helix–coil transition theory including long-range electrostatic interactions: application to...
TRANSCRIPT

Helix-Coil Transition Theory Including Long-Range Electrostatic Interactions:
Application to Globular Proteins
MAX VASQUEZ, Baker Laboratory of Chemistry, CorneU Unioersity, Ithuca, New York 14853-1301; MATTHEW R. PINCUS,
Department of Pathology, New York University Medical Center, New York, New York 10016; and HAROLD A. SCHERAGA,*
Baker Laboratory of Chemistry, Cornell University, Ithaca, New York, 14853-1301
Synopsis
An extension of the Zimm-Bragg two-state theory for the helix-coil transition in polypeptides, which takes into account the effect of peptide charge-dipole interactions on helix stability, is presented. This new theory incorporates these interactions in an expression that is parameterized on recently obtained experimental data on polypeptides for which electrostatic effects are known to influence helix content. Unlike previous two-state or multistate models, which are para- meterized on protein x-ray data, the present theoretical treatment in independent of such protein data. The theoretical model is applied to a series of peptides derived from the C-peptide of ribonuclease A, which have been the object of recent spectroscopic studies. The new theoretical approach can account for most of the structural information derived from studies of these C-peptides, and for overall average helix ptobabilities that are close in magnitude to those observed for these polypeptides in solution. An application of this new formulation for the prediction of the locations of &-helices in globular proteins from their amino acid sequence is also presented.
INTRODUCTION
Shoemaker et al.l investigated the stability of the C-peptide helix in the 13-residue N-terminal segment of bovine pancreatic ribonuclease A, and found that the high helix content could not be accounted for by the nearest-neighbor Ising model, using the Zimm-Bragg' nucleation and growth parameters u and s, determined from experiments on host-guest random copolymers of amino acids.3 They suggested that, instead, the main source of the stability of the C-peptide helix might arise from charge-dipole interactions,' similar to those proposed earlier by Blagdon and Goodman,4 and by Ihara et We subse- quently accounted for the apparent failure of the Ising model by pointing out that the values of u and s determined from random copolymers pertain to the intrinsic properties of each residue, reflecting side chain-backbone inter- actions, but not including longer range (charge-dipole) electrostatic interac- tions that are averaged out in analyzing experimental data on random copolymers.6 Therefore, in order to apply these intrinsic values of u and s properly to a specific-sequence copolymer, the values of u and s must be
*To whom requests for reprints should be addressed.
Biopolymers, Vol. 26, 351-371 (1987) 8 1987 John Wiley & Sons, Inc. CCC 0006-3525/87/030351-21$04.00

352 VASQUEZ, PINCUS, AND SCHERAGA
modified by reintroducing the long-range (charge-dipole) electrostatic interac- tions. With this modification, it is possible to retain the original formalism of the Ising model, but with altered values of s (neglecting such electrostatic effects on a), to compute helix-probability profiles. *
In this paper, we modify the Ising-model treatment of the helix-coil transition by introducing the effects of long-range (charge-dipole) electro- static interactions into the values of s. The values of the parameters characterizing the electrostatic interactions are determined by fitting the computed helix-probability profiles of C-peptide derivatives to experimental data. The revised values of s are then tested by using them to compute helix-probability profiles of globular proteins for which x-ray crystallographic data exist. In the accompanying paper,7 we apply the new treatment to several peptide analogs of the C-terminal region of cytochrome c, and show that the tendency toward formation of the a-helix conformation in these peptides correlates with their antigenicity in a T-lymphocyte proliferation assay.
ORIGINAL THEORETICAL, MODEL In the Zimm-Bragg formulation of the one-dimensional Ising model,2 each
unit (amino acid residue) can exist in one of two states-helix or coil. If we consider the case where the Occurrence of a given unit in a given conforma- tional state depends only on the state and the type of this unit and on the state, but not the type, of its nearest neighbors, the partition function for the system can be written, for a chain of N residues, as
where WA( j ) is a 2 X 2 matrix of statistical weights for the j t h amino acid, which is of type A:
In the original f~rmulat ion,~?~ the statistical weights s A ( j ) and aA( j ) at a given temperature depend only on the type A of the residue, but not on its position j , along the chain. The values of s and u have heretofore been taken from the studies of host-guest random copolymers carried out in this labora-
In this extension of the theory, however, these values of s are modified by the procedure described in the next section.**
*Alternatively, instead of Using a phenomenological treatment (i.e., Ising model), the stability of the C-peptide helix can be d i s c d in terms of pair interaction energies, using the ECEPP (Empirical Conformational Energy Program for Peptides) algorithm (Y. K. Kang, G. Nhmethy, and H. A. Scheraga, work in progreas).
**Strictly speaking, the 2 X 2 matrix formulation of the helix-coil t r a n ~ i t i o n ~ . ~ pertains to long c$ains, and the host-guest copolymer experiments3** were indeed carried out on long chains. Short chains, however, are really not treated adequately by this theory because it does not provide a sufticiently accurate treatment of end effects. For short chains (e.g., the C-peptide of fibnuclew A), it necessary to we a more elaborate theory involving an 11 X 11 matrix? Nevertheleas, we retain the 2 x 2 matrix treatment here, even for short chains, in order to make u6e of the values of u and s from host-guest copolymers3~* that, in turn, were evaluated with the 2 x 2 matrix theory.

MODIFIED HELIX-COIL TRANSITION THEORY 353
Helix probability profiles for specific-sequence copolymers can be computed with the same theory as heretofore,1°-12 but with the modified values of s. These profiles are plots of PH(i ) vs i, where PH(i) is the probability that residue i is in a helical state (independent of the state of the other N - 1 residues). PH(i ) can be expressed as
We can also compute the probability that a set A, not necessarily a contigu- ous sequence, of residues, simultaneously adopts the helical conformation, viz.,
where the symbol “I E A” means that residues I are included in set A. The quantity PH(A), as well as PH(i) , will be used in the analysis of the cytochrome c peptides.’
MODIFIED THEORETICAL MODEL
We now improve the original model by modifying the values of sA( j ) that were originally obtained from the studies of random copolymers; i.e., we now include the effects of long-range electrostatic interactions that were averaged out in the random copolymers.
We mume that the backbone of each residue has a dipole associated with it. This dipole will be aligned along the direction of the helix axis whenever the residue in question is in a helical state and will take on random orienta- tions if the residue is in a coil state. As a consequence of this behavior, the charges on the side chains of all other residues (and on the end groups) will affect the helix stability of the residue under consideration: its backbone dipole will interact with all of the charges on the molecule, with a net nonzero energy only when the residue under consideration is in a helical, i.e., aligned, state, no matter what the length of the run of helical states is. This interac- tion energy will thus affect the tendency of the backbone dipole of this residue to be aligned along the helix axis and will also modify the tendency of this residue to attain the helical state. The sign of this interaction (for a given charge) will depend on both the sign of the charge and on its position relative to the dipole. It will be negative (a stabilizing interaction) if the charge is positive and on the C-terminal side of the dipole, and positive (a destablizing interaction) if it is on the N-terminal side of the dipole; the opposite effect will be observed when the charge is negative. The magnitude of the interac- tion will decrease with increasing distance between the charge and the dipole.
These electrostatic effects (due to the charges on all of the other residues) can be taken into account by altering the original (intrinsic) host-guest values of sA( j ) ; the effect of a charge on the residue under consideration is already included in the intrinsic value3~‘*’ of sA(j) , and need not be considered explicitly in this modified treatment. Hereafter, we refer to the intrinsic value of the helix-coil stability constant of residue i as s;, and the modified value as si.

354 VASQUEZ, PINCUS, AND SCHERAGA
The true helix stability constant of residue i, si, depends on the intrinsic stability constant for this residue, s;, and on the electrostatic free energy due to the presence of charges on the other residues of the polypeptide. The true helix stability constant can then be taken as the produce of two factors, viz.,
s, = s,'exp( -AGJRT)
where AG, = (Gh - Gc)i, with G, (the electrostatic free energy of residue i in the coil state) taken as zero; Gh (= AG,) is the electrostatic free energy of residue i in the he& state, i.e., the electrostatic free energy due to the interaction of the helix dipole of residue i with the charges on the side chains of the other residues in the sequence, R is the gas constant, and T is the absolute temperature.
It is nece8881y to obtain an expression for AGi in terms of the helix dipole moment of the i th residue in a particular sequence, and the distances between the center of the dipole and the charges on the side chains of the other residues. Exact computation of the latter quantity is not possible because it requires knowledge of the exact positions of the charged side-chain atoms. These distances, however, can be approximated by making the following assumptions:
1. In order to calculate AGi, it is necessary to know the distance between backbone dipoles in the helical conformation. For this purpose, consider a two-turn helix with residue i in the middle of it; this means that residues i - 4 through i + 4 are assumed to be in the helical conformation. Hence, the distance rp between the backbone dipoles of two residues, i and j , is proportional to li - j l , i.e., is equal to [a(K)]k whereOK = li - j ( for K = 1, 2, 3, and 4, for which a ( k ) = 3.8, 2.7, 1.7, and 1.6 A, respectively. These values for a ( k ) were chosen to reproduce the average observed distances between the C" atoms in helical conformations of globular protein^.^^ Residues i - 5, i - 6, etc., and i + 5, i + 6, etc., are assumed to be in the coil state. The distances between these remote residues and residue i are, again, assumed to be proportional to I i - jl , where a( K ) is now 2.2,2.0, and 1.8 A, for k = 5,6, and 2 7, respectively. These values for a ( k ) correspond to observed distances between C" atoms averaged over all conformations in globular pr~teins. '~ Therefore, rp is a function of I i - jl only. The form of this function, viz., [ a ( k ) ] l i - j l , involves the assumption that the chain is approximately linear and does not fold back on itself significantly.
2. The average distance rN, between the charge on the side chain of residue j and the backbone dipole of residue j is estimated as 5.0 A. The distance between the charge on side chain j and the backbone dipole of residue i is then (r& + r;)'I2, where rp is assumed to be a function of li - j l only. Because of the latter assumption, it follows that the distance, and hence the electrostatic interaction energy, between the backbone dipole of residue i (in a helical state) and the charge on the side chain of any other residue j are independent of the conformation of residue j and of that of all residue between i and j .
MODIFIED HELIX-COIL TRANSITION THEORY 355
3. The interaction energy E, between a dipole p and a point charge q is given by
E = qp . r/Dlr13 = qlplcos8/DlrI2 (6)
where r is a vector from the center of the dipole to the point charge, D is the dielectric constant of the medium, and 8 is the angle between the dipole p and the vector r. Since the precise positions of the charges on the side chains of residues j relative to the backbone dipole of residue i are not known, and hence 8 is not known, the further assumption is made that the vectors p and r are roughly colinear, i.e., 8 = 0" or 180" (or cos 8 = f 1).
The value of AGi for residue i in a polypeptide segment is then approxi- mated by the expression
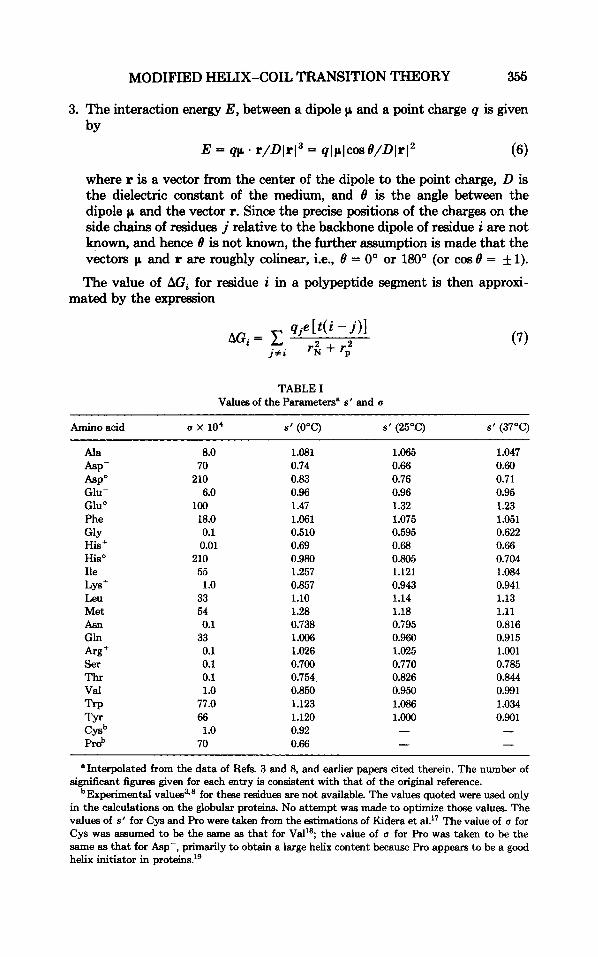
TABLE I Values of the Parametersn s' and a
Amino acid a x 10' s' (OOC) s' (25°C) s' (37°C)
Ala Asp- ASP" Glu- Glu' Phe GlY HiS+ His" Ile
Leu Met Asn Gln
Ser Thr Val np TYr Cysb Prob
LYS+
8.0 70 210
100 6.0
18.0 0.1 0.01
210 55
33 54
33
1 .o
0.1
0.1 0.1 0.1 1 .o 77.0 66 1 .o 70
1.081 0.74 0.83 0.96 1.47 1.061 0.510 0.69 0.980 1.257 0.857 1.10 1.28 0.738 1.006 1.026 0.700 0.754 0.850 1.123 1.120 0.92 0.66
1.065 0.66 0.76 0.96 1.32 1.075 0.595 0.68 0.805 1.121 0.943 1.14 1.18 0.795 0.960 1.025 0.770 0.826 0.950 1.086 1 .Ooo - -
1.047 0.60 0.71 0.95 1.23 1.051 0.622 0.66 0.704 1.084 0.941 1.13 1.11 0.816 0.915 1.001 0.785 0.844 0.991 1.034 0.901
"Interpolated from the data of Refs. 3 and 8, and earlier papers cited therein. The number of significant figures given for each entry is consistent with that of the original reference.
bExperimental v a l ~ e s ~ . ~ for these residues are not available. The values quoted were used only in the calculations on the globular proteins. No attempt was made to optimize t h a e values. The values of s' for Cys and Pro were taken from the estimations of Kidera et al?7 The value of a for Cys was assumed to be the same as that for Val'$ the value of a for Pro was taken to be the same as that for Asp-, primarily to obtain a large helix content because Pro appears to be a good helix initiator in protein^.'^

356 VASQUEZ, PINCUS, AND SCHERAGA
where e is proportional to the magnitude of the backbone dipole moment of residue i (see below) and t( i - j ) is the orientational dependence of the dipole and distance vectors (i.e., the angle 0). From assumption 3:
To gain an idea of the reasonableness of the assumptions upon which Eq. (7) is based, the value of the dielectric constant D, which has been estimated by independent methods,14-16 was computed by comparing Eqs. (6) and (7). If the angle B between the dipole p and the vector r is 0" or 180", and if the magnitude of the dipole moment p is taken as 3.5 Debye units (= 0.73 eA),14-16 then qe/r2 = qp/Dr2, or D = p/e , and a value (see below) of e = 11 (in units such that AGi is in kcal/mole when the r 's are in A, and the q j k are in electronic charge units) will correspond to a value of about 24 for the effective dielectric constant D, in agreement with the other independent estimates of this quantity.14-16 Therefore, if D is assigned a value of 24, and the peptide bond dipole is assumed to be paralled to the helix axis, the electrostatic potential created by this dipole arrangement is equivalent to that corresponding to two opposite charges of 0.73 N/1.5 N, or about +0.5 and - 0.5 elementary charge units, placed at the N- and C-termini, respectively, of the chain (cf. Ref. 14). The value of 11 taken for e was obtained by using Eqs. (5) and (7) with the original (host-guest) values of s; (and a), given in Table I, and fitting to the results of Shoemaker et al.' for C-peptide derivatives of ribonuclease A, as now discussed.
Determination of the constant e
The helix-coil transitions for the C-peptide of ribonuclease A and its homologues (see Table 11) have been studied' at OOC. These data were used to obtain the parameter e (equal to the average magnitude of the dipole moment divided by the dielectric constant) in eq. (7); i.e., the value of e was adjusted so that the computed value of (Pi), using eqs. (3), (5), and (7), reproduced the experimentally determined helix contents.
The data' on the helix contents of reference peptide I and its [Ala(5) + His] derivative (i.e., with Ala at position 5 changed to His) were used to find an appropriate value for the parameter e introduced above (see Table I1 for a description of these peptides). A helix content of about 48% was found experimentally at 0' for reference peptide I, while a value of 25% was estimated experimentally for the [Ala(5) + His] derivative.' A value of e of about 12 would reproduce the 48% result for reference peptide I, but it would greatly overestimate the helix content of the peptide with the [Ala(5) + His] substitution. On the other hand, a value of e of about 11 fits the helix content of the His(5) peptide, but underestimates the helix content of reference peptide I by a small amount. The value of e equal to 11 was thus adopted, since it gives reasonable enough agreement in both cases (see Table 111). Other factors probably d e c t the stability of the helical conformation in these peptides in water, in addition to those taken into account explicitly in this model. Also, slightly different estimates for the helix contents of some of the
MODIFIED HELIX-COIL TRANSITION THEORY 357
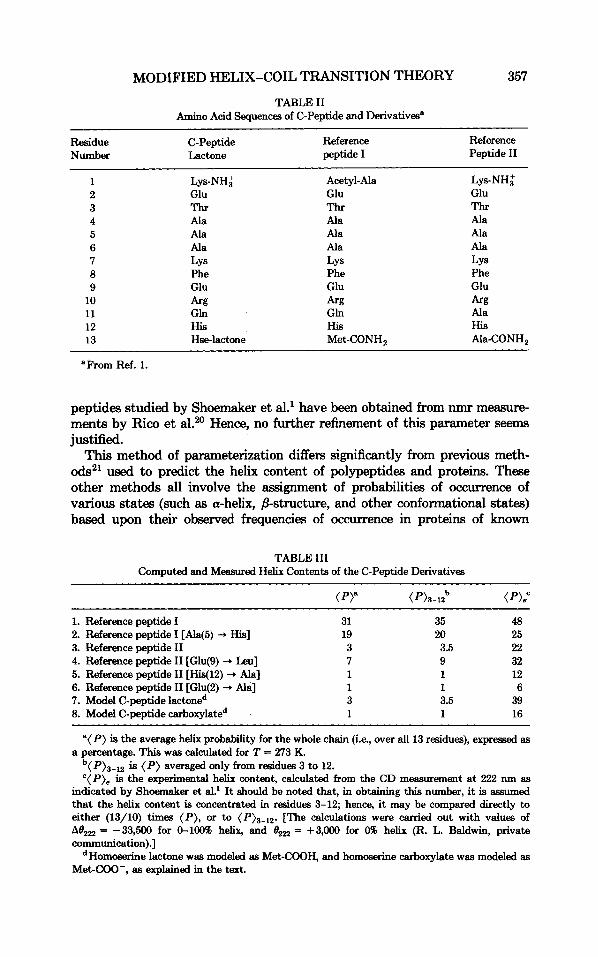
TABLE I1 Amino Acid Sequences of C-Peptide and Derivatives"
~~ ~
Residue C-Peptide Reference Reference peptide I Peptide I1 Number Lactone
1 Lys-NH; Acetyl- Ala Lys-NH; 2 Glu Glu Glu 3 Thr Thr Thr 4 Ala Ala Ala 5 Ala Ala Ala 6 Ala Ala Ala 7 LYS LYS LYS 8 Phe Phe Phe 9 Glu Glu Glu
10 k g k g k g 11 Gln Gln Ala 12 His His His 13 Hse-lactone Met-CONH, Ala-CONH,
"From Ref. 1.
peptides studied by Shoemaker et al.' have been obtained from nmr measure- ments by Rico et a1.20 Hence, no further refinement of this parameter seems justified.
This method of parameterization differs significantly from previous meth- ods" used to predict the helix content of polypeptides and proteins. These other methods all involve the assignment of probabilities of occurrence of various states (such as a-helix, /3-structure, and other conformational states) based upon their observed frequencies of occurrence in proteins of known
TABLE I11 Computed and Measured Helix Contents of the C-Peptide Derivatives
1. Reference peptide I 2. Reference peptide I [Ala(5) 3. Reference peptide I1 4. Reference peptide I1 [Glu(9) Leu] 5. Reference peptide I1 [His(l2) -D Ala] 6. Reference peptide I1 [Glu(2) -D Ala] 7. Model C-peptide lactoned 8. Model C-peptide carboxylated
His] 31 19 3 7 1 1 3 1
35 20 3.5 9 1 1 3.5 1
48 25 22 32 12 6
39 16
"( P ) is the average helix probability for the whole chain (i.e., over all 13 residues), expressed as
b ( P)a-12 is ( P ) averaged only from residues 3 to 12. ' (P) , is the experimental helix content, calculated from the CD measurement a t 222 nm as
indicated by Shoemaker et al? It should be noted that, in obtaining this number, it is assumed that the helix content is concentrated in reaidues 3-12; hence, it may be compared directly to either (13/10) times ( P ) , or to (P)3-12. [The calculations were carried out with values of At),, = -33,500 for 0-10046 helix, and O,, = +3,OOO for 0% helix (R. L. Baldwin, private communication).]
dHomoserine lactone was modeled as Met-COOH, and homoserine carboxylate was modeled as Met-COO-, as explained in the text.
a percentage. This was calculated for T = 273 K.

358 VASQUEZ, PINCUS, AND SCHERAGA
structure. The results therefore depend on the particular set of proteins selected. In addition, the computed individual residue probabilities are very small. Hence, the decision as to whether a residue is helical or nonhelical depends on whether its computed helix probability lies above a selected cutoff value.l0
In the method presented here, the intrinsic helical parameters are de- termined from studies of solutions of homopolymers and ~ ~ p ~ l y m e r ~ , ~ . * and the electrostatic effects are based on studies of model peptides where these effects are known to be important.' The computed values of such quantities as ( PH) are therefore independent of x-ray structural data on proteins. Further- more, the computed values of ( PH) are not low in value, and are in agreement with observed helix contents of medium-sized polypeptides, as discussed in the results and discussion section.
Temperature Dependence of the Helix Content
The helix content of the C-peptide was studied further22 as a function of temperature from 0" to 37°C. Attempts to use the simple temperature dependence of Eqs. (3), (5), and (7) did not reproduce the experimental valueB of AH determined in these studies. Hence, another temperature-dependent term was added to Eq. (7). The particular form that best reproduced the temperature dependence of the helix content was BIqj;.I(T - 273), where all of the terms except B, an adjustable entropic parameter, were identified above.
The full temperature-dependent expression for the electrostatic free energy then becomes
The best value of B was determined to be 0.8 by using Eqs. (3) and (9) to compute the observed value of AH for the C-peptide, which was found by CD measurements20*22 to be - 8 to - 11 kcal/mole at pH 5.5.
EVALUATION OF PARAMETERS FOR THE MODIFIED THEORETICAL MODEL
The evaluation of the two adjustable parameters, e and B, was described in the previous section. Reference peptides I (entries 1 and 2 in Table 111), rather than the C-peptide, were chosen as the basis for the parameterization of the model because of the presence in the C-peptide of a residue (the C-terminal homoserine lactone) for which the host-guest values of IJ and s' were not available. In subsequent calculations, the C-terminal residue of the C-peptide was modeled as terminally blocked methionine (i.e., as an uncharged C- terminus, CONH, or COOH) in the case of the C-peptide lactone; and as Met-carboxylate in the case of the C-peptide carboxylate. The other peptides listed in Table I1 were modeled directly from their sequences.
pH Dependence of the Helix Content
The calculations described above correspond to pH values at which Glu, Asp, Lys, Arg, and His are fully charged. Additional calculations were per- formed in which the qj 's were taken as functions of pH: this dependence was

MODIFIED HELIX-COIL TRANSITION THEORY 359
introduced in a straightforward manner by assigning suitable pK valuesz3 to the ionizable side chains, and by making qj proportional to the fraction of charged species, as derived from the ionization equilibrium equations. The dependence of the host-guest parameters s; on pH, for residues with ionizable side-chain groups, was also taken into account in these calculations: since those values are usually available for extremes of state of ionization, a simple interpolation was carried out, using the ratios of charged to uncharged species given by the ionization equilibrium expressions. These titration curves were calculated for some of the C-peptide derivatives studied by Shoemaker et al.' In particular, the effect of pH on helix stability was explored for reference peptide I. The very approximate way in which the pH dependence has been introduced, viz., the assumption that the pK 's are independent of conforma- tion, implies that the results are It.= reliable for intermediate degrees of ionization, i.e., for pH values close to any of the pK 's of the ionizable groups involved.
Temperature Dependence of Helix Content: Parameter B
There are, undoubtedly, entropic effects associated with the dipole-charge interaction in a polypeptide s ~ s t e m . ~ These and other temperature-dependent effects, such as variation of the dielectric constant with temperature and more complex solvent effects, have been included in the empirical parameter B of Eq. (9). In order to obtain a numerical value for By agreement was sought for the observed temperature dependence of the helix content of reference peptide I. The modified model gives an apparent AH for helix formation of -10 kcal/mol for reference peptide I, and around -8 kcal/mol for the C-peptide, when the pH is chosen so that all ionizable side-chain (and N-terminal, for the case of C-peptide) groups are fully charged. These apparent AH'S were calculated from van't Hoff plots under the same assumptions used by Bierzyn- ski et al.," but using the average helix probabilities computed from the present model. When the pH is taken into account explicitly in the calcula- tions, a weak dependence of AH on pH is observed; for reference peptide I, the apparent AH of helix formation is -8 kcal/mol at pH 3, goes down to - 10 kcal/mol around pH 5, and increases up to -7.5 kcal/mol at pH 8.
It should be noted that, because of the manner in which B appears in the expression for the modification of the values of s; [see Eq. (9)], the choice of the given value for B will be inconsequential unless the temperature differs from 273 K. Hence, only the results for AH reported in this section, as well as the results at higher temperatures for the cytochrome c peptides presented in the accompanying paper,' are affected by the numerical value chosen for B.
RESULTS AND DISCUSSION
Evaluation of Parameters Helix-probability profiles were computed for the peptides listed in Tables I1
and 111, using e = 11, B = 0.8, and T = 273 K, as described in the previous section. The average helix probabilities are listed in Table 111. The helix profiles are presented in Figs. 1 and 2. The modified values of s used for reference peptide I are presented in Table IV. The experimentally determined

360 VASQUEZ, PINCUS, AND SCHERAGA
I I
0.4 - ‘A -
-
-
.- Y
I a
0.2 -
0-0-0-
- 0 d 3 E Q - 0 5 10 15
Residue Number ( i ) Fig. 1. Helix-probability profiles a t 273 K for the C-peptide lactone (0), the C-peptide
carboxylate (a), reference peptide I (A), and reference peptide I with the substitution Ala(5) + His (0).
‘A -
\A
h \
O-O-O,o,O\ <\A -
// ’” -
k0
0~-~c0-0-0-0-~-o-8-~ 0’
\O
\O
s””’. .-
a
0.2
0-0-0-
O d 3 E Q - 0 5 10 15
Fig. < O . I 1. Helix-probability profiles Residue at 273 K for Number the C-peptide ( i ) lactone (0), the C-peptide
carboxylate (a), reference peptide I (A), and reference peptide I with the substitution Ala(5) + His (0).
0
I I
5 10 Residue Number ( i )
15
Fig. 2. Helix-probability profiles at 273 K for reference peptide I1 (0), and reference peptide I1 with the substitutions Glu(9) + Leu (a), His(l2) + Ala (O), and Glu(2) + Ala (0).

MODIFIED HELIX-COIL TRANSITION THEORY 361
TABLE IV Values of Parameters s' and s for Reference Peptide I
Residue" S f b SC
Ala-1 1.08 0.75 Gh-2 (-) 0.96 1.14
Ala-4 1.08 2.51
Ala-6 1.08 2.54 Lys-7 (+) 0.86 1.16
Thr-3 0.75 1 .a
Ala-5 1.08 2.21
Phe-8 1.06 0.84 Glu-9 (-) 0.96 1.84 Arg-10 (+) 1.03 1.83 Gln-11 1.01 1.15 His-12 (+) 0.69 0.65 Met-13 1.28 0.65
*For ionizable residues, the charge is given in parentheses. bunmodified values of s' at 0°C (see Table I). 'Modified values according to Eqs. (5) and (7), for the charge distribution giuen.
helix contents are also shown in Table I11 (these are calculated from the observed molar ellipticities at 222 nm, as indicated in the footnote to Table 2 of Shoemaker et a1.I). The relative changes observed by Shoemaker et al. are paralleled closely by the calculated results; the effects of substitutions in the reference peptides I and 11, as well as the general difference in behavior between the I and I1 series, are accounted for by the theoretical model.
Comparisons involving the C-peptide lactone and carboxylate (entries 7 and 8 in Table 111) indicate that the present model is unable to account for the large difference observed between the helix contents of the C-peptide lactone and reference peptide 11, as well as for the close values observed experimentally for reference peptide I and the C-peptide lactone: we would expect that the presence of two extra positive charges on the N-terminus of the C-peptide lactone should make this a much less likely peptide to attain a helical conformation than reference peptide I, especially since the only other difference in sequence, namely, Met(13) + Hse-lactone, is not expected apriori to have a major effect on helix stability. Nevertheless, the addition of a charged negative C-terminus, in going from the model C-peptide lactone to the carboxylate form, lowers the helix content, in agreement with experimen- tal observations (compare entries 7 and 8 in Table 111).
Reference peptide I1 derivatives (entries 3-6 of Table 111), in general, have lower helix contents than the reference peptide I series (entries 1 and 2 of Table 111). This trend is seen in both the experimental and calculated results.
It should be noted that the Zimm-Bragg model, without the dipole correc- tion (which is equivalent to taking e and B equal to zero), gives equally low helix contents for all peptides, and so is virtually insensitive to charged-group effects (data not shown). The modified model accounts for these effects and, with the exceptions noted above, reproduces the helix content of each peptide. As noted in the following paper' on the application of this method to cytochrome c antigenic peptides, the computed helix contents of different

362 VASQUEZ, PINCUS, AND SCHERAGA
0 5 10
PH Fig. 3. Effect of pH on the helix content of reference peptide I at T = 0 (0) and 45°C (A). The
pK 's for the ionizable side-chain groups were taken as thcwe of the terminally blocked amino acids (page 2 of Ref. 23).
cytochrome c peptides are in agreement with those observed for these peptides in mixed solvents.
pH Dependence
The results for the pH dependence of helix stability are presented in Fig. 3 and are to be compared with Fig. 2 of Shoemaker et al.' The theoretical results agree with the data of Shoemaker et al.' in the pH range of 5-10. In the low-pH region, there is a discrepancy. This is due to the much stronger helix propensity, as manifested by u and s of unionized glutamic acid compared to ionized glutamate (at O"C, s' is 1.47 for neutral Glu, whereas it is 0.96 for ionized Glu, and u is about 15 times larger for unionized Glu than for ionized Glu; see Table I); that difference is enough to overcome the stabiliza- tion due to dipole-charge interactions when (ionized) glutamate is present. These results suggest that dipole-charge interactions do not completely account for the helix content of the C-peptide and its derivatives. Some other, possibly longer range, interactions should be taken into account. The results obtained by Nieto et al.24 on the C- and S-peptides of ribonuclease have been interpreted as evidence for a salt bridge between Glu(2) and Arg(l0); this kind of interaction, which was pointed out by Maxiield and S ~ h e r a g a , ~ ~ is not taken into account by the present theoretical treatment, and so could explain the discrepancy in the pH dependence of the helix content of these peptides in the low-pH range. In this range, the salt bridge does not exist, and therefore would not be available to stabilize the helix.

MODIFIED HELIX-COIL TRANSITION THEORY 363
Summarizing Discussion
As pointed out above, application of the modified helix-coil transition theory has, with the exceptions noted, reproduced the observed helix contents of a number of polypeptides including the C-peptide series (Table 111) and the cytochrome c series, the results of which are presented in the following paper? The values of ( PH) computed by this method directly reproduce the observed values for these peptides. Other methods based on data from known protein crystal structures yield probabilities that are much lower than those observed; helix content is estimated by computing the fraction of residues with probabil- ities greater than a cutoff value that is somewhat arbitrarily defined. This approach is an “all-or-none” approach, i.e., a residue is either helical or not. Application of the method presented here gives more accurate residue prob- abilities rather than an all-or-none result. Also, as noted in the previous section, the present method is independent of any particular protein data base Selected.
However, the present method omits several effects, in particular, certain types of medium-range interactions (such as the effects of i to i + 4 interresi- due salt bridges and/or side chain-side chain hydrogen bonds) and long-range interactions. Inclusion of the effect of these medium-range interactions on the helix probabilities cannot be achieved properly with the present formalism. The present formalism indicates how to change the values of s, so that charge-dipole interactions can be taken into account. However, it does not involve changes3 in the way in which the partition function is calculated according to the original Zimm-Bragg formalism. Hence, consideration of side chain-side chain salt bridges and/or hydrogen bonds would require a change in the method for calculating the partition function, in addition to changes in the values of s. Also, inclusion of these effects would introduce a new series of adjustable parameters whose values would have to be estimated from experi- mental results; it is not clear whether the current set of experimental data on medium-sized peptides is large enough to allow such a parameterization. These questions are currently being explored in our laboratory. In the case of long-range interactions, the effects of chain folding are not taken into account in the present model since the open-chain assumption is made in the compu- tation of the charge-dipole effect, as discussed in the previous section. While these effects are not explicitly taken into account in the methods that predict regular backbone structure based on the use of protein structural data, they are included implicitly in the computed statistical weights for each residue of a particular sequence. The method presented here is expected to succeed better for open-chain oligo- and polypeptides for which no long-range interac- tions exist, but it is expected to be less accurate for proteins in which long-range interactions do exist.
Open-Chain Polypeptides
The predicted helix contents of reference peptide I (Ala 5 + His) and reference peptides I1 (entries 2,3 ,5 , and 6 in Table 111) are seen to agree with the values determined for these peptides by CD. Application of methods based on protein structural data’g*26 to the same set of peptides yields the result that the entire peptide in each case is predicted to be a-helical with little

364 VASQUEZ, PINCUS, AND SCHERAGA
TABLE V Results of Zimn-Bragg Prediction with Dipole Effect"
Protein name ntot naa noa nao noo
1. All-a class Calcium-binding parvalbumin B Cytochrome c Ferricytochrome c2 Avian pancreatic polypeptide Cytochrome c550 Cytochrome c551 Myoglobin Insulin (B chain) Hemoglobin Hemoglobin (Met) Leghemoglobin
Total for class
2. All+ class Acid proteinase Plastocyanin Acid protease Elastase a-Lytic protease Proteinase A P-nYpsin A-immunoglobin FAB (L chain) A-immunoglobin FAB (H chain) Bence- Jones immunoglobin Superoxide dismutase Concanavalin A Prealbumin
Total for class
3. a/P class Flavodoxin L-arabinose binding protein Phosphoglycerate mutase Dihydrofolate reductase Lactate dehydrogenase Alcohol dehydrogenase Adenylate kinase Rhodanase Triose phosphate isomerase Glyceraldehyde 3-phosphate
Phosphoglycerate kinase Thioredoxin reductase
dehydrogenase
Total for class
4. OL + f3 clans Cytochrome b5 Lysozyme (hen egg white) High-potential iron protein
106 101 110 34
119 80
151 28
134 146 151
1160
321 97
322 238 196 179 221 205 217 105 149 235 112
2597
136 304 228 157 327 372 192 291 245
331 413 106
3102
83 127 83
30 32 27 18 12 34
110 4
69 40 55
431
12 0 5 4 0 0 6 0 0 0 0 0 8
35
17 57 57 26 78 31 74 49 57
46 60 10
562
9 21 9
18 34 43 4
35 18 21 0
15 40 10
238
19 28 33
119 53 23 83 88 67 42 39 66 42
702
9 69 72 63
112 136 17 90 66
98 155 42
929
28 38 36
22 12 2 0
22 4 8 7
32 39 57
205
22 4 2 9 7
11 11 0 0 0 0 0 0
66
30 49 12 8
38 48 34 33 53
41 64 32
442
8 17 1
36 23 38 12 50 24 12 17 18 27 29
286
268 65
282 106 136 145 121 117 150 63
110 169 62
1794
80 129 87 60 99
157 67
119 69
146 134 22
1169
38 51 37
MODIFIED HELIX-COIL TRANSITION THEORY 365
TABLE V (Continued from the preowus page.)
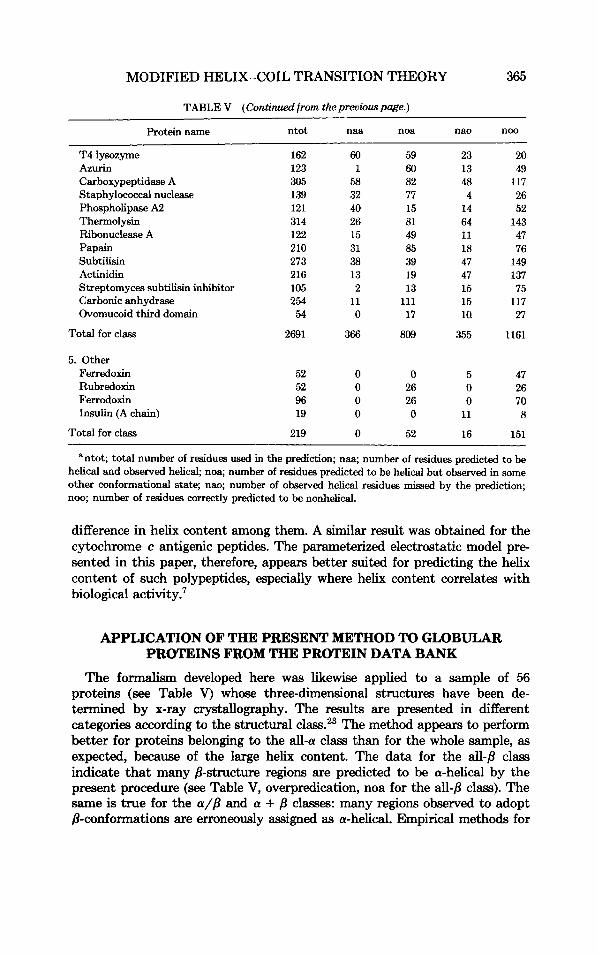
Protein name ntot naa noa nao noo
T4 lysozyme 162 60 59 23 20 Azurin 123 1 60 13 49 Carboxypeptidase A 305 58 82 48 117 Staphylococcal nuclease 139 32 77 4 26 Phospholipase A2 121 40 15 14 52 Thermolysin 314 26 81 64 143 Ribonuclease A 122 15 49 11 47 Papain 210 31 85 18 76 Subtilisin 273 38 39 47 149 Actinidin 216 13 19 47 137
Carbonic anhydrase 254 11 111 15 117 Ovomucoid third domain 54 0 17 ia 27
Total for class 2691 366 809 355 1161
Streptomyces subtilisin inhibitor 105 2 13 15 75
5. Other Ferredoxin Rubredoxin Ferrodoxin Insulin (A chain)
52 0 0 5 47 52 0 26 0 26 96 0 26 0 70 19 0 0 11 8
Total for class 219 0 52 16 151
‘ntot; total number of residues used in the prediction; naa; number of residues predicted to be helical and observed helical, noa; number of residues predicted to be helical but observed in some other conformational state; nao; number of observed helical residues missed by the prediction; noo; number of residues correctly predicted to be nonhelical.
difference in helix content among them. A similar result was obtained for the cytochrome c antigenic peptides. The parameterized electrostatic model pre- sented in this paper, therefore, appears better suited for predicting the helix content of such polypeptides, especially where helix content correlates with biological activity?
APPLICATION OF THE PRESENT METHOD TO GLOBULAR PROTEINS FROM THE PROTEIN DATA BANK
The formalism developed here was likewise applied to a sample of 56 proteins (see Table V) whose three-dimensional structures have been de- termined by x-ray crystallography. The results are presented in different categories according to the structural class.23 The method appears to perform better for proteins belonging to the all-a class than for the whole sample, as expected, because of the large helix content. The data for the all-/? class indicate that many /?-structure regions are predicted to be a-helical by the present procedure (see Table V, overpredication, noa for the all-p class). The same is true for the a//? and a + /? classes: many regions observed to adopt /?-conformations are erroneously assigned as a-helical. Empirical methods for

TAB
LE V
I Su
mm
ary o
f R
esul
ts of
App
licat
ion
of Pr
edic
tion
Sche
mes
to P
rote
ins o
f K
now
n Th
ree-
Dim
ensi
onal
Str
uctu
re
Cho
u-Fm
an"
Gam
ier e
t al?
Li
ma
Max
field
-Sch
erag
ab
Zim
m-B
ragg
' Zi
mm
-Bra
gg +
Dip
ole'
R) c:
Red
. Pred.
Pred.
Pred.
Pred.
Red
. Pred.
Pred
. Pred.
Pred
. Pr
ed.
Pred
. ad
N
ot a
d
a N
ot a
a
Not
a
a N
ot a
a
Not
a
a N
ot a
Obs
a
1275
1772
1716
1331
1542
1505
1125
1353
1948
530
1394
1084
Obs
Not
a
1577
6139
1565
6151
1142
6573
1033
6258
3924
3367
2730
4561
0
q; 0.69
0.73
0.75
0.76
0.64
0.61
9.2"
0.45
0.52
0.57
0.52
0.33
0.34
U
aDat
a tak
en f
rom
Ref
. 21
whe
re c
ompu
teriz
ed v
ersi
ons o
f th
e m
etho
ds w
ere
appl
ied
to a
set
of 62 p
rote
ins.
The
rate
s of
succ
ess o
btai
ned
by t
he o
rigin
al au
thor
s of
the
firs
t tw
o m
etho
ds l
iste
d ar
e m
uch
high
er t
han
the ones s
how
n in
Ref
. 21.
Thi
s co
uld
be d
ue t
o po
or c
ompu
ter
impl
emen
tatio
n of
thos
e sc
hem
es, a
nd/o
r in
here
nt
abig
uitie
s in
trins
ic t
o th
em t
hat m
ake
tota
l aut
omat
izat
ion
very
diff
icul
t. 8 E 2
bApp
lied t
o th
e se
t of
56 p
rote
ins l
iste
d in
Tab
le IV
(alm
ost t
he sa
me as th
ose
of R
ef. 21).
'App
lied
to th
e sa
me
set a
s fo
otno
te b
; pr
obab
ility
cut
offs
, abo
ve w
hich
a r
esid
ue w
as c
onsi
dere
d he
lical
, wer
e 0.
06 a
nd 0.20
for Z
imm
-Bra
gg a
nd Z
imn-
Bra
gg w
ith
dipo
le m
odifi
catio
n, re
spec
tivel
y. B
oth
calc
ulat
ions
wer
e ca
rrie
d ou
t at a
tem
pera
ture
of
0°C
. dP
red.
a, t
he n
umbe
r of
resi
dues
pre
dict
ed t
o be
hel
ical
; Pred. N
ot a
, the
num
ber o
f re
sidu
es p
redi
cted
to b
e no
t hel
ical
; Obs
a, t
he n
umbe
r of
resi
dues
obs
erve
d to
be
helic
al, a
nd Obs N
ot a
, the
number
of re
sidu
es ob
served in
non
helic
al st
ates
. eq
l is
the
perc
ent c
orre
ct; qi
= (naa
+ no
o)/(n
aa
+ na
o +
noo
+ noa
). q2
is th
e pr
edic
tive
pow
er d
efin
ed as
naa/
(naa
+ n
oa).
MODIFIED EELIX-COIL TRANSITION THEORY 367
I .o I I I 1 I I I I I I 1
Residue Number ( i )
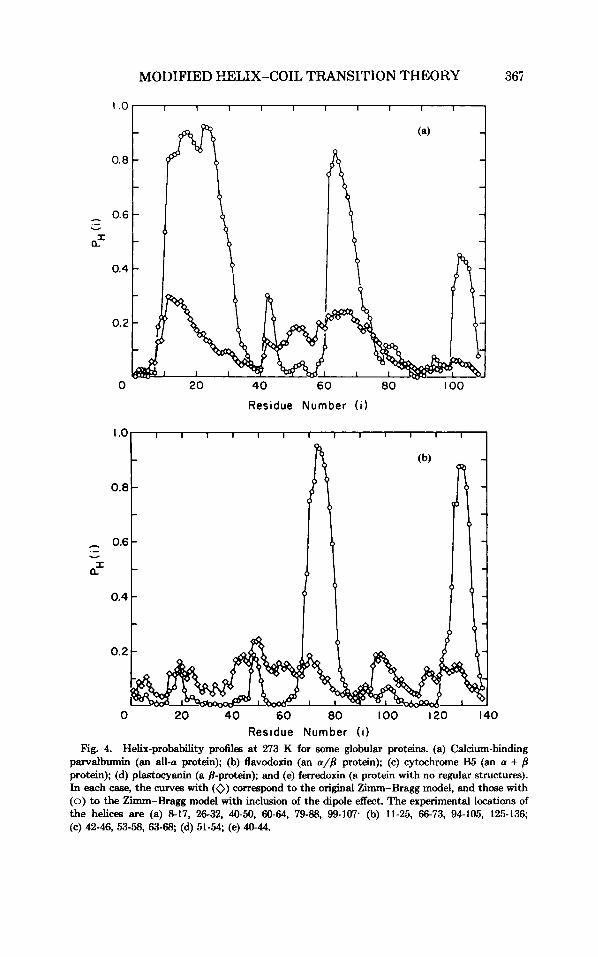
Residue Number ( i ) Fig. 4. Helix-probability profiles at 273 K for some globular proteins. (a) Calcium-binding
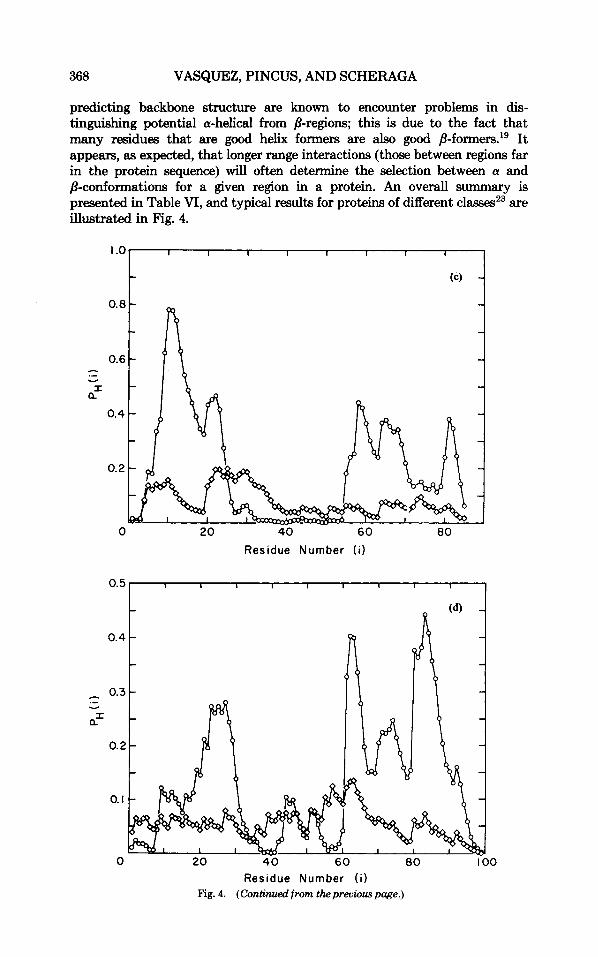
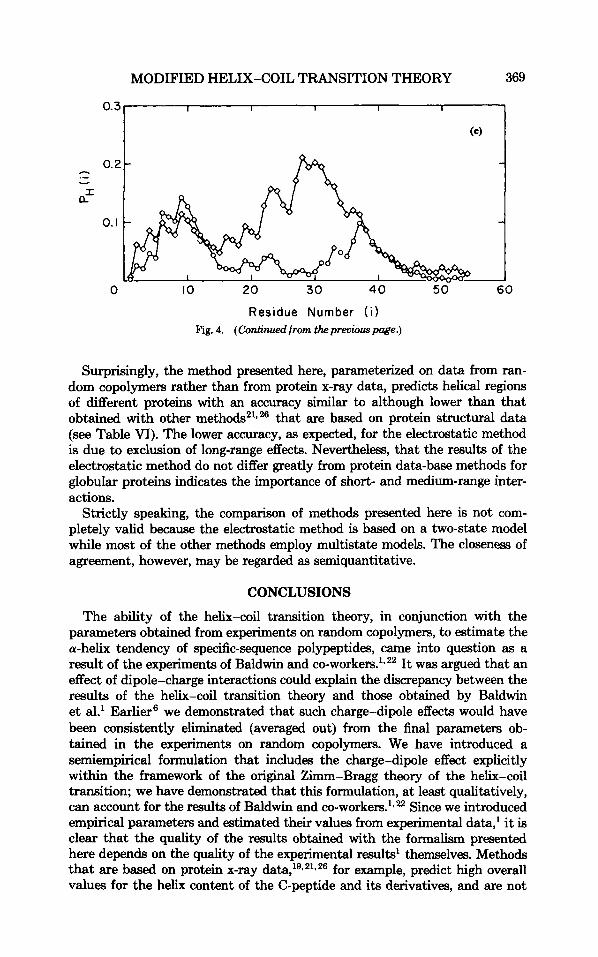
parvalbumin (an all-a protein); @) flavodoxin (an a/B protein); (c) cytochrome B5 (an a + 8 protein); (d) plastocyanin (a 8-protein); and (e) ferredoxin (a protein with no regular structures). In each case, the curves with (0) correspond to the original Zimm-Bragg model, and those with (0) to the Zimm-Bragg model with inclusion of the dipole effect. The experimental locations of the helices are (a) 8-17, 26-32, 40-50, 60-64, 79-88, 99-107. (b) 11-25, 66-73, 94-105, 125-136; (c) 42-46, 53-58, 63-68; (d) 51-54; (e) 40-44.
368 VASQUEZ, PINCUS, AND SCHERAGA
predicting backbone structure are known to encounter problems in dis- tinguishing potential a-helical from /3-regions; this is due to the fact that many residues that are good helix formers are also good /3-f0rmers.'~ It appears, as expected, that longer range interactions (those between regions far in the protein sequence) will often determine the selection between a and p-conformations for a given region in a protein. An overall summary is presented in Table VI, and typical results for proteins of different classes23 are illustrated in Fig. 4.
0.5
0.4
1 0.3
a
.- v
I
0.2
0. I
0
Residue Number ( i )
20 4 0 60 80 coo Residue Number ( i )
Fig. 4. (Continued from thepreviouspage.)
MODIFIED HELIX-COIL TRANSITION THEORY 369
0.3
0.2 .- Y
a"
0. I
0
I I 1 I I
I
10 20 30 40 5 0 60
Residue Number (i) Fig. 4. (Continued from thepreuwuspoge.)
Surprisingly, the method presented here, parameterized on data from ran- dom copolymers rather than from protein x-ray data, predicts helical regions of different proteins with an accuracy similar to although lower than that obtained with other methods21*26 that are based on protein structural data (see Table VI). The lower accuracy, as expected, for the electrostatic method is due to exclusion of long-range effects. Nevertheless, that the results of the electrostatic method do not differ greatly from protein data-base methods for globular proteins indicates the importance of short- and medium-range inter- actions.
Strictly speaking, the comparison of methods presented here is not com- pletely valid because the electrostatic method is based on a two-state model while most of the other methods employ multistate models. The closeness of agreement, however, may be regarded as semiquantitative.
CONCLUSIONS
The ability of the helix-coil transition theory, in conjunction with the parameters obtained from experiments on random copolymers, to estimate the a-helix tendency of specific-sequence polypeptides, came into question as a result of the experiments of Baldwin and co-workers.1.22 It was argued that an effect of dipole-charge interactions could explain the discrepancy between the results of the helix-coil transition theory and those obtained by Baldwin et al.' Earlier6 we demonstrated that such charge-dipole effects would have been consistently eliminated (averaged out) from the final parameters ob- tained in the experiments on random copolymers. We have introduced a semiempirical formulation that includes the charge-dipole effect explicitly within the framework of the original Zimm-Bragg theory of the helix-coil transition; we have demonstrated that this formulation, at least qualitatively, can account for the results of Baldwin and co-workers.1.22 Since we introduced empirical parameters and estimated their values from experimental data,' it is clear that the quality of the results obtained with the formalism presented here depends on the quality of the experimental results' themselves. Methods that are based on protein x-ray data,19*21926 for example, predict high overall values for the helix content of the C-peptide and its derivatives, and are not

370 VASQUEZ, PINCUS, AND SCHERAGA
sensitive to changes in charge distribution: all peptides considered in Table I11 are predicted to be equally helical. The method introduced here has also been applied to predict the location of a-helices in globular proteins; the quality of the results obtained is only slightly inferior to that of results produced by methods that rely on analyses of the structures of crystallized proteins. The effects of interactions, such as salt bridges, that appear im- portant in the series of peptides studied by Shoemaker et al.' could be taken into account by changing part of the formalism presented in this paper. We are currently evaluating the possibility of considering such effects in the context of a new formalism.
Note added in proof
We have recently completed a modification of the theory presented here, by including the effects of interactions between specific side chains (M. Vbquez and H. A. Scheraga, Biopolymers, to be submitted).
We thank Dr. R. L. Baldwin for sending us a preprint of Ref. 1. This work was supported by research grants from the National Institute of Arthritis, Diabetes,
and Digestive and Kidney Diseases of the National Institutes of Health (AM-08465), and from the National Science Foundation (DMB84-01811).
References 1. Shoemaker, K. R., Kim, P. S., Brems, D. N., Marqusee, S., York, E. J., Chaiken, I. M.,
2. Zimm, B. H. & Bragg, J. K. (1959) J. Chem. Phys. 31, 526-535. 3. Sueki, M., Lee, S., Powers, S. P., Denton, J. B., Konishi, Y. & Scheraga, H. A. (1984)
4. Blagdon, D. E. & Goodman, M. (1975) Biopolymers 14,241-245. 5. Ihara, S., Ooi, T. & Takahashi, S. (1982) Biopolymers 21, 131-145. 6. Scheraga, H. A. (1985) Proc. Natl. Acad. Sci. USA 82,5585-5587. 7. V h u e z , M., Pincw, M. R. & Scheraga, H. A. (1987) Biopolymers 26, 373-386. 8. Scheraga, H. A. (1978) Pure Appl. C h m . SO, 315-324. 9. GB N., Lewis, P. N., G6, M. & Scheraga, H. A. (1971) Macromolecules 4, 692-709.
Stewart, J. M. & Baldwin, R. L. (1985) Proc. Natl. A d . Sci. USA 82, 2349-2353.
Macromolecules 17, 148-155.
10. Lewis, P. N., GB, N., GB, M., Kotelchuck, D. & Scheraga, H. A. (1970) Proc. Natl. Acad.
11. Lewis, P. N. & Scheraga, H. A. (1971) Arch. Biochem. Biophys. 144, 576-583. 12. Lewis, P. N. & Scheraga, H. A. (1971) Arch. Biochem. Bbphys. 144,584-588. 13. Wako, H. & Scheraga, H. A. (1982) J. Protein Chem. 1, 5-45. 14. Hol, W. G. J., van Duijnen, P. T. & Berendsen, H. J. C. (1978) Nature 273, 443-446. 15. Sheridan, R. P., Levy, R. M. & Salemme, F. R. (1982) Roc. Natl. Aca. Sci. USA 79,
16. Rogers, N. K. & Sternberg, M. J. E. (1984) J. Mol. BioZ. 174, 527-542. 17. Kidera, A., Konishi, Y., Oka, M., Ooi, T. & Scheraga, H. A. (1985) J. Protein Chem. 4,
Sci. USA 65,810-815.
4545-4549.
23-55. 18. Skohick, J. & Holtzer, A. (1982) M U C F O ~ ~ ~ C & S 15, 303-314. 19. Gamier, J., Osguthorpe, D. J. & Robson, B. (1978) J. Mol. Biol. 120, 97-120. 20. Rico, M., Santoro, J., Bermejo, F. J., Herranz, J., Nieto, J. L., Gallego, E. & Jimhez,
21. K a k h , W. & Sander, C. (1983) FEBS Lett. 155, 179-182. 22. Bierzynski, A., Kim, P. S. & Baldwin R. L. (1982) Proc. Natl. Acad. Sci. USA 79,
M. A. (1986) Biopolymers 25, 1031-1053.
2470-2474.

MODIFIED HELIX-COIL TRANSITION THEORY 371
23. Schulz, G. E. & Schinner, R. H. (1979) Principles of Protein Structure, Springer-Verlag,
24. Nieto, J. L., Rico, M. Jimhez, M. A., Herranz, J. & Santoro, J. (1985) Znt. J. BWZ.
25. Maxfield, F. R. & Scheraga, H. A. (1975) Macromolec&s 8, 491-493. 26. Maxfield, F. R. & Scheraga, H. A. (1979) Biochemistry 18,697-704.
New York, chaps. 1 and 5.
Macromol. 7, 66-70.
Received April 28, 1986 Accepted August 28,1986