functional annotation of animal genomes (faang) - alan archibald
TRANSCRIPT

Function Annotation of Animal Genomes (FAANG)
Alan L. Archibald The Roslin Institute and Royal (Dick) School of Veterinary Studies
University of Edinburgh

Genotype - phenotype • Aim
– To predict outcomes • Efficacy of drug • Susceptibility to cancer • Performance of daughters of elite dairy bull • Susceptibility to nematode infections
• Discovery – From phenotype to genotype (gene)
• Prediction – From genotype to phenotype – From sequence to consequence

Phenome
Growth
Feed efficiency
Body composition
Disease resistance
Adapted from Ritchie et al. 2015 Nature Reviews Genetics 16: 85
From sequence to consequence

Value of domesticated animals
• Basic biology • Models for human biology • Agriculture
• Genetic / phenotypic diversity • Genotype to phenotype

Evolution, selection, adaptation • Domesticated animal genomes
– Wide evolutionary range • Shellfish, insects, fish, birds, mammals
• Sequence variation, function, time – Long time scales (millions of years)
• Evolution, speciation – Shorter time scales
• Domestication, signatures of (artificial) selection

Biomedical models
• Pigs, sheep, dogs, chicken,…. • Monogenic inherited diseases (coding sequence) • Genetically modified mice • Failure to recapitulate phenotype • Need better models (e.g. CF pig, HD sheep)
– i.e. understanding of genotype to phenotype

Prediction - success
• Selective breeding • Animal model (phenotypic selection) • Genomic selection (genotypic selection) • Black boxes – lack of understanding of mechanisms

Genomic selection • GS theory developed in 2001 before technology available • First 50K SNP chip (cattle) 2008; 650K in 2010 • GS implemented in all major livestock sectors in developed
world • GS is underpinning faster, more accurate and sustainable
genetic improvement • From SNPs to sequence (via imputation) • Adding knowledge of SNP effects
– Coding/non-coding; known/predicted


Successes – from association to causation
• DGAT1 – dairy cattle, milk yield
• Callipyge – sheep, muscling • MSTN – sheep, muscling • IGF2 – pigs, muscling • Noteworthy
– Regulatory sequences, epigenetics
• One gene at a time: slow, inefficient

Genome-wide • Genome projects
– International collaborative consortia – Major domesticated species – draft(y)/draughty genomes
• Unannotated genome = limited value • State of annotation
– Coding sequences, SNPs – Missing: transcript complexity, regulatory sequences

Status of annotated genomes CHICKEN PIG SHEEP COW HUMAN MOUSE
Assembly Galgal4 Sscrofa10.2 Oar_v3.1 UMD3.1 GRCh38 GRCm38.p2Database version 77.4 77.102 77.31 77.31 77.38 77.38Base Pairs 1,072,544,763 3,024,658,544 2,534,344,180 2,649,685,036 3,381,944,081 3,480,955,279Golden Path Length 1,046,932,099 2,808,525,991 2,619,054,388 2,670,422,299 3,096,649,726 2,730,871,774Genebuild by Ensembl Ensembl Ensembl Ensembl Ensembl EnsemblGenebuild method Full genebuild Full genebuild Mixed strategy build Full genebuild Full genebuild Full genebuildGenebuild last updated Dec-13 Feb-14 Dec-13 Sep-11 Aug-14 Jul-14Coding genes 15,508 21,630 20,921 19,994 20,364 22,592Small non coding genes 1,558 2,989 3,985 3,825 9,673 5,860Long non coding genes 135 14,817 5,385Pseudogenes 42 568 291 797 14,415 7,377Gene transcripts 17,954 30,585 27,099 26,740 196,345 99,934Total genes 17,066 24,754 24,906 23,819 44,854 33,837Ratio transcripts/gene 1.1 1.2 1.1 1.1 4.4 3.0

Reference genome improvement • PacBio long read technology
– Pbjelly • Sheep, cattle (Baylor); Chicken (WashU)
• de novo assembly – Goat, pig, sheep, cattle
• Disruptive technology, multiple genome(s) assemblies – Annotation - “Best in genome” – Graph visualization, but alignment tools not available

Discovering functional sequences • Evolutionary
– Sequence comparison, conservation
– 1000G, G10K,… – Genome sequence sufficient – Conserved, but what is it? – Highly variable ≠ non-
functional
• Functional, biochemical – Assay-by-sequence – *ENCODE, iHEC, Epigenome
roadmap – Expensive – Exploring 4-demensional
space (location + time) – Noise or biologically
meaningful?



ENCODE



Headlines
80.4% participates in at least one biochemical RNA- and/or chromatin-associated event in at least one cell type
95% lies within 8 kilobases (kb) of a DNA–protein interaction (as assayed by bound ChIP-seq motifs or DNase I footprints)
99% is within 1.7 kb of at least one of the biochemical events measured by ENCODE


Summary of the coverage of the human genome by ENCODE data.
Manolis Kellis et al. PNAS 2014;111:6131-6138
©2014 by National Academy of Sciences

Headlines
It is possible to correlate quantitatively RNA sequence production and processing with both chromatin marks and transcription factor binding at promoters
indicating that promoter functionality can explain most of the variation in RNA expression.

Headlines
Single nucleotide polymorphisms (SNPs) associated with disease by GWAS are enriched within non-coding functional elements, with a majority residing in or near ENCODE-defined regions that are outside of protein-coding genes.
In many cases, the disease phenotypes can be associated with a specific cell type or transcription factor.
See also Hindorff et al 2009 PNAS 106: 9362 88% of trait associated SNPs are intronic / intergenic

The complementary nature of evolutionary, biochemical, and genetic evidence.
Manolis Kellis et al. PNAS 2014;111:6131-6138
©2014 by National Academy of Sciences

Epigenetic and evolutionary signals in cis-regulatory modules (CRMs) of the HBB complex.
Manolis Kellis et al. PNAS 2014;111:6131-6138
©2014 by National Academy of Sciences

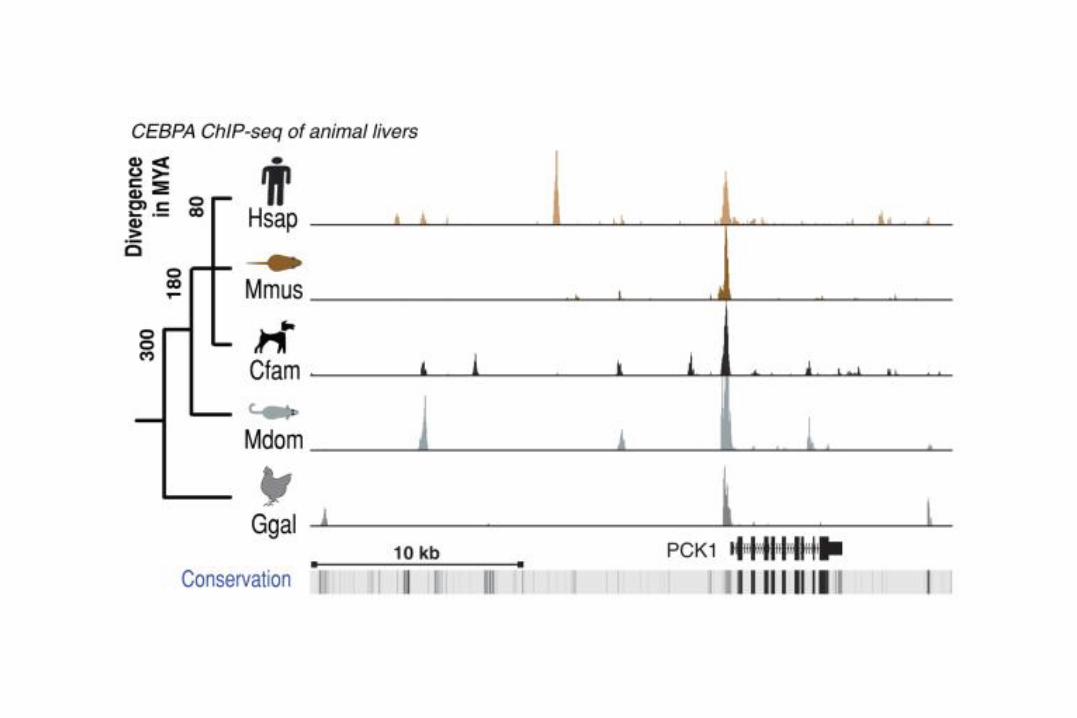



~$150 million

Functional Annotation of ANimal Genomes (FAANG)
An international collaborative programme

• By-product of biology-led research – development, differentiation, responses to perturbation
• Focus on target tissues – musco-skeletal – immune tissues
• Limited assays – DNaseI, FAIREseq, ATAC-seq – histone marks (promoters, enhancers) – Methylation (BS-seq, RRBS) – RNAseq (stranded), CAGE
Functional Annotation of ANimal Genomes (FAANG)

Virtuous cycle
Experimental data
Improved function annotation
FAANG annotation
pipeline
Re-analyse re-interpret

Transcriptomics discovery, differential expression
• From ESTs via microarrays to RNAseq • Expression atlases
• Pig: microarray (published), RNAseq (in progress) • Sheep: RNAseq (‘pilot’ data used for annotation, in progress) • Chicken: RNAseq (in progress), Avian RNA-Seq Consortium (Burt & Smith
2015) • Buffalo: RNAseq (project funded)
• Transcript complexity – Assembly from short reads challenging – Full length transcript sequences - PacBio

CHICKEN PIG SHEEP COW HUMAN MOUSEAssembly Galgal4 Sscrofa10.2 Oar_v3.1 UMD3.1 GRCh38 GRCm38.p2Database version 77.4 77.102 77.31 77.31 77.38 77.38Base Pairs 1,072,544,763 3,024,658,544 2,534,344,180 2,649,685,036 3,381,944,081 3,480,955,279Golden Path Length 1,046,932,099 2,808,525,991 2,619,054,388 2,670,422,299 3,096,649,726 2,730,871,774Genebuild by Ensembl Ensembl Ensembl Ensembl Ensembl EnsemblGenebuild method Full genebuild Full genebuild Mixed strategy build Full genebuild Full genebuild Full genebuildGenebuild last updated Dec-13 Feb-14 Dec-13 Sep-11 Aug-14 Jul-14Coding genes 15,508 21,630 20,921 19,994 20,364 22,592Small non coding genes 1,558 2,989 3,985 3,825 9,673 5,860Long non coding genes 135 14,817 5,385Pseudogenes 42 568 291 797 14,415 7,377Gene transcripts 17,954 30,585 27,099 26,740 196,345 99,934Total genes 17,066 24,754 24,906 23,819 44,854 33,837Ratio transcripts/gene 1.1 1.2 1.1 1.1 4.4 3.0

A novel gene model: complete with putative UTRs and alternate transcripts as identified by sequencing full length cDNAs derived from chicken brain using PacBio long read technology.

Sheep gene atlas Texel (ram, ewe, ewe lamb, 16d embryo) 50+ tissues per animal, whole embryo
- Samples acquired, RNA prepared Illumina paired ends (2 x 150 bp)
- > 1 Tb stranded RNAseq data Ensembl RNAseq gene models Funded 3SR, RoslinFoundation

Cerebrum Abomasum Skeletal muscle, biceps Testes, epididymis
Brain stem Rumen Skeletal muscle, longissimus dorsi
Corpus luteum, ovary, ovarian follicles
Tonsil Duodenum Skin (side/back) Uterus, cervix, placenta
Cerebellum Omentum Spleen Mammary gland
Hypothallamus Caecum Mesenteric lymph node
Pituitary gland Colon Precapuslar lymph node
Adrenal gland Rectum Peyer’s patch Whole embryo
Thyroid gland Alveolar macrophages
Bone marrow


Sheep RNAseq gene models & expression profiles

Scale warning

Functional annotation of the sheep genome • BBSRC funded 2013 – 2016
– David Hume, Alan Archibald, Mick Watson, Kim Summers, Bruce Whitelaw; Emily Clark, Iseabail Farquhar
• RNA-seq expression atlas – 2,500 tissue/cells samples (5 aliquots each) – 3 mature males, 3 mature females, embryo time course (+pregnant
ewes), lamb time course – 200 in prep for RNA-seq
• CAGE analysis for TSS and enhancers • Genome editing tests for functionality

CAGE – finding promoters and enhancers
Pig alveolar macrophages: CSF1 TSS (DAH gene)

CAGE – finding promoters and enhancers
Chicken CAGE tags from a range of tissues, visualised and quantified around the ACTA1 locus in the ZENBU browser


Adipose, muscle methylomes
• 180 samples; what is an appropriate summarising strategy? • MeDIP-seq lacks precision / resolution


H3K27me3 in sheep muscle

• By-product of biology-led research – development, differentiation, responses to perturbation
• Focus on target tissues – musco-skeletal – immune tissues
• Limited assays – DNaseI, FAIREseq, ATAC-Seq – histone marks (promoters, enhancers) – methylation – RNAseq (stranded), CAGE
Functional Annotation of ANimal Genomes (FAANG)


Data management, sharing and publication • Data hubs model (ENCODE) • Toronto Statement of pre-publication data sharing

Data Flow in FAANG: The data generated by the FAANG consortium will use standard lab protocols and be submitted to EMBL-EBI archives and their peers. The DCC will ensure the data are available to the DAG and collect the analysis results back so they can be released to the wider community and viewable within the main Genome browsers.

Screen shot of pig H3K4me3 and H3K27ac ChIP-Seq data around the POUF5F1 (OCT4) locus delivered from the Roslin Track Hub and visualised in the Ensembl genome browser.

Published 25 March 2015

Functional Annotation of ANimal Genomes
Core assays Transcription Stranded RNA-seq, total (ribo-depleted), poly A+, small RNAs Chromatin accessibility and architecture ATAC-seq CCTF (ChiP-seq) (insulator) Histone modification marks (ChIP-seq) H3K4me3 – promoters active genes, TSS H3K27me3 – genes silenced by regional modification H3K27ac – active regulatory elements (active vs inactive P & E) H3K4me1 – enhancers and other distal elements

Functional Annotation of ANimal Genomes
Additional assays Methylation Whole genome or Reduced Representation Bisulfite Sequencing Transcription Factor Binding Sites (TFBS) ChiP-seq Genome conformation Chromosme Conformation Capture (CCC) HiC
The FAANG Consortium @faangomics

Funding • USDA-NIFA – Huaijun Zhou, UCDavis • INRA – FrAgENCODE –Elisabetta Giuffra, INRA Jouy-en-Josas • ERC – Martien Groenen, Wageningen • PENCODE – Lusheng Huang, China
• BBSRC Ensembl for farmed animals (2012-15; 2015-19) • BBSRC “Establishing the infrastructure for functional
annotation of farmed animal genomes” • BBSRC “Reading and interpreting the code hidden in farm
animal genomes and epigenomes”

“Reading and interpreting the code hidden in farm animal genomes and epigenomes”
• The Roslin Institute – A Archibald, D Burt, D Hume, D Vernimmen, M Watson
• European Bioinformatics Institute – L Clarke, P Flicek
• The Genome Analysis Centre – M Caccamo, M Clark, F di Palma, D Swarbreck
• submitted 8th January 2015 • Reviews received 24 March 2015
Strategic LoLa application

“Reading and interpreting the code hidden in farm animal genomes and epigenomes”
• Chicken, cattle, pigs, sheep – adult – developmental stages
• Data Coordination Centre
• RNA-seq – PacBio, Illumina (short, long)
• CAGE • ATAC-seq • RRBS • ChIP-seq
– H3K4me3, H3K27me3, H3K27ac, H3K4me1, CCTF, TF

WG
5 training
WG
4 dissemination, com
ms.
WG1 Biological resources
WG3 Data standards, analysis, sharing
Stakeholders incl. researchers, funders, end users, public
WG2 Expt. standards
FAANG-Europe COST Action
International FAANG initiative
Submitted 24 March 2015
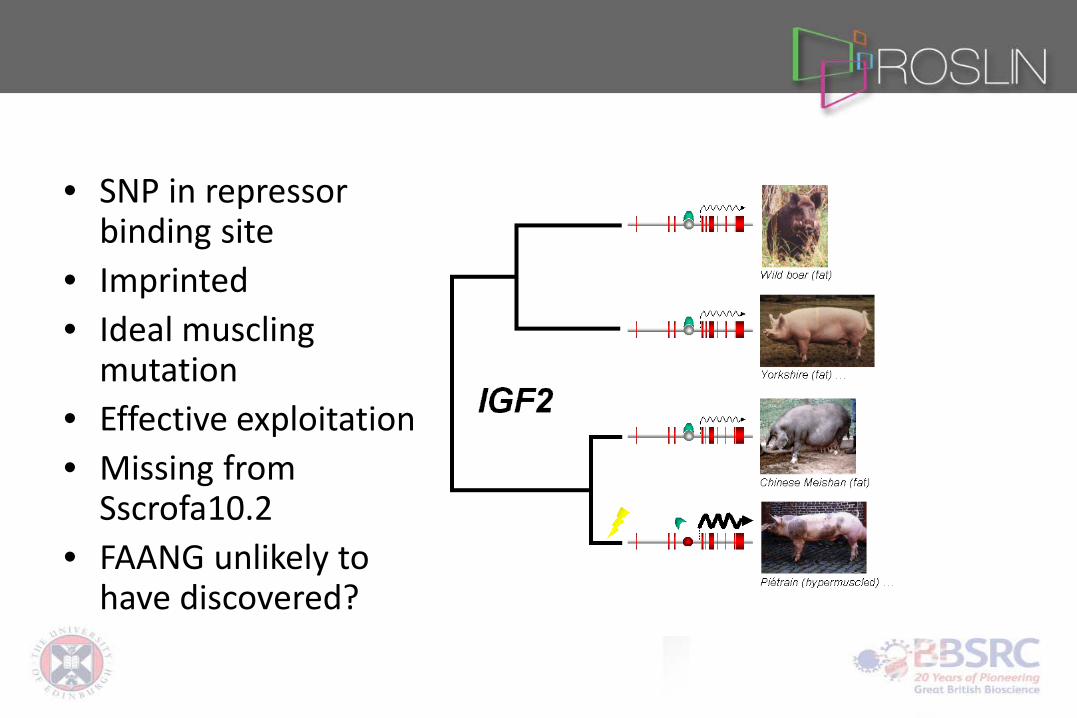
• SNP in repressor binding site
• Imprinted • Ideal muscling
mutation • Effective exploitation • Missing from
Sscrofa10.2 • FAANG unlikely to
have discovered?

Sequencing Bioinformatics
Next-generation sequencing • 6x Illumina HiSeq 2500 • 3x Illumina MiSeq Sanger sequencing • ABI 3730
Sequencing • De novo assembly, SNP discovery,
metagenomics, …. Functional genomics • RNA-seq, ChIP-seq, microarray
analyses, … Interpretation • Pathway, network analyses,.. Kit • 232 Intel Xeon 64-bit cores, 6Gb
RAM/core, 264TB high performance storage

Microarrays Genotyping
Technologies • Affymetrix • Agilent • Illumina
Applications • Gene expression • CGH
High density / custom arrays Illumina • iScan, Infinium • BeadXPress, GoldenGate • BeadChip Affymetrix • GeneTitan • Axiom

genomics.ed.ac.uk

genomics.ed.ac.uk
Jan 12, 2015 University of Edinburgh and University of Glasgow and Illumina form The Scottish Genomes Partnership
Scottish Genomes Partnership announce they will install 15 HiSeqX machines
Currently, limited to use for sequencing human samples