empirical energy models - slides - uclahelper.ipam.ucla.edu/publications/eltut/eltut_14762.pdf ·...
TRANSCRIPT
Energy model EnergyE({Ri})
Quantummechanical methods
Acc
ura
cy
Speed
Semi-empirical models
Molec
ular
Relaxati
ons
Energies
Empiricalmodels
Transit
ion
dynamics
states
Hierarchyofenergymodels
Atomicstructure{Ri}
EmpiricalEnergyModelsandForceFieldsRichardG.Hennig,UniversityofFlorida

Born-OppenheimerApproximaBon
• Theelectronsaremuchlighterthanthenuclei(mp/me≈1,800)• Hence,forgivennucleiposiDon,theelectronsareintheirgroundstateΨ0• ThisapproximaDonisknownastheBorn-OppenheimerapproximaBon• However,forsomematerialsandproperDesthisapproximaDoncanfail- Hydrogendiffusioninmaterialsbytunneling- Hydrogenphasesathightemperatureandpressure- Hedroplets- SuperconducDvity- Atomicgasesatlowtemperatures⇒Bose-EinsteincondensaDon
• TreatnucleiasclassicalparDclesandusetheBorn-OppenheimerapproximaDon
Energy model EnergyE({Ri})Atomicstructure{Ri}

Quantummechanical methods
Acc
ura
cy
Speed
Semi-empirical models
Molec
ular
Relaxati
ons
Energies
Empiricalmodels
Transit
ion
dynamics
states
Quantummechanicalmethods• Startfromthemany-bodySchrödingerequaDonandmakeapproximaDons• Examples:
Semi-empiricalmodels• FuncDonalformmoDvatedbyquantummechanics,neglecDngdifficultterms,fiUedparameters
• Examples:Tight-binding,neglectofdifferenDaloverlap
Empiricalmodels• FuncDonalformswithfiUedparameters• Examples:PairpotenDals,many-bodypotenDals, effecDvemediumpotenDals
HierarchyofEnergyModels
‣ Quantumchemistry(Hartree-Fock,Coupledclusters,configuraDoninteracDon,…)‣ Density-funcDonaltheory,GWapproximaDon,random-phaseapproximaDon‣ QuantumMonteCarlo,dynamicalmean-fieldtheory,densitymatrixrenormalizaDongroup

ChoiceoffuncBonalformofempiricalpotenBals• MoDvatedbytypesofchemicalbondsrelevantforspecificmaterial• Increasingincomplexity:pairpotenDals,pairfuncDonals,many-bodypotenDals,…
OpBmizaBonofempiricalpotenBalparameters• Least-squareopDmizaDontechniquesusingafiangandtesDngdatabase
ValidaBonstrategies• Obvious:Comparisonwithotheravailabledata• ValidaDonofenergylandscapesusingstructuresearchmethods
OpenquesBons• HowtoselecttheopDmalfuncDonalformofpotenDalforagivenmaterial?• HowtochooseanopDmaldatasetformodelparameteropDmizaDon?• HowtoidenDfytradeoffsbetweenconflicDngmodelpredicDons?
Outline

CommonFeaturesofEmpiricalPotenBals
DecomposiBoninatomicenergies• Energyisconstructedassumoflocalatomicenergies
• LocalityofatomicinteracDonsisknownasnear-sightednessinquantumchemistry
GeneralN-bodyexpansion
E =X
i
✏i(Ri)
E =X
i
�1(Ri) +X
i
X
j>i
�2(Ri,Rj) +X
i
X
j>i
X
k>j
�3(Ri,Rj ,Rk) + · · ·

PairPotenBals
Commonfeatures• ApproximaDonofonlypairwiseinteracDon• FormofpairpotenDalV(r)‣ Repulsiveatshortdistances,aUracDveatlongdistances‣ Usuallyappliedwithacutoff
• AnalyDcalformsofpotenDalsareusuallybasedonbasicphysics• PhysicalrelevanceofparametersdisappearswhenpotenDalsarefiUed• Minimalsetofparameters:energyscaleandlengthscale
FuncBonalformsofpotenBals• ManydifferentfuncDonalformshavebeendevelopedandused• Lennard-Jones,Buckingham,Morse,Coulomb,screenedCoulomb,hardsphere,Yukawa
E =12
�
i,j
V (|ri � rj |)
doi:10.1088/978-1-6817-4417-9ch4

0 0.5 1 1.5 2 2.5 3
r/!
-1.0
-0.5
0.0
0.5
1.0
V(r)/"
V (r) =A
r12� B
r6
V (r)�
=�⇥
r
⇥12� 2 ·
�⇥
r
⇥6
J.E.Lennard-Jones,Cohesion.Proc.Phys.Soc.43,461(1931).
SirJohnLennard-Jones
Lennard-JonesPotenBal
• Proposedin1931byJohnLennard-JonesatBristolUniversity• AUracDve1/r6termdescribes vanderWaalsaQracBon
• Repulsive1/r12term chosenforefficiency

• WhenexpressingT,p,andρinrenormalizedunitsallLennard-JonespotenDalsareidenDcal‣ Temperature ε/kB‣ Pressure ε/σ3
‣ Density 1/σ3
• Ifwesetlaaceparameterr0=σand cohesiveenergyEcoh=ε,then allotherproperDessuchaselasDccoefficients,melDngpointetc.aredetermined
Argon dimer
Lennard-JonesPotenBal
TheLennard-JonespotenDalisreallyonlyapplicabletonoblegases
(nobonding,onlyvanderWaalsaUracDon)
⇒ThereisonlyoneLennard-Jonesmaterial

• SomeDmes,therepulsive1/r12termoftheLennard-JonespotenDalistoosteep• TheBuckinghampotenDalemploysasorerrepulsivepotenDal
Born-Mayer/BuckinghamPotenBal
V (r) = A · exp(�B · r)� C
r6

V (r) = De
�1� e��(r�re)
⇥2
P.M.Morse,Diatomicmoleculesaccordingtothewavemechanics.II.Vibra@onallevels.Phys.Rev.34,57(1929).
MorsePotenBalforDiatomicMolecules
ProposedbyMorsein1929forthepotenDalenergyofadiatomicmolecule

V (r) = De
�1� e��(r�re)
⇥2
MorsepotenDalhighlyaccuratecomparedtoharmonic(P2)andquarDc(P4)potenDal.
Morse Potential for Stretching of C-H in CH4
Energ
y (
Kcal/
mol)
80
60
40
20
"EXACT" P2
P4
MORSE
Think about how far
from equilibrium you 0 need the potential
-0.4 -0.2 0.0 0.2 0.4 0.6 0.8 .
∆R (A)
Figure by MIT OCW.
2/1/05 Massachusetts Institute of Technology 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
BondStretchinginMethane
HowfarfromequilibriumdoweneedthepotenDaltobeaccurate?• 1kcal/molcorrespondsto43meVor503K• PotenDalneedstobeaccurateonlyclosetotheminimum

StartwithpairpotenBalswithCoulombinteracBons• BuckinghampluselectrostaDcCoulombterm
IncludepolarizaBonofions• Electricfieldfromotherionsinducesadipolemoment
• Shellmodel
‣ DescribetheioncoreandtheelectronshellseparatelyastwoparDclesconnectedbyaspring
‣ Springconstantbetweencoreandshellcorrespondstopolarizability
V (r) = A · exp�� r
B
⇥� C
r6⇧ ⌅⇤ ⌃
Buckingham
+q1 · q2
r⇧ ⌅⇤ ⌃Coulomb
PotenBalsforChargedSystems

• PairPotenDals“count”bondsbutdonottakeintoaccounttheirorganizaDon⇒Similarenergyforatriangleofthreeatomsversuschainoffour
• Tendencytoformclose-packedstructuressuchasbccandhcp- Difficulttostabilizediamond-cubicstructureofSiwithpairpotenDal- SiliconundergoesaseriesofstructuralphasetransiDons(fromtetrahedraltoβ-Dntosimplehexagonaltofcc)underpressure
- Smallenergydifferencesbetweenthesestructures- CohesiveenergynearlyindependentofcoordinaDonnumber
LimitaBonsofPairPotenBals

Silicon Crystal Phases
Compression16 GPa 36 GPa 42 GPa 79 GPa
Si(I) diamondZ=4
11 GPa 13 GPa
Si(II) −tinβZ=6
Si(V) hexagonalSi(XI) ImmaZ=6 Z=8 Z=10
Si(VI) orthorhombic Si(VII) hcpZ=12 Z=12
Si(X) fcc
Decompression
Si(XII) R8 Si(IV) hex. diamondZ=4Z=4
Si(III) BC−8Z=6β−tinSi(II)
9 GPa >480 K
Slow pressure release
Z=4
2 GPa
Fast pressure release
Si(VIII) and Si(IX) tetragonal
Under pressure silicon displays 12 crystal phases with a steady increase
of coordination and a transition from insulating to metallic.
SiliconPhases

TheassumpDonofapairpotenDaldeterminesavarietyofproperDesCrystal Ecoh/kBTm Evac/Ecoh C12/C44
Pairpoten@alLennard-Jones 13 ∼1 1Noblegases
Ar 11 0.95 1.1Kr 12 0.66 1
fccmetalsNi 30 0.31 1.2Cu 30 0.37 1.6Pd 25 0.36 2.5Ag 27 0.39 2Pt 33 0.26 3.3Au 34 0.23 3.7
LimitaBonsofPairPotenBals
Ratio between Ecoh and kBTm is about 30 in metals and 10 for pair potentials and noble
gases
Ratio between the Evac and the Ecoh is between 1/4 and 1/3 in metals and
about 1 in two-body systems (exactly 1 if relaxations are neglected)
Cauchy ratio C12/C44 = 1 for pair potentials.
Deviations in metals are common.
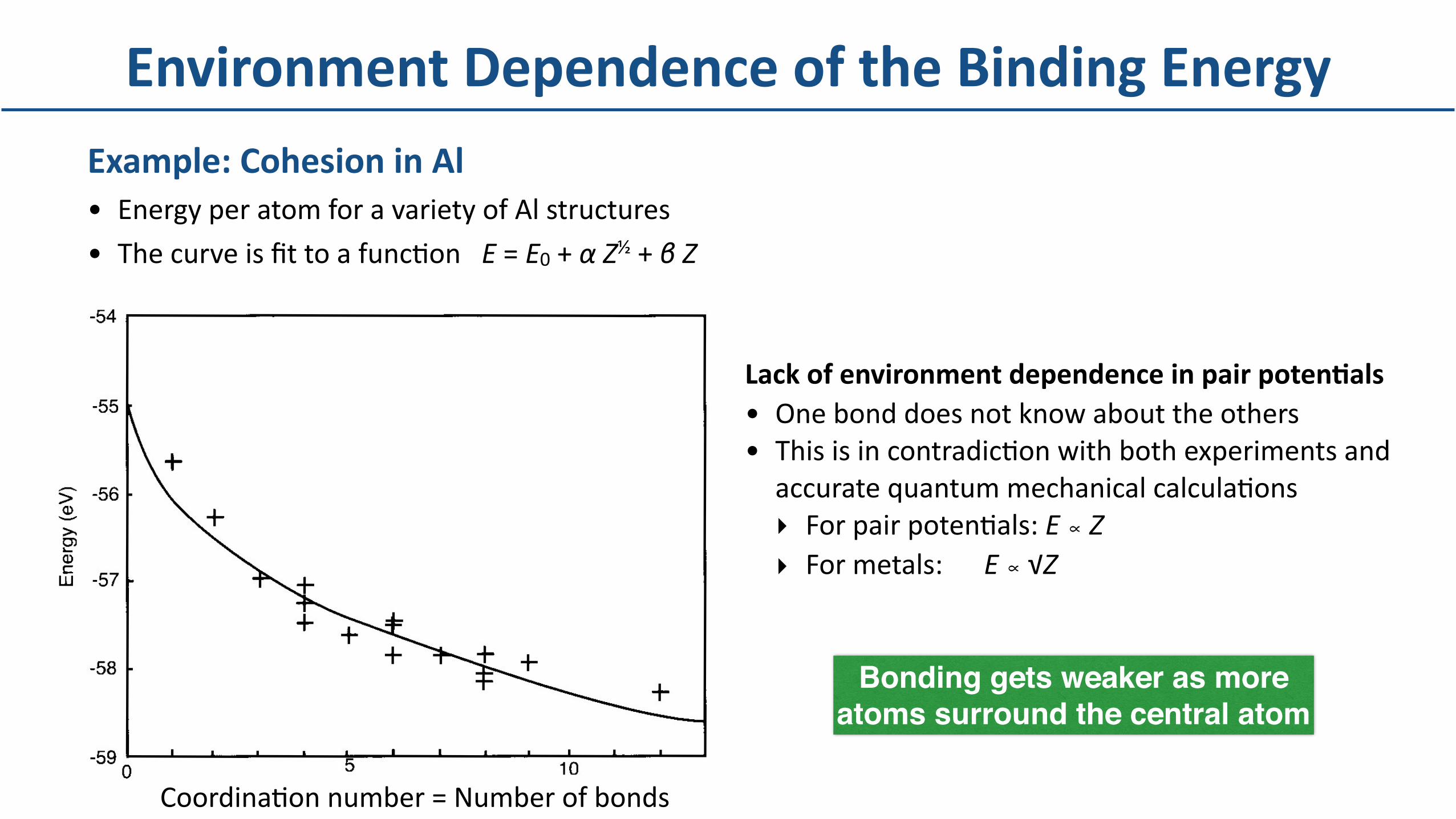
Example:CohesioninAl• EnergyperatomforavarietyofAlstructures
• ThecurveisfittoafuncDonE=E0+αZ½+βZ
EnvironmentDependenceoftheBindingEnergy
CoordinaDonnumber=Numberofbonds
LackofenvironmentdependenceinpairpotenBals• Onebonddoesnotknowabouttheothers• ThisisincontradicDonwithbothexperimentsandaccuratequantummechanicalcalculaDons‣ ForpairpotenDals:E∝Z‣ Formetals: E∝√Z
Bonding gets weaker as more atoms surround the central atom

ForpairpotenBalE∝Z• Cohesiveenergyperatom: EachAlatomhas12bondsandeachbondissharedbetweentwoatoms
• PairpotenDals–12bondsarebrokenandremovedatomisplacedinbulk
CohesiveEnergyandVacancyFormaBoninAl
�Evac = 12 · Ebond⇤ ⇥� ⌅broken bonds
� 6 · Ebond⇤ ⇥� ⌅bulk
= 6 · Ebond = Ecoh
Ecoh =12 · Ebond
2= 6 · Ebond
RemoveasingleatomandplaceitinbulkposiBonsomewhereelse
FormetalsE∝√Z• EachoftheneighboringatomschangesconfiguraDonnumberfrom12to11
• Removedatomsisagainplacedinbulk
�Evac = 12 ·�c ·⌅
12� c ·⌅
11⇥
�Evac
Ecoh= 12 ·
⇤1�
⌅11⌅12
⌅⇤ 0.5
• VacancyenergyinmetalslowerthenpredictedbypairpotenDals

Example:CohesioninSi• Cohesiveenergyhasamaximumfor4-foldcoordinateddiamondstructure• EnergyofeachbonddecreaseswithincreasedcoordinaDonnumber
0 4 8 12
Coordination Z
0
1
2
3
4
5
Ener
gy (
eV)
per bond
per atom
Silicon
Bondstrengthdependsonlocalenvironment
EnvironmentDependenceoftheBindingEnergy
• PairpotenDalscannotpredictcrystalstructuresinmetalsorcovalentsolids
• Forexample,thefcc-bccenergydifferencerequiresfour-bodyinteracDons
• Bondstrengthdependsonlocalenvironment‣ Eitherthroughangulardependencewithotherbonds‣ Orthroughdependenceonnumberofotherbonds,e.g.bond-order
• ThislimitsthetransferabilityofpairpotenDals• FiUedforoneparDcularcoordinaDonenvironmenttheycannotbeusedwithoutsignificanterrorforothercoordinaDon(e.g.fittobulkbutuseonsurface)
• Fiangtoallenvironmentssimultaneouslyonly“averages”theerror

Pair
potentials
Cluster
potentials
Pair
functionals
Cluster
functionals
Many-body
Non-linearity
HowtoOvercomeDeficienciesofPairPotenBals
• Includeeffectofotheratomsonbond• Energyasanon-linearfuncDonofcoordinaDon
• PairfuncBonalsincludeeffecDvemediumpotenDals
• Includethree-bodyterms(angulardependentforces)andfour-bodyterms
• ClusterpotenBalsincludemany-bodyterms
• ClusterfuncBonalscombineboth
E =X
i
�1(Ri) +X
i
X
j>i
�2(Ri,Rj) +X
i
X
j>i
X
k>j
�3(Ri,Rj ,Rk) + · · ·

Ecoh =�
i
Fi(�i)
⌅ ⇤⇥ ⇧Embedding energy
+12
�
i
�
j �=i
V (Rij)
⌅ ⇤⇥ ⇧Pair potential
�i =�
i �=j
f(Rij)
i j
Idea:• Energyofanatomdependsnon-linearlyonthesurroundingatoms(numberanddistance)• E=f(numberofbonds)wherefisanon-linearfuncDon⇒EnergyfuncBonals
EmbeddedAtomPotenBals(EAM)
• UseeitheranalyDcortabulatedembeddingfuncDon• TabulatedformcomputaDonallyefficient,useofcubicsplines• OrentheembeddingfuncBonisfittotheequaDonofstate(Energyversusvolume)
• Useelectrondensityasameasureofthesurroundingatoms

PhysicsConcept• Bondingenergy(embeddingenergy)duetoelectrondelocalizaDon• Aselectronsspreadoutmore,theirkineDcenergydecreases• Whenanimpurityisputintoametalitsenergyisloweredbecauseitselectronscandelocalizeintothesolid
• Theembeddingdensity(electrondensityattheembeddingsite)isameasureofthenumberofstatesavailabletodelocalizeonto ⇒Manybodyeffect
EmbeddedAtomPotenBals(EAM)
OthereffecBvemediumtheories• EAMissimilartomanyothereffecDvemediumtheories• Othertheoriesdifferinthe“non-linearity”usedorthemeasureof“embeddingdensity”• Gluemodel(Ercollesi,TosaaandParrinello)• Finnis-SinclairPotenDals• EquivalentCrystalModels(SmithandBanerjee)
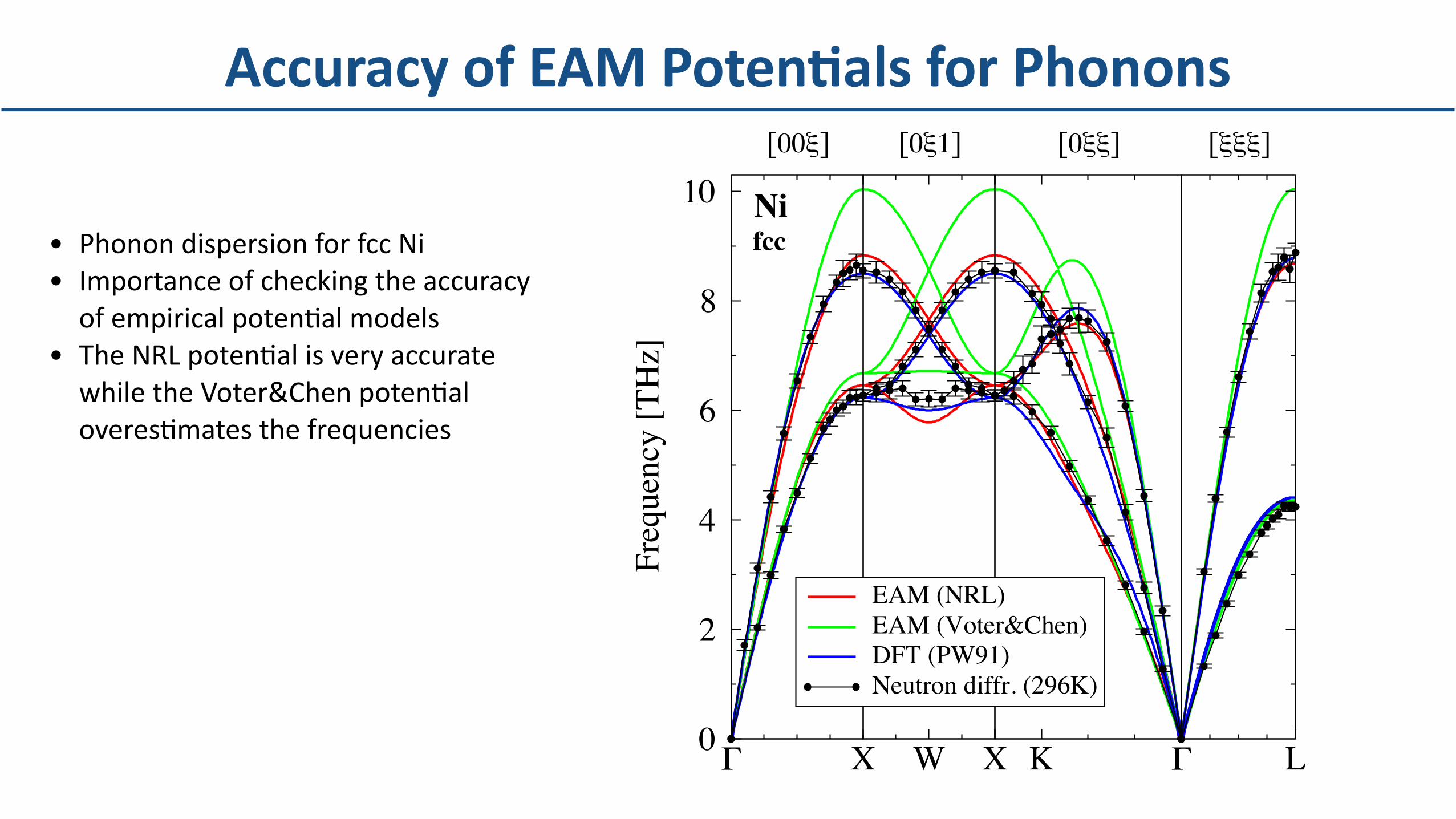
• PhonondispersionforfccNi• ImportanceofcheckingtheaccuracyofempiricalpotenDalmodels
• TheNRLpotenDalisveryaccuratewhiletheVoter&ChenpotenDaloveresDmatesthefrequencies
!0
2
4
6
8
10
Fre
quen
cy [
TH
z]
[00"]
X W
[0"1]
X K
[0""]
! L
Ni
fcc
EAM (NRL)
EAM (Voter&Chen)
DFT (PW91)
Neutron diffr. (296K)
["""]
AccuracyofEAMPotenBalsforPhonons
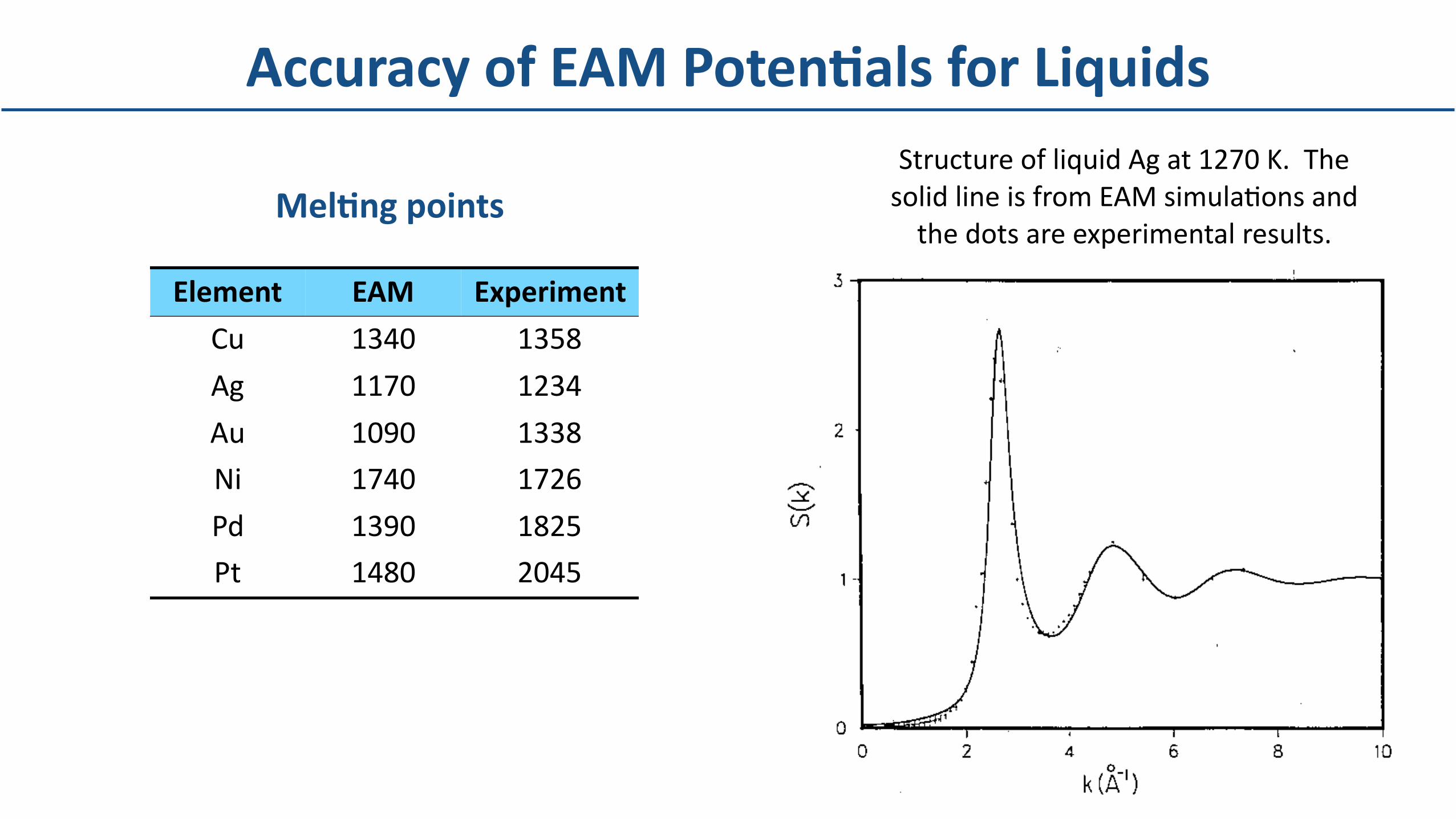
Element EAM Experiment
Cu 1340 1358
Ag 1170 1234
Au 1090 1338
Ni 1740 1726
Pd 1390 1825
Pt 1480 2045
MelBngpointsStructureofliquidAgat1270K.ThesolidlineisfromEAMsimulaDonsandthedotsareexperimentalresults.
AccuracyofEAMPotenBalsforLiquids

GrainboundaryinAl• ComparisonoftheoryandexperimentforagrainboundaryinAl.
• Thehigh-resoluDonTEMimageofDltboundaryisoverlaidwithaninsetofthesimulatedstructurepredictedbyanEAMpotenDal
AccuracyofEAMPotenBalsforGrainBoundaries
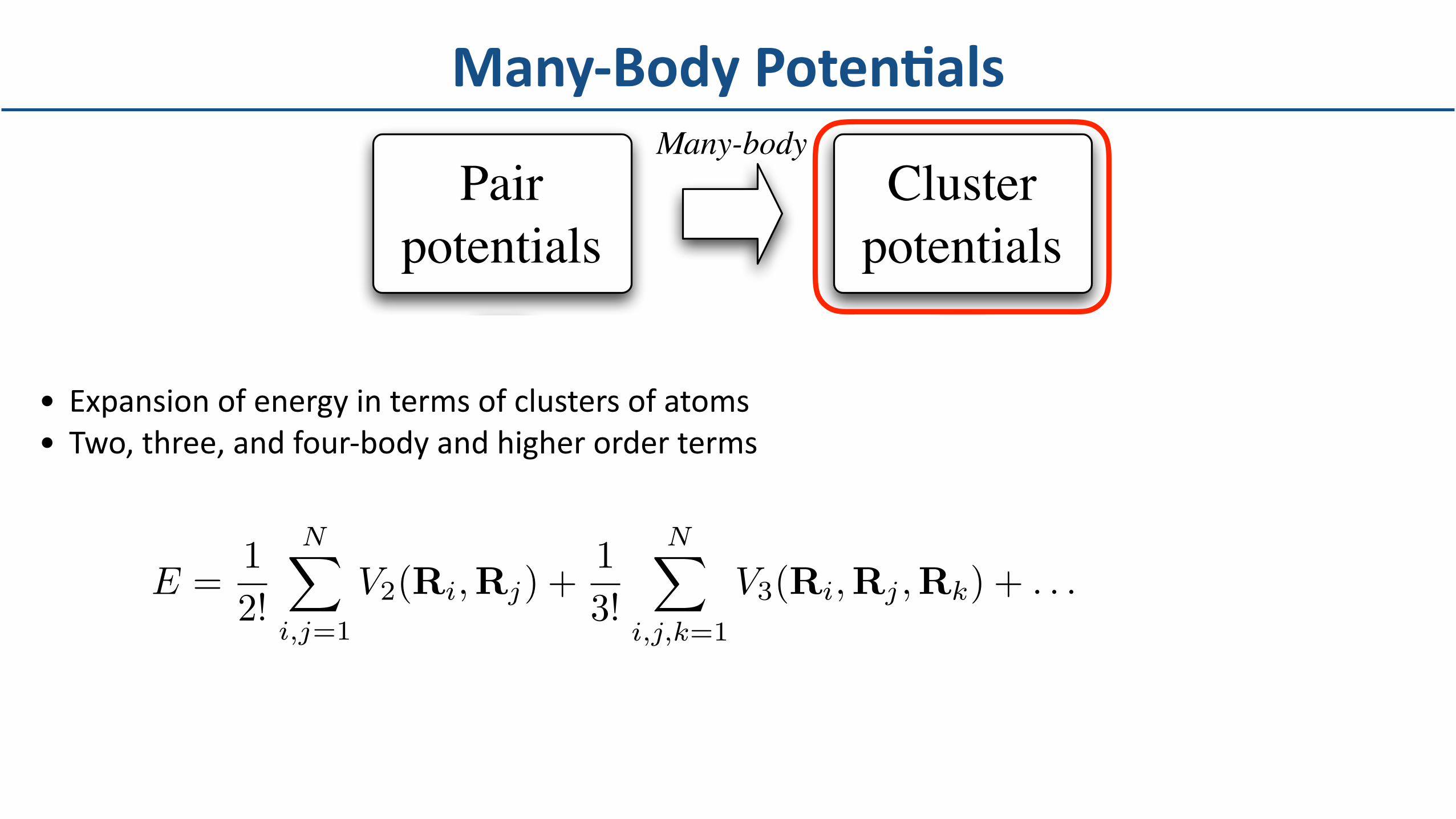
• Expansionofenergyintermsofclustersofatoms• Two,three,andfour-bodyandhigherorderterms
Pair
potentials
Cluster
potentials
Pair
functionals
Cluster
functionals
Many-body
Non-linearity
E =12!
N�
i,j=1
V2(Ri,Rj) +13!
N�
i,j,k=1
V3(Ri,Rj ,Rk) + . . .
Many-BodyPotenBals
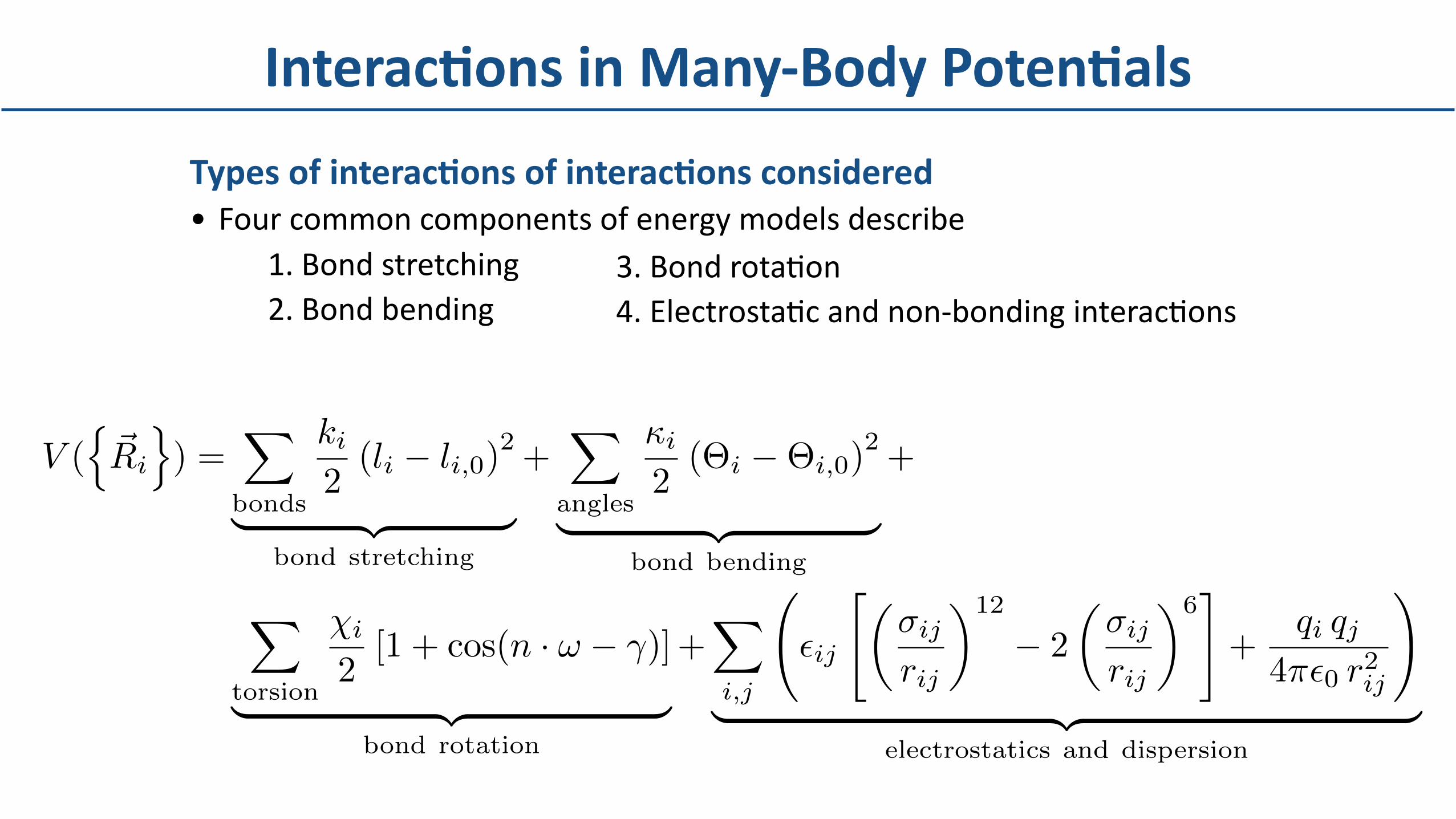
TypesofinteracBonsofinteracBonsconsidered• Fourcommoncomponentsofenergymodelsdescribe
1.Bondstretching2.Bondbending
V (
n
~Ri
o
) =
X
bonds
ki2
(li � li,0)2
| {z }
bond stretching
+
X
angles
i
2
(⇥i �⇥i,0)2
| {z }
bond bending
+
X
torsion
�i
2
[1 + cos(n · ! � �)]
| {z }
bond rotation
+
X
i,j
✏ij
"
✓
�ij
rij
◆
12
� 2
✓
�ij
rij
◆
6
#
+
qi qj4⇡✏
0
r2ij
!
| {z }
electrostatics and dispersion
InteracBonsinMany-BodyPotenBals
3.BondrotaDon4.ElectrostaDcandnon-bondinginteracDons

• SDllinger-WeberpotenDal
• ReproducesSiproperDessuchas2×1reconstrucDonofSi(100)butnot7×7reconstrucDon
V (
n
~Ri
o
) =
X
bonds
ki2
(li � li,0)2
| {z }
bond stretching
+
X
angles
i
2
(⇥i �⇥i,0)2
| {z }
bond bending
+
X
torsion
�i
2
[1 + cos(n · ! � �)]
| {z }
bond rotation
+
X
i,j
✏ij
"
✓
�ij
rij
◆
12
� 2
✓
�ij
rij
◆
6
#
+
qi qj4⇡✏
0
r2ij
!
| {z }
electrostatics and dispersion
Example:3-BodyPotenBalforSilicon• CoordinatesRi,RjandRkcanbereplacedbyRi–Rj,Rk–Rjandθijk• TetrahedralcoordinaDon:‣ Bondangleofθ0=109.5°andbonddistanceof2.35Å
• PossiblechoicesareK(θ-θ0)2orK(cosθijk+1/3)2
E =12
N⇤
i,j=1
V (Rij) +N⇤
i,j,k=1
g(Rij) g(Rkj)�
cos �ijk +13
⇥2

DisBnguishbetweenbondedandnon-bondedinteracBons
(1) EthaneH3C–CH3
- TorsionofC–Cbond- StaggeredversuseclipsedconfiguraDon hasdifferentenergy
- Requiresfour-bodypotenDal
(2) EtheneH2C=CH2
- DoublebondbetweenC=ChasdifferentstrengththansinglebondC–Cinethane
- RequiresclusterfuncDonalordifferentpotenDalsforsp,sp2,andsp3carbon
• ChangesincoordinaDonaredonebychangingthepotenDal
• Examples:AMBER,CHARMM,MM3
Vtorsion = K · cos(3�)
PotenBalsforOrganicMolecules

• Sumofmany-bodyandCoulombterms• CanhandlebondbreakingandformaDon
ReacBveForceFieldPotenBals(COMBandReaxFF)
parameterisations and development branches’ outline thecurrently employed method and available parameter sets,respectively. Comparisons with other reactive potentials availablein the literature are briefly discussed in ‘Comparison to similarmethods’. Examples of various ReaxFF applications are provided in‘Applications of ReaxFF’, with emphasis placed on demonstratingthe breadth of systems that can be modelled with the method.Finally, plans for future extensions and improvements arediscussed in ‘Future developments and outlook’.
DEVELOPMENT OF THE ReaxFF METHODHistory of ReaxFF developmentThe current functional form of the ReaxFF potential, bestdescribed in the Chenoweth et al.2 hydrocarbon combustionwork (herein referred to as 2008-C/H/O),2 has demonstratedsignificant transferability across the periodic table. It is importantto note, however, that the 2008 functional form is different fromthe original 2001 ReaxFF hydrocarbon description,1 as well as fromthe 2003 extension to silicon and silica.3 Although conceptuallysimilar to the current 2008-C/H/O functional form, the 2001hydrocarbon description employed the same dissociation energyfor C–C single, double and triple bonds. This approach wasreasonable for hydrocarbons, but could not be extended to treatSi–O single and double bonds. As such, the 2003 Si/O/H extensionrequired separate parameters describing single-, double- andtriple-bond dissociation. Furthermore a lone-pair energy term wasintroduced to handle formation and dissociation of oxygenlone-pairs. The 2003 Si/O functional form was further augmentedby a three-body conjugation term introduced to handle −NO2group chemistry in nitramines, where a triple-bond stabilisationterm was added to improve the description of terminal triplebonds. This led to the 2003–2005 ReaxFF description for the RDXhigh-energy material employed by Strachan et al.4,5 to study RDXinitiation.Since 2005, the ReaxFF functional form has been stable,
although optional additions, such as angular terms to destabiliseMg–Mg–H zero-degree angles6 or double-well angular termsnecessary for describing aqueous transition metal ions,7 haveoccasionally been added to the potential. Goddard and co-workers implemented an additional attractive van der Waals termto improve performance for nitramine crystals (ReaxFF-lg).8 Thisconcept, however, was not made transferable with previous orlater ReaxFF parameter sets. The 2005 functional form developedfor RDX is the current version of ReaxFF distributed by the vanDuin group (commonly referred to as ‘standalone ReaxFF’), as wellas integrated in the open-source LAMMPS code,9 supportedthrough Nanohub, (http://www.nanohub.org) and availablethrough the PuReMD (Purdue Reactive Molecular Dynamics)code.10–12 Apart from these open-source distributions, the ReaxFFmethod is also integrated in ADF13 (released by SCM (http://www.scm.com)) and in Materials Studio (released under license byAccelrys (http://www.accelrys.com)). The pre-2005 ReaxFF para-meter sets, including the aforementioned 2001-C/H,1 2003-Si/O,3
and 2004-Al/O14 descriptions, are not supported by any codescurated by the van Duin group or its collaborators. The materialsdescribed by the three pre-2005 ReaxFF parameterisations areequally, if not better, described by later parameterisations. The2008-C/H/O parameter set was trained against the entire 2001-C/Htraining set, while the 2010 and 2011 Si/O/H parameterisations(Fogarty et al.15 on the ‘aqueous branch’ and Neyts et al.16 on the‘combustion branch’) were validated against the full 2003-Si/O/Htraining set. Finally, the 2008-Al/H ReaxFF description, and laterapplications to aluminium oxides and aluminosilicates,17–23 fullycontain and extend the 2004-Al/O description. We are aware ofother ReaxFF implementations, often developed by individualscientists based on the 2008-C/H/O formalism. Given the
complexity of the ReaxFF functional form, it is advisable tovalidate ReaxFF implementations against the standalone ReaxFFcode prior to applying them in production-scale simulations.
Current ReaxFF methodologyThe currently implemented form of the ReaxFF potential isdescribed in detail in a recent article,24 and therefore here we onlyprovide a brief overview of the method’s central concepts. ReaxFFemploys a bond-order formalism in conjunction with polarisablecharge descriptions to describe both reactive and non-reactiveinteractions between atoms (Figure 1). This allows ReaxFF toaccurately model both covalent and electrostatic interactions for adiverse range of materials. Energy contributions to the ReaxFFpotential are summarised by the following:
Esystem ¼ Ebond þ Eover þ Eangle þ Etors þ EvdWaals þ ECoulomb
þ ESpecific: ð1Þ
Ebond is a continuous function of interatomic distance anddescribes the energy associated with forming bonds betweenatoms. Eangle and Etors are the energies associated with three-bodyvalence angle strain and four-body torsional angle strain. Eover isan energy penalty preventing the over coordination of atoms,which is based on atomic valence rules (e.g., a stiff energy penaltyis applied if a carbon atom forms more than four bonds).ECoulomb and EvdWaals are electrostatic and dispersive contributionscalculated between all atoms, regardless of connectivity andbond-order. ESpecific represents system specific terms that arenot generally included, unless required to capture propertiesparticular to the system of interest, such as lone-pair, conjugation,hydrogen binding, and C2 corrections. Full functional forms can befound in the Supplementary Information of the 2008-C/H/Opublication.2
Figure 1. (a) Overview of the ReaxFF total energy components and(b) elements currently described in available parameter sets.
The ReaxFF reactive force-fieldTP Senftle et al
2
npj Computational Materials (2016) 15011 © 2016 Shanghai Institute of Ceramics, Chinese Academy of Sciences/Macmillan Publishers Limited
parameterisations and development branches’ outline thecurrently employed method and available parameter sets,respectively. Comparisons with other reactive potentials availablein the literature are briefly discussed in ‘Comparison to similarmethods’. Examples of various ReaxFF applications are provided in‘Applications of ReaxFF’, with emphasis placed on demonstratingthe breadth of systems that can be modelled with the method.Finally, plans for future extensions and improvements arediscussed in ‘Future developments and outlook’.
DEVELOPMENT OF THE ReaxFF METHODHistory of ReaxFF developmentThe current functional form of the ReaxFF potential, bestdescribed in the Chenoweth et al.2 hydrocarbon combustionwork (herein referred to as 2008-C/H/O),2 has demonstratedsignificant transferability across the periodic table. It is importantto note, however, that the 2008 functional form is different fromthe original 2001 ReaxFF hydrocarbon description,1 as well as fromthe 2003 extension to silicon and silica.3 Although conceptuallysimilar to the current 2008-C/H/O functional form, the 2001hydrocarbon description employed the same dissociation energyfor C–C single, double and triple bonds. This approach wasreasonable for hydrocarbons, but could not be extended to treatSi–O single and double bonds. As such, the 2003 Si/O/H extensionrequired separate parameters describing single-, double- andtriple-bond dissociation. Furthermore a lone-pair energy term wasintroduced to handle formation and dissociation of oxygenlone-pairs. The 2003 Si/O functional form was further augmentedby a three-body conjugation term introduced to handle −NO2group chemistry in nitramines, where a triple-bond stabilisationterm was added to improve the description of terminal triplebonds. This led to the 2003–2005 ReaxFF description for the RDXhigh-energy material employed by Strachan et al.4,5 to study RDXinitiation.Since 2005, the ReaxFF functional form has been stable,
although optional additions, such as angular terms to destabiliseMg–Mg–H zero-degree angles6 or double-well angular termsnecessary for describing aqueous transition metal ions,7 haveoccasionally been added to the potential. Goddard and co-workers implemented an additional attractive van der Waals termto improve performance for nitramine crystals (ReaxFF-lg).8 Thisconcept, however, was not made transferable with previous orlater ReaxFF parameter sets. The 2005 functional form developedfor RDX is the current version of ReaxFF distributed by the vanDuin group (commonly referred to as ‘standalone ReaxFF’), as wellas integrated in the open-source LAMMPS code,9 supportedthrough Nanohub, (http://www.nanohub.org) and availablethrough the PuReMD (Purdue Reactive Molecular Dynamics)code.10–12 Apart from these open-source distributions, the ReaxFFmethod is also integrated in ADF13 (released by SCM (http://www.scm.com)) and in Materials Studio (released under license byAccelrys (http://www.accelrys.com)). The pre-2005 ReaxFF para-meter sets, including the aforementioned 2001-C/H,1 2003-Si/O,3
and 2004-Al/O14 descriptions, are not supported by any codescurated by the van Duin group or its collaborators. The materialsdescribed by the three pre-2005 ReaxFF parameterisations areequally, if not better, described by later parameterisations. The2008-C/H/O parameter set was trained against the entire 2001-C/Htraining set, while the 2010 and 2011 Si/O/H parameterisations(Fogarty et al.15 on the ‘aqueous branch’ and Neyts et al.16 on the‘combustion branch’) were validated against the full 2003-Si/O/Htraining set. Finally, the 2008-Al/H ReaxFF description, and laterapplications to aluminium oxides and aluminosilicates,17–23 fullycontain and extend the 2004-Al/O description. We are aware ofother ReaxFF implementations, often developed by individualscientists based on the 2008-C/H/O formalism. Given the
complexity of the ReaxFF functional form, it is advisable tovalidate ReaxFF implementations against the standalone ReaxFFcode prior to applying them in production-scale simulations.
Current ReaxFF methodologyThe currently implemented form of the ReaxFF potential isdescribed in detail in a recent article,24 and therefore here we onlyprovide a brief overview of the method’s central concepts. ReaxFFemploys a bond-order formalism in conjunction with polarisablecharge descriptions to describe both reactive and non-reactiveinteractions between atoms (Figure 1). This allows ReaxFF toaccurately model both covalent and electrostatic interactions for adiverse range of materials. Energy contributions to the ReaxFFpotential are summarised by the following:
Esystem ¼ Ebond þ Eover þ Eangle þ Etors þ EvdWaals þ ECoulomb
þ ESpecific: ð1Þ
Ebond is a continuous function of interatomic distance anddescribes the energy associated with forming bonds betweenatoms. Eangle and Etors are the energies associated with three-bodyvalence angle strain and four-body torsional angle strain. Eover isan energy penalty preventing the over coordination of atoms,which is based on atomic valence rules (e.g., a stiff energy penaltyis applied if a carbon atom forms more than four bonds).ECoulomb and EvdWaals are electrostatic and dispersive contributionscalculated between all atoms, regardless of connectivity andbond-order. ESpecific represents system specific terms that arenot generally included, unless required to capture propertiesparticular to the system of interest, such as lone-pair, conjugation,hydrogen binding, and C2 corrections. Full functional forms can befound in the Supplementary Information of the 2008-C/H/Opublication.2
Figure 1. (a) Overview of the ReaxFF total energy components and(b) elements currently described in available parameter sets.
The ReaxFF reactive force-fieldTP Senftle et al
2
npj Computational Materials (2016) 15011 © 2016 Shanghai Institute of Ceramics, Chinese Academy of Sciences/Macmillan Publishers Limited
ComputaDonalMaterials2,15011(2016)
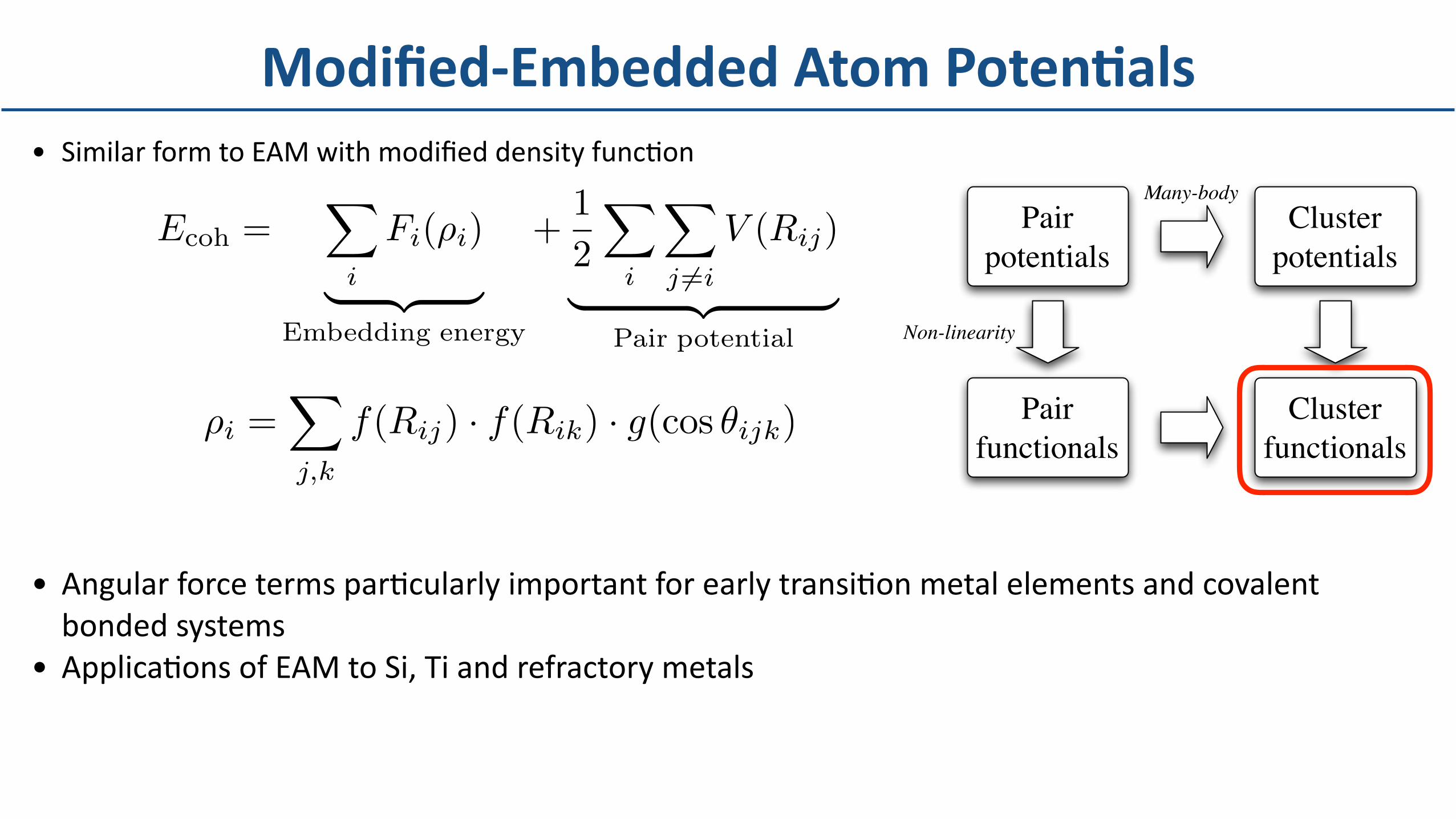
• SimilarformtoEAMwithmodifieddensityfuncDon
⇥i =�
j,k
f(Rij) · f(Rik) · g(cos �ijk)
Ecoh =�
i
Fi(�i)
⌅ ⇤⇥ ⇧Embedding energy
+12
�
i
�
j �=i
V (Rij)
⌅ ⇤⇥ ⇧Pair potential
• AngularforcetermsparDcularlyimportantforearlytransiDonmetalelementsandcovalentbondedsystems
• ApplicaDonsofEAMtoSi,Tiandrefractorymetals
Modified-EmbeddedAtomPotenBals
Pair
potentials
Cluster
potentials
Pair
functionals
Cluster
functionals
Many-body
Non-linearity
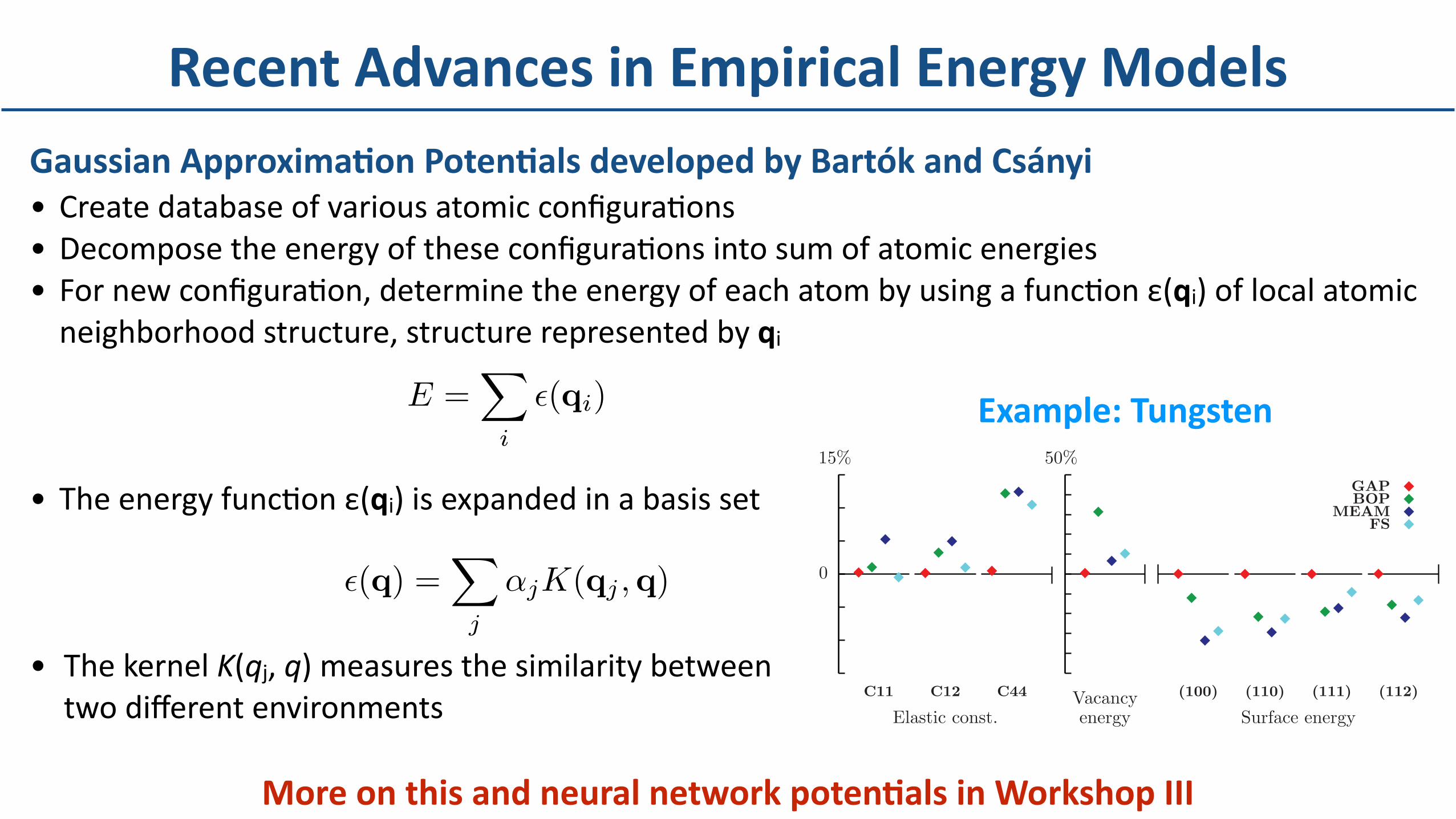
RecentAdvancesinEmpiricalEnergyModelsGaussianApproximaBonPotenBalsdevelopedbyBartókandCsányi• CreatedatabaseofvariousatomicconfiguraDons• DecomposetheenergyoftheseconfiguraDonsintosumofatomicenergies• FornewconfiguraDon,determinetheenergyofeachatombyusingafuncDonε(qi)oflocalatomicneighborhoodstructure,structurerepresentedbyqi
• TheenergyfuncDonε(qi)isexpandedinabasisset
• ThekernelK(qj,q)measuresthesimilaritybetween twodifferentenvironments
MoreonthisandneuralnetworkpotenBalsinWorkshopIII
PHYSICAL REVIEW B 90, 104108 (2014)
Accuracy and transferability of Gaussian approximation potential models for tungsten
Wojciech J. Szlachta, Albert P. Bartok, and Gabor CsanyiEngineering Laboratory, University of Cambridge, Trumpington Street, Cambridge CB2 1PZ, United Kingdom
(Received 16 May 2014; revised manuscript received 26 August 2014; published 24 September 2014)
We introduce interatomic potentials for tungsten in the bcc crystal phase and its defects within the Gaussianapproximation potential framework, fitted to a database of first-principles density functional theory calculations.We investigate the performance of a sequence of models based on databases of increasing coverage in configurationspace and showcase our strategy of choosing representative small unit cells to train models that predict propertiesobservable only using thousands of atoms. The most comprehensive model is then used to calculate propertiesof the screw dislocation, including its structure, the Peierls barrier and the energetics of the vacancy-dislocationinteraction. All software and raw data are available at www.libatoms.org.
DOI: 10.1103/PhysRevB.90.104108 PACS number(s): 65.40.De, 31.50.−x, 34.20.Cf, 71.15.Nc
Tungsten is a hard, refractory metal with the highest meltingpoint (3695 K) among metals, and its alloys are utilizedin numerous technological applications. The details of theatomistic processes behind the plastic behavior of tungstenhave been investigated for a long time, and many interatomicpotentials exist in the literature reflecting an evolution, overthe past three decades, in their level of sophistication, startingwith the Finnis-Sinclair (FS) potential [1], embedded atommodel (EAM) [2], various other FS and EAM parametrizations[3–6], modified embedded atom models (MEAMs) [7–10],and bond order potentials (BOPs) [11–13]. While some ofthese methods have been used to study other transition metals[14–16], there is renewed interest in modeling tungsten due toits many high-temperature applications—e.g., it is one of thecandidate materials for plasma facing components in the JETand ITER fusion projects [17–19].
A recurring problem with empirical potentials, due tothe use of fixed functional forms with only a few adjustableparameters, is the lack of flexibility: when fitted to repro-duce a given property, predictions for other properties canhave large errors. Figure 1 shows the basic performance ofthe BOP and MEAM, two of the more sophisticated potentialsthat reproduce the correct screw dislocation core structure,and also the simpler FS potential,1 all in comparison withresults of density functional theory (DFT). While the figureemphasizes fractional accuracy, we show the correspondingabsolute numerical values in Table I. The BOP is poor indescribing the vacancy but is better at surfaces, whereas theMEAM is the other way around. While this compromisecan sometimes be made with good judgment for specificapplications, many interesting properties, particularly thosethat determine the material behavior at larger length scales,arise from the competition between different atomic-scaleprocesses, which therefore all need to be described equallywell. For example, dislocation pinning, depinning, and climbinvolve both elastic properties and core structure, as well asthe interaction of dislocations with defects. Ways to deal withthis problem include use of multiple levels of accuracy as inquantum mechanics/molecular mechanics [20] or allowing theparameters of the potential to vary in time and space [21].
1Rescaled to the DFT lattice constant and bulk modulus.
Here we describe a milestone in a research program aimed atcreating a potential that circumvents the problem of fixed func-tional forms. The purpose of the present work is twofold. First,we showcase the power of the nonparametric database-drivenapproach by constructing an accurate potential and using itto compute atomic-scale properties that are inaccessible toDFT due to computational expense. Second, while there hasbeen vigorous activity recently in developing such models,most of the attention has been focused on the interpola-tion method and the neighborhood descriptors (e.g., neuralnetworks [22–24], Shepherd interpolation [25,26], invari-ant polynomials [27–29], and Gaussian processes [30–34]);rather less prominence was given to the question of howto construct suitable databases that ultimately determine therange of validity of the potential. Our second goal is thereforeto study what kinds of configurations need to be in a databaseso that given material properties are well reproduced. A largerdatabase costs more to create and the resulting potential isslower, but can be expected to be more widely applicable, thusproviding a tunable trade-off between transferability, accuracy,and computational cost.
In our Gaussian approximation potential (GAP) framework[30,31], the only uncontrolled approximation is the oneessential to the idea of interatomic potentials: the total energyis written as a sum of atomic energies,
E =!
i
ε(qi), (1)
with ε a universal function of the atomic neighborhoodstructure inside a finite cutoff radius as represented by the
0
15%
C11 C12 C44
50%
Elastic const.Vacancyenergy Surface energy
(100) (110) (111) (112)
GAPBOP
MEAMFS
FIG. 1. (Color online) Fractional error in elastic constants anddefect energies calculated with various interatomic potentials, ascompared to the target DFT values.
1098-0121/2014/90(10)/104108(6) 104108-1 ©2014 American Physical Society
✏(q) =X
j
↵jK(qj ,q)
E =X
i
✏(qi) Example:Tungsten

DataforopBmizaBonofpotenBalparameters• ProperDesofcrystals,defects,liquids‣ Crystalstructuresandenergydifferences‣ Laaceconstants,cohesiveenergy,equaDonofstate‣ ElasDccoefficients,phononfrequencies,andforces‣ Pointdefectstructuresandenergies,surfaceenergies,andrelaxaDon
OpBmizaBonmethods• OpDmizaDonusingsimulatedannealing,paralleltempering,geneDcalgorithms,etc.• ParametersusuallyloosetheparDcularphysicalmeaningoftheanalyDcform• ItiscrucialtotestthetransferabilityandaccuracyofthepotenDalondatathatwasnotinthefit• EmpiricalpotenDalshavethetendencytoleadtounexpectedbehaviorinpartsofphasespacesuchasenergydivergenciesandunphysicalroughnessoflandscape
• FiangapotenDalisanartformandrequiresalotofexperience
⇒Clearneedforimprovedmethods
OpBmizaBonofModelParameters

EvaluaBonofEmpiricalPotenBalsComparisonbetweenpotenBals:• MostpotenDalsresultinsimilarstaDcproperDes• Note,thattheyareorenfittostaDcproperDes• ProblemsusuallyoccurfordynamicsproperDes(forces,phonons)anddefectproperDes
Formetals• Bondenergydependsonthenumberofbondsalreadymadetoanatom
• ThiseffectisabsentinpairpotenDals,whichareenvironment-independent
• Hence,wheneverbond-breakingisinvolved,theresultofapotenDalmodelshouldbeinterpretedcauDously
Fororganicmolecules• VerygoodpotenDalshavebeenfittoC-HandC-Cbondsinvariousbondingarrangements(AMBER,CHARMM,MM3,MMFF94)
• ThesecanbeusedtomodelconformaDonalarrangementsofpolymericsystems(wherenobond-breakingisinvolved)
Foroxides• Inhighlyionicoxides,qualitaDvelyreasonableresultscanbeexpectedwithempiricalpotenDalmodels(+electrostaDcenergy)
• Accuracyismainlylimitedbytheoxygen“breathing”effect
• Themorecovalenttheoxide,themoredifficultitwillbetofindpotenDalsthatreproducethematerialsbehaviorinawiderangeofenvironments
• ShellpolarizaDonisessenDalinlowsymmetryenvironments

ShouldatleastuseatesBngdatasettoesBmateuncertainty• TesDngdatasetsofsimilarproperDesasfiangdataset• Canalsocontainmoreexpensivedatasuchas:‣ MelDngpoints‣ Thermalexpansion‣ ThermalconducDvity‣ PhasetransiDonpressures‣ Rates‣ …
• TheserequireMDsimulaDonsandarelesssuitableforfiangdatabase
ValidaBonofEmpiricalEnergyModels

ValidaBonofenergylandscapes• Goal:IdenDfyunphysicalminimawherethepotenDalfails• Strategy:ExploreconfiguraDonspaceusingglobalopDmizaDonmethods• Methods:‣ Simulatedannealing,paralleltemperingMD‣ Basinhopping‣ EvoluDonaryalgorithms‣ …
ValidaBonofEmpiricalEnergyModels

Example1:MoMEAMpotenBal• ComparisonofempiricalpotenDalwith density-funcDonaltheory
• Modifiedembedded-atommodelforMo• PerformGASPsearchwithLAMMPS• CalculateDFTenergyofnewlyfoundstructures• PotenDalreproducesthegroundstateand alllow-lyingminima
• ProvidesconfidenceinaccuracyofthepotenDal
ValidaBonofEmpiricalEnergyModels
Phys. Rev. B 85, 214121 (2012)
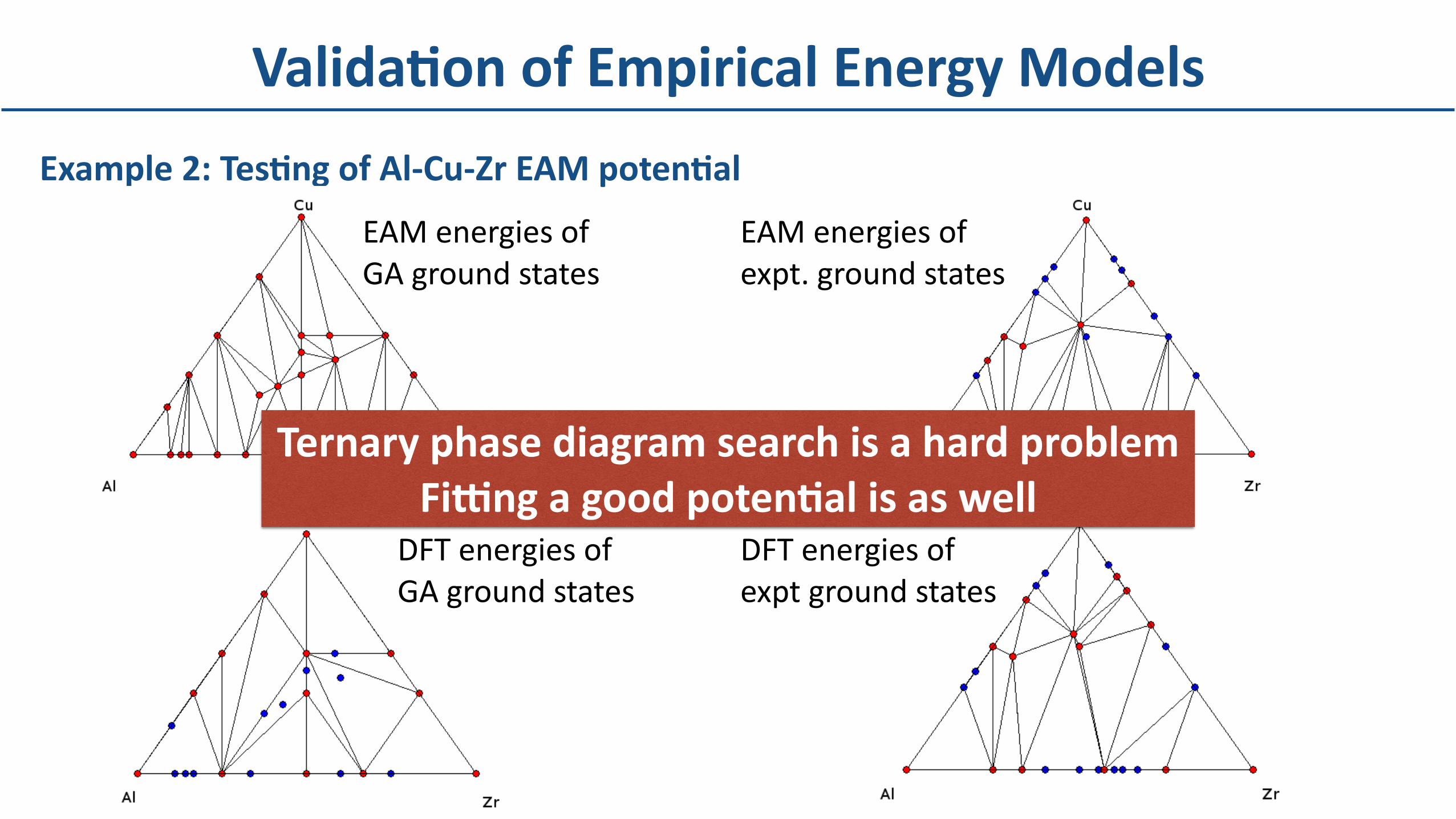
Example2:TesBngofAl-Cu-ZrEAMpotenBal
ValidaBonofEmpiricalEnergyModels
EAMenergiesof GAgroundstates
DFTenergiesofGAgroundstates
DFTenergiesof exptgroundstates
EAMenergiesof expt.groundstates
TernaryphasediagramsearchisahardproblemFidngagoodpotenBalisaswell

0 0.2 0.4 0.6 0.8 1Lithium fraction
-30
-20
-10
0
10
Form
atio
n en
ergy
(kca
l/mol
)
GA structuresGA ground states
Example2:Li-Ssystem• Threegroundstatephases:Li,Li2S,S• Limonosulfide,LiS,reportedbyThomasandJonesin1929• ReaxFFpotenDalfiangandevoluDonaryalgorithmtesDng(vanDuinetal.)• UnphysicalstructurefromfirstsearchwereusedinfittoimprovepotenDal
ValidaBonofEmpiricalEnergyModels
0 0.2 0.4 0.6 0.8 1Composition
-1.5
-1.0
-0.5
0.0
Form
atio
n En
ergy
(eV
/ato
m)
Density-funcBonaltheory ReaxFFpotenBalmodel
OpenQuesBon
HowcanweeffecDvelyemployenergymodelsassurrogatemodelsforenergylandscapes?
HowshouldweselecttheopDmalfuncDonalformofanenergymodelforagivenmaterialssystem?
HowshouldwechooseafiangdatasetsforparameteropDmizaDonthatminimizesthe
errorsonthemodelpredicDons?HowcanweidenDfytradeoffsbetweenconflicDngmodelpredicDonsforagiven
funcDonalformofthemodel?