functional nanomachines: recent advances in synthetic ...szolcsanyi/education/files/the... ·...
TRANSCRIPT

Tetrahedron Letters 59 (2018) 334–346
Contents lists available at ScienceDirect
Tetrahedron Letters
journal homepage: www.elsevier .com/locate / tet le t
Digest paper
Functional nanomachines: Recent advances in synthetic molecularmachinery
https://doi.org/10.1016/j.tetlet.2017.12.0360040-4039/� 2017 Elsevier Ltd. All rights reserved.
⇑ Corresponding author.E-mail address: [email protected] (J.D. Crowley).
James A. Findlay, James D. Crowley ⇑Department of Chemistry, University of Otago, PO Box 56, Dunedin, New Zealand
a r t i c l e i n f o
Article history:Received 31 October 2017Revised 8 December 2017Accepted 11 December 2017Available online 14 December 2017
Keywords:Molecular machineryMechanically interlocked architecturesNon-interlocked architecturesMolecular motorsNano-machines
a b s t r a c t
A mini-review: As the top-down approach for miniaturisation of technology reaches its inherent limita-tions, robust strategies to build nanoscale machinery components, which have the ability to convert aninput energy into motion, from the molecular level up, become increasingly important. Nature is cer-tainly the most proficient in the control of molecular level motion; nevertheless, many successes havebeen enjoyed in the pursuit of mimicking key aspects of nature’s molecular machines, including two stateswitches, ion pumps, unidirectional rotary motors and molecular robots that can move molecular cargo.This mini-review outlines of some of the most impressive recent examples towards this end.
� 2017 Elsevier Ltd. All rights reserved.
Contents
Mechanically interlocked architectures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 335Non-interlocked machines . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 338Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 344Acknowledgments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 345References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 345
Biological molecular machines capable of performing motionand transport (for example F1Fo adensosine triphosphate (ATP)synthase and flagella (rotary), dynein/kinesin and myosin (transla-tory)), are integral to biological systems.1 Synthetic analoguescould find use in nanoscale device applications such as robotics,2
medical diagnostics3 and medicine delivery,4 environmental reme-diation5 and high density information storage and processing6
amongst others.7 As such, synthetic chemists have taken inspira-tion from biological systems and have been investigating syntheticmolecular machinery components for more than 20 years.8
The development of synthetic molecular machinery requirescontrol over translational and/or rotary motion (the two most fun-damental movements) on command, and in a reliable and reversi-ble manner. For the most part this has been achieved usingmechanically interlocked architectures (MIAs). These interlockedsystems are held together via mechanical bonds9–11 rather that
more tradition covalent bonds. Two of the more prevalent typesof MIAs are catenanes and rotaxanes (Fig. 1). Catenanes consistof two or more macrocyclic rings linked via a mechanical bond(Fig. 1a) whereas rotaxanes are composed of a macrocyclic ringthreaded onto an axle with ‘stoppers’ blocking dethreading/disso-ciation at either end (Fig. 1b). Due to the mechanical bondinginherent in MIAs these molecules are promising structures formolecular machine-type applications because, in principle, theypermit the controlled movement and positioning of the interlockedcomponents with respect to one another. For their pioneering workon template methods for the synthesis of MIAs and the exploita-tion of some of these interlocked molecules for the developmentof functional molecular machines, Sauvage12 and Stoddart13 wereawarded the 2016 Nobel prize in Chemistry.14 As there are nowmany different template methods available for the synthesis ofMIAs,15–23 the majority of the most impressive synthetic molecularmachines to date have been MIAs. However, the generation oflarge, complicated, machine like interlocked systems can still bedifficult. Recognizing this many groups have focused on exploiting

Fig. 1. Generic cartoon representations of a [2]catenane (a) and a [2]rotaxane (b).
J.A. Findlay, J.D. Crowley / Tetrahedron Letters 59 (2018) 334–346 335
non-interlocked architectures.24,25 Indeed, Feringa’s elegant non-interlocked machine systems26–28 led to the co-award of the2016 Nobel prize.29
The field of molecular machines has been exhaustivelyreviewed a number of times in the past 10 years.30–36 Therefore,in this mini-review we make no attempt to be comprehensiverather we have focused on more recent advances (from 2015 on)in the field.
Mechanically interlocked architectures
Mechanical interlocked machines are becoming more and moresophisticated. An important feature of biological molecularmachinery is the ability to drive systems away from equilibriumstates. An elegant MIA37 mimicking characteristics of biologicalsystems such as membrane carrier proteins, which act to drive ionsor small molecules against charge and/or concentration gradi-ents,38–40 was recently reported by Stoddart and co-workers. Theartificial ‘‘molecular pump” produced was able to utilise a flashingenergy ratchet mechanism.35 The pump function was based on theformation of a rotaxane through consecutive addition of cationicmacrocycles, increasing the local concentration of small chargedmacrocycles. The structure of the pump consists of a dumbbell/thread featuring a dimethylpyridinium at one end linked to a4,40-bipyridinium unit, followed by an isopropylphenylene ‘‘stop-per” moiety (Fig. 2). The second larger 2,6-diisopropylphenyl‘‘stopper” was linked to the thread through a 1,2,3-triazole and aC11 alkyl chain. When the tetracationic macrocycles, cyclobis-(paraquat-p-phenylene) (CBPQT4+) and the dumbbell/thread com-ponents are combined in solution, they initially repel each otherdue to coulombic repulsion forces. However, upon reduction ofthe CBPQT4+ rings to CBPQT2(+�), and simultaneously the reductionof the 4,40-bipyridinium units to 4,40-bipyridinium+� units, the sta-bilising radical-radical pimerisation41 interactions promote thethreading of the macrocycles onto the cationic recognition siteforming thermodynamically stable tris-radical tricationic com-plexes. Upon oxidation of these components there are strong elec-trostatic repulsion forces, meaning they must be driven apart.However, dethreading was not observed as the coulombic repul-sion of the two cationic sections of the thread is too high. Insteadthe CBPQT4+ macrocycle was forced over the so-called energetic‘speed bump’ unit, the isopropylphenylene group, and onto theC11 alkyl chain. The threading of the CBPQT4+ rings onto the methy-lene chain was confirmed through proton nuclear Overhausereffect and diffusion ordered spectroscopy (NOESY and DOSY)experiments. Upon repeating the reduction process a secondCBPQT4+ ring was threaded onto the [2]rotaxane generating anew high energy [3]rotaxane featuring two cationic macrocycles.
Daisy chain (DC) motifs42 (Fig. 3a) have been reported a numberof times since the first examples in the late 1990’s and are recog-nised as a useful tool in the realization of synthetic molecular mus-cles/actuators.43 Coutrot and co-workers recently reported a tetra-interlocked architecture (a hetero-[4]rotaxane, Fig. 3b) which com-prised a [c2]DC linked to two [2]rotaxanes. The system wasdesigned to undergo two different reversible molecular motions
upon alteration of the pH.44 The architecture was synthesised inthe hope of generating multiple different motions within a singleMIA, something of importance if the complexity of biologicalmolecular machinery is ever to be rivaled. A pseudo-[c2]DC wasaccessed via self-sorting of a dibenzo-24-crown-8 (DB24C8)macrocycle (green) bearing an oligomethylene chain beginningwith an ammonium cationic recognition (pink) site to form a her-maphroditic dimer. A pseudo-[2]rotaxane is similarly prepared bythreading of unsubstituted DB24C8 rings onto a thread bearing analkyne at one end and a di-tert-butyl substituted anilinium (red)stopper group at the other, where the anilinium cation acts as arecognition site for the macrocycle. The oligomethylene chains ofthe pseudo-[c2]DCs are terminated by azide groups allowing themto undergo a copper-catalysed azide-alkyne cycloaddition (CuAAC)with the supplied alkyne. This process results in both the forma-tion of a tetra-interlocked architecture and simultaneously gener-ated a third unoccupied recognition site on each threadcomponent. Methylation of the 1,2,3-triazole units generatedcationic triazolium sites (blue). The DB24C8 macrocycles havethe strongest interaction with the ammonium groups as they pos-sess a +1 charge and two protons with which to hydrogen bondwith the oxygen atoms of the cycle. The N-methyltriazolium sta-tions however do not have the ability to hydrogen bond and there-fore it was rationalized the macrocycles will only reside there upondeprotonation of either the ammonium or the anilinium sites. Theanilinium sites also possess a +1 charge and two protons but aremore easily deprotonated than the ammonium sites, thereforeunder basic conditions (1M NaOH) the unsubstituted DB24C8units, initially at the anilinium stations, will be shuttled to the tri-azolium stations. NMR studies confirmed that the DB24C8 can beshuttled from the anilinium to the triazolium stations via cyclingbetween acidic (HCl) and basic (1M NaOH) conditions. However,disappointingly the authors could not find appropriate conditionsto effect the extension and contraction of the [c2]DCs componentof the hetero-[4]rotaxane machine. The model [c2]DC that doesnot feature the extra DB24C8 macrocycles was able to extendand contraction when treated with acid and base. However, theammonium stations of the hetero-[4]rotaxane machine could notbe deprotonated despite treatment with a vast excess of sodiumhydroxide. The authors suggest that a combination of steric andelectronic factors make the deprotonation impossible in their sys-tem. The work clearly shows that as MIAs become more compli-cated, control over the machine like molecular motion can bemore difficult to design.
Chiu and co-workers have reported on the first discrete cyclic[c3] and [c4]DCs, which exhibited extension and contractionmotions in two and three dimensions respectively (Fig. 4).45 These‘‘molecular muscles” were attained through synthesis of a mono-meric species featuring a series of rigid thread components sepa-rating a 2,20-bipyridine (bpy) binding site (pink) perpendicular tothe thread and a binding pocket within a pyridyl macrocycle(orange) which was co-linear to the thread. A terminal aminegroup at the opposite end of the thread to the macrocyclic unitallowed addition of a stopper unit (blue) and simultaneous forma-tion of a diphenylurea recognition site (green). Addition of anequimolar amount of Zn2+ ions to the monomer templated the for-mation of the interlocked systems. Subsequent reaction with bis(trifluoromethyl)isocyanate capped the thread component captur-ing both the [c3] and [c4] MIAs, depending on the length and flex-ibility of the linker unit. The rigidity of the threads meant that thesmaller [c2]DCs could not form, unless the linker, X, was made longenough as to allow sufficient bending of alkynyl units. The synthe-sis process generated the ‘extended’ molecular muscles as the Zn2+
ions were bound between the 2,20-bpy and macrocyclic units.Removal of the Zn2+ ions (using N,N,N0,N0-tetrakis(2-pyridyl-methyl)ethylenediamine (TPEN)) led to the formation of the con-
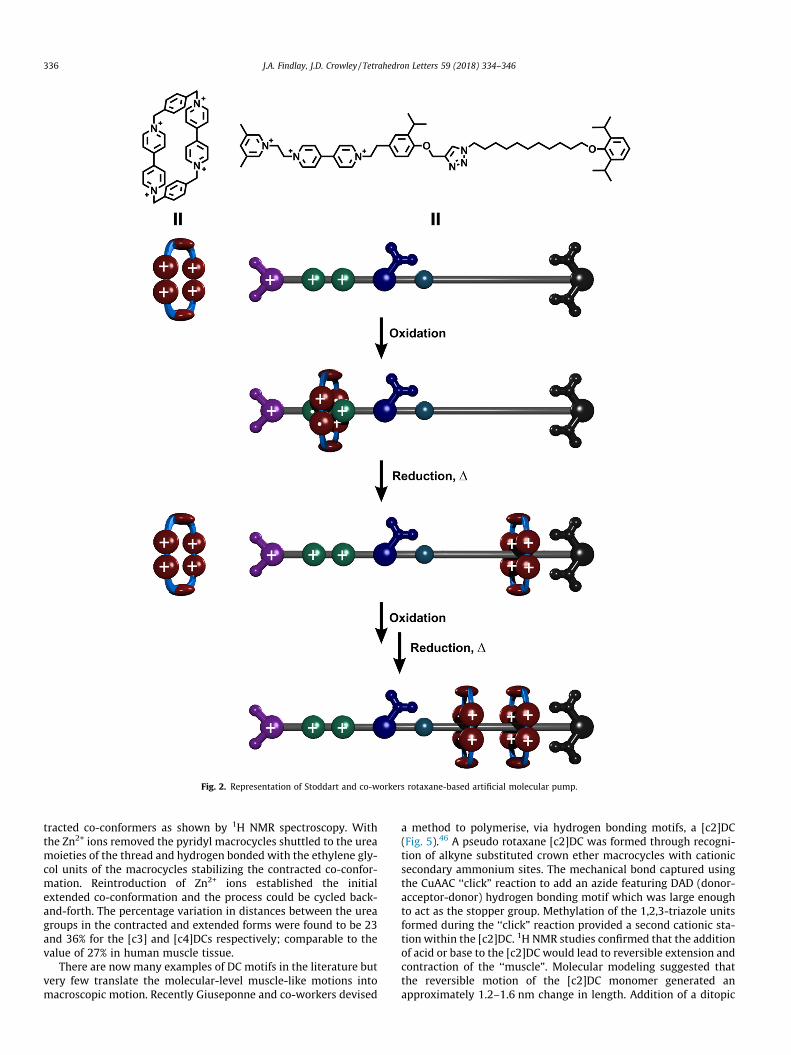
Fig. 2. Representation of Stoddart and co-workers rotaxane-based artificial molecular pump.
336 J.A. Findlay, J.D. Crowley / Tetrahedron Letters 59 (2018) 334–346
tracted co-conformers as shown by 1H NMR spectroscopy. Withthe Zn2+ ions removed the pyridyl macrocycles shuttled to the ureamoieties of the thread and hydrogen bonded with the ethylene gly-col units of the macrocycles stabilizing the contracted co-confor-mation. Reintroduction of Zn2+ ions established the initialextended co-conformation and the process could be cycled back-and-forth. The percentage variation in distances between the ureagroups in the contracted and extended forms were found to be 23and 36% for the [c3] and [c4]DCs respectively; comparable to thevalue of 27% in human muscle tissue.
There are now many examples of DC motifs in the literature butvery few translate the molecular-level muscle-like motions intomacroscopic motion. Recently Giuseponne and co-workers devised
a method to polymerise, via hydrogen bonding motifs, a [c2]DC(Fig. 5).46 A pseudo rotaxane [c2]DC was formed through recogni-tion of alkyne substituted crown ether macrocycles with cationicsecondary ammonium sites. The mechanical bond captured usingthe CuAAC ‘‘click” reaction to add an azide featuring DAD (donor-acceptor-donor) hydrogen bonding motif which was large enoughto act as the stopper group. Methylation of the 1,2,3-triazole unitsformed during the ‘‘click” reaction provided a second cationic sta-tion within the [c2]DC. 1H NMR studies confirmed that the additionof acid or base to the [c2]DC would lead to reversible extension andcontraction of the ‘‘muscle”. Molecular modeling suggested thatthe reversible motion of the [c2]DC monomer generated anapproximately 1.2–1.6 nm change in length. Addition of a ditopic
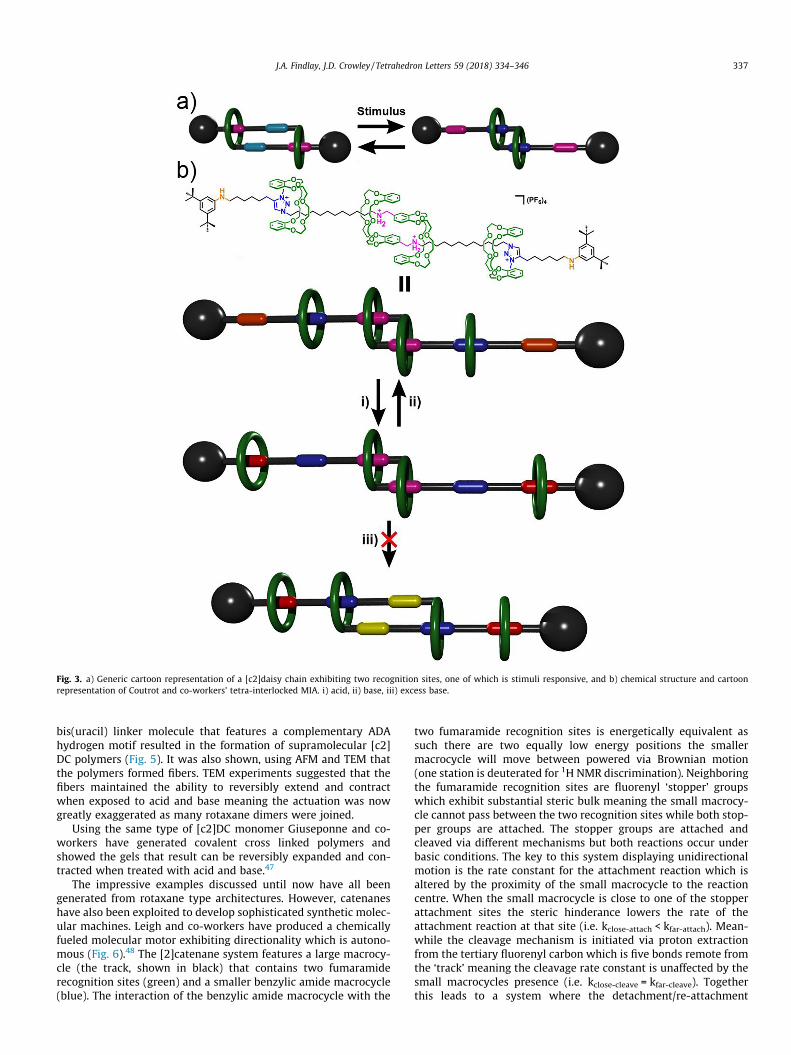
Fig. 3. a) Generic cartoon representation of a [c2]daisy chain exhibiting two recognition sites, one of which is stimuli responsive, and b) chemical structure and cartoonrepresentation of Coutrot and co-workers’ tetra-interlocked MIA. i) acid, ii) base, iii) excess base.
J.A. Findlay, J.D. Crowley / Tetrahedron Letters 59 (2018) 334–346 337
bis(uracil) linker molecule that features a complementary ADAhydrogen motif resulted in the formation of supramolecular [c2]DC polymers (Fig. 5). It was also shown, using AFM and TEM thatthe polymers formed fibers. TEM experiments suggested that thefibers maintained the ability to reversibly extend and contractwhen exposed to acid and base meaning the actuation was nowgreatly exaggerated as many rotaxane dimers were joined.
Using the same type of [c2]DC monomer Giuseponne and co-workers have generated covalent cross linked polymers andshowed the gels that result can be reversibly expanded and con-tracted when treated with acid and base.47
The impressive examples discussed until now have all beengenerated from rotaxane type architectures. However, catenaneshave also been exploited to develop sophisticated synthetic molec-ular machines. Leigh and co-workers have produced a chemicallyfueled molecular motor exhibiting directionality which is autono-mous (Fig. 6).48 The [2]catenane system features a large macrocy-cle (the track, shown in black) that contains two fumaramiderecognition sites (green) and a smaller benzylic amide macrocycle(blue). The interaction of the benzylic amide macrocycle with the
two fumaramide recognition sites is energetically equivalent assuch there are two equally low energy positions the smallermacrocycle will move between powered via Brownian motion(one station is deuterated for 1H NMR discrimination). Neighboringthe fumaramide recognition sites are fluorenyl ‘stopper’ groupswhich exhibit substantial steric bulk meaning the small macrocy-cle cannot pass between the two recognition sites while both stop-per groups are attached. The stopper groups are attached andcleaved via different mechanisms but both reactions occur underbasic conditions. The key to this system displaying unidirectionalmotion is the rate constant for the attachment reaction which isaltered by the proximity of the small macrocycle to the reactioncentre. When the small macrocycle is close to one of the stopperattachment sites the steric hinderance lowers the rate of theattachment reaction at that site (i.e. kclose-attach < kfar-attach). Mean-while the cleavage mechanism is initiated via proton extractionfrom the tertiary fluorenyl carbon which is five bonds remote fromthe ‘track’ meaning the cleavage rate constant is unaffected by thesmall macrocycles presence (i.e. kclose-cleave = kfar-cleave). Togetherthis leads to a system where the detachment/re-attachment

Fig. 4. Chiu and coworkers’ 2D and 3D cyclic [c3] and [c4]daisy chains exhibitingexpansion and contraction motions upon addition and removal of Zn2+ ions.
338 J.A. Findlay, J.D. Crowley / Tetrahedron Letters 59 (2018) 334–346
process is affected by the position of the small macrocycle and istherefore an information ratchet.35 Upon cleavage of both stoppers(loss of dibenzofulvene, orange), the re-attachment of one stopperfrom the supplied chemical fuel (9-flourenylmethoxycarbonylchloride, red) proceeds very quickly on the attachment site furthestfrom the small macrocycle, with the second attachment proceed-ing slowly while the small macrocycle occupies the close site.Shuttling of the small macrocycle to the other recognition sitethrough Brownian motion must then occur in the clockwise direc-tion as the first stopper blocks the anti-clockwise pathway. Attach-ment of the second stopper will occur faster when the macrocycleis occupying the far recognition site. This results in a 180� rotationof the small macrocycle about the track for every cleavage-attach-ment process. Under the autonomous conditions used a full unidi-rectional 360� rotation of the motor was calculated to take 12 h,thus the system is relative slow.
A more recent contribution by Leigh and co-workers alsodescribes a [2]catenane functioning as a unidirectional motormolecule.49 In this case the larger macrocycle has both a secondaryamine and a methylated triazolium station incorporated into it,acting as recognition sites for a mechanically bonded crown ether.Separating the binding sites are a disulfide stopper group and ahydrazone stopper group. When the [2]catenane is treated withacid (Cl3CCO2H), the ammonium site is protonated, becoming astrong recognition site for the crown ether, while the hydrazonebarrier undergoes exchange. This allows the small macrocycle to
move to the ammonium station via the hydrazone site. When thesystem is then treated with base the amine becomes deprotonatedand the triazolium station becomes the preferred binding site.However, now under basic conditions the hydrazone stopper groupis kinetically inert, while the disulfide barrier becomes dynamic,leading to migration of the small macrocycle to the triazolium site,completing a 360� rotation. Each repeated chemical fuel inputcycle allows for, mostly (87%) unidirectional rotary motion.
Non-interlocked machines
While many of the most impressive synthetic molecular machi-nes to date have been MIAs, they tend come along with varioussynthetic challenges. As such other workers have looked to exploitnon-interlocked systems to design functional synthetic machines.While many groups have contributed to the area, as outlined inthe introduction the pioneering work of Feringa and co-workersshowed that suitably designed overcrowded alkenes could be usedto generate light-driven unidirectional molecular motors and leadto Prof. Feringa sharing the 2016 Nobel prize in Chemistry.29 Fer-inga and co-workers have devised and reported a number oflight-driven unidirectional rotary motor molecules comprisingovercrowded alkenes.
These rotary motors exhibit helicity (P or M) and/or stereocen-tres (R or S), with these features largely credited with being the keyfactors in generating unidirectional rotation. These molecularmotors are able to undergo E/Z isomerisation upon irradiation withultraviolet light, followed by thermal helix inversion (THI). Theseconsecutive processes, allow for overall unidirectional rotationwhere the central chirality of the rotor determines the directionof rotation. In 2015 Feringa and co-workers reported a ‘third gen-eration’ molecular motor (Fig. 7) comprising two overcrowdedalkenes such that the central carbon atom is no longer chiral butmaintains a pseudo-asymmetry due to the possibility of either anE or Z alkene either side of the molecule.50 When this motor wasirradiated with 365 nm light at 173 K, one of the two alkenesunderwent photochemical E/Z isomerization, and then subsequentTHI of that alkene upon warming to room temperature. All of theisomers (Fig. 7) of the achiral motor were readily separated usingsupercritical fluid chromatography. Then, the R(Z,M),(E,P) isomerwas taken through a reaction cycle. 1H NMR and CD experimentsshowed that this process gave a mixture of two isomers after one‘cycle’ performed on a single isomer (e.g. one ‘cycle’ produced boththe r,E,E and r,Z,Z isomers starting from pure R(Z,M),(E,P) Fig. 7).This confirmed that for one irradiation and THI cycle just onealkene of a rotor would isomerise, resulting in approximately onequarter rotation relative to the central ‘stator’, followed by a onehalf rotation in the same direction upon THI. The rotor can there-fore undergo repeated unidirectional rotations upon irradiationwith UV light at room temperature. Despite the described systemhaving no selectivity for which alkene of the rotor undergoes iso-merism in a given ‘cycle’, this work demonstrates that chirality isnot necessary for unidirectional rotation; however some form ofasymmetry seems necessary.
More recently Feringa and co-workers reported a molecularmotor capable of locked synchronous unidirectional rotation, mov-ing in an analogous manner to the Moon around the Earth, as onlyone side of the Moon always faces us.51 The motor design is basedon the second generation overcrowded alkene motors, with a fluo-rene stator and an indanyl rotor with an appended naphthalenemoiety (Fig. 8). In order to achieve locked synchronous rotation,two energetic barriers had to be balanced; the barrier to biarylrotation (BR) of the naphthalene rotor had to be sufficiently highsuch that a negligible amount occurred, whilst that of the THI mustbe low enough to occur at reasonable temperatures. This allowed

Fig. 5. a) The two components of Giuseppone and co-workers’ stimuli responsive polymer, b) cartoon representation of polymerised [c2]DC through complementaryhydrogen bonding motifs as stopper units.
J.A. Findlay, J.D. Crowley / Tetrahedron Letters 59 (2018) 334–346 339
unidirectional rotation of the naphthalene moiety about the statorthrough the photochemically driven cycles without allowing thenaphthalene to rotate such that the opposite face of the biaryl rotorwas facing the stator. The mode of rotation was supported by Den-sity Functional Theory (DFT) calculations and confirmed primarilyby circular dichroism and variable temperature 1H NMR combinedwith Eyring analyses.
Giuseponne and co-workers incorporated Feringa’s second gen-eration molecular motor29 into a polymer gel and showed that irra-diation leads to coiling/contraction of the gel network.52 Thecontraction was confirmed using AFM microscopy and small-angleX-ray scattering (SAXS) experiments. The light driven process leadto the collapse of the gel but it was not reversible.52 Giuseponneand co-workers have gone on to show that by incorporating a lightsensitive modulator subunit within the ‘‘motor-gel” they can con-tract and then expand the gel network using different wavelengthsof light.53
Very recently, light driven molecular motors developed by Fer-inga and co-workers were adapted by Tour et al. for the purpose ofmechanically opening cell membranes.54 The nanomotors weresynthesised with peptide sequences appended on each side ofthe stator unit. Two different peptides were utilised, one chosenfor its selective interaction with particular transmembrane recep-tors of PC-3 human prostate cancer cells, and the other with theprotein GRP78, which targets receptors on PC-3 human prostatecancer cells. A variety of other nanomotors were synthesised ascontrol models, including two with different pendant fluorophoreson the stator to track their movement, two with smaller molecularsizes, one with a stator but no rotor, and one with a significantlyslower motor. UV-light activation of the nanomotors with fluo-rophores in the presence of live cells, caused nanomechanicalinduced entry into the cells, as evidenced by fluorescence imaging.One of the smaller control nanomotors was shown to significantlyincrease cell necrosis upon UV exposure while the other did so to alesser extent, presumably due to lessened cell membrane interac-tion. The control nanomotors which had no rotor, and which
underwent very slow rotations did not increase the rate of celldeath upon UV exposure. Additionally, the authors irradiated thepeptide substituted nanomotors in the presence of the PC-3 cellsand also untargeted control cells. The motors bearing sequencesknown to interact with the protein GRP78, showed increasednecrosis of PC-3 cells with no increase for the control cells duringUV irradiation, however the other peptide substituted motorsshowed no selectivity. This work demonstrates how carefullydesigned molecular machines could, one day, selectively interactwith cells within living organisms and be used to kill diseases withminimal harm to the host organism.
While the non-interlocked systems above have generated uni-directional rotation of small molecular entities exploiting light asthe stimuli, there are few examples where this was achievedthrough use of chemical stimulus.30–35 Feringa and co-workershave also reported a unidirectional molecular motor based on aredox cycle of palladium, driven by chemical input.55 The motormolecule itself possesses a sterically crowded CAC bond betweentwo aryl groups due to the ortho-substituents which have dualfunctions in hindering rotation of the bond, but also specificinteractions with palladium (Fig. 9). The lower stator group(red) possesses a chiral sulfur atom (taking the lone pair of elec-trons into consideration), which itself, has a dual function as italso acts as a directing group. The system exists as two atropiso-mers (S, M) and (S, P), where the S denotes the chirality of the sul-fur atom and M and P denote the chirality about the biaryl CACbond. The system was shown to act as a molecular motor asthe ‘upper’ aryl group (blue) could be rotated through chemicalstimulus. Upon addition of palladium(II) to the (S, M) atropiso-mer, the sulfur atom binds the palladium ion, acting as a directinggroup for the CAH insertion of the palladium in the ortho positionof the ‘upper’ aryl group. Before CAH insertion, the barrier torotation about the biaryl CAC bond was significant, but oncethe palladium(II) complex forms, the barrier is greatly reduced.Furthermore, it was shown the (R, M) isomer (note the palladiumtakes precedence, therefore changing the stereochemical
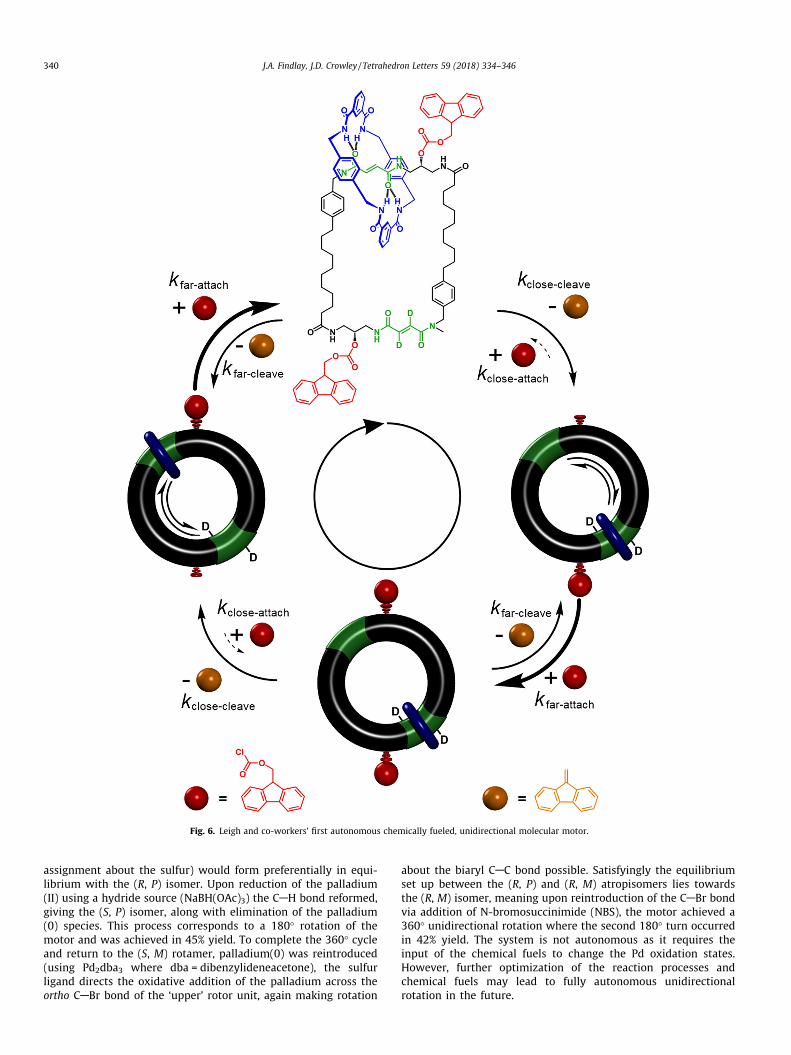
Fig. 6. Leigh and co-workers’ first autonomous chemically fueled, unidirectional molecular motor.
340 J.A. Findlay, J.D. Crowley / Tetrahedron Letters 59 (2018) 334–346
assignment about the sulfur) would form preferentially in equi-librium with the (R, P) isomer. Upon reduction of the palladium(II) using a hydride source (NaBH(OAc)3) the CAH bond reformed,giving the (S, P) isomer, along with elimination of the palladium(0) species. This process corresponds to a 180� rotation of themotor and was achieved in 45% yield. To complete the 360� cycleand return to the (S, M) rotamer, palladium(0) was reintroduced(using Pd2dba3 where dba = dibenzylideneacetone), the sulfurligand directs the oxidative addition of the palladium across theortho CABr bond of the ‘upper’ rotor unit, again making rotation
about the biaryl CAC bond possible. Satisfyingly the equilibriumset up between the (R, P) and (R, M) atropisomers lies towardsthe (R, M) isomer, meaning upon reintroduction of the CABr bondvia addition of N-bromosuccinimide (NBS), the motor achieved a360� unidirectional rotation where the second 180� turn occurredin 42% yield. The system is not autonomous as it requires theinput of the chemical fuels to change the Pd oxidation states.However, further optimization of the reaction processes andchemical fuels may lead to fully autonomous unidirectionalrotation in the future.
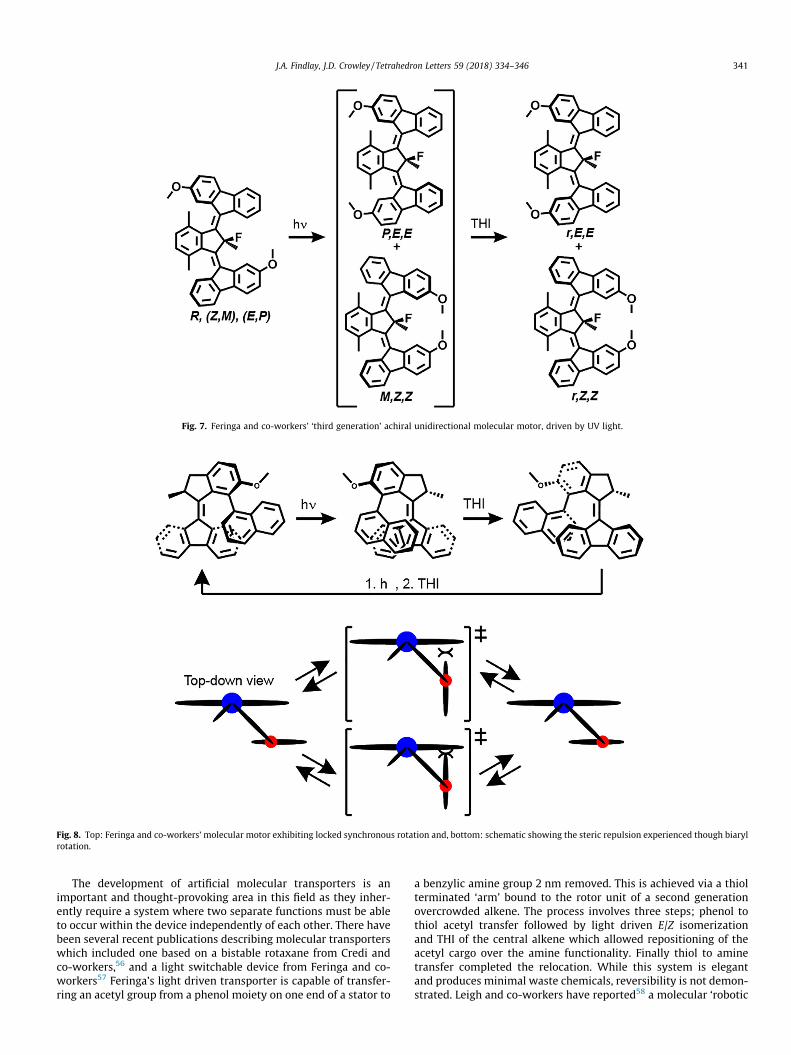
Fig. 7. Feringa and co-workers’ ‘third generation’ achiral unidirectional molecular motor, driven by UV light.
Fig. 8. Top: Feringa and co-workers’ molecular motor exhibiting locked synchronous rotation and, bottom: schematic showing the steric repulsion experienced though biarylrotation.
J.A. Findlay, J.D. Crowley / Tetrahedron Letters 59 (2018) 334–346 341
The development of artificial molecular transporters is animportant and thought-provoking area in this field as they inher-ently require a system where two separate functions must be ableto occur within the device independently of each other. There havebeen several recent publications describing molecular transporterswhich included one based on a bistable rotaxane from Credi andco-workers,56 and a light switchable device from Feringa and co-workers57 Feringa’s light driven transporter is capable of transfer-ring an acetyl group from a phenol moiety on one end of a stator to
a benzylic amine group 2 nm removed. This is achieved via a thiolterminated ‘arm’ bound to the rotor unit of a second generationovercrowded alkene. The process involves three steps; phenol tothiol acetyl transfer followed by light driven E/Z isomerizationand THI of the central alkene which allowed repositioning of theacetyl cargo over the amine functionality. Finally thiol to aminetransfer completed the relocation. While this system is elegantand produces minimal waste chemicals, reversibility is not demon-strated. Leigh and co-workers have reported58 a molecular ‘robotic

Fig. 9. Feringa and co-workers’ chemically driven unidirectional molecular motor.
342 J.A. Findlay, J.D. Crowley / Tetrahedron Letters 59 (2018) 334–346
arm’, able to move a cargo from one platform to another, selec-tively and reversibly through manipulation of pH (Fig. 10). The sta-tor, with an aldehyde ‘platform’ (red and orange) at either end, waslinked to the ‘robotic arm’ (pink) through a central quinolone unit(purple) via a hydrazone linkage to form a pH-responsive rotor unitdeveloped by Aprahamian and co-workers.59 The rotor may adoptone of two conformers where either the pyridine (E isomer) or theester (Z isomer) of the ‘arm’ will participate in hydrogen bondingwith the hydrazone proton along with the quinolone nitrogen onthe stator. Control over this E/Z isomerisation is achieved throughselective protonation of several functionalities in the system,allowing control over the position of the ‘arm’ over one of thetwo platforms. 1H NMR analysis allowed the loading, transport,and unloading processes to be followed conveniently as one ofthe aldehyde platforms was deuterated in order to distinguishwhich of the platforms the cargo was bound to, or if it was boundonly to the ‘arm’. The cargo (black) was initially shown to be able tobe transported from the deuterated station to the other in threesteps and 79% yield. The first of which is kinetic disulphide forma-tion between the end of the robotic arm and the cargo. This macro-cyclisation was followed by treatment with 70 eq. of triflic acidwhich protonates the hydrazone switch, along with facilitatingacyl hydrazone exchange, allowing for detachment of the cargofrom the station and for rotation of the robotic arm placing thecargo on the other station (red). NMR spectroscopy indicated thatthis step actually produces a 17:83 mixture of the transporter withthe cargo on the first (orange) platform to the transporter with thecargo on the red platform. The cargo can then be detached from therobotic arm upon reduction of the disulphide bond, simultaneouslyrestoring the switch. The reverse process could be completed in85% efficiency over the three associated steps. Firstly oxidationwith iodine reforms the disulphide linkage. Treatment with 5 eq.of triflic acid allowed for the return of >90% of the cargo to the first
station, and subsequent reduction of the disulphide bond releasesthe cargo from the robotic arm.
More recently Leigh and co-workers have modified and repur-posed the design of the transporter in order to achieve stereodiver-gent synthesis through mechanical manipulation of a substrate.60
This programmable molecular machine bears a terminal alkeneon the ‘robotic arm’, which allows an a,b-unsaturated aldehydesubstrate to be attached, and then positioned over one of two ‘sta-tions’ which feature chiral activating sites of opposite handedness(prolinol silyl ethers). An iminium- or enamine-promoted reactionis able to be initiated through condensation of the substrate withan activating site. The choice of station at each of the asymmetrictandem additions of a nucleophile and an electrophile to the a,b-unsaturated aldehyde determines the stereochemistry of the even-tually output product. The same organocatalysis process (withoutthe mechanical directionality) typically only allows access to thesyn diastereoisomers, whereas this programmable control allowsselective production of any of the four stereoisomeric products,including the anti-isomers.
The vast majority of synthetic machine systems are fullyorganic in design.30–35 However, metal ions offer different stereo-chemical preferences to carbon and can also provide interestingredox and/or photophysical properties. As such more and moremetal containing machine systems are beginning to appear in theliterature.12,36
Sauvage and co-workers have exploited the coordination pref-erence of metal ions to affect the synthesis and controlled molec-ular motion of MIAs, most notably catenanes.12,36 Similar largeamplitude motions in non-interlocked architectures are far lesscommon. Sauvage and co-workers reported a non-interlockedarchitecture where a 78-membered macrocycle (Fig. 11) was ableto be contracted and elongated through either the choice of metalion, or through the electrochemistry of copper ions.61 The largemacrocycle exhibits two bidentate diphenylphenanthroline (dpp)binding pockets opposite one another and two terpyridine (terpy)binding pockets oriented perpendicularly to the dpp sites. Additionof one equivalent of Cu(I) ions resulted in formation of the tetrahe-dral bis-dpp copper(I) complex and thus a ‘figure-of-eight’ shapedentity. Addition of Fe(II) ions results in the octahedral bis-terpyiron(II) complex where the macrocycle has compressed in the per-pendicular dimension. Both metal ions could be removed (KCN)and replaced by the other reversibly and quantitatively. Utilisingthe differing coordination geometry preferences of Cu(I) (tetrahe-dral) and Cu(II) (5 or 6 coordinate) ions, it was also shown thatthe reversible electrochemistry of copper ions would affect thesame orthogonal contraction/elongation process as the copper ionsswitched between the different ligand moieties upon oxidationand reduction (Fig. 11b).
Several groups have examined using metals ion as ‘‘molecularball bearings” in machine like systems. Designs based on includingmetallocenes, carboranes and lanthanide bis-porphyrins andpiano-stool type complexes have all been examined.62,63
Rapenne and co-workers have developed a ruthenium basedrotor which can perform unidirectional rotation in both clockwiseand anticlockwise directions, through use of a scanning tunnelingmicroscope (STM) tip (Fig. 12).64 The rotor consists of a tripodalbase (blue) able to bind to a gold surface through thiol linkages(red). On top of the tripodal stator a ruthenium ion (dark red) isbound through three coordinating nitrogen atoms. The remainingcoordination sites are occupied by a cyclopentadienyl ring rotorunit (dark red) forming a ‘half-sandwich’ complex. Four of the fivelinear ‘arms’ on the cyclopentadienyl ring are terminated withredox active ferrocene units (orange), while the fifth is truncated,terminating in a methyl group. The rotor unit will rotate freely inboth directions at sufficiently high temperatures, but this rotationslows at lower temperatures.
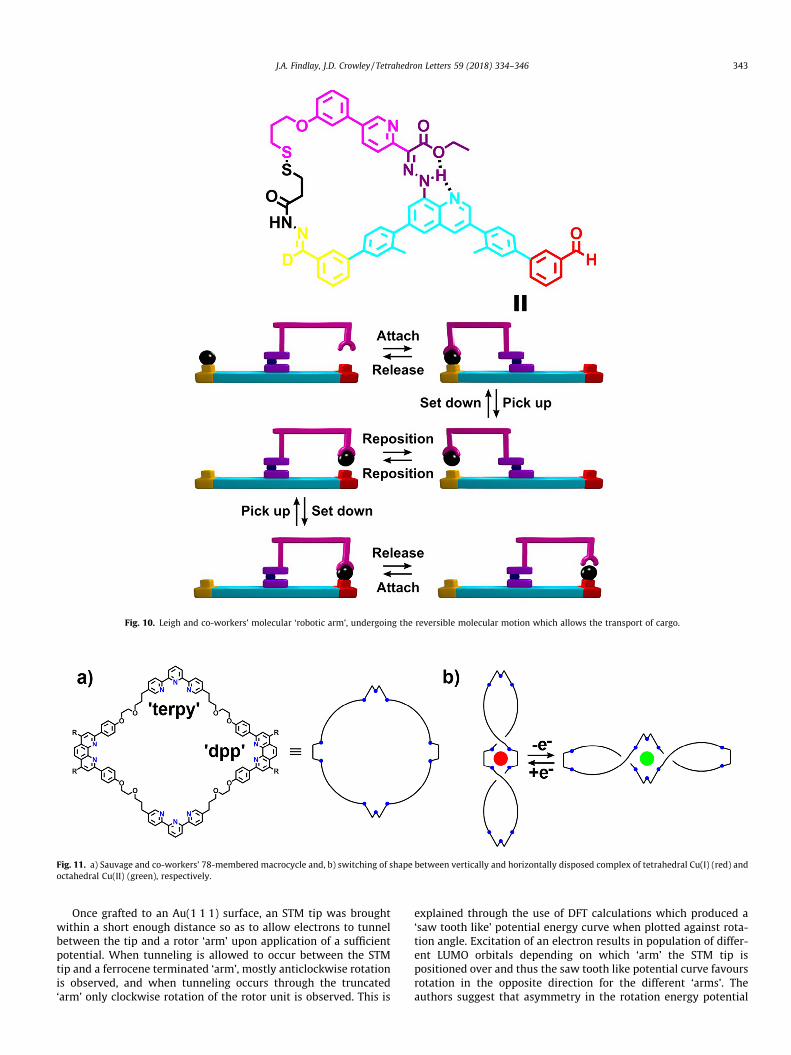
Fig. 10. Leigh and co-workers’ molecular ‘robotic arm’, undergoing the reversible molecular motion which allows the transport of cargo.
Fig. 11. a) Sauvage and co-workers’ 78-membered macrocycle and, b) switching of shape between vertically and horizontally disposed complex of tetrahedral Cu(I) (red) andoctahedral Cu(II) (green), respectively.
J.A. Findlay, J.D. Crowley / Tetrahedron Letters 59 (2018) 334–346 343
Once grafted to an Au(1 1 1) surface, an STM tip was broughtwithin a short enough distance so as to allow electrons to tunnelbetween the tip and a rotor ‘arm’ upon application of a sufficientpotential. When tunneling is allowed to occur between the STMtip and a ferrocene terminated ‘arm’, mostly anticlockwise rotationis observed, and when tunneling occurs through the truncated‘arm’ only clockwise rotation of the rotor unit is observed. This is
explained through the use of DFT calculations which produced a‘saw tooth like’ potential energy curve when plotted against rota-tion angle. Excitation of an electron results in population of differ-ent LUMO orbitals depending on which ‘arm’ the STM tip ispositioned over and thus the saw tooth like potential curve favoursrotation in the opposite direction for the different ‘arms’. Theauthors suggest that asymmetry in the rotation energy potential
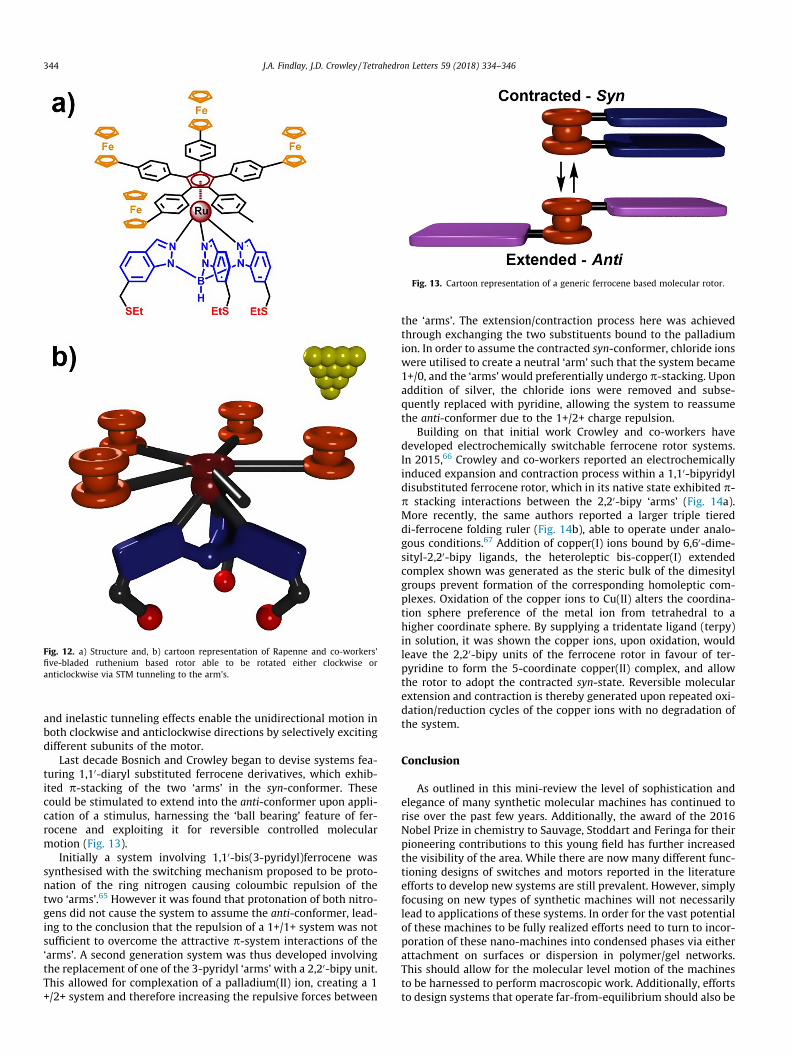
Fig. 12. a) Structure and, b) cartoon representation of Rapenne and co-workers’five-bladed ruthenium based rotor able to be rotated either clockwise oranticlockwise via STM tunneling to the arm’s.
Fig. 13. Cartoon representation of a generic ferrocene based molecular rotor.
344 J.A. Findlay, J.D. Crowley / Tetrahedron Letters 59 (2018) 334–346
and inelastic tunneling effects enable the unidirectional motion inboth clockwise and anticlockwise directions by selectively excitingdifferent subunits of the motor.
Last decade Bosnich and Crowley began to devise systems fea-turing 1,10-diaryl substituted ferrocene derivatives, which exhib-ited p-stacking of the two ‘arms’ in the syn-conformer. Thesecould be stimulated to extend into the anti-conformer upon appli-cation of a stimulus, harnessing the ‘ball bearing’ feature of fer-rocene and exploiting it for reversible controlled molecularmotion (Fig. 13).
Initially a system involving 1,10-bis(3-pyridyl)ferrocene wassynthesised with the switching mechanism proposed to be proto-nation of the ring nitrogen causing coloumbic repulsion of thetwo ‘arms’.65 However it was found that protonation of both nitro-gens did not cause the system to assume the anti-conformer, lead-ing to the conclusion that the repulsion of a 1+/1+ system was notsufficient to overcome the attractive p-system interactions of the‘arms’. A second generation system was thus developed involvingthe replacement of one of the 3-pyridyl ‘arms’ with a 2,20-bipy unit.This allowed for complexation of a palladium(II) ion, creating a 1+/2+ system and therefore increasing the repulsive forces between
the ‘arms’. The extension/contraction process here was achievedthrough exchanging the two substituents bound to the palladiumion. In order to assume the contracted syn-conformer, chloride ionswere utilised to create a neutral ‘arm’ such that the system became1+/0, and the ‘arms’ would preferentially undergo p-stacking. Uponaddition of silver, the chloride ions were removed and subse-quently replaced with pyridine, allowing the system to reassumethe anti-conformer due to the 1+/2+ charge repulsion.
Building on that initial work Crowley and co-workers havedeveloped electrochemically switchable ferrocene rotor systems.In 2015,66 Crowley and co-workers reported an electrochemicallyinduced expansion and contraction process within a 1,10-bipyridyldisubstituted ferrocene rotor, which in its native state exhibited p-p stacking interactions between the 2,20-bipy ‘arms’ (Fig. 14a).More recently, the same authors reported a larger triple tiereddi-ferrocene folding ruler (Fig. 14b), able to operate under analo-gous conditions.67 Addition of copper(I) ions bound by 6,60-dime-sityl-2,20-bipy ligands, the heteroleptic bis-copper(I) extendedcomplex shown was generated as the steric bulk of the dimesitylgroups prevent formation of the corresponding homoleptic com-plexes. Oxidation of the copper ions to Cu(II) alters the coordina-tion sphere preference of the metal ion from tetrahedral to ahigher coordinate sphere. By supplying a tridentate ligand (terpy)in solution, it was shown the copper ions, upon oxidation, wouldleave the 2,20-bipy units of the ferrocene rotor in favour of ter-pyridine to form the 5-coordinate copper(II) complex, and allowthe rotor to adopt the contracted syn-state. Reversible molecularextension and contraction is thereby generated upon repeated oxi-dation/reduction cycles of the copper ions with no degradation ofthe system.
Conclusion
As outlined in this mini-review the level of sophistication andelegance of many synthetic molecular machines has continued torise over the past few years. Additionally, the award of the 2016Nobel Prize in chemistry to Sauvage, Stoddart and Feringa for theirpioneering contributions to this young field has further increasedthe visibility of the area. While there are now many different func-tioning designs of switches and motors reported in the literatureefforts to develop new systems are still prevalent. However, simplyfocusing on new types of synthetic machines will not necessarilylead to applications of these systems. In order for the vast potentialof these machines to be fully realized efforts need to turn to incor-poration of these nano-machines into condensed phases via eitherattachment on surfaces or dispersion in polymer/gel networks.This should allow for the molecular level motion of the machinesto be harnessed to perform macroscopic work. Additionally, effortsto design systems that operate far-from-equilibrium should also be
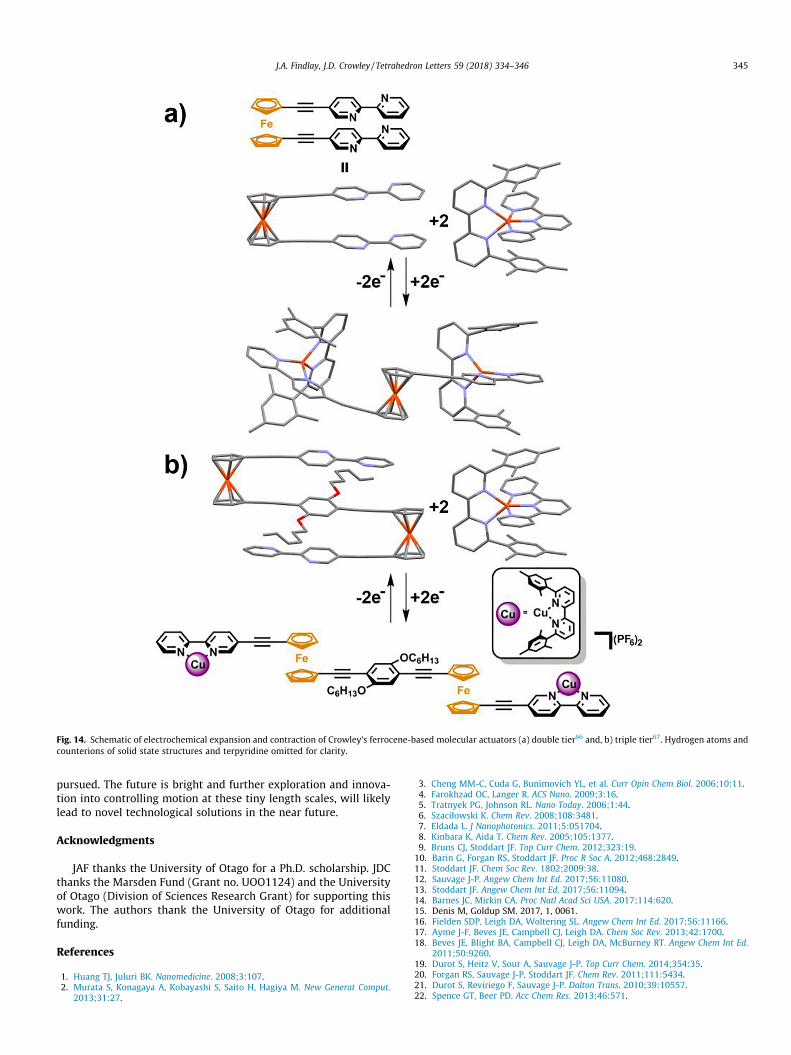
Fig. 14. Schematic of electrochemical expansion and contraction of Crowley’s ferrocene-based molecular actuators (a) double tier66 and, b) triple tier67. Hydrogen atoms andcounterions of solid state structures and terpyridine omitted for clarity.
J.A. Findlay, J.D. Crowley / Tetrahedron Letters 59 (2018) 334–346 345
pursued. The future is bright and further exploration and innova-tion into controlling motion at these tiny length scales, will likelylead to novel technological solutions in the near future.
Acknowledgments
JAF thanks the University of Otago for a Ph.D. scholarship. JDCthanks the Marsden Fund (Grant no. UOO1124) and the Universityof Otago (Division of Sciences Research Grant) for supporting thiswork. The authors thank the University of Otago for additionalfunding.
References
1. Huang TJ, Juluri BK. Nanomedicine. 2008;3:107.2. Murata S, Konagaya A, Kobayashi S, Saito H, Hagiya M. New Generat Comput.
2013;31:27.
3. Cheng MM-C, Cuda G, Bunimovich YL, et al. Curr Opin Chem Biol. 2006;10:11.4. Farokhzad OC, Langer R. ACS Nano. 2009;3:16.5. Tratnyek PG, Johnson RL. Nano Today. 2006;1:44.6. Szaciłowski K. Chem Rev. 2008;108:3481.7. Eldada L. J Nanophotonics. 2011;5:051704.8. Kinbara K, Aida T. Chem Rev. 2005;105:1377.9. Bruns CJ, Stoddart JF. Top Curr Chem. 2012;323:19.10. Barin G, Forgan RS, Stoddart JF. Proc R Soc A. 2012;468:2849.11. Stoddart JF. Chem Soc Rev. 1802;2009:38.12. Sauvage J-P. Angew Chem Int Ed. 2017;56:11080.13. Stoddart JF. Angew Chem Int Ed. 2017;56:11094.14. Barnes JC, Mirkin CA. Proc Natl Acad Sci USA. 2017;114:620.15. Denis M, Goldup SM. 2017, 1, 0061.16. Fielden SDP, Leigh DA, Woltering SL. Angew Chem Int Ed. 2017;56:11166.17. Ayme J-F, Beves JE, Campbell CJ, Leigh DA. Chem Soc Rev. 2013;42:1700.18. Beves JE, Blight BA, Campbell CJ, Leigh DA, McBurney RT. Angew Chem Int Ed.
2011;50:9260.19. Durot S, Heitz V, Sour A, Sauvage J-P. Top Curr Chem. 2014;354:35.20. Forgan RS, Sauvage J-P, Stoddart JF. Chem Rev. 2011;111:5434.21. Durot S, Reviriego F, Sauvage J-P. Dalton Trans. 2010;39:10557.22. Spence GT, Beer PD. Acc Chem Res. 2013;46:571.

346 J.A. Findlay, J.D. Crowley / Tetrahedron Letters 59 (2018) 334–346
23. Crowley JD, Goldup SM, Lee A-L, Leigh DA, McBurney RT. Chem Soc Rev.2009;38:1530.
24. Leigh DA, Lewandowska U, Lewandowski B, Wilson MR. Top Curr Chem.2014;354:111.
25. von Delius M, Leigh DA. Chem Soc Rev. 2011;40:3656.26. Pollard MM, Meetsma A, Feringa BL. Org Biomol Chem. 2008;6:507.27. Feringa BL. J Org Chem. 2007;72:6635.28. Browne WR, Feringa BL. Nat Nanotechnol. 2006;1:25.29. Feringa BL. Angew Chem Int Ed. 2017;56:11060.30. Kassem S, van Leeuwen T, Lubbe AS, Wilson MR, Feringa BL, Leigh DA. Chem Soc
Rev. 2017;46:2592.31. Wang Y, Frasconi M, Stoddart JF. ACS Cent Sci. 2017;3:927.32. Cheng C, Stoddart JF. ChemPhysChem. 2016;17:1780.33. Kay ER, Leigh DA. Angew Chem Int Ed. 2015;54:10080.34. Erbas-Cakmak S, Leigh DA, McTernan CT, Nussbaumer AL. Chem Rev.
2015;115:10081.35. Kay ER, Leigh DA, Zerbetto F. Angew Chem Int Ed. 2007;46:72.36. Frédéric N, Vincent D, Jean-Pierre S. Chem Lett. 2014;43:964.37. Cheng C, McGonigal PR, Schneebeli ST, et al. Nat Nanotechnol. 2015;10:547.38. Vinothkumar KR, Henderson R. Q Rev Biophys. 2010;43:65.39. Shi Y. Annu Rev Biophys. 2013;42:51.40. Ragazzon G, Baroncini M, Silvi S, Venturi M, Credi A. Beilstein J Nanotechnol.
2015;6:2096.41. Cheng C, Cheng T, Xiao H, et al. J Am Chem Soc. 2016;138:10214.42. Rotzler J, Mayor M. Chem Soc Rev. 2013;42:44.43. Bruns CJ, Stoddart JF. Acc Chem Res. 2014;47:2186.44. Waelès P, Riss-Yaw B, Coutrot F. Chem Eur J. 2016;28:6837.45. Chang J-C, Tseng S-H, Lai C-C, Liu Y-H, Peng S-M, Chiu S-H. Nat Chem. 2016.
advance online publication.
46. Goujon A, Du G, Moulin E, et al. Angew Chem Int Ed. 2016;55:703.47. Goujon A, Lang T, Mariani G, et al. J Am Chem Soc. 2017;139:14825.48. Wilson MR, Sola J, Carlone A, Goldup SM, Lebrasseur N, Leigh DA. Nature.
2016;534:235.49. Erbas-Cakmak S, Fielden SDP, Karaca U, et al. Science. 2017;358:340.50. Kistemaker JCM, Štacko P, Visser J, Feringa BL. Nat Chem. 2015;7:890.51. Štacko P, Kistemaker JCM, van Leeuwen T, Chang M-C, Otten E, Feringa BL.
Science. 2017;356:964.52. Li Q, Fuks G, Moulin E, et al. Nat Nanotechnol. 2015;10:161.53. Foy JT, Li Q, Goujon A, et al. Nat Nanotechnol. 2017;12:540.54. García-López V, Chen F, Nilewski LG, et al. Nature. 2017;548:567.55. Collins BSL, Kistemaker JCM, Otten E, Feringa BL. Nat Chem. 2016;8:860.56. Schaefer C, Ragazzon G, Colasson B, La Rosa M, Silvi S, Credi A. ChemistryOpen.
2016;5:120.57. Chen J, Wezenberg SJ, Feringa BL. Chem Commun. 2016;52:6765.58. Kassem S, Lee ATL, Leigh DA, Markevicius A, Sola J. Nat Chem. 2016;8:138.59. Aprahamian I. Chem Commun. 2017;53:6674.60. Kassem S, Lee ATL, Leigh DA, Marcos V, Palmer LI, Pisano S. Nature.
2017;549:374.61. Niess F, Duplan V, Sauvage J-P. J Am Chem Soc. 2014;136:5876.62. Scottwell SØ, Crowley JD. Chem Commun. 2016;52:2451.63. Stefak R, Sirven AM, Fukumoto S, Nakagawa H, Rapenne G. Coord Chem Rev.
2015;287:79.64. Perera UGE, Ample F, Kersell H, et al. Nat Nanotechnol. 2013;8:46.65. Crowley JD, Steele IM, Bosnich B. Chem Eur J. 2006;12:8935.66. Scottwell SØ, Elliott ABS, Shaffer KJ, et al. Chem Commun. 2015;51:8161.67. Scottwell SØ, Barnsley JE, McAdam CJ, Gordon KC, Crowley JD. Chem Commun.
2017;53:7628.