enantioselective total syntheses of lyconadins a–e through a...
TRANSCRIPT

Enantioselective Total Syntheses of Lyconadins A–E through a Palladium-
Catalyzed Heck-Type Reaction

The lycopodium alkaloids(石松属生物碱) are a structurally diverse group of natural products containing more than 300 alkaloids isolated from 54 Lycopodium species.Huperzine A could inhibit acetylcholinesterase, improve efficiency for learning and memory in animals, and therefore show potential in treatment of Alzheimer’s disease.Lyconadin A exhibited micromolar cytotoxicity
against murine lymphoma L1210 and human epidermoid carcinoma KB cells, while lyconadins A and B could enhance the mRNA expression for nerve growth factor in 1321N1 human astrocytoma cells.
Total Syntheses of Lyconadins A–E
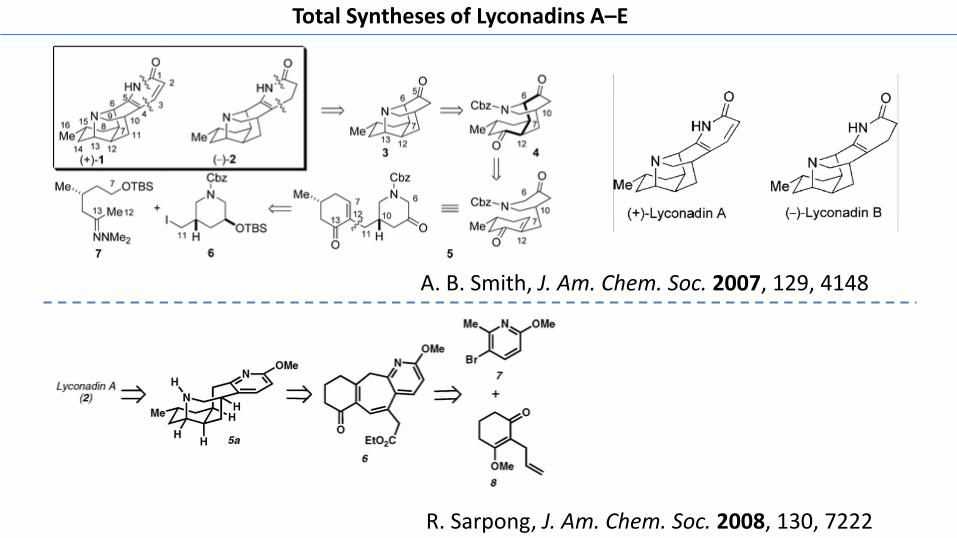
A. B. Smith, J. Am. Chem. Soc. 2007, 129, 4148
R. Sarpong, J. Am. Chem. Soc. 2008, 130, 7222
Total Syntheses of Lyconadins A–E

T. Fukuyama, J. Am. Chem. Soc. 2011, 133, 418
Total Syntheses of Lyconadins A–E

Lyconadin C
S. P. Waters, Org. Lett. 2013, 15, 4226
Total Syntheses of Lyconadins A–E

M. D. Shair, J. Am. Chem. Soc. 2014, 136, 13442
Total Syntheses of Lyconadins A–E
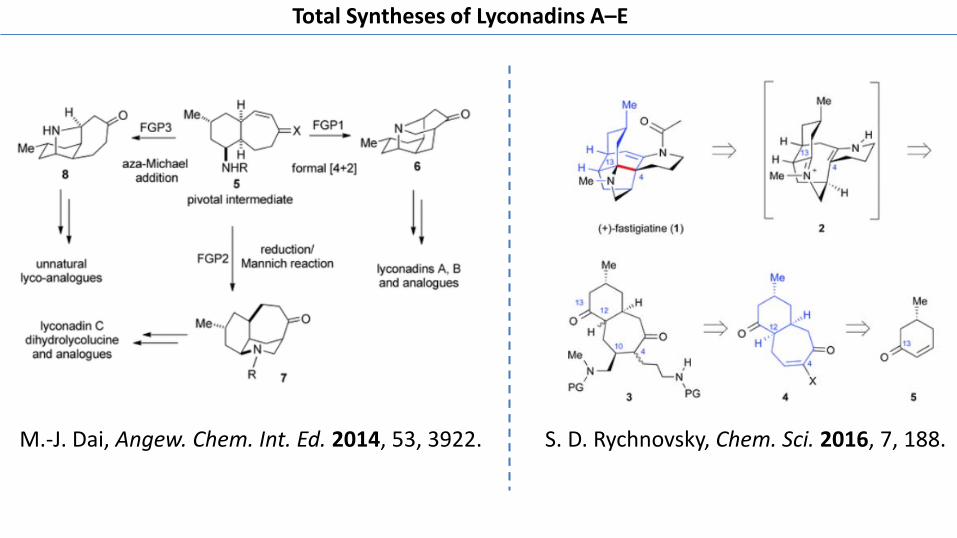
M.-J. Dai, Angew. Chem. Int. Ed. 2014, 53, 3922. S. D. Rychnovsky, Chem. Sci. 2016, 7, 188.
Total Syntheses of Lyconadins A–E


Total Syntheses of Lyconadins A–E

Total Syntheses of Lyconadins A–E

Total Syntheses of Lyconadins A–E
Total Syntheses of Lyconadins A–E

Chem. Eur. J. 2011, 17, 626
Total Syntheses of Lyconadins A–E

Chem. Eur. J. 2013, 19, 87
Total Syntheses of Lyconadins A–E
Total Syntheses of Lyconadins A–E

Total Syntheses of Lyconadins A–E

Chem. Commun., 2009, 1574
Total Syntheses of Lyconadins A–E

J. Org. Chem. 1990, 55, 1736
Total Syntheses of Lyconadins A–E

German Edition: DOI: 10.1002/ange.201912948Alkaloid SynthesisInternational Edition: DOI: 10.1002/anie.201912948
Enantioselective Total Syntheses of Lyconadins A–E througha Palladium-Catalyzed Heck-Type ReactionJiayang Zhang+, Yangtian Yan+, Rong Hu, Ting Li, Wen-Ju Bai, and Yang Yang*
Abstract: A novel palladium-catalyzed Heck-type reaction ofthiocarbamates has been designed to construct bridged seven-membered-ring systems that are otherwise challenging toprepare. Taking advantage of this newly developed method,enantioselective syntheses of lyconadins A–E (1–5), lycope-curine (6), and dehydrolycopecurine (7) have been realized ina divergent fashion. Our synthetic strategy also features anintramolecular cyclization of a N-chloroamine to forge theC6�N bond, a transannular Mannich-type reaction of a cyclicnitrone to stitch the C4 and C13 together, and a cycloconden-sation to deliver the (dihydro-)pyridone motif.
Introduction
The lycopodium alkaloids are a structurally diverse groupof natural products containing more than 300 alkaloidsisolated from 54 Lycopodium species.[1, 2] These naturalproducts display encouraging biological activities. For exam-ple, recent studies revealed that some lycopodium alkaloidslike huperzine A could inhibit acetylcholinesterase, improveefficiency for learning and memory in animals, and thereforeshow potential in treatment of Alzheimer�s disease.[2, 3] Inaddition, lyconadin A exhibited micromolar cytotoxicityagainst murine lymphoma L1210 and human epidermoidcarcinoma KB cells, while lyconadins A and B could enhancethe mRNA expression for nerve growth factor in 1321N1human astrocytoma cells.[4] Structurally, lyconadins A and B(1 and 2) possess a pentacyclic skeleton, whereas lyconadin C(3) features a tetracyclic ring system, and all containa (dihydro-)pyridone motif (Figure 1).[5] Lyconadins D andE (4 and 5),[6] lycopecurine (6), dehydrolycopecurine (7),[7]
and fastigiatine (8)[8] share a cage-shaped tetracyclic framethat highlights two contiguous quaternary centers at the C4and C13 positions, with the fifth ring either fused onto thebridged N or the N atom outside of the tetracycle. Notably,lyconadins D and E (4 and 5) are the first new fastigiatine-type alkaloids from L. complanatum. With favorable bioac-
tivity and impressive structural complexity, these lycopodiumalkaloids has served as attractive targets in the chemistrycommunity.[9] Remarkable total synthetic triumphs have beenachieved by Smith (1, 2),[10c,d] Sarpong (1),[10a, b] Fukuyama (1–3),[10e, f] Waters (3),[10g] Dai (1, 3),[10j,k] Shair (1, 2, 6–8),[10h, i]
Rychnovsky (8),[10l] but total syntheses of lyconadins D and E(4, 5) has not been realized. Considering the structurediversity of alkaloids 1–8, a divergent synthetic strategy[11]
that may address all of their constructions seems tempting yetquite challenging.
Careful structure analysis of lycopodium alkaloids 1–7revealed that all of them share a 6/6/7-azatricyclic skeleton(Figure 1), signifying the possibility of a divergent design. Toacquire all needed structural skeletons of the seven alkaloids(Scheme 1), we planned to explore the potential reactivity ofthe pivotal intermediate 11: a) Oxidizing the alcoholic moietyof 11 to the resulting carboxylic acid followed by pyridoneformation would afford lyconadin C (3); b) Converting theamine group of 11 into the corresponding N-chloroaminederivative followed by C6 intramolecular nucleophilic attackmight provide tetracycle 9, which bears the core structure oflyconadins A and B (1, 2); c) Forming the C4–C13 bond byuse of a transannular Mannich-type reaction highlightsnitrone chemistry to give the strained tetracycle 10, theprecursor of lycopodium alkaloids 4–7. Our key intermediate11 features a bridged seven-membered-ring system, which isalso found other biologically active natural products.[8, 12] Asa result, construction of this bridged seven-membered-ringsystem is clearly the central challenge in the synthesis of thesenatural products. Previous methods to construct such versatilescaffolds[13] include Michael addition,[10c] aza-Michael addi-tion,[10j, 12] Mannich reaction,[10a,l, 13a] and ring expansion.[10e,g]
Nevertheless, a general protocol to access the bridged seven-
Figure 1. Lycopodium alkaloids 1–8.
[*] J. Zhang,[+] Y. Yan,[+] R. Hu, T. Li, Prof. Dr. Y. YangHubei Key Laboratory of Natural Medicinal Chemistry and ResourceEvaluation, School of PharmacyHuazhong University of Science and Technology13 Hangkong Road, Wuhan, Hubei, 430030 (China)E-mail: [email protected]
Dr. W.-J. BaiDepartment of Chemistry, Stanford UniversityStanford, CA 94305-5080 (USA)
[+] These authors contributed equally to this work.
Supporting information and the ORCID identification number(s) forthe author(s) of this article can be found under:https://doi.org/10.1002/anie.201912948.
AngewandteChemieResearch Articles
&&&& � 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. Int. Ed. 2020, 59, 2 – 9� �
These are not the final page numbers!

membered-ring system by employing a transition-metal-catalyzed cyclization is scarce. To fill this gap, we proposedto develop a palladium-catalyzed Heck-type reaction ofa thiocarbamate tethered enone 13 to synthesize lactam 12which could be easily transformed to the key intermediate 11.
Electron-rich alkene-tethered carbamoyl derivatives havebeen exploited in palladium-catalyzed Heck-type reactions toconstruct the corresponding lactams (Figure 2a),[14] but theuse of electron-deficient alkenes such as enones as theacceptor olefin in such transformations is extremely rare. To
the best of our knowledge, there is a single report by Huang etal., wherein a stoichiometric amount of palladium catalystwas needed and the transformation process was a conjugativeaddition instead of a regular Heck-type reaction (Fig-ure 2b).[15] In this context, we report the first example ofpalladium-catalyzed Heck-type reaction of thiocarbamateswith electron-deficient alkenes to construct the correspond-ing bridged seven-membered-ring systems (Figure 2c), as wellas its application for the enantioselective syntheses oflycopodium alkaloids 1–7 in a divergent manner.
Results and Discussion
Our preparation of the Heck reaction precursors 13commenced with a Diels–Alder reaction of the known enone14 and diene 15,[10g,l] followed by ketone reduction usingDIBAL-H and acetylation with Ac2O (Scheme 2). Using
a one-pot protocol, cis-decalin 16 was isolated in 63 % yieldand 8:1 diastereoselectivity at the C13 position. Reduction ofthe C13-ketone reduction was necessary as otherwise the C12stereocenter would epimerize during the next transformation.Subjecting the obtained cis-decalin 16 to in situ generateddibromocarbene triggered a ring expansion, smoothly afford-ing bromo-enone 17, which in the next step underwenta Suzuki–Miyaura coupling to provide enone 19 in moderateyield.[16] The subsequent generation of the kinetic C5-TBSenol ether proved challenging as unselective deprotonationoccurred at both kinetically and thermodynamically stablepositions (C6 and C11 positions, respectively) under most softor hard enolization conditions, along with enolization of theC13-OAc motif. Eventually, we found that treatment of enone19 with excess TBSOTf and LDA generated the enol of theC5-ketone under kinetic control, alongside the enol of theC13-OAc. Nevertheless, the C13-TBS ketene acetate was
Scheme 1. Retrosynthetic analysis of alkaloids 1–7. PMB =p-methoxy-benzyl.
Figure 2. Heck-type reaction of carbamoyl derivatives.
Scheme 2. Preparation of Heck reaction precursor 13. DIBAL-H= diiso-butylaluminum hydride, LDA = lithium diisopropylamide, TBS = t-bu-tyldimethylsilyl, 9-BBN= 9-borabicyclo[3.3.1]nonane,PMB= p-methoxy-benzyl, DMP= Dess-Martin periodinane, TEA = triethylamine,PNB= p-nitrobenzyl .
AngewandteChemieResearch Articles
&&&&Angew. Chem. Int. Ed. 2020, 59, 2 – 9 � 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.org
These are not the final page numbers! � �

reductively cleaved using DIBAL-H, and the afforded alcoholwas oxidized to the ketone by employing Dess–Martinperiodinane. Starting from enone 14, over 10 grams of ketone20 could be easily prepared with only four column chroma-tography purification steps. Reductive amination of ketone 20followed by acylation with phenyl carbonochloridothioate[15a]
and concomitant removal of the TBS group delivered theexpected Heck reaction precursors 13.
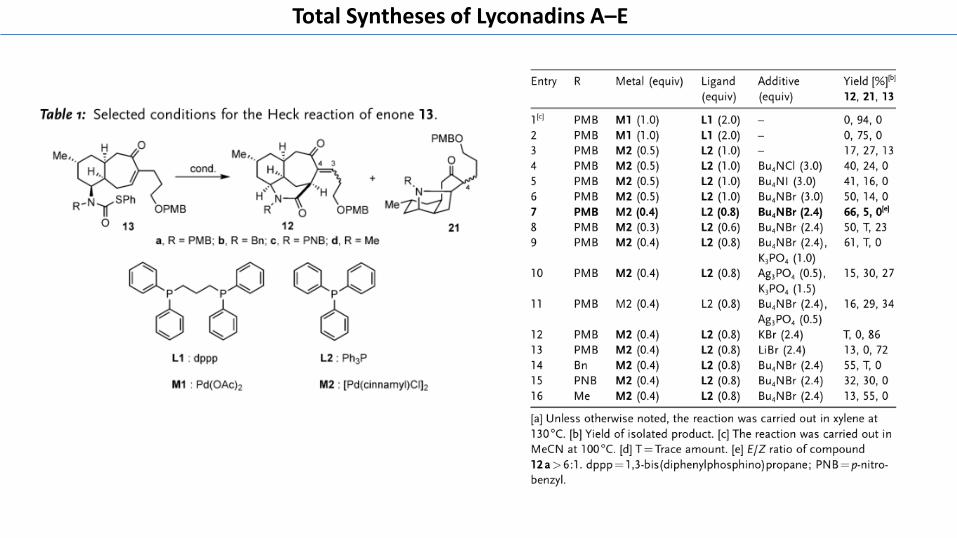
We then investigated the key Heck-type reaction for theconstruction of the azatricyclic lactam 12 (Table 1). In spite ofthe abundant examples of using the Heck reaction toconstruct complex bridge skeletons,[17] the correspondingHeck-type cyclization of a thiocarbamate tethered enone toform a lactam–enone product has been barely studied. Whenwe extended the previously described method developed byHuang et al.[15a] to our cyclization reaction, to our surprise,decarbonylative cyclization product 21a was solely observed(entries 1 and 2). This decarbonylative pathway is supposed tobe inhibited by addition of a chelating diphosphine ligandsuch as dppp. Therefore, we speculated that the dppp ligand
wasn�t effectively chelating to the palladium species in oursystem, which might be explained by the steric hindrance ofour substrate. Accordingly, other palladium species andphosphine ligands were screened, and we found that use of[Pd(cinnamyl)Cl]2 and the less bulky ligand Ph3P could giveus the desired product 12 a albeit in low yield (entry 3) (formore details, see the Supporting Information, Table S1). Toour delight, addition of tetrabutylammonium salts[18] effec-tively suppressed the production of the decarbonylation sideproduct and use of Bu4NBr gave the best results (entries 4–6).Extensive investigation showed that lowering the catalystloading could further reduce the formation of the decarbon-ylation side product at the cost of the conversion (entries 7and8). The best combination (0.4 equiv [Pd(cinnamyl)Cl]2,0.8 equiv PPh3, 2.4 equiv Bu4NBr) effectively provided thelactam 12a in 66 % yield and > 6:1 E/Z configuration(entry 7), and its E-geometry was established by NOESYexperiments. Other attempts including Takemoto�s condi-tions,[14a–c] didn�t significantly improve the conversion orselectivity (entries 9–11). It is worth mentioning that intro-ducing a halide scavenger such as Ag3PO4
[14b] to the afore-mentioned optimized condition was detrimental, indicatingthat the Br� of Bu4NBr played an important role (entry 11).Other Br� sources such as KBr and LiBr were inferior,suggesting that the tetrabutylammonium cation was alsoessential for this transformation (entries 12 and13). Similarly,substrate 13b, containing an electron-rich benzyl N-protect-ing group, provided the expected product in a comparable55% yield (entry 14), whereas the electron-deficient p-nitro-benzyl substrate 13c gave a relative low 32% yield of thedesired product 12c (entry 15). This result demonstrated thatelectron-rich N-protecting groups favor this transformation.Of note, the less sterically hindered methylamine substrate13d gave the desired product 12 d only in a meager 13 % yield(entry 16), suggesting that the size of the N-protecting groupsmight be also critical for this Heck cyclization.
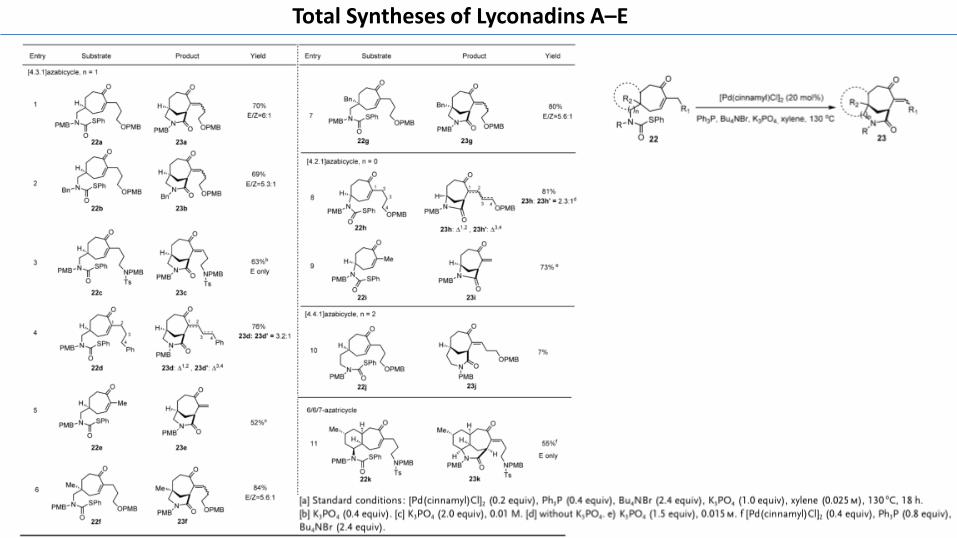
Encouraged by these results, we extended this novelHeck-type reaction to construct other bridged seven-mem-bered-ring systems (Table 2). Pleasingly, when a less stericallyhindered substrate was employed, the catalyst loading couldbe further lowered to 0.2 equivalents. Of note, addition ofK3PO4 as base gave better reaction yield, implying that theBrønsted acid HBr might be generated in the catalytic cycleand needs to be neutralized by K3PO4. Gratifyingly, bicyclo-[4.3.1] lactams 23 a–g were obtained in 52–84% yield fromthiocarbamates 22a–g. Interestingly, when the phenyl-teth-ered substrate 22d was used, a double-bond migration (relay-Heck) product 23d’ was observed besides the desired Heckproduct 23d. The relatively lower yield of 23e was largelyattributed to dimerization of the cyclization product 23e. Toour delight, substrates 22 f,g, which bear an all-carbon-substituted quaternary center, smoothly participated in theHeck-type reaction, providing 23 f,g with excellent yields.Other bridge system such as bicyclo[4.2.1] lactams 23h,i couldalso be realized in excellent yields, whereas [4.4.1]azabicycle23j was isolated in meager yield. Of note, substrate 22kcontaining a sulfonamide motif in the side chain was alsoamenable to this reaction, affording the 6/6/7-azatricyclicproduct 23k in 55 % yield with exclusive E-geometry.
Table 1: Selected conditions for the Heck reaction of enone 13.
Entry R Metal (equiv) Ligand(equiv)
Additive(equiv)
Yield [%][b]
12, 21, 13
1[c] PMB M1 (1.0) L1 (2.0) – 0, 94, 02 PMB M1 (1.0) L1 (2.0) – 0, 75, 03 PMB M2 (0.5) L2 (1.0) – 17, 27, 134 PMB M2 (0.5) L2 (1.0) Bu4NCl (3.0) 40, 24, 05 PMB M2 (0.5) L2 (1.0) Bu4NI (3.0) 41, 16, 06 PMB M2 (0.5) L2 (1.0) Bu4NBr (3.0) 50, 14, 07 PMB M2 (0.4) L2 (0.8) Bu4NBr (2.4) 66, 5, 0[e]
8 PMB M2 (0.3) L2 (0.6) Bu4NBr (2.4) 50, T, 239 PMB M2 (0.4) L2 (0.8) Bu4NBr (2.4),
K3PO4 (1.0)61, T, 0
10 PMB M2 (0.4) L2 (0.8) Ag3PO4 (0.5),K3PO4 (1.5)
15, 30, 27
11 PMB M2 (0.4) L2 (0.8) Bu4NBr (2.4),Ag3PO4 (0.5)
16, 29, 34
12 PMB M2 (0.4) L2 (0.8) KBr (2.4) T, 0, 8613 PMB M2 (0.4) L2 (0.8) LiBr (2.4) 13, 0, 7214 Bn M2 (0.4) L2 (0.8) Bu4NBr (2.4) 55, T, 015 PNB M2 (0.4) L2 (0.8) Bu4NBr (2.4) 32, 30, 016 Me M2 (0.4) L2 (0.8) Bu4NBr (2.4) 13, 55, 0
[a] Unless otherwise noted, the reaction was carried out in xylene at130 8C. [b] Yield of isolated product. [c] The reaction was carried out inMeCN at 100 8C. [d] T = Trace amount. [e] E/Z ratio of compound12a>6:1. dppp = 1,3-bis(diphenylphosphino)propane; PNB=p-nitro-benzyl.
AngewandteChemieResearch Articles
&&&& www.angewandte.org � 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. Int. Ed. 2020, 59, 2 – 9� �
These are not the final page numbers!

The applicability of this newly developed method wasimmediately demonstrated by our syntheses of lyconadins A–E (1–5), lycopecurine (6), and dehydrolycopecurine (7)(Scheme 3). With 1.2 grams of lactam 12a in hand, weperformed an amide reduction using modified Nagashimaconditions[19] to chemoselectively deliver the intermediate 24(Scheme 3). Subsequent olefin reduction and double PMBcleavage with Pd(OH)2/H2 released the free amine 11, whichwas then masked with a Boc group with a one-pot sequence.
Subjecting the obtained alcohol 25 to Tempo and bis-(acetoxy)iodobenzene (BAIB) resulted in a carboxylic acidthat further cyclocondensed with ammonium acetate andacetic acid under refluxing conditions to give a dihydropyr-idone intermediate. Treatment of this intermediate withexcess MnO2 at elevated temperatures formed the expectedpyridone ring, which upon Boc removal with TFA provided(�)-lyconadin C (3).
Table 2: Synthesis of a bridged seven-membered-ring system via Heck-type reaction.
[a] Standard conditions: [Pd(cinnamyl)Cl]2 (0.2 equiv), Ph3P (0.4 equiv), Bu4NBr (2.4 equiv), K3PO4 (1.0 equiv), xylene (0.025m), 130 8C, 18 h.[b] K3PO4 (0.4 equiv). [c] K3PO4 (2.0 equiv), 0.01 M. [d] without K3PO4. e) K3PO4 (1.5 equiv), 0.015m. f [Pd(cinnamyl)Cl]2 (0.4 equiv), Ph3P (0.8 equiv),Bu4NBr (2.4 equiv).
AngewandteChemieResearch Articles
&&&&Angew. Chem. Int. Ed. 2020, 59, 2 – 9 � 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.org
These are not the final page numbers! � �

Next, we turned our attention to lyconadins A and B (1-,2) (Scheme 3). Likewise, reducing the olefin motif andcleaving two PMB groups freed the secondary amine 11. Wethen focused on the construction of tetracycle 9 via a key C6�N bond-forming reaction of amine 11. A previous ap-proach,[10a,b,i] relying on a double-deprotonation/oxidationstrategy to stitch the C6 and N atoms, proved fruitless inour hands, due to the presence of the hydroxy motif of amine11. Masking this primary alcohol of 11 with TBS followed bytreatment with LHMDS and I2 under anhydrous and cryo-genic conditions successfully formed the C6�N bond, but thepoor efficiency of this tactic (three steps from 11, low overallyield) motivated us to look for more productive approach.Accordingly, we converted this secondary amine 11 into thecorresponding N-chloroamine compound 26 using NCS, andsubsequently subjected 26 to basic conditions to furnish thetetracyclic compound 9 in 80 % yield and 5:1 dr. Comparedwith the previously reported anhydrous operation andcryogenic conditions to form the C�N bond, our non-oxidative method was not sensitive to water or air, andsucceeded in an open flask at ambient temperature. Jonesoxidation of tetracycle 9 followed by cyclocondensation
successfully afforded lyconadin B (2). Similarly, MnO2 oxida-tion of the dihydropyridone ring smoothly delivered (+)-ly-conadin A (1).
Syntheses of lyconadins D and E (4, 5), lycopecurine (6),and dehydrolycopecurine (7) were then pursued. O-Bzprotection of compound 25 followed by N-Boc cleavageprovided free amine 27 (Scheme 3). The following C4–C13bond formation proved challenging. Previously, a transannularMannich-type reaction of an imine intermediate was em-ployed to achieve this goal, but we proposed that use ofa nitrone derivative[20] might be beneficial due to its improvedstability and easy preparation from its amine precursor viaoxidation.[20d] To our surprise, the intramolecular Mannich-type reaction of cyclic nitrones with carbonyl-derived nucle-ophiles has been barely studied. To test our idea, the cyclicnitrone intermediate 28 was prepared by oxidation of thestarting amine 27 with Na2WO4/urea peroxide.[21] Next,a variety of Brønsted acids, such as l-proline, acetic acid,benzoic acid, and 4-toluene sulfonic acid, failed to forge theC4–C13 bond. To our delight, introduction of PPTS in thepresence of l-proline at elevated temperature productivelyfulfilled the desired transformation in 71 % yield, establishing
Scheme 3. Enantioselective syntheses of lyconadins A–E (1–5), lycopecurine (6), and dehydrolycopecurine (7). Tempo = 2,2,6,6-tetramethyl-1-piperidinyloxy, BAIB= bis-(acetoxy)iodobenzene, NCS= N-chlorosuccinimide, PPTS = pyridinium p-toluenesulfonate ,UHP = urea peroxide.
AngewandteChemieResearch Articles
&&&& www.angewandte.org � 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. Int. Ed. 2020, 59, 2 – 9� �
These are not the final page numbers!

the vicinal quaternary centers at the C4 and C13 positions.The structure of the obtained cyclization product 29 wasconfirmed by 2D NMR experiments (COSY, HSQC, HMBC,and NOESY). Surprisingly, a C9-nitrone motif arose in theaforementioned Mannich-type reaction, presumably via airoxidation. Nevertheless, nitrone reduction followed by ben-zoate ester cleavage delivered secondary amine 10, Shair�slate-stage intermediate, which could be readily converted intodehydrolycopecurine (7) and lycopecurine (6) in two andthree steps, respectively.[10i] Meanwhile, Jones oxidation ofcompound 10 followed by cyclocondensation led to theformation of the last ring, affording (+)-lyconadin E (5) in58% yield over two steps. Ultimately, reductive amination oflyconadin E (5) with HCOH and NaCNBH3 installed the N-methyl motif, smoothly yielding (+)-lyconadin D (4). Thestructure of (+)-lyconadin D (4) was confirmed by X-raycrystallographic analysis.[22] All spectral data of our syntheticcompounds 1–5 were identical with that of the correspondingnatural products.
Conclusion
In conclusion, careful structural analysis of the seeminglydiverse lycopodium alkaloids 1–7 revealed their resemblance,enabling us to design a divergent strategy to access all of thesealkaloids. To fulfill this goal, a novel palladium-catalyzedHeck-type reaction of thiocarbamates was designed to con-struct bridged seven-membered-ring systems that were oth-erwise challenging to prepare. Taking advantage of thismethod, the key intermediate 11 was conveniently preparedon gram scale. By fully exploring the reactivity potential ofthis common intermediate 11, we achieved C6–N and C4–C13bond formations through an open-flask intramolecular cycli-zation of a N-chloroamine and a transannular Mannich-typereaction of a cyclic nitrone, respectively. With these discov-eries, total syntheses of (+)-lyconadins D and E (4, 5) wereachieved for the first time, as well as enantioselective totalsyntheses of lyconadins A–C (1–3) and formal syntheses oflycopecurine (6) and dehydrolycopecurine (7). More impor-tantly, our newly developed palladium-catalyzed Heck-typereaction provides an expedient way to access a challengingbridged ring system that is widely seen in other bioactivenatural products and therefore may accelerate their synthesis.
Acknowledgements
We thank Prof. Qian Wan and Dr. Qian Zhao for helpfuldiscussions, and Xiaodi Yang from the Shanghai University ofTraditional Chinese Medicine for X-ray crystallographicanalysis. We acknowledge financial support from the NationalNatural Science Foundation of China (21602066), the Funda-mental Research Funds for the Central University(2016YXSM139), and the Academic Frontier Youth TeamProject of HUST.
Conflict of interest
The authors declare no conflict of interest.
Keywords: alkaloids · Heck-type reactions · natural products ·total synthesis · transannular cyclization
[1] For reviews, see: a) W. A. Ayer, L. S. Trifonov in The Alkaloids:Chemistry and Pharmacology, Vol. 45 (Eds.: G. A. Cordell, A.Brossi), Academic Press, New York, 1994, pp. 233 – 266; b) X.Ma, D. R. Gang, Nat. Prod. Rep. 2004, 21, 752 – 772; c) J.Kobayashi, H. Morita in The Alkaloids; Chemistry and Biology,Vol. 61 (Ed.: G. A. Cordell), Academic Press, New York, 2005,pp. 1 – 57; d) Y. Hirasawa, J. Kobayashi, H. Morita, Heterocycles2009, 77, 679 – 729.
[2] J.-T. Cheng, Z.-J. Zhang, X.-N. Li, L.-Y. Peng, H.-R. Luo, X.-D.Wu, Q.-S. Zhao, Nat. Prod. Bioprospect. 2016, 6, 279 – 284.
[3] a) X.-L. He, K. C. Garcia, Science 2004, 304, 870 – 875; b) E. S.Olafsd�ttir, E. S. Halldorsdottir, N. M. Pich, S. Omarsdottir,Natural Products (Eds.: K. G. Ramawat, J. M. M�rillon), Spring-er-Verlag, Berlin Heidelberg, 2013, pp. 1239 – 1262.
[4] For isolation, see: a) J. Kobayashi, Y. Hirasawa, N. Yoshida, H.Morita, J. Org. Chem. 2001, 66, 5901 – 5904; b) K. Ishiuchi, T.Kubota, T. Hoshino, Y. Obara, N. Nakahata, J. Kobayashi,Bioorg. Med. Chem. 2006, 14, 5995 – 6000.
[5] For isolation, see: K. Ishiuchi, T. Kubota, H. Ishiyama, S.Hayashi, T. Shibata, J. Kobayashi, Tetrahedron Lett. 2011, 52,289 – 292.
[6] For isolation, see: K. Ishiuchi, T. Kubota, H. Ishiyama, S.Hayashi, T. Shibata, K. Mori, Y. Obara, N. Nakahata, J.Kobayashi, Bioorg. Med. Chem. 2011, 19, 749 – 753.
[7] For isolation, see: a) W. A. Ayer, B. Altenkirk, N. Masaki, S.Valverde-Lopez, Can. J. Chem. 1969, 47, 2449 – 2455; b) W. A.Ayer, N. Masaki, Can. J. Chem. 1971, 49, 524 – 527; c) J. C.Braekman, C. Hootele, W. A. Ayer, Bull. Soc. Chim. Belg. 1971,80, 83 – 90.
[8] For isolation, see: a) R. V. Gerard, D. B. MacLean, R. Fagianni,C. J. Lock, Can. J. Chem. 1986, 64, 943 – 949; b) R. V. Gerard,D. B. MacLean, Phytochemistry 1986, 25, 1143 – 1150.
[9] For a review, see: M. Saha, R. G. Carter, Synlett 2017, 28, 2212 –2229.
[10] For synthetic examples, see: a) A. Bisai, S. P. West, R. Sarpong, J.Am. Chem. Soc. 2008, 130, 7222 – 7223; b) S. P. West, A. Bisai,A. D. Lim, R. R. Narayan, R. Sarpong, J. Am. Chem. Soc. 2009,131, 11187 – 11194; c) D. C. Beshore, A. B. Smith, J. Am. Chem.Soc. 2007, 129, 4148 – 4149; d) D. C. Beshore, A. B. Smith, J. Am.Chem. Soc. 2008, 130, 13778 – 13789; e) T. Nishimura, A. K.Unni, S. Yokoshima, T. Fukuyama, J. Am. Chem. Soc. 2011, 133,418 – 419; f) T. Nishimura, A. K. Unni, S. Yokoshima, T.Fukuyama, J. Am. Chem. Soc. 2013, 135, 3243 – 3247; g) X.-Y.Cheng, S. P. Waters, Org. Lett. 2013, 15, 4226 – 4229; h) B. B.Liau, M. D. Shair, J. Am. Chem. Soc. 2010, 132, 9594 – 9595;i) A. S. Lee, B. B. Liau, M. D. Shair, J. Am. Chem. Soc. 2014, 136,13442 – 13452; j) Y. Yang, C. W. Haskins, W.-D. Zhang, P. L. Low,M.-J. Dai, Angew. Chem. Int. Ed. 2014, 53, 3922 – 3925; Angew.Chem. 2014, 126, 4003 – 4006; k) Y. Yang, M.-J. Dai, Synlett 2014,25, 2093 – 2098; l) R. A. Samame, C. M. Owens, S. D. Rychnov-sky, Chem. Sci. 2016, 7, 188 – 190.
[11] For selected examples of divergent total synthesis, see: a) J. M.Richter, Y. Ishihara, T. Masuda, B. W. Whitefield, T. Llamas, A.Pohjakallio, P. S. Baran, J. Am. Chem. Soc. 2008, 130, 17938 –17954; b) J. Willwacher, N. Kausch-Busies, A. F�rstner, Angew.
AngewandteChemieResearch Articles
&&&&Angew. Chem. Int. Ed. 2020, 59, 2 – 9 � 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.org
These are not the final page numbers! � �

Chem. Int. Ed. 2012, 51, 12041 – 12046; Angew. Chem. 2012, 124,12207 – 12212; c) J.-P. Krieger, G. Ricci, D. Lesuisse, C. Meyer, J.Cossy, Angew. Chem. Int. Ed. 2014, 53, 8705 – 8708; Angew.Chem. 2014, 126, 8849 – 8852; d) Y. Yang, Y. Bai, S.-Y. Sun, M.-J.Dai, Org. Lett. 2014, 16, 6216 – 6219; e) P.-W. Tan, J. Seayad, D. J.Dixon, Angew. Chem. Int. Ed. 2016, 55, 13436 – 13440; Angew.Chem. 2016, 128, 13634 – 13638; f) X.-M. Chen, H.-J. Zhang, X.-K. Yang, H.-Q. Lv, X.-R. Shao, C. Tao, H.-F. Wang, B. Cheng, Y.Li, J.-J. Guo, J. Zhang, H.-B. Zhai, Angew. Chem. Int. Ed. 2018,57, 947 – 951; Angew. Chem. 2018, 130, 959 – 963; g) Z.-H.Huang, J. Huang, Y.-Z. Qu, W.-B. Zhang, J.-X. Gong, Z. Yang,Angew. Chem. Int. Ed. 2018, 57, 8744 – 8748; Angew. Chem.2018, 130, 8880 – 8884; h) C.-X. Zhuo, A. F�rstner, J. Am. Chem.Soc. 2018, 140, 10514 – 10523; i) P.-L. Wang, Y. Gao, D.-W. Ma, J.Am. Chem. Soc. 2018, 140, 11608 – 11612.
[12] A. Saito, N. Kogure, M. Kitajima, H. Takayama, Org. Lett. 2019,21, 7134 – 7137.
[13] For selected examples of the construction of bridged seven-membered rings: a) J. I. Halliday, M. Chebib, P. Turner, M. D.McLeod, Org. Lett. 2006, 8, 3399 – 3401; b) G. Barbe, D. Fiset,A. B. Charette, J. Org. Chem. 2011, 76, 5354 – 5362; c) C. Chen, J.Hu, J.-H. Su, X.-F. Tong, Tetrahedron Lett. 2014, 55, 3229 – 3231.
[14] For representative examples of Pd0-catalyzed lactam formation,see: a) Y. Yasui, H. Takeda, Y. Takemoto, Chem. Pharm. Bull.2008, 56, 1567 – 1574; b) H. Kamisaki, T. Nanjo, C. Tsukano, Y.Takemoto, Chem. Eur. J. 2011, 17, 626 – 633; c) H. Kamisaki, Y.Yasui, Y. Takemoto, Tetrahedron Lett. 2009, 50, 2589 – 2592.
[15] a) S.-P. Luo, L.-D. Guo, L.-H. Gao, S. Li, P.-Q. Huang, Chem.Eur. J. 2013, 19, 87 – 91; b) L.-D. Guo, X.-Z. Huang, S.-P. Luo, W.-S. Cao, Y.-P. Ruan, J.-L. Ye, P.-Q. Huang, Angew. Chem. Int. Ed.2016, 55, 4064 – 4068; Angew. Chem. 2016, 128, 4132 – 4136.
[16] Y. Ochi, S. Yokoshima, T. Fukuyama, Org. Lett. 2016, 18, 1494 –1496.
[17] a) A. V. Cheprakov, I. P. Beletskaya, Chem. Rev. 2000, 100,3009 – 3066; b) A. E. Sollewijn Gelpke, J. J. N. Veerman, M. S.Goedheijt, P. C. J. Kamer, P. W. N. M. van Leeuwen, H. Hiem-
stra, Tetrahedron 1999, 55, 6657 – 6670; c) P. R. Brooks, M. G.Vetelino, C. G. Bashore, K. Bianco, A. C. Flick, J. W. Coe,Tetrahedron Lett. 2011, 52, 953 – 954; d) G. Tasic, V. Maslak, S.Husinec, N. Todorovic, V. Savic, Tetrahedron Lett. 2015, 56,2529 – 2532.
[18] a) T. Jeffery, J. Chem. Soc. Chem. Commun. 1984, 19, 1287 –1289; b) S. Babu, R. C. Larock, Tetrahedron Lett. 1987, 28, 5291 –5294.
[19] a) Y. Motoyama, M. Aoki, N. Takaoka, R. Aoto, H. Nagashima,Chem. Commun. 2009, 1574 – 1576; b) Y. Nakayama, Y. Maeda,M. Kotatsu, R. Sekiya, M. Ichiki, T. Sato, N. Chida, Chem. Eur. J.2016, 22, 3300 – 3303; c) H.-Y. Shi, I. N. Michaelides, B. Darses, P.Jakubec, Q. N. N. Nguyen, R. S. Paton, D. J. Dixon, J. Am. Chem.Soc. 2017, 139, 17755 – 17758; d) Y. Chen, W.-H. Zhang, L. Ren,J. Li, A. Li, Angew. Chem. Int. Ed. 2018, 57, 952 – 956; Angew.Chem. 2018, 130, 964 – 968.
[20] a) P. Merino, T. Tejero, Synlett 2011, 14, 1965 – 1977; b) C. M.Dombrowski, E. N. Maxwell, C. L. Safran, O. A. Akomah, C. W.Downey, Eur. J. Org. Chem. 2013, 5716 – 5720; c) Q.-Q. Cheng, J.Yedoyan, H. Arman, M. P. Doyle, J. Am. Chem. Soc. 2016, 138,44 – 47; d) V. G. Lisnyak, T. Lynch-Colameta, S. A. Snyder,Angew. Chem. Int. Ed. 2018, 57, 15162 – 15166; Angew. Chem.2018, 130, 15382 – 15386.
[21] B. Bradshaw, C. Luque-Corredera, J. Bonjoch, Org. Lett. 2013,15, 326 – 329.
[22] The absolute configuration was determined by X-ray crystallo-graphic analysis. CCDC 1910195 (the synthetic (+)-lycona-din D) contains the supplementary crystallographic data for thispaper. These data can be obtained free of charge from TheCambridge Crystallographic Data Centre.
Manuscript received: October 10, 2019Revised manuscript received: November 18, 2019Accepted manuscript online: November 19, 2019Version of record online: && &&, &&&&
AngewandteChemieResearch Articles
&&&& www.angewandte.org � 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. Int. Ed. 2020, 59, 2 – 9� �
These are not the final page numbers!

Research Articles
Alkaloid Synthesis
J. Zhang, Y. Yan, R. Hu, T. Li, W.-J. Bai,Y. Yang* &&&— &&&
Enantioselective Total Syntheses ofLyconadins A–E through a Palladium-Catalyzed Heck-Type Reaction
Divergent strategy : The bridged seven-membered-ring system 11, which wasprepared in a novel palladium-catalyzedHeck-type reaction, was the commonprecursor in the enantioselective synthe-ses of lyconadins A–E (1–5), lycopecurine(6), and dehydrolycopecurine (7), lyco-podium alkaloids with encouraging bio-logical activities.
AngewandteChemieResearch Articles
&&&&Angew. Chem. Int. Ed. 2020, 59, 2 – 9 � 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.org
These are not the final page numbers! � �