european industrial pharmacy issue 26 (september 2015)
DESCRIPTION
European Industrial Pharmacy is the electronic journal of the European Industrial Pharmacists Group (EIPG). The journal contains articles, news and comments of special interest to pharmaceutical scientists and executives working in the European pharmaceutical and allied industries. It is independently managed, has a European Editorial Advisory Board and allows the voices of Industrial Pharmacists to be communicated to as wide an audience as possible.TRANSCRIPT

ISSUE 26 • SEPTEMBER 2015www.industrialpharmacy.eu
www.eipg.eu
features4 FIRST-IN-CLASS INNOVATION IN THE PIPELINE FOR
TREATMENT OF MUSCULAR DYSTROPHYThis article examines the current Duchenne musculardystrophy therapeutics market and trends in thedevelopmental pipeline, with a specific focus on the first-in-class molecules that will potentially transform futuremuscular dystrophy treatment.by Angel Wong
8 CHILD RESISTANCE IN PHARMACEUTICALPACKAGINGThis article highlights the critical role of innovation in childresistant packaging.by Hung Le
11 GETTING TO HIGHER QUALITY PROCESSES SOONER– NEW GUIDE HELPS YOU MAKE A BETTER CHOICEOF BIOLOGICAL MATERIALSTo help remove the complexity from choosing materialsthat compose cell-based medicinal products, ‘PAS157:2015 Evaluation of Materials of Biological Origin Used inthe Production of Cell-Based Medicinal Products – Guide’was developed by BSI, the Cell Therapy Catapult and thecell therapy industry.by Patrick Ginty and Ben Sheridan
15 IN SILICO CLINICAL TRIALS: DREAM ORCERTAINTY?This September, the Avicenna Consortium, tasked by theEuropean Commission to investigate how computermodelling might mitigate the cost of clinical trials, releasedits Roadmap ‘In Silico Clinical Trials: How ComputerSimulation Will Transform The Biomedical Industry’.by Marco Viceconti, Adriano Henney, Edwin Morley-Fletcher andMartina Contin
18 700 GÉNÉRIQUES RETIRÉS DU MARCHÉ:CONCRÈTEMENT, ÇA CHANGE QUOI?Mme Isabelle Adenot, President of the French Conseilnational de l’Ordre des Pharmaciens, talks to journalistHugo Jalinière, of the French periodical Sciences et Avenir,on the impact of the EU-wide withdrawal from the marketof 700 medicinal products, following an EU-wide suspensionof their marketing authorisations.by Hugo Jalinière, for Sciences et Avenir
regulars3 EDITORIAL COMMENT22 REGULATORY REVIEW24 BOTTLED BROWN25 NEWS FROM THE EIPG26 EVENTS
europeanINDUSTRIALPHARMACY

2 european INDUSTRIAL PHARMACY September 2015 • Issue 26
europeanINDUSTRIALPHARMACY
September 2015ISSN 1759-202X
MANAGING EDITORSue Briggs
PRODUCTIONDave Johnson
SUBSCRIPTIONSJill Monk
EDITORIAL BOARDMichael AnisfeldClaude FarrugiaMichael Gamlen
Linda HakesJohn Jolley
European Industrial Pharmacyis published four times a year by:
Euromed CommunicationsPassfield Business Centre,
Lynchborough Road, Passfield,Liphook, Hampshire GU30 7SB
Tel: +44 (0)1428 752222Fax: +44 (0)1428 752223
Email:[email protected]
www.eipg.eu/eipg-journal
Indexed by:Scopus & Embase
Views expressed in European IndustrialPharmacy are those of the contributors
and not necessarily endorsed by thePublisher, Editor, Editorial Board, or by ourcorporate sponsors who accept no liabilityfor the consequences of any inaccurate or
misleading information
©2015 Euromed Communications
europeanINDUSTRIALPHARMACYdiscussion group:
www.pharmweb.net/gmp.html
european INDUSTRIAL PHARMACYis the official publication of the European IndustrialPharmacists Group (Groupement des Pharmaciens del’Industrie en Europe) www.eipg.eu
Cover photo: Muscle fibres (see First-in-Class Innovation in the Pipeline forTreatment of Muscular Dystrophy onpage 4).
associate editors
Belgium: Philippe Bollen
Bulgaria: Valentina Belcheva
Czech Republic: Ales Franc
Finland: Anni Svala
France: Jean-Pierre Paccioni
Germany: Armin Hoffmann
Great Britain: Shilpa Gohil, Janet Halliday
Greece: Ioannis Nikolakakis
Hungary: Sylvia Marton
Ireland: Stan O'Neill
Italy: Piero Iamartino
Latvia: Inta Saprovska, Anita Senberga
Malta: Claude Farrugia
Netherlands: Amon Wafelman
Norway: Wenche Gordon
Spain: Beatriz Artalejo
Sweden: Marianne Andersson
Switzerland: Valter Gianesello, Maurizio Battistini

The journey andthe goalAn oft-cited oriental storydescribes a student who asked histeacher, “How long will it take meto master your discipline?” Theteacher replied, “Ten years.” Thestudent said, “If I work twice ashard, how long will it take then?”“Twenty years.” answered theteacher. Again the student said,“But if I work really hard, night andday, how long will it take me?”The teacher responded, “Thirtyyears.” The student was confused,“How is it that if I work harder, you say that it will takelonger?” The teacher replied, “Because when youkeep one eye on the goal, you only have one eyewith which to find the way.”
The moral of the story, namely, that the journey ismore important than the destination, is perhapsdifficult to apply in the pharmaceutical industry. Howelse, if not by keeping in mind that the goal ofmaintaining the highest standards of good practicesis to guarantee patient safety, does one find thecourage and justification to face the consequences ofsuspending the marketing authorisations of hundredsof medicinal products due to flawed studies? As I penthis message, the first draft of the DelegatedRegulation laying down detailed rules for the safetyfeatures appearing on the outer packaging ofmedicinal products for human use has just beenmade public. It is a regulation that will require achallenging journey to put into practice, and howelse, if not by keeping in mind the goal of protectingpatients from the dangers of falsified medicinalproducts, does one convince all players to embark,
without delay, on the steps neededto implement the requirements ofthis important regulation within theestablished timeframes?
Similarly, consider the panic ofGreek patients unable to purchasetheir medicines at the height of theeconomic crisis but a few weeksago, or the frustration of Frenchpatients, hearing once more“Désolé mais votre médicament estindiqué manquant”, due to drugshortages that, according to theAgence Nationale de Sécurité duMédicament et des Produits deSanté, have risen by ten times in the
last 6 years. How else, if not than by keeping our eyeon the goal of solving this perennial challenge in theprovision of healthcare, can one continue to bringtogether all stakeholders at a European level, to gobeyond the limited, albeit well-meaning, initiatives ofthe European Commission as outlined in its recentresponse to the European Parliament?
Yes, the goal is important. However, this does not inany way diminish neither the importance of thejourney that needs to be undertaken, nor thechallenges, trials and tribulations that all concernedexperience in reaching that goal. Therefore, in thewords of Lord Alfred Tennyson, “Oh yet we trust thatsomehow good will be the final goal of ill”.
Professor Claude FarrugiaVice-President Communications, EIPG
3european INDUSTRIAL PHARMACY September 2015 • Issue 26
editorial
We are sad to announce the death of Arthur Dewelde, industrial pharmacist, technical director
Pfizer Jette and Lieutenant-Colonel in the pharmacy reserve of Belgium. Through his professional
drive, he shaped the aims, objectives and culture of the European Industrial Pharmacists’ Group.
Following years of representations to the Commission alongside other healthcare professions, he
ensured that pharmacy had its own professional directive appropriate to the industrial
pharmacists as well as other areas of pharmacy of that period. We wish to extend our
condolences to his friends and family.

Muscular dystrophy is a group ofheritable, genetic neuromusculardisorders characterised byprogressive muscle weakness anddegeneration. The most commontypes of muscular dystrophy areDuchenne muscular dystrophy(DMD) followed by its milder form,Becker muscular dystrophy (BMD).Both diseases are caused bymutations in the single DMD geneon the X chromosome, and as arecessive disease, this means it isexclusive to males.
Dystrophin, which is the proteinproduct of DMD, is a rod-shapedcytoplasmic protein expressedprimarily in skeletal muscles andcardiac muscles. It associates withvarious proteins, including α-dystrobrevin and β-dystroglycan,to form the dystrophin-associatedprotein complex (DAPC) at thesarcolemma (cell membrane of amuscle fibre cell). The DAPCfunctions as the structural linkbetween the actin cytoskeleton andextracellular matrix, which is crucialfor normal muscle function duringmuscle contraction and relaxation.
Destabilisation of the sarcolemmal
a DAPC, which can arise frommutations in DMD, leads toincreased susceptibility to musclefibre damage and necrosis. MostDMD-causing mutations shift thereading frame by one or two basepairs, resulting in the addition ofincorrect amino acids to thepolypeptide. A nonsense mutation,for instance, leads to a prematurestop codon, causing earlytermination of translation and theproduction of truncated, non-functional dystrophin.
Symptoms such as delay in age ofwalking generally arise in boys afterthe age of five. By late childhood,they typically lose the ability towalk, and may develop lifethreatening cardiac and respiratorycomplications during their lateteens as heart and lung musclesweaken. Despite similar symptoms,the disease progression in BMD isslower, as it is caused by non-frameshifting mutations which retainpartial protein function ofdystrophin.
It has become increasinglyapparent that dystrophin deficiencyis not the only factor driving disease
progression, particularly whenmuscle fibre degeneration has beenlong regarded as a multifacetedprocess. Following sarcolemmaldefects, secondary pathologicalprocesses that may contribute tothe hallmarks of DMD/BMD includemechanical stress, deregulatedcalcium homeostasis, impairedvascular adaptation andinflammation. Ultimately, increasedmuscle necrosis, coupled with thefailure to repair damaged muscles,leads to replacement of musclefibres by adipose and connectivetissues, namely fibrosis.
Current treatmentOver the past few decades, genericglucocorticoid treatment,specifically prednisone anddeflazacort, remains the mainstay ofpharmacological treatment of DMDand BMD. They are glucocorticoidreceptor agonists which act bysuppressing transcription ofinflammatory genes via activation ofNF-KB. In clinical studies,prednisone and deflazacort wereshown to offer similar symptomaticbenefits in improving musclestrength, delaying the loss of abilityto walk and stabilising pulmonaryfunction in DMD patients1,2.Although the exact mechanism thatgives rise to the clinical benefitremains unclear, they are thought toupregulate expression of muscle-specific target genes via thecalcineurin/nuclear factor of theactivated T-cells pathway, such asutrophin (an autosomal homologueof dystrophin), which has astructural role similar to dystrophin3.
Like other glucocorticoids,prednisone and deflazacort areassociated with significant adverseevents, including weight gain andincreased risk of vertebral fractures.Studies showed that deflazacort isassociated with less weight gainand more preservation of bonemass, making it the preferabletreatment over prednisone inpatients with pre-existing weightissues4.
Apart from muscle weakness,secondary complicationscomprising respiratory,cardiovascular and orthopaedicissues are managed with
european INDUSTRIAL PHARMACY September 2015 • Issue 26
FIRST-IN-CLASS INNOVATIONIN THE PIPELINE FORTREATMENT OF MUSCULARDYSTROPHYby Angel Wong
Duchenne research has gained momentum recentlywith the first innovative therapy to treat the
underlying cause of a nonsense mutation in the DMDgene. The exceptionally high level of innovation anddiversity in the pipeline will open up numerousopportunities for more novel products to thrive, creatingan encouraging outlook for the vast majority of musculardystrophy patients, as well as companies and investorsin the market.
Angel Wong is a Senior Analyst for business intelligence provider GBI Research. She has aparticular interest in molecular cell biology and immunology. Angel holds a BSc inBiochemistry from the University of Nottingham and an MSc in Management fromLoughborough University.
4
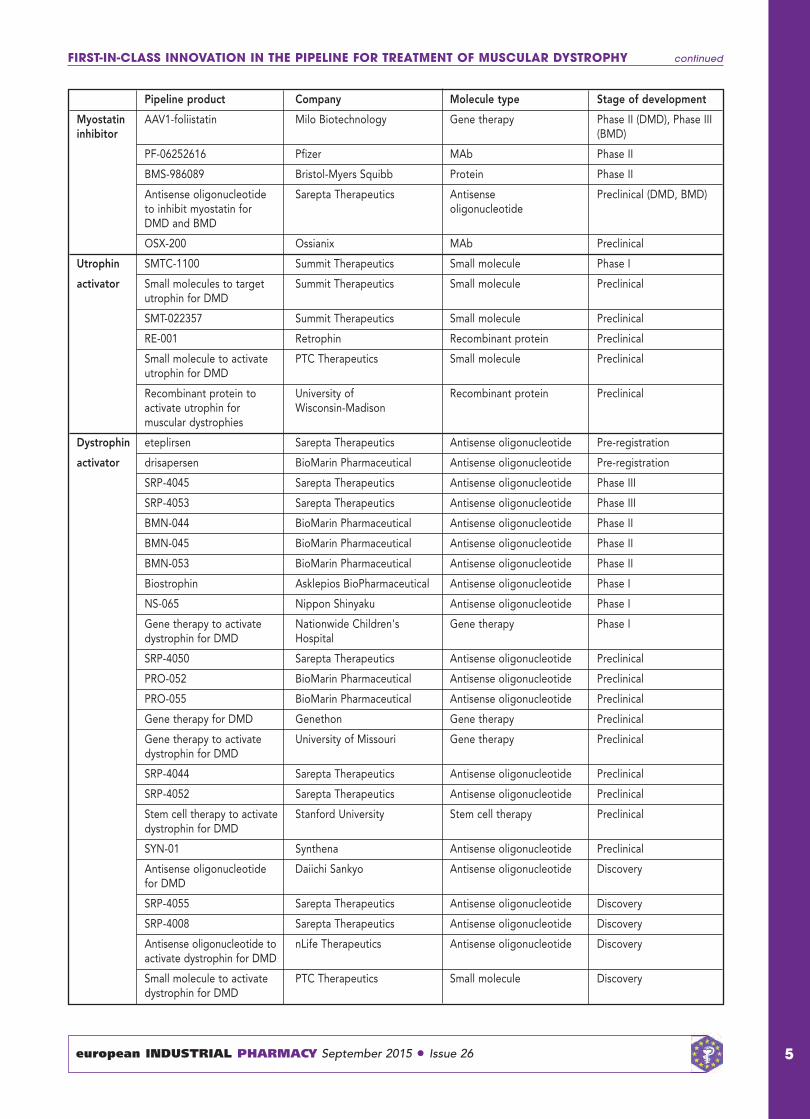
FIRST-IN-CLASS INNOVATION IN THE PIPELINE FOR TREATMENT OF MUSCULAR DYSTROPHY continued
5european INDUSTRIAL PHARMACY September 2015 • Issue 26
Pipeline product Company Molecule type Stage of development
Myostatin AAV1-foliistatin Milo Biotechnology Gene therapy Phase II (DMD), Phase III inhibitor (BMD)
PF-06252616 Pfizer MAb Phase II
BMS-986089 Bristol-Myers Squibb Protein Phase II
Antisense oligonucleotide Sarepta Therapeutics Antisense Preclinical (DMD, BMD)to inhibit myostatin for oligonucleotideDMD and BMD
OSX-200 Ossianix MAb Preclinical
Utrophin SMTC-1100 Summit Therapeutics Small molecule Phase I
activator Small molecules to target Summit Therapeutics Small molecule Preclinicalutrophin for DMD
SMT-022357 Summit Therapeutics Small molecule Preclinical
RE-001 Retrophin Recombinant protein Preclinical
Small molecule to activate PTC Therapeutics Small molecule Preclinicalutrophin for DMD
Recombinant protein to University of Recombinant protein Preclinicalactivate utrophin for Wisconsin-Madisonmuscular dystrophies
Dystrophin eteplirsen Sarepta Therapeutics Antisense oligonucleotide Pre-registration
activator drisapersen BioMarin Pharmaceutical Antisense oligonucleotide Pre-registration
SRP-4045 Sarepta Therapeutics Antisense oligonucleotide Phase III
SRP-4053 Sarepta Therapeutics Antisense oligonucleotide Phase III
BMN-044 BioMarin Pharmaceutical Antisense oligonucleotide Phase II
BMN-045 BioMarin Pharmaceutical Antisense oligonucleotide Phase II
BMN-053 BioMarin Pharmaceutical Antisense oligonucleotide Phase II
Biostrophin Asklepios BioPharmaceutical Antisense oligonucleotide Phase I
NS-065 Nippon Shinyaku Antisense oligonucleotide Phase I
Gene therapy to activate Nationwide Children's Gene therapy Phase Idystrophin for DMD Hospital
SRP-4050 Sarepta Therapeutics Antisense oligonucleotide Preclinical
PRO-052 BioMarin Pharmaceutical Antisense oligonucleotide Preclinical
PRO-055 BioMarin Pharmaceutical Antisense oligonucleotide Preclinical
Gene therapy for DMD Genethon Gene therapy Preclinical
Gene therapy to activate University of Missouri Gene therapy Preclinicaldystrophin for DMD
SRP-4044 Sarepta Therapeutics Antisense oligonucleotide Preclinical
SRP-4052 Sarepta Therapeutics Antisense oligonucleotide Preclinical
Stem cell therapy to activate Stanford University Stem cell therapy Preclinicaldystrophin for DMD
SYN-01 Synthena Antisense oligonucleotide Preclinical
Antisense oligonucleotide Daiichi Sankyo Antisense oligonucleotide Discoveryfor DMD
SRP-4055 Sarepta Therapeutics Antisense oligonucleotide Discovery
SRP-4008 Sarepta Therapeutics Antisense oligonucleotide Discovery
Antisense oligonucleotide to nLife Therapeutics Antisense oligonucleotide Discoveryactivate dystrophin for DMD
Small molecule to activate PTC Therapeutics Small molecule Discoverydystrophin for DMD

combinations of pharmacologicaland non-pharmacologicalinterventions.
With the widespread use ofglucocorticoids and improvedmanagement of respiratorycomplications, the life expectanceof DMD patients has markedlyextended to 27 years, from that of19 years in untreated patients. MostBMD patients receiving optimisedtreatment can live into mid-to-lateadulthood, or even a near normallifespan.
A recent breakthrough in DMDresearch is marked by theconditional approval of PTCTherapeutics’ Translarna (ataluren)in the European Union for thetreatment of DMD, caused by anonsense mutation in DMD,accounting for 10–15% of all DMDcases. It is thought to interact withthe ribosomal translationalmachinery to enable ribosomalreadthrough of the premature stopcodon on the dystrophin messengerRNA (mRNA), thereby restoring thesynthesis of full-length dystrophin.The conditional approval was basedon the Phase IIb trial, whereTranslarna was shown to slow therate of the loss of ambulation anddisease progression in ambulantpatients with nonsense DMDmutation5. The safety profile wasshown to be favourable, with mildand transient gastrointestinaladverse events reported6. However,data from the Phase III confirmatorytrial that is currently underway willbe required for full approval forDMD.
Despite the approval of thebreakthrough treatment Translarnawhich is only beneficial to 10–15%of the DMD population, high unmetneeds for disease-modifyingtreatments remain in most patientsegments, given the wide spectrumof DMD mutations identified in theDMD population. Additionally,unfavourable adverse events limitthe long-term use ofglucocorticoids, although it can beused as a symptomatic treatment inpatients regardless of their geneticmutation. The DMD/BMD market isstill highly under-served and,
therefore, presents opportunities forbreakthrough therapies. There ismarket opportunity not only in thenon-ambulant patient segmentalthough the critical lack ofvalidated clinical trial endpointsremains a challenge, but also fordisease-modifying treatmentsintended to treat the vast majorityof DMD/BMD patients
First-in-class innovation in thepipelineThe DMD and BMD pipeline issmall (88 molecules) but containsproducts targeting a diverse rangeof molecular targets, such as DMD,utrophin, myostatin, integrin α7β1and extracellular matrix proteins.Analysis also reveals a high level ofinnovation in the pipeline, withidentification of 46 first-in-classproducts acting on 13 uniquetargets. The majority of first-in-classmolecules are in preclinicaldevelopment, while the late stage isdominated by products withestablished targets. The mostcommon first-in-class moleculartarget is DMD, targeted by 24pipeline products. These drugcandidates, mostly exon-skippingcompounds, are developed tocorrect the disrupted reading frameof the dystrophin mRNA andproduce an internally truncated butfunctional protein. Their presence inall developmental stages indicates astrong focus on personalisedtreatments. Of which, six specifictherapies are currently indevelopment for exon 51, which isapparent in 13% of all DMDpatients, and by far the largestsubpopulation identified comparedwith other specific exon deletions.Drisapersen (GSK and BioMarinPharmaceuticals) and eteplirsen(Sarepta Therapeutics) are the twoexon-skipping therapies currently inpre-registration for the treatment ofDMD. Another approach underearly-stage investigation is DMDreplacement via gene therapy.However, discrepant preclinicalfindings and the lack of efficacy inclinical trials, due to issues withcellular immunity and insufficienttransgene expression in humans,
leaves substantial challenges forgene therapy to become asuccessful treatment.
The rest of the pipeline focuseson alleviating the secondarymanifestations of the diseaseswithout correcting the defectiveDMD, such as modulation ofproteins that interact with theDAPC, and inhibition of fibrosis.These therapies are designed topromote muscle growth, sustainmuscle regeneration and reducefibrosis in DMD/BMD patientsregardless of the genetic mutations,therefore, holding high potential forwidespread use.
Many proteins have been shownto interact with the DAPC, of whicha few are suggested to act througha functional compensatorymechanism in the absence offunctional dystrophin. Utrophin,which has interchangeable functionswith dystrophin, is among the mostpromising first-in-class targets.Animal models of DMDdemonstrated that the utrophinmodulator SMT C1100 resulted insignificant functional improvementin muscle strength and resistance tomuscle fatigue by restoring theactin cytoskeleton-sarcolemma linklost in dystrophic muscles, withouthaving unfavourable side effects7.SMT C1100 is currently one of thesix utrophin modulators underinvestigation and has progressedthe furthest (Phase I) in the pipeline.
Transforming growth factor (TGF)-βsignalling, which plays a crucial rolein many processes including tissuerepair, is of particular interest inDMD due to its role in promotingpathological fibrosis. One of thefirst-in-class targets that intervene inthis pathway is myostatin, a TGF-βsuperfamily member known toregulate muscle growth and fibrosis.It negatively regulates musclegrowth by binding to activinreceptor type IIb localised on themuscle cell surface, andsubsequently promotestranscription of pro-fibrotic genesthrough activation of the Smadtranscription complex. Myostatininhibition in mdx dystrophic micewas shown to reduce fibrosis and
FIRST-IN-CLASS INNOVATION IN THE PIPELINE FOR TREATMENT OF MUSCULAR DYSTROPHY continued
6 european INDUSTRIAL PHARMACY September 2015 • Issue 26

improve muscle regeneration,alongside the benefits of increasingmuscle mass and force in vivo,despite a lack of effect on musclenecrosis8,9. Experimentalapproaches, including myostatin-neutralising antibodies and genedelivery of an antagonist, haveprogressed to human trials, butthere have been setbacks due toefficacy issues and adverse immuneresponse in clinical trials. Anapproach currently beinginvestigated in earlier stages isknockdown of myostatin expressionthrough antisense oligonucleotide-mediated exon skipping, whichprevents synthesis of functionalmyostatin. Additional to itsimprovement in muscle mass invivo10, the low risk of provokingimmune response makes it anattractive anti-fibrotic approach toslow disease progression in DMD.
Poor disease prognosis andsignificant unmet needs in themarket has been the driving forcefor the high level of first-in-classinnovation. The diverse array offirst-in-class targets in development
which, closely aligned to thedisease pathophysiology, presents apositive outlook for the futuremarket. However, despite theirpotential disease-modifying effects,growing evidence suggests thatcorrecting the mutated DMD genealone is insufficient to cure thedisease, creating combinationopportunities for first-in-classmolecules targeting secondarydisease pathways to preservemuscle function and transform thefuture treatment algorithm.
References1 Yılmaz Ö, Karaduman A and Topalo�lu H.
Prednisolone therapy in Duchennemuscular dystrophy prolongs ambulationand prevents scoliosis. European Journalof Neurology 2004;11:541–544.
2 Houde S, Filiatrault M, Fournier A, et al.Deflazacort use in Duchenne musculardystrophy: an 8-year follow-up. PediatricNeurology 2008;38:200–206.
3 Michel RN, Chin ER, Chakkalakal JV, et al.Ca2+/calmodulin-based signalling in theregulation of the muscle fiber phenotypeand its therapeutic potential viamodulation of utrophin A and myostatinexpression. Applied Physiology, Nutrition,and Metabolism 2007;32:921–929.
4 McAdam LC, Mayo AL, Alman BA, et al.The Canadian experience with long-termdeflazacort treatment in Duchennemuscular dystrophy. Acta Myologica2012;31(1):16–20.
5 Bushby K, Finkel R, Wong B, et al.Ataluren treatment of patients withnonsense mutation dystrophinopathy.Muscle & Nerve 2014;50:477–487.
6 Finkel RS, Flanigan KM, Wong B, et al.Phase 2a study of ataluren-mediateddystrophin production in patients withnonsense mutation Duchenne musculardystrophy. PLoS One 2013;8(12):e81302.
7 Tinsley JM, Fairclough RJ, Storer R, et al.Daily treatment with SMTC1100, a novelsmall molecule utrophin upregulator,dramatically reduces the dystrophicsymptoms in the mdx mouse. PLoS One2011;6(5):e19189.
8 Bogdanovich S, Krag TO, Barton ER, et al.Functional improvement of dystrophicmuscle by myostatin blockade. Nature2002;420(6914):418–421.
9 Wagner KR, McPherron AC, Winik N, andLee SJ. Loss of myostatin attenuatesseverity of muscular dystrophy in mdxmice. Annals of Neurology2002;52:832–836.
10 Malerba A, Kang JK, McClorey G, et al.Dual myostatin and dystrophin exonskipping by morpholino nucleic acidoligomers conjugated to a cell-penetrating peptide is a promisingtherapeutic strategy for the treatment ofDuchenne muscular dystrophy. MolecularTherapy-Nucleic Acids 2012;1:e62.
FIRST-IN-CLASS INNOVATION IN THE PIPELINE FOR TREATMENT OF MUSCULAR DYSTROPHY continued
7european INDUSTRIAL PHARMACY September 2015 • Issue 26
����������������������������������� ������������������ !+�, �'��!�!�$�)-�*,�*$1�"'-*&�$�'��, ��� �*%���-,!��$����$, ��*����!�&��+�'�!�,1���������
� ��"'-*&�$�(*'.!��+����-*'(��&��'*-%��'*�(-�$!+ !&��'*!�!&�$�(�(�*+���!,'*!�$+��&��*�.!�/+�'&�+-�"��,+�, �,��'.�*��$$��+(��,+�'��, �(�*�&,�*�$��&��( �*%���-,!��$�+�!�&��+���', �(*��,!��$��&��+�!�&,!�!��!&�$-�!&��
� +,�*!$!+�,!'&�,�� &!)-�+ ���������������� !+'$�,'*�,�� &'$'�1� .�$!��,!'& ������������������������������������� �+�(,!��(*'��++!&�� %!�*'�!�$���,��,!'&�%�, '�+��������� (��#��!&�� $1'( !$!+�,!'& ������������������������������� �$��&*''%���+!�&� �!',�� &'$'�1 ������������������������������ (*�+�*.�,!.�+� ����,�+,!&������������������������������������ (*'��++��!$,*�,!'&
� ������������������������������������� ������������������ �$+'�'&,�!&+����'-&,+�'��, ��+�!�&,!�!��%��,!&�+��+1%('+!���&��/'*#+ '(+-&��*�, ���-+(!��+�'��, �������
�*��*�'&$!&���,������ ������ ������������*��%�!$��(-�$!+ �*��-*'%���'%%-&!��,!'&+��'%� ��$���������
������������0���������� ������
over 25 years of advancing pharmaceutical and healthcare sciences
2012 Volume 17 Number 2
European Journal
of Parenteral
& Pharmaceutical
Sciences
over 25 o v e r 2 5 pharmaceutical andp h a r m a c healthcare sciencesh e a l t h c a
years of advancing y e a r rs of advancing f advancing a d v a n c i n gceutical andc e u t i c a a n dare sciencesa r e s c i e n c e s

“Packaging Matters,” a recentinternational packaging satisfactionstudy, commissioned by WestRock,found that the packaging usageexperience is extremely importantto consumers’ overall experiencewith a product. Study resultsshowed that 37% of consumershave purchased something againbecause of packaging functionality.However, if consumers find theexperience of using medicationpackaging frustrating, they are notinclined to take the medication asoften as they should, delaying refillor repurchase.
What are consumers looking for intheir medicine packaging,ultimately? The following packagingattributes were ranked as mostimportant.
1) Keeps the product safe
2) Prevents spilling, leaking orbreaking
3) Keeps the productfresh/effective
4) Easy to reclose or reseal
5) Easy to get the right amountout and Designed to keep meand my family safe (tie)
The study found 34% ofconsumers strongly agreed that“packaging designed to keep theproduct safe and/or protect me andmy family” would make them morelikely to purchase products fromthat manufacturer or brand. Thestudy also noted the importance ofease of use. This tells us thatinnovation in child-resistant (CR)
packaging, to ensure it is both user-friendly for adults and resistant forchildren, can improve consumersatisfaction and boost sales.
There are two approaches to CRpackaging: (1) the use of strengthand force to access medication, or(2) the use of cognitive abilities,such as sequential motions like apushing then pulling to accessmedication. A subsequentconsumer preference studyconducted by WestRock found thatwhen using the latter approach,there is sometimes a learning curveor educational element that isneeded, but the overall openingexperience is much better.Ultimately, this study affirmed thatconsumers preferred cognitive-based approaches, and packagedesigns were modified based onthese findings.
Developing innovative packagingstarts with understanding that thebest designs emerge from thestrongest insights, which almostalways come from the consumer.One example is WestRock’s CRnasal pump that was developed inpreparation for new ConsumerProduct Safety Commission (CPSC)regulations in the United States. InDecember 2012, the CPSC passed anew regulation requiring CRpackaging for any over-the-counteror prescription drug productcontaining the equivalent of 0.08mgor more of an imidazoline in asingle package. This “specialpackaging” must be designed orconstructed to be significantlydifficult for children under 5 years ofage to open or obtain a toxic orharmful amount of the substancewithin a reasonable time, and notdifficult for normal adults to useproperly. The affected productsincluded commonly used eye dropsand nasal sprays, which can causeserious adverse reactions in childrenwhen accidentally ingested.
During the development of thenasal pump, WestRock tested threeconcepts with consumers andrefined the preferred design basedon their feedback. Specific featuresthat were incorporated into the finalpackage, the HiMark® CR NasalPump (see Figure 1), as a resultincluded the following.
8 european INDUSTRIAL PHARMACY September 2015 • Issue 26
CHILD RESISTANCE INPHARMACEUTICALPACKAGINGby Hung Le
The best solutions in child-resistance come from adeep understanding of consumer insights and a
dedication to innovation. In order to achieve both childsafety and a positive consumer experience, insightsshould be gathered and applied to packaging design andthe development of any communications or instructionsaccompanying the packaging.
Hung Le ([email protected]) is Vice President of Innovation Engagement forWestRock’s Home, Health and Beauty Group. An engineer by training, Hung leveragesa deep understanding of the development and commercialisation of consumerpackaging to lead the creation of package design concepts that are connected tobusiness, brands and consumers. Hung previously served as Vice President of GlobalDesign for WestRock, as well as Senior Director of Innovation and Product LineManagement for WestRock Healthcare ([email protected]).

• On-pack visual and tactilecues that communicate howto use the CR package.
• Use of a simple range ofmotions to open thepump, without requiringextreme precision,making the packagetruly senior-friendly.
• Audible indication, sothe product locks andseals tightly without anywiggling.
• Integration of the child-resistance feature intothe pump versus theover cap; this ensuresthe highest level ofprotection for childrenby preventing accessto the medication evenif the over cap is leftoff or lost.
These modifications to designyielded a consumer-preferredpackage that meets the needs ofboth children and seniors, includingflexibility and cognitive skills.
A second example is WestRock’sDosepak® Express with Optilock®
technology design (see Figure 2).Optilock technology is a locking
mechanism that transformsadherence packaging byenabling a significantly smallerCR package. It was developedbased on insights fromexperts in the field of patientmedication adherence,customers and consumers.
Dosepak Express withOptilock technology is partof WestRock’s line of provenadherence-enhancingsolutions. It includes acalendared medicationblister and flat panels on theouter carton, providing aformat for readablemedication information andlinks or QR codes thatconnect to additional
adherence programs. Overall,the package incorporates insights-based innovations.
• An integrated calendar thatallows patients to easily trackand monitor their medications,increasing their adherence.
• Its small size is discreet andportable, making it convenient
to use and easily fitting intopatients’ lifestyles.
It is important to keep consumerinsights at the heart of innovationand remember that when consumershave a positive experience, it canhelp create a better overallhealthcare experience, including thepossibility of improving adherenceto medication regimens. That is whywhen choosing a packagingsupplier, drug manufacturers shouldconsider the benefits of using CRpackaging that not only minimisesrisk, but is also based on consumerinsights and matches consumerpreferences.
CR packaging is critical because ithas the potential to improve and, incertain situations, even save lives.Poisoning is the third leading causeof unintentional injury deaths inchildren in the European Region –3000 deaths per year1,2, accordingto the World Health Organization.Non-fatal poisonings are even morenumerous and an important causeof ill health and long-lastingdisability. One of the mostsuccessful ways to prevent theunintentional poisoning of childrenis through CR medicationpackaging3 – which helps preventchildren from gaining access tomedications, even after they havetheir hands on the package.However, that package will onlywork if consumers understand howto use it and welcome theinnovation, adding even greatervalue to deep consumer insight(s).
While CR regulations have existedin the United States and theEuropean Union (EU) for quite some
CHILD RESISTANCE IN PHARMACEUTICAL PACKAGING continued
european INDUSTRIAL PHARMACY September 2015 • Issue 26 9
Figure 1: The HiMark® CR NasalPump.
Figure 2: WestRock’s Dosepak® Express with Optilock® technology design.

time, these requirements vary inother countries around the worldand are surprisingly non-existent inmany. Most would agree, however,that since accidental childpoisonings are a global problem,the responsibility to consider CR
packaging for medication goesbeyond regulations. Clearly, CRpackaging is the new best practiceor standard of excellence, rooted inthe desire to keep children safe. If amedication toxicity issue warrantsthe use of CR packaging in one
region, then drug manufacturers areresponsible and smart to considerintegrating CR packaging in otherlocations as well – even if formalguidelines have yet to be put inplace.
Research demonstrates that CRpackaging saves lives, and manyleaders in the space are choosingnot to wait until mandates are inplace to implement change. Forthose leaders, change starts rightnow.
References1.Sethi D, Towner E, Vincenten J, et al.European Report on Child InjuryPrevention. Geneva, Switzerland: WorldHealth Organization; 2008.http://www.euro.who.int/__data/assets/pdf_file/0003/83757/E92049.pdf (Accessed 6May 2015).
2.The European Child Safety Alliance. TheChild Safety Report Card Europe 2012.How Saftey Conscious are EuropeanCountries Towards Children. Birmingham,UK: The European Child Safety Alliance.http://www.childsafetyeurope.org/publications/info/child-safety-report-cards-europe-summary-2012.pdf (Accessed 6 May 2015).
3.Peden M, Oyegbite K, Ozanne-Smith J, etal. (Eds) World Report on Child InjuryPrevention. Geneva, Switzerland: WorldHealth Organization; 2008.http://apps.who.int/iris/bitstream/10665/43851/1/9789241563574_eng.pdf
CHILD RESISTANCE IN PHARMACEUTICAL PACKAGING continued
european INDUSTRIAL PHARMACY September 2015 • Issue 2610
esruieqrestatSedtniUheTonsitaulegre sheT.soductprhiT0791ofActgickP
pharorfnggiackpaRCofe ushetesrPon soiPhetbyedtaitniie erwons
tiedlhi
lcaiceutampharon ieventr
i
hiT.0791ofActnggiackaPonispoilantdeiccaheterov
atsngitestRCshaUEheTtcaifitcerbut,onsitaulegr
hetshae tatsrbeemmUE.yrountc
tennmgoverhet,nahiCn I
o te ponsesrn iedsspasawwalshiolehhousc ioxtbyendrlchiofngoni
niUheto tralimise rathatdsndaraalbyedruieqrnotyllarnegesion it
uieqroductprneimertdeo ttghirhe
tnsio tnsapltithatedtcandiishat
n erconcngiowgr.slcaichemd ol
estatSedtdualivndiich aE.wathatn istenemr
RCeuttit
gover,ranehetn istenemruieqr
onitaocisAsnggiackaP ssi
Lah,tleaHofyrtsniiMheTgeuro ton itdaenmecomrorfo torsecurprabeyam
plaon edsbae,urutfr mrhaPe snehiC
hitofflhabeon ngiguratoreprd ues
nced nnouayltecenre rafelWnd aborLaon itcaiedmorfnggiackpaRCofe usge
.tenopmelvdecyipollamor
lcaiceutam.evitaitniishi
tsrifstinced -everch hiwn,paaJn ion
nnouah tleaHofertsniiMheTheT.andiIn hitiwesseasdite urposexofon ieventpr
conto tmaogrprlonaitnaanced nnounghitnyamon ocus flliwmaogrprhe
onispoid lchisauch s,sorctafksiro
c onichrolrconthetngudincli,sng
.ngoni
Further informationGlobal CR trends
Industrial
Pharmaceutical
Microbiology
Standards & Controls
Editors
Geoff Hanlon
Tim Sandle
2015 edition
Expert Guidance on IndustrialPharmaMicrobiologyEdited by Geoff Hanlon and Tim Sandle
This new second edition is an excellentreference work for those working in industrial
pharmaceutical microbiology. It covers allaspects of this complex subject with
contributions from many leading figures in thefield and is highly recommended
European Journal of Parenteral and
Pharmaceutical Sciences
This book is essential for every pharmaceutical laboratory: scientific, topical and practical.
Pharmig
order online at www.euromedcommunications.comOr contact the publishers: email: [email protected];
Tel: +44 (0)1428 752222; Fax: +44 (0)1428 752223.
Industrial Pharmaceutical Microbiology:Standards and Controlsprovides clear, practical and up‐to‐date guidance for handling virtually everycompliance and operational challenge associated with pharmaceuticalmicrobiology.
Expert Advice
In over 600 pages and 25 chapters a team of twenty four internationalauthorities answer all your questions concerning regulatory expectationsin areas such as microbiological audits, rapid microbiological methods,conducting risk assessments, both proactive in terms of minimisingcontamination, and reactive in terms of addressing microbial data deviation;and also ensuring that processes meet “quality by design” principles.
International Applications
Connect instantly with regulations and current best practices on everythingfrom disinfectants to sterility testing; environmental monitoring to hazardanalysis; and from pharmaceutical processes to biological indicators. All ofthis is developed from an international perspective, where differentregulations are compared and contrasted together with insightfulcommentary as to best practices.

11european INDUSTRIAL PHARMACY September 2015 • Issue 26
BackgroundCell-based medicinal products arecomplex and their therapeuticbenefits are often derived frommechanisms that are not fullyunderstood, thus making itchallenging to demonstrate thesafety and efficacy of these
medicinal products. As such, duringthe manufacture of these products,there is a strong requirement tominimise both sources of risk andprocess variability in order tomaximise the quality of the finalproduct. The choice of biologicalmaterials used in the production of
cell-based medicinal products has astrong impact on importantbiological characteristics of the finalproduct, such as viability, purity andpotency, but can also have anadverse effect on product variabilityand also be potential sources ofcontamination.
The BSI has worked with the CellTherapy Catapult, as well as a widerrange of cell therapy experts, tocreate a guide with the intention ofhelping those people looking todevelop high quality cell-basedmedicinal products. The guideintends to help these developersselect the biological materials insuch a way that reduces theprobability of minimising bothvariation in product characteristicsand the introduction of unwantedagents, thus potentially reducingthe barriers to eventual successfulcommercialisation of the products.
This guide, with the title PAS157:2015 Evaluation of Materials ofBiological Origin Used in theProduction of Cell-Based MedicinalProducts – Guide, is a documentthat supports the evaluation ofbiological materials used in theproduction of cell-based medicinalproducts for human use.
What does the PAS cover?The PAS covers all biologicalmaterials that come into contactwith the cellular active substanceduring the manufacturing processfor cell-based medicinal products,such as those found in cell culturemedia components, e.g. cytokines,animal-derived sera, proteins, etc.The scope of the knowledgecontained within the documentincludes guidance on the following.
• Identifying, assessing andcontrolling risks associated withmaterials of biological origin.
• The evaluation of all materials ofbiological origin.
• Applying the fundamentalprinciples of risk managementto both materials of human andanimal origin, and also toreagents derived from a widerrange of biological sources.
• Legislation for developers ofcell-based medicinal products
GETTING TO HIGHERQUALITY PROCESSESSOONER – NEW GUIDEHELPS YOU MAKE ABETTER CHOICE OFBIOLOGICAL MATERIALSby Patrick Ginty and Ben Sheridan
The BSI (British Standards Institution), the UK'sNational standards body, has worked collaboratively
with the Cell Therapy Catapult and a wider group of celltherapy experts, to develop PAS 157:2015 Evaluation ofMaterials of Biological Origin Used in the Production ofCell-Based Medicinal Products – Guide. This documentsupports the evaluation of biological materials used inthe production of cell-based medicinal products forhuman use. It will help developers of high quality cell-based medicinal products to select the biologicalmaterials in a consistent manner. The aim is to reduce theprobability of minimising both variation in productcharacteristics and the introduction of unwanted agents,thus potentially reducing the barriers to eventualsuccessful commercialisation of the products. Thebackground and key benefits of PAS 157:2015 arediscussed, along with any previous guidance in this areaas well as methods in which to mitigate risks associatedwith procurement of biological materials.
Patrick Ginty is currently working as the Regulatory Affairs Manager at Cell TherapyCatapult Ltd in London. He gained his PhD in tissue engineering and drug delivery in2005 and has since spent 10 years working in both industry and academia pursuing acareer in the regulation of cellular therapies and medical devices. During this time, hehas received a certification from the Regulatory Affairs Professionals Society, hasworked on over 20 different cell therapy products, and gained over 25publications/patents in regenerative medicine and cellular therapy.
Ben Sheridan leads on the development of standards to underpin innovation in highvalue manufacturing at the BSI. He spent a number of years at the National PhysicalLaboratory, supporting the long-term development of measurement researchprogrammes in support of emerging technologies, and before this worked in thesemiconductor industry.

in both the European Union(EU) and the US.
It is important that we also identifywhat the PAS is NOT intended todo. It does not cover the selection,assessment or control of cellularactive substances, nor startingmaterials as defined in Directive2001/83/EC and excipients.Additionally, it also does not coverbiological materials that are used inthe development of any otherbiological medicinal product.
Previous guidanceThis is not the first guidancepublished by the BSI that is relevantto developers of cell therapyproducts. Recent years have seenthe following documents alsopublished by the BSI incollaboration with cell therapyexperts.
• PAS 83 Developing Human Cellsfor Clinical Applications in theEuropean Union and the UnitedStates of America – Guide. Thisdocument gives a description ofthe development pathway andrelevant regulatory regimes inthe EU and US that is applicable
to cell-based medicinalproducts.
• PAS 84 Cell Therapy andRegenerative Medicine –Glossary. This is a glossary ofterms for use in the cell therapyand regenerative medicineindustry.
• PAS 93 Characterization ofHuman Cells for ClinicalApplications – Guide. This is aguidance document helpingdevelopers of productscontaining human cells forclinical applications tocharacterise their products andprocesses.
A fundamental part of PAS 157 isdeciding whether or not a particularapplication is in or out of scope ofthe guidance. The documentcontains Figure 1, which helps usersmake this assessment.
What is in PAS 157?
Quality considerations forbiological materials used in theproduction of cell-basedmedicinal products There are a number of considerations
related to the quality of biologicalmaterials that are used in theproduction of cell-based medicinalproducts, and it is theseconsiderations that provide thecontext to the PAS. For example, theuse of biological materials bringswith it the potential for thetransmission of adventitious agents,such as the risk of transmissiblespongiform encephalopathy (TSE)from human or bovine material.Therefore, when sourcing a newmaterial for use in a manufacturingprocess, a number of key questionsshould be asked. For example, ifusing bovine material, is the materialsourced from non-TSE countries suchas Australia or New Zealand?
It is equally important to ensurethat the manufacturing processused to produce the material doesnot introduce any risk ofcontamination with adventitiousagents, i.e. processing in anenvironment where materials ofanimal or human origin are present.In cases where there is no suitablealternative to a material of humanor animal origin, it is important toknow that the supplier has takensteps to eliminate risk and that they
GETTING TO HIGHER QUALITY PROCESSES SOONER continued
12 european INDUSTRIAL PHARMACY September 2015 • Issue 26
Figure 1: Decision chart demonstrating the rationale applied to the scope of PAS 157. *Starting material orexcipient as defined in Directive 2001/83/EC and, therefore, out of scope of PAS 157.

13european INDUSTRIAL PHARMACY September 2015 • Issue 26
have tested the material incompliance with international andregional regulatory requirements.
Equally, it is valuable to know ifany downstream purification orinactivation measures have beentaken by the supplier of thematerial. Beyond the mitigation ofsafety risks, it is also essential toknow if the material in questionprovides the functionality that isrequired for its intended use.Therefore, it is important to know ifthe material has been used forother similar processes and/or thatthe supplier can provide assurancesover the consistency of the qualityof the material. One such method isto ask the supplier to provide adetailed specification and evidencethat the material is manufacturedunder a recognised quality system.Equally, the developer should makesure that they carry out anycharacterisation, the level of whichbeing dependent on the criticalityof the material and the potentialimpact on final product quality if thematerial does not meet itsspecification.
Applicable regulatoryrequirements and other sourcesof guidancePAS 157 contains a great deal ofuseful information that is intendedto help developers to develop highquality processes and products. Forexample the difference in regulatoryrequirements in the EU and USrelating to biological materialsselection is highlighted, including,for example, the potential for usinga Drug Master File in the US, butnot in the EU.
One major source of confusionthat can occur is the differing usageof terminology in the EU and theUS, and this is highlighted in PAS157, for instance, when using theterms ‘raw materials’ and ‘ancillarymaterials’. The intention is to makeit clear to the developer wherediffering usage of terminologyoccurs, and to go some way tohelping the reader deal with thissituation.
The document also contains moredetailed information relating to the
existence in the EU and US ofspecific regulatory requirementsand guidance applicable to themanufacture of cell-based medicinalproducts. It highlights recurringthemes that occur within eachterritory, and also the use of therisk-based classification system usedfor ancillary materials in the US.
Manufacturers of biologicalmaterials available on the marketcan make various qualitydeclarations about the materials inquestion, and it is important thatthe product developer is aware ofwhat these mean, and thelimitations of what usefulinformation can be inferred fromsuch statements. PAS 157 identifiescommonly used examples of suchquality declarations and explainstheir meanings/implications fromboth a technical and regulatoryperspective.
Evaluating supplied materials andmitigating potential risksMost developers of cell-basedmedicinal products will beprocuring biological materials froma third party supplier. Each of thesesuppliers will have their own qualitymanagement regimes whendemonstrating control over theirprocesses, and there is, therefore,no single means of using suppliedinformation to evaluate thematerials and establishing potentialrisks associated with them. PAS 157identifies certain criteria that can beused to evaluate such suppliedmaterials, and also steps that canbe taken to mitigate risks that areidentified during this process.
The document also givesparticular guidance on how toundertake an audit of a biologicalmaterials supplier, and also on howto use all of the informationgathered to form the basis of a riskassessment.
CharacterisationWhilst there is shared responsibilityfor sufficient characterisation todemonstrate the quality of abiological material between thesupplier and the developer, oncethe developer accepts the material,
the responsibility for control andcharacterisation of the material lieswith the developer themselves. PAS157 guides the developer on thekinds of characteristics they willneed to be aware of. Commonly,this may include testing to establishthe identity, purity and biologicalactivity of the material in question,in addition to more simplisticmeasurements such as pH andosmolality.
Changes to materials PAS 157 demonstrates that changesto the supply of biological materialsgives rise to certain responsibilitiesto the developer to ensure theirprocesses remain acceptable to theregulator. Changes in quality of thebiological material can lead toadverse consequences on thequality of the final product, andthese changes can be as a result ofchanges to the manufacturingprocess, the manufacturing site, or ifthe supplier stops manufacturingthe product altogether.
The document guides thedeveloper in how to manage suchpotential changes in advance ofthem happening, particularlythrough dialogue with the supplier,and also in establishing otherpotential sources of the material.
How was PAS 157developed?A PAS is developed to address aparticular market need. This may bea request from an individual sponsorfor a standardisation document thatserves an emergent market,technology, service or public policyinterest. The approach used in thedevelopment of a PAS is a means ofquickly introducing a standardisedapproach. It often acts as the basisfor further development towardsmore formal standardisation at aEuropean or international levelthrough international standardsbodies, such as the InternationalStandards Organization or theEuropean Committee forStandardization.
The true value of a standardisationdocument is that if it is oftenoptimised, it is more widely
GETTING TO HIGHER QUALITY PROCESSES SOONER continued

GETTING TO HIGHER QUALITY PROCESSES SOONER continued
14 european INDUSTRIAL PHARMACY September 2015 • Issue 26
adopted. As such, a PAS is generallynot only applicable within the UK,nor is its development modelrestricted to UK stakeholders. Theprocess for developing a PAS isdescribed in Figure 2. The work isled by a technical author, who isassisted by a Steering Group and aReview Panel.
In the case of PAS 157, thetechnical author was Patrick Gintyfrom the Cell Therapy Catapult, andthe Steering Group consisted ofrepresentatives from the following.
• Cell Medica
• CellData Services
• Cell Therapy Catapult
• GlaxoSmithKline (GSK)
• Consulting on AdvancedBiologicals Limited
• Miltenyi Biotec
• National Institute for BiologicalStandards and Control (NIBSC)
• Roslin Cells
• University College London
Further reading1 British Standards Organisation. PAS 83
Developing Human Cells for ClinicalApplications in the European Union
and the United States of America –Guide. London, UK: BSI; 2012.
2 British Standards Organisation. PAS 84Cell Therapy and RegenerativeMedicine – Glossary. London, UK: BSI;2012.
3 British Standards Organisation. PAS 93Characterization of Human Cells forClinical Applications – Guide. London,UK: BSI; 2011.
4 European Directorate for the Quality ofMedicines and HealthCare. EuropeanPharmacopoeia 8th Edition. Chapter 5-2-12. Raw materials for the productionof cell-based and gene therapymedicinal products (draft). Strasbourg,France: EDQM, Council of Europe; 2014.
Copyright © 2012 BSI. All rights reserved. 1
PAS development process
Public Consultation
(incl. SG & RP) ~4 weeks
SG Review ~2 weeks
SG Ed. Review ~1 week
PAS Publication
SG Meeting
SG Meeting
Draft 1
Draft 2
Draft 3
Draft 0.1 “base document”
Domain Research
Kick-off Meeting
Standard scheme Amend scheme to reflect what is in the contract – 4 versions available (see next slides)
Draft 0 “skeleton document”
SG Meeting
dSAP
sab“
SG
rptnemopleved
1 0.aft Dr”tnemuocdes
g neetiMSG
sseocr
1 aft Dr
.EdGSwevieRk ee1
onitacilbuPSAP
on
teleks“
SG
0 aft Dr
”tnemuocdont
g neetiMSG
g neetiMSG
Dr
eetiMSG
2 aft Dr
k eew1~
g n
3 aft Dr
omD
kciK
hcraeseRniaom
gniteeMfofoff-kk-
SG wevieRkseew2~
onCl. SG(inc~
c libPuonitatluson
) & RPl. SGkseew4~
eetiMSG
g n
darStansdnemA
I.SB2102©thgiyropC
eme schd wtcelfero temehcs
.devresersthgirllA
artoncehtnisitahw
avasonisrev4–tca
dilstxenees(elbalia
)sed
1
Figure 2: PAS development process. SG: Steering Group; RP: Review Panel.
Clean Air and Containment ReviewThe journal to enhance your knowledge of cleanroom, clean air and containment technology
• Learn about different aspects of these technologies from clearly written articles by experts• Keep up to date on standards with regular updates by standards committee members• Read about innovations• Understand the jargon• Become an expert yourself
To subscribe, or for more information including contents lists for all previous issues, visit www.cleanairandcontainment.com

15european INDUSTRIAL PHARMACY September 2015 • Issue 26
IntroductionThe Roadmap is the culmination of2 years’ work involving contributionsfrom over 500 international expertsat five events held across Europe. Itpromises huge cost reductions inmedical product developmentthrough the use of computersimulation.
With the release of the Roadmap,the Avicenna project officially comesto an end. However, to ensure theideas contained are taken forwardand making its recommendations
become a reality, a new organisationhas been formed: the AvicennaAlliance – the Association forPredictive Medicine. This firstpartnership of pharmaceuticalindustrialists, medical devicemanufacturers, academicresearchers and regulatory expertswill aim to revolutionise thehealthcare industry through the useof computer simulation.
This September, the AvicennaConsortium, tasked by the EuropeanCommission to investigate how
computer modelling might mitigatethe cost of clinical trials, released itsRoadmap: ‘In Silico Clinical Trials:How Computer Simulation WillTransform The Biomedical Industry’.This article describes the processand the future of the Avicennavision.
The RoadmapApproaching the investigation ofthe potential impact of in silicoclinical trials, the AvicennaConsortium brought together arange of experts from around theworld – academics, clinicians,industrialists, patients’ advocates,computer experts, regulators andsafety experts. Over 2 years, theconsortium organised five events indifferent European cities,conducted dozens of experts’interviews, and used state-of-the-artonline technologies to collecthundreds of individual opinions andalign them around agreedstatements, in order to arrive at aconsensus on how to introducecomputer simulation into the clinicaltrials process.
Why go to such efforts to discusscomputer simulation in clinical trials?Professor Marco Viceconti,Coordinator of the Avicenna Projectexplains: “Developing a newmedical product, whether it is a newmedicine or a new medical device,is becoming prohibitively expensiveand can take anything up to 12years. The latest industry figuresestimate that the costs ofdeveloping and bringing a newmedicine to market are approachingUS $3 billion. All analysts agree thatthis situation is unsustainable, andthat the biomedical industry mustquickly find alternative and moreeffective ways to develop and assessnew medical products.”
IN SILICO CLINICALTRIALS: DREAM ORCERTAINTY?by Marco Viceconti, Adriano Henney, Edwin Morley-Fletcher andMartina Contin
The astronomical cost of bringing medical products tomarket has stalled the development of drugs,
crippling healthcare budgets and, for rare diseases,making new treatments the preserve of the rich.However, this September the Avicenna Consortium,tasked by the European Commission to investigate howcomputer modelling might mitigate the cost of clinicaltrials, released its Roadmap – ‘In Silico Clinical Trials:How Computer Simulation Will Transform The BiomedicalIndustry’.
Marco Viceconti is Professor of Biomechanics in the Department of MechanicalEngineering at the University of Sheffield, UK and Professor Associate at theDepartment of Human Metabolism. He is the Scientific Director of the University ofSheffield’s Insigneo Institute for in silico Medicine, a joint initiative between theUniversity of Sheffield and the Sheffield Teaching Hospital NHS Foundation Trust.
Adriano Henney, Director of Obsidian Biomedical Consulting Ltd, worked for 13 yearsin one of the top five multinational pharmaceutical companies before becomingProgramme Director of the German Virtual Liver Network, the largest VirtualPhysiological Human (VPH) project running in Europe (http://www.virtual-liver.de). DrHenney has directed this major German national flagship research programmefocusing on modelling human liver physiology since 2010.
Edwin Morley-Fletcher is President of Lynkeus srl, Italy and has over 25 years’experience in ebusiness. In the last 10 years, he has progressively focused oninformation and communications technology for health, managing some of the largestVPH projects, including MD-Paedigree, one of the three integrated projects funded incall 9.
Martina Contin worked as Communication Manager in various VPH initiatives at theVPH Institute, Belgium, before taking the helm of the not-for-profit organisation thatcoordinates all VPH research worldwide. The organisation aims to ensure the VPH isfully realised, universally adopted and effectively used.

IN SILICO CLINICAL TRIALS: DREAM OR CERTAINTY? continued
The huge cost of developing newmedical products has manyconsequences. Countries withuniversal healthcare struggle toafford the best new products, and incountries with private healthcareonly the wealthy can afford the besttreatments. Many conditions areignored by the biomedical industrybecause they cannot return thesignificant investment needed forclinical trials – because the groupsof people affected by the conditionsare too small (e.g. SanfilippoSyndrome) or too poor (e.g. Chagasdisease).
The Avicenna Consortiumbelieves computer simulation is onesolution that will contribute torevolutionising the clinical trialsprocess. In all other industrialsectors, computer simulation playsan essential role in the design andassessment of new products. Givenrecent advances in medical imagingand other analytical technologies,the Avicenna Consortium asked ifwe have collected enoughknowledge to start using computersimulation in the development ofmedical products. Could we use thecomputer simulation of individualpatients’ bodies in the developmentor regulatory evaluation of a
medicinal product, medical deviceor medical intervention? If we could,what would be the barriers to itswidespread adoption?
In its conclusions, the AvicennaRoadmap states that, in the opinionof the Consortium and thecontributors to the AvicennaResearch and TechnologicalRoadmap, the use of individualisedcomputer simulation in thedevelopment or regulatoryevaluation of a medicinal product,medical device or medicalintervention (generally referred toas “in silico clinical trials”) is one ofthe most important strategicpriorities in biomedical andtechnological research, if we wantto make the development andassessment of new biomedicalproducts simpler, cheaper, fasterand safer, whilst at the same timeminimising those activities such asanimal or human experimentationthat pose ethical concerns. Itrecommends that all public andprivate research funding agenciesacross the world do the following.
a)Acknowledge the significantsocioeconomic relevance thatresearch and technologicaldevelopment, assessment and
adoption of in silico clinical trialstechnologies pose. Themounting needs of universalhealthcare provision indeveloped countries exceed ourability to innovate quickly andefficiently, and in silicoapproaches are the bestpossible route to address thoseneeds.
b)Progressively increase theexpenditure in this area in thenext 5 years, so that by 2020 atleast 1% of the total public andprivate expenditure inbiomedical research anddevelopment worldwide(estimated as US $268 billion in2012) is dedicated to thedevelopment and adoption ofin silico clinical trialstechnologies used to translatebiomedical research discoveriesinto new products and servicesmore quickly, safely andefficiently. This should beinitiated with a dedicatedprogramme in the 2016/17European Commission WorkProgramme of the EuropeanUnion Horizon 2020programme, with a budget of atleast €50 million per year.
16 european INDUSTRIAL PHARMACY September 2015 • Issue 26

c) Ensure that such public andprivate research andtechnological developmentfunding is dedicated in equalparts to the core scientific andtechnological development ofpredictive models, to their pre-clinical and clinical validation,including the necessaryregulatory science aspects, andto support their early adoptionin industrial and regulatorypractice.
As the Avicenna consensusprocess has demonstrated, in aglobalised economy the discourseof in silico clinical trials mustdevelop globally. Thus, it isrecommended that all agenciesremove as many barriers as possibleto developing and prototypingthese approaches, and activelysupport pre-competitive researchand technological developmentacross international boundaries.
The consensus process adopted inthe project has shown that there is astrong will to ensure these messagesare acted upon, and it has resultedin the first tangible step beingtaken: the creation of the AvicennaAlliance – the Association forPredictive Medicine. This will be anassociation of industry and researchorganisations who have acommercial or research interest in in
silico medicine. While there is agreat number of policies that impacton the in silico market, there iscurrently no one organisation that isdedicated to this field. The AvicennaAlliance will advocate for thecreation of policy and regulatoryenvironment that is favourable tothe uptake of in silico models andpromote the interests of itsmembers in this field.
The Avicenna Consortium hasbroadened its focus to promote insilico medicine in general, i.e. usingcomputer simulation to revolutionisehealthcare. The Consortium hasbeen urging the EuropeanCommission to support researchthat will allow clinical trials to be runin computer simulations, reducingthe need for animal testing andhelping to cut down theastronomical cost of testing newmedical products. This pressureappears to have been effective, asthe early drafts of the 2016/17European Commission WorkProgramme indicate support for insilico clinical trials.
As the Avicenna Consortiumcomes to an end, the newly foundedAvicenna Alliance will take up thecause of in silico medicine, as thefirst platform for research andindustry organisations with aninterest in in silico medicine toimpact the political environment for
the emerging in silico market.Following a recommendation fromthe European Commission, theAvicenna Alliance will work towardsdrafting and submitting a proposalto form a public–private partnershipwhich will raise the profile of in silicomedicine, create new fundingstreams for research, and provideevidence of genuine impact onhealthcare.
The time is now, the challenge ishuge; only if we all work togetherwill we be able to address andovercome that challenge.
FundingThe project has received fundingfrom the European Union’s SeventhFramework Programme for research,technological development anddemonstration under grantagreement no 611819.
Reference1 Chakma J, Sun GH, Steinberg JD, et al.
Asia's ascent – global trends in biomedicalR&D expenditures. N Engl J Med 2014Jan 2;370(1):3–6. doi:10.1056/NEJMp1311068.
For further informationAvicenna website: http://avicenna-isct.com
Avicenna Alliance website:http://www.avicenna-alliance.org
IN SILICO CLINICAL TRIALS: DREAM OR CERTAINTY? continued
17european INDUSTRIAL PHARMACY September 2015 • Issue 26
CALL FOR ARTICLESDear Colleague
We hope you enjoy the European Industrial Pharmacy and find it both usefuland informative.
We are currently seeking new articles for future issues of the journal and wouldlike to invite you to contribute an article or review paper on any aspect ofindustrial pharmacy to the journal. All issues of European Industrial Pharmacyare indexed by both Scopus and Embase and thus are available through thelistings for any other industrial pharmacist internationally.
Please contact the Managing Editor, Sue Briggs ([email protected]) forfurther information or submissions.

Vendredi 21 août 2015, 700médicaments génériques à traversl'Europe ont été retirés des officines.Un retrait des autorisations de misesur le marché décidé en juillet 2015par l'Agence européenne dumédicament (EMA). Une décisionprise après plus d'un an passé àréévaluer quelque 1000 génériquesdont les tests de bioéquivalencemenés en Inde avaient révélé desentorses aux bonnes pratiquescliniques. Ces tests sont conçus
pour évaluer la conformitépharmacologique du génériqueavec le médicament princeps. Uneprocédure dont nous vous
expliquions les tenants et lesaboutissants à l'annonce del'Agence européenne dumédicament.
Mais concrètement, comment cesretraits vont-ils se traduire pour lespatients concernés? "En France, cen'est vraiment pas un souci”,explique Isabelle Adenot,présidente du Conseil national del'Ordre des Pharmaciens (CNOP). “Ily a des alternatives pour chacun desgénériques retirés du marché. Parailleurs, il y a déjà eu une premièrealerte il y a quelques mois, à la suitede laquelle les laboratoiresconcernés avaient agi", précise-t-elle, confirmant ainsi les dire del'Agence nationale de sécurité dumedicament (ANSM) qui avaitprévenu qu'en France, ces retraitsn'auraient pas d'impact. Par ailleurs,"sur cette liste de 700 génériquesretirés, la France n'est concernée
que par une infime partie, unetrentaine je crois", ajoute-t-elle.
La présidente du CNOP voitmême un motif de satisfaction dansces retraits successifs l'année passée(25 génériques retirés en décembre2014, puis 8 en janvier 2015 et, donc,700 en août 2015): "Je me réjouid'une chose: c'est que les grandesagences du médicament (Etats-Unis,Australie, Europe, Canada...)fonctionnent main dans la main et sefont mutuellement confiance. Ce quime rassure, c'est que le mode decontrôle a fonctionné. Tout n'est pasparfait bien évidemment, mais cetépisode montre que les génériquessont surveillés de la même façonque les princeps; et c'est plutôtrassurant", fait-elle valoir. Une façonpeut-être légitime mais optimiste devoir les dysfonctionnementssurvenus dans les tests de
bioéquivalence menés en Inde.Dans le reste de l'Europe, certains
pays sont néanmoins touchés à plusgrande échelle par ces retraits: "EnAngleterre ou en Allemagne, cesont entre 100 et 200 médicamentsqui sont concernés", précise IsabelleAdenot. Mais même là, "lapossibilité de continuer à recevoirun des traitements retirés estenvisageable sur dérogation sijamais il existait des cas sansalternative", ajoute-t-elle. Lesdysfonctionnements constatés dansles tests de bioéquivalence nemettant pas en jeu une éventuelledangerosité des produits.
18 european INDUSTRIAL PHARMACY September 2015 • Issue 26
700 GÉNÉRIQUES RETIRÉSDU MARCHÉ:CONCRÈTEMENT, ÇACHANGE QUOI?by Hugo Jalinière, for Sciences et Avenir
Isabelle Adenot, présidente du Conseil national del'Ordre des Pharmaciens, revient pour Sciences etAvenir sur les conséquences du retrait massif demédicaments génériques du marché européen.
On the 21 August, an EU-wide suspension of the marketing authorisations of 700medicinal products entered into effect, and the products withdrawn from themarkets. Mme Isabelle Adenot, President of the French Conseil national de l’Ordredes Pharmaciens, a member of EIPG, was interviewed by the journalist HugoJalinière, of the French periodical Sciences et Avenir, on the impact of thiswithdrawal. european Industrial Pharmacy, by kind permission of M. Jalinière andSciences et Avenir, is pleased to bring to its readers a reprint of the article thatappeared on the periodical website sciencesetavenir.fr, reporting the interview.
� Il y a des alternatives pourchacun des génériques
retirés du marché” – Isabelle Adenot �
�Cet épisode montre queles génériques sont
surveillés de la même façonque les princeps"
– Isabelle Adenot �
Visit the website: www.industrialpharmacy.eu for PharmaTV andQuality by Design videos, Regulatory Review, Financial Pharma News
and other current items concerning Industrial Pharmacy
www.industrialpharmacy.eu

A unique and comprehensive guide to ensureregulatory compliance and success inpharmaceutical regulatoryinspections
Edited by Madhu Raju SagheeQuality Assurance, Micro Labs, and Director of PHSS, India
Foreword by Peter D. SmithVice President, Strategic Compliance,PAREXEL Consulting, USA
Foreword– Peter D. Smith
Preface– Madhu Raju Saghee
1 Basic Concepts of Global GMPRequirementsby Tim Sandle and Madhu RajuSaghee
2 FDA Drug Regulation andEnforcementby Seth Mailhot
3 System Based Approach toInspectionsby David Barr and Tim Sandle
4 Preparing for PreapprovalInspectionsby Ron Johnson
5 Effectively Managing and SurvivingFDA Inspectionsby John Avellanet
6 Guide for Successful EUInspection Managementby Siegfried Schmitt and Nabila Nazir
7 Regulatory Requirements ofJapanese GMP Inspectionsby Yoshikazu Hayashi
8 Preparing and Management ofInternational Inspectionsby Andreas Brutsche and Tim Sandle
9 Handling and Responding to PostInspectional Observationsby Tim Sandle, Madhu Raju Sagheeand David Barr
10 Preparing for RegulatoryInspections of Sterile Facilities:The Focal Pointsby Tim Sandle
11 Preparing for RegulatoryInspections of API Facilities: TheFocal Pointsby Siegfried Schmitt and RichardEinig
12 Optimizing your RegulatoryComplianceby Mark Tucker
PHARMACEUTICAL
REGULATORY
INSPECTIONS
“EVERY INSPECTION A SUCCESS STORY –
A PRACTICAL GUIDE TO MAKE IT HAPPEN"
EDITED BY MADHU RAJU SAGHEE
order online at www.euromedcommunications.comOr contact the publishers: email: [email protected];
Tel: +44 (0)1428 752222; Fax: +44 (0)1428 752223.
In over 500 pages and twelve chapters thisunique book provides a focussed account ofregulatory issues from pre-approvalinspections and the inspection itself to post-inspection and maintaining compliance. This isa book that every pharmaceutical company willwish to study before and during any inspectionprocess to ensure a successful outcome.
Complete Remit
The book is a fully detailed and practical guidecontaining advice and insight to help anypharmaceutical organisation prepare for GMPInspections, understand key regulatory issuesand review inspectorate trends and findings.
Expert Advice
The authors, with a wealth of regulatoryexperience behind them, express their viewsand provide useful and practical tips forsucceeding in vital regulatory inspections
International Applications
The book includes chapters covering FDAInspections, EU Inspections, JapaneseInspection and International Inspectionprocesses.

maharP
ma | onsult GlobalmaCharP|
onsult Global
thoyespeciallespecially
e vs arantonsultC
Global
who are hoping tse tho
intaopere arhco healtal tite v
HealGlobal
seasverade oo trwho are hoping t
stekal mariceutmacg in all pharin
th TrHeal
, gement sy manaualitg, qinaudits
th Trainingth Tr
ems tsygement s
aining
onsult
SK & NGpanies (omcional Ptinahip mult-cblue
, Middle EaAS, UopeEurhcum of healtrtpecslienth citg winkorW
.idervog praininrtonsultional cional cttnanaererintint
maChar. Plopmentevdeaininr, tionibutrtof dis
peciall, esyloballgpanies lookomcC
v
lopmentevdeoducpr
vererSSictacprmancorfper
oducof prg tlpinhe
oper
e
hect, bio)istarvoSK & Naliceutmacharional Pom, frsiat and As, Middle Ea
es inicve serarullhe fs toss acrlient
e pokpoky and besy and besancanconsult is an maC
t oducg and prainins eahe ary in tpeciall
e tao operg tinpanies look
e on
e pliancomy cortegula, rlopmentt oduct prpecialis, siontartegist r
e on icicvvom adom adge frge frananes res ricic.emstsyg sintaheir operom te fr
evitt effecost ce and besmanchge hivhieo acs and ttoduc
oliotfheir porlop tevo deg tg in all phar
,
s operlienth cs in whictekmaropro needs and apprmade ty seranconsultsonalised cper
t it prhapanies in tomher ctolaims it differonsult cmaCharP
landshertehe Nzil and TaBranbul, Riytlhi, Ises in Dgiefinbr
ional ctide navoill pry wpanomcont tional frtnaerhe intOn t
. egilancivomacpharhains and y cor medicines supplf
gement sy manag, q
an his c. Tetas operhe o te ttiaopr
-ailore ticvy serides a vot it pr
om s frlaims it differ. lands
, Dubai, adhanbul, Riye enceronfional c
he ear this yont t
hains and y
.uko.cyunittlobal-opporg
icvy seranconsulta char. P)AEUh (of healt, and L)ASFDies (ithorutA)savitcApanies (omc
Julphar UE-ups (tarts
paniesomo ce ticsonsult offerma Charsertal Minisoc, and L
yortegula, R)ico gener) tAJulphar UE
y HealtunittOpporGlobal |sue 01Is
g tinpanies lookomor cF. ey sitortaor labor
g a nesioninommiso cion tsolutiontpecXP insy Gortegulaa ricve audit serpliancombe c
87e 2015arhcy Healt
e tao operg t
y ortacw fg a ney enkur, or a tion
e vivo sures tic
AD
VE
RTO
RIA
L

88 Global Opportunity Healthcare 2015 | Issue 01 global-opportunity.co.uk
overseas, it provides the opportunities to register products through the National Regulatory Authority provided by its associate organisations in the Indian Gulf States and Latin America, providing a unique global networking opportunity.
Firms might be given advice on the development of their medical products (either large or small molecule) or medical devices to meet the latest international regulatory requirements by its various affiliates. Leading local agents can be proposed to distribute products through local representation provided by PharmaConsult’s associate organisations in the Indian Gulf States and Latin America, providing a unique Global networking opportunity.
Client’s staff are provided with the opportunity to develop their skills and competencies though comprehensive training services which can include online training, or onsite training solutions, comprising of over 40 online eLearning, and 25 Workshop courses. PharmaConsult is the only training provider who can translate courses in to the language appropriate to the region, so that all employers will be able to access training, which encourages a lifelong approach to learning and career development.
In addition the company will be populating automated Quality Management software to ensure the product and process training will be provide to those persons as appropriate which will be documented in accordance with GXP regulatory requirements.
Training PharmaConsult is an International Training provider of high quality courses based on the latest published technical and regulatory requirements. Method of training delivery is by:Online training: There are 40 eLearning and webinars on 8 specialist topics. These include: Pharmaceutical Product Development, Clinical Research, Pharmaceutical Product Registration, Pharmaceutical Quality Management
| Pha
rmaC
onsu
lt G
lobal
Pharma | PharmaConsult Global
Further informationwww.pharmaconsultglobal.com
systems, Pharmaceutical Manufacture, Pharmacovigilance, Pharmacoeconomics and Pharmacy Practice containing current issues (contain only new regulatory and technical requirements) Presentation (including video/graphics with minimum PowerPoint slides) and include Course notes (copy of slides, reading material, &case studies), candidate Assessment Questionnaire, and CPD Accredited Proficiency certificate. Professional Training courses are provided over 6-12month periods consisting of a combination of 2-4 day workshops held at regional centres, and online eLearning, conferences and webinars.
Certificate/diploma qualification courses for membership of the professional Group available are;
‘Services range from advice on product registration, specialist product development, regulatory compliance auditing, quality management systems for medicines supply chains and pharmacovigilance.’
Product development: The major focus of the program is to develop the knowledge and understanding for teams to devise a good regulatory strategy for continuous project assessment to promote regulatory and PharmacoEconomic acceptance of the developed product. Course Leader: Professor Luigi Martini – Kings College University London Chartered Quality Institute Certificate in Quality Management:This Certificated course establishes the principals of quality management systems allowing use of well-known quality tools and risk assessment techniques. It provides an ideal basis for any person employed in Healthcare, wishing to take the exams for a Certificate in Quality Management Practices (QMP), providing the first step to qualifying for entry on the CQI Diploma Couse and becoming a CQI Chartered Quality Professional.Medicines Procurement and Supply Chain management: This introductory qualify course establishes the wide range of causes for medicine supply shortages, dealt with by government agencies, medicines’ regulatory authorities and manufacturing organisations. And provides strategies for managing changes in procurement practices, avoiding falsified medicines and defective medicines entering the supply chain (such as insistence on World Health Organization prequalification status or registration with a stringent regulatory authority) may invalidate a previous supplier).Workshops: A range of 25 specialist subjects relevant to the region and/or company for a basis for presentation, which can vary in duration from half a day up to five days, public seminars are usually are of 2-4 days in duration, and always in small groups so as to allow for detailed discussion on the topic in question.
‘PharmaConsult is the only training provider who can translate courses in to the language appropriate to the region, so that all employers will be able to access training, which encourages a lifelong approach to learning and career development.’
King’s College London is in discussion with PharmaConsult Global to create a new Certificate course in Biopharmaceutics which will comprise 5 modules on: Clinical Pharmacology Clinical Assessment Product Discovery Product Development Manufacturing.
The course will be available from Summer 2015. Further details of this important new concept in CPD will be available onwww.pharmaconsultglobal.com or contact [email protected] to register your interest in the course.

22 european INDUSTRIAL PHARMACY September 2015 • Issue 26
regulatory reviewThe current review period hasseen a number of changes inthe regulation of medicinesand regulatory guidance inthe EU, International marketsand the USA. Incidents arereported of medicines beingsuspended in the EU becauseof flawed studies and also ofcompanies being fined forserious breaches of goodmanufacturing practice(GMP) in the manufactureand supply of products in theUK.
USARequest for Quality MetricsThis draft guidance includes anexplanation of how the Food andDrug Administration (FDA) intendsto collect data and use qualitymetrics to help ensure that theirpolicies and practices continue tosupport continuous improvementand innovation in the pharmaceuticalmanufacturing industry.
These metrics can be used by theFDA: to help develop complianceand inspection policies andpractices, such as risk-basedinspection scheduling of drugmanufacturers; to improve theAgency’s ability to predict and,therefore, possibly mitigate, futuredrug shortages; and to encouragethe pharmaceutical industry toimplement state-of-the-art,innovative quality managementsystems for pharmaceuticalmanufacturing.
Assessment and Control ofDNA Reactive (Mutagenic)Impurities in Pharmaceuticals As a result of chemical synthesis orsubsequent degradation, impuritiesreside in all drug substances/drugproducts. International Conferenceon Harmonisation (ICH) Q3A andQ3B provide guidance forqualification and control for themajority of the impurities. Limitedguidance is provided, however, forthose impurities that are DNAreactive. This guidance provides apractical (safety and quality riskmanagement) framework that is
applicable to the identification,categorisation, qualification andcontrol of mutagenic impurities tolimit potential carcinogenic risk frommutagenic impurities that reside orare reasonably expected to reside infinal drug substance or product,taking into consideration theintended conditions of human use.
Reportable CMC Changes forApproved Drug and BiologicProducts This draft guidance addresses thelack of clarity with respect to whatchemistry, manufacturing, andcontrols (CMC) information in amarketing application constitutes anestablished condition or a“regulatory commitment” that, ifchanged following approval,requires reporting to the FDA. Suchclarification, should lead to a betterunderstanding that certain CMCchanges can be made solely underthe Pharmaceutical Quality Systemwithout the need to report to theFDA. The result should be a moreeffective post-approval submissionstrategy over the lifecycle of theproduct by the regulated industry.
Size, Shape, and OtherPhysical Attributes of GenericTablets and CapsulesGeneric drug products are requiredto be both pharmaceutically andtherapeutically equivalent to areference listed drug (RLD). TheFDA is also concerned thatdifferences in physical characteristics(e.g. size and shape of the tablet orcapsule) may affect patientcompliance and acceptability ofmedication regimens or could leadto medication errors. The FDArecommends that generic drugmanufacturers consider physicalattributes when they develop qualitytarget product profiles for theirgeneric product candidates.
EuropeConcept paper on newguidance for importers ofmedicinal productsThe GMP/GDP [good distributionpractice] Implementation Working
Group agreed to draft a specificguidance for import authorisationholders. This document most likelywould take the form of a new annex(annex 21). The scope of the projectwill focus on importation activitiesnot addressed in detail in thecurrent GMP guide and annexes.
Fast track routes for medicinesthat address unmet medicalneedsThe European Medicines Agency(EMA) has revised its guidelines onthe implementation of acceleratedassessment and conditionalmarketing authorisation, two keytools in European legislation toaccelerate patients’ access tomedicines that address unmetmedical needs.
EMA confirmsrecommendation to suspendmedicines over flawed studiesThe EMA originally suspended anumber of medicines for whichauthorisation in the EU was primarilybased on clinical studies conductedat GVK Biosciences in Hyderabad,India. Following a re-examinationrequested by marketingauthorisation holders for seven ofthe medicines concerned. Around700 pharmaceutical forms andstrengths of medicines studied atthe Hyderabad site remainrecommended for suspension. Foraround 300 other pharmaceuticalforms and strengths, sufficientsupporting data from other sourceshad been provided; these medicineswill therefore remain on the marketin the EU.
Medicines and HealthcareProducts Regulatory Agency(MHRA)Companies sentenced forsupplying hospitals withdefective pre-filled syringesA major healthcare company and asister company that sold a range ofready-to-use pharmaceuticalproducts it manufactured have beensentenced for supplying hospitalswith defective pre-filled syringesthat in one case contributed to the

23european INDUSTRIAL PHARMACY September 2015 • Issue 26
death of a diabetic patient afterbeing treated with a batch ofintravenous insulin syringes thatactually contained no insulin. Thefaulty syringes were supplied byFresenius Kabi Ltd as a licencedwholesaler for Calea UK Ltd, whichmanufactured the product.
Both companies, based at thesame address in Runcorn, Cheshire,were fined at Sheffield Crown Courtafter being prosecuted by theMHRA.
These two incidents followed aseries of previous inspections by theMHRA officials that highlighteddeficiencies. Fresenius Kabi Ltd wasfined a total of £500,000 plus £5900in costs after pleading guilty tobreaching Sections 64(1) and 67(2) ofthe Medicines Act 1968. Calea UKLtd was fined £50,000 with £5900costs after pleading guilty to similarbreaches.
MHRA Inspectorate blogThis blog is aimed at organisationsthat are inspected by the MHRA andneed to keep up to date with thelatest thinking and guidelines. It will
give the MHRA inspectors a chanceto speak directly to theorganisations they inspect and getfeedback from them on topics theywould like to hear more about.
MHRA helps to future-proofmammalian cell culturemanufacturing facilityFujifilm involved the MHRA early onto meet regulatory expectations toreduce risk of any late or costlychanges. They wanted to include asmuch feedback from the MHRA intothe design of the facility as possible.
InternationalICHICH Q7 Q&As Experience gained with theimplementation of the ICH Q7Guideline since its finalisation in2000 shows that uncertaintiesrelated to the interpretation of somesections exist. Technical issues withregard to GMP of activepharmaceutical ingredients (APIs)and also in context with other newICH Guidelines are addressed, in
order to harmonise expectationsduring inspections, to removeambiguities and uncertainties, andalso to harmonise the inspections ofboth small molecules and biotechAPIs.
Pharmaceutical InspectionCooperation Scheme (PIC/S)Strengthening of internationalregulatory cooperation in thefield of GMPThe aim is to encourage PIC/Smembers to accept inspectionfindings on a voluntary basis, byrelying on mutual trust andconfidence building, based on thePIC/S accession process.
For further information on theseand other topics, we suggest yourefer to the websites of relevantregulatory bodies and to currentand past editions of “GMP ReviewNews” published by EuromedCommunications. To subscribe tothis monthly news service [email protected]
REGULATORY REVIEW continued
PharmacoVigilanceRevıew
Journal on drugsafety issues
Editor – Rob Begnett
This quarterly journal providesinformed comment and analysis ofinternational pharmaceuticalregulations relating to the safe use ofmedicines and medicinal devices. It
also carries reviews of current methods of pharmacovigilance.
Order online at www.euromedcommunications.com
Or email: [email protected]
Tel: +44 (0)1428 752222 Fax: +44 (0)1428 752223
PharmacoVigilanceRevıewSupporting the safe use of
medicines and medical devices
Managing Reference SafetyInformation
The EU centralised applicationfrom a pharmacovigilance andrisk management prospective
Post-authorisation aggregatesafety reporting: the new PSUR
New European pharmacovigilancelegislation – an adequateresponse to current challenges?
Volume 7 Number 3/4 Nov 2013

24 european INDUSTRIAL PHARMACY September 2015 • Issue 26
Target over heartUtah, USA has a problem. Citizenscannot obtain drugs required forcapital punishment by lethalinjection. The alternative firingsquad (five anonymous policemarkspersons) fire four live bulletsand one blank onto a target pinnedover the heart. Lethal injectionscontain chemicals, commonplace inindustrial pharmacy but so elderlythat they have grown whiskers.Sodium thiopental was first usedclinically in 1934; pentobarbitone in1930; curare, akin to the potentiallylethal muscle relaxant pancuronium,in 1948. Humphry Davy discoveredpotassium chloride in 1817.
Apparently some injections were“botched”. Observers noted thecondemned took hours to die. AnAmerican manufacturer, such asHospira for thiopental, will notsupply such medicaments toprisons. Moreover, the EU, under“torture regulations” (CouncilRegulation (EC) No 1236/2005) bansexport of certain drugs. That isdespite lethal injection beingdeemed not a medical procedure.That by-passes doctors’ concerns.
At that point, Brown’s nosetwitched. I felt like a small pigdetecting a whiff of mouth-wateringtruffle underneath forest litter. Whatbrutalising and barbaric nonsenseand delicious ironies did my snoutdetect?
Way to goMy starting reflection was that, had Ibeen a particularly undesirablecitizen of Utah condemned to death,
my preferred exit would be byefficiently-administered injection. I remember experiencingintravenous barbiturates asprecludes to gaseous generalanaesthesia. I drifted into sleep. Iflonger than desired, there could beworse ways to exit. It seemspreferable to the Gaelic Guillotine(fraction of a second), hanging(second or two) or electrocution(dozens of seconds). Maybe thatexposes my bias because medicineshave been my life. In salad days,dispensing wonderful-smellingBrompton cocktail with heroin,honey, brandy, chloroform water andso forth, I reflected that, if ever Irequired it, I hoped some pharmacistwould be available to compound.That would be more human than theclinically efficient MST andOramorph – and more tasty.
I then wondered whether I wouldbe willing to certify a batch of lethalinjections fit for release onto the EUmarket, as a qualified person;Netherlands citizens use similarcocktails for euthanasia. I have leftthat fray but, were I still involved, myanswer would probably be “Yes”.Indeed, I would strain every sinew toensure that it was made, as ourAmerican cousins might say, “realgood”.
Medicine safetyThose reflections deposited mewithin the Medicines andHealthcare Products RegulatoryAgency (MHRA) website. Thatinformed “what you need to know”about medicine safety. Before
licensing, medicines should be“thoroughly trialled on thousands ofpeople”, the “advantages” must“outweigh the disadvantages oftaking the medicine” and, for thepeople taking the medicine(condemned criminals), “do themost good for the least harm”. Isensed a mismatch between thoseaims and the prisons in Utahcontaining human beings whomsome staff labelled as “monsters”.For example, I am unaware ofthousands of clinical trials testing“superdoses” of lethal agentsresulting in death. In fairness,package inserts and label excludethat outcome.
Scrutiny of the MHRA websiteexposed it as glaringlyanthropocentric. Humans (not say,salmon; they require medicines,also: I have made them) are itstaken-for-granted reference species.It is fascinating that octopi aredeemed so intelligent that the UKconsiders them honoraryvertebrates for the purposes ofanimal cruelty laws. Presently, I amingesting for bronchitis – thankful tobe a patient in the rich West –clarithromycin. Hopefully, that is“harmless” to my mammalian cellsbut will kick-the-ass of Haemophilusinfluenzae, Streptococcuspneumoniae and pyogenes and anyother unfriendly interlopers.
I end with a final twitch inviewpoint. Most of humankindaccepts the scientific evidence basethat capital punishment is ineffective.
Malcolm E Brown
bottled brown

25european INDUSTRIAL PHARMACY September 2015 • Issue 26
European Medicines Agency(a) EIPG comments on the ConceptPaper for Guideline on TopicalProducts have been submitted tothe European Medicines Agency.For a copy of the comments, seethe EIPG website under “News”from the Bureau.
(b) EIPG comments on the DraftGuideline on the Chemistry of ActiveSubstances are being consolidatedby Georgina Gal (Hungary) andMarianne Anderson (Sweden).
European CommissionA draft Commission DelegatedRegulation has been published onthe Unique Identifier and includesthe details of the rules for the safetyfeatures appearing on the outerpackaging of medicines. It will apply3 years from publication of the finaltext.
The 34 pages of main document and5 pages of annexes can be found athttp://ec.europa.eu/growth/tools-databases/tbt/en/search/?tbtaction=search.detail&num=306&Country_ID=EU&dspLang=EN&BASDATEDEB=&basdatedeb=&basdatefin=&baspays=EU&basnotifnum=306&basnotifnum2=306&bastypepays=EU&baskeywords
If any reader wishes to submitcomments on the draft, they shouldbe sent to Piero Iamartino([email protected]).
News from the EuropeanParliamentA new section under “News” hasbeen added to the EIPG websitecalled "European ParliamentPharma Watch". It collectsquestions and answers, statements,etc at the European Parliament onmatters relating to thepharmaceutical industry. Seehttp://eipg.eu/eurparlpharm/
EIPG awardsFellow of EIPGThe purpose of this award is torecognise individuals who haveexhibited strong leadershipinternationally, have distinguishedthemselves in the pharmaceuticalsciences and/or practice of industrialpharmacy, who have contributed tothe advancement of pharmaceuticalsciences and/or practice of industrialpharmacy, and who have served theEIPG. Nominations for next year’saward should be submitted by anyEIPG Full Member Association notlater than 1 January 2016.
EIPG Emerging IndustrialPharmacist AwardThe purpose of this award is torecognise significant intellectualcontributions by emerging industrialpharmacists within EIPG thatpromote state of the art in industrialpharmacy and the pharmaceuticalsciences. Nominations for the awardmay be submitted by any EIPG FullMember not later than 1 January ofthe year in which the award is to bemade. At the time of nomination,the nominee should be a personwho is within 10 years of havingstarted a career in industrialpharmacy.
EducationA new publication on the Phar-Inproject has been published: AEuropean competence frameworkfor industrial pharmacy practice inbiotechnology. Pharmacy2015;3(3):101–128;doi:10.3390/pharmacy3030101
A new course on PersonalisedMedicine prepared by Drs ClaireThomson and Felicity Sartain isavailable on the PharmaConsultwebsite. This can be found athttp://www.pharmaconsultglobal.com/course_detail.html?id=31
MOGLYNET ProjectEIPG has been invited to join as apartner in an important Europeaneducational project addressed tomaster students willing to achieve adoctorate level degree. Thisproject, called MOGLYNET, involvesfive European universities (Milan,Antwerp, Leiden, Aberdeen andBarcelona) and the participation ofa few industrial partners andprofessional association partners,who have offered their support interms of consultancy, training andassistance to the development ofthe research and educationalactivities of 12 doctoratecandidates.
In particular, EIPG will be involvedin the recruitment phase of studentsfor this project and in the assistancefor setting up appropriate trainingcourses promoting an integratedknowledge between universityresearch and pharmaceuticalindustrial expectations. Anni Svala,EIPG Vice-President Education andCareers and Finnish delegate, is incharge of representing EIPG in thisproject.
European PharmaceuticalStudents AssociationTwo further successful webinarshave been held, each with anaudience of 50 participants. Thespeaker on PharmacovigilanceAudits and Quality Systems wasSusanna Heinonen, PV manager,Algol Pharmaceuticals, and theImportance of ContinuingProfessional Development waspresented by Claire Johnson, EBI,LifeTrain.
Jane Nicholson, Executive DirectorEIPG [email protected]
news from the EIPG

eventsSEPTEMBER13–14 September 2015 –Birmingham, UKAnnual Conference 2015www.rpharms.com
15–16 September 2015 – Munich,GermanyPharmaceutical Freeze DryingTechnologyhttps://europe.pda.org
28–30 September 2015 – LasVegas, NV, USA2015 Pharma EXPOwww.ispe.org
28–30 September 2015 –Washington, DC, USA2015 PDA/FDA JointRegulatory Conferencewww.pda.org
29 September–3 October 2015 –Düsseldorf, Germany75th FIP World Congress ofPharmacy and PharmaceuticalSciences 2015www.fip.org
OCTOBER5–7 October 2015 –Loughborough, UKAPS 20th AnniversaryInternational PharmaceuticalPhotostability Conference 2015www.apsgb.co.uk
6–7 October 2015 – Amsterdam,The NetherlandsPharmaceutical Cold & SupplyChain Logisticshttps://europe.pda.org
7–8 October 2015 – London, UKPharma Compliance Europe 2015www.terrapinn.com
8 October 2015 – London, UKWhat’s new in the approval andconduct of clinical trials inEurope?www.jpag.org
13–15 October 2015 – Madrid, SpainCPhI Worldwidewww.cphi.com
14–15 October 2015 – Dublin,IrelandBioProduction 2015http://www.informa-ls.com
21 October 2015 – Cambridge,UKMIBio 2015: Stability ofbiopharmaceuticals – Frommolecular interactions tosuccessful productswww.apsgb.co.uk
21–23 October 2015 –Washington, DC, USA16th Annual PharmaceuticalRegulatory and ComplianceCongresshttp://pharmacongress.com
22 October 2015 – London, UKAPS Stimulating AntimicrobialInnovationwww.apsgb.co.uk
26–28 October 2015 –Hyderabad, India 4th International Summit onGMP, GCP & Quality Controlhttp://gmp-gcp-quality-control.pharmaceuticalconferences.com
NOVEMBER3–4 November 2015 – Vienna,AustriaThe Universe of Pre-filledSyringes and Injection Deviceshttps://europe.pda.org
4–6 November 2015 –Amsterdam, The Netherlands18th APIC/CEFIC EuropeanConference on ActivePharmaceutical Ingredientswww.gmp-compliance.org
8–11 November 2015 –Philadelphia, PA, USA2015 ISPE Annual Meetingwww.ispe.orge
9–10 November 2015 – Basel,SwitzerlandWorld Biosimilar Conferencewww.terrapinn.com
10–11 November 2015 –Dusseldorf, GermanyPharmaLab Congress 2015www.pharmalab-congress.de/ple_home.html
11 November 2015 – Basel,SwitzerlandHPAPI World Congress www.terrapin.com
11–13 November 2015 – Geneva,Switzerland6th Annual World Orphan DrugConferencewww.terrapin.com
17–18 November 2015 –Copenhagen, DenmarkOutsourcing/ContractManufacturinghttps://europe.pda.org
25–26 November 2015 – Berlin,Germany10th Qualified Person Forumwww.gmp-compliance.org
DECEMBER1–2 December 2015 – Berlin,GermanyVaccineshttps://europe.pda.org
8 and 10 December 2015 –London, UKGDP Symposiumwww.gov.uk/government/news/mhra-symposiums-good-manufacturing-practice-gmp-and-good-distribution-practice-gdp
9 and 11 December 2015 –London, UKGMP Symposiumwww.gov.uk/government/news/mhra-symposiums-good-manufacturing-practice-gmp-and-good-distribution-practice-gdp
10 December 2015 – London, UKMaximising Productivity inPharmaceutical QC and StabilityTestingwww.jpag.org
european INDUSTRIAL PHARMACY September 2015 • Issue 2626