high throughput sequencing: informatics & software aspects gabor t. marth boston college biology...
TRANSCRIPT

High throughput sequencing:informatics & software aspects
Gabor T. MarthBoston College Biology Department
BI543 Fall 2013January 29, 2013

Traditional DNA sequencing

Genetics of living organisms
DNA
Chromosomes

Radioactive label gel sequencing

Four-color capillary sequencing
~1 Mb ~100 Mb >100 Mb ~3,000 Mb
ABI 3700 four-color sequence trace

Individual human resequencing

Next-generation DNA sequencing

New sequencing technologies…
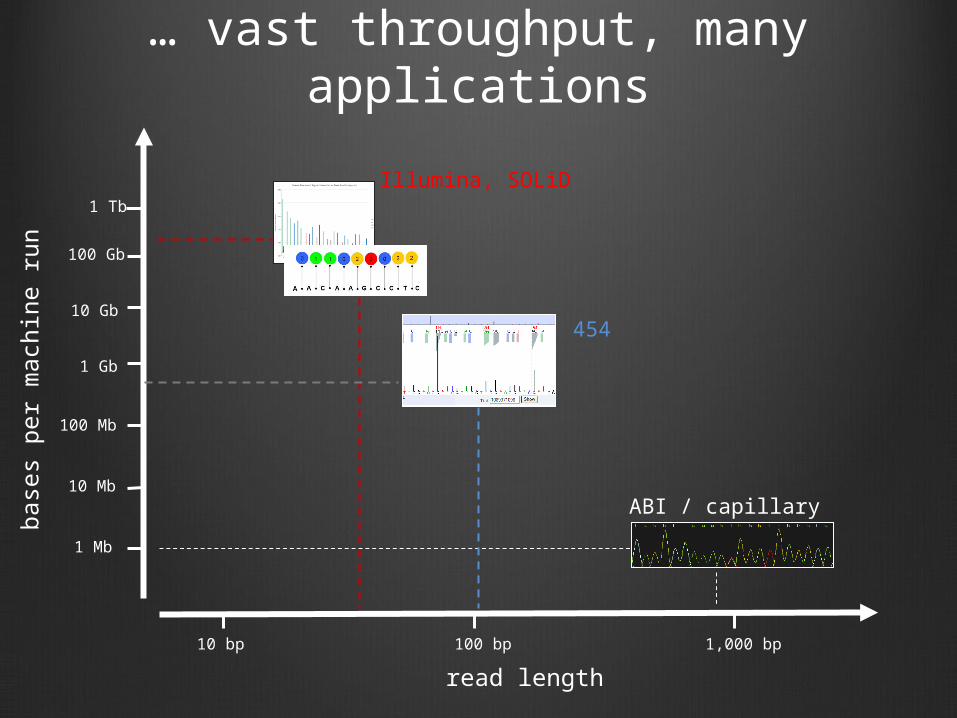
… vast throughput, many applications
read length
base
s per
mach
ine r
un
10 bp 1,000 bp100 bp
1 Gb
100 Mb
10 Mb
10 Gb
Illumina, SOLiD
ABI / capillary
454
1 Mb
100 Gb
1 Tb

DNA ligation DNA base extension
Church, 2005
Sequencing chemistries

Template clonal amplification
Church, 2005

Massively parallel sequencing
Church, 2005

Chemistry of paired-end sequencing
Double strand DNA is folded into a bridge shape then separated into single strands. The end of each strand is then sequenced.
(Figure courtesy of Illumina)

Paired-end reads
• fragment amplification: fragment length 100 - 600 bp• fragment length limited by amplification efficiency
• circularization: 500bp - 10kb (sweet spot ~3kb)• fragment length limited by library complexity
Korbel et al. Science 2007

Features of NGS data
Short sequence reads100-200bp25-35bp (micro-reads)
Huge amount of sequence per runUp to gigabases per run
Huge number of reads per runUp to 100’s of millions
Higher error as compared with Sanger sequencing
Error profile different to Sanger

Application areas of next-gen sequencing

Application areas• Genome resequencing
• variant discovery• somatic mutation detection• mutational profiling
• De novo assembly
• Identification of protein-bound DNA• chromatin structure• methylation• transcription binding sites
• RNA-Seq• expression• transcript discovery
Mikkelsen et al. Nature 2007
Cloonan et al. Nature Methods, 2008

SNP and short-INDEL discovery

Structural variation detection
• structural variations (deletions, insertions, inversions and translocations) from paired-end read map locations
• copy number (for amplifications, deletions) from depth of read coverage

Identification of protein-bound DNA
genome sequence
aligned reads
Chromatin structure (CHIP-SEQ)(Mikkelsen et al. Nature 2007)
Transcription binding sites. (Robertson et al. Nature Methods, 2007)

Novel transcript discovery (genes)
Mortazavi et al. Nature Methods
• novel exons• novel transcripts containing known exons

Novel transcript discovery (miRNAs)
Ruby et al. Cell, 2006

Expression profiling
aligned reads
aligned reads
Jones-Rhoads et al. PLoS Genetics, 2007
gene gene
• tag counting (e.g. SAGE, CAGE)• shotgun transcript sequencing

De novo genome sequencing
assembled sequence contigs
short reads
longer reads
read pairs
Lander et al. Nature 2001

The informatics of sequencing

Re-sequencing informatics pipeline
REF
(ii) read mapping
IND
(i) base calling
IND(iii) SNP and short INDEL calling
(v) data viewing, hypothesis generation
(iv) SV callingGigaBayesGigaBayes

The variation discovery toolbox
• base callers
• read mappers
• SNP callers
• SV callers
• assembly viewers

Raw data processing / base calling
Trace extraction
Base calling
• These steps are usually handled well by the machine manufacturers’ software
• What most analysts want to see is base calls and well-calibrated base quality values

Sequence traces are machine-specific
Base calling is increasingly left to machine manufacturers

…where they give you the cover on the box
Read mapping…
Is like a jigsaw puzzle…

Some pieces are easier to place than others…
…pieces with unique features
pieces that look like each other…

Repeats multiple mapping problem
Lander et al. 2001

Paired-end (PE) reads
fragment length: 100 – 600bp
Korbel et al. Science 2007
fragment length: 1 – 10kb
PE reads are now the standard for whole-genome short-read sequencing

Mapping quality values
0.8 0.19 0.01

SNP calling

SNP calling: what goes into it?
sequencing errortrue polymorphism
Base qualities
Base coverage
Prior expectation

Bayesian SNP calling
Siablevarall
]T,G,C,A[S ]T,G,C,A[SiiiorPr
iiorPr
i
iiorPr
i
NiorPrNiorPr
NN
iorPr
i Ni
N
N
N )S,...,S(P)S(P
)R|S(P...
)S(P
)R|S(P...
)S,...,S(P)S(P)R|S(P
...)S(P)R|S(P
)SNP(P
1
1
1
1 11
11
11
AAAAA
CCCCC
TTTTT
GGGGG
polymorphic permutation
monomorphic permutationBayesian
posterior probability
Base call + Base quality Expected polymorphism rate
Base composition Depth of coverage

http://bioinformatics.bc.edu/~marth/PolyBayes
Marth et al., Nature Genetics, 1999
• First statistically rigorous SNP discovery tool• Correctly analyzes alternative cDNA splice forms
The PolyBayes software

SNP calling (continued)
P(G1=aa|B1=aacc; Bi=aaaac; Bn= cccc)P(G1=cc|B1=aacc; Bi=aaaac; Bn= cccc)P(G1=ac|B1=aacc; Bi=aaaac; Bn= cccc)
P(Gi=aa|B1=aacc; Bi=aaaac; Bn= cccc)P(Gi=cc|B1=aacc; Bi=aaaac; Bn= cccc)P(Gi=ac|B1=aacc; Bi=aaaac; Bn= cccc)
P(Gn=aa|B1=aacc; Bi=aaaac; Bn= cccc)P(Gn=cc|B1=aacc; Bi=aaaac; Bn= cccc)P(Gn=ac|B1=aacc; Bi=aaaac; Bn= cccc)
P(SNP)
“genotype probabilities”
P(B1=aacc|G1=aa)P(B1=aacc|G1=cc)P(B1=aacc|G1=ac)
P(Bi=aaaac|Gi=aa)P(Bi=aaaac|Gi=cc)P(Bi=aaaac|Gi=ac)
P(Bn=cccc|Gn=aa)P(Bn=cccc|Gn=cc)P(Bn=cccc|Gn=ac)
“genotype likelihoods”
Pri
or(
G1,.
.,G
i,..,
Gn)
-----a----------a----------c----------c-----
-----a----------a----------a----------a----------c-----
-----c----------c----------c----------c-----

Insertion/deletion (INDEL) variants
These variants have been on the “radar screen” for decades
Accurate automated detection is difficultDifferent mutation mechanisms
Often appear in repetitive sequence and therefore difficult to align
Often multi-allelic
Deleted allele has no base quality values

Alignment methods became more refined
Original alignment
After left realignment
After haplotype-aware realignment

Medium length INDELs still a problem
Guillermo Angel

Structural variation detection
Feuk et al. Nature Reviews Genetics, 2006

Structural variant detection (cont’d)

Detection Approaches
Read Depth: good for big CNVs
Sample Reference
Lmap
read
contig
• Paired-end: all types of SV
• Split-Readsgood break-point resolution
• deNovo Assembly~ the future
SV slides courtesy of Chip Stewart, Boston College

SV detection – resolution
Expected CNVsKaryotype
Micro-arraySequencing
Rela
tive n
um
bers
of
even
ts
CNV event length [bp]

Standard data formats
Reads: FASTQ
Alignments: SAM/BAM
Variants: VCF

Tools for analyzing & manipulating 1000G data
• samtools: http://samtools.sourceforge.net/• BamTools: http://sourceforge.net/projects/bamtools/• GATK: http://www.broadinstitute.org/gsa/wiki/index.php/The_Genome_Analysis_Toolkit
• VCFTools: http://vcftools.sourceforge.net/• VcfCTools: https://github.com/AlistairNWard/vcfCTools
Alignments: SAM/BAM
Variants: VCF