haemophilia: diagnosis* - who3-4)_429... · haemophilia: carrier detection and prenatal diagnosis...
TRANSCRIPT

Haemophilia: strategies for carrier detection andprenatal diagnosis*I.R. Peake,1 D.P. Lillicrap,2 V. Boulyjenkov,3 E. Briet,4 V. Chan,5 E.K. Ginter,6E.M. Kraus,7 R. Ljung,8 P.M. Mannucci,9 K. Nicolaides,10 & E.G.D. Tuddenham1l
In 1977 WHO published in the Bulletin a Memorandum on Methods for the Detection of HaemophiliaCarriers. This was produced following a WHO/WFH (World Federation of Haemophilia) Meeting ofInvestigators in Geneva in November 1976, and has served as a valuable reference article on the gene-tics of haemophilia. The analyses discussed were based on phenotypic assessment, which, at that time,was the only procedure available.
The molecular biology revolution in genetics during the 1980s made enormous contributions to ourunderstanding of the molecular basis of the haemophilias and now permits precise carrier detection andprenatal diagnosis. WHO and WFH held a joint meeting on this subject in February 1992 in Geneva.This article is the result of these discussions.
Assessment of the problemThe carrier state in haemophiliaClinical and genetic considerations. Carriers ofhaemophilia usually inherit their abnormal factorVIII or factor IX gene from one of their parents.Since they have a second normal X-chromosomal
* Based on the report of a WHO/WFH meeting in Geneva,10-12 February 1992 (unpublished document No. WHO/HDP/WFH/92.4).1 Department of Medicine and Pharmacology, Royal HallamshireHospital, Sheffield, England.2 Department of Pathology, Richardson Laboratory, Queen'sUniversity, Kingston, Ontario, Canada.3 Hereditary Diseases Programme, World Health Organization.1211 Geneva 27, Switzerland. Requests for reprints should besent to this address.4 Haemostasis and Thrombosis Centre, University Hospital,Leiden, Netherlands.5 University of Hong Kong, Department of Medicine, QueenMary Hospital, Hong Kong.6 Institute of Clinical Genetics, National Research Centre ofMedical Genetics, Moscow, Russian Federation.7 New England Haemophilia Centre, The Medical Centre ofCentral Massachusetts Memorial, Worcester, MA, USA.8 Department of Paediatrics, Malmo General Hospital, Malmo,Sweden.9 A Bianchi Bonomi Haemophilia & Thrombosis Centre, Milan,Italy.10 Harris Birthright Research Centre for Fetal Medicine, King'sCollege Hospital School of Medicine & Dentistry, London,England." Clinical Research Centre, Harrow, Middlesex, England.
Reprint No. 5402
gene from the other parent the clotting factor level isaround 50% of normal, which is generally sufficientfor normal haemostasis.
Symptoms of bleeding do occur, however, incarriers if their clotting factor level is in the range ofmild haemophilia, below 40%. This may be due tohomozygosity, Turner's syndrome, other chromo-somal abnormalities, extreme lionization, or theco-inheritance of a variant von Willebrand factorallele (i.e., von Willebrand's disease Normandy).With the increasing success of patient associations,the chances of carriers marrying patients might beexpected to rise, but until now, homozygosity hasbeen distinctly rare, just as the unlikely coincidenceof Turner's syndrome and carriership. Occasionaltrue heterozygous carriers with low clotting factorlevels due to extreme lionization, however, areknown in all major haemophilia centres. Within theperspective of this article, this is relevant for invas-ive procedures for prenatal diagnosis and for themanagement of delivery.
Anxiety about the risk of haemophilia affectingtheir offspring is the reason why possible carriersseek advice. The first step in this genetic counsellingprocedure is to find out why the consultand thinksthat she might carry the gene. This may lead to anyof three conclusions: she is not a carrier, she is anobligatory carrier, she is a possible carrier.
Carriership is excluded if haemophilia occurs inthe paternal family without her father himself beingaffected. Carriership is obligatory if her father hashaemophilia or if she has maternal relatives withhaemophilia as well as an affected child. In thesesituations each newborn son has a 50% chance of
Bulletin of the World Health Organization, 71 (3/4): 429-458 (1993) © World Health Organization 1993 429

I.R. Peake et al.
being affected. If the consultand has more than oneson with haemophilia without other affected rela-tives, she may be either a true heterozygote, or amosaic (1-4). In the case of somatic or germlinemosaicism the recurrence risk of the disease in sub-sequent newborn sons depends on the proportion ofova carrying the abnormal gene, which is difficult toestablish. Carriership is possible if the consultandhas affected relatives on the maternal side and noaffected children, or if she has one affected son andno other affected relatives. In the last case, there arefour possibilities: the consultand may have inheritedthe gene through the silent maternal line, whichmakes her a true heterozygote; she may be a mosaic;the affected son may have received the abnormalgene from a mutation in the single ovum that heoriginated from; or, the affected son may himself bea somatic mosaic with a large proportion of factorVIII- or IX-producing cells carrying the mutation(5).
After this first step, the genetic counsellor pro-ceeds with rigorous pedigree analysis, clotting factorassays, and DNA studies in order to minimize uncer-tainty and set the stage for decision-making aboutprenatal diagnosis. These phases are describedbelow.
Psychological considerations. Every woman whoconsiders carrier testing for haemophilia, has a mul-titude of sociological and psychological influenceswhich might affect her perceptions of the personalimplications of possible carrier status. Someinfluences may be created by the ethnic and/or reli-gious background of the individual. A particularsociety's perception of a woman who carries thegene for haemophilia may certainly influencewhether or not a woman at risk chooses to betested. Decisions to undergo carrier testing mayalso be dependent upon the degree of anxiety abouthaemophilia and its complications, as well as theavailability and safety of treatment. Women wholive in countries where treatment for haemophilia isinadequate, may be very interested in carrier detec-tion and prenatal diagnosis, and therefore a moreagressive approach to testing may be successful.
Preconceived notions about the clinical aspectsof the disease will be formed as a result of the degreeof, and content of contact with male haemophilicrelatives. Women who have male relatives with moreclinically severe disease, with inhibitors, or AIDS,may be more inclined to seek out carrier testing. Thisis especially true if individuals have been able toclosely observe the effects of the disease on them-selves and their family over time. Women who arerelated to those with milder clinical disease, or whohave more distantly related haemophilic relatives,
may have different perceptions regarding carriertesting. In the case of mild clinical disease in thefamily, women may see no need for carrier testingbecause of perceptions that haemophilia will not havea large personal impact. Those with more distantlyrelated haemophilic relatives may be eager to pursuetesting because of a perceived low risk, but may beunprepared if positive results are obtained.
It is important that formal counselling be donebefore laboratory tests are even considered, in orderto resolve conflicts that may exist between awoman's desire to learn of her carrier status and theimplications of possible results. It should be empha-sized that the results of carrier testing may also bethat the woman is not a carrier of haemophilia. Theanxieties a woman might have regarding genetic test-ing may also be complicated by the type of testingavailable and the requirements of such, as well as thereliability of the tests involved. The possibility ofinconclusive test results, or results that are not highlyaccurate may deter some women from choosing to betested.
Many women who choose to undergo carriertesting may perceive themselves as either carriers ornon-carriers prior to actual laboratory testing. Thisperception may influence how an individual assimi-lates the results of her laboratory testing; it is there-fore an important issue to discuss during counselling.Those who assume that they are non-carriers will besupported by negative results, but may feel shockand surprise if given positive results. Those whoassume that they are carriers may be relieved whengiven negative results, but they may also feel guiltyfor "escaping" their family's genetic burden, andthey may have difficulty coping with their new statusif life decisions have been made based on theirassumptions of carrier status. Women who receivepositive results will need a great deal of support, asthey then must deal with the reality of their carrierstatus within the context of their family and theirsociety.
Prenatal diagnosis
Clinical and genetic considerations. In families atrisk of having a child with haemophilia, assessmentof carrier status and counselling regarding prenataldiagnosis should ideally be carried out beforeconception. Pregnant carriers requesting prenataldiagnosis should be counselled as to the avail-able options, including the techniques for fetaltissue sampling, their limitations, and potentialcomplications. If the fetus is affected, the optionsof (i) continuing with the pregnancy and either keep-ing or adopting their child, and (ii) terminating thepregnancy, are reviewed.
WHO Bulletin OMS. Vol 71 1993430

Haemophilia: carrier detection and prenatal diagnosis
Diagnostic centres. Prenatal diagnosis should beundertaken in centres with full genetic, haemotologi-cal and obstetric expertise.Diagnostic tests. In the early days of prenatal diagno-sis of the haemophilias (until the mid 1980s) thepolicy was to perform amniocentesis at 16 weeksfollowed by fetal blood sampling at 20 weeks forphenotypic diagnosis in male fetuses. Subsequently,with the application of recombinant DNA techniquesto the analysis of placental biopsy material, parentswere offered the advantage of first trimester diagno-sis, with the additional benefit that, in the presenceof a male fetus, only one invasive test was required.However, fetal blood sampling for phenotypicdiagnosis is still the preferred method in patientspresenting in the second trimester of pregnancy, andit is necessary for those patients who are notinformative for any of the available DNA probes,those requiring confirmation where normality is basedon a linked probe, and those who have sporadichaemophilia or lack key relatives. Second trimesterdiagnosis may also be the preferred option forthose patients wishing to avoid invasive testing forfemale fetuses because fetal sexing can now be per-formed by ultrasonography at 16-20 weeks.Termination of pregnancy. Traditionally, terminationof pregnancy in the first trimester was performedunder general anaesthesia by dilatation of the cervixand evacuation of the uterine contents. In the secondtrimester, termination involved induction of labourand delivery of the fetus. Recently with the morewidespread uptake of second trimester dilatation andevacuation, one of the potential advantages of firsttrimester diagnosis (less traumatic termination) maynot be valid.
Psychological consideration. Prenatal diagnosis ofthe haemophilias holds a multitude of psychologicalconsiderations for the women and her partner as wellas the wider family and the community as a whole.Parental anxieties arise from: (i) the risks of havingan affected child with lifelong morbidity, (ii) attend-ing a hospital and having a potentially painful invasi-ve test, (iii) miscarrying as a result of invasive test-ing, (iv) receiving an abnormal result, (v) making adecision on whether to continue or terminate anaffected pregnancy, and (vi) undergoing a termina-tion, with its potential short-term and long-termcomplications. The parents are also subjected tofurther, either real or perceived, pressures. The widerfamily, especially affected members, close friends,and even their doctors may hold strong viewsconcerning quality of life and attitudes to termina-tion. Prevailing, cultural, religious and moral valueswithin a society may impose additional stresses onthe parents.
Genetic diagnosisAssessment of carrier statusFamily data: risk assessment. A pedigree of thefamily has to be carefully drawn with accurate infor-mation on the males being affected with haemophiliaor not. If more than one haemophiliac exists or hasexisted in the family, the case is familial. If the hae-mophiliac is the only known case in the family it isconsidered as isolated. These two types of familieshave to be discussed separately.
Famiiial cases. Haemophilia A and B are X-linked,recessive disorders. The segregation probabilities arethus 50% for a carrier female to transmit the X-linkedgene to each child, male or female, while the haemo-philic male will have only normal sons and carrierdaughters.
Study of the pedigree alone will identify somefemales as obligate carriers. An obligate carrier isdefined as a woman who:
has a father who is a haemophiliac (with the rare
exception of him being a somatic mosaic);has more than one haemophilic son (identicaltwins excluded) or one haemophilic son and a
daughter who has given birth to a haemophilicson;
has a haemophilic son and a well documentedhaemophiliac on the maternal side of the family.Obligate carriers have a probability of 1.0 for
carriership and need no further investigation.Females in the pedigree who are not obligate
carriers are to be considered as possible carriers. If aconsultand is a possible carrier her probability ofcarriership calculated from pedigree data should bedone in two steps. In the first step only informationfrom the pedigree anterior to her is used. One goesback in the pedigree from the consultand to thenearest maternal relative who is a haemophiliac or
an obligate female carrier. For each step verticalor horizontal in the pedigree from this person, take0.5 and multiply these factors together to arrive atthe probability of the consultand being a carrier.
This is illustrated in the pedigree in Fig. 1.1:2 is an obligate carrier since she has given birth toa son with haemophilia and a daughter who has a sonwith haemophilia. II:4 is one step horizontal to thehaemophiliac (or one step vertical to the obligatecarrier) and thus has a probability of 0.5 for carrier-ship. III:2 is one step horizontal and one step verticalto the haemophiliac (or two steps vertical to the obli-gate carrier) and thus has a probability for carriershipof 0.5 x 0.5 = 0.25.
If the consultand has male descendants herprobability for carriership has to be modified by
WHO Bulletin OMS. Vol 71 1993 431

I.R. Peake et al.
Fig. 1. Example of a pedigree for calculating anteriorprobabilities of carriership.
1 2
1 12 3 14 5
2 3
1 2Iv D
taking these into consideration. If the consultand hashealthy male descendants her probability of being acarrier is diminished.
The probability for 111:2 being a carrier accord-ing to the anterior family history is 0.25. She hasgiven birth to two healthy sons. The next step is tocalculate the likelihood that I11:2 would have twonormal sons, separately for the two possible casesthat she is or is not a carrier. If she is a carrier thelikelihood that her 2 sons would be healthy is(0.5)2 = 0.25, since each son has a probability of 0.5of not receiving the abnormal gene. If she is not acarrier the likelihood that her two sons would benormal is 12 = 1. These two likelihoods expressed asodds for carriership gives (0.5)2:12, i.e., 0.25:1.
In order to modify the probability of 0.25obtained from the anterior pedigree by taking intoconsideration the odds obtained from the pedigreeof the descendants, the former probability must beexpressed as odds; 0.25:0.75 = 1:3. The odds in theanterior and descendant pedigree is multiplied toarrive at the final odds for carriership obtained frompedigree data; 0.25 x 1:1 x 3 = 0.25:3. The odds forcarriership can then be transformed again into aprobability according to the formula; odds a:bcorresponds to the probability according to theformula; P = a/(a+b); 0.25/(0.25 + 3) = 0.08.Expressed in words, the fact that 111:2 has given birthto two healthy sons reduces her probability of carrier-ship from 0.25 to 0.08.
These calculations are illustrated in Table 1 withthe general formula applying to a consultand havingn sons. With one normal son (n=1), the final proba-bility is reduced from 0.25 to 0.14; with two normalsons (n=2), it is reduced to 0.08, etc.
Isolated cases. An isolated case of haemophilia mayresult from transmission of the haemophilia genethrough asymptomatic females in whom the gene hasremained undetected; from a new mutation in themother, resulting in her being a carrier, or a newmutation in the haemophiliac (= true de novomutation). The existence of somatic mosaicism andgermline mosaicism has also to be taken into con-sideration (2, 3).
The true proportion of de novo mutations willdepend upon the mutation rate in males versusfemales (v/u). If it is higher in males, a high propor-tion of mothers of isolated cases will be carriers. If itis higher in females, many isolated haemophiliacswill be the result of true de novo mutations. The sexratio of mutation frequencies in haemophilia has notbeen definitively established. Most studies show ahigher mutation frequency in the male than in thefemale. In haemophilia A, v/u has been estimated as5.0 (6), 9.6 (7) and 3.1 (8), respectively, in threerecent studies. In haemophilia B it has been estima-ted as 11 (9). Even if these figures are cautiouslyinterpreted, most mothers of isolated haemophiliacsare carriers. For practical purposes one can approxi-mate the genetic probability to be 0.85 for carriershipin mothers of an isolated case.
Phenotypic assessment. Haemophilia A and B areassessed by phenotype on the basis of the following.
(1) Glossary of terms* Factor VIII:C (factor VIII coagulant activity)-the coagulant activity of factor VIII as assessedfrom the normalizing effect on the activated partialthromboplastin time (APTT) of plasma containingless than the 1% of the normal factor VIII:C concen-tration.
Table 1: Calculating the probabilities for carriershipProbability or odds
Information
Anterior to 111:2
Carrier- Noncarrier- Carrier:non-ship ship carrier
0.25 0.75Descendants
of 111:2Anterior and (0.5)nl/(0.5)n+3 3/(0.5)n+3
descendants
1:3(0.5)n:1
(0.5)n:3
WHO Bulletin OMS. Vol 71 1993432

Haemophilia: carrier detection and prenatal diagnosis
* Factor VIII:Ag (factor VIII antigen) - the factorVIII protein as assessed by immunoassays.o VWF:Ag (von Willebrand factor antigen) - thevon Willebrand factor protein as assessed by immu-noassays.o Factor IX:C (factor IX coagulant activity) - thecoagulant activity of factor IX as assessed from thenormalizing effect on the APTT of plasma contain-ing less than 1% of the normal factor IX:C concen-tration.* Factor IX:Ag (factor IX antigen) - the factor IXprotein as assessed by immunoassays.o Substrate plasma is plasma devoid of either factorVIII:C or IX:C and used in the coagulation assay.* Test plasma is the plasma sample taken from anindividual to be tested.o Local working standard plasma is a pool of plas-ma used as a "control" plasma; it should be calibra-ted in international units (IU).
(2) Laboratory standardsFactor VIII:C. All factor VIII:C estimations must beassayed by comparison with an international stan-dard and expressed as international units (IU) offactor VIII coagulant activity. One IU of VIII:Cis by definition the factor VIII coagulant activity inone millilitre dof "fresh normal human plasma". TheIU is defined by the "International Standard forBlood Coagulation Factor VIII Plasma Human" (atpresent, 90/550), which is available from the NationalInstitute for Biological Standards and Control,London, England.
In the local laboratory, pooled citrated plasmafrom at least 20 healthy donors, having an age andblood group distribution comparable to those of thelocal population from which test subjects are drawn,may be used as the working standard. Every newbatch of this local standard plasma has to be calibra-ted against the international standard plasma or an"intermediate" standard which has been calibratedagainst the international standard. In calibration, thestandard plasma should be tested three times intriplicate and a conversion factor should be calcu-lated for local U VIII:C/ml to IU VIII:C/ml. A newworking standard should be prepared every 2-3months to minimize inaccuracy due to deteriorationin storage. The standard must be stored at -70 'C.
Factor VIII.:Ag. The local working standardcalibrated against the international standard, asdescribed above, is to be used as standard.
VWF:Ag. The local working standard should beprepared according to the guidelines above and cali-brated against the international standard for VWF (at
present, 5th British Standard for Blood CoagulationFactors, plasma human 91/516).
Factor IX:C. The local working standard shouldbe prepared according to the guidelines above andcalibrated against the "International Standard forBlood Coagulation Factor IX Human Plasma" (atpresent, the 5th British Standard for Blood Coagula-tion Factors, plasma human 91/516). This standardmay also be used for Factor IX:Ag measurements.
(3) Sampling of bloodBoth carriers and controls should be in good healthat the time of sampling since inflammatory states,liver and other diseases, and certain drugs mayinfluence the concentrations of the coagulation fac-tors.
The syringes and tubes used should be of plasticor siliconized glass. Vacuum tubes may be used. Theanticoagulant should be 0.11 or 0.13 mol sodiumcitrate, 1 volume to 9 volumes of blood. The tubesshould be turned upside down immediately 2-3times after sampling and centrifugation for at least20 minutes (at 2000 g) should be performed as soonas possible. The plasma should not be haemolysed.If plasma is not analysed immediately it should befrozen at -70 °C and not stored for more than afew months.
The results of VIII:C and VWF determinationswill not be confounded if carriers are pregnant untilthe 22nd week of gestation or are taking oral contra-ceptives at the time of blood sampling (10-12). Theage (haemophilia A and B) and blood group (haemo-philia A) have to be considered in both carriers andcontrols (13, 14). The data on the effect of age onIX:C concentrations are ambiguous; no effect wasfound by Graham et al. (15), whereas Orstavik et al.(16) in a population of twins found a significantlyhigher value in the senior twins. Oestrogen-containing drugs, like oral contraceptives, result inhigher concentrations of both IX:C and IX:Ag (17).
(4) Coagulant assaysVIII:C should be measured by a one-stage clottingassay or chromogenic substrate method.
The one-stage method is based on the testsample's ability to correct the APPT in plasmawhich has a <1% VIII:C (18). The chromogenic sub-strate assay for measuring VIII:C has been shown tohave a correlation of 0.92-0.98 to one-stage clottingassays (19). In a comparison between differentVIII:C assays, the chromogenic substrate assay wasfound to have the highest precision (20).
VIII:Ag can be measured by various immunora-diometric (IRMA) or enzyme-linked immunosorbent(ELISA) assays (21). The local experience with the
WHO Bulletin OMS. Vol 71 1993 433

I.R. Peake et al.
different methods may favour one over the other, butthere is no universal advantage of using VIII:Aginstead of VIII:C or vice versa.
VWF:Ag can be measured quantitatively withtwo different methods: electroimmunoassay (EIA)(22) and IRMA or ELISA (23). EIAs are performedby the "rocket technique" (24); they are as satisfactorya method as IRMA or ELISA in the detection ofcarriers of haemophilia A and the experience atthe local laboratory should decide (25).
IX:C can be measured by one-stage or two-stageassays based on the same principles as VIII:C assays.The same precautions concerning sampling, workingstandard plasma, international standard plasma, andtest plasma are applicable to IX:C assays. A chromo-genic substrate method, which uses a factor Xa sub-strate, has been described based on the conversion offactor IX in plasma to IXa by addition of a semipurefactor XIa in the presence of Calcium (26).
IX:Ag can be measured by various immunologi-cal assays, the most reliable being IRMA and ELISA(27, 28). When these techniques were introduced itwas found that haemophilia B could be classifiedinto many subgroups according to the amount ofIX:Ag present. CRM+ (cross-reacting material posi-tive) had normal IX:Ag, CRMR had reducedamounts, and CRM- undetectable IX:Ag.
The average concentrations of IX:C or IX:Agare lower in haemophilia B- carriers than in non-carrier women. Both concentrations are influencedby the random inactivation of one of the X-chromo-somes (Lyon-phenomenon) (29). Haemophilia B+carriers also have lower concentrations of IX:C,though their IX:Ag may vary widely as a result ofthe Lyon-phenomenon (30). No consensus exists onthe most effective way of classifying carriers of dif-ferent types of haemophilia B. Measurement ofIX:Ag offers only limited predictive improvement,mainly in haemophilia B+ families according toKasper et al. (31) and Pechet et al. (32). Orstavik etal. (30), on the other hand, found that IX:Ag was ofdiscriminant value in both haemophilia B- and B+families. In another study the most efficient way ofclassifying haemophilia B- carriers was univariatediscrimination based on IX:Ag. For haemophilia B+carriers bivariate linear discriminant analysis usingboth IX:C and IX:Ag gave the best results (15). Inthis paper univariate linear discriminant analysis isadvocated for haemophilia B, using IX:C measure-ments (see below).
(5) Odds ratios based on laboratory dataOn average, carriers of haemophilia A or B haveabout 50% of the normal levels of factor VIII or IX.Due to considerable overlap between the levels incarriers and normal women it is usually not possible
to establish carrier status on these laboratory data inan unambiguous way. Therefore, the laboratory dataare used to calculate an odds ratio favouring carrier-ship: an odds ratio "X" means that the laboratoryfindings in the consultand are X-times more likely tobe found in a carrier than in a non-carrier. The sub-sequent use of Bayes' rule allows one to combinethis odds ratio with the probability of carriershipderived from the pedigree analysis and to obtain a"final" probability of carriership. In the 1977 WHOMemorandum (33) four ways were described to cal-culate odds from laboratory data. Currently, how-ever, the preferred method for haemophilia A isbivariate linear discriminant analysis based on factorVIII:C and von Willebrand factor antigen measure-ments accommodating the effects of age and ABOblood group (25). For haemophilia B, univariatelinear discriminant analysis is advocated using factorIX:C measurements and applying a correction for theuse of oral contraceptives (17, 34). Although theestimation of factor IX antigen levels may be advan-tageous in some cases (30), the routine application ofthis assay does not seem to be justified (31, 32, 34).
Both in haemophilia A and B, laboratory dataare obtained in reference groups of carriers and non-carriers using exactly the same methods, reagentsand standards as used for prospective consultands.Also the subjects used for reference purposes shouldbe as similar as possible in all respects to the consul-tands. This used to require the recruitment of about30 obligatory carriers and an equal number of non-related but very similar women. Since DNA techno-logy now allows one to definitely prove or disprovecarriership one may use the laboratory data on pre-viously diagnosed consultands for reference pur-poses. At all times, however, one should be awarethat age, blood type, severity of haemophilia, preg-nancy, use of oral contraceptives, and probablyother factors may exert an influence on the outcomeof the laboratory tests. Consequently it is important tonote whether the reference subjects and prospec-tive consultands are similar in these respects. Further-more, the laboratory data usually need to be trans-formed, such that the distributions of the data forthe reference groups are normal or at least non-skewed.
In Tables 2 and 3 we have provided one approachfor haemophilia A using the universal discriminant(25), which obviates the need to study a referencegroup of carriers, and one approach for haemophiliaB, applicable to both CRM-positive and CRM-nega-tive forms (17). Considering the primary position ofDNA analysis in carrier diagnosis we feel that thesestraightforward approaches should be sufficient inmost cases. The calculations can be easily carried outin a spreadsheet type computer program.
WHO Bulletin OMS. Vol 71 1993434

Haemophilia: carrier detection and prenatal diagnosis
Table 2: Haemophilia A, bivariate universal discriminantanalysis. This method requires factor VlIl:C and von Wille-brand factor antigen reference data on non-carriers only.Data are transformed using the natural logarithm and theeffects of age and ABO blood type are accommodated (25)
Enter for consultand:
a = age in years.
p = ABO-bloodtype: 0 for 0, 1 for non-0.
y= VWF:Ag in lU/ml.
8 = VIIL:C in lU/ml.
x = Genetic probability of carriership (fraction of 1).
Enter for normal reference group:
Rx= mean of Ln transformed VWF:Ag levels in lU/ml.
gy= mean of Ln transformed VlIl:C levels in lU/ml.
Compute for consultand:
x = ln(y) - gx
y= In(6) -lyCompute the coefficients for the modified discriminants:
a = -0.0955 - 0.0156a + 0.000196a2 + 0.0298f
b = 0.649 - 0.00184a + 0.0000314a2 + 0.1173
Compute the predicted means of the discriminants forcarriers and normals:
c = -0.391 - 0.00571a + 0.0001a2 - 0.0648,B
d = -0.0347 - 0.00171 a + 0.0000473a2 + 0.0754,B
and compute the odds ratio:
e = ax + by
f= 4.28 (e - c)
g= 7.97 (e- d)h = 0.623 + 0.5 (f + g) (f- g)
LR = exp (-h), the odds ratio favouring carriershipa
Compute the final probability of carriership:
Pc = nLR / (tLR + 1 - t).a Note: exp (-h) denotes 'e', the base of natural logarithms,raised to the power of -h.
(6) Combining pedigree and laboratory dataThe pedigree probability of carriership (P) has to betransformed to the corresponding odds (a:b) accor-ding to the formula a:b = P:(l-P). The probabilityP = aI(a + b). In familial haemophilia the anteriorpedigree itself supplies a probability of the con-sultand being a carrier. If descendants exist, one canmodify the anterior probability with the odds of beinga carrier derived from the descendant pedigree. Inthis method, the anterior probability is first trans-formed to odds before combining the two (Table 4).
Table 3: Haemophilia B, univariate linear discriminantanalysis. Factor IX:C levels are required for both the car-rier and non-carrier reference groups assuming that thereference subjects do not use oral contraceptives. The fac-tor IX levels of the consultand need to be corrected by afactor of 0.75 if she does. In this example the data aretransformed by the square root to obtain symmetrical distri-butions
Enter for consultand:
a = Factor IX:C in lU/mi.
0 = Oral contraceptive use: 0 for no, 1 for yes.
X = Genetic probability of carriership (fraction of 1).
Enter for the carrier and non-carrier reference groups:
R,= mean of square-root-transformed factor IX:C levels ofthe carriers in lU/mi.
sc= standard deviation of square-root-transformed factorIX:C levels of the carriers in lU/mI.
-,= mean of square-root-transformed factor IX:C levels ofthe non-carriers in lU/ml.
an = standard deviation of square-root-transformed factorIX:C levels of the non-carriers in lU/ml.
Compute transformed and corrected factor IX:C forconsultand
a = [(1 - 0.250)a]112Compute the odds ratio:
b = 1/(2a 2)c = 1/(2ay 2)d- (a-p)2e- (a-n4)2f= bd- ce
g= exp (f)a
LR = ,nl (goc), the odds ratio favouring carriership
Compute the final probability of carriership:
Pc = tLRI(tLR + 1 - p).a Note: exp (f) denotes "e', the base of natural logarithms,raised to the power of f.
The odds of being a carrier from the laboratorydata, as calculated above, is multiplied with the pedi-gree odds to arrive at a combined odds. This is then
Table 4: Combining pedigree and laboratory data
Odds from pedigree 1: 3Odds from the laboratory data 10: 1Combinedodds 1 x10 :3x1 = 10:3Final probability 10/(10 + 3) = 0.77
WHO Bulletin OMS. Vol 71 1993 435

I.R. Peake et al.
again transformed into a probability by the formulaP = a/(a + b). The example in Table 4 illustrates thecalculations.
Genotypic assessment. Haemophilia A and B canbe assessed by genetic linkage using intragenic poly-morphism analysis.
(1) Genes encoding factor VIII andfactor IXThe factor VIII gene is situated at the telomeric endof the long arm of the X chromosome at band Xq28.The gene encompasses 186 kbp of genomic DNA(approximately 0.1% of the DNA sequence on the Xchromosome) and comprises 26 exons ranging insize from 69 bp to 3.1 kbp (35). The factor IX geneis situated at band Xq27, approximately 40 mega-bases centromeric of the factor VIII locus on the Xchromosome. The gene is approximately 33.5 kbplong, comprises 8 exons and encodes an mRNA of1.4 kbp (36).
(2) Genetic linkageThe mutations that result in haemophilia A and Bare heterogeneous and in most instances involvechanges of single nucleotides (37, 38). These twofactors in two large genes have made the directdetection of haemophilic mutations a difficult chal-lenge. As a result, many laboratories continue toassess the genetic status of potential carriers ofhaemophilic mutations through the use of indirectgenetic markers of the factor VIII and factor IXgenes. These genetic polymorphisms representnatural variations of the genome sequence whichoccur in the general population and which can beused as convenient landmarks to track mutant genesthrough families (Fig. 2 and 3).
When the polymorphisms occur within the geneof interest (intragenic polymorphisms), the like-lihood of the polymorphic marker becoming un-linked from the mutation through genetic recombina-tion is related to the size of the gene. To date, thereare no reports of intragenic recombination events ineither haemophilia A or B and thus one can assumethat genetic diagnoses based on the analysis of anintragenic polymorphism in these conditions isextremely accurate.
(3) Types of sequence variation within the factorVIII and factor IX genesIn order to be able to track individual copies of thefactor VIII and factor IX genes, it is essential to haveavailable polymorphic sequences which will differ-entiate between the two gene copies present infemales. The presence of heterozygosity or informa-tiveness for a polymorphism is a prerequisite whichmust be satisfied if genotypic studies are to be effec-
Fig. 2. The use of two polymorphisms in segregationanalysis of a haemophilia A family. The two markersshown are the multi-allelic intron 13 CA repeat and theextragenic DXS15 polymorphism which is located approxi-mately 5 recombination units distant from the factor VIIIgene. The haemophilia A mutation is segregating with theintron 13 "6" allele in this family. With this information inmind, the potential carrier female 111.3 is identified as a non-carrier. The coincident analysis of the DXS15 markershows that a recombination event has occurred betweenthis locus and the factor Vil gene in the unaffected male111.2. In this individual, analysis with this extragenic markeralone would have incorrectly predicted that he had inheritedthe haemophilia A mutation.
1
5
1
1II
2
6 - 2 F.VIII CA rpt
2 -1 DXS1 5
4
52
2 6 6-51 2 2-1
1 2 3
6 5 5-5
Ill
2 2 1 - 2
tive. In addition, as alluded to above, the polymor-phic sequence should either be within the genewhich the disease is segragating or close enough toensure that the possibility for genetic recombinationbetween the polymorphism and the disease mutationis minimal. The final point to re-emphasize is thatalthough some polymorphisms may be in allelicassociation (linkage disequilibrium) with particularmutations, the polymorphisms themselves representphenotypically neutral sequence variation that isfound in the general population.
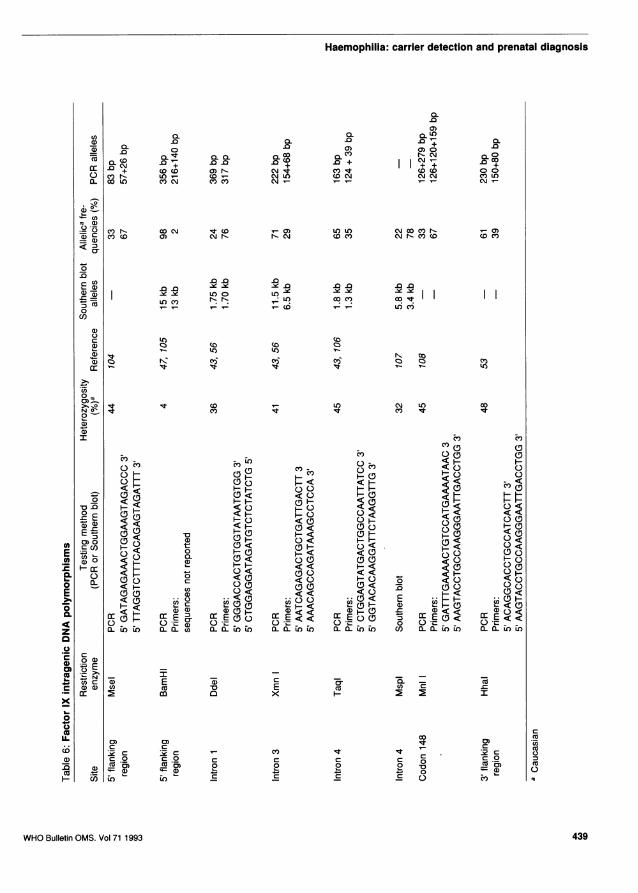
Two types of polymorphic sequence exist withinthe haemophilia genes (Tables 5 and 6). The mostfrequent and simple examples are the bi-allelicpolymorphisms resulting from single nucleotide sub-stitutions which either create or abolish restric-
WHO Bulletin OMS. Vol 71 1993436
2 3
Haemophilia: carrier detection and prenatal diagnosis
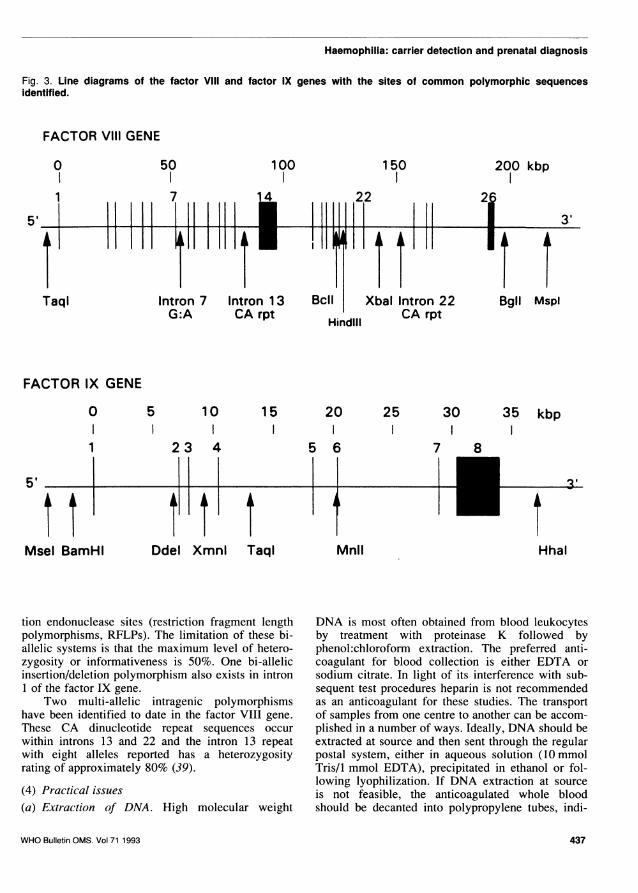
Fig. 3. Line diagrams of the factor Vlill and factor IX genes with the sites of common polymorphic sequencesidentified.
FACTOR Vil GENE
B
150
22
cil Xbal IntronfPA Pr%
Hindlil
FACTOR IX GENE
0
1
5,-
Msel BamHI
5 10
23 4
11 1
15
Ddel XmnI Taql
20 25 30
7 8
Mnil
35 kbp
t~3 I
Hhal
tion endonuclease sites (restriction fragment lengthpolymorphisms, RFLPs). The limitation of these bi-allelic systems is that the maximum level of hetero-zygosity or informativeness is 50%. One bi-allelicinsertion/deletion polymorphism also exists in intron1 of the factor IX gene.
Two multi-allelic intragenic polymorphismshave been identified to date in the factor VIII gene.These CA dinucleotide repeat sequences occurwithin introns 13 and 22 and the intron 13 repeatwith eight alleles reported has a heterozygosityrating of approximately 80% (39).
(4) Practical issues(a) Extraction of DNA. High molecular weight
DNA is most often obtained from blood leukocytesby treatment with proteinase K followed byphenol:chloroform extraction. The preferred anti-coagulant for blood collection is either EDTA orsodium citrate. In light of its interference with sub-sequent test procedures heparin is not recommendedas an anticoagulant for these studies. The transportof samples from one centre to another can be accom-
plished in a number of ways. Ideally, DNA should beextracted at source and then sent through the regularpostal system, either in aqueous solution (10 mmolTris/l mmol EDTA), precipitated in ethanol or fol-lowing lyophilization. If DNA extraction at sourceis not feasible, the anticoagulated whole bloodshould be decanted into polypropylene tubes, indi-
WHO Bulletin OMS. Vol 71 1993
50 1000
11
Taql
I iii
Intron 7G:A
200 kbp
2 M
Bgll MsplIntron 1 3CA rpt
22t, rpL
3'
11I I
437
7

I.R. Peake et al.
CO)
a)~~~~~~~~C0) M~~~~~)0.
A2 m co r- .0~~~~~~~~~~~~~~~.
~~~~~~~~~~~~~~~~~~~CUIt0coCaco
a) .0) CM Ir- .0
CO
It + + .CVcc< N~~~~~~~+ CZ)0) +<
~~~~II 0< 0 ~~~~~~~~~~~~~~~~~~~~*0)~~C Ic(0(0 (0(coi
CM 00 CD~~~~~~~~~~~~~~~~~~~~~~~~~~~~'0) 0)CDcC
0~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~.m .0 C0(lU)-
aa) be b C-e_l )m
oi'~~~~~~~~~~~~~~ I I~~~~~~~~~~~C ~c6 i CMj~CMCj10
a)
0~~~~~~~~~~~C)0
0) 00 C\ cj0
U)~~~~~~~~~~~0~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
0 C 0 C DCC < <CD
C 0 0<~~~~~~0
<0 <0) 0 < < < <00 0
0 < 0 0 ( co~000CC 0 0 1:1: 00 00 0)c
<E < < < o' HO H
0 0 co < CML .~
*P0- . < <0 <<0 CU c
a)'~~~~~~'~~~"~~~PS~~~~ ~~~'(iI- a)PC(D" *coma.~~~~~~~~~~~''-'~~~~~~. 0 oL '-0 'o( o _
a)~~~~~~~~~~~~~~~~~~~~~~~~~( ) ( )<ICLca)0C 0a
0a)a)
a) CD cCD
0)C
CcisC 0N(JC%a) C c C CCU)
CIS 'a~) 0 0 0 0 coCU 2 2 20 0 0F- CDir) C Cc C C
WHO Bulletin OMS. Vol 71 1993
Haemophilia: carrier detection and prenatal diagnosis
CY)Ul) 0 0 0LL)a) 0 ccC
C-C oCoC -cn r-.cm~a~~~~ n; ~~~~~~~~ + I~~.+C
oCw N O)P- cmI'l(0t(oD000a. co Lo cncm cn m CM ~ ~ ~ ~ ~ \JNCM)~
a)-
c)n - co N ~ (-) LOL() cm 00c r- '-0(CC c 0) N-Nc Wc cm rN -( )0 (0
0~
CU)
a1)C Ct)~~L (0l l~ U RCi(q
U)
00
0)
(0 CON Ct) (0~~~~~~~C))a)~~~~~~~~~~~~~~~~~~~~~~~~~~~~~C
a)~~~~~~~~~~~EC")~~~~~~~~~~~~~~~~~~~~~~~~C
CD0 H<(Do :~~~~< ~ ~ <
<~~~~~~~~~~CD 0~~~~~~~~~~<0 <C'CDE CD CD 0<
< L)~~~C<D
0~~~~~~CD C0C< o DD 0H
~HE < H«HCD 0Ca)0.0 < 0< D< <<<- 0
EL CD C CD <0 HO 0 <0 <O <H C CDC(D C< ~ 0 0E CDD > < < <0 C< D
< <~~_ C << E0 C:o 0C <
~~.U)Ca) E ~~~~~~~~CL -L 0 .-
0 (
co C No C
0~~~~~~~~~~~~~~~~~~~~~~~~~~~~~0CiC
aD C0 CO C C CCZCO
ala~) a)2 85
(DCa 0 La)H Lo En C C -v
WHO Bulletin OMS. Vol 71 1993 439

I.R. Peake et al.
vidually packaged in plastic or polystyrene contain-ers, and sent on dry ice via courier service to the test-ing laboratory. Whole blood samples can either bestored at -70 °C without additional manipulations orleukocyte pellets can be prepared prior to storage.In most instances using standard extraction proto-cols, between 200 and 500 ,ug of DNA will be ob-tained from a 10 ml blood sample. Abbreviated DNAextraction protocols have been proposed in the prep-aration of material for the polymerase chain reactionand these methods will result in material of adequatequality in most instances.
(b) Analysis of polymorphic sequences. The twomolecular genetic techniques used to identify DNApolymorphisms are those of Southern blotting (40)and the polymerase chain reaction (PCR) (41). Theformer method has been in use for more than a decadeand involves a relatively labour-intensive schedulecomprising the capillary transfer of endonuclease-digested DNA fragments from an agarose gel to amembrane support and the subsequent probing of themembrane with a radiolabelled DNA fragment repre-senting the sequence of interest. Studies utilizingSouthern analysis require a minimum of 5 gg of highmolecular weight DNA for testing and take at leastfive to seven days to produce results. The polymer-ase chain reaction has now replaced Southern blot-ting in many instances. This technique utilizes syn-thetic oligonucleotide primers to select for speci-fic sequences of interest which are then amplified invitro to produce a targeted product which is presentin more than a millionfold its original concentration.The power of this method has resulted in severaladvantages including the ability to work with verysmall starting quantities of DNA (less than 1 jg),increased simplicity and biosafety (non-radioisotopicmethods), and the completion of tests within 48hours.
Most of the polymorphisms in the factor VIIIand factor IX genes can now be studied through theanalysis of DNA which has been amplified in vitroby PCR (42, 43). Assessment of the XbaI polymor-phic genotype by PCR is complicated by the co-amplification of homologous sequences adjacent to,but outside of, the factor VIII gene. This complexitycan be resolved by Southern analysis (44).
Following DNA extraction, the PCR amplifica-tion of these various sequences takes approximatelythree hours, after which the amplified products aredigested with the appropriate restriction enzyme andanalysed by polyacrylamide gel electrophoresis. Aswith all PCR studies, the complication of samplecontamination with extraneous DNA must be guar-ded against. Most laboratories perform their PCRstudies in a designated clean area and use a dedica-
ted set of equipment (including positive displace-ment pipettes) and supplies to minimize the risk ofDNA carry-over. In addition, the inclusion of a "noDNA template" blank tube in each experiment pro-vides a further safeguard against this problem. Final-ly, although the endonuclease digestion of most PCRproducts will proceed to completion uneventfully,the inclusion of previously genotyped PCR amplifiedsamples in each test run ensures that all componentsof the endonuclease reaction have been added andare functional. In addition, the inclusion of an inva-riant endonuclease site within the amplified fragmentfurther assists in evaluating the digestion process.(5) Testing strategy(a) Haemophilia A. The strategy for polymorphismanalysis in any particular family must take intoaccount factors including the site of the polymor-phism, the heterozygosity rating of the marker, andthe ethnic origin of the family (see below).
The recently described intron 13 CA repeatpolymorphism appears to be informative in approxi-mately 80% of females and thus represents the logi-cal starting point for analysis of factor VIII polymor-phisms. This sequence can be amplified in a"multiplex" PCR with the other, Intron 22, CArepeat and the two sequences analysed simultaneous-ly. The fact that these sequences require the use ofradiolabelled amplification primer and electrophore-tic separation of the products on a DNA sequencinggel may, however, result in some laboratories reser-ving the analysis of this marker for those cases inwhich the two other frequent BclI and XbaI poly-morphisms are uninformative. Fortunately, these fourmarkers are not in allelic association (linkage dis-equilibrium) and more than 95% of females will beinformative for one or more of these polymorphisms.The remaining families should be tested with theBglI marker and with the intron 7 polymorphismwhich will be informative in approximately 10% offemales who are homozygous for the absence of theBclI polymorphic site.(b) Haemophilia B. The combined use of the TaqI,XmnI, DdeI, HhaI, MnlI and MseI polymorphismswill provide informative results in approximately90% of females in haemophilia B genotype testing.Therefore, there will still be about 10% of familieswith haemophilia B in whom intragenic polymor-phism analysis is uninformative and where eitherlinked extragenic markers will have to be investiga-ted or direct mutation detection will be necessary.
(6) Advantages of genotype assignment with poly-morphism testingWhere a prior family history of haemophilia exists,and an intragenic polymorphism is informative,
WHO Bulletin OMS. Vol 71 1993440

Haemophilia: carrier detection and prenatal diagnosis
diagnostic results with an error rate of less than 1%are attainable. The methods are straightforward,rapid and inexpensive to perform. Thus, in manyfamilies requesting genetic diagnosis of haemophilia,the use of intragenic polymorphism analysis repre-sents the diagnostic strategy of choice.
(7) Limitations to the use ofpolymorphism testing inhaemophiliaAlthough in many instances, the use of an informa-tive intragenic polymorphism will provide highlyaccurate genetic diagnosis of haemophilia, there are,nonetheless, some limitations to this diagnostic stra-tegy (45). All of these drawbacks relate to the factthat the haemophilic mutation itself is not identifiedby these methods.
(8) Requirement forfamily samplingPolymorphism linkage analysis requires the partici-pation of a minimum number of key individuals froma haemophilic family. At least one affected maleshould be available for testing to identify the poly-morphic allele which is associated with the mutationin the family requesting diagnosis. With the recentcatastrophe of HIV infection in the haemophiliapopulation, this initial requirement may be compro-mised by early deaths of haemophilic males. How-ever, even in the instances where all affected malesin the family are deceased, the recovery of DNAfrom stored pathological samples for analysis by PCRstill makes polymorphism testing feasible.
In addition to getting all appropriate familymem-bers to agree to participate in genetic testing, itis also vital that all stated family relationships (parti-cularly paternity) are correct.
(9) Families with an isolated affected haemophiliacSporadic cases of haemophilia comprise 30-50% ofthe total haemophilic population. In these families,because polymorphism analysis does not identify thehaemophilic mutation directly, it is not possible toascertain at which level of the pedigree the mutationarose. In fact, past studies have indicated thatapproximately 85% of mothers of isolated haemo-philiacs are carriers but in individual diagnosticcases, unless the coagulation studies of the motherare strongly suggestive of her being a carrier, it isprobably unwise to attempt the diagnosis of a hae-mophilic allele by polymorphism testing. Therefore,in these families, one is often left with the option ofusing polymorphism studies to exclude transmissionof the haemophilic mutation.
(10) Requirement for heterozygosity and possibilityof genetic recombinationAs detailed above, for polymorphism linkage studies
to yield useful information, one must be able to dif-ferentiate between the two X chromosomes in keyfemales through the presence of polymorphic hetero-zygosity. In the study of haemophilia A with intra-genic markers, this requirement is now achieved inmore than 95% of families due in large part to theintron 13 CA repeat polymorphism. In the analysisof the factor IX gene where a similarly multi-allelicsequence does not exist, some 10% of families willstill require diagnostic studies with linked extragenicmarkers to achieve informative results. In these latterstudies, the possibility of genetic recombination be-tween the polymorphic site and the haemophilicmutation adds an additional uncertainty to the preci-sion of genetic diagnosis.
Ethnic variation in frequency of polymorphisms,linked polymorphisms and linkage disequilibrium.Factor IX polymorphisms. In Caucasians, the use ofsix intragenic RFLP sites allows linkage of the genein approximately 80% of families (46). These sitesare Taq I, Xmn I, Dde I, Msp I, BamH I as well asthe residue 148 (Thr/Ala) base change (Mnl I).However, there is marked ethnic variation in theincidence of heterozygosity for these sites. In Blacks,both the 5' BamH I and the intron 3 BamH I sites areuseful, with heterozygosity rates of 0.46 and 0.22respectively (47). In comparison, informativity atthese sites is rare in the Caucasian populations (47,48). The incidence of heterozygosity for RFLP sitesof different ethnic groups are given in Table 7.Orientals, such as Chinese (49), Japanese (50), Phili-pinos (51) as well as Malays (52), were found tohave a low incidence or absence of the RFLPs listedabove. The only RFLPs informative within thesepopulations are those detected by HhaI and therecently described MseI RFLP (104), which has beenshown to be informative in Thai populations.
Linked intergenic polymorphisms. An Sst I RFLP atlocus DXS99 can be detected by probe pX58dIHc(54). This polymorphism gives rise to two alleles of5.9 kb and 8.8 kb respectively, with a frequency of0.43 for the former. The polymorphic locus DXS99is mapped to Xq26-q27 and tightly linked to the fac-tor IX gene. The precise genetic distance betweenthis locus and the factor IX gene has yet to be deter-mined, but thus far, no recombination has beendetected in 39 informative meiosis, giving a lodscore of 9.79 at E = 0.0, with 95% confidence limitof e = 0-0.06. Since this marker and the FIX loci donot appear to be in linkage disequilibrium (54), theanalysis of this Sst I RFLP at DXS99 in conjunctionwith the intragenic sites in the factor IX gene shouldincrease the diagnostic efficiency to more than 90%of females at risk.
WHO Bulletin OMS. Vol 71 1993 441

I.R. Peake et al.
CY)CY)
0 CDI o
II 0O0
r-a _ rcm a0D N N- 0)
') CO C ) D 0CO - 0C
CD
'tJ CD
co cm olo o o
~~~0M0)0o 166no6a
0
06
0
0
00
<D CMC') C')0 0
CN N CN0 0 0
L_ONN_P-co'-'-CoL
0.CO
C')
6
0)6
CC)CM
NC
IEcoCO
O.06;
Co
0cD
6
00
a)
0a)
C'0
0 C' CD0 0 0
CD)0)
'F,
o0
_ ~CO
C') 'r- CD0 0) 00 0o
0
C')C')
Cuco
7E
coCO
0C')
Ci
0
I
C')N
CD
coI'V)
CD
-t0+
CC6D
0
CD
0
0a-
C)
a00C0I a)0
cnC.)
C
C')0
-CI ..
CD
coa
0
Cu0.S
-rco C6
C; n0
SD
.oUL
WHO Bulletin OMS. Vol 71 1993
I-
I I
CM00
C)0) 06 6
C')o 6
0f) 0- 0)o o0a) Co O~
N No o6 6
CC) lt co0) 0 0)666
CO
6ov OD_ C0
coN
j 0H =a.
0 0)
0
0.co 0
Co
0D-
to
0.c>j., 0a)
co 0D
<:
.c6
0D
E
00>4
CD.o?
co 7 cr
2 =a)
(D
m I N
0.c
6 Z
D0.
co
Cu 0.
o <"-
75
0 (E-UC0.00
a.
0Ooo
)0
)0
N
CM0_ z
c;(
0
0 0
0) CCM
0) 0
0w 0
0>
0 00o
co(D't66Co o
CD lq-t CN0 0
CD CD
0)0 C 0
0 0 0)CM
0 0N
0o 0
0)0D0 0)-0 0
0
0)
0
._
0cCL
-
0
E
C
0
0
0CD
0
0
._
0
0
0?0N
0
E
._
EL0..
0
06
06
06
K,
0 0
0C q L( CO0)0 CD C'
-+ +
uLC)co C')CM cmN N
0
-
0
lCCr-
C\j
0)
IC
mEcocoIn)
442
-c ) 'aCu E 'a a)HX a 2
/

Haemophilia: carrier detection and prenatal diagnosis
Linkage disequilibrium offactor IX polymorphisms.In Caucasians, the Taq I, Xmn I, Msp I and Mnl IRFLPs show marked linkage disequilibrium (allelicassociation), thus the use of all four sites would onlyincrease the diagnostic efficiency to 55% as opposedto 45% when using the Taq I site alone (52, 55, 56).However, Dde I and 3' Hha I polymorphisms showedmuch less disequilibrium and the combined use ofthese two sites and the Taq I site increases the hetero-zygosity rate to almost 76% (53, 55).
In the American Blacks, the linkage disequili-brium between Taq I and Msp I is less marked.While the intragenic BamH 1+ alleles and Msp I-alleles showed disequilibrium, the 5' BamH I, Dde Iand Xmn I sites appeared to be in equilibrium. Thecombined use of these latter sites showed an obser-ved frequency of heterozygosity of 87% for Blackfemales (versus c. 60% in Caucasians) (48, 57).
No linkage disequilibrium was observed be-tween the Hha I locus at the 3' end of the factor IXgene and the other intragenic loci (53); thus it is like-ly that this polymorphic marker will be extremelyuseful for factor IX carrier testing. This has certainlybeen the case for Orientals (49), who lack heterozy-gosity for the common intragenic RFLP sites. TheMseI polymorphism also shows minimal linkagedisequilibrium with the other polymorphisms withinthe factor IX gene.
Factor VIII polymorphisms. As with factor IX, theincidence of factor VIII RFLPs differ significantly invarious racial groups (57, 58); thus before a prenataldiagnosis programme can be instituted in a particularregion, the RFLPs for that population should bestudied, to decide the most suitable sites for use.
Table 8 summarizes the incidence of Bcl I,Xba I, Bgl I, Hind III and MspI polymorphisms inthe factor VIII gene in various ethnic groups. Thepositive incidence of Bgl I polymorphism is higherin Chinese than in other races. Of particular interestis that in American Blacks, the rates of the (+) sitefor Bcl I and Hind III polymorphisms are the reverseof what is observed in other ethnic groups (57).Table 8 also shows the female heterozygosity rate.The Bcl I dimorphism is more informative in Medi-terraneans, Indians and Japanese (42-47%) com-pared to, Caucasian, American Blacks, Chinese andMalays (31-39%). The Hind III and Xba I RFLPsshowed similar heterozygosity -in the various groupstested, whilst the Bgl I RFLP is most useful in Ame-rican Blacks (38%), but useless in Chinese (0%).
Linked intergenic polymorphisms. The physical map-ping of the q28 region of the X chromosome hasrevealed that the loci DXS52 and DXS15 are 1-2Mb centromeric to the factor VIII gene. The highly
o
0
0
c
0
0._
C
0
E
0
0
c
0
U._
0
oF
CD
._
E.5
0D0
08
0*
IL
0)aB
>,ocD
ii
co0)00.s.eDr
0)
.c).
.< :
mI ^
E :
0)
< N=
0).
0D)
st I N.
co
a90
0s
cCIOCAU)o:3ct
>10:=L-U)a) 04-.0) .I>1
0.
00)
(1) a
C.)a:_ CoO a)
>Co
CD N
~l()
o
F
O' LO
0
C;
os *o
0 60
oo o
d -<o
8 0
4 .co0 0
'-) N 0
, CD cmN
66 6C r6a a .i .
C')C')d
U)C')6
lw- 0) N- C')N N N N0 0 0 0
coC')0
C't)c0
6 o
co
o oo o
0)10
N _ o 6. 06 6 6 6 6 6 CD -
"1-
0
o oC') No o
0)
N C')NM N
0,0,0
o w
6 6iI'- 1- C0)-l;t U. 9 lcy)a a a a
c t ur
N 0 N aC .C\ * +CM C (
I
U) U) 0 N_ 0 0 atI
- _ _
C U) C 0 0,NiL N + 4 C
D -x0 0) U
WHO Bulletin OMS. Vol 71 1993
LO)
0, 07) LO CY) 0C') CMN lq 0co 6 c6 6 6
N
CM)am(AQ
°. o3
c
Oo It
443

I.R. Peake et al.
polymorphic St 14 probe detects a polymorphismwithin the cluster MN12, cpX67, and DX13 (59).The two extragenic RFLPs, Bgl II/DX13 (60) andTaq VISt 14 (61), are both closely linked to the hae-mophilia A locus. The Bgl II RFLP detected by theDX13 probe shows two alleles, 5.8 kb (allele 1) and2.8 kb (allele 2) respectively. The heterozygosityrating for this polymorphism varies between ethnicgroups from 0.30 (Japanese) to 0.5 (Caucasians), andthe frequency of recombination with factor VIII isapproximately 4.5%.
The Taq I RFLP detectable with the St 14.1probe gives two independent systems of alleles (61).System I has eight alleles (1 to 8) ranging from 6.6to 3.4 kb in length, and system II has two alleles,"A" (5.5 kb) and "B" (4.1 and 1.4 kb, respectively).
Caution should be exercised when using inter-genic linked probes for diagnosis because of the pos-sibility of meiotic recombination (62, 63). With theSt 14.1 probe, reports from world literature sugges-ted a genetic disease of 3 cM, and this should betaken into account during genetic counselling. Theuse of the DX 13 probe would be even more prone toerror, as the cross-over rate with the factor VIII geneis thought to be about 4.5% (64).
Linkage disequilibrium of factor VIII polymor-phisms. There is a strong linkage disequilibrium be-tween the intron 18 Bcl I, intron 19 Hind III andintron 25 Bgl I sites (57, 63). Thus little additionalinformation will be gained in using more than one ofthese three sites. In contrast, the Xba I site in intron22 is often informative in females who are homo-zygous for the Bcl I site (65). Even though theBcl I and Xba I sites are not in complete linkageequilibrium, with a disequilibrium coefficient of0.0722-0.1627 in various ethnic groups reported, thecombined use of these two sites would significantlyincrease the informativeness of 79% in Japanese,64-69% in Caucasians, and 52% in Chinese femalesrespectively (58, 65, 66). In certain populations, mul-tiple Xba I polymorphisms have been described, e.g.,Chinese (44) and Canadian (67). Although theseother polymorphisms may well be non-factor VIIIsequences which are detected by the factor VIIIintron 22 probe (p482.6), they are closely linked tothe factor VIII gene and the combined use of all theXba I and Bcl I RFLPs would increase the detectionrate to 67% in Chinese.
Due to the highly polymorphic nature of the fourintragenic (two CA repeat polymorphisms, Bcl I andXba I) and one extragenic (Taq I/St 14.1) polymor-phisms, carrier detection or prenatal diagnosis shouldbe possible in 96-100% of females at risk (58, 66,68).
Direct mutation detection in the haemophilias.Linkage analysis for carrier detection and prenataldiagnosis of haemophilia has widely appreciatedbenefits including rapidity, relative technical simpli-city, wide availability and definitive diagnosticresults in a high proportion of cases. However, it hascertain inherent drawbacks and limitations whichhave been previously described.
In principle, all these defects of linkage analysismay be circumvented by identifying the specificmutation in a given kindred. It is then sufficientmerely to check the putative carrier or fetus at riskfor the relevant defect. In addition, identification ofmutations provides scientifically interesting informa-tion that may give clues on structure and function offactor VIII and IX, on mechanisms of mutation, oron phenotype/genotype aspects such as the risk ofinhibitor formation.
The obvious drawbacks of mutation analysis inhaemophilia A and B are that the two genes involvedare large and complex and the mutations thereinextremely heterogenous, as expected for X-linkedsublethal disorders. However, methods for rapidscreening of large regions of DNA for small lesionshave been developed in the past 5 years which nowmake this approach feasible in expert laboratories.These methods were first applied to the smaller gene,factor IX, with impressive results in several centres.For example, a project to identify all the mutationscausing haemophilia B in the United Kingdom popu-lation is now well advanced with successful identifi-cation of a causative gene lesion in 160 out of 161patient DNAs analysed so far (Giannelli, personalcommunication). The methods used for haemophiliaB analysis comprise Southern blotting with a fulllength cDNA probe which detects deletions down toabout 1.5 kb. This yields diagnostic information inabout 1% of cases. For more detailed analysis, enzy-matic amplification of each exon is performed usingthe polymerase chain reaction (PCR) followed bychemical cleavage mismatch detection (CCD) (55).A positive signal from CCD is confirmed by directsequencing.
Mutation analysis in haemophilia A has to copewith the fact that the 26 exons of the factor VIII geneare distributed over 186 kbp of genomic DNA andexon 14 is over 3 kbp in length. Southem blottingafter digestion of genomic DNA with the enzymeTaqI yields diagnostic information in c. 5% ofcases due to the somewhat higher incidence oflarge deletions in haemophilia A and the occurrenceof mutation hotspots in 5 TaqI sites. Currently threescreening methods are being used to screen enzy-matically amplified exons and flanking regions of thefactor VIII gene. Higuchi and colleagues (69, 70)have applied denaturing gradient gel electrophor-
WHO Bulletin OMS. Vol 71 1993444

Haemophilia: carrier detection and prenatal diagnosis
esis (DGGE) and reported on their results in 29 mildor moderate cases and 30 severe cases. Forty-fiveoligonucleotide primer sets were required to amplify99% of the coding region and 41 of 50 splice junc-tions. The disease-producing mutation was found in25 out of 29 mild or moderate cases (85%) but in only16 of 30 severe cases (53%). Even allowing for incom-plete coverage of splice junctions there is a clearimplication that a high proportion of mutationscausing severe haemophilia A lie with the previouslyaccepted essential regions of the factor VIII gene.This highly interesting finding has been confirmedusing an alternative approach (see "Developments indirect defect detection", below). Consequently untilthese unidentified mutations are located there isgoing to be a limitation on the effectiveness of evendirect defect detection, underlining the continuingimportance of polymorphism-based genotype assaysand phenotype assays in carrier and fetal diagnosis.
Another powerful and technically simplerapproach to mutation screening (compared to CCDand DGGE) is based on the property of single stran-ded DNA to form self-associated loop structures thatare highly sequence dependent. The conformationadopted strongly influences the rate of migration ofsingle-stranded DNA in non-denaturing polyacryla-mide gel electrophoresis, enabling detection ofsequence polymorphism (or mutation). The screen-ing method exploiting these phenomena is calledSingle Stranded Conformational Polymorphism(SSCP) analysis. A comparison of SSCP with CCDfor mutation detection in factor VIII exons 1 to 14has yielded essentially identical sensitivity (Tudden-ham, unpublished observations), so this method maybecome more widely utilized. Finally, direct sequen-cing of amplified DNA is used to identify any specificmutation.
Data - haemophilia A. A compilation of mutationsin the factor VIII gene updated to August 1991 hasbeen published in Nucleic acids research (37); 81different point mutations, 6 insertions, 7 small dele-tions, and 60 large deletions are catalogued. Informa-tion where available is also provided for F.VIII coag-ulant and antigen level, clinical severity and inhibitorstatus. A unique number has been assigned to eachpatient for future identification. Thirty-eight percentof point mutations are located in CpG dinucleotides.This frequency is biased by screening with TaqIwhich identifies five such sites, but an unbiasedestimate from Higuchi's data (69) is similar at 32%.Recurrent mutation in CpG dinucleotides hasoccurred in at least 16 sites, where identity bydescent can be excluded on RFLP haplotype or ex-treme geographical separation. Recurrence at non-CpG sites also occurs, for example at Arg 2307Leu,
found in American, German and Japanese patients.This intemational database will be updated
annually and published in Nucleic acids research.New cases for inclusion should be submitted to Dr S.Antonarakis, The Johns Hopkins University, Balti-more, USA or Dr E. Tuddenham, MRC, Harrow,England. The haemophilia A database has been sub-mitted to the Genome Database (Welch MedicalLibrary, 1830 East Monument Street, Baltimore, MD21205, USA) and will be accessible from there byelectronic means. In future as the number of patientssuccessfully analysed increases, the only practicalmeans of updating and retrieval will be via com-puterized databases, organized nationally and inter-nationally. Appropriate security of informationaccess will be built into these systems.
Data-haemophilia B. A compilation of point muta-tions, short deletions and insertions in the factor IXgene has been produced by Giannelli et al. (3rd edi-tion, 1992) published in Nucleic acids research (38).
The data tabulated includes levels of factor IXclotting activity and antigen, inhibitor status but notclinical severity; 574 mutants are listed, representing278 different molecular alterations. These are distri-buted across the entire coding region from the signalpeptide to codon 412, only four residues before theStop codon. Eleven examples of mutations in thepromoter region giving rise to the Leyden phenotypeare recorded. Thirty examples of mutations affectingRNA processing have been observed. This informa-tion has now been incorporated into the EMBL data-base, providing on-line access to updates.
Application of polymorphism analysis and directdefect detection in developed and developingcountries. Developed countries. Developments inmolecular genetics mean that it is now possible toidentify practically all the functional mutations in thefactor IX gene in patients with haemophilia B and asignificant number within the factor VIII gene inpatients with haemophilia A (see above). The tech-niques used are specialized and require staff withmolecular biology experience, and several groupswithin Europe and North America have establishedlaboratories where these analyses are performedupon request. They also provide a follow-up servicefor subsequent carrier status assessment and prenataldiagnosis.
In these countries the identification of the speci-fic defect will increasingly replace family studiesbased on gene tracking of polymorphisms (seeabove). Although polymorphism analysis, particular-ly when based on PCR technology is extremelysimple to perform, the problems related to non-infor-mative family members, non-paternity and sporadic
WHO Bulletin OMS. Vol 71 1993 445

I.R. Peake et al.
disease mean that where the technology is available,defect detection techniques are preferable. Therecently reported cases of germline mosaicism inhaemophilia present problems for family studiesbased on either approach. It is also expected thatincreased technological advances will make defectdetection easier and more applicable to the routinelaboratory. However, without these advances it isprobable that polymorphism-based gene tracking willstill remain important in those laboratories wheremolecular biology experience for defect detection isnot available.
Developing countries. It is unlikely that direct defectdetection will be available in the foreseeable futurein many countries where the identification and treat-ment of the disease itself is only in its infancy.However, it should be pointed out that genetic analy-sis of families with patients with haemophilia, eventhough the treatment of the haemophiliac may not beoptimal, can still be of assistance to the family byidentifying carriers and preventing the birth of affec-ted individuals. Where resources are scarce it is pro-bable that the least expensive techniques of genetracking which will involve, at the moment, poly-morphism analysis will be the only ones applicable.It is, therefore, important that such techniques shouldbe made as simple as possible and their applicationin developing countries is something that may welladvance quite rapidly in the next decade under theguidance of the World Federation of Haemophilia.
Current situationPrenatal diagnosisObstetric techniques. Ultrasonography. All preg-nant women should be offered a detailed ultrasoundexamination for (i) confirmation of fetal viability andgestational age, (ii) the diagnosis of multiplepregnancies, and (iii) exclusion of both major mal-formations but also smaller defects, which may leadto the diagnosis of an underlying chromosomalabnormality or genetic haemophilia. Ultrasonogra-phy at 16-20 weeks will reliably diagnose the fetalsex. In the presence of a female fetus, invasive fetaltesting can be avoided.
Fetal blood sampling. In the 1970s and early 1980sthe method of fetal blood sampling was fetoscopy,which involved the introduction into the amnioticcavity of an endoscope (3 mm in diameter). Bloodvessels in the chorionic plate or umbilical cord werevisualized and punctured to provide pure fetal blood(71). The patients were usually hospitalized and theprocedure was carried out under heavy sedation. Thetechnique was confined to very few centres and
under heavy sedation. The technique was confined tovery few centres and in the best hands the procedure-related risk of fetal death was 2-5%. More recently,improvements in imaging by ultrasonography havemade fetoscopic guidance unnecessary and fetalblood can be obtained by ultrasound-guided punctureof an umbilical cord vessel (cordocentesis) or thefetal heart (cardiocentesis). The preferred method iscordocentesis, which involves the ultrasound-guidedinsertion of a 20 or 22 gauge needle through thematemal abdomen and into an umbilical cord vessel(72, 73). The procedure is carried out on an out-patient basis and no matemal sedation or anaesthesiais required. Several centres throughout the worldhave now developed considerable expertise in thistechnique. Matemal complications are negligible.The risk of fetal death following cordocentesis isapproximately 1-2%. The risks are higher when themother is obese, the placenta is posterior, and thegestation at the time of sampling 16-19 weeks ratherthan 20-21 weeks.
Chorion villus sampling (CVS). Placenta tissue canbe successfully obtained from as early as six weeks'gestation by the transabdominal or transcervicalentry of aspiration cannulas, biopsy forceps, orneedles of variable sizes (74, 75). More than200 000 procedures have now been performedthroughout the world and the technique is carriedout in many centres. However, the recent report onthe possible association between CVS at less than10 weeks and fetal limb abnormalities is likely toconfine its application to pregnancies beyond thisgestation (76). Furthermore, the European MRC trialhas demonstrated a significantly higher risk of fetaldeath after CVS than after second trimester amnio-centesis (77). Despite these limitations, CVS has theadvantage of providing prenatal diagnosis in thefirst trimester rather than at 18-20 weeks as withtraditional amniocentesis.
Amniocentesis. In the context of prenatal diagnosisof haemophilia, amniocentesis is used for fetalsexing and DNA analysis. When amniocentesis wasfirst performed, it was limited to 16 weeks onwards,because at earlier gestations there was a high failurerate in obtaining amniotic fluid. However, during thelast five years several studies have established thefeasibility of early amniocentesis at 10-14 weeks(78). Furthermore, the original apprehension aboutthe smaller cellular content of amniotic fluid has notbeen substantiated; although the number of amnioticfluid cells at 10 weeks is smaller than at 16 weeks,the number of viable cells is the same. At amniocen-tesis, a 20 or 22 gauge needle is guided by ultra-sound into the amniotic cavity and 10 ml of fluid is
WHO Bulletin OMS. Vol 71 1993446

Haemophilia: carrier detection and prenatal diagnosis
aspirated. Cell culture and cytogenetic analysis aresuccessful in 98% of the cases and results can beavailable within 2-3 weeks (78). For diagnosis ofhaemophilia, DNA can be extracted from the cellseither directly after separation or after culture.
Collection, preparation and storage of prenatalsamples. Fetal blood. Meticulous care, consistencyand speed are critical in collecting the diagnosticblood samples. All the necessary tubes are preparedbefore cordocentesis. Three successive fetal bloodsamples are aspirated into l-ml plastic syringes. Thefirst sample is collected into a heparin tube for (i)immediate cell size analysis and (ii) subsequentKleihauer-Betke testing, to confirm its fetal origin.
The subsequent two samples are delivered intosix polystyrene precipitin tubes, exactly to the 500 glmark without airlocks or bubbling. The polystyrenetubes contain accurate volumes of buffered-citrateanticoagulant; in two tubes the citrate to fetal bloodratio is 1:1 and in three it is 1:9. The tubes are heldsideways between the forefinger and thumb andjerked vigorously 2-3 times to mix. The rest ofeach sample is collected into heparin tubes for (i) cellsize analysis and determination of haematocrit, whichare compared with the result of the first sample, toensure that all samples are fetal and not contamin-ated by amniotic fluid, and (ii) immunoradiometricassay of VIII:Ag or IX:Ag in the supernatant plasma.All tubes containing fetal samples are capped,placed in ice, and taken to the laboratory for assay,where they are left undisturbed for one hour.
Amniotic fluid. Amniotic fluid (1O ml) is collectedinto a sterile container, and kept at 4 °C until furtheranalysis. The sample is divided into two halves, oneto be used for direct DNA analysis and the secondfor cell culture to confirm the initial result. Theamniocytes are separated by centrifugation in Eppen-dorf tubes and PCR is performed on the DNA, whichis extracted using proteinase K and phenol, followedby ethanol precipitation.
Chorionic villi. Samples are aspirated into culturemedium and examined under a dissecting microsco-pe for any contamination with decidua, in which casethe tissue must be separated immediately after col-lection. The chorionic villi are noted for a centralvascular core and budding cytotrophoblast, unlikedecidua which are more sheet-like and less vascular.The chorionic villi are weighed in a pre-weighedEppendorf tube; the minimum required is 6 mg,although 60-80 mg of wet tissue is usually obtained.
Measurements on fetal blood. Both factors VIII andIX are measured in all cases to provide an addedcheck on the validity of the samples. In addition,
factor V is measured as an indicator of possibleconsumption or activation of VIII (79). When thefetus is at risk of haemophilia B, factor X is alsomeasured.
Prenatal diagnosis of haemophilia A and B by DNAanalysis. The effective use of DNA analysis for pre-natal diagnosis of haemophilia involves prior plan-ning and coordination between the testing laboratoryand an obstetrician specialist in fetal medicine.
The use of PCR-based genetic testing has great-ly simplified the prenatal diagnostic strategies usedin screening for haemophilia (42). The ability to per-form PCR studies on less than 1 ,ug of DNA makesthis technique especially useful in prenatal analysiswhere the yield of fetal tissue for testing may belimited. There has now been considerable experi-ence with the use of PCR-based prenatal testingfor both polymorphism linkage analysis andmore recently for direct mutation detection in thehaemophilias.
Analysis of chorionic villus (CV) tissue. The extrac-tion of DNA from both CV material and amniocytesis best carried out by alternative, "gentler" methodsto those used for blood extractions. All routine pre-cautions to avoid sample contamination with extra-neous DNA must be strictly adhered to and the pos-sibility of maternal contamination of the CV tissuemust also be kept in mind. Most laboratories willperform two sets of tests on the CV DNA. The firstinvolves sexing of the fetus through the use ofamplification primers corresponding to the regionsZFX and ZFY on the X and Y chromosome respec-tively. The DNA is then amplified for haemophiliagenotyping using either one of the previouslydetailed intragenic polymorphisms or one of themethods used for direct mutation analysis. Diagnosticstudies are achievable within 48 hours of receipt ofthe CV tissue.
Amniocyte DNA analysis. Two options are availablein testing cells obtained at amniocentesis. Direct ana-lysis of DNA extracted from amniocytes spun downfrom 10 ml of amniotic fluid is now feasible andavoids the two-week hiatus before sufficient quanti-ties of cultured amniocytes are available for analysis.Once again, in light of the small quantities of fetalcells available for amplification, PCR contaminationmust be guarded against using techniques previouslyalluded to in this document. The strategy for DNAtesting is identical to that outlined for CVS-baseddiagnoses.
Genetic counselling services for haemophiliaThe individual or individuals involved in geneticcounselling for haemophilia should hat'e proficiency
WHO Bulletin OMS. Vol 71 1993 447

I.R. Peake et al.
in the following areas: haemophilia and haemophiliacare, human genetics, prenatal diagnosis, moleculargenetics, and interpersonal communication and coun-selling. Proficiency may be gained through specificmedical/clinical genetics or genetic counsellingtraining programmes, coursework or laboratoryexperience. If all of the aforementioned aspects can-not be covered in the training of one individual, thena team approach to counselling is recommended sothat expertise may be available in all areas. When ateam approach is utilized, one individual shouldcoordinate activities of the team, and act as an inter-face between the patient and team members. Liaisonand communication between professional colleaguesis highly recommended, especially between thosewho perform and interpret laboratory tests and thosewho provide clinical information to the patients andtheir families.
The aims of genetic counselling services shouldbe (1) to provide patients with sufficient informationto make informed choices regarding carrier testingand prenatal diagnosis, and (2) to provide psychoso-cial support to the patient throughout the process oftesting. Information should be given in a non-directivemanner prior to the time of actual testing, so thatpatients and their families can make choices regard-ing their medical care.
All potential carriers should be tested early inlife to determine factor VIII or IX activity levels.This should be done for safety reasons alone, to de-tect individuals who may have bleeding problems incertain situations. Then, at the age of informed con-sent, carrier testing should be offered to all femalerelatives of haemophilic males. The individualundergoing testing, should be aware of the purposesof the test and its implications. It is preferable thatcarrier testing be completed before a potential carrierbecomes pregnant. Contact can be made withfemales at risk directly, or through their male rela-tives. An accurate family history should be obtained,and the specific clinical diagnosis of haemophilicmales within the family should be ascertained.Knowledge of the severity of disease is extremelyimportant when counselling family members regar-ding reproductive risks. If a potential carrier is notfamiliar with the clinical presentation of haemophi-lia, then various methods of introduction to thedisease should be considered, such as audiovisualmaterials, selected reading, and/or personal inter-views with affected individuals and their families.
Technical aspects of both phenotypic and geno-typic carrier testing should be discussed with eachpotential carrier, as well as the accuracy and limita-tions of the tests. Individuals at risk should be infor-
med that the most accurate type of testing availableat this time is genotypic testing, and that directdefect detection is preferred if it is available. If geno-typic testing is to be done, family counselling shouldbe considered so that all family members may beinformed of the nature of the test, and the possibleknowledge that will be gained upon completion oftesting. All females with negative carrier test resultsshould carefully be advised of the accuracy of thediagnosis, especially in the case of phenotypic test-ing, or when using linked polymorphic probes.
Women who have positive carrier test resultsshould be advised of all of their reproductiveoptions, including adoption and in vitro fertilizationwhere applicable. They should also be aware of thecurrent treatment options and prognosis for anindividual with haemophilia in their area. If theyconsider having a child of their own, all prenataltesting options should be discussed, as well as theappropriate methodology for termination of anaffected pregnancy. Risks for each testing procedureshould be given as accurately as possible, taking intoconsideration the testing facilities which would beavailable to the patient. Counselling and psychologi-cal support should be available to the patient duringall aspects of prenatal testing.
Present situation in developed anddeveloping countries
Only recently have methods for prenatal diagnosis ofhaemophilia moved from the research laboratory tothe clinical setting. Because of this, they are atpresent only available in developed countries, usual-ly in academic settings.
Developed countries. There is very little informationinternationally on the availability, organization andproficiency of centres that provide these diagnosticservices. The information available stems frompublications or communications at meetings. Asexamples, the situation in Italy and the USA, twocountries where the provision of prenatal diagnosisof haemophilia appears to be arranged with differentstrategies will be described.
Italy, a country with a population of 56 million,has registered 2036 patients with severe and moderatehaemophilia A and B, i.e., those that are most likelyto be concerned with prenatal diagnosis. The distri-bution of the diagnostic services is regional, withunits established in Milan and Genoa (NorthernItaly), Rome (Central Italy), Naples (Southern Italy)and Cagliari (located on Sardinia). With the excep-tion of the latter, these diagnostic units are establish-
WHO Bulletin OMS. Vol 71 1993448

Haemophilia: carrier detection and prenatal diagnosis
ed within comprehensive haemophilia centres andenlist the integrated cooperation of a coagulationlaboratory that provides phenotypic diagnosis, anobstetric service that performs techniques such asultrasound, amniocentesis and chorionic villus sam-pling, and a laboratory of molecular biology thatperforms DNA analysis. Delivery to the patient ofdiagnostic information and genetic counselling areprovided by genetic counsellors belonging to thestaff of the haemophilia centres. This situation is par-ticularly advantageous for the families that are regu-larly followed at such centres, because the stressfulsituation of prenatal diagnosis is dealt with by thesame staff who attend to haemophiliacs in their day-to-day care. This regional organization has devel-oped spontaneously in Italy, around haemophiliacentres which chose to develop DNA techniques forresearch purposes. There is no state or regionalrecognition of the diagnostic services, which arefunded with research money provided by the hospi-tals or universities where the haemophilia centresare established.
The regional organization of Italy, based onconveniently sized and located units providing thecoordinated services needed for prenatal diagnosis,contrasts with the organization of other countriessuch as the USA. In this country with a population of250 million, and approximately 20 000 haemophi-liacs, very few haemophilia centres (for instance, inSan Francisco and Chapel Hill, NC) have establishedtheir own services of prenatal diagnosis, spanningfrom counselling and obstetric procedures to DNAdiagnosis. Most centres enlist the help of obstetricunits (not necessarily located in the same hospital) toperform chorionic villus sampling or amniocentesisand then send DNA samples to institutions (such asthe Mayo Clinic, Johns Hopkins University and theUniversity of North Carolina at Chapel Hill) thathave a large experience in molecular biology ofcongenital coagulation disorders. This system hassome appeal, because these large units acquire con-siderable proficiency by analysing a large numberof DNA samples. On the other hand, it has the dis-advantage of being somewhat fragmented, in thatthe consultands are referred from a comprehensivehaemophilia centre (not always the centre where reg-ular care is provided) to an obstetric unit for chori-onic villus sampling or amniocentesis (not always inthe same State), with the actual DNA diagnosiseventually carried out in a totally different institution.
Developing countries. Recent initiatives in primaryhealth care are reducing infant mortality, so thatcongenital disorders are inevitably beginning to be
recognized in developing countries. The burden ofhandicapping genetic diseases such as haemophilia isheavier in developing countries, because the infra-structure and services needed to assist the handicap-ped are usually primitive or non-existent. Because ofthe lack or deficiency of the social support system,the heavy burden of chronic disease for the indivi-dual and the family further emphasizes the desirabili-ty of prevention. This is particularly evident forconditions such as haemophilia, which requiresexpensive therapeutic measures. Hence, there is aconsiderable demand for prenatal diagnosis of hae-mophilia and allied disorders in developing coun-tries. As a result of this demand, there are manyfamilies at risk that seek genetic diagnostic testing indeveloped countries, but obviously these servicescan only be afforded by the richest. Local diagnosticfacilities are practically non-existent, with the excep-tion of a few academic centres where research inter-est in molecular biology of coagulation defects hasprompted the application of DNA techniques to car-rier detection and prenatal diagnosis of haemophilia.The current availability of comprehensive haemophi-lia centres in developing countries is too scanty toenvisage the rapid development of a network ofgenetic services as feasible and reasonable. On theother hand, it must be realized that knowledgespreads faster than technology, which is expensive.This will create significant problems in developingcountries, because knowledge about haemophilia andthe corresponding expectations are fast out-distancing the ability to provide modem therapy.
Role of the World Federation of Haemophilia(WFH)Despite the fact that the WFH is the only inter-national organization that has the institutional goalof striving for the improvement of the well-beingof haemophilic patients, the Federation has as yet noinformation on the availability and organization ofunits for prenatal diagnosis in member countries(National Member Organization, NMO). It is, there-fore, recommend that the medical advisory board orthe medical secretaries of the WFH, in collaborationwith WHO, take action to gather this importantinformation, by questionnaires or other methods ofsurvey. This input is essential to prepare a registry ofdiagnostic units and plan further action. The ongoingdesignation of a few Intemational HaemophiliaTraining Centres of the WFH as WHO CollaboratingCentres on Haemophilia, should provide a networkwhere staff from developing countries that choose todevelop genetic services for prenatal diagnosis ofhaemophilia can be trained.
WHO Bulletin OMS. Vol 71 1993 449

I.R. Peake et al.
Future considerationsGenetic diagnosis and management ofhaemophiliaThe post-HIV situation. Although the impact ofHIV-I infection on the haemophilic populationdiffers widely from country, this threatening infectionis an important cause of death among haemophiliacs.The death of a large number of haemophiliacs fromAIDS has important implications on the feasibilityof carrier detection or prenatal diagnosis in hae-mophilic families at risk. The currently availablediagnostic methods based on recombinant DNA tech-nology rely heavily on the availability of the DNAfrom at least one affected member of the haemo-philic families. This need materializes not onlywhen RFLPs are employed to track the abnormalallele, as occurs most often in the diagnosis of hae-mophilia A, but also when the disease-causing DNAdefect is searched for directly, as occurs more andmore frequently in haemophilia B.
Unfortunately, because of HIV infection, thesestudies cannot be carried out in some families at riskbecause the critically affected family members havedied, so that their DNA is not available for examina-tion. On the other hand, blood for DNA extraction,and the DNA itself, is stable if stored under properconditions and may be used for studies many yearslater. To avoid the aforementioned pitfalls that makecarrier detection and prenatal diagnosis often impos-sible, the World Federation of Haemophilia hasrecently recommended that samples of blood suitablefor DNA extraction be obtained from all personswith haemophilia A and B, particularly from thoseaffected by HIV infection or other potentially lethalconditions. A technique suitable for processing andstoring DNA samples has also been recommended(80). These samples, stored at the haemophiliacentres locally or nationally, will provide valuablematerial for future studies and represent a resourcethat will be increasingly important as DNA analysisprovides a better understanding of the molecularbasis of haemophilia.
It is therefore recommended that WHO shouldalso suggest the organization of the storage of DNAsamples from patients with haemophilia and alliedcongenital coagulation disorders to all member coun-tries.
Developments of RFLP detection techniques. Geneticdiagnosis of haemophilia A and B can be performedmost effectively by direct detection of the mutationitself. However, as mentioned above, there are techni-cal and other reasons which limit the application ofthis form of diagnosis. Thus, diagnosis at the DNA
level often relies on the familial segregation oflinked restriction fragment length polymorphisms(RFLPs). With the development of in-vitro DNAamplification using the polymerase chain reaction(PCR) convenient, non-radioactive detection methodshave become available. As alluded to above, thisform of testing is now in widespread use for bothfactor VIII and factor IX intragenic and extragenicRFLPs (42, 52, 53, 57). Another approach that wouldobviate the need for restriction enzyme digestion isPCR followed by differential hybridization withsequence-specific oligonucleotide probes. Theseprobes can be linked to horse-radish peroxidase anda colour detection system used instead of radio-isotopes (81).
A more recent development is the use of immo-bilized sequence-specific oligonucleotide probes forgenetic analysis of PCR-amplified DNA (82). Unlikethe former method of immobilized DNA, where eachprobe requires a separate hybridization, this methodwould enable simultaneous screening of a sample fora number of RFLPs at an amplified locus.
Developments in direct defect detection. Analysisof large multiexonic genes on an exon-by-exon basisbecomes increasingly laborious and time-consumingas the number of exons increases. Although the fac-tor IX gene with 8 exons is tractable by presentscreening methods, the 26 exons of factor VIII stretchresources of time, personnel and laboratory equip-ment to the limits so that only a few laboratorieshave taken up the challenge of mutation analysis inhaemophilia A. The recent discovery that smallamounts of processed mRNA for many if not allgenes are present in tissues that do not normallyexpress those genes promises to greatly speed up theanalysis of genes whose normal tissue of specificexpression is inaccessible.
"Ectopically transcribed" mRNA can be isolatedfrom peripheral blood lymphocytes, reverse tran-scribed to produce cDNA, and then enzymaticallyamplified with specific primers. The PCR productsrepresenting regions of processed mRNA can then bescreened with chemical cleavage mismatch detec-tion, and sequenced to identify mutations (83). Preli-minary experience with this approach suggests thatmutations, including those affecting mRNA proces-sing, can be successfully identified in most cases ofmoderate or mild haemophilia A and in up to 60% ofcases of severe haemophilia A (Giannelli, personalcommunication).
The speed and power of this method are not indoubt but the degree to which the technology can betransferred from its originator (highly expert) labora-tory to other groups remains to be established. Also
WHO Bulletin OMS. Vol 71 1993450

Haemophilia: carrier detection and prenatal diagnosis
the problem of unidentified mutations in severe casesof haemophilia A remains, although a preliminaryreport from this same group suggests that a defect inthe processing of intron 22 from the factor VIIImRNA may be responsible for some of these addi-tional mutations (84).
Advances in prenatal diagnostic techniques.Advances in molecular genetics, such as DNAamplification of a single cell by PCR, have stimula-ted research into the feasibility of genetic diagnosisin the pre-implantation embryo and in fetal cells inthe maternal circulation.
(i) Pre-implantation genetic diagnosisThe potential advantages of pre-implantation
diagnosis are (1) avoidance of repeated terminationsin couples at high risk for affected offspring, and(2) correction of the disease by such measures asgene therapy. Possible approaches for pre-implanta-tion diagnosis include biopsy from the polar body,the 6-8-cell embryo at day 3, or the blastocyst atdays 5-6.
Polar body biopsy. Demonstration of theaffected allele in the sample implies that the primaryoocyte carries the normal allele. However, the possi-bility of recombination should be considered, whichis up to 50% for genes near the telomeres. Further-more, PCR on single cells can fail to produce suffi-cient DNA for diagnosis in approximately 20% ofcases. Of the 83 cases in which this method wasattempted (including 28 at risk of haemophilia), onlyone successful pregnancy was achieved, and in thiscase diagnosis by CVS demonstrated the fetus to beaffected (85, 86).
Early cell embryo biopsy. This procedure whichinvolves either the aspiration or herniation of onecell from the 6-8-cell embryo does not result inobvious abnormal development of fetuses in cattleand mice, although, in mice, where like humans, thepre-implantation period is relatively short, the viabi-lity of the embryos is reduced. In one human study,38 embryos underwent biopsy; 30 had two pronuclei(normal), and 8 had either 3 or no pronuclei. In 27 ofthe 30 normal embryos, morphological developmentwas assessed and 10 (37%) developed into blasto-cysts; this percent is similar to that of unmanipulatedembryos (87, 88).
Therefore, it appears that removal of one cellfrom an eight-cell embryo does not affect embryodevelopment or the success rate of establishing aviable pregnancy. However, as with polar body biop-sy, material from one cell is not always sufficient forsuccessful PCR amplification. As an alternative toPCR, fluorescence in situ hybridization may beapplied. However the latter technique is less suitable
for diagnosis of unique sequence DNA mutations.Blastocyst biopsy. This technique involves slit-
ting the zona and allowing 10-30 cells to extrude.The herniated cells are cut off and can be used forreplicate assays. Although the frequency of hatchingis apparently not affected by the biopsy, no blastocystshave yet been transferred back to the mother (89).(ii) Recovery offetal cells from maternal blood
The possibility of recovering fetal cells frommaternal blood was first raised by Walknowska et al.(110) who found male metaphases in the blood ofpregnant women. More recently, the presence offetal cells in the maternal circulation has been con-firmed by the combined use of (1) monoclonal anti-bodies against fetal specific antigens on syncytio-trophoblasts or HLA-A2 antigen-positive lympho-cytes from HLA-A2 antigen-negative women or trans-ferrin receptor and glycophorin-A on erythroblasts,(2) flow cytometry, to isolate or enrich fetal cells,and (3) PCR amplification of Y chromosomespecific sequences (90-93).
Gene therapy-a reality? Haemophilias A and B areconsidered suitable targets for development of genetherapy on the following grounds:* The relevant genes have been cloned.* Tight regulation of expression may not be essen-tial.* A modest increase in circulating levels of factorVIII or IX would greatly improve the clinical bleed-ing tendency, e.g., 0% to 10% convert from severe tomild bleeding.* Haemophilia is a lifelong condition with severeeffects on the sufferer.* Treatment by replacement of deficient factor isnot entirely satisfactory, since it involves frequentinjections of highly expensive replacement productsthat are not free of all risks.
However haemophilia is not amongst the veryfirst genetic disorders to be targeted for gene therapyas it is not invariably fatal and the best present treat-ment is successful in controlling the major clinicalsymptoms, and in achieving a relatively normal lifefor sufferers.
The basic concept of gene therapy is straight-forward. It is proposed that a normally functioninggene be transferred into somatic cells of the recipi-ent, such that those cells make and continue to makesufficient gene product to correct the inherited defect.In the case of haemophilia A or B, either factor VIIIor IX must find its way into the circulation fromwhichever cell type is chosen for gene transfer,. ina correctly modified form, at a rate sufficient toraise the circulating level of clotting factor. The prob-
WHO Bulletin OMS. Vol 71 1993 451

I.R. Peake et al.
lem may therefore be conveniently considered underseveral headings.
(i) Somatic cell type targetedfor gene transferIn vivo factor VIII is synthesized by hepatocytes andby some as yet unidentified cell type in the lymphoidsystem, particularly the spleen. However, ectopicproduction might be satisfactory, e.g., in haemato-poietic cells. Thus, target cells under considerationinclude endothelial cells, hepatocytes, fibroblasts,myoblasts, and bone marrow stem cells.
(ii) Vector for DNA transferSince a highly efficient transfer system is essential todeliver DNA to a large number of cells into whichthe DNA should be stably integrated, retroviruseshave attracted most attention. The retrovirus is modi-fied so that essential coding sequences required forpackaging and therefore infectivity are replaced byCDNA inserts representing a therapeutic gene.
Packaged virus is obtained using a cell linewhich contains defective viral DNA producing thedeleted packaging proteins but no infectious viralRNA. Vector design is undergoing continuous devel-opment at the present time, but a limitation of retro-virus is that only about 7 kb of inserted DNA can beeffectively packaged. Therefore a modified factorVIII gene must be used, lacking the B domain, whichis dispensable for normal function. The retroviruses'life-cycle leads to stable integration of viral DNAbut only into actively replicating cells, which pre-cludes the use of hepatocytes as target host cells.Therefore, alternative vectors have been developedincluding adenoviruses. The maximum size of foreigngene insert in these viruses is 5 kb. Non-virus-basedtransfer methods are also being developed.
(iii) Animal modelsRelevant animal models for stable DNA transfer,integration and expression are needed. As regardshaemophilia, dog models of haemophilia A and Bare available (94, 95), and at least one colony of hae-mophilic cats is being maintained. No mouse modelof haemophilia exists but it is now feasible to pro-duce mouse lines defective in any specified genesequence by targeted homologous recombination inembryonic stem cells. Subsequent recovery of fertilechimeric adults with the modified genotype presentamong their gametes is obtained by injecting tar-geted ES cells into morula-stage embryos. Furtherbreeding produces mice with the targeted gene dis-rupted in all their cells, provided that the phenotypeis compatible with embryonic development. By thismeans many new strains of mice representing humanhereditary defects have been produced.
(iv) Gene transfer experiments involving factors VIIIor IXFactor IX. Modified retrovirus containing humanfactor IX coding sequence has been transferred intohepatoma cell lines, and murine rat and human fibro-blasts. The majority of the factor IX protein expres-sed by these cells was correctly gamma-carboxylatedand functional in vitro. Canine factor IX has beenexpressed in dog fibroblasts and bovine endothelialcells. When fibroblasts secreting human factor IXwere transplanted into syngeneic mice, human factorIX could be detected in their circulation for twoweeks until an immunological response occurred.However, the vector was inactivated by an epigeneticmechanism regardless of the immune response toforeign protein (96).Factor VIII. Functional B-domainless human factorVIII has been secreted from human fibroblasts trans-fected with a retroviral vector containing B-domain-deleted factor VIII CDNA. When transferred tonormal mice these cells could be recovered aftertwo months and shown to still synthesize factorVIII. However, no factor VIII was detected in therecipient mice, probably due to the short half-lifeof human factor VIII in this animal (97).(v) General conclusionsAlthough considerable progress has been madetowards developing gene therapy, at present (Sep-tember 1992) no satisfactory experiments in animalmodels have been described. Clearly there is a needto use conspecific factor in a relevant animal modelbefore the success or otherwise of gene therapy canbe judged.
The nearest approach has been reached with dogfactor IX in dog fibroblasts in vitro. Presumablythese will be tested in haemophilic dogs in the nearfuture. It is certain that extensive efforts to validategene therapy in haemophilia will be undertaken overthe next 10 to 20 years with some considerablechance of ultimate success. Should such pro-cedures be shown to be safe, efficacious and reason-ably cheap, then gene therapy might become the thera-py of choice-especially in developing countrieswhere other means of support are too expensive tobe widely available. Conversely, if the price ofrecombinant factor VIII were to fall very consider-ably and especially if the oral route became feasi-ble, then gene therapy would be less attractive.
Conclusions and recommendationsRecent advances in molecular genetic procedureshave resulted in a realization that accurate DNA-based carrier and prenatal diagnosis in haemophilia
WHO Bulletin OMS. Vol 71 1993

Haemophilia: carrier detection and prenatal diagnosis
can be successfully achieved in many countries. Theprovision of these accurate methods combined withthe availability of professional counselling andobstetric procedures performed by specialists infetal medicine is seen as the ultimate goal for theeffective genetic control of haemophilia. The roleof WHO and WFH in encouraging the provision ofsuch facilities was recognized.
Techniques for specific mutation detection arenow available, particularly in relation to haemophiliaB, and developments in this area should be activelyencouraged in order to make such proceduressimpler and more applicable to routine analysis. Itwas however accepted that at present in most casesdiagnoses will be based on DNA polymorphismanalysis.
Differences between the cultural, social and eco-nomic situations in different countries will signifi-cantly affect the application of genetic proceduresand the ethnic variation in frequency in many of thepolymorphic markers will also result in varyingefficiency of such procedures. Services for carrierdetection and prenatal diagnosis of haemophiliashould be established in the places where they willbe used. This will take into account local ethnicand other influences.
The participants agreed that the present situationwith regard to pre-implantation diagnosis of haemo-philia and gene therapy, while offering exciting pos-sibilities, was unlikely to affect significantly therequirements for genetic analysis procedures in theforeseeable future.
Future research into the feasibility of non-inva-sive fetal testing and gene therapy for haemophiliashould be encouraged.
Recommendations1. There should be a multidisciplinary approach tothe prenatal diagnosis of haemophilia involvingexperts in the fields of fetal medicine, genetic coun-selling, haemophilia care and molecular genetics.2. Potential carriers of haemophilia should be testedfor clotting factor deficiencies early in childhood toassess the potential for clinical bleeding problems.These same potential haemophilia carriers shouldsubsequently be offered carrier diagnosis after theage of consent and preferably prior to pregnancy.3. Initial risk assessment for carriership should bebased on pedigree analysis. Subsequent carrier tes-ting by phenotypic analysis should include, wherepractical, the calibration of all reference materialused in assays against approved intemational stan-dards, and the final probability of carriership shouldbe based on the combined likelihoods obtained frompedigree and laboratory analysis.
4. Genotypic analysis offers the most accuratemethod of carrier detection. In haemophilia, this willmost often involve the use of linked polymorphicmarkers to follow the inheritance of a haemophilicgene within a pedigree. This assessement should ini-tially involve the study of the most informative intra-genic markers with reference to the ethnic origin ofthe family being tested. Where circumstances permit,the genetic diagnosis of haemophilia should be basedon the direct identification of the disease-causingmutation in the factor VIII or factor IX gene.5. DNA should be acquired and stored locally ornationally from all haemophiliacs to facilitate futuregenetic diagnoses. National Haemophilia Societiesshould be actively involved in this process.6. Family members undergoing counselling shouldbe made familiar with the limitations of laboratorytesting and thus be able to provide informed consent.The counselling process should be coordinated by asingle well-trained individual, who should be fami-liar with the concepts of haemophilia care andclinical/molecular genetics. The genetic counsellingprocess must also include a critical evaluationof the type of haemophilia in male relatives of theconsultand (haemophilia A or B, its severity, CRMstatus, etc.).7. All professionals involved in providing geneticservices to haemophiliacs should attain appropriatelevels of training. In this regard an intemationalregistry of potential training centres should beestablished and maintained by WFH. Some ofthese centres may be considered, after due process,as WHO Collaborating Centres.8. WHO/WFH should conduct an intemational sur-vey to assess the facilities available for prenataldiagnosis of haemophilia, and subsequently an inter-national registry of such facilities should be estab-lished.9. WHO/WFH should support the existing inter-national data bases for specific haemophilia muta-tions and encourage the development of new tech-nologies to identify such mutations. Where possible,all genetic information should be added to NationalHaemophilia Registries.
ResumeH6mophilie: strat6gies de d6pistage desconductrices et de diagnostic pr6natalEn 1977, I'OMS a publie dans le Bulletin unmemorandum sur les methodes de d6pistage desconductrices de I'hemophilie. Ce m6morandum
WHO Bulletin OMS. Vol 71 1993 453

I.R. Peake et al.
avait et6 pr6pare a l'occasion d'une r6unionOMS/FMH (F6deration mondiale de l'H6mophilie)qui s'est tenue a Geneve en novembre 1976, etconstitue une r6ference en matiere de g6netiquede I'hemophilie. Les analyses examinees 6taientbas6es sur I'evaluation phenotypique qui, a1'6poque, 6tait la seule methode disponible.
Grace aux r6cents progres de la genetiquemol6culaire, on sait maintenant qu'il est possible,dans de nombreux pays, d'obtenir avec precision,par analyse de I'ADN, un diagnostic de 1'etat deconductrice de la maladie et un diagnostic pr6na-tal de l'hemophilie. On estime que la possibilite dedisposer de ces methodes, associ6es a un conseildispens6 par des professionnels qualifies et a desm6thodes obstetricales appliqu6es par des sp6-cialistes en m6decine fcetale, est le but aatteindre si on d6sire un contr6le g6netique effica-ce de l'h6mophilie. Le r6le d'encouragement del'OMS et de la FMH dans ce domaine a 6tereconnu.
II existe maintenant des techniques de d6pis-tage sp6cifique des mutations, notamment pourlhemophilie B, et les progres dans ce domainedoivent etre activement encourages de faqon asimplifier ces m6thodes et les rendre davantageapplicables en routine. 11 est toutefois reconnuque, actuellement, dans la plupart des cas les dia-gnostics seront encore bas6s sur I'analyse dupolymorphisme de I'ADN.
Les diff6rences culturelles, sociales et 6cono-miques d'un pays a l'autre modifient de faconsensible I'application des m6thodes g6n6tiques, etles variations ethniques de la fr6quence des mar-queurs du polymorphisme entraineront 6galementdes diff6rences d'efficacit6 des m6thodes d'analy-se. Les services de depistage des conductrices etde diagnostic prenatal de l'hemophilie devront etrecre6s sur les lieux memes ou ils seront utilis6s.On tiendra pour cela compte des facteurs eth-niques locaux et d'autres facteurs pertinents.
Les participants sont convenus que la situa-tion actuelle en ce qui concerne le diagnostic delhemophilie avant implantation de l'ceuf et la the-rapie genique, bien qu'offrant des perspectivesextremement int6ressantes, n'est pas assez avan-c6e pour modifier sensiblement les besoins enanalyses gen6tiques dans un avenir previsible.
Les recherches sur la faisabilit6 d'un d6pista-ge foetal non invasif et d'une therapie genique delhemophilie devront etre encouragees.
Recommandations1. Le diagnostic pr6natal de l'hemophilie devraitfaire l'objet d'une approche multidisciplinaire impli-
quant des experts en medecine faetale, conseilgenetique, soins aux h6mophiles et gen6tiquemol6culaire.
2. Les conductrices potentielles de lhemophiliedevront faire l'objet d'un examen des facteurs decoagulation des l'enfance afin d'6valuer le risquede problemes h6morragiques d'ordre clinique. Cesmemes femmes devront par la suite se voir pro-poser un diagnostic lorsqu'elles auront atteintl'age du consentement et de preference avantd'entamer une grossesse.
3. L'6valuation initiale du risque d'etre conductricede la maladie devra etre basee sur I'analyseg6n6alogique. Les tests ult6rieurs par analyseph6notypique devront comporter, lorsque ces tech-niques sont disponibles, I'talonnage de toutes lessubstances de r6ference utilis6es par rapport auxetalons internationaux approuves, et la probabilit6finale d'etre conductrice devra etre fond6e sur lesprobabilit6s group6es obtenues par I'analysegen6alogique et par les examens de laboratoire.
4. L'analyse g6notypique constitue la m6thode laplus exacte de d6tection de l'tat de conductrice.Dans l'h6mophilie, cette technique fera souventappel a des marqueurs li6s du polymorphismeafin de suivre la transmission d'un gene del'h6mophilie au sein d'une g6n6alogie. Cette 6va-luation commencera par l'tude des marqueursintrag6niques les plus informatifs compte tenu del'origine ethnique de la famille. Lorsque les cir-constances le permettent, le diagnostic g6n6tiquede lhemophilie sera bas6 sur l'identification direc-te de la mutation provoquant la maladie dans legene codant pour le facteur VIII ou le facteur IX.
5. On devra se procurer, et conserver a l'6chelonlocal ou national, de I'ADN de tous les h6mo-philes, afin de faciliter les diagnostics g6n6tiquesfuturs. Les soci6t6s nationales de l'h6mophiliedevront participer activement a ce processus.
6. Les membres de la famille qui regoivent unconseil g6n6tique devront etre inform6s deslimites des analyses de laboratoire et, par cons6-quent, etre en mesure de fournir un consentement6claire. Le processus de conseil devra etre coor-donne par un professionnel dOment forme, quidevra etre familiarise avec les principes des soinsaux h6mophiles et avec la g6n6tique clinique etmol6culaire. Le processus de conseil g6n6tiquedoit 6galement comprendre une 6valuation cri-tique du type d'h6mophilie chez les sujets de sexemasculin appartenant a la famille du consultant
WHO Bulletin OMS. Vol 71 1993454

Haemophilia: carrier detection and prenatal diagnosis
(h6mophilie A ou B, degre de gravite, presenceou absence du CRM, etc.).
7. Tous les sp6cialistes participant aux activitesde conseil g6n6tique aux h6mophiles devrontavoir un niveau de formation approprie. Pour cela,il conviendra de creer un registre international descentres de formation potentiels, registre qui seratenu a jour par la FMH. Certains de ces centrespourront, apres avoir ete dOment evalues, etrepressentis comme centres collaborateurs del'OMS.
8. L'OMS et la FMH devront proc6der a uneenquete internationale pour evaluer les labora-toires qui pratiquent le diagnostic de l'hemophilie,et par la suite creer un registre international deces laboratoires.
9. L'OMS et la FMH devront soutenir les basesde donnees internationales consacrees aux muta-tions specifiques de l'h6mophilie et encourager ledeveloppement de nouvelles technologies permet-tant d'identifier ces mutations. Si possible, toutesles donn6es g6netiques obtenues devront etreincorporees dans les registres nationaux del'hemophilie.
References1. Gitschier, J. Maternal duplication associated with
gene deletion in sporadic hemophilia. Am. j. hum.genet., 43: 274 (1988).
2. Gitschier, J. et al. Mosaicism and sporadic hae-mophilia: implications for carrier detection. Lancet,1: 274 (1989).
3. Higuchi, M. et al. A somatic mosaic for hemophi-lia A detected at the DNA level. Mol. biol. med., 5:23 (1988).
4. Brocker-Vriends, A.H.J.T. et al. Somatic origin ofinherited hemophilia A. Hum. genet., 85: 288(1 990).
5. Taylor, S.A.M. et al. Somatic mosaicism andfemale to female transmission in a kindred withhemophilia B (factor IX deficiency). Proc. NatlAcad. Sci. USA, 88: 39 (1991).
6. Brocker-Vriends, A.H.J.T. et al. Sex ratio of themutation frequencies in haemophilia A: coagula-tion assays and RFLP analysis. Hum. genet., 86:139 (1990).
7. Winter, R.M. et al. A maximum likelihood estimateof the sex ratio of mutation rates in haemophilia A.Hum. genet., 64: 156 (1983).
8. Rosendaal, F.R. et al. Sex ratio of the mutationfrequencies in haemophilia A: estimation andmeta-analysis. Hum. genet., 86: 139 (1990).
9. Montandon, A.J. et al. Direct estimate of the hae-mophilia B (factor IX deficiency) mutation rate andof the ratio of the sex-specific mutation rates inSweden. Hum. genet., 89: 319 (1992).
10. McCallum, C.J. et al. Factor Vil levels and bloodgroup antigens. Thromb. haemost., 50: 757(1983).
11. Hoyer, L.W. et al. Detection of haemophilia car-riers during pregnancy. Blood, 60: 1407 (1982).
12. Stableforth, P. et al. Effect of oral contraceptiveson factor Vil clotting activity and factor VIII rela-ted antigen in normal women J. clin. pathol., 28:498 (1975).
13. Simpson, N.E. & Biggs, R. The inheritance ofChristmas Factor. Br. j. haematol., 8: 191 (1962).
14. Graham, J.B. et al. Carrier detection in haemo-philia A: a comparative international study. I. Thecarrier phenotype. Blood, 67: 1554 (1986).
15. Graham, J.B. et al. Statistical study of genotypeassignment (carrier detection) in hemophilia B.Thromb. res., 15: 69 (1979).
16. Orstavik, K.H. et al. Factor Vil and factor IX in atwin population: evidence for a major effect ofABO locus on factor Vil levels. Am. j. hum.genet., 37: 89 (1985).
17. Briet, E. et al. Oral contraception and the detec-tion of carriers in haemophilia B. Thromb. res., 13:379 (1978).
18. Over, J. Quality control of the one-stage factorVil (VIII:C) assay in the coagulation laboratory.Scand J. haematol., 33 (Suppl. 41): 89 (1984).
19. Rosen, S. Assay of Factor VIII:C with a chromo-genic substrate. Scand. j. haematol., 86: 139(1984).
20. Mikaelsson, M. & Oswaldsson, U. Standardisa-tion of VIII:C assays: a manufacturer's view. In:Monograph on Factor VIII concentrates. Scand. j.haematol., 19 (in press).
21. Peake, I.R. & Bloom, A.L. Immunoradiometricassay of procoagulant factor VIII antigen in plas-ma and serum and its reduction in haemophilia.Lancet, 1: 473 (1978).
22. Zimmerman, T.S. et al. Detection of carriers ofclassic haemophilia using an immunologic assayfor antihaemophilic factor (factor VIII). J. clin.invest., 50: 255 (1971).
23. Ruggeri, Z.M. et al. Immunoradiometric assay offactor Vil related antigen, with observations in 32patients with von Willebrand's disease. Brit. j. hae-matol., 33: 221 (1976).
24. Laurell, C.B. Quantitative estimation of protein byelectrophoresis in agarose gel containing antibo-dies. Anal. biochem., 15: 45 (1966).
25. Green, P.P. et al. Carrier detection in haemophiliaA: a cooperative international study. II. The effica-cy of a universal discriminant. Blood, 67: 1560(1986).
26. van Dieijen-Visser, M.P. et al. Use of chromo-genic peptide substrates in the determination ofclotting factors 11, VIl, IX and X in normal plasmaand in plasma of patients treated with oral contra-ceptives. Haemostasis, 12: 241 (1982).
27. Roberts, H.R. et al. Genetic variants of haemo-philia B: detection by means of specific PTC inhi-bitor. J. clin. invest., 47: 360 (1968).
28. Holmberg, L. et al. Prenatal diagnosis of haemo-philia B by an immunoradiometric assay of factorIX. Blood, 56: 397 (1980).
WHO Bulletin OMS. Vol 71 1993 455

I.R. Peake et al.
29. Lyon, M.F. Sex-chromatin and gene action in themammalian X-chromosome. Am. j. hum. genet.,13: 135 (1962).
30. Orstavik, K.H. et al. Detection of carriers of hae-mophilia B. Br. j. haematol., 42: 293 (1979).
31. Kasper, C.K. et al. Hemophilia B: characterisationof genetic variants and detection of carriers.Blood, 50: 351 (1977).
32. Pechet, L. et al. Relationship of factor IX antigenand coagulant in haemophilia B patients and car-riers. Thromb. haemost., 40: 465 (1978).
33. Akhmeteli, M.A. et al. Methods for the detectionof haemophilia carriers: a Memorandum. Bulletinof the World Health Organization, 55: 675 (1977).
34. Graham, J.B. et al. Statistical methods for carrierdetection in the hemophilias. In: Bloom, A.L. ed.The hemophilias, Edinburgh, Churchill Livingstone,1982, p. 156.
35. Gitschier, J. et al. Characterization of the humanfactor Vil gene. Nature, 312: 326 (1984).
36. Yoshitake, S. et al. Nucleotide sequence of thegene for human factor IX (antihaemophilic factorB). Biochem. j., 24: 3736 (1985).
37. Tuddenham, E.G.D. et al. Haemophilia A: data-base of nucleotide substitution, deletions, inser-tions and rearrangements of the factor VIII gene.Nucleic acids res., 19: 4821 (1991).
38. GianneIli, F. et al. Haemophilia B: database ofpoint mutations and short additions and deletions.Second edition. Nucleic acids res., 19 (Suppl.):2193 (1991).
39. Lalloz, M.R.A. et al. Haemophilia A diagnosis byanalysis of a hypervariable dinucleotide repeatwithin the factor VIII gene. Lancet, 338: 207(1991).
40. Southern, E.M. Detection of specific sequencesamong DNA fragments separated by gel electro-phoresis. J. mol. biol., 98: 503 (1975).
41. Saiki, R.K. et al. Primer-directed enzymatic ampli-fication of DNA with a thermostable DNA poly-merase. Science, 239: 487 (1988).
42. Kogan, S.C. et al. An improved method of pre-natal diagnosis of genetic diseases by analysisof amplified DNA sequences. Application to hemo-philia A. New Engl. j. med., 317: 985 (1987).
43. Bowen, D.J. et al. Facile and rapid analysis ofthree DNA polymorphisms within the human factorIX gene using the polymerase chain reaction. Br.j. haematol., 77: 559 (1991).
44. Chan, V. et al. Multiple Xbal polymorphisms forcarrier detection and prenatal diagnosis of haemo-philia A. Br. j. haematol., 73: 497 (1989).
45. Graham, J.B. et al. Carrier detection in the hae-mophilias. Some limitations of the DNA methods.Blood, 66: 759 (1985).
46. Winship, P.R. & Brownlee, G.G. Diagnosis ofhaemophilia B carriers using intragenic oligonu-cleotide probes. Lancet, 2: 218 (1986).
47. Zhang, M. et al. The factor IX BamHl polymor-phism: T-to-G transversion at the nucleotidesequence-561. The BamHI/MSPI haplotypes inblacks and Caucasians. Hum. genet., 82: 283(1989).
48. Driscoll, M.C. et al. A second BamHl DNA poly-
morphism and haplotype association in the factorIX gene. Blood, 72: 61 (1988).
49. Chan, V. et al. Molecular defects in haemophiliaB: detection by direct restriction enzyme analysis.Br. j. haematol., 79: 63 (1991).
50. Kojima, T. et al. Possible absence of commonpolymorphisms in coagulation factor IX gene inJapanese. Blood, 69: 349 (1987).
51. Scott, C.R. et al. Hemophilia B: population dif-ferences in RFLP frequencies useful for carrierdetection. Am. j. hum. genet., 41: O.A262 (1987)(abstr.).
52. Graham, J.B. et al. The varying frequencies offive DNA polymorphisms of X-linked coagulantfactor IX in eight ethnic groups. Am. j. hum.genet., 49: 537 (1991).
53. Winship, P.R. et al. Detection of polymorphismsat cytosine phosphoguanadine dinucleotides anddiagnosis of haemophilia B carriers. Lancet, 1:631 (1989).
54. Mulligan, L. et al. A DNA marker closely linked tothe factor IX (haemophilia B) gene. Hum. genet.,75: 381 (1987).
55. Giannelli, F. Factor IX. In: Bailliere's Clinicalhematology, Volume 2. London, W.B. Saunders,1989, p. 821.
56. Winship, P.R. et al. Carrier detection in haemo-philia B using two further intragenic restrictionfragment length polymorphisms. Nucleic acidsres., 12: 8861 (1984).
57. Graham, J.B. et al. The utility of a HindlIl poly-morphism of factor Vil examined by rapid DNAanalysis. Br. j. haematol., 76: 75 (1990).
58. Chan, V. et al. Restriction fragment length poly-morphisms associated with factor VIlI:C gene inChinese. Hum. genet., 79: 128 (1988).
59. Kenwrick, S. & Gitschier, J. A contiguous 3 Mbphysical map of Xq 28 extending from the colourblindness locus to DXS15. Am. j. hum. genet., 45:873 (1989).
60. Harper, K. et al. A clinically useful DNA probelinked to haemophilia A. Lancet, 2: 6 (1984).
61. Oberle, I. et al. Genetic screening for hemophiliaA (classic hemophilia) with a polymorphic DNAprobe. New Engl. j. med., 312: 682 (1985).
62. Driscoll, M.C. et al. Recombination between factorVIII:C gene and St14 locus. Lancet, 2: 279 (1986).
63. Janco, R.L. et al. Carrier testing strategy in hemo-philia A. Lancet, 1: 148 (1986).
64. Peake, I.R. Registry of DNA polymorphism withinor close to the human factor VIII and factor IXgenes. For the Factor Vil and Factor IX Subcom-mittee of the Scientific and Standardization Com-mittee of the International Society of Thrombpsisand Haemostasis. Thromb. haemost., 67: 277(1992).
65. Wion, K.L. et al. A new polymorphism in the factorVIII gene for prenatal diagnosis of haemophilia A.Nucleic acids res., 14: 4535 (1986).
66. Suehiro, K. et al. Carrier detection in Japanesehemophilia A by the use of three intragenic andtwo extragenic factor Vil DNA probes: a study of24 kindreds. J. lab. clin. med., 112: 314 (1988).
67. Lillicrap, D.P. et al. Variation of the non-factor VIII
456 WHO Bulletin OMS. Vol 71 1993

Haemophilia: carrier detection and prenatal diagnosis
sequences detected by a probe from intron 22 ofthe factor VIII gene. Blood, 75: 139 (1990).
68. Pecorara, M. et al. Hemophilia A: carrier detectionand prenatal diagnosis by DNA analysis. Blood,70: 531 (1987).
69. Higuchi, M. et al. Molecular characterization ofmild-to-moderate hemophilia A: detection of themutation in 25 of 29 patients by denaturing gra-dient gel electrophoresis. Proc. Natl Acad. Sci.USA, 88: 8307 (1991).
70. Higuchi, M. et al. Molecular characterization ofsevere hemophilia A suggests that about half themutations are not within the coding regions andsplice junctions of the factor Vil gene. Proc. NatlAcad. Sci. USA, 88: 7405 (1991).
71. Rodeck, C.H. & Campbell, S. Sampling pure fetalblood in the second trimester of pregnancy. Br.med. j., 2: 728 (1978).
72. Daffos, F. et al. Fetal blood sampling during preg-nancy with use of a needle guided by ultrasound: astudy of 606 consecutive cases. Am. j. obstet.gynecol., 153: 655 (1985).
73. Nicolaides, K.H. et al. Ultrasound-guided sam-pling of umbilical cord and placental blood toassess fetal well-being. Lancet, 1: 1065 (1986).
74. Hahneman, N. & Mohr, J. Genetic diagnosis inthe embryo by means of biopsy of extra amnioticmembrane. Bull. Europ. Soc. Hum. Genet., 2: 23(1968).
75. Brambati, B. et al. Genetic diagnosis before theeighth gestational week. Obstet. gynecol., 77: 318(1 991).
76. Firth, H.V. et al. Severe limb abnormalities afterchorion villus sampling at 55-66 days gestation.Lancet, 337: 762 (1991).
77. Medical Research Council European trial of cho-rion villus sampling. Lancet, 337: 1491 (1991).
78. Byrne, D. et al. Randomized study of early amnio-centesis versus chorionic villus sampling at 10-13weeks gestation. Technical and cytogenetic com-parison of 650 patients. Ultrasound obstet. gyne-col., 1: 52 (1991).
79. Mibashan, R.S. et al. Prenatal diagnosis of hemo-static disorders. In: Alter, B.P., ed. Perinatalhematology. Edinburgh, Churchill-Livingstone,1989, p. 64.
80. Peake, I.R. Blood samples for DNA extraction andstorage. Hemophilia world, 7: 12 (1991).
81. Saiki, R.K. et al. Diagnosis of sickle cell anemiaand beta thalassemia with enzymatically amplifiedDNA and non-radioactive allele-specific oligo-nucleotide probes. New Engl. j. med., 319: 537(1988).
82. Saiki, R.K. et al. Genetic analysis of amplifiedDNA with immobilized sequence-specific oligo-nucleotide probes. Proc. Natl Acad. Sci. USA, 86:6230 (1989).
83. Naylor, J.A. et al. Detection of three novel muta-tions in two haemophilia A patients by rapidscreening of whole essential region of factor VIIIgene. Lancet, 337: 635 (1991).
84. Naylor, J.A. et al. Screening ectopic transcripts ofthe factor Vil gene by chemical mismatch detec-tion rapidly locates mutations in haemophilia A.
International Society of Haematology Proceedings,August 1992, vol. 39 (abstract).
85. Verlinsky, Y. et al. Analysis of the first polar body:preconception genetic diagnosis. Hum. reprod., 5:826 (1990).
86. Verlinsky, Y. Biopsy of human gametes. In: Ver-linsky, Y. & Kuliev, A., ed. Preimplantation gene-tics. London, Plenum Press, 1991, p. 39.
87. Handyside, A.H. et al. Pregnancies from biopsiedhuman preimplantation embryo sexed by DNAamplification. Nature, 344: 768 (1990).
88. Handyside, A.H. et al. Biopsy of human preim-plantation embryos and sexing by DNA amplifica-tion. Lancet, 1: 347 (1989).
89. Dokras, A. et al. Trophectoderm biopsy in humanblastocystes. Hum. reprod., 5: 821 (1990).
90. Mueller, U.W. et al. Isolation of fetal trophoblastcells from peripheral blood of pregnant women.Lancet, 336: 179 (1990).
91. Iverson, G.M. et al. Detection and isolation of fetalcells from maternal blood using the fluorescence-activated cell sorter (FACS). Prenat. diagn., 1: 61(1981).
92. Bianchi, D.W. et al. Isolation of fetal DNA fromnucleated erythrocytes in maternal blood. Proc.Natl Acad. Sci. USA, 87: 3279 (1990).
93. Wantel, S.S. et al. Fetal cells in the maternal cir-culation: isolation by multiparameter flow cytometryand confirmation by PCR. Hum. reprod., 6: 1466(1991).
94. Giles, A.R. et al. A canine model of hemophilic(Factor VIII:C deficiency) bleeding. Blood, 60: 727(1982).
95. Evans, J.P. et al. Molecular cloning of a cDNAencoding canine factor IX. Blood, 74: 207 (1989).
96. Palmer, T.D. et al. Genetically modified skin fibro-blasts persist long after transplantation but gra-dually inactivate introduced genes. Proc. NatlAcad. Sci. USA, 88: 1330 (1991).
97. Hoeben, R.C. et al. Towards gene therapy inhemophilia A: efficient transfer of factor VIIIexpression vector into somatic cells. Thromb.haemost., 65: 727 (1991) (abstr.).
98. Kenwrick, S. et al. A Taql polymorphism adjacentto the factor Vil gene (F8C). Nucleic acids res.,19: 2513 (1991).
99. Kogan, S. & Gitschier, J. Mutations and a poly-morphism in the factor VIII gene discovered bydenaturing gradient gel electrophoresis. Proc. NatlAcad. Sci. USA, 87: 2092 (1990).
100. Ahrens, P. et al. A new HindlIl restriction fragmentlength polymorphism in the haemophilia A locus.Hum. genet., 76: 127 (1987).
101. Lalloz, M.R.A. et al. Haemophilia A diagnosis byanalysis of a novel dinucleotide tandem repeatsequence within the factor VIII gene. Br. j. haema-tol., 80: 3a (1992) (abstr.)
102. Antonarakis, S.E. et al. Hemophilia A: detectionof molecular defects and of carriers by DNA analy-sis. New Engl. j. med., 313: 842 (1985).
103. Youssoufian, H. et al. Mspl polymorphism in the3' flanking region of the human factor VIII gene.Nucl. acids res., 15: 6312.(1987).
104. Winship, P.R. & Peake, I.R. A clinically useful
WHO Bulletin OMS. Vol 71 1993 457

I.R. Peake et al.
RFLP in the 5' flanking region of the factor IXgene. Br. j. haematol., 80: 26a (1992) (abstr.).
105. Hay, C.W. et al. Use of a BamHl polymorphism inthe factor IX gene for the determination of haemo-philia B carrier status. Blood, 67: 1508 (1986).
106. Giannelll, F. et al. Characterisation and use of anintragenic polymorphic marker for detection of car-riers of haemophilia B (factor IX deficiency). Lan-cet, 1: 239 (1984).
107. Camerino, G. et al. A new Mspl polymorphism inthe haemophilia B locus. Hum. genet., 71: 79 (1985).
108. Graham, J.B. et al. The MaImo polymorphism offactor IX: establishing the genotypes by rapid ana-lysis of DNA. Blood, 73: 2104 (1989).
109. Van-der-Water, N.S. et al. Restriction fragmentlength polymorphisms associated with the factorVil and factor IX genes. J. med. genet., 28: 171(1 991).
110. Walknowska, J. et al. Practical and theoreticalimplications of fetal/maternal lymphocyte transfer.Lancet, 1: 1 1 19-1 122 (1969).
WHO Bulletin OMS. Vol 71 1993