enantioselective synthesis of heterocyclic compounds mediated by
TRANSCRIPT

Issue ICHC-20 ARKIVOC 2006 (vii) 186-206
ISSN 1424-6376 Page 186 ©ARKAT
Enantioselective synthesis of heterocyclic compounds mediated by organoselenium reagents
Marcello Tiecco,* Lorenzo Testaferri, Luana Bagnoli, Francesca Marini, Claudio Santi,
Andrea Temperini, Catalina Scarponi, Silvia Sternativo, Raffaella Terlizzi, and Cristina Tomassini
Dipartimento di Chimica e Tecnologia del Farmaco, Sezione di Chimica Organica, Università di
Perugia, Italy E-mail: [email protected]
Abstract The syntheses of several types of enantiopure or enantiomerically enriched heterocyclic compounds by electrophilic additions, nucleophilic substitutions and radical additions ring closure reactions, promoted by chiral non racemic as well as by achiral organoselenium reagents, are presented and discussed. Keywords: Diselenides, organoselenium reagents, selenonium ions, selenones, cyclization reactions, intramolecular radical additions
Table of Contents Introduction 1. Syntheses and Reactions of Organoselenium Reagents 2. Cyclization Reactions by Formation of Carbon Oxygen Bonds 3. Cyclization Reactions by Formation of Carbon Nitrogen Bonds 4. Competition between Oxygen and Nitrogen 5. Cyclization Reactions by Formation of Carbon Carbon Bonds 6. References Introduction Owing to the great importance of heterocyclic compounds as final products or as reaction intermediates, several different methods are currently used for their construction. Among the various kinds of ring forming reactions, those based on the reaction of an electrophilic reagent

Issue ICHC-20 ARKIVOC 2006 (vii) 186-206
ISSN 1424-6376 Page 187 ©ARKAT
with an alkene holding a suitably positioned nucleophilic group are certainly very useful. The term of cyclofunctionalization is generally employed to describe this kind of process which can be promoted by several electrophilic reagents. The increasing popularity gained in recent years by selenium reagents to effect ring closure reactions, not only by electrophilic additions but also by nucleophilic substitutions and by radical addition reactions, is due to the easy availability of the reagents, to the numerous chemical manipulations which can be effected on the selenium moiety before or during its removal, and to the mild reaction conditions required in the various steps. 1. Syntheses and Reactions of Organoselenium Reagents Selenium promoted cyclization reactions thus provide an easy access to a wide variety of heterocyclic compounds in general and to those containing oxygen and/or nitrogen heteroatoms in particular.1 Scheme 1 illustrates the possible modes of cyclization depending on the relative position of the nucleophile and of the double bond in the starting alkene.
NuH
Se
R1
Nu
SeR
R1
NuR1
SeR
NuHR1
R
6-endo-trig
+ 5-exo-trig
RSeX
RSeX
5-endo-trig
+
NuH R1
Nu R1
SeRNuH R1
SeR4-exo-trig
Nu
R1
SeR
Scheme 1. Cyclofunctionalization reactions The deselenenylation of the final products is an important process since in most cases the removal of the selenium group can be used to effect other cyclization reactions. As indicated in Scheme 2, the reaction with tri-n-butyl tin hydride affords a carbon radical which can directly abstract a hydrogen atom or it can add to a multiple bond and then abstract a hydrogen atom. If

Issue ICHC-20 ARKIVOC 2006 (vii) 186-206
ISSN 1424-6376 Page 188 ©ARKAT
the tri-n-butyl allyl tin is employed the carbon radical add to the double bond and the allyl derivative is obtained. By oxidation a selenoxide is obtained which easily gives rise to the classical syn elimination to produce an alkene. Much less known are the deselenenylation reactions which occur by substitution after conversion of the ArSe group into a good leaving group such as a selenonium ion or a selenone. In most of these processes the organoselenium reagent can be recovered as a diselenide.
Nu
Nu
SeAr
Nu
SeArO
Nu
NuNu
Nu
H
Nu
SeArOO
Nu
+
Nu
SeArPhSe
Scheme 2. Deselenenylation reactions. Perhaps the most important aspect of organoselenium chemistry is due to the fact that several chiral non racemic diselenides have been introduced in recent years.2-9 Starting from these diselenides a large number of heterocyclic compounds have been obtained in an enantiomerically pure form. Examples of the diselenides which have been described in the literature since 1992 are indicated in Scheme 3. The possibility of disposing of optically pure reagents is a peculiar and unique property of organoselenium reagents. Enantioselective cyclofunctionalization reactions promoted by other commonly used electrophilic reagents have not been discovered so far. The chiral non racemic electrophilic organoselenium reagents can be prepared in situ from the corresponding diselenides by treatment with bromine, thionyl chloride, bromine and silver triflate or with ammonium persulfate. In the cases of the selenenyl triflates10 and sulfates,11 the anions are scarcely nucleophilic and this is a considerable advantage for the regio and stereoselectivity of the reactions. A common characteristic of all these diselenides is the close proximity of an heteroatom (oxygen, nitrogen) to the selenium atom. On the basis of crystal structure determinations, NMR investigations and theoretical calculations it has been suggested that an orbital interaction occurs between the heteroatom lone pair and the low-lying Se-Y antibonding orbital.5e,f This interaction will force the chiral center to come closer to the reaction center and this will produce a better transfer of chiral information in the selenenylation reaction (Scheme 4).

Issue ICHC-20 ARKIVOC 2006 (vii) 186-206
ISSN 1424-6376 Page 189 ©ARKAT
OSe)2
NR2
Se)2
OH
Se)2
OH
Se)2OMe
N
Se)2
NPh
Tomoda 1992 Uemura 1994 Tomoda 1994 Deziel 1994
Back 1995 Wirth 1995 Wirth 1995 Wirth 1998 Wirth-Santi 1999
Se)2NR2
Se)2R
Ph
Ph
O
O
OO
N Se)2
O
OSe)2
NMe2
Fe
Scheme 3. Chiral non racemic diselenides. With the expectation that this interaction could be more important, and hence the transfer of chirality more efficient, if the heteroatom close to the selenium atom were a sulfur atom12 instead of an oxygen or a nitrogen atom we have synthesized the sulfur containing diselenide 1 indicated in Scheme 4. In the present case the conversions into the selenenyl bromide and chloride afforded crystalline products which could be analysed by X-Ray. As expected the distances between the selenium and the sulfur atom (2,470 and 2,344 A in the bromide and chloride respectively) were much shorter than the sum of the Van der Waals radii of selenium (2,0 Ǻ) and sulfur (1,85 Ǻ).
Se
R
X
YSe)2
R
X SCH3
Se)2
SCH3
Se)2OMe
Y = Br, OTf, OSO3H, etc.X = OH, OR', NR'2 O
Se)2
1 2 3
Scheme 4. Sulfur containing diselenides. As a matter of fact these electrophilic reagents, as well as those deriving from the methoxy derivative 2 gave rise to extremely enantioselective reactions.13 These two diselenides were extensively used in our laboratory. We have also employed the easily available camphor diselenide 3 which was introduced by Back.6 In our hands the reactions effected starting from 3 were not greatly stereoselective but they presented the important advantage which consists in the easy separation by column chromatography of the two diastereomeric reaction products. The ring closure reactions which are described below take place by formation of carbon-oxygen, carbon-nitrogen or carbon-carbon bonds. With some nucleophilic groups a competition

Issue ICHC-20 ARKIVOC 2006 (vii) 186-206
ISSN 1424-6376 Page 190 ©ARKAT
between the formation of C-O or C-N bonds is observed. The asymmetric syntheses of heterocyclic compounds were effected either starting from enantiopure organoselenium reagents and achiral substrates or starting from achiral organoselenium reagents and enantiopure substrates. 2. Cyclization Reactions by Formation of Carbon Oxygen Bonds The first examples refer to the cyclofunctionalizations of alkenols 4 (Selenoetherification), promoted by the sulfur containing diselenide 1, to afford tetrahydrofuran derivatives 5 (Scheme 5). The diastereoselectivity is very good in every case (from 91:9 to 96:4). Only in the case of the formation of a quaternary stereogenic centre the diastereomeric ratio (D.r.) is slightly lower.12,13,14 The two diastereoisomeric selenium containing reaction products could not be separated and the reductive deselenenylation afforded tetrahydrofurans 6 with an ee equal to the D.r. Under the same conditions the cyclofunctionalizations of alkenoic acids 7 (Selenolactonization) afforded seleno γ-lactones 8 and then the γ-lactones 9 with similarly good diastereoselectivities (Scheme 5).12,13,14
S h 5 S l h ifi i d S l l i i R i
+
OH R
SeAr*
TfO - O ROH RO R
SeAr*
- 30 °C
Ar*SeOTf
4 5 6
Ar*SeOTf
- 30 °CO R
SeAr*
OOH RO O RO
TfO -OH RO
SeAr*+
97 8 Scheme 5. Selenoetherification and selenolactonization reactions Tetrahydrofuran derivatives were also obtained in a completely different way. In a recent work we have described the conjugate additions of selenium containing enolates to enones.15 In this case we used the camphor diselenide 3. These reactions were highly diastereoselective and only the syn adducts 10 were obtained. As indicated in Scheme 6, in the case of the phenyl derivative 11 the reduction of the carbonyl group afforded the camphor seleno alcohol 12. Treatment with phenylselenenyl triflate gave the selenonium ion 13. The diselenide is an extremely good leaving group and therefore the OH group gave rise to an intramolecular stereospecific SN2 substitution to afford the enantiopure tetrahydrofuran derivative 14. The displaced mixed diselenide disproportionates and the camphor diselenide can thus be recovered.

Issue ICHC-20 ARKIVOC 2006 (vii) 186-206
ISSN 1424-6376 Page 191 ©ARKAT
OR* =
O
Ph
CO2MeR1 SeR*
NaBH4MeOH OH
Ph
CO2MeR1 SeR*
PhSeOTf
OH
Ph
CO2MeR1 SeR*
SePh+TfO -
O
Ph
CO2MeR1
+ R*SeSeR* + PhSeSePh R1 = Ph 58% , ee > 98% = Me 61%, ee > 98%
R1
O
R2 R1
O R2
SeR*
CO2Me
TiCl4
OMe
O*RSe +
CH2Cl2, -78°C
TiCl4, Et3N
S h 6 E i l i S h i f T i b i d T h d f
10
11 12 13
14 Scheme 6. Enantioselective synthesis of trisubstituted tetrahydrofurans An interesting example of the in situ formation of the OH group is represented by the one-pot double cyclization of bis-allyl ketones 15 promoted by camphor selenenyl sulfate and water (Scheme 7).16 The first intermediate is the product of the seleno hydroxylation of one of the two double bonds and the formed hydroxy group acts as the internal nucleophile in the double cyclization. The final products are therefore the seleno substituted perhydro[2,3-b]furans 16. The four possible enantiopure diastereoisomers 17-20 were all formed and were separated by column chromatography. The deselenenylation of the first two compounds afforded the two enantiomeric methyl derivatives 21 and 22 in an optically pure form and the third isomer gave the meso compound 23. Strangely enough the forth diastereoisomer 20, which was present in minute amounts, afforded the same meso derivative 23.
O R H2O R OOH .. : O OHR
R = Ph, Me
SeR*
R*Se+X-
*RSe
O OPh
H
O OPh
H
O OPh
H
O OPh
H
O OPh
H
O OPh
H
O OPh
H
CfSe SeCfSeCfCfSe CfSe SeCf SeCfCfSe
O OR
H
SeR**RSe15 16
17 18 19 20
21 22 23
Scheme 7. Enantioselective synthesis of perhydrofuro[2,3-b]furans

Issue ICHC-20 ARKIVOC 2006 (vii) 186-206
ISSN 1424-6376 Page 192 ©ARKAT
The starting products for the synthesis of enantiopure oxazolines were prepared from the asymmetric acetamido selenenylation of alkenes promoted by camphor selenenyl sulfate in acetonitrile and water.17 The reaction steps are indicated in Scheme 8. The acetamido selenenylation of alkenes occurs with moderate to poor facial selectivity (D.r. from 65:35 to 80:20). However, the two diastereomeric addition products 24 can be easily separated by column chromatography and can be converted into enantiomerically pure oxazolines by PhSeOTf promoted deselenenylation. Once again selenonium ions 25 are produced as intermediates and the stereospecific intramolecular substitution by the oxygen atom affords the enantiopure trans oxazolines 26. The same procedure applied to the minor isomers affords the enantiomeric oxazolines. The same procedure applied to thioacetamido selenides afforded enantiopure thiazolines. Using a method described in the literature18 the trans oxazolines 26 can be converted into cis oxazolines 29 by ring opening with dilute hydrochloric acid to give 27 and transformation of the OH group into the chlorosulfite 28 which can suffer intramolecular substitution with inversion of configuration at the carbon atom. Thus, all the possible four stereoisomers of 4,5- bis-substituted oxazolines could be obtained in an enantiomerically pure form.
. +R
H
RH
N
R*Se
COH
Me+R*SeX
R
RH
H MeCNH2O
MajorR
H
RH
NHCOMe
R*SeMinor
HR
HR
NHCOMe
R*Se
24
24 (Major)ee > 98%
TfO-
PhSeOTf -PhSeSeR*NO
Me
R HH RRH
RH
NH
R*Se
O
Me
SePh
+
25 26
.
SOCl2
RH
RH
NH
O
O
Me
SOCl
NO
Me
R HH R
NHCOMeHO
R HH R
NO
Me
R HH R
HCl 2%
26 27 28 29
Scheme 8. Synthesis of enantiopure oxazolines All the examples reported so far refer to cyclization reactions by formation of carbon-oxygen bond using enantiopure organoselenium reagents. Some examples will now be presented in which enantiopure heterocyclic compounds are produced from the reactions of achiral organoselenium reagents (PhSeX) with enantiopure substrates. The first example refers again to the synthesis of tetrahydrofurans.

Issue ICHC-20 ARKIVOC 2006 (vii) 186-206
ISSN 1424-6376 Page 193 ©ARKAT
The opening of the commercially available (R)-styrene oxide 30 with phenylselenium anions affords the hydroxy selenide 31. The reaction with tributyl allyl tin gives rise to the alkenol 32 (Scheme 9). Cyclization of 32 promoted by phenyl selenenyl sulfate gave a mixture of two diastereomeric tetrahydrofurans 33 and 34 which could be easily separated by column chromatography. Deselenenylation with tributyl tin hydride gave the 2-methyl-5-phenyl tetrahydrofurans 35 and 36. Starting from the (S)-styrene oxide ent-30 the enantiomeric compounds ent-35 and ent-36 were obtained.19
OPhSePh
29%
OPhSePh
47%AIBN, C6H6,
SnBu3
OHPh
.
OPh
OPh92%
NaBH4
(PhSe)2O
PhOHPh
PhSe
80 °C
PhSeOSO3H
R
30 3132
33 35
34 36
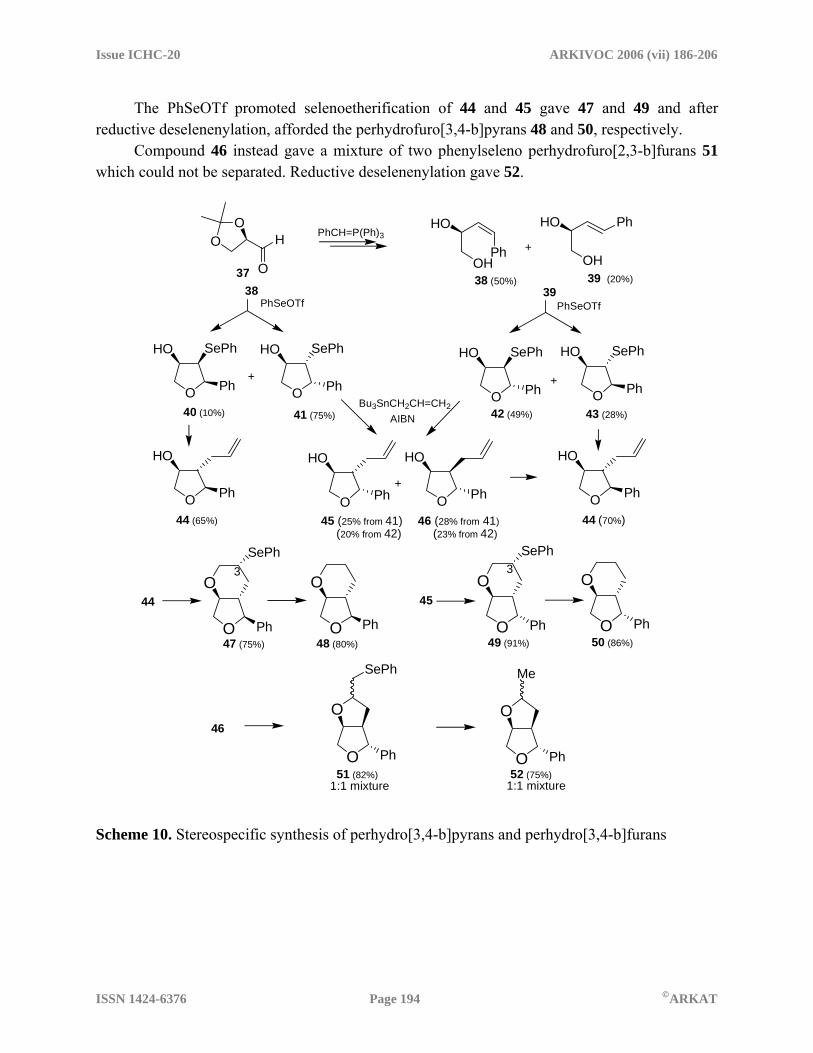
Scheme 9. Synthesis of enantiopure 2-methyl-5-phenyl tetrahydrofurans Using a similar procedure and starting from the (1R,2R)-1-phenylpropylene oxide (the opening of the epoxide was regiospecific) four diastereomeric 2,5-dimethyl-3-phenyl tetrahydrofurans were isolated. Starting from the SS epoxide the four enantiomeric compounds were prepared. Thus all the possible eight isomers of 2,5-dimethyl-3-phenyl tetrahydrofurans were prepared in an enantiomerically pure form.19 An example of the combined use of the reactivity of organoselenium reagents to promote cyclization reactions is reported in Scheme 10 which refers to the synthesis of perhydrofuro[3,4-b]pyrans and of perhydrofuro[3,4-b]furans.20 The ylide deriving from benzyl bromide was made to react with the D-glyceraldehyde acetonide 37. A mixture of the cis and trans alkenols, 38 and 39, was obtained. These were easily separated by column chromatography. The cis isomer could also be easily isomerized to the trans compound. The PhSeOTf promoted selenoetherification of 38 afforded the phenylseleno tetrahydrofurans 40 and 41, whereas the cyclization of 39 gave the products 42 and 43. The four compound were obtained in pure form after column chromatography. In each of these four compounds the PhSe group was replaced by the allyl group by treatment of tributyl allyl tin. Compounds 40 and 43 gave the same radical intermediate and hence the same reaction product 44. The introduction of the allyl group was stereoselective and the less crowded compound was obtained as the sole reaction product. Compounds 41 and 42 gave the same radical intermediate and hence the same mixture of compounds 45 and 46 which were separated by column chromatography.
Issue ICHC-20 ARKIVOC 2006 (vii) 186-206
ISSN 1424-6376 Page 194 ©ARKAT
The PhSeOTf promoted selenoetherification of 44 and 45 gave 47 and 49 and after reductive deselenenylation, afforded the perhydrofuro[3,4-b]pyrans 48 and 50, respectively. Compound 46 instead gave a mixture of two phenylseleno perhydrofuro[2,3-b]furans 51 which could not be separated. Reductive deselenenylation gave 52.
OO
PhCH=P(Ph)3
38 (50%) 39 (20%)
HO
PhOH
HO
OH
PhH
O
O
HO SePh
Ph
40 (10%) 41 (75%) 42 (49%) 43 (28%)
+ +O
HO SePh
PhO
HO SePh
PhO
HO SePh
Ph
O Ph
HO
44 (70%)O Ph
HO
45 (25% from 41)(20% from 42)
+O Ph
HO
46 (28% from 41)(23% from 42)
O Ph
HO
44 (65%)
+
38 3937
PhSeOTf PhSeOTf
Bu3SnCH2CH=CH2
AIBN
45
46
44
O
O
SePh
Ph47 (75%)
O
O
Ph O
O
SePh
Ph O
O
Ph
O Ph
O
SePh
O Ph
O
Me
3 3
48 (80%) 49 (91%) 50 (86%)
51 (82%) 52 (75%)1:1 mixture 1:1 mixture
Scheme 10. Stereospecific synthesis of perhydro[3,4-b]pyrans and perhydro[3,4-b]furans

Issue ICHC-20 ARKIVOC 2006 (vii) 186-206
ISSN 1424-6376 Page 195 ©ARKAT
3. Cyclization Reactions by Formation of Carbon Nitrogen Bonds The first examples of the selenium promoted cyclization of alkenes containing internal nitrogen nucleophiles 53 were reported by Danishetsky21 who demonstrated that with the primary amino group the cyclization reaction does not take place. As indicated in Scheme 11, the reaction occurs easily to give 54 only if an electron-withdrawing group is linked to the nitrogen atom. Uemura22 showed that if the nitrogen atom is that of an imidate 55 the cyclization occurs easily provided the PhSeBr is employed. This is because the bromine anion effects an SN2 reaction attacking the methyl group of 56 and allowing the lactam 57 to act as the leaving group. Finally, in a series of papers De Kimpe23 demonstrated that the nitrogen of imines 58 is sufficiently nucleophilic to trap the intermediate seleniranium ion and give the cyclic iminium ion 59 which was reduced to 60 with sodium borohydride.
.
NHR
NSePh
R
PhSeX
R = CO2Et, COR, SO2ArR = H No Reaction
53 54
.PhSeBr
NO
R SePhNO
Me R55 57
58 59 60
PhSeBrNR
NR SePh
NaBH4
+
+
56
NO
RMe SePhBr -
Br -+NR SePh
N
SePh
R Br -
Scheme 11. Selenocyclization by formation of C-N bonds We have found that successful cyclization reactions can be effected starting from O-allyl oximes 61 (Scheme 12). The reaction with phenylselenenyl sulfate gave the iminium ions 62 which can be reduced with sodium borohydride or they can be hydrolyzed to give the isoxazolidine 63 and the starting ketones.24

Issue ICHC-20 ARKIVOC 2006 (vii) 186-206
ISSN 1424-6376 Page 196 ©ARKAT
61 R = Me, Ph
ON R
R1 R2
PhSeS H2O+
R1
R2OO
NH
SePh
R
6362
ON
R2R1
SePh
R
O
H
H
+O
NR2R1
SePh
R
OH2+
ON
R2R1
SePh
R+HSO4
-
Scheme 12. Synthesis of isoxazolidines by cyclization of O-allyl oximes. The asymmetric version of these reactions was effected starting from our sulfur containing diselenide 1. From the oximes 64 a mixture of the two iminium salts 65 and then of the isoxazolidines 66 was obtained with the diastereomeric ratios reported in Scheme 13. The best results were obtained in the case in which R was a phenyl.25
R = Me, Et, Ph. R1 = n-Pr. D.r. from 88:12 to 96:4
.
ON R
R1 R1
O
R1R1
H2O ++ ON R
SeAr*
H
Minor
ON R
SeAr*
H
Major64 65 66
ON R
SeAr*
R1 R1
+
Scheme 13. Asymmetric synthesis of isoxazolidines from O-allyl oximes Very recently we have described26 the interesting asymmetric synthesis of the azidoselenides 67. As illustrated in Scheme 14 these compounds were obtained starting from alkenes and using the methoxy substituted sulfur containing diselenide 2.
. ++Ar*SeOTfR
RH
H NaN3
30 °CMajor
RH
RH
N3
R*SeMinor
HR
HR
N3
R*Se
67 Scheme 14. Asymmetric azidoselenenylation of alkenes

Issue ICHC-20 ARKIVOC 2006 (vii) 186-206
ISSN 1424-6376 Page 197 ©ARKAT
With the exception of the reaction of cyclohexene (D.r = 61:39) in all the other cases the diastereomeric ratios were extremely good being equal or greater than 95:5. The azido selenides thus obtained are very useful and versatile intermediates because they have two groups, the azido and the arylseleno, which can be easily transformed into several different functional groups. The first example reported here refers to the asymmetric synthesis of aziridines indicated in Scheme 15.26 The azido group of compounds 68 was first reduced and the resulting amine was protected by benzoylation. The so formed benzoyl amido selenide 69 was oxidized with an excess of m-chloroperbenzoic acid to afford the corresponding selenone 70. The nitrogen anion 71 was generated by treatment with a strong base and the aziridine 72 was then formed by intramolecular SN2 substitution of the selenone group. The product 73, deriving from the elimination of the intermediate selenoxide, was also isolated. In both reaction products the ee (94%) corresponded to the diastereomeric ratio (97:3) of the starting products.
C4H9SeR*
N3
C3H71. m-CPBA, -60°C2. t-BuOK
C4H9
N
C4H9
COPh
SeR*
NHCOPh
C4H9 C3H7
NHCOPh
C4H9 C3H7
1. Ph3P, THF2. H2O, 50°C
3. PhCOCl ,Et3N, THF, 0°C
(86%)
(42%)(21%)
+
SeO2R*
NHCOPh
C4H9 C3H7
SeO2R*
N
C4H9 C3H7
COPh SN2
D. r. 97:3
e.e. 94%
68 69 70
71 72 73
Scheme 15. Asymmetric synthesis of aziridines. The azido selenide derived from styrene 74 was also treated with methyl acetylene dicarboxylate to afford 75 and after deselenenylation the selenium free triazole 76 in excellent yield and with an ee of 94% (Scheme 16).26
CO2CH3
CO2CH3
Ph
N3
SeAr*
Ph3SnHAIBN
C6H6 reflux(-)-R
NN N
Ph
CO2CH3
CO2CH3NN N
Ph
CO2CH3
CO2CH3
SeAr*74
75 76
Scheme 16. Asymmetric synthesis of triazoles.

Issue ICHC-20 ARKIVOC 2006 (vii) 186-206
ISSN 1424-6376 Page 198 ©ARKAT
Nitrogen containing heterocyclic compounds were also prepared starting from enantiopure substrates and achiral selenium reagents. The examples reported in Schemes 17 and Scheme 18 concern again cyclization reactions by intramolecular substitution of the selenone group. In the first Scheme the various intermediates 78, 79, 80 and 81 for the conversion of epoxides 77 into 1,3-oxazolidin-2-ones 82 and 83 are indicated.27
.
R =
CH=CHCOCO2MeCN
TrOBnO
TBOPSORCONH
(PhCH2)2NBocNH
RO OH
RSePh
PhCONCO O
R
NH
O
SePh
COPh
(PhSe)2
NaBH4 K2HPO4
MCPBA
O
R
NCOPh
O
SeO
O Ph
-..
RNCOPh
OO
Acetone
K2CO3
O
R
NH
O
SeO
O Ph
COPh
RNH
OO
LiOH
77 78 7980
81 82 83
Scheme 17. Synthesis of enantiopure substituted 1,3-oxazolidin-2-ones. These reactions were carried out with a variety of substituents R. Starting from the 1-phenylpropylene oxide 4,5-substituted 1,3-oxazolidin-2-ones were obtained in good yields. The same displacement reaction was also employed to form the four membered azetidines 90.28 The starting products were two commercially available aminoacids 84 which were converted into the epoxide 85. The various intermediates 86, 87, 88 and 89 are indicated in Scheme 18.
NHBoc
ROH
SePhR
NH2
O
OHNHBoc
RO
PhSeSePhNaBH4, EtOH
40 °C
1. H+ 2. NsCl 3. DHP
KOH
NHNs
ROTHP
SePh
NHNs
ROTHP
SePh
O OK2HPO4, THF
MCPBA
NNs
R OH e.e.>98%
R = CH3CH2 PhCH2
84 85 86
87 88
89 90
..N
ROTHP
SePh
O O
Ns
Scheme 18. Synthesis of enantiopure azetidines.

Issue ICHC-20 ARKIVOC 2006 (vii) 186-206
ISSN 1424-6376 Page 199 ©ARKAT
The syntheses of enantiopure pyrrolidines and perhydropirrolizines were effected starting from Boc protected β-amino tosylate 91 (Scheme 19).28
NHBocPh
PhSe
AIBN, C6H6, 80 °C
SnBu3
NHBocPhNHBocPh
TsOPhSeSePh PhSeSePh, (NH4)2S2O8
CF3SO3H, MeCN, r.t.Column Chrom.
NaBH4
+
+NPhSePhH
(48%)
PhSeOTf
AIBN, C6H6, 80 °C
SnBu3NPh
PhSeNPhH PhSe
NPh
91 92 93
94 96 98 99
95 97 100 101
(32%)
NPhSePhH
PhSeOTf
AIBN, C6H6, 80 °C
SnBu3
PhSe
NPhNPhH
NPh
PhSe
Scheme 19. Synthesis of enantiopure perhydropyrrolizidines. The tosyl group was substituted by phenylselenium anions to give 92 and the seleno group was replaced by the allyl group in the usual way. The cyclization of the resulting alkenyl amine 93 was promoted by phenylselenenyl sulfate and the two diastereomeric reaction products 94 and 95 were separated by column chromatography. During chromatography the protecting group was removed. Finally the reductive deselenenylation afforded the 2-methyl-5-phenyl pyrrolidines. Alternatively, the phenylseleno group can be replaced by the allyl and the resulting compound 96 and 97 can be treated with PhSeOTf to afford the bicyclic perhydropyrrolizidines. In the first case the cyclization afforded the two diastereoisomers 98 and 99 which were separated by column chromatography. In the second case compound 100 was obtained as a single product. The forth diastereoisomer 101 was not formed probably for steric reasons. 4. Competition between Oxygen and Nitrogen Sometimes the internal nucleophile is an ambident group and the trapping of the seleniranium ion can occur either with the oxygen or with the nitrogen atom. In some cases the course of the reaction depends on the experimental conditions employed. Thus a competition between oxygen and nitrogen can be observed. Some years ago we observed that the phenylselenenyl sulfate promoted cyclization of alkenyl hydroxamic acid: when the reaction was carried out at low temperature (20 °C for R=Me and -20°C for R=Ph), N-hydroxy imidates were isolated.29 If the reaction mixture was warmed up, or the reactions was directly carried out at higher temperature (50 °C for R=Me and 20°C for R=Ph) the reaction products were the N-hydroxy γ-lactams. Clearly the imidates are the products

Issue ICHC-20 ARKIVOC 2006 (vii) 186-206
ISSN 1424-6376 Page 200 ©ARKAT
deriving from the kinetic control and are formed reversibly, whereas the lactams are the products deriving from the thermodynamic control of the reaction.
102
103
104NH ROOH
O Et
SeAr*
NOH
N Ph
SeAr*
OOH
R = Et
R = Ph
X
Yield = 75%D.r. 92:8
Yield = 60%D.r. 85:15
TfO -NH ROOH
SeAr*+
- 50 °C
Ar*SeOTf
Scheme 20. Enantioselective synthesis of N-hydroxy imidates and γ-lactam. The asymmetric version of this reaction, carried out with 102 and the selenenyl triflate deriving from the sulfur containing diselenide 1, gave different results (Scheme 20).30 The reactions were carried out at -50°C and afforded the N-hydroxy imidate 103 in the case of R=Et and the N-hydroxy γ-lactam 104 in the case of R=Ph. The increase of the reaction temperature did not produce any change in the composition of the reaction mixtures. With this selenenylating agent therefore the formation of the N-hydroxy imidate 103 is not reversible. An interesting case is observed in the case of cyclization of the oximes. The first examples were reported by Gallagher31 in the cyclization of allenic oximes promoted by silver tetrafluoborate. It was observed that the nature of the reaction products depended on the geometry of the starting oximes. The Z isomers gave 1,2-oxazines and the more stable E isomers gave cyclic nitrones. Under these conditions alkenyl oximes did not react. It was later observed by Grigg32 and by ourselves33 that the cyclization of alkenyl oximes can be effected using electrophilic phenyselenium reagents. It was observed that starting from a mixture of the two isomeric oximes the 1,2-oxazines were only present in traces. The asymmetric version of these cyclizations was carried out using the triflate of the methoxy substituted sulfur containing diselenide 2 (Scheme 21).34 The cyclization reaction of the alkenyl oximes 105 can take place in different ways depending on the heteroatom which acts as the nucleophile and on the carbon atom of the seleniranium ion which is attacked. A 6-exo cyclization by the oxygen atom would afford the 1,2-oxazines 106, whereas a 5-exo cyclization by the nitrogen atom would afford the five membered cyclic nitrones 107. A 6-endo cyclization by the nitrogen atom would afford the six membered cyclic nitrones 108. These product are observed only in the case in which R1 = Ph. In every case the reactions occur with a Markovnikov regioselectivity.

Issue ICHC-20 ARKIVOC 2006 (vii) 186-206
ISSN 1424-6376 Page 201 ©ARKAT
N
Me
O
H
N
Me
O
H
NO
SeAr*
Me
N
SeAr*
Me
O +-
NO
R
SeAr*6-exo
5-exo
6-endo
-+N
R
O
SeAr*
-N
R
SeAr*Ph
O +
Ar*SeOTf
CH2Cl2N
R
R1O
H
* *N
R
R1O
H
SeAr*+
R = Ph, R1 = HR = Me, R1 = HR = Ph, R1 = PhR = Me, R1 = Ph
105
106
107
108
Scheme 21. Asymmetric Synthesis of 1,2-oxazines and cyclic nitrones. In the case of the phenyl derivative (R=Ph, R1=H) the cyclization reactions afforded reaction products which reflected the geometry of the oximes. The Z isomer gave the 1,2-oxazine and the E isomer gave the cyclic nitrone. On the contrary, in the case of the methyl derivative (R=Me, R1=H) the ratio of the two reaction products did not reflect the ratio of the starting oximes. Using a 2:1 mixture of the Z:E isomers after 7 hours the 1,2-oxazine was formed in 18% yields and the cyclic nitrone in 72% yield. After 17 hours the reaction mixture is constituted only by the nitrone. Thus, the formation of the nitrone is largely preferred. From the results of some parallel experiments we concluded that this behavior is due to the fact that, under the conditions employed, the two isomeric starting oximes interconvert and that the formation of the 1,2-oxazine is a reversible process (Scheme 21). Thus, the entire process is shifted towards the formation of the thermodynamically more stable nitrone. 5. Cyclization Reactions by Formation of Carbon Carbon Bonds Cyclization reactions of alkenes containing internal carbon nucleophiles are not very common and have been mainly employed for the formation of homocyclic compounds. The results which are presented here refer to cyclization reactions in which the ring forming process is the addition of a carbon radical to a carbon oxygen double bond. These reactions were effected starting from enantiopure substrates and achiral organoselenium reagents.

Issue ICHC-20 ARKIVOC 2006 (vii) 186-206
ISSN 1424-6376 Page 202 ©ARKAT
Additions of carbon radicals generated from selenides to carbon-carbon double bonds have been largely investigated. These reactions occur easily and have been employed by Engman35 to produce a large number of oxygen and nitrogen containing heterocyclic compounds. Similar cyclizations by addition to carbon-oxygen double bond are more difficult. The alkoxy radicals can in fact suffer rapid β-scission reactions. Thus these radical cyclizations can be reversible and the trapping of the cyclic alkoxy radical is not always easy. However, substituents along the forming ring facilitate the trapping of cyclic products. As a matter of fact a successful cyclization has been recently reported by Uchiyama36 for the synthesis of the hydroxy perhydrofuro[2,3-b]furans. Our first experiments on the radical cyclization reaction were effected in order to synthesize some enantiopure 3-hydroxy tetrahydrofuran derivatives.28 As indicated in Scheme 22, the starting product was compound 109 which was prepared in satisfactory yields from the intermolecular alkoxy selenenylation of styrene with phenylselenenyl triflate and propyl (S)-lactate. This ester was reduced with DIBAL to afford the corresponding aldehyde 110. The phenyl selenium group was then removed by treatment with tributyl tin hydride and AIBN in refluxing toluene. The carbon radical thus produced 111 gave rise to the addition reaction to the carbon oxygen double bond and produced the cyclic alkoxy radical 112. However, the rate of hydrogen abstraction to afford the desired product 113 was lower than that of the β-scission and the secondary carbon radical 114 was obtained. Hydrogen abstraction gave the observed reaction product 115.
Ph O
CO2Pr
b: DIBAL-H, toluene, -78°C, 3hc: Bu3SnH, AIBN, refl. toluene, 3h
Ph O
CHO
Ph O
O.
a: PhSeOTf in THF
Ph+ COOPr
MeHO Me Me
Me Ph O Me
O
Ph O Me
O
a b c
Ph O
OH.
Me
.
109 110
111 112 114 115
113
.Ph O
O
Me
PhSe PhSe
.
Scheme 22. Attempted synthesis of 2-methyl-3-hudroxy-5-phenyl tetrahydrofurans In order to decrease the rate of the β-scission process we used a starting product in which the β-scission would afford a primary carbon radical. Thus, as indicated in Scheme 23, we

Issue ICHC-20 ARKIVOC 2006 (vii) 186-206
ISSN 1424-6376 Page 203 ©ARKAT
started from the commercially available R-(+)-glycidol (e.e. = 98%) 116. The ring opening of the epoxide with phenyl selenium anions was regiospecific and gave the β-hydroxy selenide 117.28
O
PhSe OMeOa
O
OH
O
OH
+
d
OH
PhSeb, c e
93% 98% 74%
b: Imidazole, TBDPSCl, DMF, R.T.c: NaH, C6H6, BrCH2CO2Me, 40°Cd: DIBAL-H, toluene, -78°C, 3he: Bu3SnH, AIBN, refl. toluene, 3h
O
PhSe OHO
O
OH.OH OH
O
O.a: PhSeSePh, NaBH4 in EtOH
f: Bu4NF, THF, R.T.
f
OH OH OR RO
OR RO
116 117 118 119
120 121 122 123 Scheme 23. Synthesis of enantiopure β-hydroxy tetrahydrofurans The primary OH group was then selectively protected and the resulting product was treated with methyl bromoacetate. The ester thus obtained 118 was then reduced with DIBAL to afford the corresponding seleno aldehyde 119. Treatment with tributyl tin hydride and AIBN afforded the carbon radical 120 which added to the carbonyl group. As anticipated, in this case the trapping of the alkoxy radical 121 by the tributyl tin hydride was faster than the β-scission and the desired hydroxy tetrahydrofurans 122 and 123 were obtained as an almost equimolecular mixture of the two enantiopure diastereoisomers which were easily separated by column chromatography.
Bn N
PhSe OMeO
NBn
OH
NBn
OH
+
Bn NHTos
PhSe
88% 80%
94%
Bn N
PhSe OH
Bn
124
N Tos
Tos
Tos Tos Tos
Bn N
OH
Tos
.125 126
127 128 129 130 Scheme 24. Synthesis of enantiopure β-hydroxy pyrrolidines

Issue ICHC-20 ARKIVOC 2006 (vii) 186-206
ISSN 1424-6376 Page 204 ©ARKAT
On the basis of the experience gained from these experiments we have then applied a similar reaction sequence to the commercially available N-tosyl benzyl aziridine 124 (e.e. = 98%) in order to produce enantiopure hydroxy pyrrolidines 129 and 130 passing through the reaction intermediates 125, 126, 127 and 128 (Scheme 24).28 The reaction proceeded as expected and the final products were an almost equimolecular mixture (46% yield, D.r. 53:47) of the two enantiopure diastereoisomers which were separated by column chromatography. In conclusion the results described above demonstrate that organoselenium reagents, both chiral non racemic and achiral, can be successfully employed to effect the synthesis of different types of enantiomerically enriched or in most cases enantiopure heterocyclic compounds. The examples which have been described concern the preparation of very simple compounds. However the methodologies employed are of general application and can also be used to the synthesis of much more complex molecules using properly substituted starting products. Organoselenium reagents, in fact, require very mild experimental conditions which are compatible with most of the organic functional groups. Acknowledgements Financial support from Ministero dell’Istruzione, dell’Università e della Ricerca, MIUR, National Projects PRIN2003, FIRB2001, Consorzio CINMPIS, Bari and University of Perugia is gratefully acknowledged.
References 1. (a) Tiecco, M. Electrophilic Selenium; Selenocyclizations In Topics in Current Chemistry:
Organoselenium Chemistry: Modern Developments in Organic Synthesis; Wirth, T. Ed.; Springer-Verlag: Heidelberg; 2000, pp 7-54. (b) Organoselenium Chemistry- A Practical Approach; Back, T. G. Ed.; Oxford: New York 2000. (c) Wirth, T. Angew Chem. Int. Ed. 2000, 39, 3742.
2. (a) Tomoda; S.; Iwaoka; M. Chem. Lett. 1988, 1895. (b) Tomoda, S.; Iwaoka, M.; Yakushi, K.; Kawamoto, A.; Tanaka, J. J. Phys. Org. Chem. 1988, 1,179. (c) Tomoda, S.; Iwaoka, M. J. Chem. Soc., Chem. Commun. 1988, 1283. (d) Tomoda, S.; Fujita, K.; Iwaoka, M. J. Chem. Soc., Chem. Commun. 1990, 129. (e) Tomoda, S.; Fujita, K.; Iwaoka, M. Chem Lett 1992, 1123. (f) Tomoda, S; Fujita, K; Iwaoka, M. Phosphorus Sulfur 1997, 67, 247.
3. (a) Déziel, R.; Goulet, S.; Grenier, L.; Bordeleau, J.; Bernier, J. J. Org. Chem. 1993, 58, 3619. (b) Déziel, R.; Malenfant, E. J. Org. Chem. 1995, 60, 4660. (c) Déziel, R.; Malenfant, E.; Bélanger, G. J. Org. Chem. 1996, 61, 1875. (d) Déziel, R.; Malenfant, E.; Thibault, C.; Fréchette, S.; Gravel, M. Tetrahedron Lett. 1997, 38, 4753.

Issue ICHC-20 ARKIVOC 2006 (vii) 186-206
ISSN 1424-6376 Page 205 ©ARKAT
4. (a) Nishibayashi, Y.; Singh, J. D.; Uemura, S.; Fukuzawa, S. Tetrahedron Lett. 1994, 35, 3115. (b) Nishibayashi, Y.; Srivastava, S. K.; Takada, H.; Fukuzawa, S.; Uemura, S. J. Chem. Soc., Chem. Commun. 1995, 2321. (c) Nishibayashi, Y.; Singh, J. D.; Fukuzawa, S..; Uemura, S. J. Org. Chem. 1995, 60, 4114. (d) Fukuzawa, S.; Takahashi, K.; Kato, H.; Yamazaki, H. J. Org. Chem. 1997, 62, 7711.
5. (a) Fujita, K.; Iwaoka, M.; Tomoda, S. Chem. Lett. 1994, 923. (b) Fujita, K.; Murata, K.; Iwaoka, M.; Tomoda, S. J. Chem. Soc., Chem. Commun. 1995, 1641. (c) Fujita, K.; Murata, K.; Iwaoka, M.; Tomoda, S. Tetrahedron Lett. 1995, 36, 5219. (d) Fujita, K.; Murata, K.; Iwaoka, M.; Tomoda, S. Tetrahedron 1997, 53, 2029. (e) Iwaoka, M.; Tomoda, S. J. Am. Chem. Soc. 1996, 118, 8077. (f) Komatsu, H.; Iwaoka, M.; Tomoda, S. J. Chem. Soc., Chem. Commun. 1999, 205.
6. (a) Back, T. G.; Dyck, B. P.; Parvez, M. J. Chem. Soc., Chem. Commun. 1994, 515. (b) Back, T. G.; Dyck, B. P.; Parvez, M. J. Org. Chem. 1995, 60, 703. (c) Back, T. G.; Dyck, B. P. J. Chem. Soc., Chem. Commun. 1996, 2567. (d) Back, T. G.; Nan, S. J. Chem. Soc., Chem. Commun. 1998, 3123. (e) Back, T. G.; Dyck, B. P.; Nan, S. Tetrahedron 1999, 55, 3191.
7. (a) Wirth, T. Angew. Chem., Int. Ed. 1995, 34, 1726. (b) Wirth, T.; Fragale, G. Chem. Eur. J. 1997, 3, 1894.
8. Fragale, G.; Neuburger, M.; Wirth, T. J. Chem. Soc., Chem. Commun. 1998, 1867. 9. Santi, C.; Fragale, G.; Wirth, T. Tetrahedron: Asymmetry 1998, 9, 3625. 10. (a) Murata. S.; Suzuki, T. Chem. Lett. 1987, 849. (b) Murata, S.; Suzuki, T. Tetrahedron
Lett. 1987, 28, 4297. (c) Murata, S.; Suzuki, T. Tetrahedron Lett. 1987, 28, 4415. 11. (a) Tiecco, M.; Testaferri, L.; Tingoli, M.; Bartoli, D. Tetrahedron Lett. 1989, 30, 1417. (b)
Tiecco, M.; Testaferri, L.; Tingoli, M.; Bagnoli, L.; Marini, F. J. Chem. Soc., Perkin Trans. 1 1993, 1989.
12. Tiecco, M.; Testaferri; L.; Bagnoli, L.; Marini, F.; Temperini, A.; Tomassini, C.; Santi, C. Tetrahedron Lett. 2000, 41, 3241.
13. Tiecco, M.; Testaferri, L.; Santi, C.; Tomassini, C.; Marini, F.; Bagnoli, L.; Temperini, A. Chem. Eur. J. 2002, 8, 1118.
14. Tiecco M.; Testaferri, L.; Marini, F.; Sternativo, S.; Bagnoli, L.; Santi, C.; Temperini, A. Tetrahedron: Asymmetry 2001, 12, 1493.
15. Tiecco M.; Testaferri, L.; Marini, F.; Sternativo, S.; Santi, C.; Bagnoli, L.; Temperini, A. Eur. J. Org. Chem. 2005, 543.
16. Tiecco, M.; Testaferri, L.; Bagnoli, L.; Scarponi, C.; Purgatorio, V.; Temperini, A.; Marini, F.; Santi, C. Tetrahedron: Asymmetry 2005, 16, 2429.
17. Tiecco, M.; Testaferri, L.; Santi, C.; Tomassini, C.; Marini, F.; Bagnoli, L.; Temperini, A. Eur. J. Org. Chem. 2000, 3451.
18. Bannard, R. A. B.; Gibson, N. C. C.; Parkkari, J. H. Can. J. Chem. 1971, 49, 2064. 19. Tiecco, M.; Testaferri, L.; Bagnoli, L.; Purgatorio, V.; Temperini, A.; Marini, F.; Santi, C.
Tetrahedron: Asymmetry 2004, 15, 405.

Issue ICHC-20 ARKIVOC 2006 (vii) 186-206
ISSN 1424-6376 Page 206 ©ARKAT
20. Tiecco, M.; Testaferri, L.; Bagnoli, L.; Terlizzi, R.; Temperini, A.; Marini, F.; Santi, C.; Scarponi, C. Tetrahedron: Asymmetry 2004, 15, 1949.
21. Danishefsky; S.; Webb II; R.R. Tetrahedron Lett. 1983, 24, 1357. 22. (a) Toshimitsu, A.; Terao, K.; Uemura, S. J. Chem. Soc., Chem. Commun. 1986, 530. (b)
Terao, K.; Toshimitsu, A.; Uemura, S. J. Chem. Soc., Perkin Trans. 1 1986, 1837. (c) Toshimitsu, A.; Terao, K.; Uemura, S. J. Org. Chem. 1987, 52, 2018.
23. (a) De Kimpe, N.; Boelens, M. J. Chem. Soc., Chem. Commun. 1993, 916. (b) De Smaele, D.; De Kimpe, N. J. Chem. Soc., Chem. Commun. 1995, 2029.
24. (a) Tiecco, M.; Testaferri, L.; Tingoli, M.; Bagnoli, L. J. Chem. Soc., Chem. Commun. 1995, 235. (b) Tiecco, M.; Testaferri, L.; Tingoli, M.; Bagnoli, L.; Santi, C. Tetrahedron 1995, 51, 1277.
25. Tiecco, M.; Testaferri, L.; Marini, F.; Sternativo, S.; Bagnoli, L.; Santi, C.; Temperini, A. Tetrahedron: Asymmetry 2001, 12, 3053.
26. Tiecco, M.; Testaferri, L.; Santi, C.; Tomassini, C.; Marini, F.; Bagnoli, L.; Temperini, A. Angew. Chem., Int. Ed. 2003, 42, 3131.
27. Tiecco, M.; Testaferri, L.; Temperini, A.; Bagnoli, L.; Marini, F.; Santi, C. Chem. Eur. J. 2004, 10; 1752.
28. Unpublished results from this laboratory. 29. Tiecco, M.; Testaferri, L.; Tingoli, M.; Marini, F. J. Chem. Soc., Chem. Commun. 1994,
221. 30. Tiecco, M.; Testaferri, L.; Marini, F.; Sternativo, S.; Bagnoli, L.; Santi, C.; Temperini, A.
Tetrahedron: Asymmetry 2001, 12, 1493. 31. (a) Lathbury, D.; Gallagher, T. J. Chem., Soc. Chem. Commun. 1986, 1017. (b) Lathbury,
D.; Shaw, R. W.; Bates, P. A.; Hursthouse, M. B.; Gallagher, T. J. Chem. Soc., Perkin Trans. 1 1989, 2415.
32. Grigg, R.; Hadjisoteriou, M.; Kennewell, P.; Markandu, J. J. Chem. Soc., Chem. Commun. 1992, 1537.
33. Tiecco, M.; Testaferri, L.; Tingoli, M.; Bagnoli, L.; Marini, F. J. Chem. Soc., Perkin Trans. 1 1993, 1989.
34. Tiecco, M.; Testaferri, L.; Bagnoli, L.; Purgatorio, V.; Temperini, A.; Marini, F.; Santi, C. Tetrahedron: Asymmetry 2001, 12, 3297.
35. (a) Ericsson, C.; Engman, L. Org. Lett. 2001, 3, 3459. (b) Berlin, S.; Ericsson, C.; Engman, L. Org. Lett. 2002, 4, 3. (c) Gupta, V.; Besev, M.; Engman, L. Tetrahedron Lett. 1998, 39, 2429. (d) Berlin, S.; Engman, L. Tetrahedron Lett. 2000, 41, 3701.
36. Uchiyama, M.; Hirai, M.; Nagata, M.; Katoh, R.; Ogawa, R.; Ohta, A. Tetrahedron Lett. 2001, 42, 4653.