enzymes that hydrolyze fungal cell wall polysaccharides · tutes about 85 % of the protein and...
TRANSCRIPT

THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 246, No. 22, Issue of November 25, pp. 6722-6736, 1971
Printed in U.S.A.
Enzymes That Hydrolyze Fungal Cell Wall Polysaccharides
II. PURIFICATION AND PROPERTIES OF MYCODEXTRANASE, AN ENDO-a-~-(1 -+ 4) GLUCANASE FROM PENICILLIUM MELINII”
(Received for publication, June 18, 1971)
K. K. TTJNG,$ A. ROSENTHAL, AND J. H. NORDINS
From the Department of Biochemistry, University of Massachusetts, Amherst, Massachusetts 01002
SUMMARY
A specific fungal ol-(1 --j 4)-glucanase, mycodextranase, useful for the large scale production of nigerose, has been purified to homogeneity in high yield from the culture filtrate of Penicillium melinii QM 1931. It has been resolved into fast and slow forms; designations based on their differing electrophoretic mobilities at pH 4.5. The fast form consti- tutes about 85 % of the protein and enzymatic activity. Both forms hydrolyze the glucan nigeran (mycodextran) and cer- tain oligosaccharides of nigeran yielding nigerose and the tetrasaccharide, O-a-D-glUCOp~aUOSyl-(1 -+ 3)-O-a-D-ghICO-
pyranosyl-(1 -+ 4)-0-ar-D-glucopyranosyl-(1 + 3)-D-glucose, as sole end products. Glucose is not produced.
Sedimentation equilibrium studies indicate that fast and slow enzymes both have molecular weights of 40,000 f 2,000 daltons. They have similar but not identical amino acid compositions both being rich in serine, threonine, glutamic and aspartic acids, but have a low content of cysteine and tryptophan. Both mycodextranases contain associated car- bohydrate. The ultraviolet absorption spectra, urea sensi- tivities, and pH activity profiles for both forms are practically identical. In addition both forms display maximum catalytic activities at 70” and are fully active in 40 % dimethyl sulfoxide. Of a number of general enzyme inhibitors tested, only Ag+, Hg+, ZV-chlorosuccinimide, and N-bromosuccinimide inac- tivated the fast enzyme.
Some aspects of the action pattern and mechanism of the fast form have been examined in detail. It operates via an endo-multichain mechanism; both end products are formed at a constant rate during hydrolysis but nigerose does not arise from tetrasaccharide. Competitive inhibition studies show that the fast enzyme preferentially binds oligosaccha- rides with alternating cr-(1 -+ 3)- and ar-(1 --f 4)-glucosidic linkages, and experiments with certain oligosaccharides of nigeran give results consistent with a model in which the sub-
* This research was supported by Grant GB 7774 from the National Science Foundation and bv a grant from the Herman Frasch Foundation. Paper I of thii series is Reference 8.
$ Some of the results presented in this paper were submitted by K. K. Tune in martial fulfillment of the reauirements for the degree of Dictor-of Philosophy of the Department of Biochem- istry, University of Massachusetts, Amherst, Massachusetts 01002. A preliminary report, of this work has been published (1).
$ To whom all inquires should be directed.
&ate-binding site holds 8 glucopyranose units with a cata- lytic site located midway in this site.
The enzyme has been used to study the fine structure of nigeran and the general structure of isolichenin. In the former case this has been done by identifying and quantitat- ing the linkages present at its reducing and nonreducing ter- mini. The data obtained are used to postulate a general mechanism for the biosynthesis of nigeran.
Nigeran (mycodextran), an unbranched polyglucan with alter- nating ~(1 4 4)- and a-(1 + 3).linkages (24), is a constituent of the hyphal wall matrix of various Aspergillus species (5-7). It is hydrolyzed by an endo-or-n-(1 + 3)-glucanase (8) and, after partial acid hydrolysis, by other ~(1 -+ 3).glucosidases (9) yielding glucose as a major product. It is also hydrolyzed in a very specific manner by the fungal endo-cr-n-(1 -+ 4)-glucanase, mycodextranase. This carbohydrase, first reported by Reese and Mandels (6), attacks only the ~(1 + 4).linkages of nigeran, giving nigerose and the tetrasaccharide,’ O-ol-n-glucopyranosyl- (1 -+ 3)-0-cr-D-glucopyranosyl-(1 + 4).O-cr-n-glucopyranosyl- (1 -+ 3)-n-glucose (10) as sole reaction products. It shows no demonstrable activity toward either a homo-a-(1 - 4).glucan (amylose) or a homo-oc-(1 + 3).glucan (pseudonigeran), but its specificity makes it useful in studying the location and quantity of nigeran in fungal cell walls (7) and for the large scale prep- aration of nigerose (11). Since the enzyme is active in 40% dimethyl sulfoxide, an agent capable of solubilizing nigeran, this combination makes it useful in preparing an ascending series of nigeran oligosaccharides in high yields (11).
An interesting aspect of the enzyme’s action is its rigid dual products specificity. Although endo-amylases have been shown to produce a’ preferential distribution of two end products among a total of four to seven (la), an endo-multiple attack mechanism (12) has been shown to be responsible for the observed distribu- tion. Data presented here indicate multiple att#ack does not occur during mycodextranase-catalyzed hydrolysis of nigeran, however.
1 The tetrasaccharide referred to in this paper is O-cr-D-gluco- pyranosyl-(1 ---t 3)-0-or-n-glucopyranosyl-(1 + 4)-O-a-n-glyco- pyranosyl-(1 + 3)-D-glucose.
6722
by guest on July 9, 2018http://w
ww
.jbc.org/D
ownloaded from

Issue of November 25, 1971 K. K. Tung, A. Rosenthal, and J. H. Nordin 6723
This communication details the resolution and purification of two forms of mycodextranase from Penicillium melinii &?\;I 1931. These are designated as the fast and slow forms; an assignment based on electrophoretic mobility at pH 4.5. Selected properties of the two forms of the enzyme have been examined. Experi- ments have been conducted with the fast enzyme to study its action pattern and unusual dual products specificity. A reason- able model for the size of the substrate-binding site is proposed to explain the enzyme’s affinity for certain oligosaccharides and products formed from cert’ain nigeran oligosaccharides. This direct experimentation has been used to test Perlin’s hypothesis (13) regarding the specificity of carbohydraycs as it applies to mycodextranase.
MATERISLS AND METHODS
Cultures of Aspergillus luchuensis QM 873 and P. melinii QM 1931, isolichenin, and a preparation of pure maltotriose were a gift from Dr. E. T. Reese, United States Army Natick Labora- tories, Natick, Massachusetts. Nigeran was isolated (14) from A. luchuensis QM 873 while nigerose, and nigeran tetrasaccharide were prepared enzymatically from nigeran (1 I). [‘%]Nigeran was isolated from Aspergillus niger NRRL 326 cultured (7) from spore innoculum on uniformly labeled [14C]glucose. The product had a specific activity of 12,000 cpm per mg. 3H-Labeled nigeran alcohol was obtained by reduction of the polysaccharide with NaB3H4 followed by purification via repeated reprecipitation from water. It had a specific activity of 25,000 cpm per mg. A mixture of the trisaccharides of nigeran, T1 (O-cu-n-glucopyrano- syl-(1 -+ 3)-0-cY-n-glucopyranosyl-(1 + 4)-n-glucose) and T? (O- a-n-glucopyranosyl-(1 -+ 4)-0-a-n-glycopyranosyl-(1 + 3)-n- glucose) (4) was prepared by partial acid hydrolysis of tetrasac- charide followed by paper chromatography in Solvent, System 2.
The hexasaccharide (n-glucopyranosyl-(1 3 3)-n-glucopyranosyl-
(1 % 4)-n-glucopyranosyl-(1 9 3)-n-glucopyranosyl-(1 4 4)-
n-glucopyranosyl-(1 4 3)-glucose) and octasaccharide, n-gluco-
pyranosyl- (1 4 3) -n-glucopyranosyl- (1 % 4) -n-glucopyranosyl-
(1 % 3) -n-glucopyranosyl- (1 % 4) -n-glucopyranosyl- (1 -% 3)-
D-ghcOpyranOSyl-(1 % 4)-D-glUCOpyraDOSyl-(1 % 8).ghCOSe,
were prepared in pure form by the mycodextranase digestion of dimethyl sulfoxide-solubilized nigeran (1 I). Chemically and radiochemically pure3H-labeled hexa- and octasaccharide alcohols were prepared by reduction of the parent oligosaccharide with KB3H4 (15) and purification by paper chromatography in Solvent System 2. Acid hydrolysis of portions of the tritiated reduced oligosaccharides in 1 N HsS04 for 12 hours was followed by paper chromatography in Solvent System 4, which completely separates sorbitol and glucose. All radioactivity was found in the areas corresponding to sorbitol in both hydrolysates. Other carbo- hydrates used were available in the IaboraCory.
Reagents for gel electrophoresis were purchased from Eastman and ampholites from LKB Instruments Inc., Stockholm, Sweden. Coomassie brilliant blue R250 stain was obtained from Biolab Inc. Bromphenol blue was purchased from National Biochemi- cal Company. Sephadex was obtained from Pharmacia and Bio-Gel resins from Bio-Rad Laboratories, Inc., Richmond, California. Previously coated cellulose thin layer plates were purchased from Brinkman Instruments, Inc., Westbury, New York. Amberlite resins were purchased from Mallinckrodt
Chemical Works. All the other chemicals used were of reagent grade.
Deionization of sugar solutions was performed with a mixed bed resin column (1 x 10 cm) consisting of Amberlite IR-120 (H+) and Xmberlite IR-45 (OH-). All sugar solutions were concentrated under reduced pre.jsure at 40”. Chromatography was carried out at room temperature on ‘Whatman No. 1 paper or previously coated cellulose thin layer plates with the following solvent systems: Solvent System 1: isopropyl alcohol-water-acetic acid (54:8: 18 v/v (6)) ; Solvent System 2: nitromethane-water- ethanol (35:23:41 v/v (12)) ; Solvent System 3: nitromethane- acetic acid-water-ethanol (35: 10 :23 : 41 v/v) ; Solvent System 4: methyl ethyl ketone-acetic acid-l ivr H3BOs (9 : 1: 1 v/v) ; Solvent System 5: ethyl acetate-pyridine-water (2:1:2 v/v (16)); and Solvent System 6: water-saturated phenol (16). Paper electro- phoresis in molybdate buffer was run following the procedure of Bourne, Hutson, and Weigel (17). Sugars were detected by the silver nitrate method of Trevelyan, Proctor, and Harrison (18) or with aniline phthalate (19). Reducing power of sugars was determined by the methods of Somogyi (20) and Park and Johnson (21) while total carbohydrate was estimated by the phenolsulfuric acid method (22). Protein was determined quantitatively by the biuret procedure (23) and the method of Lowry et al. (24) with crystalline bovine serum albumin as a standard, and qualitatively by absorbance at 280 nm.
Radioactivity measurements were made in a Packard Tri-Garb liquid scintillation spectrometer. Each counting vial contained sample (0.1 ml) dissolved in scintillation fluid containing 10 g of 2,5-diphenyloxazole (PPO) and 0.5 g of 1,4-bis[2-(5-phenyl- oxazolyl)]benzene (POPOP) in a mixture of 1 liter of ethanol and 1.5 liters of toluene. Prior to counting, polysaccharides were routinely hydrolyzed in 1 N HCl and neutralized to insure their solubility in the scintillation fluid.
Ultracentrifugation Methods
A Spinco model E analytical ultracentrifuge epuipped with a phot.oelectric scanner was used for all sedimentation studies. The minimum molecular weight analysis in guanidine hydro- chloride and the nat,ive enzyme molecular weight analyses were performed in a double sectored cell with a 12.mm light path length. Concentration dependence of the molecular weights was investigated with a multichannel cell described by Yphantis (25). Determination of molecular weights was performed by the meniscus depletion method of Yphantis (25) with initial prot,ein concentrations of 0.1 to 0.3 mg per ml depending on the experi- ment All runs were performed for a minimum of 60 hours which was sufficient, to insure equilibrium Protein concentrations were measured directly from an extrapolation of the protein-free meniscus which comprised at least one-third of the solution depth. The accuracy of this base-line was checked by overspeed centrifugation at 40,000 rpm. The protein partial specific volume of 0.725 ml per g was used as an average value reported for glycoproteins (26). Minimum molecular weight analysis was performed essentially according t,o the procedure of Tanford, Kawahara, and Lapnje (27).
Gel Electrophoresis
Polyacrylamide gel electrophoresis was carried out at pH 8.3 according to the method Ornstein and Davis (28) and at pH 4.5 by the method of Reisfeld, Lewis, and Williams (29) with 5 ma of current being applied to each tube for 4 hours. After the
by guest on July 9, 2018http://w
ww
.jbc.org/D
ownloaded from

Mycodextranase Vol. 246, No. 22
runs, gels were visualized with Coomassie blue according to Charambach et al. (30). For relating the positions of protein bands to mycodextranase, two identical gels were electrophoreti- tally treated at the same time. One gel was stained immediately after the experiment and the other cut into 0.25.cm sections and each section placed in a test tube with 0.5 ml of 0.5 M acetate buffer, pH 4.5. The gel segments were macerated and 0.5 ml of 0.05 M acetate buffer containing 4 mg of nigeran in suspension was added. The tubes were then incubated at 50” for 30 min. The reducing power of aliquots of each tube was measured by the Somogyi procedure (20). Isoc,lectric focusing in acrylamide gel was performed by a modification of the technique of Kaplan and Foster (31) followed by a unique method of ampholine removal. Gels were prepared with acrylamide solutions which had a 2ol, final concentration of Ampholine, pH 5 to 7. Fifty microliters of the mycodextranases (1 mg per ml) or hemoglobin (5 mg per ml) were analyzed. Focusing of mycodestranases was carried out at 4” for 67 hours with a potential of 15 vo1t.s per tube. (Hemoglobin focused in 18 hours.) pH was determined in uniform slices of one set of gels. Another set of unstained gels to be stained for protein was placed in 10% trichloroacetic acid for 2 hours and then transferred to a destainer containing 77;) acetic acid. These gels were subjected to a current of 50 to 75 ma per tube for 4 hours to remove ampholines and were then placed in 0.259; Coomassie blue in 10% methanol for 2i hours. Gels were destained electrolytically at 50 to 75 ma per tube for 8 hours with 7% acetic and in lOTi> methanol v/v.
Chemical Analyses
-1mino acid analyses were performed with a Spinco model 20 analyzer. To prepare the enzymes for analyses, samples were precipitated wit.h 5% (final concentrat.ion) trichloroacetic acid solution and the pellets washed t,wice with an ethanol-ether mixture (3:l v/v), twice with ether, and finally dried under vacuum at room temperature. Both mycodextranases were hydrolyzed under argon in 6 N HCl at 100”. Portions of the fast enzyme were analyzed after 23, 53, and 75 hours of hydrolp- sis, and 22- and 36.hour samples of slow enzyme were examined. Compositions were determined by extrapolation to zero time to correct for destruction during hydrolysis. Cysteic acid content of the fast enzyme was determined by the method of Hirs (32) and tryptophan by the procedure of Spande and Witkop (33). Analyses for carbohydrate in mycodextranases were carried out as follows. Cold 2OG/, trichloroacet,ic acid solution was added to 1 ml of chilled protein solution containing 1 mg of fast or slow mycodextranase until the final concentration of trichloroacetic acid was 5yc. The turbid solution was allowed to stand over- night at 4”. The precipit,ated protein was collected by centrifu- gation at 5,000 x g and the pellet washed once with 5% tri- chloroacetic acid. The content of carbohydrat,e and protein in the precipitate were estimated on suitable suspensions of the precipitate by the phenolsulfuric acid method and the method of Lowry et al. (24), respectively. The remainder of the sample was suspended in 1 ml of 1 N H&O4 and heated in a sealed vial at 100” overnight. The hydrolysate was neutralized with RaC03, centrifuged, and the concentrated supernatant was subject,ed to cellulose thin layer chromatography in Systems 4 and 5 (ascending) and visualization with aniline phthalate.
Enzyme Assays
Assay l-This assay for mycodextranase is based on the reducing power produced by the end products of enzymatic
hydrolysis. The standard enzyme assay mixture contained 50 pmoles of acetate buffer, pH 4.5; 4 mg of nigeran (suspended in solution); and up to 0.03 unit of enzyme in a final volume of 1 ml. Incubations were conducted at 50” for 10 min and were stopped by adding 1 ml of Somogyi (20) alkaline copper reagent. l’rod- uct formation was found to be linear with respect to enzyme concentration within this range of enzyme. One unit of enzyme is defined as that amount catalyzing the formation of 1 pmole of glucose-reducing equivalent per min under the conditions de- scribed above.
Assay %--It is based on the release of water-soluble products from 3H-labeled nigeran alcohol. The incubation mixture coil- sisted of O.lyO [3H]nigeran suspension (25,000 cpm per mg) in 0.5 ml of 0.05 M acetate buffer (pII 4.5) with up to 0.05 unit of enzyme. lfter incubation for 10 rnin at 50” the reaction was stopped by the addition of 1 ml of cold (0”) absolute alcohol and immersion of the reaction vessel in an ice bath. After 15 min the reaction mixture was centrifuged at 0” for 10 min at 6,000 x g to remove unreacted [3H]nigeran alcohol and the supernatant was then filtered through a Millipore filter with a hypodermic syringe fitted with a Swinney adaptor to remove traces of insolu- ble polysaccharide. The clear filtrate, 0.5 ml, was then analyzed for radioactivity by liquid scintillation spectrometry. The amount of water-soluble products released were linear over the above range of enzyme concentrations used for this time period.
The specificity of mycodextranase was examined by t’esting a mixture of the fast and slow forms with a variety of carbohy- drates. Samples of 1 mg were incubated with 0.25 unit of enzyme in a final volume of 0.25 ml of 0.02 M acetate buffer, pH 4.5, for 24 hours at 40”. Reaction mixtures were analyzed by paper chromat#ography in Solvent Systems 1 and 3 followed by visualization with alkaline silver nitrate.
Since two mycodextranase activities were isolated from P. me&ii culture filtrates, single spore cultures2 were utilized to produce enzyme in one experiment to establish that both forms arise from a single genome. Spores of P. melinii were suspended in 1 ml of 0.05% sodium lauryl sulfate solution.‘ The diluted solution, 0.1 ml, was mixed with 10 ml of warm nutrient agar and poured into a Petri dish. A number of these Petri dishes were incubated at room temperature for 16 to 20 hours and germinated single spore hyphal extensions were located by light microscopy. After further incubation to insure that no other vegetative growth was present around the germinated spore, the hyphal extension was carefully isolat,ed and its further vegetative growth and sporulation allowed to occur. This culture was then used as a source for enzyme production.
Multiplicity of Attack
The multiplicity of attack (12) of mycodextranase was deter- mined by a modification of the procedure of Robyt and French (34) with the method of Park and Johnson (21) to measure re- ducing power. Nigeran, 20 mg, was dissolved in 8 ml of dimethyl sulfoside and the solution diluted by the addition of 10 ml of 0.1 M acetate buffer, pH 4.5, and 2 ml of water. After equilib- rium of the solution at 50” in a water bath, 0.1 unit of fast mycodestranase was introduced and the solution mixed rapidly. Two-milliliter aliquot,s of react.ion mixture were removed peri- odically and 4 ml of ice-cold 95y0 ethanol was immediately added to each aliquot to stop mycodextranase activity. This also had
2 We wish to thank Dr. Sara Faultz of the Botany Department, University of Massachusetts for help in preparing the single spore cultures.
by guest on July 9, 2018http://w
ww
.jbc.org/D
ownloaded from

Issue of November 25, 1971 K. K. Tung, A. Rosenthal, and J. H. Nordin 6725
the effect of precipitat’ing any remaining nigeran. The resulting suspension was mixed thoroughly and 2 ml of this mixture were used for measurement of total reducing power (RJ. The remain der of each suspension was centrifuged at 6,000 x g for 10 min at 0” and the supernatant carefully filtered through a Millipore filter with a hypodermic syringe fitted with a Swinney adaptor. The clear filtrate was used for determination of the reducing power of the filtrate (Rf). The reducing power of the precipitate (R,) w-as then determined directly on the precipitate. The number of multiple attacks is defined as the ratio (r) of Rt:R, (34). In this experiment a nigeran solut.ion without enzyme was used as a control. Determination of whether the enzyme operates by an endo- or exomechanism was made by the method of Tung and Nordin (35).
Dual Products Specificity
[“C]Nigeran was used to monitor the enzymatic production of nigerose and tetrasaccharide. Two milligrams of [YJnigeran (10,750 cpm per mg) were suspended in a final volume of 2 ml of 0.05 RI acetate buffer, pH 4.5, containing 0.2 unit (0.2 ml) of fast mycodextranase. Aliquots of 0.2 ml of the reaction mixture were removed periodically and heated in a boiling water bath for 5 min to destroy enzymatic activity. The cooled mixture was then applied as a l-inch streak on a sheet of Whatman No. 1 chromatography paper along with appropriate standards. The chromatograms were developed in Solvent System 2 for 24 hours. After thorough drying of the papers, the origins and areas cor- responding to glucose, nigerose, and tetrasaccharide standards were excised from the paper and eluted with water. after evaporation of the samples to dryness, they were dissolved in 0.4 ml of water. Of each sample, 0.2 ml was analyzed for radio- activity by liquid scintillation spectrometry.
Measurement 01 Inhibition Constants of Oligosaccharides
Certain oligosaccharides were tested for their ability to inhibit mgcodextranase. Of [3H]nigeran alcohol (25,000 cpm per mg), 0.025 and 0.5 mg were incubated with various concentrations of oligosaccharides and 0.02 to 0.05 unit of fast mycodextranase in 1 ml of 0.05 M acetate buffer, pH 4.5, at 50” for 10 min. No detectable hydrolysis of inhibitors occurs under these conditions. Relative velocities of the reactions were measured by enzyme Assay 2. A plot of the reciprocal of the relative reaction velocity versus the molar concentration of inhibitor yielded a straight line for each of the two concentrations of [3H]nigeran used. The intercept of the two lines was used to calculate the K; for the oligosaccharide (36).
Digestion of 311-labeled Hezasaccharide Alcohol
31-i-Labeled hexasaccharide alcohol (0.16 pmole, 8.7 x lo4 cpm per pmole) was incubated with and without 0.11 unit of purified fast enzyme in a final volume of 0.7 ml of 0.05 M acetate buffer, pH 4.5, at 50” for 60 min. Analyses of the reaction mixtures were carried out as described below for the %labeled octasaccharide alcohol.
Digestion of 3H-labeled Octasaccharide Alcohol
[31-I]Octosaccharide alcohol (5.8 x lo5 cpm per pmole), 0.36 pmole, was incubated at 50” for 30 min with and without 0.11 unit of purified fast enzyme in a final volume of 0.7 ml of 0.05 M
acetate buffer, ~1-1 4.5. The reaction mixtures were applied directly as streaks on separate sheets of Whatman No. 1 paper.
Appropriate marker standards were applied on both sides of the reaction mixture and chromatograms developed in Solvent System 2 for 15 to 17 hours. This solvent resolves the disaccha- ride, tetrasaccharide, hexasaccharide, and octasaccharide frac- tions from etlch other but does not separate nigerose from nigeri- to1 or tetrasaccharide from its alcohol, etc. After visualization of the marker strips, appropriate areas were excised from the paper and eluted with water. Identical’sections of paper were cut from corresponding locations on a blank sheet also developed in the same solvent. Appropriate aliquots of each sample in a known volume were analyzed in duplicat,e for total carbohydrate by the phenolsulfuric acid assay and for radioactivity by liquid scintillation spectrometry. Appropriate corrections for blank carbohydrate and radioactivity values have been made in all data presented.
E$ect of Chemical lnhibitors and Metal Ions
In these experiments 0.05 unit of fast mycodextranase was previously incubated with a final concentration of 1 X 10e3 M
metal ion or inhibitor in a final volume of 0.5 ml for 30 min at 25”. All metal ions, NaCN, L-cysteinc, EDTA, and NazS were previously incubated with the enzymes in 0.05 M acetate buffer, pH 4.5; NBS3 and NCS in 0.05 M acetate buffer, pH 4.0, and N-ethylmaleimide and iodoacetic acid in 0.02 RI phosphate buffer, pH 6.0. After preliminary incubation, 0.5 ml of 0.4% nigeran suspension in 0.1 M acetate buffer, pH 4.5, was added to each sample and enzyme activities measured by Assay 1. Blanks containing no enzyme were used with each potential inhibitor to correct for any effect on the reducing power assay.
RESULTS
Production and Purijication of Mycodextranase-To prepare crude mycodextranase, P. melinii spores were germinated in a Trichoderma salts medium (37). Two hundred milliliters of medium containing 2.25% glucose were used in a 500-ml Erlen- meyer flask with rotary shaking at room temperature. The hyphal suspension resulting from 7-days growth was homoge- nized under sterile conditions and used to inoculate 10 litrrs of the same medium. The culture was grown at 25-26” in a New Brunswick fermentor with an aeration rate of 3 liters per min and agitation with stirring at 150 rpm. After 2 days, 1 liter of sterilized growth medium (minus glucose) containing 11 g of suspended nigeran was introduced aseptically into the fermentor. The enzyme activity was monitored periodically and culture growth stopped when the activity reached a plateau of 0.3 to 0.4 unit per ml of culture medium which usually occurred 5 days after addition of nigeran.
Under similar conditions, glucose, nigerose, maltose, and amylose failed to cause enzyme production. Nigeran tetrasac- charide and acid-degraded (soluble) nigeran present in the medium resulted in the production of minimal amount of activity but neither gave unitage comparable to nigeran. Different concentrations of nigeran were tested for enzyme production and it was found that 0.1% was satisfactory for routine use.
First Ethanol Precipitation-The culture medium was filtered through four layers of cheesecloth to remove the bulk of the hyphal mass and the pH of the filtrate was adjusted to 6.0 by the addition of 1 N acetic acid. The filtrate was then cooled to 3” and 2 volumes of chilled ( -10”) 95yc ethanol was added
3 The abbreviations used are: NBS, N-bromosuccinimide: NCS, N-chlorosuccinimide.
by guest on July 9, 2018http://w
ww
.jbc.org/D
ownloaded from
6726 Mycoclextranase Vol. 246, No. 22
gradually to the slowly stirring filtrate in a carboy. The result- ing turbid solution was allowed to stand at 4” overnight. A flocculent precipitate containing t,he enzyme was left undisturbed while the supernatant was carefully siphoned off. The precipi- tate was collected by centrifugation at 5000 X g for 10 min at 0” and was dissolved in 100 ml of 0.02 M citrate-phosphate buffer, pH 6.0. ,4ny insoluble material was removed by centrifugation.
Second Ethanol Precipitation--To 100 ml of enzyme solution from the first ethanol precipitation was added 65 ml of chilled (-loo) 95y0 ethanol (0.65 volume). The resulting suspension was centrifuged at 5000 x g for 10 min at 0” and the precipit,ate discarded. another S5 ml of chilled ( -10”) 95%) ethanol was added (0.85 volume of original solution) to the supernatant solu- tion, the precipitate collected by centrifugation and dissolved in 20 ml of 0.02 M citrate phosphate buffer, pH 4.2. Further purification was carried out at 4”.
SE-Xephadex Column Chromatography--An SE-Sephadex C-25 column (2.6 x 30 cm) wa-; equilibrated wibh 0.02 M citrate phos- phate buffer, pH 4.2. Twenty milliliters of enzyme solution
t SO8
A I \ , 1 Kenzqne activity / 1 36
FRACTION NUMBER
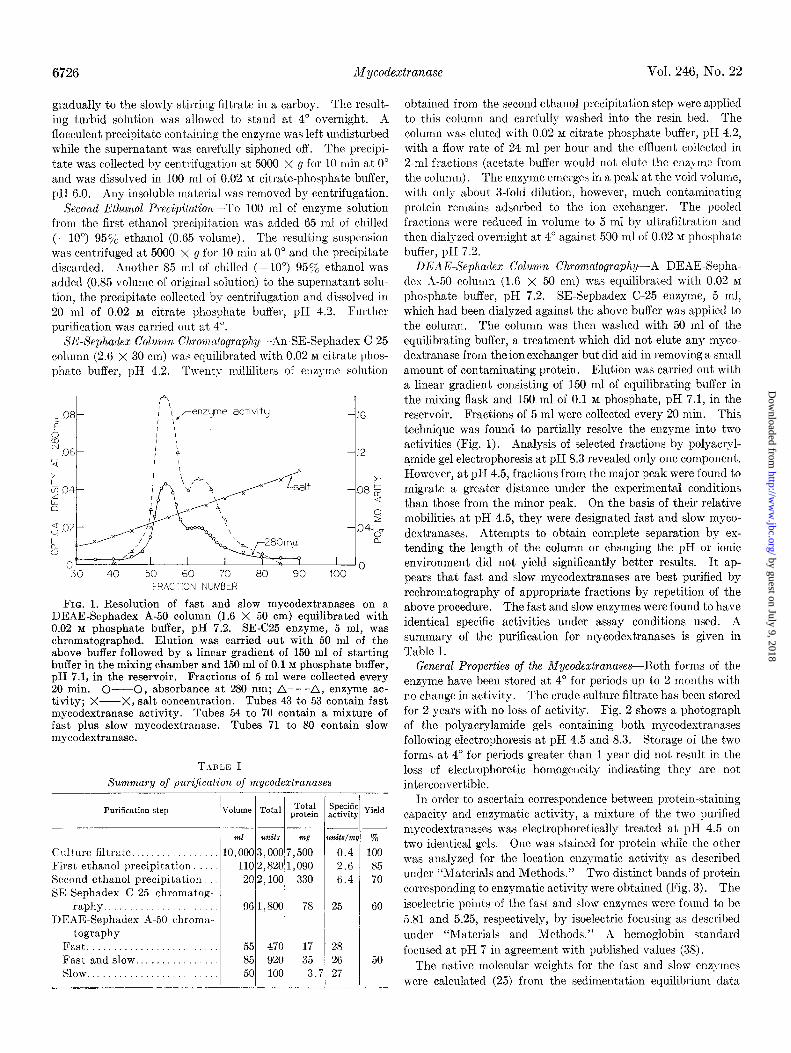
FIG. 1. Resolution of fast and slow mycodextranases on a DEAE-Sephadex A-50 column (1.6 X 50 cm) equilibrated with 0.02 M phosphate buffer, pH 7.2. SE-C25 enzyme, 5 ml, was chromatographed. Elution was carried out with 50 ml of the above buffer followed by a linear gradient of 150 ml of starting buffer in the mixing chamber and 150 ml of 0.1 M phosphate buffer, pH 7.1, in the reservoir. Fractions of 5 ml were collected every 20 min. C----O, absorbance at 280 nm; A-- -A, enzyme ac- tivity; X--X, salt concentration. Tubes 43 to 53 contain fast mycodextranase activity. Tubes 54 to 70 contain a mixture of fast plus slow mycodextranase. Tubes 71 to 80 contain slow mycodextranase.
T-IDLE I Summary of puriftcafion of mycodextranases
Purification step
ml units ng
Culture filtrate.. 10,000 3,000 7,500 First ethanol precipitation. 110 2,820 1,090 Second ethanol precipitation. 202,100 330 SE-Sephadex C-25 chromatog-
raphy. 96 1,800 78 DEAE-Sephadex A-50 chroma-
tography Fast......................... 55 470 17 Fast and slow.. 85 920 35 Slow.. 50 100 3.7
units/mq 70
0.4 100
2.6 85 6.4 70
25 60
28 26 50 27
obtained from t,he second ethanol precipitation step were applied to this column and carefully washed into the resin bed. The column was eluted with 0.02 M citrate phosphate buffer, pH 4.2, with a flow rate of 24 ml per hour and the effluent collected in 2-ml fractions (acetate buffer would not elute the enz? me from the column). The enzyme emerges in a peak at the void volume, with only about 3-fold dilution, however, much contaminating protein remains adsorbed to the ion exchanger. The pooled fractions were reduced in volume to 5 ml by ultrafiltration and then dialyzed overnight at 4” against 500 ml of 0.02 M phosphate buffer, pI-I 7.2.
DEAE-Sephadex Column Chromatography-A DEAE-Sepha- des X-50 column (1.6 x 50 cm) was equilibrated with 0.02 M
phosphate buffer, pH 7.2. SE-Sephadex C-25 enzyme, 5 ml, which had been dialyzed against the above buffer was applied to the column. The column was then washed with 50 ml of the equilibrating buffer, a treatment which did not elute any myco- dextranase from the ion exchanger but did aid in removing a small amount of contaminating protein. Elution was carried out with a linear gradient consisting of 150 ml of equilibrating buffer in the mixing flask and 150 ml of 0.1 M phosphate, pH 7.1, in the reservoir. Fractions of 5 ml were collected every 20 min. This technique was found to partially resolve the enzyme into two activities (Fig. 1). Analysis of selected fractions by polyacryl- amide gel electrophoresis at pH 8.3 revealed only one component. However, at pH 4.5, fractions from the major peak were found to migrate a greater distance under the experimental conditions than those from the minor peak. On the basis of their relative mobilities at pH 4.5, they were designated fast and slow myco- dextranases. Attempts to obtain complete separation by ex- tending the length of the column or changing the pH or ionic environment did not yield significantly better results. It ap- pears that fast and slow mycodextranases are best purified by rechromatography of appropriate fractions by repetition of the above procedure. The fast and slow enzymes were found to have identical specific activities under assay conditions used. A summary of the purification for mycodextranases is given in Table I.
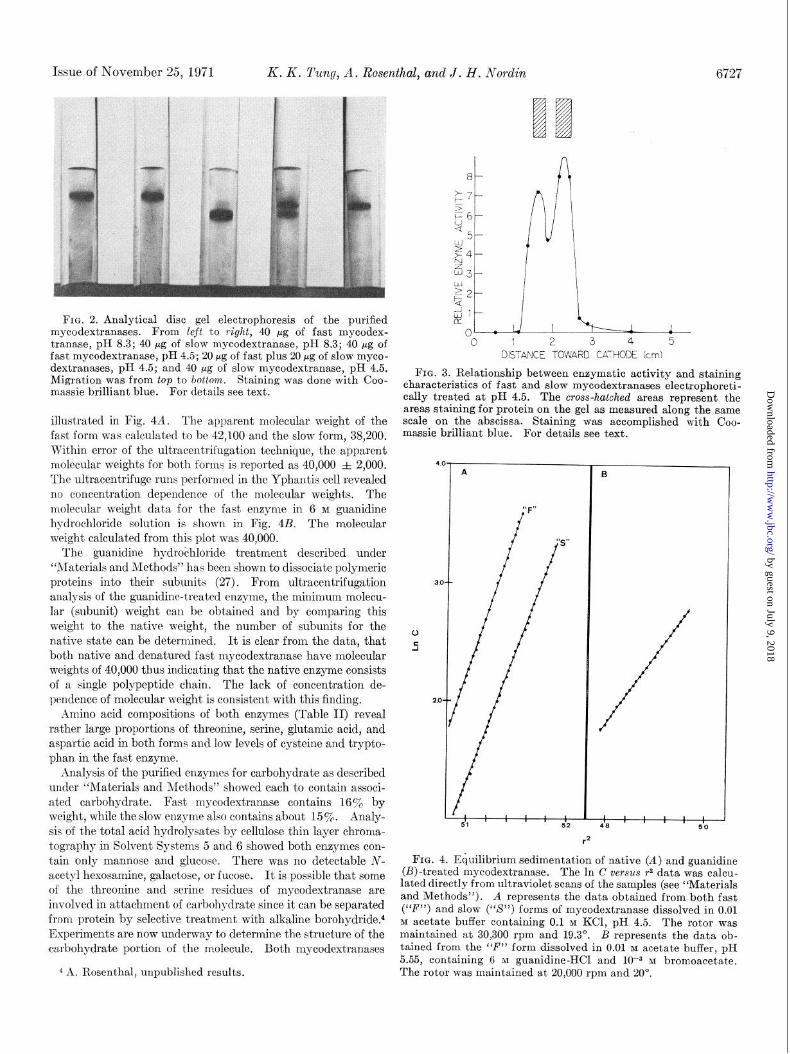
General Properties of the Mycodextranases-Both forms of the enzyme have been stored at 4” for periods up to 2 months with no change in activity. The crude culture filtrate has been stored for 2 years with no loss of activity. Fig. 2 shows a photograph of the polyacrylamide gels containing both mycodestranases following electrophoresis at pI-1 4.5 and 8.3. Storage of the two forms at 4” for periods greater than 1 year did not result in the loss cf electrophoretic homogeneity indicating they are not interconvertible.
In order to ascertain correspondence between protein-staining capacity and enzymatic activity, a mixture of the two purified mycodextranases was electrophoretically treated at pH 4.5 on two identical gels. One was stained for protein while the other was analyzed for the location enzymatic activity as described under ‘Waterials and Methods.” Two distinct bands of protein corresponding to enzymatic activity were obtained (Fig. 3). The isoelectric points of the fast and slow enzymes were found to be 5.81 and 5.25, respectively, by isoelectric focusing as described under “Materials and Methods.” A hemoglobin standard focused at pH 7 in agreement with published values (38).
The native molecular weights for the fast and slow enzymes were calculated (25) from the sedimentation equilibrium data
by guest on July 9, 2018http://w
ww
.jbc.org/D
ownloaded from
Issue of November 25, 1971 K. K. Tung, A. Rosenthal, and J. H. Nordin 6727
FIG. 2. Analytical disc gel electrophoresis of the purified mycodextranases. From left to right, 40 pg of fast mycodex- tranase, pH 8.3; 40 fig of slow mycodextranase, pH 8.3; 40 pg of fast mycodextranase, pH 4.5; 20 fig of fast plus 20 pg of slow myco- dextranases, pH 4.5; and 40 pg of slow mycodextranase, pH 4.5. Migration was from top to bottom. Staining was done with Coo- massie brilliant blue. For details see text.
illustrated in Fig. 4A. The apparent molecular weight of the fast form was calculated to be 42,100 and the slow form, 38,200. Within error of the ultracentrifugation technique, the apparent molecular weights for both forms is reported as 40,000 =t 2,000. The ultracentrifuge runs performed in the Yphantis cell revealed no concentration dependence of t’he molecular weights. The molecular weight data for the fast enzyme in 6 M guanidine hydrochloride solution is shown in Fig. 4B. The molecular weight calculated from this plot was 40,000.
The guanidine hydrochloride treatment described under “Materials and Methods” has been shown to dissociate polymeric proteins into their subunits (27). From ultracentrifugation analysis of the guanidine-treated enzyme, the minimum molecu- lar (subunit) weight can be obtained and by comparing this weight to the native weight, the number of subunits for the native state can be determined. It is clear from the data, that both native and denatured fast mycodextranase have molecular weights of 40,000 thus indicating that the native enzyme consists of a single polypeptide chain. The lack of concentration de- pendence of molecular weight is consistent with this finding.
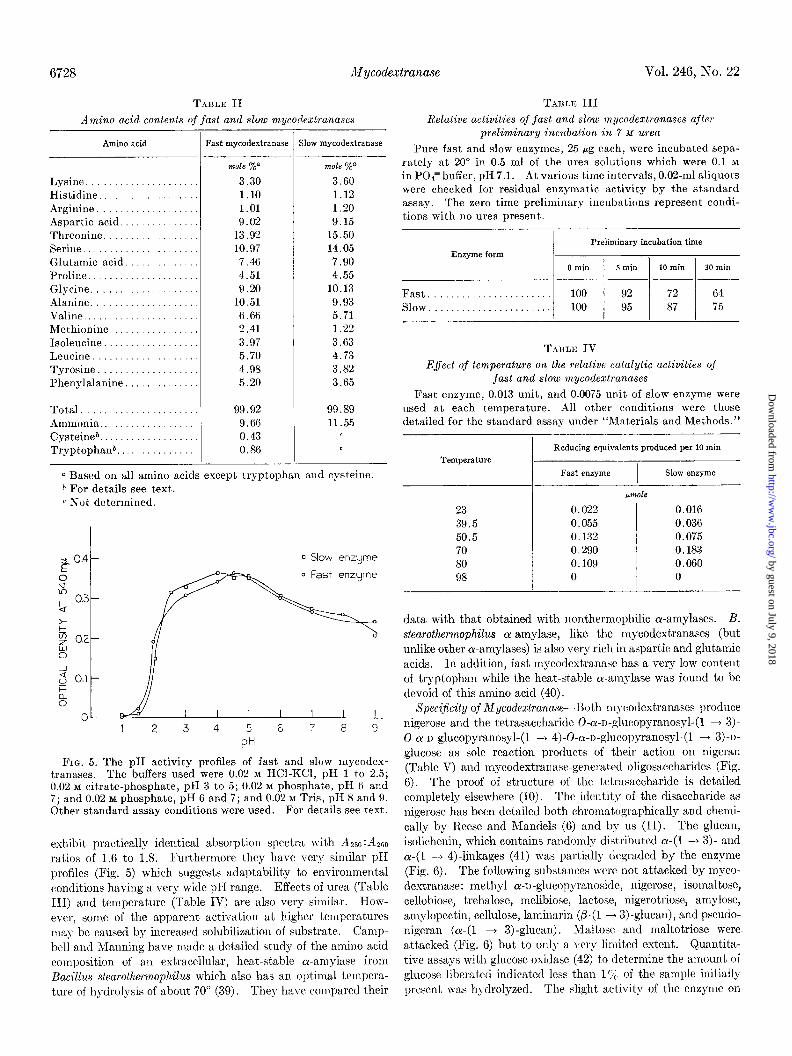
Amino acid compositions of both enzymes (Table II) reveal rather large proportions of threonine, serine, glutamic acid, and aspartic acid in both forms and low levels of cysteine and trypto- phan in the fast enzyme.
analysis of the purified enzymes for carbohydrate as described under “Materials and Methods” showed each to contain associ- ated carbohydrate. Fast mycodextranase contains 16% by weight, while the slow enzyme also contains about 15y0. Analy- sis of the total acid hydrolysates by cellulose thin layer chroma- tography in Solvent Systems 5 and 6 showed both enzymes con- tain only mannose and glucose. There was no detectable N- acetyl hexosamine, galactose, or fucose. It is possible that some of the threonine and serine residues of mycodextranase are involved in attachment of carbohydrate since it can be separated from protein by selective treatment with alkaline borohydride? Experiments are now underway to determine the stjructure of t,he carbohydrate portion of the molecule. Both mycodextranases
DISTANCE TOWARD CATHODE km)
FIG. 3. Relationship between enzymatic activity and staining characteristics of fast and slow mycodextranases electrophoreti- tally treated at pH 4.5. The cross-hatched areas represent the areas staining for protein on the gel as measured along the same scale on the abscissa. Staining was accomplished with Coo- massie brilliant blue. For details see text.
40
3c
0
5
20
A
51 52
B
de r2
FIG. 4. Equilibrium sedimentation of native (A) and guanidine @)-treated mycodextranase. The In C versus r2 data was calcu- lated directly from ultraviolet scans of the samples (see “Materials and Methods”). A represents the data obtained from both fast (“F”) and slow (“S’) forms of mycodextranase dissolved in 0.01 M acetate buffer containing 0.1 M KCl, pH 4.5. The rotor was maintained at 30,300 rpm and 19.3”. B represents t’he data ob- tained from the “F” form dissolved in 0.01 M acetate buffer, pH 5.55, containing 6 M guanidine-HCI and lo+ M bromoacet’ate. ^̂ . _̂ ̂The rotor was maintained at 20,Ouu rpm and YU'. 4 A. Rosenthal, unpublished results.
by guest on July 9, 2018http://w
ww
.jbc.org/D
ownloaded from
6728 il4ycodextranase Vol. 246, No. 22
TABLE II TABLE III Amino acid contents of fast and slow mycoclextranases Relative activities of fast and slow rnycodextranases after
preliminary incubation in Y M Urea Pure fast and slow enzymes, 25 pg each, were incubated sepa-
rately at 20” in 0.5 ml of the urea solutions which were 0.1 M in POP buffer, pH 7.1. At various time intervals, 0.02-ml aliquots were checked for residual enzymatic activity by the standard assay. The zero time preliminary incubations represent condi- t,ions with no urea present,.
Amino acid Fast mycodextranase Slow mycodextranase
Lysine. ................... Histidine. ................. Arginine .................. Aspartic acid. ............ Threonine ................. Serine ............ ...... Glutamic acid. ............ Proline .................... Glycine. .................. Alanine. .................. Valine .................... Methionine ................ Isoleucine ................. Leucine ................... Tyrosine .................. Phenylalanine .............
mole %” mole Yo”
3.30 3.GO 1.10 1.12 1.01 1.20 9.02 9.15
13.92 15.50 10.97 14.05 7.46 7.90 4.51 4.55 9.20 10.13
10.51 9.93 6.Gti 5.71 2.41 1.22 3.97 3.63 5.70 4.73 4.98 3.82 5.20 3.65
Total. .... ............... Ammonia. ................. Cysteineb .................. Tryptophanb ...............
99.92 9.66 0.43 0.86
99.89 11.55
-
- - D Based on all amino acids except tryptophan and cysteine. * For details see text. c Not determined.
0 Slow enzyme
1 2 3 4 5 6 7 8 9 PH
FIG. 5. The pH activity profiles of fast and slow mycodex- tranases. The buffers used were 0.02 hf HCl-KCl, pH 1 to 2.5; 0.02 M citrate-phosphate, pH 3 to 5; 0.02 M phosphate, pH 6 and 7; and 0.02 M phosphate, pH G and 7; and 0.02 M Tris, pH 8 and 9. Other standard assay conditions were used. For details see text.
exhibit practically identical absorption spectra with Am:Am ratios of 1.6 to 1.8. Furthermore they have very similar pK profiles (Fig. 5) which suggests adaptability to environmental conditions having a very wide pI-I range. Effects of urea (Table III) and temperature (Table IV) a.re also very similar. How- ever, some of the apparent activation at higher temperatures may be caused by increased solubilization of substrate. Camp bell and Manning have made a detailed study of the amino acid composition of an extracellular, heat-stable cr-amylase from Bacillus stearothermophilus which also has an optimal tempera- ture of h\-drolysis of about 70” (39). They have corn.pared their
Enzyme form Preliminary incubation time
0 min 5 min 10 min 30 min
Fast....................... 100 92 72 64 Slow. . . . . 100 95 87 75
TABLE IV Effect of temperature on the relative catalytic activities of
fast and slow mycodextranases Fast enzyme, 0.013 unit, and 0.0075 unit of slow enzyme were
used at each temperature. All other conditions were those detailed for the standard assay under “Materials and Methods.”
Temperature
23 39.5 50.5 70 80 98
Reducing equivalents produced per 10 min
Fast enzyme I
Slow enzyme -
~rnOl‘3
0.022 0.016 0.055 0.036 0.132 0.075 0.290 0.183 0.109 0.060 0 0
data with that obtained with nonthermophilic oc-amylases. B. stearothermophilus cr-amylase, like the mycodextranases (but unlike other cu-anzylases) is also very rich in aspartic and glutsmic acids. In addition, fast mycodextranasc has a very low content of tryptophan while the heat&able ol-amylase was found to be devoid of this am.ino acid (40).
Specijkity of Mycodextranase--130th mycodextranases produce nigerose and the tetrasaccharide 0-a-r-glucopyranosyl-(1 --) 3)- 0-cr-n-glucopyranosyl-(1 --) 4)-0-c-I-glucopyranosyl-(1 + 3)-n- glucose as sole reaction products of their action on nigeran (Table V) and mycodextranase-generated oligosaccharides (Fig. 6). The proof of structure of the tetrasaccharide is detailed completely elsewhere (10). The identity of the disaccharide as nigerose has been detailed both chromatographically and chemi- cally by Reese and Mandels (6) and by us (11). The glucan, isolichenin, which contains randomly distributed a-(1 + 3). and a-(1 --f 4).linkages (41) was partially degraded by the enzyme (Fig. 6). The following substances were not attacked by myco- dextranase: methyl cY-n-glucopyranoside, nigerose, isornaltose, cellobiose, trehalose, melibiosc, lactose, nigerotriose, amylose, amylopectin, cellulose, laminarin (p-(1 + 3)-glucan), and pseudo- nigeran (a- (1 + 3) -glucan) . YIaltose and maltotriose were attacked (Fig. 6) but to only a very limited extent. Quantita- tive assays with glucose oxidase (42) to determine the amount, of glucose liberated indicated less than l’;, of the sample initially present was hydrolyzed. The slight activity of the enzyme on
by guest on July 9, 2018http://w
ww
.jbc.org/D
ownloaded from

Issue of November 25, 1971 K. K. Tung, A. Rosenthal, and J. H. Nordin 6729
TABLE v Nobilities of various carbohydrates subjectecl to papeT
chromatograph
Compound
Nigerose Glucose Maltose Nigerotriose Maltotriose O-u-u-glucopyranosyl,3a-u-glnco-
pyranosyl,4-glucose 0-cu-o-glucopyranosyl,4ol-ogluco-
pyranosyl,3-glucose 0-a-u-glucopyranosy1,3a-D-gluco-
pyranosyl, 401-n-glucopyranosyl, 3- glucose
Products released by hydrolysis of nigeran by :
Fast enzyme
Slow enzyme
RNigerose
Solvent system 1
~-
1.00 1.10 0.86 0.76 0.59 0.65
0.65
0.48
-
_
1.00 1.00 0.48 0.6G 1.00 1.00
0.48 0.66
Solvent system 3
1.00 1.20 0.90
0.75
0.75
0.66
OE
G . V f?
L-2
I I I
STDS. TRI HEXA NIGERAN srbs
TETRA OCTA ISOLICHENIN
FIG. 6. Chromatography of hydrolysis products resulting from mycodextranase action on various oligo- and polysaccharides. Incubations of 0.5 to 1 mg of various substrate were carried out with 0.05 unit of enzyme overnight at 50” in a final volume of 0.5 ml of 0.05 M acetate buffer, pH 4.5. Portions of each sample were chromatographed in Solvent System 2. Standards: A, B, and C, octa-, hexa-, and tetrasaccharides of nigeran; D, nigerose; E, glucose; F, maltotriose; G, nigerotriose; H, maltose. Sub- strates: Tri, (a mixture of T1 and Tz trisaccharides), tetra, heza, and octa all refer to specific nigeran oligosaccharides whose struc- tures are defined under “Materials and Methods.”
these two substances is not unespected if the mechanism pro- posed below for mycodestranase is correct. Based on the known structures of nigeran (3, 4), the tetrasaccharide obtained by the mycodextranase digestion of nigeran (10) and from the specificity
0 20 40 60 SO 100 120 TIME OF INCUBATION (min.1
FIG. 7. Dual products specificity of mycodextranase. [%I- Nigeran was used to monitor the relative rates of production of nigerose and tetrasaccharide. Applied to the chromatogram at each time interval were 1960 counts (0.2 ml) and one-half of the material eluted from the various areas was counted. “Total counts” equals the sum of the counts eluted from the origin, tetra- saccharide, and nigerose areas of the chromatogram after each time interval (for details of the experiment see text).
data above, it is clear that mycodextranase preferentially hydro- lyzes cu-(1 + 4)-glucosidic linkages adjacent to glucopyranose units linked a-(1 + 3). This postulate was strengthened by additional studies. While tetrasaccharide is a very poor sub- strate for mycodextranase, incubation of excess mycodextranase with tetra,saccharide as described in Fig. 6 does result in the slow release of nigerose but not glucose. Similarly, incubation with a mixture of T1 and TJ trisaccharides results in the liberat’ion of traces of nigerose and glucose. Maltose is not produced.
Mechanism of Mycodextrunase-The results df [14C]nigeran digestion by fast mycodextranase is shown in Fig. 7. With the exception of the 3- and 5min samples, the sum of the counts in nigerose, tetrasaccharide, and the origin material equals the total counts present in the incubation mixtures. The 2-hour values, which represent complete digestion of the polymer indicates complete transfer of counts from polymer into nigerose and tetraeaccharide. The remainder of this strip of chromat.ography paper was also analyzed and showed no radioactivity in any other area. This confirmed results obtained in other experiments by visual examination of developed chromatograms. The zero time and 3-min origin material samples showed losses of radio- activity but this was attributed to the fact that they contain essentially undegraded nigeran and losses of material probably resulted from some of the polysaccharide failing to elute from the paper with water. One other important result of this experi- ment, is that nigerose and tetrasaccharide are produced simul- taneously in constant proport,ion by mycodextranase action since the molar ratio of tetrasaccharide to disaccharide remained at approximately 1.5 throughout the course of the digestion. In a control experiment run under identical conditions only 5% of the tetrasaccharide incubated with mycodextranase was con- verted to nigerose after 2 hours of incubation. These results show quite clearly that nigerose does not arise from tetrasaccha- ride.
by guest on July 9, 2018http://w
ww
.jbc.org/D
ownloaded from
6730 Mycodextranase Vol. 246, No. 22
4 y0.5mg nigeran/ml
0.1 0.2 0.3 0.4 05 0.6 MICROMOLES OF END PRODUCTS RELEASED
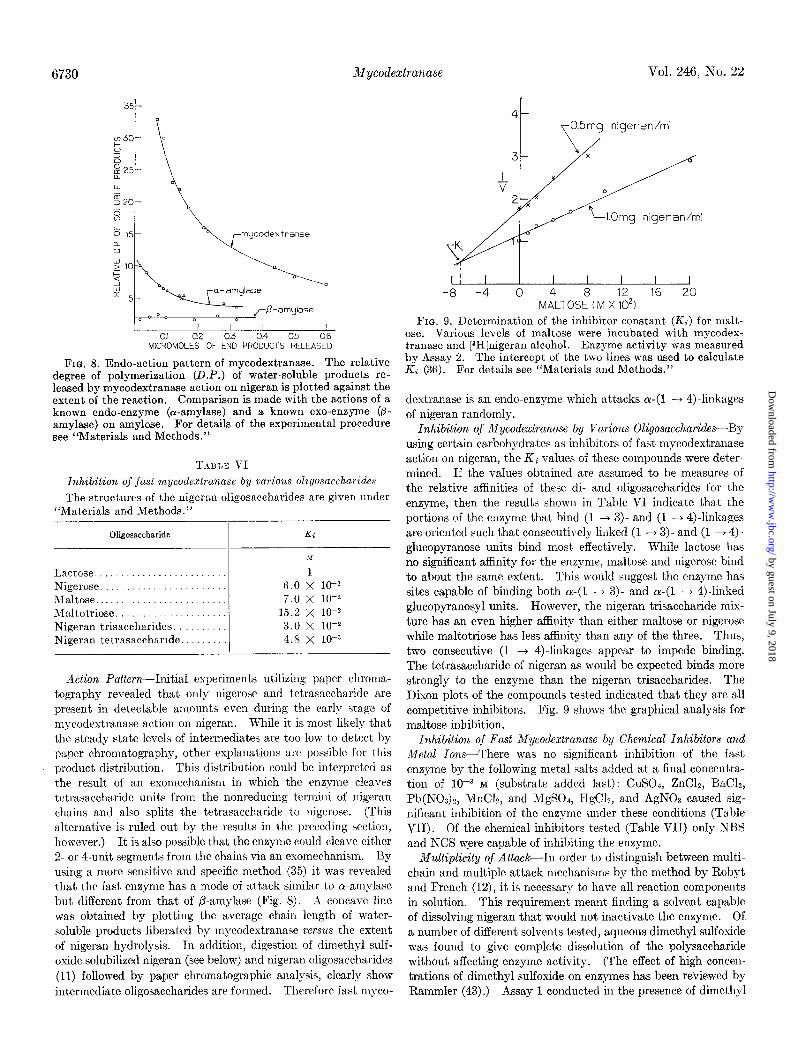
FIG. 8. Endo-action pattern of mycodextranase. The relative degree of polymerization (D.P.) of water-soluble products re- leased by mycodextranase action on nigeran is plotted against the extent of the reaction. Comparison is made with the actions of a known endo-enzyme (or-amylase) and a known exo-enzyme (p- amylase) on amylose. For details of the experimental procedure see “Materials and Methods.”
T.~ULE VI
Inhibition of just mycodextranase by va,rious oligosaccharides
The structures of the nigeran oligosaccharides are given under “Materials and Methods.”
Oligosaccharide Ki
Lactose ......................... Nigerose ........................ Maltose ......................... Maltotriose. .................... Nigeran trisaccharides. ......... Nigeran tetrasaccharide .........
1 6.0 X lo- 7.0 x 10-Q
15.2 X lo-* 3.0 x 10-t 4.8 x 10-S
Action Pa,ttern-Initial experiments utilizing paper chroma- tography revealed that only nigerose and tetrasaccharide are present in detectable amounts even during the early stage of mycodextranase action on nigeran. While it is most likely that the steady state levels of intermediates are too low to detect by paper chromatography, other explanations are possible for t,his product distribution. This distribution could be interpreted as the result of a.11 exomechanism in which the enzyme cleaves tetrasaccharide units from the nonreducing tcrmini of nigeran chains and also splits the tetrasaccharide to nigerose. (This alternat,ive is ruled out by the results in the preceding section, however.) It is also possible that the enzyme could cleave either 2. or 4-unit segments from the chains via an exomechanism. By using a more sensitive and specific method (35) it was revealed that the fast enzyme has a mode of att,ack similar to ol-amylase but different from that of P-amylase (Fig. 8). Ai concave line was obtained by plotting the average chain length of water- soluble products liberated by mycodextranase versus the extent of nigeran hydrolysis. In addition, digestion of dimethyl sulf- oxide solubilized nigeran (see below) and nigeran oligosaccharides (11) followed by paper chromatographic analysis, clearly show intermediate oligosaccharides are formed. Therefore fast myco-
I I I 1 I I I I -8 -4 0 4 8 12 16 20
MALTOSE (M X lo21
FIG. 9. Determination of the inhibitor constant (IL) for malt- ose. Various levels of maltose were incubated with mycodex- tranase and [aH]nigeran alcohol. Enzyme activity was measured by Assay 2. The intercept of the two lines was used to calculate Ki (36). For details see “Materials and Methods.”
dextranase is an endo-enzyme which attacks a-(1 -+ 4)-linkages of nigeran randomly.
Inhibition of Mycoclextranase by Various Oligosaccharicles-By using certain carbohydrates as inhibitors of fast mycodextranase action on nigeran, the Ki values of these compounds were deter- mined. If the values obtained are assumed to be measures of the relative affinities of these di- and oligosaccharides for the enzyme, then the results shown in Table VI indicate that the portions of the enzyme that bind (1 + 3)- and (1 -+ 4)linkages are oriented such that consecutively linked (1 + 3) and (1 + 4)- glucopyranose units bind most. effectively. While lactose has no significant affinity for the enzyme, maltose and nigerose bind to about the same extent. This would suggest the enzyme has sites capable of binding both cr-(1 + 3)- and a-(1 + 4)linked glucopyranosyl units. However, the nigeran trisaccharide mix- ture has an even higher affinity than either maltose or nigerose while maltotriose has less affinity than any of the three. Thus, two consecutive (1 --f 4)linkages appear to impede binding. The tetrasaccharide of nigeran as would be expected binds more strongly to the enzyme than the nigeran trisaccharides. The Dixon plots of the compounds tested indicated that they are all competitive inhibitors. Fig. 9 shows the graphical analysis for maltose inhibition.
Inhibition of Fast Mycodextranase by Chemical Inhibitors and
Metal Ions-There was no significant inhibition of the fast enzyme by the following metal salts added at a final concentra- tion of lop3 M (substrate added last): CuSO+ ZnClz, BaCL, Pb(NO&, MnCL, and MgS04, ITgCL, and AgN03 caused sig- nificant inhibition of the enzyme under these conditions (Table VII). Of the chemical inhibitors tested (Table VII) only NBS and NCS were capable of inhibiting the enzyme.
Multiplicity of &a&-In order to distinguish between multi- chain and multiple at,tack mechanisms by the method by Robyt and French (12), it is necessary to have all reaction components in solution. This requirement meant finding a solvent capable of dissolving nigeran that would not inactivate the enzyme. Of. a number of different solvents tested, aqueous dimethyl sulfoxide was found to give complete dissolution of the polysaccharide without affecting enzyme activity. (The effect of high concen- trations of dimethyl sulfoxide on enzymes has been reviewed by Rammler (43).) Assay 1 conducted in the presence of dimethyl
by guest on July 9, 2018http://w
ww
.jbc.org/D
ownloaded from
Issue of November 25, 1971 K. K. Tuny, A. Rosenthal, and J. H. No&in 6731
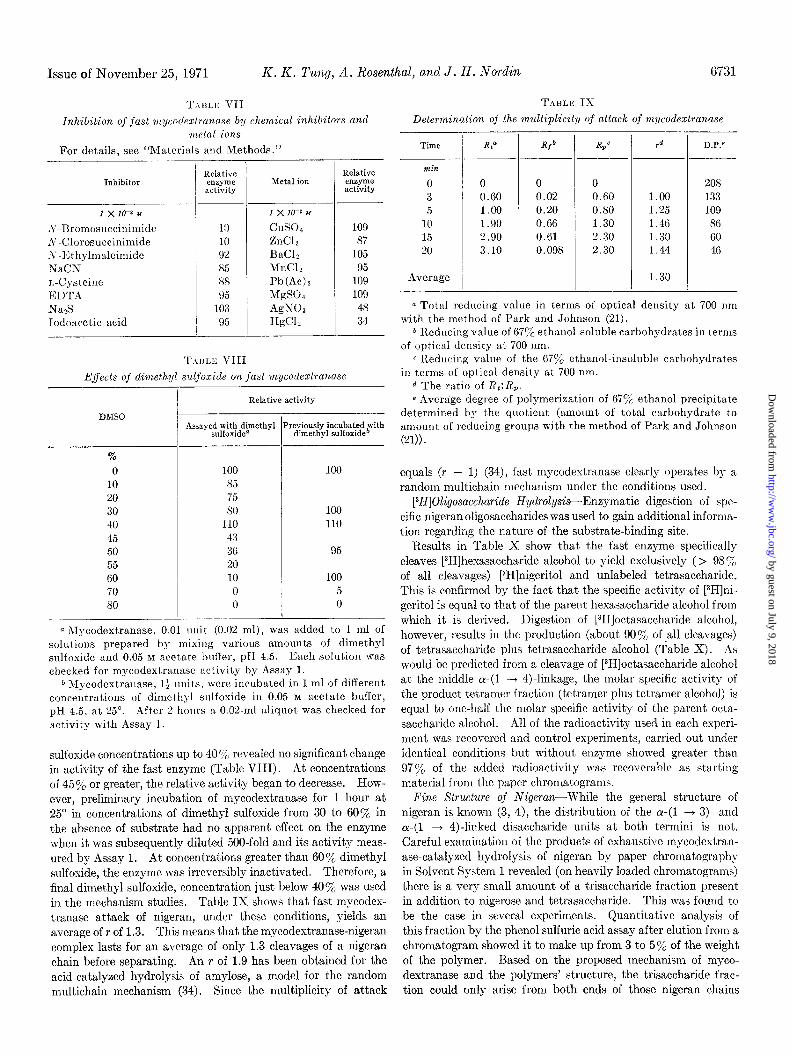
T.\IILK VII TABLE IX
Inhibition of fast mycotlezlranuse b:y chemical inhibitors and Determination of the multiplicity of attack of mycodextranase metal ions
For details, see “Materials and Methods.”
Inhibitor
1 x 10-3 M
S-Bromosuccinimide A-Clorosuccinimide ,Y -I<; t hylmaleimide XaCN r,-Cysteine El)TA
-T Nan!, Iodoncetic acid
Relative enzyme activity
-
Metal ion
1 x 10-z M
cuso4
ZnC12 BaCI, MnCl? Pb(Ac)n MgS04 AgNOs H&I,
Relative enzyme activity
109 87
105 95
109 109
48 34
E.ffects of dimethyl sdfoxide on fast mycodextranase
DMSO
%
0 10 20 30 40 45 50 55 GO 70 so
/ -
T
I- _I. I
Relative activity
Assayed with dimethyl Previously incubated with sulfoxide” dimethyl sulfoxid@
100 100 85 75 80 100
110 110 43 36 95 20 10 100 0 5 0 0
a hIycodextranase, 0.01 rulit (0.02 ml), was added to 1 ml of solutions prepared by mixilrg various amount,s of dimethyl sulfoxide and 0.05 M acetate bufler, pH 4.5. Each solution was checked for mycodextranase activity by Assay 1.
0 Mycodextranase, l+ lmits, were incltbated in 1 ml of different concentrations of dimethyl slllfoxide in 0.05 b~ acetate buffer, pH 4.5, at 25”. After 2 horus a 0.02.ml aliquot was checked for act,ivity with Assay 1.
sulfoxide concentrations up to 40 ‘;‘c revealed no significant change in activity of the fast enzyme (Table VIII). At concentrations of 45% or greater, the relative activity began to decrease. How- ever, preliminary incubation of mycodestranase for 1 hour at 25“ in concentrations of dimethyl sulfoxide from 30 to 60% in the absence of substrate had no apparent effect on the enzyme when it was subsequently dilut,ed 500.fold and its activity meas- ured by Assay 1. At concentrations greater than 60% dimethyl sulfoxide, the enzyme was irreversibly inactivated. Therefore, a final dimethyl sulfoxide, concentration just below 40% was used in the mechanism studies. Table IX shows that fast mycodex- tranase attack of nigeran, under these conditions, yields an
average of r of 1.3. This means that the mycodextranase-nigeran complex lasts for an average of only 1.3 cleavages of a nigeran chain before separating. An r of 1.9 has been obtained for the acid-catalyzed hydrolysis of amylose, a model for the random multichain mechanism (34). Since the multiplicity of attack
Time
min
0 3 5
10 15 20
Average
4
/
RP
0 0 0 O.GO 0.02 O.GO 1.00 0.20 0.80 1.90 0.66 1.30 2.90 0.61 2.30 3.10 0.098 2.30
R/b -
-I-
r d
1.00 1.25 1.46 1.30 1.44
1.30
D.P.e
208 133 109 86 60 46
a Total redllcing value in terms of optical density at 700 nm with the method of Park and Johnson (21).
* Reducing value of G7Y’ ethanol-soluble carbohydrates in terms of optical density at 700 nm.
c I’ledllcing valrle of the 67% ethanol-insoluble carbohydrates in terms of optical density at 700 nm.
d The ratio of XL:&. e Average degree of polymerization of 670y0 ethanol precipitate
determined by the quotient (amount of total carbohydrate to amount of reducing groups with the method of Park and Johnson
(21)).
equals (r - 1) (34), fast mycodextranase clearly operates by a
random multichain mechanism under the conditions used.
[3H]Oligosaccharide Hydrolysis-Enzymatic digestion of spc- cific nigeranoligosaccharides was used to gain additional informa- tion regarding the nature of the substrate-binding site.
Results in Table X show that the fast enzyme specifically cleaves [3H]hexasaccharide alcohol to yield exclusively ( > 989; of all cleavages) [3H]nigeritol and unlabeled tetrasaccharide. This is confirmed by the fact that the specific activity of [3H]ni- geritol is equal to that of the parent hexasaccharide alcohol from which it is derived. Digestion of [3H]octasaccharide alcohol, however, results in the production (about 90% of .a11 cleavages) of tetrasaccharide plus tetrasaccharide alcohol (Table X). As would be predicted from a cleavage of [3H]octasaccharide alcohol at the middle ~(1 ---f 4)-linkage, the molar specific activity of the product tetramer fraction (tetramer plus tetramer alcohol) is equal to one-half the molar specific activity of the parent octa- saccharide alcohol. All of the radioactivity used in each experi- ment was recovered and cont.rol experiments, carried out under identical conditions but without enzyme showed greater than 97% of the added radioactivity was recoverable as starting material from the paper chromatograms.
Fine Structure of Nigeran-While the general structure of nigeran is known (3, 4), the distribution of the ~(1 + 3)- and ~(1 + 4)-linked disaccharide units at both termini is not. Careful examination of the products of exhaustive mycodextran- ase-catalyzed hydrolysis of nigeran by paper chromatograph? in Solvent System 1 revealed (on heavily loaded chromatograms) there is a very small amount of a trisaccharide fraction present in addition to nigerose and tetrasaccharide. This was found to be the case in several experiments. Quantitative analysis of this fraction by the phenol sulfuric acid assay after elution from a chromatogram showed it to make up from 3 to 5yo of the weight of the polymer. Based on the proposed mechanism of myco- dextranase and the polymers’ structure, the trisaccharide frac- tion could only arise from both ends of those nigeran chains
by guest on July 9, 2018http://w
ww
.jbc.org/D
ownloaded from

T.\IILIC x
l’rotlucts derived from mycoclextranase digestion of 3H-labeled hexa-
and octasaccharide alcohols
-I- Substrate
Product
~- 1. cm/ cpm/
The two types of chains (A & B) are present in pmole cpm pmole j.lntole CM /mole about equal amounts (see Results).
Nigerose p111s lligeri- Only type B
to1 0.18 15,001 3 _t <O.Ol 19,000 chains can yield Tl trisaccharides from their re-
x 10
Tetrasaccharide plus ducing ends as a result of mycodextranase action.
tetrasaccharide al- cohol. 0.19 148 0 O.G( il 74,000 2.6
2. Chains in various stages of construction
x 105 (degradat'on) having ~(1~4) linkages at their Hexasaccharide plus
hexasaccharide al- non-reducing termini (A2 and Bl) represent 25% of
cohol* <O.Ol ,322 <O.Ol %JOO/ Hexasaccharide al- I
the polymer population as isolated.
cohol (control) 0.10 14,37c 9 FIG. 10. Fine structure of nigeran chains.
.
x 10
Octasaccharide alco- I
T.\I<LK XI ho1 <O.Ol 5,400~
Octassarharide alco- Proclucis derived from th,e enzymatic digcation of “H-labeled nigeran
alcohol and L4C-uni.forml~/ labeled nigerana hol (Control). 0.3; 72 10,0005.8
x 102 Product [WlNigeran L- -
a Micromoles of each product, not glucose equivalents, as -
determined by the phenolsulfuric acid method. * Hexasaccharide alcohol only, in the case of digestion of [3H]-
Origin ................................ 900 46
hexasaccharide alcohol. Tetrasaccharide (alcohol)h ......... 11,000 3,480 Trisaccharide (alcohols) b ............ 11,300 156
................ having terminal (Y-(1 + 4)-linkages (Fig. 10). A portion of this
Nigerose (alcoholj *. 4,000 1,251 GllIcose
fraction was reduced wit,h NaB3H4. Following deionization, (Sorbitol)* .._._.... ..:I.‘.:.: 11
500
TYPO B [3H]Hexasaccharide alcohol [3HlOctasaccharide alcohol
(8.9 X 104 cpm/@mle) (5.8 X 105 cpm/mole) GP~~4GP~,3GP~,4~G~l,3Gp1,4Gpl~l,3Gpl,4Gpl,3Gpl,4~ Bl
Total hmlnt’ radio- ipecific To+ Specific
activity activity $$~~y activity
GP~,3G~l,4~Gp~,3Gpl,4Gpl~l,3Gpl,4Gpl,3Gpl,4~ B2
the mixture was resolved by molybdate electrophoresis (17). This procedure revealed, as expected, the presence of two differ- a For details of procedures llsed see “Materials-and Methods.”
ent trisaccharide alcohols. These had mobilities identical with h Products from [3H]nigeran.
T1 and T1 trisaccharide alcohol standards prepared both from ̂ ̂partial acid hydrolysates of nigeran (4) and nigeran tetrasaccha- from the mechanism of mycodestrana~e. Since the prol)ortion ride. Based on radioactivity, T1 trisaccharide accounted for of T1 [3H]trisaccharide (459;)), reflects the relative percentage of 75% of the total fraction. The fact that 25cl, of the trisaccharide chains bearing maltosyl units at the reducing terminus, about fraction was the Tz trisaccharide indicates that 25yh of all chains one-half of the chains have a-(1 + 4).linkages at the reducing have or-(1 ---f 4).linkages between the nonreducing terminal and end as isolated (Fig. IO). the penultimate glucose units, under the steady state condition Isolichenin is au unbranched a-glucnn found together with of their isolation from the hyphal wall. Further supporting fi-glucan lichenin in Iceland moss, C&aria islandica. Chemical evidence that these trisaccharides only arise from the ends of the studies (41,44) have shown that the ratio of a-(1,4)- to a-(1,3)- chains, ca,me from an experiment in which separate 2-mg samples linkages in isolichenin is 55:45 and that it contains sequences of of NaB3H4-reduced nigeran and ‘%-uniformly labeled nigeran one to two cr-(1 + 3).linked glucose moieties flanked by a- were digested with 0.1 unit of mycodextranase in .05 M acetate (1 + 4).linkages. This conclusion is based on the fact that T1 buffer, pH 4.5, at 50” overnight and a portion of each reaction and Tz trisaccharides have been isolated from partial hydrolysates mixture chromatographed in Solvent System 1. Analysis of (44) and that a Smith degradation of isolichenin yielded 38% product,s was carried out essentially as described for other oligo- glucosyl erythritol, 430/, nigerosyl erythritol, and only 59; of a saccharides and results are shown in Table XI. While 3yG of compound presumed to be nigerotriosyl erythritol (41). It was
the weight of the digest is trisaccharides (%-monitor), this same concluded that a sequence of 3 a-(1 + 3)-linked units was an fraction contains 459; of t,he 3H-counts. The tritium-labeled important structural feature of this polysaccharide. Initial trisaccharide fraction was resolved by molybdate electrophoresis experiments with mycodestranasa indicated that isolichcnin is and all of the radioactivity was found to migrate with the T1 hydrolyzed as evidenced by the liberation of reducing groups trisaccharide alcohol. The area corresponding to the Tz alcohol measured by method of Park and Johnson (21). An examination standard counted background. This distribution is predicted of the products of the digestion by paper chromatography in
Type A
GPl,3GPl,4G~l,3Gp1,4~Gpl,3Gp1,4Gpl~l,3~ A1
GP~,4G~~,3Gp1,4~Gpl,3Gpl,4Gpl~l,3~ A2
6732 l!dycodextranase Vol. 246, No. 22
by guest on July 9, 2018http://w
ww
.jbc.org/D
ownloaded from

Issue of November 25, 1971 K. K. Puny, A. Rosenthal, and J. H. Nodin 6733
Solvrnt System 2 showed compounds with mobilities identical with nigerose and nigerotriosc, and alho three spots corresponding to higher oligosaccharidcs (Fig. 6). The compound migrating more slow-ly than nigerosc had a mobility identical with nigero- triosc but different from mnlt,otriose and T1 aud Tf trisaccharides. Furthermore, iligeran tttrasacchnride was not present in the isolichcuin hydrolysatc. l%ased on the action pattern of ~nyco-
.destrallase, the polysncch:aride does Ilot, therrfore, contain any segments with 3 consecutive nigeiose units joined by two ~(1 ,4)- linkages (nigeran hesusaccharide) or nonreducing terminal seg- nmlts equivalent to a nigeran pentasaccharide with a 1,3-linkage at the nonreducing terminal, but does contain a majority of segmruts with one or two consecutive ~(1 + 3)-linkages sur- rounded with ~(1 4 4)Jinkages. These results essentially confirm those obtained with the Smith degradation (41). The :tbst,nce of nigerotetraosc from the enzymatic hydrolysates makes it unlikely that there are any segments containing 4 or-(1 + 3)- linked urlits and the absence of glucose in these hydrolysates is again consistent with the action pattern of mycodextranase. The larger fragments of the hydrolysis mixture ww not examined further.
UISCUSSION
The evidence presented regarding the mode of action of myco- destranase suggests certain similarities to that of lysoz) me. Situdits by Phillips showed that the lysozyme-binding site can accolllmodate, in a specific manner, a hexasaccharide unit COE sisting of alternating molecules of A-acetyl glucosamine and N-ncrtyl muramic acid joined in p-(1 + 4)-glucosidic linkage (45). Results of the l)rcscnt studies indicate mycodextrauase prefers to bind an octasaccharide unit of nigeran. While the hexose units are identical in this substrate, the linkages alternate. Studies with the hesasaccharide hexa-N-acetyl chitohexaose indicate that a specific linkage is preferentially hydrolyzed by lysozyme. Cleavage of that linkage produces a disaccharide from the reducing end of the hesasaccharide (46). Hydrolysis of t,hc hexasaccharide alcohol of nigeran by mycodestranase also produces a disaccharide from the reducing end of t.hat substrate. Thus chemical studies with both enzymes indicate that the bind- ing sit,cs accommodntc 4 hc~sosyl units adjacent to the cleavage site.
Of the two general ~l:~+cs of glycanases, endo-enzymes (e.g. a-amylase) hydrolyze their I)olysaccharide substrates randomly while cso-enzymes (e.g. fi-amylasr) remove end products se- quelltially from the nonreducing tcrmini of the polymer chains (34). Each type could possibly eshibit one of three types of mechanisms during digestion of their substrates: Mechanism 1, multiple attack, the enzyme substrate contact esists until several attacks have been made on the polysaccharide chain before another chain is attacked; Mechanism 2, multichain mrchanism, wherein the enzyme-substrate complex lasts for a period of time sufficicilt to allow the clravage of only one linkage in the poly- snrcharide chain; Mechanism 3, air& chain attack (an extreme case of multiple attack), once the enzyme-substrate complex has been formed, the enzyme c~omplrt~ly hydrolyzes one entire poly- sacchnride chain before it attacks another. A41though Mecha- nisnls 1 and 2 have btcn obsclrtd with a number of glyranascs, none operating by a single chain mechanism has been reported. While endo enzymes usually operate via a multichain mechanism, Robyt and French propobrtl an endo-multiple attack mechanism to explain the partial dual products specificity of the CIK~O-a-
amylase of Bacillus subtilis (12). A primary random endo cleavage of the amylose chain occurs and the substrate then “slides” over the binding site and a second bond is broken libtr- sting an oligosaccharide (this concept has been proven unequivo- cally with model substrates (47)). Therefore, a single random endo-cat,alytic event is followed by a series of “eso” attacks on the new nonreducing terminal ends of the chain. The Ilumber of such scoondary attacks and the degree of polymerizatflion of the products is a function of both the “multiplicity” of attack and the size of the bindiug site. The product distribution would reflect these catalytic processes. Although mycodextranase displays estremely rigorous dual products specificity, the results presented in this paper do not support an endo-multiple attack mechanism of mycodestranase, but do support au endo-multi- chain mechanism (Fig. 8 and Table IX). The lack of glucose production by mycodextranase, an unusual feature for an cndo- glucanasc, is undoubtedly caused by both the restriction iml)osed on mycodestranase by nigeran’s st,ructure and the enzyme’s specificity for ~(1 + 4).linkages.
On the basis of data in Tables VI and X, a 4-, 6-, or 8-u& binding site was considered for mycodextranase. -4 site larger than that required to bind 8 glucose units does not seem likel! since the enzyme preferentially hydrolyzes the bond 4 units from t,he nonreducing terminal in both hesa- and octasaccharide alcohols. Furthermore, the results of these experiments suggest a binding site that positions 4 glucose units to the left of the catalytic site. Represent&ions of the three various sized bind- ing sites are shown in Fig. 11, A, B, and C. The locations of all possible catalytic sites for each type of binding site arc rcprc- sentcd by the lower case letters and cu,rl:ed arrows (see Fig. 11 for details).
In order to interpret these results and postulate a mechanism, some assumptions have been made. (a) The enzyme cleaves only (1 + 4)-linkages (this is documented by specificity evidence prc- sented above). (b) There is only one catalytic site in each sub- strate-binding site. (c) Th e substrate only fits on the binding site in one dir&ion (i.e. its reducing terminus ifi always to the right in the diagrams as of Fig. 11). (d) Any portion of the binding site that, accommodates 2 hexose residues separated by an ~(1 4 3).linkage cannot accommodate 2 glucose residues linked a-(1 + 4) (see Fig. 11). (e) The more glucose units posi- tioned in the binding site, the more reactive will be that particular enzymt-substrate (ES) complex (12, 46). For esample, if the structure of the enzyme’s binding site is as presented in Fig. 11, C and D, it is assumed that catalysis will proceed almost esclu- sively via the type C complex since more of the substrate molecule is in contact with the binding site in this case. This assump- tioll is consistent with the rigid specificity of end product distri- but,ion shown by mycodextranase. Considering each possible clcavngc site independently, a 4-unit site with a type ‘(a” cata- lytic site (Fig. lln) could provide for both the observed product and radioactivity distributions observed with [311]hexasaccharide and octasaccharide alcohols and not facilitate the hydrolysis of tetl,as:rcchnride. h type “b” site would be expected to permit hydrolysis of tetrasaccharide. Furthermore, this cleavage site would, when the binding site was filled, give a product distribu- tion from hydrolysis of [3H]hesasaccharide alcohol that was a mixture of [3H]tetrasaccharide alcohol and t3H]nigeritol since this substrate can be positioned in two ways which will lead to cataly- sis with a type “b” site. These possibilities are inconsistent with
by guest on July 9, 2018http://w
ww
.jbc.org/D
ownloaded from

6734 Mycodextranase Vol. 246, No. 22
A Four Unit Site
B Six Unit Site
c Eight Unit Site
D
FIG. 11. Models of proposed substrate-binding sites of myco- dextranase. The curved arrows and accompanying lower case letters represent hypothetical cleavage (catalytic) sites. 4, reducing
terminal gIycosyI unit; O--O, D-gIucopyranosyI-(1 -% 4)-D-
/'I glucopyranosyl; 0 0, D-glucopyranosyl-(1 3 3)-D-glucopyran- osyl. The specificity of the binding site for alternating a-(1+4)- and a-(1+ 3)Jinked glycopyranosyl units is indicated by the two different shapes of the binding site coincident to the two different linkage representations.
the experimental observation (Table X). Similarly a type “c” cleavage site would be predicted to give only [3H]tetrasaccharide alcohol as a product of [3H]hexasaccharide alcohol hydrolysis. There are three ways to position the [3H]octasaccharide alcohol in the 4-unit site and still fill it completely. However, one of these positions will not yield a cleavage via a type “a” cleavage site. The data show that the central ~(1 + 4)linkage of [3H]octa- saccharide alcohol is highly susceptible to cleavage while the other two are not. Since mycodextranase is an endo enzyme that hydrolyzes nigeran ~(1 + 4)linkages randomly, there is no apparent requirement for a free 4’.hydroxy group on the glucose unit 4 units removed from a type “a” catalytic site. Therefore, one would not expect a 4-unit site to discriminate between the two catalytically productive arrangements of [3H]octasaccharide alcohol which could fill the site. However, the fact that such a preference exists further weakens any argument for a 4-unit binding site.
A g-unit site as pictured in Fig. 11B with a type “b” cleavage site will, when filled, account for the results obtained in the digestion of [3H]hexasaccharide and octasaccharide alcohols since the position of the cleavage site is such that [3H]nigeritol and tetrasaccharide will be the sole products of 13H]hexasaccharide hydrolysis and tetrasaccharide and [3H]tetrasaccharide alcohol the products of [3H]octasaccharide hydrolysis. However, when this sized binding site is completely filled, cleavage sites “a” and “d” are unable to yield any products. A type “c” site is not favored since the enzyme does not produce any [3H]tetrasaccha- ride alcohol by its action on the [3H]hexasaccharide alcohol (Table X). For a type “c” site to give the observed distribution of products from [3H]hexasaccharide alcohol the binding site would have to be only partially filled during catalysis. Similarly, sites ‘la” and “d” would only be expected to yield the observed product distribution when partially filled. Since there are two
ways in which ]3H]octasaccharide alcohol can be positioned in the 6-unit binding site with a type “b” cleavage site and since the enzyme’s endo-multichain mechanism does not suggest the need for a free 4’-hydroxy group 4 units removed from the catalytic site (in order to have a catalytic event), it would be expected that this site would provide for an equal distribution of 13H]tetrasaccharide alcohol and [3H]nigeritol from [3H]octasac- charide alcohol. This is not the result obtained since 98yG of the cleavages occur at the central ~(1 + 4)-linkage of the [3H]octasaccharide alcohol. An S-unit binding site with a type “c” cleavage site (Fig. 11C) will explain the hydrolysis data with tetra, hexa, and octasaccharides. Such a binding site with type “a” or “e” cleavage sites would lcad to no catalytic events when the binding site is filled and type “d” would give a product distribution which differs from that observed with [3H]hexa- and octasaccharide alcohols. A type “b” cleavage site could give the observed product distribution from [31-I]hexasaccharide alco- hol but it cannot provide for the observed distribution with [3H]octasaccharide alcohol. Therefore, an R-unit binding site with a cleavage site that attacks the center (Y-(1 + 4)-linkage of the octasaccharide appears most likely.
Product specificity of the [3H]hexasaccharide alcohol hydrolysis also suggest that the enzyme requires more than a nigerosyl unit (viz. a tetrasaccharide unit) to the left of the cleavage site for catalysis to occur but does not require more than a nigerosyl unit to the right of the cleavage site. The fact that [3H]octasaccha- ride alcohol hydrolysis yields very little [3H]nigeritol argues against a specific requirement for more than 4 hexosyl units being bound to the left of the enzyme’s cleavage site during catalysis. I f 6 units were required, it is predicted that [3H]nigeritol would be a significant product with this model com.pound. Therefore, a site binding 10 units or more seems unlikely on the basis of the results obtained. The remote possibility of two identical but independent cleavage sites on the same binding site (e.g. “c” and “d” together on an &unit site) is ruled out since this would have to result in an equimolar distribution of [3H]tetramer alcohol and [3H]nigeritol front [3H]hexasaccharide alcohol hy- drolysis. The fact that tetrasaecharide is a very poor substrate for the enzyme is probably caused by the fact that a normal positioning in the substrate-binding site provides for no catalysis by a type “c” cleavage site. Occasional complexes probably form such that the tetrasaccharide’s ~(1 + 4).linkage is positioned opposite the type “c” site and cleavage can occur.
Based on studies with a number of p-(1 + 3). and p-(1 + 4). glycanases, Perlin summ.arized some general observations re- garding their action patterns (13). The endo-fi-glycanases studied were found not to be specific for the linkages which they split but for the glycosidic linkage to the right of a special disac- charide (the reducing end of the molecule is visualized as being at the right hand terminus). In general, the nature of the sub- stitut,ion on the glycosyl unit being cleaved determines the specificity of the glycanase. With model substrates, Perlin found a cellulase capable of cleaving any linkage to the right of a cellobiosyl unit provided the glucose unit bearing the new potential reducing group had its 4’-hydroxy group substituted with a P-linked unit. These observations led Reese and Mandels to propose that because of the product distribution with myco- dextranase, instead of a disaccharide unit dictating specificity a tetrasaccharide unit probably specifics the linkage to be cleaved (6). The data presented in Table X support this t,heory. There is only one ~(1 + 4).linkage at the right of a tetra-
by guest on July 9, 2018http://w
ww
.jbc.org/D
ownloaded from

Issue of November 25, 1971 K. K. l’ung, A. Rosenthal, and J. H. Nordin 6735
saccharide unit in [3H]hexasaccharide alcohol but two to the right of the disaccharide unit at its nonreducing terminus. Since nigeritol is the sole radioactive hydrolysis product, the ~(1 -+ 4)-linkage to the right of the tetrasaccharide unit is the only one susceptible to cleavage in this molecule. However, because the enzyme will not hydrolyze amylose or pseudonigeran, consecutive ~(1 ---f 4) and ~(1 + 3).glycosydic linkages are also required in the substrate for significant binding and cleavage to occur.
Nigeran could possibly be synthesized by two mechanisms. The first would occur by the addition of single glucose units with alternating linkages, probably via two different transferases. The second would occur via insertion of a preformed disac- charide unit (e.g. maltosyl or nigerosyl) in each synthetic step but with the other linkage separating them (48). The simplest mechanism that will explain this observed distribution involves the transfer of single glucosyl units to the growing chain by two synthetases each specific for joining glucosyl units via either a 1,4- or a 1 ,3-linkage but each requiring the presence of the alternate linkage between the glucosyl units immediately penultimate to the new linkage being synthesized in the acceptor chain. If donation of previously formed disaccharide units (either maltosyl or nigerosyl) to the growing chain were to occur via a single transferase, then one would predict that all chains would have the same disaccharide unit at the reducing end. The observed distribution of terminal 1,3- and 1,4-linkages would require an extra set of enzymes for the synthesis of the alternate chains if it were to occur by the latter route. The observed distribution of maltosyl and nigerosyl moieties at the reducing term.inus of nigeran (about 1: 1) would indicate that de no00 initiat.ion of chains occurs with either a 1,3- or 1,4- linkage being formed. Since glgcanases do not degrade polysac- charides from the reducing end and nigeran in vivo is buried in the cell wall matrix (7), it is assumed that the structures at the reducing end of the molecule reflect the pattern of their synthesis only.
Mycodextranase has proven to be an extremely useful tool for a number of specific biochemical applications. It has been used for analyzing the content of nigeran in fungal cell walls, without prior separation of the polymer from the remainder of the mycelial material (7), and for the preparation in gram quantities of nigerose, tetrasaccharide, and a homologous series of nigeran oligosaccharides varying in degree of polymerization of 2 glucose units (11). Coupled with the use of the controlled periodate oxidation and acetolysis techniques developed by Saier and Ballou (15), it should be possible to generate from nigeran oligosaccharides a family of oligosaccharides with an odd number of glucose units since t,his technique only removes the reducing terminal unit from an oligosaccharide. For example, the tetra- saccharide could serve as a starting material for the generation of the trisaccharide 0-c-n-glucopyranosyl-(1 -+ S)-O-oc-n-glyco- pyranosyl-(1 + 4)n-glucose in far higher yield than would be possible by acid hydrolysis. For use in these various applica- tions there is no reason to separate the fast and slow isozymes since their action patterns are identical.
REFERENCES
I. TUNG, K. K., Fed. Proc., 28, 904 (1968). 2. BARICER, S. A., BOURNE, E. J., AND STACEY, hl., Chem. Ind.
(London), 756 (1952).
3. BARKER, S. A.. BOURNE, E. J., AND STACEY. M.. J. Chem. Sot.. 3084 (i953).
I ,
4. BARICER, S. A., BOURNE, E. J., O’MANT, 1). M., AND STACEY, M., J. Chem. Sot., 2448 (1957).
5. YUIL~, J. L., Chem. Ind. (London), 755 (1952). 6. REESE, E. T., AND MANDELS, M., Can. J. Microbial., 10, 103.
(1964). 7. TUNG, K. K., AND NORDIN, J. H., Biochem. Biophus. I&s.
Commun., 28, 519 (1967). ̂ I
8. HASEGAWA. S.. NORDIN. J. H.. .~XD KIRICWOOD. S.. J. Biol. Chem., 244, i460 (1969). ’
~3 I
9. HUTSON, D. H., AND MANNERS, D. J., Biochem. J., 94, 783 (1965).
10. TUNG, K. K., AND NORDIN, J. H., Biochim. Biophus. Acta, 168, 154 ‘(1968):
- 1 .
11. TUNG, K. K., AND NORDIN, J. H., Anal. Biochem., 38, 164 (1970).
12. R~BYT,. J. F., AND FRENCH, D., Arch. Biochem. Biophys., 100, 451 (1963).
13. PERLIN, A. S., in E. T. REESE, (Editor), Advances in enzymic hydrolysis of cellulose and related materials, Macmillan Company, New York, 1963, p. 185.
14. BARKER, S. A., Methods in carbohydrate chemistry, Vol. 5, Academic Press, New York, 1965, p. 165.
15. SAIER, M., AND B~LLOU, C., J. Biol: Chem., 243, 4319 (1968). 16. BLOCK, R. J., DURRUM, E. L., AND ZWEIG, G., Paper chroma-
tography and paper electrophoresis, Academic Press, New York, 1958, p.176. _
17. BOURNE, E. J., HUTSON, D. H., AND WEIGEL, J. Chem. Sot., 34 (1961).
18. TR&E~YAN, W. E., PROCTOR, D. P., AND HARRISON, J. S.,
19.
20. 21.
22.
23.
24.
25. 26.
27.
28.
29.
30.
31. 32.
33.
34.
35.
36.
37.
38.
39.
40.
Nature, 166, 444 (1950). BOBBIT, J. M., Thin layer chromatography, Reinhold Book
Corporation, New York, 1963, p. 84. SOMOGYI, M., J. Biol. Chem., 160,69 (1945). PARIC. J. T.. AND JOHNSON, M. J., J. Biol. Chem.. 181. 149
(1949). ’ ,
DUBOIS, M., GILLIS, K. A., HAMILTON, J. K., REBERS, P. A., AND SMITH. F., Anal. Chem., 28, 350 (1956).
GORNALL, A: G., BARDAWILL; C. J., AND DAVID, M. M., J. Biol. Chem., 177, 751 (1949).
LOWRY, 0. H., ROSEBROUGH, N. J., FARR, A. L., AND RANDALL, R. J., J. Biol. Chem., 193, 265 (1951).
YPHAN& D. A., Biochemistry, 3, 297 (1964). SOBER. H. A. (Editor). Handbook of biochemistru. Chemical
Rubber Company, Cleveland, Ohio, 1968, p. C-36. TANFORD, C., KAWAHARA, K., AND LAPNJE, S., J. Biol. Chem.,
241, 1921 (1966). ORNSTEIN, L., AND DAVIS, B. J., Ann. N. Y. Acad. Sci., 121,
404 (1964). REISFELD, R. A., LEWIS, W. J., AXD WILLIAMS, D. E., Nature,
196, 281 (1962). CHARAMBACH, A., REISFELD, 1~. A., WYCICOFF, M., AND ZAC-
CHARI, J., Anal. Biochem., 20, 150 (1967). KAPLAN, L. J., AND FOSTER, J., Biochemistry, 10, 630 (1971). HIRS, C. H. W., in C. H. W. HIRS (Editor), Methods in en-
zymology, Vol. XI, Academic Press, New York, 1967, p. 59. SPANDE, T. F., AND WITI<OP, B., in C. H. W. HIRS (Editor),
Methods in enzymology, Vol. XI, Academic Press, New York, 1967, p. 498.
ROBYT, J. F., AND FRENCH, D., Arch. Biochem. Biophys., 122, 8 (1967).
TUNG, K. K., AND NORDIN, J. H., Anal. Biochem., 29, 84 (1969).
DIXON, M., AND WEBB, E. C., Enzymes, Academic Press, New York, 1964, p. 329.
MANDELS, M., PARRISH, F. W., AND REESE, E. T., J. Bacterial., 83, 400 (1962).
SPECTOR, W. S. (Editor), Handbook of biological data, W. B. Saunders Company, Philadelphia, 1961, p. 24.
CAMPBELL, L. L., .~ND MANNING, G. B., J. Biol. Chem., 236, 2962 (1961).
MANNING, G. B., AND CAMPUELL, L. L., J. Biol. Chem., 236, 2952 (1961).
by guest on July 9, 2018http://w
ww
.jbc.org/D
ownloaded from

6736 Mycodextranase Vol. 246, No. 22
41. FLEMING, M., AND MANNERS, D. J., Biochem. J., 100, 24p 45. PHILLIPS, D. C., Proc. Nut. Acad. Sci. U. S. A., 67, 484 (1967). (1966). 46. RUPLEY, R. U., AND GATES, V., Proc. Nat. Acad. Sci. U. S. A.,
42. HUGETT, A. S., AND NIXON, D. A., Lczncet, 368 (1957). 67, 496 (1967). 43. RAMMLER, D. H., Ann. N. Y. Acad. Sci., 141,291 (1967). 47. ABDULLAH, M., FRENCH, D., AND ROBYT, J. F., Arch. Biochem. 44. PEAT, S., WHELAN, W. J., TURVEY, J. R., AND MORGAN, K., Biophys., 114, 595 (1966).
J. Chem. SOL, 623 (1961). 48. ROBBINS, P. W., AND WRIGHT, A., Science, 168, 1536 (1967).
by guest on July 9, 2018http://w
ww
.jbc.org/D
ownloaded from

K. K. Tung, A. Rosenthal and J. H. NordinGLUCANASE FROM PENICILLIUM MELINII
4)→-d-(1 αAND PROPERTIES OF MYCODEXTRANASE, AN ENDO-Enzymes That Hydrolyze Fungal Cell Wall Polysaccharides: II. PURIFICATION
1971, 246:6722-6736.J. Biol. Chem.
http://www.jbc.org/content/246/22/6722Access the most updated version of this article at
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/246/22/6722.full.html#ref-list-1
This article cites 0 references, 0 of which can be accessed free at
by guest on July 9, 2018http://w
ww
.jbc.org/D
ownloaded from