fast identification and statistical evaluation of segmental homologies in comparative maps peter...
Post on 21-Dec-2015
212 views
TRANSCRIPT


Fast identification and statistical evaluation of segmental
homologies in comparative maps
Peter Calabrese1, Sugata Chakravarty2 and Todd Vision3
1Department of Mathematics, University of Southern California; Departments of 2Operations Research and 3Biology
University of North Carolina at Chapel Hill
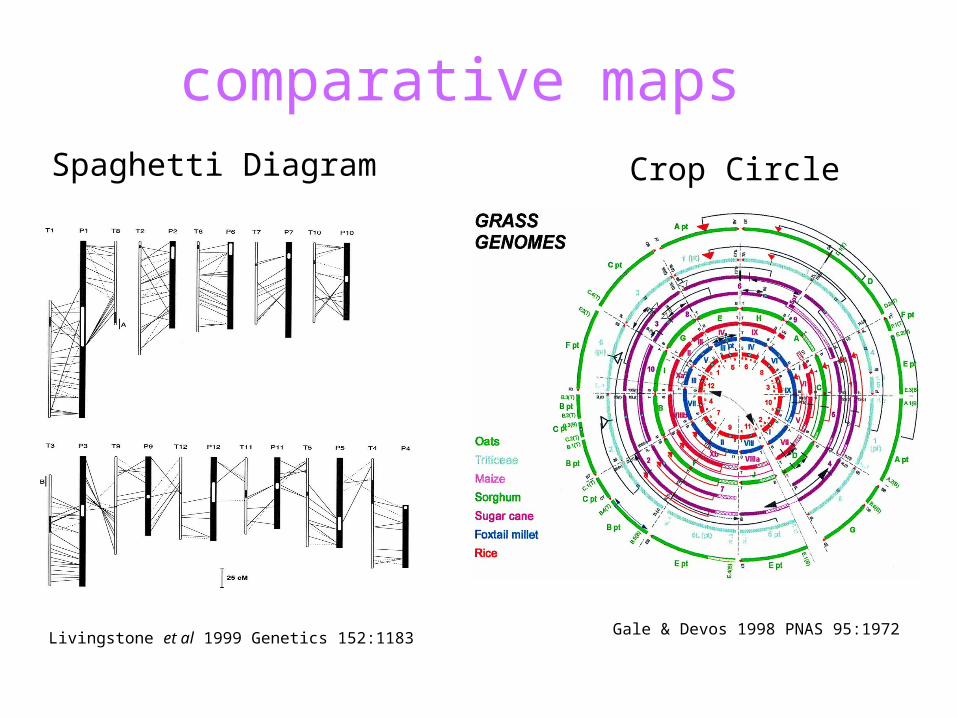
comparative maps Spaghetti Diagram Crop Circle
Livingstone et al 1999 Genetics 152:1183Gale & Devos 1998 PNAS 95:1972

some terms
• Feature: a gene or some other marker
• Segment: a string or substring of features
• Homology: descent from a common ancestor
• Block: a pair of segments that are putatively homologous. These are what we seek!

local genome alignment
• Consider each chromosome to be a string of features
• Assign common letters to homologous features • Identify segments sharing multiple pairs of
common letters• Differences from DNA/protein alignment
– high frequency of gaps relative to matches– inversions may occur within the alignment
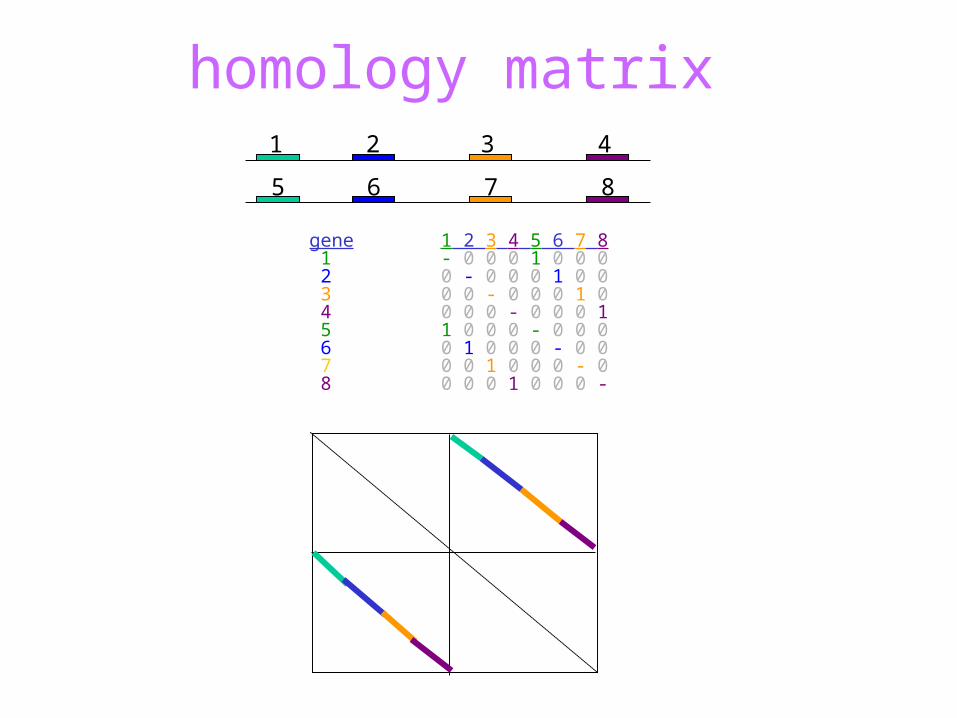
homology matrix
gene 1 2 3 4 5 6 7 8 1 - 0 0 0 1 0 0 0 2 0 - 0 0 0 1 0 0 3 0 0 - 0 0 0 1 0 4 0 0 0 - 0 0 0 1 5 1 0 0 0 - 0 0 0 6 0 1 0 0 0 - 0 0 7 0 0 1 0 0 0 - 0 8 0 0 0 1 0 0 0 -
1 2 3 4
5 6 7 8

duplication and multiplication
there is not necessarily a one-to-one alignment

genome rearrangements
inversion reciprocal translocation
homologous segments may be small

Bancroft (2001) TIG 17, 89 after Ku et al (2000) PNAS 97, 9121
Non-homologous features may be abundant within homologous segments

We must allow some non-colinearityin marker order between segmental
homologs
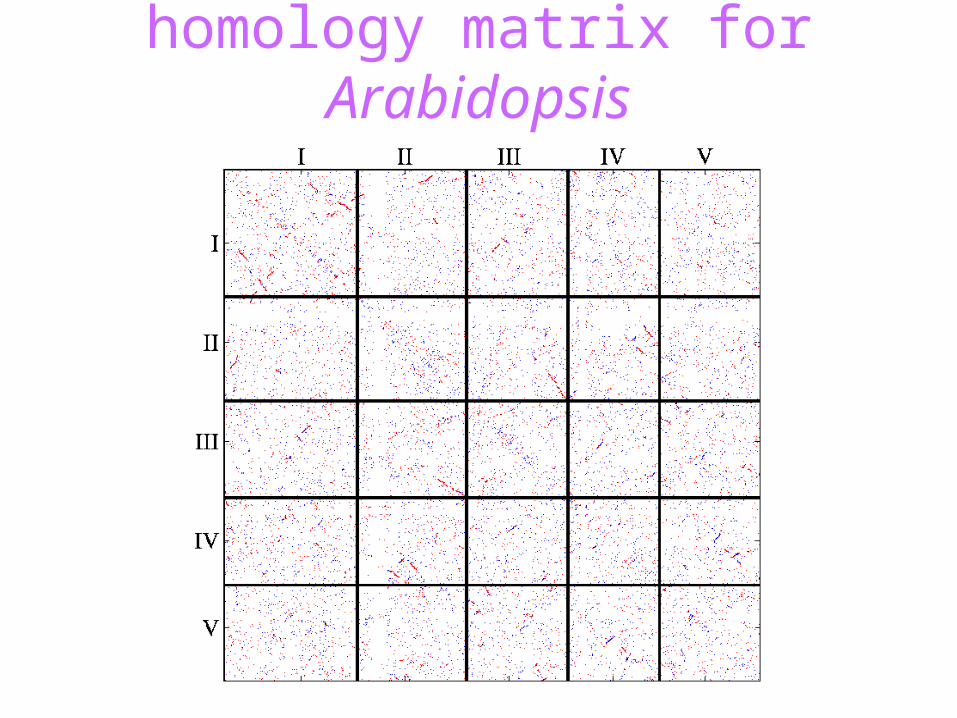
homology matrix for Arabidopsis

going beyond eyeballing
• LineUp – Hampson et al (2003)– Designed for genetic maps with error
• ADHoRe – Van der Poele (2002)– Designed for unambiguous marker order data
• Both provide automatic detection of blocks• For statistics, both employ permutation tests
– Computationally intensive– p-values are approximate

FISH:Fast Identification of Segmental Homology
• Block identification– Dynamic programming provides speed and
optimality guarantee– Can be generalized to multiple alignments
• Statistical assessment– Null model of duplication and transposition– Closed-form equation for calculating p-
values (i.e. no permutation testing)

from homology matrix to graph
• nodes ()– represent dots in the homology
matrix

from homology matrix to graph
• nodes ()– represent dots in the homology
matrix
• edges ()– connect nodes with nearest
neighbors– are unidirectional– have an associated distance– must be shorter than some
threshold

from homology matrix to graph
• nodes ()– represent dots in the homology matrix
• edges ()– connect nodes with nearest neighbors– are unidirectional– have an associated distance– must be shorter than some threshold
• paths ()– traverse shortest available edges– can be efficiently computed– can be considered candidate blocks

null model
• Within a genome: homologies are due to the duplication of individual features followed by insertion into a (uniformly) random position
• Between genomes: homologies are due to the above process plus the transposition of randomly chosen features into randomly chosen positions.

computing neighborhood size
• h = # nodes / # cells in matrix• n = # cells in neighborhood• Prob(neighborhood has 1 node)
p = 1 – (1-h)n
• Threshold distance for p=T under Manhattan distance (x+y)dT = 0.5 + sqrt[(log(1-p)/log(1-h)+0.25]
• T is analogous to a gap parameter– small T: few false positive edges, short blocks– large T: more false positive edges, longer blocks

neighborhood geometry

blocks of nearest neighbors

block statistics
• Chen-Stein Theorem: number of blocks with k nodes is approximately Poisson
• Expected number of blocks = cpu
• Conservative matrix-wide p-value
Prob(X k) < 1 – e –cpu
where c is the # of cells in the matrix and pu = h(nh)k-1

identifying blocks
• Let edge from i to j have weight wij =1
• Initialize: score of block terminating at i Si = 0
• Recursion for block scores Sj = max(Si + wij) i such that j Ti
• Dynamic programming can be used to find all maximally extended blocks

simulation experiment
k obs stderr upbound lowbound2 45.8 0.06 47.6 40.13 2.28 0.02 2.39 1.784 0.113 0.003 0.120 0.0795 0.006 0.001 0.006 0.0046 0.0003 0.0002 0.0003 0.0002
How often are blocks of size k observed under the null model compared with
expectation?

FISH v.1.0
• http://www.bio.unc.edu/faculty/vision/lab/FISH– source code – compiled executables– documentation– sample data
• Adjustable parameters (e.g. T)• Reports statistics on blocks• Is fast and memory-efficient

applications
• Automated pairwise alignment of genome maps as part of Phytome project
• Prediction of gene content in regions of unsequenced genomes
• Studies of genome evolution, especially duplication and gene order rearrangement

future work
• Biologically motivated neighborhood geometries (ADHoRe)
• Non-discrete marker positions (LineUp)– Genetic versus physical maps– Map uncertainty
• Robustness to deviation from null model (permutation tests)
• Extension to homologies among 3 or more segments

Thanks!
Sugata Chakravarty
Peter Calabrese
U.S. National Science Foundation

http://www.bio.unc.edu/vision/faculty/lab/FISH

Ghosts
Simillion, Vandepoele, Van Montagu, Zabeau, Van de Peer (2002) PNAS 99, 13627