high throughput sequencing: technologies & applications
DESCRIPTION
High Throughput Sequencing: Technologies & Applications. Michael Brudno CSC 2431 – Algorithms for HTS University of Toronto 06/01/2010. High Throughput Sequencers. 100 Gb. AB/SOLiDv3, Illumina/GAII short-read sequencers. (10+Gb in 50-100 bp reads, >100M reads, 4-8 days). - PowerPoint PPT PresentationTRANSCRIPT

High Throughput Sequencing: Technologies & Applications
Michael BrudnoCSC 2431 – Algorithms for HTS
University of Toronto 06/01/2010

High Throughput Sequencers
read length
ba
ses
pe
r m
ach
ine
ru
n
10 bp 1,000 bp100 bp
1 Gb
100 Mb
10 Mb
10 Gb
AB/SOLiDv3, Illumina/GAII
short-read sequencers
ABI capillary sequencer
454 GS FLX pyrosequencer
(100-500 Mb in 100-400 bp reads,
0.5-1M reads, 5-10 hours)
(10+Gb in 50-100 bp reads,
>100M reads, 4-8 days)
1 Mb
(0.04-0.08 Mb in 450-800 bp reads,
96 reads, 1-3 hours)
100 Gb
From Gabor Marth, BCFrom Gabor Marth, BC
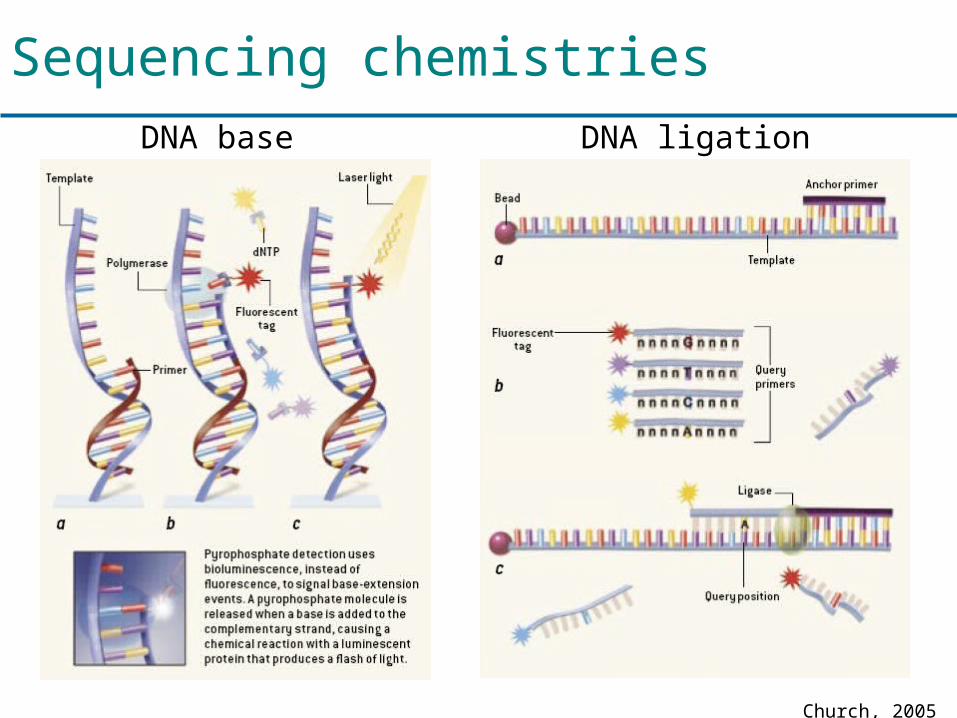
DNA ligation DNA base extension
Church, 2005
Sequencing chemistries

Massively parallel sequencing
Church, 2005

Features of HTS data
• Short (for now) sequence reads–200-400bp: 454 (Roche)–35-100bp Solexa(Illumina), SOLiD(AB)
• Huge amount of sequence per run–Up to 10s of gigabases per run
• Huge number of reads per run–Up to 100’s of millions
• Higher error (compared with Sanger)–Different error profile

The Raw Data
• Machine Readouts are different
• Read length, accuracy, and error profiles are variable.
• All parameters change rapidly as machine hardware, chemistry, optics, and noise filtering improves

454 Pyrosequencer error profile
• multiple bases in a homo-polymeric run are incorporated in a single incorporation test the number of bases must be determined from a single scalar signal the majority of errors are INDELs
• error rates are nucleotide-dependent

Illumina/Solexa base accuracy
• Error rate grows as a function of base position within the read
• A large fraction of the reads contains 1 or 2 errors

3’ 5’
N N N T G z z z
3’ 5’
N N N G A z z z
3’ 5’
N N N A T z z z
2-base, 4-color: 16 probe combinations
● 4 dyes to encode 16 2-base combinations● Detect a single color indicates 4 combinations & eliminates 12 ● Each color reflects position, not the base call● Each base is interrogated by two probes● Dual interrogation eases discrimination
– errors (random or systematic) vs. SNPs (true polymorphisms)
A C G T
A
C
G
T
2nd Base
1st
Bas
e
0
0
0
0
1
1
1
1
2
2
2
2
3
3
3
3
AB SOLiD System dibase sequencing

The decoding matrix allows a sequence of transitions to be converted to a base sequence, as long as one of two bases is known.
A C G T
A
C
G
T
2nd Base
1st
Bas
e
0
0
0
0
1
1
1
1
2
2
2
2
3
3
3
3
AA AC AC AA AG AT AA AG AG CC CA CA CC CT CG CC CT CT GG GT GT GG GA GC GG GA GA TT TG TG TT TC TA TT TC TC
A A C A A G C C T C C C A C C T A A G A G G T G G A T T C T T T G T T C G G A G
10 01 2 3 0 2 2
10 01 2 3 0 2 2
4Possible
Sequences
Converting dibase (color) into letters

A C G G T C G T C G T G T G C G T
A C G G T C G T C G T G T G C G TNo change
A C G G T C G C C G T G T G C G TSNP
A C G G T C G T C G T G T G C G T Measurementerror
SOLiD error checking code

SOLiD Error rate & QVs
0
5
10
15
20
25
30
35
40
0 5 10 15 20 25 30 35 40
Position on Read
Measured QV
0.00%
1.00%
2.00%
3.00%
4.00%
5.00%
6.00%
7.00%
8.00%
9.00%
10.00%
Error rate

Pacific Biosystems (PacBio)


Current and future application areas
De novo genome sequencing
Short-read sequencing will be (at least) an alternative to microarrays for:
• DNA-protein interaction analysis (CHiP-Seq)• novel transcript discovery• quantification of gene expression• epigenetic analysis (methylation profiling)
Genome re-sequencing: somatic mutation detection, organismal SNP discovery, mutational profiling, structural variation discovery
DELSNP
reference genome

What’s in it for us?
VISION/Graphics Machine Learning String Algorithms
Systems DatabasesHuman-Computer
Interaction
Image ManagementBase calling
Probabilistic Models Variant Calling
Read MappingAssembly
Data StorageCloud Computing
Data ManagementData Integrity
Data representation for Biologists

Fundamental informatics challenges
1. Interpreting machine readouts – base calling, base error estimation
3. Dealing with non-uniqueness in the genome: resequenceability
2. Alignment of billions of reads
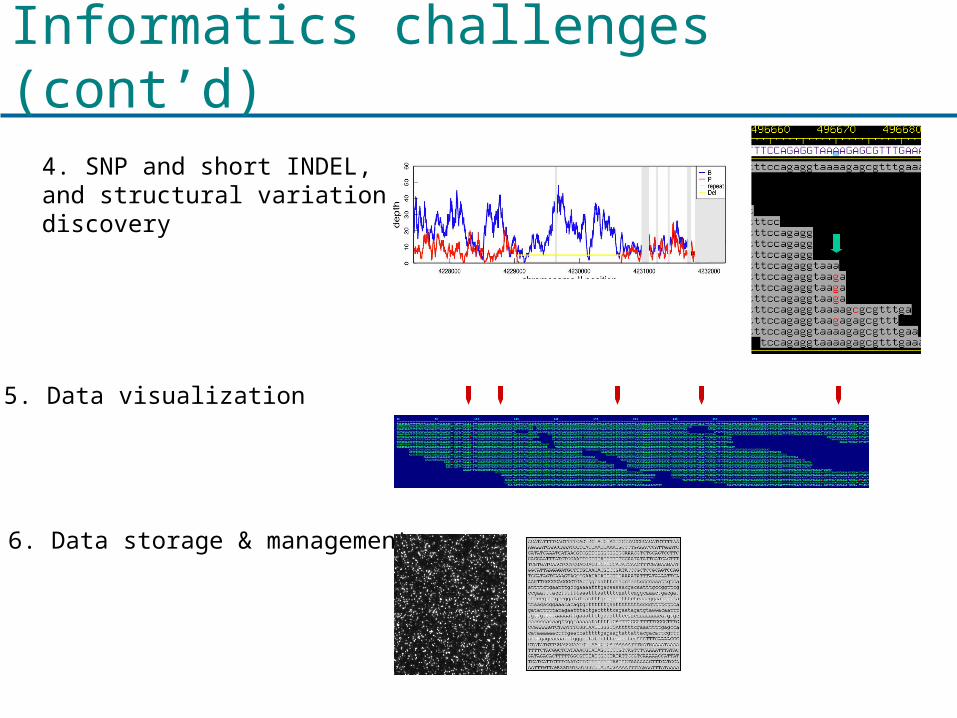
Informatics challenges (cont’d)
5. Data visualization
4. SNP and short INDEL, and structural variation discovery
6. Data storage & management

High Throughput Sequencing: Technologies & Applications
Questions?

• Fast Mapping Algorithm
- Spaced seed hashing
- Vectored (very fast) Smith Waterman
- Handles micro insertions/deletions
• Specialized algorithm for aligning color-space (AB SOLiD) reads
• Computes p-values (and other statistics)
SHRiMP: SHort Read Mapping Package

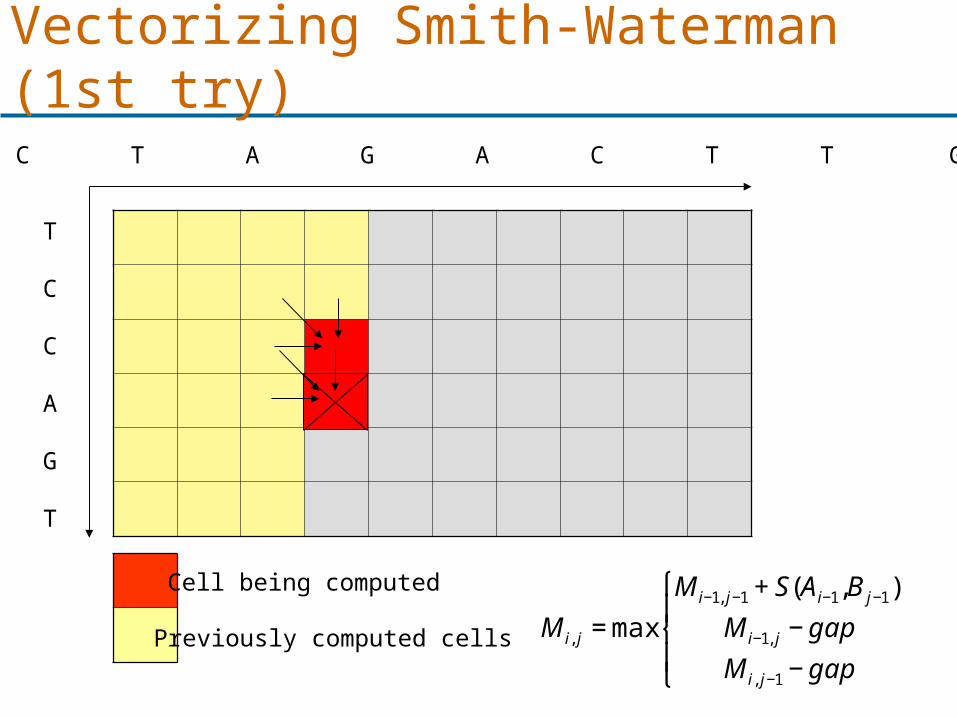
Cell being computed
Previously computed cells
A C T A G A C T T G
T
C
C
A
G
T
€
M i, j = max
M i−1, j−1 + S(Ai−1,B j−1)
M i−1, j − gap
M i, j−1 − gap
⎧
⎨ ⎪
⎩ ⎪
Regular Smith-Waterman

Fast Local Alignment
BLAST FASTAAGTGCCCTGGAACCCTGACGGTGGGTCACAAAACTTCTGGA
AGTGACCTGGGAAGACCCTGAACCCTGGGTCACAAAACTC
AGTGCCCTGGAACCCTGACGGTGGGTCACAAAACTTCTGGA
AGTGACCTGGGAAGACCCTGAACCCTGGGTCACAAAACTC
Altschul et al 1990 Pearson 1987

Genome
Rea
ds
SHRiMP Hashing
• SHRiMP uses spaced seeds
• Vectored Smith-Waterman

• Modern computers provide with capacity for performing same operation on several elements (SIMD)
• Can we take advantage of vectorized instruction in Smith-Waterman?
6
3
9
1
4
8
4
5
+ = max =
10
11
13
6
6
3
9
1
4
8
4
5
6
8
9
5
Vectored Instructions
Cell being computed
Previously computed cells
A C T A G A C T T G
T
C
C
A
G
T
€
M i, j = max
M i−1, j−1 + S(Ai−1,B j−1)
M i−1, j − gap
M i, j−1 − gap
⎧
⎨ ⎪
⎩ ⎪
Vectorizing Smith-Waterman (1st try)

Current
Previous
Penultimate
A C T A G A C T T G
T
C
C
A
G
T
€
M i, j = max
M i−1, j−1 + S(Ai−1,B j−1)
M i−1, j − gap
M i, j−1 − gap
⎧
⎨ ⎪
⎩ ⎪ Wozniak, 1997
Vectorizing Smith-Waterman (Wozniak)

+
-
-
-
+
Current
Previous
Penultimate
A C T A G A C T T G
T
C
C
A
G
T
A C T A G A C T T G
T G A C C T
+ - - - +
Vectorizing Smith-Waterman (SHRiMP)
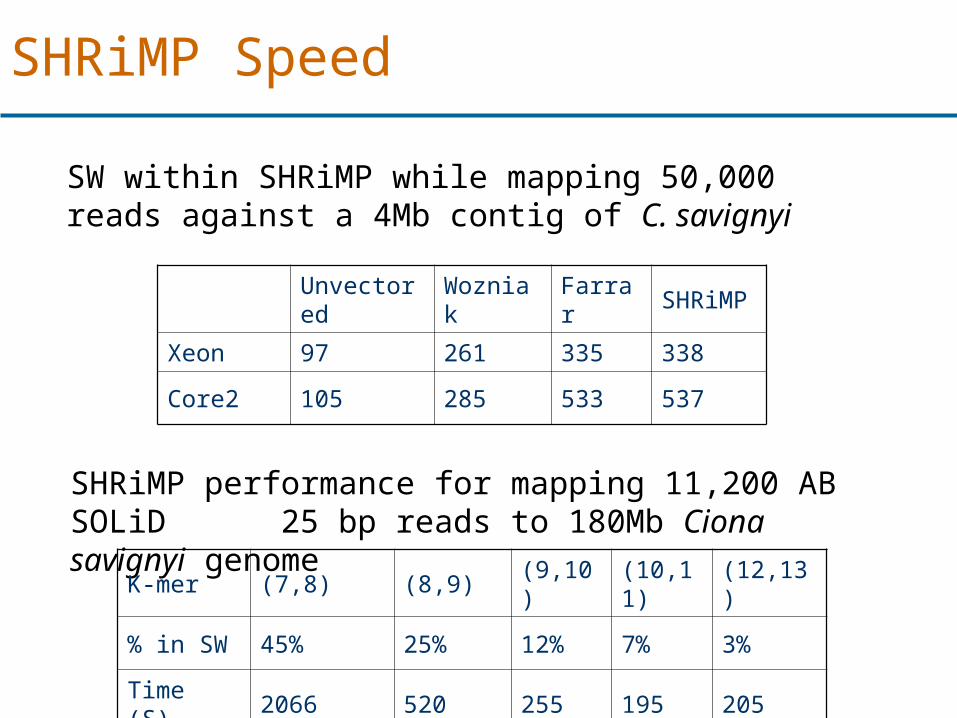
Unvectored Wozniak Farrar SHRiMP
Xeon 97 261 335 338
Core2 105 285 533 537
SW within SHRiMP while mapping 50,000 reads against a 4Mb contig of C. savignyi
SHRiMP Speed
SHRiMP performance for mapping 11,200 AB SOLiD 25 bp reads to 180Mb Ciona savignyi genome
K-mer (7,8) (8,9) (9,10) (10,11) (12,13)
% in SW 45% 25% 12% 7% 3%
Time (S) 2066 520 255 195 205
A G
C T
0 0
0 0
1 1
2
2
3 3
A C G T
A 0 1 2 3
C 1 0 3 2
G 2 3 0 1
T 3 2 1 0
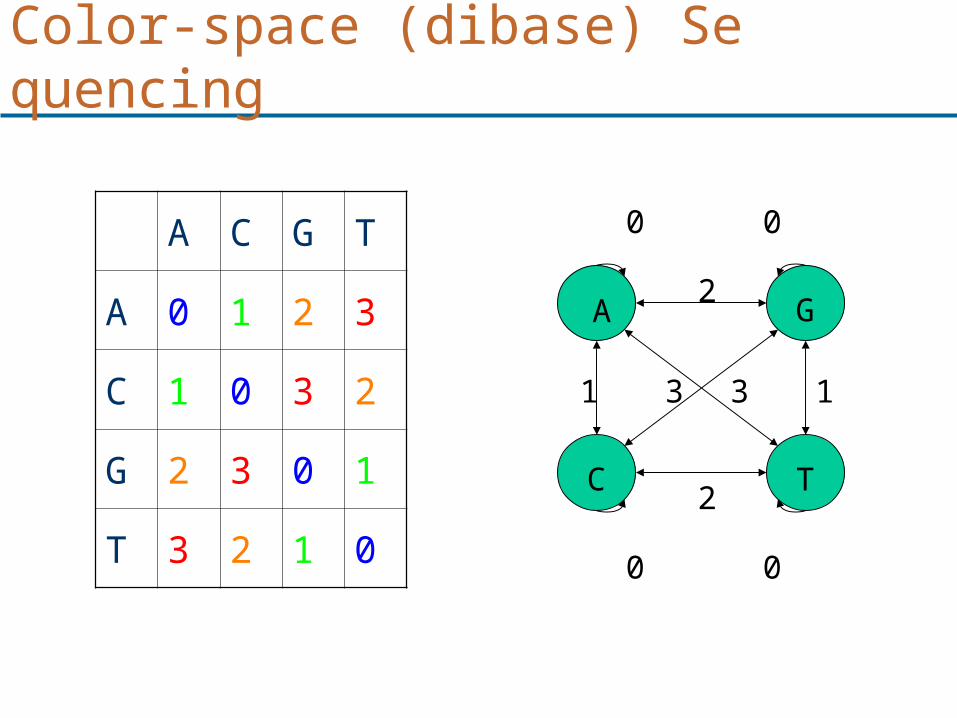
Color-space (dibase) Sequencing

G: TTGAGTTATGGAT 012210331023 R: 012120331023 TTGACTTATGGAT
SNPs
TGAGTT 12210 TGACTT 12120TGAATT 12030TGATTT 12300
Mapping reads in Color-space
INDELS
TGAGTTA 122103
TGA-TTA 12-303
TGAGTTTA 1221003
TGAGTATA 1221333

Mapping reads in Letter Space
A G
C T
0 0
0 0
1 1
2
2
3 3
G: TGACTTATGGAT ||||| TTGAGTCGCAAGC CCAGACTATGGATR: 012212331023
|||||||

SOLiD Translations
• Given the following read, there are 4 translations (we need an initial base):
0 1 2 2 3 3 1 0 2
A A C T C G C A A G
C C A G A T A C C T
G G T C T A T G G A
T T G A G C G T T C

SOLiD Translations
• Reads begin with a known primer (‘T’)– The translation is: T T G A G C G T T C
0 1 2 2 3 3 1 0 2
A A C T C G C A A G
C C A G A T A C C T
G G T C T A T G G A
T T G A G C G T T C

SOLiD Translations
0 1 0 2 3 3 1 0 2
A A C C T A T G G A
C C A A G C G T T C
G G T T C G C A A G
T T G G A T A C C T
• What if we had a sequencing error?– The right translation was: T T G A G C G T T C

Colour-space Smith-Waterman
• Think of 4 SW matrices stacked above one another
• If we have 1 read error, but otherwise perfect match, we’ll use 2 matrices
Genome
Read
Frame 1 Frame 2 Frame 3 Frame 4
Letter

Combined Color/Letter Space SW
A G
C T
0 0
0 0
1 1
2
2
3 3
A C
3
2
A C
G T
T G
C A
C A
T G
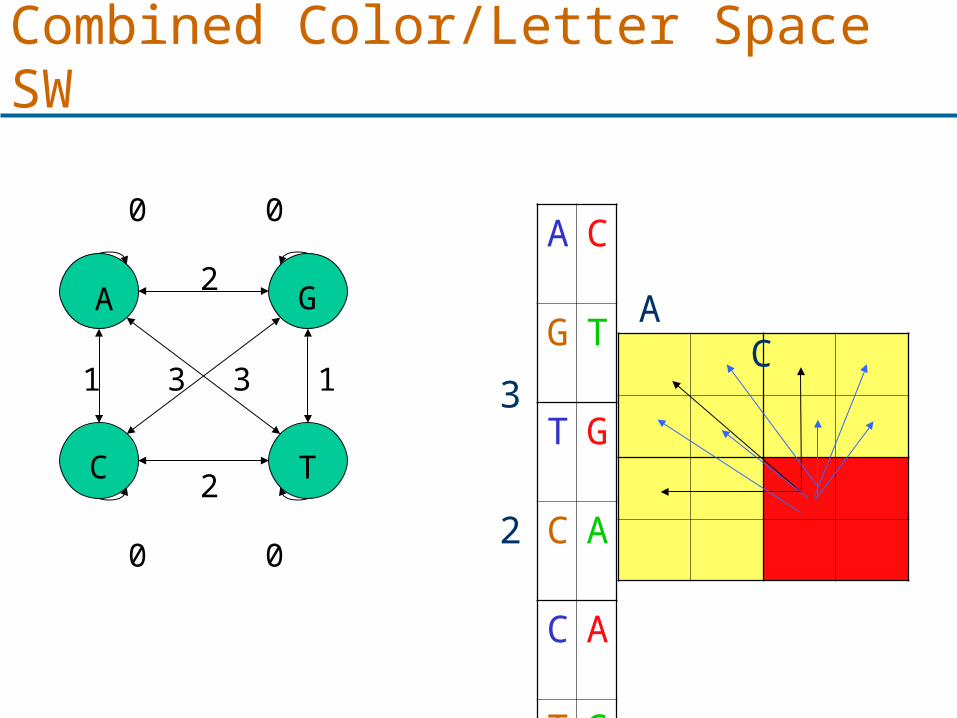
Combined Color/Letter Space SW
A G
C T
0 0
0 0
1 1
2
2
3 3
A C
3
2
A C
G T
T G
C A
C A
T G

G: 1123724 TA-ACCACGGTCACACTTGCATCAC 1123701 || |||||||||| |||X|||||||T: TACACCACGGTCAGACTtGCATCACR: 0 T0311101130121221211313211 24
p<.05
p<.01
Reads mapped
20% 9%
SNP rate
.039 .024
Indel rate
.004 .003
Error rate
.024 .020
SHRiMP on Ciona savignyi
• C. savignyi is a chordate with a very large SNP rate (5%)
• Mapped 22 million AB SOLiD reads to the reference C. savignyi genome (6 hours on 200 CPUs).

• Fast mapping of short reads to a genome
-- Handles indels & color-space reads
-- Easy to parallelize
-- Small memory footprint
• Computation of p-values & other statistics for hits
• Publicly available & free
SHRiMP Summary

Acknowledgments
Stephen Rumble UofT
Phil Lacroute
Anton Valouev
Arend Sidow
http://compbio.cs.toronto.edu/shrimp
FUNDING: NSERC, CFI, NIH
Stanford

Acknowledgments
Stephen Rumble UofT
Phil Lacroute
Anton Valouev
Arend Sidow
http://compbio.cs.toronto.edu/shrimp
FUNDING: NSERC, CFI, NIH
Stanford

• SNP discovery
• Error correction with letter & color reads (assembly)
• Can fix errors without (explicit) overlap
• Don’t just do everything in color space!
Why is color-space good?
R1: 0 TAGACCACGGTCACACTTGCATCAC 24 || |||||||||| |||X|||||||T: TACACCACGGTCAGACTtGCATCACR2: 0 T0311101130121221211313211 24
T: TACACCACGGTCAGACTTGCATCACR1: T0311101130121221211013211 24R2: T2113013122121101321103111 24R3: T2212110132110311121130131 24

What are structural variations?
Various examples of structural variations
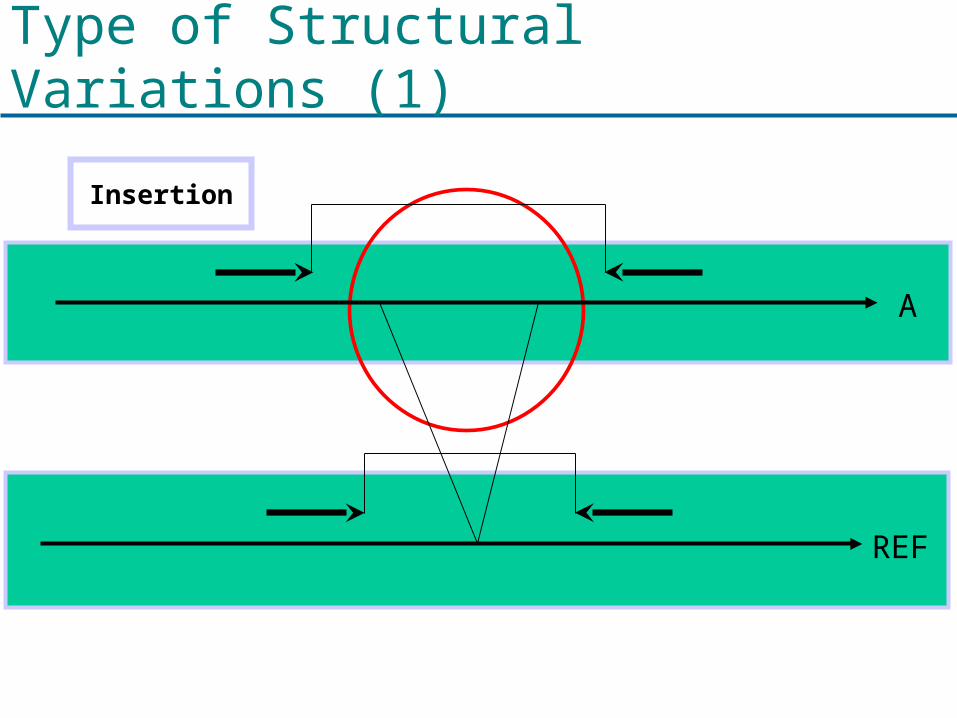
Type of Structural Variations (1)
Insertion
A
REF

Type of Structural Variations (2)
Deletion
A
REF

Type of Structural Variations (3)
Inversion
A
REF
5’ 3’
5’ 3’
5’3’
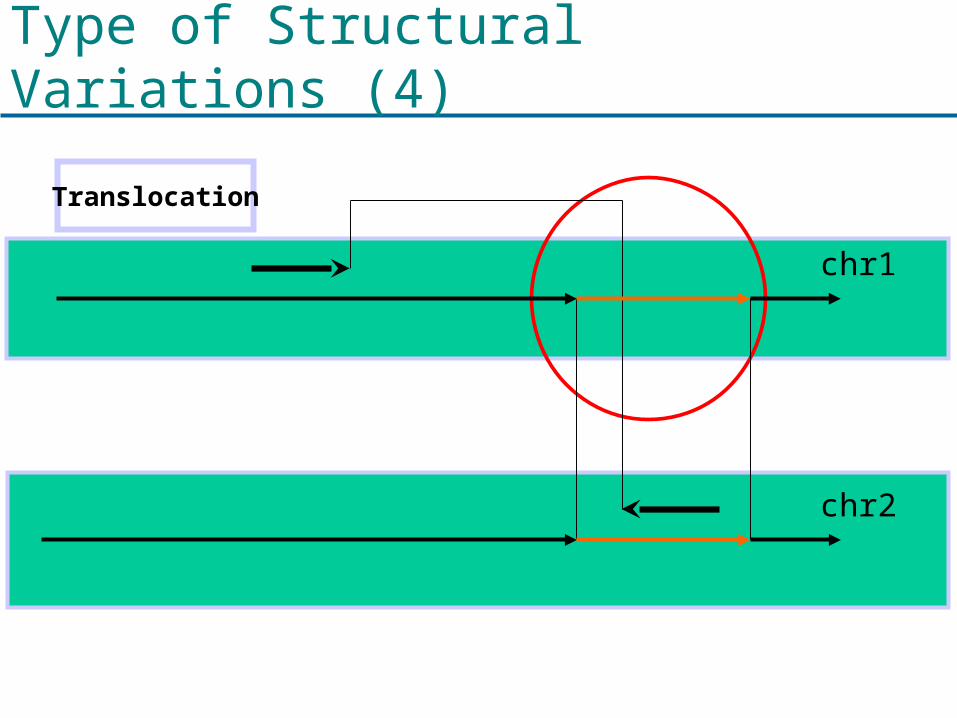
Type of Structural Variations (4)
Translocation
chr1
chr2

Clone-end Sequencing Approaches
1. “Fine-scale structural variation of the human genome” [Tuzun et al, 2005]
• Mapping matepairs onto the reference genome • If mappings of matepairs are not consistent, then there exist structural variations.
2. “Paired-End mappings Reveals Extensive Structural Variation in the Human Genome”
[Korbel et al, 2007]
• Proposed high-throughput and massive paired end mapping technique• Detailed types of structural variations

Motivation
Tuzun & Korbel used scores which are combinationof several factors. (e.g. length, identity, quality of the sequences, concordance)
Reads can map to many locations on the genome. How do we choose between them?

Probabilistic Framework (1)
p(Y): distribution of mapped distances of “uniquely mapped” matepairs of various sizes
We play with p(Y) to describe our probabilistic framework

Probabilistic Framework (2)
Insertion
μY = (s+r)
P(Xi, Xj|ins=r) = P(Xi|ins=r)P(Xj|ins=r)P(Xi|ins=r) = 1 - P(μY - δ ≤Y≤μy+ δ)
where δ= |μY- (s+r)|, s = mapped distance
μy - δ
p(Y)

Probabilistic Framework (3)
Deletion
μY = (s-r)
P(Xi, Xj|del=r) = P(Xi|del=r)P(Xj|del=r)P(Xi|del=r) = 1 - P(μY - δ ≤ Y ≤μy+ δ)
where δ= |μY- (s-r)|, s = mapped distance
μy - δ
p(Y)
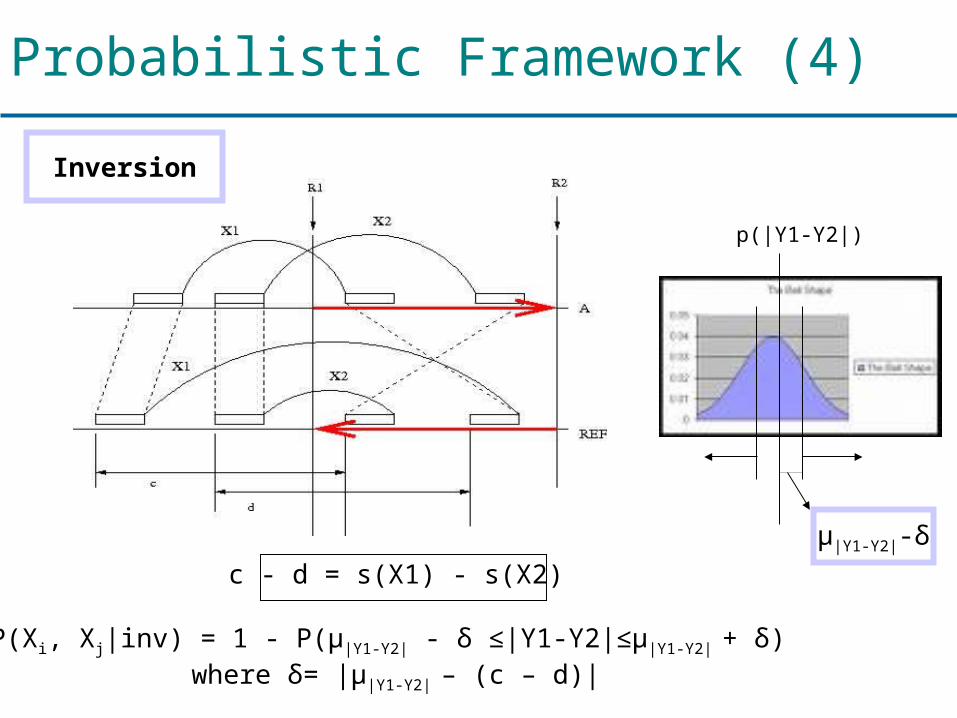
Probabilistic Framework (4)
c - d = s(X1) - s(X2)
P(Xi, Xj|inv) = 1 - P(μ|Y1-Y2| - δ ≤|Y1-Y2|≤μ|Y1-Y2| + δ) where δ= |μ|Y1-Y2| – (c – d)|
μ|Y1-Y2|-δ
p(|Y1-Y2|)
Inversion

Probabilistic Framework (5)
μ|Y1-Y2|-δ
(c – a) – (d – b) = s(X1) - s(X2)
P(Xi, Xj|trans) = 1 - P(μ|Y1-Y2| - δ ≤ |Y1- Y2| ≤μ|Y1-Y2| + δ) where δ= |μ|Y1-Y2| – (c – a) – (d – b) |
p(|Y1-Y2|)
Translocation

Remove veryRemove verysimilar mappingssimilar mappingsRemove veryRemove verysimilar mappingssimilar mappings
Flow of our Framework (1)1. Preprocessing step
Remove Remove short mappingsshort mappingsRemove Remove short mappingsshort mappings
Make all possible Make all possible combinations of combinations of
mappingsmappings
Make all possible Make all possible combinations of combinations of
mappingsmappings
Discard concordantDiscard concordantmatepairs matepairs
Discard concordantDiscard concordantmatepairs matepairs
Remove invalid Remove invalid strands (-,+) strands (-,+) Remove invalid Remove invalid strands (-,+) strands (-,+)
Get top K Get top K mappings mappings Get top K Get top K mappings mappings
Mask Mask repeatsrepeatsMask Mask repeatsrepeats

Flow of our Framework (2)2. Clustering
3. Finding structural variations
Do hierarchical clustering for each structural variationDo hierarchical clustering for each structural variation(Insertion, Deletion, Inversion, Translocation)(Insertion, Deletion, Inversion, Translocation)
Do hierarchical clustering for each structural variationDo hierarchical clustering for each structural variation(Insertion, Deletion, Inversion, Translocation)(Insertion, Deletion, Inversion, Translocation)
Find a (locally) Find a (locally) optimal optimal configurationconfiguration
Find a (locally) Find a (locally) optimal optimal configurationconfiguration
Learn parametersLearn parametersfor the objective for the objective functionfunction
Learn parametersLearn parametersfor the objective for the objective functionfunction
Find initial Find initial configurationconfigurationFind initial Find initial configurationconfiguration

X2
Hierarchical Clustering (1)
(ex) Insertion
A
REF
• A cluster is a set of maped locations explaining the same structural variant
•Linkage distance is D(X1, X2) = - ln P(X1, X2|C)
X1
X1X2
C={X1, X2}
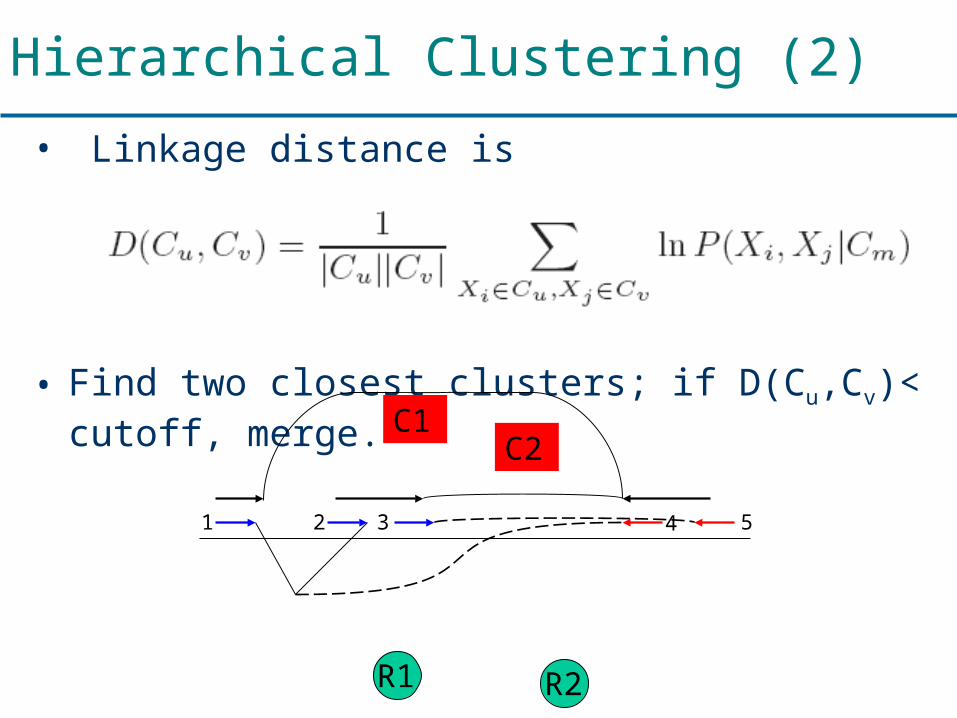
Hierarchical Clustering (2)
• Linkage distance is
• Find two closest clusters; if D(Cu,Cv)< cutoff, merge.
R1 R2
C2C1
1 2 3 4 5

Find a Unique Mapped Location
Assign matepairs to unique mapped locations (and hence unique clusters).
R1 R2
C2C1 C2C1
R2R1
1 2 3 4 5
M1,4 M2,4 M3,5

Which Location is Best?
• We define a objective Function J(ω)
– ƒ1 corresponds to BLAT hit scores
– ƒ2 corresponds to the probability
– ƒ3 corresponds to the size of clusters

Finding the “Best” Location
• Find the initial configuration greedily.– Assign matepairs to clusters starting with
those with fewest mapped locations
• Learn parameters for objective function J(ω).– We used hill climbing search to maximize
the log likelihood of P(ω|λi).
• Finally, find a configuration, locally maximizing J(ω) using hill climbing search.

Clustering Results
We started with ~2,984,000 matepair• ~93% were uniquely mapped• ~94% had a concordant position (mapped at ± 2)
Through the clustering procedure we found (FDR 0.05)
• 795 Insertion clusters (691 had a uniquely mapped read)• 1289 Deletion clusters (1120)• ~200 Inversion clusters (~150)• 164 Translocation (cross-chromosome) cluster
(all were required to have a uniquely mapped read)

Example Deletion

Agreement with Previous Results
We have comparedAll of the correlations (besides the one) are significant (p-values < 0.001 via Monte Carlo)
Type All Tuzun Levy Korbel DGV-All
Insertion 795(691) 50(36)/139 109(101)/319 1(1)/34 209(169)/2216
Deletion 1289(1120) 84(70)/102 194(188)/344 275(236)/742 539(446)/4697
Inversion ~200(~150) 198(46)/56 N/A 67(55)/105 111(87)/164

Translocations
• 47% of the translocations were close to the centromeres
• She et al. predicted up to 200 interchromosomal rearrangement events near centromeres per million years. The two donors are ~0.2 million years apart
• These could also be mis-assemblies.
Distance to centromere
<106 (106, 4.5*106] >4.5*106
<106 38 36 19
(106,4.5*106] 3 3
>4.5*106 65

Summary (Structural Variation)
• Introduced a probabilistic framework for finding structural variants that does not rely on ab initio mapping of matepairs to genomic positions.
• Isolated hundreds of insertions, deletions, and inversions between the reference public human genome and the JCVI donor.
• These results show statistically significant correlation with previous variation studies
• About 2/3 of the structural variants we isolate is not found in the Database of Genomic Variants

What about Copy Number Variants?
• Copy Number Variants are the result of duplications and deletions of large genomic segments
• Currently mainly found using microarray technology (ROMA, CGH)
• There is no algorithm for CNV finding with short reads (?)
• Goal: predict the number of times a certain segment appears in the genome

A Little Bit of Math
0
0.05
0.1
0.15
0.2
0.25
0.3
0 1 2 3 4 5 6
Let C = #reads / length of genome
Let i be a read
Let xi be # of times it was sampled.
Assembled genome should contain every read about xi / C times.
For example, let C = 3, xi = 7

More formally
Let n = number of reads, N = length of the genome
• The probability Pi that the read i was sampled xi times given that it appears in the genome gi times is
• We want to maximize the likelihood that all of the reads were sampled from the genome:
• However there is an additional constraint
€
Pi =
€
Pii
∏

The additional constraint…
0
0.05
0.1
0.15
0.2
0.25
0.3
0 1 2 3 4 5 6
0
0.05
0.1
0.15
0.2
0.25
0.3
0 1 2 3 4 5 6
0
0.05
0.1
0.15
0.2
0.25
0.3
0 1 2 3 4 5 6
ATCGGCACTG
GATCGGCACT
TATCGGCACT
g1 + g2 = g3
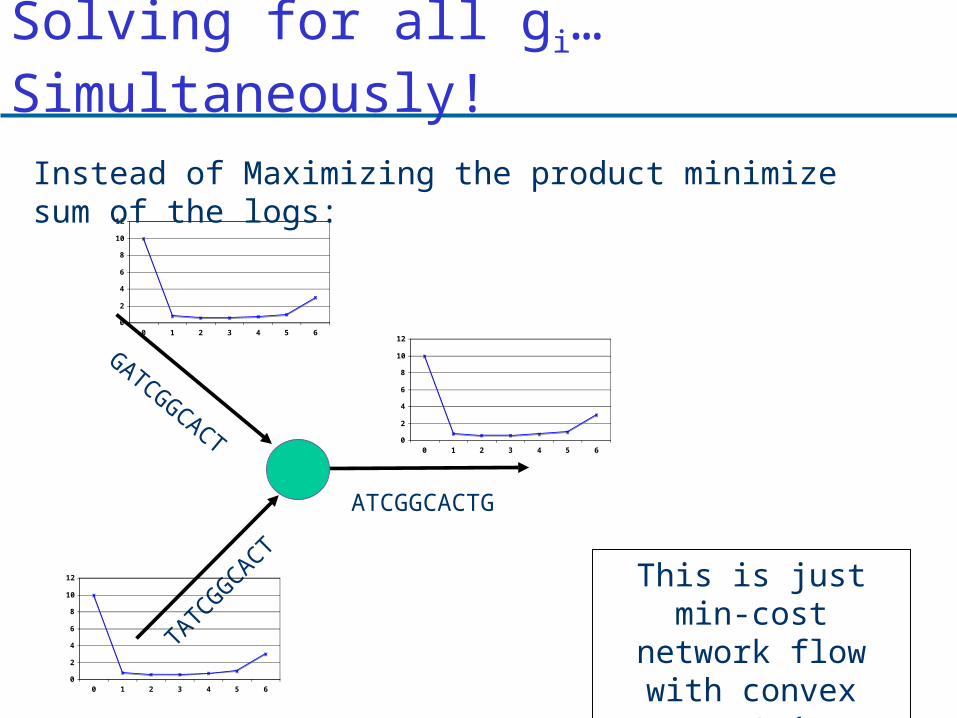
Solving for all gi… Simultaneously!
ATCGGCACTG
GATCGGCACT
TATCGGCACT
This is just min-cost network flow with
convex costs!
0
2
4
6
8
10
12
0 1 2 3 4 5 6
0
2
4
6
8
10
12
0 1 2 3 4 5 6
0
2
4
6
8
10
12
0 1 2 3 4 5 6
Instead of Maximizing the product minimize sum of the logs:

Copy Count Prediction Results
• Simulated reads from E.Coli bacteria (4.5Mb)
• How to scale this to Human???
C Copy-Count Error
-2 -1 0 +1 +2 +3
50x 4 397 3.9 M 170 18 6
75X 0 7 4.3 M 22 0 0
100X 0 2 4.5 M 6 0 0
200X 0 0 4.5 M 4 0 0

Discovering Variation
• SHRiMP -- SHort Read Mapping Package– Computes p-values & other statistics– Specialized Color-space alignment
• Algorithm for Structural Variation Discovery– Will it scale to short reads?
• A model for Copy Count Prediction– Works well with reads from E. coli, but how to scale to
Human?