fabrication and hemocompatibility of cell outer membrane mimetic surfaces on chitosan by layer by...
TRANSCRIPT

Fsb
Ma
Nb
a
AA
KHLPCE
1
lpptbtamtc
fmM((
0d
Colloids and Surfaces B: Biointerfaces 85 (2011) 48–55
Contents lists available at ScienceDirect
Colloids and Surfaces B: Biointerfaces
journa l homepage: www.e lsev ier .com/ locate /co lsur fb
abrication and hemocompatibility of cell outer membrane mimeticurfaces on chitosan by layer by layer assembly with polyanionearing phosphorylcholine groups
ing Gonga, Yan-Bing Wanga, Ming Lia, Bi-Huang Hub, Yong-Kuan Gonga,∗
Key Laboratory of Synthetic and Natural Functional Molecule Chemistry of Ministry of Education, College of Chemistry and Materials Science,orthwest University, Xi’an, Shaanxi 710069, PR ChinaCollege of Oceanography, Hainan University, Haikou, Hainan 570228, PR China
r t i c l e i n f o
rticle history:vailable online 5 November 2010
eywords:emocompatible surfaceayer by layer assemblyhosphorylcholine
a b s t r a c t
Three random copolymers poly(2-methacryloyloxyethyl phosphorylcholine-co-methacrylic acid) (PMAs)were synthesized by free radical polymerization of 2-methacryloyloxyethyl phosphorylcholine (MPC)and methacrylic acid (MA) with different monomer ratios under monomer-starved conditions. The syn-thesized PMA polyanions were assembled on chitosan (CS) film surfaces via electrostatic interactions.Using layer by layer (LbL) assembly with PMA polyanion and chitosan polycation, PMA/CS multilayer thinfilms with phosphorylcholine groups on the outer surfaces were fabricated. The modified surfaces were
hitosanlectrostatic interaction
characterized by dynamic contact angle (DCA), X-ray photoelectron spectroscopy (XPS) and atomic forcemicroscopy (AFM). Hemocompatibility of the surfaces was estimated by protein adsorption and plateletadhesion measurements. The results indicated that cell outer membrane mimetic structures were formedon the modified surfaces with PMA as the outermost layer, and the hemocompatibility of the modifiedsurfaces was significantly improved. This facile method of fabricating cell outer membrane mimetic sur-faces may have potential applications in the fields of hemocompatible coatings, drug delivery, and tissue
engineering.. Introduction
Chitosan (CS) is a polycationic biopolymer obtained by the alka-ine deacetylation of chitin, one of the most abundant naturalolysaccharides from crustacean shells. With numerous desirableroperties such as biodegradability, antibacterial, and non-toxicityo human tissues [1–3], chitosan has emerged as a promisingiomedical material with potential applications as a scaffold inissue engineering [3,4], as a hemodialysis membrane [2,5] ands a coating for coronary stents [6]. However, as a stand-aloneembrane or coating, chitosan is susceptible to biofouling and
hrombus formation, limiting its use in hemodialysis and endovas-ular devices.
To prepare hemocompatible surfaces, phosphorylcholine (PC)unctional groups were attached to chitosan surfaces by several
ethods, including bonding to surface amino groups throughichael addition of 2-methacryloyloxyethyl phosphorylcholine
MPC) [7–9], reaction with 2-chloro-1,3,2-dioxaphosphospholaneCOP) [10,11], and grafting of phosphorylcholine dichloride [12].
∗ Corresponding author. Tel.: +86 29 8830 2109.E-mail address: [email protected] (Y.-K. Gong).
927-7765/$ – see front matter © 2010 Elsevier B.V. All rights reserved.oi:10.1016/j.colsurfb.2010.10.049
© 2010 Elsevier B.V. All rights reserved.
Furthermore, modification of chitosan through amino groups byreductive amination of phosphorylcholine-glyceraldehyde (PC-CHO) [13] or conjugation with zwitterionic PC through theAtherton–Todd reaction [14] afforded water soluble PC function-alized chitosans. As anticipated, PC-modified chitosan surfacesshowed improved hemocompatibility in blood clotting and plateletadhesion assays, attributed to the cell outer membrane mimeticstructures formed on the surfaces [15–17].
In this communication we propose another approach to pre-pare hemocompatible surfaces by attaching phosphorylcholine(PC) groups to chitosan through layer-by-layer (LbL) assemblyusing a polyanion containing phosphorylcholine (PC) groups byelectrostatic adsorption [18,19]. LbL assembly involves consec-utive adsorption of polyanions and polycations by alternatelydepositing polyanions and polycations to fabricate multilayer thinfilms/surfaces [19,20]. The LbL deposition technique has beenemployed to build biocompatible surfaces [21–23].
Combining the advantages of LbL technique such as simplicity
and easy thickness control with the effectiveness of cell outer mem-brane mimetic structure approaches, we report a facile method forthe preparation of hemocompatible surfaces by modifying chitosansurface with cell outer membrane mimetic structure using LbLassembly of poly(2-methacryloyloxyethyl phosphorylcholine-co-
aces B
motmu
2
2
mWprc2rbPb
2p
cptrsNwrafaia1
sas
M. Gong et al. / Colloids and Surf
ethacrylic acid) (PMA) and chitosan. The structure and stabilityf these surfaces were investigated by X-ray photoelectron spec-roscopy (XPS), dynamic contact angle (DCA) and atomic force
icroscopy (AFM). The hemocompatibility of the surfaces was eval-ated by platelet adhesion and protein adsorption experiments.
. Materials and methods
.1. Materials
Chitosan with a degree of deacetylation of 94% (averageolecular weight of 330,000 Da) was purchased from Xi’anolsen Biotechnology Company. 2-Methacryloyloxyethyl phos-
horylcholine (MPC) was synthesized according to the methodeported by Ishihara et al. [24]. Methacrylic acid (MA) was pur-hased from Aldrich Chemical Co. and used as received. The initiator,2′-azoisobutyronitrile (AIBN, from Shanghai Chemical Co.) wasecrystallized from methanol. Bovine serum albumin (BSA) andovine plasma fibrinogen (Fg) were purchased from Sigma–Aldrich.latelet-rich plasma (PRP) was prepared using fresh blood providedy a healthy donor according to a previously reported method [12].
.2. Synthesis of poly(2-methacryloyloxyethylhosphorylcholine-co-methacrylic acid) (PMA)
The random copolymer PMA was synthesized by free-radicalopolymerization of MPC and MA. The two monomers with molarroportions of 30, 50 and 70% MPC were dissolved in a solvent mix-ure of isopropanol (iPA) and tetrahydrofuran (THF) (35:5) (v/v),espectively. After addition of 1 wt% AIBN in THF to the monomerolution over 3–4 h using a “monomer-starved” method [25] under2 protection and with agitation at 80 ◦C, the reaction mixtureas refluxed and stirred for 20 h to complete the polymerization
eaction. The resulting polymer products were purified by dialysisgainst deionized water (6000-MW-cutoff) for 48 h at 25 ◦C. Afterreeze-drying, the PMA samples were labeled as PMA30, PMA50,nd PMA70 according to their MPC monomer molar percentagen the feed of 30, 50 and 70%, respectively. The actual percent-
ges of MPC unit in PMA30, PMA50 and PMA70 determined byH NMR were 28%, 54% and 77%, respectively. MALDI TOF masspectra showed m/z peaks at 7100–7400, suggesting that the aver-ge molecular weights of PMAs were greater than 7000 Da. Theynthesis scheme is shown in Fig. 1.H2C C H2C C
C C
CH3 CH3
O O
AI
iPA/TH
O
O
OH
O
P
O
O O
N
CH3
H3C CH3
Fig. 1. Synthesis scheme of poly(MPC-c
: Biointerfaces 85 (2011) 48–55 49
2.3. Surface modification of chitosan film
Chitosan (CS) film was prepared according to our previous work[12]. Briefly, a 0.5 wt% solution of CS was prepared by dissolving CSin 1% (v/v) aqueous acetic acid. After filtration, a clean glass coverslip was immersed upright into the solution to attach the CS. Thecover slip was gently removed from the solution, and the adheredCS coating was dried in vacuo at 40 ◦C for 24 h. Finally the CS filmwas thoroughly rinsed with de-ionized water and dried at 30 ◦Cunder vacuum.
Solutions of PMA (1.0 mg/mL in 0.14 M aqueous NaCl) and CS(1.0 mg/mL in 0.10 M acetic acid containing 0.14 M NaCl) wereprepared separately. PMA coated monolayer films were preparedby the following procedure. CS films were immersed in the PMAsolution for 15 min at room temperature, followed by rinsingwith 0.14 M NaCl solution and deionized water to remove weaklyadhered PMA [26]. Multilayer build-up was accomplished bysequentially dipping the CS substrate into the two solutions (alter-nating between CS and PMA), followed by adsorbing for 15 min andwashing with 0.14 M NaCl solution. After rinsing thoroughly withdeionized water and freeze drying, a cell outer membrane mimeticsurface (PMA as the outermost layer) was finally obtained.
2.4. Characterization of the modified surfaces
2.4.1. Dynamic contact angle measurementThe hydrophilicity and hydrophobicity of the modified sur-
faces were characterized by a highly sensitive dynamic contactangle technique with a DCAT 21 tensiometer from DataphysicsInstruments. Dynamic contact angles, including advancing (�adv)and receding (�rec) angles, can provide information on the surfacehydrophilicity/hydrophobicity and group reorientation [27,28].The technique, which is based on the Wilhelmy plate method,requires samples presenting identical front and back surfaces.These were obtained by coating square films on both sides withpolyelectrolyte CS or PMA as the outermost layer. In each case, aminimum of three specimens was analyzed to ensure reproducibil-ity. Deionized water (Millipore system, 18 M�) was used as probe
liquid. The water surface tension was determined before and afterthe analysis of each surface. A dry specimen was attached to theelectrobalance via a clip and the stage carrying a beaker of waterwas automatically raised and lowered (10 mm/min) to allow waterto contact the specimen. A dwell time of 60 s was allowed betweenH2C C
H2C C
C C
CH3 CH3
O Om n
BN
F,
O
O
OH
O
P
O
O O
N
CH3
H3C CH3
o-MA) random copolymer (PMA).

5 aces B: Biointerfaces 85 (2011) 48–55
sva
2
mwmucih
2
fit
2
tssSe
2
ds
2
iwiokCpcpbcttt
2
dicfiTprfiwP
the deposition of CS on the CS–PMA surface. Third, the large –NH2signal on the CS–PMA surface suggested that the PMA coated layerwas less than 8 nm in thickness, while the–N+(CH3)3 signal of theCS–PMA–CS surface indicated that the outer CS layer was less than
Table 1
0 M. Gong et al. / Colloids and Surf
ample immersion and withdrawal. Analysis of the resulting forceersus distance curves (loops) yields the advancing and recedingngles (�adv and �rec).
.4.2. X-ray photoelectron spectroscopy measurementsThe elemental composition of the modified surfaces was deter-
ined by X-ray photoelectron spectroscopy (XPS). The XPS spectraere recorded with an Axis Ultra, Kratos instrument (UK) usingonochromatic Al K� radiation (150 W, 15 KV, 1486.6 ev). The vac-
um in the spectrometer was kept at 10−9 Torr. All spectra wereollected at an electron take-off angle of 70◦ from the surface. Bind-ng energies were calibrated relative to the C1S peak (284.8 ev) fromydrocarbons adsorbed on the surface of the samples.
.4.3. Atomic force microscopy (AFM) measurementsThe topography of the surfaces was observed using an atomic
orce microscope (SPA-400, Seiko Instruments Inc.). The AFMmages were recorded in tapping mode in air using a silicon can-ilever at ambient temperature.
.4.4. Stability in PBSThe modified chitosan films were immersed in PBS buffer solu-
ion (pH 7.4). After being immersed for 10, 40, and 60 days, theamples were rinsed with deionized water and freeze-dried. Theurface stability was verified mainly by dynamic contact angles.urface chemical composition was also determined by XPS for sev-ral surfaces.
.4.5. Stability in airThe multilayer modified chitosan films were stored in room con-
itions for 2 months. The dynamic contact angles before and aftertorage were compared to show stability in air.
.4.6. Protein adsorptionIn vitro single protein adsorption experiments were performed
n phosphate-buffered saline (PBS, pH = 7.4). The sample surfacesere first equilibrated with the PBS solution for 2 h, and then
mmersed in a solution of bovine serum albumin (BSA) (4.5 mg/mL)r fibrinogen (Fg) (0.3 mg/mL) at 37 ◦C. The adsorption system wasept in a humidified water jacketed incubator equilibrated with 5%O2 in air for 2 h. After being rinsed with PBS three times, the sam-les were transferred to a beaker filled with 2 mL of PBS solutionontaining 1 wt% of sodium dodecyl sulfate (SDS). The adsorbedrotein was completely desorbed by sonication for 20 min. Theicinchoninic acid (BCA) method was used to determine the con-entration of protein in the SDS solution [29,30], and this was usedo calculate the amount of protein on the sample surface. At leasthree replicate samples were analyzed to ensure reproducibility ofhe measurements.
.4.7. Platelet adhesionThe platelet adhesion test was performed under static con-
itions [31,32]. The sample surfaces were first equilibrated bymmersion in PBS for 2 h, then 20 �L of fresh PRP was applied at theenter of the samples, followed by incubation at 37 ◦C in a humidi-
ed water jacketed incubator equilibrated with 5% CO2 in air for 2 h.he samples were then rinsed with phosphate buffer solution (PBS,H 7.4) three times to remove the weakly adherent platelets. Theemaining adherent platelets were fixed in 2.5 wt% glutaraldehydeor 1 h. The fixed samples were then rinsed with PBS and deion-zed water several times. After freeze-drying at −50 ◦C, the samplesere observed with a scanning electron microscope (Quanta 200,hilips-FEI, Netherlands) and the images were recorded.
Fig. 2. Contact angles of CS and PMA adsorbed CS (CS–PMA) surfaces. Bars representSD (n = 3).
3. Results and discussion
3.1. Fabrication of PMA modified surfaces
The partial N-acylation of the CS and hydrogen bonding betweenthe polysaccharide chains increased the surface hydrophobicity ofthe CS films. When hydrophilic PMA polyanion was immobilizedon the CS polycation surface by electrostatic adsorption, a decreasein contact angle should be observed. As shown in Fig. 2, both theadvancing and receding contact angles of PMA modified surfacesdecreased significantly. �adv decreased by at least 37% from 84.5◦ onCS to 48◦, 49◦ and 53◦ on the PMA30, PMA50 and PMA70 modifiedmonolayer surfaces, respectively, and �rec decreased by at least 41%from 12◦ on CS to 4◦, 5◦ and 7◦ on the PMA30, PMA50 and PMA70surfaces, respectively. These large changes in the contact anglesindicate that the fabrication of the CS–PMA monolayer surfaces wassuccessful.
This conclusion is further supported by the surface elemen-tal composition data listed in Table 1 from XPS survey spectra. Aphosphorus concentration of 2.7% on the CS–PMA30 surface sug-gests that a 3–5 nm PMA30 layer was adsorbed on the CS surface.When the CS–PMA30 surface was covered by a layer of positivelycharged CS by LbL deposition, the surface phosphorus concen-tration decreased significantly to 1.4%, suggesting that a 3–5 nmCS layer was deposited (CS–PMA30–CS). The layer thickness wasestimated by comparing the bulk atomic concentration with themeasured one and assuming a maximum detection depth of 9 nmat a take off angle of 70◦ for polymers [33]. No silicon signal wasdetected in the XPS survey spectra, suggesting that the films coatedon the glass substrate were thicker than 10 nm.
The high resolution XPS spectra shown in Fig. 3 gave additionalinformation on the surface modifications. First, the new signals forP2p at 133 eV and –N+(CH3)3 at 402.8 eV showed the presence ofPC groups on the CS–PMA30 surface. Second, the decreased peakareas of P2p and –N+(CH3)3 on the CS–PMA30–CS sample confirmed
Surface elemental composition of CS and the LbL modified films measured by XPSsurvey scan.
Surface Atomic concentration (%)
C O N P P/C
CS 57.7 29.3 13.0 0 0CS–PMA30 62.5 30.1 4.7 2.7 4.3CS–PMA30–CS 61.6 31.1 5.9 1.4 2.3

M. Gong et al. / Colloids and Surfaces B: Biointerfaces 85 (2011) 48–55 51
spect
9wr
Fodnalaaoealto
3
w
FawB
Fig. 3. High resolution C1s, N1s, O1s and P2p XPS
nm. Fourth, the degree of deacetylation of the chitosan of 94%as confirmed by the –NCOCH3 peak area of 6% in the N1s high
esolution spectrum of the CS surface.Multilayers of CS/PMA were also fabricated by LbL deposition.
ig. 4 indicates that the deposition of PMA30 on CS surfaces as theutermost layer (odd numbers) resulted in a sharp decrease of theynamic contact angles, while for CS as the outermost layer (evenumbers) the contact angles increased. The variation of the contactngle with layer number showed clear alternation of the outermostayer between PMA30 and CS. The remarkable decrease of contactngles on the deposition of PMA (polyanion bearing PC groups)s the outermost layer indicates an increase in the hydrophilicityf the CS membrane. Based on both the contact angle and surfacelemental composition data, we conclude that all of the monolayernd multilayer modified CS surfaces with PMA as the outermostayer formed cell outer membrane mimetic structures, in whichhe zwitterionic PC groups were oriented to the surface/interfacef the polymer layer.
.2. Stability of PMA modified surfaces
The stability of CS–PMA monolayer surfaces in PBS solutionas investigated for up to 60 days. As shown in Fig. 5, �adv of the
ig. 4. Dynamic contact angle change with the number of coated layers of PMA30nd CS. Even numbers represent membranes with CS as the outermost layer,hereas odd numbers represent membranes with PMA30 as the outermost layer.ars represent SD (n = 3).
ra of the CS, CS–PMA, and CS–PMA–CS surfaces.
CS–PMA30 surface was stable in PBS solution for 40 days at roomtemperature. We know that self assembled monolayers formedby electrostatic interaction are very thin. XPS data (Table 1) sug-gest a thickness of 3–5 nm. A small loss or dissolution of PMApolymer from the CS surface would change the surface contactangles dramatically. As shown in Fig. 5, �adv of the CS–PMA50 andCS–PMA70 surfaces increased dramatically in 40 days. Further-more, slow hydrolysis or degradation of the CS substrate in PBSsolution is also suggested by the contact angle data. Therefore, thestable contact angles of the CS–PMA30 surface indicate that theelectrostatically assembled cell outer membrane mimetic structurewas stable.
The high stability of the CS–PMA30 surface was mainly due tothe strong electrostatic interaction between the positively chargedchitosan and negatively charged PMA. It is clear that the morecharges the polyions bear, the stronger the interaction between theoppositely charged polyelectrolytes. On the other hand, the zwit-terionic PC group does not enhance the attachment of PMA to CS,but weakens the interaction by “dragging” PMA molecules into thewater phase. Since PMA30 has both the largest amount of negativecharges and the smallest amount of hydrophilic PC groups among
the three PMA polymers, it forms the most stable assembly on CS(CS–PMA30) with a cell outer membrane mimetic structure. Con-sidering that complete prevention of protein adsorption requiresat least 25% of the PC moiety in the polyanion [21], PMA with aFig. 5. Advancing contact angle changes of CS–PMA monolayer surfaces with timeimmersed in PBS solution. Bars represent SD (n = 3).
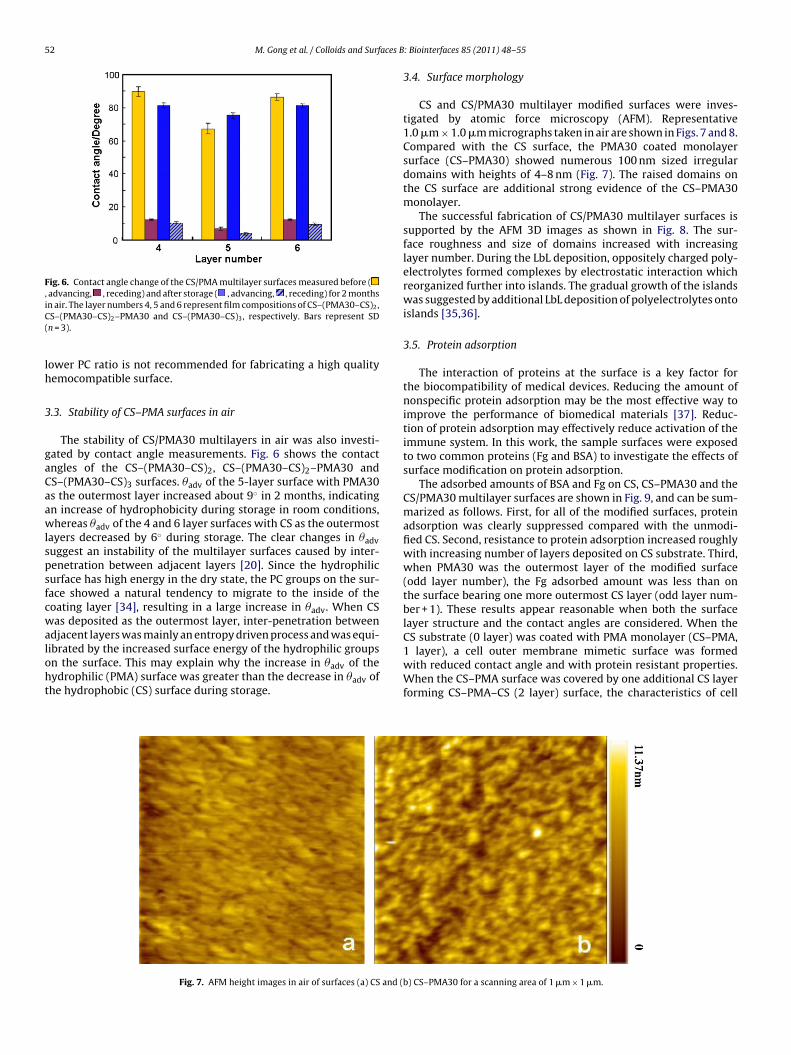
52 M. Gong et al. / Colloids and Surfaces B
Fig. 6. Contact angle change of the CS/PMA multilayer surfaces measured before (, advancing, , receding) and after storage ( , advancing, , receding) for 2 monthsiC(
lh
3
gaCaawlspsfcwaloht
CS substrate (0 layer) was coated with PMA monolayer (CS–PMA,
n air. The layer numbers 4, 5 and 6 represent film compositions of CS–(PMA30–CS)2,S–(PMA30–CS)2–PMA30 and CS–(PMA30–CS)3, respectively. Bars represent SDn = 3).
ower PC ratio is not recommended for fabricating a high qualityemocompatible surface.
.3. Stability of CS–PMA surfaces in air
The stability of CS/PMA30 multilayers in air was also investi-ated by contact angle measurements. Fig. 6 shows the contactngles of the CS–(PMA30–CS)2, CS–(PMA30–CS)2–PMA30 andS–(PMA30–CS)3 surfaces. �adv of the 5-layer surface with PMA30s the outermost layer increased about 9◦ in 2 months, indicatingn increase of hydrophobicity during storage in room conditions,hereas �adv of the 4 and 6 layer surfaces with CS as the outermost
ayers decreased by 6◦ during storage. The clear changes in �advuggest an instability of the multilayer surfaces caused by inter-enetration between adjacent layers [20]. Since the hydrophilicurface has high energy in the dry state, the PC groups on the sur-ace showed a natural tendency to migrate to the inside of theoating layer [34], resulting in a large increase in �adv. When CSas deposited as the outermost layer, inter-penetration between
djacent layers was mainly an entropy driven process and was equi-
ibrated by the increased surface energy of the hydrophilic groupsn the surface. This may explain why the increase in �adv of theydrophilic (PMA) surface was greater than the decrease in �adv ofhe hydrophobic (CS) surface during storage.Fig. 7. AFM height images in air of surfaces (a) CS and (
: Biointerfaces 85 (2011) 48–55
3.4. Surface morphology
CS and CS/PMA30 multilayer modified surfaces were inves-tigated by atomic force microscopy (AFM). Representative1.0 �m × 1.0 �m micrographs taken in air are shown in Figs. 7 and 8.Compared with the CS surface, the PMA30 coated monolayersurface (CS–PMA30) showed numerous 100 nm sized irregulardomains with heights of 4–8 nm (Fig. 7). The raised domains onthe CS surface are additional strong evidence of the CS–PMA30monolayer.
The successful fabrication of CS/PMA30 multilayer surfaces issupported by the AFM 3D images as shown in Fig. 8. The sur-face roughness and size of domains increased with increasinglayer number. During the LbL deposition, oppositely charged poly-electrolytes formed complexes by electrostatic interaction whichreorganized further into islands. The gradual growth of the islandswas suggested by additional LbL deposition of polyelectrolytes ontoislands [35,36].
3.5. Protein adsorption
The interaction of proteins at the surface is a key factor forthe biocompatibility of medical devices. Reducing the amount ofnonspecific protein adsorption may be the most effective way toimprove the performance of biomedical materials [37]. Reduc-tion of protein adsorption may effectively reduce activation of theimmune system. In this work, the sample surfaces were exposedto two common proteins (Fg and BSA) to investigate the effects ofsurface modification on protein adsorption.
The adsorbed amounts of BSA and Fg on CS, CS–PMA30 and theCS/PMA30 multilayer surfaces are shown in Fig. 9, and can be sum-marized as follows. First, for all of the modified surfaces, proteinadsorption was clearly suppressed compared with the unmodi-fied CS. Second, resistance to protein adsorption increased roughlywith increasing number of layers deposited on CS substrate. Third,when PMA30 was the outermost layer of the modified surface(odd layer number), the Fg adsorbed amount was less than onthe surface bearing one more outermost CS layer (odd layer num-ber + 1). These results appear reasonable when both the surfacelayer structure and the contact angles are considered. When the
1 layer), a cell outer membrane mimetic surface was formedwith reduced contact angle and with protein resistant properties.When the CS–PMA surface was covered by one additional CS layerforming CS–PMA–CS (2 layer) surface, the characteristics of cell
b) CS–PMA30 for a scanning area of 1 �m × 1 �m.

M. Gong et al. / Colloids and Surfaces B: Biointerfaces 85 (2011) 48–55 53
MA30
otec4wtasls
3
i
FP
Fig. 8. AFM 3D images of the LbL modified surfaces. (a) CS–PMA30; (b) CS–(P
uter membrane mimetic surface were decreased as shown byhe increased �adv, resulting in increased adsorption of Fg. How-ver, Fg adsorption on the 3-layered surface was further decreasedompared to that on the 1 layer surface and the increase on thelayer surface was insignificant. We conclude that Fg adsorptionas suppressed by both the cell outer membrane mimetic struc-
ure and increasing multilayer number. On the other hand, BSAdsorption did not show the same variations as Fg with surfacetructure, but decreased monotonically with increasing depositedayer number and showed the lowest adsorption after 3 layer depo-ition.
.6. Platelet adhesion
It is well known that when a material contacts blood, proteinsnstantaneously adsorb onto the surface and deform. Subsequently,
ig. 9. Protein adsorption on the surfaces modified with different numbers ofMA/CS layers. Bars represent SD (n = 3).
–CS)1–PMA30; (c) CS–(PMA30–CS)2–PMA30; (d) CS–(PMA30–CS)4–PMA30.
platelets adhere, are activated, and aggregate. The adhesion andaggregation of platelets play a significant role in thrombus for-mation [38]. Therefore, the extent of platelet adhesion and themorphology of the adhered platelets are considered to be early indi-cators of the thrombogenicity of biomaterials in contact with blood[39,40].
Fig. 10 shows the adhesion of platelets to CS, CS–PMA30 and theCS/PMA30 multilayer surfaces. Compared with the CS surface, thenumbers of platelets adhered to the CS–PMA30 and the CS/PMA30multilayer surfaces were significantly reduced, demonstrating theeffectiveness of the cell outer membrane mimetic surface in sup-pressing platelet adhesion, activation and aggregation. Plateletadhesion on the modified surfaces did not show fluctuation withsurface structure change.
It is well documented that Fg is the key plasma protein mediat-ing platelet adhesion under static conditions [41,42]. The adsorbedamount of Fg is an important factor influencing platelet adhe-sion. The results from Figs. 9 and 10 suggest that when Fgadsorption from single protein in buffer decreased to less than0.6 �g/cm2 platelet adhesion was effectively suppressed on themodified surfaces. This threshold value of Fg adsorption appearsmuch higher than the value that has been shown to cause sig-nificant platelet adhesion on poly(dimethylsiloxane) surfaces [43].This difference may be due to the conformational difference of theadsorbed Fg [44]. Our data indicate that the PMA30 coated sur-faces reduced Fg adsorption, and more importantly that they didnot change the protein conformation as much as other materialsmay have.
In summary, the data from contact angle, XPS, AFM, proteinadsorption and platelet adhesion experiments provided strong evi-dence for the successful fabrication of cell outer membrane mimeticsurfaces/interfaces with indications of excellent hemocompatibil-ity. In addition the cell outer membrane mimetic surfaces fabricated

54 M. Gong et al. / Colloids and Surfaces B: Biointerfaces 85 (2011) 48–55
F –PMAC
bhf
4
sececPtispPmdo
A
dfC
R
[[
[
[
[[[[[[[[
[
[
[[
[[[
[[[
ig. 10. Scanning electron micrographs of platelet adhesion on: (a) CS; (b) CSS–(PMA30–CS)2–PMA30.
y electrostatic interaction were stable for up to 40 days. It seems,owever, that development of a more stable biomimetic sur-
ace/interface for specific applications is still a challenge.
. Conclusions
A facile method of fabricating cell outer membrane mimeticurfaces was developed based on electrostatic assembly of poly-lectrolytes bearing phosphorylcholine groups on an oppositelyharged polymer surface. Water contact angles, XPS surfacelemental analysis, and AFM morphology data confirmed the suc-essful fabrication of these cell outer membrane mimetic surfaces.rotein adsorption and platelet adhesion investigations suggestedhat the hemocompatibility of the modified surfaces were signif-cantly improved. More specifically, Fg adsorption showed clearurface structure dependence, while BSA adsorption was sup-ressed mainly by the thickness or number of layers deposited.latelet adhesion was effectively suppressed on the cell outerembrane mimetic surface when the adsorbed amount of Fg, as
etermined in single protein experiments, was less than the thresh-ld value of 0.6 �g/cm2.
cknowledgements
This work was supported by the National Natural Science Foun-ation of China (No. 20774073, No. 20974087) and a open fundrom Key Laboratory of Synthetic and Natural Functional Moleculehemistry of Ministry of Education (No. KF09008).
eferences
[1] M. Thanou, B.I. Florea, M. Geldof, H.E. Junginger, G. Borchard, Biomaterials 23(2002) 153.
[2] W.-C. Lin, T.-Y. Liu, M.-C. Yang, Biomaterials 25 (2004) 1947.[3] J.C. Shiu, M. Ho, S. Yu, A. Chao, Y. Su, W. Chen, Z. Chiang, W.P. Yang, Carbohydr.
Polym. 79 (2010) 724.
[[[
[[
30; (c) CS–PMA30–CS; (d) CS–(PMA30–CS)1–PMA30; (e) CS–(PMA30–CS)2; (f)
[4] G.R. Ragetly, D.J. Griffon, H.-B. Lee, Y.S. Chung, J. Mater. Sci. Mater. Med. 21(2010) 2479.
[5] S. Sagnella, K. Mai-Ngam, Colloids Surf. B 42 (2005) 147.[6] S. Meng, Z.-J. Liu, L. Shen, Z. Guo, L.-L. Chou, W. Zhong, Q.-G. Du, J.-B. Ge, Bio-
materials 30 (2009) 2276.[7] A.-P. Zhu, S.-Q. Wang, Y.-L. Yuan, J. Shen, J. Biomater. Sci. Polym. Ed. 13 (2002)
501.[8] J.-P. Xu, X.-L. Wang, D.-Z. Fan, J. Ji, J.-C. Shen, J. Appl. Polym. Sci. 255 (2008)
538.[9] A.-P. Zhu, B. Shan, Y.-L. Yuan, J. Shen, Polym. Int. 52 (2003) 81.10] J.-P. Xu, J. Zhang, J. Shen, J. Appl. Polym. Sci. 88 (2003) 489.11] S. Meng, Z.-J. Liu, W. Zhong, Q.-H. Wang, Q.-G. Du, Carbohydr. Polym. 70 (2007)
82.12] P.-B. Huangfu, M. Gong, C.-F. Zhang, S. Yang, J. Zhao, Y.-K. Gong, Colloids Surf.
B 71 (2009) 268.13] M.J. Tiera, X.-P. Qiu, S. Bechaouch, Q. Shi, J.C. Fernandes, F.M. Winnik, Biomacro-
molecules 7 (2006) 3151.14] R. Zeng, H. Fu, Y.-F. Zhao, Macromol. Rapid Commun. 27 (2006) 548.15] T. Nakaya, Y.-J. Li, Prog. Polym. Sci. 24 (1999) 143.16] J. Watanabe, K. Ishihara, Colloids Surf. B 65 (2008) 155.17] M. Gong, S. Yang, S.-P. Zhang, Y.-K. Gong, Prog. Chem. 20 (2008) 1628.18] J. Desbrieres, V. Babak, Soft Matter 6 (2010) 2358.19] L.-H. Ma, C.-S. Liu, Colloids Surf. B 75 (2010) 448.20] G. Decher, Science 277 (1997) 1232.21] A. Reisch, J.-C. Voegel, E. Gonthier, G. Decher, B. Senger, P. Schaaf, P.J. Mesini,
Langmuir 25 (2009) 3610.22] F. Tristan, G. Palestino, J.-L. Menchaca, E. Perez, H. Atmani, F. Cuisinier, G. Ladam,
Biomacromolecules 10 (2009) 2275.23] P. Kujawa, G. Schmauch, T. Viitala, A. Badia, F.M. Winnik, Biomacromolecules 8
(2007) 3169.24] K. Ishihara, T. Ueda, N. Nakabayashi, Polym. J. 22 (1990) 355.25] A.L. Lewis, P.D. Hughes, L.C. Kirkwood, S.W. Leppard, R.P. Redman, L.A. Tolhurst,
P.W. Stratford, Biomaterials 21 (2000) 1847.26] D.-G. Yu, C. Jou, W.-C. Lin, M. Yang, Colloids Surf. B 54 (2007) 222.27] K.G. Tingey, J.D. Andrade, Langmuir 7 (1991) 2471.28] Y.-K. Gong, F. Mwale, M.R. Wertheimer, F.M. Winnik, J. Biomater. Sci. Polym.
Ed. 15 (2004) 1423.29] Y. Xu, M. Takai, K. Ishihara, Biomacromolecules 10 (2009) 267.30] T. Goda, T. Konno, M. Takai, T. Moro, K. Ishihara, Biomaterials 27 (2006) 5151.31] Y.M. Chen, M. Tanaka, J.P. Gong, K. Yasuda, S. Yamamoto, M. Shimomura, Y.
Osada, Biomaterials 28 (2007) 1752.
32] D.-G. Wang, J. Ji, C.-Y. Gao, G.-H. Yu, L.-X. Feng, Biomaterials 22 (2001) 1549.33] Y.-K. Gong, F.M. Winnik, Acta Chim. Sin. 63 (2005) 643.34] S. Yang, S.-P. Zhang, F.M. Winnik, F. Mwale, Y.-K. Gong, J. Biomed. Mater. Res.84A (2008) 837.35] K.-F. Ren, Y.-X. Wang, J. Ji, Q.-K. Lin, J.-C. Shen, Colloids Surf. B 46 (2005) 63.36] R. Advincula, M. Park, A. Baba, F. Kaneko, Langmuir 19 (2003) 654.

aces B
[[
[
[
[
M. Gong et al. / Colloids and Surf
37] H. Chen, L. Yuan, W. Song, Z.-K. Wu, D. Li, Prog. Polym. Sci. 33 (2008) 1059.
38] L.-J. Yu, X. Wang, X.-H. Wang, X.-H. Liu, Surf. Coat. Technol. 128–129 (2000)484.39] M. Tanaka, A. Mochizuki, N. Ishii, T. Motomura, T. Hatakeyama, Biomacro-
molecules 3 (2002) 36.40] Q. Yang, Z.-K. Xu, Z.-W. Dai, J.-L. Wang, M. Ulbricht, Chem. Mater. 17 (2005)
3050.
[
[
[
: Biointerfaces 85 (2011) 48–55 55
41] Y.-G. Wu, F.I. Simonovsky, B.D. Ratner, T.A. Horbett, J. Biomed. Mater. Res. 74A
(2005) 722.42] C.J. Nonckreman, S. Fleith, P.G. Rouxhet, C.C. Dupont-Gillain, Colloids Surf. B 77(2010) 139.
43] H. Chen, W. Song, F. Zhou, Z.-K. Wu, H. Huang, J.-H. Zhang, Q. Lin, B. Yang,Colloids Surf. B 71 (2009) 275.
44] B. Sivaraman, R.A. Latour, Biomaterials 31 (2010) 832.