establishing and applying critical quality attributes ... · establishing and applying critical...
TRANSCRIPT

1 www.fda.gov
ISCT Liaison Meeting
October 19, 2016
Tom Finn, Ph.D.
Product Reviewer
Office of Tissues and Advanced Therapies
FDA/CBER
Establishing and Applying Critical
Quality Attributes During the
Product Development Lifecycle

2
Topics
• Product terminology
• CQA and CPP and how they are
developed and used during the
product lifecycle
• Considerations for demonstrating
product comparability after a
manufacturing change
3
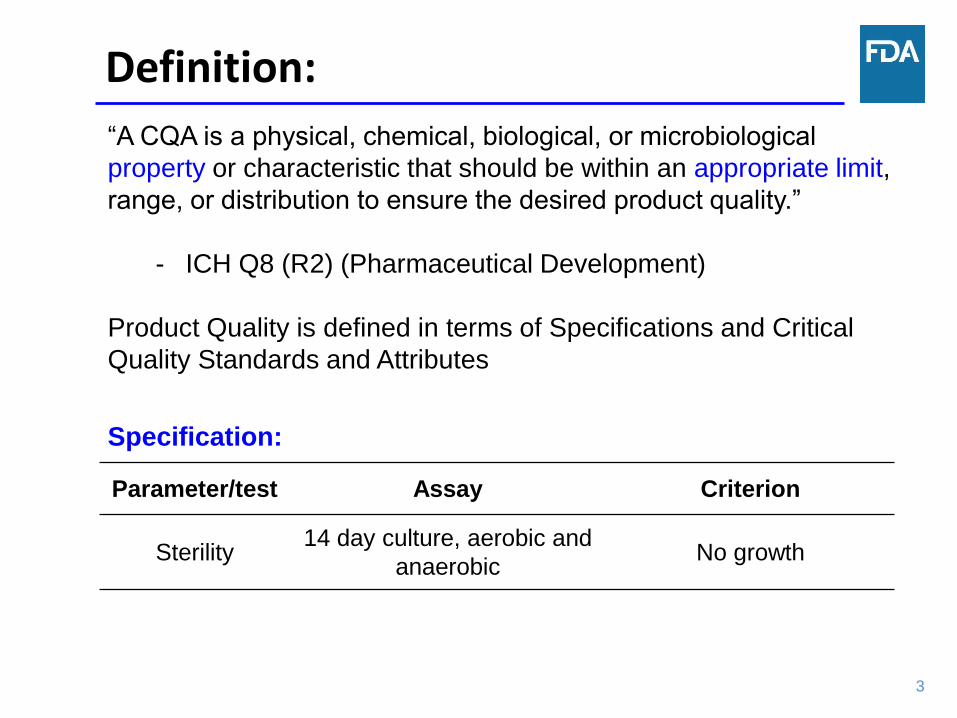
“A CQA is a physical, chemical, biological, or microbiological
property or characteristic that should be within an appropriate limit,
range, or distribution to ensure the desired product quality.”
- ICH Q8 (R2) (Pharmaceutical Development)
Product Quality is defined in terms of Specifications and Critical
Quality Standards and Attributes
Definition:
Parameter/test Assay Criterion
Sterility 14 day culture, aerobic and
anaerobic No growth
Specification:

4
Definition:
specifications Full product characterization
“Specifications are critical quality standards (CQAs) that are
proposed and justified by the manufacturer and approved by
regulatory authorities… Specifications are chosen to confirm the
quality of the DS and DP rather than to establish full
characterization, and should focus on those characteristics
found to be useful in ensuring
the safety and efficacy
of the DS and DP.”
- ICH Q6B and Q11
CQA

5
BLA Phase III Phase II Phase I Preclinical Research
Clinical efficacy
Potency
Preclinical efficacy
But efficacy data is not usually obtained until late in product development,
long after CQAs have been established
• You should make use of preclinical and clinical data whenever
available to adjust CQA and CPP parameters. This is easier to do if
two phase 3 studies are done- revise CQA prior to conducting
confirmatory trial.
• Although not required until phase 3, we recommend you develop a
potency assay as early as possible.
The more reflective CQA are of clinical safety
and efficacy, the easier it is to evaluate the
consequences of a manufacturing change
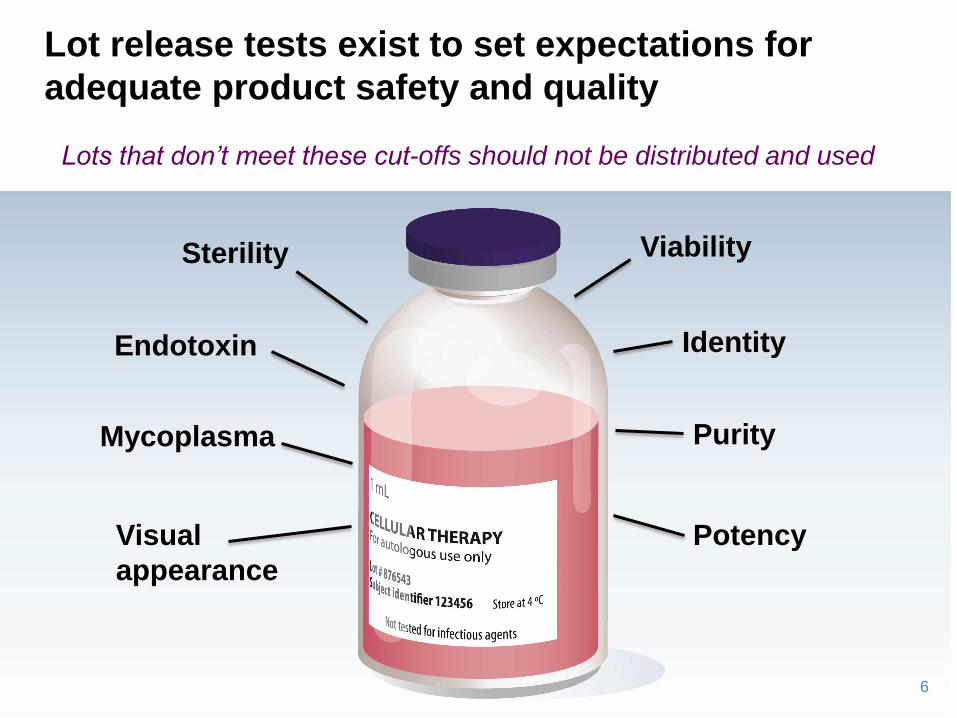
Sterility
Endotoxin
Mycoplasma
Viability
Identity
Purity
Visual
appearance
Potency
Lots that don’t meet these cut-offs should not be distributed and used
Lot release tests exist to set expectations for
adequate product safety and quality
6
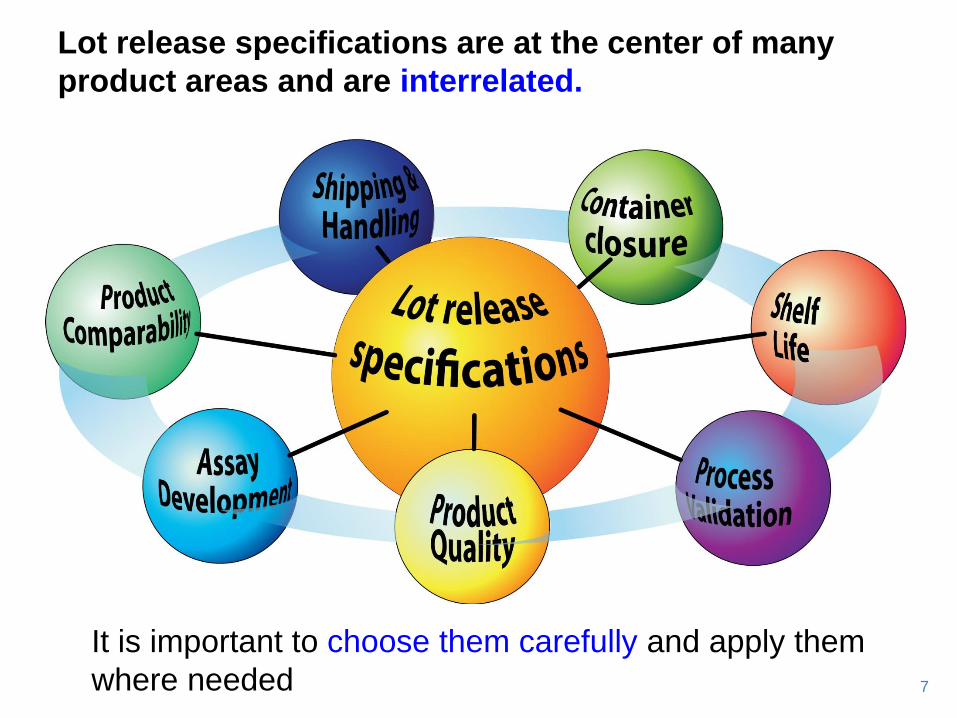
It is important to choose them carefully and apply them
where needed
Lot release specifications are at the center of many
product areas and are interrelated.
7
8
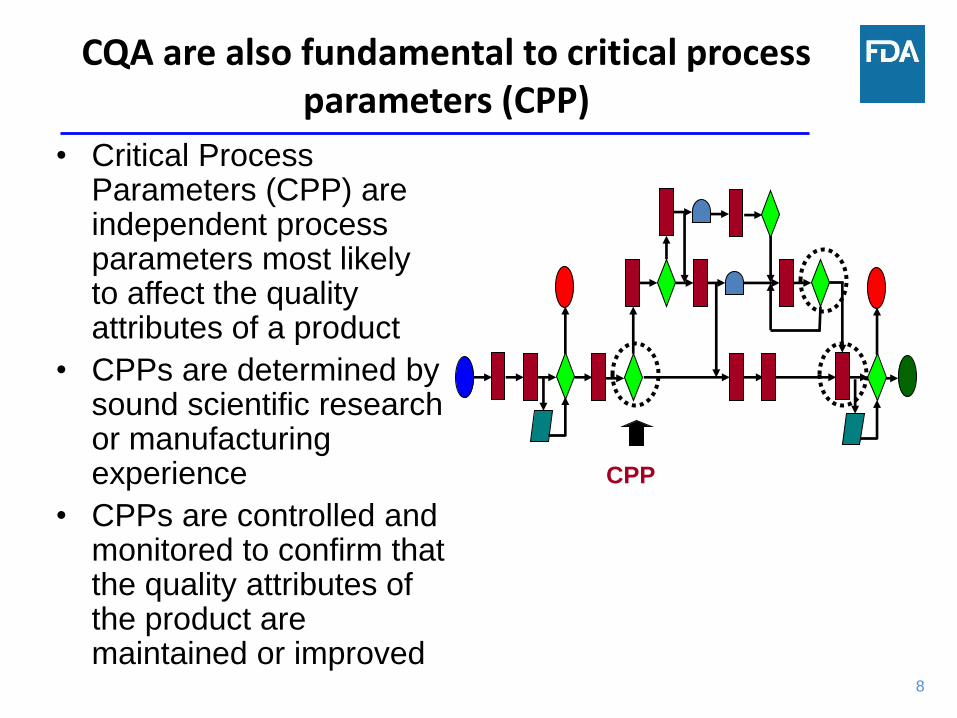
CQA are also fundamental to critical process parameters (CPP)
• Critical Process Parameters (CPP) are independent process parameters most likely to affect the quality attributes of a product
• CPPs are determined by sound scientific research or manufacturing experience
• CPPs are controlled and monitored to confirm that the quality attributes of the product are maintained or improved
CPP
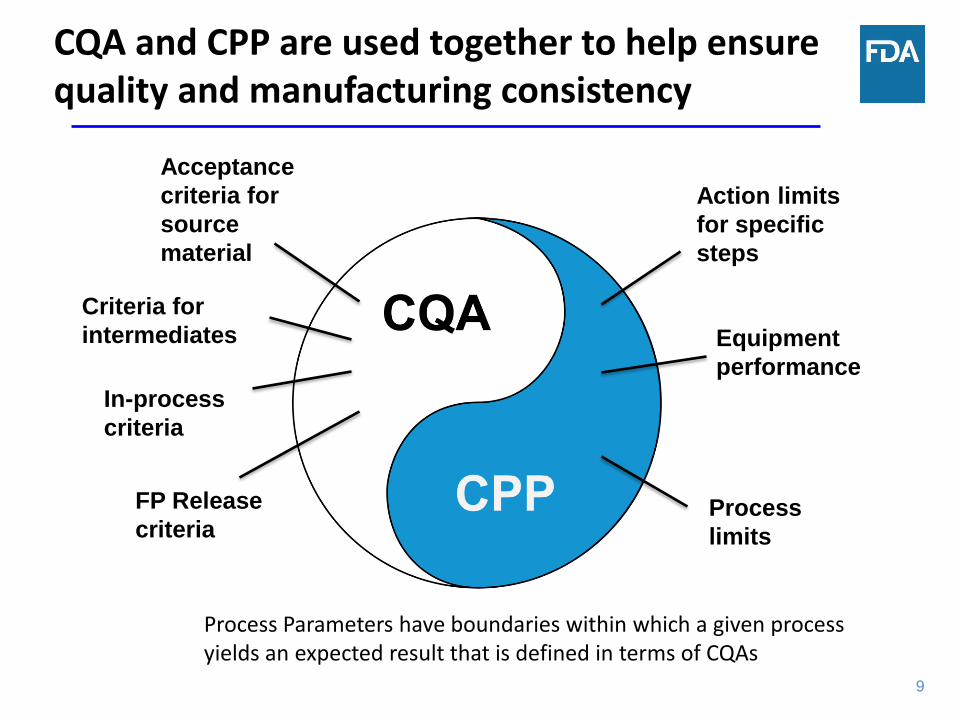
9
CQA and CPP are used together to help ensure quality and manufacturing consistency
In-process
criteria
Process
limits
Acceptance
criteria for
source
material
FP Release
criteria
Equipment
performance
Action limits
for specific
steps
Criteria for
intermediates
Process Parameters have boundaries within which a given process yields an expected result that is defined in terms of CQAs
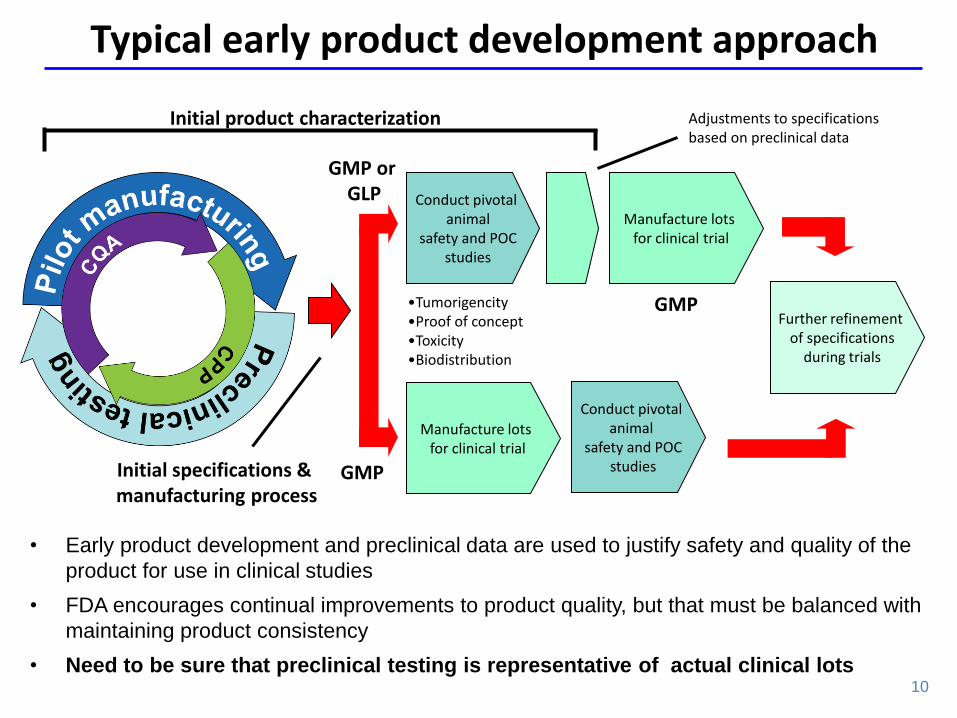
• Early product development and preclinical data are used to justify safety and quality of the
product for use in clinical studies
• FDA encourages continual improvements to product quality, but that must be balanced with
maintaining product consistency
• Need to be sure that preclinical testing is representative of actual clinical lots
Typical early product development approach
Initial product characterization
Initial specifications & manufacturing process
Conduct pivotal animal
safety and POC studies
Manufacture lots for clinical trial
Further refinement of specifications
during trials
Adjustments to specifications based on preclinical data
•Tumorigencity •Proof of concept •Toxicity •Biodistribution
Manufacture lots for clinical trial
Conduct pivotal animal
safety and POC studies
GMP or GLP
GMP
GMP
10

11
• Proposed mechanism of action
• What properties or characteristics of the product (i.e.,
quality attributes) are likely to achieve the intended action
in the patient?
• What manufacturing steps are critical and how would you
measure and ensure they were successful? -identify
process parameters
• What safety concerns are associated with this type of
product
• What undesirable properties do you want to minimize?
• What labeling claims do you want to make based on
identified attributes and/or process parameters?
• Assay suitability & qualification
Factors important for establishing
CQA and CPP
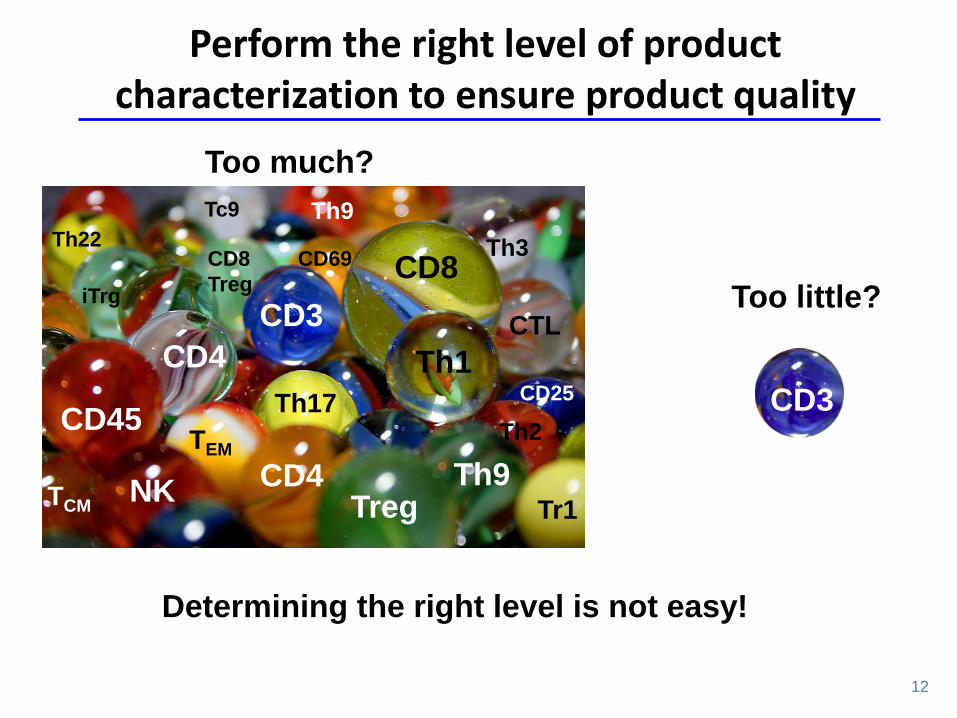
CD3
Too little?
Too much?
Perform the right level of product characterization to ensure product quality
Determining the right level is not easy!
CD3
CD8
CD3
CD45
Th1
Treg CD4
CD4
NK
Th2
Th9
CTL
CD8
Treg
Th3
Th17
Tr1
TEM
TCM
Th9 Th22
iTrg
Tc9
CD25
CD69
12

Impractical
Too much
emphasis on a
single attribute
Appropriate attributes,
criteria too low Appropriate attributes,
appropriate criteria
Focus on critical quality attributes
13

Common issues with choosing product
release specifications
• Specifications not capturing key product attributes
• Criteria inconsistent with manufacturing experience
• Lack of supportive data or rationale
• Product characterization that does not take into account
cellular impurities that might interfere with the activity of
the product, or present a safety concern
• Criteria set for a very wide range – could add variability
to clinical trial outcomes
• Misinterpretation or over-interpretation of data
14

15
Assays are sometimes qualified/validated under ideal or
best case conditions, and may factor only one variable at
a time. This can lead to overconfidence of an assay. Real
world use may involve:
Consider assay variability and “worst case” in designing assays and setting criteria
• Different QC analysts
• Different batches of reagents
• Different equipment
• Samples held for different
lengths of time
• Different interpretation of
procedures due to vague
SOPs
• Subjective parameters (such
as flow cytometry gates,
background cut offs, dilutions,
etc.)
Assay variability can confound efforts to demonstrate manufacturing consistency, comparability, or stability.

16
CQA and CPP are not meant to be static- they should
be continually evaluated and revised as needed
Carved in stone Continually upgrading
• Changes to CQA could include either revising existing criteria, or adding or
removing a specification (as supported by product characterization data)
• But since these have tremendous impact, revise cautiously!
• Additional product characterization data may indicate a better way of
ensuring quality
• Clinical outcome data may provide clues as to what product properties are
the most important
• Additional manufacturing experience may guide CPP and CQA

17
Major manufacturing changes

A little planning up front can help avoid problems later
Think in advance about: • Donor eligibility of source
material • Cell bank qualification • Cell bank capacity • Logistical issues for
products with short shelf lives
• Scale up needs • Second source for custom
or critical materials • Qualification & validation
18

19
It is easier to accommodate manufacturing
changes at earlier developmental stages
• Product knowledge should increase with stage of development
(identity, stability, potency, manufacturing, consistency/product
comparability, etc.)
• Consider manufacturing changes that might be needed to
accommodate larger trials and commercial production
• Manufacturing changes can be implemented at any stage, but
the potential impact of a manufacturing change can increase
the farther you are along in the product lifecycle.
Phase 1 & 2 may be a good time to implement a
major manufacturing change prior to conducting
pivotal phase 3 studies. However, for these
phases manufacturing is often “on autopilot”

20
Phase 3 is a little like commercial
manufacturing on training wheels
• Should be using as close to the commercial process as
is feasible for registration studies
• Potency should be in place
• Critical Quality Attributes (CQA) should
be identified and appropriate assays in
place
• Additional stability data should be
collected
• Well defined CPPs should be in place:
Phase 3 is critical for demonstrating
manufacturing consistency
• But some details are still being
worked out to prepare for
commercial production

Depending on the
issue or the study,
there may be the
need to extend an
analysis to
product properties
beyond lot release
values
It is important to
understand where
the gaps exist so
that an
appropriate
characterization
can be done
As important as lot release specifications
are, they alone only provide a partial
assessment of quality
21

Situations where additional product
characterization and analysis may be needed
• Process qualification and validation studies (to
demonstrate manufacturing consistency)
– Additional in-process and final product attributes, yield
• Comparability studies after a major manufacturing change
(e.g. new process step, new facility, new critical reagent,
etc.)
– Additional measures of identity, potency, purity, etc.
– Yield
• Stability studies (not all lot release tests are stability
indicating- you need to evaluate each one)
– Genetic stability and identity of cell lines
– Evaluate apoptosis in addition to viability
– Additional measures of potency 22

23
Goalpost
CQA are often used as goalposts
Applications for lot release
• Establish a lower limit for key
attribute
• Maximum limit of an impurity
• Establish an allowable range

24
There are advantages to targeting narrow versus wide tolerances for
specifications
Narrower tolerances make it easier to assess comparability
Size matters
Goalpost Narrow tolerances Wide tolerances
Need to have a very good
understanding of your
process and product, with
sufficient control points
Difficult to rely on just lot release
specifications to show consistency and
comparability

25
You should aim like this…
CQA as a goalpost
Not this…
When a product has substantial inherent variability you need to
consider what you are targeting
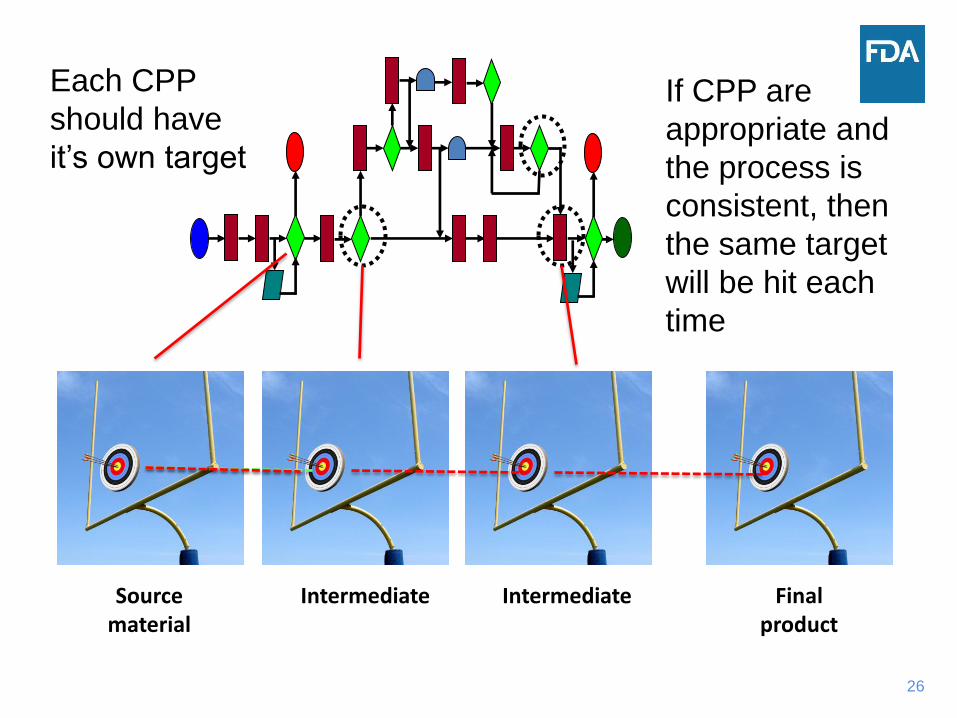
26
Each CPP
should have
it’s own target
If CPP are
appropriate and
the process is
consistent, then
the same target
will be hit each
time
Source material
Intermediate Intermediate Final product

Designing meaningful comparability studies
• Perform risk assessment to establish scope- what is most likely to be
affected and to what degree?
• Consider what are the most sensitive parameters to test
• What assumptions are you making?
• Leverage what you already know from product development
• Where does known variability exist and how will you try to control for
that?
• Justify # of samples, types of samples, number of tests, and type of
analysis
• What limits does the study design place on interpretation?
• How will you analyze the data?
27

28
• First show the current process is consistent
• Where possible use a split manufacturing
approach to factor out source material
variability- this allows for a head-to-head
comparison of the new method compared to
the existing method
• Criteria should be predetermined
• Understand and incorporate worst case
Additional thoughts

What to include in a manufacturing
change IND amendment
• Clear description of what you are changing and why
• Change control- what is the scope of the change and what was
impacted
• A summary of your risk assessment
• Whether this is intended to be a temporary fix or a long-term
solution
• Comparability study, including:
– Design of the study (with justification for sample size)
– Justify relevant CQAs and test methods
– Risk assessment identifying the type and level of impact
– Rationale for acceptance criteria
– How the study was executed
– Data demonstrating an acceptable level of product
comparability
29

30 www.fda.gov
• Think carefully about what you are expecting clinically of your
product and work backwards
• Think of all the CMC parameters that are relying on CQA and
CPP and factor those into your specifications and action limits
• Reset/Refine your release specifications and your “goalposts”
after you’ve identified key sources of variability in your process
and have taken steps to control them
• Choose assays that are suitable for assuring product quality, with
adequate sensitivity and specificity. Factor assay variability into
your specifications
• First show your existing process is consistent, then show after a
manufacturing change your product is comparable
• CQA and CPP should be continually evaluated and revised as
needed based on multiple identified attributes and process
parameters
• Manufacturing changes are inevitable, but they are easier to
accommodate early in product development, so plan ahead
Take home messages

OTAT Contact Information
For product questions please contact:
Tom Finn at [email protected]
Regulatory Questions: Contact the Regulatory Management Staff in OTAT at [email protected] or by calling (240) 402-8361
OTAT Learn Webinar Series: http://www.fda.gov/BiologicsBloodVaccines/NewsEvents/ucm232821.htm
www.fda.gov

CBER website:
http://www.fda.gov/BiologicsBloodVaccines/default.htm
Phone: 1-800-835-4709 or 301-827-1800
Consumer Affairs Branch (CAB)
Email: [email protected]
Phone: 301-827-3821
Manufacturers Assistance and Technical Training Branch (MATTB)
Email: [email protected]
Phone: (240) 402-8010
Follow us on Twitter
https://www.twitter.com/fdacber
Public Access to CBER
www.fda.gov