filippo tesi phd completa - [email protected]/2736/1/filippo_zanetti.pdf ·...
TRANSCRIPT

1
INDEX
ABSTRACT ......................................................................... 7
RIASSUNTO ....................................................................... 9
INTRODUCTION .............................................................. 11
Prions and prion diseases ........................................................................ 11 The cellular prion protein (PrPC) ........................................................... 12 The scrapie prion protein (PrPSc) and its conversion from PrPC 15 PrPC-PrPSc conversion: the gain-of-function hypothesis ......... 15 PrPC-PrPSc conversion: the loss-of-function hypothesis .......... 16
Biological functions of PrPC ..................................................................... 17 Oxidative stress .......................................................................................... 21 ROS mediated oxidative damage ......................................................... 24 PrPC and oxidative stress ......................................................................... 25 The Prion Protein and muscular tissue .............................................. 27 The heart and ischemia/reperfusion injury ...................................... 28 Ischemia and reperfusion injury ....................................................... 29 Functional and metabolic modifications in ischemic heart ..... 29 Reversible and non-reversible injury .............................................. 30 The reperfusion injury and the ROS damage .............................. 30 Ischemic Preconditioning ..................................................................... 32
MATERIALS AND METHODS ............................................. 35
Mouse models .............................................................................................. 35 The Langendorff model and protocols for the perfusion of isolated hearts ............................................................................................. 36 Perfusion Protocols .................................................................................... 37 H2O2 titration ................................................................................................ 39 Estimation of LDH release ....................................................................... 39 Preparation of mitochondria from mouse hearts ........................... 41 SDS-PAGE and Western blotting .......................................................... 42 Sample preparation ............................................................................... 42 Deglycosylation with peptide N-glycosidase F ............................ 42 SDS-PAGE .................................................................................................. 42 Western blotting ..................................................................................... 43 Antibodies .................................................................................................. 43 Estimation of tropomyosin oxidation .............................................. 44
In situ superoxide detection .................................................................. 44 Enzymatic activity assays ....................................................................... 46 Superoxide dismutase activity assay ............................................. 46 Catalase activity assay ......................................................................... 46

2
AIMS AND RATIONALE.................................................... 47
RESULTS ......................................................................... 49
Evaluation of the myocardial damage induced by I/R protocols in isolated hearts with different PrPC levels ...................................... 50 Hearts isolated from PrPC-OE mice are protected against loss of viability induced by post-ischemic reperfusion ...................... 50 The over-expression of PrPC reduces the degree of oxidative stress caused by post-ischemic reperfusion ................................ 52
PrPC performs anti-oxidant functions in the heart ......................... 55 The absence of PrPC enhances the protective effects of ischemic preconditioning...................................................................... 55 PrPC protects the heart from non-ischemic oxidative injury .. 58
Evaluation of the expression and/or activity of proteins involved the oxidative response, in hearts with different PrPC levels ................................................................................................................ 62 The enzymatic activity of CAT is diminished in PrPC–KO hearts .......................................................................................................... 62 Hearts with different PrPC levels have no difference in superoxide dismutase activities and expression ........................ 63 p66Shc expression is increased in PrPC–KO hearts ..................... 65
The fate of PrPC during and after the ischemic and oxidative challenges ...................................................................................................... 69 PrPC levels are decreased after I/R, but not after ischemia alone, in WT and PrPC-OE hearts ...................................................... 69 PrPC levels are preserved when I/R is preceded by IPC .......... 71 PrPC levels are largely reduced after perfusion with H2O2 ...... 72 Which is the fate of myocardial PrPC during post-ischemic reperfusion, or perfusion with H2O2? .............................................. 73
CONCLUSIONS AND PERSPECTIVES ................................ 77
BIBLIOGRAPHY .............................................................. 85
AKNOWLEDGEMENTS .................................................... 101

3
ABBREVIATIONS
.HO-: hydroxil radical
.O2-: superoxide anion
a.a.: aminoacids
ab: antibody
ADP: adenosine diphosphate
AMP: adenosine monophosphate
Asn: asparagine
ATP: adenosine triphosphate
BCA: bicinchoninic acid (protein assay)
bpm: beats per minute
BSA: bovine serum albumin
BSE: bovine spongiform encephalophathy
CAM: cell adhesion molecules
cAMP: cyclic adenosine monophosphate
CAT: catalase
CJD: Creutzfeldt Jacob Disease
CK: creatine-kinase
CNS: central nervous system
CWD: chronic wasting disease
Cys: cystein
DCB: disulphide cross-bridges
DMSO: dimethyl sulfoxide
DNA: deoxyribonucleic acid
Dpl: Doppel
DTT: dithiothreitol
ECL: Enhanced ChemiLuminescence
ECM: extra cellular matrix
EDTA: ethylenediaminetetraacetic acid
EGTA: ethylene glycol tetraacetic acid
ER: endoplasmic reticulum
Erk: extracellular signal-regulated kinases
FFI: fatal familial insomnia

4
GAGs: glycosamminoglycans
GPI: glycosyl-phosphatidylinositol
GSH: glutathione
GSS: Gerstmann-Sträussler-Scheinker
H2O2: hydrogen peroxide
HE: hydroethidium
His: histidine
HRP: horse radish peroxidase
I/R: ischemia/reperfusion
IPC: ischemic preconditioning
IS: isolation solution
KO: knock-out
LDH: lactate dehydrogenase
Mab: monoclonal antibody
MAPK: mitogen-activated protein kinases
MMPs: matrix metallo-proteases
MPG: N-2-mercaptopropionyl-glycine
mRNA: messenger ribonucleic acid
MW: molecular weight
NAD+: nicotinamide adenine dinucleotide (oxidized form)
NADH: nicotinamide adenine dinucleotide (reduced form)
NEM: N-ethylmaleimide
NMR: Nuclear Magnetic Resonance
NO: nitric oxide
NP-40: nonyl phenoxylpolyethoxylethanol
OE: over-expressing
ON: over-night
OONO-: peroxynitrite
ORF: open reading frame
Pab: polyclonal antibody
PB: perfusion buffer
PBS: phosphate buffer saline
PBS-T: phosphate buffered saline Tween-20
PC: phosphate-creatine
PI3K: phosphoinositide 3-kinases
PK: proteinase K
PKA: protein kinase A

5
PKC: protein kinase C
PM: plasma membrane
PNGase-F: peptide N-glycosidase F
Prnp: prion protein gene
PrP: prion protein
PrPC: cellular prion protein
PrPSC: scrapie prion protein (infective isoform of PrP)
RNA: ribonucleic acid
ROS: reactive oxygen species
RT: room temperature
SB: sample buffer
SDS: sodium dodecyl-sulphate
SDS-PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis
SERCA: sarco/endoplasmic reticulum Ca2+-ATPase
SOD: superoxide dismutase
SOD1: Cu/Zn SOD
SOD2: Mn SOD
SP: signal peptide
STI1: stress inducible factor 1
TEMED: tetramethylethylenediamine
Tg: transgenic
Tm: tropomyosin
TNF: tumor necrosis factor
Tris: tris(hydroxymethyl)aminomethane
TSE: transmissible spongiform encephalopathies
Tyr: tyrosine
w/v: weight/volume
WT: wild type

6

7
ABSTRACT
The elusive function of the cellular prion protein (PrPC) hampers the
understanding of the molecular mechanism at the basis of prion diseases, and
the development of suitable therapeutic protocols. Use of cell model systems,
and genetically modified animals, have nevertheless suggested a number of
potential roles for the protein, ranging from protecting against oxidative
stress to cell differentiation. Because we now know that muscle is involved in
PrPC pathophysiology, we have considered intact heart paradigms for the in
situ study of the cell-protecting function of PrPC.
Isolated muscle organs retain the cell native environment and are also more
suitable to experimental designs than whole animals. Accordingly, by taking
advantage of mice expressing different PrPC amounts (wild type (WT), knock-
out (KO) and overexpressors (OE)), the protection of PrPC against cell
oxidative injuries was investigated in isolated hearts subjected to
ischemia/reperfusion and perfusion protocols that involve oxidative stress. In
line with the putative capability of PrPC to antagonize oxidative injury and cell
death mechanisms, our prediction was that hearts from PrPC-KO adult mice
manifest an overt phenotype after ischemic challenge, resulting in
exacerbation of heart oxidative damage. Conversely, PrPC-OE mice should
demonstrate a higher resistance over reactive oxygen species (ROS)
production.
We found that PrPC-OE hearts were more protected from the damage induced
by post-ischemic reperfusion than WT and PrPC-KO hearts, as indicated by
reduced cell death and decreased oxidation of myofibrillar protein and
accumulation of ROS. We then reasoned that, if indeed PrPC acts as an
antioxidant, absence of PrPC should increase the effect of ischemic
preconditioning (IPC), in contrast to the less evident protection in hearts from
PrPC-OE mice. Our data on hearts subjected to IPC nicely fitted with this
prediction, given that IPC led to a strong decrease of damage in PrPC-KO
hearts, an intermediate protection in WT hearts, and no significant effect in
PrPC-OE hearts. We also applied protocols of non-ischemic oxidative injury, by
subjecting isolated hearts to perfusion with hydrogen peroxide. Such
treatment was associated with a significantly larger myocardial cell loss and

8
myofibrillar oxidative damage PrPC-KO hearts, compared to hearts from WT
and PrPC-OE mice.
We then investigated the possible modulation by PrPC of proteins involved in
the oxidative stress response. We performed enzymatic activity assays on
catalase (CAT) and mitochondrial and cytosolic superoxide dismutase (SOD):
we found a decrease in CAT activity in PrPC-KO hearts with respect to PrPC-
expressing counterparts, whereas no major variation in the
activity/expression of SOD was registered among the different PrPC-
genotypes. In addition, we found increased levels of both total and
mitochondrial p66Shc, a protein involved in oxidative stress-mediated
apoptosis, in hearts lacking PrPC. This unprecedented and intriguing finding
demands further investigations in the future.
This data thus supports both the value of the in situ muscle paradigm for
studying the physiologic function of PrPC, and the role of PrPC against
oxidative insults and cell damage.

9
RIASSUNTO
L’esatto ruolo che la proteina prionica cellulare (PrPC) svolge nella fisiologia
della cellula è ancora incerto, e questo impedisce la comprensione dei
meccanismi che stanno alla base delle malattie da prione e lo sviluppo di
opportune strategie terapeutiche. Studi condotti su modelli cellulare e animali
geneticamente modificati, tuttavia, hanno suggerito che PrPC possa svolgere
un ruolo protettivo nei confronti dello stress ossidativo e di segnali di morte
cellulare. PrPC è particolarmente abbondante nel sistema nervoso centrale, ma
è espressa a livelli elevati anche nei tessuti muscolari. Inoltre, recenti
evidenze hanno correlato la proteina alla fisiopatologia muscolare. Per questo
motivo, abbiamo orientato la nostra ricerca sull’utilizzo di cuori isolati e
perfusi, un paradigma sperimentale innovativo per lo studio in situ delle
funzioni protettive della PrPC.
Rispetto alle cellule in coltura, i cuori isolati hanno il vantaggio di mantenere
l’ambiente cellulare di origine e sono inoltre modelli più adatti dell’animale
intero per le manipolazioni sperimentali. Di conseguenza, servendoci di topi
wild-type (WT) e geneticamente modificati, esprimenti differenti quantità di
PrPC (knock-out (KO) e sovra-esprimenti (OE) la proteina), abbiamo verificato
la putativa funzione antiossidante di PrPC servendoci di cuori isolati sottoposti
a protocolli di ischemia/riperfusione (I/R), o di perfusione, che implicano lo
stress ossidativo. La nostra previsione, in linea con la putativa capacità di PrPC
di contrastare l’insulto ossidativo ed i meccanismi di morte cellulare, era che i
cuori espiantati da topi PrPC-KO e da topi PrPC-OE, e sottoposti a protocolli di
I/R, manifestassero, rispettivamente, una maggiore e minore sensibilità al
danno rispetto alla controparte WT.
Quello che abbiamo rilevato è che i cuori PrPC-OE sono più resistenti al danno
indotto dalla riperfusione post-ischemica rispetto a cuori WT e PrPC-KO, come
indicato dalla riduzione di morte cellulare, ossidazione di proteine miofibrillari
ed accumulo di specie reattive dell’ossigeno (ROS). Abbiamo quindi ipotizzato
che, se realmente PrPC agisse come un agente anti-ossidante, l’assenza della
proteina avrebbe potuto aumentare la protezione conferita dall’utilizzo di un
protocollo di pre-condizionamento ischemico (IPC), il cui meccanismo si basa
sulla produzione di piccole quantità di ROS. Questa ipotesi si è dimostrata

10
corretta, dato che il protocollo di IPC svolge un forte ruolo protettivo nei cuori
PrPC-KO, uno intermedio nei WT, e nessun effetto nei cuori PrPC-OE. Abbiamo
inoltre applicato protocolli basati su un tipo di danno ossidativo non
ischemico, perfondendo i cuori isolati con perossido di idrogeno. Tale
trattamento produce una maggiore morte cellulare ed una maggiore
ossidazione delle proteine miofibrillari nei cuori PrPC-KO, paragonati a quelli
WT e PrPC-OE.
Abbiamo infine ipotizzato un possibile ruolo di PrPC nella modulazione
dell’attività/espressione di proteine coinvolte nella risposta agli stimoli
ossidativi. A tal fine, abbiamo testato l’attività, in cuori non perfusi, di alcuni
enzimi scavenger di ROS, tra cui catalasi (CAT) e superossido dismutasi
(SOD) mitocondriale e citosolica. Mentre abbiamo osservato una riduzione
significativa dell’attività di CAT nei cuori PrPC-KO rispetto a quelli esprimenti
PrPC, l’espressione e l’attività delle SOD non sono risultate differenti nei tre
genotipi di PrPC. Da sottolineare, infine, che è stato dimostrato un aumento
dell’espressione di p66Shc, una proteina coinvolta nella mediazione di segnali
pro-apoptotici, nei cuori privi PrPC. Tale osservazione, assolutamente inedita,
meriterà ulteriori approfondimenti futuri.
I nostri risultati supportano dunque sia il valore del nuovo modello
sperimentale in situ per lo studio della funzione fisiologica di PrPC, sia il
coinvolgimento della proteina nelle difese contro lo stress ossidativo ed il
danno cellulare.

11
INTRODUCTION
The prion protein (PrP) was discovered while trying to identify an elusive
etiological agent of a group of rare fatal neurodegenerative disease, anatomo-
pathologically defined transmissible spongiform encephalopathies (TSEs).
Such etiological agent, later termed prion, was found in patients and animals
affected by TSEs. Prions were found in β-amyloid aggregates that were mainly
composed by an aberrant conformer (PrPSc) of the cellular prion protein
(PrPC). PrPC is a highly conserved cell surface sialo-glycoprotein,
physiologically expressed – in a non-aggregated form – in all mammalian
tissues, particularly in the central nervous system (CNS). While the
implication of PrPSc in the onset and transmission of TSEs is now well
recognised, the mechanisms of prion-associated neurodegeneration and the
physiologic role of PrPC are still largely elusive.
Prions and prion diseases
TSEs can be of infectious, genetic, or sporadic nature and are characterized
by neurodegeneration and protein aggregation (Prusiner, 1998). These
diseases include Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-
Scheinker (GSS), fatal familial insomnia (FFI), and kuru in humans, scrapie in
sheep, chronic wasting disease (CWD) in cervids and the bovine spongiform
encephalopathy (BSE), also known as “mad cow disease”. Human TSEs can
affect subjects at distinct age groups, with a variety of motor or cognitive
symptoms, and although their prevalence is relatively low (one case per
million per year in western countries), they are still incurable and invariably
fatal (Knight and Will, 2004).
In 1967, J.S. Griffith proposed the idea that a sole protein, without the action
of nucleic acid, could “replicate”, thus spreading biological information in other
organisms (Griffith, 1967). This proposal was confirmed by several studies
demonstrating that the transmissible agent resisted doses of radiation that
easily inactivated both viruses and bacteria (Alper, 1967), and the profile of
sensitivity of the infectious agent to various chemicals differed from both
viruses and viroids, suggesting that it might not depend on nucleic acids to

12
propagate (Bellinger-Kawahara et al., 1987). On the basis of those
observations, Stanley Prusiner demonstrated that a protein unusually
resistant to proteolysis was required for infectivity of diseased brain extracts
(Prusiner et al., 1984), whereas no compelling evidence supported the need
for other components, especially nucleic acids. This and other findings
brought him to reconsider the Griffith’s hypothesis of the sole protein in the
“prion hypothesis”, were the newly coined term prion (the acronym for
“proteinaceous infectious particle”) indicates this novel pathogen (Prusiner
1998). This hypothesis contributed the assignment of the Nobel Prize in
Medicine to Prusiner in 1997. According to this hypothesis, the TSE
pathogenesis would not be determined by a common infectious agent
(bacteria, virus), but it would be caused by a conformational conversion of
PrPC into the aberrant isoform PrPSc, where the suffix “Sc” stands for scrapie,
the first “prion disease” to be historically (18th century) identified (Fig. 3). At
the basis of Prusiner’s hypothesis is the PrPSc putative capacity to catalyze the
pathological structural conversion of the physiologically expressed PrPC. PrPSc
could this way accumulate in the nervous tissue - due to its high resistance to
degradation - through an auto-catalytic process not mediated by nucleic
acids.
In the light of these observations, prions are unique elements in the world of
proteins, able to transmit a biological function, a property known only for
nucleic acids. This hypothesis was subsequently supported by the discovery of
prions in yeast and fungi, acting as heritable protein-based genetic elements
that cause biologically important phenotypic changes without any underlying
nucleic acid modification (Uptain and Lindquist, 2002).
The cellular prion protein (PrPC)
PrPC (Figs. 1 and 2) is a sialo-glycoprotein of about 210 aminoacids (a.a.) in
mammals, having a molecular weight of 35-36 kDa. It is probably present in
all vertebrates, which express the protein in all tissues, and particularly in the
CNS.
The prion protein gene (Prnp) was identified in 1986 (Basler et al., 1986). It is
well conserved among mammalian species, and in humans it is localized in
the short branch of chromosome 20, in the position 20p12.1 (Sparkes et al.,
1986). The gene is composed by three exons and no alternative splicing is

13
present, the open reading frame (ORF) being contained in the third exon only.
For this reason the origin of the two PrP isoforms (PrPC and PrPSc) from an
alternative splicing event was excluded. In humans the ORF codify for a 253
a.a. long polypeptide that is subsequently processed in the endoplasmatic
reticulum (ER). In the ER, the N-terminal signal peptide (a.a. 1-22) and the
sequence for a glycosyl-phosphatidylinositol (GPI) anchor docking in the C-
terminus (a.a. 231-253) are removed, and the N-glycosilation process on two
asparagine residues (Asn181 and Asn197) begins. In the Golgi apparatus,
glycans are processed by the removal of mannose residues and the addition
of complex oligosaccharidic chains. The mature protein then moves along the
secretory pathway, to be eventually delivered to the plasma membrane (PM).
PrPC is located extracellularly, bound to the external leaflet of the PM through
the GPI moiety. Like other GPI-anchored proteins, PrPC is enriched in
sphingolipid- and cholesterol-abundant membrane microdomains, known as
detergent-resistant membranes, or rafts (Taylor and Hooper, 2006),
putatively considered centres for signal transduction events (Kabouridis,
2006). PrPC half-life is of about six hours (Caughey et al., 1989), and during
its turnover it is internalized, to be then either recycled to the PM, or
degraded in acidic compartments (Vey et al., 1996; Peters et al., 2003).
FIGURE 1. Scheme of PrPC structure. The Signal peptide (SP, a.a. 1-22) is removed in the
mature form, as well as a.a. 231-253 (TM2) for the binding of the glycosyl-phosphatidylinositol
anchor (GPI). a.a. 51-91 contain a sequence of eight aminoacids repeated five-six folds. a.a. 104-
126 contain a putative transmembrane region. In the blue boxes α-helix (A, B, C) and β-strands
positions are indicated. B and C α-helix contain the glucidic (CHO) branches bound to the Asn
residues and the disulphide cross-bridge (Cys179-Cys210).

14
PrPC is highly conserved among mammals, with a 89% identity and a 97%
homology between human and murine protein sequences. The tri-dimensional
structure of recombinant PrPs from different species has been resolved by
nuclear magnetic resonance (Riek et al., 1996). It contains an N-terminal
flexible, random coiled sequence of about 100 a.a., and a C-terminal globular
domain of about another 100 a.a.. The globular domain is arranged in three
α-helices, interspersed with an anti-parallel β-pleated sheet formed by two
short β-strands. This structure is stabilized by a single disulfide bond between
cysteine residues 179 and 214 (human sequence). The N-terminal domain of
the protein has not a well-defined secondary structure, and contains five
repetitions of sequences of eight aminoacids (PHGGGWGQ) (octarepeats) that
can coordinate up-to six copper ions (Brown et al., 1997a). A hydrophobic
region, located between the octarepeat region and the first α-helix (a.a. 106-
126) is considered a possible trans-membrane domain, and exerts neurotoxic
functions (Forloni et al., 1993). Notably, despite the low sequence identity
between PrPC in chicken, turtle, frog, or fish, and the mammalian proteins, the
major structural features of PrPC are remarkably preserved in those non-
mammalian species, suggesting evolutionarily conserved functions of the
protein.
FIGURE 2. Tridimensional structure of the prion protein. The three α-helix and the two
short β-strands, composing the structured domain of PrPC, are shown in the figure. As
represented in the figure PrPC is anchored by a GPI extension to the plasma-membrane.

15
The scrapie prion protein (PrPSc) and its conversion from PrPC
PrPC and its aberrant isoform share the same aminoacidic sequence, the same
covalent structure and undergo the same post-translational modifications. The
two isoforms, however, have a different content of secondary structure. The
α-helix and β-strands content of PrPC is about 30% and 3%, respectively.
PrPSc maintains the α-helix portion, while the β-strands percentage is much
higher (45%), due to a remarkable conversion from random coil to β-
structure (Pan et al., 1993; Safar et al., 1993). The different conformation
confers to PrPSc different physico-chemical and biological properties, such as
detergent insolubility and propensity to aggregate, resistance to proteolytic
digestion, the ability to self-propagate in a host-organism, and neurotoxic
potentials (Caughey et al., 1991; Prusiner, 1984). In particular, the presence
of proteinase K (PK)-resistant PrP in tissue extracts is often taken as a proof
of prion infection. The conversion of PrPC into PrPSc can be initiated
spontaneously, as occurring in sporadic or genetic TSEs, or induced by the
challenge of exogenous prions in a host organism, as in the case of the
infectious forms. The mechanisms of prion-induced neurotoxicity are,
however, still debated. (For a recent review on prion properties and the
putative mechanisms of prion toxicity, see Aguzzi and Calella, 2009).
PrPC-PrPSc conversion: the gain-of-function hypothesis
Although several models have been proposed to account for the formation of
PrPSc aggregates, the basic proposal is that, following either infection with
PrPSc or conversion of PrPC into PrPSc associated with certain mutations
thought to destabilize the protein (Cohen et al., 1994), binding of PrPSc to
PrPC leads to further conversion, thus resulting in accumulation of PrPSc at the
expense of the normal PrPC. This hypothesis is consistent with the progressive
nature of all variants of the prion diseases, as well as with the resistance of
Prnp knockout mice to prion infection (Steele et al., 2007; Weissmann and
Flechsig, 2003). It is also thought to underlie the predominant sporadic forms,
in which pathogenesis might start with spontaneous conversion of a fraction
of PrPC by hitherto unknown reasons (Fornai et al., 2006). It is believed that
accumulation of PrPSc is the main pathogenic event leading to
neurodegeneration. PrPSc, as well as the PrPC 106–126 fragment (PrPC 105–
125 in the murine sequence), known as the neurotoxic peptide, induce cell

16
death both in vitro and in vivo (Ettaiche et al., 2000). These data are taken as
evidence that prion diseases are gain-of-function consequences of the
formation of PrPSc (Collins et al., 2004).
PrPC-PrPSc conversion: the loss-of-function hypothesis
Despite compelling evidence for conformational conversion in the course of
the diseases, it is still not clear what leads to the accumulation and
cytotoxicity of the pathological conformer. For example, although it is widely
assumed that accumulation of PrPSc causes neurodegeneration, systematic
examination of the brains of deceased patients revealed no spatial correlation
between neuronal apoptosis and deposits of PrPSc (Chrétien et al., 1999;
Dorandeu et al., 1998). Accumulated PrPSc within PrPC-expressing tissue
grafted into the brains of Prnp-knockout mice does not damage the
neighboring PrPC-null tissue (Brandner et al., 1996), and progressive
accumulation of PrPSc in glial cells around PrPC-null neurons does not induce
cell death in the knockout neurons, also arguing against a direct cytotoxic
effect of PrPSc (Mallucci and Collinge, 2004).
Moreover, subclinical forms of prion diseases have been observed in
experimentally or naturally infected animals that harbor high levels of
infectivity and PrPSc but are asymptomatic during a normal life-span (Race
and Chesebro, 1998; Hill et al., 2000). Conversely, wild-type mice inoculated
with PrPSc of bovine spongiform encephalopathy showed no detectable PK-
resistant PrP in the brain despite the presence of neurological symptoms and
neuronal death (Lasmézas et al., 1997). These conditions were observed not
only in animals but also in humans. FFI or GSS with substitution of valine for
alanine at residue 117 (A117V) revealed striking clinical manifestations but
little or undetectable PK-resistant PrP (Collinge et al., 1990; Medori et al.,
1992). Thus the pervasive gain-of-toxic-function hypothesis is still unproven,
and current models assume that PrPSc propagates at the expense of depletion
of PrPC (Weissmann, 1999), which warrants an examination of the hypothesis
that loss of function of PrPC (Samaia and Brentani, 1998), or of neurochemical
systems associated with PrPC, contributes to the pathogenesis of TSEs. Critical
appraisal of loss-of-function components in prion diseases is, nonetheless,
hampered by the controversies surrounding the physiological functions of
PrPC.

17
FIGURE 3. Ribbon drawing of the NMR structure model of the PrPC and of the
hypotetical structure of PrPSc. The α-helical regions are shown in green, β-strands blue, and
the unstructured regions in yellow. To be noted the conversion from the prevalent α-helical
structure, in PrPC (on the left), to the β-enriched structure, in PrPSc (on the right) (Cohen et al.,
1999).
Biological functions of PrPC
Despite the intimate involvement of PrPC in the onset of TSEs, its function in
cell physiology remains enigmatic. A plausible conceptual obstacle to this issue
is the lack of serious alterations in lifespan, development, or behavior of
genetically modified mice with the targeted (also post-natal) disruption of the
Prnp gene (Büeler et al., 1992; Manson, 1994; Mallucci et al., 2002). Recently,
however, mild vacuolar brain degeneration was observed in PrPC-KO mice with
FVB genotype. These animals show no prion-like clinical manifestation but
sensorimotor deficits are clearly evident long before the vacuolization stage
(Nazor et al., 2007). The sensorimotor phenotype also occurs in a GSS Tg
model, 2-3 months before the disease manifestation, highlighting the
possibility that the prion pathogenic mechanism involves the progressive loss
of PrPC function. To account for the absence of obvious PrPC-KO-associated
phenotypes, another current hypothesis proposes that PrPC deficiency provokes
subtle changes, whose manifestation needs, however, defined cell stress
conditions (reviewed in Steele et al., 2007). This notion has recently been
supported in vivo, in that PrP-less mice show a defective response to
hematopoietic cell depletion (Zhang et al., 2006). This result is particularly

18
relevant since it is the first example in which the combination of stress
conditions and analysis of extra-neuronal cells provides a clear insight on PrPC
function.
The search for the true physiologic role of PrPC is further confounded by the
unrealistic plethora of possible functions that have been ascribed to the
protein. Indeed, the extensive research devoted in last years to this issue, by
means of several cellular and animal models, has resulted in the proposition
that PrPC may play multiple, sometime contrasting, cellular actions. To name a
few of them, the involvement in copper metabolism and in defense
mechanisms against oxidative and apoptotic challenges, and a role in cell
adhesion, migration, proliferation, differentiation and death, possibly by
interacting with extracellular partners, or by taking part in multi-component
signaling complexes at the cell surface. A brief review of the major putative
PrPC’s functions is reported in the following paragraphs. (For comprehensive
reviews of PrP’s structure and functional properties, see Aguzzi et al., 2008;
Linden et al., 2008).
A large body of evidence supports the concept that PrPC is involved in
pathways protecting cells from different injuries. PrPC is able to protect cells
from (apoptotic) death induced by diverse stimuli, including serum deprivation
(Kim et al., 2004), Bax overexpression (Bounhar et al., 2001), TNF-α (Diarra-
Mehrpour et al., 2004), and anisomycin (Zanata et al., 2002). PrPC is also able
to counteract the neurodegeneration induced by N-terminal and N-proximal
PrPC deletion mutants, and by the ectopic expression in the brain of the PrPC
paralogue Doppel (Dpl), a PrPC-like protein lacking the whole unstructured N-
terminus. Indeed, these truncated PrPs induce a specific cerebellar
neurodegeneration, with demyelination and apoptotic neuronal death, only
when expressed in mice with a PrP-null genotype, and these dramatic
phenotypes are partially or totally abrogated by reintroduction of a functional
full-length PrP transgene (Shmerling et al., 1998; Moore et al., 1999; Moore et
al., 2001; Rossi et al., 2001; Radovanovich et al., 2005; Li et al., 2008). The
neuroprotective potentials of PrPC have been further underscored by studies on
ischemic brain injury in rodents. PrPC is up-regulated after cerebral ischemia,
and this correlates with a reduced damage severity (Weise et al., 2004; Shyu
et al., 2005). Accordingly, adenovirus-mediated PrPC overexpression reduces
infarct size and neurological impairment in rat brain (Shyu et al., 2005), while
– conversely – a more severe ischemic brain injury is observed in PrPC-KO mice

19
(McLennan et al., 2004; Spudich et al., 2005; Weise et al., 2006; Mitteregger
et al., 2007; Steele et al., 2009).
PrPC was also implicated in cell adhesion, recognition and differentiation,
possibly through the interaction with cell adhesion molecules (CAMs),
responsible for cell growth and differentiation (Hansen et al., 2008), and other
extra-cellular matrix (ECM) proteins, and the activation of downstream
signalling pathways. A case in point is the interaction, both in cis- and trans-
configurations, with the neuronal adhesion protein N-CAM (Schmitt-Ulms et al.,
2001) that led to neurite outgrowth (Santuccione et al., 2005). N-CAM belongs
to the CAM superfamily, which can not only mediate adhesion of cells, or link
ECM proteins to the cytoskeleton, but also, following homo- or heterophylic
interactions, act as a receptor to transduce signals ultimately resulting in
neurite outgrowth, neuronal survival and synaptic plasticity (Hansen et al.,
2008). Another example is the binding of PrPC to laminin, an heterotrimeric
glycoprotein of the ECM, which induced neuritogenesis together with neurite
adhesion and maintenance (Graner et al., 2000a, b), but also learning and
memory consolidation (Coitinho et al., 2006). Further, it has been described
that PrPC interacts with the mature 67 kDa-receptor (67LR) (and its 37 kDa-
precursor) for laminin, and with glycosamminoglycans (GAGs), each of which is
involved in neuronal differentiation and axon growth (Caughey et al., 1994;
Rieger et al., 1997; Gauczynski et al., 2001; Hundt et al., 2001; Pan et al.,
2002). More recently, Hajj et al. (2007) have reported that the direct
interaction of PrPC with another ECM protein, vitronectin, could accomplish the
same process, and that the absence of PrPC could be functionally compensated
by the overexpression of integrin, another laminin receptor (McKerracher et al.,
1996). Incidentally, the latter finding may provide a plausible explanation for
the absence of clear phenotypes in mammalian PrP-null paradigms. By
exposing primary cultured neurons to recombinant PrPs, others have shown
that homophylic trans-interactions of PrPCs are equally important for neuronal
outgrowth (Chen et al., 2003; Kanaani et al., 2005), including the formation of
synaptic contacts (Kanaani et al., 2005). Finally, it has been demonstrated that
the binding of PrPC with the secreted co-chaperone stress-inducible protein 1
(STI1) stimulated neuritogenesis (Lopes et al., 2005). However, this same
interaction had also a pro-survival effect, as did the interaction of PrPC with its
recombinant form (Chen et al., 2003).

20
More recently, by using zebrafish as an experimental paradigm, a lethal
developmental phenotype linked to the absence of PrPC was unravelled.
Zebrafish expresses two PrPC isoforms (PrP1 and PrP2) that, similarly to
mammalian PrPCs, are glycosylated and attached to the external side of the
plasma membrane through a glycolipid anchor. PrP1 and PrP2 are, however,
expressed in distinct time frames of the zebrafish embryogenesis. Accordingly,
the knockdown of the PrP1, or PrP2, gene very early in embryogenesis
impaired development at different stages (Málaga-Trillo et al., 2009). By
focusing on PrP1, this study showed that the protein was essential for cell
adhesion, and that this event occurred through PrP1 homophilic trans-
interactions and signaling. This comprised activation of the Src-related tyrosine
(Tyr) kinase p59fyn, and, possibly, Ca2+ metabolism, leading to the regulation
of the trafficking of E-cadherin, another member of CAMs superfamily. It was
also reported that overlapping PrP1 functions were performed by PrPCs from
other species, while the murine PrPC was capable to replace PP1 in rescuing, at
least in part, the PrP1-knockdown developmental phenotype. Apart from
providing the long-sought proof for a vital role of PrPC, the demonstration that
a mammalian isoform corrected the lethal zebrafish phenotype strongly
reinforces the notion of a functional interplay of PrPC with CAMS or ECM
proteins, and cell signaling, to promote neuritogenesis and neuronal survival.
The most sensible hypothesis for the multifaceted behaviour of PrPC is that the
protein participates in signal transduction centres at the cell surface, as already
suggested for other GPI-anchored proteins (Simons and Ikonen, 1997).
Accordingly, several putative partners of PrPC have been proposed (recently
reviewed in Aguzzi et al., 2008). If one assumes that these interactions are all
functionally significant, the most immediate interpretation of this “sticky”
behavior entails that PrPC acts as a scaffolding protein in different
ECM/membrane protein complexes. Each complex could then activate a specific
signaling pathway depending on the type and state of the cell, the expression
and glycosylation levels of PrPC, and availability of extra- and/or intra-cellular
signaling partners. In line with this proposition, several intracellular effectors of
PrPC-mediated signalling events have been proposed, including p59fyn,
mitogen-activated kinases (MAPK) Erk1/2, PI3K/Akt, and cAMP-PKA.
For example, it has been shown – by antibody-mediated cross-linking of PrPC –
that activation of the protein converged to Erk1/2 through p59fyn signalling
(Mouillet-Richard et al., 2000; Schneider et al., 2003). A PrPC-dependent

21
activation of p59fyn (Kanaani et al., 2005; Santuccione et al., 2005), and
Erk1/2 (but also of PI3K and cAMP-PKA) (Chen et al., 2003), was evident in
other neuronal cell paradigms, and, consistent with the almost ubiquitous
expression of PrPC, in non-neuronal cells such as Jurkat and T cells (Stuermer
et al., 2004). In addition, it has been proposed that the interaction of PrPC with
STI1 can either lead to neuritogenesis, through the activation of the ERK1/2
pathway, or promote neuronal survival, by impinging on the cAMP/PKA
pathway (Lopes et al., 2005). Interestingly, this is not the only example
reporting that engagement of PrPC activates simultaneously two independent
pathways. In fact, possibly after trans-activating the receptor for the epidermal
growth factor, the antibody-mediated clustering of PrPC was shown to impinge
on both the Erk1/2 pathway, and on a protein (stathmin) involved in
controlling microtubule dynamics (Monnet et al., 2004). It must also be noted
that, in line with the alleged role of PrPC in mediating signal transduction
events, perturbations of the ERK1/2 (Spudich et al., 2005) and Akt (Weise et
al., 2006) signalling pathways have been reported upon ischemic challenge in
PrPC-KO brains with respect to the WT counterparts, with consequent increased
post-ischemic caspase-3 activation, and exacerbation of neuronal damage.
(Spudich et al., 2005; Weise et al., 2006).
In conclusion, regardless of the still uncertain molecular and cellular
mechanisms, a mosaic of experimental data is accumulating that convincingly
assign to PrPC benign functions. This also reinforces the notion that a clear
PrPC-less phenotype, which is probably masked by compensative systems in
normal circumstances, could emerge under specific stress conditions, and that
a loss of function of PrPC may cause, or take part to, prion-induced
neurodegeneration.
Given that the proposed role of PrPC in protecting cells against oxidative injury
is central to our work, the basic principles of oxidative stress, and the putative
anti-oxidant functions of PrPC, will be briefly described in the next sections.
Oxidative stress
Oxidative stress is caused by an imbalance between the production of reactive
oxygen species (ROS) and the ability of a biological system to readily detoxify
the reactive intermediates, and/or easily repair the resulting damage. All
forms of life maintain a reducing environment within their cells. This reducing

22
environment is preserved by enzymes that maintain the reduced state
through a constant input of metabolic energy. Disturbances in this normal
redox state can cause toxic effects through the production of peroxides and
free radicals that damage all components of the cell, including proteins, lipids,
and DNA. Some of the less reactive of these species (such as superoxide) can
be converted by oxido-reduction reactions with transition metals or other
redox cycling compounds (including quinones) into more aggressive radical
species that can cause extensive cellular damage (Valko et al., 2005) (Fig. 4).
SUPEROXIDE ANION
HYDROGEN PEROXIDE
HYDROXYL RADICAL
2H+ 2H+
SUPEROXIDE ANION
HYDROGEN PEROXIDE
HYDROXYL RADICAL
2H+ 2H+
FIGURE 4. Intermediate compounds formation in the reduction of the oxygen into
water.
Most of these oxygen-derived species are produced at a low level by normal
aerobic metabolism and the damage they cause to cells is constantly repaired.
However, under severe levels of oxidative stress, the damage causes ATP
depletion, causing the cell to simply fall apart.
One source of reactive oxygen under normal conditions in humans is the
leakage of activated oxygen from the electron transport chain in mitochondria
during oxidative phosphorylation. In the electron transport chain, electrons
are passed through a series of proteins via redox reactions, with each
acceptor protein along the chain having a greater reduction potential than the
previous. The last destination for an electron along this chain is an oxygen
molecule. Normally, oxygen is reduced to produce water. However, in about
0.1-2% of electrons passing through the chain, oxygen is instead prematurely
and incompletely reduced to give the superoxide radical (�O2-), an event most
well documented for Complex I and Complex III. This radical can act both as
an oxidant and as reducer. As a reducer it reacts with cytochrome c and
metallic ions, while as an oxidant it reacts with cathecolamines and leuco-
flavins. Another important radical is hydrogen peroxide (H2O2). However,
toxicity is seldom mediated by a direct effect of H2O2, except at high
concentrations. Instead, the H2O2 is a precursor of highly oxidizing, tissue-

23
damaging radicals. H2O2 reacts with Fe2+ ions to form the hydroxyl radical
(�OH-), by the Fenton reaction. This intermediate can be originated also by the
reaction of �O2- with H2O2 in the Haber-Weiss reaction. Other sources of �OH
-
are high energy electromagnetic radiations over-exposure, or the Fe-
independent reaction between �O2- and nitric oxide (Fig. 5) (Beckman et al.,
1996).
FIGURE 5. Generation of �OH-.
ROS play important roles in cell signalling, a process termed redox signalling.
Thus, to maintain proper cellular homeostasis, a balance must be struck
between reactive oxygen production and consumption. The best studied
cellular antioxidants are the enzymes superoxide dismutase (SOD), catalase
(CAT), and glutathione peroxidise (Fig. 6). Less well studied (but probably
just as important) enzymatic antioxidants are the peroxiredoxins and the
recently discovered sulfiredoxin. Other enzymes that have antioxidant
properties (though this is not their primary role) include paraoxonase,
glutathione-S transferases, and aldehyde dehydrogenases.
Fenton’s Reaction
Peroxinitrate Reaction
Haber-Weiss Reaction
Fe2+ + H2O2 → Fe3+ + ·OH + OH−
.O2- + H2O2 → OH + HO- + O2
NO+ .O2-→ OONO-
ONOO- + H → ONOOH
ONOOH → �OH + NO2
Fenton’s Reaction
Peroxinitrate Reaction
Haber-Weiss Reaction
Fe2+ + H2O2 → Fe3+ + ·OH + OH−
.O2- + H2O2 → OH + HO- + O2
NO+ .O2-→ OONO-
ONOO- + H → ONOOH
ONOOH → �OH + NO2

24
FIGURE 6. Reactions of two scavengers of ROS: Superoxide dismutase and catalase.
It is now widely accepted that oxidative stress might be important in
neurodegenerative diseases including ALS, Parkinson's disease, Alzheimer's
disease, Huntington's disease, and – probably – also prion diseases. Moreover
it is thought to be linked to certain cardiovascular diseases, since oxidation of
low-density lipoproteins in the vascular endothelium is a precursor to plaque
formation. Oxidative stress also plays a role in the ischemic cascade due to
oxygen reperfusion injury following hypoxia. This cascade includes both
strokes and heart attacks.
ROS mediated oxidative damage
ROS, in particular �OH-, can oxidize all the biological macromolecules. The
major portion of long term effects is inflicted by damage on DNA (Evans et
al., 2004). Iron ions involved in the Fenton reaction show an electrostatic
affinity for DNA. As a consequence the oxygen radicals produced in such
reaction could form nearby DNA, thus provoking its oxidation. DNA oxidation
can lead to helix scission and production of several hydroxilated bases. In this
situation reparation enzymes could not prevent from degradation and these
modifications, in the years, could bring to carcinogenesis and aging. Protein
modification is another destructive aspect of oxidative stress. The principles
that regulate protein oxidation have been established by Garrison and co-
workers (Rodgers et al., 1968; Peterson et al., 1969). Oxidized groups, found
in the modified proteins, include charbonilic, catecholic and hydroperossidic
groups. Oxidized proteins can not be repaired but can be proteolized by a
Superoxide Dismutase Reaction
Catalase Reaction
2 H2O2 → 2 H2O + O2
2 .O2- + 2H → H2O2 + O2
Superoxide Dismutase Reaction
Catalase Reaction
2 H2O2 → 2 H2O + O2
2 .O2- + 2H → H2O2 + O2

25
family of enzymes called proteases, and replaced. Proteases can undergo
oxidation themselves, however, determining an accumulation of damaged
proteins.
FIGURE 7. Scheme of biological ROS reaction.
PrPC and oxidative stress
It is clearly established that oxidative stress can induce apoptosis (Hampton et
al., 1998). The mechanism leading to neuronal demise in TSEs is unknown, but
it is a common opinion that, as is the case for other neurodegenerative
disorders, it may be related – at least in part – to a deregulation of the defence
mechanisms against oxidative stress.
Besides its possible generic neuroprotective and antiapoptotic functions, many
reports ascribe to PrPC a role in the control of copper homeostasis and – most
importantly –anti-oxidant potentials (see Milhavet and Lehmann, 2002 for a
review). Cells selected for the resistance to copper toxicity or oxidative stress
show increased levels of PrPC, associated with increased activity of anti-oxidant
enzymes, such as SOD and glutathione peroxidase (Brown et al., 1997a). In
addition, it has been found that primary neurons from PrPC-KO mice are more
sensitive to the oxidative challenge than WT neurons (Brown et al., 1997b;
White et al., 1999; Brown et al., 2002). This may be related to a decreased
activity of glutathione reductase (White et al., 1999) and Cu/Zn SOD (SOD1)
(Brown et al., 1997b). Accordingly, decreased total SOD activity, together with
BiologicalBiological DamageDamageO2
-
H2O2
RO2.
HO.
O2 ROO.
Enzyme Enzyme inactivationinactivation
PeroxidationPeroxidation
Fenton ReactionFenton ReactionHabeHabe--Weiss ReactionWeiss Reaction
DNA DNA degradationdegradationEnzyme inactivation Enzyme inactivation
RadicalsRadicals
PeroxidationPeroxidation
Fenton ReactionFenton Reaction
BiologicalBiological DamageDamageO2
-
H2O2
RO2.
HO.
O2 ROO.
Enzyme Enzyme inactivationinactivation
PeroxidationPeroxidation
Fenton ReactionFenton ReactionHabeHabe--Weiss ReactionWeiss Reaction
DNA DNA degradationdegradationEnzyme inactivation Enzyme inactivation
RadicalsRadicals
PeroxidationPeroxidation
Fenton ReactionFenton Reaction

26
increased protein oxidation and lipid peroxidation, was reported in different
brain regions from PrPC-KO mice compared to WT brains (Klamt et al., 2001).
Importantly, reduced SOD1 activity and resistance to oxidative stress was also
reported in cultured PrPC-KO skeletal myocytes (Brown et al., 1998), indicating
that the anti-oxidant potentials of PrPC may also be significant for non-neuronal
cell types. In line with this, reduced CAT activity and increased oxidative
damage to proteins and lipids were observed in PrPC-KO cardiac and skeletal
muscles with respect to the WT counterparts. It is also important to mention
the brain metals perturbations and altered anti-oxidant activities, associated
with oxidative damage, reported in prion-infected brains, supporting the
hypothesis that a loss of the anti-oxidant functions of PrPC in the course of
prion pathogenesis is relevant for the neurodegenerative process (Wong et al.,
2001). Although the proposition that PrPC possesses an intrinsic SOD1 activity
(Brown et al., 1999) is strongly disputed (Hutter et al., 2003; Jones et al.,
2005), the possibility that the protein serves to the delivery of copper ions to
intracellular cupro-enzymes, such as SOD1, is intriguing (Brown and Besinger,
1998). Indeed, PrPC is able to bind with high affinity Cu2+ through the His-rich
N-proximal octarepeats domain (see above) (Kramer et al., 2001), and – to a
lesser extent – also through a C-proximal site (Cereghetti et al., 2001), and
Cu2+ binding promotes PrPC internalisation (Pauly and Harris, 1998). An
alternative explanation entails that PrPC might serve as a detoxifying agent
that buffers Cu2+ at the synaptic cleft, where improperly high Cu2+
concentrations are extremely harmful, and PrPC is particularly abundant. In line
with these propositions is the finding that brain microsomes from WT mice
have a 15-fold higher Cu2+ concentration per gram of wet weight than the
PrPC-KO counterpart, whereas serum from PrPC-KO mice contained almost
twice as much copper ions as WT mice (Prince and Gunson, 1998).
Besides enhancing the activity of ROS-scavenging systems, PrPC might as well
control the cellular production/accumulation of ROS. Mitochondria are believed
to be among the major intracellular sources of ROS, and an increase of
superoxide formation has been recently reported in brain mitochondria from
PrPC-KO mice with respect to WT mice (Paterson et al., 2008).
Given that in this study we have selected the cardiac muscle as an
experimental paradigm in which studying the biologic functions of PrPC, we
will now briefly describe some of the evidences that have related PrPC patho-
physiology to muscular tissues, the functional properties of the myocardium,

27
and the injury induced by ischemia/reperfusion (I/R) events, all of which are
instrumental to the understanding of our experimental approach.
The Prion Protein and muscular tissue
Also extra-neural tissues can be affected by prions, e.g. cardiac and skeletal
muscles that naturally express substantial levels of PrPC (Miele et al., 2003;
Massimino et al., 2006). PrPSc accumulates in the skeletal muscle of individuals
(humans and animals) naturally, or experimentally, affected by TSEs (Bosque
et al., 2002; Glatzel et al., 2003; Andreoletti et al., 2004; Thomzig et al.,
2004; Angers et al., 2006; Peden et al., 2006), but also of transgenic (Tg)
mouse models of some inherited TSEs, showing specific muscular pathological
changes. An example is Tg mice expressing the murine homologue of a nine-
octapeptide insertional mutation (PG14), where necrotic fibers and
accumulation of a PrPSc-like form in the skeletal muscle were observed (Chiesa
et al., 2001). Recently, a primary myopathy has been found in a Tg mouse
with muscle-specific 40 fold-overexpression of PrPc, together with abnormal
processing of the protein (Huang et al., 2007). Notably, dilated
cardiomyopathy (Ashwath et al., 2005) and skeletal muscle myositis,
accompanied by PrPSc-rich inclusion bodies (Kovacs et al., 2004), have been
described in two cases of sporadic CJD.
Two different interpretations of TSE-associated myopathies are possible, which,
however, are not necessarily mutually exclusive. The first one envisages that
loss of functional PrPC, due to its continuous conversion into PrPSc, is the major,
or at least a concurrent, cause of muscle damage (Bianchin et al., 2005). In
line with this, stand the higher levels of oxidative stress of both skeletal and
cardiac muscles, and the diminished tolerance to physical exercise of PrPC-KO
mice (Nico et al., 2005; Klamt et al., 2001). On the other hand, given that
myocytes respond to stress conditions with increasing PrPC expression (Sarkozi
et al., 1994; Zanusso et al., 2001), one cannot exclude that, consequent to
injury, availability of higher amounts of PrPC favors the formation and
accumulation of PrPSc in extra-neural organs. Whatever the explanation, the
established capacity of cells to respond to injuries with increasing PrPC levels
(see also Marciano et al., 2004; Shyu et al., 2005a), highlights once again the
possible role of PrPC as major defendant against cell insults, also in non-
neuronal tissues.

28
The heart and ischemia/reperfusion injury
The heart is a muscular organ found in all vertebrates that is responsible for
pumping blood throughout the blood vessels by repeated, rhythmic
contractions. The mammalian heart is derived from embryonic mesoderm
germ-layer cells.
The heart lies in the anterior part of the body cavity, above the diaphragm and
behind the sternum, dorsal to the gut. The heart has four chambers, two
superior atria and two inferior ventricles, communicating by means of two
valves: tricuspid and mitral atrio-ventricular valves. Oxygen-deprived blood
from the vena cava enters the right atrium of the heart and flows into the right
ventricle, from which it is pumped through the pulmonary semilunar valve into
the pulmonary arteries which go to the lungs. Pulmonary veins return the now
oxygen-rich blood to the heart, where it enters the left atrium before flowing
into the left ventricle. Then, oxygen-rich blood from the left ventricle is
pumped out via the aorta, and on to the rest of the body.
The heart is enclosed in a double-walled sac called the pericardium. The heart
is composed of three layers, all of which are rich with blood vessels. The
superficial layer, called the visceral layer, the middle layer, called the
myocardium, and the third layer which is called the endocardium.
The heart is effectively a syncytium, a meshwork of cardiac muscle cells
interconnected by contiguous cytoplasmic bridges. This relates to electrical
stimulation of one cell spreading to neighboring cells. A region of the human
heart called the sinoatrial node, or pacemaker, sets the rate and timing at
which all cardiac muscle cells contract. The impulses also pass to another
region of specialized cardiac muscle tissue, a relay point called the
atrioventricular node, located in the wall between the right artrium and the
right ventricle.
The heart rate (50-80 bpm in humans, ∼500 bpm in mice) is the principal
cause of variation in the cardiac output (blood volume pumped per minute) and
it is regulated by: cardiac muscle automaticity, energetic and chemical factors,
extra-cardiac nervous control; the latter can be inhibitory, through the vagus
nerve fibers and acetylcolin mediator, or stimulatory, through the release by
sympathetic nervous system of catecholamines, such as norepinephrine.

29
Ischemia and reperfusion injury
Ischemic heart deseases arise when there is an imbalance between the
myocardial oxygen demand and blood supply. Ischemia usually progresses
from hypoxia and result in a condition in which the heart is unable to maintain
its rate of cellular oxidation leading to metabolic inbalances. These changes are
initially of a reversible nature (stunning), however, if oxygen is deprived for an
extended period of time, these changes progressively become more severe,
leading to tissue damage and eventually irreversible injury (or infarction).
Furthermore, the severity and progression of ischemia is not solely determined
by the extent of oxygen deprivation but by any other factors including the
relative accumulation of toxic metabolites and ionic imbalances. The reduction
in blood supply during ischemia also limits the removal of these
metabolites/catabolites further contributing to the severe metabolic injury
(Hearse, 1998).
Functional and metabolic modifications in ischemic heart
In the first 8-10 seconds of ischemia, the available oxygen is consumed and
functional and metabolic changes occur (Allen et al., 1990; Allen and Orchard,
1987). These alterations include:
• Contractile activity arrest: the myocardial ATP and phosphate creatine
(PC) supply is able to sustain a short number of contractions. Moreover, in
the early phases, there is an increased accumulation of phosphate and H+
ions, which, linked to a reduced pH, are the causes of the reduced
contractility (Lee et al., 1991);
• Conversion of aerobic metabolism into anaerobic: hypoxia occurring
during ischemia accelerates anaerobic glycolisis;
• Anaerobic glycolisis is able to maintain a sufficient rate of ATP; this is due
to the reduced ATP demand occurring in the first minutes of the ischemic
event. PC is used by the creatine-kinase (CK) to phosphorilate ADP and its
level is rapidly decreased. Ohterwise, the enzyme myokinase can convert
ADP into ATP;
• AMP produced by myokinaseis dephosphorilated by adenosine which is
then degradated to inosin. The diffusion of these nucleotides in the

30
extracellular matrix lead to a reduction in the intracellular nucleotides
pool.
At the beginning of the ischemic period the rate of anaerobic glycolisis is
maximum and determines a high accumulation of lactate (Braasch et al.,
1968; Jennings, 1987). After 60-90 seconds the lactate and reduced
nucleotides accumulation and the diminished pH reduce the rate of the
anaerobic glycolisis. This condition lasts for 40-60 minutes, then the glycolisis
arrests. Other than the lactate accumulation, glycolisis arrest leads to
glucose-6-phosphate, glycerol-3-phosphate and glucose-1-phosphate
accumulation, which leads to an increased osmolarity and edema.
Reversible and non-reversible injury
The salvage of ischemic tissue is most successful when interventions are
initiated as soon as possible (within 15 minutes from the beginning) after
vessel occlusion, resulting in the restoration of blood flow (reperfusion) to the
affected myocardium.
Ischemic damage can be irreversible after 40-60 minutes of ischemia; the ATP
content is reduced to the 90% and glycolisis arrests, reduced co-enzymes
cannot be re-oxidized. In the cell important alterations can be observed in
mitochondria and sarcolemma. Mitochondria undergo swelling, the cristae
appear disorganized and lipids and denaturated protein accumulate in the
matrix. This cellular membrane aberrations are termed “blebs” and are
normally associated to apoptotic cell death. The cause for the blebs formation
is the disorganization and disruption of the cytoscheleton.
The reperfusion injury and the ROS damage
Reperfusion injury refers to damage to tissue caused when blood supply
returns to the tissue after a period of ischemia. The reperfusion injury is
caused by:
• Rapid normalization of tissue pH;
• Rapid normalization of tissue osmolality;
• Re-energisation;

31
• Oxygen radical generation (Hearse, 1991).
The damage includes:
• Lethal arrhytmias and death of the cells previously weakened by ischemia;
• Myocardial stunning, a mechanical disfunction persisting during reperfusion,
even in the absence of histological damage or metabolic disfunctions. It
consists in a setting of the post-ischemic blood pressure at a lower rate
with respect to the pre-ischemic rate. The higher accumulation of Ca2+ ions
contributes to myocardial stunning;
• Loss of cellular proteins (Demaison et al. 1999, Lemasters et al., 1995;
Reimer et al., 1979).
The reduction of the ATP content and pH during ischemia leads to tissue
acidification that activates the Na+/H+ exchanger, in order to restore the
physiological pH. ATP depletion and phosphate ions increase inhibit the Na+/K+-
ATPase, thus leading to an accumulation of Na+ ions in the cell. With reduction
of the Na+ gradient and membrane depolarization, the Na+/Ca2+ exchanger is
turned into its "reverse mode," which leads to cytosolic accumulation of Ca2+;
this accumulation during reperfusion is the cause of activation of degradation
enzymes such as phospholipases, proteases and nucleases, which in turn
damage the myocardial tissue. This effect is demonstrated by the loss of ionic
homeostasis, mitochondrial swelling and de-energization, release of cytosolic
enzymes in the coronary effluent such as the lactate dehydrogenase (LDH) and
the CK, routinely used as markers of myocardial cell death. Once the plasma-
membrane integrity is loss, the cell cannot recover.
Even if the Ca2+ imbalance is an important mediator, ROS generation plays a
major role in I/R damage. Vanden-Hoek and collegues (1997) demonstrated
that the mitochondrial respiratory chain is an important source of ROS in the
act of reperfusion. ROS could directly damage mitochondrial proteins
determining a drop in the mitochondrial potential leading to the activation of
cell death pathways (Levraut et al., 2003). Moreover ROS could affect ionic
pumps at the plasma-membrane level, that together with the ATP deprivation
lead to ionic imbalance.

32
FIGURE 8. Ischemia and Reperfusion damage.
Ischemic Preconditioning
Ischemic preconditioning (IPC) is based on brief episodes of I/R given before a
long I/R episode, which trigger adaptive mechanisms that protect the
myocardium from oxidative injury. In 1986 Murry and coworkers exposed the
myocardium to a ‘preconditioning protocol’ with repetitive short episodes of
regional ischemia. Surprisingly, they found that this protocol induced increased
tolerance to a subsequent long-lasting ischemic period (Sommerschild et al.,
2002). In their study the myocardial infarct size after a 40-min period of
ischemia was reduced from 29% without IPC to 7% with IPC. The cellular basis
for this adaptation is not identified in detail. Different plausible hypotheses
have been suggested and several mechanisms are probably involved.
Hausenloy and Yellon (2006) identified a signalling mechanism based on two
phases: an early phase and a late phase. In the early phase the cellular
memory is related to translocation of PKC from cytosol to cellular membranes.
This causes a more rapid activation of PKC during the prolonged ischemic
period (Downey et al., 1994). After a certain time, PKC will re-translocate to
cytosol, and the memory will disappear. During the late phase of
preconditioning it is believed that cellular memory is related to synthesis of
proteins (Marber et al., 1997). The cells will need some time to produce new

33
proteins, therefore it needs a while before protection occurs. Synthesis of heat-
shock proteins has been also implicated in the mechanisms of IPC (Housenloy
and Yellon, 2006).

34

35
MATERIALS AND METHODS
Mouse models
In this study we have used 3 to 4 month-old male congenic (FVB) mice
expressing different PrPC amounts:
• FVB wild type (WT) (Harlan, Milano).
• Knock-out (KO) for the gene codifying for PrPC on an almost pure FVB
genetic background (F10 mouse line, kindly provided by Dr. G. Mallucci,
MRC Prion Unit, University of Leicester, UK).
• A transgenic mouse line over-expressing (OE) PrPC 3 to 4-folds the natural
levels (TG37 line, kindly provided by prof. J. Collinge, Imperial College,
London, UK).
PrPC-KO mice derived from Zurich I PrPC-KO mice, originally obtained by the
group led by Charles Weissman on a hybrid genotype SV129X/C57-Bl6
(Büeler et al., 1992). These mice have been cross-bred for ten generations in
hemizygosis with FVB WT (PrP+/+) mice. The PrP+/- littermates (N10) have
been inter-bred to generate the PrPC-KO line (F10) with an almost pure
(theoretically >99.9%) FVB genotype (Mallucci et al., 2002). PrPC-OE TG37
transgenic mice have been obtained by transgenesis on PrPC-KO F10 mice
(Mallucci et al., 2002).
All aspects of animal care and experimentation were performed in accordance
with the Guide for the Care and Use of Laboratory Animals published by
European and Italian (D.L. 116/92) laws concerning the care and use of
laboratory animals. The Authors’ Institution has been acknowledged by the
Italian Ministry of Health for the use of mice for experimental purposes, and
the experimental protocols were approved by the Ethical Committee of the
University of Padova.

36
The Langendorff model and protocols for the perfusion of isolated
hearts
Mice were weighed and then anaesthetized with an intraperitoneal injection of
Zoletil 100 (30 mg/kg body weight). Hearts were rapidly excised from the
thorax and placed in a cold (4°C) bicarbonate buffer (see below). Under an
illuminated magnifier, the aortic opening was immediately cannulated and tied
on a stainless steel blunt needle. Perfusion was performed in the non-
recirculating Langendorff mode (Langendorff, 1985). In the Langendorff
preparation, the heart is perfused in a retrograde (reverse) fashion,
maintained at the desired flow rate by a pump, with a nutrient rich,
oxygenated solution. The pressure of the solution causes the aortic valve to
shut and the perfusate is then forced into the coronary vessels, from these to
the right atrium and then to the right ventriculm. The perfusate is then sent
out from the pulmonary arteries (Fig. 1). This may allow the isolated heart to
beat for up to several hours.
The hearts were perfused with bicarbonate buffer gassed with 95% O2-5%
CO2 (pH 7.4) (perfusion buffer, PB). The bicarbonate buffer contained (in mM)
115.0 NaCl, 4.75 KCl, 2.15 KH2PO4, 25.0 NaHCO3, 0.65 MgSO4, 1.69 CaCl2,
and 11 glucose (Barbato et al., 1996). The PB was warmed at 37°C through a
water-jacketed glass cylinder/heat exchanger system with a warming/cooling
bath, and was circulated (at a constant flow rate of 5 ml/min) by a water
pump. The tissue temperature was maintained at 37°C by suspending the
heart in a water-jacketed chamber (Fig. 2).

37
FIGURE 1. Scheme of circulation of the perfusion buffer in an isolated heart subjected
to the Langendorff mode of perfusion.
FIGURE 2. Langendorff perfusion setup. The red arrow indicates the isolated organ.
Perfusion Protocols
At the beginning of the experiment, hearts were subjected to a 5 min-
stabilization period by perfusion with PB, a sufficient time period to wash
hearts from blood. During this step the heart beating and the coronary out-
flow were constantly monitored. When beating and/or coronary out-flow were
absent or interrupted, hearts were discarded. Hearts were then subjected to
the following perfusion protocols.
(i) Ischemia/Reperfusion (I/R), consisting of 40 min of global ischemia
(achieved by stopping the coronary flow), followed by 15 min of reperfusion
with the PB (gradually re-established at the flow-rate of 5 ml/min) (Fig. 3A).
The experimental significance of the I/R protocol is described in details in the
Introduction.
(ii) IPC protocol. A general description of the protocol, the significance, and
the effects of IPC is given in the Introduction. In our case, IPC consisted of
three episodes of 5 min of ischemia and 5 min of reperfusion, followed or not
by the previously described I/R protocol (Fig. 3B).

38
(iii) Perfusion with H2O2. This protocol was aimed at challenging hearts with a
non-ischemic oxidative injury, i.e. at probing the effects of oxidative stress in
hearts that were not previously weakened by a prolonged period of global
ischemia. To this purpose, isolated hearts were perfused for 15 or 30 min with
PB containing 1mM H2O2, immediately after the equilibration step (Fig. 3C).
Because of the high instability of H2O2, titration of the stock solution (10 M) was
always checked spectrophotometrically before each experiment (see below).
A
B
C
FIGURE 3. Schematic description of the perfusion protocols for I/R (A), IPC (B), and
perfusion with H2O2 (C).
When necessary, N-2-mercaptopropionyl-glycine (MPG, 1 mM) was added to
the PB. Control (normoxic) hearts were subjected to the 5 min-stabilization
protocol only.
For subsequent analyses, 5 ml-samples of the coronary effluent were
collected at 1 min-intervals during post-ischemic reperfusion, or perfusion

39
with H2O2, as previously described (Di Lisa et al., 2001; Carpi et al., 2009). At
the end of the experiments, hearts were quickly removed from the set-up,
dried with Whatman blotting paper to remove all PB residues, and cut into two
parts that were weighed separately. One part, homogenized (50 mg/ml) in PB
containing 0.5% (w/v) Triton X-100, was used to evaluate the release of LDH
(see below). The other part was flash-frozen, and stored in liquid nitrogen for
further biochemical/histochemical analyses.
H2O2 titration
For H2O2 titration we first diluted a 10 M stock solution to 100 mM. Then, 30, 60,
and 90 µl of the 100 mM solution were further diluted to a final volume of 3 ml.
The spectrophotometrical measurements were performed in quartz cuvettes at a
wavelength of 240 nm (Spectrophotometer Cary 50 Bio, Varian). Absorbance
readouts for each H2O2 diluition had to be comparable to those reported in the
following table. When this was not the case, the H2O2 stock solution was replaced.
Aliquots Absorbance Concentration
30 µl 0.4 1 mM
60 µl 0.8 2 mM
90 µl 1.2 3 mM
Estimation of LDH release
To assess the loss of heart viability after post-ischemic reperfusion, or during
perfusion with H2O2, we evaluated the release of the soluble enzyme LDH in
the coronary effluent, as an index of myocardial cell death (Di Lisa et al.,
2001). To this end, LDH activity was evaluated in both the coronary effluent,
collected during the (re)perfusion period, and the corresponding heart
homogenate. LDH activity was then expressed as the percentage activity in
the coronary effluent over the total (i.e., effluent + heart homogenate)
activity (Carpi et al., 2009; Schluter et al., 1991):
100(%)hom
×+
=ogenateeffluent
effluenteffluent LDHLDH
LDHLDH

40
FIGURE 4. Reaction mediated by LDH. LDH catalyzes the interconversion of pyruvate and
lactate with concomitant interconversion of NADH and NAD+.
Taking advantage of the functional properties of LDH, which catalyzes the
interconversion of pyruvate and lactate, with concomitant interconversion of
NADH and NAD+ (Fig. 4), LDH activity was determined spectrophotometrically
(Multiskan Ex. Lab System) by measuring the oxidation of NADH (absorption at
340 nm), according to a classic procedure (Bergmeyer et al., 1974; Di Lisa et
al., 2001). The essay was performed (at room temperature, RT) into 96 wells
plates. Briefly, the following reaction solution (180 µl), prepared just before
use, was dispensed into each well: 81.3 mM Tris/HCl, pH 7.2, 203.3 mM NaCl
(TRIS/NaCl solution), 240 µM NADH. Then, either homogenate (diluted 1:5 or
1:10 in Tris/NaCl solution) or coronary effluent samples (40 µl) were added to
each well. A first reading was performed as a blank. Reaction was then
started by adding 50 µl of 10 mM pyruvate (in Tris-NaCl solution) to the
reaction mixture, and the reaction kinetics were monitored for 4 min. LDH
activity is directly proportional to the oxidation of NADH, which can be easily
extrapolated by the Lambert-Beer equation (A = ε x C x L, where A is the
absorbance, ε is the molar extinction coefficient, C is the molar concentration
of NADH, L is the path length).
In I/R protocols, the total activity of LDH released in the coronary effluent at
the end of reperfusion was calculated according to the formula:
∑=
=15
1iieffluent LDHLDH
(in which LDHi is the LDH activity in the coronary effluent collected at the ith
min of reperfusion)

41
For perfusion with H2O2, the cumulative activity of LDH released in the
coronary effluent during the perfusion time-period was calculated according to
the formula:
∑=
=n
ii
neffluent LDHLDH
1
(in which LDHneffluent is the LDH activity in the coronary effluent after n min of
perfusion, being n=1-15/30)
Preparation of mitochondria from mouse hearts
Mice were killed by cervical dislocation and the hearts were quickly removed
and placed in ice-cold isolation solution (IS) containing 250 mM sucrose, 0.1
mM EGTA, 10 mM Tris/HCl, pH 7.4. Mitochondria were isolated within 30 min
after tissue dissection. Briefly, hearts were minced and washed several times
in IS to eliminate blood, homogenized by an Ultra-Turrax homogenizer in 3 ml
of IS, and then centrifuged at 900�g (4°C, 10 min) to remove the cell debris.
The supernatant was then centrifuged at 8000�g (4°C, 10 min). The cytosolic
fraction (supernatant) was kept for enzymatic activity assays and Western
blot analyses. The mitochondrial fraction (pellet) was resuspended either in a
buffer containing 50 mM potassium-phosphate (pH 8.0) and 0.5% Triton X-
100, for enzymatic assay, or in a sample buffer (SB) containing 2% (w/v)
sodium dodecyl-sulphate (SDS), 5% (w/v) glycerol, 125 mM Tris–HCl (pH
6.8), for Western blot analyses. The cytosolic fraction was diluted with 4 part
of methanol and the proteins precipitated by incubation over-night (ON) at -
80°C, then centrifuged at 8000�g (4°C, 30 min); the pellet was resuspended
in the SB for Western blot analyses. The protein content was measured using
a BCA protein assay kit (Thermo Scientific, Rockford, IL, USA).

42
SDS-PAGE and Western blotting
Sample preparation
Heart biopsies, stored in liquid nitrogen, were cut into 10 µm cryosections at -
25°C, then added with 1 ml of ice-cold phosphate-buffered saline (PBS), pH
7.2 containing 0.5 mM EDTA. Just before use, the solution was stirred under
vacuum to reduce the oxygen tension. To avoid protein degradation the entire
procedure was performed at 4°C. The suspension was then vortexed and
centrifuged (10 min) at 12000·g (Eppendorf Centrifuge 5417 R). The resulting
pellet was homogenized in SB by means of a Teflon potter, and the proteins
were denatured by 10 min boiling. When SDS-PAGE was performed under
reducing conditions, dithiothreitol (DTT, 100 mM) was added to the SB.
Conversely, when SDS-PAGE was performed under non-reducing conditions,
N-ethylmaleimide (NEM, 1mM) was added to the SB, to avoid artefacts due to
the oxidation of thiol groups in vitro. The total protein content of
homogenates was determined by the BCA protein assay kit.
Deglycosylation with peptide N-glycosidase F
For glycan removal with peptide N-glycosidase F (PNGase-F), proteins (40 µg)
denatured in SB were diluted five-fold in deglycosylation buffer (20 mM
sodium phosphate, 0.8% NP-40, 1.2% β-mercaptoethanol, 30 mM EDTA, and
1% (w/v) Triton X-100, pH 8.0). The protein suspension was then incubated
under agitation (24 h, 37°C) in the absence or in the presence of PNGase-F
(Roche) (2.5 U). Proteins were then diluted in SB and resolved by SDS-PAGE.
SDS-PAGE
SDS poly-acrylamide gel electrophoresis was performed in the mini gel format
(7 cm gel size), using the BioRad Protean III electrophoresis system (BioRad,
Hercules, CA, USA). The acrylamide concentration was always set to 4%
(w/v) in stacking gels, while – in separating gels – it was either 12% (for PrPC
and tropomyosin (Tm) immunoblotting), 8% (for p66Shc) or 15% (for SOD1
and Mn SOD (SOD2)). The following amounts of total proteins were loaded
into each lane: 15 µg for PrPC; 50 µg for SOD1 and 30 µg for SOD2, from
cytosolic or mitochondrial fractions, respectively; 50 and 20 µg for p66Shc,
from total heart homogenates or mitochondrial fractions, respectively; 5 µg

43
and 50 µg for the monomeric and the dimeric form of Tm, respectively (see
below).
Western blotting
After SDS-PAGE, proteins were electrophoretically transferred onto 0.45 µm
pore-size nitrocellulose membranes (Bio-Rad) (350 mA, 1 h, 4°C) in a buffer
containing 25 mM Tris, 192 mM glycine, 0.03% SDS and 20% methanol. The
electro-blotting efficiency was always checked by staining with Red Ponceau S
(Sigma). Unspecific binding sites on nitrocellulose membranes were blocked
by incubation (1h, RT) in PBS containing 0.1% (w/v) Tween-20 (PBS-T) and
either 3% (w/v) bovine serum albumine (BSA) (for PrPC immunodetection), or
5% (w/v) non-fat dry milk (for tropomyosin, p66Shc, SOD1 and SOD2).
Membranes were then incubated (ON, 4°C) with the desired antibody diluted
in PBS-T containing either 1%, 3% BSA or 5% non-fat dry milk. After
extensive washing with PBS-T, membranes were incubated (1h, RT) with
horseradish peroxidase-conjugated anti-mouse or anti-rabbit secondary
antibodies (Santa Cruz Biotechnology), diluted (1:4000) in PBS-T containing
either 1% BSA or 5% non-fat dry milk. Immunoreactive bands were visualized
using a chemiluminescence detection system (ECL, Pierce), and acquired by a
digital Kodak imaging workstation, checking that exposure times were within
the linear range of detection. Densitometry was performed on the digitalized
images of immunoblots using the Kodak 1D 3.5 computer program (Kodak,
New Haven, USA). The intensity values are the result of the sum of each pixel
intensity composing the analyzed area, considered in each lane, subtracted
for the background of the same area.
Antibodies
For Western blot analyses, the following mono- (M) and polyclonal (P)
antibodies (ab) were used (dilutions are indicated in parenthesis). Anti-PrP
mouse Mab 8H4 (1:7000) (a kind gift of Dr. M.S. Sy, Case Western
University, Cleveland, OH), raised against the human (173–185) sequence;
anti-tropomyosin mouse Mab CH1 (1:2000) (Sigma); anti-SOD1 rabbit Pab
(1:1000) (Abcam); anti-SOD2 rabbit Pab (1:2000) (Sigma); anti-Shc rabbit
Pab (1:2000) (BD Transduction Laboratories).

44
Estimation of tropomyosin oxidation
Western blot analyses were also used to quantify the oxidation of Tm, i.e.,
the relative presence in the heart homogenates of Tm dimers resulting from
the formation of disulphide cross-bridges (DCB). Of consequence, SDS-PAGE
for Tm immunodetection were always run under non-reducing conditions.
Given that, under the used conditions, the monomeric, reduced form of Tm
was always predominant over the oxidized (dimeric) form, two gels were run
in parallel for each set of samples. In one gel, to be blotted for evaluating the
immuno-intensity of the Tm dimers, 50 µg of total proteins were loaded onto
each well. Instead, in the other gel, which was used for evaluating the
immuno-signal of the Tm monomers, 5 µg of total proteins were loaded onto
each well. Quantitative analysis of the degree of tropomyosin oxidation was
performed on the densitometric values of the bands detected in immunoblots.
In particular, the intensity of the approx. 80 kDa band, indicative of Tm
dimers, therefore reflecting the formation of inter-molecular DCB, was
normalized to the band intensity of the corresponding Tm monomer (Fig. 5).
FIGURE 5. Schematic representation of the reversible oxidation of Cys residues leading to homodimers formation (cross-linking).
In situ superoxide detection
Tissue staining with hydroethidium (HE, Sigma) was used to evaluate the
accumulation of the �O2- in heart cryosections (Oudot et al., 2006). In the
presence of superoxide, HE is converted into the fluorogenic molecule 2-
hydroxyethidium, which becomes fluorescent upon binding to nuclear DNA
(Zielonka et al., 2008) (Fig. 5). Freshly prepared heart cryosections (10 µm)

45
were incubated (30 min) in a light-protected humidified chamber at 37°C in
the presence of HE (10 µM in DMSO), then rinsed twice with PBS, and covered
with a coverslip. The fluorescence images were acquired, with a constant
exposure time, by means of an Olympus IMT-2 inverted microscope, equipped
with a Xenon lamp and a 12-bit digital cooled CCD camera (Micromax,
Princeton Instruments, Monmouth Junction, NJ, USA). Fluorescence was
detected with 510–560 nm excitation and 590 nm emission filters. Automatic
computer-based analysis was performed with the same fluorescence threshold
for all sections. Four fields were randomly selected for each tissue section,
and the mean of their fluorescence was normalized to the intensity of a
reference sample, considered as 100%.
FIGURE 6. Proposed mechanism of 2-hydroxyethidium (2-OH-E+) formation from the reaction between hydroethidium (HE) and �O2
-.

46
Enzymatic activity assays
Superoxide dismutase activity assay
Cu/Zn SOD (SOD1) and Mn SOD (SOD2) activities were measured on the
cytosolic and mitochondrial fractions, respectively, prepared as described
previously. SOD activity was determined by quantifying
spectrophotometrically the inhibition of xanthine/xanthine oxidase–induced
cytochrome c reduction (McCord and Fridovich, 1969). The reaction mixtures
contained 50 mM potassium phosphate buffer (pH 8.0), 0.1 mM EDTA, 50 µM
cytochrome c, 100 µM xanthine and 50 µg of cytosolic proteins or 5 µg of
mitochondrial proteins (previously diluted in the reaction medium), for SOD1
and SOD2, respectively. Reactions were performed in plastic cuvettes at
25°C, and were started by adding xanthine oxidase (0.1 U/ml). Cytochrome c
reduction rate was determined by the slope of absorbance increase at a
wavelength of 550 nm (Spectrophotometer Cary 50 Bio, Varian). The inhibition
caused by heart homogenates was normalized to that afforded by 1 U of
purified SOD (Sigma-Aldrich). Data were then reported as percentage of the
mean value of WT samples.
Catalase activity assay
To determine CAT activity, tissues were homogenized in 50 mM phosphate
buffer (pH 7.0), 0.5% (w/v) Triton X-100 by means of a Teflon potter, and
the resulting suspension was centrifuged at 3000�g (10 min, 4°C) to discard
tissue debris. The supernatant was used for the enzymatic assay, after
determining the total protein content by the BCA protein assay kit. CAT
activity was assayed by measuring the consumption of H2O2 through the
absorbance decrease at a wavelength of 240 nm (Aebi, 1984)
(Spectrophotometer Cary 50 Bio, Varian). The reaction medium contained 50
mM phosphate buffer (pH 7.0) and 215 µg of total proteins. Reactions,
performed in a quartz cuvette at 25°C, were started by adding 10 mM H2O2.
Measurements were calibrated by means of a standard curve, generated by
using known amounts of purified CAT (Sigma-Aldrich), and then data were
expressed as percentage of the mean value obtained with WT hearts.

47
AIMS AND RATIONALE
The major purpose of this work was to contribute to the understanding of the
physiologic functions of PrPC. This issue is relevant not only for cell biology per
se, but especially for elucidating possible mechanisms of TSE-related
neurodegeneration, and the future development of suitable therapeutic
strategies. As clearly detailed in the Introduction, the richness of data on the
putative PrPC activities has not resulted in a comprehensive picture. This is
ascribable to the fact that use of cell (mainly neuronal) models have
contributed overwhelming – often confused – insights into the cellular and
molecular events which PrPC may come to play in, whereas animal models –
with few exceptions – have provided no concrete advancement in the
understanding of the true physiologic importance of the protein. In the
attempt to fill the gap between cell models and whole animals, in this work we
have used, for the first time in the prion field, an intact isolated organ – the
heart – to probe the function of PrPC against cell damage and, more
specifically, oxidative stress. The rationale of this choice relies on the notion
that PrPC is abundantly expressed in the heart, and on recent findings
correlating PrPC pathophysiology to the skeletal and cardiac muscles. In
addition, isolated hearts has the advantage of retaining the cell properties and
physiological environment, while being by far more amenable to experimental
manipulations than live animals. Thus, we have subjected hearts isolated from
mice with a different PrP genotype (WT, PrPC-KO and PrPC-OE) to different
perfusion protocols, that allowed us to specifically probe different myocardial
properties, ranging from the response to post-ischemic reperfusion damage,
which involves a broad set of physiologic parameters, to ischemic
preconditioning and non-ischemic oxidative insult. In the attempt to provide
explanation for our ex-vivo data, we have also analysed the efficiency of some
myocardial pro- and anti-oxidant systems in hearts with different PrPC levels,
and monitored the cell fate of PrPC in hearts subjected to the different stress
conditions.

48

49
RESULTS
As detailed previously, we used in our experiments WT FVB mice and
genetically modified congenic mice. These included PrPC-KO mice and a
transgenic mouse line (OE) reported to overexpress PrPC three-to-four fold
the natural level. The amounts of PrPC in hearts isolated from the different
used strains were routinely assessed by Western blot analysis (Fig. 1). Such
experiments showed that, though to a lesser extent than in the brain (lane 1),
PrPC could nonetheless be readily identified in WT hearts (lane 3), and that
higher amounts of the protein were present in the heart of PrPC-OE mice (lane
4) (OE to WT PrPC ratio being 3.0 ± 0.2, n = 3). Significantly, in hearts from
either WT and PrPC-OE mice, PrPC was found mainly in the mature, di-
glycosylated form. Conversely, no immunosignal was present in PrPC-KO
hearts (lane 2).
WTKO OE
35.5
29
MW (kDa)
D
M
U
WT Brain
1 2 3 4
WTKO OE
35.5
29
MW (kDa)
D
M
U
WT Brain WTKO OE
35.5
29
MW (kDa)
D
M
U
WT Brain
1 2 3 4
FIGURE 1. Immunodetection of PrPC in the heart of the different mouse strains. Heart
samples from 4 month-old WT, PrPC-KO and PrPC-OE male mice were homogenized and proteins
(15 µg per lane) were resolved on a 12% SDS-PAGE gel under non-reducing conditions,
electroblotted onto a nitrocellulose membrane, and then probed with anti-PrP Mab 8H4. For
comparison, a WT brain homogenate (5 µg of proteins) was also loaded in the gel. The PrPC
immunosignal is readily appreciable in the heart from WT mice, and is significantly increased in
the PrPC-OE heart. Conversely, no signal is evident in the KO sample. Arrows on the right indicate
the different PrPC glycoforms, i.e., un-glycosylated (U), mono-glycosylated (M) and di-
glycosylated (D). Molecular mass standards are reported on the left. Shown blot is representative
of 3 independent experiments that yielded comparable results.

50
Evaluation of the myocardial damage induced by I/R protocols in isolated hearts with different PrPC levels
Hearts isolated from PrPC-OE mice are protected against loss of viability
induced by post-ischemic reperfusion
A by now long tradition of experimental cardiology has established that
prolonged periods of no-flow ischemia in isolated hearts cause a complex set
of myocardial dysfunctions, including impairment of mitochondria, ATP
depletion, pH changes and ion dyshomeostasis. However, it is also accepted
that the major cause of injury in ischemic hearts is the oxidative damage that
follows the massive ROS production occurring after the re-establishment of
the coronary flow (Zweier et al., 1987; Vanden-Hoek et al., 2000).
This notion prompted us to assess whether PrPC was capable to protect the
myocardium from post-ischemic reperfusion. To this end, we first subjected
hearts –isolated from mice expressing different amounts of PrPC - to a 40
min-period of ischemia (I) followed by 15 min of reperfusion (R); then, we
monitored the occurrence of cell death by quantifying the release of LDH in
the coronary effluent over the entire reperfusion time-period. As shown in the
bar diagram of Fig. 2, and in Table 1 (first line), we found that the I/R
protocol produced ∼27% loss of cell viability in WT samples, and,
unexpectedly, that a similar value pertained to PrPC-KO hearts. In contrast,
PrPC-OE hearts had a significantly reduced cell death (∼ 22%).
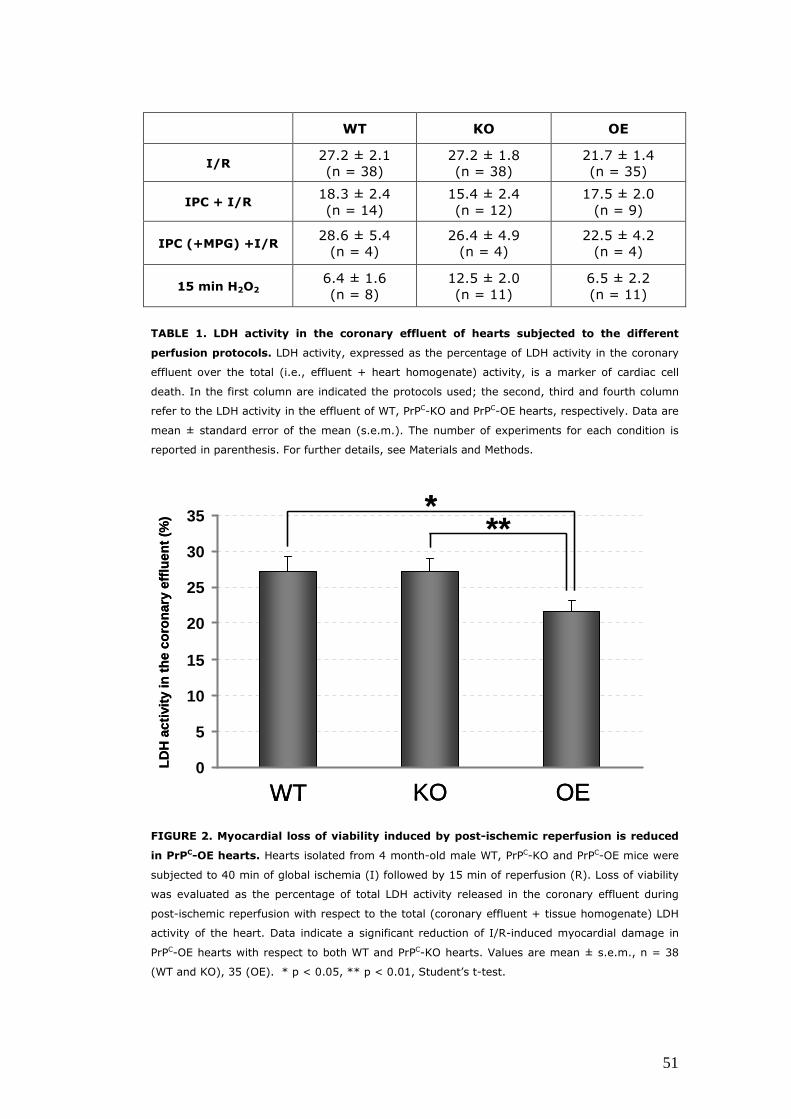
51
WT KO OE
I/R 27.2 ± 2.1 (n = 38)
27.2 ± 1.8 (n = 38)
21.7 ± 1.4 (n = 35)
IPC + I/R 18.3 ± 2.4 (n = 14)
15.4 ± 2.4 (n = 12)
17.5 ± 2.0 (n = 9)
IPC (+MPG) +I/R 28.6 ± 5.4 (n = 4)
26.4 ± 4.9 (n = 4)
22.5 ± 4.2 (n = 4)
15 min H2O2 6.4 ± 1.6 (n = 8)
12.5 ± 2.0 (n = 11)
6.5 ± 2.2 (n = 11)
TABLE 1. LDH activity in the coronary effluent of hearts subjected to the different
perfusion protocols. LDH activity, expressed as the percentage of LDH activity in the coronary
effluent over the total (i.e., effluent + heart homogenate) activity, is a marker of cardiac cell
death. In the first column are indicated the protocols used; the second, third and fourth column
refer to the LDH activity in the effluent of WT, PrPC-KO and PrPC-OE hearts, respectively. Data are
mean ± standard error of the mean (s.e.m.). The number of experiments for each condition is
reported in parenthesis. For further details, see Materials and Methods.
0
5
10
15
20
25
30
35 ***
WT KO OE
LDH
act
ivity
in th
e co
rona
ry e
fflue
nt (
%)
0
5
10
15
20
25
30
35 ***
WT KO OEWT KO OE
LDH
act
ivity
in th
e co
rona
ry e
fflue
nt (
%)
FIGURE 2. Myocardial loss of viability induced by post-ischemic reperfusion is reduced
in PrPC-OE hearts. Hearts isolated from 4 month-old male WT, PrPC-KO and PrPC-OE mice were
subjected to 40 min of global ischemia (I) followed by 15 min of reperfusion (R). Loss of viability
was evaluated as the percentage of total LDH activity released in the coronary effluent during
post-ischemic reperfusion with respect to the total (coronary effluent + tissue homogenate) LDH
activity of the heart. Data indicate a significant reduction of I/R-induced myocardial damage in
PrPC-OE hearts with respect to both WT and PrPC-KO hearts. Values are mean ± s.e.m., n = 38
(WT and KO), 35 (OE). * p < 0.05, ** p < 0.01, Student’s t-test.

52
The over-expression of PrPC reduces the degree of oxidative stress caused by
post-ischemic reperfusion
Since excess production of ROS is the main feature of post-ischemic
reperfusion, we monitored the oxidative stress in the different hearts
subjected to I/R using two alternative methods. With the first, we evaluated
the oxidation of tropomyosin (Tm), a reliable index of oxidative damage of
cardiac contractile proteins (Canton et al., 2004; 2006), which we quantified
by assessing the amount of dimers that are generated by inter-molecular
disulphide cross bridges following the oxidation of the only Cys residue
present in each Tm molecule (Canton et al., 2006; for details see Fig. 5 in the
Materials and Methods section). As illustrated in Fig. 3, reporting data from
immunoblot experiments (under non reducing conditions) of heart samples
with an anti-Tm antibody, the degree of Tm dimers (of ∼80 kDa) produced by
the I/R injury was considerably reduced (by ∼40%) in PrPC-OE hearts
compared to the other mouse strains.

53
WT KO OE Cntr
80 Oxidized tropomyosin(dimer)
40Tropomyosin(monomer)
MW (kDa)WT KO OE Cntr
80 Oxidized tropomyosin(dimer)
40Tropomyosin(monomer)
MW (kDa)
0
20
40
60
80
100
120
140
WT KO OE
Nor
mal
ized
ban
d in
tens
ity (
%)
**
0
20
40
60
80
100
120
140
WT KO OEWT KO OE
Nor
mal
ized
ban
d in
tens
ity (
%)
***
FIGURE 3. Oxidation of tropomyosin after I/R injury is decreased in PrPC-OE hearts.
Hearts from mice with different PrPC levels, subjected to I/R (40 min/15 min), were homogenized,
proteins resolved onto 12% SDS-PAGE under non-reducing conditions, and then electro-blotted
onto nitrocellulose membranes. Tm oxidation, consequent to post-ischemic reperfusion, was
evaluated, by Western blotting with the anti-Tm Mab CH1, as the appearance of a high molecular
weight (∼80 kDa) immuno-reactive band due to the formation of inter-molecular disulphide cross
bridges. To better appreciate both the oxidized and the reduced (monomeric, ∼40 kDa) forms of
Tm, each set of samples was always run in double, by loading 5 µg and 50 µg of total proteins
into each lane for the detection of the Tm monomers and dimers, respectively (see Materials and
Methods for further details). In the upper panel, the results of a Western blot analysis,
representative of one out of five independent experiments, are reported. A sample of a normoxic
heart (Cntr) from a WT mice was also loaded into the gel as a negative control. In the lower
panel, the results of the densitometric analysis are reported. Values (mean ± s.e.m., n = 5 for
each PrPC genotype) are expressed as percentage of the WT samples. Tm oxidation after I/R
injury is largely reduced in PrPC-OE with respect to WT and PrPC-KO hearts. * p < 0.05, Student’s
t-test.

54
We then evaluated ROS accumulation in I/R-treated hearts by subjecting
tissue cryosections to the HE staining assay. HE reacts specifically with the
superoxide anion (�O2-), generating the fluorogenic 2-hydroxyethidium
product that can be easily detected and quantified by fluorescence microscopy
(Oudot et al., 2006). In line with the previous results, heart cyosections from
PrPC-OE mice were significantly less fluorescent than the WT and PrPC-KO
counterparts (Fig. 4). No fluorescent signal was detected in cyosections of
normoxic hearts, or when cryosections were treated with recombinant SOD
before the HE-staining procedure (data not shown).
KOWT OEKOKOWTWT OEOE
0
20
40
60
80
100
120
140
WT KO OE
****
Nor
mal
ized
fluo
resc
ence
inte
nsity
(%
)
0
20
40
60
80
100
120
140
WT KO OEWT KO OE
****
Nor
mal
ized
fluo
resc
ence
inte
nsity
(%
)
FIGURE 4. �O2- accumulation in cardiomyocytes after I/R injury is decreased in PrPC-OE
hearts. Tissue cryosections of hearts with different PrPC levels, subjected to I/R (40 min/15 min),
were processed for HE staining to quantify superoxide accumulation. In the upper panel,
fluorescence micrographs of HE-stained cryosections from WT, PrPC-KO and PrPC-OE mice,
representative of five independent experiments, are reported. In the lower panel, the quantitative
analysis of fluorescence intensities is shown. Values (mean ± s.e.m., n = 5 for each PrPC
genotype) are expressed as percentage of the WT samples. Accumulation of �O2- after I/R injury

55
is largely reduced in PrPC-OE with respect to WT and PrPC-KO hearts. * p < 0.05 and *** p <
0.001, Student’s t-test.
As mentioned, the considerable amounts of ROS produced in the reperfusion
step are fatal to the viability of myocardial cells, especially because cells get
undermined during the ischemic period. The aggressiveness of the entire I/R
protocol, involving a very complex set of myocardial dysfunctions, could thus
explain why, only when over-expressed, PrPC elicited an appreciable
protection over cell death. Accordingly, this may also explain why no
difference of cell death could be detected between PrPC-KO hearts and hearts
that express normal amounts of PrPC. For the same reasons, data acquired
with two different markers of oxidative stress, i.e., oxidation of myofibrillar
proteins and accumulation of �O2-, suggest that one, albeit perhaps not the
only, possible reason of the enhanced viability of I/R PrPC-OE hearts is the
capacity of PrPC to counteract the production, and/or accumulation, of ROS.
PrPC performs anti-oxidant functions in the heart
The absence of PrPC enhances the protective effects of ischemic
preconditioning
To explore in more details the direct involvement of PrPC against ROS, hearts
were subjected to ischemic preconditioning (IPC). IPC consists of a short
sequence of I/R brief episodes preceding a long ischemic period, which
significantly protects the heart from the damage induced by post-ischemic
reperfusion (Pain et al., 2000). The protective effects of IPC have been
ascribed to the production of sub-lethal amounts of ROS during the
preconditioning phase (Pain et al., 2000), which would trigger defence
mechanisms, and eventually counteract the large burst of ROS generated at
the onset of post-ischemic reperfusion (Vanden-Hoek et al., 1998).
Accordingly, known anti-oxidant compounds are able to diminish or suppress
the beneficial effect of IPC (Liu et al., 1998; Pain et al., 2000). We thus
reasoned that, were indeed PrPC part of the cell apparatus protecting against
the production and/or accumulation of ROS, the benefits of IPC should have
been found to inversely correlate with the levels of PrPC. This is precisely what
we observed after treating hearts with 3 cycles of short I/R episodes (5 min/5

56
min), followed by the standard I/R challenge (40 min/15 min). As reported in
Fig. 5 (white bars) and Table 1 (second line), the protection - estimated by
the relative reduction of LDH release in the coronary effluent - was maximal
(∼45%) in the absence of PrPC, intermediate (∼30%) in WT hearts, and totally
abrogated in PrPC-OE hearts. Interestingly, the degree of cardioprotection by
IPC in WT hearts is comparable to that afforded by PrPC overexpression in the
absence of IPC (Fig. 5, compare white (WT) and grey (OE) bars).
Next, we investigated if this observed difference in IPC efficiency could be
specifically ascribed to the anti-ROS potential of PrPC. We found that this was
the case for two reasons. One was that the presence, during IPC, of the
potent free radical-scavenger N-2-mercaptopropionyl-glycine (MPG)
completely abolished the preconditioning protective effects, irrespective of the
PrPC genotype (Fig. 5, red bars and Table 1, third line). The other was the
demonstration that, during IPC, the heart content of ROS was dependent on
PrPC expression. Indeed, Fig. 6 reports that the PrPC-KO heart, stained with
HE at the end of the preconditioning step, has more abundant ROS compared
to WT hearts. Further, a slight, though not significant, reduction of ROS was
observed in PrPC-OE hearts with respect to the WT counterpart.

57
0
5
10
15
20
25
30
35
WT KO OE
**
****
LDH
act
ivity
in th
e co
rona
ry e
fflue
nt (
%)
0
5
10
15
20
25
30
35
WT KO OE0
5
10
15
20
25
30
35
WT KO OE
****
********
LDH
act
ivity
in th
e co
rona
ry e
fflue
nt (
%)
FIGURE 5. PrPC reduces the protective effect of IPC on myocardial cell loss. Isolated
hearts were subjected to I/R without (grey bars) or with (white bars and red bars) a protocol of
IPC before the step of prolonged no-flow ischemia. Ischemic preconditioning (IPC) (3 cycles of 5
min of ischemia followed by 5 min of reperfusion) was run in the absence (white bars) or in the
presence (red bars) of 1 mM MPG in the perfusion buffer. The protection afforded by IPC was
evaluated as a decrease in the release of LDH during post-ischemic reperfusion. The beneficial
effect of IPC is maximal (∼45%) in PrPC-KO, intermediate (∼30%) in WT and non-significant in
PrPC-OE hearts. In the presence of MPG, the effect of IPC is completely abolished, irrespective of
PrPC expression levels. Values are mean ± s.e.m.; grey bars: n = 38 (WT and KO), 35 (OE);
white bars: n = 14 (WT), 12 (KO), 9 (OE); red bars: n = 4 (WT, KO, OE). * p < 0.05, ** p <
0.01, Student’s t-test. Other experimental details are as in the legend to Fig. 2.

58
KO OEWT KOKOKO OEOEOEWTWTWT
0
50
100
150
200
WT KO OE
Nor
mal
ized
fluo
resc
ence
inte
nsity
(%
)
** ***
0
50
100
150
200
WT KO OEWT KO OE
Nor
mal
ized
fluo
resc
ence
inte
nsity
(%
)
** ***
FIGURE 6. PrPC reduces ROS production during IPC. Isolated hearts were subjected to the
IPC protocol (3 cycles of 5 min of ischemia followed by 5 min of reperfusion) and then analysed
for the accumulation of �O2- by means of the HE-staining assay. In the upper panel, fluorescence
micrographs of HE-stained cryosections from WT, PrPC-KO and PrPC-OE mice, representative of
four independent experiments, are reported. In the lower panel, the quantitative analysis of
fluorescence intensities is shown. Values (mean ± s.e.m., n = 4 for each PrPC genotype) are
expressed as percentage of the WT samples. Accumulation of the �O2- after IPC is considerably
increased in PrPC-KO hearts with respect to the PrPC-expressing counterparts. ** p < 0.01 and
*** p < 0.001, Student’s t-test. Other experimental details are as in the legend to Fig. 5.
PrPC protects the heart from non-ischemic oxidative injury
To corroborate further the anti-oxidant potential of PrPC, we used a protocol
based on non-ischemic oxidative damage, i.e. perfusion with H2O2. In this
experimental paradigm, isolated hearts, after the equilibration step, were
perfused for 15 or 30 min with a buffer containing 1mM H2O2. Though severe,
this means of oxidative challenge is conceptually simpler that the I/R protocol
in that it allows to test the anti-ROS potential of myocardial cells that have
not been weakened by a previous ischemic challenge. In fact, at difference
from the cell death by post-ischemic reperfusion, most of which occurred

59
immediately after the re-establishment of the coronary flow, we observed
that, initially, the myocardial damage by H2O2 was small, but that it worsened
progressively with the duration of perfusion (Fig. 7). Importantly, with this
protocol the myocardial damage, during the first 15 min of perfusion, was
constantly more pronounced in PrPC-KO hearts than in the PrPC-expressing
hearts, the difference eventually becoming significant at the 14th and 15th min
of the perfusion period ((6.4 ± 1.6)% in WT, (12.5 ± 2.0)% in PrPC-KO, (6.5
± 2.2)% in PrPC-OE, at the end of the 15-min perfusion time-period) (see also
table 1, fourth line).
When perfusion with H2O2 was prolonged up to 30 min, the differences in
myocardial cell death between PrPC-KO and WT hearts disappeared, while
PrPC-OE hearts still resulted significantly more protected. This might be due to
PrPC downregulation during the first phase of the perfusion period (see Fig. 17
and the Conclusions and Perspectives section).
As expected, ROS are the main actors of the myocardial damage by H2O2,
given that loss of cell viability of PrPC-KO hearts after 15 min of perfusion,
was drastically reduced by the presence of MPG in the perfusion medium (Fig.
7, black line). In line with these results, we observed that the formation of Tm
dimers in hearts treated with H2O2 for 15 min was higher in the absence,
than in the presence, of PrPC (Fig. 8), indicating an increased degree of
myofibrillar protein oxidation.
Thus, in line with the results obtained with the IPC protocol, this data proves
once again the anti-ROS capacity of PrPC, and the protection that the protein
exerts on the myocardium against cell death and oxidative damage.

60
24 25 26 27 28 29 30
LDH
act
ivity
in th
e co
rona
ry e
fflue
nt (
%)
**
**
0
10
20
30
40
50
60
70
80
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
t (min)
**
= WT
= KO = OE
= KO + MPG
24 25 26 27 28 29 30
LDH
act
ivity
in th
e co
rona
ry e
fflue
nt (
%)
**
**
0
10
20
30
40
50
60
70
80
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
t (min)
**
= WT
= KO = OE
= KO + MPG
FIGURE 7. PrPC protects from H2O2-induced myocardial cell damage. Isolated hearts were
subjected to perfusion (15 or 30 min) with a buffer containing 1 mM H2O2. Myocardial cell loss
was evaluated as the cumulative LDH activity measured in the coronary effluent (for further
experimental details see Materials and Methods). After 14 and 15 min of perfusion, myocardial
cell viability is significantly increased in PrPC-KO hearts (red) with respect to WT (blue) and PrPC-
OE (green) hearts. When MPG (1 mM) is included in the perfusion medium, the incidence of cell
death in PrPC-KO hearts (black) is reduced to the level of PrPC-expressing hearts. Instead, at 26-
29 min of perfusion, myocardial cell death is significantly decreased in PrPC-OE hearts, while no
difference persists between WT and PrPC-KO hearts. Values are mean ± s.e.m., n = 12 (WT), 15
(KO and OE), 3 (KO + MPG) for the 15 min perfusion protocol, n = 4 for each genotype. * p <
0.05, Student’s t-test.
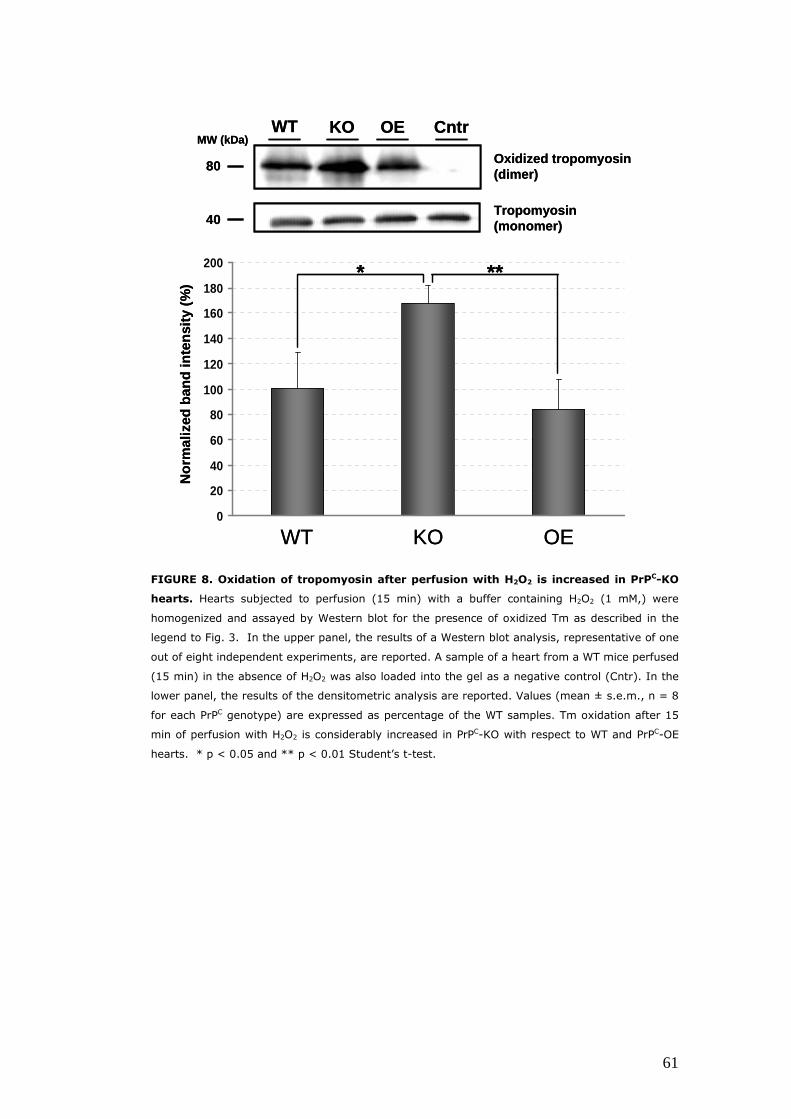
61
80 Oxidized tropomyosin(dimer)
40Tropomyosin(monomer)
WT KO OE CntrMW (kDa)
80 Oxidized tropomyosin(dimer)
40Tropomyosin(monomer)
WT KO OE CntrMW (kDa)
0
20
40
60
80
100
120
140
160
180
200
WT KO OE
* **
Nor
mal
ized
ban
d in
tens
ity (
%)
0
20
40
60
80
100
120
140
160
180
200
WT KO OE
** ****
Nor
mal
ized
ban
d in
tens
ity (
%)
FIGURE 8. Oxidation of tropomyosin after perfusion with H2O2 is increased in PrPC-KO
hearts. Hearts subjected to perfusion (15 min) with a buffer containing H2O2 (1 mM,) were
homogenized and assayed by Western blot for the presence of oxidized Tm as described in the
legend to Fig. 3. In the upper panel, the results of a Western blot analysis, representative of one
out of eight independent experiments, are reported. A sample of a heart from a WT mice perfused
(15 min) in the absence of H2O2 was also loaded into the gel as a negative control (Cntr). In the
lower panel, the results of the densitometric analysis are reported. Values (mean ± s.e.m., n = 8
for each PrPC genotype) are expressed as percentage of the WT samples. Tm oxidation after 15
min of perfusion with H2O2 is considerably increased in PrPC-KO with respect to WT and PrPC-OE
hearts. * p < 0.05 and ** p < 0.01 Student’s t-test.

62
Evaluation of the expression and/or activity of proteins involved the
oxidative response, in hearts with different PrPC levels
As detailed in the Introduction, since the discovery that PrPC had the
capability to bind copper at physiologic concentrations, a large number of
studies have been focussed on its possible (direct or indirect) role in the cell
defence against oxidative insults. Although the concept that PrPC possesses an
intrinsic SOD1-like activity is strongly disputed (and probably unrealistic), the
capability of the protein to modulate the expression and/or the activity of
different anti-oxidant systems is well documented (for detailed reviews see
Brown and Sassoon, 2002).
In light of these notions, we have tested the hypothesis that the different
capability to respond to the oxidative insult by hearts with different PrPC levels
might be due to variations in their endogenous anti-oxidant resources.
The enzymatic activity of CAT is diminished in PrPC–KO hearts
To unravel the molecular mechanisms at the basis of the PrPC-related
cardioprotection, we first compared the activity of CAT in hearts from WT,
PrPC-KO and PrPC-OE mice, not subjected to perfusion protocols. The
importance of CAT in the cellular detoxification from H2O2 is well recognized,
and reduced levels (by ~30%) of CAT activity have been already reported in
PrPC-KO hearts with respect to WT hearts (Klamt et al., 2001). We observed a
significant decrease (∼15%) of CAT activity in the hearts of PrPC-KO mice
compared to the PrPC-expressing counterparts. Conversely, no significant
difference was detected between WT and PrPC-OE hearts (Fig 9). This data
may contribute to justify the lower oxidative damage observed in PrPC-
expressing hearts subjected to perfusion with H2O2. Unfortunately, we were
not able to detect CAT in Western blot analyses, possibly due to the low
expression levels of the protein in the cardiac tissue (Ishikawa et al., 1986).
Of consequence, it remains an open question if PrPC modulates CAT activity,
or the myocardial content of the enzyme.
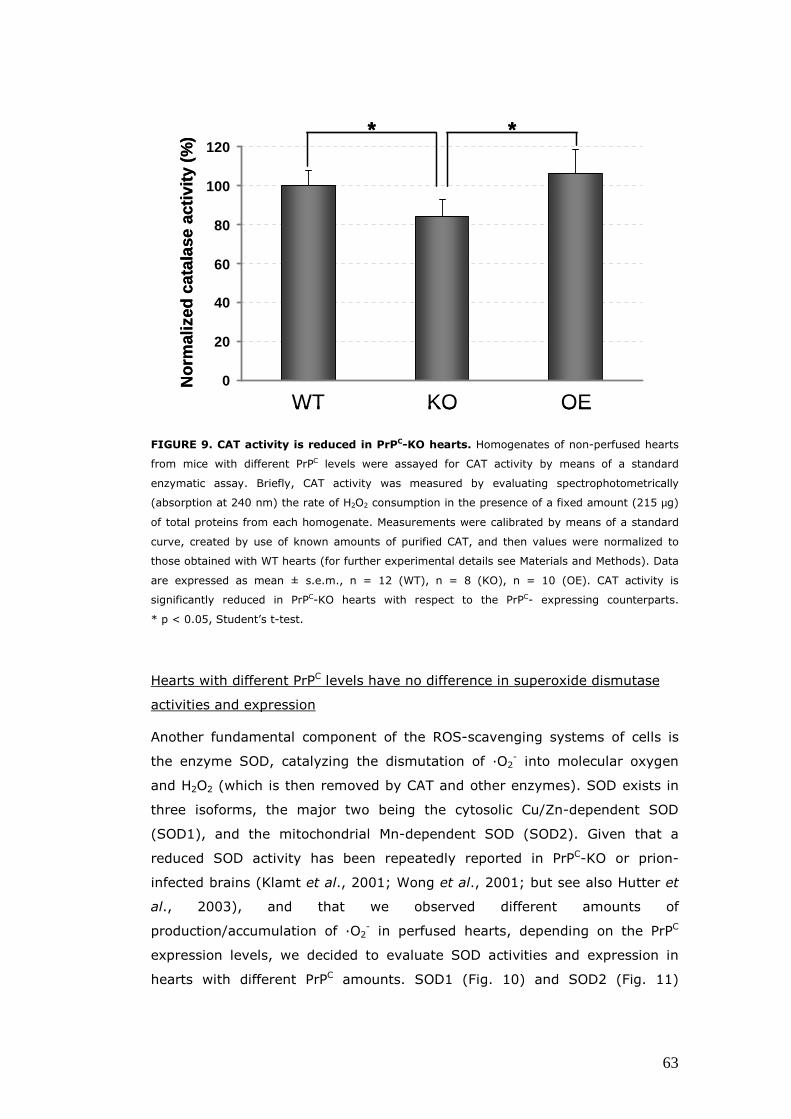
63
0
20
40
60
80
100
120
WT KO OE
* *
Nor
mal
ized
cata
lase
activ
ity(%
)
0
20
40
60
80
100
120
WT KO OE0
20
40
60
80
100
120
WT KO OE
** **
Nor
mal
ized
cata
lase
activ
ity(%
)
FIGURE 9. CAT activity is reduced in PrPC-KO hearts. Homogenates of non-perfused hearts
from mice with different PrPC levels were assayed for CAT activity by means of a standard
enzymatic assay. Briefly, CAT activity was measured by evaluating spectrophotometrically
(absorption at 240 nm) the rate of H2O2 consumption in the presence of a fixed amount (215 µg)
of total proteins from each homogenate. Measurements were calibrated by means of a standard
curve, created by use of known amounts of purified CAT, and then values were normalized to
those obtained with WT hearts (for further experimental details see Materials and Methods). Data
are expressed as mean ± s.e.m., n = 12 (WT), n = 8 (KO), n = 10 (OE). CAT activity is
significantly reduced in PrPC-KO hearts with respect to the PrPC- expressing counterparts.
* p < 0.05, Student’s t-test.
Hearts with different PrPC levels have no difference in superoxide dismutase
activities and expression
Another fundamental component of the ROS-scavenging systems of cells is
the enzyme SOD, catalyzing the dismutation of �O2- into molecular oxygen
and H2O2 (which is then removed by CAT and other enzymes). SOD exists in
three isoforms, the major two being the cytosolic Cu/Zn-dependent SOD
(SOD1), and the mitochondrial Mn-dependent SOD (SOD2). Given that a
reduced SOD activity has been repeatedly reported in PrPC-KO or prion-
infected brains (Klamt et al., 2001; Wong et al., 2001; but see also Hutter et
al., 2003), and that we observed different amounts of
production/accumulation of �O2- in perfused hearts, depending on the PrPC
expression levels, we decided to evaluate SOD activities and expression in
hearts with different PrPC amounts. SOD1 (Fig. 10) and SOD2 (Fig. 11)
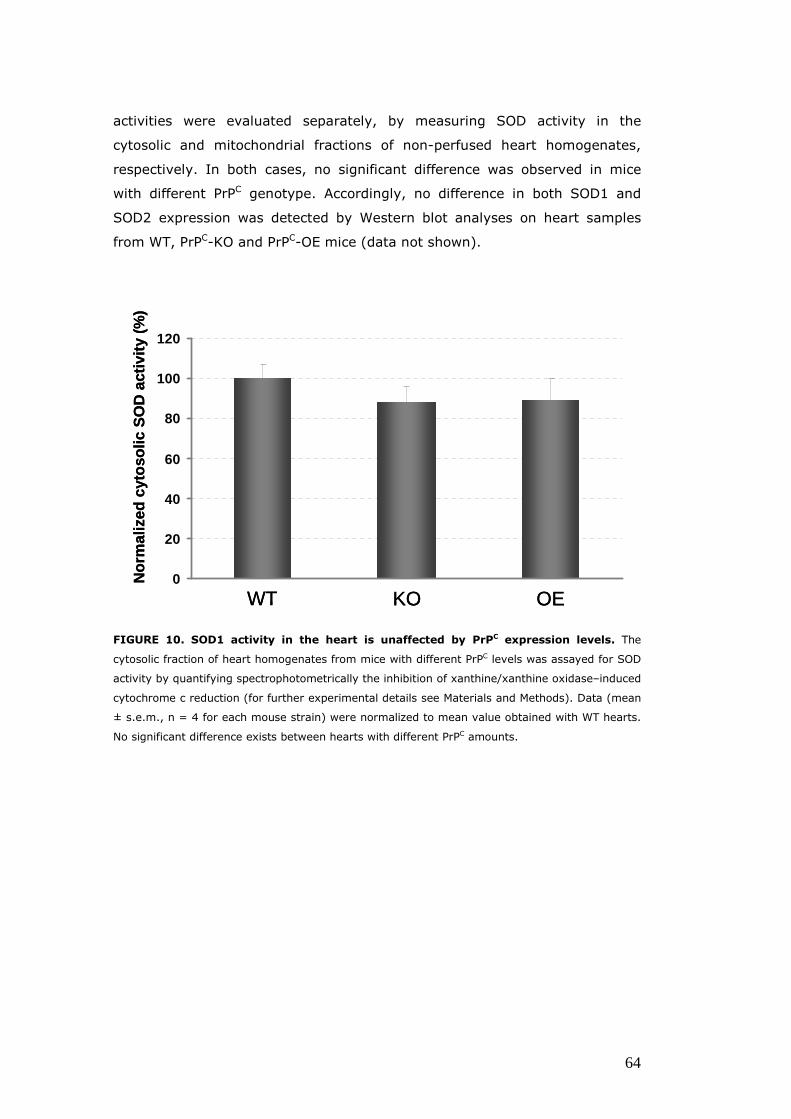
64
activities were evaluated separately, by measuring SOD activity in the
cytosolic and mitochondrial fractions of non-perfused heart homogenates,
respectively. In both cases, no significant difference was observed in mice
with different PrPC genotype. Accordingly, no difference in both SOD1 and
SOD2 expression was detected by Western blot analyses on heart samples
from WT, PrPC-KO and PrPC-OE mice (data not shown).
0
20
40
60
80
100
120
WT KO OE
Nor
mal
ized
cyto
solic
SO
D a
ctiv
ity(%
)
0
20
40
60
80
100
120
WT KO OEWT KO OE
Nor
mal
ized
cyto
solic
SO
D a
ctiv
ity(%
)
FIGURE 10. SOD1 activity in the heart is unaffected by PrPC expression levels. The
cytosolic fraction of heart homogenates from mice with different PrPC levels was assayed for SOD
activity by quantifying spectrophotometrically the inhibition of xanthine/xanthine oxidase–induced
cytochrome c reduction (for further experimental details see Materials and Methods). Data (mean
± s.e.m., n = 4 for each mouse strain) were normalized to mean value obtained with WT hearts.
No significant difference exists between hearts with different PrPC amounts.

65
0
20
40
60
80
100
120
140
Nor
mal
ized
mito
chon
dria
lSO
D a
ctiv
ity(%
)
WT KO OE0
20
40
60
80
100
120
140
Nor
mal
ized
mito
chon
dria
lSO
D a
ctiv
ity(%
)
WT KO OEWT KO OE
FIGURE 11. SOD2 activity in the heart is unaffected by PrPC expression levels. The
mitochondrial fraction of heart homogenates from mice with different PrPC levels was assayed for
SOD activity. Data (mean ± s.e.m., n = 4 for each mouse strain) were normalized to mean value
obtained with WT hearts. No significant difference exists between hearts with different PrPC
amounts. Other experimental details are as in the legend to Fig. 10.
p66Shc expression is increased in PrPC–KO hearts
A very recent work has demonstrated that the protein p66Shc significantly
contributes to mitochondrial ROS formation and myocardial damage caused in
the heart by I/R challenge (Carpi et al., 2009). p66Shc is a splice variant of
p52Shc/p46Shc, two cytoplasmic adaptor proteins involved in the propagation of
intracellular signals from activated tyrosine kinases to Ras. Rather than
functioning as an adaptor protein, p66Shc is mainly involved in the intracellular
pathways that regulate ROS metabolism and apoptosis (Migliaccio et al.,
1997). A fraction of p66Shc localizes within the mitochondrial inter-membrane
space where it oxidizes reduced cytochrome c, contributing to ROS formation
and apoptosis (Cosentino et al., 2008; for an illustrated mechanism of p66Shc
functions see Conclusions and Perspectives, Fig. 1). Accordingly, mice ablated
for p66Shc expression display strongly reduced ROS formation and damage,
increased resistance to apoptotic stimuli, increased life-span (Migliaccio et al.,
1999), and, as anticipated, reduced myocardial damage upon I/R injury
(Carpi et al., 2009).

66
We therefore asked whether PrPC might influence the expression of p66Shc in
the heart. Surprisingly, by subjecting total heart homogenates to immunoblot
analyses with an antibody directed against all Shc isoforms, we observed a
significant 2.5-fold increase in the levels of p66Shc in PrPC-KO hearts with
respect to the PrPC-expressing counterparts (Fig. 12). Conversely, no
significant difference was observed, in the different mouse strains, in the
cardiac levels of p52Shc/p46Shc (data not shown).
The increased levels of p66Shc in PrPC-KO hearts were also reflected by a
larger mitochondrial content of the protein. Indeed, when probing the
mitochondrial fraction of heart homogenates for the presence of p66Shc, a
significant (∼60%) increase of the protein was observed in the absence of PrPC
with respect to PrPC-expressing hearts (Fig. 13).
Taken together, these results nicely fit with the data shown previously, and
may open new perspectives in the study of PrPC patho-physiology.

67
66
OEKOWTMW (kDa)
66
OEOEKOKOWTWTMW (kDa)
0
50
100
150
200
250
300
WT KO OE
Nor
mal
ized
ban
d in
tens
ity (
%)
** *
0
50
100
150
200
250
300
WT KO OEWT KO OE
Nor
mal
ized
ban
d in
tens
ity (
%)
**** **
FIGURE 12. p66Shc expression is increased in PrPC-KO hearts. Non-perfused hearts from
mice with different PrPC levels, were homogenized, proteins (50 µg) were resolved onto 8% SDS-
PAGE under reducing conditions, and then electro-blotted onto nitrocellulose membranes. p66Shc
expression was evaluated by Western blotting with an anti-Shc Pab, as the abundancy of the 66
kDa immuno-reactive band. In the upper panel, the results of a Western blot analysis,
representative of at least nine independent experiments, are reported. In the lower panel, the
results of the densitometric analysis are reported. Values (mean ± s.e.m., n = 9 (WT), 10 (KO)
and 11 (OE)) are expressed as percentage of the WT samples. p66Shc expression is largely
increased in PrPC-KO hearts compared to WT and PrPC-OE hearts. * p < 0.05 and ** p < 0.01,
Student’s t-test.

68
OEKOWT
66
MW (kDa)OEOEKOKOWTWT
66
MW (kDa)
0
20
40
60
80
100
120
140
160
180
200 **
Nor
mal
ized
ban
d in
tens
ity (
%)
WT KO OE
*
0
20
40
60
80
100
120
140
160
180
200 **
0
20
40
60
80
100
120
140
160
180
200 **
Nor
mal
ized
ban
d in
tens
ity (
%)
WT KO OEWT KO OE
**
FIGURE 13. Mitochondrial p66Shc levels are increased in PrPC-KO hearts. The
mitochondrial fractions from homogenates of non-perfused hearts were probed for the presence
of p66Shc. In the upper panel, the results of a Western blot analysis, representative of at least five
independent experiments, are reported. In the lower panel, the results of the densitometric
analysis are reported. Values (mean ± s.e.m., n = 6 (WT), 5 (KO and OE)) are expressed as
percentage of the WT samples. p66Shc presence is largely increased in PrPC-KO hearts compared
to WT and PrPC-OE hearts. * p < 0.05 and ** p < 0.01, Student’s t-test. Other experimental
details are as in the legend to Fig. 12, except that 20 µg of proteins were loaded into the gels.

69
The fate of PrPC during and after the ischemic and oxidative
challenges
It is well known that during I/R the levels of myocardial proteins may vary.
This up- or down-regulation may be due to inflammatory injury, with
endothelial dysfunction and neutrophil accumulation into the myocardium, as
well as generation of free radicals and cytokines, that, in turn, can activate
proteolysis pathways. Otherwise, the activation of the complement system,
which leads to inflammatory injury, can trigger cellular activation and protein
synthesis (Maroko et al., 1978; Lefer et al., 1994). In this scenario, the
expression of proteins involved in protective pathways may be crucial for cell
survival. Given that PrPC plays a role in the heart cell defences, we decided to
measure its expression during and after the used perfusion protocols.
PrPC levels are decreased after I/R, but not after ischemia alone, in WT and
PrPC-OE hearts
The finding that PrPC-OE hearts, but not WT hearts, were less damaged by I/R
injury than the PrPC-KO counterpart led us to hypothesize that the physiologic
expression levels of PrPC were not sufficient to counteract the insult. This also
prompted us to check the actual levels of PrPC at the end of the entire I/R
protocol in PrPC-expressing hearts. Interestingly, we found that after the I/R
challenge the expression of PrPC, assessed by Western blotting analyses, was
decreased by ∼20% in WT and ∼35% in PrPC-OE hearts with respect to the
corresponding non-perfused hearts (Fig. 14).

70
MW (kDa)
D
MU
35.5
29
WTCNTR
WTIPC+I/R
OECNTR
OEIPC+I/R
1 2 3 4
MW (kDa)
D
MU
35.5
29
WTCNTR
WTIPC+I/R
OECNTR
OEIPC+I/R
1 2 3 4
D
MU
35.5
29
WTCNTR
WTIPC+I/R
OECNTR
OEIPC+I/R
1 2 3 4
0
50
100
150
200
250
300
350
WT CNTR OE CNTRWT I/R OE I/R
Nor
mal
ized
band
inte
nsity
(%)
**
*
0
50
100
150
200
250
300
350
WT CNTR OE CNTRWT I/R OE I/R
Nor
mal
ized
band
inte
nsity
(%)
****
**
FIGURE 14. The expression of PrPC is decreased after the I/R challenge. Hearts from WT
and PrPC-OE mice, subjected, or not (CNTR), to I/R (40 min/15 min), were probed for the
expression of PrPC by Western blot analysis with anti-PrP Mab 8H4. In the upper panel, the results
of a Western blot analysis, representative of at least three independent experiments, are
reported. Arrows on the right indicate the different PrPC glycoforms, while on the left molecular
mass standards are reported. In the lower panel, the results of the densitometric analysis are
reported. Data (mean ± s.e.m., n = 3 for WT and PrPC-OE CNTR hearts, n = 6 for WT and n = 7
for PrPC-OE I/R-treated hearts) are expressed as percentage of the mean value of WT CNTR
hearts. PrPC expression after the I/R protocol is significantly reduced in both WT and PrPC-OE
hearts (lanes 2 and 4 of the Western Blot, dark bars of the diagram) with respect to the
corresponding untreated hearts (lanes 1 and 3 of the Western Blot, light bars of the diagram).
* p < 0.05 and ** p < 0.01, Student’s t-test. Other experimental details are as in the legend to
Fig. 1.
It was therefore reasonable to speculate that the reduction in myocardial PrPC
content, observed upon I/R in WT hearts, although not dramatic, was

71
nevertheless sufficient to cause a significant loss of the PrPC-dependent
protective functions. However, when probing the PrPC levels at the end of the
ischemic time-period (40 min), no difference was observed in both WT and
PrPC-OE hearts with respect to the untreated counterparts (Fig. 15). Thus,
when reperfusion starts and the destructive burst of ROS occurs, both WT and
PrPC-OE hearts still contain their original PrPC reservoir.
D
MU
35.5
29
MW (kDa)
WTCNTR
WTI 40’
OECNTR
OEI 40’
1 2 3 4
D
MU
35.5
29
MW (kDa)
WTCNTR
WTI 40’
OECNTR
OEI 40’
1 2 3 4
D
MU
35.5
29
MW (kDa)
WTCNTR
WTI 40’
OECNTR
OEI 40’
D
MU
35.5
29
MW (kDa)
WTCNTR
WTI 40’
OECNTR
OEI 40’
1 2 3 4
FIGURE 15. The expression PrPC is unchanged after 40 min of ischemia. Hearts from WT
and PrPC-OE mice, subjected, or not (CNTR), to a 40-min ischemia (I 40’), were assayed for the
expression of PrPC by Western blot analysis with anti-PrP Mab 8H4. The reported Western blot is
representative of three independent experiments, which yielded comparable results. PrPC
expression after the ischemic time-period is not appreciably changed in both WT and PrPC-OE
hearts (lanes 2 and 4) with respect to the corresponding untreated counterparts (lanes 1 and 3).
Other experimental details are as in the legends to Fig. 1 and 14.
PrPC levels are preserved when I/R is preceded by IPC
In view of the previous results we could conclude that the reduction in
myocardial PrPC content, observed after the I/R protocol, occurs during the
reperfusion step. Given that IPC protects hearts from the ROS challenge
caused by post-ischemic reperfusion, we next asked if IPC could also protect
the myocardium from the I/R-induced loss of PrPC. We found that indeed this
was the case, as the levels of the protein were preserved when both WT and
PrPC-OE hearts were subjected to I/R after the application of the IPC stimulus
(Fig. 16).

72
MW (kDa)
D
MU
35.5
29
WTCNTR
WTIPC+I/R
OECNTR
OEIPC+I/R
1 2 3 4
MW (kDa)
D
MU
35.5
29
WTCNTR
WTIPC+I/R
OECNTR
OEIPC+I/R
1 2 3 4
D
MU
35.5
29
WTCNTR
WTIPC+I/R
OECNTR
OEIPC+I/R
1 2 3 4
FIGURE 16. PrPC levels are preserved when I/R is preceded by IPC. Hearts from WT and
PrPC-OE mice, subjected, or not (CNTR), to I/R (40 min/15 min) preceded by IPC (3 cycles of 5
min I/5 min R) (IPC+I/R), were probed for the expression of PrPC by Western blot analysis with
anti-PrP Mab 8H4. The reported Western blot is representative of three independent experiments,
which yielded comparable results. PrPC expression after the IPC+I/R is not appreciably changed in
both WT and PrPC-OE hearts (lanes 2 and 4) with respect to the corresponding untreated
counterparts (lanes 1 and 3). Other experimental details are as in the legends to Fig. 1 and 14.
PrPC levels are largely reduced after perfusion with H2O2
To investigate further if oxidative stress is a major player in the loss of
myocardial PrPC during post-ischemic reperfusion, we analysed PrPC
expression in hearts subjected to perfusion (15 min) with H2O2 (1 mM). We
found that, in both WT and PrPC-OE hearts, PrPC levels were dramatically
reduced (by ∼50%) at the end of the perfusion period, compared to non-
perfused hearts (Fig. 17).

73
D
MU
35.5
29
MW (kDa)
WTCNTR
WTH2O2
OECNTR
OEH2O2
1 2 3 4
D
MU
35.5
29
MW (kDa)
WTCNTR
WTH2O2
OECNTR
OEH2O2
D
MU
35.5
29
MW (kDa)
WTCNTR
WTH2O2
OECNTR
OEH2O2
1 2 3 41 2 3 4
0
50
100
150
200
250
300
350
WT H2O2 OE H2O2WT CNTR OE CNTR
Nor
mal
ized
band
inte
nsity
(%)
***
**
0
50
100
150
200
250
300
350
WT H2O2 OE H2O2WT CNTR OE CNTR
Nor
mal
ized
band
inte
nsity
(%)
******
****
FIGURE 17. Perfusion with H2O2 produces a drastic loss of myocardial PrPC. Hearts from
WT and PrPC-OE mice, subjected, or not (CNTR), to perfusion (15 min) with H2O2 (1 mM), were
probed for the expression of PrPC by Western blot analysis with anti-PrP Mab 8H4. In the upper
panel, the results of a Western blot analysis, representative of at least three independent
experiments, are reported. In the lower panel, the results of the densitometric analysis are
reported. Data (mean ± s.e.m., n = 3 for WT and PrPC-OE CNTR hearts, n = 7 for WT and n = 9
for PrPC-OE H2O2-treated hearts) are expressed as percentage of the mean value of WT CNTR
hearts. PrPC expression after perfusion with H2O2 is largely reduced in both WT and PrPC-OE
hearts (lanes 2 and 4 of the Western Blot, dark bars of the diagram) with respect to the
corresponding untreated hearts (lanes 1 and 3 of the Western Blot, light bars of the diagram).
** p < 0.01 and *** p < 0.001 Student’s t-test. Other experimental details are as in the legend
to Fig. 1 and 14.
Which is the fate of myocardial PrPC during post-ischemic reperfusion, or
perfusion with H2O2?
The expression of the prion protein is significantly reduced after the injury by
post-ischemic reperfusion and perfusion with H2O2. Both protocols largely
include the production of ROS as a prime mediator of damage. Oxidative

74
stress in the heart is known to trigger the activation of matrix metallo-
proteases (MMPs), responsible for the proteolysis of several matrix proteins
(for a review, see Schulz 2007), and PrPC might be a target of MMPs (Parkin
et al., 2004). In addition, it has been reported that mature PrPC may
physiologically undergo two distinct endo-proteolytic cleavage events,
generating two membrane-bound C-terminal fragments, named C1 and C2, of
approx. 18 and 20 kDa, respectively, which bear both the glycosylation sites
(Chen et al., 1995; Vincent et al., 2001; Mangè et al., 2004). Interstingly, the
cleavage event generating the C2 fragment, occurring next to the octapeptide
repeat region, appears to be mediated by ROS (McMahon et al., 2001), and
seems to play a role in the cellular response to oxidative stress (Watt et al.,
2005). In light of these notions, we decided to verify if the reduction in
myocardial PrPC content was a consequence of increased proteolytic cleavage.
The C1 and C2 fragments are not easily distinguishable from one another, or
from the full-lenght protein, in Western blot analysis of crude homogenates,
as they carry the high molecular weight oligo-saccharidic chains. However,
they become easily appreciable after removal of glycans with the de-
glycosylating enzyme PNGase-F (Massimino et al., 2005). Therefore, we
analysed the PrPC expression pattern in heart homogenates treated with
PNGase-F. Our results indicated that, after either I/R (Fig 18, upper panel) or
perfusion with H2O2 (Fig 18, lower panel), the amount of C1 and C2 were not
significantly increased in both WT and PrPC-OE hearts compared to the
respective non-perfused hearts. From these data, we can conclude that I/R-
or H2O2-induced oxidative stress did not increase the endo-proteolytic
cleavage of PrPC.
Another possible explanation for the myocardial loss of PrPC entails that the
protein is released from the plasma membrane of cardiomyocytes into the
perfusion buffer. However, no detectable PrPC was never found in methanol-
precipitated protein samples from the coronary effluent of perfused hearts
(data not shown). In conclusion, PrPC is probably degraded, either in the
extracellular matrix, or after internalization into cells, but this issue demands
further elucidation.

75
35.5
+ +- -WT I/R
PNGaseF
WT CNTR
29
20
D
M
U
+ +- -OE I/ROE CNTR
MW (kDa)
1 3 52 4 6 7 8
18
C 2
C 1
35.5
+ +- -WT I/R
PNGaseF
WT CNTR
29
20
D
M
U
+ +- -OE I/ROE CNTR
MW (kDa)
1 3 52 4 6 7 8
18
35.5
+ +- -WT I/R
PNGaseF
WT CNTR
29
20
D
M
U
+ +- -OE I/ROE CNTR
MW (kDa)
1 3 52 4 6 7 8
18
C 2
C 1
35.5
+ +- -WT H2O2
PNGaseF
WT CNTR
29
20
D
M
U
+ +- -OE H2O2OE CNTR
MW (kDa)
1 3 52 4 6 7 8
18
C 2
C 1
35.5
+ +- -WT H2O2
PNGaseF
WT CNTR
29
20
D
M
U
+ +- -OE H2O2OE CNTR
MW (kDa)
1 3 52 4 6 7 8
18
35.5
+ +- -WT H2O2
PNGaseF
WT CNTR
29
20
D
M
U
+ +- -OE H2O2OE CNTR
MW (kDa)
1 3 52 4 6 7 8
18
C 2
C 1
FIGURE 18. The endo-proteolytic pattern of PrPC is unaffected by I/R and perfusion
with H2O2. Homogenates of WT and PrPC-OE hearts, subjected, or not (CNTR), to I/R (40 min/15
min) (upper panel), or perfusion (15 min) with H2O2 (1 mM) (lower panel), were treated (+), or
not (-), with the sugar-removing enzyme PNGase-F, and then probed for PrPC expression by
Western blot analysis with anti-PrP Mab 8H4. In both panels, the results of a Western blot,
representative of three independent experiments, are reported. After either I/R or H2O2-
treatment, a decrease in full-length PrPC levels is observed, whereas no significant difference is
evident in the amounts of the C1 and C2 endo-proteolytic PrPC products, at around 18 and 20
kDa, respectively, in both WT and PrPC-OE hearts. Other experimental details are as in the legend
to Fig. 1 and 14. To note that the differences between the de-glycosylated full-length PrPC band in
perfused and non-perfused PrPC-OE hearts might not be readily appreciable because blots have
been over-exposed in the attempt to visualize better the cleavage products.

76

77
CONCLUSIONS AND PERSPECTIVES
A wealth of evidence implicates PrPC in cell protective mechanisms. Most of
these data are, however, merely circumstantial, and – with rare exceptions –
do not support any physiologic significance for the putative PrPC function. For
example, cells deprived of PrPC were shown to be more susceptible to
oxidative and apoptotic injury, and decreased anti-oxidant defences and
increased oxidative damage have been repeatedly reported in the brain and
other tissues of PrPC-KO mice. These mice, however, do not succumb to
oxidative overload, nor display gross signs of physiologic disturbances, and
apparently live (under normal conditions) a regular and joyful life. The most
sensible justifications for these findings are that either PrPC is in fact
dispensable for life, or it becomes necessary only in yet unidentified
situations.
Quite surprisingly, the biologic functions of PrPC have never been verified in
isolated organs. These systems maintain the cell native environment, but they
are also more amenable than whole animals to experimental manipulations
aimed at elucidating the molecular and cellular mechanisms by which proteins
exert their functions. Recent findings have highlighted a role for PrPC in
protecting from ischemic brain injury. Given that oxidative stress is a prime
mediator of hypoxia-induced cell damage in both heart and brain tissues
(Dröge, 2002), this suggested an intriguing parallelism between heart and
brain with respect to PrPC protective activity. These are the basic reasons for
our choice of using perfused hearts as intact organ paradigms where to verify
the putative antagonism of PrPC over ischemic injury by ROS.
The use of different perfusion protocols gave us the possibility to differentiate
the insults imposed to cardiomyocytes. We firstly used an I/R protocol, which
is the most renowned, and perhaps physiologically relevant, experimental
paradigm of isolated hearts. Contrary to the most simplistic expectations, and
despite the large number of experiments performed, a perfusion protocol
consisting of 40 min ischemia followed by 15 min reperfusion failed to
underscore any significant difference in myocardial cell death between WT and
PrPC-KO mice. Instead, we observed a significant reduction of myocardial

78
damage in PrPC-OE hearts. This data nicely correlates with the reduced
superoxide accumulation and myofibrillar protein oxidation observed in PrPC-
OE hearts. It is also important to underline that overexpression of PrPC,
naturally occurring at different days after focal cerebral ischemia and hypoxia,
or mediated by adenoviral delivery of a PrP transgene, was demonstrated to
protect from ischemic brain injury (Weise et al., 2004; Shyu et al., 2005).
However, it is not easy to rationalize the fact that, in our I/R protocol, the
absence of PrPC does not result in any appreciable effect. As already
mentioned, post-ischemic reperfusion injury is the result of a complex set of
functional and metabolic modifications (Allen et al., 1990; Allen and Orchard,
1987). Thus we can speculate that the physiologic PrPC levels are not
sufficient to counteract the burst of ROS, which occurs at the onset of
reperfusion, in hearts that have been previously weakened by a prolonged
ischemic event.
Accordingly, when a strong oxidative challenge, i.e. perfusion (15 min) with 1
mM H2O2, is given without ischemia, a PrPC-KO-related phenotype becomes
clearly evident. It has to be pointed out that such a high H2O2 dose was
administrated in order to take the oxidative processes to their maximal
extent. This should override the endogenous antioxidant defences of the
cardiac myocytes (Canton et al., 2004), and elicit the sole contribute of PrPC
against the oxidative insult. In this case, a significant increase of myocardial
cell loss and protein oxidation is observed in the absence of PrPC compared to
PrPC-expressing hearts. In addition, the lack of differences between WT and
PrPC-OE hearts after 15 min of perfusion argues in favour of the hypothesis
that the natural levels of the protein are sufficient to protect the myocardium
from a simple, though strong, oxidative insult. Intriguingly, the PrPC
protective effects against the H2O2 insult is further corroborated by the results
obtained when prolonging the perfusion up to 30 min. Indeed, it is quite fair
to speculate that the strong downregulation of PrPC, observed after 15 min of
perfusion with H2O2 (see Fig. 17), abrogates the beneficial effects of the
protein in WT hearts. Of consequence, a more long-lasting protocol abolishes
the differences between WT and PrPC-KO hearts, while highlighting a
significantly reduced cell loss in PrPC-OE hearts, in which PrPC is still
appreciably detectable at the 15th min of perfusion.
PrPC’s antioxidant properties were further supported by the results obtained
with ischemic preconditioning. Though the protective mechanisms at the basis

79
of IPC have not been completely unravelled, the evidence of an involvement
of small amount of ROS produced during the intermittent short reperfusion
time periods has been widely documented (Pain et al., 2000; Vanden-Hoek et
al., 1998; see the Introduction for further details). In our experimental
paradigm, the extent of IPC-induced cardioprotection inversely correlates with
the levels of PrPC, being most remarkable in PrPC-KO hearts. It is thus
reasonable to conclude that increased ROS production during IPC in hearts
lacking PrPC triggers more powerfully the adaptive response that protects the
myocardium from the subsequent I/R insult. In line with this, decreased ROS
accumulation was observed in the PrPC-KO myocardium at the end of IPC,
while the addition of an anti-oxidant in the perfusion medium completely
abrogates IPC’s beneficial effect in PrPC-KO hearts, further supporting the
anti-oxidant potential of the protein.
The protection given by PrPC over injury by ROS has been already suggested
by several works. Evidence of a modulation of the cellular antioxidant
defences by PrPC, not only in the CNS but also in other tissues (Brown et al.,
1997c; White et al., 1999; Klamt et al., 2001), and of a putative direct
involvement in the scavenging of ROS have been reported (Brown et al.,
1997c). In particular, CAT activity was found significantly reduced in hearts
form PrPC-KO C57BL/6J mice with respect to the WT counterpart (Klamt et al.,
2001). CAT decomposes H2O2 to H2O and oxygen, and is important to prevent
the Fenton and Haber-Weiss reactions, which generate the highly reactive,
and tissue-damaging, hydroxyl radical. Our results support what previously
reported, in that a significant reduction of CAT activity was observed in PrPC-
KO compared to PrPC-expressing hearts. CAT is a very efficient enzyme,
bearing one of the highest turnover numbers of all enzymes. Of consequence
it can be physiologically relevant even when expressed at very low levels. We
were not able to detect myocardial CAT by Western blot analyses, indicating
that it is indeed scarcely expressed in the heart. Of consequence, we cannot
conclude if the observed reduction of CAT activity in the absence of PrPC is
ascribable to a decreased expression of the enzyme. The effects of PrPC on
SOD activity are strongly disputed. While some research groups have reported
a decreased SOD activity in PrPC-less paradigms (Brown et al., 1997c;
Milhavet et al., 2002; Wong et al., 2001; Klamt et al. 2001), or even an
intrinsic SOD activity of the recombinant PrP (Brown et al., 1999), others
have categorically rejected such a possibility (Waggoner et al., 2000; Hutter

80
et al., 2003). Our results support this second possibility, given that no
significant difference was seen in the activity of both cytosolic SOD1 and
mitochondrial SOD2 in hearts with different PrPC genotypes. Importantly,
following the work by Hutter and colleagues, we assessed the SOD activity by
means of xanthine/xanthine oxidase-based assay (Okado-Matsumoto and
Fridovich, 2001). This assay may be more sensitive and less error-prone than
the NBT-method mainly used in other studies. Indeed, the NBT-based assay
was shown to potentially interfere with xanthine oxidase (Ukeda et al., 1997)
and resulted in poor reproducibility (Hutter et al., 2003).
At present, however, we cannot conclude that the protection from oxidative
injury exerted by PrPC has to be ascribed to the unique modulation of CAT
activity, being this enzyme not particularly abundant in the heart (see also
Ishikawa et al., 1986). Nevertheless, it is possible that, under conditions of
extreme oxidative stress, which in most cases involve GSH depletion, CAT
becomes important in providing cytoprotection (Jones et al., 1978; Kang et
al., 1996). Previous studies performed on cerebellar granule neurons cultures
showed a higher susceptibility of PrPC-KO cells to H2O2 administration, not
ascribable to altered CAT activity, but rather linked to a reduced activity of
GSH reductase (White et al., 1999). To assess whether, or not, CAT activity
could be crucial in the detoxification from H2O2 in our paradigm, further
experiments, also addressing the activity of the GSH-linked systems in hearts
with different PrPC levels, are demanded.
Another novel and important insight came from our experiments on p66Shc
expression. As previously mentioned, p66Shc is a splice variant of
p52Shc/p46Shc, two cytoplasmic adaptor proteins involved in Ras signalling.
p66Shc, a fraction of which localizes within the mitochondrial inter-membrane
space where it oxidizes reduced cytochrome c, regulates ROS metabolism
(Migliaccio et al., 1997) (for an illustrated mechanism of p66Shc see Fig. 1).
Following the mitochondrial re-localization of p66Shc, part of the respiratory
chain electron flow is therefore diverged to the production of remarkable
amounts of H2O2, corresponding to approximately one third of the total
intracellular H2O2 pool (Giorgio et al., 2005). The biological significance of this
effect is underscored by the fact that mouse-derived p66Shc−/− cells
accumulate significantly less markers of oxidative stress (Giorgio et al.,
2005). Thereby, this molecule plays an important role in mediating oxidative
damage and apoptosis. For this reason, for the first time in the field of prion

81
biology, we measured the expression of p66Shc in hearts with different PrPC
genotype. Our results clearly demonstrated a significantly higher expression
of p66Shc in heart homogenates of PrPC-KO hearts with respect to the WT and
PrPC-OE counterparts, also reflected by an increased mitochondrial content of
the protein. This further supports the possible involvement of PrPC in
preventing oxidative stress and mitochondria-mediated apoptosis. Moreover a
role for p66Shc in apoptotic and necrotic myofibers death due to ischemia-
reperfusion injury has been repeatedly demonstrated (Migliaccio et al., 1999;
Napoli et al., 2003; Zaccagnini et al., 2004), and – most importantly – it has
been recently reported that p66Shc plays a major role in ROS mediated
myocardial damage consequent to post-ischemic reperfusion (Carpi et al.,
2009). It would be of importance to verify the presence of p66Shc after IPC in
hearts with different PrPC levels, being already demonstrated that a down-
regulation of this protein occurs in preconditioning-mediated protection of
human neuroblastoma cells (Andoh et al., 2001). Another important
correlation between p66Shc and PrPC could be ascribed to the finding that
dilated cardiomyopathy, which is associated with increased levels of oxidative
stress–mediated cell death, was observed in TSE-affected individuals, where
the physiological content of PrPC is diminished due to its conversion in the
pathological isoform (Ashwath et al., 2005). Indeed, the same pathology was
correlated to an increased p66Shc expression (Cesselli et al., 2001). It would
be very interesting to assess if, in the absence of PrPC, increased levels of
p66Shc also pertains to other cell types, in particular neurons, in which PrPC
should mainly perform its physiologic functions. Importantly, the expression
levels and mitochondrial translocation of p66Shc should be evaluated in TSE-
affected brains, in order to understand if a correlation exists between PrPC
depletion and p66Shc-mediated oxidative stress in prion diseases.

82
FIGURE 1. p66Shc is part of the oxidative stress-induced apoptosis mechanism. Free
radicals activate protein kinase C-β isoform to induce Ser36 phosphorylation of the p66Shc, allowing
transfer of the protein from the cytosol to the mitochondrion. In the mitochondrion, p66Shc binds
to a complex which includes members of the TIM-TOM import system. Pro-apoptotic stimuli
destabilize the p66Shc-mtHsp70 complex and lead to the release of p66Shc in its monomeric form.
Once activated, p66Shc oxidizes cytochrome C and catalyzes the reduction of O2 to H2O2. This
latter event triggers the opening of the mitochondrial permeability transition pore, with
subsequent increase of mitochondrial membrane permeability to ions, solutes and water, swelling
and disruption of the organelle, and consequent release of pro-apoptotic factors into the cytosol
(Cosentino et al., 2008).
Thus, we have provided strong evidence that PrPC plays a role in the
myocardial defence against both I/R-mediated, and non-ischemic, oxidative
damage, possibly by modulating the expression of pro- and anti-oxidant
systems. This function may be physiologically relevant, given that it is
reflected by increased cell survival in PrPC-expressing intact hearts. But how
can a protein that is located extracellularly impinge on processes, such as
ROS production and scavenging and protein expression, which are mainly
intracellular? In this context, it is important to remember that several lines of
evidence support the possibility that PrPC takes part to multi-component

83
signal transduction complexes at the cell surface. Accordingly, several
putative functional partners of PrPC, and different signalling pathways in which
the protein may come to play, have been proposed (for exhaustive reviews
see Aguzzi et al., 2008; Linden et al., 2008; Sorgato et al., 2009). The I/R
damage involves a large number of dysfunction correlated to the alteration of
different signalling pathways. Ionic imbalance, e.g. calcium overload, is one of
the major causes of death in the I/R model, implicated in a large variety of
processes, ranging from activation of calpains, to mitochondria mediated
apoptosis (Garcia-Dorado et al., 2009). The expression of proteins involved in
Ca2+ handling seems important in protecting from this damage. SERCA over-
expression, for example, has been shown to result in several beneficial effects
in animal models, such as a diminished occurrence of arrhythmias during
ischemia-induced Ca2+ overload (del Monte et al., 2004). It will be therefore
important to measure the expression of such proteins in hearts with different
PrPC genotypes, most importantly in light of the evidences that correlate Ca2+
homeostasis to PrPC (Sorgato and Bertoli, 2009), and the finding that PrPC
positively modulates SERCA expression (Brini et al., 2005). Other signalling
pathways that could link PrPC to cardioprotection are those involving Akt or
the MAP kinase Erk1/2. Indeed, it has been shown that, in a model of
ischemic brain injury, the absence of PrPC results in reduced and increased
activation of Akt and Erk1/2, respectively, both events resulting in enhanced
post-ischemic caspase-3 activity, and exacerbation of brain damage (Spudich
et al., 2005; Weise et al., 2006). On the contrary, overexpression of PrPC
decreases early post-ischemic Erk1/2 activation and protects against focal
cerebral ischemia (Weise et al., 2008). Importantly, it has been documented
that Akt protects hearts from I/R injury (Miao et al., 2000; Fujio et al., 2000).
In this same context, it is of value to underscore the recent finding that the
activation of the MEK-Erk1/2 pathway in cardiac myocytes promotes the
phosphorylation of p66Shc at Ser36, required for the mitochondrial re-
localization of the protein (Obreztchikova et al., 2006). This would be
consistent with our data, whereby the absence of PrPC might increase the
mitochondrial p66Shc pool, through increased activation of Erk1/2. Further
analyses need to be performed to validate these hypotheses.
Another intriguing finding of this work is the substantial reduction of
myocardial PrPC observed after I/R and perfusion with H2O2 in PrPC-expressing
hearts. It is well known that during ischemia and reperfusion the levels of

84
myocardial proteins may vary, as a consequence of altered protein synthesis
and/or degradation. It is also recognized that the generation of free radicals
can activate proteolytic pathways. At the end of the I/R protocol we observed
a significant decrease of PrPC levels in both WT and PrPC-OE mice. We asked if
this reduction might account for the lack of difference between WT and PrPC-
KO hearts in I/R protocols. These seems not to be the case, given that after
the 40 min ischemia time period, no reduction of myocardial PrPC content was
seen with respect to non-perfused hearts. Thus, at the beginning of
reperfusion, WT hearts still contained the natural amounts of PrPC. This result,
together with the loss of PrPC upon perfusion with H2O2, suggests that this
event might be due to ROS-dependent degradation of the protein. This
possibility is also supported by the finding that PrPC levels are preserved when
I/R is preceded by the anti-oxidant activity of IPC. PrPC may undergo two
distinct endo-proteolytic cleavage events, one of which is mediated by ROS
and may play a role in the cellular response to oxidative stress (Watt et al.,
2005). We did not observe, however, any significant increase in the
myocardial content of the two C-terminal PrPC fragments, C1 and C2,
following I/R or H2O2 administration. It is therefore likely, although not
conclusive, that PrPC is subjected to unspecific degradation, and/or reduced
synthesis, due to myocardial metabolic impairments under the imposed
insults. Further studies are requested to understand if these events may be
physiologically relevant.
In conclusion, the novel approach, based on the use of an extra-neural tissue,
i.e. intact hearts, gave us the possibility to verify, for the first time, the
putative antioxidant and cell-protective function of PrPC in isolated organs,
suggesting that this activity of PrPC might be physiologically important under
specific stress conditions. This work allows us to confirm previous findings,
achieved by different models, and opens new interesting insights in the field
of PrPC physiology.

85
BIBLIOGRAPHY
• Aebi H. Catalase in vitro. Methods Enzymol. 1984;105:121-6.
• Aguzzi A, Baumann F, Bremer J. The prion's elusive reason for being. Annu Rev Neurosci. 2008;31:439-77.
• Aguzzi A, Calella AM. Prions: protein aggregation and infectious diseases. Physiol Rev. 2009 Oct;89(4):1105-52.
• Allen DG, Orchard CH. Myocardial contractile function during ischemia and hypoxia. Circ Res. 1987 Feb;60(2):153-68.
• Allen KL, Busza AL, Proctor E, Williams SR, Van Bruggen N, Gadian DG, Crockard HA. Restoration of energy metabolism and resolution of oedema following profound ischaemia. Acta Neurochir Suppl (Wien). 1990;51:171-3.
• Alper T, Does the agent of scrapie replicate without nucleic acid? Nature. 1967 May 20;214(5090):764-6.
• Andoh T, Lee SY, Chiueh CC. Preconditioning regulation of bcl-2 and p66shc by human NOS1 enhances tolerance to oxidative stress. Faseb J. 2000;14:2144–2146.
• Andréoletti O, Simon S, Lacroux C, Morel N, Tabouret G, Chabert A, Lugan S, Corbière F, Ferré P, Foucras G, Laude H, Eychenne F, Grassi J, Schelcher F. PrPSc accumulation in myocytes from sheep incubating natural scrapie. Nat Med. 2004 Jun;10(6):591-3.
• Angers RC, Browning SR, Seward TS, Sigurdson CJ, Miller MW, Hoover EA, Telling GC. Prions in skeletal muscles of deer with chronic wasting disease. Science. 2006 Feb 24;311(5764):1117.
• Ashwath ML, Dearmond SJ, Culclasure T, Prion-associated dilated cardiomyopathy. Arch Intern Med. 2005 Feb 14;165(3):338-40.
• Barbato R, Menabo R, Dainese P, Carafoli E, Schiaffino S, Di Lisa F. Binding of cytosolic proteins to myofibrils in ischemic rat hearts, Circ. Res. 78 (1996) 821–828.
• Basler K, Oesch B, Scott M, Westaway D, Walchli M, Groth DF, McKinley MP, Prusiner SB, Weissmann C. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell. 1986 Aug 1;46(3):417-28.
• Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996 Nov;271(5 Pt 1):C1424-37.
• Bellinger-Kawahara C, Diener TO, McKinley MP, Groth DF, Smith DR, Prusiner SB. Purified scrapie prions resist inactivation by procedures that hydrolyze, modify, or shear nucleic acids. Virology. 1987 Sep;160(1):271-4.
• Bergmeyer HU, Bernt E. in: H.U. Bergmeyer (Ed.), Lactate Dehydrogenase. Weinheim, 1974, pp. 607–612

86
• Bianchin MM, Walz R, Brentani RR, Martins VR. Dilated cardiomyopathy and Creutzfeldt-Jakob disease: evidence for a role of
cellular prion protein in the heart? Arch Intern Med. 2005 Jul 25;165(14):1663-4.
• Bosque PJ, Ryou C, Telling G, Peretz D, Legname G, DeArmond SJ, Prusiner SB. Prions in skeletal muscle. Proc Natl Acad Sci U S A. 2002 Mar 19;99(6):3812-7.
• Bounhar Y, Zhang Y, Goodyer CG, LeBlanc A. Prion protein protects human neurons against Bax-mediated apoptosis. J Biol Chem. 2001 Oct 19;276(42):39145-9.
• Braasch W, Gudbjarnason S, Puri PS, Ravens KG, Bing RJ. Early changes in energy metabolism in the myocardium following acute
coronary artery occlusion in anesthetized dogs. Circ Res. 1968 Sep;23(3):429-38.
• Brandner S, Raeber A, Sailer A, Blättler T, Fischer M, Weissmann C, Aguzzi A. Normal host prion protein (PrPC) is required for scrapie spread within the central nervous system. Proc Natl Acad Sci U S A. 1996 Nov 12;93(23):13148-51.
• Brini M, Miuzzo M, Pierobon N, Negro A, Sorgato MC. The prion protein and its paralogue Doppel affect calcium signaling in Chinese
hamster ovary cells. Mol Biol Cell. 2005 Jun;16(6):2799-808.
• Brown DR, Besinger A. Prion protein expression and superoxide dismutase activity. Biochem J. 1998 Sep 1;334 ( Pt 2):423-9.
• Brown DR, Qin K, Herms JW, Madlung A, Manson J, Strome R, Fraser PE, Kruck T, von Bohlen A, Schulz-Schaeffer W, Giese A, Westaway D, Kretzschmar H. The cellular prion protein binds copper in vivo. Nature. 1997a; Dec 18-25;390(6661):684-7.
• Brown DR, Sassoon J. Copper-dependent functions for the prion protein. Mol Biotechnol. 2002 Oct;22(2):165-78.
• Brown DR, Schmidt B, Groschup MH, Kretzschmar HA. Prion protein expression in muscle cells and toxicity of a prion protein
fragment. Eur J Cell Biol. 1998 Jan;75(1):29-37.
• Brown DR, Schmidt B, Kretzschmar HA, Effects of oxidative stress on prion protein expression in Pc12 cells. Int. J. Dev. Neurosci. 1997b, 146:104-12.
• Brown DR, Schulz-Schaeffer WJ, Schmidt B, Kretzschmar HA, Prion protein-deficient cells show altered response to oxidative stress
due to decreased SOD-1 activity. Exp. Neurol. 1997c, 146: 104-12
• Brown DR, Wong BS, Hafiz F, Clive C, Haswell SJ, Jones IM. Normal prion protein has an activity like that of superoxide dismutase.
Biochem J. 1999 Nov 15;344 Pt 1:1-5.
• Büeler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 1992; 356, 577-82.
• Canton M, Neverova I, Menabò R, Van Eyk J, Di Lisa F. Evidence of myofibrillar protein oxidation induced by postischemic reperfusion in

87
isolated rat hearts. Am J Physiol Heart Circ Physiol. 2004 Mar;286(3):H870-7.
• Canton M, Skyschally A, Menabò R, Boengler K, Gres P, Schulz R, Haude M, Erbel R, Di Lisa F, Heusch G. Oxidative modification of tropomyosin and myocardial dysfunction following coronary
microembolization. Eur Heart J. 2006 Apr;27(7):875-81.
• Carpi A, Menabò R, Kaludercic N, Pelicci P, Di Lisa F, Giorgio M. The cardioprotective effects elicited by p66(Shc) ablation demonstrate
the crucial role of mitochondrial ROS formation in ischemia/reperfusion
injury. Biochim Biophys Acta. 2009 Jul;1787(7):774-80.
• Caughey B, Brown K, Raymond GJ, Katzenstein GE, Thresher W. Binding of the protease-sensitive form of PrP (prion protein) to sulfated
glycosaminoglycan and congo red J Virol. 1994 Apr;68(4):2135-41. Erratum in: J Virol 1994 Jun;68(6):4107.
• Caughey B, Race R, Chesebro B. Comparative sequence analysis, in vitro expression and biosynthesis of mouse PrP. Prog Clin Biol Res. 1989;317:619-36.
• Caughey B, Raymond GJ. The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-
sensitive. J Biol Chem. 1991 Sep 25;266(27):18217-23.
• Cereghetti GM, Schweiger A, Glockshuber R, Van Doorslaer S. Electron paramagnetic resonance evidence for binding of Cu(2+) to the
C-terminal domain of the murine prion protein. Biophys J. 2001 Jul;81(1):516-25.
• Cesselli D, Jakoniuk I, Barlucchi L, Beltrami AP, Hintze TH, Nadal-Ginard B, Kajstura J, Leri A, Anversa P. Oxidative stress-mediated cardiac cell death is a major determinant of ventricular
dysfunction and failure in dog dilated cardiomyopathy. Circ Res. 2001 Aug 3;89(3):279-86.
• Chen S, Mangé A, Dong L, Lehmann S, Schachner M. Prion protein as trans-interacting partner for neurons is involved in neurite
outgrowth and neuronal survival. Mol Cell Neurosci. 2003 Feb;22(2):227-33.
• Chiesa R, Pestronk A, Schmidt RE, Tourtellotte WG, Ghetti B, Piccardo P, Harris DA. Primary myopathy and accumulation of PrPSc-like like molecules in peripheral tissues of transgenic mice ex
pressing a prion protein insertional mutation. Neurobiol Dis. 2001 Apr;8(2):279-88.
• Chrétien F, Dorandeu A, Adle-Biassette H, Ereau T, Wingertsmann L, Brion F, Gray F. A process of programmed cell death as a mechanisms of neuronal death in prion diseases. Clin Exp Pathol. 1999;47(3-4):181-91.
• Cohen FE, Pan KM, Huang Z, Baldwin M, Fletterick RJ, Prusiner SB. Structural clues to prion replication. Science. 1994 Apr 22;264(5158):530-1.
• Cohen FE. Protein misfolding and prion diseases. J Mol Biol. 1999 Oct 22;293(2):313-20.

88
• Coitinho AS, Freitas AR, Lopes MH, Hajj GN, Roesler R, Walz R, Rossato JI, Cammarota M, Izquierdo I, Martins VR, Brentani RR. The interaction between prion protein and laminin modulates memory
consolidation. Eur J Neurosci. 2006 Dec;24(11):3255-64.
• Collinge J, Owen F, Poulter M, Leach M, Crow TJ, Rossor MN, Hardy J, Mullan MJ, Janota I, Lantos PL. Prion dementia without characteristic pathology. Lancet. 1990 Jul 7;336(8706):7-9.
• Collins SR, Douglass A, Vale RD, Weissman JS. Mechanism of prion propagation: amyloid growth occurs by monomer addition. PLoS Biol. 2004 Oct;2(10):e321.
• Cosentino F, Francia P, Camici GG, Pelicci PG, Lüscher TF, Volpe M. Final common molecular pathways of aging and cardiovascular disease: role of the p66Shc protein. Arterioscler Thromb Vasc Biol. 2008 Apr;28(4):622-8.
• del Monte F, Lebeche D, Guerrero JL, Tsuji T, Doye AA, Gwathmey JK, Hajjar RJ. Abrogation of ventricular arrhythmias in a model of ischemia and reperfusion by targeting myocardial calcium
cycling. Proc Natl Acad Sci U S A. 2004 Apr 13;101(15):5622-7.
• Demaison L, Moreau D, Martine L, Chaudron I, Grynberg A. Myocardial ischemia and in vitro mitochondrial metabolic efficiency. Mol Cell Biochem. 1999 Apr;194(1-2):291-300.
• Di Lisa F, Menabò R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of
mitochondrial and cytosolic NAD+ and is a causative event in the death
of myocytes in postischemic reperfusion of the heart. J Biol Chem. 2001 Jan 26;276(4):2571-5.
• Diarra-Mehrpour M, Arrabal S, Jalil A, Pinson X, Gaudin C, Piétu G, Pitaval A, Ripoche H, Eloit M, Dormont D, Chouaib S. Prion protein prevents human breast carcinoma cell line from tumor necrosis
factor alpha-induced cell death. Cancer Res. 2004 Jan 15;64(2):719-27.
• Dorandeu A, Wingertsmann L, Chrétien F, Delisle MB, Vital C, Parchi P, Montagna P, Lugaresi E, Ironside JW, Budka H, Gambetti P, Gray F. Neuronal apoptosis in fatal familial insomnia. Brain Pathol. 1998 Jul;8(3):531-7.
• Downey JM, Cohen MV, Ytrehus K, Liu Y. Cellular mechanisms in ischemic preconditioning: the role of adenosine and protein kinase C. Ann N Y Acad Sci. 1994 Jun 17;723:82-98.
• Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002 Jan;82(1):47-95.
• Ettaiche M, Pichot R, Vincent JP, Chabry J. In vivo cytotoxicity of the prion protein fragment 106-126. J Biol Chem. 2000 Nov 24;275(47):36487-90.
• Evans MD, Dizdaroglu M, Cooke MS. Oxidative DNA damage and disease: induction, repair and significance. Mutat Res. 2004 Sep;567(1):1-61.

89
• Forloni G, Angeretti N, Chiesa R, Monzani E, Salmona M, Bugiani O, Tagliavini F. Neurotoxicity of a prion protein fragment. Nature. 1993 Apr 8;362(6420):543-6.
• Fornai F, Ferrucci M, Gesi M, Bandettini di Poggio A, Giorgi FS, Biagioni F, Paparelli A. A hypothesis on prion disorders: are infectious, inherited, and sporadic causes so distinct? Brain Res Bull. 2006 Mar 31;69(2):95-100.
• Fujio Y, Nguyen T, Wencker D, Kitsis RN, Walsh K. Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-
reperfusion injury in mouse heart. Circulation. 2000 Feb 15;101(6):660-7.
• Garcia-Dorado D, Agulló L, Sartorio CL, Ruiz-Meana M. Myocardial protection against reperfusion injury: the cGMP pathway.
Thromb Haemost. 2009 Apr;101(4):635-42.
• Gauczynski S, Peyrin JM, Haïk S, Leucht C, Hundt C, Rieger R, Krasemann S, Deslys JP, Dormont D, Lasmézas CI, Weiss S. The 37-kDa/67-kDa laminin receptor acts as the cell-surface receptor for
the cellular prion protein. EMBO J. 2001 Nov 1;20(21):5863-75.
• Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M, Pinton P, Rizzuto R, Bernardi P, Paolucci F, Pelicci PG. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that
trigger mitochondrial apoptosis. Cell. 2005 Jul 29;122(2):221-33.
• Glatzel M, Abela E, Maissen M, Aguzzi A. Extraneural pathologic prion protein in sporadic Creutzfeldt-Jakob disease. N Engl J Med. 2003 Nov 6;349(19):1812-20.
• Graner E, Mercadante AF, Zanata SM, Forlenza OV, Cabral AL, Veiga SS, Juliano MA, Roesler R, Walz R, Minetti A, Izquierdo I, Martins VR, Brentani RR Cellular prion protein binds laminin and mediates neuritogenesis. Brain Res Mol Brain Res. 2000b Mar 10;76(1):85-92.
• Graner E, Mercadante AF, Zanata SM, Forlenza OV, Cabral AL, Veiga SS, Juliano MA, Roesler R, Walz R, Minetti A, Izquierdo I, Martins VR, Brentani RR. Cellular prion protein binds laminin and mediates neuritogenesis. Brain Res Mol Brain Res. 2000a Mar 10;76(1):85-92.
• Graner E, Mercadante AF, Zanata SM, Martins VR, Jay DG, Brentani RR. Laminin-induced PC-12 cell differentiation is inhibited following laser inactivation of cellular prion protein. FEBS Lett. 2000b Oct 6;482(3):257-60.
• Griffith, J.S. Self-replication and scrapie. Nature. 1967 215(105):1043-4.
• Hajj GN, Lopes MH, Mercadante AF, Veiga SS, da Silveira RB, Santos TG, Ribeiro KC, Juliano MA, Jacchieri SG, Zanata SM, Martins VR. Cellular prion protein interaction with vitronectin supports axonal growth and is compensated by integrins. J Cell Sci. 2007 Jun 1;120(Pt 11):1915-26.
• Hampton MB, Orrenius S. Redox regulation of apoptotic cell death. Biofactors. 1998;8(1-2):1-5.

90
• Hansen SM, Berezin V, Bock E. Signaling mechanisms of neurite outgrowth induced by the cell adhesion molecules NCAM and N-
cadherin. Cell Mol Life Sci. 2008 Nov;65(23):3809-21.
• Hausenloy DJ, Yellon DM. Survival kinases in ischemic
preconditioning and postconditioning. Cardiovasc Res. 2006 May 1;70(2):240-53.
• Hearse DJ. Free radicals and the heart. Bratisl Lek Listy. 1991 Mar-Apr;92(3-4):115-8.
• Hearse DJ. Ischemia at the crossroads? Cardiovasc Drugs Ther. 1988 May;2(1):9-15.
• Hearse DJ. Myocardial protection during ischemia and reperfusion. Mol Cell Biochem. 1998 Sep;186(1-2):177-84.
• Hill AF, Joiner S, Linehan J, Desbruslais M, Lantos PL, Collinge J. Species-barrier-independent prion replication in apparently resistant
species. Proc Natl Acad Sci U S A. 2000 Aug 29;97(18):10248-53.
• Huang S, Liang J, Zheng M, Li X, Wang M, Wang P, Vanegas D, Wu D, Chakraborty B, Hays AP, Chen K, Chen SG, Booth S, Cohen M, Gambetti P, Kong Q. Inducible overexpression of wild-type prion protein in the muscles leads to a primary myopathy in
transgenic mice. Proc Natl Acad Sci U S A. 2007 Apr 17;104(16):6800-5.
• Hundt C, Peyrin JM, Haïk S, Gauczynski S, Leucht C, Rieger R, Riley ML, Deslys JP, Dormont D, Lasmézas CI, Weiss S. Identification of interaction domains of the prion protein with its 37-
kDa/67-kDa laminin receptor. EMBO J. 2001 Nov 1;20(21):5876-86.
• Hutter G, Heppner FL, Aguzzi A. No superoxide dismutase activity of cellular prion protein in vivo. Biol Chem. 2003 Sep;384(9):1279-85.
• Ishikawa T, Akerboom TPM, Sies H. Target Organ Toxicity. CRC Press Inc., Boca Raton FL, 1986; 129–143.
• Jennings RB. Treatment of acute myocardial ischemia. West J Med. 1987 Jul;147(1):62-4.
• Jones DP, Thor H, Andersson B, Orrenius S. Detoxification reactions in isolated hepatocytes. Role of glutathione peroxidase,
catalase, and formaldehyde dehydrogenase in reactions relating to N-
demethylation by the cytochrome P-450 system. J Biol Chem. 1978 Sep 10;253(17):6031-7.
• Jones S, Batchelor M, Bhelt D, Clarke AR, Collinge J, Jackson GS. Recombinant prion protein does not possess SOD-1 activity. Biochem J. 2005 Dec 1;392(Pt 2):309-12.
• Kabouridis PS. Lipid rafts in T cell receptor signaling. Mol Membr Biol. 2006 Jan-Feb;23(1):49-57.
• Kanaani J, Prusiner SB, Diacovo J, Baekkeskov S, Legname G. Recombinant prion protein induces rapid polarization and development
of synapses in embryonic rat hippocampal neurons in vitro. J Neurochem. 2005 Dec;95(5):1373-86.

91
• Kang YJ, Chen Y, Epstein PN. Suppression of doxorubicin cardiotoxicity by overexpression of catalase in the heart of transgenic
mice. J Biol Chem. 1996 May 24;271(21):12610-6.
• Kim BH, Lee HG, Choi JK, Kim JI, Choi EK, Carp RI, Kim YS. The cellular prion protein (PrPC) prevents apoptotic neuronal cell death and
mitochondrial dysfunction induced by serum deprivation. Brain Res Mol Brain Res. 2004 Apr 29;124(1):40-50.
• Klamt F, Dal-Pizzol F, Conte da Frota ML JR, Walz R, Andrades ME, da Silva EG, Brentani RR, Izquierdo I, Fonseca Moreira JC. Imbalance of antioxidant defense in mice lacking cellular prion protein.
Free Radic Biol Med. 2001 May 15;30(10):1137-44.
• Knight RS, Will RG. Prion diseases. J Neurol Neurosurg Psychiatry. 2004 Mar;75 Suppl 1:i36-42.
• Kovacs GC, Linleck-Pozza E, Chimelli L, Araujo AQ, Gabbai AA, Strobel T, Glatzel M, Aguzzi A, Budke H, Creutzfeld-Jacob and inclusion body myositis: abundant disease-associated prion protein in
muscle. Ann Neurol. 2004 Jan;55(1):121-5.
• Kramer ML, Kratzin HD, Schmidt B, Römer A, Windl O, Liemann S, Hornemann S, Kretzschmar H. Prion protein binds copper within the physiological concentration range. J Biol Chem. 2001 May 18;276(20):16711-9.
• Langendorff, O. Untersuchungen am uber lebenden Sangetier Herzen. Pflüegers Archiv. 1985; 61 : 291-322.
• Lasmézas CI, Deslys JP, Robain O, Jaegly A, Beringue V, Peyrin JM, Fournier JG, Hauw JJ, Rossier J, Dormont D. Transmission of the BSE agent to mice in the absence of detectable abnormal prion
protein.Science. 1997 Jan 17;275(5298):402-5.
• Lee JA, Allen DG. Mechanism of acute ischemic contractile failure of heart. Role of intracellular calcium. J Clin Invest. 1991 Aug;88(2):361-7.
• Lefer AM, Weyrich AS, Buerke M. Role of selectins, a new family of adhesion molecules, in ischaemia-reperfusion injury. Cardiovasc Res. 1994 Mar;28(3):289-94.
• Lemasters JJ, Thurman RG. The many facets of reperfusion injury. Gastroenterology. 1995 Apr;108(4):1317-20.
• Levraut J, Iwase H, Shao ZH, Vanden Hoek TL, Schumacker PT. Cell death during ischemia: relationship to mitochondrial depolarization
and ROS generation. Am J Physiol Heart Circ Physiol. 2003 Feb;284(2):H549-58.
• Li Y, Mei GY, Jiang HY, Wang GR, Tian C, Chen C, Wang X, Wang KX, Han J, Dong XP. The construction and identification of the PRNP vectors with ubiquitin or the lysosome-targeting signal. Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi. 2008 Dec;22(6):419-21.
• Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR, Physiology of the prion protein. Physiol Rev. 2008 Apr;88(2):673-728.

92
• Liu H, Cala PM, Anderson SE. Ischemic preconditioning: effects on pH, Na and Ca in newborn rabbit hearts during Ischemia/Reperfusion. J Mol Cell Cardiol. 1998 Mar;30(3):685-97.
• Lopes MH, Hajj GN, Muras AG, Mancini GL, Castro RM, Ribeiro KC, Brentani RR, Linden R, Martins VR. Interaction of cellular prion and stress-inducible protein 1 promotes neuritogenesis and
neuroprotection by distinct signaling pathways. J Neurosci. 2005 Dec 7;25(49):11330-9.
• Málaga-Trillo E, Sempou E. PrPs: Proteins with a purpose: Lessons from the zebrafish. Prion. 2009 Jul;3(3):129-33.
• Mallucci G, Collinge J. Update on Creutzfeldt-Jakob disease. Curr Opin Neurol. 2004 Dec;17(6):641-7.
• Mallucci GR, Ratte S, Asante EA, Linehan J, Gowland I, Jefferys JG, Collinge J. Post-natal knockout of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration.
EMBO J. 2002 Feb 1;21(3):202-10
• Mangé A, Béranger F, Peoc'h K, Onodera T, Frobert Y, Lehmann S. Alpha- and beta- cleavages of the amino-terminus of the cellular prion protein. Biol Cell. 2004 Mar;96(2):125-32.
• Manson JC, Clarke AR, Hooper ML, Aitchison L, McConnell I, Hope J. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol. 1994 Apr-Jun;8(2-3):121-7.
• Marber M. Viewpoint: stunning and longterm perfusion-contraction matching are clinically indistinguishable components of clinical
hibernation and separation, even if practical, is unlikely to matter. Basic Res Cardiol. 1997;92 Suppl 2:26-9.
• Marciano PG, Brettschneider J, Manduchi E, Davis JE, Eastman S, Raghupathi R, Saatman KE, Speed TP, Stoeckert CJ Jr, Eberwine JH, McIntosh TK. Neuron-specific mRNA complexity responses during hippocampal apoptosis after traumatic brain injury. J Neurosci. 2004 Mar 24;24(12):2866-76.
• Maroko PR, Carpenter CB, Chiariello M, Fishbein MC, Radvany P, Knostman JD, Hale SL. Reduction by cobra venom factor of myocardial necrosis after coronary artery occlusion. J Clin Invest. 1978 Mar;61(3):661-70.
• Massimino ML, Ballarin C, Bertoli A, Casonato S, Genovesi S, Negro A, Sorgato MC. Human Doppel and prion protein share common membrane microdomains and internalization pathways. Int J Biochem Cell Biol. 2004 Oct;36(10):2016-31.
• Massimino ML, Ferrari J, Sorgato MC, Bertoli A. Heterogeneous PrPC metabolism in skeletal muscle cells. FEBS Lett. 2006 Feb 6;580(3):878-84.
• McCord JM, Fridovich I. The utility of superoxide dismutase in studying free radical reactions. I. Radicals generated by the interaction
of sulfite, dimethyl sulfoxide, and oxygen. J Biol Chem. 1969 Nov 25;244(22):6056-63.

93
• McKerracher L, Chamoux M, Arregui CO. Role of laminin and integrin interactions in growth cone guidance. Mol Neurobiol. 1996 Apr;12(2):95-116.
• McLennan NF, Brennan PM, McNeill A, Davies I, Fotheringham A, Rennison KA, Ritchie D, Brannan F, Head MW, Ironside JW, Williams A, Bell JE. Prion protein accumulation and neuroprotection in hypoxic brain damage. Am J Pathol. 2004 Jul;165(1):227-35.
• McMahon HE, Mangé A, Nishida N, Créminon C, Casanova D, Lehmann S. Cleavage of the amino terminus of the prion protein by reactive oxygen species. J Biol Chem. 2001 Jan 19;276(3):2286-91.
• Medori R, Montagna P, Tritschler HJ, LeBlanc A, Cortelli P, Tinuper P, Lugaresi E, Gambetti P. Fatal familial insomnia: a second kindred with mutation of prion protein gene at codon 178. Neurology. 1992 Mar;42(3 Pt 1):669-70.
• Miao W, Luo Z, Kitsis RN, Walsh K. Intracoronary, adenovirus-mediated Akt gene transfer in heart limits infarct size following
ischemia-reperfusion injury in vivo. J Mol Cell Cardiol. 2000 Dec;32(12):2397-402.
• Miele G, Alejo Blanco AR, Baybutt H, Horvat S, Manson J, Clinton M. Embryonic activation and developmental expression of the murine prion protein gene. Gene Expr. 2003;11(1):1-12.
• Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402:309 –313.
• Migliaccio E, Mele S, Salcini AE, Pelicci G, Lai KM, Superti-Furga G, Pawson T, Di Fiore PP, Lanfrancone L, Pelicci PG. Opposite effects of the p52shc/p46shc and p66Shc splicing isoforms on the EGF
receptor-MAP kinase-fos signalling pathway. EMBO J. 1997 Feb 17;16(4):706-16.
• Milhavet O, Lehmann S. Oxidative stress and the prion protein in transmissible spongiform encephalopathies. Brain Res Brain Res Rev. 2002 Feb;38(3):328-39.
• Mitteregger G, Vosko M, Krebs B, Xiang W, Kohlmannsperger V, Nölting S, Hamann GF, Kretzschmar HA. The role of the octarepeat region in neuroprotective function of the cellular prion protein.Brain Pathol. 2007 Apr;17(2):174-83.
• Monnet C, Gavard J, Mège RM, Sobel A. Clustering of cellular prion protein induces ERK1/2 and stathmin phosphorylation in GT1-7
neuronal cells. FEBS Lett. 2004 Oct 8;576(1-2):114-8.
• Moore RC, Lee IY, Silverman GL, Harrison PM, Strome R, Heinrich C, Karunaratne A, Pasternak SH, Chishti MA, Liang Y, Mastrangelo P, Wang K, Smit AF, Katamine S, Carlson GA, Cohen FE, Prusiner SB, Melton DW, Tremblay P, Hood LE, Westaway D. Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J Mol Biol. 1999 Oct 1;292(4):797-817.
• Moore RC, Mastrangelo P, Bouzamondo E, Heinrich C, Legname G, Prusiner SB, Hood L, Westaway D, DeArmond SJ, Tremblay P.

94
Doppel-induced cerebellar degeneration in transgenic mice. Proc Natl Acad Sci U S A. 2001 Dec 18;98(26):15288-93.
• Mouillet-Richard, S., Ermonval, M., Chebassier, C., Laplanche, J.L., Lehmann, S., Launay, J.M., Kellermann, O. Signal transduction through prion protein. Science, 2000 289, 1925-1928.
• Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986 Nov;74(5):1124-36.
• Napoli C, Martin-Padura I, de Nigris F, Giorgio M, Mansueto G, Somma P, Condorelli M, Sica G, De Rosa G, Pelicci P. Deletion of the p66Shc longevity gene reduces systemic and tissue oxidative stress,
vascular cell apoptosis, and early atherogenesis in mice fed a high-fat
diet. Proc Natl Acad Sci U S A. 2003;100:2112–2116.
• Nazor KE, Seward T, Telling GC. Motor behaviour and neuropathological deficits in mice deficient form normal prion protein
expression. Biochim Biophys Acta. 2007 Jun;1772(6):645-53.
• Nico PB, Lobão-Soares B, Landemberger MC, Marques W Jr, Tasca CI, de Mello CF, Walz R, Carlotti CG Jr, Brentani RR, Sakamoto AC, Bianchin MM. Impaired exercise capacity, but unaltered mitochondrial respiration in skeletal or cardiac muscle of
mice lacking cellular prion protein. Neurosci Lett. 2005 Nov 4;388(1):21-6.
• Obreztchikova M, Elouardighi H, Ho M, Wilson BA, Gertsberg Z, Steinberg SF. Distinct signaling functions for Shc isoforms in the heart. J Biol Chem. 2006 Jul 21;281(29):20197-204.
• Okado-Matsumoto A, Fridovich I. Assay of superoxide dismutase: cautions relevant to the use of cytochrome c, a sulfonated tetrazolium,
and cyanide. Anal Biochem. 2001 Nov 15;298(2):337-42.
• Oudot A, Martin C, Busseuil D, Vergely C, Demaison L, Rochette L. NADPH oxidases are in part responsible for increased cardiovascular superoxide production during aging. Free Radic Biol Med. 2006 Jun 15;40(12):2214-22.
• Pain T, Yang XM, Critz SD, Yue Y, Nakano A, Liu GS, Heusch G, Cohen MV, Downey JM. Opening of mitochondrial K(ATP) channels triggers the preconditioned state by generating free radicals. Circ Res. 2000 Sep 15;87(6):460-6.
• Pan KM, Baldwin M, Nguyen, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE, Conversion of alpha-helices into beta sheet features in the formation of scrapie prion
protein. Proc. Natl. Acad. Sci. USA 1993, 90: 10962-6.
• Pan T, Wong BS, Liu T, Li R, Petersen RB, Sy MS. Cell-surface prion protein interacts with glycosaminoglycans. Biochem J. 2002 Nov 15;368(Pt 1):81-90.
• Parkin ET, Watt NT, Turner AJ, Hooper NM. .Dual mechanisms for shedding of the cellular prion protein. Biol Chem. 2004 Mar 19;279(12):11170-8.

95
• Paterson AW, Curtis JC, Macleod NK. Complex I specific increase in superoxide formation and respiration rate by PrP-null mouse brain
mitochondria. J Neurochem. 2008 Apr;105(1):177-91.
• Pauly PC, Harris DA Copper stimulates endocytosis of the prion protein. J Biol Chem. 1998 Dec 11;273(50):33107-10
• Peden AH, Ritchie DL, Head MW, Ironside JW. Detection and localization of PrPSc in the skeletal muscle of patients with variant,
iatrogenic, and sporadic forms of Creutzfeldt-Jakob disease. Am J Pathol. 2006 Mar;168(3):927-35.
• Peters PJ, Mironov A Jr, Peretz D, van Donselaar E, Leclerc E, Erpel S, DeArmond SJ, Burton DR, Williamson RA, Vey M, Prusiner SB. Trafficking of prion proteins through a caveolae-mediated endosomal pathway. J Cell Biol. 2003 Aug 18;162(4):703-17.
• Peterson DB, Holian J, Garrison WM. Radiation chemistry of the alpha-amino acids. Gamma radiolysis of solid cysteine. J Phys Chem. 1969 May;73(5):1568-72.
• Prince RC, Gunson DE. Prions are copper-binding proteins. Trends Biochem Sci. 1998 Jun;23(6):197-8.
• Prusiner SB. Prions. Proc. Natl. Acad. Sci. 1998, 95, 13363-13383.
• Prusiner SB. Prions: novel infectious pathogens. Adv Virus Res. 1984;29:1-56.
• Race R, Chesebro B. Scrapie infectivity found in resistant species. Nature. 1998 Apr 23;392(6678):770.
• Radovanovic I, Braun N, Giger OT, Mertz K, Miele G, Prinz M, Navarro B, Aguzzi A. Truncated prion protein and Doppel are myelinotoxic in the absence of oligodendrocytic PrPC. J Neurosci. 2005 May 11;25(19):4879-88.
• Reimer KA, Jennings RB. The “wavefront phenomenon” of myocardial ischemic cell death. II. Trensmural progressin of necrosis
within the framework of ischemic deb size (myocardium at risk) and
collateral flow. Lab Invest. 1979 Jun;40(6):633-44.
• Rieger R, Edenhofer F, Lasmezas CI, Weiss S. The human 37-kDa laminin receptor precursor interacts with the prion protein in
eukaryotic cells. Nat Med. 1997 Dec;3(12):1383-8.
• Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wüthrich K. NMR structure of the mouse prion protein domain PrP(121-321). Nature. 1996 Jul 11;382(6587):180-2.
• Rodgers MA, Sokol HA, Garrison WM. The radiation-induced “hydrolysis” of the peptide boned. J Am Chem Soc. 1968 Jan 31;90(3):795-6.
• Rossi D, Cozzio A, Flechsig E, Klein MA, Rülicke T, Aguzzi A, Weissmann C. Onset of ataxia and Purkinje cell loss in PrP null mice inversely correlated with Dpl level in brain. EMBO J. 2001 Feb 15;20(4):694-702.

96
• Safar J, Roller PP, Gajduesek DC, Gibbs CJ Jr., Thermal stability and conformational transitions of scrapie amyloid (prion) protein
correlate with infectivity. Protein Sci. 1993, 2: 2206-16.
• Samaia HB, Brentani RR. Can loss-of-function prion-related diseases exist? Mol Psychiatry. 1998 May;3(3):196-7.
• Santuccione A, Sytnyk V, Leshchyns’ka I, Schachner M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate
p59Fyn and to enhance neurite outgrowth. J Cell Biol. 2005 Apr 25;169(2):341-54.
• Sarkozi E, Askanas V, Engel WK. Abnormal accumulation of prion protein mRNA in muscle fibres of patients with sporadic inclusion-body
myositis and hereditary inclusion-body myopathy. Am J Pathol. 1994 Dec;145(6):1280-4.
• Schluter KD, Schwartz P, Siegmund B, Piper HM. Prevention of the oxygen paradox in hypoxic-reoxygenated hearts. Am J Physiol. 1991 Aug;261(2 Pt 2):H416-23.
• Schmitt-Ulms G, Legname G, Baldwin MA, Ball HL, Bradon N, Bosque PJ, Crossin KL, Edelman GM, DeArmond SJ, Cohen FE, Prusiner SB. Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J Mol Biol. 2001 Dec 14;314(5):1209-25.
• Schneider B, Mutel V, Pietri M, Ermoval M, Mouillet-Richard S, Kellermann O. NADPH oxidase and extracellular regulated kinases1/2 are targets of prions protein signalling in neuronal and nonneuronal
cells. Proc Natl Acad Sci U S A. 2003 Nov 11;100(23):13326-31.
• Schulz R. Intracellular targets of matrix metalloproteinase-2 in cardiac disease: rationale and therapeutic approaches. Annu Rev Pharmacol Toxicol. 2007;47:211-42.
• Shmerling D, Hegyi I, Fischer M, Blättler T, Brandner S, Götz J, Rülicke T, Flechsig E, Cozzio A, von Mering C, Hangartner C, Aguzzi A, Weissmann C. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell. 1998 Apr 17;93(2):203-14.
• Shyu WC, Lin SZ, Chiang MF, Ding DC, Li KW, Chen SF, Yang HI, Li H. Overexpression of PrPC by Adenovirus-mediated gene targeting reduces ischemic injury in a stroke rat model. J Neurosci. 2005 Sep 28;25(39):8967-77.
• Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997 Jun 5;387(6633):569-72.
• Sommerschild HT, Kirkebøen KA. Preconditioning endogenous defence mechanisms of the heart. Acta Anaesthesiol Scand. 2002 Feb;46(2):123-37.
• Sorgato MC, Bertoli A. From cell protection to death: may Ca2+ signals explain the chameleonic attributes of the mammalian prion
protein? Biochem Biophys Res Commun. 2009 Feb 6;379(2):171-4.
• Sorgato MC, Peggion C, Bertoli A. Is, indeed, the prion protein a Harlequin servant of "many" masters? Prion. 2009 Oct;3(4):202-5.
• Sparkes RS, Simon M, Cohn VH, Fournier RE, Lem J, Klisak I, Heinzmann C, Blatt C, Lucero M, Mohandas T. Assignment of the

97
human and mouse prion protein genes to homologous chromosomes.
Proc Natl Acad Sci U S A. 1986 Oct;83(19):7358-62.
• Spudich A, Frigg R, Kilic E, Kilic U, Oesch B, Raeber A, Bassetti CL, Hermann DM. Aggravation of ischemic brain injury by prion protein deficiency: role of ERK-1/-2 and STAT-1. Neurobiol Dis. 2005 Nov;20(2):442-9.
• Steele AD, Lindquist S, Aguzzi A. The prion protein knockout mouse: a phenotype under challenge. Prion. 2007 Apr;1(2):83-93.
• Steele AD, Zhou Z, Jackson WS, Zhu C, Auluck P, Moskowitz MA, Chesselet MF, Lindquist S. Context dependent neuroprotective properties of prion protein (PrP).Prion. 2009 Oct 16;3(4).
• Stuermer CA, Langhorst MF, Wiechers MF, Legler DF, Von Hanwehr SH, Guse AH, Plattner H. PrPc capping in T cells promotes its association with the lipid raft proteins reggie-1 and reggie-2 and
leads to signal transduction. FASEB J. 2004 Nov;18(14):1731-3.
• Taylor DR, Hooper NM. The prion protein and lipid rafts. Mol Membr Biol. 2006 Jan-Feb;23(1):89-99.
• Thomzig A, Schulz-Schaeffer W, Kratzel C, Mai J, Beekes M. Preclinical deposition of pathological prion protein PrPSc in muscles of
hamsters orally exposed to scrapie. J Clin Invest. 2004 May;113(10):1465-72.
• Ukeda H, Maeda S, Ishii T, Sawamura M. Spectrophotometric assay for superoxide dismutase based on tetrazolium salt 3'--1--
(phenylamino)-carbonyl--3, 4-tetrazolium]-bis(4-methoxy-6-
nitro)benzenesulfonic acid hydrate reduction by xanthine-xanthine
oxidase. Anal Biochem. 1997 Sep 5;251(2):206-9.
• Uptain SM, Lindquist S. Prions as protein-based genetic elements. Annu Rev Microbiol. 2002;56:703-41.
• Valko M, Morris H, Cronin MT. Metals, toxicity and oxidative stress. Curr Med Chem. 2005;12(10):1161-208.
• Vanden Hoek TL, Becker LB, Shao Z, Li C, Schumacker PT. Reactive oxygen species released from mitochondria during brief
hypoxia induce preconditioning in cardiomyocytes. J Biol Chem. 1998 Jul 17;273(29):18092-8.
• Vanden Hoek TL, Shao Z, Li C, Schumacker PT, Backer LB. Mitochondrial electron transport can become a significant source of
oxidative injury in cardiomyocytes. J Mol Cell Cardiol. 1997 Sep;29(9):2441-50.
• Vey M, Pilkuhn S, Wille H, Nixon R, DeArmond SJ, Smart EJ, Anderson RG, Taraboulos A, Prusiner SB. Subcellular colocalization of the cellular and scrapie prion proteins in caveolae-like membranous
domains. Proc Natl Acad Sci U S A. 1996 Dec 10;93(25):14945-9.
• Vincent B, Paitel E, Saftig P, Frobert Y, Hartmann D, De Strooper B, Grassi J, Lopez-Perez E, Checler F. The disintegrins ADAM10 and TACE contribute to the constitutive and phorbol ester-
regulated normal cleavage of the cellular prion protein. J Biol Chem. 2001 Oct 12;276(41):37743-6.

98
• Waggoner DJ, Drisaldi B, Bartnikas TB, Casareno RL, Prohaska JR, Gitlin JD, Harris DA. Brain copper content and cuproenzyme activity do not vary with prion protein expression level. J Biol Chem. 2000 Mar 17;275(11):7455-8.
• Watt NT, Taylor DR, Gillott A, Thomas DA, Perera WS, Hooper NM. Reactive oxygen species-mediated beta-cleavage of the prion protein in the cellular response to oxidative stress. J Biol Chem. 2005 Oct 28;280(43):35914-21.
• Weise J, Crome O, Sandau R, Schulz-Schaeffer W, Bähr M, Zerr I. Upregulation of cellular prion protein (PrPC) after focal cerebral ischemia and influence of lesion severity. Neurosci Lett. 2004 Nov 30;372(1-2):146-50.
• Weise J, Doeppner TR, Müller T, Wrede A, Schulz-Schaeffer W, Zerr I, Witte OW, Bähr M. Overexpression of cellular prion protein alters postischemic Erk1/2 phosphorylation but not Akt
phosphorylation and protects against focal cerebral ischemia. Restor Neurol Neurosci. 2008;26(1):57-64.
• Weise J, Sandau R, Schwarting S, Crome O, Wrede A, Schulz-Schaeffer W, Zerr I, Bähr M. Deletion of cellular prion protein results in reduced Akt activation, enhanced postischemic caspase-3 activation,
and exacerbation of ischemic brain injury. Stroke. 2006 May;37(5):1296-300.
• Weissmann C, Flechsig E. PrP knock-out and PrP transgenic mice in prion research. Br Med Bull. 2003;66:43-60.
• Weissmann C. Molecular genetics of transmissible spongiform encephalopathies. J Biol Chem. 1999 Jan 1;274(1):3-6.
• White AR, Collins SJ, Maher F, Jobling MF, Stewart LR, Thyer JM, Beyreuther K, Masters CL, Cappai R. Prion protein-deficient neurons reveal lower glutathione reductase activity and increased
susceptibility to hydrogen peroxide toxicity. Am J Pathol. 1999 Nov;155(5):1723-30.
• Wong BS, Brown DR, Pan T, Whiteman M, Liu T, Bu X, Li R, Gambetti P, Olesik J, Rubenstein R, Sy MS. Oxidative impairment in scrapie-infected mice is associated with brain metals perturbations
and altered antioxidant activities. J Neurochem. 2001 Nov;79(3):689-98.
• Zaccagnini G, Martelli F, Fasanaro P, Magenta A, Gaetano C, Di Carlo A, Biglioli P, Giorgio M, Martin-Padura I, Pelicci PG, Capogrossi MC. p66ShcA modulates tissue response to hindlimb ischemia. Circulation. 2004;109:2917–2923.
• Zanata SM, Lopes MH, Mercadante AF, Hajj GN, Chiarini LB, Nomizo R, Freitas AR, Cabral AL, Lee KS, Juliano MA, de Oliveira E, Jachieri SG, Burlingame A, Huang L, Linden R, Brentani RR, Martins VR. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 2002 Jul 1;21(13):3307-16.
• Zanusso G, Vattemi G, Ferrari S, Tabaton M, Pecini E, Cavallaro T, Tomelleri G, Filosto M, Tonin P, Nardelli E, Rizzuto N, Monaco S. Increased expression of the normal cellular isoform of prion protein

99
in inclusion-body myositis, inflammatory myopathies and denervation
atrophy. Brain Pathol. 2001 Apr;11(2):182-9.
• Zhang CC, Steele AD, Lindquist S, Lodish HF. Prion protein is expressed on long-term repopulating hematopoietic stem cells and is
important for their self-renewal. Proc Natl Acad Sci U S A. 2006 Feb 14;103(7):2184-9.
• Zielonka J, Vasquez-Vivar J, Kalyanaraman B. Detection of 2-hydroxyethidium in cellular systems: a unique marker product of
superoxide and hydroethidine. Nat Protoc. 2008;3(1):8-21.
• Zweier JL, Flaherty JT, Weisfeldt ML. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc Natl Acad Sci U S A. 1987 Mar;84(5):1404-7.

100

101
AKNOWLEDGEMENTS
Prof. MC Sorgato and Prof. Di Lisa for giving me the possibility to do this
research. Dr. Alessandro Bertoli, whose competence has been fundamental for
my work. Dr. Roberta Menabò, Dr. Andrea Carpi, , Dr. Maria Lina Massimino,
Dr. Cristian Lazzari, Dr. Roberto Stella, Dr. Caterina Peggion, Dr. Luca
Rizzetto.
My sweetheart Ilaria, my Parents who supported me during these years, all
my friends.