gxp and cgxp in bio/pharmaceutical industry
TRANSCRIPT

GxP and cGxP in Bio/Pharmaceutical Industry
Prof. Dr. Basavaraj K. Nanjwade M. Pharm., Ph. D
Department of PharmaceuticsKLE University College of Pharmacy
BELGAUM – 590010, Karnataka, IndiaE-mail: [email protected]
Cell No: 00919742431000
28 March 2011 1Department of Pharmaceutics

GxP
• The bio/pharmaceutical industry has created its own language and GxP is one of many acronyms that we all tend to use.
• While this may seem “elementary” to some of you, many people may not know what this means.
• G = Goodx (variable replaced with Manufacturing, Clinical,
Laboratory, Storage, Distribution and Review) P = Practice
2Department of Pharmaceutics28 March 2011

GxP
• As you can see, GxP is used as short-hand form for referring to the regulations established by the United States Food and Drug Administration which are published in the Code of Federal Regulations.
• Sometimes people refer to the “GCPs” which specifically regards the rules that govern clinical trials vs. product manufacturing (GMPs) or laboratory regulations (GLPs).
• Together, these are known collectively as the “predicate rules” that govern a wide spectrum of regulatory obligations across this diverse industry.
3Department of Pharmaceutics28 March 2011

GxP
• GxP is also where citations emanate from (typically) as regards FDA inspections.
• When a regulation is cited, the title tells you where it is published.
For example: 21 CFR 312.2
Means:21 = Title 21CFR = Code of Federal Regulations312.2 (312 = part and 2 =section)
4Department of Pharmaceutics28 March 2011

Lifecycle Requirements
5Department of Pharmaceutics28 March 2011

GxP
• “GxP” is a collective term for the Good Practice quality guidelines and regulations used in many fields, encompassing such internationally-recognized standards as GMP, GCP, GLP, GSP, GDP and GRP.
• GxP guidelines are designed to ensure that products are safe, meet their intended use and, in regulated industries such as drugs, food, medical devices and cosmetics, adhere to quality processes during manufacturing, control, storage and distribution.
6Department of Pharmaceutics28 March 2011

GxP• GxP is a general term for Good Practice quality
guidelines and regulations. These guidelines are used in many fields, including the pharmaceutical and food industries.
• The titles of these good practice guidelines usually begin with "Good" and end in "Practice", with the specific practice descriptor in between.
• GxP represents the abbreviations of these titles, where x (a common symbol for a variable) represents the specific descriptor.
7Department of Pharmaceutics28 March 2011

Core GXP Information
8Department of Pharmaceutics28 March 2011

Regional Harmonization Initiatives
9Department of Pharmaceutics28 March 2011

GxP
10Department of Pharmaceutics28 March 2011

List of GxP’s in Pharmaceuticals
1. GMP – (Good manufacturing Practice)
2. GCP – (Good Clinical Practice)
3. GLP – (Good Laboratory Practice)
4. GSP – (Good Storage Practice)
5. GDP – (Good Distribution practice)
6. GRP – (Good Review Practice)
11Department of Pharmaceutics28 March 2011

Purpose of GxP
• The purpose of the GxP quality guidelines is to ensure a product is safe and meets its intended use.
• GxP guides quality manufacture in regulated industries including food, drugs, medical devices and cosmetics.
The most central aspects of GxP are:
1. Traceability: the ability to reconstruct the development history of a drug or medical device.
2. Accountability: the ability to resolve who has contributed what to the development and when.
12Department of Pharmaceutics28 March 2011

Regulators
13Department of Pharmaceutics28 March 2011

GMP – (Good Manufacturing Practice)
28 March 2011 Department of Pharmaceutics 14

What is GMP ?
• GMP is that part of Quality assurance which ensures that the products are consistently manufactured and controlled to the Quality standards appropriate to their intended use
• A set of principles and procedures which, when followed by manufacturers for therapeutic goods, helps ensure that the products manufacture will have the required quality.
28 March 2011 Department of Pharmaceutics 15

Good Manufacturing Practices
• A basic tenet of GMP is that quality cannot be tested into a batch of product but must be built into each batch of product during all stages of the manufacturing process.
• It is designed to minimize the risks involved in any pharmaceutical production that cannot be eliminated through testing the final product.
- Some of the main risks are unexpected contamination of products, causing damage to health or even death
- In correct labels on containers, which could mean that patient receive the wrong medicine.
- Insufficient or too much active ingredient, resulting in ineffective treatment or adverse effects.
28 March 2011 Department of Pharmaceutics 16
GMP
28 March 2011 Department of Pharmaceutics 17
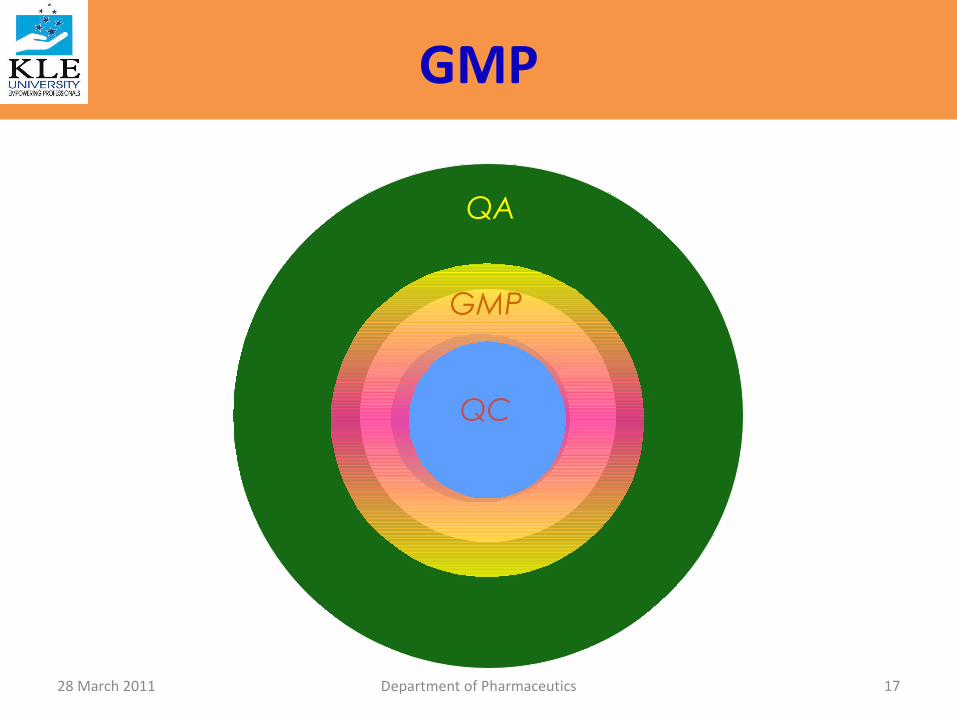
QC
GMP
QA

GMP
• GMP is the magic key that opens the door of the Quality
• In matter of GMP, swim with the current and
in matter of Quality stand like a rock!
28 March 2011 Department of Pharmaceutics 18

GMP
28 March 2011 Department of Pharmaceutics 19
GMP
Is that part of Quality
Assurance aimed at
ensuring that products are
consistently manufactured
to a quality appropriate to
their intended use

GMP guidelines
• GMP as per Schedule “M”• GMP as per WHO• GMP as per MCA now known as MHRA• GMP as per TGA• GMP as per US FDA• GMP as per ICH guidelines
28 March 2011 Department of Pharmaceutics 20

GMP guidance documents
• EU Good Manufacturing Practice (GMP) Guidelines, Volume 4 of “The rules governing medicinal products in the European Union”
• US FDA current Good Manufacturing Practice (cGMP) for finished pharmaceuticals, 21 CFR, 210 and 211
• WHO Good Manufacturing Practices for pharmaceutical products, Annex 4 to WHO Technical Report Series, No. 908, 2003
21Department of Pharmaceutics28 March 2011

GMP
• GMP in solid dosage forms• GMP in semisolid dosage forms• GMP in Liquid orals• GMP in Parenterals Production• GMP in Ayurvedic medicines• GMP in Bio technological products• GMP in Nutraceuticals and cosmeceuticals
28 March 2011 Department of Pharmaceutics 22

API Manufacturing Process
23Department of Pharmaceutics28 March 2011

Secondary Manufacturing Dosage Forms
24Department of Pharmaceutics28 March 2011

Secondary Manufacturing Process - Tablets
25Department of Pharmaceutics28 March 2011
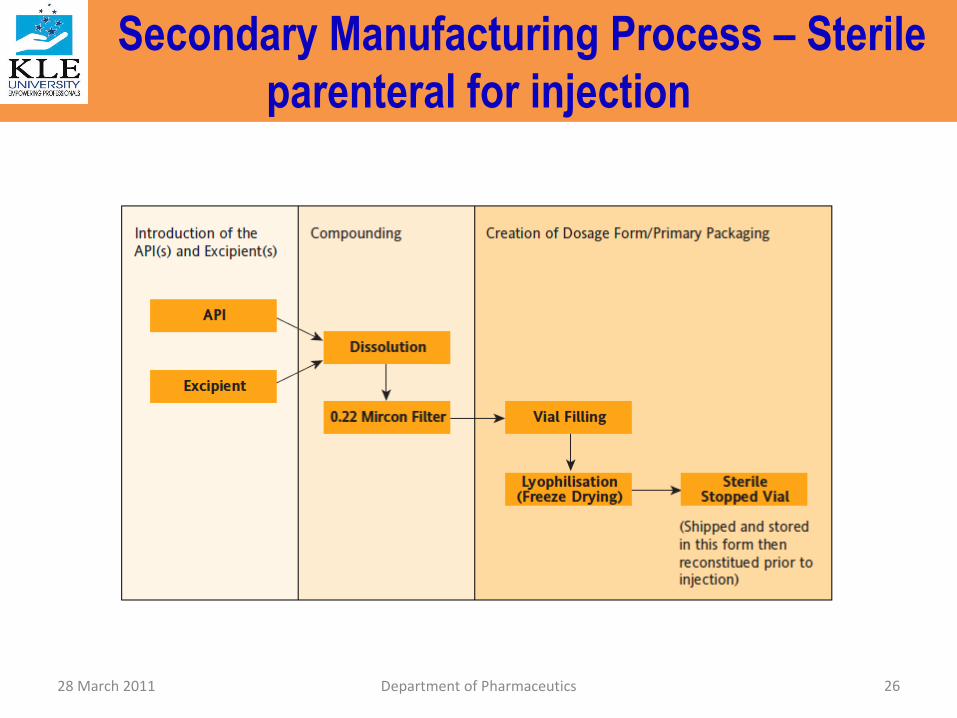
Secondary Manufacturing Process – Sterile parenteral for injection
26Department of Pharmaceutics28 March 2011

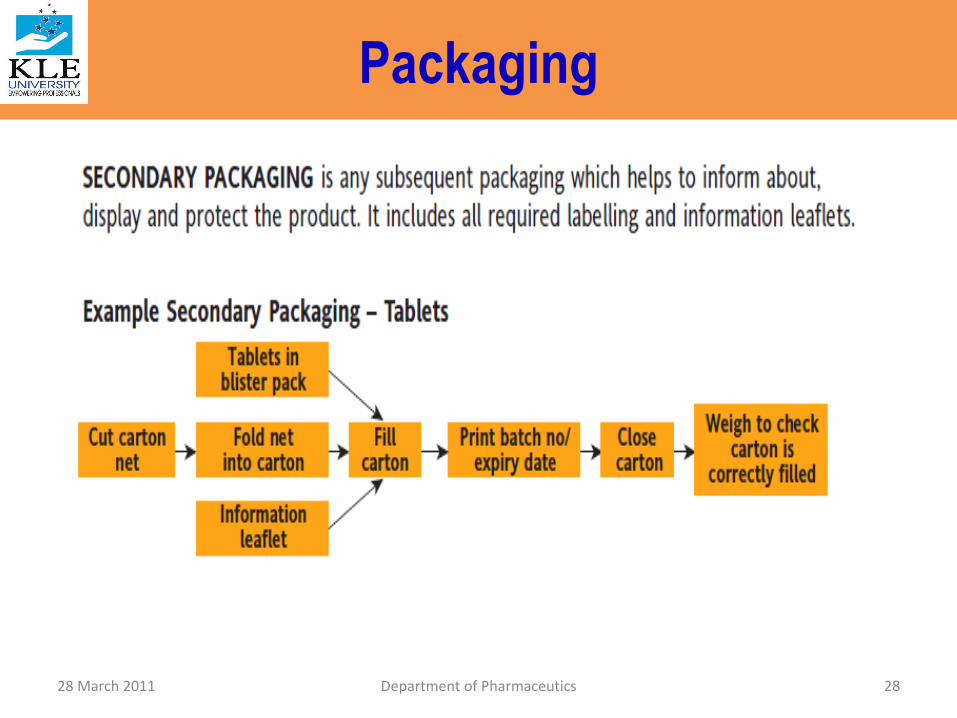
Packaging
27Department of Pharmaceutics28 March 2011
Packaging
28Department of Pharmaceutics28 March 2011

Packaging
29Department of Pharmaceutics28 March 2011
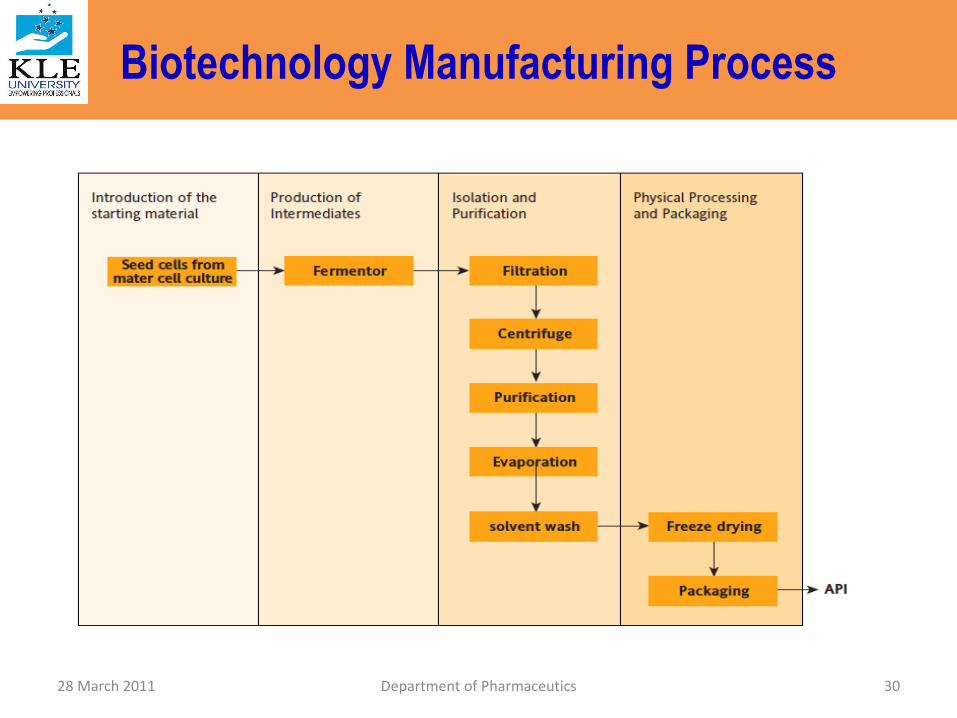
Biotechnology Manufacturing Process
30Department of Pharmaceutics28 March 2011

Ten Principles of GMP
1. Design and construct the facilities and equipments properly
2. Follow written procedures and Instructions3. Document work4. Validate work5. Monitor facilities and equipment6. Write step by step operating procedures and work on
instructions7. Design ,develop and demonstrate job competence8. Protect against contamination9. Control components and product related processes 10. Conduct planned and periodic audits 28 March 2011 Department of Pharmaceutics 31

Beyond GMP
• Reduce pollution - Zero discharge
• Adaptation of environment friendly methods
• Consideration for better and healthier life tomorrow
• Consideration of ethics in life
• One should begin with end in mind otherwise it will be the beginning of the end
28 March 2011 Department of Pharmaceutics 32

Cost of effective GMP
• In fact Cost benefits – positive cost benefits of GMP/QA
• Good plant lay out, Smooth work flows, Efficient documentation systems, well controlled process, good stores lay outs and stores records- These are Good manufacturing practices
• Reduction in work in process and inventory holding costs
• Avoidance of cost of Quality failure ( cost of waste, of rework, of recall, of consumer compensation and of loss of company reputation
28 March 2011 Department of Pharmaceutics 33

Cost / Benefit analysis
• GMP is not an “On-cost”. • It is not even “Just free”• It is a contribution to profit• Good manufacturing Practice is also Good
management Practice leading to Good Manufacturing Profit
• GMP is central and basic and has cost benefits ( not to be considered as extrinsic or imposed upon manufacturing activities)
28 March 2011 Department of Pharmaceutics 34

Cost / benefit analysis
28 March 2011 Department of Pharmaceutics 35
• Cost of quality = Cost of A – Cost of B- Payback from C = Profit
A B CStaff Scrap Improved moraleTraining Rework MotivationSystems Complaints Faster throughputDocumentation Chaos Higher productivityEquipment Lost sales Increased salesMaintenance Recalls lower inventoryCalibration Closedown SamplingTestingIn process controlValidationAuditing

GCP – (Good Clinical Practice)
28 March 2011 Department of Pharmaceutics 36

What It Is GCP
• An international ethical & scientific quality standard for designing, conducting, recording & reporting human clinical studies
– EU
– Japan
– US
• Applies to registration studies that may have an impact on safety & welfare of human subjects
28 March 2011 Department of Pharmaceutics 37

GCP Participating Parties
• IRB/Ethics Committee
• Investigators
• Sponsor
• Regulatory Authorities
28 March 2011 Department of Pharmaceutics 38

GCP Key Documents
• Investigator Brochure
• Study Protocol
• Informed Consent Document
28 March 2011 Department of Pharmaceutics 39

GCP Principles
1. Studies in accordance with Declaration of Helsinki; consistent with GCP & applicable regulatory requirements
2. Studies initiated & continued only if anticipated benefits outweigh risks
3. Rights, safety & welfare of human subjects take priority over interests of science & society
4. Available non-clinical & clinical info on product adequate to support study
28 March 2011 Department of Pharmaceutics 40

GCP Principles
5. Studies scientifically sound; described in clear, detailed protocol
6. Study in compliance with IRB/EC approved protocol
7. Medical care given to subjects is the responsibility of qualified medical professional(s)
8. Individuals conducting studies qualified by education, training & experience
9. Freely given informed consent obtained from every subject prior to study participation
28 March 2011 Department of Pharmaceutics 41

GCP Principles
10. Study information recorded, handled & stored to allow accurate reporting, interpretation & verification
11. Confidentiality of subject records protected in accordance with applicable regulatory requirements
12. Investigational products manufactured, handled & stored in accordance with GCP & used in accordance with approved protocol
13. Systems/procedures implemented to assure quality of study
28 March 2011 Department of Pharmaceutics 42

IRB/EC Roles & Responsibilities
To safeguard study subjects’ rights & welfare by:
• Evaluation/disposition of study proposal
• Evaluation of proposed subject consent materials
• Evaluation of emergency use consent methodology
• Evaluation of investigator qualifications
• Ongoing review of study progress (at least yearly)
• Evaluation of proposed subject compensation plans
28 March 2011 Department of Pharmaceutics 43

IRB/EC Composition & Operations
• Membership has qualifications & experience to evaluate science, medical aspects & ethics of proposed study
– ≥ 5 members
– ≥ 1 member whose primary interest in nonscientific
– ≥ 1 member independent of institution or study site
• Written SOPs & records
• Decisions rendered at announced meetings with quorum in attendance
28 March 2011 Department of Pharmaceutics 44

IRB/EC Composition & Operations
• Only members participating in review should vote
• Investigator may provide info on study, but should not be involved in review or vote
• Nonmembers with expertise in special areas may be invited to assist with review (but cannot vote)
28 March 2011 Department of Pharmaceutics 45

IRB/EC Procedures
• Document group membership & qualifications
• Schedule meetings & notify members
• Conduct initial & ongoing review of studies
• Determine ongoing review frequency
• Provide expedited review of minor study changes, in accordance with regulatory requirements
• Specify that no subject should be enrolled in study prior to IRB/EC approval
28 March 2011 Department of Pharmaceutics 46

IRB/EC Procedures
• Specify that no deviations from protocol should be initiated without prior IRB/EC approval
– Emergency situations require immediate notification of IRB/EC after the fact
• Specify that Investigator should promptly report:
– Protocol deviations
– Changes increasing subject risk or study procedures
– Serious and unexpected adverse events
28 March 2011 Department of Pharmaceutics 47

IRB/EC Procedures
• Notify Investigator promptly of:
– Study-related decisions
– Reason for decisions
– Procedures for appeal of decisions
28 March 2011 Department of Pharmaceutics 48

IRB/EC Required Records
• Relevant records maintained ≥ 3 yr after study completion
• Records available for review by regulatory authorities
28 March 2011 Department of Pharmaceutics 49

IRB/EC What is Reviewed
• Investigator Brochure or Report of Prior Investigations
• Study protocol & amendments
• Investigator qualifications
• Informed consent documents, including subject recruiting tools
• Other written information provided to subjects
• Subject compensation plans
• Adverse events
• Protocol deviations28 March 2011 Department of Pharmaceutics 50

IRB/EC When Reviews Occur
• Prior to study initiation at site
• At least yearly during study
• During study, as necessitated by:
– Changes in protocol, consent documents, etc.
– Changes in study investigator
– Reports of serious or unanticipated device-related adverse events
• At study completion or termination
28 March 2011 Department of Pharmaceutics 51

Investigator Roles & Responsibilities
• Qualified to conduct study
• Have adequate resources to conduct study
• Provide medical care to study subjects
• Regular communication with IRB/EC reviewing study
• Compliance with study protocol
• Maintenance of investigational product accountability
• Compliance with study randomization & unmasking procedures
• Provide informed consent to study subjects28 March 2011 Department of Pharmaceutics 52

Investigator ResponsibilitiesAppropriate Qualifications
• Training & experience demonstrated via:
– Medical license
– CV
– Specialized study training
– GCP training
• If study responsibilities delegated, need a list of qualified persons to whom responsibilities are delegated
28 March 2011 Department of Pharmaceutics 53

Investigator ResponsibilitiesAdequate Resources
• Suitable staff & good methods for keeping them apprised
• Suitable facilities
• Appropriate patient population
– Access to disease or condition
– Volume of patients with disease or condition
28 March 2011 Department of Pharmaceutics 54

Investigator ResponsibilitiesRequired Records & Reports
• Essential regulatory document file(s)– Protocol & amendments
– Approved informed consent documents
– Product accountability documentation
– Investigator qualifications & agreements
– IRB correspondence
– Study delegation list
– Subject screening/enrollment logs
– Study monitoring reports
– Calibration/maintenance logs
– Memos to file
28 March 2011 Department of Pharmaceutics 55

SponsorRoles & Responsibilities
• Study quality assurance
• Appropriately qualified medical personnel to advise on study
• Utilization of qualified personnel in study design & operations
• Study management, data handling & record keeping
• Investigator selection & training
• Definition/allocation of study responsibilities28 March 2011 Department of Pharmaceutics 56

SponsorRoles & Responsibilities
• Facilitation of communications between Investigators
• Study compensation (investigators and/or subjects) & financing
• Regulatory authority notification/submission
• Confirmation of IRB/EC review/approval
• Investigational product information
• Investigational product manufacturing, packaging, labeling & coding
• Investigational product supply & handling28 March 2011 Department of Pharmaceutics 57

SponsorRoles & Responsibilities
• Record access
• Ongoing safety evaluation & reporting
• Serious/unanticipated adverse event reporting
• Study monitoring
• Study noncompliance procedures
• Study termination or suspension notification
• Study reports
28 March 2011 Department of Pharmaceutics 58

SponsorRoles & Responsibilities
• Sponsor may transfer responsibilities to CRO
– Transfer must be documented in writing
– Sponsor still has ultimate responsibility for study quality and data integrity
28 March 2011 Department of Pharmaceutics 59

Study Protocol Components
• General administrative info
• Background
• Study purpose & objectives
• Study design
• Subject eligibility requirements
• How subjects will be treated
• How safety & efficacy will be assessed
• Sample size justification & statistical analysis methods28 March 2011 Department of Pharmaceutics 60

Study Protocol Components
• How data will be captured & maintained
• Monitoring procedures
• Proposed informed consent document
28 March 2011 Department of Pharmaceutics 61

Informed Consent DocumentComponents
• Statement that study involves “research” & product “experimental” (if applicable)
• Study purpose
• Number of expected study subjects to be enrolled
• Study treatment(s) & probability for random assignment
• Study exams & procedures for duration of trial
• Subject’s responsibilities
• Foreseeable risks to subject (embryo, fetus, nursing infant)
28 March 2011 Department of Pharmaceutics 62

Informed Consent DocumentComponents
• Expected benefits
• Alternatives procedures or therapies & associated risk/benefit
• Compensation available in event of study-related injury or sickness
• Anticipated payments to subject for study participation
• Anticipated expenses to subject for study participation
• Statement that participation is voluntary28 March 2011 Department of Pharmaceutics 63

Informed Consent DocumentComponents
• Description of extent to which confidentiality can be assured
• Commitment to keep subject apprised on new information that may affect subject’s willingness to participate in study
• Contact info for questions re: subject rights; trial-related adverse events
• Circumstances under which subject’s participation may be terminated
28 March 2011 Department of Pharmaceutics 64

Investigator BrochureWhat It Is
A compilation of clinical & non-clinical data on the product that is relevant to the product’s study in
humans
Necessary for Investigator & IRB/EC review to assess the risks/benefits associated with study
28 March 2011 Department of Pharmaceutics 65

Investigator BrochureComponents
Product formulation summary
Introduction/background info regarding product & investigational plan
• Investigational product physical, chemical & pharmaceutical properties & formulation
• Non-clinical studies
• Human clinical studies
• Summary of data & guidance for Investigator28 March 2011 Department of Pharmaceutics 66

Good Clinical PracticeReference Documents & Links
• ICH - E6: Guideline for Good Clinical Practice
• 21 CFR 50 - Informed Consent
• 21 CFR 56 - Institutional Review Board
• http://www.ich.org/cache/compo/276-254-1.html
• http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm
28 March 2011 Department of Pharmaceutics 67

GLP – (Good Laboratory Practice)
28 March 2011 Department of Pharmaceutics 68

What It Is GLP
• Describes good practices for non-clinical lab studies that support research or marketing approvals for FDA-regulated products
28 March 2011 Department of Pharmaceutics 69

GLP General Requirements
• Appropriately qualified personnel
• Adequate resources
• Appropriate procedures for:
– Sanitation, health precautions, clothing
– Test protocol development, test methods
– Data analysis, report development
• Appropriately qualified study director
• Quality assurance function
28 March 2011 Department of Pharmaceutics 70

GLP Facilities Requirements
• Suitable size, construction, segregation
– Animal care
– Animal supplies
– Test & control products maintained in a secure area
– Operating “suite”
– Specimen & data storage
28 March 2011 Department of Pharmaceutics 71

GLP Equipment Requirements
• Appropriately designed
• Adequate thru-put capacity
• Appropriately located
• Routinely maintained & calibrated
28 March 2011 Department of Pharmaceutics 72

GLP Standard Operating Procedures
• Animal room prep
• Animal care
• Receipt, ID, storage, handling, mixing & sampling of test & control articles
• Test system observations
• Lab tests
• Handling of moribund or dead animals
• Necropsy or postmortem exams of animals
28 March 2011 Department of Pharmaceutics 73

GLP Standard Operating Procedures
• Collection & ID of specimens
• Histopathology
• Data handling, storage & retrieval
• Equipment maintenance & calibration
• Transfer, proper placement & ID of animals
28 March 2011 Department of Pharmaceutics 74

GLP Reagents & Solutions
• Adequate labeling
– Identity
– Concentration
– Storage requirements
– Expiration date
28 March 2011 Department of Pharmaceutics 75

GLP Test & Control Articles
• Adequate characterization
• Proper receipt, storage, distribution
• When mixed with a carrier, adequate methods to confirm
– Mixture uniformity
– Article concentration
– Article stability
28 March 2011 Department of Pharmaceutics 76

GLP Study Implementation
• Written, approved protocol indicating test objectives & methods
• Study conducted in accordance with protocol
• Study monitoring to confirm protocol compliance
• Appropriate labeling of specimens by test system, study, nature & collection date
• Records of gross findings from postmortems available to pathologist for specimen histopathology
28 March 2011 Department of Pharmaceutics 77

GLP Study Implementation
• Standard data capture/recording requirements
– Legibility
– Permanence
– Accountability
– Changes
28 March 2011 Department of Pharmaceutics 78

GLP Records & Reports
• Final report of results
• Study records & data methodically archived to facilitate expedient retrieval
– Study documents
– Raw data
– Specimens
– Protocols
– QA inspections
– Personnel training & qualifications
– Calibration & maintenance records28 March 2011 Department of Pharmaceutics 79

GLP Records & Reports
• Records retention (shortest of):
– ≥ 2 yr after FDA marketing clearance
– ≥ 5 yr after data submitted to FDA in support of marketing application
– ≥ 2 yr after Sponsor decision not to proceed with marketing application
– Wet specimens hold as long as viable
• Records transferable with written FDA notification
28 March 2011 Department of Pharmaceutics 80

GLP Facility Disqualification
• Grounds for disqualification:
– Failure to comply with regulations &
– Noncompliance adversely affects study validity &
– Previous regulatory actions have been unsuccessful in modifying facility operations
28 March 2011 Department of Pharmaceutics 81

GSP – (Good Storage Practice)
28 March 2011 Department of Pharmaceutics 82

GSP – (Good Storage Practice)
1. Glossary
2. Personnel
3. Premises and facilities
4. Storage requirements
5. Returned goods
6. Dispatch and transport
7. Product recall
83Department of Pharmaceutics28 March 2011

1. Glossary
a. Active pharmaceutical ingredient
b. Contamination
c. Cross-contamination
d. Excipient
e. Expiry date
f. Labelling
28 March 2011 Department of Pharmaceutics 84

1. Glossary
g. Packaging material
h. Pharmaceutical product
i. Production
j. Retest date
k. Storage
l. Supplier
28 March 2011 Department of Pharmaceutics 85

2. Personnel
• At each storage site (e.g. that of a manufacturer, distributor, wholesaler, community or hospital pharmacy) there should be an adequate number of qualified personnel to achieve pharmaceutical quality assurance objectives.
• National regulations on qualifications should be followed.
28 March 2011 Department of Pharmaceutics 86

2. Personnel
• All personnel should receive proper training in relation to good storage practice, regulations, procedures and safety.
• All members of staff should be trained in, and observe high levels of, personal hygiene and sanitation.
• Personnel employed in storage areas should wear suitable protective or working garments appropriate for the activities they perform
28 March 2011 Department of Pharmaceutics 87

3. Premises and facilities
a. Storage areas
b. Storage conditions
c. Monitoring of storage conditions
28 March 2011 Department of Pharmaceutics 88

4. Storage requirements
a. Documentation: written instructions and records
b. Labeling and containers
c. Receipt of incoming materials and pharmaceutical products
d. Stock rotation and control
28 March 2011 Department of Pharmaceutics 89

5. Returned goods
• Returned goods, including recalled goods, should be handled in accordance with approved procedures and records should be maintained.
• All returned goods should be placed in quarantine and returned to saleable stock only after this has been approved by a nominated, responsible person following a satisfactory quality re-evaluation.
• Any stock reissued should be so identified and recorded in stock records. Pharmaceuticals returned from patients to the pharmacy should not be taken back as stock, but should be destroyed.
28 March 2011 Department of Pharmaceutics 90

6. Dispatch and transport
• Records for dispatch should be retained, stating at least:
— the date of dispatch;
— the customer’s name and address;
— the product description, e.g. name, dosage form and
strength (if appropriate), batch number and quantify;
— the transport and storage conditions.
• All records should be readily accessible and available on request.
28 March 2011 Department of Pharmaceutics 91

7. Product recall
• There should be a procedure to recall from the market, promptly and effectively, pharmaceutical products and materials known or suspected to be defective.
28 March 2011 Department of Pharmaceutics 92

GDP – (Good Distribution practice)
28 March 2011 Department of Pharmaceutics 93

GDP – (Good Distribution practice)
• GDP governs the proper distribution of medicinal products for human use and regulates the movement of products from the manufacturers’ premises (or other central point) to the end user (or other intermediate point).
94Department of Pharmaceutics28 March 2011

GDP – (Good Distribution practice)
1. Principle
2. Personnel
3. Documentation
4. Premises and equipment
5. Deliveries to customers
6. Returns
7. Self inspection
8. Provision of information to Member States in relation to wholesale activities
28 March 2011 Department of Pharmaceutics 95

1. Principle
• Policy ensures that products released for distribution are of the appropriate quality.
• In addition to this, the quality system should ensure that the right products are delivered to the right addressee within a satisfactory time period.
• A tracing system should enable any faulty product to be found and there should be an effective recall procedure.
28 March 2011 Department of Pharmaceutics 96

2. Personnel
• He should fulfil his responsibilities personally.
• Person should be appropriately qualified: although a degree in Pharmacy is desirable, the qualification requirements may be established by the Member State on whose territory the wholesaler is located.
28 March 2011 Department of Pharmaceutics 97

2. Personnel
• Key personnel involved in the warehousing of medicinal products should have the appropriate ability and experience to guarantee that the products or materials are properly stored and handled.
• Personnel should be trained in relation to the duties assigned to them and the training sessions recorded.
28 March 2011 Department of Pharmaceutics 98

3. Documentation
• All documentation should be made available on request of competent authorities
a. Orders
b. Procedures
c. Records28 March 2011 Department of Pharmaceutics 99

4. Premises and equipment
• Premises and equipment should be suitable and adequate to ensure proper conservation and distribution of medicinal products.
a. Receipt
b. Storage
28 March 2011 Department of Pharmaceutics 100

5. Deliveries to customers
• Deliveries should be made only to other authorised wholesalers or to persons authorised to supply medicinal products to the public in the Member State concerned.
• In case of emergency, wholesalers should be in a position to supply immediately the medicinal products that they regularly supply to the persons entitled to supply the products to the public.
28 March 2011 Department of Pharmaceutics 101

5. Deliveries to customers
• Medicinal products should be transported in such a way that :
a) Their identification is not lost;
b) They do not contaminate, and are not contaminated by, other products or materials;
c) Adequate precautions are taken against spillage, breakage or theft;
d) They are secure and not subjected to unacceptable degrees of heat, cold, light, moisture or other adverse influence, nor to attack by microorganisms or pests.
28 March 2011 Department of Pharmaceutics 102

6. Returns
a. Returns of non-defective medicinal products
b. Emergency plan and recalls
c. Counterfeit medicinal products
d. Special provisions concerning products classified as not for sale
28 March 2011 Department of Pharmaceutics 103

7. Self inspection
• Self-inspections should be conducted 9and recorded) in order to monitor the implementation of and compliance with this guideline.
28 March 2011 Department of Pharmaceutics 104

8. Provision of information to Member States in relation to wholesale activities
• Wholesalers wishing to distribute or distributing medicinal products in Member State(s).
• Where appropriate, the competent authorities of this (these) other Member State(s) will inform the wholesaler of any public service obligation imposed on wholesalers operating on their territory.
28 March 2011 Department of Pharmaceutics 105

Guidance documents deal with GDP• WHO Good Distribution Practice, Annex 5 to Technical Report
Series, No. 937, 2006
• Health Canada Guidelines for Temperature Control of Drug Products during Storage and Transportation, 2005
• Irish Medicines Board Guide to Control and Monitoring of Storage and Transportation Temperature Conditions for Medicinal Products and Active Substances, 2006
• USP chapter <1079> Good Storage and Shipping Practice
• EU Guidelines on Good Distribution Practice of Medicinal Products for Human Use (94/C 63/03)
106Department of Pharmaceutics28 March 2011

GRP – (Good Review Practice)
28 March 2011 Department of Pharmaceutics 107

GRP – (Good Review Practice)
• A good review practice (GRP) is a documented best practice within CDER that discusses any aspect related to the process, format, content, and/or management of a product review.
• GRPs are developed over time as superior practices based on CDER’s collective experience to provide consistency to the overall review process of new products.
• GRPs are developed to improve the quality of reviews and review management.
28 March 2011 Department of Pharmaceutics 108

GRP – (Good Review Practice)
• GRPs improve efficiency, clarity, and transparency of the review process and review management.
• GRPs are expected to be adopted by review staff as standard processes through supervisor mentoring, implementation teams, and formal training when necessary.
• Developing GRPs is an attempt to identify, collect, enhance, implement, and adopt may of these best practices as documented and standardized GRPs that can be shared among all review division
28 March 2011 Department of Pharmaceutics 109

− Quality — Consistent implementation of GRPs by review staff will enhance the quality of reviews, the review process, and the resultant regulatory action.
− Efficiency — GRPs will improve the efficiency of the review process through standardization.
− Clarity — GRPs support clarity throughout the review process, including critical review and decision activities that must be completed before a regulatory decision is made.
28 March 2011 Department of Pharmaceutics 110
GRPs Fundamental Values

GRPs Fundamental Values
− Transparency — Developing and documenting GRPs ensures that our review processes are readily available in one location via the Internet (through CDER’s Web site) to sponsors and the public.
− Consistency — By offering a consistent approach and only deviating from it when appropriate (after supervisory concurrence), GRPs help reviewers achieve consistency with their reviews and provide standard review processes across divisions and offices.
28 March 2011 Department of Pharmaceutics 111

cGxP
28 March 2011 Department of Pharmaceutics 112

cGxP
• A "c" or "C" is sometimes added to the front of the acroynm.
• The preceding "c" stands for "current."
• For example, cGMP is an acronym for "current Good Manufacturing Practices." cGMP is the most well known example of a GxP.
• The term GxP is only used in a casual manner, to refer in a general way to a collection of quality guidelines.
113Department of Pharmaceutics28 March 2011

cGxP
• What does cGxP stand for?
• Current Good X Practice (FDA compliance; X can mean: Manufacturing, Clinical, Laboratory, Storage, Distribution, Review Pharmaceutical)
114Department of Pharmaceutics28 March 2011

28 March 2011 Department of Pharmaceutics 115
Thank youCell No: 00919742431000
E-mail: [email protected]