familial cardiomegaly
TRANSCRIPT

J. clin. Path. (1959), 12, 355.
FAMILIAL CARDIOMEGALYBY
G. GARRETT,* W. J. HAY, AND A. G. RICKARDSFrom the Royal Lancaster Infirmary, Lancaster
(RECEIVED FOR PUBLICATION SEPTEMBER 18, 1957)
In 1949 William Evans gave the name offamilial cardiomegaly to what he believed to be a"distinct syndrome having a definite clinical,cardiographic, and pathological pattern." Theessential features were the familial incidenceof cardiomegaly without obvious cause and amarked tendency to arrhythmia and heart blockwith associated palpitation, giddiness, andsyncope. Death may be sudden or rapid due tothe development of left ventricular failure. In theelectrocardiogram the QRS complexes are oftenexceptionally wide and the T waves inverted.Conspicuous myocardial fibrosis and hypertrophyof the remaining muscle fibres are the strikingpathological findings. Both the degree ofconduction defect and the prognosis seem todepend on the extent of myocardial fibrosis andcardiac enlargement.
Evans referred to two previous accounts ofobscure cardiomegaly occurring in members ofthe same family published in the case records ofthe Massachusetts General Hospital in 1942 andby Addarii (1943) and Addarii, Martini, Mahaim,and Winston (1946). Subsequently cases havebeen published by Davies (1952), Parsons (1952),De Matteis and Ozzano (1954), Campbell andTurner-Warwick (1956), Gaunt and Lecutier(1956), and Paulley, Jones, Green, and Kane(1956). Among these, however, are included casesof cardiomegaly with no supporting familyhistory and others where there is good reasonto believe that the cardiac hypertrophy wassecondary to acquired valvular disease, congenitalcardiac malformations, or toxoplasmosis. In ourview the diagnosis of familial cardiomegalyshould only be made when other causes of cardiacenlargement have been excluded and when thereis a clear supporting family history. Moreover,in the present state of knowledge, it seems wiseonly to include those cases where the diagnosishas been established by necropsy study in at leastone affected member of the family. The reportsof seven affected families (15 cases) comply with*Present address: Department of Clinical Pathology, Manchester
Royal Infirmary.
these criteria and include eight necropsy studies.Only these cases are included in Table I and thesubsequent discussion.The purpose of this paper is to present a further
example of a family with cardiomegaly, the threeaffected members of which died suddenly. Post-mortem studies were made in all three casesalthough in one details of the necropsy findingsare no longer available.
Case HistoriesCase 1.-G. D., a man aged 30 years at death, came
home from work one evening in April, 1956, in hisusual good health, and while playing with his childrenin the garden suddenly collapsed and died. Therewas no history of rheumatic fever or other seriousillness in the past. Eight months before his death hesustained a slight injury to the right groin withhaematoma formation in the spermatic cord. Hiswife thought that he was slow to pick up after thataccident, but he eventually recovered, and his mother,who was with him on the day before he died,remarked that she thought he was in excellent health.At no time was he heard to complain of dyspnoea,chest pain, faintness, or palpitation.Necropsy performed by one of us (A. G. R.) on the
instructions of the coroner 21 hours after deathrevealed the following abnormalities.The body was that of a well-built young male with
a previous appendicectomy scar; significant macro-scopic findings were confined to the thoracic cavity.The heart was markedly enlarged (weight 500 g.), thehypertrophy mostly affecting the left ventricle, whichin parts was nearly 2 cm. thick and appeared at firstsight to be of the usual hypertensive type. On closerinspection it was seen that the outer part of themyocardium consisted of pale fibrous tissue almostgiving the impression of an outer shell surroundingthe inner layer (Fig. 1). Strands of fibrous tissuecould be seen penetrating the deeper aspects of themyocardium, but the predominantly peripheraldistribution was striking. The compact outer rim waswell demarcated from the rest of the myocardiumexcept in the region of the interventricular septumwhere its discrete pattern became replaced by diffusefibrotic mottling. The trabeculae carnae, papillarymuscles, and chordae tendineae showed no evidenceof fibrosis. The right ventricle and both auricles
copyright. on D
ecember 3, 2021 by guest. P
rotected byhttp://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.12.4.355 on 1 July 1959. Dow
nloaded from

G. GARRETT, W. J. HAY, anid A. G. RICKARDS
TABLE IMAIN CLINICAL FEATURES
AgeAuthor's ()
No. Author Case Age SexNo. at
Death----A_ , i l,X4|
FamilyRelationship
I1l (21) M Brother with con-gestive failure.Sister with car-diomegaly A-V
I (?) M
I (21) M
I (18) M
2 20 M
3 43 F
UIU6CK anaI conl-gestive failure
Brother of Case 3
Brother of Case 2.Mother diedC.C.F. and syn-cope
Son of Case 6.Brother of Case5
Son of Case 6.Brother of Case4
Mother of4 and 5.Three of her sib-lings believed
Symptoms Arrhythmias Heart Block
Dyspnoea,fatigue,congestivefailure
Dyspnoea,syncope
Dyspnoea,fatigue,syncope,congestivefailure
Giddiness.paroxysmaltachycardia
Giddiness
Syncope
affected -
8 (16) M Son of Case 8
p. 42 F Mother of Case 7 Paroxysmal,tachycardiawith occa-sionalsyncope
I (51) M Brother of Case I I Dyspnoea,syncope
2 23 F' Daughter of Case Arthralgia,1t1 slight
dyspnoea3 (531) F Mother of Case 10. Syncope,
Sister of Case 9 dyspnoea
(23) M Son of Case 13 Syncope,palpitation,dyspnoea,severe chestpain
2 (29) F Mother of Case 12l Dyspnoea,Eight of her 12 congestivesiblings died of failure,heart disease syncopeunder 40 years
3 (36) M Son of Case 15 Syncope
4 66 F Mother of Case 14. Faintness,Her sister, fatigue,mother, three dyspnoeaaunts, and grand-mother died ofheart disease
Complete A-Vblock
Auricularfibrillation
Auricular fibril-lation, parox-ysmal tachy-cardia
Paroxysmaltachycardia,extrasystoles
Auricular fibril-lation, parox-ysmal auricu-lar tachycardia
Auricular fibril-lation, extra-systoles
Sinus brady-cardia
Paroxysmaltachycardia
Auricularfibrillation
Paroxysmaltachycardia,ventricularextrasystoles
Complete A-Vblock, branclibundle block
Complete A-Vblock, branchbundle block
Branch bundleblock
Prolonged P-Rinterval,branch bundleblock
Comple:e A-Vblock
Length ofHistory Necropsyfrom IFindingsOnset of
Symptoms
6 months Yes
9 ,.
15 years ,,
2 tooiths
6 ,,
12 years
412 *s
2 months
20
Yes
Yes
3 ,,
Complete A-Vblock, branclhbundle block
Prolonged P-R,branch bundleblock
Branch bundleblock
Sinus and nodal Varying degreesbradycardia, of A-V block,nodal or ven- branch bundletricular extra- blocksystoles
Paroxysmal Branch bundleauricular blockflutter,sinusbradycardia
4 years -
3 years Yes
I year
6 months Yes
40 years
appeared unaffected, and the pericardium andendocardium were also macroscopically normal. Theaorta merely showed slight atheroma of its first part,and there was no evidence of coarctation. The lungswere engorged and the trachea and bronchi containeda large amount of mucus; all the other organs,including the brain and spinal cord, appeared normalto the naked eye.
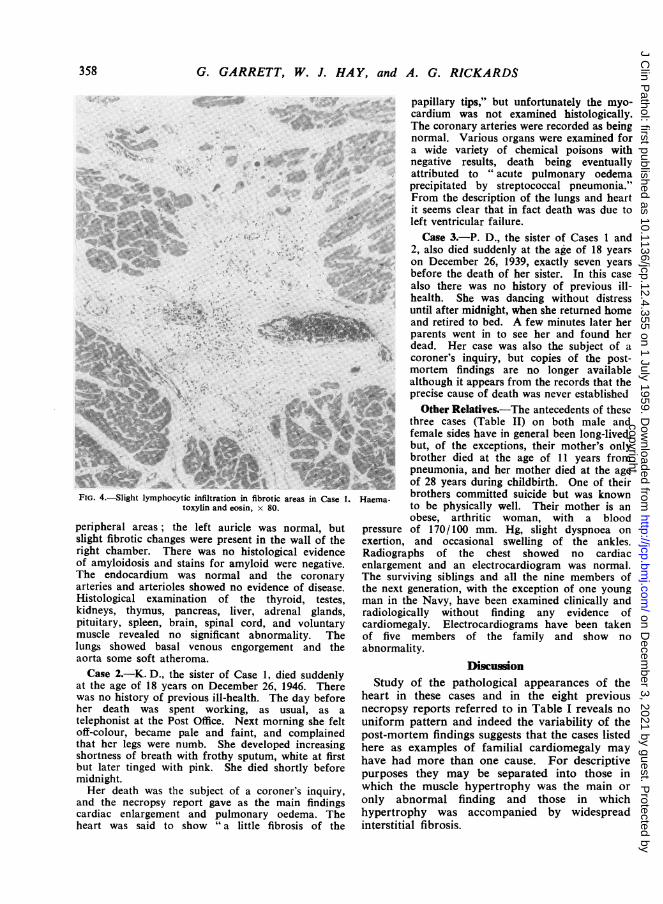
Microscopic examination of the left ventricleshowed marked hypertrophy of individual musclefibres associated with gross fibrosis of the
myocardium distributed peripherally and correspond-ing to the area seen with the naked eye. The fibrousstrands tended to separate the muscle fibres intobundles (Fig. 2) and many of these isolated lobules ofmuscle showed degenerative changes similar to thoseseen in early cell death following ischaemia (Fig. 3);slight infiltration with lymphocytes was seen inoccasional areas of fibrosis (Fig. 4). The rightventricle showed a similar pattern of interstitialfibrosis, although of lesser extent, and its distributionwas more irregular and was not confined to the
356
Case recordsof M.G.Hosp.(1942)
Addarrii et al.(t943)
Addarii et al.((1946)
Evans (1949)
Gaunt andLecut ier(1956)
1,* .
Campbell andTurner-Warwick(1956)
,. ..1
Ap
2
2
3
4
5
6
7
8
9
10
11
12
13
14
15
...:,t-
copyright. on D
ecember 3, 2021 by guest. P
rotected byhttp://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.12.4.355 on 1 July 1959. Dow
nloaded from

FAMILIAL CARDIOMEGALY
FIG.
cv,
12"IN~~~~~~*.*
~~~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ w
tFG.
FIG. 3
FIG. I.-Cross section of left ventricular wall showing peripheralfibrotic mottling in Case 1.
FIG. 2.-Trabeculated fibrosis of the left ventricle in Case 1. Haema-toxylin and eosin, x 60.
FIG. 3.-Degenerative change in muscle fibres surrounded by fibroustissue in Case 1. Haematoxylin and eosin, x 150.
357
*4::::4,
copyright. on D
ecember 3, 2021 by guest. P
rotected byhttp://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.12.4.355 on 1 July 1959. Dow
nloaded from
G. GARRETT, W. J. HAY, and A. G. RICKARDS.......... ... ..
FIG. 4.-ight Iymphocytic infiltration in fibrotic areas in Ca
toxylin and eosin, x 80.
peripheral areas; the left auricle was normal, tslight fibrotic changes were present in the wall ofright chamber. There was no histological eviderof amyloidosis and stains for amyloid were negatiThe endocardium was normal and the coronaarteries and arterioles showed no evidence of diseaHistological examination of the thyroid, testkidneys, thymus, pancreas, liver, adrenal glanipituitary, spleen, brain, spinal cord, and voluntamuscle revealed no significant abnormality. Tlungs showed basal venous engorgement and taorta some soft atheroma.Case 2.-K. D., the sister of Case 1, died sudder
at the age of 18 years on December 26, 1946. Th4was no history of previous ill-health. The day befcher death was spent working, as usual, astelephonist at the Post Office. Next morning she foff-colour, became pale and faint, and complainthat her legs were numb. She developed increasishortness of breath with frothy sputum, white at fibut later tinged with pink. She died shortly bef(midnight.Her death was the subject of a coroner's inqui
and the necropsy report gave as the main findircardiac enlargement and pulmonary oedema. Theart was said to show "a little fibrosis of t
papillary tips," but unfortunately the myo-cardium was not examined histologically.The coronary arteries were recorded as beingnormal. Various organs were examined fora wide variety of chemical poisons withnegative results, death being eventuallyattributed to " acute pulmonary oedemaprecipitated by streptococcal pneumonia."From the description of the lungs and heart
41it seems clear that in fact death was due toleft ventricular failure.
Case 3.-P. D., the sister of Cases 1 and2, also died suddenly at the age of 18 years
* on December 26, 1939, exactly seven yearsbefore the death of her sister. In this casealso there was no history of previous ill-
= ; health. She was dancing without distressuntil after midnight, when she returned homeand retired to bed. A few minutes later herparents went in to see her and found her
. dead. Her case was also the subject of at coroner's inquiry, but copies of the post-
mortem findings are no longer availablealthough it appears from the records that the
17INK precise cause of death was never establishedX>* Other Relatives.-The antecedents of these
three cases (Table II) on both male and¶! < e:^ xv female sides have in general been long-lived,
but, of the exceptions, their mother's only- brother died at the age of 11 years from
pneumonia, and her mother died at the age>>}R<z of 28 years during childbirth. One of their
,se 1. Haema- brothers committed suicide but was knownto be physically well. Their mother is anobese, arthritic woman, with a blood
but pressure of 170/100 mm. Hg, slight dyspnoea onthe exertion, and occasional swelling of the ankles.ice Radiographs of the chest showed no cardiacve. enlargement and an electrocardiogram was normal.iry The surviving siblings and all the nine members ofLse. the next generation, with the exception of one youngtes, man in the Navy, have been examined clinically andds, radiologically without finding any evidence ofiry cardiomegaly. Electrocardiograms have been taken'he of five members of the family and show nothe abnormality.
Discussionnlrye Study of the pathological appearances of theDre heart in these cases and in the eight previousa necropsy reports referred to in Table I reveals no
Felt uniform pattern and indeed the variability of theled post-mortem findings suggests that the cases listedng here as examples of familial cardiomegaly mayrst have had more than one cause. For descriptiveire purposes they may be separated into those inry, which the muscle hypertrophy was the main or
ngs only abnormal finding and those in which'he hypertrophy was accompanied by widespreadthe interstitial fibrosis.
358
copyright. on D
ecember 3, 2021 by guest. P
rotected byhttp://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.12.4.355 on 1 July 1959. Dow
nloaded from

FAMILIAL CARDIOMEGALY
TABLE IIFAMILY TREE
(11)4
38
3 9 6 21 12 20 6 8 5
4 t Affected ( ) Age at death
Only two cases fall into the first category,namely, Cases 1 and 14 (Table I). In the formercase the heart weighed 600 g. and was grosslydilated, being described by Dr. Tracy Mallory as
"one of the largest hearts from the point ofview of capacity that we have ever seen." Allthe chambers shared in the hypertrophy, butthere was no other microscopic abnormality ofthe myocardium. In Campbell and Turner-Warwick's case the heart weighed 1,050 g., chieflydue to hypertrophy of the left ventricle whichmeasured up to 25 mm. in thickness. Histologic-ally there was simple hypertrophy of musclefibres some of which showed central vacuolation;the nature of the vacuolar material was notestablished, but stains for fat and glycogen were
negative.The remaining cases belong to the second
fibrotic group (Cases 2, 3, 4, 7, 9, and 12 in TableI and our own Case 1). Moderate or markedcardiac hypertrophy was present in all, the lowestheart weight recorded being 737 g. (Case 9)and the highest 1,134 g. (Case 4). Extensivemyocardial fibrosis was present in all cases
although the precise distribution of the fibroustissue is not always recorded. In most the fibrosisaffected both right and left ventricular walls, butin few was there any reference to the histological
appearance of the auricles. In no case has therebeen found any macroscopic or microscopicevidence of disease of the coronary arteries.Occasional focal collections of chronic inflam-matory cells have been noted in two cases (Cases12 and 3) similar to the occasional foci present inthe first case described here; Case 3 also showedpatchy necrosis of muscle fibres similar to thatoccurring in our Case 1. Two of the cases in thisfibrotic group (4 and 9) have shown unusuallymarked deposits of glycogen within the musclefibres, although similar in other respects to theremainder of the group.
Consideration of these pathological typessuggests a fundamental difference between thoseshowing extensive myocardial fibrosis and thosein which the pathological picture is one of simplehypertrophy. Despite the not dissimilar clinicalfeatures it seems unlikely that these are differentstages of the same disease process or indeed thatthey have any aetiological relationship. The twotypes have not been found in the same family.The picture of the fibrotic type appears to be
that of replacement fibrosis and survival hyper-trophy. Previous episodes of myocarditis whichmight account for this picture have naturally beensought in case histories. Rheumatic infection isrecorded in only three cases (11, 12, and 13), in
359
copyright. on D
ecember 3, 2021 by guest. P
rotected byhttp://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.12.4.355 on 1 July 1959. Dow
nloaded from

G. GARRETT, W. J. HA Y, and A. G. RICKARDS
two of which the diagnosis was doubtful; Aschoffnodes were not found in the only case which cameto necropsy. In the remaining cases there is nohistory of illness suggesting Fiedler's or othernon-rheumatic myocarditis.
Paulley, Jones, Green, and Kane (1956) have,however, described several cases of fibroticcardiomegaly which they attributed, on the basisof positive serological reactions, to toxoplasmosis.In one family, at least three of the four casescoming to necropsy showed a fibrotic cardio-megaly similar to that described above, althoughatypical in that the left ventricle was notpredominantly affected and mural thrombus andembolism was a marked feature. Moreover, twoof the four cases had enlarged firm spleens.Clinically their cases bore less resemblance tofamilial cardiomegaly, the absence of syncopebeing particularly noteworthy. Paulley et al.(1956) were unable to demonstrate toxoplasmabodies in the myocardium of their cases andpositive serological reactions cannot be consideredas conclusive evidence of active toxoplasmabodies from a case of obscure myocarditis inwhich evidence of generalized toxoplasmosis waslacking, but toxoplasma infection may well havebeen the aetiological factor in the cases of Paulleyet al. In view of their findings we obtainedsamples of serum for toxoplasma antibodytitration from a brother, son, and wife of ourCase 1 and in no instance was a significantantibody titre recorded.The finding of glycogen deposits in the muscle
fibres in Cases 4 and 9 raises the question as towhether some cases of familial cardiomegaly maybe due to a disorder of glycogen metabolism. Ina few cases of Von Gierke's disease, glycogeninfiltration has been confined to the heart, so-called "cardiomegalia glycogenica," but diSant'Agnese, Andersen, and Mason (1950), whohave recently reviewed this subject, consider that afatal termination within the first year of life isinvariable in this condition, and, moreover, thosecases of glycogen storage disease in which theheart is involved do not progress to myocardialfibrosis. Russell (1948), in trying to assess thesignificance of the presence of muscle glycogendeposits in post-mortem tissue, examinedspecimens of muscle from 11 necropsy cases takenat varying intervals after death; although she wasable to demonstrate glycogen in the muscle fibresthe infiltration was scanty compared with thatfound in the first case described by Evans (1949).The exact significance of these glycogen depositsis obscure, but the mere presence of glycogenwithin the myocardium does not provide a basis
for the diagnosis of glycogen disease. In the lightof our present knowledge it seems more reason-able to regard the deposits as a non-specificinfiltration which may complicate the pathologicalpattern of the fibrotic form of familialcardiomegaly.A further possible aetiological factor is the
curious association between cardiomegaly andtwo familial neurological conditions, Friedreich'sataxia and progressive muscular dystrophy. Theassociation of myocarditis with Friedreich's ataxiawas first recorded by Pitt (1886-7). Severalreports later appeared in the Continentalliterature and in 1946 Russell reported fourexamples of the condition with post-mortemdetails; not infrequently the neurological lesionis accompanied by a chronic progressivemyocarditis of fibrotic type which closelyresembles that found in the fibrotic form offamilial cardiomegaly. It was Russell's opinionthat the heart muscle is destroyed through a focal,piecemeal, coagulative necrosis as a result ofwhich the fibres are ultimately replaced bycollagenous tissue, the surviving muscle under-going a compensatory hypertrophy. Thisdescription could well be applied to thehistological appearances in the cases of familialcardiomegaly now described, although, as Evans(1949) comments, " the relation of this conditionto Friedreich's disease would be more definitelyestablished if instances of each were met with inone family." Roth (1948) described a familyaffected by Friedreich's disease and peronealmuscular dystrophy in which there was a highincidence of heart disease, but there was noevidence to suggest that they were cases offamilial cardiomegaly. Some 30 cases of cardiacinvolvement have been reported in progressivemuscular dystrophy and the subject has recentlybeen reviewed by Bevans (1945) and Storstein andAustarheim (1955). The post-mortem appear-ances of the heart in these cases seem to show avery variable picture; sometimes the hearts areatrophic and in other cases marked hypertrophyis found. Macroscopically fatty infiltration andintracellular vacuolation of the fibres has beendescribed; in several cases a patchy fibrosis,often maximal in the outer myocardium, has beenpresent. This association of hereditary neuro-pathies and fibrotic cardiomegaly has suggestedto some that the cardiopathies in the familial andneuropathic varieties may have a commonaetiology which may lie in an as yet undetectedinborn error of metabolism. Support for this" chemical theory " of diffuse cardiac fibrosis isreinforced by the knowledge that diffuse
360
copyright. on D
ecember 3, 2021 by guest. P
rotected byhttp://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.12.4.355 on 1 July 1959. Dow
nloaded from

FAMILIAL CARDIOMEGALY
myocarditis may follow the ingestion of a varietyof chemical compounds including arsenicals,sulphur, and sulphonamides (Elster, Horn, andTuchman, 1955), and recently McAllen (1955) hasobserved severe myocardial changes, includingwidespread fibrosis, supervene in two cases inwhich there was prolonged depletion in potassiumabsorption. These human lesions were strikinglysimilar to the cardiac lesions found in potassium-deficient animals.
In conclusion it must be admitted that in spiteof the numerous theories of origin, the aetiologyof familial cardiomegaly still remains obscure.
SummaryA family with cardiomegaly is described in
which the three affected members died suddenlywhen apparently in good health and weresubmitted to necropsy.The clinical and pathological features of these
and previously published cases of familialcardiomegaly are discussed and the possibleaetiology of the condition considered.
Our thanks are due to Mr. L. G. Powell, H.M.Coroner for South Westmorland, for makingavailable previous post-mortem reports.
REFERENCESAddarii, F. (1943). Boll. Sci. med., 21, 41.
Martini, L., Mahaim, I., and Winston, M. (1946). Cardiologia(Basel), 11, 36.
Bevans, M. (1945). Arch. Path. (Chicago), 40, 225.Campbell, M., and Turner-Warwick, M. (1956). Brit. Heart J., 18,
393.Case Records of Massachusetts General Hospital (1942). New Engl.
J. Med., 226, 158.Davies, L. G. (1952). Brit. Heart J., 14, 206.De Matteis, F., and Ozzano, T. (1954). Minerva med. (Torino), 45,
(1), 1549.di Sant'Agnese, P. A., Andersen, D. H., and Mason, H. H. (1950).
Pediatrics, 6, 607.Elster, S. K., Horn, H., and Tuchman, L. R. (1955). Amer. J. Med.,
18,900.Evans, W. (1949). Brit. Heart J., 11, 68.Gaunt, R. T., and Lecutier, M. A. (1956). Ibid., 18, 251.McAllen, P. M. (1955). Ibid., 17, 5.Parsons, P. J. (1952). Med. J. Aust., 2, 435.Paulley, J. W., Jones, R., Green, W. P. D., and Kane, E. P. (1956).
Brit. Heart J., 18, 55.Pitt, G. N. (1887). Guy's Hosp. Rep., 44, 369.Roth, M. (1948). Brain, 71, 416.Russell, D. S. (1946). J. Path. Bact., 58, 739.- (1948). Quoted by Evans (1949).Storstein, O., and Austarheim, K. (1955). Acta med. scand., 150,431.
2K
361
copyright. on D
ecember 3, 2021 by guest. P
rotected byhttp://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.12.4.355 on 1 July 1959. Dow
nloaded from