genetics journal club cara skraban, md clinical genetics fellow february 12, 2015
TRANSCRIPT

Genetics Journal Club
Cara Skraban, MDClinical Genetics Fellow
February 12, 2015

JAMA Neurology Feb 2015

Myasthenia Gravis
• Autoimmune disorder of neuromuscular transmission• Characterized by muscle fatigability• Typically mediated by antibodies against nicotinic
acetylcholine receptors (AChRs) or against related proteins at NM junction– Muscle-specific tyrosine kinase (MuSK)– Lipoprotein receptor-related protein 4– Agrin
• Bimodal affected populations– Young women– Older men

Myasthenia Gravis

Genetics Factors of MG
• HLA locus is the most strongly associated risk factor for disease
• Previous GWAS studies– Major histocompatibility complex class II– Protein tyrosine phosphatase nonreceptor type 22 (PTPN22)– TNFAIP3 interacting protein 1 (TNIP1)
• Gene studies have suggested association of cytotoxic T-lymphocyte-associated protein 4 gene (CTLA4)
• Patients often have a family history of autoimmune disease
• 5% of patients have a positive family history of MG following an AD inheritance

Patients
• Patients attending MG clinics at 14 centers throughout North America (972 patients)– Diagnosed by a neurologist specializing in MG– Onset of symptoms after 18 yo– Non-Hispanic white race– Diagnosed clinically and confirmed with anti-AChR antibodies– Samples collected using Oragene DNA Saliva Collection kits
• Control Cohort (1977 patients)– Downloaded genotype data from dbGAP– Neurologically normal individuals– Matched for race and ethnic group, not age and sex

Replication Cohort
• 423 Italian patients with AChR-positive MG• 467 Italian neurologically normal controls• Matched to the case cohort for race/ethnic
group but not for age or sex• Blood samples collected
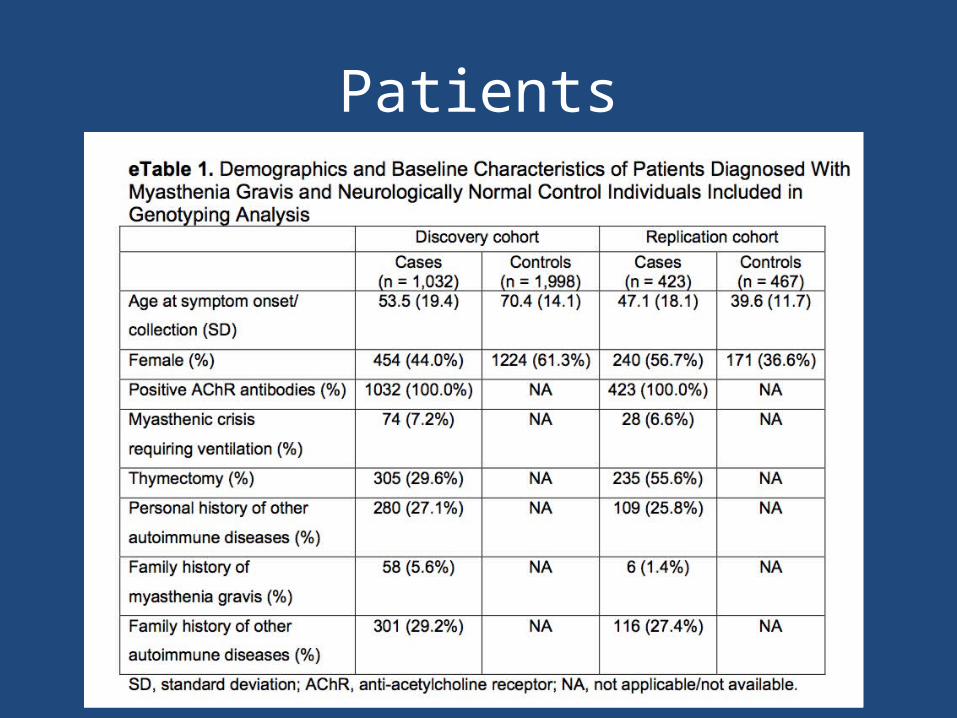
Patients

Genome-wide Genotyping
• Genotyped in the Laboratory of Neurogenetics, National Institute of Aging, using HumanOmniExpress BeadChips (Illumina)– Assay 730,525 SNPs across the genome
• Control cohort previously genotyped at the Center for Inherited Disease Research at Hopkins on HumanOmni1-Quad BeadChips (Illumina)
• Analyses were confined to the 677,673 autosomal SNPs that were common to both chips

Genotyping Bias• To exclude possibility of genotyping bias arising from different
sources of DNA, they compared from two patients: – whole genome genotyped data (Illumina) generated using paired DNA
samples extracted from blood– DNA extracted from saliva using Oragene DNA Saliva Collection system– Concordance rate >99.99%– None of discordant SNPs were located within significantly associated
loci• Exclude genotyping bias from using amplified DNA, they
compared from 94 samples:– Sanger sequencing data generated using DNA samples that were
amplified– Data generated using genomic, unamplified DNA– Concordance rate 100% for both rs601006 and rs9271850

Genotyping in the Replication Cohort
• RS231770, rs4263037, rs9270986– Taqman genotyping assays – Scanned on an ABI 7900HT Real-Time PCR
• Rs601006 and rs9271850– Sequencing using Big-Dye Terminator version 3.1
sequencing kit– Run on an ABI 3730xl DNA analyzer– Analyzed with Sequencher software and Mutation
Surveyor

Statistical Analysis: Genome-wide Association
• Statistical analyses were performed using R statistical software
• Standard quality-control procedures; Exclusion of the following:– SNP call rates of less than 95%– Non-European ancestry– Cryptic relatedness- identity-by-descent > 0.1– Minor allele frequency <0.01 in the control cohort– Hardy-Weinberg equilibrium P < 0.001 in the
control cohort

Imputation
• Markov chain-based Haplotyper to impute genotypes– Imputed by a two-stage design– Confirmed accuracy of imputation for most associated
SNPs for the 972 MG patients• Taqman genotyping for rs231770• Sanger sequencing for rs601006 and rs9271850• High concordance for all: 99.8%, 98%, 100%
• 8,114,394 SNPs available for analysis• 513,081 genotyped SNPs• 7,601,313 imputed SNPs

Statistical Analysis Continued
• P values calculated using logistic regression modeling– First two principle components used as covariates
to compensate for any residual population stratification.
– Principle components were generated using Genome-wide Complex trait analysis software package implementation of eigenstrat
– Threshold of 5.0 x 10-8 for genome wide significance after Bonferroni correction

Probability Analysis and Heritability Estimates
• Density estimation was used to generate posterior probabilities of developing MG based on sex and age
• Genome-wide Complex Trait Analysis– Used to compare each case series to control individuals
(all cases, early-onset, late-onset)– Compared two separate sets of SNPs
• All genotyped SNPs• Only those within 1 MB from the loci identified as genome-
wide significant in the discovery phase• Only SNPs passing quality control were used to evaluate the
heritability
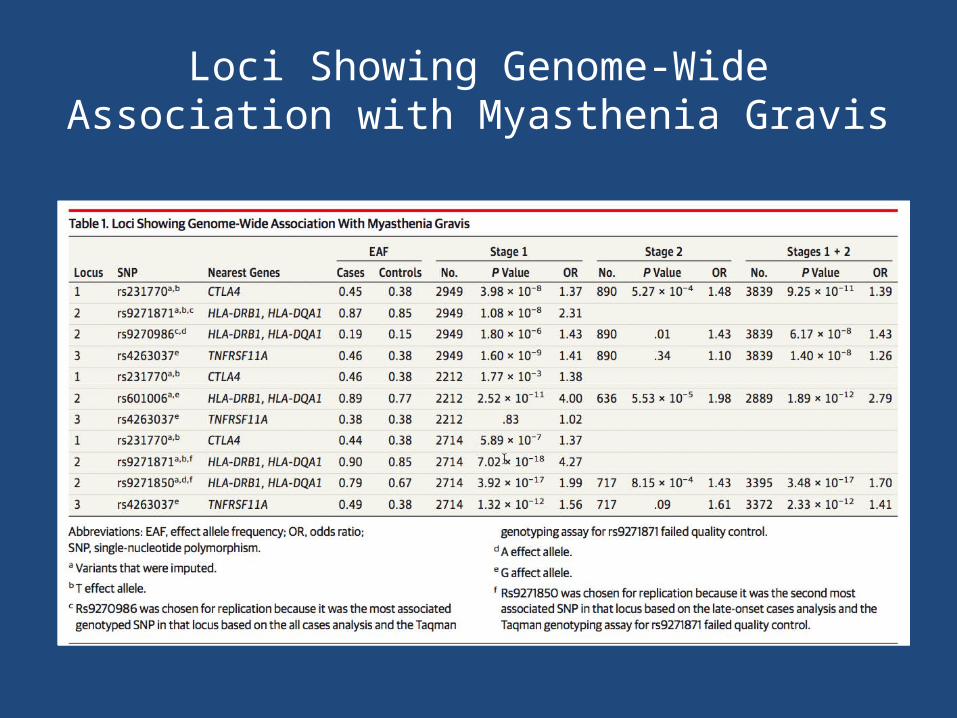
Loci Showing Genome-Wide Association with Myasthenia Gravis

Quartile-Quartile Plot
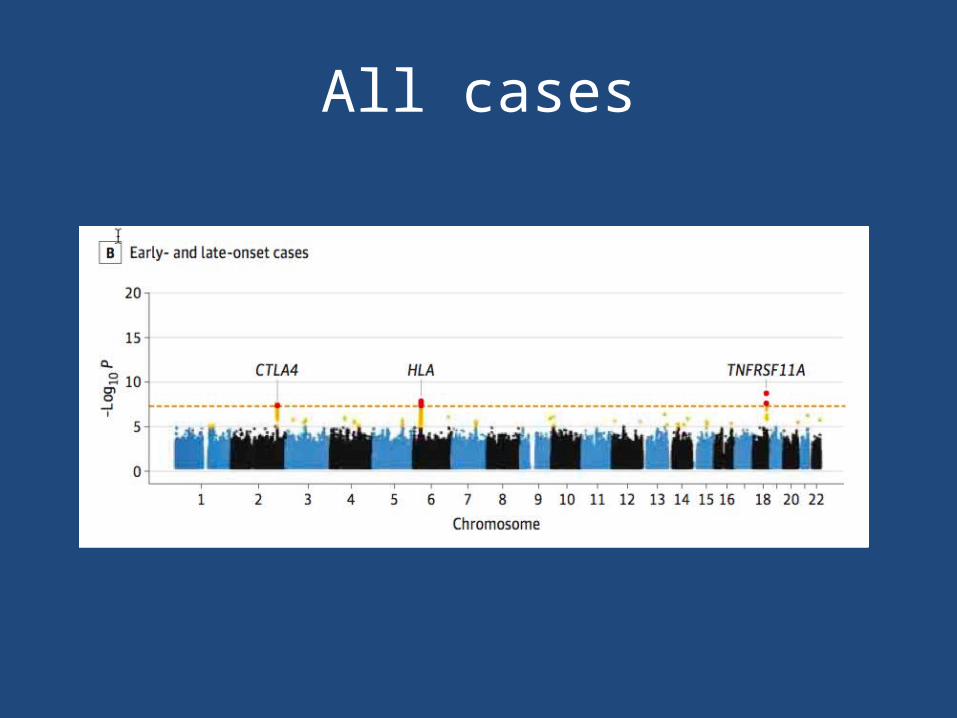
All cases
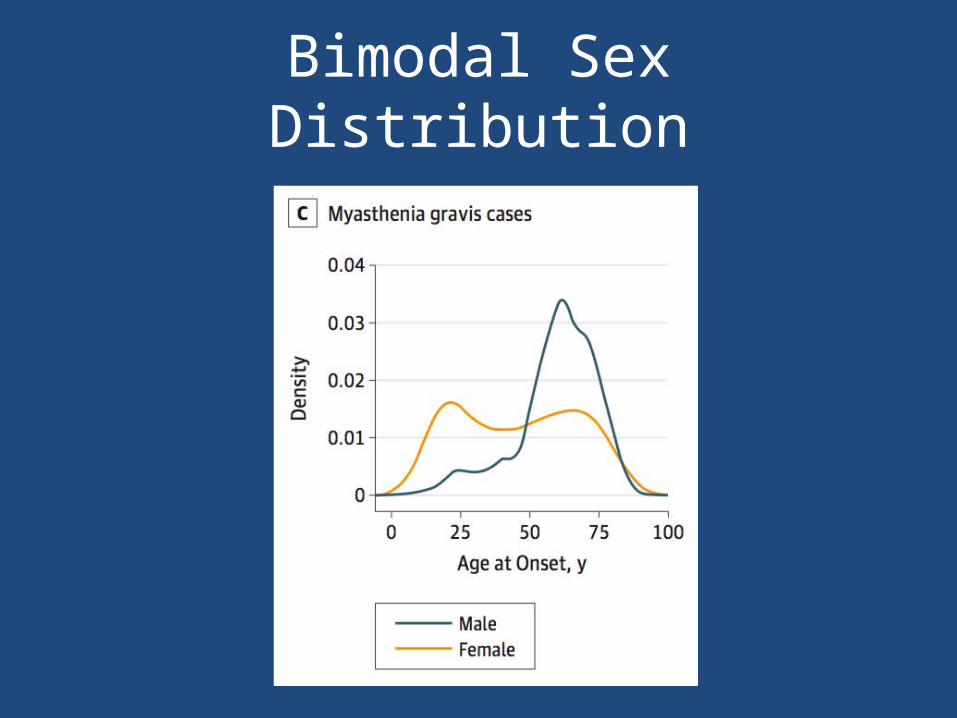
Bimodal Sex Distribution
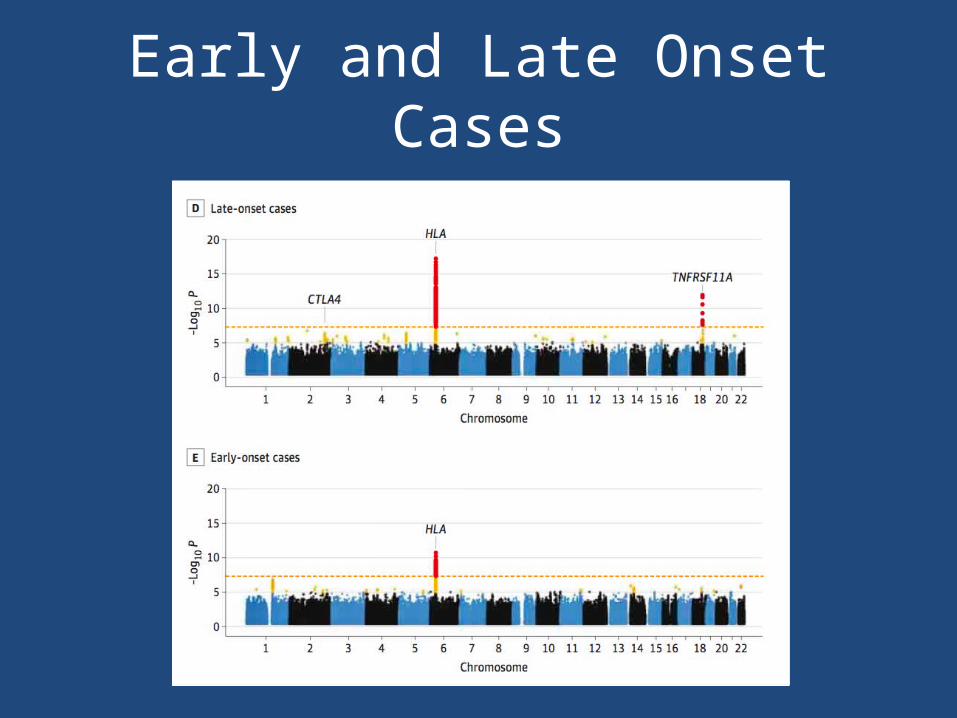
Early and Late Onset Cases

Replication Cohort
• 3 SNPs from the risk loci identified in the overall cohort for genotyping in the replication cohort of 423 Italian AChR antibody-positive MG cases and 467 controls.
• Strongest signals– rs9270986 in the intergenic region between HLA-
DRB1 and HLA-DQA1– rs231770 located 3.3 kb upstream of CTLA4

Combined Analysis
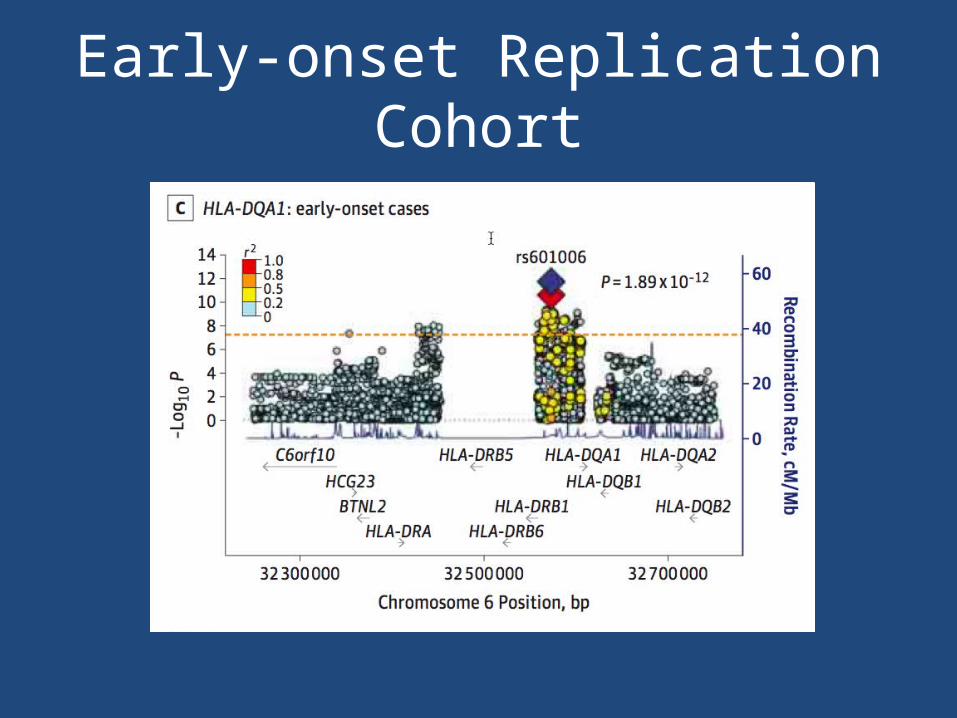
Early-onset Replication Cohort

Late-onset Replication Cohort


Summary of Results
• Overall case-control cohort– CTLA4 (rs231770) – HLA-DQA1 (rs9271871)– TNFRSF11A (rs4263037)
• Replicated for CTLA4 and HLA-DQA1 in the Italian cohort

Summary of Results
• Early and late-onset disease have distinct, but overlapping, genetic architecture – Genetic variation within TNFSRF11A locus drives
susceptibility to disease among older cases– Different haplotypes across the same HLA region on
chromosome 6 were identified in early and late-onset cases
– CTLA4 exerts significant effect regardless of age at symptom onset, suggesting it plays a central role in generating the aberrant autoimmune response that leads to neuromuscular junction dysfunction
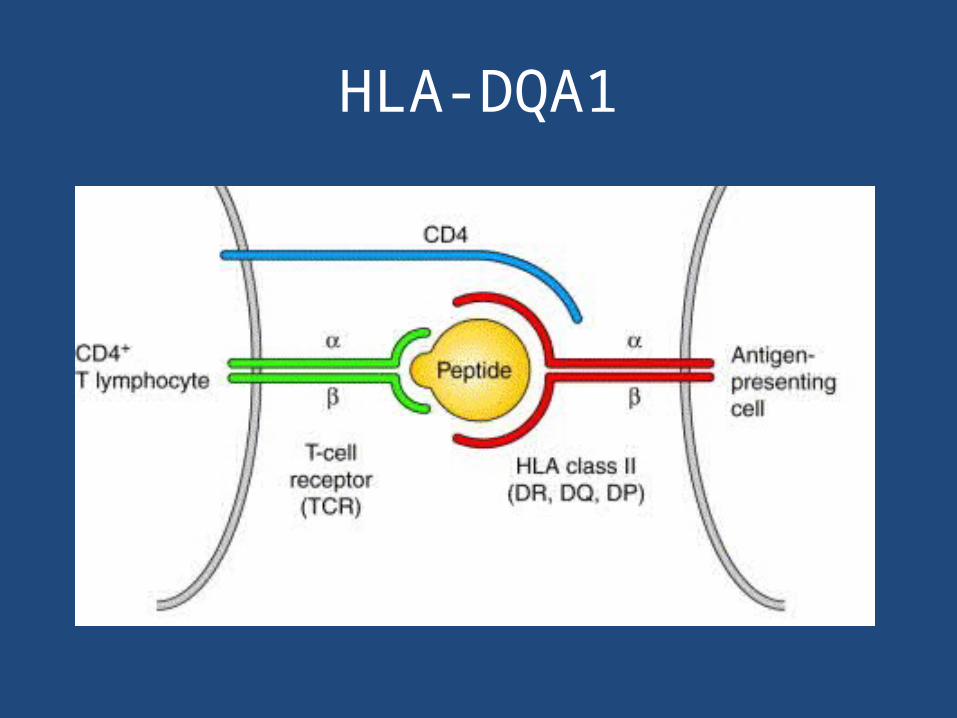
HLA-DQA1

CTLA4

TNFSF11A
• 4.5-kDa receptor activator of nuclear factor-K B expressed on the surface of antigen-presenting dendritic cells.
• Important regulator of the interaction between T cells and dendritic cells that is essential for immune surveillance and regulation of specific immunity

Study Limitations
• Sample size• Possible population stratification• Lack of age and sex match controls• AChR antibody positive only patients (85%
MG)• Different results from previous studies– Different signal in MHC region– No association of PTPN22 and TNIP1