global post market surveillance - cbinet.com post market surveillance 3. post market clinical...
TRANSCRIPT

1
Global Post market Surveillance
Patrick Caines, Ph.D.
Baxter Healthcare
15 June 2016

2
Agenda
Definition of an effective Post market surveillance System
Regulatory Requirements
Components
• Surveillance information sources
• Investigation and Analysis
• Action System and Outputs
‾ MDR, MDV, AE, FAR, BPDR
• Management Reviews & Dissemination
Monitoring Health of Post market surveillance system
Best Practices
Questions & Discussion Case Studies

Global Post Market Surveillance
3
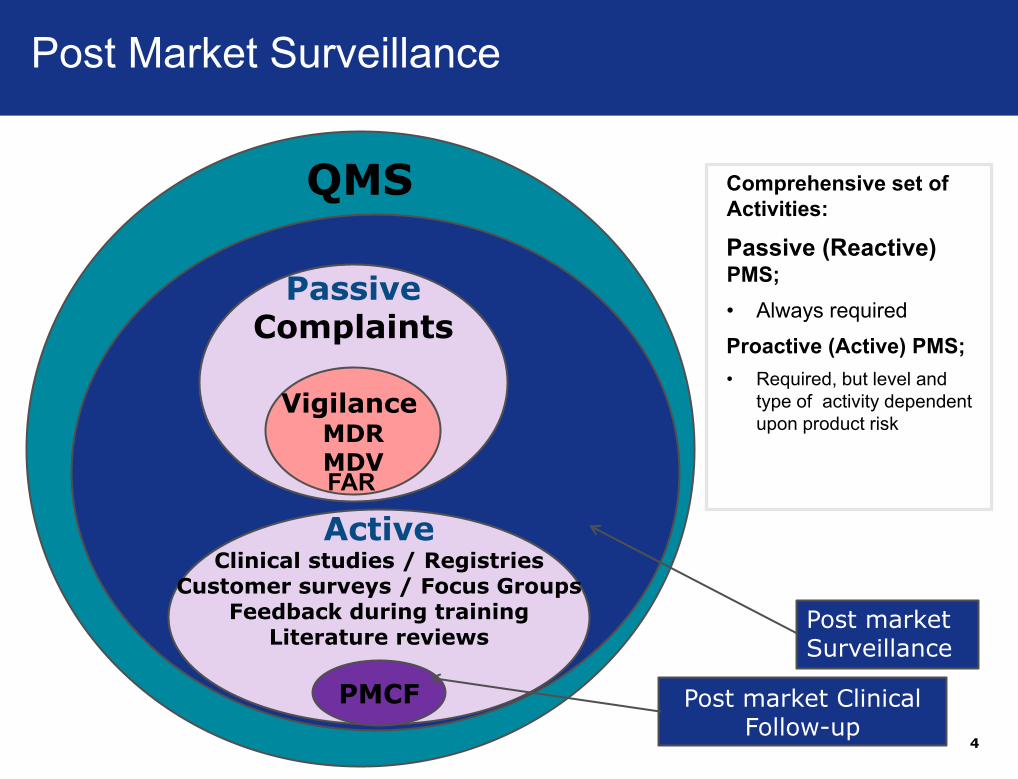
Post market Clinical Follow-up
4
Comprehensive set of
Activities:
Passive (Reactive) PMS;
• Always required
Proactive (Active) PMS;
• Required, but level and
type of activity dependent
upon product risk
PassiveComplaints
VigilanceMDRMDV
ActiveClinical studies / Registries
Customer surveys / Focus GroupsFeedback during training
Literature reviews
QMS
PMCF
Post market Surveillance
FAR
Post Market Surveillance
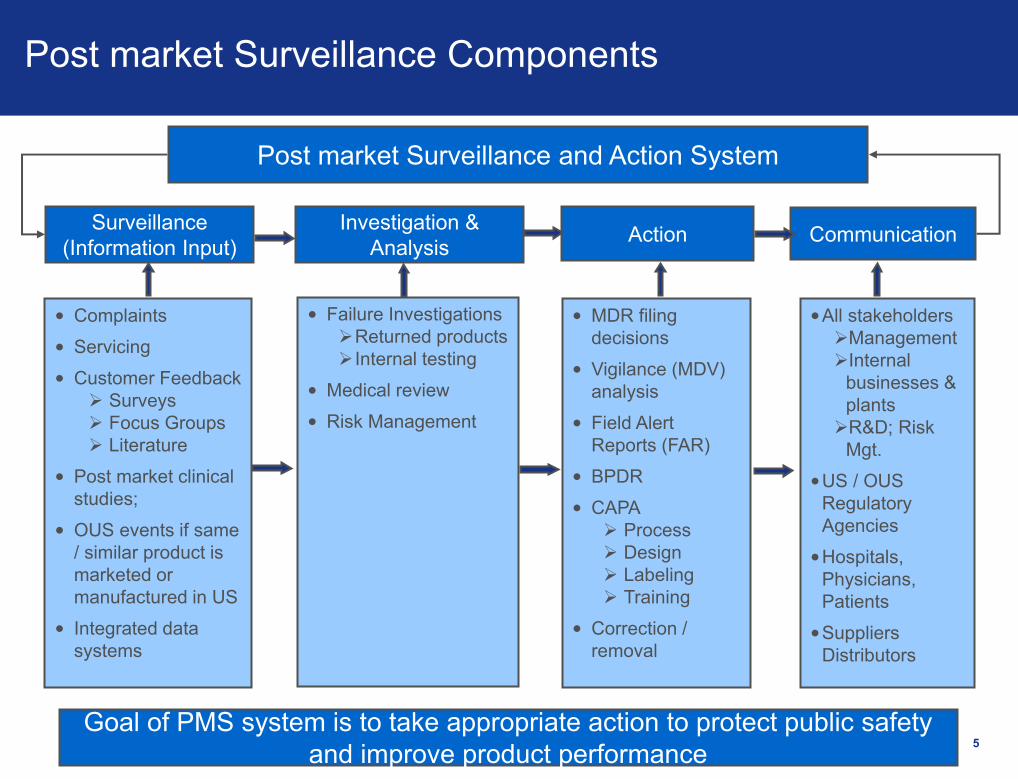
5
Post market Surveillance Components
Surveillance
(Information Input)
•All stakeholders
Management
Internal
businesses &
plants
R&D; Risk
Mgt.
•US / OUS
Regulatory
Agencies
•Hospitals,
Physicians,
Patients
•Suppliers
Distributors
Investigation &
AnalysisAction Communication
Post market Surveillance and Action System
• MDR filing
decisions
• Vigilance (MDV)
analysis
• Field Alert
Reports (FAR)
• BPDR
• CAPA
Process
Design
Labeling
Training
• Correction /
removal
• Failure Investigations
Returned products
Internal testing
• Medical review
• Risk Management
• Complaints
• Servicing
• Customer Feedback
Surveys
Focus Groups
Literature
• Post market clinical
studies;
• OUS events if same
/ similar product is
marketed or
manufactured in US
• Integrated data
systems
Goal of PMS system is to take appropriate action to protect public safety
and improve product performance

US FDA Regulatory Framework
6
21 Code of Federal Regulations Part 820. Current Good Manufacturing Practice
(cGMP) for design, manufacture, packaging, labeling, storage, installation, and
servicing of all finished devices intended for human use.• Part 806: Reports of Corrections and Removals
• Part 803: Medical Device Reporting
• Part 801: Labeling
• Part 314: Applications for FDA Approval to Market a New Drug
• Part 11: Electronic Records; Electronic Signatures
Reports on Accidental Radiation Occurrences or Device Defects (21 CFR 1003.10 and
1002.20)
Medical Device Tracking as ordered by FDA (21 CFR 821)
Post-Market Surveillance for Class II and Class III medical devices (21 CFR 822 – FDCA
§522 – “Section 522 Studies”)
Post-Approval Studies – conditions of PMA, HDE, PDP approval, such as participation in a
registry (21 CFR 814.82)
Guidance:‾ Section 522 Guidance – April 2006
‾ Post-Approval Studies Guidance – June 2009
‾ Draft Recalls and Enhancements Guidance – February 2013
‾ Draft MDR Guidance – July 2013
‾ Balancing Premarket and Post-Market Data Collection for Devices subject to Premarket Approval –
April 2014

7
EU Regulatory Requirements vs. Guidance
• Notified bodies look for confirmation that the MEDDEV guidance is implemented.
• Assurance that the legally binding regulations are met.
• NCA have specifically instructed Notified Bodies to focus on adequacy of the vigilance process and expect full implementation of MEDDEV 2.12 rev 8 requirements for post market clinical follow-up, post market plan, and review, trending.
Legally Binding Non-Binding (Voluntary)
Regulations Standards (e.g. QMS - ISO 13485)
MDD - 93/42/EEC*
AIMD – 90/385/EEC*
IVDD – 98/79/EEC*
* (As transposed into Natl. Legislation)
MEDDEV
(Vigilance - MEDDEV 2.12-1 rev 7;
2.12-1 rev 8
PMS – MEDDEV 2.12-2
EC Reps – MEDDEV 2.5/10)

ISO Standards for Post-Market Surveillance
ISO 13485:2012 - Medical devices -- Quality management systems --
Requirements for regulatory purposes
‾ Procedures to collect information from various sources such as users, service personnel,
training personnel, incident reports and customer feedback.
‾ ISO 13485:2003 – Section 8.2 “gain experience from the post-production phase, the
review of this experience shall form part of the feedback system”.
ISO 14971:2007
– Establish, document and maintain a PMS system to collect information in production and
post-production
– “… appropriate methods are in place to obtain relevant production and post-production
information. The results for this review shall be recorded as the risk management report
and included in the risk management file.”
ISO 14971:2009, Annex F.7
– “The risk management plan should include documentation of decisions, based on risk
analysis, about what sort of post-market surveillance is appropriate for the device, for
example, whether reactive surveillance is adequate or whether proactive studies
are needed. ”
8

ISO Standards for Post-Market Surveillance
ISO 14971:2012 - Medical devices — Application of risk management to
medical devicesMethods of obtaining relevant post-production information
Established quality management system procedures (for example ISO
13485:2003)
ISO 14971:2009, Annex F.7 “The risk management plan should include
documentation of decisions, based on risk analysis, about what sort of post-market
surveillance is appropriate for the device, for example, whether reactive
surveillance is adequate or whether proactive studies are needed. ”
‾ Post market surveillance data required:
‾ Correction on current products
‾ Inputs to future designs
Risk Management Plan should also define what type of post-market surveillance is
appropriate for the device
‾ Reactive surveillance
‾ Proactive studies
9

MDD 93/42 EEC; MEDDEV 2.12-2; Annexes II,IV,V,VI,VII; Annex X 1.1c Manufacturer must institute and keep up to date a systematic procedure
to review experience gained in post-production phase; Implement
appropriate means to apply any necessary corrective action.
– Identification and investigation of residual risks associated with the use of medical
devices placed on the market.
– These residual risks should be investigated and assessed in the post-market phase
through systematic Post-Market Clinical Follow-up (PMCF) studies.
– PMCF studies are performed on a device within its intended use/purpose(s)
according to the instructions for use.”
– Where PMCF, as part of the post-market surveillance plan for the device, is not
deemed necessary, this must be fully justified and documented
Manufacturer must have procedures to gather clinical data post-market on
all devices, analyze the data, and take action as needed.
– Clinical evaluation and documentation must be actively updated with data obtained
from post-market surveillance.
10
MEDDEV requirements (guidance)

11
MEDDEV requirements (guidance)
• Ensure the Product development process includes establishing Post market Surveillance Plan and a Post Market Surveillance Review
• Post Market Surveillance Plans: Evaluate type of post market surveillance needed – Passive
(complaints) and/or Active.• Passive PMS (complaints, MDR, Vigilance always required)• Active PMS – Required, but type of activity dependent upon product risk
Describe the product specific methods by which information is collected, processed, and evaluated for possible relevance to safety and efficacy.
May be implemented as part of the Risk Management Plan; Responsibilities typically fall with the Risk Management Documentation owners, depts. that submit for licensure to NB.
• Post Market Surveillance Review: Periodic product reviews and periodic Risk-benefit reanalysis

Key Stakeholders
US Food Drug Administration
Competent Authority (CA) - responsible for monitoring vigilance data and taking
appropriate action
EU Commission Home page lists Vigilance Contact Points within CAs
http://ec.europa.eu/health/medicaldevices/links/vigilance_contact_points_en.htm
Notified Body (NB) - responsible for reviewing QMS / Technical documentation to
ensure compliance with legislation (re: vigilance)
Vigilance shall be forwarded in standardized format (form in MEDDEV 2.12/1/Rev. 6)
Vigilance data shall be stored in European Database (EUDAMED)
Manufacturer - responsible for notifying Competent Authorities (CA) of incidents
immediately upon learning (per MEDDEV 2.12-1: without delay – 2 / 10 / 30d) – May be
done through EC Rep
12

Key Points so far…
13
Post market surveillance is the active and passive monitoring of marketed
product performance, safety and effectiveness.
It supports the product lifecycle and executed correctly enables
continuous improvement and manufacture of safe and effective products.
It is required by good manufacturing practices and global drug and
medical device regulations and standards.
External stakeholders include US FDA, outside US country Ministries of
Health, Competent Authorities, and Notified bodies.

Key Points so far…
14
Post market surveillance consists of four sub-systems:
1) Intake from surveillance sources;
2) Investigation and analysis of safety signals;
3) global regulatory reporting and escalation as required to CAPA and
field action processes, and
4) Dissemination of product performance information to internal and
external stakeholders.
Post market surveillance has tight linkage with other quality system
elements including design control, CAPA, risk management, and field
action.
Global regulations require continued monitoring of product post-production
risk-benefit and updating of risk management files with post market
information.

15
Surveillance Information Sources
• Consider how is the company structured – Organizational Alignment
Multiple manufacturing sites, businesses, divisions
Complaint processing site vs. investigation site
‾ Complaint Record Accessibility
‾ Complaints Not maintained at Manufacturing Site
‾ Call centers
• Who are the designated complaint handling units
• Reporting – Central team; Regulatory Affairs; Local Units
• Electronic systems and flow of information. Consider:
‾ Service Systems;
‾ Complaint Handling Systems; Electronic vs. paper
‾ Translations and Time zones
‾ Record availability

16
Surveillance Information Training
• Customers, Sales Force, Field Service, Affiliates, Distributors; 3rd parties
Are roles, responsibilities, accountabilities for reporting complaints and adverse
events in a timely fashion, undertaking the necessary follow-up; when req’d;
parts returns etc. clearly understood and documented by all parties?
Training
− Do all employees know where and how to report Complaints and Adverse
events?
− Complaint and Adverse event (pharmacovigilance) staff trained on the
products, use, safety signals, and the regulation.
− Document complaints and adverse events so that they are easy to follow
and understand – internal and external stakeholders
− Training records for company employees service; sales force; documenting
they have been appropriately trained in complaint handling and adverse
events.
Quality agreements between Manufacturing sites. Pharmacovigilance Unit,
Complaint handling unit.
Quality agreements with Affiliates and Distributors.

17
Surveillance Information Sources
• Field Service reports must be reviewed for complaint information
and MDR reportability 21 C.F.R. § 820.200(c)
Train field service staff to recognize and report complaints
Distributors and 3’rd parties providing service
• Establish process for capturing and reviewing service records
Quality of information received and required follow-up
A request for routine service is not always a complaint vs unplanned service
(corrective repair)
‾ Service reports for routine service requests (e.g. general maintenance)
typically are not complaints and do not require same level of investigation
Out of Box vs. post-installation failures
Open vs. closed service records; review of incremental information added to
service records
Trending of service records; component replacement – feeders into the
complaint system.

Must be both Patient- and Product-Centric
Patient–Related Questions What was the patient’s condition prior, during, and after the use of the device or
drug?
Did or would the patient require medical or surgical intervention related to an issue associated with the use of the device?
What medication did the patient require prior to and subsequent to the adverse event?
Did the patient require return visits to a physician or health care provider to monitor healing after the adverse event?
Product-Related Questions How and why was Company made aware of this event?
What other experience has Company learned about the use of this device in the same or similar circumstances?
What have past Company investigations revealed about the use of this device?
What is the severity and frequency of reported complaints associated with this device?
Has there been any change to the manufacturing of, or materials used in the manufacturing of, the device, even ones meant to improve quality?
18
Investigation & Analysis

19
Investigation & Analysis
Sample & Device returns
Consistent policy on when to request sample return
Make it easy for customers and field to return device
Shipping hazardous materials
May have to decontaminate locally before device shipped back
Non-destructive processes; should be validated;
Use technology – e.g. digital images sent to investigation site etc.,

20
Action System & Outputs
Regulatory Reporting
Manufacturing Device Reports (5 - 30 day time frame)
Vigilance Reports (10 - 30 day time frame)
Field Alert Reports (3-day time frame)
Escalation: Corrective & Preventive Action (CAPA)
Process
Design
Labeling
Training
Non-conformances
Risk Management
Correction / removal
Field actions
Recalls
Safety notices

21
Medical Device Reports (MDRs)
• Applies to Devices; Within 30 - work days of becoming aware of an event; OR
Within 5 – work days of becoming an aware of an event that meets the 5-day
requirement.
• 21 CFR. §803.50(a) Subpart E Manufacturer Reporting Requirements
“ . . . You must report to us no later than 30 calendar days after the day you receive or
otherwise become aware of information, from any source, that reasonably suggests that a
device that you market: May have caused or contributed to a death or serious injury; or
Has malfunctioned and this device or a similar device that you market would be likely to cause
or contribute to a death or serious injury, if the malfunction were to recur.”
5-day reports for an event designated by FDA or an event that requires remedial action to
prevent an unreasonable risk of substantial harm to the public health.
• 21 CFR. § 820.200(c): Servicing
“….Each manufacturer who receives a service report that represents an event which must be
reported to FDA under part 803 of this chapter shall automatically consider the report a
complaint and shall process in accordance with the requirements of § 820.198.
Service reports need to be evaluated for MDR reporting and those that meet the MDR criteria
should be processed as complaints.
Action System & Outputs - MDRs

22
MDR Reporting Challenges
Conducting robust and timely investigations;
Timely reporting
MDR Reportability
‾ Adverse Events Occurring Outside the U.S.
‾ Events from clinical trials
‾ Events that are the result of user error; off-label use; abnormal use
‾ Events that are within labeled frequency
‾ Discontinued product
Clear, consistent documentation;

23
Vigilance Reporting Challenges
Challenges:
• Differing global regulatory requirements
• Timeframes for reporting differ by country and / or region
• Implementing MDD / EU / MEDDEV Requirements
• Field actions - Reliance on Affiliates, Distributors, 3’rd parties
• Maintaining accurate install base and product listing
Solutions:
• Be aware of current and emerging regulations
• Use country-specific Decision trees and standard forms documenting reporting
decision
• Establish process for updating decision matrices and electronic systems
• Clarify authority to make reportabilty decisions at local / country level and ensure
local / country personnel are adequately trained
• Local language and provisions for translation
• Ensure consistent record content

Adverse Events
An adverse event can be any unfavorable and unintended sign (for example, an
abnormal laboratory finding), symptom, or disease temporally associated with the use
of a medicinal product, whether or not considered related to this medicinal product.*
*ICH: International Conference of Harmonization
Note: This concerns medicinal / biological products but does not include devices.
(MDRs and product complaints relate to devices).
• Event has to be evaluated to determine if it is serious or non-serious
• Pharmacovigilance reporting also includes:
‾ Lack of efficacy
‾ Transmission of infectious agents
‾ Medication interactions
‾ Medication error
‾ Abuse
‾ Overdose
‾ Off label use
‾ Administration during pregnancy or lactation
24

25
Applies to drug products; 21 CFR Part 314.81 Applications for FDA Approval to
Market a New Drug; Other Postmarketing Reports
NDA--Field alert report (FAR). The applicant shall submit information of the following
kinds about distributed drug products and articles to the FDA district office that is
responsible for the facility involved within 3 working days of receipt by the
applicant…..
(i) Information concerning any incident that causes the drug product or its
labeling to be mistaken for, or applied to, another article.
(ii) Information concerning any bacteriological contamination, or any significant
chemical, physical, or other change or deterioration in the distributed drug
product, or any failure of one or more distributed batches of the drug product to
meet the specification established for it in the application.
Note: Adverse events (AE) also apply to drug products; (Pharmacovigilance)
Field Alert Reports (FAR)

26
Applies to distributed biologic products; 21 CFR Part 600.14 (b) Reporting of Biological Product
Deviations by Licensed Manufacturers; 21 CFR Part 606.171 (b) Reporting of Product
Deviations by Licensed Manufacturers, Unlicensed Registered Blood Establishments, and
Transfusion Services
You should report a biological product deviation as soon as possible but you must report at a
date not to exceed 45-calendar days from the date you, your agent, or another person who
performs a manufacturing, holding, or distribution step under your control, acquire information
reasonably suggesting that a reportable event has occurred.
Biological Product Deviation Reports (BPDRs): ….”You must report any event, and information
relevant to the event, associated with the manufacturing, to include testing, processing,
packing, labeling, or storage, or with the holding or distribution, of a licensed biological product,
if that event meets all the following criteria:
(1) Either:
(i) Represents a deviation from current good manufacturing practice, applicable regulations,
applicable standards, or established specifications that may affect the safety, purity, or
potency of that product; or
(ii) Represents an unexpected or unforeseeable event that may affect the safety, purity, or
potency of that product; and
(2) Occurs in your facility or another facility under contract with you; and
(3) Involves a distributed biological product.
Biological Product Deviation Reports (BPDRs)

Key Points so far…
27
Surveillance inputs include active and passive sources.
Investigations must be both product and patient-centric with adequate documented
justification and rationales (including medical rationales where needed) to support
actions taken or not.
Phamarcovigilance is required for monitoring of adverse events and drug safety
signals. It is required by good manufacturing practices and global drug and
medical device regulations and standards.
The PV information augments information collected during drug development and
provides a more comprehensive real-world use of the drug or device.
Pharmacovigilance utilizes a signal management process to provide
recommended actions, as applicable, including but not limited to label updates, PV
Risk Management Plan updates and/or escalation for FCA consideration.
FARs (drug) MDRs, MDVs (devices) and BPDRs (biologic products) are other
required regulatory reports. Regulators use this information to monitor and trend
product safety and performance and protect public health.

Ship Hold
28
One Approach to Field Action Decision Making
Complaints &
Complaint Trending
PM Studies &
Registries
Product Testing
Inspections
Suppliers
MedicalHealth Hazard
EvaluationRegulatory
Recommends Field Action / Recall
QualityEscalates Issue
Event descriptionInvestigationRoot Cause
Field Action Committee
Senior Mgmt. Decision Makers
No Field Action
CAPA
Field Action Execution
CAPA
Escalation
Product Inquiry Report

Timely & Accurate information Exchange Internal & External
stakeholders:
US & Outside US Regulatory Agencies
Hospitals, Physicians, Patients
Site, Business Unit, Regional and Exec Management Reviews
Businesses; Manufacturing plants; R&D; Risk Mgt.
Local, Regional & Corporate quality boards
Suppliers, Distributors, other 3rd parties
Typical information disseminated/discussed:
Serious AE; Reportable events;
Product Trends
System performance efficiency and effectiveness metrics
Relevant CAPAs; Projects/initiatives
Investigations Status & Aging
Industry trends and new/changing regulations
User feedback
Procedural changes/improvements
29
Dissemination & Management Reviews

Systematic process for reviewing post market surveillance system and provide timely feedback
• Product Performance Metrics
Product AE, complaints, MDRs Trending
System reliability trending
Top 10 As Reported and As Analyzed Codes
Top Complaint Products
Unfavorable Trends
• System Efficiency Metrics
Late Regulatory Reports & Late Entered Complaints
Feeder to CAPA / NCE process
Complaint and Investigation Cycle Times vs. Targets
Complaint and Investigation Aging
Total Complaints & Investigations Entered and Closed
• System Effectiveness Metrics
Independent Review of Complaint Files and Documentation
Review MDR / MDV reportable and non-reportable decisions
Use queries and filters to identify files with high risk of incorrect decision e.g. patient harm codes with non-reportable decisions; MDV with no MDR;
Monitor results to identify if need for systemic fixes or additional training required30
Post market surveillance Product & System Monitoring

Key Points so far…
31
Action Systems and Outputs include global regulatory reports, escalation
to the CAPA and filed action systems, and dissemination of product safety
and performance signal to internal and external stakeholders.
The Corrective and Preventive (CAPA) is an important quality system
element designed to address root causes of product safety and
performance concerns and prevent their recurrence.
A systematic process for reviewing post market surveillance outputs
should include review of product performance and both system efficiency
and effectiveness metrics and feed into management reviews.

Best Practices - Effective Post market Surveillance
1. Establish procedures and systems to assure complaint and adverse
event information from all sources are entered into the complaint and
adverse event handling systems within defined timeframes
Establish procedures and train all company employees and agents to recognize and report complaints and adverse events within a defined timeframe:
Customers to call centers; Customers to Sales Reps; Distributors; 3rd
parties; Service
Customer feedback – Literature; focus groups, training sessions, social media; Post market clinical studies; registries (condition of approval and “marketing” studies.
Clearly outline linkages between business units, sales and marketing, design assurance, R&D, clinical, risk management, manufacturing, with clearly defined roles and responsibilities
32

Best Practices - Effective Post market Surveillance
2. Develop Tools to facilitate collection and documentation of complaint
information.
• Develop Tools for collection and documentation of comprehensive
complaint adverse information e.g. product specific complaint
questionnaires; electronic links on company homepage.
• Take proactive steps to maximize likelihood of sample and device return.
Communicate to customers importance of device return. Provide clear
instructions and processes to facilitate return.
• Balance Patient-centric vs. Product-centric information collection:
− Use Product / therapy-specific Complaint Notification Forms
What happened? When did it happen? Was the patient / user injured?
Severity of Injury?
If injury or suspected injury, follow-up to determine patient outcome
Device / Lot / Batch #? Is the device available for return?
Is the device past shelf-life? Reprocessed?
33

Best Practices - Effective Post market Surveillance
3. Ensure reporting procedures and processes fully comply with
regulatory requirements in all regions where products are marketed
Evaluate all complaints and adverse events.
Regulatory – AE, FAR, MDR, MDV, BPDR reportability
Need for further investigation and root cause determination
Balance information needed with timely decision-making; If in doubt
report on-time and supplement when additional information becomes
available.
Use decision trees and examples to facilitate consistent decision-making;
What objective evidence supports the “non-reporting” decision?
Medical review and escalation process for safety issues and serious events &
Assessment of various clinical scenarios: e.g.
‾ Treatment/Therapy not achieved
‾ Significance of delay in treatment
‾ Medical Intervention
34

Best Practices - Effective Post market Surveillance
4. Establish and Maintain Strong Linkage with Risk Management
Leverage Risk Management information to aid in assessment and making reporting decisions.
Establish formal requirement for a post market surveillance plans and review (Post market surveillance policy).
‾ Clearly outline passive (reactive) and active post market surveillance; sources; mechanisms for flow-down into the post market surveillance system.
35

36
Questions & Case Study
Discussion

Case Study 1 – Uneeda Device Inc.
• Uneeda Device, Inc., a U.S. manufacturer of cardiovascular medical
devices is a Division of their parent holding company Uneedmore
Devices, located in Vietnam.
• The global sales offices of Uneedmore warehouse, sell, distribute and
service Uneeda products internationally, within their own country, as
well as have an established network of sales representatives to
provide products and services throughout the world.
• Uneeda Device, Inc. has provided training to the global sales staff
about the importance and timeliness of notification of reported
complaints, urgency of any investigation and subsequent MDR
reporting requirements in the United States.
37

Case Study 1 – Uneeda Device Inc.
• Uneedmore’s global sales offices have frequent employee turn-over and
has had some difficulty with language barriers in communicating with the
U.S. manufacturer.
• During a routine quarterly visit to a remote client a global sales
representative learned of a patient injury associated with the U.S.
manufactured product. They also learned that one of their sales
representatives had been present and observed the surgical procedure at
which this injury occurred. The nature of the injury would not require any
vigilance filing in the foreign country where it occurred.
• Uneedmore’s sales representative returned to his office after his two week
client road trip and left immediately for a 1 week vacation.
• Upon his return, he sent the injury information to Uneeda in the U.S. for
their evaluation and review
38

Case Study 1 Questions
1. Should Uneedmore Devices employees report such events to
Uneeda?
Yes; Uneeda needs to be clear who is the designated complaint unit -All employees, affiliates, distributors need to be trained on identifying a complaint, required information, where to send the information and timeframes for reporting the information.
2. Uneeda Device first learned of the injury upon receipt of the sales rep.
report. Is that the “Become Aware Date”?
No! The firm’s become aware date is the date when the firm firstbecame aware of the incident; this is the date the sales representatives had been present and observed the surgical procedure at which this injury occurred.
39

Case Study 1 Questions
3. At what point was the Become Aware Date reached?
The date the sales representatives had been present and observed the surgical procedure at which this injury occurred.
4. Will any subsequent MDR filing, if needed, be late?
There is a potential for a late MDR. The firm has 30 days after the day the firm first became aware to report the MDR. Given the sales rep took 3 weeks to report the complaint, the firm has only a short time left to complete the investigation and file the MDR if needed.
40

Case Study 1 Questions
5. What steps can be taken to reduce this problem from recurring?
The firm needs to ensure all company personnel – Sales reps, Service, all employees know how to identify a complaint, what information is minimally required, where to report the complaint.
Roles, responsibilities need to be clearly documented between the parent company and its Divisions.
The Firm needs to clarify and document:
Who holds the registration for the product?
Who manufactures the product?
Investigation site?
Designated Complaint Handling Unit?
Who is responsible for reporting MDRs and Vigilance reports?
41

Case Study 2 “Uneeda Inc.”
• “Uneeda Inc.” received a report that a patient who was being monitored at
the Nursing station via its “Far signal” telemetry device, incorrectly showed
Vfib, leading to the Nurse applying a defibrillator to the patient even though
the defibrillator displayed a non-shockable rhythm. This resulted in a
patient injury. The device is approved and cleared in the US and EU.
• The “Uneeda Inc.” telemetry device User manual warns that the device
could pick up ambient noise which can mimic Vfib, and requires users to
re-assess the patient before treatment.
• The firm concluded this was not a MDR reportable event due to the fact
that the “The telemetry device did not malfunction”. The firm documented
the event as abnormal use as the User knowingly, and beyond the control
of the manufacturer, applied defibrillation despite a visible normal rhythm
being obtained from the defibrillator.
42

Case Study 2 Questions
1. Did the firm make a correct decision not to report this event as a MDR?
− No! ISO differentiates between Use error and Abnormal use, however, FDA
does not and Use errors may be reportable as MDRs. In this case, the Use
error led to a patient injury and is reportable as a MDR
43

FDA Guidance – July 2013
2.5 What is meant by “caused or contributed” to a death or serious injury? This means that a death or serious injury was or may have been attributed to a medical device or that a medical device was or may have been a factor in a death or serious injury, including events occurring as a result of [21 CFR 803.3]:Contains Nonbinding Recommendations Draft – Not for Implementation 7
1. Failure; 2. Malfunction; 3. Improper or inadequate design; 4. Manufacture; 5. Labeling; or 6. User error.
2.6 What is device “user error” and why do you want to know about events involving user error? We consider a device “user error” to mean a device-related error or mistake made by the person using the device. The error could be the sole cause of an MDR reportable event, or merely a contributing factor. Such errors often reflect problems with device labeling, the user interface, or other aspects of device design. Thus, FDA believes these events should be reported in the same way other adverse events a device causes or contributes to should be reported. This is especially important for devices used in non-health care facility settings.

45
•ISO 14971 2007 2.27 Use error: A use error is an act or omission that results
in a medical device response that is either not expected by the user or unintended by
the manufacturer. Use errors include slip-ups, lapses, and mistakes.
•MEDDEV 2.12-1 4.20 Use Error: Act or omission of an act, that has a different
result to that intended by the Manufacturer or expected by the Operator of the Medical
Device.
4.1 Abnormal Use: Act or omission of an act by the Operator
or User of a Medical Device as a result of conduct which is beyond any means of risk
control by the Manufacturer. Reference: EN IEC 60601-1-6
•MEDDEV 2.12-1rev 4 Misuse: Intentional use of a device contrary to its intended
purpose according to the labeling, instructions for use and or in the promotional
material related to the device.
Definitions

Case Study 2 Questions
2. What is the most important thing the firm is required to do to in this
situation to be fully compliant with the regulations?
− Ensure that the decision not to report this serious injury was made by a
person qualified to make a medical judgment and the decision and rationale
documented.
− Additionally, the firm could improve the process for handling this customer
complaint by incorporating reviews of non-reporting decisions, including
routine medical review of all SI and death events and ensuing investigations
must be both patient-centric and device centric and must be timely.
46

Case Study 3 “MedRight Inc.”
• “MedRight Inc.” is preparing for a Notified Body inspection. The firm
reviewed a major nonconformance received by one of its competitors from
the same Notified Body. The firm’s product risk profile and markets are
similar to that of its competitor.
• The nonconformance issued by the Notified Body reads as follows:
“Post Market surveillance does not meet the requirements of the amended MDD… the current procedure does not define relative inputs of the post market procedure including inputs to design process, risk management and clinical reviews.”
“Documented evidence that post market surveillance plans are based on the outputs of risk management… could not be provided.”
“Documented evidence that vigilance reports are filed on otherwise not reportable complaints, when trends exceed a pre-set level could not be provided.” (section 4.18 of MEDDEV 2.12-1 rev 6)
Questions:
1. What actions should MedRight Inc, take to avoid a similar non-
conformance?
47

Case Study 3 “MedRight Inc.”
Firm should prepare and be ready to provide documentation (and
examples) showing:
• System/process for conducting Post Market Surveillance Reviews (PMSR)
(Procedures, templates; examples showing use..)
• Information on Clinical Evaluations and Post Market Surveillance Planning as
part of Risk Management.
• Clearly defined linkages between business units, sales and marketing, design
assurance, R&D, clinical, risk management, with clearly defined roles and
responsibilities (Post market surveillance Policy)
• Process established and procedure updated to meet the MEDDEV guidance
and include:
− How to identify trends
− How to report trends
− How to document reported trends
− Clearly defined roles and responsibilities
48

Case Study 4 “MedRight Inc.”
• “MedRight Inc.” is conducting clinical trials under an IDE for its new intra-
arterial blood pressure monitoring catheter. The catheter uses an ancillary
device, the firm’s “Tail wind” pressure gauge which is marketed world-wide.
• The blinded study has been in progress for six months, and as a result of
accidental un-blinding, the study coordinator noticed reports that the “Tail
wind” device was showing erroneous results and having difficulty
calibrating.
• At a meeting with the “MedRight Inc.” Design team, the Clinical Trials VP,
and the R&D VP, it was decided these events did not compromise the
study, the accidental un-blinding was documented, and the study
continued.
49

Case Study 4 Questions
1. What gaps exist in the “MedRight Inc.” process and why is the firm at
compliance risk?
− The firm did not evaluate reports on the marketed device for MDR reportability.
2. What should have “MedRight Inc.” done with the information from its
clinical trial?
− The events on the marketed device should be fed into the complaint process for
investigation and evaluation for MDR reportability.
3. How could the firm improve the process for handling clinical trials and
ensure adequate linkages with post-market surveillance?
− The firm should consider instituting controls that before starting a clinical trial, an
assessment is done to determine if marketed devices are to be used, and if
complaint management plans are required.
− Additionally, process, procedures established and training of Design, R&D,
medical etc. that all clinical studies require review for use of marketed devices.
50

Case Study 5 “Healthy for Life, Inc”
• A Sales Representative for “Healthy for Life Inc.,” which located in Brazil,
and is a major distributor of Uneeda Inc.’s medical products in Latin
America, received a customer complaint on the “Uneeda Inc.” patient
monitor. “Uneeda Inc.” determined the event to be reportable as a MDR.
“Uneeda Inc.” is the legal US manufacturer and owns the design.
Questions:
1. Who should have been responsible (“Healthy for Life Inc.” or “Uneeda
Inc.) for making the MDR report?
− Uneeda Inc.
2. What else must “Uneeda Inc.” do to ensure it fully meets its regulatory
responsibilities?
− Quality agreement between the two firms outlining responsibilities for
investigating complaints and reporting complaint information.
− Documentation that the distributor was trained to identify and report complaints
in a timely fashion.
51