extra supporting information - royal society of · pdf filesupporting information ... e used...
TRANSCRIPT

Supporting Information
Unusual thermogelling behaviour of poly[2‐(dimethylamino)ethyl methacrylate] (PDMAEMA)‐based polymers polymerized in bulk
Jason Yujie Zhenga, Mein Jin Tanb, Praveen Thoniyotb and Xian Jun Loha,b,*
(a) Department of Materials Science and Engineering, National University of Singapore, 9 Engineering Drive 1, Singapore 117576, Singapore.
(b) Institute of Materials Research and Engineering (IMRE), 3 Research Link, Singapore 117602, Singapore.
Email: [email protected]‐star.edu.sg
1. Materials
Monomers 2‐(dimethylamino)ethyl methacrylate (DMAEMA) and methyl methacrylate (MMA) were supplied
by Sigma‐Aldrich, and purified by passing through a chromatography column filled with inhibitor removers
before use. Azobisisobutyronitrile (AIBN), tetrahydrofuran (THF), toluene (anhydrous) and hexane were
obtained from Sigma‐Aldrich and used as received.
2. Synthesis of P(DMAEMA‐ran‐MMA) Copolymers by FRP
2.1 Bulk copolymerization of DMAEMA and MMA
Random copolymers of DMAEMA and MMA (Table 1) were synthesized by free radical polymerization using
AIBN as the initiator. As an example, the synthesis of copolymer DM2 is described. The monomers, DMAEMA
(22.172 g, 141.04 mmol), MMA (1.570 g, 15.68 mmol) and AIBN (0.128 g, 0.78 mmol) were introduced into a
250 ml 2‐necked round bottom flask equipped with a reflux condenser and a stir bar. The mixture was
degassed by bubbling nitrogen for 15 min to remove oxygen, which could potentially inhibit the free radical
polymerization reaction. The flask was heated in an oil bath at 70 °C for 24 h under nitrogen flow with
continuous stirring. The crude copolymers were dissolved in THF before it was precipitated dropwise in ten‐
fold excess of cold hexane. The samples were subsequently dried under vacuum overnight at 40 °C. The
purified polymer was in the form of a white powder. Polymer yields were as high as up to 82.4% for polymer
DM5 = 19.3 g.
1H‐NMR (CDCl3) of P(DMAEMA‐ran‐MMA) DM3: δ (ppm) 0.88‐1.05 ((C‐CH3) of the backbone of P(DMAEMA)),
1.25 ((C‐CH3) of the backbone of P(MMA))1.82‐1.90, 1.43 ((C‐CH2) of the backbone of P(MMA)), ((C‐CH2) of the
backbone of P(DMAEMA)), 2.33 ((N‐CH3) of P(DMAEMA)), 2.60 ((N‐CH2) of P(DMAEMA)), 3.57 ((O‐CH3) of
P(MMA)), 4.07 (‐(CH2‐O‐C=O) protons of P(DMAEMA)).
2.2 Solution copolymerization of DMAEMA and MMA
Similarly, random copolymers of DMAEMA and MMA (Table 2) were synthesized by a procedure similar to the
bulk copolymerization described above, except that 25 ml of anhydrous toluene was used as the solvent for
the synthesis.
Electronic Supplementary Material (ESI) for RSC Advances.This journal is © The Royal Society of Chemistry 2015

Summary of Polymers Synthesized
a Calculated from H
1‐NMR results.
e Based
on solubility in
aqueo
us 10 wt % solution.
hFrom UV‐Vis m
easurements of 1 wt % aqueo
us solutions
b,cAs determ
ined
from GPC.
f As determined
from tube inversion test and rheo
logical m
easuremen
ts.
dPDI: Poly dispersity index.
g As determined
from rheo
logical m
easuremen
ts
Table S1. R
esults from the bulk polymerization of P(DMAEM
A‐ran‐M
MA) at 70°C
Composition of Copolymer
Copolymer Characteristics
Copolymer
Theo
retical m
olar ratio
([DMAEM
A]:[M
MA])
Actual m
olar ratioa
([DMAEM
A]:[M
MA]))
Molar %
[AIBN]
Mwb
(kDa)
Mnc
(kDa)
PDId
Water
Solubility
e
Thermogelling
Capability
f
T gelg
(°C)
LCST
h
(°C)
PD1
100:0
100:0
0.495
285
62
4.58
38.1
33.0
DM1
95:5
94.1:5.9
0.495
208
63
3.28
36.3
32.1
DM1a
95:5
95.2:4.8
0.495
135
43
3.15
36.8
33.4
DM1b
95:5
94.5:5.5
0.495
104
35
2.96
37.5
34.5
DM1c
95:5
94.7:5.3
0.495
81
26
3.11
38.5
35.8
DM2
90:10
91.5:8.52
0.495
141
81
2.19
26.3
31.4
DM3
90:10
92.1:7.9
0.495
316
81
3.91
34.6
32.5
DM4
90:10
91.0:9.0
0.248
435
148
2.93
25.5
33.2
DM5
85:15
87.4:12.6
0.495
211
68
3.10
26.9
27.9
DM6
50:50
57.7:42.3
0.495
88
44
2.00
‐ ‐

a Calculated from H
1‐NMR results.
e Based
on solubility in
aqueo
us 10 wt % solution.
b,cAs determined
from GPC.
f As determined
from tube inversion test and rheo
logical m
easuremen
ts.
dPDI: Poly dispersity index.
Table S2. R
esults from the polymerization of P(DMAEM
A‐ran‐M
MA) in Toluen
e at 70°C
Composition of Copolymer
Copolymer Characteristics
Copolymer
Theo
retical m
olar ratio
([DMAEM
A]:[M
MA])
Actual m
olar ratioa
([DMAEM
A]:[M
MA]))
Molar %
[AIBN]
Mwb
(kDa)
Mnc
(kDa)
PDId
Water
Solubility
e
Thermogelling
Capability
f
PD2
100:0
100:0
0.495
153
70
2.19
DMT0
95:5
94.9:5.1
0.495
138
62
2.23
DMT0a
95:5
94.7:5.3
0.495
97
40
2.43
DMT0b
95:5
94.6:5.4
0.495
68
32
2.11
DMT0c
95:5
95.3:4.7
0.495
41
21
1.97
DMT0d
95:5
94.6:5.4
0.495
294
156
1.88
DMT1
90:10
84.3:15.7
0.495
48
29
1.65
DMT2
90:10
93.3:6.7
0.005
89
38
2.34
DMT3
80:20
74.3:25.7
0.495
160
82
1.95
DMT4
70:30
62:38
0.495
116
63
1.84
DMT5
60:40
44.6:55.4
0.495
88
50
1.76
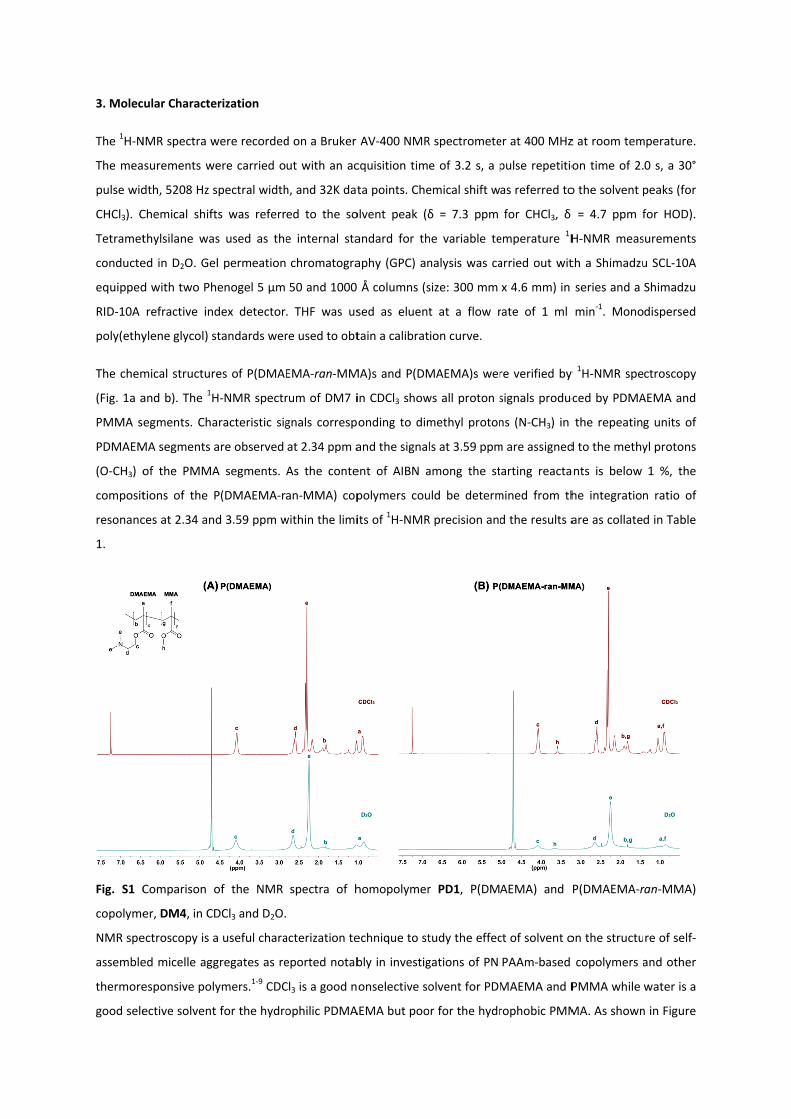
3. Molec
The 1H‐N
The mea
pulse wid
CHCl3). C
Tetramet
conducte
equipped
RID‐10A
poly(ethy
The chem
(Fig. 1a a
PMMA s
PDMAEM
(O‐CH3) o
composit
resonanc
1.
Fig. S1 C
copolyme
NMR spe
assemble
thermore
good sele
cular Characte
NMR spectra w
surements w
dth, 5208 Hz s
Chemical shift
thylsilane wa
ed in D2O. Ge
d with two Ph
refractive ind
ylene glycol) s
mical structur
and b). The 1H
egments. Cha
MA segments a
of the PMMA
tions of the P
ces at 2.34 an
Comparison o
er, DM4, in CD
ectroscopy is a
ed micelle agg
esponsive pol
ective solvent
erization
were recorded
were carried o
spectral width
ts was referr
s used as the
el permeation
henogel 5 µm
dex detector
standards wer
res of P(DMA
H‐NMR spectr
aracteristic sig
are observed
A segments.
P(DMAEMA‐r
d 3.59 ppm w
of the NMR
DCl3 and D2O.
a useful chara
gregates as re
ymers.1‐9 CDC
t for the hydro
d on a Bruker
ut with an ac
h, and 32K dat
ed to the so
e internal sta
chromatogra
50 and 1000
. THF was us
re used to obt
EMA‐ran‐MM
rum of DM7 i
gnals corresp
at 2.34 ppm a
As the conte
an‐MMA) cop
within the limi
spectra of h
acterization te
eported notab
Cl3 is a good n
ophilic PDMA
AV‐400 NMR
cquisition tim
ta points. Che
olvent peak (δ
andard for th
aphy (GPC) an
Å columns (s
sed as eluent
tain a calibrat
MA)s and P(DM
in CDCl3 show
onding to dim
and the signal
ent of AIBN a
polymers cou
its of 1H‐NMR
homopolymer
echnique to st
bly in investig
onselective so
AEMA but poo
R spectromete
e of 3.2 s, a p
emical shift w
δ = 7.3 ppm
he variable te
nalysis was ca
size: 300 mm
t at a flow r
ion curve.
MAEMA)s wer
ws all proton s
methyl proton
ls at 3.59 ppm
among the st
uld be determ
R precision and
r PD1, P(DM
tudy the effec
gations of PNI
olvent for PDM
or for the hydr
er at 400 MHz
pulse repetitio
as referred to
for CHCl3, δ
emperature 1H
arried out wit
x 4.6 mm) in
rate of 1 ml
re verified by
signals produc
ns (N‐CH3) in
m are assigned
arting reacta
mined from th
d the results a
AEMA) and
ct of solvent o
PAAm‐based
MAEMA and P
rophobic PMM
z at room tem
on time of 2.
o the solvent p
= 4.7 ppm f
H‐NMR meas
th a Shimadzu
series and a
min‐1. Mono
y 1H‐NMR spe
ced by PDMA
the repeatin
d to the methy
ants is below
he integration
are as collate
P(DMAEMA‐r
on the structu
copolymers a
PMMA while
MA. As shown
mperature.
0 s, a 30°
peaks (for
for HOD).
surements
u SCL‐10A
Shimadzu
dispersed
ctroscopy
AEMA and
g units of
yl protons
1 %, the
n ratio of
d in Table
ran‐MMA)
re of self‐
and other
water is a
n in Figure
1b, in CDCl3, the peaks due to PDMAEMA and PMMA were sharp and well‐defined. In D2O, the PDMAEMA
peaks remain relatively sharp but the PMMA peaks are collapsed and broadened to an extent that they are
almost undetectable. This evidently shows that molecular motion of PMMA is greatly reduced in water,
indicating that a core‐corona micelle structure made up of a hydrophobic PMMA core and a PDMAEMA corona.
However, it should be recognised that all the PDMAEMA peaks of PDMAEMA homopolymer in CDCl3 are
suppressed in D2O (Fig. 1a) due to the amphiphilic nature of PDMAEMA itself. Therefore, this suggests our
synthesized PDMAEMA homopolymer could potentially aggregate to form micelles.9
3.2 Variable Temperature 1H‐NMR Studies
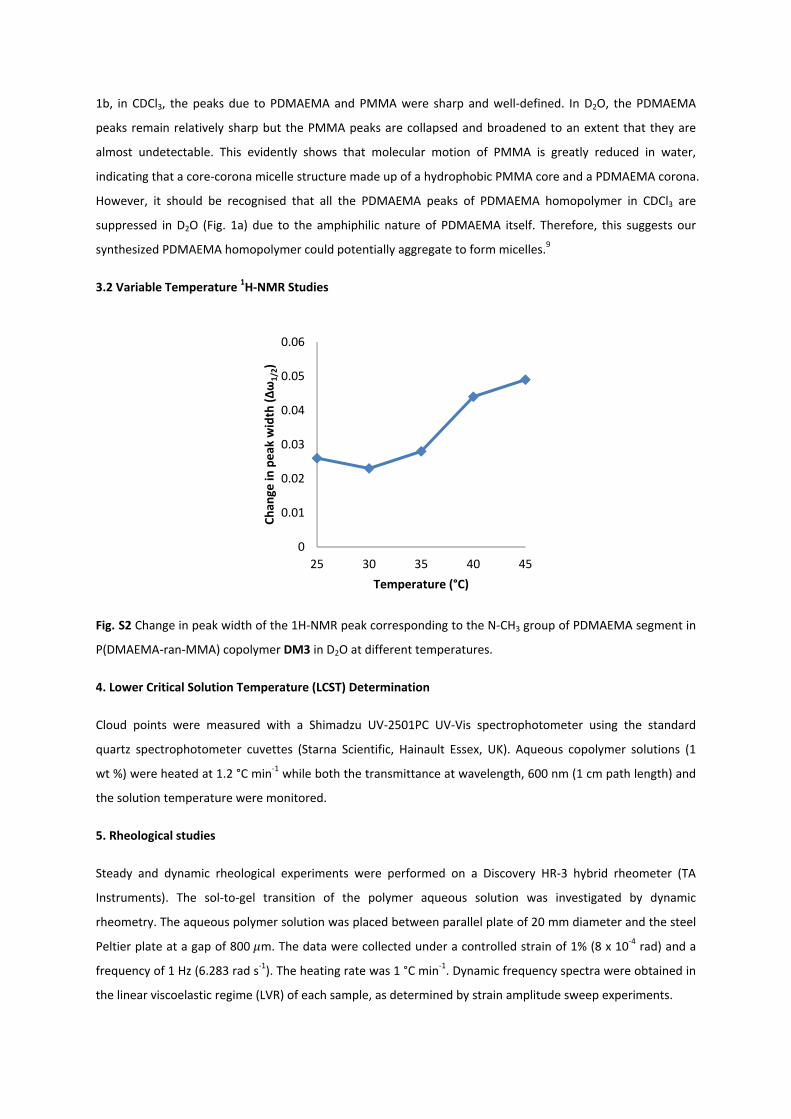
Fig. S2 Change in peak width of the 1H‐NMR peak corresponding to the N‐CH3 group of PDMAEMA segment in
P(DMAEMA‐ran‐MMA) copolymer DM3 in D2O at different temperatures.
4. Lower Critical Solution Temperature (LCST) Determination
Cloud points were measured with a Shimadzu UV‐2501PC UV‐Vis spectrophotometer using the standard
quartz spectrophotometer cuvettes (Starna Scientific, Hainault Essex, UK). Aqueous copolymer solutions (1
wt %) were heated at 1.2 °C min‐1 while both the transmittance at wavelength, 600 nm (1 cm path length) and
the solution temperature were monitored.
5. Rheological studies
Steady and dynamic rheological experiments were performed on a Discovery HR‐3 hybrid rheometer (TA
Instruments). The sol‐to‐gel transition of the polymer aqueous solution was investigated by dynamic
rheometry. The aqueous polymer solution was placed between parallel plate of 20 mm diameter and the steel
Peltier plate at a gap of 800 m. The data were collected under a controlled strain of 1% (8 x 10‐4 rad) and a
frequency of 1 Hz (6.283 rad s‐1). The heating rate was 1 °C min‐1. Dynamic frequency spectra were obtained in
the linear viscoelastic regime (LVR) of each sample, as determined by strain amplitude sweep experiments.
0
0.01
0.02
0.03
0.04
0.05
0.06
25 30 35 40 45
Chan
ge in
peak width (Δω
1/2)
Temperature (°C)

Fig. S3 Rheological profile of copolymer, DM4 (10 wt %) aqueous solution as a function of temperature.
6. Transmission Electron Microscopy (TEM)
The samples were imaged on a JEOL JEM‐2010F FasTEM field emission transmission electron microscope
operated at 100 kV. Samples were prepared for TEM by directly depositing one drop of sample solution (10 mg
mL‐1) containing 0.1 wt % phosphotungstic acid (PTA) onto copper grids, which were coated in advance with
supportive Formvar films and carbon (Agar Scientific). The samples were kept in an oven for 12 h for drying at
25 °C before TEM imaging.
References
1. V. P. Nam Nguyen, N. Kuo and X. J. Loh, Soft Matter, 2011, 7, 2150‐2159. 2. X. J. Loh, Z.‐X. Zhang, Y.‐L. Wu, T. S. Lee and J. Li, Macromolecules, 2009, 42, 194‐202. 3. B. Jeong, Y. H. Bae and S. W. Kim, Macromolecules, 1999, 32, 7064‐7069. 4. B. Jeong, Y. Han Bae and S. Wan Kim, Colloids and Surfaces B: Biointerfaces, 1999, 16, 185‐193. 5. B. Jeong, C. F. Windisch, M. J. Park, Y. S. Sohn, A. Gutowska and K. Char, The Journal of Physical
Chemistry B, 2003, 107, 10032‐10039. 6. B. H. Lee, Y. M. Lee, Y. S. Sohn and S.‐C. Song, Macromolecules, 2002, 35, 3876‐3879. 7. X. J. Loh, S. H. Goh and J. Li, Biomacromolecules, 2006, 8, 585‐593. 8. A. Durand, D. Hourdet and F. Lafuma, The Journal of Physical Chemistry B, 2000, 104, 9371‐9377. 9. X. J. Loh, Journal of Applied Polymer Science, 2013, 127, 992‐1000. 10. X. J. Loh, S. H. Goh and J. Li, The Journal of Physical Chemistry B, 2009, 113, 11822‐11830. 11. J.‐h. Ma, C. Guo, Y.‐l. Tang and H.‐z. Liu, Langmuir, 2007, 23, 9596‐9605.
0.4
4
40
400
20 25 30 35 40 45
Modulus (Pa)
Temperature (°C)
G'
G"
tan (δ)
Gel Point
CGT = 29.5 °C