force&inducedreversalof&eliminations:+stressed
TRANSCRIPT

Force-‐Induced Reversal of -‐Eliminations: Stressed Disulfide Bonds in Alkaline Solution
Przemyslaw Dopieralski,* Jordi Ribas-‐Arino,* Padmesh Anjukandi, Martin Krupicka, and Dominik Marx* Dr. P. Dopieralski, Dr. J. Ribas-‐Arino, Dr. P. Anjukandi, Dr. M. Krupicka, Prof. Dr. D. Marx Lehrstuhl für Theoretische Chemie, Ruhr-‐Universität Bochum 44780 Bochum (Germany) E-‐mail: [email protected] [email protected] [email protected] Dr. P. Dopieralski Permanent address: Faculty of Chemistry, University of Wroclaw Joliot-‐Curie 14, 50-‐383 Wroclaw (Poland) Dr. J. Ribas-‐Arino Permanent address: Departament de Química Física and IQTCUB Universitat de Barcelona Av. Diagonal 645, 08028 Barcelona (Spain) Dr. M. Krupicka Current address: Max-‐Planck-‐Institut für Chemische Energiekonversion Stiftstrasse 34–36, 45470 Mülheim an der Ruhr (Germany)

2
Abstract
Understanding the impact of tensile forces on disulfide bond cleavage is not only
crucial to the breaking of cross-‐linkers in vulcanized materials such as strained
rubber, but also to the regulation of protein activity by disulfide switches. By using
ab initio simulations in the condensed phase, we investigated the response of
disulfide cleavage by β-‐elimination to mechanical stress. We reveal that the rate-‐
determining first step of the thermal reaction, which is the abstraction of the β-‐
proton, is insensitive to external forces. However, forces larger than about 1 nN
were found to reshape the free-‐energy landscape of the reaction so dramatically
that a second channel is created, where the order of the reaction steps is reversed,
turning β-‐deprotonation into a barrier-‐free follow-‐up process to C−S cleavage. This
transforms a slow and force-‐independent process with second-‐order kinetics into
a unimolecular reaction that is greatly accelerated by mechanical forces.

3
The impact of tensile forces on the kinetics and reaction mechanisms of disulfide
bond reductions has been intensely investigated in recent years.1-‐15 Much of the
current interest in biochemistry stems from the increasingly acknowledged key
role played by the reduction of so-‐called “disulfide switches”, commonly found
in protein regions of pronounced mechanical strain, in the regulation of the activity
of many proteins.14-‐18 Orthogonal to this function in living organisms is the force‐
induced breaking of sulfur‐based cross‐linkers in vulcanized rubber, which is
essential not only in waste rubber recycling (an urgent economic and
environmental task faced by industry worldwide)19-‐21 but also in defining the
mechanical fatigue properties of rubber.22, 23
In landmark single‐molecule experiments in the context of the covalent
mechanochemistry24-‐28 of disulfide bond breaking, Fernandez and co-‐workers
probed the cleavage of such bonds in aqueous alkaline solutions as a function of
applied tensile force.3 These force-‐clamp atomic force microscopy (AFM)
experiments revealed a “mechanochemical switch” at a force of about 0.5 nN above
which the accelerating effect of tensile force on the reaction rate was considerably
diminished.3 In a recent computational study,12 we showed that the reaction
probed in these experiments is S−S bond cleavage triggered by a nucleophilic
attack of an OH− ion onto one of the sulfur atoms by an SN2 mechanism. Upon
studying the response of this reaction pathway to tensile stress, we disclosed that
the mechanochemical switch originates from a force‐induced conformational
rearrangement of the disulfide bridge, which drives the disulfide into a
conformation that is shielded against nucleophilic attack. Our subsequent work
compared the force response of the SN2 mechanism, which is preferred at low
mechanical stress, to other scenarios becoming relevant in the high-‐force regime.29
When it comes to disulfide bond cleavage reactions, it is well known that under
certain circumstances,30-‐32 disulfides can undergo β -‐elimination in alkaline
solution. This requires investigations into the response of this more complex
reaction pathway to mechanical stress to achieve a detailed understanding of the
mechanochemistry of disulfide bridges. Herein, based on ab initio molecular

4
dynamics studies in the condensed phase enhanced with metadynamics
sampling,33 we explore how the free‐energy landscape associated with the β‐
elimination channel gets transformed under the action of external tensile stress.
Unexpectedly, we disclose significant force-‐induced changes in the mechanism at
intermediate forces, which reveals a novel reaction channel aside from the slow
and force-‐insensitive bimolecular process in terms of a greatly accelerated
unimolecular process.
The model system introduced here to explore the mechanical response of the β‐
elimination scenario greatly exceeds our previously studied minimal diethyl
disulfide model in complexity12, 29 (Figure 1, inset), and the model compound was
hence solvated in a spacious bulk water periodic supercell that also hosts the
attacking fully solvated OH− (aq). In particular, the disulfide model employed
herein exhibits one of its sulfur atoms linked to a large organic moiety in which the
β‐carbon atom of the disulfide is bonded to two fragments excised from the
protein backbone used in the AFM experiments.3 This ensures that the acidity of
the β-‐proton (see Figure 1, inset) is properly represented, whereas the far‐
distant sulfur site is saturated using a methyl group. Two so-‐called collective
variables (CVs) were introduced to span the free‐ energy landscape that
characterizes β‐elimination (see the Supporting Information for details): The first
one is the coordination number, CNC−S, between the carbon and sulfur atoms of the
C−S bond (denoted by CV1 in Figure 2). CV2 is the difference in the coordination
number between the β-‐hydrogen and the carbon atom to which it is linked in the
disulfide molecule, CNC−Hβ, and the coordination number of the β-‐hydrogen atom
with respect to the oxygen atom of the hydroxide ion, that is, CNC−Hβ – CNC−Hβ -‐ OH-‐
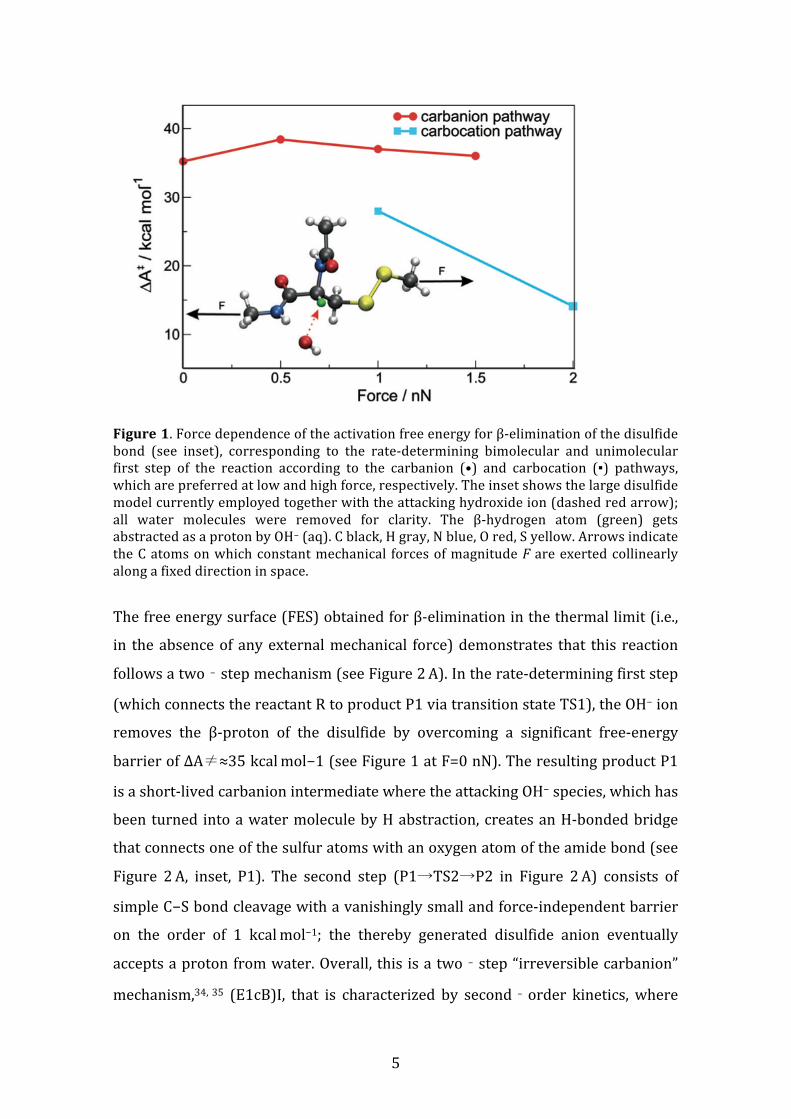
5
Figure 1. Force dependence of the activation free energy for β-‐elimination of the disulfide bond (see inset), corresponding to the rate-‐determining bimolecular and unimolecular first step of the reaction according to the carbanion (•) and carbocation (▪) pathways, which are preferred at low and high force, respectively. The inset shows the large disulfide model currently employed together with the attacking hydroxide ion (dashed red arrow); all water molecules were removed for clarity. The β-‐hydrogen atom (green) gets abstracted as a proton by OH− (aq). C black, H gray, N blue, O red, S yellow. Arrows indicate the C atoms on which constant mechanical forces of magnitude F are exerted collinearly along a fixed direction in space.
The free energy surface (FES) obtained for β-‐elimination in the thermal limit (i.e.,
in the absence of any external mechanical force) demonstrates that this reaction
follows a two‐step mechanism (see Figure 2 A). In the rate-‐determining first step
(which connects the reactant R to product P1 via transition state TS1), the OH− ion
removes the β-‐proton of the disulfide by overcoming a significant free-‐energy
barrier of ΔA≠≈35 kcal mol−1 (see Figure 1 at F=0 nN). The resulting product P1
is a short-‐lived carbanion intermediate where the attacking OH− species, which has
been turned into a water molecule by H abstraction, creates an H-‐bonded bridge
that connects one of the sulfur atoms with an oxygen atom of the amide bond (see
Figure 2 A, inset, P1). The second step (P1→TS2→P2 in Figure 2 A) consists of
simple C−S bond cleavage with a vanishingly small and force-‐independent barrier
on the order of 1 kcal mol−1; the thereby generated disulfide anion eventually
accepts a proton from water. Overall, this is a two‐step “irreversible carbanion”
mechanism,34, 35 (E1cB)I, that is characterized by second‐order kinetics, where

6
the first bimolecular step—involving the conjugate base and yielding carbanion
P1—is rate‐determining and essentially irreversible given that the second step is
very fast. Indeed, this finding is in agreement with general expectations for the
purely thermal reaction as electron‐withdrawing groups in the β-‐position, such
as the CONH2 group in our case, shift β -‐eliminations towards the E1cB
mechanism.35 Moreover, strong bases and high base concentrations also favor
E1cB,35 which is certainly the case for our aqueous alkaline solution.
Figure 2. A) Free-‐energy landscape for β-‐elimination (see Figure 1) in the thermal limit (i.e., at F=0 nN) where only the carbanion pathway is operating. B) Force-‐transformed

7
free-‐energy landscape for β-‐elimination (see Figure 1) at a constant force of F=1 nN where the carbocation channel is energetically preferred over the still operating carbanion pathway. Red and blue arrows indicate the carbanion and carbocation pathways, respectively, whereas R, Pn, and TSn denote reactants, intermediates/products, and transition states, respectively, for which representative snapshots sampled from ab initio metadynamics are shown.
Having successfully deciphered the thermal β-‐elimination reaction mechanism,
we focused on the effect of finite mechanical tensile forces on this process. This
investigation was performed by means of isotensional metadynamics sampling28,36
carried out in the presence of different, but constant external forces (see Figure 1).
For each fixed value of the external force, the isotensional metadynamics
simulations lead to a different so‐called “force‐transformed FES”,36 which
provides the proper reaction landscape including the mechanical distortion.28
At a force of 0.5 nN, the entire mechanism remains unchanged when compared to
the purely thermally activated process (see the corresponding force-‐transformed
FES in the Supporting Information, Figure S3). Interestingly, however, the barrier
ΔA≠ for the rate-‐determining first step remains roughly constant according to
Figure 1, which means that the external force does not result in any acceleration of
the overall reaction. Larger forces do not lead to a reduction of this free-‐energy
barrier either (Figure 1). Our simulations thus clearly reveal that the rate-‐
determining step of the (E1cB)I process, that is, β-‐proton abstraction, is fully
decoupled from the applied mechanical force. Other examples of force-‐insensitive
chemical processes whose reaction coordinates are orthogonal to the mechanical
coordinates can be found in Refs. 5 and 37-‐39.
A dramatic change of scenario is observed upon reaching an intermediate tensile
force of 1 nN. Indeed, Figure 2 B discloses that the force-‐transformed FES enables a
different reaction channel: First, the carbocation P3 is obtained by cleaving the C−S
bond without involvement of the base, OH−(aq), which only subsequently abstracts
the β-‐proton (R→P3→P2); the disulfide anion generated after the first step
eventually stabilizes itself by abstracting a proton from the solvent. The activation
free energy of the rate‐determining first step at F=1.0 nN, ΔA≠≈28 kcal mol−1
according to Figure 1, is much smaller than that associated with the rate‐

8
determining first step of the (E1cB)I process, which is 37 kcal mol−1 for β‐H
abstraction by OH− (aq) at 1 nN. However, the generation of carbocations in
aqueous solution appears to be an utmost unrealistic scenario. Indeed, our
simulations show that no carbocations are generated in the limit of the thermally
activated reaction (i.e., at F=0 nN, see Figure 2 A), and that their generation is also
not feasible at moderate forces below approximately 1 nN (see Figure 1). So, why
is carbocation generation enabled by sufficiently large mechanical forces?
Mechanistic analysis of the novel scenario shows that the same final product P2 as
before is obtained, yet by a vastly different mechanism. It was classified to be a
two-‐step E1 process35 as the rate-‐determining first step leads, in a unimolecular
process (R→P3), to a carbocation that readily loses the β-‐proton (see P3→P2 in
Figure 2 B), therefore leading to first‐order kinetics. The picture underlying this
scenario is that the internal strain generated by mechanically stretching the
molecule is so large that the covalent bond is simply ripped apart (R→P3) before
any other process occurs. The nascent carbocation is short‐lived and stabilizes
itself quickly after its generation by detaching the β-‐proton into the basic
solution. This view is supported by the observation that the rate‐determining R→
P3 process, and thus the entire E1 pathway, is very responsive to tensile forces:
The activation free energy, ΔA≠, is reduced to half from about 28 kcal mol−1 at 1
nN to approximately 14 kcal mol−1 at 2 nN. Moreover, Mulliken population analysis
(see Figure S1) reveals that the α-‐carbon atom involved in the C−S bond cleavage
of P3 intermittently acquires a positive partial charge. Last but not least,
reference‐coupled cluster calculations of C−S bond breaking using a reduced
model embedded in continuum water provide no support for a homolytic diradical
mechanism instead of the heterolytic ionic pathway involving the reported
carbocationic intermediate. In conclusion, the E1 reaction channel favored at
sufficiently large forces not only implies a significant acceleration of a hitherto
force‐insensitive reaction, but also a force‐induced change from a bimolecular
to a unimolecular process and from second-‐order to first-‐order reaction kinetics.

9
Given the vastly different reaction channel that is preferred at forces beyond 1 nN,
our final analysis addresses the nature of the elementary β-‐proton abstraction
step. Figure 3 nicely reveals that C−S cleavage (dark‐brown line) occurs
after/before β-‐H abstraction by OH− (green lines) in the carbanion/carbocation
mechanism depicted in panels B/D at F=1.0 nN. The OH− (aq) hydration shell (red
line) is more flexible in the carbanion scenario, whereas mostly two water
molecules remain hydrogen‐bonded along the carbocation pathway (see panels
A/C, respectively). Moreover, the β-‐proton-‐abstracting OH− (aq) forms a stable
hydrogen bond to one of the NH groups of the reactant molecule (blue line). The
fact that this H-‐bond is broken shortly after the β-‐proton is abstracted, but largely
locked before, suggests that this hydrogen bond keeps the nucleophile at the
reaction center. Indeed, such an hydrogen‐bonded intermediate state (HBIS) has
been observed for base‐catalyzed peptide hydrolysis.38 Here, only a slight
migration of OH− from such an HBIS configuration to the C−Hβ group is required to
achieve the elementary elimination step.
Figure 3. Elementary elimination step involving b-‐proton abstraction by OH-‐ (aq) at a constant force of F = 1 nN along the carbanion and carbocation reaction channels (top and bottom, respectively). The left panels depict configuration snapshots of the reactive complex when OH-‐ (aq) is about to abstract the b-‐proton (green dotted line) and to break

10
its stabilizing hydrogen bond to H-‐N-‐(red dotted line) together with a contour plot of the corresponding spatial distribution functions of the oxygen site of OH in the N-‐H···Hb plane sampled shortly before, during, and after Hb abstraction and thus H2O formation. The bulk water environment present in the simulations is not shown. The panels on the right show in their lower parts the evolution of the following distances along the two pathways: dark brown: C-‐S distance; blue and green lines: distance from the O site of OH-‐ to the H sites of the H-‐N-‐ and Hβ-‐C-‐ groups, respectively; red line: number of water molecules hydrogen-‐ bonded to OH-‐ (aq). The top parts show the coordination of OH-‐ (aq) with respect to the H sites of the H-‐N-‐ and Hβ –C-‐ groups using again blue and green lines, where the final jumps to zero and unity correspond to the termination of the HBIS (see text) and to β-‐proton abstraction, respectively; a simple distance criterion of R<2.4 Å was used to visualize the hydration and coordination numbers. Note that OH-‐ (aq) changes to H2O (aq) along the red line shortly before the green and blue lines cross.
Acknowledgements
We are grateful to partial financial support from the Deutsche Forschungsgemeinschaft (Reinhart Koselleck Grant “Understanding Mechanochemistry”, MA 1547/9) and Cluster of Excellence “RESOLV”, EXC 1069)), the Alexander von Humboldt Stiftung (Humboldt Fellowship to J.R.‐A.), and the Spanish Government (Ramón y Cajal Fellowship to J.R.‐A.), as well as by the National Science Center Poland (2014/13/B/ST4/05009), and the Ministry of Science and Higher Education Poland (627/STYP/9/20l4) for fellowships to P.D. We also gratefully acknowledge the Gauss Centre for Supercomputing (GCS) for providing computing time for a GCS Large Scale Project on JUQUEEN at the Jülich Supercomputing Centre (JSC) as well as additional computational support by HPC‐RESOLV, BOVILAB@RUB, the Rechnerverbund NRW, the Wroclaw Supercomputer Center (WCSS), the Galera‐ACTION Cluster, and the Academic Computer Center in Gdańsk (CI TASK).

11
References
[1] A. P. Wiita, S. R. K. Ainavarapu, H. H. Huang, J. M. Fernandez, Proc. Natl. Acad. Sci. USA 2006, 103, 7222 – 7227.
[2] A. P. Wiita, R. Perez-Jimenez, K. A. Walther, F. Gräter, B. J. Berne, A. Holmgren, J.M. Sanchez-Ruiz, J.M. Fernandez, Nature 2007, 450, 124 – 127.
[3] S. Garcia-Manyes, J. Liang, R. Szoszkiewicz, T. L. Kuo, J. M. Fernandez, Nat. Chem. 2009, 1, 236 – 242.
[4] J. Liang, J. M. Fernandez, ACS Nano 2009, 3, 1628 – 1645.���
[5] T. J. Kucharski, Z. Huang, Q. Z. Yang, Y. Tian, N. C. Rubin, C. D. Concepcion, R. Boulatov, Angew. Chem. Int. Ed. 2009, 48,
7040 – 7043 ; Angew. Chem. 2009, 121, 7174 – 7177.���
[6] W. Li, F. Gräter, J. Am. Chem. Soc. 2010, 132, 16790–16795.
[7] M.F.Iozzi, T.Helgaker, E.Uggerud, J.Phys.Chem.A2011,115,2308 – 2315.���
[8] S. Keten, C.-C. Chou, A. C. van Duin, M. J. Buehler, J. Mech. Behav. Biomed. Mater. 2012, 5, 32 – 40.
[9] F. Hofbauer, I. Frank, Chem. Eur. J. 2012, 18, 16332 – 16338.
[10] I. Popa, P. Kosuri, J. Alegre-Cebollada, S. Garcia-Manyes, J. M. Fernandez, Nat. Protoc. 2013, 8, 1261 – 1276.
[11] Y. Tian, T. J. Kucharski, Q. Z. Yang, R. Boulatov, Nat. Commun. 2013, 4, 2538.
[12] P. Dopieralski, J. Ribas-Arino, P. Anjukandi, M. Krupicka, J. Kiss, D. Marx, Nat. Chem. 2013, 5, 685 – 691.
[13] Y. Li, A. Nese, K. Matyjaszewski, S. S. Sheiko, Macromolecules, 2013, 46, 7196 – 7201.
[14] P. Anjukandi, P. Dopieralski, J. Ribas-‐Arino, D. Marx, PLoS One 2014, 9,
e108812.
[15] B. Zhou, I. B. Baldus, W. Li, S. A. Edwards, F. Gräter, Biophys. J. 2014, 107, 672
– 681.
[16] P. J. Hogg, Trends Biochem. Sci. 2003, 28, 210 – 214.
[17] J. Ahamed, H. H. Versteeg, M. Kerver, V. M. Chen, B. M. Mueller, P. J. Hogg, W.
Ruf, Proc. Natl. Acad. Sci. USA 2006, 103, 13932– 13937.
[18] M. A. Wouters, S. W. Fan, N. L. Haworth, Antioxid. Redox Signaling 2010, 12, 53
– 91.

12
[19] X. Guo, D. Xiang, G. Duan, P. Mou, Waste Manage. 2010, 30, 4 –10.
[20] X. Zhang, Z. Lu, D. Tian, H. Li, C. Lu, J. Appl. Polym. Sci. 2013, 127, 4006 – 4014.
[21] H. Rust, US8957119 B2, 2015.
[22] W. V. Mars, A. Fatemi, Rubber Chem. Technol. 2004, 77, 391 –412.
[23] B. N. J. Persson, O. Albohr, G. Heinrich, H. Ueba, J. Phys. Condens. Matter 2005,
17, R1071 – R1142.
[24] M. K. Beyer, H. Clausen-‐Schaumann, Chem. Rev. 2005, 105, 2921 – 2948.
[25] M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White, J. S.
Moore, Chem. Rev. 2009, 109, 5755 – 5798.
[26] A. L. Black, J. M. Lenhardt, S. L. Craig, J. Mater. Chem. 2011, 21, 1655– 1663. [27] Z. Huang, R. Boulatov, Chem. Soc. Rev. 2011, 40, 2359– 2384. [28] J. Ribas-‐Arino, D. Marx, Chem. Rev. 2012, 112, 5412– 5487. [29] P. Dopieralski, J. Ribas-‐Arino, P. Anjukandi, M. Krupicka, D. Marx, unpublished results. [30] M. Friedman, J. Agric. Food Chem. 1999, 47, 1295– 1319. [31] S. Cohen, C. Price, J. Vlasak, J. Am. Chem. Soc. 2007, 129, 6976– 6977. [32] B. Lagrain, K. de Vleeschouwer, I. Rombouts, K. Brijs, M. Hendrickx, J. Delcour, J. Agric. Food Chem. 2010, 58, 10761– 10767. [33] D. Marx, J. Hutter, Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods, Cambridge University Press, Cambridge, 2009. [34] F. G. Bordwell, Acc. Chem. Res. 1972, 5, 374– 381. [35] M. B. Smith, March's Advanced Organic Chemistry: Reactions, Mechanisms and Structure, 7th ed., Wiley, Hoboken, 2013. [36] P. Dopieralski, J. Ribas-‐Arino, D. Marx, Angew. Chem. Int. Ed. 2011, 50, 7105– 7108; [37] J. Ribas-‐Arino, M. Shiga, D. Marx, Chem. Eur. J. 2009, 15, 13331– 13335. [38] F. Xia, A. Baranowska, S. Cheng, F. Gräter, J. Phys. Chem. B 2011, 115, 10126– 10132. [39] R. Groote, B. M. Szyja, F. A. Leibfarth, C. J. Hawker, N. L. Doltsinis, R. P. Sijbesma, Macromolecules 2014, 47, 1187– 1192.