ethanol 2g: development of a methodology to evaluate the ... · ethanol 2g: development of a...
TRANSCRIPT

Ethanol 2G: Development of a methodology to evaluate
the morphology of lignocellulosic substrates
Ana Sofia Brazão Borrego
Thesis to obtain the Master of Science Degree in
Chemical Engineering
Supervisors: Dr. Damien Hudebine
Dr. Nadège Charon
Prof. João Carlos Moura Bordado
Examination Committee
Chairperson:
Supervisor:
Member of the Committee:
Prof. Sebastião Manuel Tavares Silva Alves
Prof. João Carlos Moura Bordado
Dr. Maria Margarida Pires dos Santos Mateus
September 2015

This page was intentionally left blank.

You have to learn the rules of the game.
And then you have to play better than anyone else.
Albert Einstein (1879-1955)

This page was intentionally left blank.

I
ACKNOWLEDGMENTS
I appreciate this awesome chance that IFP Energies nouvelles provided me. Damien and Nadège,
the achievement of this work will not be possible without your significant collaboration. I’m grateful for
the continuous learning that you gave me, the challenges, the confidence and all suggestions. You
made this internship an enriching experience. Also a special thanks to Marie-Olive that supported me
in my first days and always cared about my good being. I extend my gratitude to the excellent people
from Elbaite (Serge, Amandine, Karine). A particular thanks to Michel for his availability and good
humor. Generally, the support from both R12 and R05 was indispensable.
I want to thank deeply to the person that made possible that this opportunity took part of my life.
Prof. Filipa Ribeiro: thank for your teaching during last years and dedication to allow this extraordinary
experience to me and to my colleagues. Also an word to Joana Fernandes and Vitor Costa that
welcomed us in IFP in the best way. To my supervisor from Instituto Superior Técnico, Prof. João
Bordado, I thanks for the given suggestions and the final revision of this work.
This period of time made me grow up professionally but also personally. I’m grateful to the
portuguese community that integrated us (Ruben, Sonia, Leonor, Max, Leonel, Mafalda,…). I also had
the opportunity to meet people that encouraged me to learn French language and costumes (Fabien,
Mathieu, Swetan,…). The coffees and lunches together were also important, a word to Larissa, Alexis
and Raido. To my office partners, Rami, Romain, and Laure, that taught me the first words in french.
To all my friends from the university (particularly, Mariana, Filipe and Bernardo) and my friends from
ever (Ana Marta, Ana Luísa, Sara, …). People, you are incredible! Thanks for sharing extraordinary
moments with me.
A huge thank to my family in Lyon: Loios, Solange, Casinhas, David, Catarina, Joana, Diogo. For
the sharing of experiences during these six months, the adventures in the trips, the everyday dinners
together, every moments, always together. Thank you all for the friendship.
I reserve this last paragraph to express my biggest acknowledgments. To whom that told me (and
still remember me every time) to work and put all my best qualities in everything that I do. The people
who deserve all my respect: Mãe, Pai, Mano. Pedro, I thank you for believing me and for your
unconditional support.

II
This page was intentionally left blank.

III
RESUMO
Este trabalho teve como objectivo o desenvolvimento de um método que permite caracterizar a
área superficial disponível de substratos linhocelulósicos e relacioná-la com a reactividade desses
substratos na hidrólise enzimática.
Numa primeira fase, foi efectuado um estudo bibliográfico extensivo que permitiu identificar os
métodos disponíveis para o propósito. Uma técnica baseada na exclusão molecular de solutos foi
proposta com o objectivo de determinar o volume acessível nos materiais linhocelulósicos a
moléculas sonda de diferentes tamanhos. Foram exploradas duas abordagens distintas baseadas na
utilização de um substrato saturado ou seco. A segunda abordagem não existe na literatura e foi
adaptada com sucesso a partir da primeira.
A metodologia utilizando um substrato seco foi testada com uma celulose comercial (Alphacel
C40) e palha de trigo (nativa e pré-tratada a 160 oC, lavada e não lavada). Também foi proposta uma
equação modelo que descreve a distribuição de poros por tamanhos. Foi feito um estudo completo
com a Alphacel, no entanto, é necessário mais estudo sobre os restantes substratos. A técnica
caracteriza-se pelo longo tempo de espera (1 dia por molécula sonda, 10 dias por substrato). Para
solucionar este problema, foram sugeridas diversas optimizações neste trabalho.
A metodologia proposta é reprodutível e foi validada para a Alphacel. Este trabalho deverá ser
completado com a aplicação do método na caracterização de outros substratos pré-tratados, com o
objectivo de obter uma base de comparação da eficiência dos pré-tratamentos.
PALAVRAS-CHAVE:
Biocombustíveis; Etanol 2G; Linhocelulose; Área acessível; Exclusão de solutos.

IV
This page was intentionally left blank.

V
ABSTRACT
This work focused on the development of a methodology that allows to characterize the available
surface area of lignocellulosic substrates and to relate it with their reactivity on enzymatic hydrolysis.
Firstly, an extended literature review was done on the methods used for this purpose. A method
based on solute exclusion was proposed and aimed to measure the accessible volume of
lignocellulosic materials by using chemical probes of different sizes. Two approaches were explored
based on a saturated or a dried substrate. The second method does not exist in literature and was
adapted with success from the first one.
The methodology using a dried substrate was tested using a commercial cellulose (Alphacel C40)
and wheat straw (native and pretreated at 160 oC, non-washed and washed). A model equation was
also proposed in order to describe pore size distribution. A complete study was done on Alphacel but
more studies are still required for the other substrates. The main drawback of this technique is its long
experimental standby time (1 day per probe, 10 days per substrate). To solve this issue, several
optimizations were suggested in this work.
The methodology proposed is reproducible and was validated for Alphacel. The present work shall
be completed with the characterization of other pretreated substrates, in order to provide a basis to
compare pretreatment’s effectiveness.
KEYWORDS:
Biofuels; Ethanol 2G; Lignocellulose; Available area; Solute-exclusion.

VI
This page was intentionally left blank.

VII
Table of contents
Acknowledgments .................................................................................................................................... I
Resumo .................................................................................................................................................. III
Abstract .................................................................................................................................................... V
Table of contents ................................................................................................................................... VII
List of Symbols and Abbreviations ......................................................................................................... IX
List of Figures ......................................................................................................................................... XI
List of Tables ........................................................................................................................................ XIII
1 Introduction ....................................................................................................................................... 1
2 Literature Review ............................................................................................................................. 3
Energy Context .................................................................................................................... 3
Lignocellulosic Biomass ...................................................................................................... 4
Production Processes of Ethanol 2G ................................................................................... 6
Pretreatment, the first key decision ............................................................................. 7
Enzymatic hydrolysis, the bottleneck of the process ................................................. 10
Process configurations .............................................................................................. 12
Measurement of Porosity and Surface Area ..................................................................... 14
BET method using nitrogen adsorption ..................................................................... 14
Mercury porosimetry .................................................................................................. 15
Simons’ stain ............................................................................................................. 16
Solute exclusion technique ........................................................................................ 17
Size-exclusion chromatography – SEC ..................................................................... 18
Other techniques ....................................................................................................... 18
Solute Exclusion Technique .............................................................................................. 19
State of the art ........................................................................................................... 19
Comparison of protocols ............................................................................................ 21
Conclusion and aim of the study ....................................................................................... 22
3 Pretreatment of the substrate ......................................................................................................... 23
Thermochemical pretreatment ........................................................................................... 23
Acid hydrolysis ................................................................................................................... 25
Enzymatic hydrolysis ......................................................................................................... 26

VIII
4 Method to determine the pore volume ........................................................................................... 29
Materials and methods ...................................................................................................... 29
Methodology for substrate in saturated form ..................................................................... 30
Water retention value method – WRV ....................................................................... 30
Methodology for substrate in dried form ............................................................................ 33
Probe solutions analysis .................................................................................................... 34
5 Results of Substrate Porosity ......................................................................................................... 39
Determination of pore volume ........................................................................................... 39
Saturated substrate method ...................................................................................... 39
Dried substrate method ............................................................................................. 40
Pore volume distribution .................................................................................................... 43
Determination of specific surface area .............................................................................. 54
Summary and discussion .................................................................................................. 55
6 Conclusions and Future Prospects ................................................................................................ 63
7 References ..................................................................................................................................... 65
8 Appendix ........................................................................................................................................ 71
Results from enzymatic hydrolysis .................................................................................... 71
Probe molecules ................................................................................................................ 71
Results from water retention value method ....................................................................... 72
Calibration curves (refractometry) ..................................................................................... 74
Saturated substrate methodology – Alphacel .................................................................... 75
Dried substrate methodology – Alphacel ........................................................................... 76
Dried substrate methodology – Non-washed native wheat straw ..................................... 78
Dried substrate methodology – Washed native wheat straw ............................................ 83
Dried substrate methodology – Wheat straw pretreated at 160 °C and washed .............. 85
Pore volume distributions for dried substrate methodology .............................................. 86

IX
List of Symbols and Abbreviations
Symbol Description Units
1G First-generation [ ]
2G Second-generation [ ]
AFM Atomic force microscopy [ ]
AV Average value *
BET Brunauer-Emmett-Teller [ ]
CBM Cellulose-binding module [ ]
Ceq Equilibrium concentration g/100mL or %w/v
Cf Concentration of the final solution g/100mL or %w/v
Cg Concentration in glucose of the final solution g/L
Ci Concentration of the initial solution g/100mL or %w/v
CLSM Confocal laser scanning microscopy [ ]
D Diameter of probe molecule Å
DSC Differential scanning calorimeter [ ]
s Substrate porosity [ ]
FSP Fiber saturation point mL or mL/g mds
H Humidity %
ID Sample identification [ ]
k Constant of pore volume distribution curves 1/Å
mds Mass of dried substrate g
mf Final mass of substrate g
mi Initial mass of substrate g
ms Mass of substrate g
mss Mass of saturated substrate g
mtotal Total mass of the mixture g
nD Refractive index at 20°C [ ]
nD’ Refractive index at 20°C with correction [ ]
NMR Nuclear magnetic resonance [ ]
nprobe Amount of probe in solution mol
PEG Polyethylene glycol [ ]
s Bulk density of the substrate g/mL
SD Standard deviation *
SE Solute exclusion [ ]
SEC Size-exclusion chromatography [ ]
SEM Scanning electron microscopy [ ]
SSA Specific surface area m2/g
*depends on the measured parameter

X
Symbol Description Units
SHF Separate hydrolysis and fermentation [ ]
SSF Simultaneous saccharification and fermentation [ ]
t Time h
T Temperature °C
TEM Transmission electron microscopy [ ]
TGC Fluorescent protein [ ]
Va Accessible pore volume mL or mL/g mds
Ve Exterior volume mL or mL/g mds
Vi Inaccessible pore volume mL or mL/g mds
Vi,max Maximum inaccessible pore volume mL or mL/g mds
Vp Pore volume mL
Vsol Volume of solution mL
Vsol,i Volume of initial probe solution mL
WRV Water retention value g/g or %

XI
List of Figures
Figure 2-1: Projected world energy-related CO2 emissions (Mton/year) [5]............................................ 3
Figure 2-2: Evolution in consumption of biofuels in transportation sector, in EU28 [7]. .......................... 4
Figure 2-3: Arrangement of the mainly constituents of lignocellulosic biomass in the cell wall [12]. ...... 5
Figure 2-4: Scheme from vegetal cells to glucose monomer – adapted from [17, 18]............................ 6
Figure 2-5: Biocatalysed-production of fuel ethanol from lignocellulosic biomass [20]. .......................... 7
Figure 2-6: Categories of pretreatment methods for lignocellulosic biomass – according to [10, 21, 22].
................................................................................................................................................................. 8
Figure 2-7: Simplified scheme of the impact of pretreatment on biomass [22]. .................................... 10
Figure 2-8: SSF in relation to other process options [26]. ..................................................................... 12
Figure 2-9: Scheme of ethanol 2G production process in SHF configuration – adapted from [17]. ..... 12
Figure 2-10: Schematic representation of an SSF process [26]. .......................................................... 13
Figure 2-11: Gas adsorption models [31]. ............................................................................................. 14
Figure 2-12: Mercury intrusion porosimetry [34].................................................................................... 15
Figure 2-13: Example of light microscope image of Simons' stained mechanical pulp fibers [38]........ 16
Figure 2-14: Representation of the accessibility to the pores of a substrate using solute exclusion [43].
............................................................................................................................................................... 17
Figure 2-15: Layout for the size-exclusion system proposed by Yang and his co-workers [46]. .......... 18
Figure 2-16: Schematic illustration of pore distribution curve to solute excluded from the pores [53]. . 20
Figure 2-17: General scheme of the different steps to perform solute exclusion. ................................. 21
Figure 3-1: Pilot unit U868 for thermochemical pretreatment of lignocellulosic substrates. ................. 23
Figure 3-2: Samples obtained by different severities of acid pretreatment. .......................................... 24
Figure 3-3: Yield in dry substrate after pretreatment. ............................................................................ 25
Figure 3-4: Schematic representation of two step acid hydrolysis. ....................................................... 25
Figure 3-5: Glucostat used to measure the concentration in glucose of the samples. ......................... 27
Figure 3-6: Glucose yield on enzymatic hydrolysis (Appendix 8.1). ...................................................... 27
Figure 4-1: Scheme of the methodology for substrate in saturated form. ............................................. 30
Figure 4-2: Evolution of mass substrate during drying, for Avicel PH101 (Table 8-4, Appendix 8.3). .. 32
Figure 4-3: Evolution of mass substrate during drying, for Alphacel C40 (Table 8-6, Appendix 8.3). .. 32
Figure 4-4: Results from determination of WRV for Avicel PH101 (Table 8-7, Appendix 8.3). ............. 33
Figure 4-5: Results from determination of WRV for Alphacel C40 (Table 8-8, Appendix 8.3). ............. 33
Figure 4-6: Scheme of the methodology for substrate in dried form. .................................................... 34
Figure 4-7: Refractometer used to measure the refractive index of solutions. ..................................... 34
Figure 4-8: Sample recovered after decantation and prepared to analyze in refractometer. ............... 35
Figure 4-9: Linear correlation between refractive index and concentration for PEG 35000 (02/07/2015).
............................................................................................................................................................... 35
Figure 4-10: Linear correlation between refractive index and concentration for PEG 35000
(10/07/2015). ......................................................................................................................................... 36
Figure 4-11: Evolution of calibration curves for PEG 200 (between 28/05/2015 and 15/07/2015). ...... 36
Figure 4-12: Examples of calibration curves for the different probes. ................................................... 38

XII
Figure 5-1: Distribution of values for water retention method, for Alphacel (Table 8-10, Appendix 8.5).
............................................................................................................................................................... 39
Figure 5-2: Calibration curve and results for experiment SE02 (Table 8-10, Appendix 8.5). ............... 40
Figure 5-3: Scheme of a porous substrate and penetration of molecules. ........................................... 41
Figure 5-4: Expected increasing in accessible porous volume by type of substrate. ............................ 42
Figure 5-5: Example of a series of samples in a trial. ........................................................................... 43
Figure 5-6: Pore volume distribution for pulp fibers exposed to different conditions [40]. .................... 44
Figure 5-7: Scheme representative of different levels of porosity. ........................................................ 45
Figure 5-8: Pore volume distribution for Alphacel (Table 8-21, Appendix 8.10). .................................. 46
Figure 5-9: Pore volume distribution for Alphacel (Table 8-21, Appendix 8.10). .................................. 47
Figure 5-10: Pore volume distribution of celluloses (Table 5-2) [19]. .................................................... 47
Figure 5-11: Pore volume distribution for non-washed native wheat straw (Table 8-22, Appendix 8.10).
............................................................................................................................................................... 48
Figure 5-12: Pore volume distribution for non-washed native wheat straw (Table 8-22, Appendix 8.10).
............................................................................................................................................................... 48
Figure 5-13: Pore volume distribution for non-washed native wheat straw, with refractive index
correction – all points included (Table 8-23, Appendix 8.10). ............................................................... 49
Figure 5-14: Pore volume distribution for non-washed native wheat straw, with refractive index
correction. (Table 8-23, Appendix 8.10) ................................................................................................ 50
Figure 5-15 : Pore volume distribution for washed native wheat straw (Table 8-24, Appendix 8.10). .. 51
Figure 5-16: Pore volume distribution for washed native wheat straw, with refractive index correction
(Table 8-25, Appendix 8.10). ................................................................................................................. 51
Figure 5-17: Pore volume distribution for washed and pretreated wheat straw (Table 8-26, Appendix
8.10). ...................................................................................................................................................... 52
Figure 5-18: Pore volume distribution for Alphacel and different wheat straw products. ...................... 53
Figure 5-19: Pore volume distribution for pulp fibers – zoom of Figure 5-6 [40]. .................................. 53
Figure 5-20: Schematic representation of the structural features of the cellulose particle surface [19].
............................................................................................................................................................... 54
Figure 5-21: Accessible pore volume of corn stover, measured by solute exclusion [42]. ................... 55
Figure 5-22: Influence of substrate quantity in final concentration of probe. ........................................ 57
Figure 5-23: Influence of the ratio mass of substrate by volume of solution in final concentration of
probe. ..................................................................................................................................................... 58
Figure 5-24: Experimental issue on stirring. .......................................................................................... 59
Figure 5-25: Comparison between a native and a pretreated wheat straw samples, after stirring. ...... 59
Figure 5-26: Comparison between a washed (1’) and a non-washed (1) native wheat straw
supernatants. ......................................................................................................................................... 60
Figure 5-27: Pore volume distribution for Alphacel (Table 8-21, Appendix 8.10). ................................ 61
Figure 8-1: Correlation obtained for PEG probes by power curve. ....................................................... 72

XIII
List of Tables
Table 2-1: Composition of the different components in lignocellulosic biomasses – adapted from [15]. 5
Table 2-2: Preparation conditions for substrates and probe solutions. ................................................. 21
Table 3-1: Operating conditions of the pretreatment. ............................................................................ 24
Table 3-2: Results from acid hydrolysis. ............................................................................................... 26
Table 4-1: Molecular weights and solution diameters of probes used (Appendix 8.2).......................... 29
Table 4-2: Initial conditions of substrates to water retention value method. ......................................... 31
Table 4-3: Linear correlations between refractive index and concentration for PEG 200 (Figure 4-11).
............................................................................................................................................................... 36
Table 5-1: Exterior and maximal pore volume determined for the substrates. ..................................... 42
Table 5-2: Fiber saturation point by solute exclusion technique, from literature. .................................. 43
Table 5-3: Fiber saturation point from different pretreatments. ............................................................. 44
Table 5-4: Types of porosity in solids [61]. ............................................................................................ 45
Table 5-5: Example of exterior and total porous volume determination, for Alphacel (Table 8-11). ..... 46
Table 5-6: Contribution of soluble components for refractive index measurements. ............................ 51
Table 5-7: Results of specific surface area from N2 gas adsorption, in this work. ................................ 54
Table 5-8: Specific surface area from literature, for wheat straw, by N2 gas adsorption. ..................... 55
Table 5-9: Example of concentrations of probe (Appendix 8.6; Appendix 8.7). .................................... 56
Table 5-10: Influence of substrate quantity in final concentration of probe. .......................................... 57
Table 5-11: Day work plan to performed one experiment with the dried substrate methodology. ........ 60
Table 5-12: Day work plan to performed two experiments by dried substrate methodology. ............... 61
Table 8-1: Glucose yield from enzymatic hydrolysis for the pretreated samples. ................................. 71
Table 8-2: Solution molecular diameters of probes from literature [40, 53]. ......................................... 71
Table 8-3: Drying of substrate WRV1, for Avicel PH101. ...................................................................... 72
Table 8-4: Drying of substrate WRV2, for Avicel PH101. ...................................................................... 72
Table 8-5: Drying of substrate WRV3, for Avicel PH101. ...................................................................... 73
Table 8-6: Drying of substrate WRV4, for Alphacel C40. ...................................................................... 73
Table 8-7: Results of WRV for Avicel PH101. ....................................................................................... 73
Table 8-8: Results of WRV for Alphacel C40. ....................................................................................... 73
Table 8-9: Linear correlations between refractive index and concentration of probe. .......................... 74
Table 8-10: Results from saturated substrate methodology, for Alphacel. ........................................... 75
Table 8-11: Results from dried substrate methodology, for Alphacel (part 1). ...................................... 76
Table 8-12: Results from dried substrate methodology, for Alphacel (part 2). ...................................... 77
Table 8-13: Results from dried substrate methodology, for non-washed native wheat straw (part 1). . 78
Table 8-14: Results from dried substrate methodology, for non-washed native wheat straw (part 2). . 79
Table 8-15: Results from dried substrate methodology, for non-washed native wheat straw (part 3). . 80
Table 8-16: Results from dried substrate methodology, for non-washed native wheat straw – nD
correction (part 1). ................................................................................................................................. 81
Table 8-17: Results from dried substrate methodology, for non-washed native wheat straw – nD
correction (part 2). ................................................................................................................................. 82

XIV
Table 8-18: Results from dried substrate methodology, for washed native wheat straw. ..................... 83
Table 8-19: Results from dried substrate methodology, for washed native wheat straw – nD correction.
............................................................................................................................................................... 84
Table 8-20: Results from dried substrate methodology, for wheat straw pretreated at 160 °C and
washed. ................................................................................................................................................. 85
Table 8-21: Pore volume distribution data for Alphacel. ....................................................................... 86
Table 8-22: Pore volume distribution data for non-washed native wheat straw. ................................... 86
Table 8-23: Pore volume distribution data for non-washed native wheat straw – nD correction. ......... 87
Table 8-24: Pore volume distribution data for washed native wheat straw. .......................................... 87
Table 8-25: Pore volume distribution data for washed native wheat straw – nD correction. ................ 87
Table 8-26: Pore volume distribution data for washed and pretreated wheat straw. ............................ 87

1
1 INTRODUCTION
Due to the energy context in the XXI century, fossil fuels have started to be replaced and
renewable alternatives are being seriously considered. In this field, the interest for second-generation
fuels obtained from lignocellulosic biomass has increased: one possible way to that is using this
biomass to produce ethanol from a biological way (pretreatment, enzymatic hydrolysis, fermentation,
distillation). However, the technology costs are still an obstacle, particularly in the pretreatment step.
In order to improve the digestibility of the lignocellulosic biomass, i.e. degradation of cellulose and
hemicelluloses, a pretreatment of the substrate has to be performed. This will increase the total yield
of monomeric sugars in the hydrolysis step. Afterwards, an understanding about the role of each
enzymatic family (cellobiohydrolases, endoglucanases, -glucosidases) is needed to improve
enzymatic hydrolysis.
Since porosity and surface area are reported as key-parameters in the mentioned process, the
main goal of this study was to ascertain methodologies suitable to characterize lignocellulosic
substrates. More particularly, elaborate a methodology that allows to determine the morphology of a
given substrate expeditiously. With this, a basis to compare pretreatment’s effectiveness can be
established, as well, to relate it with the reactivity of the substrate in hydrolysis.
⃝ ⃝ ⃝
Hence, a bibliographic study was done to explain general concepts about biofuels and its insertion
in the current energy context. Particularly, the second-generation is discussed. Then, a review about
the different methodologies available to characterize the substrates surface area was done (second
chapter).
Before to proceed for the essence of the work, a previous work was done in order to prepare the
substrates and is explained in chapter three. Subsequently, for the chosen technique (solute
exclusion), preliminary assays were performed to define the adequate conditions of experimentation.
The materials and methodologies used are described in chapter four. Using a statistical treatment, the
data obtained from the measurement of pore distribution is reported in chapter five and deliberated.
Lastly, the main conclusions obtained by this work are presented and discussed. To complete this
study, future prospects and suggestions were proposed.

2
This page was intentionally left blank.

3
2 LITERATURE REVIEW
Energy Context
In 2008 [1], no one had a definitive answer for the question “when will lignocellulosic ethanol
become economically viable?”. In 2013 [2], the world’s largest cellulosic ethanol production facility
opened at Crescentino, Italy, and currently, cellulosic ethanol is being produced on commercial scale
in Europe, USA and Brazil.
Current energy context
Today fossil fuels are the dominant energy sources, meeting more than 80% of the world’s energy
demand and is set to grow by 37% by 2040 (IEA, 2014). Nevertheless, fossil fuels are non-renewable
and their reserves are limited: at the current consumption rates, the supply of petroleum, natural gas,
and coal will only be able to last for another 45, 60, and 120 years, respectively (IEA, 2013).
Furthermore, the carbon dioxide represents 77% of greenhouse gas emissions and this
tremendous amount of emissions has been released essentially from fossil fuels combustion [3] –
Figure 2-1. This resulted in elevating the atmospheric CO2 concentration. Consequently, renewable
alternatives should be seriously considered. The recent energy independence and climate change
policies encourage development and utilization of renewables such as bioenergy, solar and wind
energy [4].
Figure 2-1: Projected world energy-related CO2 emissions (Mton/year) [5].
In this context, the EU supported the utilization of renewable energies proposing a replacement of
5.75%, by 2010, of the classical fuels by substitute products, as biofuels, (2003/30/CE), as well, fixing
a goal of 10% incorporation of renewable energies in the total of automobile fuels to 2020
(2009/28/CE). Recently, EU countries have agreed on a new renewable energy target of at least 37%
by 2030 [6]. By over the years, it is visible an increasing in the consumption of biofuels – Figure 2-2.

4
Figure 2-2: Evolution in consumption of biofuels in transportation sector, in EU28 [7].
Nowadays, there is utmost of alternative energy resources which are cheap, renewable and limit
pollution. Biomass is inserted in the context of renewables and is being considered as an important
resource all over the world. Actually, biomass it is the fourth largest source of energy after coal,
petroleum and natural gas, providing about 14% of the world’s primary energy consumption [8].
Second generation (2G) biofuels
It is known that renewables will continue to play a key role in reducing the greenhouse gas
emissions and their sources are abundant in the world. By this, the history of ethanol as a biofuel
dates back to the early days of the automobile era [8]. It is expected that biofuels can provide up to
27% of world transportation fuel, in 2050 (IEA, 2011).
Currently, the first-generation (1G) biofuels are already in the market in industrial scale, and the
second-generation (2G) is emerging, being extensively researched in the past two decades [9, 10].
Still, to minimize the adverse impacts, they must be produced in a sustainable way.
In contrast to ethanol 1G made from food crops, cellulosic ethanol is manufactured from non-food
plant materials (such as agricultural residues or energy crops). This lignocellulosic feedstock is
abundant, however it consists in a complex and very resistant structure (cellulose, hemicelluloses and
lignin) that needs to be broken down into simple sugars before fermentation and distillation.
Lignocellulosic Biomass
Cellulose, hemicelluloses and lignin are the three mainly constituents of lignocellulosic biomass
(along with small amounts of proteins, pectin, extractives and ash). These polymers, which are
associated each other, compose a complex and very resistant structure – Figure 2-3.
Cellulose is a glucose polymer and the mainly constituent of lignocellulosic biomass. It consists of
parts with a crystalline structure and parts with an amorphous structure [9, 11]. This polymer is a linear
chain of D-glucose subunits linked by -1,4 glycosidic bonds [11, 12].
Hemicelluloses are composed by different sugars like pentoses (C5 sugars such as xylose and
arabinose), hexoses (C6 sugars such as glucose, mannose and galactose) and acid sugars. These

5
components serve as a connection between the lignin and the
cellulose fibers and give the whole cellulose-hemicellulose-
lignin network more rigid [11].
Lignin is, after cellulose and hemicelluloses, one of the
most abundant polymers in nature and this main purpose is to
give the plant structural support [11]. It fills the spaces in the
cell wall between cellulose and hemicelluloses [12]. In this
way, lignin provides further strength to plant cell walls, but
hinders the enzymatic hydrolysis of carbohydrates [13].
As said above, plant cell walls are composed mostly of
lignocellulose which is the most abundant organic material on
Earth [8, 12, 14], being available from different sources –
Table 2-1. The composition of lignocellulosic biomass directly
depends on its origin:
Table 2-1: Composition of the different components in lignocellulosic biomasses – adapted from [15].
Lignocellulosic
biomass
Cellulose
(%w/w)
Hemicelluloses
(%w/w)
Lignin
(%w/w)
Wheat straw 33 23 17
Corn cob 45 35 15
Newspapers 40-55 25-40 18-30
Miscanthus 45 30 21
The nature of lignocellulosic material makes the pretreatment (section 2.3.1) a crucial step [16] due
to the physical and chemical barriers caused by the close association of the main components.
Figure 2-3: Arrangement of the mainly constituents of lignocellulosic biomass in the cell wall [12].

6
Cellulose, a complex substrate
The complex material named cellulose presents various levels of organization – Figure 2-4. Three
levels can be distinguished [17]: macroscopic scale, that are the cellulose particles, nanometric scale,
corresponding to the microfibers scale, and molecular scale that corresponds to the cellulose chain.
Figure 2-4: Scheme from vegetal cells to glucose monomer – adapted from [17, 18].
There are different morphologic cellulose parameters which develop an important role in the
reactivity of the substrate during hydrolysis. By this, the variety in physico-chemical characteristics
reveals the requirement of pretreatment technologies to help in the rapid and efficient conversion of
carbohydrate polymers into fermentable sugars [13, 19].
The main parameters include the crystallinity, the surface area, the degree of swelling, the degree
of polymerization and the size of the particles. These parameters are exploited later (section 2.3.2),
with a discussion of their influence in enzymatic hydrolysis step.
Production Processes of Ethanol 2G
Inversely to the production processes of first-generation ethanol, the sugars are not directly
accessible to fermentation in second-generation. The production of ethanol from lignocellulosic
material consists of mainly five different steps, namely, pretreatment, (enzymatic) hydrolysis,
fermentation, product separation, and post-treatment of the liquid fraction – Figure 2-5.

7
Figure 2-5: Biocatalysed-production of fuel ethanol from lignocellulosic biomass [20].
This process involves three main categories of costs: the costs of the feedstock, the costs of sugar
preparation, and the costs of ethanol production. Among these categories, conversion of cellulosic
components into fermentable sugars is the major technological and economical bottleneck [4].
Nevertheless, the tremendous technological advances in converting lignocellulosic biomass into
simple sugars, specifically in feedstock pretreatment and industrial enzymes preparation, eventually
realized the commercial-scale production of 2G ethanol in 2013 [4].
The next section approaches the pretreatment step, in order to clarify the different types of
treatments, as well, the applicability of each one. After, the enzymatic hydrolysis is discussed, due to
its character referred as the bottleneck of the process. Then, the main process configurations available
to produce ethanol from lignocellulosic material are presented.
Pretreatment, the first key decision
Generalizing, pretreatments have as a goal to improve the digestibility of the lignocellulosic
biomass. Otherwise, the pretreatment is required to improve the rate of production and total yield of
monomeric sugars in the hydrolysis step [11]. Also, the choice of pretreatment method avoids the
degradation of sugars derived from hemicelluloses and minimizes the formation of inhibitors for
subsequent fermentation steps [13].
The pretreatment methods can be divided into different categories: physical, physico-chemical,
chemical, biological, electrical, or a combination of these – Figure 2-6.

8
Figure 2-6: Categories of pretreatment methods for lignocellulosic biomass – according to [10, 21, 22].
PRETREATMENT
PHYSICAL
milling
chipping
grinding
extrusion
microwave oven
electron beam irradiation
PHYSICO-CHEMICAL
steam explosion
wet oxidation
liquid hot water (LHV)
ammonia fiber explosion (AFEX)
ammonia recycle percolation
aqueous ammonia
organosolv
CO2 explosion
CHEMICAL
alkali
dilute acid
concentrated acid
organic solvents
ozonolysis
oxidative delignification
wet oxidation
BIOLOGICAL
fungal
bio-organosolv
ELECTRICAL HYBRIDS

9
Physical pretreatment allows to increase the accessible surface area, as well the pore size of
lignocellulosic materials. Also, the crystallinity and degree of polymerization of cellulose is reduced
using this pretreatment method, increasing the reactivity of the substrate in enzymatic hydrolysis. This
method of fragmentation can greatly increase the accessible surface area [19], depending on the
porosity and cellulose particle size. However, this mechanical method is unattractive due to its high-
energy requirement and capital costs [10, 21].
Chemical pretreatment employs different chemical agents (such as acids, alkalis, ozone, among
others). This method has become one of the most promising to improve the biodegradability of
cellulose, by removing lignin and/or hemicelluloses, and decreasing the degree of polymerization and
crystallinity of cellulose. However, there are limitations associated to this method: it requires lower-cost
chemical reagents [13], and they must be recovered to make the pretreatment economically feasible
[10]. Following this, inorganic acids, such as H2SO4 or HCl, have been preferably used [21] and dilute
acid pretreatment (along with steam explosion) is one of the most widely studied method.
Physico-chemical pretreatment consists in a combination of the last methods and allows to dissolve
hemicelluloses and makes modifications in lignin structure, which provides an improved accessibility of
the cellulose for hydrolytic enzymes. The type of combination selected depends on the process
conditions and the solvents used that affect the physical and chemical properties of the biomass [10].
These methods are considerably more effective than physical and the steam explosion is the most
studied method of this type [21].
Biological pretreatment is mostly associated with the action of fungi that are capable of producing
enzymes to degrade lignin, hemicelluloses and polyphenols present in the biomass [10]. In
comparison with the pretreatments described above, the main advantages are that requires low
energy and the yield in desired products is high. However, the process is very slow and requires
careful control of growth conditions and large space to perform [21], limiting its application at industrial
level.
These methods have been investigated and reviewed by several researchers and authors [10, 11,
13, 23] and, among all, chemical pretreatment has been proven to be a promising one.
Due to the context of this study, the next section focus on the dilute acid pretreatment, since it was
the method used to prepare the substrates in this work.

10
Chemical pretreatment – dilute acid
Acid pretreatment particularly enhances the hydrolysis [13] and dilute acid pretreatment is one of
the oldest, simplest and frequently employed technique in biofuel production due to its efficiency [8,
23]. Additionally, H2SO4 and HCl have been preferably used for biomass pretreatment [21].
The treatment consists in adding a quantity of acid to the biomass (between 0.2 %w/w to 2.5
%w/w, depending if it is diluted or concentrated) and continuous stirring at temperatures between 130
°C and 210 °C [11].
In dilute acid treatment, the firm structure of the lignocellulosic materials is cracked, followed by the
removal of hemicelluloses – Figure 2-7, which increases the porosity and enzymatic digestibility of the
cellulose.
Figure 2-7: Simplified scheme of the impact of pretreatment on biomass [22].
This method has been successfully developed, achieving high reaction rates that can improve
significantly the subsequent process of cellulose hydrolysis [21]. The advantage of this type of
pretreatment is the solubilization of hemicelluloses and by this, cellulose will be more easily accessible
for the enzymes.
Still, there is a risk of formation of degradation products [11, 21] which is in many cases lost for the
conversion to ethanol. The authors point out that the realization of dilute acid pretreatment at low
temperatures (around 121 °C [11]) allows avoiding the degradation of sugars to furfural and
hydroxymethylfurfural (HMF), but the sugars yields are also lower.
Enzymatic hydrolysis, the bottleneck of the process
As said before, in enzymatic hydrolysis, cellulose is converted into glucose with the action of
enzymes. Particularly, to produce ethanol 2G, a cocktail of enzymes [17] is used mainly produced by a
fungus named Trichoderma reesei.
It is reported by various authors that suitable pretreatment methods enhance the enzymatic
hydrolysis and the key parameter in ethanol production is the digestibility of biomass to produce
sugars.
Various factors affect that digestibility: crystallinity, moisture content, degree of polymerization,
available surface area, size of particles, swelling degree. The more mentioned and studied in literature
are presented below, even if the relative importance of these factors is still unclear.

11
Lignin and hemicelluloses content
As can be seen is Figure 2-3, most of the lignin is concentrated between the outer layers of the
cellulose and hemicelluloses fibers, providing rigidity to the complex, but other part is intertwined with
them. Hence, lignin content and nature significantly affect the hydrolysis of biomass. Also, it is
reported that lignin can bind with cellulase enzyme resulting in less availability of the enzyme for
hydrolysis [9].
Lignocellulosic biomass typically contains 55-75 %w/w cellulose plus hemicelluloses [9]. So, the
removal of hemicelluloses in the pretreatment will significantly improve the hydrolysis and increase the
availability of ex-cellulose sugars for ethanol production. Theoretically, fractionation of any biomass
species allows to solubilize the majority of the hemicelluloses into the solution, and leaves the
cellulose fraction intact [10, 21].
Surface area and pore volume
The specific surface corresponds to the available surface by mass unit and takes in account the
porosity of the solid. It is reported by several authors (for example, [19, 24, 25]) that the glucose yield
from enzymatic hydrolysis depends mostly on the surface area available to the enzyme, regardless of
pretreatment method.
This parameter has been identified as a particularly important factor in the rate of enzymatic
deconstruction, particularly, in the early stages of the hydrolysis [23]. Essentially, increasing the
accessible surface area increases the amount of cellulases that can react with the cellulosic substrate,
resulting in an increment of the hydrolysis rate. The same authors claim that the pore surface area is
the limiting parameter in the hydrolysis reaction.
Crystallinity and degree of polymerization
Cellulose is a polymer of glucose and its arrangement results in a structure with amorphous and
crystalline parts. The amorphous configuration makes the substrate available to reaction; on the other
hand, regarding crystalline parts, this provides a protection against enzymatic attack and solubilization
in water [9]; then, crystallinity directly affects digestibility.
The polymerization degree cannot be considered isolated since it is intimate linked with crystallinity
and accessible surface area. If pretreatment cleaves the internal cellulose bonds, then enzymes can
easily attack the cellulose chains, since the degree of polymerization is lower.
Degree of swelling
The cellulose structure is deeply influenced by the presence of water. More, the chains of cellulose
are linked by hydrogen bonds and the insertion of water molecules can change the arrangement of its
structure. So, this property is important since enzymatic hydrolysis takes place in aqueous medium.

12
Process configurations
There are some types of configurations [26] possible to produce ethanol 2G: SHF (Separate
Hydrolysis and Fermentation), SSF (Simultaneous Saccharification and Fermentation), SSCF
(Simultaneous Saccharification and co-Fermentation), CBP (Consolidated Bioprocessing) and SoSF
(Solid State Fermentation). As can be seen in Figure 2-8, the variances can be described as a
modification of the SSF process. Olofsson and his co-authors suggest that this can be seen as a move
of the “classical” SSF process in the direction of other process options, resulting in new “hybrid”
processes, which will be improved for the feedstock and the enzymes used.
Figure 2-8: SSF in relation to other process options [26].
Separate Hydrolysis and Fermentation – SHF
This process is the conventional method where the hydrolysis is carried out in a period and
fermentation process after then, using two separate reactors. With this, it is allowed to first produce the
simple sugars and then ferment them.
This configuration comprises four main steps [17] – Figure 2-9: pretreatment, to make cellulose and
hemicelluloses accessible to enzymes; hydrolysis, to convert cellulose into glucose with the combined
action of enzymes; fermentation, to transform sugars into ethanol in an aqueous medium; and
distillation, to recover the ethanol from water.
Figure 2-9: Scheme of ethanol 2G production process in SHF configuration – adapted from [17].

13
The main advantage of this configuration is the possibility to obtain optimal conditions of pH and
temperature in each step. However, glucose produced during biomass saccharification is an inhibitor
of the reaction. Additionally, cellobiose represents also an inhibitor of cellulases [16, 27]. The
accumulation of these components strongly constrains the activity of cellulases.
Simultaneous Saccharification and Fermentation – SSF
This process is an alternative to the first presented, and consists in performing the enzymatic
hydrolysis simultaneously with the fermentation, as the name indicates, in a single reactor. In this way,
enzyme and yeast are put together, and glucose released by the action of cellulases is rapidly
converted into ethanol by the fermenting organism [16, 27] – Figure 2-10.
The principal benefit of this configuration, reported by various authors [16, 27, 28], is the higher
yield of ethanol obtained due to the removal of inhibitors (glucose, cellobiose by fermentation) from the
reaction medium. This continuous removal will minimize the depression of enzyme activity, because
low residual sugars (inhibitors of cellulases) are eliminated. Moreover, there is a reduction in
investment costs [27] in SSF over the sequential process, since less reactors are required.
Furthermore, the presence of ethanol in the culture broth helps to avoid undesired microbial
contamination [16]. All these advantages result in an increased rate of saccharification compared with
separate hydrolysis.
On the other hand, the principal drawback is the need to find favorable operating conditions for
both the enzymatic hydrolysis and the fermentation [26, 17]. This inconvenient will decrease the
reaction temperature because of the micro-organisms: this makes an effect of temperature between
30-35 °C and the optimal temperature is 50 °C for hydrolysis step, which decreases the catalytic
performance of the enzymes [17]. Accordingly, a compromise must be found in this process.
Furthermore, the yeast cannot be reused in an SSF process due to the problems of separating the
yeast from the lignin after fermentation. Recirculation of enzymes is equally difficult once the enzymes
bind to the substrate, although a partial desorption can be obtained after addition of surfactants [26].
Figure 2-10: Schematic representation of an SSF process [26].

14
Measurement of Porosity and Surface Area
Once enzymatic hydrolysis depends mostly on the surface area available to enzymes [19, 29], this
result is significant as it provides a common basis for the comparison of pretreatment’s effectiveness.
Due to this, measurement of porosity has been frequently used to determine the accessible area and
this section focus on a review of the multiple techniques that could be used to this purpose.
BET method using nitrogen adsorption
One of the classic techniques to measure the specific surface area is the Brunauer-Emmett-Teller
(BET) method using nitrogen adsorption. This method is based on the principle of the physical
adsorption of N2 molecules on the internal surface of a solid by weak interaction forces [30].
The procedure associated with a volumetric method is the most widely used [30]. In this technique,
samples are pretreated by applying some combination of heat, vacuum, and/or flowing gas to remove
adsorbed contaminants [31]. After this, known amounts of nitrogen gas pass readily through a cell
containing the solid to analyze, allowing the gas to condense on the surface and the equilibrium
pressure is measured. The quantity of gas that condenses is determined from the drop pressure after
the sample was exposed to the gas [29, 30].
After the experiment, the next step consists in applying a model in order to convert the isotherm
into a surface area, in this case, the BET model in which multiple layers of gas may adsorb to the
surface – Figure 2-11.
Figure 2-11: Gas adsorption models [31].
Regarding the probe utilized, nitrogen as a very small molecule (approximately 0.11 Å [32]) forms a
monolayer on all surfaces and its uptake provides a good measure of total surface area. However,
since enzymes are larger molecules (cellulose size range extends from 24 to 77 Å [33]), its access into
most pores will be denied. So, the total surface area potentially available for enzymatic attack is
considerably lower than that available to nitrogen [19, 25, 29].

15
Mercury porosimetry
This methodology is based on the behavior of non-wetting liquids and on the use of the Washburn
equation [30]. Using this technique a pore size distribution can be obtained.
Similar to nitrogen adsorption, the samples are dried and degassed. Then they are introduced into
a chamber and surrounded by mercury with pressure. Gradually increasing the pressure, mercury is
forced into the pores – Figure 2-12. This increasing of pressure is required because a liquid which
does not wet a solid cannot enter a pore spontaneously [30]. So, inversely to nitrogen adsorption,
mercury only can penetrate the pores if it is forced by a pressure.
Figure 2-12: Mercury intrusion porosimetry [34].
In this technique, the injected volume at a given pressure is equal to a cumulative volume in the
pores. It is assumed [25, 30] that the larger pores are most easily accessible, and, therefore, closer to
the surface, and the others are distributed in an ordered way. Then, there is an inversely proportional
relationship between the pressure required and the size of the pores.
Depending on the shape of the pores, different hysteresis loops may be encountered, but the
intermediate shapes are usually obtained experimentally [30], and these can yield information on the
geometry of the pores.
Mercury porosimetry allows the pore size analysis to be undertaken over a wide range of meso and
macro-pore widths [29] and can determine a broader pore size distribution more quickly and
accurately than other methods, offering a wide range of data (e.g. total pore volume, total pore surface
area, permeability). By its limits of detection, this method can be inappropriate for cell wall studies.
On the other side, the measurements require prior drying of the substrate [29]. Additionally,
because of the substantial volume of mercury retained in the pores, and the possible effect of the
crushing or structural collapse of the solid, this method is considered to be destructive [30].

16
Simons’ stain
An alternative approach for examining pore size employs direct dyes [29] for estimating total
available surface area of lignocellulosic substrates. The original method was developed by Simons, in
1950, using two color differential stain: orange and blue dyes. According to the same author, Direct
Blue 1 and Direct Orange 15 are preferred [35].
Dyes are well known to be sensitive probes to characterize cellulose fine structure, and direct dyes
are particularly appropriate because they are physically adsorbed on cellulose [36]. Then, with this
method it is possible to determine the accessibility of the probes into the interior structure of fibers.
When lignocellulosic biomass is treated with a mixed solution of the direct dyes, the blue dye
enters all the pores with a diameter larger than approximately 1 nm, while the orange dye only adsorbs
in the larger pores size (more than 5 nm) [37, 35, 38]. Additionally, when the pore size is large enough
for the orange dye to penetrate, the fiber adsorbs preferentially this one because of its stronger affinity.
Figure 2-13: Example of light microscope image of Simons' stained mechanical pulp fibers [38].
Consequently, for fibers with a wide pore size distribution range, the color of the stained fiber will
depend on the ratio of surface area accessible to orange dye and to the surface area that is
accessible to blue dye but is not accessible to orange. Fibers that appear green, for example, clearly
have significant amounts of both small and large pores [35] – Figure 2-13.
The ratio of adsorbed orange and blue dye is the value used to estimate the amount of large pores
to small pores and subsequently cellulose accessibility in lignocellulosic biomass for enzymatic
hydrolysis [37].
Normally, this technique is combined with NMR method and/or microscopy [35, 39]. Meng and his
co-authors used this approach in order to probe biomass porosity and thus access to cellulose
accessibility. However, it is not a method suitable for any rapid characterization of water-swollen
cellulose materials [36].

17
Solute exclusion technique
This method was the first used to determine the cell wall porosity by Stone and Scalan [40], in
1968, and nowadays is widely used to investigate the pore characteristics of the lignocellulosic
substrates [29]. It is based on the measurement of accessibility to the pores of a set of probe
molecules, such as dextran [14] or other non-interacting probe molecules.
With this technique, it is possible to obtain directly measurements of porosity of a substrate (or the
total amount of water inside the cell wall). The advantage of this technique lies in the fact that it can be
directly applicable to wet materials [19, 41, 42]. This point is significant since water removal from non-
rigid porous materials, such as biomass, often produces the collapse, partial or total, of the internal
structure of the substrate [14, 41].
The procedure consists in adding a solution of known concentration of a probe into a substrate,
previously saturated in water (by water retention value methodology – described later in this section).
The probe solution will be then diluted by the water contained in the initial substrate and as a result,
the substrate pore size and volume distribution can be determined.
Therefore, the driving force of this method is the concentration of probe in solution, once the
system tends to reach the equilibrium. In this way, the probe will penetrate into the pores, and a part of
water is excluded.
Figure 2-14: Representation of the accessibility to the pores of a substrate using solute exclusion [43].
Regarding the scheme above (Figure 2-14), if all pores are accessible to the probe molecule, then
all water in the initial substrate will contribute to the dilution (case I). As progressively larger molecules
are used (cases II and III), some of the smaller pores and, finally, all of the pores become inaccessible
to the probe molecules and unavailable for dilution of the solution.
Resuming, the measured concentration of the probe molecule in the final substrate mixture
depends on the pore size and volume distribution [43] and so, the accessible pore volume of the
substrate can be determined using a set of solutions with different molecule sizes.

18
Size-exclusion chromatography – SEC
With the same fundamental than solute exclusion, the size-exclusion chromatography can be
applied to measure specific pore volume and specific surface area.
In theory, SEC is a separation process in which molecules are separated on the basis of molecular
size differences. The stationary phase consists of spherical porous particles with a carefully controlled
pore size, through which the biomolecules diffuse using an aqueous buffer as the mobile phase [44].
This technique is an analytical method of choice when diminutive effects are to be followed and only
small amounts of samples are available [45].
In a methodology proposed by Yang and his co-workers, the probes of known molecular weight are
allowed to diffuse into the pore structure of the biomass substrate packed in the column, and,
subsequently, eluted to generate high resolution concentration measurements – Figure 2-15.
Figure 2-15: Layout for the size-exclusion system proposed by Yang and his co-workers [46].
Yang et al. reported an excellent reproducibility for the measurements and suggests this method as
a fast and precise technique to measure accessible pore volume and surface area in native and
pretreated lignocellulosic biomasses.
To estimate with precision the widths of the molecules measured, various studies [44, 45, 47, 48]
suggest the combination of SEC with various detectors, including: light scattering and refractive index,
multi angle laser light scattering, ultra-visible spectroscopy or viscometer.
Other techniques
Other methods are available to determine the accessible area of lignocellulosic substrates,
nevertheless, they are not inserted in the context of the current study. By this, a small review is
presented in this sub-chapter. These techniques are not less important, but generally they are used
coupled with the methods described above.
Regarding nuclear magnetic resonance, there are two techniques mostly referred in literature
related to biomass characterization: cryoporosimetry and relaxometry [29, 39]. The first, NMR
cryoporosimetry, it allows to determine pore size distribution. NMR relaxometry provides information
about the molecular mobility within a porous system. Both techniques have as advantage the non-

19
destructibility of the substrate. However, the method is expensive and requires complicated
experimentation setup.
Another technique referenced as promising [14, 29] is the adsorption of non-hydrolytic fusion
protein containing cellulose-binding module (CBM) and fluorescent protein (TGC). Quantitative
determination of cellulose accessibility to cellulose is done, based on the Langmuir adsorption of a
fusion protein. Both proteins have a very similar molecular size to that of cellulose enzymes, being this
the main advantage. In disadvantage, these proteins also bind unspecifically to lignin, and then
Simons’ stain is still the alternative preferred.
Calorimetry is a primary technique for measuring the thermal properties of materials to establish a
connection between temperature and specific physical properties of substances [49] and differential
scanning calorimeter (DSC) is a popular one. This method is commonly used for the study of
biochemical reactions, to monitor effects associated with phase transitions as function of temperature.
The distribution of cell wall material in the plant may contribute significantly to the variation in
degradability of the material. Consequently, microscopy techniques are required to visualize, measure
and quantify plant cell wall features as a result of pretreatment [50, 51]. To obtain a more complete
and detailed image of the substrate, various microscopy techniques should be combined. For example
confocal laser scanning microscopy (CLSM) [50] is referred as a method appropriated to estimate the
volume of cell wall material present in tissue sections before and after digestion. Scanning electron
microscopy (SEM) and transmission electron microscopy (TEM) have been extensively used to follow,
at high resolution, the structural changes in cell walls after biomass pretreatment [51]. Filament
organization of cell walls in native biomass has often been imaged by the atomic force microscopy
(AFM), a versatile powerful tool to study topographic, physical and chemical properties of biological
samples at nanometer scale [51].
Solute Exclusion Technique
The first part of this work intends to explore the advantages and disadvantages, as well the
applicability of a methodology to characterize the surface area of lignocellulosic substrates. From all
methodologies available in literature (described in the previous section), solute exclusion was selected
to be explored more accurately due to the adjustability in the context of this work.
In this section a review about the conditions used by different authors is done and, as result of this,
a new approach of the technique will be established in order to do experiments and, subsequently,
discuss the results obtained.
State of the art
Solute exclusion has been widely studied by several authors to investigate the pore characteristics
of lignocellulosic substrates.
In 1968, it was hypothesized by Stone and Scalan [40] that the rate of reaction between enzymes
and their substrate is dependent on the surface area which is accessible to the enzymes. To support
this hypothesis, the authors developed a technique to measure that accessibility using a solute
molecule of the same size as the enzyme. The methodology was elaborated by Van Dyke [52], in
1972.

20
For this, series of polymers, as PEG’s, with different sizes, were used as probes to determine the
inaccessible volume to the pores, being the basis for pore size measurement (as described before).
As a result of this work, many authors have used this methodology as basis to their studies, being
a good way to measure the total amount of water inside the cell wall, because it is applied to a swollen
substrate. Following this, as can be seen in Figure 2-16, a cumulative curve can be predicted and
gives the pore inaccessible volume to a given probe, as function of its diameter. The plateau of the
curve is the fiber saturation point [53], and corresponds to a probe which size is too large to fit into the
pores.
Figure 2-16: Schematic illustration of pore distribution curve to solute excluded from the pores [53].
According to the protocol mentioned, in 1986, Lin and his co-authors [53] reported an experimental
methodology employing a differential refractometer to determine the final concentration of solutions,
combined with statistical treatment of the data to estimate the porosity. This treatment consisted in
representing each point as an average of four samples (each sample analyzed 4 to 8 times).
In 1989, with a technique similar to the last author, Thompson [24] suggested a solute balance
before and after contacting the wet substrate to determine the concentration in probe. However, since
he used a series of Dextran as probes, and due to the high viscosity of the solutions prepared, as well
the variances in readings of refractometry, the author suggests the utilization of a polarimeter. Both of
these techniques have in common the long standby time in the stirring step.
Later in 1993, an expeditious and accurate simplification of Stone and Scalan technique was
developed by Gama et al. [19]. This method aimed to eliminate sources of experimental error and they
demonstrate that the external surface represents a major part of the accessibility to the enzymes in the
beginning of hydrolysis reaction.
Differences in the accessible pore volume of pretreated samples compared to untreated were
found by Ishizawa and his co-workers [42], in 2007. However, no significant difference in porosity was
observed between samples pretreated at different severities.
Recently works [14, 41] still follow the same methodology that Stone and Scalan proposed. The
authors reported an important point: new types of macro-pores can be created using this technique, as

21
well, some micro-pores are lost irreversibly. The collapse of pores can be partially recovered by re-
wetting the samples after drying and this technique can be used to reveal this phenomenon.
Comparison of protocols
Subsequently to the review done about the protocols, some of them were explored in a more
detailed way with the intention to establish an adequate technique to our intends.
An important point to take in account, previously to the technique itself, is the quantity of material
that will be required for each experiment. As can be seen in Table 2-2, the mass of substrate, ms, used
is similar for different protocols. All authors report the use of wet substrate, assuming that the
substrate is saturated in water. With the exception of Lin et al. [53], the quantity of one gram of wet
substrate is consistent.
Concerning the probe solution, regarding the ratio mass of substrate, ms, by volume of probe
solution, Vi, the values are discrepant. The same thing occurs with the concentration of initial solution,
Ci, that ranges between 0.5 – 1 %w/v. At this point, a reflection was done considering the conditions of
work (as characteristics of the substrate).
Table 2-2: Preparation conditions for substrates and probe solutions.
Replicates ms (g) Vi (mL) ms/Vi (g/mL) Ci (%w/v) Reference
4 10 20 0.5 0.9 [53]
3 1 20 0.05 0.5 or 2 [24]
4 1 11 0.09 0.7 [19]
3 0.5 – 1 1 0.5 – 1 1 [42]
Glucose was utilized by all authors, being the probe with smaller diameter (8 Å). Various PEG with
average molecular weight between 13 and 240 Å were used, except by Thompson that used a series
of Dextran. For the last, the author advises about the high viscosity of the solutions and posterior
difficulty in perform the analysis to the final solution. The authors refer also the use of some
carbohydrates with low molecular weight to complete the series of diameters (such as cellobiose and
fructose).
Once the goal is to develop a standard methodology to determine the accessible pore volume of a
substrate, it is important to attentively observe each step and deliberate about the most accurately way
to perform it. Regarding the protocols, a differentiation of three main steps can be done: stirring,
settling down and separation to analyze – Figure 2-17.
Figure 2-17: General scheme of the different steps to perform solute exclusion.

22
The first phase consists in putting in contact the substrate with the probe solution. This step can be
the key of the technique because it allows the probe molecules to enter or not into the pores. The type
of stirring diverges a lot from author to author. For Lin et al. and Thompson, an occasional mechanic
shaking is done during a long period of time, 36 hours and overnight, respectively. The same type of
technique is adopted by Ishizawa, but done manually (each 30 seconds during 2 or 3 hours). Gama
and his co-workers used an orbital shaker during a 5 hours period.
The settling down step is referred as important to avoid cellulose packing during centrifugation,
which would give rise to water removal from the pores, by Gama, suggesting a 1 hour duration.
In order to analyze the supernatants (probe solution in the end), different separation processes are
recommended. Both Lin et al. and Thompson performed a filtration, using a Buckner funnel. Gama et
al. did a centrifugation at 5000 rpm during 10 minutes to the supernatants recovered from settling
down. Similar to the last author, Ishiwaza et al. performed a centrifugation and then the supernatant is
recovered using a syringe. To assure that no particles will interfere during measurements, the same
author suggests the use of a 0.45 m nylon filter before transfer the samples into the analyzers.
The final concentration of the solutions is then measured and the pore volume determined. All the
authors suggest the use of refractometry with some modifications. Lin et al. proposed the use of a
differential refractometer. Thompson also used a differential refractometer at beginning but due to the
viscosity difficulties, he changed to a polarimeter. In the same way, Gama et al. determined the
concentration refractometrically, using HPLC. An HPLC column equipped with a refractive index
detector was used also by Ishiwaza and his-coworkers. However, there is any information about HPLC
columns specifications, being this a lack in literature.
Conclusion and aim of the study
From all techniques available, solute exclusion was chosen and explored in detail. The protocol
from Gama et al. was selected as basis to reproduce the technique, since it is the most rigorous from
the experimental point of view. The same method will be reproduced and a new approach will be
proposed. To discuss the results, an approach of statistical treatment of the data will be performed,
similar to Lin et al.. Bounding with this, a model equation will be proposed to describe pore size
distribution.
To follow up this, the next parameters will be investigated and optimized: size particle of the
substrate, ratio between mass of substrate and volume and/or concentration of initial solution, contact
time between substrate and probe solution, importance of the settling step, and time and speed of
centrifugation. Another focal point will be the repeatability and reproducibility of the experiments,
reported both as not satisfactory.
In the end of this study a methodology and a model equation will be defined. This will be an
important contribution to the understanding of the relationship between porosity of substrates and
their reactivity on enzymatic hydrolysis.

23
3 PRETREATMENT OF THE SUBSTRATE
This work has as purpose to develop a methodology that directly relates the severity of the
pretreatment with the reactivity of the substrate in terms of enzymatic hydrolysis. For this intention, an
acid pretreatment was done, at different temperatures, in order to obtain five samples with various
characteristics. Hereafter, an enzymatic hydrolysis was performed in standard conditions in order to
estimate their reactivity.
Thermochemical pretreatment
The lignocellulosic substrate should be prepared for enzymatic hydrolysis, to make cellulose
accessible to enzymes. For this, an initial step of pretreatment is frequently recommended (section
2.3.1), before the hydrolysis, which will allow increasing the reactivity of the initial substrate.
The pretreatment was done in a pilot unit that works in batch and which goal is to perform
thermochemical experiments with lignocellulosic substrates, where the treatment can be done in acid
or alkaline medium. The intention of the experiments is to study the reactivity of the substrate, as
function of the operating conditions (as temperature or residence time).
Figure 3-1: Pilot unit U868 for thermochemical pretreatment of lignocellulosic substrates.
purge valve
air refrigeration
system
nitrogen valve
stirring motor
reactor

24
As can be seen in Figure 3-1, this installation is constituted by an autoclave where the reaction
occurs. The reactor has a capacity of 2L and contains a mechanic agitation system coupled.
The unit works under controlled nitrogen pressure and temperature. This control is assured by a
PID which values are specified previously to the experiment. The temperature is measured on the top
and on the bottom of the reactor and is controlled by the alternate system of heating and refrigeration.
The substrate used was wheat straw and to respect the requirements of the unit (substrate
particles diameter between 0.5 – 4 mm), a previous grinding (from 5 to 2 mm) and sieving (between
0.71 – 2 mm) was done. The acid employed was concentrated sulfuric acid.
Five samples (example on Figure 3-2) were prepared by acid pretreatment under different
severities generated by the temperature of reaction (from 100 to 180 °C, with an increment of 20 °C).
The acid concentration, as well the residence time and stirring were maintained constant:
Table 3-1: Operating conditions of the pretreatment.
ID 1080 1081 1082 1083 1084
T (°C) 140 120 100 160 180
mds (g) 40
mtotal (g) 400
H2SO4 (%w/w) 1
t (min) 20
stirring (rpm) 3000
Figure 3-2: Samples obtained by different severities of acid pretreatment.
For all assays were determined a mass balance of 97.3±0.6 %, meaning that the majority of the
mixture has been recovered. However, regarding the figure below in terms of dry substrate, the
material when dried represents a lower quantity which decreases with the severity of the pretreatment:

25
Figure 3-3: Yield in dry substrate after pretreatment.
Since the pretreatment involves an acid component, the substrate will be in an acidic medium, and,
by this, protons can be released in hydrolysis, bringing difficulties in the pH control. Hereafter, was
applied a post-treatment of neutralization of the mixture, to obtain a pH around 4.8. This is an
important point for hydrolysis because the enzymes work in a restrict range of pH [16, 26, 27] and will
suffer denaturation if the range is not respected. Still, the pH should be constantly controlled during
hydrolysis.
Once neutralization is done, the solid was recovered by pressing. The liquid part consists in the
extractible sugars and degradation products. Both solid and liquid phases were conserved.
Following this, the liquid part was analyzed by HPLC in order to determine its composition. The
solid phase was washed to obtain a final substrate without sugars and/or degradation products.
Acid hydrolysis
To determine the glucose potential of the pretreated substrates, an acid hydrolysis was done in two
stages: a first one at 30 °C and a second one at 121 °C (NREL method), as schematized in Figure
3-4.
Figure 3-4: Schematic representation of two step acid hydrolysis.
In the first step, the acid and the substrate were added into a flask. The substrate was previously
dried to obtain a water content of 10% as maximum. Then, this mixture was put in a thermostatic bath
at 30 °C, during 60 minutes. Before the second step, deionized water was added and the mixture was
50
60
70
80
100 120 140 160 180dry
su
bs
tra
te y
ield
(%
)
temperature ( C)

26
homogenized. Then, the second hydrolysis was performed at 121 °C and 60 minutes. For this, it was
used a sterilizer working at 2 bar.
Hereafter, the effluent was subjected to a centrifugation at 4000 rpm during 20 minutes and the
supernatant was recovered to analyze. An HPLC analysis was performed, as well, measurements
using the Glucostat (glucose analyser):
Table 3-2: Results from acid hydrolysis.
ID CR1082L CR1081L CR1080L CR1083L CR1084L
T (°C) 100 120 140 160 180
HPLC Xylose (%) ~0 ~0 5.9 2.1 ~0
Glucose (%) 49.4 54.8 60.06 62.7 43.5
Glucostat Glucose (g/dl) 111 121 135 138 100
Enzymatic hydrolysis
Here cellulose was put in contact with an enzymatic cocktail under specific temperature and pH
conditions. This cocktail catalyzes the hydrolysis and usually operates with highest efficiency at
temperatures of at least 45 °C, and at a pH of 4.8 (as reported in section 2.3.2).
To be economically viable, this step should be performed at high dry substrate content, to reduce
the water consumption and also the associated equipment cost (such as, reactor volume).
The enzymes used in this study are produced by Trichoderma reesei (Genencor GC220 plus
Novozymes N188). Then, the beginning of the reaction happens when enzymes are introduced into
the flask. Samples of 1 mL were recovered at predefined times (1.5, 3, 6, 24, 48 and 72 hours) for
analyzing. Before each sampling, the temperature and the pH of the mixture was controlled and
adjusted when required (45°C<T<50°C and 4.6<pH<4.9).
To stop the reaction, the samples were put into a water bath at 90 °C, during 15 minutes. This step
denatures the enzymes. After refrigeration, the samples were centrifuged (4000 rpm) during 20
minutes.
After this, the measurement of glucose content was performed in the equipment below (Figure 3-5).
Each sample was measured twice and the final value taken is the average of both values.

27
Figure 3-5: Glucostat used to measure the concentration in glucose of the samples.
Finally, it was possible to compare the efficiency of the pretreatment by studying the reactivity of
the different pretreated substrates. Regarding the figure below, it is clear that the best yields in
glucose correspond to the sample pretreated at 160 °C. Hereupon, the pretreatment at this
temperature is the most efficient at the operating conditions described in Table 3-1.
Figure 3-6: Glucose yield on enzymatic hydrolysis (Appendix 8.1).
Higher the temperature of the pretreatment is, better is the reactivity (from 100 to 160 °C).
However, at 180 °C a loss of reactivity is observed. It is possible that the degradation products (mainly
furfural) are condensed at this temperature to form humins. This will drop the substrate area off and
reduce the reactivity.
0
10
20
30
40
50
60
0 10 20 30 40 50 60 70 80
glu
co
se y
ield
(%
)
time (h)
100 °C 120 °C 140 °C 160 °C 180 °C
rotative sampler
results reader
samples

28

29
4 METHOD TO DETERMINE THE PORE VOLUME
The main part of this work intends to develop a methodology to determine the accessible area of
the substrates. Among the techniques presented in literature, solute exclusion technique was chosen
due to its advantages and interests to the work. To define the initial conditions of the experiments, a
preliminary work was performed and is described in the current chapter.
Materials and methods
Substrates
In order to compare the samples between them and with the literature, standard celluloses were
chosen (Avicel PH101 and Alphacel C40) and wheat straw was utilized (native wheat straw, non-
washed, washed, and pretreated at 160 °C – see previous chapter).
To prepare substrates in dried form, the material was dried during a night, at 45 °C. To obtain
substrates in water saturated form, the water retention value method was used.
Probe Molecules
Several criteria are taken in account to select the probe molecules. First of all, the polymers should
represent a significant molecular weight distribution, using a range over the whole range of pore sizes,
anticipating the results. Then, concerning the hydrodynamic, they should exhibit spherical shape in
solution [40, 53]. Last, but not the least, the probes should not adsorb and/or react with the material
being analyzed.
Hereupon, it was chosen a set of probes of various molecular sizes (Table 4-1) that fits all the
criteria above: PEG’s ranging in relative molecular weight from 200 to 35000 g/mol and having
equivalent spherical diameters in solution from 13 to 170 Å. In addition, two low diameter
carbohydrates and a Dextran (with diameter similar to a PEG) were used to supplement the study.
Table 4-1: Molecular weights and solution diameters of probes used (Appendix 8.2).
Probe Molecular weight (g/mol) Diameter (Å)
Glucose 180 8
Cellobiose 342 10
PEG 200 190-210 13
PEG 600 570-630 21
PEG 1500 1300-1600 33
PEG 2000 2000 40
PEG 4000 4000 56
PEG 8000 7000-9000 84
PEG 10000 10000 90
Dextran 75000 72000 120
PEG 20000 15000-20000 130
PEG 35000 35000 170

30
Methodology for substrate in saturated form
The first experiments were performed using a previously saturated substrate, as described in the
literature. The protocol consisted in the following sequence (Figure 4-1):
a) Saturation of the substrate in water by water retention value method;
b) Preparation of probe solutions;
c) Mixing of 1 g of saturated substrate with 10 mL of probe solution 1 %w/v – orbital shaking
during 3 h, at ambient temperature;
d) Decantation during 1 h and removal of exceeding solution;
e) Centrifugation of remaining mixture, during 30 min, at 3000 rpm and recovering of
supernatants to analyze;
f) Determination of dry matter content.
Figure 4-1: Scheme of the methodology for substrate in saturated form.
Water retention value method – WRV
This method is a useful reference to evaluate the performance of cellulosic materials relative to
moisture behavior [54, 55]: it measures the water retained by a material after centrifuging under
standard conditions.
The water retention value, WRV, is defined as the ratio of water contained in the sample after
centrifuging, at a certain force and time, relative to the dry weight of the sample, and is given by:
WRV(g/g) =Wwet − Wdry
Wwet
Equation 4-1
where Wwet is the weight of the sample after centrifuging and Wdry is the absolute dry weight of the
sample [54, 56].
The norm TAPPI UM 256 was used as reference to establish the operating conditions for this
methodology. So, to measure the WRV, the substrate was first put in contact with water during a night

31
under magnetic agitation. After, one hour decantation was done and then the excess of water was
removed with a syringe and the solid was recovered into a centrifugation tube. The centrifugation
occurred at 3000 rpm, during 30 min and the solid was recovered and weighted immediately. To
determine the dry weight, the solid stayed in the oven, at 45 °C, during a night and was weighted in
the end, and Equation 4-1 was employed.
Preliminary results
The factors that may influence WRV measurements include sample weights, centrifugal time and
force, pore size of filters used, and cellulosic particle sizes [54]. So, to verify the repeatability of the
results, step a) was particularly studied using the two standard celluloses (Avicel PH101 and Alphacel
C40) under the conditions described below – Table 4-2. Then, in order to perform this step, for
samples of each cellulose were prepared: three flasks with one gram of substrate each one, and a
fourth flask with five grams of substrate. To each flask were added 100 mL of water.
Table 4-2: Initial conditions of substrates to water retention value method.
ID Avicel PH101 Alphacel C40
ms (g) 1 (x3) 5 1 (x3) 5
water (mL) 100 100
In the assay with Avicel, the decantation was performed and no clearly separation occurred
(particles in suspension were observed). The experiment was repeated three times in these conditions
and one time with 6 h decantation, always with the same observation. For Alphacel a visible
decantation occurred at each time.
Then, six different samples were obtained from the four initial flasks: three samples were prepared,
one from each flask with 1 g ms (Set A), and the other were three obtained from the fourth flask (Set
B). This intended to verify if there is an influence in preparation of WRV method, regarding the sample
weights.
To determine the WRV, the saturated substrates were put into six different aluminum cups
(approximately 1 g) and transferred to the oven, at 45 °C, in order to determine also the ideal time of
drying.
As can be seen in Figure 4-2 and Figure 4-3, for both substrates, 2 or 3 hours is the minimum
period sufficient to get a material completely dried. This conclusion reduces drastically the time of the
experiment since only 3 hours are required for these substrates, instead of one night, as reported in
literature. However, maybe this time depends on the nature of the substrate.
Regarding the mass of initial substrate, there is no apparent difference in using 1 g or 5 g to
produce saturated substrate (Appendix 8.3). However, this sentence is contradictory when standard
deviation is included in the results: the values obtained from the same initial solution (Set B) produce a
more consistent set of samples, presenting an evident lower error.
Comparing the fiber size of the substrates (Avicel PH101 – 50 m and Alphacel C40 – 120 m),
there are no significant conclusion that can be made.

32
Figure 4-2: Evolution of mass substrate during drying, for Avicel PH101 (Table 8-4, Appendix 8.3).
Figure 4-3: Evolution of mass substrate during drying, for Alphacel C40 (Table 8-6, Appendix 8.3).
Another important point concerns the filters used during centrifugation. Sometimes holes were
found after centrifugation with no apparent reason. Factors that contribute for this can be the force of
centrifugation or the quantity of sample. Actually, this problem was found only for the 5 g samples,
which leads to conclude that this could be the cause of the holes. To solve this, an equilibrium
between the mass of sample and force of centrifugation should be done, for example, reducing the
force for heavy samples. However, the time of centrifugation maybe should be high to compensate. A
simply solution will be to reduce the mass of sample to centrifuge.
In the experiments, the filters used present blank color. When the substrate in study has the same
color as the filter, during the transfer of the matter to the cup, particles of filter can be dragged. This
could have repercussions in the final results, so the use of filters with a different color is proposed.
0.4
0.6
0.8
1.0
1.2
1.4
0 1 2 3 4 5 6
ma
ss
of
su
bs
trate
(g
)
time (h)
Set A
Set B
0.4
0.6
0.8
1.0
1.2
1.4
0 1 2 3 4 5 6
ma
ss
of
su
bs
tra
te (
g)
time (h)
Set A
Set B

33
Figure 4-4: Results from determination of WRV for Avicel PH101 (Table 8-7, Appendix 8.3).
Figure 4-5: Results from determination of WRV for Alphacel C40 (Table 8-8, Appendix 8.3).
Lastly, the WRV was determined and represented graphically (Figure 4-4 and Figure 4-5), being
1.01±0.19 g/g for Avicel and 0.73±0.02 g/g for Alphacel, with a confidence interval of 95%.
With these results the repeatability for the second substrate is validated, but not for the first one. At
this point, Alphacel was chosen to do the assays with the proposed methodology.
Methodology for substrate in dried form
For the second type of experiments, the samples were previously dried and this procedure is not
described in literature. So, an adaptation of the method using a substrate in saturated form was done,
following the next steps (Figure 4-6):
a) Drying of the substrate in oven by night, at 45 °C;
b) Preparation of probe solutions;
c) Mixing of 1 g of dried substrate with 10 mL of probe solution 1 %w/v – using orbital shaking
during 3 h, at ambient temperature;
d) Decantation during 1 h and removal of exceeding solution;
e) Recovering of remaining solution to analyze.
0.6
0.8
1.0
1.2
1 2 3 4 5 6
WR
V (
g/g
)
ID sample
0.68
0.72
0.76
0.80
1 2 3 4 5 6
WR
V (
g/g
)
ID sample

34
Figure 4-6: Scheme of the methodology for substrate in dried form.
Preliminary results
Once Alphacel demonstrated to be a more adequate substrate to practice the experiments, it was
also used in dried form methodology. A first series of trials was done to observe the main experimental
barriers and find a solution for them.
Concerning the substrate drying, due to the results obtained for the other methodology, it was clear
that three hours of drying were sufficient. Even less it will be acceptable, since the substrate in this
case is not saturated, it contains only the water from the air (near 4 %w/w).
Instead of the orbital shaking, as Gama et al. refer, a magnetic agitation was performed since the
first option did not allow the mixture of the substrate with the probe solution.
Probe solutions analysis
In order to determine the concentration of probe in the final solutions, a refractometer Anton Paar:
RXA170 was used – Figure 4-7.
For a known concentration solution in the begging, the refractive index of the final solution was
determined and then converted to concentration using calibration curves.
Figure 4-7: Refractometer used to measure the refractive index of solutions.
measuring chamber
results reader

35
To avoid interferences in the results, a syringe coupled to a 0.45 m filter (Figure 4-8) was used to
ensure that no fragments were transferred to the measuring chamber.
Figure 4-8: Sample recovered after decantation and prepared to analyze in refractometer.
Due to the few quantity of supernatant obtained in the end of the experiments, the samples were
measured only one time. For the calibration curves, the solutions were prepared in a quantity sufficient
to obtain at least four measurements for each concentration.
Calibration of the refractometer
The aim of the calibration experiments was to define an equation that relates the refractive index
measurement with concentration of probe. Then, the repeatability and reproducibility of the
measurements were also tested. So, as said above, each refractive index was measured four times,
for each concentration, for the different probe molecules. Also, new solutions were prepared for each
experiment.
To convert the refractive index measurements in concentration of probe, the linear correlation
obtained for each series of data was used:
Cf =nDsample − b
m Equation 4-2
where m is the slope of the calibration curve and b is the interception with the yy axis and
corresponds to a solution with a null concentration in probe (so, water).
Calibration curves were performed using an adequate concentration range, depending on the
substrate used (between 1 %w/v and 3 %w/v). As an example, the values measured for PEG 35000
and PEG 200 are presented and discussed.
Figure 4-9: Linear correlation between refractive index and concentration for PEG 35000 (02/07/2015).
y = 0.00135x + 1.33289R² = 0.9999
1.3340
1.3350
1.3360
1.3370
1.0 1.5 2.0 2.5 3.0
nD
concentration (%w/v)
syringe
filter

36
Figure 4-10: Linear correlation between refractive index and concentration for PEG 35000 (10/07/2015).
Regarding previous figures, it was possible to verify a good linear correlation between refractive
index and concentration for PEG 35000 and the equation is the same for different days. This proves
that the assays with PEG 35000 are repeatable, as well, reproducible. But then, when other probes
were tested, a different conclusion can be made. For example, for PEG 200:
Figure 4-11: Evolution of calibration curves for PEG 200 (between 28/05/2015 and 15/07/2015).
Table 4-3: Linear correlations between refractive index and concentration for PEG 200 (Figure 4-11).
ID Date Linear correlation R2
15/07/2015 y = 0.00132x + 1.33274 0.9996
10/06/2015 y = 0.00133x + 1.33272 0.9985
28/05/2015 y = 0.00130x + 1.33272 0.9970
29/05/0215 y = 0.00126x + 1.33276 0.9975
y = 0.00135x + 1.33289R² = 1
1.3340
1.3350
1.3360
1.3370
1.0 1.5 2.0 2.5 3.0
nD
concentration (%w/v)
1.3340
1.3341
1.3342
1.3343
1.3344
1 1.05 1.1 1.15 1.2 1.25
nD
concentration (%w/v)

37
As can be seen in Table 4-3, the sensibility for this probe is very similar (same slope). Also the
coefficient R² demonstrates the good repeatability of the data obtained. Nevertheless, regarding
Figure 4-11 it is clear that there are some variations. These results are no negligible for concentration
determination.
So, two parameters are significant when calibration is performed: the probe used and the evolution
of the measurements in time. The first one is evident in the example above and, in general, this
variation will be related with experimental errors (such as preparation of the probe solution). Regarding
the evolution in time, for the same probe, it makes us to conclude that calibration should be done at
each experiment in order to avoid errors in calculations.
Then, after all calibrations, can be concluded that:
the sensibility of the method to the probes is similar (Figure 4-12);
the calibration curves should be done at each experiment;
about the number of measurements, can be settled that one or two measurements will be
sufficient due to the good repeatability observed.
All the linear correlations obtained between refractive index and concentration for all the probe
molecules are presented in Appendix 8.4.

38
Figure 4-12: Examples of calibration curves for the different probes.
PEG35000
PEG200
Glucose
Cellobiose
1.3340
1.3350
1.3360
1.3370
1.0 1.5 2.0 2.5 3.0
nD
concentration (%w/v)
PEG35000 PEG20000 PEG8000 PEG4000 PEG1500 PEG600 PEG200 Glucose Cellobiose

39
5 RESULTS OF SUBSTRATE POROSITY
For the methodologies described and the equations established, the accessible pore volume to
probe molecules into the substrate was determined. Following this, the reproducibility of the procedure
was evaluated and discussed for the different substrates. This section intends to explore the results
obtained, as well discuss about them.
Determination of pore volume
Saturated substrate method
In this method, two phases can be distinguished: the beginning of the experiment (t0), where the
concentration in solution, Ci, is assumed to be the same concentration of the probe – Equation 5-1,
and a second time (t), when the concentration will be different from the initial, Ceq, if the probe enters
inside the pores – Equation 5-2.
t0 ⇒ nprobe = Vsol ∙ Ci Equation 5-1
t ⇒ nprobe = (Vsol + Vp) ∙ Ceq Equation 5-2
By mass balance, the pore volume, Vp, can be determined by:
Vp =Vsol ∙ (Ci − Ceq)
Ceq
⇔ Ceq = Ci ∙Vsol
(Vsol + Vp) Equation 5-3
As said before, the substrate is saturated. Consequently, if the probe goes into the pores, the
concentration of the final solution, Ceq, is decreased by dilution.
In order to use the saturated substrate method, it is appropriate to determine the water retention
value. Subsequently, at each experiment, this value was calculated. As can be seen in Figure 5-1, the
method is consistent when Alphacel is used, presenting a value of 0.64±0.02 g/g.
Figure 5-1: Distribution of values for water retention method, for Alphacel (Table 8-10, Appendix 8.5).
0.60
0.62
0.64
0.66
0.68
0.70
1 6 11 16
WR
V (
g/g
)
ID
SE01 SE02 SE03 SE04 SE10

40
So, from Equation 5-3, it is expected to obtain a final concentration lower (if probe penetrates) or
equal (if nothing happens) to the initial solution.
Regarding the final concentrations for these trials (Table 8-10, Appendix 8.5) using this method the
porous volume determined is often negative. This result is obtained whenever the final concentration is
higher than the concentration of initial solution:
Ceq ≥ Ci ⇒ (Ci − Ceq) ≤ 0 ⇒ Vp ≤ 0 Equation 5-4
Figure 5-2: Calibration curve and results for experiment SE02 (Table 8-10, Appendix 8.5).
Taking the experiment on Figure 5-2 as example, the relation described by Equation 5-4 is shown
and then pore volume is negative.
The first issue that can be discussed is the possible existence of soluble compounds or
contaminants in the substrate. Chen and his co-workers [57] report a mass percentage of water-
soluble materials varied from 14 to 27% for corn samples. Among these soluble, monomeric sugars
(primarily glucose) were found to be the predominant water-soluble components. These compounds
can significantly affect the refractive index measurements. Following this, it is reported in literature [19,
57] that the removal of soluble contaminants and fine cellulose particles should be done, since they
are a source of interference during the refractometric measurements.
Additionally, sample preparation can also add significant deviation in the results. For example,
during centrifugation can occurs water removal from the pores, due to the cellulose packing [19]. After,
when samples are prepared for mixing with the probe solution, evaporation of water from the saturated
substrate can also occur.
Therefore, the cause of these unexpected results was not clear. For this reason, it was decided to
discard this first methodology.
Dried substrate method
To determine the porous volume with this second methodology, it was necessary to make
assumptions and do the calculations in two different parts.
When a dried substrate is used, after the contact with the probe solution, two situations can occur:
only water penetrates into the pores or the solution of probe penetrates into the pores, depending on
the size diameter of the probe.

41
Figure 5-3: Scheme of a porous substrate and penetration of molecules.
Following the previous reasoning, by using several probes of different sizes, it will be possible to
obtain a distribution of pore sizes. By assumption, once water is a small molecule (bond O-H lengths
approximately 1 Å [58])., then enters on the total volume of pores, Vp, However, the probe, depending
on its size, penetrates only on the volume where the pore diameter is higher than its diameter, Va (as
schematized on Figure 5-3). The difference between the total pore volume and the accessible volume
is defined as the inaccessible volume, Vi.
Furthermore, it is assumed that the probe concentration into the pores is the same of the solution
that surrounds the substrate.
Hereupon, the following equations were established:
nprobe = Vsol,i ∙ Ci Equation 5-5
nprobe = (Va + Ve) ∙ Cf Equation 5-6
Vp = Va + Vi Equation 5-7
Vsol,i = Ve + Vp Equation 5-8
This equation system cannot be solved by itself. In this way, some hypotheses were assumed in
order to determine the unknown variables.
Then, for the probe molecule with higher diameter (PEG 35000), it was assumed that this probe is
too big to penetrate inside the pores (even the biggest ones) and the accessible pore volume is
defined as equals to zero. Doing this, it was possible to determine the external volume, Ve, with
Equation 5-6. This value will be constant for all the experiments with the probes of smaller diameter
than PEG 35000. After, the total pore volume can be also determined from Equation 5-8, and, in this
case, it corresponds to the inaccessible pore volume to the probe.
On the other side, for the remaining molecules the inaccessible volume can be easily estimated
using Equation 5-9, that results from the rearranging of the equations below.
Vi =Vsol,i ∙ (Cf − Ci)
Cf
Equation 5-9
At this time, all the parameters were known, therefore the accessible volume was estimated once
the porous volume was assumed as constant.
Knowing these parameters, the pore volume was estimated for all the substrates (Table 5-1) and
the distribution is discussed in the next section.

42
Table 5-1: Exterior and maximal pore volume determined for the substrates.
Substrate 𝐕𝐞 (mL/g mds) 𝐕𝐩 (mL/g mds) Reference
Alphacel 9.17±0.03 0.77±0.04 Table 8-11
non-washed native wheat straw 9.81±0.04 0.18±0.05 Table 8-16
washed native wheat straw 9.23±0.04 0.72±0.04 Table 8-19
washed pretreated wheat straw 9.25±0.01 0.75±0.00 Table 8-20
It was expected an increment on porous volume in the order described in Figure 5-4. Regarding the
results, this relationship was verified when non-washed native wheat straw is compared with the
washed, as well, the washed pretreated. However, the value for non-washed could be probably wrong
due to the uncertainty associated to the measurements and calculations.
Then, comparing washed native with washed pretreated, the result is controversial. The porous
volume is the same if standard deviation is taken in account. One reason for this can be the difficulty
to delete the part of the soluble compounds, which still remain after washing, in the refractive index
measurement. Another cause can be related with the pretreatment effects. In this step, some pores
can be created with the treatment, but also other ones can be destroyed [59, 60]. This can result in
larger pores, or, on the other side, irreversible collapse of pores. So, even if porosity value does not
change, new pores with different sizes can be created.
Figure 5-4: Expected increasing in accessible porous volume by type of substrate.
The total pore volume determined here corresponds to the fiber saturation point, FSP. As said
previously, this value corresponds to the plateau of the distribution curve (Figure 2-16). In this work
were obtained results of the same order of magnitude than these obtained by other authors (Table
5-2).
For Alphacel, the standard cellulose, it was found a value near to Avicel PH 101. This commercial
product was studied by Gama and his-co-workers. However, it is important to retain that Gama used a
protocol with the substrate in saturated form. In the present work, dried substrate methodology was
applied.
For substrates in native form, it is not clear in literature if they were previously washed. Comparing
with the result obtained in this work, the value obtained by Thompson [24] is similar. Nevertheless, the
substrate is not the same, consequently, any conclusion can be done. Regarding other studies [59],
also the substrates used are not the same that the one used in this study. Further, the value obtained
is quite different from wheat straw (same order of magnitude).
Lastly, to the substrate pretreated at 160 °C and washed, literature refers a similar value but for
mixed hardwood. This substrate was pretreated under similar conditions than wheat straw.
non-washed
native
washed
native
pretreated
washed
+𝐕𝐩

43
Furthermore, it can be noticed that there are no significant difference in FSP between the native mixed
hardwood sample and the pretreated one. The same evidence occurred in the present study.
Table 5-2: Fiber saturation point by solute exclusion technique, from literature.
Substrate Pretreatment FSP (mL/g) Reference
Avicel PH 101 0.70
[19]
Sigmacell 100 1.09
Whatman CF 11 0.42
Crystalline cotton 0.93
Amorphous cotton 1.35
mixed hardwood
0.76
[24]
180 °C, 1 %w/w H2SO4 0.77
200 °C, 1 %w/w H2SO4 1.06
220 °C, 1 %w/w H2SO4 1.33
mixed hardwood 0.51
[59] 200 °C, 1 %w/w acid 0.92
white pine 0.49
200 °C, 1 %w/w acid 0.71
Solka Floc BW 300 1.6 [53]
Pore volume distribution
As said before, the measurements of refractive index for each sample were done only one time,
due to the restrict quantity of sample available. Then, each point was obtained from a series of four
samples for each different probe experiment – Figure 5-5. Thus, the experimental errors were
calculated as the relative standard deviation of the data considered for calculations (with a 98%
confidence interval) and are shown by the error bars in the figures along this chapter. The fifth sample
was prepared adding only water to the substrate. This sample served as control, allowing the
subtraction of possible soluble compounds that interferes in the refractive index measurements.
Figure 5-5: Example of a series of samples in a trial.
blank
sample
samples
in a trial

44
For the series of probes used and for the different substrates, it was expected to obtain a set of
points similar to what is shown in the next figure, obtained from previous studies:
Figure 5-6: Pore volume distribution for pulp fibers exposed to different conditions [40].
This data reveals that there is a plateau corresponding to a maximum inaccessible volume to the
probe or the fiber saturation point (signed on Figure 5-6 with an arrow). Also occurs a decrease of the
volume as function of the probe molecular diameter. This looks like to be independent from the
methodology or pretreatment applied to the substrate [40] – Table 5-3.
Table 5-3: Fiber saturation point from different pretreatments.
Substrate Type Pretreatment FSP (mL/g) Reference
wheat straw acid 0.72
this work 160 °C, 1 %w/w H2SO4 0.75
mixed hardwood acid
0.76
[16]
180 °C, 1 %w/w H2SO4 0.77
200 °C, 1 %w/w H2SO4 1.06
220 °C, 1 %w/w H2SO4 1.33
mixed hardwood alkaline
0.5 h, H2O2 0.87
[33] 5 h, H2O2 1.43
19 h, H2O2 1.41
mixed hardwood organosolv
10 % ethylenediamine 1.08
[33] 50 % ethylenediamine 0.96
70 % ethylenediamine 1.47

45
So, from substrate to substrate, this pore volume distribution, as function of pore diameter, can be
defined by two variables: the value of the plateau or fiber saturation point and a constant. The plateau
will indicate the total porous volume of the substrate. The constant will define the behavior of the curve
and will depend on the substrate used.
Following this reasoning, an equation was proposed with the intuit of modeling the experimental
results obtained in this work:
Vi = Vi,max(1 − e−kD) Equation 5-10
where Vi,max is the value of the plateau and is determined by using the higher molecular diameter
probe, D, when accessible volume is null. The constant, k, was determined using the minimum square
error method. Both these parameters are specific of each substrate.
The type of pores in the substrates was also determined (Table 5-4). This study reveals the type of
pores that exist, but says nothing about their location in the cell wall.
Table 5-4: Types of porosity in solids [61].
Type Pore size (nm)
microporous < 2
mesoporous 2 – 50
macropores > 50
To remind, the higher size diameter of probe molecule used in this work corresponds to PEG35000
(170 Å). By this, nothing can be concluded about the macroporosity of the substrates (when pore size
is higher than 500 Å) – example in Figure 5-7.
In summary, the type of porosity defined in this study is valid on the range of probe molecular sizes
studied (between 8 and 170 Å). Additionally, to determine the position of that pores, another type of
characterization method should be used (such as scanning electron microscopy).
Figure 5-7: Scheme representative of different levels of porosity.
To introduce the results, a briefly explanation is done about how the points were obtained. For this,
the example of Alphacel is used.

46
Alphacel C40
To obtain the first point, the larger molecule size diameter was used (PEG 20000, in this case). By
this, it was assumed that this molecule will not enter the pores and, consequently, the accessible
volume will be zero. In this way, the total porous volume of the substrate was determined.
To do that, the concentration of final solution was measured by refractometry (one measurement
by sample). Using the calibration curve, the refractive index measurement is converted in
concentration of probe. Finally, the exterior volume was determined using Equation 5-6, as well, the
total porous volume, using Equation 5-8. The final volume value was determined by the average of the
four measurements and standard deviation was also calculated.
Table 5-5: Example of exterior and total porous volume determination, for Alphacel (Table 8-11).
Sample mds (g) nD Cf (g/100mL) Ve (mL) 𝐕𝐩 (mL/g mds)
1 1.0131 1.33435 1.10 9.15 0.85
2 1.0136 1.33433 1.08 9.27 0.73
3 1.0008 1.33434 1.09 9.21 0.79
4 1.0031 1.33434 1.09 9.21 0.79
AV±SD – – – 9.17±0.03 0.77±0.04
As can be noted on Table 5-5, an error of 0.00001% on refractive index will reproduce an error of
0.1% on the value of final concentration. Furthermore, this will represent an error of almost 20% in the
total porous volume. The last value is tremendous and certainly will affect the final distribution.
At this point, the first point was obtained. In order to obtain the other points, progressively smaller
molecules were used. By this, the accessible volume to the probes was calculated using Equation 5-6,
once the external volume is constant for the substrate.
Finally, the model equation proposed can be employed and the pore size distribution was obtained.
The data for this substrate are clearly repeatable (Figure 5-8) and Equation 5-11 reflects these results.
Figure 5-8: Pore volume distribution for Alphacel (Table 8-21, Appendix 8.10).
Vi = (0.77 ± 0.04)(1 − e−0.05D) Equation 5-11
0.0
0.2
0.4
0.6
0.8
1.0
0 50 100 150 200
Inaccessib
le p
ore
vo
lum
e
(mL
/g m
ds)
diameter (Å)

47
As can be noticed, the data reveal that the pores are almost impermeable to probes with a
diameter above 50 Å. So, it can be concluded that exists micro and mesoporosity in this substrate, in
the studied range of diameters.
Another standard cellulose was tried by Gama and his co-workers, named Avicel, and the same
evidences were verified – Figure 5-10. To clearly compare the data obtained in this work with these
results, the accessible volume as function of diameter is represented on Figure 5-9:
Figure 5-9: Pore volume distribution for Alphacel (Table 8-21, Appendix 8.10).
Figure 5-10: Pore volume distribution of celluloses (Table 5-2) [19].
For the five substrates studied by Gama, it was observed a fiber saturation point that correspond to
probe molecules with a 50 Å diameter or higher.
In particular, regarding Avicel, it was found a behavior of distribution similar to Alphacel. However,
it is important to retain that Gama and his co-workers worked with a saturated substrate. In the present
work, the dried substrate methodology is used.
To conclude, the dried substrate methodology was validated for Alphacel.
0.0
0.2
0.4
0.6
0.8
1.0
0 50 100 150 200
Ac
ce
ss
ible
po
re v
olu
me
(m
L/g
md
s)
diameter (Å)
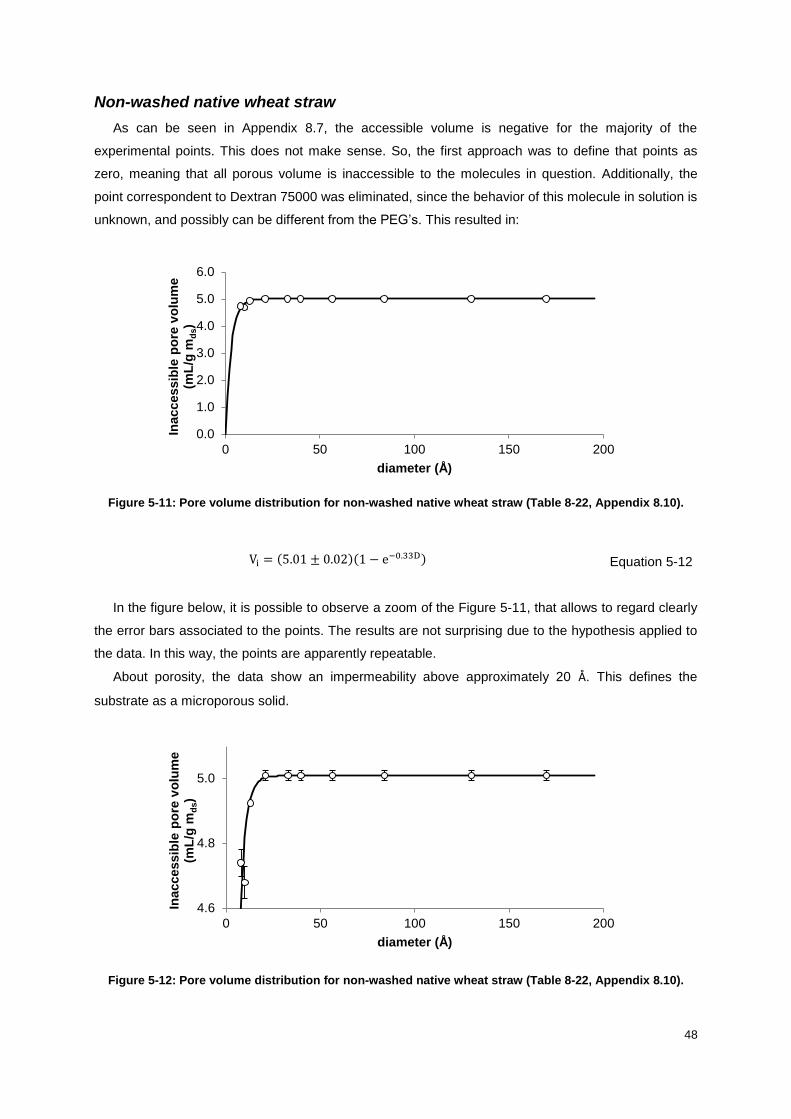
48
Non-washed native wheat straw
As can be seen in Appendix 8.7, the accessible volume is negative for the majority of the
experimental points. This does not make sense. So, the first approach was to define that points as
zero, meaning that all porous volume is inaccessible to the molecules in question. Additionally, the
point correspondent to Dextran 75000 was eliminated, since the behavior of this molecule in solution is
unknown, and possibly can be different from the PEG’s. This resulted in:
Figure 5-11: Pore volume distribution for non-washed native wheat straw (Table 8-22, Appendix 8.10).
Vi = (5.01 ± 0.02)(1 − e−0.33D) Equation 5-12
In the figure below, it is possible to observe a zoom of the Figure 5-11, that allows to regard clearly
the error bars associated to the points. The results are not surprising due to the hypothesis applied to
the data. In this way, the points are apparently repeatable.
About porosity, the data show an impermeability above approximately 20 Å. This defines the
substrate as a microporous solid.
Figure 5-12: Pore volume distribution for non-washed native wheat straw (Table 8-22, Appendix 8.10).
0.0
1.0
2.0
3.0
4.0
5.0
6.0
0 50 100 150 200
Ina
ccessib
le p
ore
vo
lum
e
(mL
/g m
ds)
diameter (Å)
4.6
4.8
5.0
0 50 100 150 200
Inaccessib
le p
ore
vo
lum
e
(mL
/g m
ds)
diameter (Å)

49
As said earlier, a blank sample was prepared and analyzed at each trial. From the experimental
point of view, this sample containing only water instead of probe solution was prepared as control and
is advised by some authors [24, 42]. The reason for this is to allow deducting the contributions of water
soluble extractives, or other contaminants, in the refractive index measurements.
Thus, for this substrate, final concentrations were corrected for the possible existence of
contaminants (directly deducted on refractive index measurements).
The correction was done by subtracting out the specific reading from blank to wheat straw sample.
For this, the average of all measurements for the blanks was calculated. Then, the refractive index
correspondent to the contribution of the soluble can be determined through:
∆soluble = nDblank,AV − nDwater Equation 5-13
where nDblank,AV and nDwater are 1.33420 and 1.33286, respectively. As can be noticed by the
value obtained, there is a strong contribution of the soluble components.
Likewise, it is possible to determine the variation associated to the presence of probe:
∆probe = nDsample − nDblank,AV Equation 5-14
where nDsample depends on the sample and probe solution.
To finish, the corrected refractive index can be calculated by:
nD′ = ∆probe − nDwater Equation 5-15
This equations were then applied and a completely different set of results was obtained, as can be
seen in the next figure:
Figure 5-13: Pore volume distribution for non-washed native wheat straw, with refractive index correction – all points included (Table 8-23, Appendix 8.10).
The new data obtained look like as expected. In a calculation of this type, there is a subtraction of
two big numbers, with a small deviation associated to each one. The result of this deduction is a small
number, with a deviation associated of its order of magnitude.
To model these results, the point correspondent to PEG 2000 was eliminated. Also, it was used the
same hypothesis than the one applied for Alphacel: once the accessible or inaccessible volume is
negative, then it turns zero. Then, for the three smallest diameters, there are not accessible volume. In
-1.5
-1.0
-0.5
0.0
0.5
1.0
0 50 100 150 200
Inaccessib
le p
ore
vo
lum
e (
mL
/g m
ds)
diameter (Å)

50
addition, a reasoning was done concerning the measured concentrations: the points with higher
deviation between them were eliminated. These data can be extensively consulted in Appendix 8.7.
Figure 5-14: Pore volume distribution for non-washed native wheat straw, with refractive index correction. (Table 8-23, Appendix 8.10)
Vi = (0.18 ± 0.05)(1 − e−0.01D) Equation 5-16
Therefore, the new result is traduced by Figure 5-14. As explained, the points with no error bars
represent an apparent repeatability, since they result from the elimination of points in order to optimize
them. This was done with a logical reasoning due to the knowledge acquired with Alphacel
experiments.
To conclude, the results with non-washed native wheat straw are not exploitable. Too much
hypotheses and approximations were applied. This seriously affected the results. Moreover, the final
result is not similar to literature data (Table 5-3).
At this point of the work, the question of the possible existence of soluble components was taken in
account. Once the mathematically correction did not result in exploitable data, another solution was
established, acting directly on the substrate preparation. This introduces the next section, where the
native wheat straw was previously washed.
Washed native wheat straw
To confirm the hypothesis that the soluble compounds affect tremendously the refractive index
measurements, the native wheat straw was previously washed with water. Due to the time available
for the trials, only three points were obtained.
The quantity of soluble was then determined using Equation 5-13. Concerning Table 5-6 it is
perfectly visible that the quantity of soluble decreases drastically with the washing. Furthermore, the
results are measured in the refractive index apparatus. As can be seen, the order of magnitude of the
contribution of these compounds is significant for non-washed wheat straw when compared with
washed. By this, the results for this substrate were extremely affected. Subsequently, the contribution
of soluble was also deducted to the results obtained for washed native wheat straw.
0.00
0.05
0.10
0.15
0.20
0.25
0 50 100 150 200
Inaccessib
le p
ore
vo
lum
e
(mL
/g m
ds)
diameter (Å)

51
Table 5-6: Contribution of soluble components for refractive index measurements.
Substrate ∆𝐬𝐨𝐥𝐮𝐛𝐥𝐞
native non-washed 0.00134
native washed 0.00014
Figure 5-15 : Pore volume distribution for washed native wheat straw (Table 8-24, Appendix 8.10).
Vi = (1.51 ± 0.03)(1 − e−0.13D) Equation 5-17
Figure 5-16: Pore volume distribution for washed native wheat straw, with refractive index correction (Table 8-25, Appendix 8.10).
Vi = (0.72 ± 0.04)(1 − e−0.04D) Equation 5-18
Concerning these figures (Figure 5-15 and Figure 5-16), the differences are evident. The first thing
that can be noted is the difference in the fiber saturation point value. By subtracting the contribution of
soluble, the plateau decreases from 1.5 to 0.7 mL/g mds, half of the first value obtained. It can be firstly
0.0
0.5
1.0
1.5
0 50 100 150 200
Ina
cc
es
sib
le p
ore
vo
lum
e
(mL
/g m
ds)
diameter (Å)
0.0
0.2
0.4
0.6
0.8
0 50 100 150 200
Inaccessib
le p
ore
vo
lum
e
(mL
/g m
ds)
diameter (Å)

52
conclude that this contribution should be subtracted. Afterwards, if the soluble are not deducted, the
distribution obtained will correspond to a pore size value higher than the real one.
Regarding porosity, a conclusion can be made, based on the behavior of model equation
proposed. A fiber saturation point was encountered at 0.7 mL/g mds, since a size diameter of
approximately 50 Å. This indicates the presence of micro and mesopores.
Comparing the corrected data of washed native wheat straw with non-washed native wheat straw.
The fiber saturation point is completely different. Actually, the total pore volume for the washed sample
is approximately four times higher than the non-washed.
Due to this evidence, the non-washed data were not considered to make conclusions. Excepting
the evidence about the soluble compounds. With this, the existence of soluble can be corroborated
and it should be taking in account.
Washed and pretreated wheat straw
The other product tried was a sample pretreated with acid, at 160 oC. After pretreatment, the
sample was subjected to a washing in order to eliminate some contaminants.
Due to limited time, only three experiments were performed using as probes PEG 35000, PEG
20000 and glucose. Among these, the results obtained for glucose were not considered in
calculations.
Figure 5-17: Pore volume distribution for washed and pretreated wheat straw (Table 8-26, Appendix 8.10).
Vi = (0.75 ± 0.00)(1 − e−0.02D) Equation 5-19
As can be seen on Figure 5-17, two points did not allow to make conclusions. Additionally, the
model equation cannot describe this distribution due to this absent of data. A value for the fiber
saturation was obtained, nevertheless, it is not clear if the total porous volume corresponds to that
value. Regarding the same figure, it is not observed the plateau. Can be supposed that the plateau will
be at high value, as result of the pretreatment. Also, no one conclusion should be made about the type
of porosity. Accordingly, at this point, more study is required on pretreated sample.
0.0
0.2
0.4
0.6
0.8
0 50 100 150 200
Inaccessib
le p
ore
vo
lum
e
(mL
/g m
ds)
diameter (Å)

53
Conclusion
At this moment is time to make a summary of all data obtained until now. Once the data for non-
washed native wheat straw was found to be not exploitable, it is not represented in this section. Then,
the results obtained were put all in the same figure and represented using a logarithmic scale:
Figure 5-18: Pore volume distribution for Alphacel and different wheat straw products.
Once there are not published studies using this new approach of solute exclusion, as well, there
are few information on wheat straw, only a preliminary comparison can be made. Recalling Stone and
Scalan work, can be noted on Figure 5-19 that the behavior of data from the present work is
comparable with their study on pulp fibers dried and reswollen.
Figure 5-19: Pore volume distribution for pulp fibers – zoom of Figure 5-6 [40].
0.0
0.2
0.4
0.6
0.8
1 10 100 1000
Inaccessib
le p
ore
vo
lum
e (
mL
/g m
ds)
diameter (Å)
alphacel
washed native
pretreated 160oC

54
Similarly to the same authors, in the current study, the plateau was observed between 0.6 and 0.8
mL/g mds. The behavior of the curve (defined by a constant) is also analogous, however, about this
none conclusion should be made, since that this behavior depends on the substrate used.
Determination of specific surface area
As previously referred, surface area available is an important parameter on enzymatic hydrolysis,
regarding the pretreatment method (section 2.3.2). Consequently, it is interesting to study the five
different samples pretreated in this work in order to compare them with native sample and to notice the
differences between them.
Though, the limitation of the applicability of this method should be retained: nitrogen adsorption
measurements provide specific surface area for a molecule that is 3200 times smaller than the
average cellulase [60]. Concerning the figure above, this affirmation turns completely clear. There, the
influence of probe size on the determination of available surface area is perceptible.
Figure 5-20: Schematic representation of the structural features of the cellulose particle surface [19].
Afterwards, this analysis was performed on the different substrates and the results can be seen on
Table 5-7. The specific surface area, SSABET, increases from the sample pretreated at 100 °C until the
sample pretreated at 180 °C. These results were predictable since it was expected an increment on
SSA with the severity of the pretreatment. Furthermore, the native sample has a SSA lower than the
other ones. Likewise, this relation was expected and translates the effectiveness of the pretreatment
on the increasing of the available surface area.
Table 5-7: Results of specific surface area from N2 gas adsorption, in this work.
Substrate ID Pretreatment SSABET (m2/g)
wheat straw
C0033 – 0.77
CR_1082L 100 °C, 20 min, 1 %w/w H2SO4 0.97
CR_1081L 120 °C, 20 min, 1 %w/w H2SO4 1.27
CR_1083L 140 °C, 20 min, 1 %w/w H2SO4 1.71
CR_1080L 160 °C, 20 min, 1 %w/w H2SO4 2.79
CR_1084L 180 °C, 20 min, 1 %w/w H2SO4 6.27

55
Some similar results on pretreated wheat straw were found in literature. In fact, the values reported
have the same order of magnitude than the ones achieved in this work even if the operating conditions
are not the same: different temperatures (between 120 and 190 °C) with different quantities of acid
(0.5 to 2 %w/w), as well, variable residence time (7 to 240 min). By this, the results are only coherent
for the higher temperatures.
Table 5-8: Specific surface area from literature, for wheat straw, by N2 gas adsorption.
Substrate Pretreatment SSABET (m2/g) Reference
wheat straw
– 3.3 [25] – 4.0
[62]
120 °C, 240 min, 1 %w/w H2SO4 5.7 140 °C, 25 min, 2 %w/w H2SO4 5.4 160 °C, 7 min, 1.5 %w/w H2SO4 5.5 170 °C, 10 min, 1 %w/w H2SO4 6.2 180 °C, 7 min, 0.5 %w/w H2SO4 7.1 190 °C, 10 min, 0.5 %w/w H2SO4 6.6
Summary and discussion
At this point, some considerations can be done. Using Alphacel, the results of size exclusion
analysis were reproducible and dried substrate methodology was validated. Regarding non-washed
and washed native wheat straw, it can be settled that the washing is significant in order to avoid
deviation on the results. For washed native wheat straw, as well, pretreated at 160 oC and washed, a
more complete study is required.
This section intends to make some general conclusions to summarize the data obtained and to
explain details that can considerably affect the experiments.
In the figure below is represented the accessible volume of untreated and pretreated corn stover,
as function of probe diameter proposed by Ishizawa et al. [42]. In parentheses are indicated the
cellulose digestibilities after seven days. The error bars represent the standard deviation of three
replicates.
Figure 5-21: Accessible pore volume of corn stover, measured by solute exclusion [42].

56
Ishizawa and his co-workers obtained the measurements of concentration of probe using an HPLC
apparatus equipped with a refractive index detector. Considering these results can be noted that the
error bars present a random and important deviation from sample to sample. This bad reproducibility
was found in the present study and is also encountered by these authors. This reinforces the
imprecision associated to a measurement of this type.
Due to this evidence, it is important to solve the problem of the measurements. So, in order to
improve the acquisition of data, a brainstorming was performed. The issue that seems to affect more
these results is the small gap between the initial and the final concentration of probe. Consequently, it
is needed to find a solution for this.
Using Equation 5-5 and Equation 5-6, the ratio between final and initial concentration of probe can
be obtained:
Cf
Ci
=Vsol,i
Va + Ve
Equation 5-20
As it is known, the volume of initial solution is already defined (Equation 5-7) and the porous
volume as well (Equation 5-8). By this, rearranging the equation, it results in:
Cf
Ci
=1
1 −Vi
Vsol,i⁄
Equation 5-21
where Vi is characteristic of the solid in study and can be expressed by:
Vi = εsolid × Vsolid Equation 5-22
where Vsolid is the volume of biomass and εsolid its porosity. Substituting this variable in Equation
5-21, the ratio of concentrations will be defined by:
Cf
Ci
=1
1 − εsolid ∙msolid
dsolid∙
1Vsol,i
Equation 5-23
The last equation represents the relation between the ratio of concentrations at the begging and at
the end of the experiment, with the variables of the trial. As can be noted on Table 5-9, it can be seen
that the final concentration can be very different regarding the substrate used. This concentration will
allow to determine the porous volume. Therefore, this measurement should be obtained accurately.
Table 5-9: Example of concentrations of probe (Appendix 8.6; Appendix 8.7).
Substrate Probe Ci (g/100mL) Cf (g/100mL)
Alphacel PEG 20000 1.00
1.10
Wheat straw 1.02
Regarding the same expression (Equation 5-23) it is evident that two parameters can be modified
from the experimental point of view in order to maximize the ratio of concentrations: the volume of the
probe solution, Vsol,i, and the mass of substrate used, msolid. Porosity and density will depend on the
substrate and cannot be modified.

57
Influence of the mass of substrate and the volume of probe
To evaluate the impact of the mass of substrate used, Equation 5-23 can be simplified:
Cf
Ci
=1
1 − K ∙ msolid
; K =εsolid
dsolid
∙1
Vsol,i
Equation 5-24
In this case, the influence of the quantity of solid is been evaluated, maintaining constant the
volume of probe solution. Supposing that the ratio between porosity and density it is one, than the
constant K will take the value of 0.1 g-1 (once the initial volume of solution is 10 mL).
Concerning this example and the results on Table 5-10, it is clear that an increase in solid mass will
allows the final concentration to increase. In this way, with the possibility of increasing the
concentration in the end, more precise results would be obtained.
Table 5-10: Influence of substrate quantity in final concentration of probe.
Increment 𝐊 ∙ 𝐦𝐬𝐨𝐥𝐢𝐝 𝟏 − 𝐊 ∙ 𝐦𝐬𝐨𝐥𝐢𝐝 𝟏
𝟏 − 𝐊 ∙ 𝐦𝐬𝐨𝐥𝐢𝐝
𝐂𝐟
2 ∙ msolid 2 0.8 1.25 ↑
6 ∙ msolid 6 0.4 2.50 ↑↑
8 ∙ msolid 8 0.2 5.00 ↑↑↑
10 ∙ msolid 10 0 - -
Nevertheless, it exists an asymptote in the increment of mass. As can be noted on Figure 5-22, at
certain point, that increasing does not make sense: thus, there is a limit to this parameter. Observing
Equation 5-23 it is perceptible that this asymptote corresponds to the point where the denominator of
the equation turns zero.
Figure 5-22: Influence of substrate quantity in final concentration of probe.
This behavior was expected. Once the mass of substrate is increased, the porous volume is also
increasing. By this, the final concentration of probe will be equal to the initial concentration or higher
than the initial concentration if not all the probe can enters the pores. Additionally, the quantity of
solution is fixed in 10 mL. In this case, if the quantity of substrate is being increased, the porous
volume is increasing as well. Consequently, this limit of concentration can happen before, since the
0
2
4
6
8
10
0 2 4 6 8 10 12
Cf(g
/100m
L)
msolid (g)
Ci

58
porous volume can equalize the volume of solution. In this situation, the final concentration will depend
totally on the probe size diameter: if the probe size is higher than all pores, than only water will
penetrate into the substrate; on the other hand if it is smaller than the porous diameter, the final
concentration will be zero. The last case is a limit case and should not be achieved because the
volume of solution will be higher than the porous volume available and the measurement will not be
correspondent to the reality.
Then, regarding Equation 5-23, it can be noted that reducing the volume in order to increase the
final concentration, is the same thing that increase the mass of substrate. By this, can be concluded
that the true parameter will be the ratio instead of the parameters by themselves. From a mathematical
point of view, if the mass of substrate and the volume of solution are considered as independent
parameters, it will results in one equation and two parameters (over-parameterization). In order to
reduce this problem, the ratio is considered as the only parameter and then there are one equation
and one parameter.
Following this, the Equation 5-23 will be simplified as:
Cf
Ci
=1
1 − K′ ∙ Z; K′ =
εsolid
dsolid
; Z =msolid
Vsol,i
Equation 5-25
Concerning the equation below, to increase final concentration, it is required an increment on the
ratio of mass of substrate and volume of probe solution. This can mean an increasing of mass of
substrate or a decreasing on the volume of probe solution.
Figure 5-23: Influence of the ratio mass of substrate by volume of solution in final concentration of probe.
In the figure above, is represented the variation of the ratio used in this work (0.1 g/mL). The graph
was obtained by multiplying the original ratio by values higher than one, in order to make the
increment. The asymptote determined corresponds to the value obtained by multiplying the ratio by
ten. As expected it was obtained the same result generated for mass influence.
Influence of the concentration of the initial solution
Hereupon, the impact of change the initial concentration was studied. For this, the mass of solid
and the initial volume of probe solution were maintained constants. The same assumption was done
with the ratio porosity by density and this resulted in:
0
2
4
6
8
10
0.1 0.3 0.5 0.7 0.9 1.1
Cf(g
/100
mL
)
msolid/Vsol,i (g/mL)

59
Cf = Y ∙ Ci ; Y =1
1 − K′′; K′′ =
εsolid
dsolid
∙msolid
Vsol,i
Equation 5-26
As can be noted by the equation above, the influence on final concentration will be proportional
with the increment in the initial concentration, with a constant that will depend on the substrate.
Accordingly, change only the initial concentration does not make sense, except for limiting the impact
of soluble in the refractive index compared to the impact of the probe.
Experimental issues
As said, the mathematic analysis performed to study the influence of the variables does not
evaluate the issues associated to the experimental part. The parameters can be modified, still the
experience with the materials in cause is determinant to make decisions.
Regarding the protocol described on section 4.3 that was the one applied in this work, some
suggestions can be done in order to optimize the experiments. These recommendations result from
the observations performed during the laboratory work.
Considering the elementary steps, the first issue is related to the mixing of substrate with probe
solution. First of all, the probe solution is added to the dried substrate (case A). Then, with the help of
a spatula, the substrate is pushed to the bottom of the flask in order to allow the contact between
probe and substrate (case B). After the stirring begins.
Figure 5-24: Experimental issue on stirring.
Figure 5-25: Comparison between a native and a pretreated wheat straw samples, after stirring.

60
As can be noted in Figure 5-24, after stirring and decantation, there is straw that ascend the flask.
This could be affect the results, since a part of substrate cannot being in contact with probe solution.
This evidence was noted for native wheat straw but it is not so visible with Alphacel and pretreated
wheat straw (Figure 5-25). To solve this problem the type of agitation should be changed.
Another important point is the washing of the substrate. In Figure 5-26, there is a visible difference
in the color of the samples if the substrate is washed or not. It was verified that there is a contribution
of soluble compounds that affects the results. A solution is to increase the initial concentration of
probe, to limit the impact of these soluble compounds. Another solution passes by the washing of the
substrate. If these solutions are not possible, the blank sample should be maintained in order to
deduce these contributions.
Figure 5-26: Comparison between a washed (1’) and a non-washed (1) native wheat straw supernatants.
Methodology optimization
Since this technique is very labor-intensive, it took some time to learn how to do it and get
consistent and reproducible data. By this, this section intends to propose a way to reduce the time of
the experiment and obtain the high number of data possible in a reduced time space.
As said before, instead of what is described in literature, an approach to dry substrate was
performed in this study. The final protocol resulted in the performing of one experiment by day, as can
be seen in next table:
Table 5-11: Day work plan to performed one experiment with the dried substrate methodology.
08H 09H 10H 11H 12H 13H 14H 15H 16H 17H
Step b)
Step c)
Step d)
Step e), Step f)
Step g)
Step h)
Results treatment
Preparation of next assay
As can be seen in the timetable above, each experiment (one experiment corresponds to one
probe) takes one entire day to be performed, concerning the experimental part, as well the treatment
of the results and the preparation of the next one. So, subsequently to the first experiments, with the
intention to obtain a high number of results in a short period of time, an adjustment was done, to try
perform two experiments in one day:

61
Table 5-12: Day work plan to performed two experiments by dried substrate methodology.
08H 09H 10H 11H 12H 13H 14H 15H 16H 17H 18H
Step b)
Step c)
Step d)
Step e), Step f)
Step g)
Step h)
Results treatment
Preparation of next assay
Making this upgrading, it was possible to obtain more data during this work. Still, this planning can
be improved. After to known how to perform the experiments, the operator will be able to perform three
different trials simultaneously. For this, step d) (settling down) should be reduced or eliminated, for
example. Another way to reduce the obtain data quickly is to use a different analyzer instead of the
refractometry apparatus. Actually, this step comprises more than 25% of the total time of the
methodology (due to the calibration).
The number of probes used can also be a point to explore. At this moment the expected behavior
of data for this method is known. Therefore, the probes can be selected more accurately and not by
trial and error experiments. Recovering the example of Alphacel:
Figure 5-27: Pore volume distribution for Alphacel (Table 8-21, Appendix 8.10).
If this trial was being repeat now, some changes should be done in order to adjust better the
experimental points to the proposed equation. First, more trials should be performed using the
smallest probe molecules, since this data revealed to be not feasible. The points referred to the probes
between 30 and 70 Å should be maintained since they define the form of the curve. Last, but not the
0.0
0.2
0.4
0.6
0.8
1.0
0 50 100 150 200
Inaccessib
le p
ore
vo
lum
e
(mL
/g m
ds)
diameter (Å)

62
least, the high size diameter molecules should be kept and maybe a bigger ones should be tried to
verify the possible existence of other type of macroporosity. By this, concerning Figure 5-27 can be
suggested a reduction of the number of probes used to define the distribution of pores. Five probes
can be enough: two to define the fiber saturation point, two to define the curve (and the constant as
result) and one smaller to better adjust the model equation.
Regarding the type of analyzer to the concentration measurements an HPLC apparatus coupled to
a refractometric detector can be used. By this, a solution containing of several probe molecules can be
used and the time of measurement of final concentration will considerably decrease.

63
6 CONCLUSIONS AND FUTURE PROSPECTS
The literature review on the available methods allowed us to explore the techniques used for the
structural characterization of lignocellulosic substrates. From this study, one method named solute
exclusion technique was selected and applied on wet substrates. Rapidly, the first experiments have
shown poor results in terms of reproducibility and a new approach was established using dry
substrates.
The present work focused on the solute exclusion technique, which was firstly performed by Stone
and Scalan, in 1968, and is still employed. Regarding the method selected, and, particularly, the
preparation of the substrate, all methods have in common the use of a water saturated substrate. This
methodology was performed using commercial products (Avivel and Alphacel) and it was found to be
not interesting due to the high uncertainties on the final results, being coherent with literature. By this,
it was discarded.
Subsequently, the new approach of the method was performed. The main difference was the use
of a dry substrate. Nevertheless, it is referred in literature an issue that involves the possible collapse
of pores during drying [60].
In despite of this risk, the dried substrate method has been tested during this training period. It was
found reproducible and was validated for a commercial product, Alphacel C40. Similar results were
found in literature [19] for other standard celluloses, such as Avicel. The technique was also applied to
wheat straw (native non-washed and washed, and pretreated at 160 oC). For these substrates a more
complete study would be required due to the low number of points obtained.
For determining the concentration of the probes after mixing, a refractive index apparatus was
used. It was found the need to eliminate contaminants that affected the measurements, especially in
the case of the wheat straw wherein the amount of soluble compounds is high. This evidence was
found by comparing native wheat straw samples after washing step or not. Following this, it is strongly
recommended the preparation of a blank sample which allows to deduct the contribution of these
compounds on the measurements. Additionally, the washing of the substrate is recommended to
minimize their contribution in the refractive index of the final solution.
To describe the pore volume distribution, a model equation was also proposed. This equation
describes the distribution of the porosity of a substrate as function of the pore diameter using two
variables: the fiber saturation point, or maximal porous volume, and a constant that depends on the
substrate. Using this equation it was possible to known the pore size distribution of various substrates.
It is proposed the use of only five or six probes for a substrate, and, applying the equation proposed it
will be possible to have a complete description of the distribution of pores.
In summary, this work proposes a new approach of the solute exclusion technique, as well, a
model equation that can describe the pore size distribution. Optimizations shall be done and the
technique must be validated for other type of substrates. It was also suggested the use of combined
characterization techniques in order to obtain complete information about accessibility. This work was
significant since it deal with the relation between substrate accessibility and enzymatic hydrolysis.

64

65
7 REFERENCES
[1] P. Halling and P. Simms-Borre, "Overview of lignocellulosic feedstock conversion into ethanol -
focus on sugarcane bagasse," 2008. [Online]. Available: www.internationalsugarjournal.com.
[Accessed 4 March 2015].
[2] European Biofuels - Technology Platform, "Cellulosic Ethanol," [Online]. Available:
http://www.biofuelstp.eu/cellulosic-ethanol.html#ce1. [Accessed 10 August 2015].
[3] "Dioxyde de Carbone," [Online]. Available: http://www.respire-asso.org/dioxyde-de-carbone-co2/.
[Accessed 15 June 2015].
[4] M. Guo, W. Song and J. Buhain, "Bioenergy and biofuels: History, status, and perspective,"
Renewable and Sustainable Energy Reviews, no. 42, pp. 712-725, 2015.
[5] International Transport Forum, "Reducing Transport Greenhouse Gas Emissions - Trends&Data,"
2010.
[6] "2030 Energy Strategy," [Online]. Available: http://ec.europa.eu/energy/node/163. [Accessed 15
June 2015].
[7] IFP Energies nouvelles, "Biofuels 2G, Biocarburants - Production/consommation par zone
géographique," [Online]. Available: https://prisme/IntranetIFP/jcms/pr2_1589617/production-/-
consommation-par-zone-geographique. [Accessed 9 April 2015].
[8] R. Saxena, D. Adhikari and H. Goyal, "Biomass-based energy fuel through biochemical routes: A
review," Renewable and Sustainable Energy Reviews, no. 13, pp. 167-178, 2009.
[9] R. B. Gupta and A. Demirbas, Gasoline, Diesel and Ethanol Biofuels from Grasses and Plants,
Cambridge University Press, 2010.
[10] S. Behera, R. Arora, N. Nandhagopal and S. Kumar, "Importance of chemical pretreatment for
bioconversion of lignocellulosic biomass," Renewable and Sustainable Energy Reviews, no. 36,
pp. 91-106, 2014.
[11] A. Hendriks and G. Zeeman, "Pretreatments to enhance the digestibility of lignocellulosic
biomass," Biosource Technology, no. 100, pp. 10-18, 2009.
[12] S. K. Ritter, "Lignocellulose: A Complex Biomaterial," Science & Technology, vol. 86, p. 15, 2008.
[13] M. Badiei, N. Asim, J. M Jahim and K. Sopian, "Comparison of Chemical Pretreatment Methods
for Cellulosic Biomass," in APCBEE Procedia 9, 2014.
[14] Q. Q. Wang, Z. He, Z. Zhu, Y.-H. Zhang, Y. Ni, X. Luo and J. Zhu, "Evaluations of Cellulose
Accessibilities of Lignocelluloses by Solute Exclusion and Protein Adsorption Techniques,"
Biotechnology and Bioengineering, vol. 109, no. 2, February 2012.
[15] Z. Anwar, M. Gulfraz and M. Irhad, "Agro-industrial lignocellulosic biomass a key to unlock the
future bio-energy: A brief review," Journal of Radiation Research and Applied Sciences, no. 7, pp.
163-173, 2014.

66
[16] E. Tomas-Pejo, J. Oliva, M. Ballesteros and L. Olsson, "Comparison of SHF and SSF Processes
From Steam-Exploded Wheat Straw for Ethanol Production by Xylose-Fermenting and Robust
Glucose-Fermenting Saccharomyces cerevisiae Strains," Biotechnology and Bioengineering, vol.
100, no. 6, pp. 1122-1131, 2008.
[17] M. Chauve, "Modélisation cinétique de l'hydrolyse enzymatique des substrats cellulosiques,"
2011.
[18] "La fabrication du papier," 2013. [Online]. Available: http://tpepapier.e-monsite.com/pages/i-de-l-
arbre-a-la-feuille-de-papier/page.html. [Accessed 2015 August 13].
[19] F. M. Gama, J. a. Teixeira and M. Mota, "Cellulose Morphology and Enymatic Reactivity: A
Modified Solute Exclusion Technique," Biotechnology and Bioengineering, vol. 43, pp. 381-387,
1994.
[20] F. Monot, "Fuels from biomass," [Online]. Available:
http://www.ifpenergiesnouvelles.com/Research-themes/Renewable-energies/Fuels-from-
biomass/Biocatalysts-one-of-IFPEN-s-expertise-field-Questions-to-Frederic-Monot-Head-of-the-
Biotechnology-Department-at-IFPEN. [Accessed 28 May 2015].
[21] O. Sanchez and C. Cardona, "Trends in biotechnological production of fuel ethanol from different
feedstocks," Bioresource Technology, no. 99, p. 5270–5295, 2008.
[22] Z. Liu and B. Fei, Sustainable Degradation of Lignocellulosic Biomass - Techniques, Applications
and Commercialization, pp. 1-14.
[23] M. Foston and A. Ragauskas, "Changes in the Structure of the Cellulose Fiber Wall during Dilute
Acid Pretreatment in Populus Studied by 1H and 2H NMR," Energy Fuels, no. 24, pp. 5677-5685,
2010.
[24] D. N. Thompson, "The effects of physical and chemical constraints on the enzymatic hydrolysis of
lignocellulosic materials," Michigan, 1989.
[25] A. Chesson, P. T. Gardner and T. J. Wood, "Cell Wall Porosity and Available Surface Area of
Wheat Straw and Wheat Grain Fractions," J Sci Food Agric, no. 75, pp. 289-295, 1997.
[26] K. Olofsson, M. Bertilsson and G. Lidén, "A short review on SSF – an interesting process option
for ethanol production from lignocellulosic feedstocks," Biotechnology for Biofuels, pp. 1-14, 2008.
[27] F. Alfani, A. Gallifuoco, A. Saporosi, A. Spera and M. Cantarella, "Comparision of SHF and SSF
processes for the bioconvertion of steam-exploded wheat straw," Journal of Industrial
Microbiology & Biotechnology, no. 25, pp. 184-192, 2000.
[28] D. Dahnum, S. O. Tasum, E. Triwahyuni, M. Nurdin and H. Abimanyu, "Comparison of SHF and
SSF processes using enzyme and dry yeast for optimization of bioethanol production from empty
fruit bunch," Energy Procedia, no. 68, pp. 107-116, 2015.
[29] X. Meng and A. J. Ragauskas, "Recent advances in understanding the role of cellulose
accessibility in enzymatic hydrolysis of lignocellulosic substrates," Current Opinion in
Biothecnology, no. 27, pp. 150-158, 2014.

67
[30] H. Toulhoat and P. Raybaud, Catalysis by transition metam sulphides, E. TECHNIP, Ed., IFP
Energies nouvelles Publications, 2013.
[31] Micromeritics Instrument Corporation, "Micromeritics Analytical Services," [Online]. Available:
http://www.particletesting.com/Services-Provided/Surface-Area.aspx. [Accessed 13 July 2015].
[32] "Dinitrogen 2D dimensions," [Online]. Available:
https://commons.wikimedia.org/wiki/File:Dinitrogen-2D-dimensions.png. [Accessed 15 August
2015].
[33] D. N. Thompson and H.-C. Chen, "Comparison of Pretreatment Methods on the Basis of Available
Surface Area," Biosource Technology, no. 39, pp. 155-163, 1992.
[34] "Particle Analytical," [Online]. Available: http://particle.dk/methods-analytical-laboratory/mercury-
porosimetry-pore-size/. [Accessed 13 July 2015].
[35] X. Yo, J. L. Minor and R. H. Atalla, "Mechanism of action of Simons’ stain," Fiber Analysis, vol. 78,
no. 6, pp. 175-180, 1994.
[36] M. Inglesby and S. Zeronian, "Direct dyes as molecular sensors to characterize cellulose
substrates," Cellulose, vol. 9, p. 19–29, 2002.
[37] R. Chandra, S. Ewanick, C. Hsieh and J. Saddler, "The Characterization of Pretreated
Lignocellulosic Substrates Prior to Enzymatic Hydrolysis, Part 1: A Modified Simons’ Staining
Technique," Biotechnology Progress, no. 24, pp. 1178-1185, 2008.
[38] M. Rusu, K. Morseburg, O. Gregersen, A. Yamakawa and S. Liukkonen, "Relation between fibre
flexibility and cross-sectional properties," BioResources, vol. 6, no. 1, pp. 641-655, 2011.
[39] X. Meng, M. Foston, J. Leisen, J. DeMartini, C. Wyman and A. Ragauskas, "Determination of
porosity of lignocellulosic biomass before and after pretreatment by using Simons' stain and NMR
techniques," Bioresource Technology, no. 144, pp. 467-476, 2013.
[40] J. E. Stone and A. M. Scallan, "A structural model for the cell wall of water-swallen wood pulp
fibers based on their accessibility to macromolecules," Cellulose Chem. Technol., no. 2, pp. 343-
358, 1968.
[41] L. Hui, Z. Liu and Y. Ni, "Characterization of high-yield pulp (HYP) by the solute exclusion
technique," Bioresource Technology, no. 100, pp. 6630-6634, 2009.
[42] C. I. Ishizawa, M. F. Davis, D. F. Schell and D. K. Johnson, "Porosity and Its Effect on the
Digestibility of Dilute Sulfuric Acid Pretreated Corn Stover," Journal of Agricultural and Food
Chemistry, no. 55, pp. 2575-2581, 2007.
[43] L. A. Lucia and O. J. Rojas, Eds., The Nanoscience and Technology of Renewable Biomaterials,
Wiley, 2010.
[44] S. Fekete, A. Beck, J.-L. Veuthey and D. Guilharme, "Theory and practice of size exclusion
chromatography for the analysis of protein aggregates," Journal of Pharmaceutical and
Biomedical Analysis, no. 101, pp. 161-173, 2014.
[45] T. Lojewki, K. Zieba and J. Lojewska, "Size exclusion chromatography and viscometry in paper

68
degradation studies. New Mark-Houwink coefficients for cellulose in cupri-ethylenediamine,"
Journal of Chromatography A, no. 1217, pp. 6462-6468, 2010.
[46] D. Yang, J.-Y. Parlange and L. P. Walker, "Revisiting Size-Exclusion Chromatography for
Measuring Structural Changes in Raw and Pretreated Mixed Hardwoods and Switchgrass,"
Biotechnology and Bioengineering, vol. 112, no. 3, pp. 549-559, 2015.
[47] A. Oliva, M. Llabrés and J. B. Fariña, "Estimation of uncertainty in size-exclusion chromatography
with a double detection system (light-scattering and refractive index)," Talanta, no. 78, pp. 781-
789, 2009.
[48] P.-T. Chen, H.-P. Chen, C.-H. Hung and S.-C. Wang, "Using Second-derivate Filters to Assist in
Width Estimations of Size Exclusion Chromatography Signal Peaks with Static Light-Scattering
Detections to Obtain More Accurate Molecular Weight," Analytical Sciences, vol. 30, pp. 1063-
1068, 2014.
[49] P. Gill, T. Moghadam and B. Ranjbar, "Differential Scanning Calorimetry Techniques: Applications
in Biology and Nanoscience," Journal of Biomolecular Techniques, no. 21, pp. 164-193, 2010.
[50] A. Travis, S. Murison, P. Perry and A. Chesson, "Measurement of Cell Wall Volume using
Confocal Microscopy and its Application to Studies of Forage Degradation," Annals of Botany, no.
80, pp. 1-11, 1997.
[51] C. Sant'Anna and W. de Souza, "Microscopy as a tool to follow deconstruction of lignocellulosic
biomass," in Current Microscopy Contributions to Advances in Science and Technology, A.
Mendez-Vilas, Ed., 2012.
[52] J. B.H. Van Dyke, "Enzymatic Hydrolysis of Cellulosic Materials - A Kinetic Study," Cambridge,
1972.
[53] J. K. Lin, M. R. Ladisch, J. A. Patterson and C. H. Noller, "Determining Pore Size Distribution in
Wet Cellulose by Measuring Solute Exclusion Using a Differential Refractometer," Biotechnology
an Bioengineering, vol. XXIX, pp. 976-981, 1987.
[54] Q. Cheng, J. Wang, J. F. McNeel and P. M. Jacobson, "Water Retention Value Measurements of
Cellulosic Materials Using a Centrifuge Technique," BioResources, vol. 5, no. 3, pp. 1945-1954,
2010.
[55] I. C. Hoeger, S. S. Nair, A. J. Ragauskas, Y. Deng, O. J. Rojas and J. Y. Zhu, "Mechanical
deconstruction of lignocellulose cell walls and their enzymatic saccharification," in Cellulose,
Springer, 2013.
[56] Q. Cheng, S. Wang, T. G. Rials and S.-H. Lee, "Physical and mechanical properties of polyvinyl
alcohol and polypropylene composite materials reinforced with fibril aggregates isolated from
regenerated cellulose fibers," in Cellulose, vol. 14, Springer, 2007, pp. 593-602.
[57] S.-F. Chen, R. A. Mowery, C. J. Scarlata and C. K. Chambliss, "Compositional Analysis of Water-
Soluble Materials in Corn Stover," Journal of Agricultural and Food Chemistry, no. 55, pp. 5912-
5918, 2007.
[58] M. Chaplin, "Water Molecule Structure," [Online]. Available:

69
http://www1.lsbu.ac.uk/water/water_molecule.html. [Accessed 2015 August 19].
[59] H. Grethlein, D. Allen and A. Converse, "A Comparative Study of the Enzymatic Hydrolysis of
Acid-Pretreated White Pine and Mixed Hardwood," Biotechnology and Bioengineering, vol. XXVI,
pp. 1498-1505, 1984.
[60] R. Neuman and L. Walker, "Solute Exclusion from Cellulose in Packed Columns: Experimental
Investigation and Pore Volume Measurements," Biotechnology and Bioresource, vol. 40, pp. 218-
225, 1992.
[61] Institut Français du Pétrole Publications, Physico-Chemical Analysis of Industrial Catalysts - A
Practical Guide to Characterisation, Editions Technip, 2001.
[62] R. Dhabhai, S. P. Chaurasia and A. K. Dalai, "Effect of pretreatment conditions on structural
characteristics of wheat straw," Chemical Engineering Communications, vol. 200, pp. 1251-1259,
2013.

70

71
8 APPENDIX
Results from enzymatic hydrolysis
Table 8-1: Glucose yield from enzymatic hydrolysis for the pretreated samples.
ID CR_1080L CR_1081L CR_1082L CR_1083L CR_1084L
T (°C) 140 120 100 160 180
t (h) Glucose yield (%)
1.5 11.2 11.4 11.4 11.1 7.1 7.3 12.1 12.8 5.4 6.3
3 15.7 16.7 15.4 15.7 8.7 8.9 17.9 19.1 7.5 9.0
6 21.4 22.6 19.8 20.4 10.7 11.3 25.2 26.4 10.3 12.7
24 33.0 36.6 29.1 29.8 14.3 15.1 40.8 42.8 17.6 22.0
48 38.7 43.1 34.3 35.4 16.3 16.8 49.6 52.6 21.8 26.7
72 42.9 47.8 37.5 38.4 17.2 18.0 52.5 53.4 24.6 29.6
t (h) AV SD AV SD AV SD AV SD AV SD
1.5 11.3 0.2 11.3 0.2 7.2 0.1 12.5 0.5 6.3 0.6
3 16.2 0.7 15.6 0.2 8.8 0.2 18.5 0.8 9.0 1.1
6 22.0 0.9 20.1 0.4 11.0 0.4 25.8 0.8 12.7 1.7
24 34.8 2.5 29.4 0.5 14.7 0.5 41.8 1.4 22.0 3.2
48 40.9 3.1 34.9 0.8 16.5 0.4 51.1 2.1 26.7 3.5
72 45.3 3.5 37.9 0.6 17.6 0.6 52.9 0.7 29.6 3.5
Probe molecules
The average molecular weight and diameter for each probe were obtained from the literature:
Table 8-2: Solution molecular diameters of probes from literature [40, 53].
Probe Molecular weight
(g/mol)
Diameter
(Ǻ)
Glucose 180 8
Cellobiose 342 10
PEG 200 190-210 13
PEG 400 285-315 18
PEG 600 570-630 21
PEG 1000 950-1050 27
PEG 1500 1300-1600 33
PEG 3500 3000-3700 50
PEG 8000 7000-9000 84
Dextran 75000 72000 120
PEG 20000 15000-20000 130

72
For the probes used, some diameters were not found, and for that one’s an equation obtained by
power curve was employed:
Figure 8-1: Correlation obtained for PEG probes by power curve.
Results from water retention value method
Table 8-3: Drying of substrate WRV1, for Avicel PH101.
ID Set A Set B
t (h) m1 m2 m3 m4 m5 m6
0 1.1984 1.2150 1.3864 0.9793 1.0289 1.1076
17.5 0.7695 0.7976 0.7568 0.6138 0.7826 0.8392
Table 8-4: Drying of substrate WRV2, for Avicel PH101.
ID Set A Set B
time (h) m1 m2 m3 m4 m5 m6
0 1.1726 1.0822 0.8832 0.9990 1.0546 1.0236
0.5 0.7352 0.6836 0.5657 0.6681 0.7020 0.6783
1 0.6612 0.5578 0.4941 0.4887 0.5452 0.5222
1.5 0.6604 0.5571 0.4932 - - -
2 - - - 0.4635 0.4896 0.4654
2.5 0.6599 0.5566 0.4930 - - -
3 - - - 0.4632 0.4882 0.4651
3.5 0.6594 0.5565 0.4931 - - -
4 - - - 0.4630 0.4880 0.4657
4.5 0.6598 0.5564 0.4931 - - -
5 - - - 0.4620 0.4873 0.4644
5.5 0.6586 0.5554 0.4918 - - -

73
Table 8-5: Drying of substrate WRV3, for Avicel PH101.
ID Set A Set B
time (h) m1 m2 m3 m4 m5 m6
0 1.0954 0.9998 1.0690 1.0555 1.0773 1.1101
17 0.5949 0.5260 0.5445 0.5960 0.6163 0.6284
Table 8-6: Drying of substrate WRV4, for Alphacel C40.
ID Set A Set B
time (h) m1 m2 m3 m4 m5 m6
0 1.1606 1.2046 1.2871 1.1842 1.3343 1.3151
0.5 0.7254 0.7481 0.8217 0.8181 0.9491 0.9941
1 0.6698 0.6894 0.7341 - - -
1.5 0.6676 0.6869 0.7302 0.6935 0.7784 0.7589
2 0.6664 0.6863 0.7290 - - -
2.5 - - - 0.6928 0.7774 0.753
3 0.6665 0.6866 0.7296
3.5 - - - 0.6916 0.7772 0.7533
4 0.6653 0.6863 0.7290 - - -
4.5 - - - 0.6933 0.7778 0.7531
5 0.6661 0.6870 0.7309 - - -
Table 8-7: Results of WRV for Avicel PH101.
ID m1 m2 m3 m4 m5 m6
mss (g) 1.1606 1.2046 1.2871 1.1842 1.3343 1.3151
mds (g) 0.6677 0.6890 0.7330 0.6949 0.7795 0.7549
WRV (g/g) 0.78 0.95 0.80 1.16 1.16 1.20
AV±SD (g/g) 1.01±0.19
Table 8-8: Results of WRV for Alphacel C40.
ID m1 m2 m3 m4 m5 m6
mss (g) 1.1726 1.0822 0.8832 0.9990 1.0546 1.0236
mds (g) 0.6586 0.5554 0.4918 0.4620 0.4873 0.4644
WRV (g/g) 0.74 0.75 0.76 0.70 0.71 0.74
AV±SD (g/g) 0.73±0.02

74
Calibration curves (refractometry)
For all the assays a new set of solutions was prepared and the calibration of the refractometer was
performed. In the next table are presented the linear correlations correspondent to the considered
assays:
Table 8-9: Linear correlations between refractive index and concentration of probe.
ID Probe Date Linear correlation R2
SE01 PEG 20000 29/04/2015 y = 0.00137x + 1.33283 0.9988
SE02 PEG 20000 29/04/2015 y = 0.00137x + 1.33283 0.9988
SE03 PEG 8000 06/05/2015 y = 0.00138x + 1.33280 0.9995
SE04 PEG 8000 06/05/2015 y = 0.00137x + 1.33281 0.9995
SE06 PEG 20000 19/05/2015 y = 0.00143x + 1.33278 0.9974
SE10 PEG 200 29/05/2015 y = 0.00126x + 1.33276 0.9975
SE12 PEG 8000 01/06/2015 y = 0.00139x + 1.33281 0.9993
SE13 PEG 10000 02/06/2015 y = 0.00143x + 1.33277 0.9977
SE14 PEG 1500 05/06/2015 y = 0.00142x + 1.33278 0.9904
SE15 PEG 600 08/06/2015 y = 0.00134x + 1.33280 0.9993
SE16 PEG 2000 08/06/2015 y = 0.00140x + 1.33280 0.9936
SE18 Cellobiose 09/06/2015 y = 0.00141x + 1.33295 0.9890
SE19 PEG 200 10/06/2015 y = 0.00133x + 1.33272 0.9985
SE21 PEG 20000 17/06/2015 y = 0.00132x + 1.33291 0.9999
SE22 PEG 20000 18/06/2015 y = 0.00138x + 1.33286 0.9995
SE23 Dextran 75000 19/06/2015 y = 0.00142x + 1.33288 0.9999
SE24 PEG 2000 22/06/2015 y = 0.00115x + 1.33314 0.9767
SE25 PEG 8000 22/06/2015 y = 0.00133x + 1.33289 0.9999
SE26 PEG 1500 23/06/2015 y = 0.00132x + 1.33289 0.9999
SE27 PEG 4000 23/06/2015 y = 0.00132x + 1.33290 0.9999
SE28 PEG 200 24/06/2015 y = 0.00121x + 1.33285 0.9999
SE29 Glucose 30/06/2015 y = 0.00134x + 1.33298 0.9993
SE30 Glucose 30/06/2015 y = 0.00141x + 1.33289 0.9999
SE31 PEG 35000 02/07/2015 y = 0.00135x + 1.33289 0.9999
SE32 PEG 35000 02/07/2015 y = 0.00135x + 1.33289 0.9999
SE33 Glucose 09/07/2015 y = 0.00142x + 1.33289 0.9986
SE34 Glucose 10/07/2015 y = 0.00144x + 1.33287 0.9973
SE35 PEG 35000 10/07/2015 y = 0.00135x + 1.33289 0.9998
SE36 PEG 200 15/07/2015 y = 0.00132x + 1.33274 0.9996
SE37 Cellobiose 15/07/2015 y = 0.00147x + 1.33288 0.9998
SE38 PEG 600 16/07/2015 y = 0.00131x + 1.33285 0.9999

75
Saturated substrate methodology – Alphacel
Table 8-10: Results from saturated substrate methodology, for Alphacel.
PE
G 2
00
00 ID SE01 mi (g) mss (g) WRV Ci (g/100mL) nD Cf (g/100mL) Vp (mL)
m1 1.0 1.0014 0.52
1.02
1.33427 1.05 -0.29
m2 1.0 1.0000 0.64 1.33429 1.07 -0.46
m3 1.0 1.0045 0.65 1.33426 1.04 -0.24
m4 1.0 1.0082 0.63 1.33428 1.05 -0.34
PE
G 2
00
00
ID SE02 mi (g) mss (g) WRV Ci (g/100mL) nD Cf (g/100mL) Vp (mL)
m1 1.0 1.0006 0.63
1.02
1.33424 1.03 -0.09
m2 1.0 1.0103 0.62 1.33424 1.03 -0.10
m3 1.0 1.0017 0.67 1.33423 1.02 -0.05
m4 1.0 1.0056 0.63 1.33428 1.05 -0.34
PE
G 8
00
0
ID SE03 mi (g) mss (g) WRV Ci (g/100mL) nD Cf (g/100mL) Vp (mL)
m1 1.0 1.0052 0.61
1.00
1.33418 1.00 0.03
m2 1.0 1.0050 0.65 1.33415 0.98 0.25
m3 1.0 1.0011 0.65 1.33416 0.99 0.17
m4 1.0 1.0010 0.61 1.33417 0.99 0.10
PE
G 8
00
0
ID SE04 mi (g) mss (g) WRV Ci (g/100mL) nD Cf (g/100mL) Vp (mL)
m1 1.0 1.0069 0.70
1.00
1.33420 1.01 -0.16
m2 1.0 1.0035 0.63 1.33419 1.01 -0.08
m3 1.0 1.0080 0.61 1.33419 1.01 -0.08
m4 1.0 1.0002 0.65 1.33419 1.01 -0.08
PE
G 2
00
ID SE10 mi (g) mss (g) WRV Ci (g/100mL) nD Cf (g/100mL) Vp (mL)
m1 1.0 1.0180 0.67
1.00
1.33401 0.99 0.10
m2 1.0 1.0190 0.67 1.33402 1.00 0.02
m3 1.0 1.0120 0.65 1.33400 0.98 0.18
m4 1.0 1.0250 0.59 1.33399 0.98 0.26

76
Dried substrate methodology – Alphacel
Results that present a significant deviation were not considered in calculations (signed with an asterisk – *). For the molecules with high diameter, when the result of determination of accessible volume is negative, is considered zero for the treatment of the data (signed with double asterisk – **).
Table 8-11: Results from dried substrate methodology, for Alphacel (part 1).
PE
G 2
00
00
ID SE06 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Ve (mL) Va (mL) Vi (mL) Ve (mL/g mds) Va (mL/g mds) Vi (mL/g mds)
m1 1.3134 1.2702 3.3% 1.0131
1.00
1.33435 1.10 9.15 0 0.85 9.03 0 0.84 *
m2 1.3343 1.2906 3.3% 1.0136 1.33433 1.08 9.27 0 0.73 9.14 0 0.72
m3 1.3051 1.2632 3.2% 1.0008 1.33434 1.09 9.21 0 0.79 9.20 0 0.79
m4 1.3190 1.2774 3.2% 1.0031 1.33434 1.09 9.21 0 0.79 9.18 0 0.79
PE
G 1
00
00
ID SE13 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.2017 1.1726 2.4% 1.0006
1.00
1.33441 1.15 -0.50 1.27 -0.50 1.27 **
m2 1.1657 1.1392 2.3% 1.0003 1.33433 1.09 -0.05 0.82 -0.05 0.82 **
m3 1.1636 1.1365 2.3% 1.0009 1.33433 1.09 -0.05 0.82 -0.05 0.82 **
m4 1.1438 1.1202 2.1% 1.0014 1.33439 1.13 -0.39 1.16 -0.39 1.16 **
PE
G 8
00
0
ID SE12 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.1576 1.1243 2.9% 1.0004
1.01
1.33430 1.07 0.18 0.59 0.18 0.59 *
m2 1.2266 1.1904 3.0% 1.0203 1.33434 1.10 -0.02 0.80 -0.02 0.78 *
m3 1.2001 1.1654 2.9% 1.0070 1.33433 1.09 0.04 0.73 0.04 0.73
m4 1.1984 1.1647 2.8% 1.0022 1.33433 1.09 0.04 0.73 0.04 0.73
PE
G 2
00
0
ID SE16 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.2017 1.1690 2.7% 1.004
1.00
1.33431 1.08 0.06 0.71 0.06 0.71
m2 1.2147 1.1789 2.9% 1.0002 1.33430 1.07 0.12 0.65 0.12 0.65
m3 1.2116 1.1770 2.9% 1.0043 1.33431 1.08 0.06 0.71 0.06 0.71
m4 1.2209 1.1868 2.8% 1.0001 1.33434 1.10 -0.12 0.90 -0.12 0.90 *

77
Table 8-12: Results from dried substrate methodology, for Alphacel (part 2).
PE
G 1
50
0
ID SE14 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.1983 1.1683 2.5% 1.0013
1.00
1.33430 1.07 0.16 0.61 0.16 0.61 *
m2 1.1885 1.1600 2.4% 1.0007 1.33431 1.08 0.10 0.68 0.10 0.68
m3 1.1860 1.1565 2.5% 1.0048 1.33431 1.08 0.10 0.68 0.10 0.67
m4 1.2042 1.1724 2.6% 1.0007 1.33431 1.08 0.10 0.68 0.10 0.68
PE
G 6
00
ID SE15 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.2216 1.1864 2.9% 1.0010
1.01
1.33424 1.07 0.20 0.58 0.20 0.57
m2 1.2259 1.1900 2.9% 1.0009 1.33424 1.07 0.20 0.58 0.20 0.57
m3 1.2198 1.1858 2.8% 1.0084 1.33423 1.07 0.26 0.51 0.26 0.51
m4 1.2332 1.1961 3.0% 1.0068 1.33424 1.07 0.20 0.58 0.20 0.57
PE
G 2
00
ID SE19 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.1925 1.1565 3.0% 1.0004
1.02
1.33413 1.06 0.39 0.39 0.39 0.38
m2 1.2022 1.1666 3.0% 1.0004 1.33413 1.06 0.39 0.39 0.41 0.38
m3 1.2075 1.1720 2.9% 1.0004 1.33412 1.05 0.46 0.32 0.48 0.32
m4 1.1951 1.1601 2.9% 1.0004 1.33413 1.06 0.39 0.39 0.41 0.38
Cell
ob
iose
ID SE18 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.1566 1.1249 2.7% 1.0003
1.00
1.33442 1.04 0.41 0.37 0.41 0.37
m2 1.1675 1.1382 2.5% 1.0026 1.33441 1.04 0.47 0.30 0.47 0.30
m3 1.1663 1.1355 2.6% 1.0074 1.33442 1.04 0.41 0.37 0.40 0.37
m4 1.1636 1.1335 2.6% 1.0035 1.33441 1.04 0.47 0.30 0.47 0.30
Glu
co
se
ID SE29 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.1842 1.1304 4.5% 1.0061
1.00
1.33437 1.01 0.70 0.07 0.70 0.07
m2 1.1708 1.1388 2.7% 1.0001 1.33437 1.01 0.70 0.07 0.70 0.07
m3 1.1641 1.1317 2.8% 1.0043 1.33437 1.01 0.70 0.07 0.70 0.07
m4 1.1706 1.1332 3.2% 1.0084 1.33437 1.01 0.70 0.07 0.69 0.07
m5 1.1791 1.1462 2.8% 1.0026 water 1.33289 -0.07 - - - -

78
Dried substrate methodology – Non-washed native wheat straw
Results that present a significant deviation were not considered in calculations (signed with an asterisk – *). When the result of determination of accessible
volume or inaccessible is negative, is considered zero for the treatment of the data (signed with double or triple asterisk, respectively).
Table 8-13: Results from dried substrate methodology, for non-washed native wheat straw (part 1).
PE
G 3
50
00
ID SE31 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Ve (mL) Va (mL) Vi (mL) Ve (mL/g mds) Va (mL/g mds) Vi (mL/g mds)
m1 1.1639 1.1160 4.1% 1.0010
1.00
1.33560 2.01 5.00 0 5.00 4.99 0 5.00
m2 1.1708 1.1196 4.4% 1.0034 1.33556 1.98 5.07 0 4.93 5.06 0 4.91 *
m3 1.1564 1.1076 4.2% 1.0002 1.33561 2.01 4.98 0 5.02 4.98 0 5.02
m4 1.1650 1.1155 4.2% 1.0021 1.33565 2.04 4.91 0 5.09 4.90 0 5.08 *
m5 1.1992 1.1473 4.3% 1.0028 water 1.33409 0.89 - - - - - -
PE
G 2
00
00
ID SE21 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.1400 1.0858 4.8% 1.0038
1.00
1.33560 2.04 -0.08 5.09 -0.08 5.07 **
m2 1.1524 1.0997 4.6% 1.0028 1.33559 2.03 -0.06 5.07 -0.06 5.06 **
m3 1.1545 1.1025 4.5% 1.0015 1.33557 2.02 -0.02 5.04 -0.02 5.03 **
m4 1.1531 1.1001 4.6% 1.0030 1.33559 2.03 -0.06 5.07 -0.06 5.06 **
m5 1.1506 1.0991 4.5% 1.0034 water 1.33416 0.95 - - - -
DE
XT
RA
N 7
50
00 ID SE23 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.1983 1.1497 4.1% 1.0004
1.01
1.33572 2.00 0.04 4.97 0.04 4.97 *
m2 1.1736 1.1270 4.0% 1.0029 1.33565 1.95 0.17 4.84 0.17 4.83
m3 1.1694 1.1223 4.0% 1.0018 1.33565 1.95 0.17 4.84 0.17 4.83
m4 1.1706 1.1225 4.1% 1.002 1.33565 1.95 0.17 4.84 0.17 4.83
m5 1.1793 1.1301 4.2% 1.0026 water 1.33418 0.92 - - - -

79
Table 8-14: Results from dried substrate methodology, for non-washed native wheat straw (part 2).
PE
G 8
00
0
ID SE25 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.1697 1.1240 3.9% 1.0051
1.00
1.33561 2.05 -0.09 5.10 -0.09 5.08 *
m2 1.1653 1.1124 4.5% 1.0007 1.33554 1.99 -0.09 4.97 -0.09 4.97 *
m3 1.1606 1.1085 4.5% 1.0016 1.33558 2.02 -0.04 5.05 -0.04 5.04 *
m4 1.1788 1.1238 4.7% 1.0049 1.33558 2.02 -0.04 5.05 -0.04 5.02 *
PE
G 4
00
0
ID SE27 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.1829 1.1326 4.3% 1.0002
1.00
1.33559 2.04 -0.08 5.09 -0.08 5.09 *
m2 1.1714 1.1208 4.3% 1.0033 1.33557 2.02 -0.04 5.05 -0.04 5.04 *
m3 1.1676 1.1161 4.4% 1.0048 1.33556 2.02 -0.02 5.04 -0.02 5.01 *
m4 1.1659 1.1153 4.3% 1.0001 1.33569 2.11 -0.26 5.27 -0.26 5.27 *
m5 1.1755 1.1234 4.4% 1.0081 water 1.33424 1.02 - - - -
PE
G 2
00
0
ID SE24 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.1598 1.1077 4.5% 1.0048
1.00
1.33551 2.06 -0.13 5.14 -0.13 5.12 *
m2 1.1832 1.1325 4.3% 1.0030 1.33554 2.09 -0.19 5.20 -0.19 5.19 *
m3 1.1660 1.1152 4.4% 1.0034 1.33558 2.12 -0.27 5.28 -0.27 5.26 *
m4 1.1585 1.1065 4.5% 1.0069 1.33559 2.13 -0.29 5.30 -0.29 5.26 *
m5 1.1638 1.1100 4.6% 1.0016 water 1.33418 0.90 - - - -
PE
G 1
50
0
ID SE26 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.1747 1.1261 4.1% 1.0039
1.00
1.33556 2.02 -0.03 5.04 -0.03 5.02 *
m2 1.1713 1.1215 4.3% 1.0008 1.33555 2.02 -0.01 5.02 -0.01 5.02 *
m3 1.1898 1.1418 4.0% 1.0014 1.33556 2.02 -0.03 5.04 -0.03 5.04 *
m4 1.1786 1.1295 4.2% 1.0059 1.33557 2.03 -0.05 5.06 -0.05 5.03 *
m5 1.1739 1.1251 4.2% 1.0077 water 1.33416 0.96 - - - -
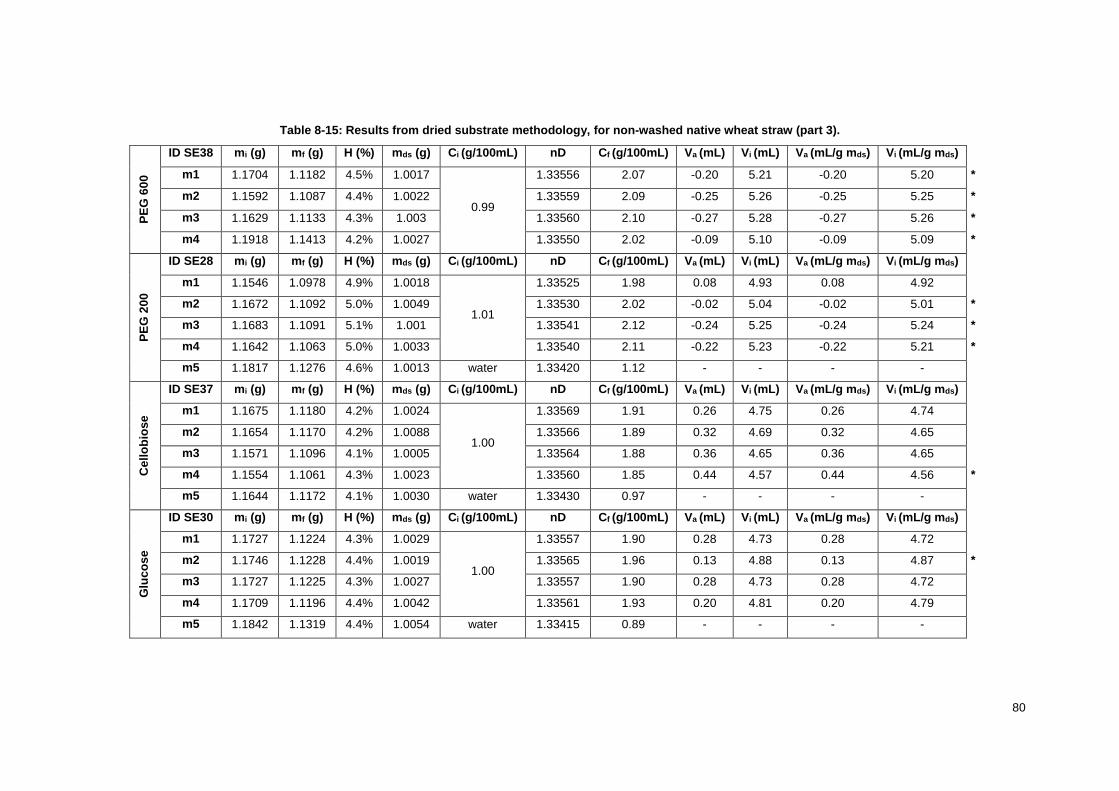
80
Table 8-15: Results from dried substrate methodology, for non-washed native wheat straw (part 3). P
EG
60
0
ID SE38 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.1704 1.1182 4.5% 1.0017
0.99
1.33556 2.07 -0.20 5.21 -0.20 5.20 *
m2 1.1592 1.1087 4.4% 1.0022 1.33559 2.09 -0.25 5.26 -0.25 5.25 *
m3 1.1629 1.1133 4.3% 1.003 1.33560 2.10 -0.27 5.28 -0.27 5.26 *
m4 1.1918 1.1413 4.2% 1.0027 1.33550 2.02 -0.09 5.10 -0.09 5.09 *
PE
G 2
00
ID SE28 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.1546 1.0978 4.9% 1.0018
1.01
1.33525 1.98 0.08 4.93 0.08 4.92
m2 1.1672 1.1092 5.0% 1.0049 1.33530 2.02 -0.02 5.04 -0.02 5.01 *
m3 1.1683 1.1091 5.1% 1.001 1.33541 2.12 -0.24 5.25 -0.24 5.24 *
m4 1.1642 1.1063 5.0% 1.0033 1.33540 2.11 -0.22 5.23 -0.22 5.21 *
m5 1.1817 1.1276 4.6% 1.0013 water 1.33420 1.12 - - - -
Cell
ob
iose
ID SE37 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.1675 1.1180 4.2% 1.0024
1.00
1.33569 1.91 0.26 4.75 0.26 4.74
m2 1.1654 1.1170 4.2% 1.0088 1.33566 1.89 0.32 4.69 0.32 4.65
m3 1.1571 1.1096 4.1% 1.0005 1.33564 1.88 0.36 4.65 0.36 4.65
m4 1.1554 1.1061 4.3% 1.0023 1.33560 1.85 0.44 4.57 0.44 4.56 *
m5 1.1644 1.1172 4.1% 1.0030 water 1.33430 0.97 - - - -
Glu
co
se
ID SE30 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.1727 1.1224 4.3% 1.0029
1.00
1.33557 1.90 0.28 4.73 0.28 4.72
m2 1.1746 1.1228 4.4% 1.0019 1.33565 1.96 0.13 4.88 0.13 4.87 *
m3 1.1727 1.1225 4.3% 1.0027 1.33557 1.90 0.28 4.73 0.28 4.72
m4 1.1709 1.1196 4.4% 1.0042 1.33561 1.93 0.20 4.81 0.20 4.79
m5 1.1842 1.1319 4.4% 1.0054 water 1.33415 0.89 - - - -

81
Table 8-16: Results from dried substrate methodology, for non-washed native wheat straw – nD correction (part 1).
PE
G 3
50
00
ID SE31 nD nD’ Cf (g/100mL) Ve (mL) Va (mL) Vi (mL) Ve (mL/g mds) Va (mL/g mds) Vi (mL/g mds)
m1 1.33560 1.33426 1.02 9.85 0 0.15 9.84 0 0.15
m2 1.33556 1.33422 0.99 10.15 0 -0.15 10.11 0 -0.15 *
m3 1.33561 1.33427 1.03 9.78 0 0.22 9.78 0 0.22
m4 1.33565 1.33431 1.06 9.51 0 0.49 9.49 0 0.49 *
PE
G 2
00
00
ID SE21 nD nD’ Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.33560 1.33426 1.03 -0.07 0.25 -0.07 0.25 **
m2 1.33559 1.33425 1.02 0.00 0.18 0.00 0.18 **
m3 1.33557 1.33423 1.00 0.15 0.03 0.15 0.03 *
m4 1.33559 1.33425 1.02 0.00 0.18 0.00 0.18 **
PE
G 8
00
0
ID SE25 nD nD’ Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.33561 1.33427 1.04 -0.20 0.38 -0.20 0.38 *
m2 1.33554 1.33420 0.99 -0.20 -0.13 -0.20 -0.13 *
m3 1.33558 1.33424 1.02 0.02 0.17 0.02 0.17
m4 1.33558 1.33424 1.02 0.02 0.17 0.02 0.17
PE
G 4
00
0
ID SE25 nD nD’ Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.33559 1.33425 1.03 -0.07 0.25 -0.07 0.25 *
m2 1.33557 1.33423 1.01 0.08 0.10 0.08 0.10
m3 1.33556 1.33422 1.00 0.15 0.03 0.15 0.03
m4 1.33569 1.33435 1.10 -0.74 0.92 -0.74 0.92 *
PE
G 2
00
0
ID SE24 nD nD’ Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.33551 1.33417 0.90 1.31 -1.13 1.30 -1.12 *
m2 1.33554 1.33420 0.93 1.00 -0.81 0.99 -0.81 *
m3 1.33558 1.33424 0.96 0.60 -0.42 0.60 -0.42 *
m4 1.33559 1.33425 0.97 0.51 -0.33 0.51 -0.33 *

82
Table 8-17: Results from dried substrate methodology, for non-washed native wheat straw – nD correction (part 2).
PE
G 1
50
0
ID SE26 nD nD’ Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.33556 1.33422 1.01 0.10 0.08 0.10 0.08
m2 1.33555 1.33421 1.00 0.17 0.01 0.17 0.01
m3 1.33556 1.33422 1.01 0.10 0.08 0.10 0.08
m4 1.33557 1.33423 1.02 0.03 0.16 0.03 0.16 *
PE
G 6
00
ID SE38 nD nD’ Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.33556 1.33422 1.05 -0.37 0.55 -0.37 0.55 *
m2 1.33559 1.33425 1.07 -0.57 0.76 -0.57 0.75 *
m3 1.33560 1.33426 1.08 -0.64 0.82 -0.64 0.82 *
m4 1.33550 1.33416 1.00 0.06 0.12 0.06 0.12
PE
G 2
00
ID SE28 nD nD’ Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.33525 1.33391 0.88 1.61 -1.42 1.60 -1.42 ***
m2 1.33530 1.33396 0.92 1.09 -0.91 1.09 -0.91 ***
m3 1.33541 1.33407 1.01 0.11 0.07 0.11 0.07 *
m4 1.33540 1.33406 1.00 0.20 -0.01 0.19 -0.01 ***
Cell
ob
iose
ID SE37 nD nD’ Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.33569 1.33435 1.00 0.19 -0.01 0.19 -0.01 ***
m2 1.33566 1.33432 0.98 0.40 -0.22 0.40 -0.22 ***
m3 1.33564 1.33430 0.97 0.54 -0.36 0.54 -0.36 ***
m4 1.33560 1.33426 0.94 0.84 -0.66 0.84 -0.66 ***
Glu
co
se
ID SE30 nD nD’ Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.33557 1.33423 0.95 0.69 -0.51 0.69 -0.51 ***
m2 1.33565 1.33431 1.01 0.10 0.08 0.10 0.08 *
m3 1.33557 1.33423 0.95 0.69 -0.51 0.69 -0.51 ***
m4 1.33561 1.33427 0.98 0.38 -0.20 0.38 -0.20 ***

83
Dried substrate methodology – Washed native wheat straw
Results that present a significant deviation were not considered in calculations (signed with an asterisk – *). For the molecules with high diameter, when
the result of determination of accessible volume is negative, is considered zero for the treatment of the data (signed with double asterisk – **).
Table 8-18: Results from dried substrate methodology, for washed native wheat straw.
PE
G 3
50
00
ID SE32 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Ve (mL) Va (mL) Vi (mL) Ve (mL/g mds) Va (mL/g mds) Vi (mL/g mds)
m1 1.6907 0.6721 60.2% 0.4999
1.01
1.33436 1.09 4.63 0 0.37 9.25 0 0.75
m2 1.6220 0.6361 60.8% 0.5000 1.33434 1.07 4.69 0 0.31 9.38 0 0.62 *
m3 1.6363 0.6451 60.6% 0.5019 1.33438 1.10 4.56 0 0.44 9.09 0 0.87 *
m4 1.1974 0.4705 60.7% 0.5007 1.33436 1.09 4.63 0 0.37 9.24 0 0.75
m5 - - - 0.4503 water 1.33286 -0.02 - - - -
PE
G 2
00
00
ID SE36 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.2021 0.4764 60.4% 0.4804
1.00
1.33436 1.09 -0.01 0.38 -0.02 0.80 *
m2 1.2086 0.4745 60.7% 0.4828 1.33434 1.07 0.05 0.32 0.11 0.67
m3 1.2223 0.4736 61.3% 0.4825 1.33437 1.09 -0.04 0.41 -0.08 0.86 *
m4 1.2308 0.4957 59.7% 0.5045 1.33433 1.07 0.08 0.29 0.17 0.58
m5 1.1954 0.4913 58.9% 0.4986 water 1.33302 0.12 - - - -
Glu
co
se
ID SE34 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.2972 0.5258 59.5% 0.5011
1.00
1.33451 1.14 -0.23 0.60 -0.46 1.20
m2 1.2914 0.5083 60.6% 0.5020 1.33451 1.14 -0.23 0.60 -0.45 1.20
m3 1.2993 0.5169 60.2% 0.5002 1.33451 1.14 -0.23 0.60 -0.46 1.20
m4 1.2731 0.2311 81.8% 0.2883 1.33451 1.14 -0.23 0.60 -0.79 2.09
m5 1.2987 0.5020 61.3% 0.5013 water 1.33303 0.10 - - - -

84
Table 8-19: Results from dried substrate methodology, for washed native wheat straw – nD correction.
PE
G 3
50
00
ID SE32 nD nD’ Cf (g/100mL) Ve (mL) Va (mL) Vi (mL) Ve (mL/g mds) Va (mL/g mds) Vi (mL/g mds)
m1 1.33449 1.33435 1.08 9.26 0 0.74 9.23 0 0.74
m2 1.33448 1.33434 1.08 9.32 0 0.68 9.27 0 0.68
m3 1.33449 1.33435 1.08 9.26 0 0.74 9.19 0 0.74
m4 1.33451 1.33437 1.10 9.13 0 0.87 9.10 0 0.87 *
PE
G 2
00
ID SE36 nD nD’ Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.33426 1.33412 1.05 0.37 0.35 0.37 0.35
m2 1.33424 1.33410 1.03 0.51 0.21 0.51 0.21 *
m3 1.33426 1.33412 1.05 0.37 0.35 0.37 0.35
m4 1.33430 1.33416 1.08 0.10 0.62 0.10 0.62 *
Glu
co
se
ID SE34 nD nD’ Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 1.33446 1.33432 1.01 0.65 0.07 0.65 0.07
m2 1.33447 1.33433 1.02 0.58 0.14 0.58 0.14
m3 1.33447 1.33433 1.02 0.58 0.14 0.58 0.14
m4 1.33450 1.33436 1.04 0.38 0.34 0.38 0.34 *

85
Dried substrate methodology – Wheat straw pretreated at 160 °C and washed
Results that present a significant deviation were not considered in calculations (signed with an asterisk – *). For the molecules with high diameter, when
the result of determination of accessible volume is negative, is considered zero for the treatment of the data (signed with double asterisk – **).
Table 8-20: Results from dried substrate methodology, for wheat straw pretreated at 160 °C and washed.
PE
G 3
50
00
ID SE35 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Ve (mL) Va (mL) Vi (mL) Ve (mL/g mds) Va (mL/g mds) Vi (mL/g mds)
m1 6.4620 1.2814 80.2% 1.0023
1.00
1.33449 1.19 8.47 0 1.53 8.45 0 1.53
m2 6.7528 1.3003 80.7% 1.0053 1.33448 1.18 8.52 0 1.48 8.47 0 1.47
m3 6.0815 1.1687 80.8% 1.0068 1.33449 1.19 8.47 0 1.53 8.41 0 1.52
m4 6.2017 1.2963 79.1% 1.0030 1.33451 1.20 8.36 0 1.64 8.34 0 1.63 *
m5 6.1016 1.3021 78.7% 1.0078 water 1.33301 0.09 - - - - -
PE
G 2
00
ID SE22 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 6.2351 1.3581 78.2% 1.0016
1.01
1.33426 1.15 0.30 1.22 0.30 1.22
m2 6.0296 1.2350 79.5% 1.0014 1.33424 1.14 0.41 1.10 0.41 1.10 *
m3 6.1902 1.2227 80.2% 1.0094 1.33426 1.15 0.30 1.22 0.29 1.21
m4 6.2415 1.1663 81.3% 1.0017 1.33430 1.18 0.07 1.45 0.07 1.44 *
m5 6.0455 1.2929 78.6% 1.0002 water 1.33301 0.20 - - - -
Glu
co
se
ID SE33 mi (g) mf (g) H (%) mds (g) Ci (g/100mL) nD Cf (g/100mL) Va (mL) Vi (mL) Va (mL/g mds) Vi (mL/g mds)
m1 6.0339 1.1702 80.6% 1.0042
1.00
1.33446 1.10 0.59 0.93 0.59 0.92
m2 6.0001 1.1951 80.1% 1.0012 1.33447 1.11 0.53 0.98 0.53 0.98
m3 6.0204 1.1364 81.1% 1.0014 1.33447 1.11 0.53 0.98 0.53 0.98
m4 6.0270 1.1631 80.7% 1.0002 1.33450 1.13 0.37 1.15 0.37 1.15 *
m5 6.0273 1.2072 80.0% 1.0006 water 1.33297 0.07 - - - -

86
Pore volume distributions for dried substrate methodology
To the standard deviation of the results, a 98% of confidence interval was applied to the data.
Results that present a significant deviation were not considered in calculations (signed with an
asterisk – *). For the molecules with high diameter, when the result of determination of accessible
volume is negative, is considered zero for the treatment of the data (signed with double asterisk – **).
Table 8-21: Pore volume distribution data for Alphacel.
Probe Diameter (Ǻ) Ve (mL/g mds) Va (mL/g mds) Vi (mL/g mds)
PEG 20000 130 9.17±0.03 0 0.77±0.04
PEG 10000 90 9.17±0.03 0 0.77±0.04
PEG 8000 84 9.17±0.03 0.04±0.00 0.73±0.00
PEG 2000 40 9.17±0.03 0.08±0.04 0.69±0.03
PEG 1500 33 9.17±0.03 0.10±0.00 0.67±0.00
PEG 600 21 9.17±0.03 0.21±0.03 0.56±0.03
PEG 200 13 9.17±0.03 0.42±0.04 0.37±0.03
Cellobiose 10 9.17±0.03 0.44±0.04 0.33±0.04
Glucose 8 9.17±0.03 0.70±0.00 0.07±0.00
Table 8-22: Pore volume distribution data for non-washed native wheat straw.
Probe Diameter (Ǻ) Ve (mL/g mds) Va (mL/g mds) Vi (mL/g mds)
PEG 35000 170 4.99±0.01 0 5.01±0.02
PEG 20000 130 4.99±0.01 0 5.01±0.02
Dextran 75000 120 4.99±0.01 0.17±0.00 4.83±0.00 *
PEG 8000 84 4.99±0.01 -0.06±0.03 5.03±0.04 *
PEG 4000 56 4.99±0.01 -0.10±0.10 5.10±0.11 *
PEG 2000 40 4.99±0.01 -0.22±0.07 5.21±0.07 *
PEG 1500 33 4.99±0.01 -0.03±0.01 5.03±0.01 *
PEG 600 21 4.99±0.01 -0.20±0.08 5.20±0.08 *
PEG 200 13 4.99±0.01 0.08±0.00 4.92±0.00
Cellobiose 10 4.99±0.01 0.31±0.05 4.68±0.05
Glucose 8 4.99±0.01 0.26±0.04 4.74±0.04

87
Table 8-23: Pore volume distribution data for non-washed native wheat straw – nD correction.
Probe Diameter (Ǻ) Ve (mL/g mds) Va (mL/g mds) Vi (mL/g mds)
PEG 35000 170 9.81±0.04 0 0.18±0.05
PEG 20000 130 9.81±0.04 0 0.18±0.05
Dextran 75000 120 9.81±0.04 0.14±0.00 0.04±0.00 *
PEG 8000 84 9.81±0.04 0.02±0.00 0.17±0.00
PEG 4000 56 9.81±0.04 0.11±0.05 0.07±0.15
PEG 2000 40 9.81±0.04 0.85±0.36 -0.67±0.36 *
PEG 1500 33 9.81±0.04 0.12±0.04 0.06±0.04
PEG 600 21 9.81±0.04 0.06±0.00 0.12±0.00
PEG 200 13 9.81±0.04 0.18±0.05 0 **
Cellobiose 10 9.81±0.04 0.18±0.05 0 **
Glucose 8 9.81±0.04 0.18±0.05 0 **
Table 8-24: Pore volume distribution data for washed native wheat straw.
Probe Diameter (Ǻ) Ve (mL/g mds) Va (mL/g mds) Vi (mL/g mds)
PEG 35000 170 8.44±0.03 0 1.51±0.03
PEG 200 13 8.44±0.03 0.29±0.00 1.21±0.01
Glucose 8 8.44±0.03 0.55±0.03 0.96±0.03
Table 8-25: Pore volume distribution data for washed native wheat straw – nD correction.
Probe Diameter (Ǻ) Ve (mL/g mds) Va (mL/g mds) Vi (mL/g mds)
PEG 35000 170 9.23±0.04 0 0.72±0.04
PEG 200 13 9.23±0.04 0.37±0.00 0.35±0.00
Glucose 8 9.23±0.04 0.60±0.03 0.12±0.04
Table 8-26: Pore volume distribution data for washed and pretreated wheat straw.
Probe Diameter (Ǻ) Ve (mL/g mds) Va (mL/g mds) Vi (mL/g mds)
PEG 35000 170 9.25±0.01 0 0.75±0.00
PEG 20000 130 9.25±0.01 0.29±0.00 0.62±0.06
Glucose 8 9.25±0.01 -0.54±0.17 1.42±0.44 *