gene finding with genemark.hmm (lukashin & borodovsky, 1997 ) cs 466 saurabh sinha
TRANSCRIPT

Gene finding with GeneMark.HMM
(Lukashin & Borodovsky, 1997)
CS 466
Saurabh Sinha

Gene finding in bacteria
• Large number of bacterial genomes sequenced (10 at the time of paper, 1997)
• Previous work: “Genemark” program identified gene as ORF that looks more like genes than non-genes.– Uses Markov chains of coding and non-coding sequence
• 5’ (starting) boundary not well predicted
– Resolution of start point ~ 100 nucleotides

Genemark.hmm
• Builds on Genemark, but uses HMM for better prediction of start and stop
• Given DNA sequence S = {b1,b2,….bL}
• Find “functional sequence” A={a1,…aL} where each ai = 0 if non-coding, 1 if coding in forward strand, 2 if coding in reverse strand
• Sounds like the Fair Bet Casino problem (sequence of coin types “fair” or “biased”)
• Find Pr(A | S) and report A that maximizes this

Functional sequence• “A” carries information about where the coding
function switched into non-coding (stop of gene) and vice versa.
• Model sequence by HMM with different states for “coding” and “non-coding”
• Maximum likelihood “A” is the optimal path through the HMM, given the sequence
• Viterbi algorithm to solve this problem


Hidden Markov Model
• In some states, choose (i) a length of sequence to emit and (ii) the sequence to emit
• This is different from the Fair Bet Casino problem. There, each state emitted exactly one observation (H or T)

Hidden Markov Model• “Typical” and “Atypical” gene states (one for
each of forward and reverse strands)
• These two states emit coding sequence (between and excluding start and stop codons) with different codon usage patterns
• Clustering of E. coli genes showed that – majority of genes belong to one cluster (“Typical”)– many genes, believed to have been “horizontally
transferred” into the genome, belong to another cluster (“Atypical”)

Hidden State Trajectory “A”
• This is similar to the “functional” sequence defined earlier– except that we have one for each state, not one for
each nucleotide
• Sequence of M hidden states ai having duration di:– A = {(a1d1), (a2d2), …. (aMdM)}– ∑di = L
• Find A* that maximizes Pr(A|S)

Formulation
• Find trajectory (path) A that has the highest probability of occurring simultaneously with the sequence S
• Maximizing Pr(A,S) is the same as maximizing Pr(A|S). Why ?
€
Pmax = P(A*,S) = max
(a1d1 )...(aM d M )
ds =L
s=1
M
∑
Pr[(a1d1)...(aM dM ),b1,b2 ...bL ]

Solution
• Maximization problem solved by Viterbi algorithm (seen in previous lecture)

Solution
maximizing over all possible trajectories

Solution
Define (for dynamic progamming):
the joint probability of a partial trajectory of m states (with thelast state being am) and a partial sequence of length l.
transition prob.
prob. of duration prob. of sequence

Solution

Parameters of the HMM
• Transition probability distributions, emission probability distributions
• Fixed a priori– What was the other possibility ?– Learn parameters from data
• Emission probabilities of coding sequence state obtained from previous statistical studies: “What does a coding sequence look like in general?”
• Emission probabilities of non-coding sequence obtained similarly

Parameters of the HMM• Probability that a state “a” has duration “d” (i.e.,
length of emission is d) is learned from frequency distribution of lengths of known coding sequences

Parameters of the HMM
•… and non-coding sequences

Parameters of the HMM• Emission probabilities of start codon fixed from
previous studies– Pr(ATG)=0.905, Pr(GTG)=0.090, Pr(TTG)=0.005
• Transition probabilities: Non-coding to Typical/Atypical coding state = 0.85/0.15

Post-processing
• As per the HMM, two genes cannot overlap. In reality, genes may overlap !
G2G1

Post-processing
• As per the HMM, two genes cannot overlap. In reality, genes may overlap !
G2G1
Will predict second gene to begin here
What about the start codon for that second gene?

Post-processing
• As per the HMM, two genes cannot overlap. In reality, genes may overlap !
G2G1
• Look for an RBS somewhere here.• Take each start codon here, and find RBS -19 to -4 bp upstream of it
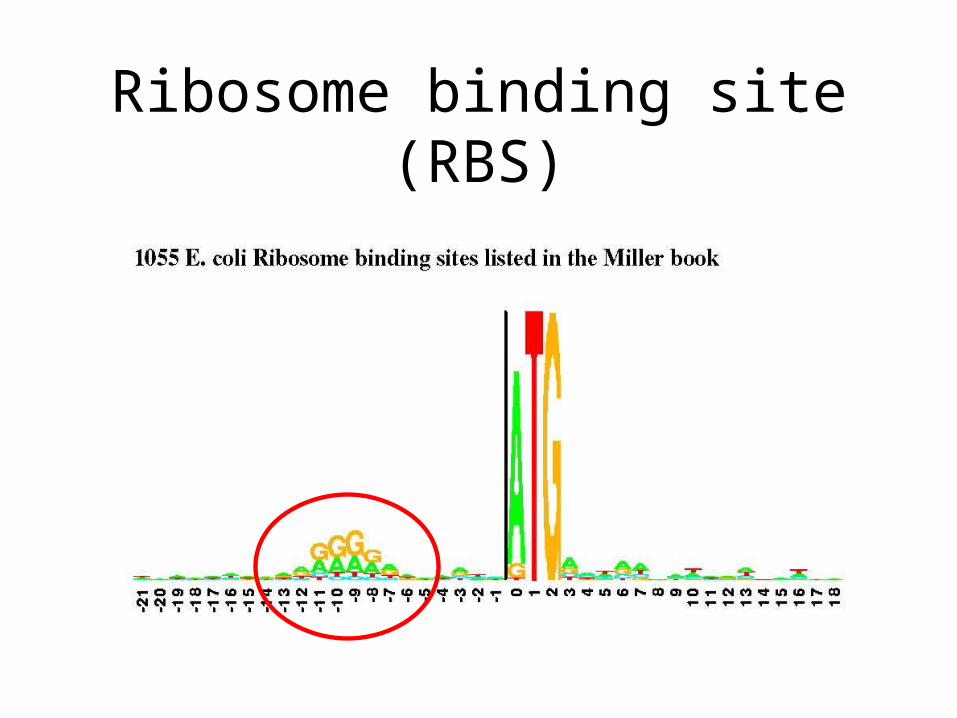
Ribosome binding site (RBS)

How to search for RBS?
• Take 325 genes from E. coli (bacterium) with known RBS
• Align them using sequence alignment
• Use this as a PWM to scan for RBS

Gene prediction in different species
• The coding and non-coding state emission probabilities need to be trained from each species for predicting genes in that species

Gene prediction accuracy
• Data set #1: all annotated E. coli genes
• Data set #2: non-overlapping genes
• Data set #3: Genes with known RBS
• Data set #4: Genes with known start positions

Results
VA: Viterbi algorithmPP: With post-processing

Results• Gene overlap is an important factor
• Performance goes up from 58% to 71% when overlapping genes are excluded from data set
• Post-processing helps a lot – 58% --> 75% for data set #1
• Missing genes: “False negatives” < 5%
• “Wrong” gene predictions: “False positives” ~8%– Are they really false positives, or are they unannotated
genes?

Results
• Compared with other programs

Results
• Robustness to parameter settings
• Alternative set of transition probability values used
• Little change in performance (~20% change in parameter values leads to < 5% change in performance)

Higher Order Markov models
• Sequence emissions were modeled by a second order Markov chain.– Pr (Xi|Xi-1, Xi-2,…X1) = Pr (Xi|Xi-1, Xi-2)
• Examined the effect of changing the “Markov order” (0,1,3,4,5)
• Even zeroth order Markov chain does pretty well.

Higher Order Markov models