germ cell tumors, hepatoblastoma & retinoblastoma
DESCRIPTION
Germ Cell Tumors, Hepatoblastoma & Retinoblastoma. Neyssa Marina, MD Professor of Pediatrics Division of Hematology-Oncology. Pediatric GCT. Rare: 2-3% of childhood malignancies Arise from pluripotent cells & composed of tissues foreign to site of origin - PowerPoint PPT PresentationTRANSCRIPT

Germ Cell Tumors, Hepatoblastoma &
Retinoblastoma
Neyssa Marina, MDProfessor of Pediatrics
Division of Hematology-Oncology
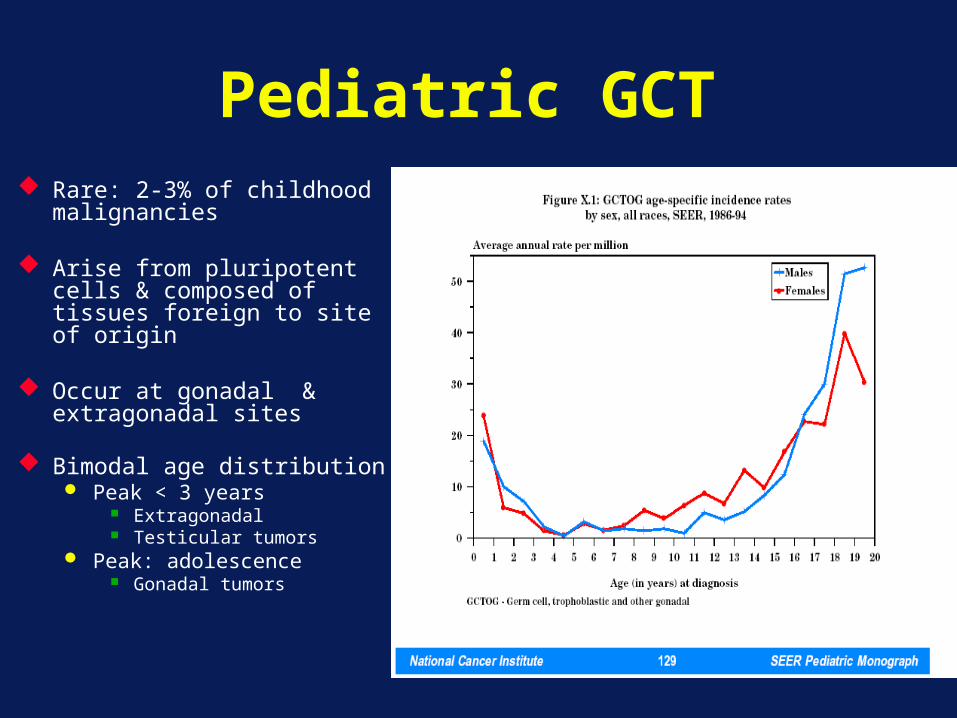
Pediatric GCT Rare: 2-3% of childhood
malignancies
Arise from pluripotent cells & composed of tissues foreign to site of origin
Occur at gonadal & extragonadal sites
Bimodal age distribution Peak < 3 years
Extragonadal Testicular tumors
Peak: adolescence Gonadal tumors

Pediatric GCT: Clinical Presentation
Depends on primary site:Ovarian: abdominal pain (may mimic acute
abdomen), palpable abdominal massTesticular: Irregular, non-tender massesExtragonadal tumors: depends on tumor
location Constipation & urinary retention for
sacrococcygeal tumors Respiratory distress for mediastinal tumors

Pediatric GCT: Laboratory Work-up
Alfa fetoprotein (AFP): elevated in yolk sac tumor and embryonal carcinoma; half-life 5-7 days
β-Human chorionic gonadotropin (β-HCG): usually synthesized during pregnancy & elevated in choriocarcinoma, embryonal carcinoma and germinomas; half-life 24-36 hours
Lactic dehydrogenase (LDH): correlate with tumor burden in patients with dysgerminoma
Placental alkaline phosphatase (PLAP): elevated in patients with dysgerminoma

Pediatric GCT: Imaging Work-up
CT scan or MRI of primary: to evaluate the extent of loco-regional disease
Chest CT: to evaluate presence of metastases
Bone scan: to evaluate for distant metastases

Pediatric GCT: StagingStage
Description
I Complete resection with normalization of tumor markers within expected half-life.
II Microscopic residual disease: persistent marker elevation; lymph nodes < 2 cms
III Gross residual disease: retroperitoneal lymph nodes > 2 cms; no extra-abdominal or visceral metastases
IV Distant metastases
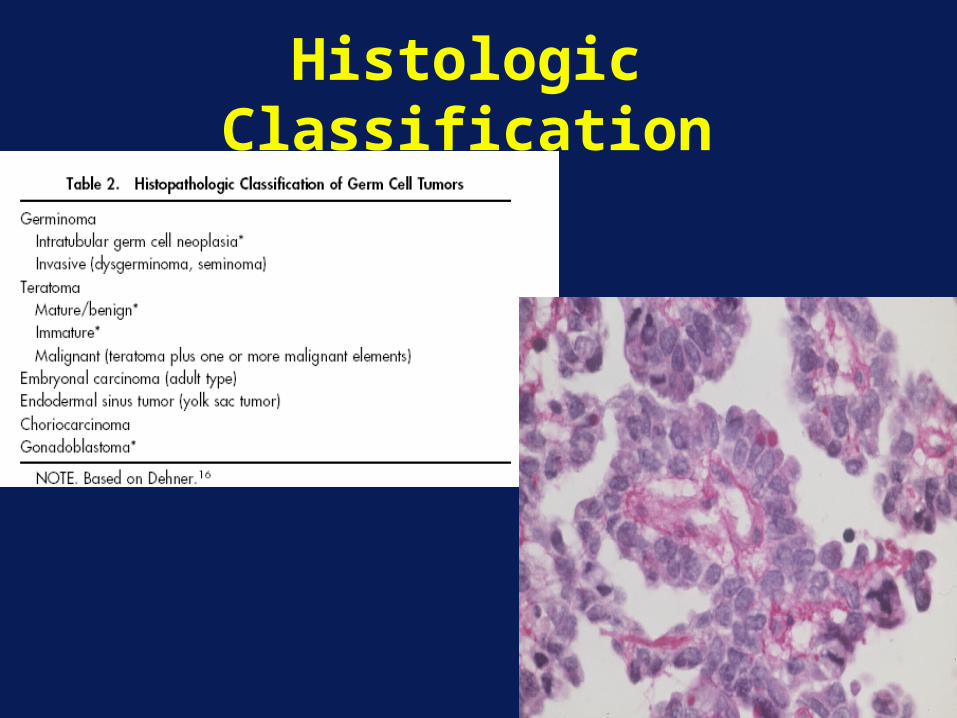
Histologic Classification

GCT: Pediatric Versus Adult
HistologicallyChildren < 4 years age: endodermal sinus
tumorAdolescents: mixed histology tumors
Genetically (Schneider, Genes, Chromosomes & Cancer 34:115, 2001)Childhood tumors: diploid & tetraploid
Gains of chromosomes (1q, 3 & 20q) & deletions 1p & 6q
Adolescent tumors: aneuploid Isochromosome 12p
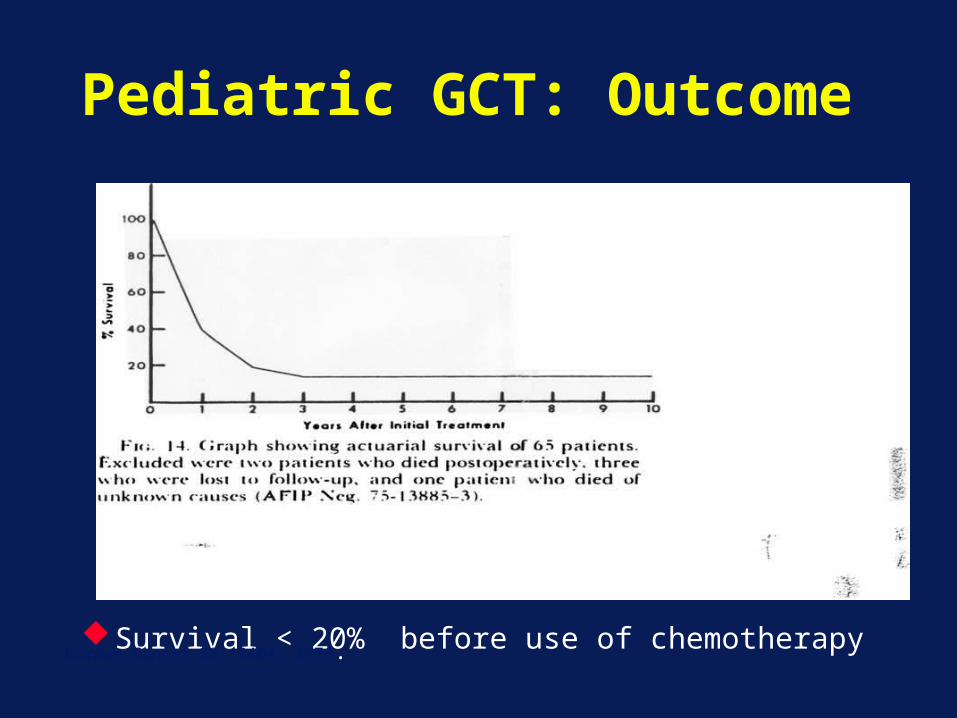
Pediatric GCT: Outcome
Survival < 20% before use of chemotherapyKurman Cancer 38: 2404, 1976.
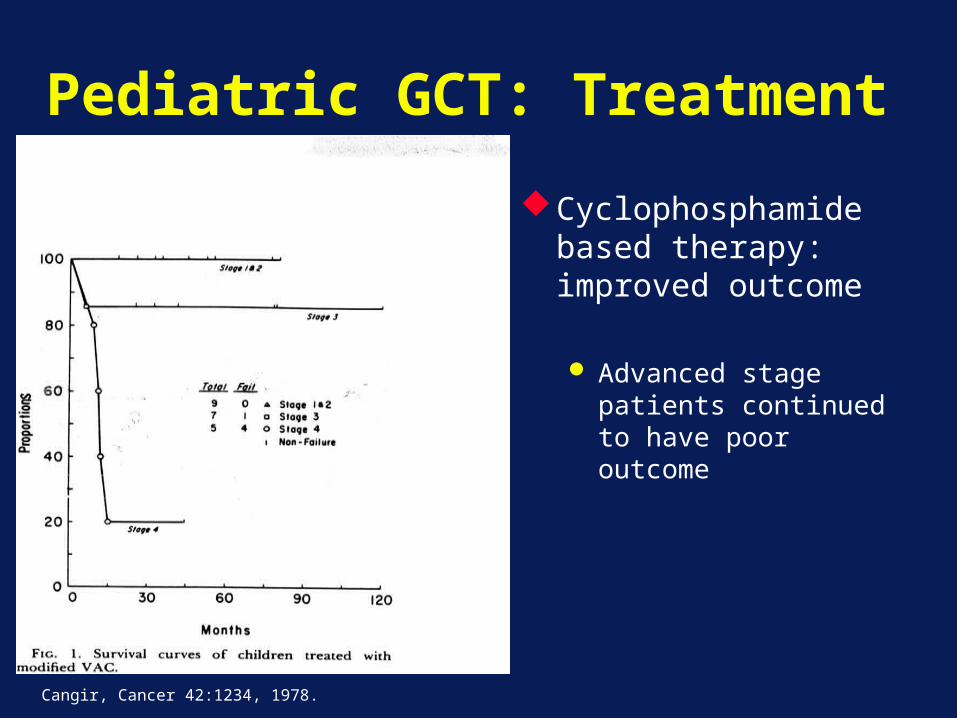
Pediatric GCT: Treatment
Cyclophosphamide based therapy: improved outcome
Advanced stage patients continued to have poor outcome
Cangir, Cancer 42:1234, 1978.
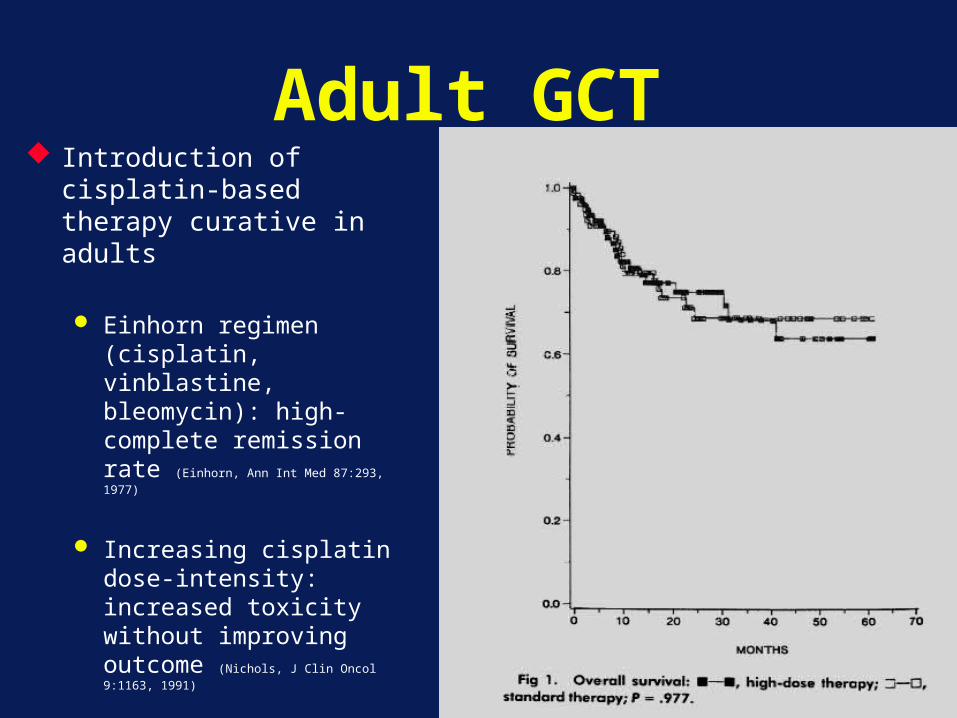
Adult GCT Introduction of cisplatin-
based therapy curative in adults
Einhorn regimen (cisplatin, vinblastine, bleomycin): high-complete remission rate (Einhorn, Ann Int Med 87:293, 1977)
Increasing cisplatin dose-intensity: increased toxicity without improving outcome (Nichols, J Clin Oncol 9:1163, 1991)

Pediatric GCT: Outcome
Although cisplatin-based therapy appeared effective in small number of pediatric patients
Significant concerns regarding pulmonary and ototoxicity prevented widespread use of this therapy
Mann, Cancer 63:1657, 1989
Pinkerton, et al. J Clin Oncol, 1986

Pediatric GCT: TreatmentBased on differences between pediatric
and adult tumors, the Pediatric Oncology Group (POG) and the Children’s Cancer Group (CCG) designed two prospective studiesLocalized gonadal GCT:
Stage I testicular: evaluate the event-free survival & overall survival following surgical resection.
Stage I/II malignant GCT: evaluate the role of surgery + PEB
Advanced GCT: Stage III/IV gonadal & stage I-IV extragonadal:
evaluate the role of cisplatin dose-intensity in a randomized trial
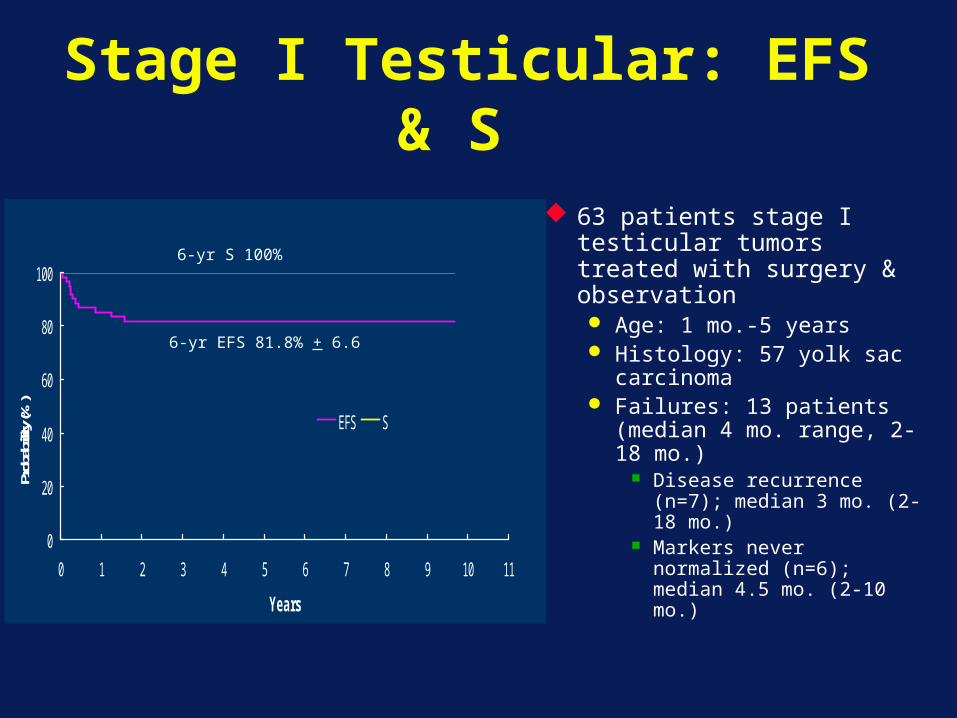
Stage I Testicular: EFS & S
0
20
40
60
80
100
0 1 2 3 4 5 6 7 8 9 10 11
Years
Prob
abilit
y (%
)
EFS S
45 37
56 46
63 patients stage I testicular tumors treated with surgery & observation Age: 1 mo.-5 years Histology: 57 yolk sac
carcinoma Failures: 13 patients
(median 4 mo. range, 2-18 mo.)
Disease recurrence (n=7); median 3 mo. (2-18 mo.)
Markers never normalized (n=6); median 4.5 mo. (2-10 mo.)
6-yr EFS 81.8% + 6.6
6-yr S 100%
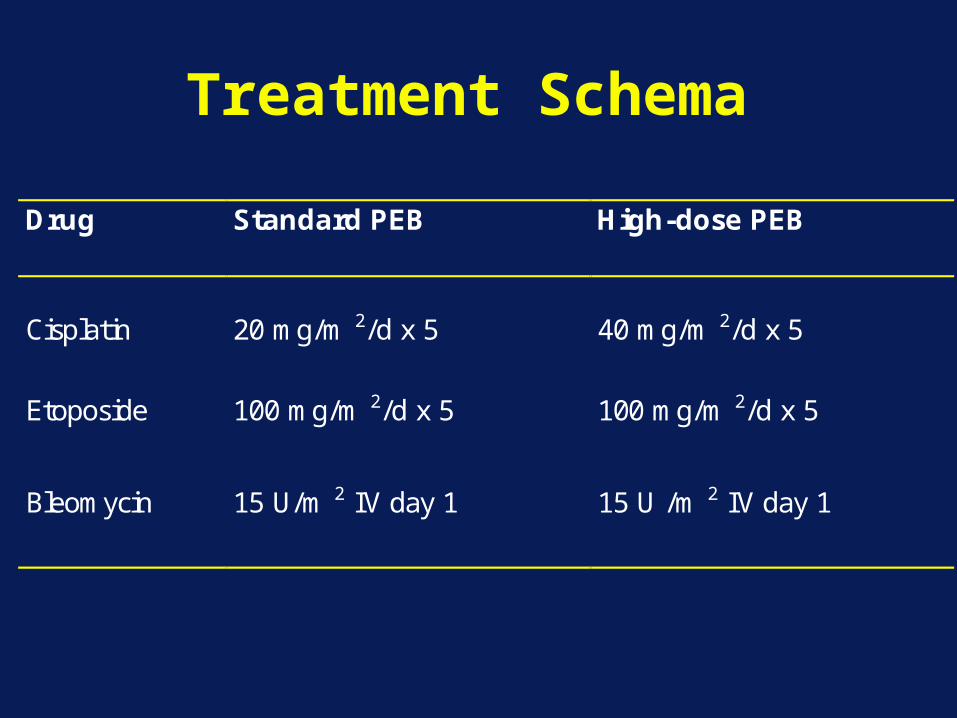
Treatment Schema
Drug Standard PEB High-dose PEB
Cisplatin
20 mg/m 2/d x 5
40 mg/m 2/d x 5
Etoposide
100 mg/m 2/d x 5
100 mg/m 2/d x 5
Bleomycin 15 U/m 2 IV day 1 15 U /m 2 IV day 1
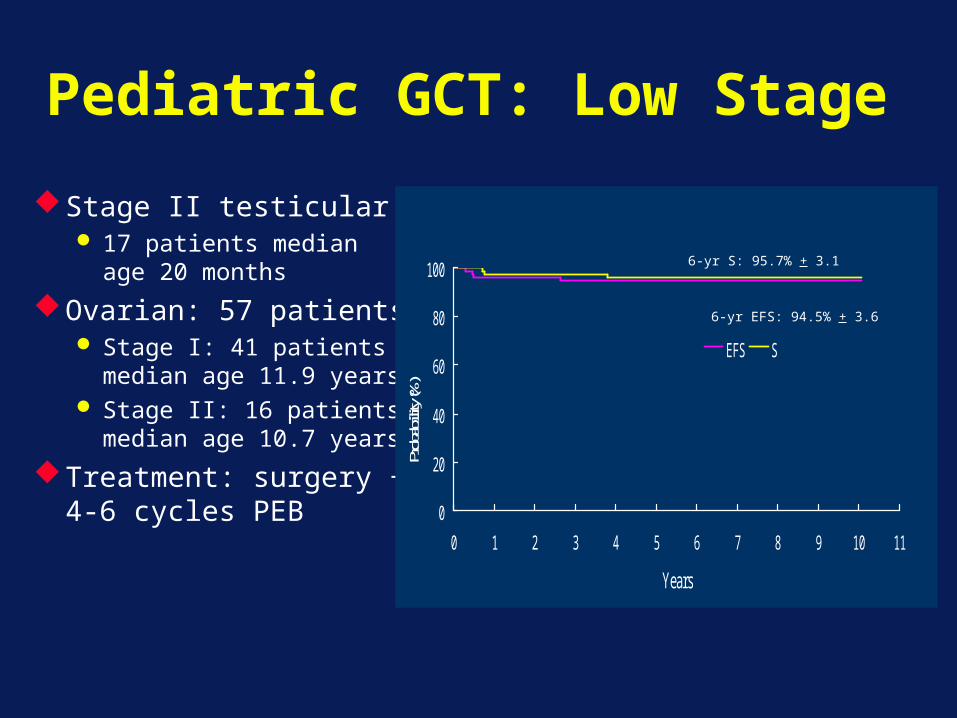
Pediatric GCT: Low Stage
Stage II testicular 17 patients median
age 20 months
Ovarian: 57 patients Stage I: 41 patients
median age 11.9 years Stage II: 16 patients
median age 10.7 years
Treatment: surgery + 4-6 cycles PEB
0
20
40
60
80
100
0 1 2 3 4 5 6 7 8 9 10 11
Years
Probab
ility (
%)
EFS S
69 52
70 536-yr S: 95.7% + 3.1
6-yr EFS: 94.5% + 3.6

Advanced GCT Study Design
Diagnosis
RANDOMIZE
Cisplatin 100 mg/m2EtoposideBleomycinPEB
Cisplatin 200 mg/m2
EtoposideBleomycinHD-PEB

Advanced Pediatric GCT: Patients
299 patients diagnosed between February 1990-1996 Median age 3.4 years (range 3 days-20 years) 183 female Primary sites
165 extragonadal tumors 134 gonadal tumors
Stage distribution: 30 stage I/II 136 stage III 133 stage IV
Following surgery patients randomized 150 patients (PEB): 67 gonadal tumors; 83 extragonadal 149 patients (HD-PEB): 67 gonadal; 82 extragonadal
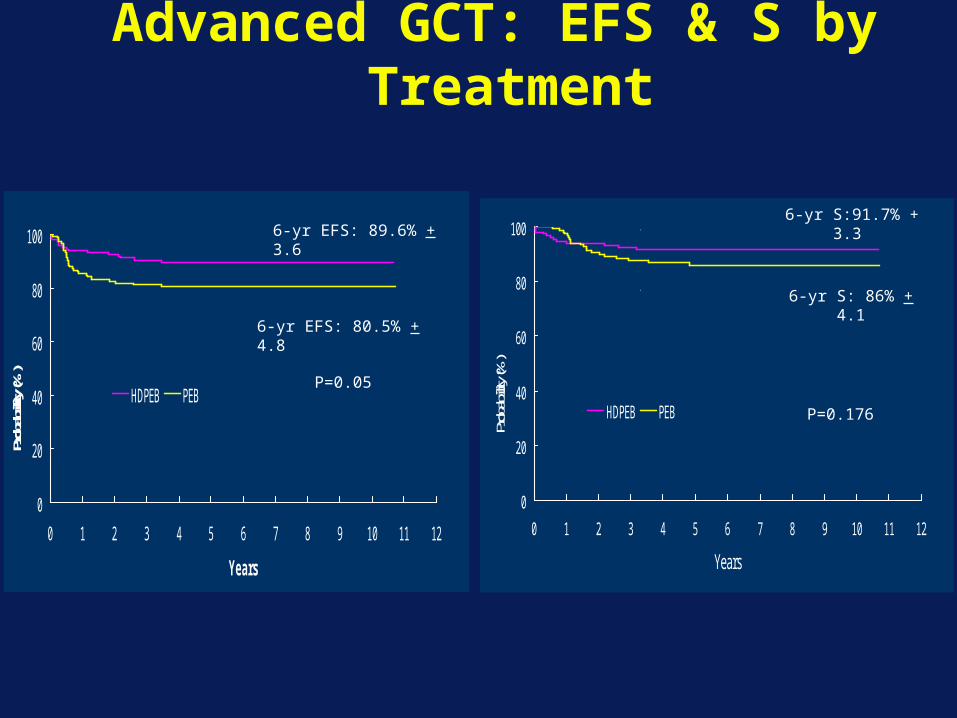
Advanced GCT: EFS & S by Treatment
0
20
40
60
80
100
0 1 2 3 4 5 6 7 8 9 10 11 12
Years
Proba
bility
(%)
HDPEB PEB
132 89
122 72
P=0.0284
6-yr EFS: 89.6% + 3.6
6-yr EFS: 80.5% + 4.8
0
20
40
60
80
100
0 1 2 3 4 5 6 7 8 9 10 11 12
Years
Proba
bility
(%)
HDPEB PEB
134 91
134 80
6-yr S:91.7% + 3.3
6-yr S: 86% + 4.1
P=0.176
P=0.05

Extragonadal GCT: Prognostic Factors
Extragonadal GCT typically considered high-risk Examine prognostic factors in a large group of patients
By multivariate Cox regression for EFS Age > 12 years: only significant prognostic factor
(p=0.002) Relative Risk 3.8
After adjusting for age, treatment was borderline significant (p=0.064)
In multivariate Cox regression for OS, the interaction of age & primary site was highly significant (p<0.0001) Patients > 12 years with thoracic tumors 5.9 times
greater risk of death than patients < 12 years or patients with any other primary

GCT: ConclusionsPatients with stage I GCT represent a low-risk group Patients with stage II-III gonadal GCT appear to be an
intermediate risk group
Patients with advanced extragonadal tumors represent a high-risk group Age > 12 years is the factor most predictive for EFS in these
patients There is a significant interaction between age and primary
site. This suggests that patients over 12 years with thoracic
tumors are biologically different.
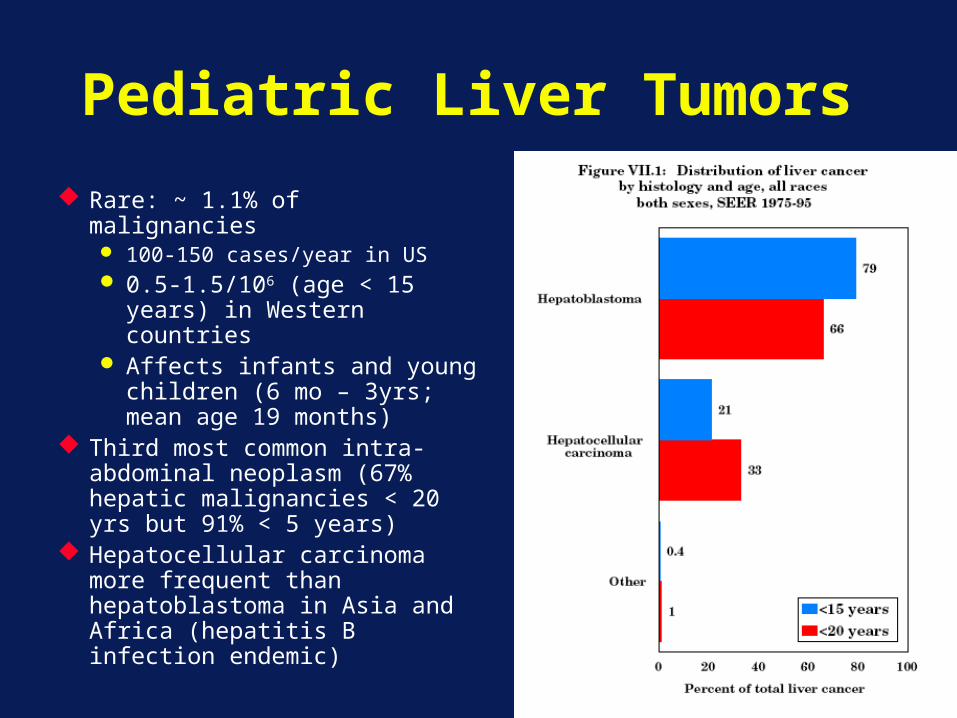
Pediatric Liver Tumors
Rare: ~ 1.1% of malignancies 100-150 cases/year in US 0.5-1.5/106 (age < 15
years) in Western countries
Affects infants and young children (6 mo – 3yrs; mean age 19 months)
Third most common intra-abdominal neoplasm (67% hepatic malignancies < 20 yrs but 91% < 5 years)
Hepatocellular carcinoma more frequent than hepatoblastoma in Asia and Africa (hepatitis B infection endemic)
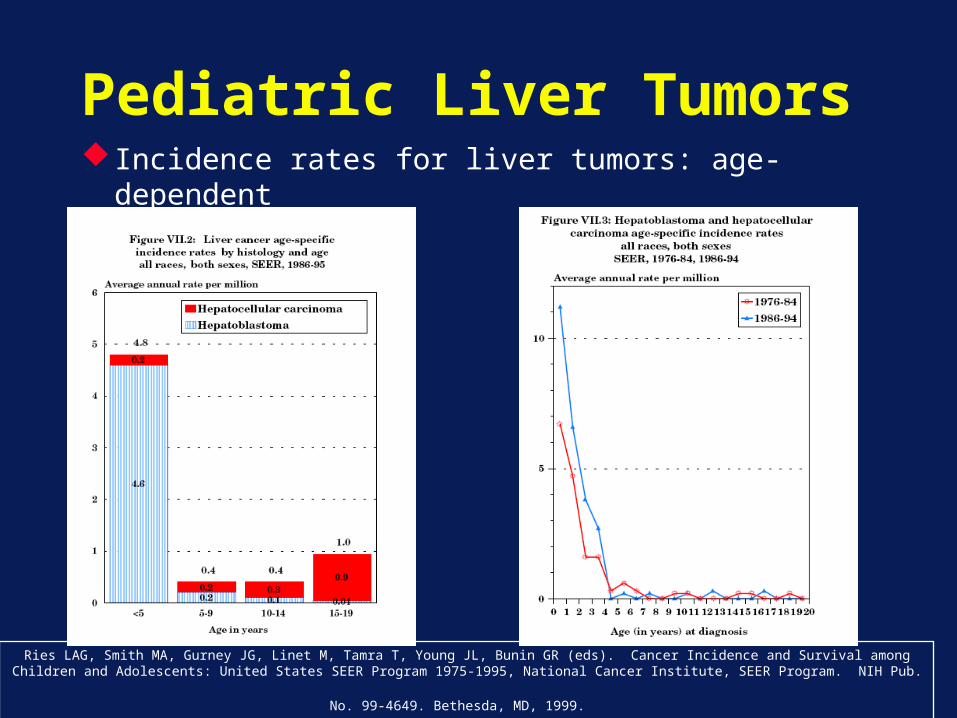
Pediatric Liver Tumors Incidence rates for liver tumors: age-dependent
Ries LAG, Smith MA, Gurney JG, Linet M, Tamra T, Young JL, Bunin GR (eds). Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975-1995, National Cancer Institute, SEER Program. NIH Pub. No. 99-4649. Bethesda, MD,
1999.

Hepatoblastoma: Risk Factors
Prematurity and low birth weight Disproportionate # of cases with BW < 2500
grams RR 15.64 for BW <1000g, 2.53 for BW 1000-1499g,
1.21 for BW 1500-2499g
Association with overgrowth syndromes: Beckwith-Wiedemann (LOH 11p15) Familial adenomatous polyposis (FAP; inactivation
of tumor suppressor gene on chromosome 5) Estimated that 1:20 cases of hepatoblastoma have FAP Lifetime risk of hepatoblastoma for children of FAP
families: 1/250 compared to 1/100,000 in general population

Hepatoblastoma: Clinical Presentation
Asymptomatic abdominal massWeight loss, anorexia, emesis, and abdominal pain
(advanced disease)Distant metastases ~ 20% of cases mostly to lung
Intraperitoneal, lymph node, brain, and local tumor thrombus
Thrombocytosis is common HB cells secrete IL-1B: induces fibroblasts/endothelial cells
to produce IL-6 hepatocyte growth factor secretion and thrombopoeitin secretion
90% of patients have elevated alpha-fetoproteinRare: hypertension in cases of renin-secreting mixed
HB or precocious puberty in HB secreting human chorionic gonadotropin

Hepatoblastoma: Histology
Derived from undifferentiated embryonal tissue/pluripotent hepatic stem cells
Differentiates into hepatocytes, biliary epithelial cells Originally, 2 subtypes recognized
Epithelial (mixture of embryonal and fetal) Mixed epithelial and mesenchymal
Later classification based on degree of differentiation Embryonal (30%) : tubular or glandular; rosettes of
elongated cells Fetal (54%) : highly differentiated; resemble normal
hepatocytes with rare mitoses; lack normal lobular architecture
Anaplastic/small cell undifferentiated type (6%) : small cells with densely stained nuclei and scant cytoplasm
Macrotrabecular (10%) : features similar to hepatocellular carcinoma

Hepatoblastoma: Relevance of Histology
Favorable histology defined: “completely resected tumor with a uniform, well-differentiated fetal component exhibiting < 2 mitoses per 10 HPF”Patients treated with surgical resection alone
All other histology is considered unfavorable and if stage II-IV, histology is considered irrelevant
Ortega et. al. J Clin Oncology, 2000

Hepatoblastoma: Work-Up
Diagnostic imaging: important role in diagnosis, staging and treatmentUltrasound: usually first test performed
Helps evaluate cystic versus solid masses
CT scan or MRI: defines the tumor extent, vascular supply, operability and distant extent of tumor
Laboratory work-up:Alfa Fetoprotein: most valuable test
Elevated in 80-90% of patients & useful for monitoring
Biologic half-life: 5-7 days

Hepatoblastoma: Staging
Critical to have agreed-upon staging allowing comparison between different studies
Early studies of hepatoblastoma showed that surgical resection is the mainstay of therapy and required for cureStaging based on surgical criteria (currently used
by German Cooperative Group, CCG, POG)
Investigators at SIOP began using preoperative chemotherapy for all patients and thus devised alternative staging system (PRETEXT)
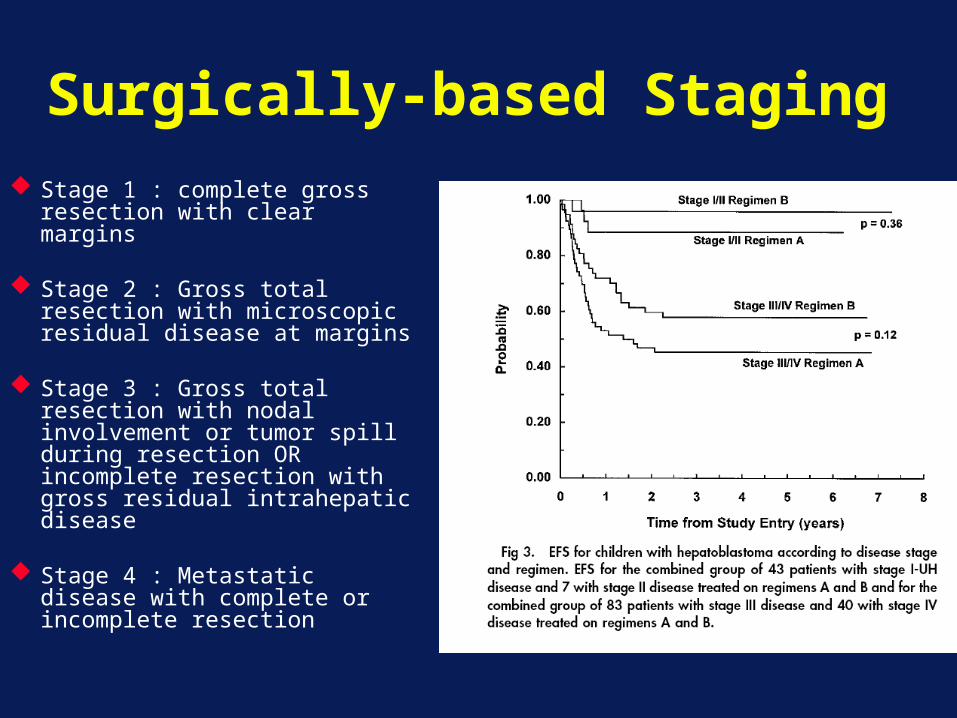
Surgically-based Staging Stage 1 : complete gross
resection with clear margins
Stage 2 : Gross total resection with microscopic residual disease at margins
Stage 3 : Gross total resection with nodal involvement or tumor spill during resection OR incomplete resection with gross residual intrahepatic disease
Stage 4 : Metastatic disease with complete or incomplete resection
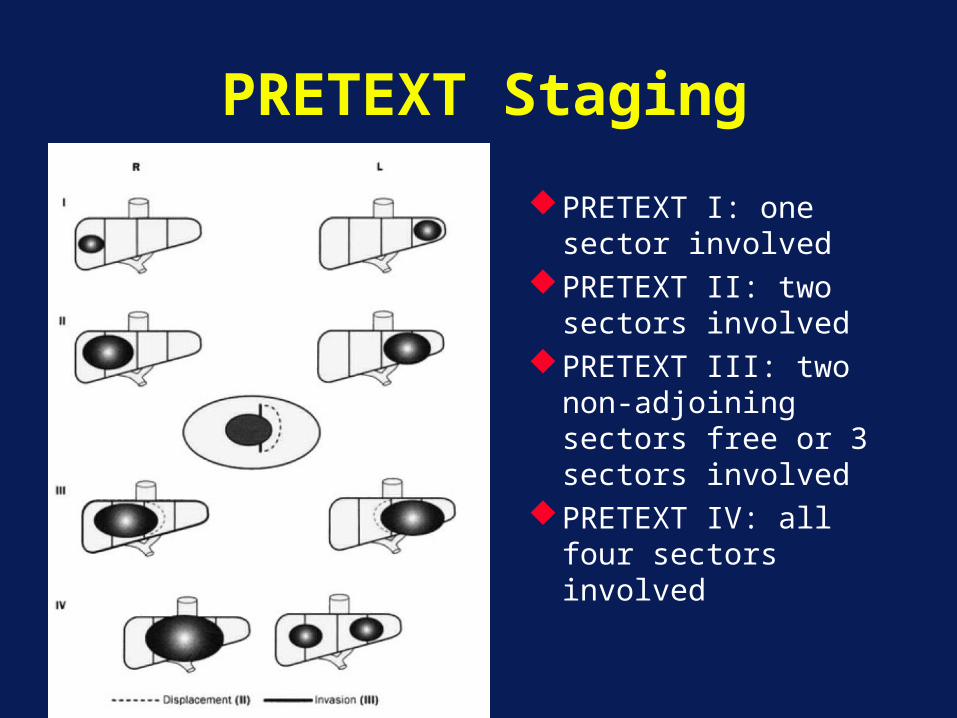
PRETEXT Staging
PRETEXT I: one sector involved
PRETEXT II: two sectors involved
PRETEXT III: two non-adjoining sectors free or 3 sectors involved
PRETEXT IV: all four sectors involved
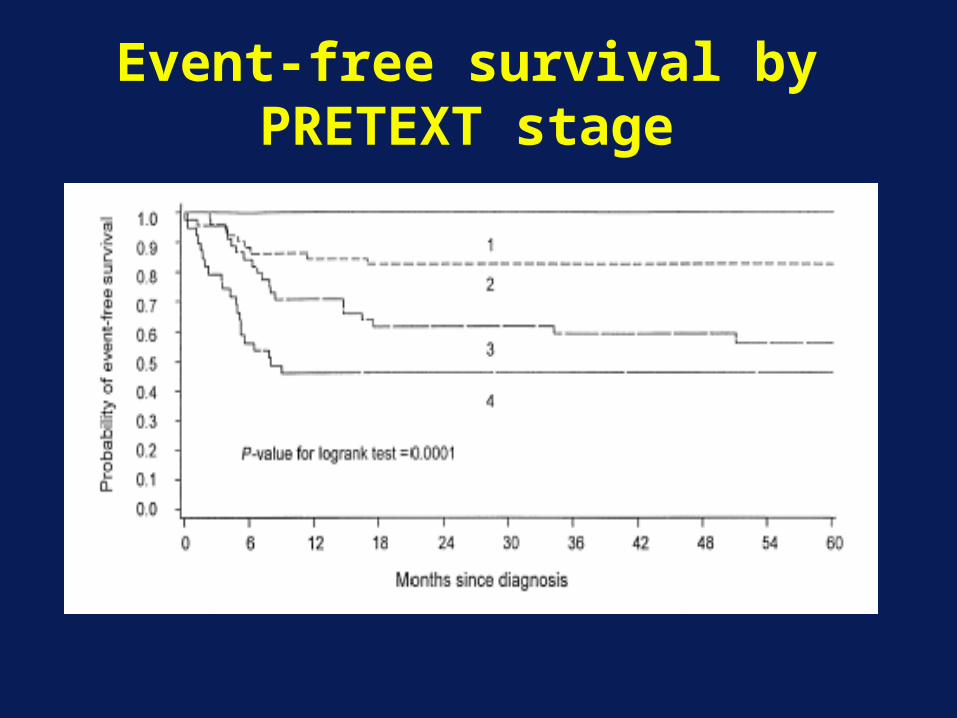
Event-free survival by PRETEXT stage
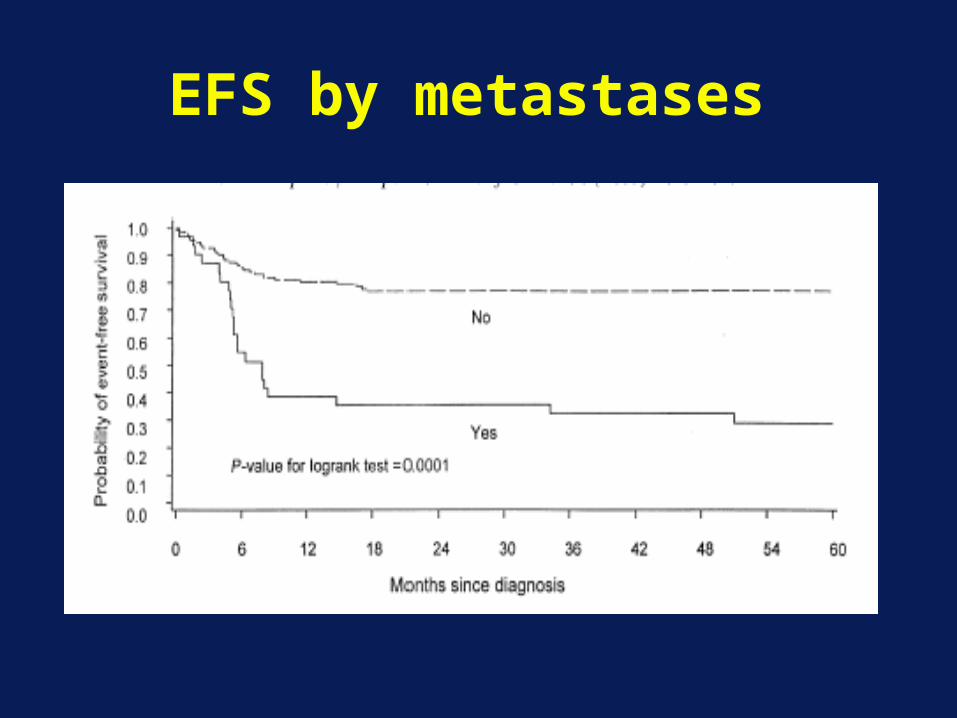
EFS by metastases

Hepatoblastoma: Treatment
Complete surgical resection: mainstay of therapyPossible at diagnosis: < 50% of patientsSurgery: curative > 90% of purely fetal
hepatoblastomas 5-year survival with surgery: < 10% other
histologies
Chemotherapy: used to convert inoperable tumors into resectable tumors
Current 5-year survival rate 75%Current objective: improve the prognosis for the
25% of patients who die of disease

New Approaches to Treatment
“New Agents”: attempt to increase response rate
Chemoembolization: Intra-arterial co-administration of chemotherapeutic and vascular occlusive agents to treat malignant diseases.
Liver Transplant: an alternative patients with unresectable disease following chemotherapy
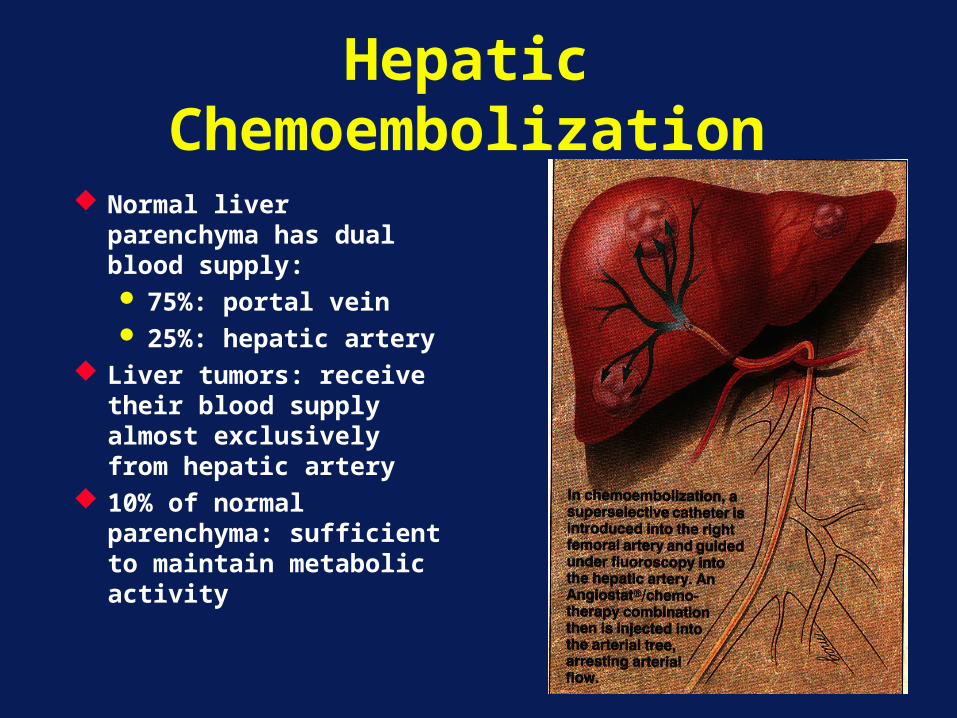
Hepatic Chemoembolization
Normal liver parenchyma has dual blood supply: 75%: portal vein 25%: hepatic artery
Liver tumors: receive their blood supply almost exclusively from hepatic artery
10% of normal parenchyma: sufficient to maintain metabolic activity
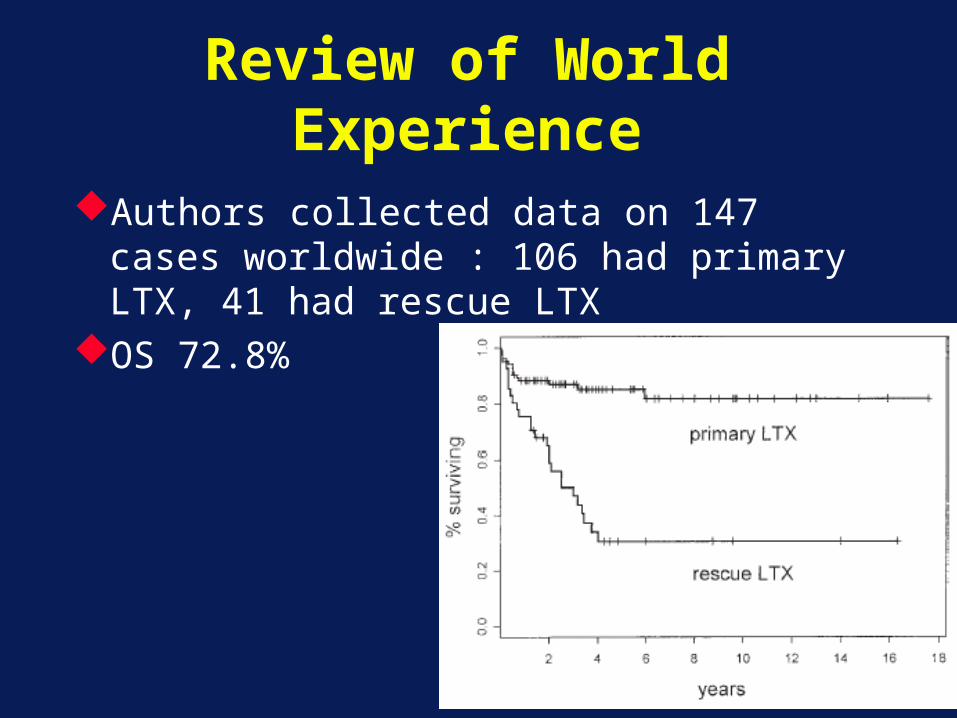
Review of World Experience
Authors collected data on 147 cases worldwide : 106 had primary LTX, 41 had rescue LTX
OS 72.8%

Hepatoblastoma: Conclusions
The addition of cisplatin-based therapy has improved the outcome for patients with hepatoblastoma Increasing the proportion of patients who can undergo
resection
Prognosis: sub-optimal for patients with unresectable tumors (following chemotherapy) and for patients with metastases Chemo-embolization and liver transplantation appear
to be promising in this subset of patients Identification of new active agents important to
attempt to decrease the number of patients with unresectable tumors following chemotherapy
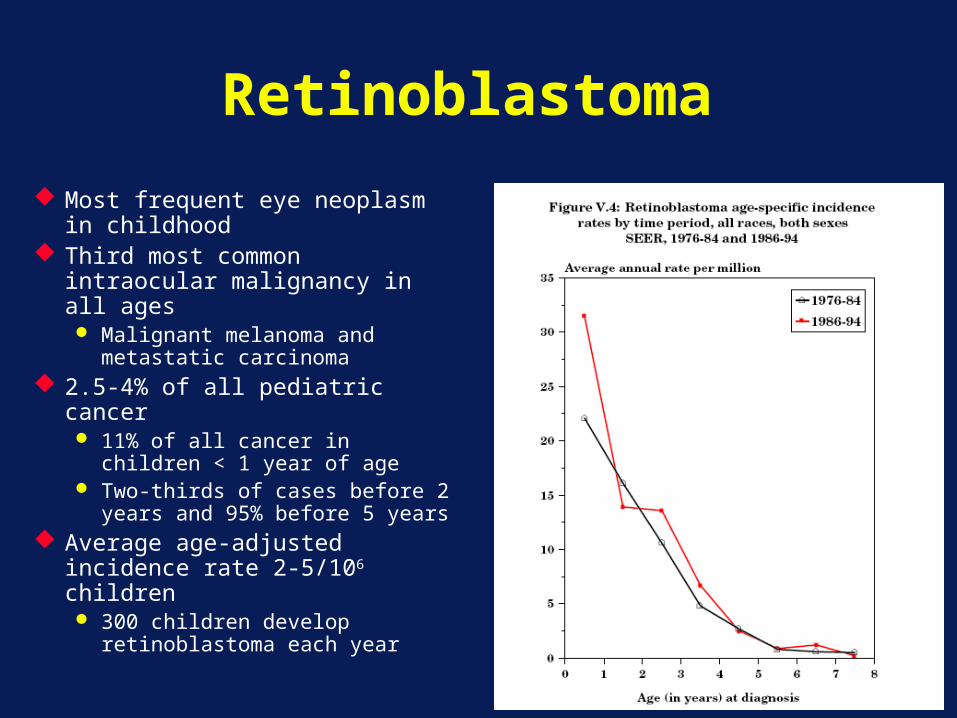
Retinoblastoma
Most frequent eye neoplasm in childhood
Third most common intraocular malignancy in all ages Malignant melanoma and
metastatic carcinoma 2.5-4% of all pediatric cancer
11% of all cancer in children < 1 year of age
Two-thirds of cases before 2 years and 95% before 5 years
Average age-adjusted incidence rate 2-5/106 children 300 children develop
retinoblastoma each year
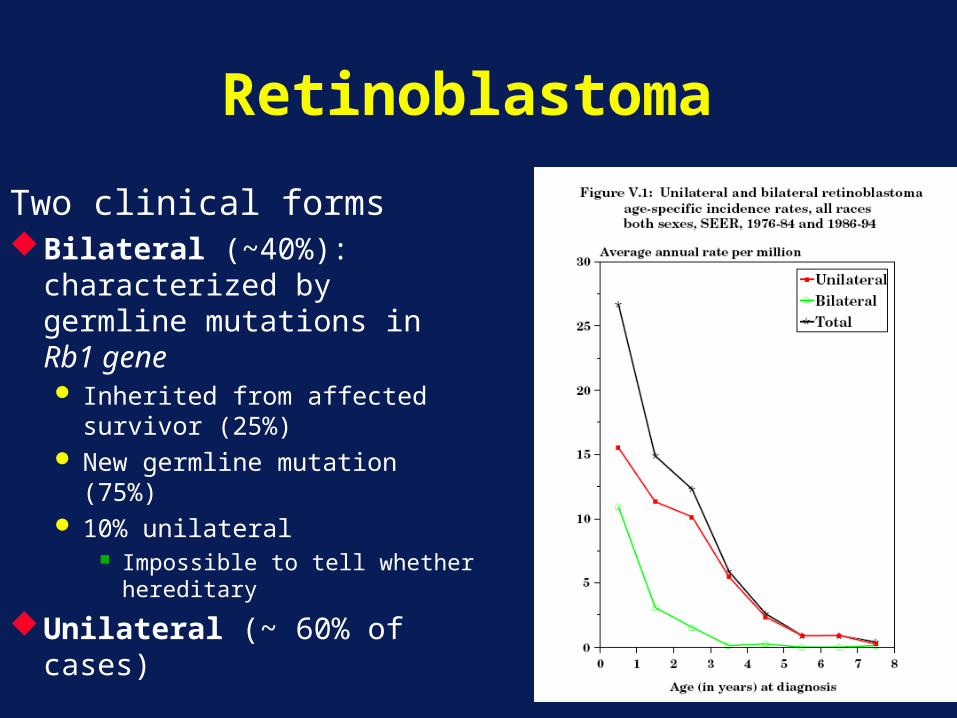
Retinoblastoma
Two clinical formsBilateral (~40%):
characterized by germline mutations in Rb1 gene Inherited from affected
survivor (25%) New germline mutation
(75%) 10% unilateral
Impossible to tell whether hereditary
Unilateral (~ 60% of cases)
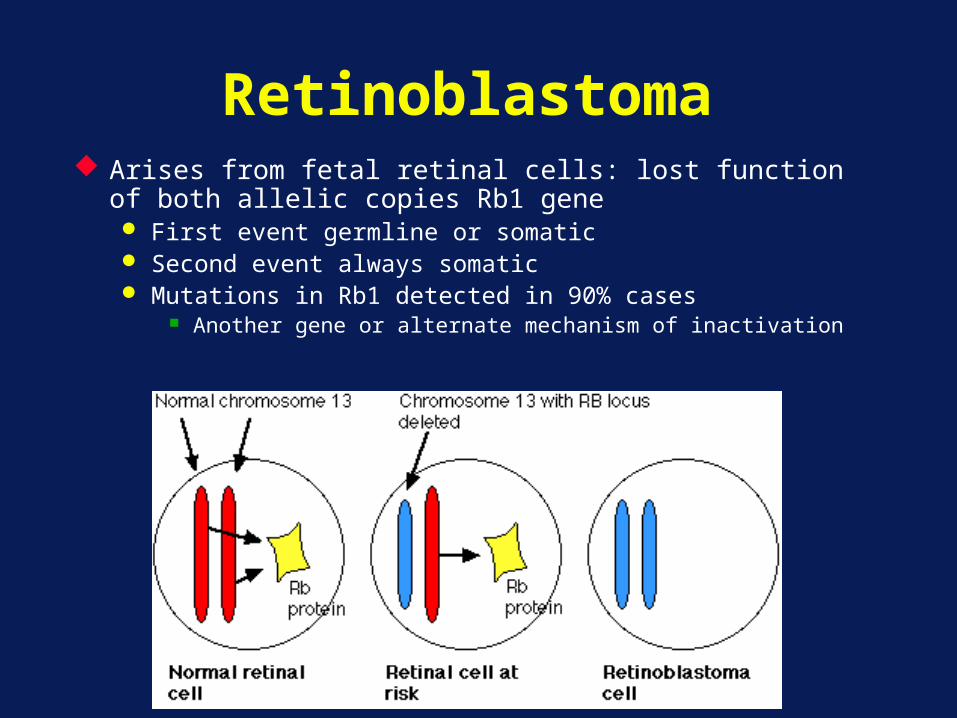
Retinoblastoma Arises from fetal retinal cells: lost function of both allelic
copies Rb1 gene First event germline or somatic Second event always somatic Mutations in Rb1 detected in 90% cases
Another gene or alternate mechanism of inactivation

Retinoblastoma
Unique tumor: genetic form predisposes to tumor development in autosomal dominant fashion (85-90% penetrance)
Majority of children acquire new mutation (15-25% positive family history)
Risk of retinoblastoma in offspring of retinoblastoma survivors Bilateral disease: 45% Unilateral disease: 2.5%
Risk of retinoblastoma in siblings: Bilateral disease: 45% Unilateral disease: 30%

Retinoblastoma: Clinical Presentation
Tumor of the young Age at presentation
correlates with laterality Bilateral < 1 year of age Unilateral: 2nd or 3rd year
of life Half of cases diagnosed
under 1 year: bilateral compared to <10% of cases diagnosed after 1 year
Most common presentation leukocoria followed by strabismus

Retinoblastoma: Evaluation
Diagnosis made without pathologic confirmation Mass protruding into the vitreous Detailed documentation of number, location & size of
tumors as well as retinal detachment, sub-retinal fluid & vitreous, sub-retinal seeds
Imaging studies aid diagnosis CT, ultrasound & MRI: important to evaluate
extraocular extensionMetastases: 10-15% of patients associated
with choroidal, scleral invasion or involvement of iris-ciliary body or optic nerve Bone marrow aspirate, CSF & bone scintigraphy to
evaluate patients with these findings
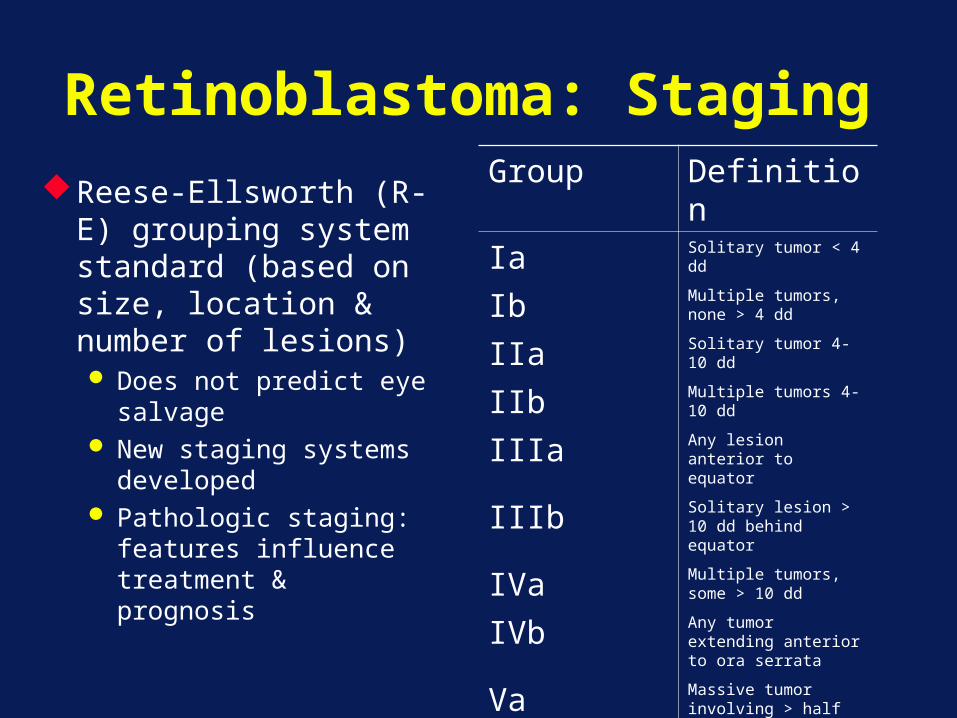
Retinoblastoma: StagingReese-Ellsworth (R-E)
grouping system standard (based on size, location & number of lesions) Does not predict eye
salvage New staging systems
developed Pathologic staging:
features influence treatment & prognosis
Group Definition
Ia Solitary tumor < 4 dd
Ib Multiple tumors, none > 4 dd
IIa Solitary tumor 4-10 dd
IIb Multiple tumors 4-10 dd
IIIa Any lesion anterior to equator
IIIb Solitary lesion > 10 dd behind equator
IVa Multiple tumors, some > 10 dd
IVb Any tumor extending anterior to ora serrata
Va Massive tumor involving > half retina
Vb Vitreous seeding

Retinblastoma: Staging
Extra retinal extension: large intraocular dimensionMetastatic risk & mortality: invasion of ocular
coats and optic nerve
Optic nerve involvement common (25-45%): impact on outcome limited to involvement beyond lamina cribosa
Choroidal involvement: up to 40% patientsExtensive < 10%: prognostic implication

Retinoblastoma: Treatment
Treatment: aims at preserving life and useful vision
Factors considered:Disease: unilateral vs. bilateralPotential for visionStaging: intra & extra ocular

Retinoblastoma: Treatment
Enucleation: large tumors filling the vitreous with no likelihood of restoring vision Ocular implant usually placed
Focal treatments: small tumors in patients with bilateral disease combined with chemotherapy
Chemotherapy: extraocular disease, intraocular disease with high-risk features and patients with bilateral disease (combined with focal therapies)
Radiotherapy: combined with focal treatment provides excellent local control Radiation predisposes to second malignancies: avoid or
delay its use

Retinoblastoma: Treatment
Outcome: excellent for unilateral disease treated with enucleation (85-90% cure) Successful chemoreduction has led to attempts at
salvaging eyes in very young children with unilateral disease
Bilateral disease: treated enucleation of eyes with advanced disease and radiation for remaining eyes Up-front chemotherapy to achieve chemoreduction
followed by aggressive focal therapy Increase in eye salvage rate & decrease and delay of
radiotherapy Best results with carboplatin, vincristine and etoposide

Retinoblastoma: Conclusion
The outcome for patients with retinoblastoma is excellent
Treatment strategies are aimed at increasing eye salvage rate and decreasing late effectsPatients with bilateral disease are at risk for
second malignancies The use of radiotherapy increases that risk
Genetic counseling is an essential part of treatment for patients with bilateral disease