 Features of BBS in zebrafish. .........................................................................83
Figure 31 Medium-dose pair-wise combination knockdowns of cep290, bbs1, bbs4 and bbs7. ................................................................................................84
Figure 32 The rd16 mice have increased body fat percentage. ......................................85
Figure 33 Increased body weight of mice with combined loss of Cep290 and Bbs4 genes. .....................................................................................................86
Figure 34 Increased body weight and higher leptin levels in Bbs4+/-;Cep290+/rd16
mice. ...............................................................................................................87
Figure 35 Attenuated leptin receptor signaling in Bbs4+/- Cep290+/rd16 mice. ................90
Figure 36 Accelerated photoreceptor degeneration with additional loss of Bbs4 alleles in Cep290rd16/rd16 mice. ........................................................................91
Figure 37 Lack of photoreceptor degeneration with additional loss of Bbs4 alleles in Cep290+/rd16 mice. ......................................................................................92
Figure 38 Impaired rhodopsin trafficking in mice with combined loss of Cep290 and Bbs4 alleles. .............................................................................................93
Figure 39 Diminished ERG responses in CEP290rd16/rd16 mice with additional loss of Bbs4 alleles. ...............................................................................................94
Figure 40 Reduced ERG b-wave and oscillatory potential of mice with combined loss of Cep290 and Bbs4 genes. .....................................................................96
Figure 41 The N-terminal region of CEP290 interacts with NPHP2. ...........................106
Figure 42 Low-level CEP290 variants are unstable and degraded by the proteasome. ...................................................................................................107
Figure 43 Physical interaction between the C-terminus of CEP290, MKS1 and SDCCAG8. ...................................................................................................108

xii
12
Figure 44 Identification of the NPHP/MKS complex. ..................................................109
Figure 45 Suppression of NPHP and MKS gene expression by RNAi. .......................110
Figure 46 Loss of INVS ciliary localization in CEP290 and SDCCAG8 depleted cells. ..............................................................................................................111
Figure 47 Quantification of INVS ciliary localization. ................................................112
Figure 48 NPHP and MKS are required for ciliary localization of TMEM67. ............113
Figure 49 Percentage of TMEM67-positive cilia in depleted cells. .............................114
Figure 50 NPHP and MKS proteins mediate ciliary localization of ARL13B. ............115
Figure 51 Summary of ARL13B ciliary localization. ...................................................116

xiii
13
LIST OF ABREVIATIONS
BBS Bardet-Biedl Syndrome
CEP290 Centrosomal protein 290
CO-IP Co-immunoprecipitation
CORS Cerebello-oculo-renal Syndrome
DRF Days post fertilization
ERG Electroretinography
IFT Intraflagellar transport
IS Inner segment
JBTS Joubert Syndrome
KV Kupffer’s vesicle
LCA Leber congenital amaurosis
MKKS McKusick-Kaufman Syndrome
MKS Meckel-Gruber Syndrome
MO Morpholino
NPHP Nephronophthisis
ONL Outer nuclear layer
OS Outer segment
PCM1 Pericentriolar material 1
Prph2 Peripherin 2
RD retinal degeneration
RPE Retinal pigmented epithelium
RT-PCR Reverse Transcriptase Polymerase Chain Reaction
SLS Senior-Loken Syndrome
TZ Transition zone
WT Wild-type

1
CHAPTER I
INTRODUCTION
Cilia and Intraflagellar Transport
Cilia are microtubule-based hair-like organelles. They project from the apical
surface into the extracellular space (Marshall and Nonaka 2006) in a number of organs,
such as kidneys and the retina of the eye (De Robertis 1956), as well as almost all cell
types of the human body. Cilia contain a basal body, transition zone, axoneme, ciliary
membrane and the ciliary tip. The transition zone has transition fibers, which in
combination with the basal body are thought to function as a gate to control the transport
of proteins into and out of the cilium (Figure 1). Cilia fall into two classes: motile and
primary cilia. Motile cilia contain a ‘9+2’ microtubule configuration where nine
microtubule doublets surround a central pair. Motile cilia function to generate flow or
movement of fluid. In contrast, primary cilia have a ‘9+0’ axonemal structure, which
lacks the central microtubule pair. Primary cilia are sensory in function and typically
exist as monocilia.
Mammalian spermatozoa contain a specialized cilium known as the sperm
flagellum. While flagella and motile cilia share many features, they have distinctive
patterns of movement (Bisgrove and Yost 2006). Flagella usually occur singly or as a
pair, whereas numerous motile cilia are found on ciliated cells, such as the cells of the
airway epithelium or the ependymal cells lining the cerebral ventricles.
Unlike other cell organelles, cilia are only observed when cells are in a quiescent
and/or differentiated state. Entry into the cell cycle for cell division requires resorption
of the primary cilium to free up the centriole for the mitotic spindle (Azimzadeh and
Bornens 2007). The presence of the cilium prevents cells from undergoing cell division,
suggesting that the cilium is a cell division checkpoint organelle (Pan and Snell 2007).

2
The cilium is a complex structure that contains over 1000 proteins. Due to the fact
that there are no transcriptional or translational machinery in the cilium, all proteins need
to be transported into and out of the cilium by means of a process known as intraflagellar
transport (IFT) (Rosenbaum and Witman 2002), which is responsible for cilia formation
and maintenance. Proteins are loaded onto IFT particles at the ciliary base and
transported along the ciliary axoneme by two main molecular motors – kinesin and
dynein motors (Rosenbaum 2002). Kinesins functions in anterograde transport towards
the ciliary tip, while dyneins facilitates retrograde transport back to the cell body (Figure
1). These motor proteins associate with IFT particles, which consist of two complexes –
IFT complex A and complex B (Cole, Chinn et al. 1993; Piperno, Siuda et al. 1998).
Based on mutant analysis and RNAi data, loss of any component of complex B leads to
the inhibition of ciliary assembly (Cole, Diener et al. 1998; Haycraft, Schafer et al. 2003),
indicating that complex B is associated with anterograde transport. IFT complex A is
involved in retrograde transport because cilia often show an accumulation of particles and
abnormal morphology in the absence of normal complex A components (Perkins,
Hedgecock et al. 1986; Tran, Haycraft et al. 2008).
CEP290 and CEP290-Related Cilia Disorders
Studies during the last decade show that primary cilia are involved in various
fundamental signaling pathways, such as sonic hedgehog, receptor tyrosine kinase, and
Wnt (Singla and Reiter 2006; Fliegauf, Benzing et al. 2007; Goetz and Anderson 2010;
Christensen, Clement et al. 2012). Consistent with their ubiquitous occurrence and
diversity of function, ciliary defects result in a range of human genetic disorders,
including Leber Congenital Amaurosis (LCA), Joubert syndrome (JBTS), Meckel-Gruber
syndrome (MKS), Senior-Loken syndrome (SLSN), Bardet-Biedl syndrome (BBS), and
nephronophthisis (NPHP), which are collectively called “ciliopathies” (Badano, Mitsuma
et al. 2006; Fliegauf, Benzing et al. 2007; Sheffield 2010).

3
One of the most interesting disease genes related with cilia disorders is CEP290,
in which mutations cause the phenotypic spectrum ranging from isolated blindness to the
lethal MKS, suggesting that CEP290 plays an important role in cilium function in various
human tissues.
Basic Characteristics of CEP290
CEP290 is a centrosomal protein of 2472 amino acids with a molecular weight of
290 kDa. It was first identified in a proteomic analysis of the human centrosome
(Andersen, Wilkinson et al. 2003). The protein is strongly conserved throughout
evolution, and contains several predicated motifs, including 13 putative coiled-coil
domains, a region with homology to SMC chromosome segregation ATPases, six KID
motifs, three tropomyosin homology domains and an ATP/GTP binding site motif A
(Figure 2) (Sayer, Otto et al. 2006). Besides its localization to the centrosome of dividing
cells and to the nucleus, the protein localizes to the basal bodies at the base of the cilia in
many different cell types, including the photoreceptor connecting cilium (Chang, Khanna
et al. 2006; Sayer, Otto et al. 2006; Valente, Silhavy et al. 2006). Expression was also
established in dendritic knobs in olfactory sensory neurons (McEwen, Koenekoop et al.
2007).
The CEP290 gene (93.2 kb) is located on chromosome 12q21.32 and has 54
exons with the start codon found in exon 2 (Figure 2). Transcription produces 14
different mRNAs, 11 alternatively spliced variants and 3 unspliced forms. There are 5
probable alternative promoters, 6 non-overlapping alternative last exons and 5 validated
alternative polyadenylation sites. So far, 112 distinct mutations have been identified
(Figure 3) (Coppieters, Lefever et al. 2010), of which three are missense (two affecting
the start codon) and one single amino acid deletion. The remaining 110 mutations are
frameshift, splice-site mutations, or nonsense mutations. These mutations are predicted to
cause loss-of-function of the resulting protein. Seventy-three mutations have been

4
reported only once, while 21 mutations occur at least twice (Valente, Brancati et al.
2008).
CEP290 Mutations and Joubert syndrome
Joubert syndrome (JBTS) is a genetically heterogeneous disorder characterized by
hypotonia, ataxia, psychomotor delay and variable occurrence of oculomotor apraxia and
neonatal breathing abnormalities (Boltshauser and Isler 1977). A group of pleiotropic
conditions, termed ‘‘Joubert syndrome related disorders’’ (JSRDs), have the
pathognomonic clinical and neuroradiological features with variable involvement of other
organs and systems, mainly the eyes and kidneys (Gleeson, Keeler et al. 2004). These
included classical (or pure) JBTS, JBTS plus retinal dystrophy, Dekabane-Arima
syndrome, COACH syndrome, oro-facio-digital VI syndrome, JBTS plus polymicrogyria
and Malta syndrome (Maria, Hoang et al. 1997). All patients diagnosed with JSRD have
developmental and intellectual disability. In the neonatal period there is an altered
respiratory pattern with episodes with hyperpnea and/or apnea. These symptoms can
range from short intervals to pronged attacks requiring assisted ventilation, and usually
improve with age. Usually they disappear around the sixth month of life.
Nearly all JSRD genes identified so far encode for proteins expressed in the
primary cilium or in the centrosome. In particular, mutations in CEP290 represent the
most common cause of JBTS associated with both renal and retinal involvement, being
responsible of approximately 50% of cerebello-oculo-renal syndrome cases. Overall, 57
patients in 44 families with the diagnosis of JSRD have been reported to have CEP290
mutations (Figure 4) (Brancati, Barrano et al. 2007; Helou, Otto et al. 2007; Perrault,
Delphin et al. 2007; Tory, Lacoste et al. 2007).
Among JSRD patients, the most common mutation is c.5668G > T in exon 41
(p.G1890X) that was identified in 10 unrelated JSRD families (Figure 4). Eight of the
nine patients homozygous for p.G1890X do not have severe LCA (as in typical CORS),

5
but present a milder retinal phenotype or even preserved vision with normal
ophthalmological testing. It has been suggested that this truncating mutation might
somehow spare the retina (Valente, Brancati et al. 2008). However, patients with
G1890X in compound heterozygosity with other mutations have JSRD with severe retinal
involvement (Brancati, Barrano et al. 2007; Cideciyan, Aleman et al. 2007), and two
cases with isolated LCA have recently been reported with compound heterozygosity for
the two recurring mutations c.2991 + 1655A > G and p.G1890X (Cideciyan, Aleman et
al. 2007).
Forty-seven distinct mutations are found in JSRD families, of which 35 (75%)
cluster in the second half of the gene (exon 28-54), and 42 (89%) are detected in exon 17-
54 (Figure 4) (Valente, Brancati et al. 2008).
CEP290 Mutations and Leber Congenital Amaurosis
Leber Congenital Amaurosis (LCA) is a severe retinal dystrophy, which causes
blindness or severe visual impairment before the age of 1 year. It has four diagnostic
clinical features: severe and early visual loss, sensory nystagmus, amaurotic pupils, and
absent electrical signals on electroretinogram (ERG) (Franceschetti and Dieterle 1954).
The worldwide incidence ranges from 1/30,000 to 1/81,000 live births (Koenekoop 2004;
Stone 2007). It accounts for 5% of all inherited retinopathies and approximately 20% of
children attending schools for the blind (Koenekoop 2004). LCA patients present various
manifest visual function and visual acuity, usually from 20/200 to light perception or
even no-light perception. To be noted, complete loss of retinal tissue in the fovea is a
prominent and frequent retinal feature found in the patients.
Fourteen LCA genes have been identified, explaining approximately 70% of the
cases. CEP290 was identified in consanguineous families by identity-by-descent (IBD)
mapping (den Hollander, Koenekoop et al. 2006). Moreover, CEP290 is the most
frequently mutated LCA gene (15%) (Figure 5). The most frequent CEP290 mutation,

6
which also shows the strongest genotype-phenotype correlation, is the intronic mutation
c.2991 + 1655A>G that creates a splice-donor site and inserts a cryptic exon in the
CEP290 mRNA between exons 26 and 27, introducing a stop codon immediately
downstream of exon 26 (Figure 5). It has been found in homozygosity or compound
heterozygosity in 56 unrelated patients with isolated LCA, while it has never been
detected in patients with other phenotypes (Cideciyan, Aleman et al. 2007; Perrault,
Delphin et al. 2007). In addition, this particular mutation is considered hypomorphic in
that some wild-type transcript is still present in homozygous affected individuals (den
Hollander, Koenekoop et al. 2006).
Of note, unlike CEP290 mutations detected in JSRD families, the 45 mutations
identified in LCA patients appear to be scattered throughout the gene rather than
clustered in the 3’ half of the gene (Figure 5) (Valente, Brancati et al. 2008).
CEP290 Mutations and Nephronophthisis
Nephronophthisis (NPHP) is an autosomal recessive cystic kidney disease and the
most frequent genetic cause for end-stage renal disease in children and adolescents
(Hildebrandt and Otto 2005). The term “nephronophthisis” derives from the Greek and
means “disintegration of nephrons”, which is one aspect of the histopathology. Key
histology findings include tubulointerstitial fibrosis, tubular atrophy and cyst formation.
The incidence of NPHP varies from 1:50,000 in Canada to about 1 in 1 million in the
USA (Wolf and Hildebrandt 2011). So far, eleven genes are known to cause NPHP (Wolf
and Hildebrandt 2011). CEP290 mutations do not represent a major cause of NPHP,
accounting for 1% of NPHP cases (Figure 6). However, about 10-15% of patients with
NPHP have late onset of retinal degeneration, which is characteristic of Senior-Loken
syndrome (Figure 6). Interestingly, the frequency of retinal degeneration in patients with
CEP290 associated NPHP is high.

7
CEP290 Mutations and Meckel-Gruber Syndrome
Meckel-Gruber Syndrome (MKS) is a neonatal lethal disorder characterized by
central nervous system malformations, bilateral cystic kidney, and postaxial polydactyly.
In some cases, cardiac abnormalities, cleft palate and situs inversus are associated with
MKS. Newborns with MKS rarely survive longer than 2 weeks. It is a rare disorder, with
a population frequency between 1:13,250 and 1:140,000 (Alexiev, Lin et al. 2006). In
addition, some patients are diagnosed as “Meckel-like syndrome” due to the absence of at
least one MKS diagnostic feature. A strong allelism has recently been described in MKS
wheretwo truncating mutations (nonsense, frame-shift or splice site mutations) in the
genes MKS1, MKS3, NPHP3, NPHP6/CEP290, and NPHP8/RPGRIP1L cause MKS,
whereas the presence of at least one missense mutation causes the milder phenotype.
Mutations in CEP290 were identified in nineteen MKS patients from eight families and
five patients from four families with Meckel-like syndrome, further expanding the
phenotypes associated with CEP290 mutations (Figure 7) (Baala, Audollent et al. 2007;
Frank, den Hollander et al. 2008).
Proposed CEP290 Function in Cilia
The clinical feature associated with CEP290 mutations indicates a role for
CEP290 in cilia. Recent work in Chlamydomonas showed that CEP290 is localized
specifically between the outer doublet microtubules and membrane of the transition zone,
and appears to function as a gatekeeper that allows specific proteins to pass, thereby
regulating protein content in the cilium (Craige, Tsao et al. 2010). This gatekeeping role
of CEP290 raises the possibility that some mutations in CEP290 in human patients
results in altered function of CEP290. In other words, mutations in CEP290 may partially
affect the function of the protein (such as a single domain) and thus limit disruption to a
subset of CEP290-mediated transport events. Such selective disruptions could give rise to
partially overlapping yet distinct phenotypes observed in human patients. In addition,

8
recent work mapped the NPHP-JBTS-MKS interactome (Sang, Miller et al. 2011), which
suggested specific underlying mechanism leading to disease progression. Mutations in
CEP290 could partially disrupt the function of the interactome or partially alter the
components of the interactome, resulting in various degrees of deficiency in some key
developmental pathways, such as sonic hedgehog signaling, Wnt signaling, or cell
polarity in specific tissues, and thus lead to a wide range of distinct phenotypes.
The presence of modifier genes could also contribute to the clinical variability of
CEP290-related diseases. One of the potential modifier genes is TMEM67, which is
involved in ciliary function and has been shown to interact genetically with cep290 in
zebrafish (Leitch, Zaghloul et al. 2008). The only BBS patient identified with a
homozygous CEP290 mutation also harbors a heterozygous mutation in TMEM67. This
could be coincidental or the mutant TMEM67 could influence the phenotype. Another
possible modifier gene is AHI1. In a recent study, a cohort of 91 unrelated LCA patients
in Belgium were screened for CEP290 mutations. Three patients were found with the
same CEP290 genotype. In addition, a heterozygous novel AHI1 mutation, p.Asn811Lys,
was found in the most severely affected patient (Coppieters, Casteels et al. 2010). Out of
five patients with CEP290-related disease and neurological involvement, a AHI1
missense variant, p.His758Pro, was found in one patient with mild mental retardation and
autism (Coppieters, Casteels et al. 2010). These results suggest the possible modifying
role of AHI1 in patients harboring CEP290 mutations.
Vision in cep290 animal models
So far, there are two naturally occurring animal model with mutations in cep290:
the rd16 mouse and a pedigree of Abyssinian cats. Both models display progressive
retinal degeneration with autosomal recessive inheritance. Interestingly, no other
abnormal phenotypes in brain or kidney are present in these two models (Chang, Khanna
et al. 2006; Menotti-Raymond, David et al. 2007). Because mutations in CEP290 can

9
lead to LCA and syndromic retinal diseases in human patients, animal models of
spontaneous retinal degeneration provide insights into pathological mechanisms of
disease progression and help in designing therapeutic strategies.
The retina is located at the back of the eye, and consists of seven alternating
layers of cells and processes. It is the responsibility of the retina, more specifically the
photoreceptors, to convert the light signal into a neural signal, which can be transmitted
to the brain via the axons of ganglion cells. There are two photoreceptor types: rods,
which mediate achromatic vision in starlight, and cones, which are important for color
vision and fine acuity in daylight. The photoreceptor is a highly polarized, light-sensing
cell with a modified cilium (the connecting cilium) that connects the photosensitive outer
segment (OS) to the inner segment (IS), where protein synthesis occurs. As a non-motile
cilium, the connecting cilium has a microtubule-based axoneme which is anchored in the
IS by the basal body (Horst, Johnson et al. 1990). The photoreceptor OS is often
recognized as a modified cilium due to the fact that the axoneme extends almost the
entire length of OS. The connecting cilium plays a key role in transport of
phototransduction proteins as well as structural components from the IS to the OS
through the process of IFT (Besharse, Baker et al. 2003; Tsujikawa and Malicki 2004;
Krock and Perkins 2008). IFT and the connecting cilium are both critical for
development, maintenance and function of the photoreceptor as approximately 10% of
the distal ends of the OS are shed daily (Luby-Phelps, Fogerty et al. 2008). Disruptions in
proper protein trafficking have been shown to cause photoreceptor degeneration and
ultimately blindness (Pazour and Rosenbaum 2002; Tsujikawa and Malicki 2004;
Deretic, Williams et al. 2005). CEP290 is involved in microtubule-associated protein
transport and has been shown to localize to the connecting cilium of the photoreceptor
cell (Chang, Khanna et al. 2006; Sayer, Otto et al. 2006), suggesting the involvement of
CEP290 in protein trafficking into the OS of the photoreceptor.

10
The rd16 mouse, which exhibits early-onset retinal degeneration with autosomal
recessive inheritance, carries a Cep290 in-frame deletion involving exons 35 to 39
(Chang, Khanna et al. 2006). The mutation causes progressive degeneration of the outer
segment of the photoreceptor cell and reduction in thickness of the outer nuclear layer at
P21 (Cideciyan, Rachel et al. 2011). At the molecular level, this mutant protein alters the
distribution of RPGR, rhodopsin and arrestin in the retina, suggesting a function of
Cep290 in protein transport across the connecting cilium (Chang, Khanna et al. 2006). In
addition, considerable thickening of the inner nuclear and plexiform layers were observed
in central retinal regions (Cideciyan, Aleman et al. 2007), suggesting that Cep290 also
functions in the inner retina.
In contrast to the rd16 mouse, the Abyssinian cat with homozygous cep290
mutations displays a late-onset retinal dystrophy. The age of onset is 12-18 months of age
in the majority of animals. A single-nucleotide polymorphism is found in intron 50 of
cep290 (IVS50 + 9T>G) which creates a strong splice donor site, resulting in a 4-bp
insertion and a frameshift in the mRNA transcript with subsequent introduction of a stop
codon and thus premature truncation of the protein (Menotti-Raymond, David et al.
2007). This animal model offers considerable promise in developing gene-based
therapies for human LCA.
Clinical Features of Bardet-Biedl Syndrome
One of the cilia related disorders is Bardet-Biedl Syndrome (BBS,
OMIM209900), which displays an autosomal recessive pattern of inheritance, and is
genetically heterogeneous. In the early 1920s, George Bardet and Arthur Biedl (Bardet,
1920; Biedl, 1922) independently described the features of BBS as a discrete clinical
syndrome. BBS is characterized by retinal degeneration, obesity, polydactyly, renal
malformation, hypogenitalism and cognitive impairment (Harnett, Green et al. 1988;
Green, Parfrey et al. 1989). Individuals are deemed affected if they have at least four out

11
of six primary features. Furthermore, BBS patients have an increased susceptibility to
hypertension, diabetes mellitus and cardiac anomalies (Harnett, Green et al. 1988; Green,
Parfrey et al. 1989; Elbedour, Zucker et al. 1994; Sheffield 2004; Sheffield 2010). In
addition, BBS is associated with several minor features not considered to be part of the
diagnostic criteria. It has been reported that asthma is highly prevalent in BBS patients
(Beales, Warner et al. 1997; Beales, Elcioglu et al. 1999). A 22-year cohort study of BBS
in Newfoundland populations reported a higher incidence of asthma, with required
hospitalization in 68% of cases (Moore, Green et al. 2005). Of note, situs inversus is also
observed in a few BBS patients (Lorda-Sanchez, Ayuso et al. 2000; Ansley, Badano et al.
2003). Other abnormalities include anosmia, hearing loss, colonic disorders and
hypothyroidism (Beales, Elcioglu et al. 1999; Kulaga, Leitch et al. 2004; Moore, Green et
al. 2005).
In addition to the inter-familial phenotypic variability described above, BBS
patients also show intrafamilial variation for expressivity of obesity, skeletal anomalies of
the extremities, hypogenitalism, short stature, paraplegia, and dental abnormalities, as
well as the course of the retinal dystrophy (Riise, Andreasson et al. 1997; Moore, Green
et al. 2005). For example, one sibling in one BBS family became simultaneously night
blind and visually impaired during daylight, while another sibling had visual problems in
daylight first and became night blind later, and the third sibling developed night blindness
followed by visual impairment in the daylight (Riise, Andreasson et al. 1997). Cherian
and coworkers reported five patients within one family: One died during the neonatal
period, two developed end stage renal failure at 14 and 15 years of age, respectively,
whereas one had a congenital duplication of the right collecting system (Cherian and Al-
Sanna'a 2009).
The pleiotropic features of BBS have significant overlap with the clinical features
of other human disorders including Alstrom Syndrome, McKusick-Kauffman syndrome
(MKKS), MKS, and JBTS. Alstrom syndrome is usually differentiated from BBS by the

12
presence of hearing loss and absence of polydactyly (Alstrom, Hallgren et al. 1959).
MKKS is characterized by urogenital and cardiac anomalies (Robinow and Shaw 1979).
Mutations in the MKKS gene were found to cause BBS in some cases (Stone, Slavotinek
et al. 2000), although the patients normally lack the obesity and retina dystrophy
characteristic of BBS. JBTS is an autosomal recessive disorder characterized by retinal
dystrophy and development delay, and the most consistent feature is a radiographic
finding of the cerebellum known as the molar tooth sign (Brancati, Dallapiccola et al.
2010). The most severe ciliopathy is MKS, which is a lethal disorder characterized by
encephalocele, central nervous system malformations, and polycystic kidneys (Mecke
and Passarge 1971). More recently, hypomorphic mutations in MKS1 have been shown to
be associated with BBS (Leitch, Zaghloul et al. 2008), while only MKS1 truncating
mutations are found in MKS. Conversely, mutation in at least three BBS genes cause
MKS-like phenotypes but not encephalocele (Karmous-Benailly, Martinovic et al. 2005),
suggesting MKS might represent a more severe variant of BBS.
Genetic Heterogeneity of Bardet-Biedl Syndrome
BBS is a genetically heterogeneous autosomal recessive disorder. The genetic
heterogeneity of BBS was first described in linkage analysis studies in isolated Bedouin
Arab populations (Kwitek-Black, Carmi et al. 1993). The increased frequency of the
recessive alleles resulting from the high rate of consanguinity in this population first
made the mapping and position cloning of BBS genes possible. To date, seventeen BBS
loci have been identified: BBS1, on 11q13 (Leppert, Baird et al. 1994); BBS2, on 16q21
(Kwitek-Black, Carmi et al. 1993); BBS3, on 3p13 (Sheffield, Carmi et al. 1994; Chiang,
Nishimura et al. 2004); BBS4, on 15q22.3 (Carmi, Rokhlina et al. 1995); BBS5, on 2q13
(Young, Penney et al. 1999); BBS6, on 20p12 (Woods, Young et al. 1999); BBS7, on
4q27 (Badano, Ansley et al. 2003); BBS8, on 14q32.11 (Ansley, Badano et al. 2003);
BBS9, on 7p14 (Nishimura, Swiderski et al. 2005); BBS10, on12q21 (Stoetzel, Laurier et

13
al. 2006); BBS11 (TRIM32), on 9q33 (Chiang, Beck et al. 2006); BBS12, on 4q27
(Stoetzel, Muller et al. 2007); BBS13 (MKS1),on 17q22 (Leitch, Zaghloul et al. 2008);
BBS14 (CEP290), on 12q21.32 (Leitch, Zaghloul et al. 2008); BBS15(WDPCP), on 2p15
(Kim, Shindo et al. 2010); BBS16 (SDCCAG8), on 1q43 (Otto, Hurd et al. 2010); BBS17
(LZTFL1), on 3p21.31 (Marion, Stutzmann et al. 2012).
Among the seventeen BBS genes, the largest proportion of pathogenic mutations
is found in BBS1 and BBS10, together accounting for about 40% of clinically diagnosed
BBS cases. The most common BBS alleles are BBS1 M390R and BBS10 C91LfsX5. In
addition, some genes appear to have greater ethnic specific frequency than others. For
example, mutations in BBS4 and BBS5 are mainly seen in patients of Middle Eastern and
North African descent (Iannaccone, Mykytyn et al. 2005; Billingsley, Deveault et al.
2011). Due to the rare nature of BBS, some of the genes, such as BBS3 and BBS8,
account for less than 5% of the BBS cases (Ansley, Badano et al. 2003; Pereiro, Valverde
et al. 2010). In some instances, BBS mutations were found in only a single family
(BBS11 and BBS14) (Chiang, Beck et al. 2006; Leitch, Zaghloul et al. 2008).
Approximately 20% of BBS families do not have an observed mutation in any of the
known BBS genes, suggesting that additional disease genes remain to be identified
(Stoetzel, Muller et al. 2007).
Although BBS is considered an autosomal recessive disease, it has previously
been proposed that a third mutated allele at a second locus is required for penetrance of
the BBS phenotype, a phenomenon known as triallelism (Hichri, Stoetzel et al. 2005).
For example, in one BBS family, a phenotypically normal individual carries two BBS2
nonsense mutations (Q59X and Y24X), while the affected sibling carries a heterozygous
BBS4 mutation (Q147X) as well as the same nonsense mutation in BBS2 (Katsanis,
Ansley et al. 2001). In contrast, Mykytyn and coworkers evaluated the inheritance of
BBS in a cohort of 43 unrelated BBS patients with two mutant BBS1 alleles, but did not
identify any additional disease causing mutations in the three other BBS genes known at

14
that time, BBS2, BBS4 and BBS6 (Mykytyn, Nishimura et al. 2003), nor did they observe
any non-penetrant individuals with two BBS1 alleles. An independent study screened 19
consanguineous Tunisian families diagnosed with BBS and found no evidence of
triallelism (Smaoui, Chaabouni et al. 2006). A similar study was undertaken in 49
unrelated BBS patients and no evidence of triallelism was detected in six genes screened
(Hjortshoj, Gronskov et al. 2010). Therefore, these studies suggest oligogenic inheritance
is not necessary in the vast majority of human cases and BBS should be considered an
autosomal recessive disorder for genetic counseling purposes.
Structure and Function of BBS Proteins
BBS1, BBS2, BBS5, and BBS7 show no significant homology to any proteins of
known function (Nishimura, Searby et al. 2001; Mykytyn, Nishimura et al. 2002; Li,
Gerdes et al. 2004). However, BBS4 and BBS8 both contain tetratricopeptide repeat
(TPR) domains, which may mediate different protein-protein interactions (Blatch and
Lassle 1999). BBS9 (also known as B1, parathyroid hormone-responsive protein) does
not appear to contain any known functional domains (Adams, Rosenblatt et al. 1999).
BBS6, BBS10 and BBS12 share homology to the chaperonin containing TCP1 family
(CCT) and have predicted chaperonin function (Stone, Slavotinek et al. 2000; Stoetzel,
Laurier et al. 2006; Stoetzel, Muller et al. 2007). These three proteins are members of the
type II chaperonin superfamily and contain chaperonin domains. BBS11 or TRIM32
(tripartite motif –containing gene 32) is a member of the TRIM protein family, which is
characterized by a RING-finger, a B-Box and a coiled-coil motif (Kudryashova,
Kudryashov et al. 2005). It has also been shown that TRIM32 is an E3 ubiquitin ligase
capable of ubiquitinating actin. LZTFL1 (BBS17) has a coiled-coil domain in its C-
terminal half, which includes a leucine-zipper domain (Seo, Baye et al. 2010).
It has been suggested that BBS proteins play a role in primary cilia function
(Table 1) (Mykytyn and Sheffield 2004; Zhang, Seo et al. 2012). Work by Nachury et al.

15
has shown that seven BBS proteins (BBS1, BBS2, BBS4, BBS5, BBS7, BBS8 and
BBS9) form a complex (the BBSome) necessary for ciliogenesis (Nachury, Loktev et al.
2007; Zhang, Nishimura et al. 2013). Interestingly, this complex localizes to non-
membranous centriolar satellites in the cytoplasm, as well as the cilia membrane.
Recruitment of the BBSome to the basal body results in the activation of Rab8 and
ultimately cilia biogenesis. Rabin8 is the GTP exchange factor for Rab8, and has been
shown to interact with the BBSome at the basal body (Nachury, Loktev et al. 2007).
Although triallelic inheritance is not frequent in BBS patients, it is proposed that
in some cases a third allele could modify the penetrance or severity of the BBS
phenotype. One possible mechanism for this is that having mutations in more than one
subunit of the BBSome would affect the stability or function of the BBSome and cause a
more severe phenotype.
The BBS proteins with chaperonin homology, BBS6, BBS10 and BBS 12, form
their own complex with six CCT proteins (CCT1-5, CCT8) (Seo, Baye et al. 2010). This
complex binds to BBS7 and facilitates the folding and/or assembly of the BBSome (Seo,
Baye et al. 2010). Knockdown of any CCT protein in this complex using siRNA in
HEK293T cells causes reduced interactions among BBS2, BBS7 and BBS9, indicating
that the CCT proteins are necessary for BBSome assembly (Seo, Baye et al. 2010).
Mutation of CEP290 (BBS14) has been found in BBS patients, suggesting that
CEP290 may have a potential epistatic effect on mutations in known BBS-associated
genes. Like BBS4, CEP290 is localized to the centrosome. BBS4 directly interacts with
pericentriolar material 1 (PCM1), a major component of centriolar satellites. PCM1 is
required for recruitment of centrosomal proteins and the organization of the microtubule
network (Dammermann and Merdes 2002), while BBS4 functions as an adaptor that
connects the p150glued subunit of dynein transport machinery with PCM1 and thus
assists the centrosomal recruitment of PCM1 (Kim, Badano et al. 2004). This model is
strongly supported by the finding that Bbs4 knockout mice exhibit mislocalization of

16
PCM1 (Kulaga, Leitch et al. 2004). Recent work by Joon Kim shows that CEP290 also
binds to PCM1 and that both are required for the ciliary localization of Rab8, a Ras-like
small GTPases required for the biogenesis of ciliary membranes (Kim, Krishnaswami et
al. 2008). In addition, CEP290 has been shown to affect formation of the BBS4-PCM1
complex in vitro (Kim, Krishnaswami et al. 2008). These results suggest that PCM1 is a
potential mediator linking CEP290 and BBS4 in some molecular pathways.
LZTFL1 has been shown to be a negative regulator of the BBSome trafficking to
the ciliary membrane. It interacts with the BBSome via BBS9 within the cytoplasm and
inhibits ciliary entry of the BBSome (Seo, Zhang et al. 2011).
Animal Models
Mus musculus
The mouse as a model organism is closely related to humans and is a powerful
tool for investigating the shared biological processes of genes. Our lab has created several
knockout mice lines (Bbs2-/-; Bbs3-/-; Bbs4-/-; Bbs6-/-) (Nishimura, Fath et al. 2004; Fath,
Mullins et al. 2005; Zhang, Nishimura et al. 2011), as well as a knock-in line
(Bbs1M390R/M390R). Analysis of these mouse models reveals that these mice recapitulate
major components of the human phenotypes except polydactyly. Interestingly, absence of
a BBS protein prevents the formation of flagella during spermatogenesis, although BBS
mutant mice do develop other motile cilia as well as a modified primary cilium (the
connecting cilium) in the photoreceptors.
In addition to the hydrocephalus (Carter, Vogel et al. 2012), all these mutant mice
become obese and have high leptin levels after about four-months of age. Leptin, one of
the hormones derived from adipose tissue, plays a crucial role in energy homeostasis.
Within the brain, the hypothalamic proopiomelanocortin (POMC) and agouti-related
protein neurons (AgRP) have been identified as major targets of leptin action. While
POMC neurons provide a strong anorexigenic effect – release of the POMC-derived

17
neuropeptides decreases food intake and body weight, AgRP neurons have a potent
orexigenic effect – secretion of AgRP increases food intake (Varela and Horvath 2012).
At the molecular level, leptin binds to the extracellular domain of leptin receptor b, which
is the long form of the leptin receptor and highly expressed in those two neurons, leading
to the activation of the Janus kinase/signal transducer and activator of transcription
(JAK/STAT) pathway (Banks, Davis et al. 2000). It is expected that obese patients would
present with lower serum leptin levels, however surprisingly, the opposite is true -- high
levels are expressed. This is believed to lead to leptin resistance. Our BBS mutant mice
also display leptin resistance, and further analysis reveals that leptin receptor signaling is
disrupted in the absence of the normal BBSome (Seo, Guo et al. 2009).
Moreover, work in our lab with Bbs2 and Bbs4 null mice has shown that these
proteins are required for maintenance of photoreceptors. Interestingly, death of the
photoreceptors is preceded by mislocalization of rhodopsin, suggesting that there is a
defect in intracellular transport in BBS mutant mice. Moreover, the photoreceptor
degeneration in Bbs4-null mice is not caused by structural defects to the connecting
cilium or basal body, but rather results from the disrupted intraflagellar transport between
the inner segment and outer segment of the photoreceptors (Abd-El-Barr, Sykoudis et al.
2007; Swiderski, Nishimura et al. 2007), indicating Bbs4 is important for the transport of
phototransduction proteins, but not for maintaining structural integrity. Additionally,
absence of BBS proteins disrupts the formation of flagella during spermatogenesis,
leading to the infertility of male mice (Chamling, Seo et al. 2013).
Danio rerio
Zebrafish have several advantages over other available animal models including:
(1) a relatively short generation time, (2) small in size compared to mice, rat, chick etc.,
and (3) transparent bodies allowing optical observation during early and late
development. Nonetheless, zebrafish are not without disadvantages. To date, no cells in a

18
zebrafish have been identified that are equivalent to mouse embryonic stem cells. Thus it
is not yet possible to generate a zebrafish knockout model, however, it is possible to
study gene knockdown using morpholino antisense oligonucleotides. At the amino acid
level, the zebrafish CEP290 and BBS proteins show high homology to the human
proteins, which suggests that the functions of these proteins may be evolutionary
conserved and important for early development. Loss of bbs genes in zebrafish generates
two prototypical defects: reduction in the size of Kupffer’s vesicle (KV) and retrograde
melanosome transport defects (Yen, Tayeh et al. 2006; Tayeh, Yen et al. 2008; Pretorius,
Baye et al. 2010). KV is a fluid-filled and ciliated organ that forms transiently at the
posterior end of the notochord at the early somite stages (approximately 12 hours post
fertilization). Monocilia (a typical 9+2 microtubule arrangement) form at the apical
membrane of the cells facing the lumen in KV of zebrafish embryos. Surgical removal of
KV results in randomization of laterality at later stages, demonstrating that KV is the
organizer region that establishes LR asymmetric patterning in zebrafish(Essner, Amack et
al. 2005). Knockdown of bbs genes in zebrafish results in a reduction of KV size to less
than width of the notochord (Tayeh, Yen et al. 2008; Pretorius, Baye et al. 2010).
The second prototypical phenotype observed in bbs morphants is delayed
trafficking of the melanosome. Zebrafish can alter their skin pigmentation by trafficking
of melanosomes within melanophores in response to visual cue and hormonal stimuli
(Marks and Seabra 2001; Barral and Seabra 2004). The melansome is a lysosome-related
organelle containing melanin, the most common light absorbing pigment in animal
kingdom. In the melanophores, the melanosome can be shuttled bidirectionally between
the cell periphery and the perinuclear region. Dispersion of melanosomes to the cell
periphery (anterograde) is kinesin directed, while dynein is required for the retraction of
the melanosomes to the perinuclear region (retrograde) (Marks and Seabra 2001; Barral
and Seabra 2004). Taking advantage of the zebrafish pigment, a time course of organelle
translocations in the whole animal can be observed to evaluate the effect of gene

19
knockdown on cellular trafficking. For example, pigment aggregation (retrograde) can be
stimulated within minutes upon treatment with epinephrine (Nascimento, Roland et al.
2003). Typically, stimulating 5-day-old embryos with epinephrine results in melanosome
transport within 1.5 minutes, while loss of bbs genes causes statistically significant delay
in melanosome transport (Yen, Tayeh et al. 2006; Tayeh, Yen et al. 2008).
Zebrafish are able to elicit a characteristic escape response when exposed to
sudden changes in light density, presumably as an adaptive response to escape the
looming predator, and this startle response can be used as an assay for vision function
(Easter and Nicola 1996). In this assay, the swimming behavior of a 5-day-old embryo
was monitored in response to 1-second block in light. The typical response for WT fish is
a distinct C-bend and a change in swimming direction. However, some BBS zebrafish
morphants present vision defects observed by a measurable loss of response to changes in
light conditions (Nishimura, Baye et al. 2010; Pretorius, Baye et al. 2010; Pretorius,
Aldahmesh et al. 2011).
Specific Aims
The overall objective of this study is to evaluate the molecular and biological
processes behind the retinal degeneration and obesity phenotypes observed in cilia
disorders with respect to CEP290 and BBS genes using animal models. In particular,
both the mouse and zebrafish have been established as models and these systems can be
used to investigate both the cellular and molecular mechanisms of retinal degeneration
and obesity in cilia disorders. We hypothesized that synergic interactions between
CEP290 and other ciliopathy genes are important for its function in normal cilia
formation and regulating ciliary localization of proteins. Specifically this project aimed
to:
1. Characterize CEP290 function and identify other specific BBS genes with
which CEP290 interacts functionally.

20
2. Explore the mechanism by which CEP290 mutations cause BBS using both the
zebrafish and mouse model systems by characterizing the genetic and physical interaction
between BBS and CEP290 proteins.
3. Extend this work to other cilia disorders through the characterization of genetic
interaction between CEP290 and genes causing other human cilia related disorders.

21
Table 1 Frequency of involvement in BBS and the function of the known BBS genes
Gene Frequency Function
BBS1 23% BBSome protein
BBS2 8% BBSome protein
BBS3/ARL6 1% GTPase
BBS4 2% BBSome protein
BBS5 <1% BBSome protein
BBS6/MKKS 6% Chaperonin complex
BBS7 2% BBSome protein
BBS8 1% BBSome protein
BBS9 6% BBSome protein
BBS10 20% Chaperonin complex
BBS11/TRIM32 < 1% E3 ubiquitin ligase
BBS12 5% Chaperonin complex
BBS13/MKS1 5% Centriole migration
BBS14/CEP290 < 1% Gate keeper
BBS15/WDPCP < 1% Ciliogenesis
BBS16/SDCCAG8 < 1% Interacts with OFD1
BBS17/LZTFL1 < 1% Negative regulator of BBSome
22
Figure 1 Cilia and intraflagellar transport. All cilia arise from a basal body. intraflagellar transport moves IFT-particles anterograde (kinesin, blue) or retrograde (dynein, red).

23
Figure 2 Schematic representation of the CEP290 gene and the CEP290 protein. Top row: The human CEP290 gene spans 93.2 kb encoding 54 exons. Translation initiation (ATG in exon 2) and termination (TAA in exon 54) codons are indicated (exons are drawn to scale with every fifth exon numbered). Bottom row: A scale representation of CEP290 protein (2479 residues, white) shows the putative protein motifs in relation to the position of the exons encoding them. Coiled-coiled domains are shown in blue. SMC, structural maintenance of chromosomes; MYO-Tail, myosin tail homology domain.

24
Figure 3 Overview of all mutations in CEP290 identified in patients with cilia-related disorder. The pie chart shows the proportion of mutations in CEP290 that result in certain ciliopathies.
JSRD38%
LCA42%
NPHP7%
MKS12%
BBS1%

25
Figure 4 Overview of all mutations identified in JBTS patients. Top row: The human CEP290 gene spans 93.2 kb encoding 54 exons. Bottom row: A scale representation of CEP290 protein (2479 residues, white) shows the putative protein motifs in relation to the position of the exons encoding them. Coiled-coiled domains are shown in blue. Mutations in CEP290 identified in 57 JSRD patients are indicated (dotted line). The position of the most frequent change detected in JSRD patients with CEP290 mutations, a G > T change in exon 41 causing premature stop codon is shown (red box). SMC, structural maintenance of chromosomes; MYO-Tail, myosin tail homology domain.

26
Figure 5 Overview of all mutations identified in LCA patients. Top row: The human CEP290 gene spans 93.2 kb encoding 54 exons. Bottom row: A scale representation of CEP290 protein (2479 residues, white) shows the putative protein motifs in relation to the position of the exons encoding them. Coiled-coiled domains are shown in blue. Mutations in CEP290, identified in approximately 15% of patients with LCA, are indicated (dotted line). The position of the most frequent change detected in LCA patients with CEP290 mutations (∼20%), an A > G change in intron 26 causing the incorporation of a cryptic exon resulting in a frameshift and premature stop codon is shown (red box). SMC, structural maintenance of chromosomes; MYO-Tail, myosin tail homology domain.

27
Figure 6 Overview of all mutations identified in NPHP patients with retinal degeneration. Top row: The human CEP290 gene spans 93.2 kb encoding 54 exons. Bottom row: A scale representation of CEP290 protein (2479 residues, white) shows the putative protein motifs in relation to the position of the exons encoding them. Coiled-coiled domains are shown in blue. Mutations in CEP290 identified in NPHP patients with retinal involvement are indicated (dotted line). SMC, structural maintenance of chromosomes; MYO-Tail, myosin tail homology domain

28
Figure 7 Overview of all mutations identified in MKS and MKS-like patients. Top row: The human CEP290 gene spans 93.2 kb encoding 54 exons. Bottom row: A scale representation of CEP290 protein (2479 residues, white) shows the putative protein motifs in relation to the position of the exons encoding them. Coiled-coiled domains are shown in blue. Mutations in CEP290 identified in MKS and MKS-like patients are indicated (dotted line). SMC, structural maintenance of chromosomes; MYO-Tail, myosin tail homology domain

29
CHAPTER II
FUNCTIONAL ANALYSIS OF CEP290 IN ZEBRAFISH
Introduction
Mutations in centrosomal protein 290 (CEP290) cause the non-syndromic
blinding disorder Leber's congenital amaurosis (LCA, OMIM 611755) as well as several
cilia-related syndromic disorders including Meckel–Gruber syndrome (MKS, OMIM
611134), Joubert syndrome (JSTS, OMIM 610188), Senor–Loken syndrome (SLSN,
OMIM 610189) and Bardet–Biedl syndrome (BBS, OMIM 209900). The CEP290 protein
is encoded by 54 exons, is 2479 amino acids in length and has 25 predicted protein
domains, motifs and localization signals (Sayer, Otto et al. 2006). Disease-causing
mutations including missense, nonsense, splicing and frame shifting changes are found
throughout the length of the gene (Frank, den Hollander et al. 2008; Coppieters, Lefever
et al. 2010). To date, although some mutations cluster in the same region, no clear
correlations between phenotype and the corresponding genotype have been identified
among CEP290 mutations and observed diseases (Coppieters, Lefever et al. 2010). The
precise role and the functional domains of this large protein remain unclear.
LCA is an early-onset blinding disorder in which patients present in infancy with
lack of a visual response, but a relatively normal appearing retina (Stone 2007; den
Hollander, Roepman et al. 2008; Pasadhika, Fishman et al. 2010). Mutations in CEP290
account for about one-third of all LCA cases, and the most common single CEP290
mutation accounts for almost half of the disease-causing variation in this gene (Perrault,
Delphin et al. 2007; Stone 2007; den Hollander, Roepman et al. 2008). This common
CEP290 allele causes a premature stop codon resulting from an intronic mutation that
disrupts normal splicing of the transcript.
cep290 knockdown in zebrafish has been previously shown to result in several
phenotypes including convergence extension defects, hydrocephalus, small eyes, kidney

30
cysts and body curvature (Leitch, Zaghloul et al. 2008; Schafer, Putz et al. 2008; Tobin
and Beales 2008). In our targeted knockdown of cep290 in zebrafish, we observed
curvature of the body axis indicative of primary cilia dysfunction. Mutation of CEP290
has been reported in a single individual with phenotypic features overlapping BBS. We
evaluated our cep290 knockdown embryos and found that they exhibited two phenotypes
shared when BBS genes are knocked down in zebrafish, specifically reduced Kupffer's
vesicle (KV) size and delayed retrograde melanosome transport (Yen, Tayeh et al. 2006;
Tayeh, Yen et al. 2008; Pretorius, Baye et al. 2010). Moreover, functional analysis of
vision revealed a reduction in visual behavior. Importantly, we were able to restore visual
responsiveness by expressing only the N-terminal region of the human CEP290 protein in
the knockdown embryos. This observation indicates that the N-terminal region of the
CEP290 gene is sufficient to suppress the visual impairment and may be a viable
treatment option for LCA patients.
Materials and Methods
Ethics Statement
The University of Iowa Animal Care and Use Committee approved all animal
work in this study.
Reverse Transcriptase–polymerase Chain Reaction
RNA was extracted from pools of wild-type embryos at the following stages: 1–
128 cells (maternal stage), 8–10 somites, 48 hpf and 5 dpf, as well as the adult retina and
whole eye. cDNA was synthesized using oligo-dT primers. Gene expression was
evaluated using the following primers whose location in the cep290 gene is depicted in
Figure 2A. β-actin expression served as a control.
• cep290—primer F1: 5′-GGAACAGGCCTTTGAAAACA-3′
• cep290—primer R1: 5′-GCAAACTTGGTCTCCAGCTC-3′

31
• β-actin—primer F: 5′-TCAGCCATGGATGATGAAAT-3′
• β-actin —primer R: 5′-GGTCAGGATCTTCATGAGGT-3′
MO Knockdown
The antisense splice site MO was designed and purchased from Gene Tools. The
MO was air pressure injected into 1–4-cell embryos at concentrations ranging from 5 to
10 ng. The efficiency of transcript knockdown in the morphant embryos, especially in
those with normal morphology, was assessed by RT–PCR (described above) using cDNA
generated from pools of cep290 morphants with only the straight body axis phenotype at
the following stages: 8–10 somites, 72 hpf and 5 dpf.
• Standard control MO: 5′-CCTCTTACCTCAGTTACAATTTATA-3′
• cep290 MO: 5′-TTGATGTGTACCAGTTGTGCTGATG-3′
Analysis of KV Size and Melanosome Transport Time
KV size was assessed in live embryos between the 6–12-somite stage of
development. Embryos with a KV smaller than the width of the notochord were recorded
as abnormal. For the melanosome transport assay, 5 dpf dark-adapted embryos were
treated with epinephrine (500 µg/ml, Sigma E4375) added to embryo medium.
Melanosome retraction was continuously monitored under the microscope, and the
quantity of time that it took the melanosomes to traffic from the periphery of the cell to
their perinuclear location was recorded. Embryos were imaged live using a stereoscope
with a Zeiss Axiocam camera.
Vision Startle Response Assay
Prior to experimentation, 5 dpf zebrafish embryos were light adapted for 1 hour,
and then the visual stimulus of a 1 second block in bright light intensity was performed
under a dissecting microscope. A positive visual response was recorded if the embryo
made an abrupt change in swimming behavior within 1 second of the light intensity

32
change. Five trials were performed spaced 30 seconds apart followed by the mechanical
stimulus of probing embryos with the tip of a blunt needle. Embryos that failed to
respond to the mechanical stimulus were not included in the analysis.
DNA Constructs
The fragments encoding the N-terminal (1–1059 amino acids) and the C-terminal
regions (1765–2479 amino acids) of human wild-type CEP290 (NCBI Reference
Sequence: NG_008417.1) were amplified from human retina cDNA (Clonetech), TA
cloned into the Gateway vector system (Invitrogen) and sequence confirmed.
Subsequently, the genes were recombined into gateway expression vectors with an N-
terminal 6× myc tag.
• CEP290—primer N-terminal F: 5′-ATGCCACCTAATATAAACTGG-3′
• CEP290—primer N-terminal R: 5′-
TCATATTTTTTTTGAAATGGAAACAATGTC-3′
• CEP290—primer C-terminal F: 5′-ATGTCTGCAACTTCTCAAAAAGAG-3′
• CEP290—primer C-terminal R: 5′-TTAGTAAATGGGGAAATTAACAGG-3′
Protein Localization and Rescue Experiments
N-terminally tagged human myc-CEP290 RNA was synthesized using the
mMessage mMachine transcription kit (Ambion) and injected into 1–2-cell embryos.
Whole-mount immunohistochemistry was performed on 50% epiboly stage embryos as
described (Westfall, Brimeyer et al. 2003) using an anti-myc antibody (9E10, Santa Cruz)
and fluorescent secondaries (Alexa Fluor 568, Molecular Probes). The centrin-eGFP was
detected by direct GFP fluorescence. For rescue experiments, the synthetic RNA was co-
injected with the cep290 MO.

33
Cell Culture and Immunofluorescence Microscopy
ARPE-19 cells were maintained in DMEM/F12 media (Invitrogen) supplemented
with 10% fetal bovine serum (FBS) and seeded on glass coverslips in 24-well plates. To
induce ciliogenesis, cells were shifted to serum-free medium 24 h after seeding and
further incubated for 48 h. Cells were fixed with methanol for 10 min at 4°C, blocked
with 5% bovine serum albumin and 3% normal goat serum and stained with indicated
primary antibodies. Alexa Fluor 488 goat anti-rabbit immunoglobulin (Ig) G (Invitrogen)
and 568 goat anti-mouse IgG (Invitrogen) were used to detect the primary antibodies.
Coverslips were mounted on Vecta-Shield mounting with DAPI (Vector Lab), and
images were taken with an Olympus 1X71 inverted microscope. Antibodies were
purchased from the following sources: mouse monoclonal antibodies against γ-tubulin
(GTU-88, Sigma) and acetylated-tubulin (T7451, Sigma); rabbit polyclonal antibody
against Myc (ab9106, Abcam).
Results
cep290 is Expressed Throughout Development
in Several Tissues
To understand the functional role of cep290 in the zebrafish, we first examined
the spatial–temporal expression pattern of the gene throughout development and in adult
tissues. Reverse transcriptase–polymerase chain reaction (RT–PCR) analysis revealed
that the cep290 transcript is inherited maternally and is present throughout all stages of
development (Figure 8). cep290 is also expressed in the adult zebrafish eye and,
specifically, in the retina (Figure 8) as well as in all other tissues examined (data not
shown).

34
Knockdown of cep290 Results in
Characteristic BBS Phenotypes
The most common human CEP290 mutation underlying LCA is an intronic
mutation, c.2991 + 1655A > G, which alters gene splicing and results in a stop at amino
acid C998X (Figure 9A) (den Hollander, Koenekoop et al. 2006). We mimicked this
LCA mutation in zebrafish by using an antisense Morpholino (MO) that disrupts the
splicing of the zebrafish cep290 transcript at the corresponding exon 25 leading to intron
inclusion (Figure 9A, hashed bar). To verify disrupted splicing in morphant embryos,
primers were designed to flank intron 25 (Figure 9A). 10-20 MO-injected embryos (8 ng
dose) with a straight body axis were pooled for RNA isolation and RT–PCR was
performed. cDNA from un-injected embryos amplified the expected wild-type transcript
(Figure 9B). cep290 MO-injected embryos have a larger transcript consistent with intron
inclusion (Figure 9B).
cep290 zebrafish morphants exhibit body curvature defects in a MO dose-
dependent manner ranging from straight to a severe C-shaped (curly) axis at 5 dpf (Figure
10). Embryos injected with the highest dose of MO (10 ng) had the greatest percentage of
curly embryos (80%), whereas only 34% of embryos injected with low-dose (5 ng) MO
presented with the curly body axis (Table 2).
Knockdown of bbs genes in zebrafish results in reduction of the size of the KV as
well as delay in retrograde melanosome transport in the skin pigment (Yen, Tayeh et al.
2006; Tayeh, Yen et al. 2008; Pretorius, Baye et al. 2010). Therefore, we examined these
cardinal features in cep290 morphant embryos.
The KV is a ciliated organ found transiently in the posterior tailbud of developing
embryos from 6–15 somites (Figure 11A). At the 8-12 somite stage, KV size was
assessed by comparing the vesicle width to the width of the developing notochord.
Embryos with a KV larger than the width of the notochord are considered normal (Figure
11B), whereas a KV that is the same size or smaller than the width of the notochord is

35
considered abnormal (Figure 11C and 11D). Our data showed that the knockdown of
cep290 causes a statistically significant percentage of embryos with abnormally sized
KVs, 20.8% at the 10 ng dose, compared with 5.6% of wild-type (Figure 11E). Moreover,
the percentage of embryos with abnormal KVs increased in a MO dose-dependent
manner.
The second phenotype assessed in zebrafish is the measurement of the rate of
melanosome movement to examine intracellular transport. Zebrafish embryos are able to
adapt to their environment by trafficking their melanosomes (pigmented vesicles) within
melanophores (cells on the surface of the fish) in response to light and/or hormonal
stimuli (Marks and Seabra 2001; Aspengren, Skold et al. 2009). The average time of
melanosome transport is assessed by treating 5 dpf dark-adapted embryos with
epinephrine to chemically stimulate movement. Dark adaptation maximally disperses the
melanosomes throughout the melanophore (Figure 12A and 12B), whereas epinephrine
treatment results in rapid retrograde movement of melanosomes to the perinuclear region
(Figure 12C). Completion of melanosome transport in wild-type embryos averages 1.58
minutes, whereas cep290 morphant embryos at all doses showed a statistically significant
delay with the 10 ng dose averaging 2.50 minutes (Figure 12D). Together, these data
indicate that the knockdown of the cep290 transcript results in the cardinal BBS
phenotypes in the zebrafish, which supports a role for CEP290 in some patients with
BBS.
N-terminus of Human CEP290 Rescues the Vision Defect
in cep290 Morphant Zebrafish
To evaluate visual function in zebrafish, we utilized a natural escape response that
is elicited when embryos are exposed to rapid changes in light intensity (Pretorius, Baye
et al. 2010). In the vision assay, visually responsive 5 dpf embryos change their
swimming direction when there is a short block in a bright light source (Figure 13A). The

36
assay is repeated five times spaced 30 s apart, and the average number of responses is
reported. Wild-type embryos respond an average of 3.53 times (Figure 13B). As a control
for the assay, the visually impaired cone-rod homeobox (crx) morphant embryos were
utilized and responded only 2.00 times. Only cep290 morphant embryos that possessed a
straight body axis were used for this behavioral assay to ensure uncompromised
locomotor activity. In contrast to wild-type and control MO-injected embryos, cep290
morphants responded only 2.46 times during the five trials (Figure 13B).
To functionally test the specific region of CEP290 in vision, rescue experiments
were performed. We divided the human CEP290 gene into an N-terminal fragment that
represents the first 1059 amino acids of the protein and a C-terminal fragment, amino
acids 1765–2479 (Fig 13C). The N-terminal construct extends over the region where the
most common LCA mutation is found, while the C-terminal construct covers the entire
region that is truncated in the rdAc cat model of late-onset retinal degeneration. We first
expressed the N-terminal and the C-terminal constructs in undifferentiated and ciliated
ARPE-19 cells and found both constructs to localize to the pericentriolar region (Figure
14 and 15). Subsequently, mRNA encoding myc-tagged fusion constructs were injected
into zebrafish embryos and we observed the pericentriolar localization of both constructs
(Figure 16). Next, we assessed if the human constructs can function in zebrafish to rescue
the MO-induced vision defect. Embryos co-injected with cep290 MO and the mRNA
encoding myc-tagged N- and C-terminal constructs were grown to 5 dpf. In the vision
assay, cep290 morphant embryos responded on average 2.46 times, whereas morphant
embryos injected with the N-terminal CEP290 construct responded 3.48 times (Figure
13B). This response was statistically the same as the wild-type response of 3.53,
suggesting rescue of the MO-induced vision defect. However, injection of the C-terminal
construct was unable to rescue the vision defect (Figure 13B). Interestingly, neither the
N-terminal nor the C-terminal construct was sufficient to rescue the KV size defect and
only the C-terminal construct showed a suppression of the melanosome transport delay

37
(Table 3). Taken together, these data indicate that the N-terminal region of the human
CEP290 gene is sufficient to rescue the cep290 MO-induced vision defect, while other
regions of the protein are needed for the normal KV morphology and function.
Discussion
CEP290 has been associated with several cilia-related disorders, but the precise
functional domains of the protein and their role in normal development need to be
determined. We observed that the cep290 transcript is present throughout all stages of
zebrafish embryo development and is enriched in the retina. Using a targeted knockdown
strategy to model the most common CEP290 mutation in LCA patients, we found body
curvature defects, KV size reduction, delayed retrograde melanosome transport and
vision impairment. We determined by using truncated CEP290 constructs that the N-
terminal region of the CEP290 protein is sufficient to rescue the vision defect observed in
cep290 knockdown embryos.
LCA patients with the c.2991 + 1655A > G mutation are hypomorphic because
some wild-type transcript is present in homozygous carriers (den Hollander, Koenekoop
et al. 2006). It is thought that these patients present with non-syndromic LCA in that
there is enough wild-type transcript present that other organ systems are unaffected (den
Hollander, Koenekoop et al. 2006). Consistent with this hypothesis, our data showed that
the penetrance of the curly body axis, as well as the severity of the KV defect and
melanosome transport delay, is MO dose-dependent.
Our finding that the expression of only the N-terminal region of the CEP290
protein is sufficient to rescue the vision defect suggests that domains within this region
are able to interact with their retina-specific binding partners to allow visual function.
The ability of the N-terminal region of the human CEP290 to rescue the vision defect
observed in cep290 knockdown zebrafish embryos seems to be inconsistent with two
existing animal models, the rd16 mouse and the rdAc cat. Mutations in these model

38
systems are found in the C-terminal region, suggesting that this region of the protein is
required for vision. Possible explanations for this inconsistency include that the mutant
Cep290 protein in the mouse and cat models is unstable and degraded, or folds in such a
manner as to affect the structure of the N-terminal region resulting in alteration of
binding properties and thus changes in protein function. It has been shown in rd16 mice
that there is a reduced amount of the mutant Cep290 protein; moreover, the mutant
Cep290 protein disrupts the localization of RPGR and rhodoposin in the photoreceptor
cells (Chang, Khanna et al. 2006).
In human patients, there are several LCA-causing mutations identified within the
N-terminal region of CEP290, including the mutation modeled in our study (den
Hollander, Koenekoop et al. 2006; Perrault, Delphin et al. 2007; Frank, den Hollander et
al. 2008; Coppieters, Lefever et al. 2010). These mutations observed in human patients
suggest that regions outside that of the C terminus have a role in regulating binding to
interacting proteins required for proper vision, which corresponds with a hypothesis that
the severity of a disease phenotype is the result of the degree to which individual
complexes are disrupted (Murga-Zamalloa, Desai et al. 2010).

39
Figure 8 Expression of zebrafish cep290. RT–PCR of the cep290 transcript in wild-type embryos showing expression at all the stages and tissues examined: maternal, 8–10 somites, 48 hpf, 5 dpf, adult retina and adult whole eye. β-actin expression served as a loading control.

40
Figure 9 cep290 gene targeting and knockdown efficacy. (A) Schematic of full-length human (white) and zebrafish (gray) CEP290 proteins. The most common CEP290 mutation in LCA patients (C998X) is noted. The location of the splice-blocking MO is depicted by the hashed bar and the flanking primers used for RT–PCR are shown with black arrows. The open reading frame for the region of interest is depicted with amino acids highlighted in gray representing the sequence from exons 25 and 26 flanking the included intron sequence that begins at amino acid 1018. Asterisks represent the stop codons. (B) RT–PCR from wild-type and cep290 morphant embryos injected with 8 ng of MO at the following stages: 8–10 somites, 72 hpf and 5 dpf. Only morphants with a straight body axis at 72 hpf and 5 dpf were used to assess the gene knockdown. The aberrantly spliced cep290 transcript in morphant embryos (intron inclusion) is present through 5 dpf. β-actin expression served as a loading control.

41
Figure 10 Gross cep290 morphant phenotypes. Live images of 5 dpf wild-type, control-injected and cep290 morphant embryos representing the range of gross morphology observed. MO-injected embryos presented with body plans that were straight, bent or curly.

42
Table 2 Percentages of embryos with body curvature defects based on MO dose
ExperimentalgroupPhenotype(percentage)
nStraight Bent Curly
Wild‐type 100 0 0 129cep290MO(5ng) 60 6 34 205cep290MO(8ng) 40 5 55 285cep290MO(10ng) 13 7 80 208

43
Figure 11 cep290 gene knockdown results in KV defects. (A) Side view of a 10-somite embryo highlighting the location of the ciliated KV (circle) in the tailbud. (B-D) Dorsal views of an average-sized KV, an abnormally reduced in KV size and an absent KV, respectively, (arrow heads) at the 10-somite stage. Embryos were scored has having an abnormal KV if the vesicle was less than the width of the notochord. (E) The percentage of wild-type and cep290 morphant embryos with abnormal KVs. MO concentration and the number of embryos (n) are noted on the x-axis. **P < 0.01, Fisher's exact test when compared with wild-type

44
Figure 12 cep290 gene knockdown results in melanosome transport delay. (A) Dorsal view of the head melanocytes of a 5 dpf dark-adapted wild-type embryo. The boxed region represents the zoomed region for B and C. (B) Dark-adapted melanocytes prior to epinephrine treatment note the dispersed melanosomes. (C) The end point, perinuclear localization of melanosomes, after epinephrine treatment. (D) Epinephrine-induced retrograde transport times for wild-type and cep290 morphants. MO concentration and the number of embryos (n) are noted on the x-axis. **P < 0.01, ANOVA with Tukey when compared with wild-type.

45
Figure 13 Rescue of the cep290 morphant vision defect. (A) Vision function was assayed in 5-day old embryos by testing embryos sensitivity to short block in light at 30 second intervals for 5 trials (adapted from easter and Nicola 1996). Selected images from a time-lapse collection before and immediately after a one second block in light. The typical response, a distinct C-bend, is scored as a positive response as shown in time period 139ms. ms, milliseconds. (B) Vision startle response asay for wild-type, cep290 morphants and rescue experiments. Experimental group and the number of embryos (n) are noted on the x-axis. The N-terminal RNA is sufficient to rescue the cep290 morphant vision defect. **P < 0.01, ANOVA with Tukey when compared with wild-type. (C) Schematic of wild-type full-length human CEP290 protein with the N-terminal construct highlighted in blue (amino acids 1–1059) and the C-terminal construct highlighted in green (amino acids 1765–2479). The location of the common LCA human mutation, C998X, is noted. Crx, cone-rod homebox gene.

46
Table 3 Compiled data set of KV, melanosome transport and vision startle response assay results for rescue experiments
Experimentalgroup KV(%) MT(min)±SE Vision(response)±SE
Wild‐type 5.2 1.51±0.07 3.53±0.17controlMO(10ng) 8.3 1.52±0.06 3.48±0.18cep290MO(8ng) 18.2++ 2.10±0.10** 2.46±0.12**cep290MO(8ng)+Nterminus(1.8ng) 22.6++ 2.05±0.10** 3.48±0.13cep290MO(8ng)+Cterminus(0.6ng) 15.4++ 1.82±0.07 2.44±0.22**
++ Fisher’s Exact test P< 0.01.
** ANOVA with Tukey P< 0.01.
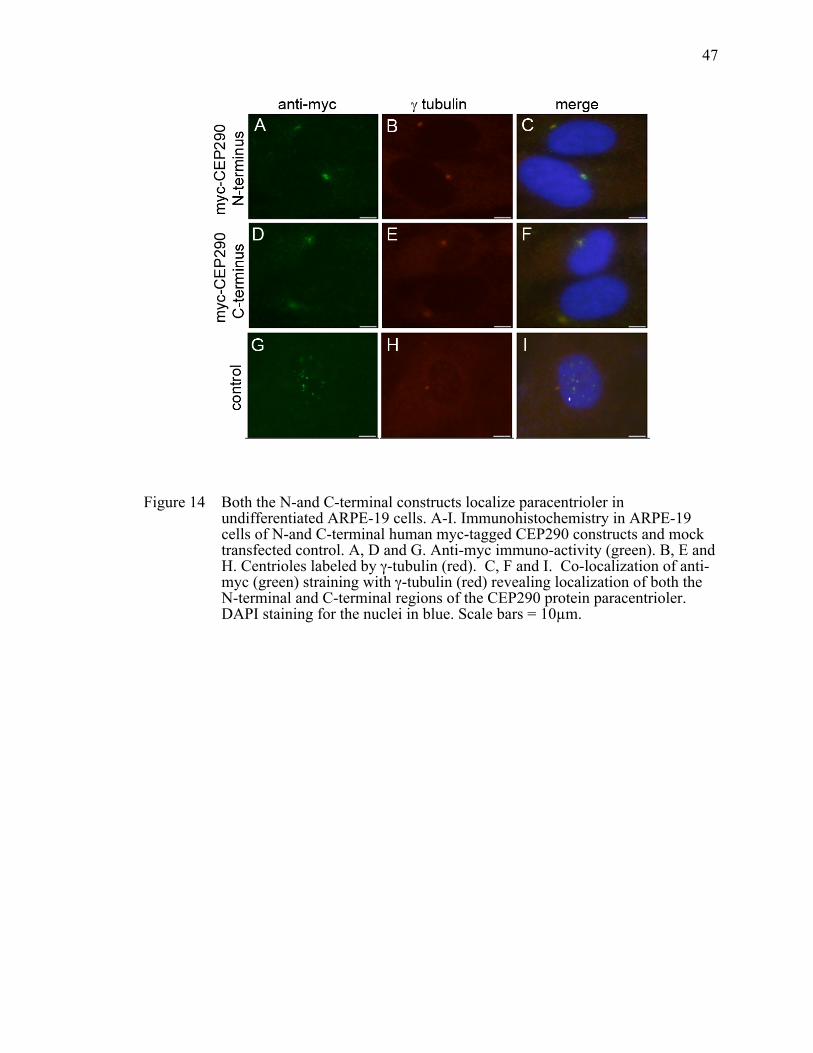
47
Figure 14 Both the N-and C-terminal constructs localize paracentrioler in undifferentiated ARPE-19 cells. A-I. Immunohistochemistry in ARPE-19 cells of N-and C-terminal human myc-tagged CEP290 constructs and mock transfected control. A, D and G. Anti-myc immuno-activity (green). B, E and H. Centrioles labeled by γ-tubulin (red). C, F and I. Co-localization of anti-myc (green) straining with γ-tubulin (red) revealing localization of both the N-terminal and C-terminal regions of the CEP290 protein paracentrioler. DAPI staining for the nuclei in blue. Scale bars = 10µm.

48
Figure 15 N-terminal and C-terminal of CEP290 specifically localize to the mature centriole in ciliated ARPE-19 cells. A-I. Immunohistochemistry in ciliated ARPE-19 cells of N-and C-terminal human myc-tagged CEP290 constructs and mock transfected control. A, D and G. Anti-myc immuno-activity (green). B, E and H. Cilia labeled by acetylated tubulin (red). C, F and I. Merge of anti-myc (green) straining with acetylated tubulin (red) revealing localization of both the N-terminal and C-terminal regions of the CEP290 protein only in the centrioles and not the cilia. DAPI staining for the nuclei in blue. Scale bars = 10µm.
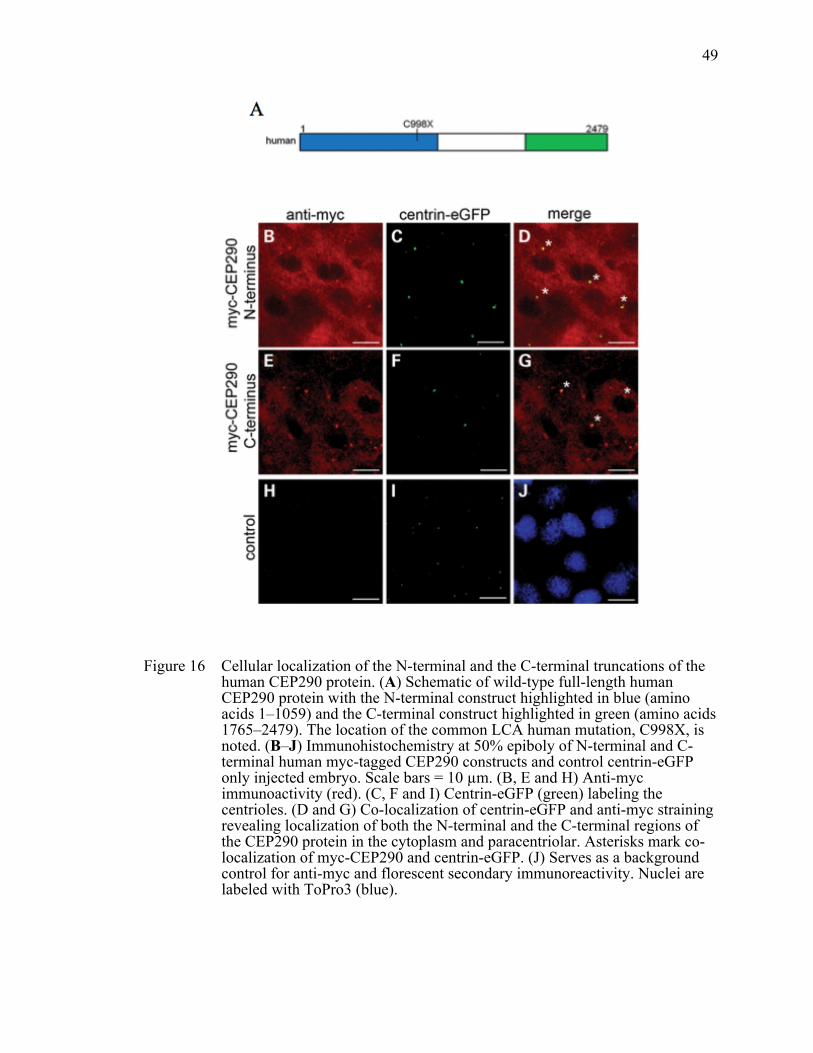
49
Figure 16 Cellular localization of the N-terminal and the C-terminal truncations of the human CEP290 protein. (A) Schematic of wild-type full-length human CEP290 protein with the N-terminal construct highlighted in blue (amino acids 1–1059) and the C-terminal construct highlighted in green (amino acids 1765–2479). The location of the common LCA human mutation, C998X, is noted. (B–J) Immunohistochemistry at 50% epiboly of N-terminal and C-terminal human myc-tagged CEP290 constructs and control centrin-eGFP only injected embryo. Scale bars = 10 µm. (B, E and H) Anti-myc immunoactivity (red). (C, F and I) Centrin-eGFP (green) labeling the centrioles. (D and G) Co-localization of centrin-eGFP and anti-myc straining revealing localization of both the N-terminal and the C-terminal regions of the CEP290 protein in the cytoplasm and paracentriolar. Asterisks mark co-localization of myc-CEP290 and centrin-eGFP. (J) Serves as a background control for anti-myc and florescent secondary immunoreactivity. Nuclei are labeled with ToPro3 (blue).

50
CHAPTER III
THE INTERACTION BETWEEN THE BBSOME
AND CEP290 IS REQUIRED FOR MEDIATING
CILIA FUNCTION
Introduction
Bardet-Biedl Syndrome (BBS, OMIM 209900) is a pleiotropic disorder
characterized by retinal degeneration, obesity, learning difficulties, polydactyly and renal
abnormalities. To date, seventeen BBS genes have been identified (BBS1-BBS12, MKS1,
CEP290, WDPCP, SDCCAG8, and LZTFL1) (Leitch, Zaghloul et al. 2008; Zaghloul and
Katsanis 2009; Otto, Hurd et al. 2010; Seo, Baye et al. 2010; Marion, Stutzmann et al.
2012) Interestingly, four BBS genes (MKS1, CEP290, WDPCP and SDCCAG8) are
primarily associated with other ciliopathies including nephronophthisis (NPHP), Joubert
syndrome (JBTS) and Meckel-Gruber syndrome (MKS), whose phenotypic components
partly overlap with those of BBS (Weatherbee, Niswander et al. 2009; Kim, Shindo et al.
2010; Sang, Miller et al. 2011; Di Gioia, Letteboer et al. 2012). Although functional
relationships between these four genes and other BBS genes are currently not well
understood, recent protein-protein interaction studies have determined an interaction
network composed of NPHP, JBTS, and MKS proteins, including CEP290 and MKS1
(Garcia-Gonzalo, Corbit et al. 2011; Sang, Miller et al. 2011; Williams, Li et al. 2011). In
this network, CEP290 has been implicated in establishing a gatekeeping function that
regulates ciliary protein trafficking (Craige, Tsao et al. 2010; Garcia-Gonzalo, Corbit et
al. 2011).
Interestingly, mutations in CEP290 result in a wide variety of distinct phenotypes,
ranging from isolated blindness (Leber Congenital Amaurosis; LCA), NPHP, JBTS, and
BBS to the lethal MKS (Coppieters, Lefever et al. 2010). It is presumed that the clinical
variability of CEP290-related diseases might be caused by second-site modifier alleles,

51
such as AHI1(Ferland, Eyaid et al. 2004) and TMEM67 (MKS3) (Leitch, Zaghloul et al.
2008). In addition, BBS is characterized by significant intra-familial phenotypic
variability and it has been proposed that in some families this phenomenon can be
explained by the presence of a third BBS gene mutation (Badano, Kim et al. 2003; Leitch,
Zaghloul et al. 2008) and/or the contribution of modifier genes (Badano, Leitch et al.
2006). However, how CEP290 mutations result in BBS, whether and how CEP290
interacts with other BBS proteins, and whether this interaction contributes to intra-
familial phenotypic variability in BBS patients remain unclear.
Here, we report physical and functional interaction between the BBSome and
CEP290 and show that the BBSome is required for the proper localization of Cep290 in
mouse photoreceptor cells. We also demonstrate the physiological relevance of the
genetic interaction between Cep290 and Bbs4 in mediating cilia function. Our studies
explore the synergic interaction between BBS genes and CEP290, and phenotypic
variability in ciliopathies.
Materials and Methods
Animal Studies
Mice were bred and maintained in standardized conditions at the University of
Iowa. The use of mice was approved by the University Animal Care and Use Committee.
Cep290 rd16/rd16 mutant mice (Chang, Khanna et al. 2006) on a BXD24 background and
Bbs4-/- mice (Mykytyn, Mullins et al. 2004) on a 129/Sv background were used to
generate double mutants on a mixed BXD24 and 129 background. Weight of mice was
measured monthly beginning at weaning, and that of double heterozygous mice was
compared with single heterozygous mice from 1 to 5 months of age using an average of
10 animals per group. Circulating leptin levels were analyzed in blood collected from
adult double heterozygous mutant mice (n =5) and control mice (n =3). Plasma was

52
obtained by centrifuging blood at 2,040 g for 8 min. Concentration of murine leptin was
measured by ELISA using a mouse leptin ELISA kit (Crystal Chem).
Antibodies, Plasmids, and Reagents
Expression vectors for BBS genes have been previously published (Seo, Baye et
al. 2010). The four CEP290 fragments encoding about 600 amino acid (1–661 amino
acids, 661–1276 amino acids, 1276–1714 amino acids and 1714–2479 amino acids) of
human wild-type CEP290 (NCBI Reference Sequence: NG_008417.1) were amplified
from human retina cDNA (Clonetech), cloned into the pSS-FS2 vector (Humbert,
Weihbrecht et al. 2012) and sequence verified. The PCR primers used for these clonings
are:
• CEP290—primer (1-661 amino acids) F: 5′-ATGCCACCTAATATAAACTG-3′
• CEP290—primer (1-661 amino acids) R: 5′-CATTTCCTTAATTGCTT-3′
• CEP290—primer (661–1276 amino acids) F: 5′-ATGCAGAAAGATCCTG-3′
• CEP290—primer (661–1276 amino acids) R: 5′-TCCACTAAACTGTC-3′
• CEP290—primer (1276–1714 amino acids) F: 5′-
GGAGCTTTACCCTTGGCAC-3′
• CEP290—primer (1276–1714 amino acids) R: 5′-
TGCTTCTTTTTGAGCCTGAAG-3′
• CEP290—primer (1714–2479 amino acids) F: 5′-GCAAATTCAAGAGCTCC-3′
• CEP290—primer (1714–2479 amino acids) R: 5′-
TTAGTAAATGGGGAAATTAACAGG -3′
Small interfering RNAs (siRNAs) were purchased from Dharmacon (ON-
TARGETplus SMARTpool) and transfected at 50 nM concentration for all experiments
with RNAiMAX (Invitrogen) following manufacturer’s protocol.
Antibodies against BBS1, BBS2 and BBS4 were described previously (Nachury,
Loktev et al. 2007). Other antibodies used were purchased from the following sources:

53
mouse monoclonal antibodies against acetylated tubulin (6–11B-1; Sigma), γ-tubulin
(GTU-88; Sigma), FLAG (M2; Sigma), GFP (3E6; Invitrogen), beta-actin (AC-15;
Sigma), IQ-motif containing B1 (IQCB1/NPHP5; Abcam), Rhodopsin (RET-P1; Santa
Cruz), and Synaptophysin (sc-55507; Santa Cruz); rabbit polyclonal antibodies against
BBS7 (Proteintech Group), BBS8 (HPA003310; Sigma), BBS9 (Sigma), CEP290
(Bethyl lab; for immunoblotting and RPE1 cell immunofluorescence microscopy),
ABCA4 (Abcam), HSPA5/GRP78 (Cell Signaling), PCM1 (HPA023374; Sigma), PDI
(P7372; Sigma), PRPH2 (18109; Proteintech Group), and Tgoln2/Tgn46 (Abcam). Anti-
Cep290 antibody used for mouse photoreceptor cell immunofluorescence microscopy
was a gift from Dr. Robert Mullins (University of Iowa).
Cell culture, Transfection and Co-Immunoprecipitation Assay
hTERT-RPE1 cells were maintained in DMEM/F12 media (Invitrogen)
supplemented with 10% FBS. Cells were transfected with siRNAs using RNAiMAX for
48 hrs and further incubated in serum-free medium for 24 hrs for ciliation. HEK293T
cells were maintained in DMEM (Invitrogen), supplemented with 10% fetal bovine
serum and penicillin/streptomycin (Invitrogen), and grown at 370C in 5% CO2. Stable cell
lines expressing FLAG-tagged BBS4 and FLAG-tagged BBS5 were generated by co-
transfecting pSS-FS-BBS4 and pSS-FS-BBS5 with pCS2-puro into HEK293T cells
followed by puromycin selection (1.5 µg/ml; Invitrogen). Cells were grown in 6-well
dishes, transfected with FLAG-tagged CEP290 or BBSome subunits using Lipofectamine
2000 (Invitrogen), and harvested after 24 hours by scraping into 500 µL ice-cold lysis
buffer (50mM HEPES, 200mM KCl, 2mM EGTA, 1mM MgCl2, 10% glycerol, 1%
Triton-X100) supplemented with protease inhibitors (Roche). Co-IP was performed as
previously described (Seo, Baye et al. 2010).

54
Quantitative Real-Time PCR
Total RNA was extracted using TRIzol Reagent (Invitrogen) following the
manufacturer’s instruction. Quantitative PCR was performed as previously described
(Seo, Baye et al. 2010). RPL19 mRNA levels were used for normalization and ΔΔCt
method (Livak and Schmittgen 2001) was used to calculate fold inductions. The PCR
products were confirmed by melt-curve analysis and sequencing.
Immunoprecipitation
Human HEK293T cells were lysed in ice-cold lysis buffer supplemented with
protease inhibitors (Roche) and spun at 20,000 x g for 15 min at 4°C. The supernatants
were incubated with antibodies against CEP290 overnight. Protein A beads (Thermo
scientific) were then added and incubated for 2 hrs. The beads were washed four times
with lysis buffer and the interactions were detected by Western blotting. TrueBlot anti-
rabbit IgG secondary antibodies (Rockland) were used to avoid detecting IgG heavy and
light chains. For the immunoprecipitation in the mouse retina, the mouse eyes were
enucleated and homogenized in ice-cold lysis buffer (50 mM HEPES, 200 mM KCl, 2
mM EDTA, 1% NP-40, 0.5 mM DTT) supplemented with protease inhibitors (Roche).
The lysate were spun at 20,000 x g for 15 min at 4°C, and the supernatants were
precleared by incubation with protein A and proteins G beads (Thermo scientific) for 2
hours. Immunoprecipitation was performed on the cleared lysates through the procedure
described above.
Immunofluorescence Microscopy
For immunofluorescence microscopy, cultured cells were seeded onto glass
coverslips in 24-well plates and transfected with siRNAs or with plasmid DNAs as
described above. Cells were cultured for 72 hours before fixation with the last 24 hours in
serum-free medium to induce ciliogenesis, and then fixed with 4% paraformaldehyde in
PBS followed by cold methanol. Samples were blocked with 5% BSA and 2% normal

55
goat serum in PBST (0.1% Triton X-100), and incubated with primary antibodies in the
blocking buffer. For immunofluorescence microscopy of retinal sections, eyes were fixed
as above and embedded and frozen in OCT. Sections (7 µm) were collected and blocked
with 5% BSA in PBST for 20 min at 25°C before incubation with primary antibody at
4°C overnight. Primary antibodies were visualized by Alexa Fluor 488 goat anti-rabbit
IgG (Invitrogen) and Alexa Fluor 568 goat anti-mouse IgG (Invitrogen). Coverslips were
mounted on VectaShield mounting medium with DAPI (Vector Lab), and images were
taken with Olympus IX71 microscope.
Photoreceptor Outer Segment Isolation
The mouse eyes were enucleated and placed in ice-cold homogenization buffer
(50% w/v sucrose in PBS, complete protease inhibitor). The retina was dissected from
enucleated eyes and then disrupted by repeated pipetting through a P1000 tip with
approximately 1.5-2.0 mm orifice followed by 30-second vortexing. The homogenate
was spun at 200 g for 3 minutes and supernatant was further spun at 13,000 g for 20 min
at 4oC. After centrifugation, supernatant was diluted 1:1 in PBS without sucrose. The
diluted OS fraction was applied on top of a 50% (w/w) sucrose/PBS cushion and
centrifuged at 13,000 g for 30 min at 4oC. The outer segment fraction was collected at the
interface and diluted with 5 volumes of PBS. Rod outer segment (ROS) was collected as
pellet after the diluted fraction was spun at 6,000 g for 10 min at 4oC. For SDS-PAGE
and Western blot analysis, the pellet was re-suspended in 50 µL of 2x LDS buffer
(Invitrogen).
Histology and Immunohistochemistry
After euthanasia by carbon dioxide inhalation, eyes were collected and immersed
in a solution of 4% paraformaldehyde in 10 mM phosphate-buffered saline (PBS; pH
7.4). After 2 to 4 hours of fixation, eyes were washed three times in 10 mM PBS
followed by infiltration and embedding in acrylamide (Johnson and Blanks 1984), and

56
then were frozen in OCT (Sakura). Cryostat sections were collected at a thickness of 7
µm, and stored at -20°C until use. Sections were stained following standard Hematoxylin
and Eosin (H&E) staining protocol.
For immunofluorescence microscopy of retinal sections, the cryosections (7 µm)
were collected and blocked with BSA in PBST for 20 min at 25°C before incubation with
primary antibody at 4°C overnight. Following washes with PBS three times, sections
were incubated in Alexa Fluor-conjugated goat anti-rabbit and goat anti-mouse secondary
antibodies for 2h at 25°C. Nuclei were counterstained with Vectashield containing DAPI.
Electroretinography (ERG) Recordings
Full field ERGs were obtained (Espion V5 system; Diagnosys LLC, Lowell, MA).
After overnight dark adaptation, the mice were anesthetized with an intraperitoneal
injection of ketamine (87.5 mg/kg) and xylazine (2.5 mg/kg). ERGs were recorded
simultaneously from the corneal surface of each eye after pupil dilation (1% tropicamide)
using gold ring electrodes (Diagnosys) referenced to a needle electrode (Roland/LKC,
Brandenburg, Germany) placed on the back of the head. Another needle electrode placed
in the tail served as ground. A drop of methylcellulose (2.5%) was placed on the corneal
surface to ensure electrical contact and to maintain corneal integrity. Body temperature
was maintained at a constant temperature of 38°C using the system's heating pad. All
stimuli were presented in a Ganzfeld (ColorDome; Diagnosys), and the mouse head and
electrode positioning were monitored on the camera attached to the system. Dim red light
was used for illumination until dark-adapted testing was completed. The a- and b-waves
were recorded with an escalating 11-step protocol starting at 0.01 cd · s · m−2 and ending
at 25 cd · s · m−2. The b-wave used for analysis was the highest amplitude b-wave
registered, typically the ninth flash (4 cd · s · m−2), and the a-wave was calculated from
the same flash.

57
The a-wave was measured from the baseline to the trough of the first negative
wave. The b-wave was measured from the trough of the a-wave to the peak of the first
positive wave, or from the baseline to the peak of the first positive wave if no a-wave was
present.
Leptin Resistance Study
Sex-, age-, and body weight-matched control (single heterozygous) and double
heterozygous mice were housed in individual cages at 6-10 weeks of age. Food was
removed 18 hours before intra peritoneal (i.p.) leptin injection. Leptin (1 µg/g body
weight; R&D Systems) or vehicle (PBS) was injected i.p. and animals were sacrificed 2
hours later by CO2 asphyxiation. Hypothalami were quickly dissected and homogenized
in the lysis buffer containing protease and phosphatase inhibitors. Protein extracts were
loaded onto 4-12% NuPAGE Bis-Tris gels and analyzed by immunoblotting.
Analysis of Kupffer’s Vesicle
Live somite staged embryos were photographed with the use of Zeiss Axiocam
Camera. Vesicles in embryos with the width smaller than that of the notochord (less than
approximately 50 µm in diameter) were considered reduced, while embryos in which
vesicles could not be morphologically identified were scored as absent.
Melanosome Transport Assay
The melanosome transport assay was performed as previously described (Yen,
Tayeh et al. 2006; Tayeh, Yen et al. 2008; Pretorius, Baye et al. 2010). Dark-adapted day
5 larvae were exposed with epinephrine (50mg/ml, Sigma, E4375) added to embryo
medium for a final concentration of 500 µg/ml. Melanosome movement was continuously
monitored under the microscope and the endpoint time was recorded when all
melanosomes in the head and the trunk were perinuclear.

58
Results
Physical Interaction Between the BBSome and CEP290
The current model suggests that CEP290 exerts its major effects at the transition
zone of cilia through interactions with other NPHP, JBTS and MKS proteins to control
ciliary gating (Craige, Tsao et al. 2010; Sang, Miller et al. 2011). However, localization
of CEP290 and BBSome components (BBS4 and BBS9) to the centriolar satellites has
been noted (Kim, Krishnaswami et al. 2008; Lopes, Prosser et al. 2011; Seo, Zhang et al.
2011), suggesting possible interactions between CEP290 and the BBSome. To test this,
we utilized stable cell lines expressing FLAG-BBS4 and FLAG-BBS5 and examined
their association with endogenous CEP290. As shown in Fig 17, endogenous CEP290
and other BBSome subunits were co-immunoprecipated with both BBS4 and BBS5,
indicating that CEP290 associates with the BBSome. Immunoprecipitaion (IP) of
endogenous CEP290 using HEK293T cell lysates and mouse retinal extracts confirmed
the interaction of the BBSome and CEP290 under more physiological conditions (Figure
18A and B). To determine the BBSome-interacting domain of CEP290, we transfected
four truncated CEP290 proteins, with each containing about 600 amino acids, into
HEK293T cells and examined their association with endogenous BBSome subunits.
Figure 19 and 20A showed that the BBSome (as represented by BBS4, BBS7, and BBS9)
binds to the N-terminal part of CEP290. Among the BBSome components, BBS4 is the
BBSome subunit that directly interacts with CEP290 (Figure 21).
It has been shown that Pericentriolar material 1 (PCM1) also localizes to the
centriolar satellite and physically interacts with BBS4 and CEP290 (Kim, Krishnaswami
et al. 2008; Lopes, Prosser et al. 2011). To test whether PCM1 mediates the interaction
between CEP290 and the BBSome, we blocked the expression of PCM1 by transfecting
small interfering RNAs (siRNAs) and evaluated the BBSome-CEP290 interaction. In this
experiment, depletion of PCM1 did not result in any significant change in the association

59
between CEP290 and the BBSome (Figure 20B), indicating that the BBSome-CEP290
physical interaction is PCM1-independent.
The Colocalization of the BBSome and CEP290
We next examined localization of the BBSome and CEP290. As previously
reported (Kim, Krishnaswami et al. 2008; Lopes, Prosser et al. 2011), CEP290 localizes
to the ciliary base (the transition zone) and centriolar satellites in hTERT-RPE1 cells
(Figure 22A). BBS4 shows extensive overlap with CEP290 in the centriolar satellite but
also localizes to the cilium, where CEP290 is not detected. BBS9, another component of
the BBSome, shows similar localization pattern (Figure 26A). These observations suggest
that the BBSome-CEP290 interaction mostly occurs in the centriolar satellites.
We next investigated the localization of Cep290 and Bbs4 in mouse retina by
immunofluorescence microscopy. Consistent with previous reports (Chang, Khanna et al.
2006), Cep290 was primarily detected in the connecting cilium of photoreceptor cells
(Figure 22B). Bbs4 was also found in the connecting cilium of photoreceptor cells. Anti-
Bbs4 staining in the Outer Nuclear Layer (ONL) is non-specific because staining is also
observed in Bbs4-null retinas (Figure 22B, right panel). To further verify the co-
localization of Cep290 and the BBSome to the photoreceptor connecting cilium, the
photoreceptor outer segments (OS) and connecting cilium were isolated from mouse
retina and probed for the presence of BBSome components and Cep290. Western blotting
results showed that the OS markers, Rhodopsin and ATP-binding cassette sub-family A,
are highly enriched in the isolated OS fraction(Illing, Molday et al. 1997), whereas
GRP78, PDI (two endoplasmic reticulum markers)(Vasireddy, Jablonski et al. 2006;
Yang, Wu et al. 2007), TGN-46 (Golgi marker)(Evans, Schwarz et al. 2010) and
synaptophysin (synaptic vesicles marker)(Brandstatter, Lohrke et al. 1996) were detected
in the whole retinal extract but not isolated OS, findings which support the high degree of
purity of outer segment isolation (Fig 23). In the isolated OS, we also found Bbs4, Bbs7

60
and Cep290 (Fig 23), supporting co-localization of the BBSome and Cep290 to the
photoreceptor OS.
Together, our data indicate that the BBSome and CEP290 interact in the centriolar
satellite in RPE1 cells and in the connecting cilium of the photoreceptor cell.
Proper Localization of CEP290 to the Centriolar Satellite and
Photoreceptor Connecting Cilium is BBSome-dependent
We next evaluated functional interactions between the BBSome and CEP290. We
first tested whether the BBSome is required for proper localization of CEP290. To this
end, we examined CEP290 localization in BBS1, BBS4 and BBS9 depleted hTERT-
RPE1 cells. Efficient depletion of BBS1, BBS4, and BBS9 by siRNA transfection was
confirmed at the protein or mRNA level (Figure 24). In control siRNA transfected cells,
CEP290 localized to centriolar satellites, particularly near the centriole (Figure 25).
BBS1, BBS4 or BBS9 knockdown resulted in a substantial decrease in the centriolar
satellite pool of CEP290. A similar finding was also observed in PCM1 knockdown cells.
These results indicate that association of CEP290 with the centriolar satellite is BBSome-
dependent. Of note is that the CEP290 pool in the transition zone and centrosomes was
not affected by PCM1 and BBS protein depletion. Interestingly, CEP290 in the
centrosome and transition zone was also not affected by microtubule depolymerization,
while centriolar satellite localization of CEP290, PCM1 and BBS4 was disturbed in the
same condition (Lopes, Prosser et al. 2011). These findings suggest that localization of
CEP290 to the transition zone and centrosomes may be independent of centriolar
satellites. Alternatively, centrosome-associated CEP290 may be stable with a slow rate of
turn over. A previous study by Craige et al. reported that BBS proteins were moderately
increased in Chlamydomonas cep290 mutant flagella (Craige, Tsao et al. 2010). In our
experiment, ciliary localization of the BBSome was not affected by CEP290 depletion
and its localization to the centriolar satellite was also not affected (Figure 26).

61
We then examined whether the BBSome is required for Cep290 localization in
mouse retina. To minimize potential secondary effects of retinal degeneration in BBS
mice, we examined Cep290 localization in young animals (P21-P30) before retinal
degeneration is apparent. At this age, the ONL thickness in Bbs1M390R/M390R and Bbs4-/-
retina is comparable to that of wild-type animals (Figure 27). In addition, OS proteins
Peripherin 2 (Prph2; also known as Rds) and the vast majority of Rhodopsin localize
properly to the OS, demonstrating that protein trafficking to the OS is not completely
disrupted. In Bbs1M390R/M390R retina, however, Cep290 was found dispersed throughout
the photoreceptor cell, including the entire OS, inner segment, and the nuclear layer
(Figure 27). A similar phenotype was also observed in Bbs4-/- mice. Consistent with the
results obtained from RPE1 cells, the Cep290 pool at the connecting cilium persisted in
BBS mutant retinas. These results suggest that the BBSome limits Cep290 to the
connecting cilium and prevents improper localization of Cep290 in other locations in
photoreceptor cells.
Synergetic Interaction Between CEP290 and BBS Genes
in vitro and in Zebrafish
Given the physical interaction between CEP290 and the BBSome and the
requirement of the BBSome for proper localization of CEP290, we examined the genetic
interaction between CEP290 and other BBS genes in vitro and in zebrafish. First, we
examined whether the interaction is required for ciliary localization of ARL13B/ JBTS8,
which is a small GTPase and localized to the cilia in primary neurons (Cantagrel, Silhavy
et al. 2008). Although knockdown of CEP290 or BBS4 did not affect the localization of
ARL13B in vitro, double depletion of CEP290 and BBS4 disrupted the localization of
ARL13B to the cilium (Figure 28 and 29), suggesting the interaction is required for its
ciliary localization.

62
Furthermore, in zebrafish, knockdown of bbs1-14 individually using morpholinos
(MO) results in KV defects and intracellular melanosome transport delay (Yen, Tayeh et
al. 2006; Tayeh, Yen et al. 2008; Pretorius, Baye et al. 2010). The KV is a transient
ciliated structure and has a role in left-right patterning (Essner, Amack et al. 2005), and
KV defects are identified as the reduction of KV size to less than the width of the
notochord (Figure 30). In addition, zebrafish are able to adapt to their surroundings
through trafficking of melanosomes in response to either light or hormonal stimulation
(Barral and Seabra 2004). In the melanosome transport assay, 5-day old zebrafish
embryos were dark adapted and treated with epinephrine to stimulate retrograde
melanosome transport (Pretorius, Baye et al. 2010) (Figure 30). Our data showed that
double knockdown of cep290 and bbs genes results in more severe KV defects and delay
in retrograde melanosome transport delay than single zebrafish morphants with equal
amount of morpholino injected (Figure 31), suggesting the synergetic interaction between
these genes.
Increased Body Weight in Mice with Combined Loss of
Cep290 and Bbs4 Genes
Next, we investigated the genetic interaction between Cep290 and BBS genes in
mouse system. To this end, we used the rd16 mice (Cep290rd16) (Chang, Khanna et al.
2006) and Bbs4 knockout (Mykytyn, Mullins et al. 2004) mouse lines. The rd16 mice
(Cep290rd16) harbor a hypomorphic allele with an internal in-frame deletion of 299
residues (amino acids 1599–1897; NP_666121) from the Cep290 protein (Chang, Khanna
et al. 2006). In humans, the phenotype of CEP290 mutations ranges from LCA, which
manifests only photoreceptor degeneration, to the neonatal lethal MKS, which involves
severe cystic kidney and neural tube patterning defects as well as photoreceptor
degeneration. Cep290rd16 homozygous mutant mice were first reported to display
photoreceptor degeneration phenotype but other components of the CEP290-associated

63
ciliopathy phenotypes such as obesity, cystic kidney, and neural tube patterning defects
were not fully investigated. Obesity is one of the cardinal features of BBS patients. To
determine whether rd16 mice develop obesity as found in BBS human patients and other
BBS mutant mice, we weighed the rd16 mice and their WT littermate controls at 4
months. The rd16 mice have minimally elevated body weight (Figure 26); however, fat
mass analysis by weighing individual fat depots revealed that rd16 mice have increased
fat mass (Figure 32).
We examined whether the phenotype of Cep290rd16 mice is affected by the
presence of Bbs4 mutations. First, we measured the body weight of progenies from
Cep290rd16 and Bbs4 null mouse crosses (Figure 33). In general, body weight correlated
with the number of mutant alleles in Cep290 and Bbs4 genes (Table 4 and 5). For
example, Cep290rd16/rd16 single homozygous mutant mice did not develop obesity (Figure
33). However, loss of one copy of Bbs4 on this background (i.e. Cep290rd16/rd16; Bbs4+/-)
resulted in a significant increase in body weight. Cep290rd16/rd16; Bbs4-/- double
homozygous mutant mice were the most obese among all genotypes examined (Figure
33). Interestingly, double heterozygous mice (Cep290+/rd16; Bbs4+/-) showed significantly
increased body weight compared with their sex-matched wild-type or single heterozygous
littermates (Figure 34). Higher blood leptin levels and leptin resistance were observed in
obese BBS mice (Davis, Swiderski et al. 2007; Rahmouni, Fath et al. 2008). Consistent
with this, Cep290+/rd16; Bbs4+/- animals showed higher blood leptin levels than wild-type
or single heterozygous littermates (Figure 34). We further compared the responsiveness
of hypothalamic anorexic circuitry to exogenous leptin by measuring phosphorylated
STAT3 upon leptin injection (Seo, Guo et al. 2009). In control single heterozygous mice,
intra-peritoneal injection of leptin results in a robust increase in STAT3 phosphorylation
(Figure 35). However, the phosphorylation level of STAT3 was significantly reduced in
double heterozygous mice injected with leptin compared to single heterozygous mice.

64
Combined, these data suggest that the obesity component of CEP290-associated
ciliopathy phenotype is affected by the presence of mutations in BBS genes.
Accelerated Retinal Degeneration in Mice with Combined
Loss of Cep290 and Bbs4 alleles
The Cep290rd16/rd16 and Bbs4-/- mice display retinal deficits (Mykytyn, Mullins et
al. 2004; Chang, Khanna et al. 2006; Eichers, Abd-El-Barr et al. 2006). Like the obesity
phenotype, mice carrying a combination of 3 or 4 mutations in Cep290 and Bbs4 genes
displayed more severe phenotypes in retinal photoreceptor cells (Fig 36). For example,
although all layers of the retina were present and appeared grossly normal with inner
segment (IS) lengths comparable with the WT and Cep290rd16/rd16 single homozygotes at
P14, OS elongation was clearly stalled and begin to regress in Cep290rd16/rd16; Bbs4+/- and
Cep290rd16/rd16; Bbs4-/- mouse retinas (Fig 36). Almost complete loss of the OS were
observed in these retinas at P21, with thinner ONL in Cep290rd16/rd16; Bbs4-/- mouse
retinas. Cep290rd16/rd16 single homozygotes also showed severe photoreceptor
degeneration at P28. Interestingly, the double heterozygous mice (Cep290+/rd16; Bbs4+/-)
do not present photoreceptor degeneration compared to the corresponding single
heterozygous mice (Cep290+/rd16; Bbs4+/+) at P14 and P30 (Figure 37), suggesting that
one copy of normal Cep290 is sufficient for normal photoreceptor maintenance, and thus
losing one copy of Bbs4 in this background does not affect the photoreceptor in the
retina.
Next, we sought to examine the effect of combined loss of Cep290 and Bbs4
alleles on the trafficking of phototransduction proteins in the retina. Immunofluorescence
microscopy analysis revealed relatively mild mis-localization of rhodopsin to the ONL in
Cep290rd16/rd16 single homozygous retinas at P21, while no mis-localization was detected
in wild-type photoreceptor cells (Figure 38). Losing one or two copies of Bbs4 in this
background further impaired rhodopsin trafficking and more rhodopsin was found in the

65
ONL and IS. Consistent with these findings, dark-adapted standard combined response
(DA-SCR) electroretinography (ERG) b-wave of Cep290rd16/rd16; Bbs4+/- and
Cep290rd16/rd16; Bbs4-/- mice at 1-month of age showed considerable decline compared
with that of Cep290rd16/rd16 and Bbs4-/- single homozygotes (Fig 39 and Table 6). In
Cep290rd16/rd16 mice, in addition to the reduction of the ERG b-wave, the oscillatory
potentials were also markedly reduced (Figure 40). This reduction was more pronounced
when one or two Bbs4 null alleles were present, suggesting more severe photoreceptor
dysfunction by loss of Bbs4 alleles in Cep290rd16/rd16 retinas. Alternatively, this reduction
in ERG b-wave may be due to more inner retina dysfunction in these animals. Together,
these data suggest that although homozygous Cep290rd16 alleles cause severe
photoreceptor degeneration in mice, loss of one or two copies of Bbs4 on this background
further disrupts retinal morphology and intercellular protein trafficking in the retina, and
thus results in accelerated photoreceptor degeneration.
Discussion
Genetic and phenotypic heterogeneity of ciliopathies may be ascribed to the
inherent complexity of the molecular mechanisms that underlie the formation and
function of the primary cilium. One possible mechanism is that a group of diverse cilia
proteins implicated in different types of ciliopathies can act collaboratively in a common
molecular pathway probably as several multisubunit protein complexes. Mutations in
each protein disrupt the partial function of a protein complex in various tissues, resulting
in distinct but overlapping symptoms in various diseases. In addition to the BBSome
previously identified, recent data reported the NPHP-JBST-MKS complex, which is
required for cilia integrity, apical organization and sonic hedgehog signaling (Sang,
Miller et al. 2011). Moreover, more than 100 unique human mutations were identified in
CEP290, giving rise to a range of phenotypes including NPHP, LCA, SLSN, JBTS, BBS
or the lethal MKS (Czarnecki and Shah 2012). Although the exact mechanism behind the

66
phenotypic variability is not clear, it is conceivable that various symptoms are caused by
different interactions between CEP290 and its interacting partners. Our data presented
here further develop the idea of the interlinked actions of cilia proteins.
The BBSome functions as a coat complex to transport membrane proteins
between plasma and ciliary membranes (Jin, White et al. 2010). Our data show that both
the BBSome and CEP290 colocalize to centriolar satellites. Moreover, CEP290 binds to
the BBSome via its N-terminal domain. Notably, depletion of the BBSome causes loss of
CEP290 localization to centriolar satellites. CEP290 has been shown to interact with
components of dynein and kinesin motor machineries (Chang, Khanna et al. 2006;
McEwen, Koenekoop et al. 2007), suggesting that CEP290 distribution is regulated by
coordinated actions between plus end- and minus end-directed microtubule motors. The
BBSome knockdown phenotype, which consists of abnormal concentric accumulation of
CEP290 rather than dispersion of CEP290, indicates that the BBSome might act to either
destabilize the interaction between CEP290 and dynein motors or to promote the
CEP290-kinesin motor interaction, helping the movement of CEP290 back to the cytosol.
Although knockdown of PCM1 also results in a similar mis-localization of CEP290, it is
very likely that the BBSome and PCM1 behave differently in regulating CEP290
localization. The BBSome may act downstream of PCM1 based on previous work that
showed that depletion of two BBS complex subunits (BBS1 or BBS5) does not effect the
localization of PCM1 and centrosomal proteins (Nachury, Loktev et al. 2007). This idea
is supported by our data showing PCM1-independent interaction between CEP290 and
the BBSome. Consistent with our data, a recent study showed that ciliary recruitment of
BBS4 and the BBSome is regulated by a mechanism involving CEP72 and CEP290 that
occurs at the level of BBS4 relocalization from centriolar satellites, and that PCM1 is not
necessary for this process (Stowe, Wilkinson et al. 2012). Taken together, the physical
interaction between CEP290 and the BBSome is important for their correct localization,
which provides further insights into the cause of the genetic heterogeneity of BBS.

67
Obesity is highly prevalent in BBS individuals (Moore, Green et al. 2005), but not
in other CEP290-related disease. Moreover, the observation that obesity is frequent in
BBS heterozygous individuals raises the possibility that carrying one mutant allele of
BBS genes predispose to obesity (Croft, Morrell et al. 1995). In our mouse model, double
heterozygous male mice (Cep290+/rd16; Bbs4+/-) show higher body weight and leptin
levels than do single heterozygous male mice. These data support a genetic interaction
between Cep290 and Bbs4. In addition, our data showed that rd16 mice have a higher fat
percent without increased body weight. However, when we crossed the rd16 mouse with
the Bbs4-null mice from another mouse strain, this obesity phenotype was not observed
in Cep290rd16/rd16 mice on this mixed genetic background, suggesting that the obesity
phenotype is strain-dependent and is possibly influenced by some unknown modifiers
from the original rd16 strain. In fact, the clinical variability of cilia-related disorders may
be explained, at least in part, by the presence of modifiers. For example, CCDC28B
(coiled-coil domain containing 28B; also named MGC1203) had been shown to
contribute to the penetrance of the BBS phenotypes (Badano, Leitch et al. 2006).
Other than increased body weight, losing additional copies of either Cep290 or
Bbs4 genes also exacerbate photoreceptor degeneration. Moreover, using morpholinos to
simultaneously knock down cep290 and three BBS genes (bbs1, bbs4 and bbs9) in
zebrafish caused more severe defects in Kupffer’s Vesicles and delay in epinephrine
induced melanosome transport (Essner, Amack et al. 2005). Both fish and mouse data
suggest a synergic interaction between these Cep290 and BBS genes.
A recent study using Mkks (also known as Bbs6) null mice and Cep290rd mice
showed a less severe phenotype or even rescue of the defects observed in sensory cells of
Cep290rd16 and Mkksko double-homozygous and triple allelic combinations (Rachel, May-
Simera et al. 2012). However, fish data suggest a more severe ciliary defect by combined
morpholino knockdown of Cep290 and Mkks (Rachel, May-Simera et al. 2012), which
indicates the cooperative effect of these two genes on zebrafish eye development.

68
Considering the requirement of Mkks for the BBSome assembly (Seo, Baye et al. 2010),
these contrasting results in these two studies are interesting. However, it is noteworthy
that in Mkks-/- mice several BBSome components (e.g. Bbs2, Bbs7, and Bbs9) are
unstable and degraded leading to failure of BBSome assembly (Seo, Baye et al. 2010),
while in Bbs4-/- mice the BBSome is formed normally except the absence of Bbs4
(Zhang, Yu et al. 2012). The presence of the BBSome (with or without BBS4) in
Cep290rd16/rd16 or Cep290rd16/rd16; Bbs4-/- mice may cause disease in these animals, while
the lack of BBSome in Cep290rd16/rd16; Mkks-/- mice may alleviate the mutant phenotype.
Another possibility that can cause the differences between the Cep290-Mkks study
and ours is the functional independence of individual domains in Cep290 and specific
loss of a protein-protein interaction in rd16 mutant mice. In the Mkksko/ko; Cep290rd16/rd16
mouse study, Cep290 was shown to interact with Mkks via its Myosin-tail homology
domain, which is deleted in the rd16 mouse (Rachel, May-Simera et al. 2012), whereas
our data show that CEP290 interacts with the BBSome via its N-terminus. Therefore, it is
possible that residual Cep290 function, i.e. the function of Cep290 through its interaction
with the BBSome, still persists in the rd16 mouse model, and additional loss of one or
two copies of BBSome genes will disrupt additional Cep290 function, resulting in the
more severe phenotypes observed in our mouse model. In addition, our data show that
both BBSome proteins and CEP290 localize to the connecting cilium in mouse retina,
and that Cep290 localization is BBSome-dependent, indicating that the BBSome is
required for normal function of CEP290 in retina. It is known that the deleted region of
Cep290 in the rd16 mouse, particularly the myosin-tail homology domain of Cep290,
performs a retina-specific function (Badano, Mitsuma et al. 2006; Chang, Khanna et al.
2006). Here our findings indicate that N-terminal of Cep290 is also required for correct
localization of OS components, key to the normal function and structure of
photoreceptors, and that this function possibly occurs through interaction with the
BBSome.

69
In summary, the data presented here demonstrate physical and genetic interactions
between BBS proteins and CEP290. Our findings show that the BBSome is required for
proper localization of CEP290 in cultured ciliated cells and in photoreceptor connecting
cilia. We also demonstrate the combined loss of 3 or 4 copies of BBS genes and CEP290
results in more severe phenotypes compared to homozygous mutation of a single gene.

70
Figure 17 The BBSome interacts with CEP290. Co-immunoprecipitation of endogenous CEP290 in FLAG-BBS4 and FLAG-BBS5 transfected HEK293 cells. Lysates from transfected and control cells were subjected to immunoprecipitation (IP) with anti-FLAG antibody and precipitated proteins were analyzed by immunoblotting with indicated antibodies. Normal mouse IgG pull-down was used as a negative control.

71
Figure 18 Interaction of endogenous CEP290 and the BBSome in HEK293T cells (A) and mouse retina (B). Lysates from HEK293T cells and mouse retina were subjected to IP using antibodies against CEP290 and precipitated proteins were analyzed by immunoblotting with indicated antibodies.

72
Figure 19 Schematic representation of the CEP290 deletion mutants. Numbers indicate expressed portions of CEP290 in amino acid positions. Known IQCB1, CC2D2A and RAB8A binding domains and the BBSome interacting region are also summarized. SMC, structural maintenance of chromosomes; MYO-Tail, myosin tail homology domain.

73
Figure 20 PCM1-independent physical interaction between CEP290 and the BBSome. (A) The BBSome binds to the N-terminal part of CEP290. CEP290 deletion mutants (FLAG-tagged) were transfected into HEK293T cells and lysates were analyzed by IP using anti-FLAG antibodies. Expression of endogenous BBS4,7,9 and NPHP5 is shown in lysates (left). NPHP5, known to interact with a CEP290 fragment spanning amino acid 696-896, was used as positive control. Untransfected cells were used as a negative control. (B) PCM1-independent interaction between the BBSome and CEP290. HEK293T cells were co-transfected with Cep_1 fragment and siRNA against PCM1. Untransfected cells were used as a negative control.

74
Figure 21 BBS4 interacts with CEP290. HA-tagged, individual BBSome components were transiently transfected with FLAG-Cep_1 construct. Lysates were subjected to IP with anti-HA antibodies.

75
Figure 22 Co-localization of CEP290 and the BBSome to centriolar satellites in cultured cells and to the connecting cilium of photoreceptor cells. (A) Co-localization of GFP-tagged BBS4 and CEP290 in RPE1 cells (yellow arrowheads). Localization of CEP290 (red) was probed with anti-CEP290 antibody in hTERT-RPE1 after 48 hours of serum withdrawal, while BBS4-GFP was probed with anti-GFP antibody. Nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI, blue). Note the variable localization of BBS4, although these cells were cultured in the same condition (from a single well). (B) Co-localization of Bbs4 and Cep290 in the mouse retina. Antibodies against Cep290 (red) and rhodopsin (green; a marker of the outer segment) were used in WT photoreceptors in the left panel, whereas in the middle and right panel Bbs4 (red) and rhodopsin (green) localizations were probed in WT and Bbs4-/- (4KO) photoreceptors. Both Cep290 and Bbs4 localize to the connecting cilium. OS, outer segment; CC, connecting cilium; IS, inner segment; ONL, outer nuclear layer.

76
Figure 23 Co-fractionation of the BBSome and Cep290 in the photoreceptor outer segment fraction. Photoreceptor outer segments (OS) were isolated from the mouse retina and probed with antibodies against multiple subcellular marker proteins, Bbs4, Bbs7 and Cep290.

77
Figure 24 Suppression of BBS gene expression by RNAi. hTERT-RPE1 cells were transfected with siRNAs as indicated and relative mRNA levels (A) and protein levels (B and C) were compared by quantitative PCR and immunoblotting, respectively. The lower band in panel C is recognized as non-specific one.

78
Figure 25 CEP290 localization in the centriolar satellite is BBSome-dependent. (A) The BBSome is required for centriolar satellite localization of CEP290 (red). RPE1 cells were transfected with siRNAs against CEP290, BBS1, BBS4, BBS9 and PCM1. Antibodies against γ-tubulin and acetylated tubulin (green) were used to mark the basal body and cilia, respectively. (B) Quantification of CEP290 mis-localization in BBSome depleted RPE1 cells. One-way ANOVA followed by Tukey post-test was used for statistical analysis. **, P<0.01 compared to Ctrl KD cells. Graphs represent mean ± SE from three independent experiments.

79
Figure 26 CEP290 is not involved in BBS9 localization to cilia. (A) BBS9 localization is CEP290-independent. RPE1 cells were transfected with siRNAs against CEP290 and PCM1 and BBS9 (red) localization was examined. Cilia and basal body are labeled with acetylated tubulin and γ-tubulin antibodies (green). (B) Suppression of CEP290 and PCM1 expression by RNAi. hTERT-RPE1cells were transfected with siRNAs as indicated and protein levels were compared by immunoblotting. (C) Reduced cilia in PCM1-depleted cells. hTERT-RPE1 cells were transfected with siRNAs against CEP290 and PCM1, and then percentage of ciliated cells is examined.

80
Figure 27 Cep290 localization to the connecting cilium is BBSome-dependent. Localization of Cep290 (green; left) in WT (top), Bbs1M390R/M390R (middle), and Bbs4-/- (bottom) mouse retinas. OS localization of Prph2 (green) is not affected in BBS mutant retinas (right panels).

81
Figure 28 Decreased ARL13B ciliary localization in BBS4 and CEP290 double knockdown cells. In ciliated RPE1 cells, ARL13B (green) localizes to cilium. Expression of indicated genes was blocked by siRNA transfection. Cilia and centrosomes were marked by acetylated tubulin and γ -tubulin antibodies (red).

82
Figure 29 Quantitation of ARL13B ciliary localization. The number of ciliated cells with ciliary ARL13B staining was counted. Results are averages of two independent experiments with at least 60 cells counted in each experiment. Error bars represent standard errors. **, P < 0.01 compared to CTRL.

83
Figure 30 Features of BBS in zebrafish. (A-C) Images of live zebrafish embryos at 8-10 somite stage. (A) Slide view of an embryo highlighting the location of the ciliated KV. (B and C) Dorsal view of a normal sized and reduced sized KV. (D-F) Epinephrine-induced melanosome transport of a wild-type 5-day old zebrafish embryo. Melanosome transport is observed in cells on the head of the embryos. Boxed region is magnified for E and F. (E) Wild-type embryos prior to treatment with epinephrine and (F) the endpoint at 1.45 minutes after treatment.

84
Figure 31 Medium-dose pair-wise combination knockdowns of cep290, bbs1, bbs4 and bbs7. Phenotypic pair-wise combination knockdowns of genetically interacting bbs genes result in increase severity and penetrance of bbs knockdown phenotpyes, including KV defect (A) and epinephrine-induced melanosome trasnport (B) when compared to low-dose bbs-MO injected embryos. The numbers of embryos were noted on the x axis and values on the top of the bars. **, P<0.01

85
Figure 32 The rd16 mice have increased body fat percentage. Comparison of (A) body weight and fat mass as assessed by weight of individual fat depots (B) between the rd16 mice and WT littermate controls. Although the mice have increased white adipose tissues like gonadal fat and retroperitoneal fat, they do not have increased brown adipose tissue (BAT). *, < 0.05 compared to wild type.
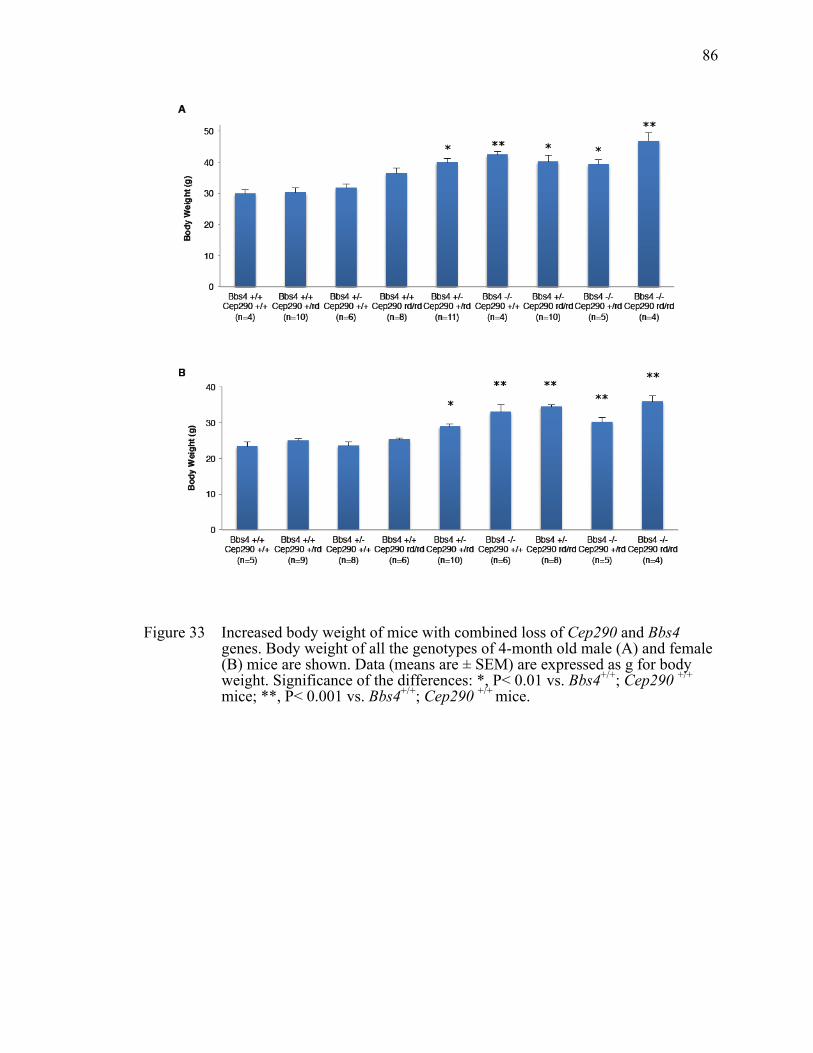
86
Figure 33 Increased body weight of mice with combined loss of Cep290 and Bbs4 genes. Body weight of all the genotypes of 4-month old male (A) and female (B) mice are shown. Data (means are ± SEM) are expressed as g for body weight. Significance of the differences: *, P< 0.01 vs. Bbs4+/+; Cep290 +/+
mice; **, P< 0.001 vs. Bbs4+/+; Cep290 +/+ mice.

87
Figure 34 Increased body weight and higher leptin levels in Bbs4+/-;Cep290+/rd16 mice. (A) Weight gain in male animals vs. age (minimum of 6 animals per group). Values are expressed as mean+SEM. By month 3, double heterozygous mutant mice are significantly heavier than single heterozygous littermates. (B) Serum leptin levels of single and double heterozygous mice. One-way ANOVA and t-test were used for statistical analysis. *, P< 0.05 vs. single heterozygous animals; **, P<0.01 vs. single heterozygous animals.

88
Table 4 Body Weight of all the genotypes (male) from 1 month to 4 month
1 month 2 month 3 month 4 month
Bbs4 +/+ Cep290 +/+ (n=4) 15.00 22.54 24.65 29.98
Bbs4 +/- Cep290 +/+ (n=6) 15.11 22.94 27.85 31.77
Bbs4 +/+ Cep290 +/rd (n=10) 16.04 22.35 25.87 30.23
Bbs4 +/- Cep290 +/rd (n=11) 18.13 25.69 34.50 39.94
Bbs4 -/- Cep290 +/+ (n=4) 12.25 27.14 34.97 42.40
Bbs4 +/+ Cep290 rd/rd (n=8) 15.56 24.09 28.44 36.52
Bbs4 -/- Cep290 +/rd (n=5) 20.85 26.45 34.17 39.43
Bbs4 +/- Cep290 rd/rd (n=10) 16.93 25.93 30.92 40.23
Bbs4 -/- Cep290 rd/rd (n=4) 12.55 27.80 37.81 46.88

89
Table 5 Body Weight of all the genotypes (female) from 1 month to 4 month
1 month 2 month 3 month 4 month
Bbs4 +/+ Cep290 +/+ (n=5) 13.06 18.47 21.89 23.35
Bbs4 +/- Cep290 +/+ (n=8) 12.35 18.63 22.30 23.54
Bbs4 +/+ Cep290 +/rd (n=9) 13.20 19.59 21.48 25.02
Bbs4 +/- Cep290 +/rd (n=10) 16.69 21.58 25.26 28.93
Bbs4 -/- Cep290 +/+ (n=6) 13.54 23.48 29.16 33.01
Bbs4 +/+ Cep290 rd/rd (n=6) 13.27 18.42 22.91 25.30
Bbs4 -/- Cep290 +/rd (n=5) 10.75 22.21 25.61 30.12
Bbs4 +/- Cep290 rd/rd (n=8) 15.07 19.46 24.39 34.50
Bbs4 -/- Cep290 rd/rd (n=4) 11.31 25.31 31.41 35.92

90
Figure 35 Attenuated leptin receptor signaling in Bbs4+/- Cep290+/rd16 mice. A) STAT3 phosphorylation upon leptin administration was reduced in double heterozygous (Bbs4+/- Cep290+/rd16) mice compared with single heterozygous (Bbs4+/- Cep290+/+ or Bbs4+/+ Cep290+/rd16) mice. Hypothalamic protein extracts were analyzed by western blotting. B) Quantification of STAT3 phosphorylation. Band intensities of phospho-STAT3 (P-STAT3) were normalized with those of total STAT3 and induction ratios were calculated by comparing with vehicle-injected samples. Data represent mean + SEM. n=5 for vehicle and n=10 for leptin. *, P< 0.05.

91
Figure 36 Accelerated photoreceptor degeneration with additional loss of Bbs4 alleles in Cep290rd16/rd16 mice. H&E stained sections of the superior central retina at the level of the optic nerve head from P14, P21 and P28 mice are shown.

92
Figure 37 Lack of photoreceptor degeneration with additional loss of Bbs4 alleles in Cep290+/rd16 mice. H&E stained sections of the superior central retina at the level of the optic nerve head from P14 and P30 mice are shown.

93
Figure 38 Impaired rhodopsin trafficking in mice with combined loss of Cep290 and Bbs4 alleles. Immunohistochemical analysis of WT, Bbs4+/+; Cep290rd16/rd16, Bbs4+/-; Cep290rd16/rd16 and Bbs4-/-; Cep290rd16/rd16 retinas at P21 with antibodies against rhodopsin.

94
Figure 39 Diminished ERG responses in CEP290rd16/rd16 mice with additional loss of Bbs4 alleles. Dark-adapted standard combined response ERG b-wave amplitudes in mice with the indicated genotypes (at age 1-month). Removing one or two Bbs4 alleles on a Cep290rd16/rd16 background results in a lower ERG response. **P<0.01 vs. Bbs4+/+; Cep290rd16/rd16 mice. Error bars are SD.

95
Table 6 Summarize of ERG data of 1-month old mice
b–wave amplitude
BBS4 -/- Cep290 rd/rd (n=6) 21.79 + 6.9A
BBS4 +/- Cep290 rd/rd (n=6) 57.61 + 14.41A
BBS4 +/+ Cep290 rd/rd (n=7) 98.06 + 18.36A
BBS4 +/- Cep290 +/rd (n=4) 527.6 + 161.39
BBS4+/+ Cep290 +/rd (n=3) 607.37 + 49.26
BBS4+/- Cep290+/+ (n=3) 821.2 + 24.84
A ANOVA with Tukey P< 0.01.

96
Figure 40 Reduced ERG b-wave and oscillatory potential of mice with combined loss of Cep290 and Bbs4 genes. Representative dark-adapted standard combined response ERG (top) and oscillatory potentials (bottom) from indicated genotypes are shown. All mice were 1 month old.

97
CHAPTER IV
INTERACTION BETWEEN CEP290 AND OTHER
CILIOPATHY PROTEINS IS REQUIRED FOR THE
CORRECT LOCALIZATION OF PROTEINS TO CILIA
Introduction
Ciliopathies exhibit considerable variations in phenotype even between siblings
with the same mutations in a single family, as well as phenotypic overlap among
clinically distinct diseases. CEP290 is one of the most representative ciliopathy genes
displaying variable expressivity and associated with multiple ciliopathies (Coppieters,
Lefever et al. 2010). For example, mutations in CEP290 are associated with Leber
congenital amaurosis (LCA), NPHP, SLSN, JBTS, BBS, and MKS. LCA is an inherited
retinal dystrophy causing severe vision losses within the first year of life. SLSN involves
cystic kidney as well as retinal degeneration. Patients with JBTS or JBTS-related disease
(JSRD) such as cerebello-oculo-renal syndrome (CORS) display cerebellar vermis
aplasia/hypoplasia in addition to renal and ocular anomalies. Finally, MKS is a neonatal
lethal disease characterized by central nervous system malformations, cystic kidney, and
polydactyly. Patients harboring the same CEP290 mutations often develop different
degrees of neurological, ocular, and renal involvement, and are clinically diagnosed with
different diseases
Other than CEP290, mutations in NPHP1, NPHP3, and NPHP8/RPGRIP1L are
linked to NPHP, JBTS and MKS (Baala, Audollent et al. 2007), which suggests it is not
uncommon that different alleles of the same gene can result in these three disorders, and
thus the associated proteins in these diseases may interact and function together in
common pathways. This idea is supported by recent work identifying the NPHP-JBTS-
MKS interactome (Sang, Miller et al. 2011). The proteins associated with this
interactome do not form a single complex. Rather, this large group of disease proteins can

98
be clustered into three biochemically and functionally distinct modules: NPHP1-4-8,
NPHP5-6, and Mks (Sang, Miller et al. 2011). Furthermore, some of these proteins have
been shown to localize to the basal body (BB)—a centriolar structure universally required
for extending the microtubule-based ciliary axoneme—or to an adjacent domain, termed
transition zone (TZ) in most cilia (NPHP1, NPHP4, CEP290, RPGRIP1L), as well as to
connecting cilia in photoreceptors (NPHP1, NPHP5, CEP290). The connecting cilia of
photoreceptors are considered to be the orthologous structure of the TZ in other cell types
(Sayer, Otto et al. 2006; Delous, Baala et al. 2007; Bergmann, Fliegauf et al. 2008;
Valente, Logan et al. 2010). In particular, CEP290 has been shown to localize to the Y-
linkers of the TZ, and its disruption in Chlamydomonas alteres the ciliary composition of
IFT components (Craige, Tsao et al. 2010). However, an obvious CEP290 orthologue is
absent from the Caenorhabditis genome, where morphologically intact Y-linkers are still
present (Czarnecki and Shah 2012); thus, the role of CEP290 and other BB/TZ-associated
ciliopathy proteins remains unclear. Here, we demonstrate that CEP290 interacts
physically with other NPHP and MKS proteins in BB/TZ, and show that this interaction
is important for the correct localization of proteins to the cilium.
Materials and Methods
DNA Constructs, Reagents and Antibodies
The fragments encoding the N-terminal (1–1059 amino acids) and the C-terminal
regions (1765–2479 amino acids) of human wild-type CEP290 (NCBI Reference
Sequence: NG_008417.1) and full-length Inversin (NCBI Reference Sequence:
NG_008316.1) were amplified from human retina cDNA (Clonetech), cloned into the
pEGFP_C3, pCS2+FLAG or pCS2+Myc and sequence confirmed. BBS3 expression
vector with FLAG tag was a gift from Dr. Zhang (University of Iowa). The CEP290
c.3176del mutation was generated by introducing the appropriate nucleotide change into

99
the N-terminal flag-tagged hCEP290 constructs using the Quick Chang II site-directed
mutagenesis kit (Stratagene).
• CEP290—primer N-terminal F: 5′-ATGCCACCTAATATAAACTGG-3′
• CEP290—primer N-terminal R: 5′-
TCATATTTTTTTTGAAATGGAAACAATGTC-3′
• CEP290—primer C-terminal F: 5′-ATGTCTGCAACTTCTCAAAAAGAG-3′
• CEP290—primer C-terminal R: 5′-TTAGTAAATGGGGAAATTAACAGG-3′
Small interfering RNAs (siRNAs) were purchased from Dharmacon (ON-
TARGETplus SMARTpool) and transfected at 50 nM concentration for all experiments
with RNAiMAX (Invitrogen) following manufacturer’s protocol.
Antibodies used were purchased from the following sources: mouse monoclonal
antibodies against acetylated tubulin (6–11B-1; Sigma), γ-tubulin (GTU-88; Sigma),
FLAG (M2; Sigma), GFP (18A11, Santa Cruz), Myc (9E10; Santa Cruz), IQ-motif
containing B1 (IQCB1/NPHP5; Abcam); rabbit polyclonal antibodies against CEP290
(Bethyl lab), SDCCAG8 (Proteintech), Inversin (Proteintech), MKS1 (sigma), NPHP4
(Proteintech), ARL13b (Proteintech), MKS3 (Sigma). MG132 was purchased from
Calbiochem (San Diego, CA).
Cell Culture, Transfection and
Co-immunoprecipitation Assay
HEK293T cells were grown in Dulbecco's modification of Eagle's medium
(DMEM) (Invitrogen) supplemented with 10% FBS (Invitrogen). For individual protein–
protein interaction studies, cells were transfected in 6-well plates with total 1.5 µg
indicated plasmids using Lipofectamine Reagent (Invitrogen). After 30 h of incubation,
cells were lysed in the lysis buffer (50mM HEPES, 200mM KCl, 2mM EGTA, 1mM
MgCl2, 10% glycerol, 1% Triton-X100) supplemented with Complete Protease Inhibitor
Mixture (Roche Applied Science). Lysates were immunoprecipitated with anti-Flag and

100
anti-GFP antibodies conjugated to agarose beads for 4 h at 4°C. Beads were washed in
the lysis buffer four times, and precipitated proteins were analyzed by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS–PAGE) and western blotting following
a standard protocol.
hTERT-RPE1 cells were maintained in DMEM/F12 media (Invitrogen)
supplemented with 10% FBS. Cells were transfected with siRNAs using RNAiMAX for
48 hrs and further incubated in serum-free medium for 24 hrs for ciliation.
Quantitative Real-time PCR
Total RNA was extracted using TRIzol Reagent (Invitrogen) following
manufacturer’s instruction. Quantitative PCR was performed as previously described
(Seo, Baye et al. 2010). Relative gene expression was calculated by the ΔΔCt method
after normalization with GAPDH. The PCR products were confirmed by melt-curve
analysis and sequencing.
Immunofluorescence Microscopy
For immunofluorescence microscopy, cells were seeded on glass coverslips in 24-
well plates and transfected with siRNAs or with plasmid DNAs as described above. Cells
were cultured for 72 hours before fixation with the last 24 hours in serum-free medium to
induce ciliogenesis, and then were fixed with cold methanol. Samples were blocked with
5% BSA and 2% normal goat serum in PBST (0.1% Triton X-100), and incubated with
primary antibodies in the blocking buffer. Primary antibodies were visualized by Alexa
488 or Alexa 568 labeled secondary antibodies (Invitrogen, Carlsbad, CA). The slides
were mounted with Vectashield mounting media containing DAPI (Vector Laboratories,
Burlingame, CA).

101
Sucrose Gradient Ultracentrifugation
Eyes from animals were lysed in lysis buffer, clarified by centrifugation,
concentrated by Amicon Ultra-15 (30 kDa). The samples were loaded on a Superose-6
10/300 GL column (GE Healthcare), and spun at 166,400 x Gavg for 13 hrs. Fractions
(~210 µl) were collected from the bottom using a 26 G needle and concentrated by
TCA/acetone precipitation. Proteins were re-suspended in equal volume of 2x SDS-
PAGE sample loading buffer and analyzed by SDS-PAGE and immunoblotting. The
column was calibrated with Gel Filtration Standard (Bio-Rad).
Results
Physical Interaction Between CEP290 and
Other Ciliopathy Proteins
We tested the hypothesis that CEP290 interacts with other ciliopathy proteins. In
transient transfection and co-immunoprecipitation assays, we found that the N-terminus
of CEP290 interacts with NPHP2/Inversin (Figure 41). Next, we transfected low levels of
the C-terminus of CEP290 with FLAG tag and probed its association with endogenous
ciliopathy proteins. As shown in Figure 43, endogenous SDCCAG8 and MKS1 were co-
immunoprecipitated with the C-terminus of CEP290, indicating that this region associates
with SDCCAG8 and MKS1. To verify the immunoprecipitation data, wild type mouse
eye extracts were applied to a Superose-6 gel filtration column, and the elution profile
was examined (Figure 44). All the proteins tested (Cep290, Mks1, Inversin, Sdccag8 and
Nphp4) were eluted in the same faction with an estimated molecular weight of 770-1,000
kDa, supporting the association between these proteins.
In addition, to further explore the N-terminus of CEP290, we generated another
CEP290 construct, which included 5 additional amino acids downstream (towards the
COOH terminus) of our previous N-terminal construct. This new construct mimicks the
mutation in CEP290 that causes cerebello-oculo-renal syndrome in human patients

102
(Valente, Silhavy et al. 2006). When we transfected this construct into HEK293T cells,
the resulting truncated protein is degraded (Figure 42), indicating that no or low levels of
CEP290 are the cause of severe compromise in the relevant tissues resulting from this
human mutation. Degradation of the truncated protein can be inhibited by MG132, a
proteasome inhibitor (Figure 42), suggesting that the ubiquitin-proteasome pathway is
involved in this degradation of this specific truncated CEP290. In contrast, PBA, a
chemical chaperone, had no effect on the degradation of the truncated protein.
Requirement of CEP290 and Other NPHP and MKS
Proteins for the Correct Localization of Ciliary Proteins
We sought to determine the functional interaction between NPHP and MKS
proteins. First, we examined whether the localization NPHP and MKS proteins is co-
dependent. Depletion of CEP290, Inversin, SDCCAG8 and MKS1 by siRNA transfection
was performed and confirmed at the mRNA level (Figure 45). Normally Inversin
localizes to the centrosome and the proximal region of the cilium (Figure 46). Depletion
of CEP290 or SDCCAG8 results in loss of ciliary localization of Inversin (Figure 46 and
47), indicating that the localization of Inversin to the cilium is CEP290 and SDCCAG8-
dependent. We also investigated the ciliary localization of other proteins by double
knockdown of specific NPHP and MKS genes in vitro. One of the proteins tested is
MKS3/TMEM67. Mutations of this gene cause MKS, as well as Joubert syndrome
(Baala, Romano et al. 2007). MKS3 localizes to the TZ and the proximal end of the
cilium (Figure 48), but simultaneous depletion of any two of three NPHP proteins
(NPHP2/Inversin, NPHP6/CEP290, NPHP10/SDCCAG8) disrupts ciliary localization,
rather than the TZ localization, of MKS3/TMEM67 (Figure 48 and 49), although single
knockdown of each of them does not affect the localization of TMEM67. In addition, we
examined the localization of ARL13B (JBST8) in double knockdown cells. ARL13B, a
small GTPase of the Arf/Arl family, specifically localizes to cilia and has been shown to

103
regulate the placement of interneurons in the developing cerebral cortex (Higginbotham,
Eom et al. 2012). Depletion of either MKS1 or Inversin in CEP290-depleted cells
decreases the amount of ARL13B entering cilia (Figure 50 and 51). Taken together, our
data indicate that interaction between CEP290 and other NPHP and MKS proteins is
required for cilia entry of some proteins (Table 7).
Discussion
Numerous cilia-related diseases are associated with developmental defects of the
central nervous system, skeleton, kidney, retina and liver suggesting that cilia play
critical roles in the development, tissue organization, and physiological function of
multiple organ systems. Many ciliopathy genes encode proteins that localize to the TZ of
the primary cilium, underscoring the role of the TZ in the generation and maintenance of
the cilia. In fact, the presence of transition fibers, Y-linkers and the necklace structure of
the TZ forms an obvious physical gate to ciliary-bound transmembrane proteins, while
acting as a barrier to exclude others. In this study, by combining genetic and biochemical
analyses, we identified a complex containing several MKS and NPHP proteins. This
complex is necessary for normal ciliary membrane composition. Furthermore, our data
supported the concept that MKS and NPHP are likely disorders of macromolecular
complexes sharing a common biological function (Sang, Miller et al. 2011), with severe
loss of complex function leading to the lethal disorders such as MKS, while less severe
impairment causing milder disorders such as NPHP. Consistent with this model, mouse
null mutants including Mks1-/-, Inv-/-, and Cep290-/- mice are lethal (Weatherbee,
Niswander et al. 2009; Sugiyama, Kohno et al. 2012), whereas the rd16 mouse, which
has an in-frame deletion in Cep290, has a less severe phenotype (Chang, Khanna et al.
2006), suggesting only partial loss of CEP290 protein function. In addition, other
functional modules/complexes including TCTN1, TCTN3 and several MKS proteins

104
have been reported to have distinct yet essential ciliary functions that are associated with
equivalent ciliopathies (Garcia-Gonzalo, Corbit et al. 2011).
We also identified several CEP290 interactors, which underscore the importance
of CEP290 in the TZ. Interestingly, CEP290 interacting proteins partner with CEP290 via
different CEP290 domains, supporting the idea that the clinical variability of CEP290-
related diseases is caused by partial loss of the complex function due to mutations in
different domains of CEP290 leading to various degrees of compromise of the TZ and
cilia function. In addition, one CEP290 mutant variant (c.3176del (p.Ile1059LysfsX6)),
which is found in one patient with cerebello-oculo-renal syndrome, creates a frameshift
starting at codon Ile1059 with the new reading frame ending in a nonsense mutation five
codons downstream (Valente, Silhavy et al. 2006). Our data show that this mutant protein
is not stable, which suggests that the more severe phenotype related to this mutation is
caused by loss of the entire CEP290 protein rather than partial compromise of its
function.
The ciliary gate has been long thought to be important in cilia function
(Rosenbaum and Witman 2002), but is only recently being studied at the molecular level
(Craige, Tsao et al. 2010). It is presumed that the BB/TZ region facilitates ciliary gate
function as a docking site for proteins destined for the cilium, as well as a diffusion
barrier. We demonstrate that ciliated cells without normal MKS or NPHP proteins lack a
normal docking cite or lack active ciliary transport. This is supported by the lack of
specific proteins in the cilia. Alternatively, ciliary proteins may enter cilia normally, but
require NPHP and MKS proteins for retention in cilia. At a minimum, our data indicate
that MKS /NPHP proteins play a role in establishing the TZ as a ciliary gate. We predict
that these TZ proteins likely function in coordination with other mechanisms (such as IFT
and the BBSome) to control ciliary composition and thus function (Craige, Tsao et al.
2010; Jin, White et al. 2010).

105
The genetic interactions we observe in vitro also likely exist between TZ genes in
mammals. For example, MKS1 and CEP290 heterozygous variants are associated with
increased phenotypic pleiotropy in some NPHP and BBS patients in which primary
disease symptoms are caused by mutations in other genes (Tory, Lacoste et al. 2007;
Leitch, Zaghloul et al. 2008). The phenotypic diversity caused by transition zone
dysfunction may relate to whether mutations disrupt ciliogenesis in addition to affecting
ciliary membrane composition. The former may cause MKS, while the latter may result
in NPHP or BBS.

106
Figure 41 The N-terminal region of CEP290 interacts with NPHP2. (A) Schematic of wild-type full-length human CEP290 protein with the N-terminal construct highlighted in blue (amino acids 1–1059) and the C-terminal construct highlighted in green (amino acids 1765–2479). The location of the common LCA human mutation, C998X, is noted. (B) FLAG or GFP-tagged CEP290 expression constructs and Myc-tagged NPHP2 were transfected into HEK293T cells, and lysates were subjected to co-IP and immunoblotting (IB). Expression of each component in the lysate is found in the bottom two panels. IPs were analyzed by SDS–PAGE and immunoblotting using the Myc antibody and either the FLAG or the GFP antibodies shown in the upper two panels. Myc-NPHP2 was only co-immunoprecipitated from cells expressing the N-terminal CEP290 fragment (top panel). FLAG-tagged BBS3 served as a negative control.
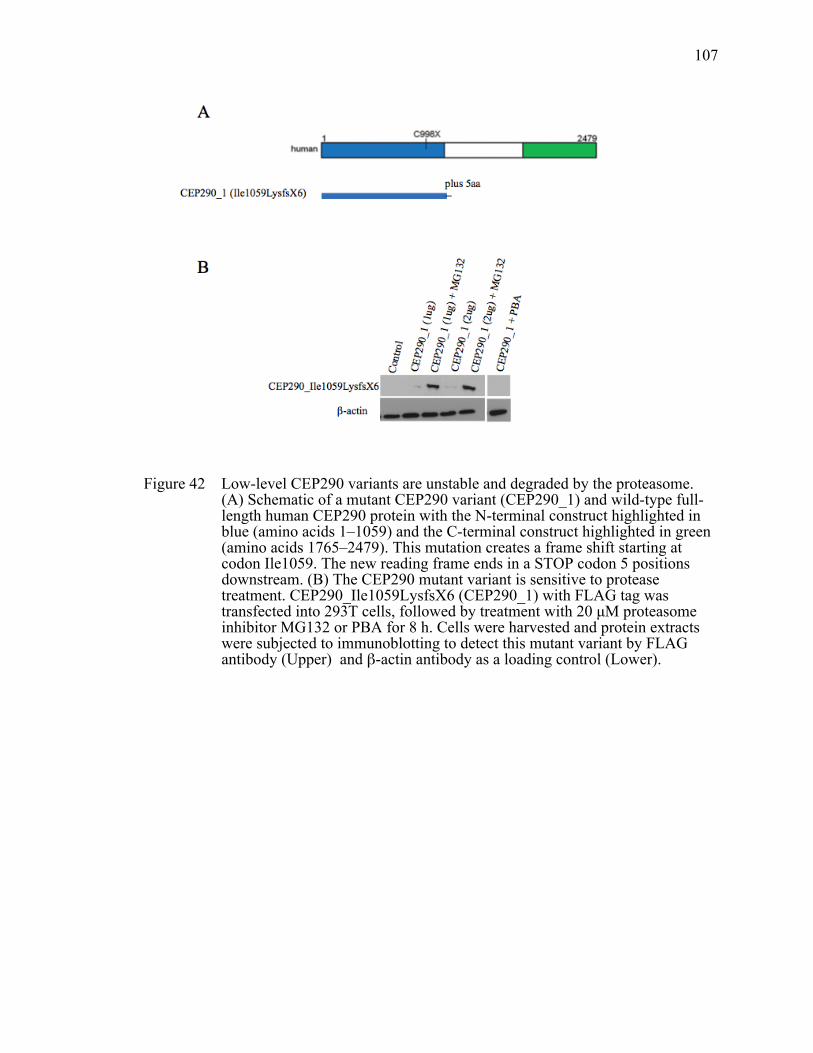
107
Figure 42 Low-level CEP290 variants are unstable and degraded by the proteasome. (A) Schematic of a mutant CEP290 variant (CEP290_1) and wild-type full-length human CEP290 protein with the N-terminal construct highlighted in blue (amino acids 1–1059) and the C-terminal construct highlighted in green (amino acids 1765–2479). This mutation creates a frame shift starting at codon Ile1059. The new reading frame ends in a STOP codon 5 positions downstream. (B) The CEP290 mutant variant is sensitive to protease treatment. CEP290_Ile1059LysfsX6 (CEP290_1) with FLAG tag was transfected into 293T cells, followed by treatment with 20 µM proteasome inhibitor MG132 or PBA for 8 h. Cells were harvested and protein extracts were subjected to immunoblotting to detect this mutant variant by FLAG antibody (Upper) and β-actin antibody as a loading control (Lower).

108
Figure 43 Physical interaction between the C-terminus of CEP290, MKS1 and SDCCAG8. HEK293T cells were transfected with FLAG-tagged N-terminus or C-terminus of CEP290 and cell lysates were subjected to immunoprecipitation (IP) using anti-FLAG antibody. Co-precipitation of endogenous MKS1 and SDCCAG8 was examined.

109
Figure 44 Identification of the NPHP/MKS complex. Wild-type mouse eye extracts were fractionated by size exclusion chromatography. Elution fractions were subjected to SDS/PAGE and immunoblotting. Membranes were probed with antibodies against Cep290, Mks1, Inversin, Nphp4, and Sdccag8.

110
Figure 45 Suppression of NPHP and MKS gene expression by RNAi. hTERT-RPE1 cells were transfected with siRNAs as indicated and relative mRNA levels were compared by quantitative PCR. Error bars represent standard errors.

111
Figure 46 Loss of INVS ciliary localization in CEP290 and SDCCAG8 depleted cells. In ciliated RPE1 cells, INVS (green) shows two distinct localization patterns: ciliary and centrosome. Cilia (red) were marked by antibodies against acetylated tubulin and γ – tubulin.

112
Figure 47 Quantification of INVS ciliary localization. The number of ciliated cells with ciliary INVS staining was counted. Knock-down results are the average of two independent experiments with at least 100 cells counted in each experiment. **, P < 0.01 compared to CTRL.

113
Figure 48 NPHP and MKS are required for ciliary localization of TMEM67. Localization of TMEM67 was probed by immunofluorescence after transfecting with indicated siRNAs into hTERT-RPE1 cells. Cilia are labeled with antibodies for acetylated tubulin and γ-tubulin (red) and TMEM67 is in green.

114
Figure 49 Percentage of TMEM67-positive cilia in depleted cells. Graphs represent average of at least two independent experiments a with minimum 60 cells counted in each experiment. Data are shown as means + SEM. **, P < 0.01 compared to CTRL.

115
Figure 50 NPHP and MKS proteins mediate ciliary localization of ARL13B. Expression of indicated genes was blocked by siRNA transfection and ciliary localization of ARL13B (green) was examined in hTERT-RPE1 cells. Cilia and centrosomes were marked by acetylated tubulin and γ – tubulin antibodies (red).

116
Figure 51 Summary of ARL13B ciliary localization. Graphs represent average of at least two independent experiments and bars, SEs. Depleted proteins are shown in the abscissa. *, P< 0.05 compared to CTRL; **, P < 0.01 compared to CTRL.

117
Table 7 Summary of KO Phenotypes in NPHP Proteins and MKS1 Knockdown Cells
siRNA Proteins showing loss of ciliary localization
CEP290 INVS
SDCCAG8 INVS
CEP290 + SDCCAG8 TMEM67 and INVS
INVS + CEP290 TMEM67 and ARL13B
INVS + SDCCAG8 TMEM67
CEP290 + MKS1 ARL13B and INVS
INVS + MKS1 TMEM67

118
CHAPTER V
SUMMARY, CONCLUSIONS AND FUTURE
DIRECTIONS
Summary
Cilia are highly conserved organelles critical for a broad array of homeostatic
mechanisms and paracrine signals. Due to their broad tissue and cellular distribution,
defects in cilia give rise to organ-specific disorders such as polycystic kidney disease as
well as broad, pleiotropic syndromes, such as Bardet-Biedl syndrome. These disorder are
collectively called “ciliopathies” (Quinlan, Tobin et al. 2008). Although the disorders are
defined by unique clinical criteria, they present many overlapping phenotypes such as
retinal degeneration, polydactyly, situs inversus, mental retardation, encephalocele, as
well as cysts in the kidney, liver, and pancreas. Although ciliopathies are thought of as
rare genetic diseases individually, when viewed collectively, their prevalence rate could
be as high as ∼1 in 2000 [based on three common disease traits: renal cysts (1 in 500
adults), retinal degeneration (1 in 3000), and polydactyly (1 in 500)] (Quinlan, Tobin et
al. 2008). Recently, CEP290 has emerged as a pan-ciliopathy gene because the
phenotypic spectrum of its mutations ranges from Leber congenital amaurosis (LCA),
Joubert syndrome (JBTS), Senior-Loken syndrome (SLSN), Bardet-Biedl syndrome
(BBS), Nephronophthisis (NPHP) to the lethal Meckel-Gruber syndrome (MKS),
suggesting the importance of this protein and comprehensive interaction with other
ciliopathy proteins. At the molecular level, CEP290 is believed to be involved in
controlling protein trafficking at the ciliary base. In Chlamydomonas reinhardtii, CEP290
localizes to the transition zone of flagella and is proposed to function as a gatekeeper
regulating flagellar protein content (Craige, Tsao et al. 2010). Recent protein-protein
interaction studies established the CEP290 interaction network at the ciliary transition
zone (Garcia-Gonzalo, Corbit et al. 2011; Sang, Miller et al. 2011). Most proteins in this

119
network are associated with ciliopathies. Although progress has been made in
determining the role of CEP290 in the cilia, gaps remain in our understanding of the
cellular function of CEP290 and its interaction with other ciliopathy proteins.
Using gene knockdown in zebrafish, it has been previously demonstrated that loss
of BBS genes results in reduced size of the ciliated Kupffer’s vesicle (KV), delays in
intracellular melanosome transport, as well as vision impairment (Yen, Tayeh et al. 2006;
Tayeh, Yen et al. 2008; Pretorius, Baye et al. 2010). These phenotypes were used to
examine the function of CEP290 in specific tissues. Knockdown of cep290 in zebrafish
results in key zebrafish features of BBS (KV defects and melanosome transport delay)
and vision impairment. Importantly, the N-terminus of human CEP290, but not the C-
terminus of human CEP290, is sufficient to rescue the vision defect in cep290 zebrafish
morphants. These data demonstrate that the N-terminus of CEP290 is required for proper
retinal function.
With the initial results that underscore the importance of the N-terminus of
CEP290, we used in vitro cells and mice to fully investigate this region of CEP290. We
found that the BBSome binds to CEP290 via its N-terminus, and that this interaction is
required for the correct localization of CEP290 in cultured ciliated cells and in
photoreceptor connecting cilium in the mouse retina. To further characterize the genetic
interaction between CEP290 and BBSome genes, we crossed rd16 mice with Bbs4-null
mice, and found that increased body weight and accelerated retinal degeneration are
associated with combined loss of Cep290 and Bbs4 genes. Interestingly, compared to
single heterozygous mice (Cep290+/rd16; Bbs4+/-+ and Cep290+/+; Bbs4+/-), double
heterozygous mice (Cep290+/rd16; Bbs4+/-) have higher body weight, higher leptin levels
and are resistant to leptin, which recapitulates the obesity phenotypes observed in other
BBS mutant mice (Rahmouni, Fath et al. 2008), However, double heterozygous mice do
not have photoreceptor degeneration, indicating phenotype-specific penetrance.
Importantly, this finding in mice indicates that some humans could be obese due to the

120
combined effect of two or more heterozygous BBS mutations without having other
features of the syndrome. In zebrafish, double knockdown of cep290 and other BBSome
genes results in more severe KV defects and delay in melanosome transport, confirming
the synergetic interaction between CEP290 and the BBSome.
Finally, we utilized in vitro studies to further investigate physical interaction of
CEP290 with other ciliopathy proteins. We found that both termini of CEP290 physically
interact with other ciliopathy proteins, suggesting the interacting proteins share a
common mechanism resulting in disease phenotypes (Table 8). Moreover, double
knockdown of CEP290 and other ciliopathy genes disrupts the cilia entry of specific
proteins in ciliated cells, indicating a role for CEP290 as a gatekeeper in the transition
zone to control entry into and out of the cilium through the interaction with other NPHP
and MKS proteins (Table 8). Additionally, these data support the idea that the
overlapping phenotypes of the ciliopathies result from compromised function of the
transition zone.
Conclusion
Ciliopathies display overlapping as well as distinct phenotypes despite the fact
that they arise from common defects in ciliary function. Recent analysis of ten
comprehensive genomic and proteomics data sets enriched for basal body and ciliary
proteins demonstrate that the cilium requires about 1000 different proteins for its function
(Gherman, Davis et al. 2006). Due to content diversity in the cilium, it is not surprising
that the cilium is involved in multiple fundamental signaling pathways including Wnt and
Hedgehog signaling (Fliegauf, Benzing et al. 2007; Goetz and Anderson 2010;
Christensen, Clement et al. 2012), as well as in the establishment and maintenance of cell
polarity (Fan, Hurd et al. 2004). This expanding view of ciliary roles aids in
understanding the phenotypic variation both within and among syndromes.

121
Ciliopathies are characterized by significant inter- and intra-familial phenotypic
variability. The former could be explained, at least in part, by the extensive genetic
heterogeneity of each disorder. So far, there are 17 BBS genes, 14 LCA genes, 11 NPHP
genes, and 7 JBTS genes identified, and the associated disease proteins have been shown
to share some functional similarities. For example, seven BBS proteins (BBS1, 2,3,5,7,8
and 9) have been shown to form a complex, called the BBSome (Nachury, Loktev et al.
2007), while three other proteins (BBS6, 10 and 12) form a separate complex with CCT
proteins to mediate the formation of the BBSome (Seo, Baye et al. 2010). In addition, the
intra-familial phenotypic variability suggests the presence of modifiers and genetic
interactions among ciliopathy genes. Indeed, modifiers and genetic interactions among
ciliopathy genes have been reported in several studies. For example, a JBTS gene Ahi1
genetically interacts with Nphp1 to modulate retinal degeneration phenotype and
functions as a modifier for ocular component in NPHP (Louie, Caridi et al. 2010).
Synergetic interactions between NPHP and MKS genes were also observed in C. elegans
(Williams, Li et al. 2011). Finally, variable expression of neurological phenotypes that
span MKS and JBTS has been reported in Tmem67 mutant mice (Abdelhamed, Wheway
et al. 2013), which suggests the contribution of modifier alleles in different genetic
backgrounds to the variable cilia related phenotypes.
Other than the mentioned phenotypic overlap among the syndromes, there is
emerging evidence suggesting that genes mutated in ciliopathies exhibit some genetic
overlap. For example, variants in BBS2, BBS4, and BBS6 have been identified in MKS or
MKS-like patients (Karmous-Benailly, Martinovic et al. 2005). Similarly, MKS1 and
MKS3 mutations have been associated with JBTS and BBS (Leitch, Zaghloul et al. 2008).
Given the phenotypic overlap between BBS and MKS, these data suggest that MKS
might represent the extreme end of the phenotypic spectrum (Leitch, Zaghloul et al.
2008). Similar findings are also observed between NPHP and MKS. Mutations in NPHP3
result in NPHP and MKS-like phenotypes in human patients (Bergmann, Fliegauf et al.

122
2008). One of the most intriguing disease genes is CEP290 because mutations in this
gene can contribute to clinically distinct ciliary disorders with an extreme phenotypic
spectrum of severity.
The overall goal of this thesis has been to characterize the function and interaction
of CEP290 in terms of the clinical variability of CEP290-related disease using zebrafish
and mouse model systems. This was accomplished by three independent projects. Firstly,
we utilized the zebrafish system to characterize the function of CEP290, and identified
the CEP290 region that is necessary for visual function. Secondly, we characterized the
synergetic interaction between CEP290 and BBSome genes. Interestingly, compared to
single heterozygous mice, increased body weight but not retinal degeneration was found
in double heterozygous mice (Cep290rd16/+; Bbs4+/-), in which one copy of Cep290 has a
hypomorphic mutation and one copy of Bbs4 is a null mutation. This finding suggests a
modifying effect of BBS alleles on CEP290-related phenotypes, and importantly
indicates that specific combinations of heterozygous alleles could contribute to individual
common human phenotypes such as obesity. Finally, we identified the genetic interaction
between CEP290 and other ciliopathy genes, demonstrating a central role for CEP290 in
CEP290 biological/cellular functions. Our research has examined the role of CEP290 in
cilia and leads to a proposed mechanism underlying the clinical variability of CEP290-
related diseases.
Future Directions
Many ciliopathy proteins are components of the BBSome, IFT complexes, and the
NPHP and MKS modules (Rosenbaum and Witman 2002; Nachury, Loktev et al. 2007;
Garcia-Gonzalo, Corbit et al. 2011; Sang, Miller et al. 2011; Williams, Li et al. 2011).
These proteins participate in specific steps or aspects of pathways that controls protein
trafficking to and from cilia. For example, while the BBSome and the IFT complexes are
involved in transporting ciliary proteins, NPHP and MKS proteins form distinct modular

123
complexes at the ciliary transition zone and function as gatekeepers. Coordinated
functions of these proteins are essential for primary cilia formation and function, and thus
mutations in each protein disrupt a specific step in a pathway, resulting in varying
severity and cellular and physiologic defects. In a study using C. elegans as a model,
synthetic genetic interactions were observed between members of the NPHP and the
MKS modules but not within each module (Williams, Li et al. 2011). The BBSome and
CEP290 also belong to different modules/complexes and we observe synthetic genetic
interaction among these complexes. In addition, recent studies in Chlamydomonas show
that cilia in cep290 mutants have abnormal accumulations and reductions of various IFT
and BBS proteins (Craige, Tsao et al. 2010). Furthermore, the BBSome has been
identified as the key player regulating IFT assembly and turnaround in cilia (Wei, Zhang
et al. 2012). It will be interesting to test whether BBS genes genetically interact with
other members of the transition zone localizing ciliopathy genes and whether these
genetic interactions contribute to the phenotypic variability and overlap among
ciliopathies.
Although the C-terminus of CEP290 is recognized as an important domain for
vision function based on two animal models (Chang, Khanna et al. 2006; Menotti-
Raymond, David et al. 2007), our mouse data demonstrated that the N-terminus of
CEP290 is also necessary for normal function and structure of photoreceptors, and that
this function likely occurs through interaction with the BBSome. This is consistent with
our zebrafish data, which show that the N-terminus of human CEP290 is sufficient to
rescue the vision defects in cep290 zebrafish morphants. Considering the genetic
interaction between CEP290 and other BBS genes, it will be interesting to evaluate
whether the N-terminus of human CEP290 can rescue the vision defect in cep290/bbs4
double zebrafish morphants. In addition, our data also supports a central role for CEP290
in the transition zone as a ciliary gatekeeper to control transport of protein into and out of
the cilium. A recent paper identifies SEPT2 as part of a diffusion barrier at the base of the

124
ciliary membrane. This diffusion barrier restricts the diffusion of ciliary membrane
proteins between the ciliary and periciliary membrane, but permits the diffusion of ciliary
transport proteins (IFT88) (Hu, Milenkovic et al. 2010). Our pilot experiments show a
physical interaction between CEP290 and SEPT2. To further investigate the mechanism
underlying the gatekeeper function of transition zone proteins, the interaction between
CEP290 and SEPT2 should be assessed.
An interesting finding in this study is that our mouse models exhibit phenotypic
features with tissue-specific penetrance. For example, Cep290rd16/rd16; Bbs4+/- mice
showed both increased body weight and more severe retinal degeneration compared to
the Cep290rd16/rd16; Bbs4+/+ mice. Moreover, Cep290rd16/rd16; Bbs4-/- double homozygous
mice show the most severe phenotypes in both body weight and retinal degeneration
among all genotypes. Of note, kidney cysts are not observed in any of the mice we
examined including Cep290rd16/rd16; Bbs4-/- double homozygous mutant mice. It is
presumed that cilia from different tissues have different functions and compositions,
perhaps reflecting different ciliogenic mechanisms. In addition to the various expression
levels of BBS proteins in different tissues, variable BBSome intermediates, which were
found when certain mutant BBS genes were ectopically overexpressed in vitro, allows for
the possibility of tissue-specific forms of the BBSome, which results in various roles for
BBS proteins in specific tissues (Zhang, Yu et al. 2012). Furthermore, CEP290 has been
shown to be involved in re-localization of BBS4 from centriolar satellites to the cilium as
well as the localization of BBS8 into the cilium in vitro (Stowe, Wilkinson et al. 2012).
Our results raise the possibility of a cell type-specific functional link between CEP290
and the BBSome. The correct localization and thus the function of CEP290 and the
BBSome may be affected by each other in a tissue-dependent manner. It will be
interesting to investigate whether CEP290 is involved in the regulation of BBSome
assembly and whether it is tissue-specific.

125
Table 8 Summary of proteins interacting with CEP290 as well as the proteins for which CEP290 serves as a gatekeeper
CEP290 interacting protein Interaction via Loss of cilia localization of
CEP290 domain Proteins without this interaction
CEP290 C- terminus INVS
INVS N-terminus TMEM67
SDCCAG8 C-terminus TMEM67, ARL13B
MKS1 C-terminus ARL13B

126
REFERENCES
Abd-El-Barr, M. M., K. Sykoudis, et al. (2007). "Impaired photoreceptor protein transport and synaptic transmission in a mouse model of Bardet-Biedl syndrome." Vision Res 47(27): 3394-3407.
Abdelhamed, Z. A., G. Wheway, et al. (2013). "Variable expressivity of ciliopathy neurological phenotypes that encompass Meckel-Gruber syndrome and Joubert syndrome is caused by complex de-regulated ciliogenesis, Shh and Wnt signalling defects." Hum Mol Genet 22(7): 1358-1372.
Adams, A. E., M. Rosenblatt, et al. (1999). "Identification of a novel parathyroid hormone-responsive gene in human osteoblastic cells." Bone 24(4): 305-313.
Alexiev, B. A., X. Lin, et al. (2006). "Meckel-Gruber syndrome: pathologic manifestations, minimal diagnostic criteria, and differential diagnosis." Arch Pathol Lab Med 130(8): 1236-1238.
Alstrom, C. H., B. Hallgren, et al. (1959). "Retinal degeneration combined with obesity, diabetes mellitus and neurogenous deafness: a specific syndrome (not hitherto described) distinct from the Laurence-Moon-Bardet-Biedl syndrome: a clinical, endocrinological and genetic examination based on a large pedigree." Acta Psychiatr Neurol Scand Suppl 129: 1-35.
Andersen, J. S., C. J. Wilkinson, et al. (2003). "Proteomic characterization of the human centrosome by protein correlation profiling." Nature 426(6966): 570-574.
Ansley, S. J., J. L. Badano, et al. (2003). "Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome." Nature 425(6958): 628-633.
Aspengren, S., H. N. Skold, et al. (2009). "Different strategies for color change." Cell Mol Life Sci 66(2): 187-191.
Azimzadeh, J. and M. Bornens (2007). "Structure and duplication of the centrosome." J Cell Sci 120(Pt 13): 2139-2142.
Baala, L., S. Audollent, et al. (2007). "Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome." Am J Hum Genet 81(1): 170-179.
Baala, L., S. Romano, et al. (2007). "The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome." Am J Hum Genet 80(1): 186-194.
Badano, J. L., S. J. Ansley, et al. (2003). "Identification of a novel Bardet-Biedl syndrome protein, BBS7, that shares structural features with BBS1 and BBS2." Am J Hum Genet 72(3): 650-658.
Badano, J. L., J. C. Kim, et al. (2003). "Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet-Biedl patients with two mutations at a second BBS locus." Hum Mol Genet 12(14): 1651-1659.
Badano, J. L., C. C. Leitch, et al. (2006). "Dissection of epistasis in oligogenic Bardet-Biedl syndrome." Nature 439(7074): 326-330.

127
Badano, J. L., N. Mitsuma, et al. (2006). "The ciliopathies: an emerging class of human genetic disorders." Annu Rev Genomics Hum Genet 7: 125-148.
Banks, A. S., S. M. Davis, et al. (2000). "Activation of downstream signals by the long form of the leptin receptor." J Biol Chem 275(19): 14563-14572.
Bardet, G. (1920) Sur un syndrome d’obesite infantile avec polydactylie et retinite pigmentaire (contribution a l’etude des forme cliniques de l’obesite hypophysaire). [PhD thesis].
Barral, D. C. and M. C. Seabra (2004). "The melanosome as a model to study organelle motility in mammals." Pigment Cell Res 17(2): 111-118.
Beales, P. L., N. Elcioglu, et al. (1999). "New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey." J Med Genet 36(6): 437-446.
Beales, P. L., A. M. Warner, et al. (1997). "Bardet-Biedl syndrome: a molecular and phenotypic study of 18 families." J Med Genet 34(2): 92-98.
Bergmann, C., M. Fliegauf, et al. (2008). "Loss of nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber-like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia." Am J Hum Genet 82(4): 959-970.
Besharse, J. C., S. A. Baker, et al. (2003). "Photoreceptor intersegmental transport and retinal degeneration: a conserved pathway common to motile and sensory cilia." Adv Exp Med Biol 533: 157-164.
Biedl, A. (1922) Ein geschwisterpaar mit adipose-genitaler dystrophie. Deutsch Med Wochenschr 48,1630
Billingsley, G., C. Deveault, et al. (2011). "BBS mutational analysis: a strategic approach." Ophthalmic Genet 32(3): 181-187.
Bisgrove, B. W. and H. J. Yost (2006). "The roles of cilia in developmental disorders and disease." Development 133(21): 4131-4143.
Blatch, G. L. and M. Lassle (1999). "The tetratricopeptide repeat: a structural motif mediating protein-protein interactions." Bioessays 21(11): 932-939.
Boltshauser, E. and W. Isler (1977). "Joubert syndrome: episodic hyperpnea, abnormal eye movements, retardation and ataxia, associated with dysplasia of the cerebellar vermis." Neuropadiatrie 8(1): 57-66.
Brancati, F., G. Barrano, et al. (2007). "CEP290 mutations are frequently identified in the oculo-renal form of Joubert syndrome-related disorders." Am J Hum Genet 81(1): 104-113.
Brancati, F., B. Dallapiccola, et al. (2010). "Joubert Syndrome and related disorders." Orphanet J Rare Dis 5: 20.
Brandstatter, J. H., S. Lohrke, et al. (1996). "Distributions of two homologous synaptic vesicle proteins, synaptoporin and synaptophysin, in the mammalian retina." J Comp Neurol 370(1): 1-10.

128
Cantagrel, V., J. L. Silhavy, et al. (2008). "Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome." Am J Hum Genet 83(2): 170-179.
Carmi, R., T. Rokhlina, et al. (1995). "Use of a DNA pooling strategy to identify a human obesity syndrome locus on chromosome 15." Hum Mol Genet 4(1): 9-13.
Carter, C. S., T. W. Vogel, et al. (2012). "Abnormal development of NG2+PDGFR-alpha+ neural progenitor cells leads to neonatal hydrocephalus in a ciliopathy mouse model." Nat Med 18(12): 1797-1804.
Chamling, X., S. Seo, et al. (2013). "Ectopic expression of human BBS4 can rescue Bardet-Biedl syndrome phenotypes in Bbs4 null mice." PLoS One 8(3): e59101.
Chang, B., H. Khanna, et al. (2006). "In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse." Hum Mol Genet 15(11): 1847-1857.
Cherian, M. P. and N. A. Al-Sanna'a (2009). "Clinical spectrum of Bardet-Biedl syndrome among four Saudi Arabian families." Clin Dysmorphol 18(4): 188-194.
Chiang, A. P., J. S. Beck, et al. (2006). "Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet-Biedl syndrome gene (BBS11)." Proc Natl Acad Sci U S A 103(16): 6287-6292.
Chiang, A. P., D. Nishimura, et al. (2004). "Comparative genomic analysis identifies an ADP-ribosylation factor-like gene as the cause of Bardet-Biedl syndrome (BBS3)." Am J Hum Genet 75(3): 475-484.
Christensen, S. T., C. A. Clement, et al. (2012). "Primary cilia and coordination of receptor tyrosine kinase (RTK) signalling." J Pathol 226(2): 172-184.
Cideciyan, A. V., T. S. Aleman, et al. (2007). "Centrosomal-ciliary gene CEP290/NPHP6 mutations result in blindness with unexpected sparing of photoreceptors and visual brain: implications for therapy of Leber congenital amaurosis." Hum Mutat 28(11): 1074-1083.
Cideciyan, A. V., R. A. Rachel, et al. (2011). "Cone photoreceptors are the main targets for gene therapy of NPHP5 (IQCB1) or NPHP6 (CEP290) blindness: generation of an all-cone Nphp6 hypomorph mouse that mimics the human retinal ciliopathy." Hum Mol Genet 20(7): 1411-1423.
Cole, D. G., S. W. Chinn, et al. (1993). "Novel heterotrimeric kinesin-related protein purified from sea urchin eggs." Nature 366(6452): 268-270.
Cole, D. G., D. R. Diener, et al. (1998). "Chlamydomonas kinesin-II-dependent intraflagellar transport (IFT): IFT particles contain proteins required for ciliary assembly in Caenorhabditis elegans sensory neurons." J Cell Biol 141(4): 993-1008.
Coppieters, F., I. Casteels, et al. (2010). "Genetic screening of LCA in Belgium: predominance of CEP290 and identification of potential modifier alleles in AHI1 of CEP290-related phenotypes." Hum Mutat 31(10): E1709-1766.

129
Coppieters, F., S. Lefever, et al. (2010). "CEP290, a gene with many faces: mutation overview and presentation of CEP290base." Hum Mutat 31(10): 1097-1108.
Craige, B., C. C. Tsao, et al. (2010). "CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content." J Cell Biol 190(5): 927-940.
Croft, J. B., D. Morrell, et al. (1995). "Obesity in heterozygous carriers of the gene for the Bardet-Biedl syndrome." Am J Med Genet 55(1): 12-15.
Czarnecki, P. G. and J. V. Shah (2012). "The ciliary transition zone: from morphology and molecules to medicine." Trends Cell Biol 22(4): 201-210.
Dammermann, A. and A. Merdes (2002). "Assembly of centrosomal proteins and microtubule organization depends on PCM-1." J Cell Biol 159(2): 255-266.
Davis, R. E., R. E. Swiderski, et al. (2007). "A knockin mouse model of the Bardet-Biedl syndrome 1 M390R mutation has cilia defects, ventriculomegaly, retinopathy, and obesity." Proc Natl Acad Sci U S A 104(49): 19422-19427.
De Robertis, E. (1956). "Morphogenesis of the retinal rods; an electron microscope study." J Biophys Biochem Cytol 2(4 Suppl): 209-218.
Delous, M., L. Baala, et al. (2007). "The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome." Nat Genet 39(7): 875-881.
den Hollander, A. I., R. K. Koenekoop, et al. (2006). "Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis." Am J Hum Genet 79(3): 556-561.
den Hollander, A. I., R. Roepman, et al. (2008). "Leber congenital amaurosis: genes, proteins and disease mechanisms." Prog Retin Eye Res 27(4): 391-419.
Deretic, D., A. H. Williams, et al. (2005). "Rhodopsin C terminus, the site of mutations causing retinal disease, regulates trafficking by binding to ADP-ribosylation factor 4 (ARF4)." Proc Natl Acad Sci U S A 102(9): 3301-3306.
Di Gioia, S. A., S. J. Letteboer, et al. (2012). "FAM161A, associated with retinitis pigmentosa, is a component of the cilia-basal body complex and interacts with proteins involved in ciliopathies." Hum Mol Genet 21(23): 5174-5184.
Easter, S. S., Jr. and G. N. Nicola (1996). "The development of vision in the zebrafish (Danio rerio)." Dev Biol 180(2): 646-663.
Eichers, E. R., M. M. Abd-El-Barr, et al. (2006). "Phenotypic characterization of Bbs4 null mice reveals age-dependent penetrance and variable expressivity." Hum Genet 120(2): 211-226.
Elbedour, K., N. Zucker, et al. (1994). "Cardiac abnormalities in the Bardet-Biedl syndrome: echocardiographic studies of 22 patients." Am J Med Genet 52(2): 164-169.

130
Essner, J. J., J. D. Amack, et al. (2005). "Kupffer's vesicle is a ciliated organ of asymmetry in the zebrafish embryo that initiates left-right development of the brain, heart and gut." Development 132(6): 1247-1260.
Evans, R. J., N. Schwarz, et al. (2010). "The retinitis pigmentosa protein RP2 links pericentriolar vesicle transport between the Golgi and the primary cilium." Hum Mol Genet 19(7): 1358-1367.
Fan, S., T. W. Hurd, et al. (2004). "Polarity proteins control ciliogenesis via kinesin motor interactions." Curr Biol 14(16): 1451-1461.
Fath, M. A., R. F. Mullins, et al. (2005). "Mkks-null mice have a phenotype resembling Bardet-Biedl syndrome." Hum Mol Genet 14(9): 1109-1118.
Ferland, R. J., W. Eyaid, et al. (2004). "Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome." Nat Genet 36(9): 1008-1013.
Fliegauf, M., T. Benzing, et al. (2007). "Mechanisms of disease - When cilia go bad: cilia defects and ciliopathies." Nature Reviews Molecular Cell Biology 8(11): 880-893.
Fliegauf, M., T. Benzing, et al. (2007). "When cilia go bad: cilia defects and ciliopathies." Nat Rev Mol Cell Biol 8(11): 880-893.
Franceschetti, A. and P. Dieterle (1954). "[Diagnostic and prognostic importance of the electroretinogram in tapetoretinal degeneration with reduction of the visual field and hemeralopia]." Confin Neurol 14(2-3): 184-186.
Frank, V., A. I. den Hollander, et al. (2008). "Mutations of the CEP290 gene encoding a centrosomal protein cause Meckel-Gruber syndrome." Hum Mutat 29(1): 45-52.
Garcia-Gonzalo, F. R., K. C. Corbit, et al. (2011). "A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition." Nat Genet 43(8): 776-784.
Gherman, A., E. E. Davis, et al. (2006). "The ciliary proteome database: an integrated community resource for the genetic and functional dissection of cilia." Nat Genet 38(9): 961-962.
Gleeson, J. G., L. C. Keeler, et al. (2004). "Molar tooth sign of the midbrain-hindbrain junction: occurrence in multiple distinct syndromes." Am J Med Genet A 125A(2): 125-134; discussion 117.
Goetz, S. C. and K. V. Anderson (2010). "The primary cilium: a signalling centre during vertebrate development." Nat Rev Genet 11(5): 331-344.
Green, J. S., P. S. Parfrey, et al. (1989). "The cardinal manifestations of Bardet-Biedl syndrome, a form of Laurence-Moon-Biedl syndrome." N Engl J Med 321(15): 1002-1009.
Harnett, J. D., J. S. Green, et al. (1988). "The spectrum of renal disease in Laurence-Moon-Biedl syndrome." N Engl J Med 319(10): 615-618.

131
Haycraft, C. J., J. C. Schafer, et al. (2003). "Identification of CHE-13, a novel intraflagellar transport protein required for cilia formation." Exp Cell Res 284(2): 251-263.
Helou, J., E. A. Otto, et al. (2007). "Mutation analysis of NPHP6/CEP290 in patients with Joubert syndrome and Senior-Loken syndrome." J Med Genet 44(10): 657-663.
Hichri, H., C. Stoetzel, et al. (2005). "Testing for triallelism: analysis of six BBS genes in a Bardet-Biedl syndrome family cohort." Eur J Hum Genet 13(5): 607-616.
Higginbotham, H., T. Y. Eom, et al. (2012). "Arl13b in primary cilia regulates the migration and placement of interneurons in the developing cerebral cortex." Dev Cell 23(5): 925-938.
Hildebrandt, F. and E. Otto (2005). "Cilia and centrosomes: a unifying pathogenic concept for cystic kidney disease?" Nat Rev Genet 6(12): 928-940.
Hjortshoj, T. D., K. Gronskov, et al. (2010). "Bardet-Biedl syndrome in Denmark--report of 13 novel sequence variations in six genes." Hum Mutat 31(4): 429-436.
Horst, C. J., L. V. Johnson, et al. (1990). "Transmembrane assemblage of the photoreceptor connecting cilium and motile cilium transition zone contain a common immunologic epitope." Cell Motil Cytoskeleton 17(4): 329-344.
Hu, Q., L. Milenkovic, et al. (2010). "A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution." Science 329(5990): 436-439.
Humbert, M. C., K. Weihbrecht, et al. (2012). "ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting." Proc Natl Acad Sci U S A 109(48): 19691-19696.
Iannaccone, A., K. Mykytyn, et al. (2005). "Clinical evidence of decreased olfaction in Bardet-Biedl syndrome caused by a deletion in the BBS4 gene." Am J Med Genet A 132(4): 343-346.
Illing, M., L. L. Molday, et al. (1997). "The 220-kDa rim protein of retinal rod outer segments is a member of the ABC transporter superfamily." J Biol Chem 272(15): 10303-10310.
Jin, H., S. R. White, et al. (2010). "The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia." Cell 141(7): 1208-1219.
Johnson, L. V. and J. C. Blanks (1984). "Application of acrylamide as an embedding medium in studies of lectin and antibody binding in the vertebrate retina." Curr Eye Res 3(7): 969-974.
Karmous-Benailly, H., J. Martinovic, et al. (2005). "Antenatal presentation of Bardet-Biedl syndrome may mimic Meckel syndrome." Am J Hum Genet 76(3): 493-504.
Katsanis, N., S. J. Ansley, et al. (2001). "Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder." Science 293(5538): 2256-2259.

132
Kim, J., S. R. Krishnaswami, et al. (2008). "CEP290 interacts with the centriolar satellite component PCM-1 and is required for Rab8 localization to the primary cilium." Hum Mol Genet 17(23): 3796-3805.
Kim, J. C., J. L. Badano, et al. (2004). "The Bardet-Biedl protein BBS4 targets cargo to the pericentriolar region and is required for microtubule anchoring and cell cycle progression." Nat Genet 36(5): 462-470.
Kim, S. K., A. Shindo, et al. (2010). "Planar cell polarity acts through septins to control collective cell movement and ciliogenesis." Science 329(5997): 1337-1340.
Koenekoop, R. K. (2004). "An overview of Leber congenital amaurosis: a model to understand human retinal development." Surv Ophthalmol 49(4): 379-398.
Krock, B. L. and B. D. Perkins (2008). "The intraflagellar transport protein IFT57 is required for cilia maintenance and regulates IFT-particle-kinesin-II dissociation in vertebrate photoreceptors." J Cell Sci 121(Pt 11): 1907-1915.
Kudryashova, E., D. Kudryashov, et al. (2005). "Trim32 is a ubiquitin ligase mutated in limb girdle muscular dystrophy type 2H that binds to skeletal muscle myosin and ubiquitinates actin." J Mol Biol 354(2): 413-424.
Kulaga, H. M., C. C. Leitch, et al. (2004). "Loss of BBS proteins causes anosmia in humans and defects in olfactory cilia structure and function in the mouse." Nat Genet 36(9): 994-998.
Kwitek-Black, A. E., R. Carmi, et al. (1993). "Linkage of Bardet-Biedl syndrome to chromosome 16q and evidence for non-allelic genetic heterogeneity." Nat Genet 5(4): 392-396.
Leitch, C. C., N. A. Zaghloul, et al. (2008). "Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome." Nat Genet 40(4): 443-448.
Leppert, M., L. Baird, et al. (1994). "Bardet-Biedl syndrome is linked to DNA markers on chromosome 11q and is genetically heterogeneous." Nat Genet 7(1): 108-112.
Li, J. B., J. M. Gerdes, et al. (2004). "Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene." Cell 117(4): 541-552.
Livak, K. J. and T. D. Schmittgen (2001). "Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method." Methods 25(4): 402-408.
Lopes, C. A., S. L. Prosser, et al. (2011). "Centriolar satellites are assembly points for proteins implicated in human ciliopathies, including oral-facial-digital syndrome 1." J Cell Sci 124(Pt 4): 600-612.
Lorda-Sanchez, I., C. Ayuso, et al. (2000). "Situs inversus and hirschsprung disease: two uncommon manifestations in Bardet-Biedl syndrome." Am J Med Genet 90(1): 80-81.

133
Louie, C. M., G. Caridi, et al. (2010). "AHI1 is required for photoreceptor outer segment development and is a modifier for retinal degeneration in nephronophthisis." Nat Genet 42(2): 175-180.
Luby-Phelps, K., J. Fogerty, et al. (2008). "Spatial distribution of intraflagellar transport proteins in vertebrate photoreceptors." Vision Res 48(3): 413-423.
Maria, B. L., K. B. Hoang, et al. (1997). ""Joubert syndrome" revisited: key ocular motor signs with magnetic resonance imaging correlation." J Child Neurol 12(7): 423-430.
Marion, V., F. Stutzmann, et al. (2012). "Exome sequencing identifies mutations in LZTFL1, a BBSome and smoothened trafficking regulator, in a family with Bardet--Biedl syndrome with situs inversus and insertional polydactyly." J Med Genet 49(5): 317-321.
Marks, M. S. and M. C. Seabra (2001). "The melanosome: membrane dynamics in black and white." Nat Rev Mol Cell Biol 2(10): 738-748.
Marshall, W. F. and S. Nonaka (2006). "Cilia: tuning in to the cell's antenna." Curr Biol 16(15): R604-614.
McEwen, D. P., R. K. Koenekoop, et al. (2007). "Hypomorphic CEP290/NPHP6 mutations result in anosmia caused by the selective loss of G proteins in cilia of olfactory sensory neurons." Proc Natl Acad Sci U S A 104(40): 15917-15922.
Mecke, S. and E. Passarge (1971). "Encephalocele, polycystic kidneys, and polydactyly as an autosomal recessive trait simulating certain other disorders: the Meckel syndrome." Ann Genet 14(2): 97-103.
Menotti-Raymond, M., V. A. David, et al. (2007). "Mutation in CEP290 discovered for cat model of human retinal degeneration." J Hered 98(3): 211-220.
Moore, S. J., J. S. Green, et al. (2005). "Clinical and genetic epidemiology of Bardet-Biedl syndrome in Newfoundland: a 22-year prospective, population-based, cohort study." Am J Med Genet A 132(4): 352-360.
Murga-Zamalloa, C. A., N. J. Desai, et al. (2010). "Interaction of ciliary disease protein retinitis pigmentosa GTPase regulator with nephronophthisis-associated proteins in mammalian retinas." Mol Vis 16: 1373-1381.
Mykytyn, K., R. F. Mullins, et al. (2004). "Bardet-Biedl syndrome type 4 (BBS4)-null mice implicate Bbs4 in flagella formation but not global cilia assembly." Proc Natl Acad Sci U S A 101(23): 8664-8669.
Mykytyn, K., D. Y. Nishimura, et al. (2003). "Evaluation of complex inheritance involving the most common Bardet-Biedl syndrome locus (BBS1)." Am J Hum Genet 72(2): 429-437.
Mykytyn, K., D. Y. Nishimura, et al. (2002). "Identification of the gene (BBS1) most commonly involved in Bardet-Biedl syndrome, a complex human obesity syndrome." Nat Genet 31(4): 435-438.

134
Mykytyn, K. and V. C. Sheffield (2004). "Establishing a connection between cilia and Bardet-Biedl Syndrome." Trends Mol Med 10(3): 106-109.
Nachury, M. V., A. V. Loktev, et al. (2007). "A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis." Cell 129(6): 1201-1213.
Nascimento, A. A., J. T. Roland, et al. (2003). "Pigment cells: a model for the study of organelle transport." Annu Rev Cell Dev Biol 19: 469-491.
Nishimura, D. Y., L. M. Baye, et al. (2010). "Discovery and functional analysis of a retinitis pigmentosa gene, C2ORF71." Am J Hum Genet 86(5): 686-695.
Nishimura, D. Y., M. Fath, et al. (2004). "Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin." Proc Natl Acad Sci U S A 101(47): 16588-16593.
Nishimura, D. Y., C. C. Searby, et al. (2001). "Positional cloning of a novel gene on chromosome 16q causing Bardet-Biedl syndrome (BBS2)." Hum Mol Genet 10(8): 865-874.
Nishimura, D. Y., R. E. Swiderski, et al. (2005). "Comparative genomics and gene expression analysis identifies BBS9, a new Bardet-Biedl syndrome gene." Am J Hum Genet 77(6): 1021-1033.
Otto, E. A., T. W. Hurd, et al. (2010). "Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy." Nat Genet 42(10): 840-850.
Pan, J. and W. Snell (2007). "The primary cilium: keeper of the key to cell division." Cell 129(7): 1255-1257.
Pasadhika, S., G. A. Fishman, et al. (2010). "Differential macular morphology in patients with RPE65-, CEP290-, GUCY2D-, and AIPL1-related Leber congenital amaurosis." Invest Ophthalmol Vis Sci 51(5): 2608-2614.
Pazour, G. J. and J. L. Rosenbaum (2002). "Intraflagellar transport and cilia-dependent diseases." Trends Cell Biol 12(12): 551-555.
Pereiro, I., D. Valverde, et al. (2010). "New mutations in BBS genes in small consanguineous families with Bardet-Biedl syndrome: detection of candidate regions by homozygosity mapping." Mol Vis 16: 137-143.
Perkins, L. A., E. M. Hedgecock, et al. (1986). "Mutant sensory cilia in the nematode Caenorhabditis elegans." Dev Biol 117(2): 456-487.
Perrault, I., N. Delphin, et al. (2007). "Spectrum of NPHP6/CEP290 mutations in Leber congenital amaurosis and delineation of the associated phenotype." Hum Mutat 28(4): 416.
Piperno, G., E. Siuda, et al. (1998). "Distinct mutants of retrograde intraflagellar transport (IFT) share similar morphological and molecular defects." J Cell Biol 143(6): 1591-1601.

135
Pretorius, P. R., M. A. Aldahmesh, et al. (2011). "Functional analysis of BBS3 A89V that results in non-syndromic retinal degeneration." Hum Mol Genet 20(8): 1625-1632.
Pretorius, P. R., L. M. Baye, et al. (2010). "Identification and functional analysis of the vision-specific BBS3 (ARL6) long isoform." PLoS Genet 6(3): e1000884.
Quinlan, R. J., J. L. Tobin, et al. (2008). "Modeling ciliopathies: Primary cilia in development and disease." Curr Top Dev Biol 84: 249-310.
Rachel, R. A., H. L. May-Simera, et al. (2012). "Combining Cep290 and Mkks ciliopathy alleles in mice rescues sensory defects and restores ciliogenesis." J Clin Invest 122(4): 1233-1245.
Rahmouni, K., M. A. Fath, et al. (2008). "Leptin resistance contributes to obesity and hypertension in mouse models of Bardet-Biedl syndrome." J Clin Invest 118(4): 1458-1467.
Riise, R., S. Andreasson, et al. (1997). "Intrafamilial variation of the phenotype in Bardet-Biedl syndrome." Br J Ophthalmol 81(5): 378-385.
Robinow, M. and A. Shaw (1979). "The McKusick-Kaufman syndrome: recessively inherited vaginal atresia, hydrometrocolpos, uterovaginal duplications, anorectal anomalies, postaxial polydactyly, and congenital heart disease." J Pediatr 94(5): 776-778.
Rosenbaum, J. (2002). "Intraflagellar transport." Curr Biol 12(4): R125.
Rosenbaum, J. L. and G. B. Witman (2002). "Intraflagellar transport." Nat Rev Mol Cell Biol 3(11): 813-825.
Sang, L., J. J. Miller, et al. (2011). "Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways." Cell 145(4): 513-528.
Sayer, J. A., E. A. Otto, et al. (2006). "The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4." Nat Genet 38(6): 674-681.
Schafer, T., M. Putz, et al. (2008). "Genetic and physical interaction between the NPHP5 and NPHP6 gene products." Hum Mol Genet 17(23): 3655-3662.
Seo, S., L. M. Baye, et al. (2010). "BBS6, BBS10, and BBS12 form a complex with CCT/TRiC family chaperonins and mediate BBSome assembly." Proc Natl Acad Sci U S A 107(4): 1488-1493.
Seo, S., D. F. Guo, et al. (2009). "Requirement of Bardet-Biedl syndrome proteins for leptin receptor signaling." Hum Mol Genet 18(7): 1323-1331.
Seo, S., Q. Zhang, et al. (2011). "A novel protein LZTFL1 regulates ciliary trafficking of the BBSome and Smoothened." PLoS Genet 7(11): e1002358.
Sheffield, V. C. (2004). "Use of isolated populations in the study of a human obesity syndrome, the Bardet-Biedl syndrome." Pediatr Res 55(6): 908-911.

136
Sheffield, V. C. (2010). "The blind leading the obese: the molecular pathophysiology of a human obesity syndrome." Trans Am Clin Climatol Assoc 121: 172-181; discussion 181-172.
Sheffield, V. C., R. Carmi, et al. (1994). "Identification of a Bardet-Biedl syndrome locus on chromosome 3 and evaluation of an efficient approach to homozygosity mapping." Hum Mol Genet 3(8): 1331-1335.
Singla, V. and J. F. Reiter (2006). "The primary cilium as the cell's antenna: signaling at a sensory organelle." Science 313(5787): 629-633.
Smaoui, N., M. Chaabouni, et al. (2006). "Screening of the eight BBS genes in Tunisian families: no evidence of triallelism." Invest Ophthalmol Vis Sci 47(8): 3487-3495.
Stoetzel, C., V. Laurier, et al. (2006). "BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus." Nat Genet 38(5): 521-524.
Stoetzel, C., J. Muller, et al. (2007). "Identification of a novel BBS gene (BBS12) highlights the major role of a vertebrate-specific branch of chaperonin-related proteins in Bardet-Biedl syndrome." Am J Hum Genet 80(1): 1-11.
Stone, D. L., A. Slavotinek, et al. (2000). "Mutation of a gene encoding a putative chaperonin causes McKusick-Kaufman syndrome." Nat Genet 25(1): 79-82.
Stone, E. M. (2007). "Leber congenital amaurosis - a model for efficient genetic testing of heterogeneous disorders: LXIV Edward Jackson Memorial Lecture." Am J Ophthalmol 144(6): 791-811.
Stowe, T. R., C. J. Wilkinson, et al. (2012). "The centriolar satellite proteins Cep72 and Cep290 interact and are required for recruitment of BBS proteins to the cilium." Mol Biol Cell 23(17): 3322-3335.
Sugiyama, N., M. Kohno, et al. (2012). "Inhibition of the p38 MAPK pathway ameliorates renal fibrosis in an NPHP2 mouse model." Nephrol Dial Transplant 27(4): 1351-1358.
Swiderski, R. E., D. Y. Nishimura, et al. (2007). "Gene expression analysis of photoreceptor cell loss in bbs4-knockout mice reveals an early stress gene response and photoreceptor cell damage." Invest Ophthalmol Vis Sci 48(7): 3329-3340.
Tayeh, M. K., H. J. Yen, et al. (2008). "Genetic interaction between Bardet-Biedl syndrome genes and implications for limb patterning." Hum Mol Genet 17(13): 1956-1967.
Tobin, J. L. and P. L. Beales (2008). "Restoration of renal function in zebrafish models of ciliopathies." Pediatr Nephrol 23(11): 2095-2099.
Tory, K., T. Lacoste, et al. (2007). "High NPHP1 and NPHP6 mutation rate in patients with Joubert syndrome and nephronophthisis: potential epistatic effect of NPHP6 and AHI1 mutations in patients with NPHP1 mutations." J Am Soc Nephrol 18(5): 1566-1575.

137
Tran, P. V., C. J. Haycraft, et al. (2008). "THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia." Nat Genet 40(4): 403-410.
Tsujikawa, M. and J. Malicki (2004). "Intraflagellar transport genes are essential for differentiation and survival of vertebrate sensory neurons." Neuron 42(5): 703-716.
Valente, E. M., F. Brancati, et al. (2008). "Genotypes and phenotypes of Joubert syndrome and related disorders." Eur J Med Genet 51(1): 1-23.
Valente, E. M., C. V. Logan, et al. (2010). "Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes." Nat Genet 42(7): 619-625.
Valente, E. M., J. L. Silhavy, et al. (2006). "Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome." Nat Genet 38(6): 623-625.
Varela, L. and T. L. Horvath (2012). "Leptin and insulin pathways in POMC and AgRP neurons that modulate energy balance and glucose homeostasis." EMBO Rep 13(12): 1079-1086.
Vasireddy, V., M. M. Jablonski, et al. (2006). "Elovl4 5-bp-deletion knock-in mice develop progressive photoreceptor degeneration." Invest Ophthalmol Vis Sci 47(10): 4558-4568.
Weatherbee, S. D., L. A. Niswander, et al. (2009). "A mouse model for Meckel syndrome reveals Mks1 is required for ciliogenesis and Hedgehog signaling." Hum Mol Genet 18(23): 4565-4575.
Wei, Q., Y. Zhang, et al. (2012). "The BBSome controls IFT assembly and turnaround in cilia." Nat Cell Biol 14(9): 950-957.
Westfall, T. A., R. Brimeyer, et al. (2003). "Wnt-5/pipetail functions in vertebrate axis formation as a negative regulator of Wnt/beta-catenin activity." J Cell Biol 162(5): 889-898.
Williams, C. L., C. Li, et al. (2011). "MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis." J Cell Biol 192(6): 1023-1041.
Wolf, M. T. and F. Hildebrandt (2011). "Nephronophthisis." Pediatr Nephrol 26(2): 181-194.
Woods, M. O., T. L. Young, et al. (1999). "Genetic heterogeneity of Bardet-Biedl syndrome in a distinct Canadian population: evidence for a fifth locus." Genomics 55(1): 2-9.
Yang, L. P., L. M. Wu, et al. (2007). "Activation of endoplasmic reticulum stress in degenerating photoreceptors of the rd1 mouse." Invest Ophthalmol Vis Sci 48(11): 5191-5198.

138
Yen, H. J., M. K. Tayeh, et al. (2006). "Bardet-Biedl syndrome genes are important in retrograde intracellular trafficking and Kupffer's vesicle cilia function." Hum Mol Genet 15(5): 667-677.
Young, T. L., L. Penney, et al. (1999). "A fifth locus for Bardet-Biedl syndrome maps to chromosome 2q31." Am J Hum Genet 64(3): 900-904.
Zaghloul, N. A. and N. Katsanis (2009). "Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy." J Clin Invest 119(3): 428-437.
Zhang, Q., D. Nishimura, et al. (2011). "Bardet-Biedl syndrome 3 (Bbs3) knockout mouse model reveals common BBS-associated phenotypes and Bbs3 unique phenotypes." Proc Natl Acad Sci U S A 108(51): 20678-20683.
Zhang, Q., D. Nishimura, et al. (2013). "BBS7 is required for BBSome formation and its absence in mice results in Bardet-Biedl syndrome phenotypes and selective abnormalities in membrane protein trafficking." J Cell Sci 126(Pt 11): 2372-2380.
Zhang, Q., S. Seo, et al. (2012). "BBS proteins interact genetically with the IFT pathway to influence SHH-related phenotypes." Hum Mol Genet 21(9): 1945-1953.
Zhang, Q., D. Yu, et al. (2012). "Intrinsic protein-protein interaction-mediated and chaperonin-assisted sequential assembly of stable bardet-biedl syndrome protein complex, the BBSome." J Biol Chem 287(24): 20625-20635.