ft-ir study of the gas-phase thermolysis of triethylgermane
TRANSCRIPT

Organometallics 1995, 14, 3507-3515 3507
FT-IR Study of the Gas-Phase Thermolysis of Triethylgermane
Philip G. Harrison* and Ashley C. Torr Department of Chemistry, University of Nottingham,
University Park, Nottingham NG7 2RD, U.K.
Received December 20, 1994@
Thermolysis of neat triethylgermane in the temperature range 628-653 K gives as products ethene, ethane, and diethylgermane in the ratio 48:14:1 and a silvery metallic film which contains some incorporated ethyl groups. Loss of triethylgermane over the whole temper- ature range follows second-order kinetics with an activation energy of 305(7) kJ mol-l. Second-order rate constants vary from 1.35 mol-l L s-l at 628 K to 12.9 mol-l L s-l a t 653 K. When the decomposition is performed in the presence of methyl iodide, the reaction occurs nearly 3 times faster. Ethene and nitrous oxide are the products when decomposition is carried out in the presence of nitric oxide, and the reaction proceeds at a 20th of the rate of the neat reaction. Decomposition in the presence of excess oxygen affords ethene, carbon monoxide, carbon dioxide, methane, water, formaldehyde, and hexaethyldigermoxane. Mechanisms involving intermediate radicals are proposed.
Introduction
Triethylgermane (TEG; (CzH&GeH) is a possible alternative precursor for the formation of germanium films by MOCVD. Previous decomposition studies have been initiated by both thermal and photolytic tech- niques.' The susceptibility of ethylgermanes toward decomposition under photolysis increases with the number of ethyl groups attached to germanium (ger- mane decomposes to only ca. 2% and triethylgermane decomposes up to 75% under the same conditions). However, little is known about the gas-phase chemistry of alkylgermanes at elevated temperatures. Compared to diethylgermane, the additional ethyl group in tri- ethylgermane produces increased thermal stability. The byproducts of the gas-phase decomposition of triethyl- germane as well as the characteristics of the resulting films have been noted. However, essentially no atten- tion has been paid to the predeposition chemistry of the precursor, even though it has been established that the structure of the precursor molecule and the conditions under which its decomposition is carried out can have a profound effect on the nature and characteristics of the resulting film. In this paper, we describe a detailed account of the chemistry of triethylgermane at elevated temperatures in the gas phase. The reactions are rationalized on the basis of initial Ge-C bond homolysis followed by subsequent radical reactions.
Experimental Section
Triethylgermane was prepared2 by the reduction of EtaGeCl using LiAlH4 (95+%, Aldrich Chemical Co. Ltd) in diethyl ether or tetrahydrofuran solvent under argon. The hydride was transferred to a conventional Schlenk line on which all
a Abstract published in Advance ACS Abstracts, June 15, 1995. ( 1) ( a ) Pola, J.; Parsons, J. P.; Taylor, R. J . Chem. SOC., Faraday
Trans. 1992, 88, 1637. (b) Stanley, A. E.; Johnson, R. A.; Turner, J . B.; Roberts, A. H. Appl. Spectrosc. 1986, 40, 374. I C ) Morancho, R.; Reynes, A.; El Boucham, J.; Sefiani, N.; Mazerolles, P. Proc. Eur. Conf. Chem. Vap. Deposition 1987, 6, 381.
(2) Mochida, K.; Yoshida, Y. Bull. Chem. SOC. Jpn. 1988, 61, 1789.
0276-7333/95/2314-3507$09.00/0
subsequent manipulations were carried out. For experiments involving neat hydride, a known pressure of hydride was admitted to the infrared cell a t ambient temperature. Mix- tures of the hydride with methyl iodide, nitric oxide, or oxygen were prepared using a gas bulb fitted with a small reservoir. A known pressure of the hydride was allowed into the bulb, which was then sealed. The hydride was then isolated in the reservoir by freezing a t 77 K. The desired pressure of the second component was admitted into the remainder of the bulb prior to the opening of the reservoir, and the two components were allowed to mix. The general methodology of the infrared experiments has been described p rev i~us ly .~
All infrared measurements were carried out using a Nicolet BOSXC spectrometer. Mass spectra were recorded using a Micromass VG 7070E mass spectrometer.
Results
Infrared Spectrum and Beer-Lambert Charac- teristics of Triethylgermane. The gas-phase infra- red spectral data of triethylgermane recorded at a pressure of 10.6 Torr at ambient temperature are listed in Table 1. For the purpose of studying quantitatively the thermal decomposition, the band centered at 2016 cm-' ( v ( G ~ - H ) ~ ~ ) was used to quantify the gas-phase abundance of triethylgermane. This band is actually a composite of two conformers4b with individual maxima at 2018 and 2012 cm-', although this does not affect the data. AH the temperature is increased (prior to a temperature sufficient to initiate decomposition), the infrared bands characteristic of triethylgermane begin to broaden but suffer no diminution of integrated area. The limits of the envelope for the v(Ge-H) band increase from 2038-1987 cm-l t o 2056-1971 cm-l as the temperature approaches that necessary to cause decomposition.
The Beer-Lambert plot (Figure 1) was constructed by allowing a known pressure of triethylgermane in the
~~ ~~ ~~ ~~~
(3) Harrison, P. G.; Ashworth, A.; Clarke, E. N.; McManus, J. J. Chem. SOC., Faraday Trans. 1990.86, 4059-4063.
( 4 ) (a) Mackay, K. M.; Watt, R. J . Organomet. Chem. 1966.6.336. (b ) Mckean, D. C.; Torto, I.; Mackenzie, M. W.; Morrison, A. R. Spectrochim. Acta 1983, 39A, 387.
0 1995 American Chemical Society
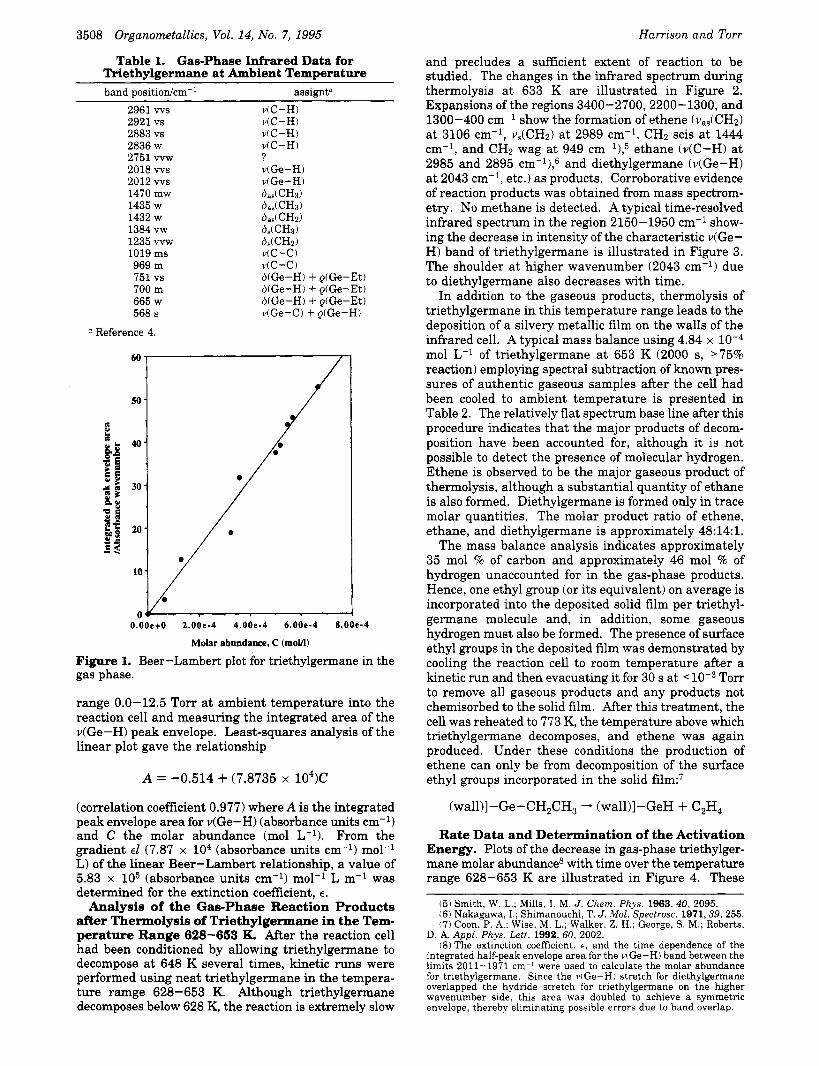
3508 Organometallics, Vol. 14, No. 7, 1995
Table 1. Gas-Phase Infrared Data for Triethylgermane at Ambient Temperature band positiodcm-' assignt"
2961 ws v(C-H) 2921 vs viC-H) 2883 vs ViC-H) 2836 w v(C-H) 2751 vvw ? 2018 ws v(Ge-H) 2012 ws v(Ge-H) 1470 mw O d C H d 1435 w b,&CHd 1432 w b d C H d 1384 vw d,(CHd 1235 w w dJCH2) 1019 ms v ( C - 0 969 m v(C-C) 751 vs b(Ge-H) + g(Ge-Et) 700 m b(Ge-H) + QiGe-Et) 665 w b(Ge-H) + Q(Ge-Et) 568 s v(Ge-C) + g(Ge-H)
Reference 4.
Harrison and Torr
O.OOe+O 2.00e-4 4.00e-4 6.00e-4 8.00e-4
Molar abundance, C (moU1)
Figure 1. Beer-Lambert plot for triethylgermane in the gas phase.
range 0.0-12.5 Torr at ambient temperature into the reaction cell and measuring the integrated area of the v(Ge-H) peak envelope. Least-squares analysis of the linear plot gave the relationship
A = -0.514 + (7.8735 x 104)C
(correlation coefficient 0.977) where A is the integrated peak envelope area for v(Ge-H) (absorbance units cm-l) and C the molar abundance (mol L-l), From the gradient €1 (7.87 x lo4 (absorbance units cm-l) mol-' L) of the linear Beer-Lambert relationship, a value of 5.83 x lo5 (absorbance units cm-l) mol-l L m-l was determined for the extinction coefficient, E .
Analysis of the Gas-Phase Reaction Products after Thermolysis of Triethylgermane in the Tem- perature Range 628-653 K. After the reaction cell had been conditioned by allowing triethylgermane to decompose at 648 K several times, kinetic runs were performed using neat triethylgermane in the tempera- ture ramge 628-653 K. Although triethylgermane decomposes below 628 K, the reaction is extremely slow
and precludes a sufficient extent of reaction to be studied. The changes in the infrared spectrum during thermolysis at 633 K are illustrated in Figure 2. Expansions of the regions 3400-2700,2200-1300, and 1300-400 cm-l show the formation of ethene (vas(CHz) at 3106 cm-l, vs(CH2) at 2989 cm-', CH2 scis at 1444 cm-', and CH;, wag at 949 ~ m - l ) , ~ ethane (v(C-H) at 2985 and 2895 cm-1),6 and diethylgermane (4Ge-H) at 2043 cm-', etc.) as products. Corroborative evidence of reaction products was obtained from mass spectrom- etry. No methane is detected. A typical time-resolved infrared spectrum in the region 2150-1950 cm-' show- ing the decrease in intensity of the characteristic 4Ge- H) band of triethylgermane is illustrated in Figure 3. The shoulder at higher wavenumber (2043 cm-l) due to diethylgermane also decreases with time.
In addition to the gaseous products, thermolysis of triethylgermane in this temperature range leads to the deposition of a silvery metallic film on the walls of the infrared cell. A typical mass balance using 4.84 x mol L-l of triethylgermane at 653 K (2000 s, '75% reaction) employing spectral subtraction of known pres- sures of authentic gaseous samples after the cell had been cooled to ambient temperature is presented in Table 2. The relatively flat spectrum base line after this procedure indicates that the major products of decom- position have been accounted for, although it is not possible to detect the presence of molecular hydrogen. Ethene is observed to be the major gaseous product of thermolysis, although a substantial quantity of ethane is also formed. Diethylgermane is formed only in trace molar quantities. The molar product ratio of ethene, ethane, and diethylgermane is approximately 48:14:1.
The mass balance analysis indicates approximately 35 mol % of carbon and approximately 46 mol % of hydrogen unaccounted for in the gas-phase products. Hence, one ethyl group (or its equivalent) on average is incorporated into the deposited solid film per triethyl- germane molecule and, in addition, some gaseous hydrogen must also be formed. The presence of surface ethyl groups in the deposited film was demonstrated by cooling the reaction cell to room temperature after a kinetic run and then evacuating it for 30 s at Torr to remove all gaseous products and any products not chemisorbed to the solid film. After this treatment, the cell was reheated t o 773 K, the temperature above which triethylgermane decomposes, and ethene was again produced. Under these conditions the production of ethene can only be from decomposition of the surface ethyl groups incorporated in the solid film:7
(wall)l-Ge-CH,CH, - (wall)l-GeH + C,H,
Rate Data and Determination of the Activation Energy. Plots of the decrease in gas-phase triethylger- mane molar abundances with time over the temperature range 628-653 K are illustrated in Figure 4. These
( 5 ) Smith, W. L.; Mills, I. M. J . Chem. Phys. 1963, 40, 2095. (6) Nakagawa, I.; Shimanouchi, T. J . Mol. Spectrosc. 1971,39, 255. ( 7 ) Coon, P. A,; Wise, M. L.: Walker, 2. H.; George, S. M.; Roberts,
D. A. Appl . Phys. Lett. 1992, 60, 2002. 18) The extinction coeficient, E , and the time dependence of the
integrated half-peak envelope area for the vt Ge-H) band between the limits 2011-1971 cm-' were used to calculate the molar abundance for triethylgermane. Since the VI Ge-H) stretch for diethylgermane overlapped the hydride stretch for triethylgermane on the higher wavenumber side, this area was doubled to achieve a symmetric envelope, thereby eliminating possible errors due to band overlap.
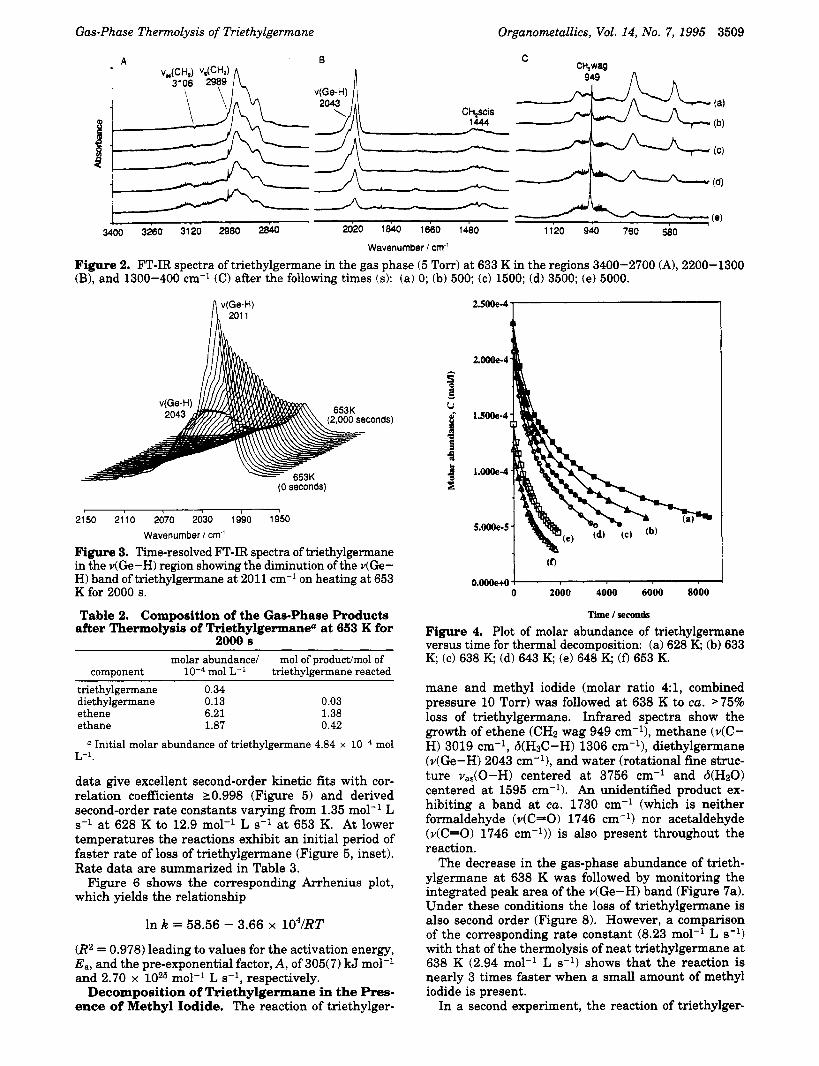
Gas-Phase Thermolysis of Triethylgermane Organometallics, Vol. 14, No. 7, 1995 3509
A C
(a)
(b)
(C)
(d) >- 1
4
T .
A_. 300 3280 3120 2980 2840 2020 1640 1660 1480 1120 940 760 580
Wavenumber / cm"
Figure 2. FT-IR spectra of triethylgermane in the gas phase (5 Torr) at 633 K in the regions 3400-2700 (A), 2200-1300 (B), and 1300-400 cm-l (C) after the following times (SI: (a) 0; (b) 500; (c) 1500; (d) 3500; (e) 5000.
v(Ge-H) 2011 I 2.5OOe-4
(0 seconds)
I I 6
2150 2110 2070 2030 1990 1950 Wavenumber / cm-'
Figure 3. Time-resolved FT-IR spectra of triethylgermane in the v(Ge-H) region showing the diminution of the v(Ge- H) band of triethylgermane at 2011 cm-' on heating at 653 K for 2000 s.
Table 2. Composition of the Gas-Phase Products after Thermolysis of Triethylgermanea at 653 K for
2000 8
molar abundance/ mol of productlmol of component lo-* mol L-' triethylgermane reacted
triethylgermane 0.34 diethylgermane 0.13 0.03 ethene 6.21 1.38 ethane 1.87 0.42
Initial molar abundance of triethylgermane 4.84 x mol L-1.
data give excellent second-order kinetic fits with cor- relation coefficients 20.998 (Figure 5) and derived second-order rate constants varying from 1.35 mol-' L s-l a t 628 K to 12.9 mol-' L s-l at 653 K. At lower temperatures the reactions exhibit an initial period of faster rate of loss of triethylgermane (Figure 5 , inset). Rate data are summarized in Table 3.
Figure 6 shows the corresponding Arrhenius plot, which yields the relationship
Ink = 58.56 - 3.66 x 104/RT
(R2 = 0.978) leading to values for the activation energy, E,, and the pre-exponential factor,A, of 305(7) kJ mol-l and 2.70 x loz5 molW1 L s-l, respectively.
Decomposition of Triethylgermane in the Pres- ence of Methyl Iodide. The reaction of triethylger-
I o.o0oe+o4 ' ' ' ' ' ' '
0 2000 4000 6000 8000
Time I seconds
Figure 4. Plot of molar abundance of triethylgermane versus time for thermal decomposition: (a) 628 K, (b) 633 K (c) 638 K (d) 643 K (e) 648 K (0 653 K.
mane and methyl iodide (molar ratio 4:1, combined pressure 10 Torr) was followed at 638 K to ca. '75% loss of triethylgermane. Infrared spectra show the growth of ethene (CH2 wag 949 cm-l), methane (4C- H) 3019 cm-l, d(H3C-H) 1306 cm-'1, diethylgermane (v(Ge-H) 2043 cm-'), and water (rotational fine struc- ture v,,(O-H) centered at 3756 cm-' and d(H20) centered at 1595 cm-'1. An unidentified product ex- hibiting a band at ca. 1730 cm-' (which is neither formaldehyde (v(C-0) 1746 cm-') nor acetaldehyde (v(C=O) 1746 cm-l)) is also present throughout the reaction.
The decrease in the gas-phase abundance of trieth- ylgermane at 638 K was followed by monitoring the integrated peak area of the v(Ge-H) band (Figure 7a). Under these conditions the loss of triethylgermane is also second order (Figure 8). However, a comparison of the corresponding rate constant (8.23 mol-' L s-l) with that of the thermolysis of neat triethylgermane at 638 K (2.94 mol-' L s-l) shows that the reaction is nearly 3 times faster when a small amount of methyl iodide is present.
In a second experiment, the reaction of triethylger-

3510 Organometallics, Vol. 14, No. 7, 1995 Harrison and Torr
16000
: 12000 -
Y - 8000
4000 7 I 0 2000 4000 6000 8000
Time I s Figure 5. Second-order rate plots for the thermal decom- position of triethylgermane: (W 628 K, (0) 633 K, (A) 638 K (A) 643 K (a) 648 K, (0) 653 K.
Table 3. Second-Order Rate Constant and Arrhenius Data for the Thermolysis of
Triethylgermane in the Temperature Range 628-653 K
second-order rate constant, correlation
tempiK kdmol-l L s-1 coeff 628 633 638 643 648 653
1.48 0.993 2.22 0.995 3.15 0.998 4.21 0.998 8.26 0.998
14.5 0.994
mane with excess methyl iodide (molar ratio 1:4, combined pressure 35 Torr) was followed. In this case, extensive decomposition of triethylgermane had already occurred by the time a temperature of 638 K was attained, and total decomposition was achieved after only 400 s at this temperature (Figure 7b). Products are methane (dC-H) 3019 cm-', G(H3C-H) 1306 cm-'1, ethene (CHZ wag 949 cm-'1, water (rotational fine structure vas(O-H) centered at 3756 cm-l and G(Hz0) centered at 1595 cm-'1, and trace amounts of methanol (4C-0) 1032 cm-l) together with residual methyl iodide. No diethylgermane is formed. Unlike when neat triethylgermane decomposes thermally or when only small amounts of methyl iodide are present, the decomposition of triethylgermane with excess methyl iodide proceeds by first-order kinetics with a rate constant of 3.76 x s-l (Figure 9). In a similar experiment carried out at 553 K (molar ratio 1:4, combined pressure 25 Torr), the product distribution was essentially the same as a t 638 K and the loss of triethylgermane was again first order with a rate constant of 8.71 x 10-5 s-',
Decomposition of Triethylgermane in the Pres- ence of Nitric Oxide. The reaction of triethylgermane and nitric oxide (1:l molar ratio, combined pressure 12
0.00152 0.00154 0.00156 0.00158 0.00160
1/T (1IK) Figure 6. Arrhenius plot for the thermal decomposition of triethylgermane in the temperature range 628-653 K.
0 1000 2000 3000 4000 5000
Time I seconds
Figure 7. Comparison of the reaction rate profiles at 638 K (a) for a 4:l molar ratio mixture of triethylgermane and methyl iodide; (b) for a 1:4 molar ratio mixture of trieth- ylgermane and methyl iodide; (c) for neat triethylgermane.
Torr) gave ethene (CHZ wag 949 cm-'1 and nitrous oxide as products, but no diethylgermane. The fate of the germanium is uncertain due to the poor quality of the spectra. At large absorption in the 4GeOGe) region is observed, although unequivocal assignment is not pos- sible. Decomposition of triethylgermane under these conditions is much slower than for neat triethylger- mane. Whereas neat triethylgermane is almost totally decomposed after 1700 s, in the presence of nitric oxide only ea. 10% of triethylgermane has reacted after 3300 s (Figure 10) and the consumption of the germane follows second-order kinetics with a rate constant of 0.71 mol-' L s-l (Figure 11).
Decomposition of Triethylgermane in the Pres- ence of Oxygen. Infrared spectra (Figure 12) show that triethylgermane is totally absent from a mixture of triethylgermane and oxygen (1:lO molar ratio, com-

Gas-Phase Thermolysis of Triethylgermane Organometallics, Vol. 14, No. 7, 1995 3511
0 IO00 2000 3000 4000 5000
Time I seconds
Figure 8. Second-order rate plots for the thermal decom- position at 638 K of (a) a 4:l molar ratio mixture of triethylgermane and methyl iodide and (b) neat triethyl- germane.
I -10.5
0 100 200 300 400 500
Time I seconds
Figure 9. First-order rate plot for the thermal decomposi- tion at 638 K of a 1:4 molar ratio mixture of triethylger- mane and methyl iodide.
bined pressure 44 Torr) by the time a temperature of 643 K is reached. Identified products are ethene (CH2 wag 949 cm-'1, carbon monoxide (v(C0) 2144 cm-'1, carbon dioxide (~as(CO2) 2349 cm-', d(C02) 667 cm-'1, methane (v(C-H) 3019 cm-l, G(H3C-H) 1306 cm-'1, water (rotational fine structure vdO-H) centered at 3756 cm-' and d(H20) centered at 1595 cm-'1, and formaldehyde (~as(CH2) 2843 cm-l, vS(CH2) 2783 cm-', v(C=O) 1746 cm-l, CH:! scis 1500 cm-', e(CH2) 1249 cm-'h9 Even though decomposition of triethylgermane is complete, subsequent reactions occur as time elapses a t 643 K. A second carbonyl compound (not identified) with a band at 1730 cm-' is formed, and substantial band growth below 1400 cm-l is also observed, as is the formation of hexaethyldigermoxane (Table 4). Carbon
(9) Harvey, K. B.; Ogilvie, J. F. Can. J. Chem. 1962, 40, 85.
'"I
0 1000 2000 3000 4000
Time I seconds
Figure 10. Comparison of the reaction rate profiles at 653 K (a) for a 1:l molar ratio mixture of triethylgermane and nitric oxide and (b) for neat triethylgermane.
21500
20500 - " c - ii 5
19500
18500
Time I seconds
Figure 11. Second-order rate plot for the thermal decom- position at 638 K of a 1:l molar ratio mixture of triethyl- germane and nitric oxide.
monoxide and carbon dioxide continue to grow continu- ously up to 2000 s, after which time the abundance of both remains approximately constant.
Discussion
Product and kinetic data for the thermal decomposi- tion of triethylgermane under a variety of conditions are summarized in Table 5.
Thermolysis of neat triethylgermane in the temper- ature range 628-653 K gives as gaseous products ethene, ethane, and diethylgermane in a typical molar ratio of 48:14:1. Simultaneously, a silvery metallic coating is deposited on the walls of the reaction cell, which from the mass balance contains ca. 33% carbon. The overall stoichiometry may be represented by the equation

3512 Organometallics, Vol. 14, No. 7, 1995 Harrison and Torr
3i00 3400 2900 2400 1900 1400 900 <bo
Wavenumber 1 cm"
Figure 12. FT-IR spectra of a 1:lO molar ratio mixture of triethylgermane and oxygen in the gas phase at 643 K a h r (a) 0 s, (b) 300 s, (c) 1000 s, (d) 2200 s, and (e) 2900 s.
Table 4. Comparison of Infrared Data (1600-400 cm-') for HexaethyldigermoxaxW and the Product from the Thermolysis of Triethylgermane in the
Presence of Excess Oxygen (Eta&)zO product assignt 1451 (m) 1414 (m) 1374 (w) 1218 (w)
1016 (m) 962 (w) 855 (vs) 699 ( s ) 582 ( s ) 536 (w)
1464 (m) 1420 (m) 1385 (m) 1202 (w) 1066 (s ) 1031 (SI 988 ( m ) 891 (vs) 699 (s ) 600 (s) 572 (m)
O1 Cross, R. J.; Glockling, F. J. Organomet. Chem. lW, 3, 146.
Et,GeH - C2H4 + C,H, + Et,GeH, + (HxGeEtJGe),,,id
Loss of triethylgermane follows second-order kinetics over the whole of the temperature range studied, and rate constants vary from 1.35 mol-l L s-l at 628 K to 12.9 mol-l L s-l a t 653 K. From the corresponding Arrhenius plot, the activation energy for the decomposi- tion of triethylgermane under these conditions is 305(7) kJ mol-'.
Similar product distributions have been observed for the decomposition of triethylgermane under photolytic conditions. Irradiation (excimer laser photolysis at 193 nm) of the ethylgermanes Et,GeHr-, (n = 1-3) results in the formation of large quantities of ethane and ethene together with small quantities of ethylgermanes with a lower number of ethyl groups in the molecule.la The dominant gaseous product from photolysis of triethyl- germane was ethane, and the ethane to ethene product ratio was (2.8-3.8):l. Trace amounts of n-butane and diethylgermane are also formed, and in addition to these products, a dark film was obtained on the inner surface of the photolysis reactor. Only very slight decomposi- tion occurred with a barely visible deposition of the germanium during the Con-laser-induced photolysis of triethylgermane (irradiation of the maximum vapor pressure for 15 s at 1075 cm-', 100 W/cm2 output power), although germane, ethene, and hydrogen were observed as gas-phase products.1b
Thermal decomposition of triethylgermane under dynamic conditions in a cold-wall CVD reactor has been shown to start at a minimum temperature of 623 K and also affords ethene and hydrogen as the gas-phase products.lc The mechanism proposed involved succes- sive elimination of ethene molecules and finally hydro- gen:
Et,GeH - Et,GeH, + C2H4
Et,GeH, - EtGeH, + C2H4
EtGeH, - Ge + C2H4 + 2H2
Products resulting from radicals (eg . methane, propane, butane) were not observed.
In the present study, when a small amount of methyl iodide (TEGMeI 4: 1 molar ratio) is present as a radical initiator, decomposition of triethylgermane is again second order but occurs a t a faster rate at the same temperature ( x 2.8 at 638 K). Under these conditions the gas-phase products are ethene, methane, diethyl- germane, and water. When the decomposition is per- formed at 638 K in the presence of excess methyl iodide (1:4 molar ratio), the reaction is faster and becomes first order with respect to loss of triethylgermane and the constitution of the gas phase changes somewhat. Ob- served gas-phase products comprise methane, ethene, water, and trace methanol (together with residual unreacted methyl iodide), but no diethylgermane is formed. The presence of water and methanol in the gas phase indicates that adsorptioddesorption reactions of methyl radicals must be occurring at the surface of the cell wall, since this is the only source of oxygen available in the system. The reaction is still first order when the decomposition is performed at 553 K using the same 1:4 molar ratio of reactants but is much slower (0.023 x rate at 638 K), and now the gaseous products comprise only methane, water, and methanol.
Nitric oxide usually inhibits radical reactions, func- tioning as a radical scavenger. The reaction of triethyl- germane and nitric oxide (1:l molar ratio) at 653 K is second order with respect to loss of triethylgermane and slower (0.05 x rate of neat decomposition at 653 K). Gaseous products are ethene and nitrous oxide together with residual nitric oxide.
Thus, since the decomposition of triethylgermane is promoted by methyl iodide, a radical reaction promoter, and inhibited by nitric oxide, a radical reaction inhibitor, it would appear that the mechanism involves a gas- phase radical process. A concerted molecular elimina- tion of ethene from triethylgermane cannot be unequivo- cally excluded as a parallel reaction pathway, although the observation that no diethylgermane is formed serves to exclude this possibility. Some ethene is, however, formed by an elimination process from ethyl groups incorporated in the film at 773 K, which may or may not involve radicals.
In the gas phase, the observed data indicate that the thermolysis of triethylgermane proceeds by a radical chain mechanism, the component steps of which are shown in Scheme 1. For the initiation step we prefer that involving Ge-C bond homolysis (step 1) rather than Ge-H homolysis
Et,GeH - Et,Ge* + H' ( la)

Gas-Phase Thermolysis of Triethylgermane
since Ge-C bond energies are ca. 20 kJ mol-l lower (D(Me3Ge-Me) = 320 kJ mol-', D(Me3Ge-H) = 344 kJ mol-l, D(H3Ge-H) = 346 kJ mol-l)lo.ll and also the A factor for reaction 2 would be expected to be ca. 100 times lower than that for (1). Subsequent steps involve hydrogen abstraction from triethylgermane at germa- nium by ethyl radicals and hydrogen atoms to generate the Et3Ge' radicallo (steps 2 and 3). Although /.?-hydro- gen abstraction from tetraethylgermane by benzoyl radicals giving the 'CHzCHzGeEt3 radical has been observed,12 the relative C-H (441 kJ mol-')lo and Ge-H (344 kJ mol-l)lo bond energies suggest that hydrogen abstraction at germanium will be highly favored. Since we have no information concerning the evolution of molecular hydrogen, it is not possible to estimate the relative extents of hydrogen abstraction by hydrogen atoms (step 2) compared to abstraction by ethyl radicals (step 3), although it might be expected that the former is more important. Further propagation reactions involve the stepwise homolytic loss of ethyl ligands from ethylgermyl radicals (steps 4-71.
Organometallics, Vol. 14, No. 7, 1995 3513
Scheme 1. Proposed Radical Mechanism for the Thermolysis of Triethylgermane
F Ge + Et* EtGe* ,
'' (EtGe)sol,d (1 0)
That ethene is formed in sigmficantly greater amounta than ethane indicates that step 8 occurs extensively. Step 8 will be fast under these conditions (reported values of log 4s-l in the range 12.9-13.58 and E in the range 159-175 kJ m ~ l - ' ) . ' ~ J ~ With a half-life of <1 s at 628 K, this step is probably be the principal process responsible for removing ethyl radicals from the system. If this is indeed the case, hydrogen atoms would be generated in significant quantities, and there- fore hydrogen abstraction (which would be more facile than by ethyl radicals) would be expected to be extensive (step 3). Ethene could be formed by the elimination of ethene from the 'CHzCH2EtzGeH radical resulting from abstraction at the /.?-carbon of the ethyl group, analogous to that reported12 to occur for the 'CHzCHzGeEt3 radical:
Et,GeH + Et' - 'CH,CH,Et,GeH + Et,GeH + H - 'CH,CH,Et,GeH + H,
Initiation: 'CH,CH,Et,GeH - CZH4 + Et,HGe'
Et3GeH - Et,HGe' + Et' (1)
Propagation:
Et,GeH + Et' - Et,Ge' + C2H6 (2)
Et,GeH + H - Et,Ge' + H, (3)
Et,Ge' - Et,Ge: + Et' (4)
Et,Ge: - EtGe' + Et' ( 5 )
Et,HGe' - EtHGe: + Et' (6)
EtHGe: - HGe' + Et' (7)
Et' - C,H, + H (8)
Termination:
H 2% inactive products (9)
Mass balance data indicate significant carbon and hydrogen incorporation in the deposited film, and evolu- tion of ethene can be detected by the thermal decom- position of the film at 773 K. Approximately one ethyl group per germanium atom appears to be incorporated in the deposited film. It is, therefore, relatively unlikely that loss of the last ethyl ligand from germanium (path a in eq 10) takes place in the gas phase. Rather, it would appear that condensation from the gas phase takes place at this stage, giving a highly ethylated germanium film (path b in eq 10).
However, as stated above, we consider C-H abstraction to be unimportant in the case of triethylgermane.
Chain termination is by hydrogen atom deactivation at the cell wall (step 9). Since hydrogen atom elimina- tion from ethyl radicals (step 8) is extensive, the flux of hydrogen atoms reaching the cell walls is likely to be much greater than the flux of ethyl groups, although impingement of the latter at the cell walls will also result in their removal from the system. Hydrogen atoms are known to interact strongly with surfaces and surface-bound organic species.15J6 Particularly relevant to the present discussion is the formation of surface I-Ge-H monohydride species upon adsorption of atomic hydrogen on germanium(ll1) surfaces at 500 K.17
The diethylgermane observed may be formed by either a combination reaction between H atoms and EtzHGe' radicals or by hydrogen abstraction from tri- ethylgermane by a EtzHGe' radical:
H + Et,HGe' - Et,GeH,
Et,GeH + Et,HGe' - Et,GeH, + Et,Ge
The identity reaction
Et,GeH + Et,Ge' - Et,Ge' + Et,GeH
(10) Jackson, R. A. J. Organomet. Chem. 1979, 166, 17. (11) Clarke, K. B.; Griller, D. Organometallics 1991, 10, 746. (12) Razuvaev, G. A.; Vyazankin, N. S.; Gladyshev, E. N. Dokl. Akad.
(13) Louchs, L. F.; Laidler, K. J. Can. J. Chem. 1967, 45, 2795. (14) Trenwith, A. B. J . Chem. SOC., Faraday Trans. 2, 1986,82,457
(15) Cheng, C. C.; Lucas, S. R.; Gutleben, H.: Choyke, W. J.; Yates,
(16) Lutterloh, C.; Schenk, A.; Biener, J.; Winter, B.; Kuppers, J .
(17) Crowell, J. E.; Lu, G. J . Electron Spectrosc. Relat. Phenom.
Nauk SSSR 1963, 153. 104.
and references therein.
J. T., Jr. Surf. Sci. Lett. 1992, 273, L441.
Surf. Sci. 1994, 316, L1039.
1990,54-55, 1045.

3514 Organometallics, Vol. 14, No. 7, 1995 Harrison and Torr
Table 5. Summary of Reaction Data for the Pyrolysis of Triethylgermane under a Variety of Conditions temp ( KM order wt
reactants molar ratio pressure (Torr) products loss of Et3GeH rate constanta Et3GeH 628-65315 CzH4, C2H6, EtpGeHz 2 1.48-14.5 EtaGeH + Me1 4: 1 638110 C2H4, CH4, EtzGeHn, H2O 2 8.23
1:4 638135 CzH4, CH4, H20, MeOH 1 3.76 10-3 1:4 553125 CzH4, CH4, H20, MeOH 1 8.71 x 10-E
Et3GeH + NO 1:l 653/12 CzH4, N2O 2 0.71 Et3GeH + 0 2 1:lO 643144 C2H4, CO, CO2, CH4, H20,
HCHO, (Et3Ge)zO Second-order units mol-1 L s-l, first-order units SKI.
almost certainly occurs, although it is not detectable in the present experimental arrangement.
According to Scheme 1, the rate of loss of triethylger- mane from the system is given by the expression
-d[Et,GeHl/dt = k,[Et,GeHI + k,[Et'l[Et,GeHl + k,[Hl[Et,GeHl (11)
If (9) is the sole termination step, at steady state [Et'l and [HI are given by the expressions
[H'I = (k,/k,)[Et,GeHI (12)
[Et'] = (kl/kg)[Et,GeHl{(k3/ka~[Et3GeHl + kdk8) (13)
which, after substitution in eq 11 affords the rate equation
-d[Et,GeHYdt =
(k,k~ka)[Et,GeH12{(k3/kg)[Et,GeHl + l } + (klk3/kg)[Et,GeH12 (14)
Since ks[EtsGeHYkg << 1, this expression reduces to
-d[Et,GeH]/dt = kl[k,/ka + k,/kg1[Et,GeH12 (15)
which shows second-order dependence of the rate on triethylgermane, as observed experimentally. The com- plex rate constant term involves, besides the rate constant for the initiation process, the rate constants for processes involving hydrogen atoms (steps 3 and 9) and ethyl radicals (steps 2 and 8). In the initial stages of reaction at lower temperatures, the first term in eq 14 is significant, and an apparent enhancement in rate is observed. The overall activation energy, Eact, for the process is determined experimentally to be 305(7) kJ mol-'; however, the complexity of the rate constant term in eq 15 precludes discussion of the component contri- butions to the Arrhenius parameters.
In the presence of methyl iodide the rate of decom- position of triethylgermane increases, but the kinetic dependence changes from second order a t low methyl iodide abundance to first order at high methyl iodide abundance. Significant quantities of methane are formed, and hence hydrogen abstraction by methyl radicals
Et,GeH + Me' - Et,Ge' + CH, (16)
is now important in addition to the other reactions in Scheme 1. At low abundances of methyl iodide this results in an enhancement of the rate of initiation; i.e., the rate of initiation is now given by kl[EtsGeHI +
Scheme 2. Mechanism for the Formation of Nitrous Oxide in the Reaction of Triethylgermane
with Nitric Oxide R3Ge* + NO - R3GeON:
1.2 3 R3GeON:NOGeR3 - R3Ge* + R3GeO* + N 2 0
\ Et3GeH / R3Ge* + R3GeOH
x2. -H20 1 R3GeOGeR3
R = H, Et
kls[EtsGeHI[MeIl. However, at high abundances of methyl iodide a very large concentration of methyl radicals is produced; these radicals are able to effectively scavenge EtsGe' radicals before any further ethyl group elimination can take place (step 17).
Et,Ge' + Me' - Et,GeMe (17)
Hence, the rate of loss of triethylgermane is now given by the expression
-d[Et,GeHYdt = k,,[Et,GeHI[Me'I
which, since [Me'] is very large and therefore constant, can be rewritten as
- d[E t,GeHYdt = k ,,,[E t,GeHl
thereby rationalizing the change to first-order kinetics with loss of triethylgermane.
Reaction of triethylgermane with nitric oxide produces nitrous oxide and ethene. The formation of nitrous oxide most probably takes place via scavenging of germy1 radicals by NO to form R3GeON: intermediates, which then dimerize. Nitrous oxide is formed by elimination from the intermediate dimer (Scheme 2). Similar behavior has been observed in the reaction of trimethylsilyl radicals with nitric oxide to give nitrous oxide.18 Although it was not possible to unequiv- ocally distinguish any germanium-containing prod- ucts, it is most likely that an organodigermoxane is formed.
In the presence of excess oxygen the decomposition of triethylgermane is very fast. Reaction is complete by the time a temperature of 643 K is attained, affording as products ethene, carbon monoxide, carbon dioxide,
118) Nay, M. A.; Woodall, G. N. C.; Strausz, 0. P.; Gunning, H. E. J. Am. Chem. SOC. 1966.87, 179.

Gas-Phase Thermolysis of Triethylgermane
methane, water, formaldehyde, and hexaethyldigermox- ane: Et,GeH + 100, - C,H, + CO + CO, +
CH, + H,O + H,CO + (Et,Ge),O
Although kinetic data were not obtained in this present case, the mechanism most probably parallels that determined for the autoxidation of trimethylgermane in the temperature range 493-533 K.19 Over a wide molar ratio of reactants (Me3GeH:Oz ratio in the range 1:l to 1:31) loss of MesGeH follows second-order kinetics, but the reaction was found to be independent of oxygen abundance. The products of the reaction are hexa- methyldigermoxane (MesGeOGeMes) and water, and no other products are apparent. The principal features of the mechanism are (i) an initial abstraction of the hydridic hydrogen atom from germanium, (ii) reaction of the trimethylgermyl radical thus formed with oxygen, giving the (trimethylgermy1)peroxyl radical, MeaGeOO', (iii) conversion into trimethylgermyl hydroperoxide, MesGeOOH, by further H abstraction from MesGeH, (iv) 0-0 bond fission to form (trimethylgermy1)oxyl (MesGeO') and hydroxyl radicals, (v) propagation of the reaction by H abstraction from MesGeH, forming the intermediate MesGeOH and water, and (vi) self-con- densation of two MesGeOH molecules to form hexa- methyldigermoxane. Analogous processes are expected to take place in the present case with triethylgermane. The formation of products of more exhaustive oxidation is a consequence of the harsher reaction conditions.
(19) Harrison, P. G.; Podesta, D. M. Organometallics 1994,13, 1569.
Organometallics, Vol. 14, No. 7, 1995 3515
It is pertinent to note that the reaction of triethylger- mane with ozone at 195 K generates triethylgermanium hydrotrioxide, EtsGeOOOH, which at higher tempera- tures eliminates oxygen to give EtsGeOH and subse- quently ( E t 3 G e ) ~ 0 . ~ ~
Conclusions
1. Triethylgermane decomposes by a free-radical mechanism in the temperature range 628-653 K by second-order kinetics with an activation energy of 305(7) kJ mol-l, giving as gaseous products ethene, ethane, and diethylgermane in a molar ratio of 48:14:1. Second- order rate constants vary from 1.35 mol-l L s-l at 628 K to 12.9 mol-' L s-l at 653 K. 2. In the presence of small amounts of methyl iodide
the rate is enhanced and the kinetics are still second order, but with excess methyl iodide the kinetics change to first order. 3. Nitric oxide inhibits the decomposition, but the
presence of oxygen accelerates the reaction substan- tially.
Acknowledgment. We thank the SERC for support in the form of a studentship (to A.C.T.). We thank the reviewers for some helpful suggestions.
OM940971L
120) Koenig, M.; Barrau, J.; Hamaida, N. B. J . Organomet. Chem. 1988,356, 133.