gsk - us postmarketing commitments€¦ · us postmarketing commitments ... designed to address...
TRANSCRIPT

US Postmarketing Commitments – January 28, 2013
Page 1 of 20
TRADE NAME (generic name)
DUE DATE (m/d/yyyy) STATUS
EXPLANATION OF STATUS
DESCRIPTION
ADVAIR DISKUS (fluticasone propionate and salmeterol xinafoate inhalation powder)
June 28, 2017
Ongoing PMR 1750-1 A randomized, double-blind, 26-week, active-controlled clinical trial comparing Advair Diskus (fluticasone propionate and salmeterol xinafoate inhalation powder) and fluticasone propionate inhalation powder to evaluate the risk of serious asthma outcomes (hospitalizations, intubation, death) in 11,700 adult and adolescent patients 12 years of age and older with persistent asthma.
ADVAIR DISKUS (fluticasone propionate and salmeterol xinafoate inhalation powder)
June 28, 2017
Ongoing PMR 1750-2 A randomized, double-blind, 26-week, active-controlled clinical trial comparing Advair Diskus (fluticasone propionate and salmeterol xinafoate inhalation powder) and Flovent Diskus (fluticasone propionate inhalation powder) to evaluate the risk of serious asthma outcomes (hospitalizations, intubation, death) in 6200 pediatric patients 4 to 11 years of age with persistent asthma.
ADVAIR HFA (fluticasone propionate and salmeterol xinafoate) Inhalation Aerosol
December 31, 2007
Released PMC Deferred pediatric study under PREA for the treatment of asthma in pediatric patients ages greater than 4 to less than 12.
alli (orlistat) 4/30/2011 Submitted PMC Conduct a label comprehension study of the Alli Drug Facts label to evaluate how consumers understand and interpret the new warning statement related to potential liver injury.
Altabax Ointment (retapamulin)
12/31/2008 Submitted
PMR Deferred pediatric study under PREA for the treatment of impetigo in pediatric patients ages 2 months to 9 months.
Arixtra (fondaparinux) 5/31/2009 Delayed Study report submitted in Supplement S-027; did not fulfill the PMR.
PMR Deferred pediatric study under PREA for the treatment of acute deep vein thrombosis when administered in conjunction with warfarin sodium in pediatric patients ages birth to 16 years.
Arixtra (fondaparinux) 5/31/2009 Delayed Study report submitted in Supplement S-027; did not fulfill the PMR.
PMR Deferred pediatric study under PREA for the treatment of acute pulmonary embolism when administered in conjunction with warfarin sodium in pediatric patients ages birth to 16 years.

US Postmarketing Commitments – January 28, 2013
Page 2 of 20
TRADE NAME (generic name)
DUE DATE (m/d/yyyy) STATUS
EXPLANATION OF STATUS
DESCRIPTION
Arranon (nelarabine) Injection
12/31/2016 Ongoing Subpart H-1; Accelerated Approval Submit the results of the proposed phase III trial (AALL0434) to be conducted by the Children's Oncology Group to demonstrate nelarabine's clinical benefit.
ARZERRA (ofatumumab)
06/30/2014 Ongoing PMC-1 under 21 CFR 601-70 To submit a final report for ongoing clinical trial OMB110911, entitled, "A Phase III Open-label, Randomized, Multicenter Trial of Ofatumumab Added to Chlorambucil versus Chlorambucil Monotherapy in Previously Untreated Patients with Chronic Lymphocytic Leukemia" which is intended to verify the clinical benefit of ofatumumab through demonstration of a clinically meaningful effect on progression-free survival.
ARZERRA (ofatumumab)
03/31/2010 Submitted PMR under Section 505(o) To develop a validated, sensitive, and accurate assay for the detection of an immune response (binding antibodies) to ofatumumab, including procedures for accurate detection of antibodies to ofatumumab in the presence of ofatumumab levels that are expected to be present in the serum or plasma at the time of patient sampling.
ARZERRA (ofatumumab)
12/31/2013 Ongoing PMR-3 under Section 505(o) To conduct an assessment of anti-drug antibody (ADA) response to ofatumumab with a validated assay (required in PMC 2) capable of sensitively detecting ADA responses in the presence of ofatumumab levels that are expected to be present at the time of patient sampling. ADA response will be evaluated in at least 300 patients, including ofatumumab-treated patients enrolled in clinical trial OMB110911. The final report will include information on the level of ofatumumab in each patient’s test sample at each sampling time point.

US Postmarketing Commitments – January 28, 2013
Page 3 of 20
TRADE NAME (generic name)
DUE DATE (m/d/yyyy) STATUS
EXPLANATION OF STATUS
DESCRIPTION
ARZERRA (ofatumumab)
12/31/2012 Submitted PMR-4 under Section 505(o) To conduct clinical trial OMB112855, a trial of QTc intervals in patients who have been administered ofatumumab: QTc assessments will be performed in patients who have failed at least one fludarabine-containing regimen (at least two cycles) and failed at least one alemtuzumab-containing regimen (a minimum of at least 12 administrations) or who are considered inappropriate for treatment with alemtuzumab due to lymphadenopathy with at least one lymph node> 5 cm and requiring therapy and who receive the dose and schedule of ofatumumab per the approved prescribing information. The number of patients evaluated for QTc interval changes will be at least 12. For the QTc assessments, ECGs will be collected in triplicate at baseline, at steady-state ofatumumab concentrations, periodically on-therapy (e.g., every 3 months), and at the end of treatment. The final report will be a comprehensive combined report of the results (including primary data) of clinical trial OMB112855 and of the sub-trial assessing QTc intervals in OMB110911 (see below).
ARZERRA (ofatumumab)
12/31/2012 Submitted PMR-5 under Section 505(o) To conduct an assessment of QTc intervals as a sub-trial in clinical trial OMB110911. The total number of patients in OMB110911 with evaluable ECG measurements will be at least 50 (25 per treatment arm). For the QTc assessments, ECGs will be collected in triplicate at baseline, at steady-state ofatumumab concentrations, periodically on-therapy (e.g., every 3 months), and at the end of treatment. The final report will be a comprehensive combined report of the results (including primary data) of the sub-trial assessing QTc intervals in OMB110911 and of clinical trial OMB112855.
US Postmarketing Commitments – January 28, 2013
Page 4 of 20
TRADE NAME (generic name)
DUE DATE (m/d/yyyy) STATUS
EXPLANATION OF STATUS
DESCRIPTION
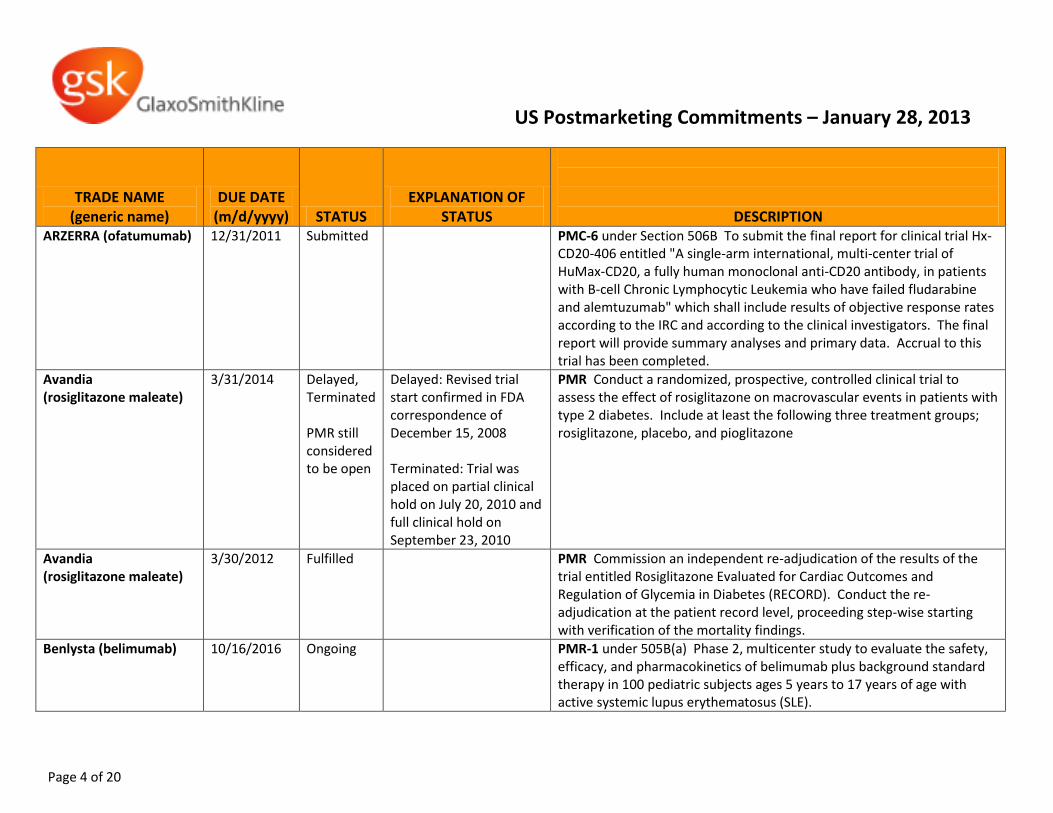
ARZERRA (ofatumumab)
12/31/2011 Submitted PMC-6 under Section 506B To submit the final report for clinical trial Hx-CD20-406 entitled "A single-arm international, multi-center trial of HuMax-CD20, a fully human monoclonal anti-CD20 antibody, in patients with B-cell Chronic Lymphocytic Leukemia who have failed fludarabine and alemtuzumab" which shall include results of objective response rates according to the IRC and according to the clinical investigators. The final report will provide summary analyses and primary data. Accrual to this trial has been completed.
Avandia (rosiglitazone maleate)
3/31/2014 Delayed, Terminated PMR still considered to be open
Delayed: Revised trial start confirmed in FDA correspondence of December 15, 2008 Terminated: Trial was placed on partial clinical hold on July 20, 2010 and full clinical hold on September 23, 2010
PMR Conduct a randomized, prospective, controlled clinical trial to assess the effect of rosiglitazone on macrovascular events in patients with type 2 diabetes. Include at least the following three treatment groups; rosiglitazone, placebo, and pioglitazone
Avandia (rosiglitazone maleate)
3/30/2012 Fulfilled PMR Commission an independent re-adjudication of the results of the trial entitled Rosiglitazone Evaluated for Cardiac Outcomes and Regulation of Glycemia in Diabetes (RECORD). Conduct the re-adjudication at the patient record level, proceeding step-wise starting with verification of the mortality findings.
Benlysta (belimumab)
10/16/2016 Ongoing PMR-1 under 505B(a) Phase 2, multicenter study to evaluate the safety, efficacy, and pharmacokinetics of belimumab plus background standard therapy in 100 pediatric subjects ages 5 years to 17 years of age with active systemic lupus erythematosus (SLE).

US Postmarketing Commitments – January 28, 2013
Page 5 of 20
TRADE NAME (generic name)
DUE DATE (m/d/yyyy) STATUS
EXPLANATION OF STATUS
DESCRIPTION
Benlysta (belimumab)
1/31/2013 Pending PMR-2 under 505(o) Develop improved immunogenicity assays that are less sensitive to product interference that are capable of detecting human anti-human antibodies (HAHA) in the presence of belimumab at ranges that would be expected to occur in patients receiving both high and low doses.
Benlysta (belimumab) 4/30/2019 Pending PMR-3 under 505(o) Conduct a pregnancy registry to evaluate pregnancy outcomes for women exposed to Benlysta (belimumab) during pregnancy.
Benlysta (belimumab) 9/30/2014 Released PMR-4 under 505(o) Conduct a randomized clinical trial to evaluate the effects of Benlysta (belimumab) treatment on host response to therapeutic vaccines. B cell-dependent antigens (e.g., pneumococcal polysaccharide vaccine) and T cell-dependent antigens (e.g., tetanus toxoid) will be evaluated.
Benlysta (belimumab) 5/31/2023 Released PMR-5 under 505(o) Conduct a randomized, placebo-controlled clinical trial with Benlysta (belimumab) in 5000 patients with active, autoantibody-positive systemic lupus erythematosus to evaluate Benlysta’s long term safety profile including adverse events of special interest (e.g., mortality, malignancy, serious and opportunistic infections and depression/suicidality).
Benlysta (belimumab) 10/31/2017 Pending PMC-6 under 506B Conduct a randomized, controlled clinical trial in patients with lupus nephritis to evaluate the efficacy and safety of Benlysta (belimumab).
Benlysta (belimumab) 1/31/2018 Pending PMC-7 under 506B Conduct a randomized, controlled clinical trial to evaluate the efficacy and safety of Benlysta (belimumab) in African-American patients with SLE.
Benlysta (belimumab) 12/31/2016 Ongoing PMC-8 under 506B Submit a final report for the long-term, open-label, continuation trial LBSL99.

US Postmarketing Commitments – January 28, 2013
Page 6 of 20
TRADE NAME (generic name)
DUE DATE (m/d/yyyy) STATUS
EXPLANATION OF STATUS
DESCRIPTION
Benlysta (belimumab) 12/31/2015 Ongoing PMC -9 under 506B Submit a final report for the long-term, open-label, continuation trial C1066.
Benlysta (belimumab) 10/31/2015 Ongoing PMC-10 under 506B Submit a final report for the long-term, open-label, continuation trial C1074.
Benlysta (belimumab) 12/31/2019 Ongoing PMR-14 under 505(o) Conduct a one-year randomized, placebo controlled clinical trial with Benlysta (belimumab) in 5000 patients with active, autoantibody-positive systemic lupus erythramatosus to evaluate Benlysta’s long term safety profile including adverse events of special interest (e.g., mortality, malignancy, serious and opportunistic infections and depression (suicidality)).
BEXXAR (tositumomab and iodine I 131 tositumomab)
05/09/2008 Released PMC-1 under 21 CFR 601.70 To conduct an open-label efficacy trial of Rituximab versus the Bexxar® therapeutic regimen in patients with lymphoma who have received at least one, and no more than two, prior chemotherapy regimens, and who are appropriate candidates for systemic therapy (Study CCBX001-049). The primary objective of this study is demonstration of a longer event-free survival in patients treated with the Bexxar® therapeutic regimen as compared to those receiving Rituximab.
BEXXAR (tositumomab and iodine I 131 tositumomab)
09/29/2008 Released PMC-3 under 21 CFR 601.70 To conduct a single-arm, open-label, multicenter, Phase 2 trial evaluating the pharmacokinetics, safety, and efficacy of retreatment with the Bexxar® therapeutic regimen in patients who have had duration of response of at least 6 months in the studies CCBX-001-049 and CCBX001-053. The primary objective of the study (Study CCBX001-054) is to compare the pharmacokinetics associated with retreatment and with initial treatment. In addition, the study will assess the efficacy of retreatment with the Bexxar® therapeutic regimen.
US Postmarketing Commitments – January 28, 2013
Page 7 of 20
TRADE NAME (generic name)
DUE DATE (m/d/yyyy) STATUS
EXPLANATION OF STATUS
DESCRIPTION
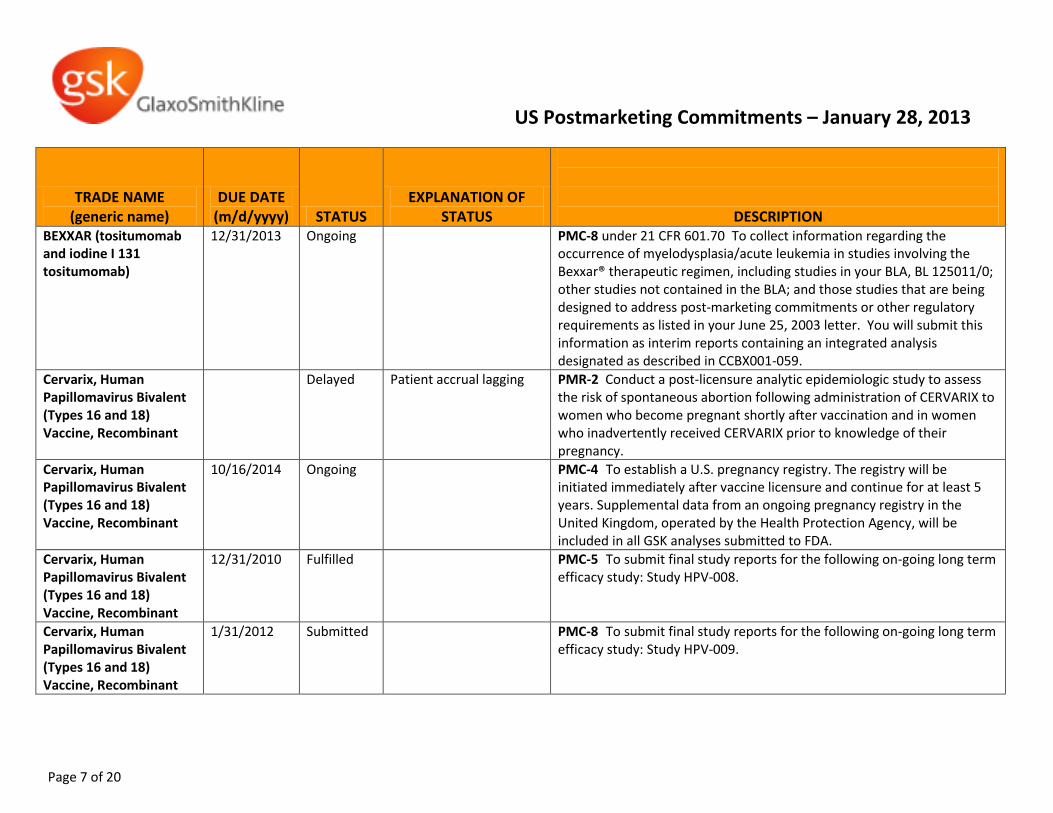
BEXXAR (tositumomab and iodine I 131 tositumomab)
12/31/2013 Ongoing PMC-8 under 21 CFR 601.70 To collect information regarding the occurrence of myelodysplasia/acute leukemia in studies involving the Bexxar® therapeutic regimen, including studies in your BLA, BL 125011/0; other studies not contained in the BLA; and those studies that are being designed to address post-marketing commitments or other regulatory requirements as listed in your June 25, 2003 letter. You will submit this information as interim reports containing an integrated analysis designated as described in CCBX001-059.
Cervarix, Human Papillomavirus Bivalent (Types 16 and 18) Vaccine, Recombinant
Delayed Patient accrual lagging PMR-2 Conduct a post-licensure analytic epidemiologic study to assess the risk of spontaneous abortion following administration of CERVARIX to women who become pregnant shortly after vaccination and in women who inadvertently received CERVARIX prior to knowledge of their pregnancy.
Cervarix, Human Papillomavirus Bivalent (Types 16 and 18) Vaccine, Recombinant
10/16/2014 Ongoing PMC-4 To establish a U.S. pregnancy registry. The registry will be initiated immediately after vaccine licensure and continue for at least 5 years. Supplemental data from an ongoing pregnancy registry in the United Kingdom, operated by the Health Protection Agency, will be included in all GSK analyses submitted to FDA.
Cervarix, Human Papillomavirus Bivalent (Types 16 and 18) Vaccine, Recombinant
12/31/2010 Fulfilled PMC-5 To submit final study reports for the following on-going long term efficacy study: Study HPV-008.
Cervarix, Human Papillomavirus Bivalent (Types 16 and 18) Vaccine, Recombinant
1/31/2012 Submitted PMC-8 To submit final study reports for the following on-going long term efficacy study: Study HPV-009.

US Postmarketing Commitments – January 28, 2013
Page 8 of 20
TRADE NAME (generic name)
DUE DATE (m/d/yyyy) STATUS
EXPLANATION OF STATUS
DESCRIPTION
Cervarix, Human Papillomavirus Bivalent (Types 16 and 18) Vaccine, Recombinant
12/31/2011 Delayed Low enrollment; study plan revised. Clinical study report will be submitted by 03 March 2015.
PMC-9 To submit final study reports for the following on-going long term efficacy study: Study HPV-015.
Cervarix, Human Papillomavirus Bivalent (Types 16 and 18) Vaccine, Recombinant
9/30/2011 Submitted PMC-10 To submit final study reports for the following on-going long term efficacy study: Study HPV-023.
Cervarix, Human Papillomavirus Bivalent (Types 16 and 18) Vaccine, Recombinant
12/31/2015 Ongoing PMC-12 To submit final study reports for the following on-going long term efficacy study: Study HPV-040.
Cervarix, Human Papillomavirus Bivalent (Types 16 and 18) Vaccine, Recombinant
3/31/2015 Released PMC Conduct an observational study in a U.S. managed care organization to evaluate the incidence of new onset autoimmune disease among at least 50,000 CERVARIX recipients 9 through 25 years of age.
Cervarix, Human Papillomavirus Bivalent (Types 16 and 18) Vaccine, Recombinant
10/31/2011 Fulfilled PMC Submit the final study report for study HPV-029.
Cervarix, Human Papillomavirus Bivalent (Types 16 and 18) Vaccine, Recombinant
10/31/2011 Fulfilled PMC Submit the final study report for study HPV-030.
FLUARIX, Influenza Virus Vaccine
6/30/2011 Fulfilled PMC Establish a pregnancy registry to prospectively collect data on spontaneously-reported exposures to Fluarix during pregnancy.

US Postmarketing Commitments – January 28, 2013
Page 9 of 20
TRADE NAME (generic name)
DUE DATE (m/d/yyyy) STATUS
EXPLANATION OF STATUS
DESCRIPTION
FLUARIX Quadrivalent March 2014 Ongoing PMR (PREA): Pediatric study (deferred study under PREA): Efficacy study to evaluate prevention of disease caused by influenza A subtype viruses and type B viruses contained in Fluarix Quadrivalent, in pediatric patients ages 6 months to 35 months of age.
FLUARIX Quadrivalent April 30, 2013 – protocol submission
Ongoing PMC: To establish a pregnancy registry to prospectively collect data on spontaneously-reported exposures to Fluarix® Quadrivalent during pregnancy. A protocol for this pregnancy registry will be submitted by April 30, 2013. The pregnancy registry will be established by August 30, 2013 and annual reports will be submitted with the periodic safety update reports (PSURs) for Fluarix® Quadrivalent. When the registry has collected data on the outcomes specified in the protocol for five years, GSK will submit a full study report 18 months from submission of the fifth annual PSUR. After submission of the registry report, GSK will continue enrolling in the registry pending CBER review of the report and determination that the registry can be discontinued.
FluLaval, Influenza Virus Vaccine
6/30/2011 Submitted PMC Conduct a study to evaluate the immunogenicity and safety of FluLaval compared to Fluzone (Sanofi-Pasteur, Inc.) administered to children 3 to 17 years of age in the US.
FluLaval, Influenza Virus Vaccine
3/31/2014 Ongoing PMC PREA: Conduct an ongoing efficacy study of the seasonal influenza vaccine Flu Q QIV, administered to children 3 to 8 years of age.
FluLaval, Influenza Virus Vaccine
3/31/2014 Ongoing PMR Accelerated Approval, 21 CFR 601.41 Complete ongoing Study FLU Q-QIV-006, a non-influenza vaccine comparator-controlled clinical endpoint study of FLU Q-QIV (quadrivalent seasonal influenza vaccine produced using the FluLaval process) in persons 3 to 8 years of age.
Hiberix, Haemophilus b Conjugate Vaccine (Tetanus Toxoid Conjugate)
12/31/2013 Ongoing PMC-1 To conduct Study Hib-097, a comparative safety and immunogenicity clinical trial of primary and booster immunization with Hiberix® relative to U.S. licensed control vaccines.

US Postmarketing Commitments – January 28, 2013
Page 10 of 20
TRADE NAME (generic name)
DUE DATE (m/d/yyyy) STATUS
EXPLANATION OF STATUS
DESCRIPTION
Hiberix, Haemophilus b Conjugate Vaccine (Tetanus Toxoid Conjugate)
12/31/2013 Ongoing PMC-2 Deferred pediatric study (Study Hib-097) under PREA for the prevention of invasive disease caused by H. influenzae type b in pediatric patients ages 6 weeks to 14 months.
Horizant (gabapentin enacarbil) Extended-Release Tablets*
06/2017 Pending PMR 1588-1 Conduct a PK/PD study in adolescents ages = 13 years to 17 years with moderate to severe symptoms of primary Restless Legs Syndrome. Final Report Submission.
Horizant (gabapentin enacarbil) Extended-Release Tablets*
10/2024 Pending PMR 1588-2 Conduct a double-blind, randomized, placebo-controlled, parallel group efficacy and safety evaluation trial in adolescents = 13 years to 17 years with moderate to severe symptoms of primary Restless Legs Syndrome. Final Report Submission.
Horizant (gabapentin enacarbil) Extended-Release Tablets*
07/2025 Pending PMR 1588-3 Conduct a long-term safety study of adolescents ages =13 years to 17 years with moderate to severe symptoms of primary Restless Legs Syndrome. The study must provide a descriptive analysis of safety data in pediatric patients during at least 12 months of continuous treatment with gabapentin enacarbil at individualized doses in association with the study described in PMR #1588-2. Final Report Submission.
Horizant (gabapentin enacarbil) Extended-Release Tablets*
06/2022 Pending PMR 1588-4 Conduct a driving study in adolescent patients of legal driving age who have Restless Legs Syndrome, using diphenhydramine as active control. Final Report Submission.
Horizant (gabapentin enacarbil) Extended-Release Tablets*
10/2011 Fulfilled PMR 1588-5 An in vitro study to evaluate the potential for gabapentin enacarbil and gabapentin to be inhibitors of CYP2C8 and CYP2B6. Final Report Submission.
Horizant (gabapentin enacarbil) Extended-Release Tablets*
06/2011 Fulfilled PMR 1588-6 An in vitro dissolution study to evaluate alcohol dose dumping using the final dissolution method, and evaluate different concentrations of alcohol up to 40% (0, 5, 10, 20, and 40%). Final Report Submission.

US Postmarketing Commitments – January 28, 2013
Page 11 of 20
TRADE NAME (generic name)
DUE DATE (m/d/yyyy) STATUS
EXPLANATION OF STATUS
DESCRIPTION
Horizant (gabapentin enacarbil) Extended-Release Tablets*
02/2012 Submitted PMR 1588-7 A simulated driving trial in healthy adult subjects treated with 600 mg gabapentin enacarbil that includes active comparator and placebo arms. Final Report Submission.
Horizant (gabapentin enacarbil) Extended-Release Tablets*
09/2015 Pending PMR 1588-8 A simulated driving trial in healthy adult subjects treated with an appropriate dose of gabapentin enacarbil determined in PMC 1588-12 that includes active comparator and placebo arms. Final Report Submission.
Horizant (gabapentin enacarbil) Extended-Release Tablets*
11/2012 Submitted PMR 1588-9 An adequate, randomized, double-blind, placebo- and moxifloxacin-controlled trial to evaluate the effect of gabapentin enacarbil on cardiac repolarization in healthy adult subjects. Final Report Submission.
Horizant (gabapentin enacarbil) Extended-Release Tablets*
04/2012 Submitted PMR 1588-10 A clinical drug-drug interaction trial to evaluate the pharmacokinetic and the pharmacodynamic interaction between gabapentin enacarbil and morphine. Final Report Submission.
Horizant (gabapentin enacarbil) Extended-Release Tablets*
06/2011 Fulfilled PMC 1588-11 Develop a dosage form that will allow for a 300 mg dose that could be taken once daily in patients with severe renal impairment, including patients on hemodialysis. Final Report Submission.
Horizant (gabapentin enacarbil) Extended-Release Tablets*
02/2015 Ongoing PMC 1588-12 Conduct a randomized, placebo-controlled, double-blind, parallel-group clinical trial of gabapentin enacarbil at 300 mg/day, 450 mg/day and 600 mg/day in patients with moderate to severe symptoms of RLS. Final Report Submission.
Kinrix, Diphtheria and Tetanus Toxoids and Acellular Pertussis Vaccine Adsorbed and Inactivated Poliovirus Vaccine
7/31/2011 Fulfilled PMC Conduct a randomized, open label, comparative trial, designed primarily to evaluate the immunogenicity of KINRIX when given concomitantly with varicella vaccine. The trial will be conducted in approximately 400 children 4-6 years of age who previously received three doses ofDTaP using INFANRIX® and/or PEDIARIX® and a fourth dose ofDTaP using INFANRIX®.

US Postmarketing Commitments – January 28, 2013
Page 12 of 20
TRADE NAME (generic name)
DUE DATE (m/d/yyyy) STATUS
EXPLANATION OF STATUS
DESCRIPTION
Lamictal (lamotrigine) Tablets
07/31/2013 Ongoing PMR Deferred pediatric study under PREA for the maintenance treatment of Bipolar I disorder in pediatric patients ages 10 to 17 years. Final Report Submission.
Lamictal (lamotrigine) Chewable Dispersible Tablets
07/31/2013 Ongoing PMR Deferred pediatric study under PREA for the maintenance treatment of Bipolar I disorder in pediatric patients ages 10 to 17 years. Final Report Submission.
Lamictal ODT (lamotrigine) Orally Disintegrating Tablets
07/31/2013 Ongoing PMR Deferred pediatric study under PREA for Lamictal as Maintenance treatment of Bipolar I disorder in pediatric patients ages 10 to 17 years. Final Report Submission.
Menhibrix, Meningococcal Groups C and Y and Haemophilus b Tetanus Toxoid Conjugate Vaccine
12/15/2016 Ongoing PMC-1 To conduct a Phase IIIb open-label administration (laboratory personnel will be blinded to treatment), parallel-group, controlled, multicenter study to evaluate concomitant administration of MenHibrix with rotavirus, 13-valent pneumococcal conjugate and hepatitis A vaccines administered according to a US recommended vaccine schedule.
POTIGA (ezogabine) Tablets, CV
05/2018 Pending PMR Conduct a prospective, randomized, placebo-control, double-blinded efficacy/safety trial of Potiga (ezogabine) in children >12 years old. Final Report Submission.
POTIGA (ezogabine) Tablets, CV
11/2019 Ongoing PMR Conduct a long-term open label extension study of ezogabine in children >12 years old. Final Report Submission
US Postmarketing Commitments – January 28, 2013
Page 13 of 20
TRADE NAME (generic name)
DUE DATE (m/d/yyyy) STATUS
EXPLANATION OF STATUS
DESCRIPTION
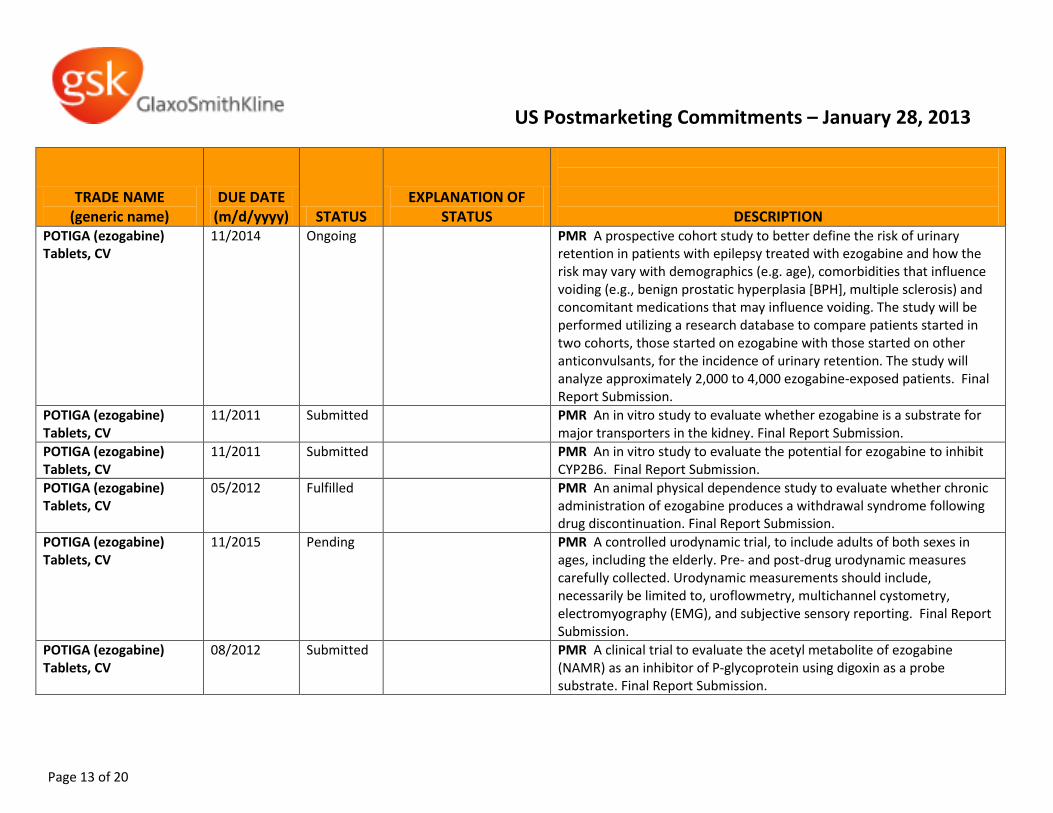
POTIGA (ezogabine) Tablets, CV
11/2014 Ongoing PMR A prospective cohort study to better define the risk of urinary retention in patients with epilepsy treated with ezogabine and how the risk may vary with demographics (e.g. age), comorbidities that influence voiding (e.g., benign prostatic hyperplasia [BPH], multiple sclerosis) and concomitant medications that may influence voiding. The study will be performed utilizing a research database to compare patients started in two cohorts, those started on ezogabine with those started on other anticonvulsants, for the incidence of urinary retention. The study will analyze approximately 2,000 to 4,000 ezogabine-exposed patients. Final Report Submission.
POTIGA (ezogabine) Tablets, CV
11/2011 Submitted PMR An in vitro study to evaluate whether ezogabine is a substrate for major transporters in the kidney. Final Report Submission.
POTIGA (ezogabine) Tablets, CV
11/2011 Submitted PMR An in vitro study to evaluate the potential for ezogabine to inhibit CYP2B6. Final Report Submission.
POTIGA (ezogabine) Tablets, CV
05/2012 Fulfilled PMR An animal physical dependence study to evaluate whether chronic administration of ezogabine produces a withdrawal syndrome following drug discontinuation. Final Report Submission.
POTIGA (ezogabine) Tablets, CV
11/2015 Pending PMR A controlled urodynamic trial, to include adults of both sexes in ages, including the elderly. Pre- and post-drug urodynamic measures carefully collected. Urodynamic measurements should include, necessarily be limited to, uroflowmetry, multichannel cystometry, electromyography (EMG), and subjective sensory reporting. Final Report Submission.
POTIGA (ezogabine) Tablets, CV
08/2012 Submitted PMR A clinical trial to evaluate the acetyl metabolite of ezogabine (NAMR) as an inhibitor of P-glycoprotein using digoxin as a probe substrate. Final Report Submission.

US Postmarketing Commitments – January 28, 2013
Page 14 of 20
TRADE NAME (generic name)
DUE DATE (m/d/yyyy) STATUS
EXPLANATION OF STATUS
DESCRIPTION
POTIGA (ezogabine) Tablets, CV
11/2022 Pending PMR Conduct a prospective, randomized, placebo-control, double-blinded efficacy /safety trial of Potiga (ezogabine) for the treatment of partial-onset seizures in children aged 2 years to <12 years old. Final Report Submission.
POTIGA (ezogabine) Tablets, CV
05/2026 Pending PMR Conduct a prospective, randomized, placebo-control, double-blinded efficacy/safety trial of Potiga (ezogabine) for the treatment of partial-onset seizures in children aged 1 month to <2 years old. This study must also utilize endpoints and assessment methods best suited to this pediatric age group. Final Report Submission.
Promacta (eltrombopag) Tablets .
11/30/2019 Delayed Study start date goal was November 2009. Study is active, but no subjects have been enrolled to date.
PMR-3 To develop and maintain a prospective, observational pregnancy exposure registry study conducted in the United States that compares the pregnancy and fetal outcomes of women exposed to Promacta (eltrombopag) Tablets during pregnancy to an unexposed control population. The registry will detect and record major and minor congenital anomalies, spontaneous abortions, stillbirths, elective terminations, adverse effects on immune system development, platelet number and function, neoplasm formation, bone marrow reticulin formation, thrombotic events, and any serious pregnancy outcomes. These events will also be assessed among infants through at least the first year of life
Promacta (eltrombopag) Tablets
11/30/2019 Delayed Study start date goal was November 2009. The study start date has passed but no subjects have been enrolled.
PMR-6 To conduct a milk-only lactation study in the subset of women enrolled in the pregnancy registry who choose to breastfeed their infants. This study will be designed to detect the presence and concentration of Promacta (eltrombopag) Tablets in breast milk and, when feasible, in the blood of infants. The study will include a symptom diary for mothers to record any adverse effects in the breastfeeding infants.

US Postmarketing Commitments – January 28, 2013
Page 15 of 20
TRADE NAME (generic name)
DUE DATE (m/d/yyyy) STATUS
EXPLANATION OF STATUS
DESCRIPTION
Promacta (eltrombopag) Tablets
August 2013 Ongoing PMR Conduct a PK trial to evaluate the effect of boceprevir and telaprevir on eltrombopag PK and the effect of eltrombopag on boceprevir and telaprevir PK in healthy adult subjects.
Promacta (eltrombopag) Tablets
01/31/2014 Ongoing PMR-5 To conduct trial TRA105325 entitled, "EXTEND (Eltrombopag extended dosing study): an extension study of eltrombopag olamine in adults, with idiopathic thrombocytopenic purpura (ITP), previously enrolled in an eltrombopag study." The protocol for this trial was previously submitted to FDA and the study is currently active. The protocol will be modified to include performance of bone marrow examinations prior to the initiation of Promacta (eltrombopag) Tablets, following 12 months of Promacta (eltrombopag) Tablets therapy as well as following the completion of 24 months of Promacta (eltrombopag) Tablets therapy; enrollment will continue until these data are obtained from at least 150 patients. An interim report will contain, in addition to any other items, results of bone marrow evaluations for patients who have completed bone marrow evaluations at baseline and following 12 months of Promacta (eltrombopag) Tablets therapy.
(raxibacumab) To be determined should an event occur
Pending Conduct a field study to evaluate the efficacy, pharmacokinetics, and safety of raxibacumab use for Bacillus anthracis in the United States. (To be conducted only if an anthrax incident occurs)
(raxibacumab) 10/01/2017 Pending Conduct a Phase 4 study to evaluate the effect of raxibacumab on immunogenicity of anthrax vaccine.
Requip XL (ropinirole) Extended-Release Tablets
7/2012 Ongoing FDA acknowledged new target date of March 2015 for submission of study report.
PMR-1 Conduct a fixed-dose, placebo-controlled, double-blinded study that examines multiple doses in early Parkinson's disease. The trial should identify a range of doses inclusive of the lowest effective dose and the lowest maximally effective therapeutic dose. Final Report Submission.

US Postmarketing Commitments – January 28, 2013
Page 16 of 20
TRADE NAME (generic name)
DUE DATE (m/d/yyyy) STATUS
EXPLANATION OF STATUS
DESCRIPTION
Requip XL (ropinirole) Extended-Release Tablets
07/2012 Ongoing FDA acknowledged new target date of April 2015 for submission of study report.
PMR-2 Conduct a fixed-dose, placebo-controlled, double-blinded study that examines multiple doses in late Parkinson's disease. The trial should identify a range of doses inclusive of the lowest effective dose and the lowest maximally effective therapeutic dose. Final Report Submission.
Requip XL (ropinirole) Extended-Release Tablets
07/2009 Submitted PMC-3 Evaluate whether ropinirole is a P-gp substrate and/or inducer for major CYP enzymes (e.g., CYP3A4) and, if so, any drug-drug interaction potential through either mechanism. This can be accomplished through a comprehensive literature review or by conducting an in vitro study. Final Report Submission.
Rotarix
3/31/2012 Delayed Delayed due to slow accrual; new due date agreed with CBER is 12/31/2015.
PMR-1 Conduct a large-scale observational post-licensure safety study in the US to assess the potential serious risk of intussusception and other serious adverse effects (specifically Kawasaki disease, hospitalizations due to acute lower respiratory tract infections, and convulsions) in recipients of approximately 44,000 Rotarix vaccinated subjects.
Sorilux Foam (calcipotriene 0.005%)
6/30/2013 Pending PMR Deferred pediatric study under PREA for the topical treatment of plaque psoriasis in pediatric patients ages 12 through 16. A Pharmacokinetic/Pharmodynamic trial of calcipotriene foam under maximal use conditions in 20 evaluable pediatric subjects with plaque psoriasis age 12 through 16 years. Evaluate the effect of the product on calcium metabolism in all subjects.
Sorilux Foam (calcipotriene 0.005%)
9/30/2013 Pending PMR Deferred pediatric study under PREA for the topical treatment of plaque psoriasis in pediatric patients ages 2 through 11. A Pharmacokinetic/Pharmodynamic trial of calcipotriene foam under maximal use conditions in 25 evaluable pediatric subjects with plaque psoriasis age 2 through 11 years. Evaluate the effect of the product on calcium metabolism in all subjects.

US Postmarketing Commitments – January 28, 2013
Page 17 of 20
TRADE NAME (generic name)
DUE DATE (m/d/yyyy) STATUS
EXPLANATION OF STATUS
DESCRIPTION
Sorilux Foam (calcipotriene 0.005%)
6/30/2013 Pending PMR Deferred pediatric study under PREA for the topical treatment of plaque psoriasis in pediatric patients ages 2 through 11. A vehicle-controlled trial of the safety and efficacy of calcipotriene foam in 100 evaluable pediatric subjects with plaque psoriasis age 2 through 11 years. Evaluate the effect of the product on calcium metabolism in all subjects.
Sorilux Foam (calcipotriene 0.005%)
6/2015 Ongoing PMR A Pharmacokinetics/ Pharmacodynamics trial of Sorilux Foam, 0.005% under maximum use conditions in 20 evaluable pediatric subjects with plaque psoriasis of the scalp and body age 12 years to 16 years and 11 months. Evaluate the effect of the product on calcium metabolism in all subjects.
Sorilux Foam (calcipotriene 0.005%)
12/2019 Ongoing PMR A vehicle-controlled trial of the safety and efficacy of Sorilux Foam, 0.005% in 150 evaluable pediatric subjects with plaque psoriasis of the scalp and body age 2 years to 11 years and 11 months. Pharmacokinetic/Pharmacodynamic parameters will be evaluated in a subset of at least 25 evaluable subjects under maximum use conditions. Evaluate the effect of the product on calcium metabolism in all subjects.
Treximet (sumatriptan / naproxen sodium)
04/2011 Delayed Final report submitted by due date, then withdrawn at request of FDA. Final report will be included in future sNDA.
PMR Conduct a controlled effectiveness study of Treximet™ for the acute treatment of migraine attacks with and without aura in pediatric patients ages 12 years to 17 years. Final Report Submission.
Treximet (sumatriptan / naproxen sodium)
04/2011 Delayed Final report submitted by due date, then withdrawn at request of FDA. Final report will be included in future sNDA.
PMR Conduct a long-term open label safety study in pediatric patients with migraine ages 12 years to 17 years. Final Report Submission.

US Postmarketing Commitments – January 28, 2013
Page 18 of 20
TRADE NAME (generic name)
DUE DATE (m/d/yyyy) STATUS
EXPLANATION OF STATUS
DESCRIPTION
Tykerb (lapatinib) Tablets
Original 03/31/2016; Revised 05/31/2018
Ongoing Subpart H-1 Accelerated Approval (S-7) A randomized trial comparing lapatinib in combination with trastuzumab and an aromatase inhibitor versus trastuzumab in combination with an aromatase inhibitor versus lapatinib in combination with an aromatase inhibitor in postmenopausal women with hormone receptor positive metastatic breast cancer that overexpresses the HER2 receptor.
Verdeso Foam (desonide 0.05%)
12/01/2011 Fulfilled PMC The applicant commits to conducting a study to determine the photocarcinogenic potential of Verdeso (desonide) Foam.
Votrient (pazopanib) Tablets
Original 07/31/2012; Revised 06/28/2013
Ongoing PMR-1 Examine the safety of dose modification of pazopanib and patient rechallenge with pazopanib following hepatotoxicity. This examination should include at least 1,500 treated patients and may be derived from ongoing or completed trials, including VEG108844, VEG110727, and VEG110665.
Votrient (pazopanib) Tablets
Original 12/31/2010; Revised 06/28/2013
Delayed Completion of the trial is delayed due to a slower than expected rate recruiting patients, a higher than expected drop-out rate, and a higher than expected number of patients who had insufficient data to determine disease progression. FDA determined there was good cause for the delay on January 5, 2012.
PMR-2 Examine the cardiotoxicity, clinical cardiac events and changes in ejection fraction in your ongoing trial VEG108844.
US Postmarketing Commitments – January 28, 2013
Page 19 of 20
TRADE NAME (generic name)
DUE DATE (m/d/yyyy) STATUS
EXPLANATION OF STATUS
DESCRIPTION
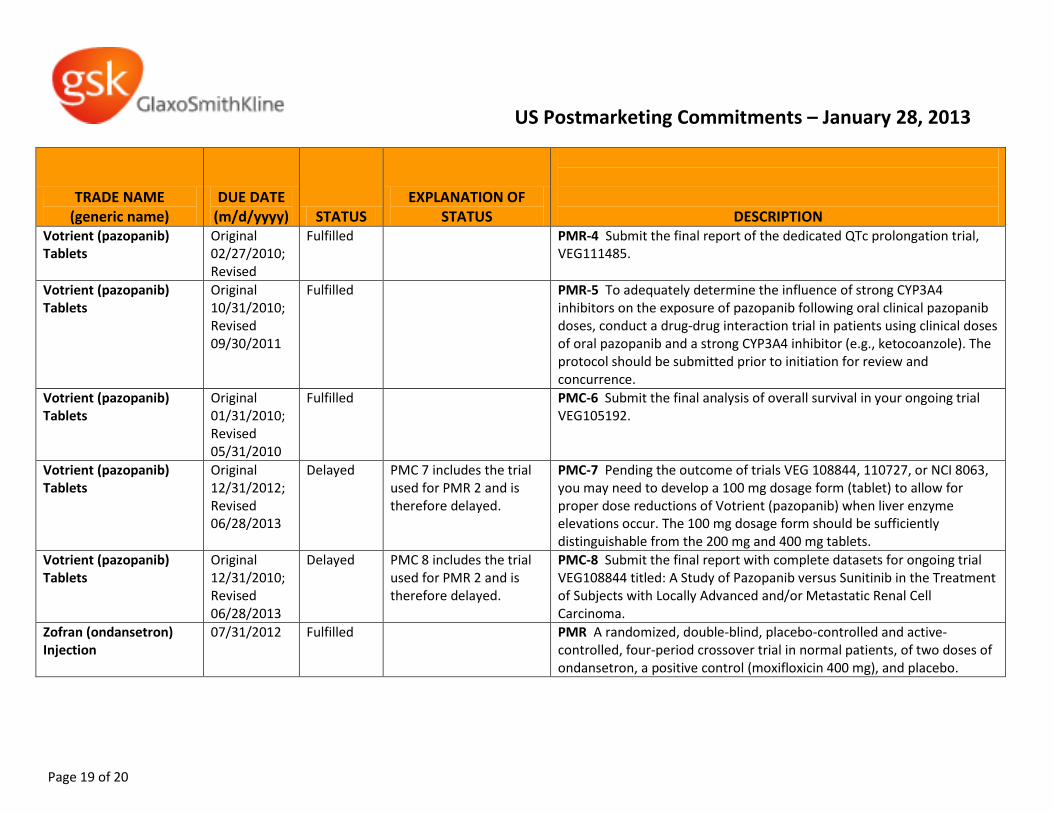
Votrient (pazopanib) Tablets
Original 02/27/2010; Revised
Fulfilled PMR-4 Submit the final report of the dedicated QTc prolongation trial, VEG111485.
Votrient (pazopanib) Tablets
Original 10/31/2010; Revised 09/30/2011
Fulfilled PMR-5 To adequately determine the influence of strong CYP3A4 inhibitors on the exposure of pazopanib following oral clinical pazopanib doses, conduct a drug-drug interaction trial in patients using clinical doses of oral pazopanib and a strong CYP3A4 inhibitor (e.g., ketocoanzole). The protocol should be submitted prior to initiation for review and concurrence.
Votrient (pazopanib) Tablets
Original 01/31/2010; Revised 05/31/2010
Fulfilled PMC-6 Submit the final analysis of overall survival in your ongoing trial VEG105192.
Votrient (pazopanib) Tablets
Original 12/31/2012; Revised 06/28/2013
Delayed PMC 7 includes the trial used for PMR 2 and is therefore delayed.
PMC-7 Pending the outcome of trials VEG 108844, 110727, or NCI 8063, you may need to develop a 100 mg dosage form (tablet) to allow for proper dose reductions of Votrient (pazopanib) when liver enzyme elevations occur. The 100 mg dosage form should be sufficiently distinguishable from the 200 mg and 400 mg tablets.
Votrient (pazopanib) Tablets
Original 12/31/2010; Revised 06/28/2013
Delayed PMC 8 includes the trial used for PMR 2 and is therefore delayed.
PMC-8 Submit the final report with complete datasets for ongoing trial VEG108844 titled: A Study of Pazopanib versus Sunitinib in the Treatment of Subjects with Locally Advanced and/or Metastatic Renal Cell Carcinoma.
Zofran (ondansetron) Injection
07/31/2012 Fulfilled PMR A randomized, double-blind, placebo-controlled and active-controlled, four-period crossover trial in normal patients, of two doses of ondansetron, a positive control (moxifloxicin 400 mg), and placebo.

US Postmarketing Commitments – January 28, 2013
Page 20 of 20
TRADE NAME (generic name)
DUE DATE (m/d/yyyy) STATUS
EXPLANATION OF STATUS
DESCRIPTION
Zyban (bupropion hydrochloride) Sustained-Release Tablets Wellbutrin SR (bupropion hydrochloride) Sustained-Release Tablets Wellbutrin (bupropion hydrochloride) Tablets
04/2017 Ongoing PMR A Phase 4, Randomized, Double-Blind, Active and Placebo-Controlled, Multicenter Study Evaluating the Neuropsychiatric Safety and Efficacy of 12 Weeks Varenicline Tartrate 1mg BID and Bupropion Hydrochloride 150mg BID for Smoking Cessation in Subjects With and Without a History of Psychiatric Disorders. Final Report Submission.
*NDA transferred to a new sponsor in November 2012.