hiv: cell binding and entry - cold spring...
TRANSCRIPT

HIV: Cell Binding and Entry
Craig B. Wilen1, John C. Tilton2, and Robert W. Doms1
1Department of Microbiology, University of Pennsylvania, Philadelphia, Pennsylvania 191042Department of General Medical Science, Center for Proteomics and Bioinformatics, Case WesternReserve University, Cleveland, Ohio 44106
Correspondence: [email protected]
The first step of the human immunodeficiency virus (HIV) replication cycle—binding andentry into the host cell—plays a major role in determining viral tropism and the ability ofHIV to degrade the human immune system. HIV uses a complex series of steps to deliverits genome into the host cell cytoplasm while simultaneously evading the host immuneresponse. To infect cells, the HIV protein envelope (Env) binds to the primary cellular recep-tor CD4 and then to a cellular coreceptor. This sequential binding triggers fusion of the viraland host cell membranes, initiating infection. Revealing the mechanism of HIVentry has pro-found implications for viral tropism, transmission, pathogenesis, and therapeutic interven-tion. Here, we provide an overview into the mechanism of HIV entry, provide historicalcontext to key discoveries, discuss recent advances, and speculate on future directions inthe field.
HIV ENTRY FUNDAMENTALS
HIVentry, the first phase of the viral replica-tion cycle, begins with the adhesion of
virus to the host cell and ends with the fusionof the cell and viral membranes with subsequentdelivery of the viral core into the cytoplasm.The intricate series of protein–protein interac-tions that ultimately results in virus infectioncan be divided into several phases, some ofwhich are essential and others that may serveto modulate the efficiency of the process. First,virions must bind to the target cell, with thisbeing mediated either by the viral envelope(Env) protein or host cell membrane proteinsincorporated into the virion with any one of anumber of various cell attachment factors.Attachment can be relatively nonspecific, with
Env interacting with negatively charged cell-surface heparan sulfate proteoglycans (Saphireet al. 2001), or can result from more specificinteractions between Env and a4b7 integrin(Arthos et al. 2008; Cicala et al. 2009) or patternrecognition receptors such as dendritic cell–specific intercellular adhesion molecular 3-grabbing non-integrin (DC-SIGN) (Geijten-beek et al. 2000; reviewed in Ugolini et al.1999). HIV attachment to the host cell via anyof these factors likely brings Env into close prox-imity with the viral receptor CD4 and corecep-tor, increasing the efficiency of infection (Fig. 1)(Orloff et al. 1991). However, attachment fac-tors differ from receptors in that they are notessential, and although they augment infectionin vitro, their physiologic role in vivo remainsunclear.
Editors: Frederic D. Bushman, Gary J. Nabel, and Ronald Swanstrom
Additional Perspectives on HIV available at www.perspectivesinmedicine.org
Copyright # 2012 Cold Spring Harbor Laboratory Press; all rights reserved; doi: 10.1101/cshperspect.a006866
Cite this article as Cold Spring Harb Perspect Med 2012;2:a006866
1
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
on August 19, 2018 - Published by Cold Spring Harbor Laboratory Press http://perspectivesinmedicine.cshlp.org/Downloaded from
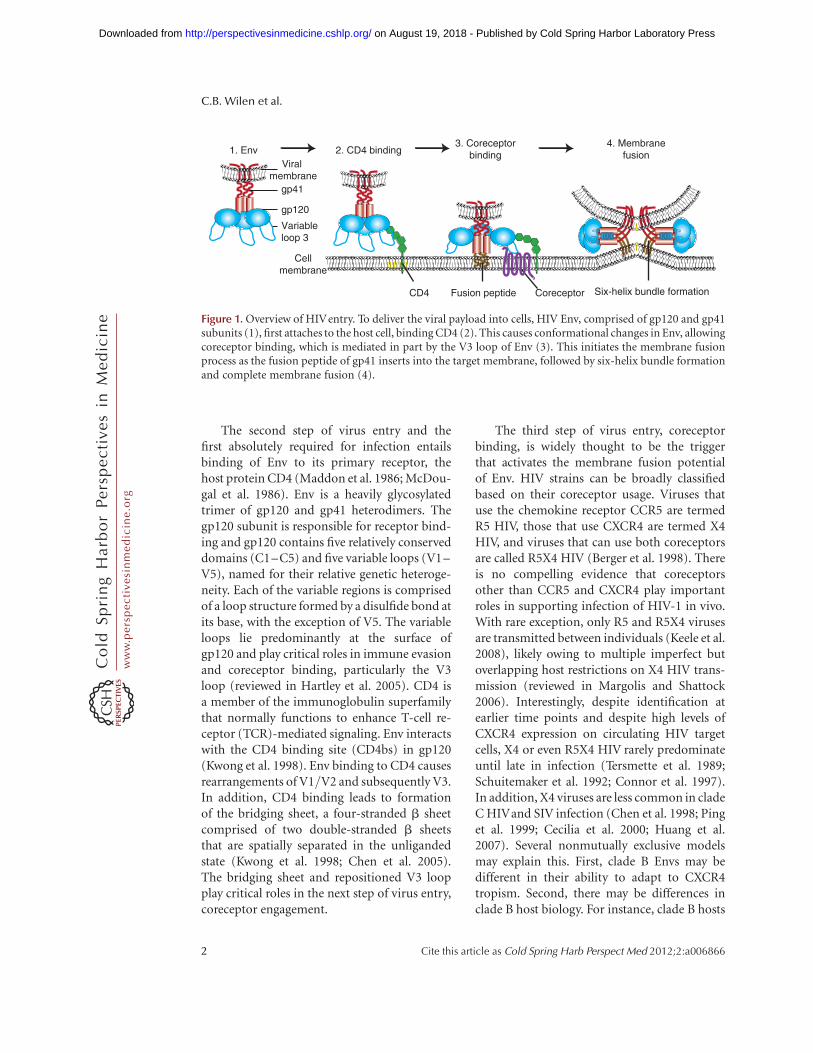
The second step of virus entry and thefirst absolutely required for infection entailsbinding of Env to its primary receptor, thehost protein CD4 (Maddon et al. 1986; McDou-gal et al. 1986). Env is a heavily glycosylatedtrimer of gp120 and gp41 heterodimers. Thegp120 subunit is responsible for receptor bind-ing and gp120 contains five relatively conserveddomains (C1–C5) and five variable loops (V1–V5), named for their relative genetic heteroge-neity. Each of the variable regions is comprisedof a loop structure formed by a disulfide bond atits base, with the exception of V5. The variableloops lie predominantly at the surface ofgp120 and play critical roles in immune evasionand coreceptor binding, particularly the V3loop (reviewed in Hartley et al. 2005). CD4 isa member of the immunoglobulin superfamilythat normally functions to enhance T-cell re-ceptor (TCR)-mediated signaling. Env interactswith the CD4 binding site (CD4bs) in gp120(Kwong et al. 1998). Env binding to CD4 causesrearrangements of V1/V2 and subsequently V3.In addition, CD4 binding leads to formationof the bridging sheet, a four-stranded b sheetcomprised of two double-stranded b sheetsthat are spatially separated in the unligandedstate (Kwong et al. 1998; Chen et al. 2005).The bridging sheet and repositioned V3 loopplay critical roles in the next step of virus entry,coreceptor engagement.
The third step of virus entry, coreceptorbinding, is widely thought to be the triggerthat activates the membrane fusion potentialof Env. HIV strains can be broadly classifiedbased on their coreceptor usage. Viruses thatuse the chemokine receptor CCR5 are termedR5 HIV, those that use CXCR4 are termed X4HIV, and viruses that can use both coreceptorsare called R5X4 HIV (Berger et al. 1998). Thereis no compelling evidence that coreceptorsother than CCR5 and CXCR4 play importantroles in supporting infection of HIV-1 in vivo.With rare exception, only R5 and R5X4 virusesare transmitted between individuals (Keele et al.2008), likely owing to multiple imperfect butoverlapping host restrictions on X4 HIV trans-mission (reviewed in Margolis and Shattock2006). Interestingly, despite identification atearlier time points and despite high levels ofCXCR4 expression on circulating HIV targetcells, X4 or even R5X4 HIV rarely predominateuntil late in infection (Tersmette et al. 1989;Schuitemaker et al. 1992; Connor et al. 1997).In addition, X4 viruses are less common in cladeC HIVand SIV infection (Chen et al. 1998; Pinget al. 1999; Cecilia et al. 2000; Huang et al.2007). Several nonmutually exclusive modelsmay explain this. First, clade B Envs may bedifferent in their ability to adapt to CXCR4tropism. Second, there may be differences inclade B host biology. For instance, clade B hosts
2. CD4 binding3. Coreceptor
binding4. Membrane
fusion1. Env
CD4
gp120
gp41
Variableloop 3
Coreceptor Six-helix bundle formationFusion peptide
Viral membrane
Cellmembrane
Figure 1. Overview of HIVentry. To deliver the viral payload into cells, HIV Env, comprised of gp120 and gp41subunits (1), first attaches to the host cell, binding CD4 (2). This causes conformational changes in Env, allowingcoreceptor binding, which is mediated in part by the V3 loop of Env (3). This initiates the membrane fusionprocess as the fusion peptide of gp41 inserts into the target membrane, followed by six-helix bundle formationand complete membrane fusion (4).
C.B. Wilen et al.
2 Cite this article as Cold Spring Harb Perspect Med 2012;2:a006866
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
on August 19, 2018 - Published by Cold Spring Harbor Laboratory Press http://perspectivesinmedicine.cshlp.org/Downloaded from

may have mitigated neutralizing antibody orcytotoxic T lymphocyte responses against X4HIV compared with R5 HIV. Finally, clade Bhosts most often live in developed countriesand may face different environmental stressesincluding fewer or different chronic coin-fections, which may increase target cell CCR5expression. Elucidating the mechanism of co-receptor switch is a critical next step because ithas implications for disease progression andtherapy with HIV entry inhibitors.
A fourth step of virus entry is movement ofthe virus particle to the site where productivemembrane fusion occurs. A series of recentstudies has shown that a number of virusesusurp cellular transport pathways to reach spe-cific destinations that are either needed forinfection or that make entry more efficient,and that HIV might likewise use the host cellmachinery to reach sites where membranefusion can occur (Lehmann et al. 2005; Coyneand Bergelson 2006; Sherer et al. 2010). Someviruses have been shown to “surf” along thecell surface, moving from distal sites of attach-ment to more proximal regions of the cellbody where virus entry occurs. Retroviruses,including HIV, have been shown to use thisprocess on some cell lines (Lehmann et al.2005; Sherer et al. 2010). In addition, HIVmay need to be internalized by the host cell’sendocytic machinery for productive membranefusion to occur, as is discussed in a later section(Miyauchi et al. 2009).
The fifth and final step of virus entry ismembrane fusion mediated by Env. Coreceptorbinding induces exposure of the hydrophobicgp41 fusion peptide, which inserts into thehost cell membrane. This tethers the viral andhost membranes, allowing the fusion peptideof each gp41 in the trimer to fold at a hingeregion, bringing an amino-terminal helicalregion (HR-N) and a carboxy-terminal helicalregion (HR-C) from each gp41 subunit togetherto form a six-helix bundle (6HB) (Chan et al.1997; Weissenhorn et al. 1997). Because theHR-N domain is in close proximity to thehost cell membrane owing to the fusion peptide,and the HR-C domain is in close proximityto the viral membrane owing to the gp41
transmembrane domain, formation of the 6HBis the driving force that brings the opposingmembranes into close apposition, resultingin the formation of a fusion pore (reviewedin Melikyan 2008). Whether one or multipleHIV Env trimers are needed for complete mem-brane fusion is not yet clear. In summary, core-ceptor binding unlocks the potential energy ofthe gp41 fusion complex resulting in 6HB for-mation, opening and stabilization of the mem-brane fusion pore, and subsequent delivery ofthe viral contents into the host cell cytoplasm.
DISCOVERY OF THE HIV RECEPTORS
In 1981, several years before the discovery ofHIV, Gottlieb and colleagues (1981) reportedCD4þ T-cell decline in four men who presentedwith pneumocystis pneumonia and mucosalcandidiasis, among other opportunistic infec-tions. Three years later, it was shown that HIVpreferentially infects CD4þ T cells (Klatzmannet al. 1984) and that infection is potently inhib-ited by CD4-specific antibodies (reviewed inSattentau and Weiss 1988). CD4 was thenshown to coimmunoprecipitate with Env (Mc-Dougal et al. 1986) and CD4 expression couldrescue infection in some nonpermissive cells(Maddon et al. 1986). However, CD4 transfec-tion into mouse cells rescued binding of virusto the cell surface but not membrane fusion orvirus infection, suggesting that there were otherrequired cofactors (Maddon et al. 1986).
Although the discovery of CD4 as the pri-mary HIV receptor occurred shortly after theonset of the epidemic, it took more than a dec-ade to discover the first coreceptor. In 1993,CD26 was reported as the elusive HIV corecep-tor (Callebaut et al. 1993); however, this waslater disproved by several groups (Lazaro et al.1994; Stamatatos and Levy 1994). In 1995,Feng and colleagues conclusively identifiedCXCR4 as a major HIV coreceptor by the useof an expression cloning strategy. A critical find-ing of this study was that CXCR4, then termedfusin, functioned as a coreceptor for what hadbeen termed T-cell line tropic strains of HIVbut not for virus strains that could infect humanmacrophages but that failed to enter T-cell lines
HIV: Cell Binding and Entry
Cite this article as Cold Spring Harb Perspect Med 2012;2:a006866 3
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
on August 19, 2018 - Published by Cold Spring Harbor Laboratory Press http://perspectivesinmedicine.cshlp.org/Downloaded from

(Feng et al. 1995, 1996). The seminal discoveryof CXCR4 as a G-protein-coupled receptor(GPCR) in combination with the identificationof the inhibitory effect of the b chemokinesCCL3 (MIP-1a), CCL4 (MIP1b), and CCL5(RANTES) (Cocchi et al. 1995) on some virusisolates led to the simultaneous and rapid dis-covery of CCR5 as the coreceptor for macro-phage-tropic virus strains by five differentgroups (Alkhatib et al. 1996; Choe et al. 1996;Deng et al. 1996; Doranz et al. 1996; Dragicet al. 1996).
The importance of the viral coreceptors forHIV infection in vivo was shown by the discov-ery of a 32 base-pair deletion in ccr5, termedccr5D32, which has an allelic frequency of�10% in Caucasians (Dean et al. 1996; Liuet al. 1996; Samson et al. 1996). The D32 muta-tion results in a premature stop codon in the sec-ond extracellular loop of CCR5 and subsequentretention of the mutant protein in the endoplas-mic reticulum. Homozygosity for this polymor-phism results in profound resistance to HIVinfection, although several D32 homozygoteshave been infected with X4 viruses (Balottaet al. 1997; O’Brien et al. 1997; Theodorouet al. 1997). In addition, heterozygosity conferspartial protection to infection (Dean et al. 1996;Samson et al. 1996) and disease progression(Dean et al. 1996; Huang et al. 1996).
Elucidating the mechanism of HIVentry hasdirectly translated into therapeutic benefit. Cur-rently, there are two FDA-approved entry inhib-itors, enfuvirtide and maraviroc, whereas othersare in various stages of development. In 2003,enfuvirtide became the first licensed entryinhibitor; it is a 36-residue-long peptide whosesequence is based on that of the HR-C in gp41.As a result, enfuvirtide behaves much like HR-Cin that it binds to the HR-N prehairpin inter-mediate and inhibits 6HB formation and subse-quent membrane fusion (Wild et al. 1992,1993). Although enfuvirtide is a highly specificand effective membrane fusion inhibitor (Lale-zari et al. 2003; Lazzarin et al. 2003), its use hasbeen limited because it must be injected owingto its lack of oral bioavailability. Recently,protease-resistant D-peptide fusion inhibitorshave been developed that also prevent 6HB
formation, which may overcome this limitation(Eckert et al. 1999; Welch et al. 2007, 2010). Inaddition to enfuvirtide, the CCR5 inhibitormaraviroc has been approved for clinical use.Maraviroc is a small-molecule allosteric inhibi-tor that binds within the CCR5 transmembranecavity resulting in conformational changes inthe extracellular loop domains of the chemo-kine receptor that interact with Env (Dorr et al.2005). Similar CCR5 small-molecule inhibitorsare in various stages of testing (reviewed in Til-ton and Doms 2009).
KEY RECENT ADVANCES
Our understanding of the HIV entry processis derived largely from structural and in vitrostudies. As the field has evolved, there is nowincreased emphasis on placing the now ratherwell-understood membrane fusion reaction ina cellular context, asking where and when virusentry takes place as well as how virus particlesare transferred between cells. Increased struc-tural detail continues to provide insight intothe entry process and suggests targets for small-molecule inhibitors and neutralizing anti-bodies. Finally, attempts to recapitulate theccr5D32 phenotype have been developed withsome being brought forward to early-stage clin-ical development (Perez et al. 2008).
New Structural Information
A full understanding of the HIV entry processrequires detailed structural information. Thestructure of CD4 alone and in complex with agp120 core fragment has been solved for HIV(Kwong et al. 1998; Huang et al. 2005) and sim-ian immunodeficiency virus (SIV) (see onlineMovie 1 at www.perspectivesinmedicine.org)(Chen et al. 2005). The structure of the postfu-sion 6HB in gp41 has also been determined (seeonline Movie 2 at www.perspectivesinmedicine.org) (Chan et al. 1997; Weissenhorn et al. 1997).What has been lacking is a structure of the nativeEnv trimer and the HIV coreceptors. However,Wu et al. (2010) recently described five inde-pendent structures of CXCR4 bound to two dif-ferent small-molecule antagonists, which have
C.B. Wilen et al.
4 Cite this article as Cold Spring Harb Perspect Med 2012;2:a006866
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
on August 19, 2018 - Published by Cold Spring Harbor Laboratory Press http://perspectivesinmedicine.cshlp.org/Downloaded from

given insight into both the tertiary and quater-nary structure of the native protein (Fig. 2).First, both chemokines and Env have beenreported to engage CCR5 and CXCR4 in a two-site model with the chemokine receptor aminoterminus as site one and the extracellular loops(ECLs), particularly ECL2, as site two. Al-though the orientation of the CXCR4 amino-terminal domain could not be solved owing tostructural flexibility, the crystal structure pro-vides high-resolution insight into the ECL2binding site. Second, all five structures portrayCXCR4 as a homodimer (see online Movie 3at www.perspectivesinmedicine.org), which isconsistent with biochemical studies that havesuggested CXCR4 exists as an oligomer in thehost cell membrane (Babcock et al. 2003).Although the implications of CXCR4 dimeriza-tion remain unclear for HIV infection, it mayexplain the dominant phenotype of a carboxy-terminal CXCR4 human mutation that resultsin WHIM syndrome, which is characterizedby warts, hypogammaglobulinemia, infections,and myelokathexis (retention of neutrophils inthe bone marrow) (Hernandez et al. 2003).Finally, the identified homodimer interfacemay represent a novel CXCR4 or potentially
CCR5 drug target, because CCR5 and CXCR4have been reported to heterodimerize in vivo(Sohy et al. 2007, 2009). Further structural stud-ies are needed to better define the precise inter-actions of Env and coreceptor and to assess themechanisms of signaling and heterodimeriza-tion with other chemokine receptors.
Where Does Virus Entry Occur?
The entry of viruses into cells is controlled inboth time and space, with these parametersbeing regulated by host cell factors that serveto unlock the membrane fusion potential ofviral membrane proteins. Many viruses requiredelivery by the host cell into an acidic, intracel-lular compartment where low pH triggers mem-brane-fusion-inducing conformational changes(reviewed in Marsh and Helenius 2006). HIVentry does not require low pH; instead it istriggered by receptor engagement (Stein et al.1987). The fact that HIV does not require lowpH for cellular entry does not imply that fusionoccurs at the cell surface. In fact, no spatialinformation is provided by the triggering mech-anism. Despite this, it was often assumed thatHIV fuses at the cell surface owing to several
Figure 2. Model of gp120 engagement of CD4 and CXCR4. Recent structural studies have enhanced our under-standing of the molecular interactions between gp120 (cyan) and its receptors. Here, CD4 (green) and CXCR4(purple), shown as monomers for clarity, are shown simultaneously binding to gp120. (A) Lateral view. (B) Topview. However, the number of CD4 and coreceptor molecules required to interact with Env to mediate produc-tive fusion remains unknown. (C) Gp120 has two key interactions with coreceptor. (1) The base of the V3 loopbinds to the amino-terminal domain of the coreceptor, whereas the tip of the V3 loop binds to the second extrac-ellular loop (ECL2). Although both interactions are important, viral strains differ on their dependency of eachinteraction. (Structural model generated by Wu et al. 2010.)
HIV: Cell Binding and Entry
Cite this article as Cold Spring Harb Perspect Med 2012;2:a006866 5
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
on August 19, 2018 - Published by Cold Spring Harbor Laboratory Press http://perspectivesinmedicine.cshlp.org/Downloaded from

observations (reviewed in Uchil and Mothes2009). First, Env expression on the cell surfacecan mediate cell-to-cell fusion, indicating notonly that Env is the only viral membraneprotein needed to elicit fusion but that lowpH is clearly not required. Second, very earlystudies on HIVentry showed that lysomotropicagents, which increase endosomal pH, do notinhibit HIV infection (McClure et al. 1988).Third, inhibiting endocytosis of CD4 in celllines by mutating its cytoplasmic domain doesnot affect HIV infection (Maddon et al. 1988).Together, these studies show that HIV entry isnot pH dependent, but they provide no defini-tive information as to whether fusion occursat the cell surface or from within endocyticvesicles, albeit in a pH-independent fashion.
The question of where HIV-membranefusion occurs has recently been reexamined(Miyauchi et al. 2009). By combining lipidand content mixing assays with single virion flu-orescent imaging, Miyauchi et al. tracked thelocation of virus membrane fusion in HeLa cellsoverexpressing CD4, CCR5, and CXCR4. Theyfound that whereas lipid mixing can occur atthe cell surface, content mixing only occurredin intracellular perinuclear compartments andthus concluded that complete fusion requiresendocytosis. Whether this is always the caseremains to be determined because the geneticvariability of HIV and the diverse cell types itcan infect make generalization difficult.
An interesting question is whether the siteof entry matters; with regard to the use ofentry inhibitors, probably not: Both coreceptorantagonists and fusion inhibitors block virusinfection in vitro and in vivo, and neutralizingantibodies clearly function as well. However,the site of entry is more likely to have an impacton the likelihood of a productive infectionactually occurring. For instance, after cellularattachment, HIV can actively surf along the cel-lular membrane from filopodia or microvilli tothe cell body. This actin-dependent processrequires receptor engagement and serves toenhance infection efficiency. Surfing towardthe cell body may have several favorable conse-quences for the virus. First, it may facilitateendocytic HIV uptake. Second, it may bring
the virus to a membrane region that has higherlevels of coreceptor or important downstreamsignaling molecules (McDonald et al. 2003).Third, it may allow the fusion event to occurcloser to the nucleus, which is the ultimate tar-get of HIV. Thus, the site of initial HIV attach-ment is likely random; however, HIV hijacksthe cellular machinery to traverse the cell mem-brane to a more favorable site of entry, be it atthe plasma membrane or endosome, which ulti-mately serves to augment infection efficiency(Lehmann et al. 2005).
Cell–Cell Transfer and the VirologicalSynapse
In vitro, the rate-limiting step of virus infectionis attachment to the host cell. In vivo, newlyproduced virions may well encounter an imme-diately adjoining, uninfected cell. In some cases,transfer of virus from one cell to another is aspecialized process, as in the case of dendriticcells (DCs), which are professional antigen-presenting cells (APCs) that scavenge theperiphery, sampling antigen. They are com-monly found in the mucosa and thus may beencountered by HIV during transmission. Onantigen binding, DCs migrate to the lymphnodes, process, and present the antigen to Tcells to trigger an adaptive immune response.DCs are relatively resistant to productive HIVinfection owing to a combination of low CD4and coreceptor expression, host restriction fac-tors, postintegration HIV transcription blocks,and other unknown factors (Bakri et al. 2001).However, they express a diverse range of attach-ment factors that facilitate the internalizationand processing of pathogens before antigen pre-sentation. HIV, along with viruses (Igakura et al.2003; Yang et al. 2004), can take advantage ofthis pathway to augment infection efficiencyand dissemination (Fig. 3) (reviewed in Piguetand Steinman 2007).
Cameron et al. (1992) first showed that DCscould catalyze HIV infection of coculturedCD4þ T cells without themselves getting pro-ductively infected. Each DC can bind up to sev-eral hundred virions (McDonald et al. 2003)most likely via a C-type lectin such as DC-SIGN
C.B. Wilen et al.
6 Cite this article as Cold Spring Harb Perspect Med 2012;2:a006866
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
on August 19, 2018 - Published by Cold Spring Harbor Laboratory Press http://perspectivesinmedicine.cshlp.org/Downloaded from

(Geijtenbeek et al. 2000; Turville et al. 2002).After binding, the virions are endocytosedinto a trypsin-resistant compartment (Geijten-beek et al. 2000), and then after DC binding toa T cell, internalized virus migrates to theDC:T-cell interface (McDonald et al. 2003)where it encounters the T-cell membrane form-ing the infectious synapse, analogous to theimmunologic synapse that forms on MHC–TCR binding (reviewed in Vasiliver-Shamiset al. 2010). In addition to efficiently concen-trating and presenting HIV at the site of T-cellcontact, the infectious synapse is characterizedby recruitment of CD4, CCR5, and CXCR4.
Recent advances in electron microscopy haveenabled 3D (three-dimensional)-structural stu-dies of the infectious synapse that have shedlight on this mechanism (Felts et al. 2010).DCs produce membranous protrusions thatengulf the surrounding extracellular environ-ment, trapping virions in a surface-accessiblebut protected compartment. It remains unclear
as to whether this occurs before or after virionbinding and whether it is Env induced. WhenCD4þT cells contact DCs, they extend filopodia,enriched for CD4 and coreceptor, into the inva-ginated DC compartments that contain boundvirions (Fig. 3). Together, the efficient bindingof HIV, relocalization to the point of CD4þ
T-cell contact, and the recruitment of the requi-site HIVentry receptors promote HIV infectionat the infectious synapse (McDonald et al. 2003;Hubner et al. 2009).
A Novel Attachment Factor: a4b7 Integrin
Although cell-to-cell transmission of HIV aug-ments infection efficiency, the mechanism ofvirological synapse formation remains unclear.a4b7 integrin has been reported to bindgp120; induce activation of LFA-1 (aLb2 in-tegrin), which contributes to formation of theimmunologic synapse (reviewed in Bromley etal. 2001); and subsequently augment infection
Figure 3. Model of DC-mediated transinfection of CD4þ T cells. (A) DCs capture and concentrate virions intrypsin-resistant surface-accessible compartments. (B) CD4þ T cells, containing membrane protrusions,bind DCs. (C) The CD4þ T-cell protrusions invade the virus-containing compartments and efficiently bindHIV. (D) Virus then migrates toward the cell body to initiate infection. (Figure reproduced from Felts et al.2010; reprinted, with permission, from Proceedings of the National Academy of Sciences # 2010.)
HIV: Cell Binding and Entry
Cite this article as Cold Spring Harb Perspect Med 2012;2:a006866 7
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
on August 19, 2018 - Published by Cold Spring Harbor Laboratory Press http://perspectivesinmedicine.cshlp.org/Downloaded from

efficiency in vitro (Arthos et al. 2008; Cicalaet al. 2009).
a4b7 is a heterodimeric protein comprisedof an a4 and b7 subunit that when expressed onCD4þ T cells facilitates homing to the gut andother mucosal tissues. Its activation and expres-sion are up-regulated by retinoic acid in vitro,which may also be locally secreted by mucosalDCs in vivo. The discovery ofa4b7 as an attach-ment factor is of particular interest because HIVdisrupts the integrity of the mucosal barrier andpreferentially depletes gut CD4þ T cells, whichare more activated and express higher levels ofCCR5 than peripheral CD4þ T cells. a4b7 isthought to bind an LDV (Leu-Asp-Val) tripep-tide motif on the second variable loop (V2) ofgp120, with this resulting in lymphocyte func-tion-associated antigen 1 (LFA-1) activation.In addition, a4b7 colocalizes with CD4 andCCR5 at the virological synapse, which may fur-ther enhance infection. Blockade of a4b7 withmonoclonal antibodies or a peptide delays rep-lication of HIV in vitro, further supporting itsrole in HIV infection (Cicala et al. 2009). Futurework is needed to assess whether there are pro-tective effects of inhibiting HIV-a4b7 interac-tions in vivo, and to validate this novelattachment factor as a therapeutic target.
FUTURE DIRECTIONS SIGNALING
Signal Transduction Mediated by HIV Env
HIV Env has the capacity to mediate signaltransduction cascades through CD4 and core-ceptors, although the physiological importanceon viral entry, replication, and pathogenesisremains controversial. Recent discoveries haveprovided evidence that signaling does play animportant role under certain circumstances.In this section, we briefly review what is knownabout Env signaling through its receptor andcoreceptors and highlight some of the recentdiscoveries in the field.
Signal Transduction through CD4
During encounters between CD4þ T cells andAPCs, the CD4 molecule acts to enhance signal-ing through the TCR as it engages a cognate
peptide bound to major histocompatibilitycomplex class II (pMHC). Recruitment of theSrc-family protein tyrosine kinase Lck to theimmunological synapse through an interac-tion with the cytoplasmic tail of CD4 resultsin phosphorylation of immunoreceptor-tyro-sine-based activation motifs (ITAMs) presenton the CD3g, CD3d, CD31, and TCR-z subu-nits of the TCR complex and interactions witheffector molecules (Love and Hayes 2010; Pad-han and Varma 2010). Doubly phosphorylatedITAMs on the TCR-z subunit recruit the Syk-family protein tyrosine kinase ZAP-70, whichin turn phosphorylates the scaffold proteinsLAT and SLP-76 that recruit many additionalsignaling proteins, eventually resulting inT-cell activation and proliferation (reviewed inLove and Hayes 2010).
Although HIV has been shown to signalthrough CD4 on binding (Hivroz et al. 1993;Briant et al. 1998) and to increase the activityof Lck (Juszczak et al. 1991), it remains unclearwhether signaling is essential for infectivity.Although multiple early studies with truncatedforms of CD4 indicated that signaling throughCD4 was not required for HIVentry (Benkiraneet al. 1994; Tremblay et al. 1994), most of theseassays were performed on cell lines and usingcell-free virus infection. Several more recentstudies have shown a potential role for Env-CD4signaling in the context of cell–cell spread at theinfectious synapse, which as noted before is aspecialized junction between cells that resem-bles the immunological synapse between APCsand T cells. One of the key features of the immu-nological synapse is that T cells stop migrationthrough the lymph node or target tissue to allowsustained interaction with the APCs. HIV gp120was able to arrest the migration of primary acti-vated CD4þ T cells in the presence of ICAM-1and spontaneously induce the formation of avirological synapse (Vasiliver-Shamis et al.2008). Signal transduction through CD4 andLck were subsequently found to be responsiblefor depletion of cortical F-actin underneaththe virological synapse using a planar mem-brane model system, allowing transfer of theviral core from the plasma membrane to thenucleus (Vasiliver-Shamis et al. 2009). These
C.B. Wilen et al.
8 Cite this article as Cold Spring Harb Perspect Med 2012;2:a006866
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
on August 19, 2018 - Published by Cold Spring Harbor Laboratory Press http://perspectivesinmedicine.cshlp.org/Downloaded from

studies suggest that Env signaling through CD4may play a role in HIVentry under physiologicalconditions.
Signal Transduction through CCR5and CXCR4
CCR5 and CXCR4 are both members of theseven-transmembrane-spanning family of het-erotrimeric GPCRs. This family of proteinsis characterized by an extracellular amino-terminal domain, seven membrane-spanningdomains that form three extracellular and threeintracellular loops, and a cytoplasmic tail do-main. The amino terminus (site one) and threeECLs (site two) together form the bindingpocket for the cognate chemokines, whichappear to attach to their receptor and transmitsignals in a two-site binding process (reviewedin Clark-Lewis et al. 1995). The intracellularloops and cytoplasmic tail bind to the heterotri-meric G proteins that in turn mediate effectorfunctions.
The ability of HIV to signal through the che-mokine receptors has been documented sinceshortly after their identification as coreceptors.Early studies tested the requirement for HIVto signal through coreceptor during entry intocells by inhibiting Gai subunits through pertus-sis toxin (PTX) (Cocchi et al. 1996) or by creat-ing chemokine mutants that abolished theability to mobilize intracellular Ca2þ, a key sec-ond messenger in signal transduction pathways(Alkhatib et al. 1997; Farzan et al. 1997; Goslinget al. 1997). Neither of these interventionsblocked the ability of HIV to enter cells, leadingto the conclusion that signaling was dispensablefor viral infection and replication in target cells.During the next decade, a multitude of studiesexamined the requirement for coreceptor sig-naling during HIV entry, with sometimes con-tradictory results (reviewed in Wu and Yoder2009). Often, experiments using cell lines oractivated primary cells would indicate thatsignaling was not required for entry, whereasexperiments with resting primary cells wouldindicate a role for signaling. Because the major-ity of T cells in lymphoid tissues are in rest-ing or nondividing states—particularly before
infection with HIV—examining the role for sig-naling under these conditions is particularlyrelevant.
Several recent studies have indicated thatHIV depends on chemokine receptor signal-ing for efficient infection of target cells. First,Yoder and colleagues (2008) examined the abil-ity of X4 HIV to infect resting CD4þ T cellsand found that HIV-mediated, Gai signalingthrough CXCR4 was required for entry. Thispathway triggered the activation of a cellularactin-depolymerizing molecule, cofilin, alteringcortical actin dynamics near the cell surfaceand facilitating viral fusion. Moreover, the studysuggested a role for cofilin in movement of theviral preintegration complex (PIC) toward thenucleus. In contrast to the resting cell modelused in this study, most activated and cyclingCD4þ T cells disassemble cortical actin withoutthe requirement of cofilin. Although this studywas limited to CXCR4 viral entry, activationof cofilin may also be required for CCR5 virusesto enter resting, nondividing memory cells,such as the majority of CD4þ T cells presentin the lamina propria of the gut.
A second series of experiments from Har-mon and colleagues (2010) showed that HIValso signals through the Gaq subunit, resultingin phospholipase C and Rac activation. Racand the tyrosine kinase Abl then become linkedto the Wave2 complex through the adapter pro-teins Tiam-1 and IRSp53, promoting Arp2/3-dependent actin nucleation and polymeriza-tion. Blocking activation of the Wave2 complexwith small interfering RNAs (siRNAs) or Ablkinase inhibitors arrested HIVentry at the hem-ifusion stage. Together, these experiments sug-gest a critical role for envelope-coreceptorsignaling-induced actin remodeling duringHIV entry, particularly in the case of restingCD4þ T cells.
The role of Env-mediated signaling throughCD4 and coreceptor in the HIV entry processremains incompletely defined, but it appearsincreasingly likely that there are essential rolesfor signal transduction with physiologicallyrelevant conditions and cell types. A commontheme among these studies is the necessity forsignaling in the reorganization of cytoskeletal
HIV: Cell Binding and Entry
Cite this article as Cold Spring Harb Perspect Med 2012;2:a006866 9
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
on August 19, 2018 - Published by Cold Spring Harbor Laboratory Press http://perspectivesinmedicine.cshlp.org/Downloaded from

actin, which may be involved in several key viralprocesses including “surfing,” passage throughthe cortical actin barrier near the plasma mem-brane, and movement of the viral PIC into thenucleus.
CONCLUDING REMARKS
HIV entry is an active process that involveshijacking various components of the cellularmachinery. Although Env engagement of CD4and coreceptor are the most critical events ofthe entry process, viral surfing, endocytosis,cell-to-cell transmission, and receptor-medi-ated signaling likely play a role in enhancinginfection efficiency by overcoming varioushost restrictions. Such factors likely affect theefficiency of transmission host cell tropism,and disease progression and are thus criticalareas of future investigation. Although numer-ous questions remain, the most crucial is howcan we better exploit our knowledge of HIVentry for therapeutic gain? Maraviroc and enfu-virtide have shown the efficacy of inhibitingentry, but novel therapeutic targets are neededand these likely represent the host moleculescoopted by HIV.
ACKNOWLEDGMENTS
C.B.W., J.C.T., and R.W.D. were supported bygrants T32 AI000632, F32 1F32AI077370, andR01 AI 040880, respectively. We thank BeiliWu, Ray Stevens, and Sriram Subramaniamfor the use of figures and PDB (Protein DataBank) files.
REFERENCES
Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE,Murphy PM, Berger EA. 1996. CC CKR5: A RANTES,MIP-1a, MIP-1b receptor as a fusion cofactor formacrophage-tropic HIV-1. Science 272: 1955–1958.
Alkhatib G, Locati M, Kennedy PE, Murphy PM, Berger EA.1997. HIV-1 coreceptor activity of CCR5 and its inhibi-tion by chemokines: Independence from G protein sig-naling and importance of coreceptor downmodulation.Virology 234: 340–348.
Arthos J, Cicala C, Martinelli E, Macleod K, Van Ryk D, WeiD, Xiao Z, Veenstra TD, Conrad TP, Lempicki RA, et al.2008. HIV-1 envelope protein binds to and signals
through integrina4b7, the gut mucosal homing receptorfor peripheral T cells. Nat Immunol 9: 301–309.
Babcock GJ, Farzan M, Sodroski J. 2003. Ligand-independ-ent dimerization of CXCR4, a principal HIV-1 corecep-tor. J Biol Chem 278: 3378–3385.
Bakri Y, Schiffer C, Zennou V, Charneau P, Kahn E, Ben-jouad A, Gluckman JC, Canque B. 2001. The maturationof dendritic cells results in postintegration inhibition ofHIV-1 replication. J Immunol 166: 3780–3788.
Balotta C, Bagnarelli P, Violin M, Ridolfo AL, Zhou D, Ber-lusconi A, Corvasce S, Corbellino M, Clementi M, ClericiM, et al. 1997. Homozygous D32 deletion of the CCR-5chemokine receptor gene in an HIV-1-infected patient.AIDS 11: F67–F71.
Benkirane M, Jeang KT, Devaux C. 1994. The cytoplasmicdomain of CD4 plays a critical role during the early stagesof HIV infection in T-cells. EMBO J 13: 5559–5569.
Berger EA, Doms RW, Fenyo EM, Korber BT, Littman DR,Moore JP, Sattentau QJ, Schuitemaker H, Sodroski J,Weiss RA. 1998. A new classification for HIV-1. Nature391: 240.
Briant L, Robert-Hebmann V, Acquaviva C, Pelchen-Mat-thews A, Marsh M, Devaux C. 1998. The protein tyrosinekinase p56lck is required for triggering NF-kB activationupon interaction of human immunodeficiency virus type1 envelope glycoprotein gp120 with cell surface CD4.J Virol 72: 6207–6214.
Bromley SK, Burack WR, Johnson KG, Somersalo K, SimsTN, Sumen C, Davis MM, Shaw AS, Allen PM, DustinML. 2001. The immunological synapse. Annu Rev Immu-nol 19: 375–396.
Callebaut C, Krust B, Jacotot E, Hovanessian AG. 1993. Tcell activation antigen, CD26, as a cofactor for entry ofHIV in CD4þ cells. Science 262: 2045–2050.
Cameron PU, Freudenthal PS, Barker JM, Gezelter S, InabaK, Steinman RM. 1992. Dendritic cells exposed to humanimmunodeficiency virus type-1 transmit a vigorous cyto-pathic infection to CD4þ T cells. Science 257: 383–387.
Cecilia D, Kulkarni SS, Tripathy SP, Gangakhedkar RR, Para-njape RS, Gadkari DA. 2000. Absence of coreceptorswitch with disease progression in human immunodefi-ciency virus infections in India. Virology 271: 253–258.
Chan DC, Fass D, Berger JM, Kim PS. 1997. Core structureof gp41 from the HIV envelope glycoprotein. Cell 89:263–273.
Chen Z, Gettie A, Ho DD, Marx PA. 1998. Primary SIVsmisolates use the CCR5 coreceptor from sooty mangabeysnaturally infected in West Africa: A comparison of core-ceptor usage of primary SIVsm, HIV-2, and SIVmac.Virology 246: 113–124.
Chen B, Vogan EM, Gong H, Skehel JJ, Wiley DC, HarrisonSC. 2005. Structure of an unliganded simian immunode-ficiency virus gp120 core. Nature 433: 834–841.
Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD,Wu L, Mackay CR, LaRosa G, Newman W, et al. 1996. Theb-chemokine receptors CCR3 and CCR5 facilitate infec-tion by primary HIV-1 isolates. Cell 85: 1135–1148.
Cicala C, Martinelli E, McNally JP, Goode DJ, Gopaul R,Hiatt J, Jelicic K, Kottilil S, Macleod K, O’Shea A, et al.2009. The integrin a4b7 forms a complex with cell-surface CD4 and defines a T-cell subset that is highly
C.B. Wilen et al.
10 Cite this article as Cold Spring Harb Perspect Med 2012;2:a006866
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
on August 19, 2018 - Published by Cold Spring Harbor Laboratory Press http://perspectivesinmedicine.cshlp.org/Downloaded from

susceptible to infection by HIV-1. Proc Natl Acad Sci 106:20877–20882.
Clark-Lewis I, Kim KS, Rajarathnam K, Gong JH, Dewald B,Moser B, Baggiolini M, Sykes BD. 1995. Structure-ac-tivity relationships of chemokines. J Leukoc Biol 57:703–711.
Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC,Lusso P. 1995. Identification of RANTES, MIP-1 a, andMIP-1 b as the major HIV-suppressive factors producedby CD8þ T cells. Science 270: 1811–1815.
Cocchi F, DeVico AL, Garzino-Demo A, Cara A, Gallo RC,Lusso P. 1996. The V3 domain of the HIV-1 gp120 enve-lope glycoprotein is critical for chemokine-mediatedblockade of infection. Nat Med 2: 1244–1247.
Connor RI, Sheridan KE, Ceradini D, Choe S, Landau NR.1997. Change in coreceptor use correlates with diseaseprogression in HIV-1–infected individuals. J Exp Med185: 621–628.
Coyne CB, Bergelson JM. 2006. Virus-induced Abl and Fynkinase signals permit coxsackievirus entry through epi-thelial tight junctions. Cell 124: 119–131.
Dean M, Carrington M, Winkler C, Huttley GA, Smith MW,Allikmets R, Goedert JJ, Buchbinder SP, Vittinghoff E,Gomperts E, et al. 1996. Genetic restriction of HIV-1infection and progression to AIDS by a deletion alleleof the CKR5 structural gene. Hemophilia Growth andDevelopment Study, Multicenter AIDS Cohort Study,Multicenter Hemophilia Cohort Study, San FranciscoCity Cohort, ALIVE Study. Science 273: 1856–1862.
Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, BurkhartM, Di Marzio P, Marmon S, Sutton RE, Hill CM, et al.1996. Identification of a major co-receptor for primaryisolates of HIV-1. Nature 381: 661–666.
Doranz BJ, Rucker J, Yi Y, Smyth RJ, Samson M, Peiper SC,Parmentier M, Collman RG, Doms RW. 1996. A dual-tropic primary HIV-1 isolate that uses fusin and theb-chemokine receptors CKR-5, CKR-3, and CKR-2b asfusion cofactors. Cell 85: 1149–1158.
Dorr P, Westby M, Dobbs S, Griffin P, Irvine B, MacartneyM, Mori J, Rickett G, Smith-Burchnell C, Napier C,et al. 2005. Maraviroc (UK-427,857), a potent, orallybioavailable, and selective small-molecule inhibitor ofchemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimi-crob Agents Chemother 49: 4721–4732.
Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Naga-shima KA, Cayanan C, Maddon PJ, Koup RA, Moore JP,et al. 1996. HIV-1 entry into CD4þ cells is mediatedby the chemokine receptor CC-CKR-5. Nature 381:667–673.
Eckert DM, Malashkevich VN, Hong LH, Carr PA, Kim PS.1999. Inhibiting HIV-1 entry: Discovery of D-peptideinhibitors that target the gp41 coiled-coil pocket. Cell99: 103–115.
Farzan M, Choe H, Martin KA, Sun Y, Sidelko M, MackayCR, Gerard NP, Sodroski J, Gerard C. 1997. HIV-1 entryand macrophage inflammatory protein-1b-mediatedsignaling are independent functions of the chemokinereceptor CCR5. J Biol Chem 272: 6854–6857.
Felts RL, Narayan K, Estes JD, Shi D, Trubey CM, Fu J, Hart-nell LM, Ruthel GT, Schneider DK, Nagashima K, et al.2010. 3D visualization of HIV transfer at the virological
synapse between dendritic cells and T cells. Proc NatlAcad Sci 107: 13336–13341.
Feng Y, Zhang F, Lokey LK, Chastain JL, Lakkis L, EberhartD, Warren ST. 1995. Translational suppression by trinu-cleotide repeat expansion at FMR1. Science 268: 731–734.
Feng Y, Broder CC, Kennedy PE, Berger EA. 1996. HIV-1entry cofactor: Functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science272: 872–877.
Geijtenbeek TB, Kwon DS, Torensma R, van Vliet SJ, vanDuijnhoven GC, Middel J, Cornelissen IL, Nottet HS,KewalRamani VN, Littman DR, et al. 2000. DC-SIGN,a dendritic cell-specific HIV-1-binding protein thatenhances trans-infection of T cells. Cell 100: 587–597.
Gosling J, Monteclaro FS, Atchison RE, Arai H, Tsou CL,Goldsmith MA, Charo IF. 1997. Molecular uncouplingof C-C chemokine receptor 5-induced chemotaxis andsignal transduction from HIV-1 coreceptor activity. ProcNatl Acad Sci 94: 5061–5066.
Gottlieb MS, Schroff R, Schanker HM, Weisman JD, Fan PT,Wolf RA, Saxon A. 1981. Pneumocystis carinii pneumoniaand mucosal candidiasis in previously healthy homosex-ual men: Evidence of a new acquired cellular immunode-ficiency. N Engl J Med 305: 1425–1431.
Harmon B, Campbell N, Ratner L. 2010. Role of Abl kinaseand the Wave2 signaling complex in HIV-1 entry ata post-hemifusion step. PLoS Pathog 6: e1000956. doi:10.1371/journal.ppat.1000956
Hartley O, Klasse PJ, Sattentau QJ, Moore JP. 2005. V3:HIV’s switch-hitter. AIDS Res Hum Retroviruses 21:171–189.
Hernandez PA, Gorlin RJ, Lukens JN, Taniuchi S, Bohinjec J,Francois F, Klotman ME, Diaz GA. 2003. Mutations in thechemokine receptor gene CXCR4 are associated withWHIM syndrome, a combined immunodeficiency dis-ease. Nat Genet 34: 70–74.
Hivroz C, Mazerolles F, Soula M, Fagard R, Graton S,Meloche S, Sekaly RP, Fischer A. 1993. Human immuno-deficiency virus gp120 and derived peptides activateprotein tyrosine kinase p56lck in human CD4 T lympho-cytes. Eur J Immunol 23: 600–607.
Huang Y, Paxton WA, Wolinsky SM, Neumann AU, Zhang L,He T, Kang S, Ceradini D, Jin Z, Yazdanbakhsh K, et al.1996. The role of a mutant CCR5 allele in HIV-1 trans-mission and disease progression. Nat Med 2: 1240–1243.
Huang CC, Tang M, Zhang MY, Majeed S, Montabana E,Stanfield RL, Dimitrov DS, Korber B, Sodroski J, WilsonIA, et al. 2005. Structure of a V3-containing HIV-1 gp120core. Science 310: 1025–1028.
Huang W, Eshleman SH, Toma J, Fransen S, Stawiski E, Pax-inos EE, Whitcomb JM, Young AM, Donnell D, Mmiro F,et al. 2007. Coreceptor tropism in human immunodefi-ciency virus type 1 subtype D: High prevalence ofCXCR4 tropism and heterogeneous composition of viralpopulations. J Virol 81: 7885–7893.
Hubner W, McNerney GP, Chen P, Dale BM, Gordon RE,Chuang FY, Li XD, Asmuth DM, Huser T, Chen BK.2009. Quantitative 3D video microscopy of HIV transferacross T cell virological synapses. Science 323: 1743–1747.
HIV: Cell Binding and Entry
Cite this article as Cold Spring Harb Perspect Med 2012;2:a006866 11
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
on August 19, 2018 - Published by Cold Spring Harbor Laboratory Press http://perspectivesinmedicine.cshlp.org/Downloaded from

Igakura T, Stinchcombe JC, Goon PK, Taylor GP, Weber JN,Griffiths GM, Tanaka Y, Osame M, Bangham CR. 2003.Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science 299:1713–1716.
Juszczak RJ, Turchin H, Truneh A, Culp J, Kassis S. 1991.Effect of human immunodeficiency virus gp120 glyco-protein on the association of the protein tyrosine kinasep56lck with CD4 in human T lymphocytes. J Biol Chem266: 11176–11183.
Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, PhamKT, Salazar MG, Sun C, Grayson T, Wang S, Li H, et al.2008. Identification and characterization of transmittedand early founder virus envelopes in primary HIV-1infection. Proc Natl Acad Sci 105: 7552–7557.
Klatzmann D, Barre-Sinoussi F, Nugeyre MT, Danquet C,Vilmer E, Griscelli C, Brun-Veziret F, Rouzioux C, Gluck-man JC, Chermann JC, et al. 1984. Selective tropism oflymphadenopathy associated virus (LAV) for helper-inducer T lymphocytes. Science 225: 59–63.
Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hen-drickson WA. 1998. Structure of an HIV gp120 envelopeglycoprotein in complex with the CD4 receptor and aneutralizing human antibody. Nature 393: 648–659.
Lalezari JP, Henry K, O’Hearn M, Montaner JS, Piliero PJ,Trottier B, Walmsley S, Cohen C, Kuritzkes DR, Eron JJ,et al. 2003. Enfuvirtide, an HIV-1 fusion inhibitor, fordrug-resistant HIV infection in North and South Amer-ica. N Engl J Med 348: 2175–2185.
Lazaro I, Naniche D, Signoret N, Bernard AM, Marguet D,Klatzmann D, Dragic T, Alizon M, Sattentau Q. 1994.Factors involved in entry of the human immunodefi-ciency virus type 1 into permissive cells: Lack of evidenceof a role for CD26. J Virol 68: 6535–6546.
Lazzarin A, Clotet B, Cooper D, Reynes J, Arasteh K, NelsonM, Katlama C, Stellbrink HJ, Delfraissy JF, Lange J, et al.2003. Efficacy of enfuvirtide in patients infected withdrug-resistant HIV-1 in Europe and Australia. N Engl JMed 348: 2186–2195.
Lehmann MJ, Sherer NM, Marks CB, Pypaert M, Mothes W.2005. Actin- and myosin-driven movement of virusesalong filopodia precedes their entry into cells. J CellBiol 170: 317–325.
Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R,MacDonald ME, Stuhlmann H, Koup RA, Landau NR.1996. Homozygous defect in HIV-1 coreceptor accountsfor resistance of some multiply-exposed individuals toHIV-1 infection. Cell 86: 367–377.
Love PE, Hayes SM. 2010. ITAM-mediated signaling by theT-cell antigen receptor. Cold Spring Harb Perspect Bioldoi: 10.1101/cshperspect.a002485.
Maddon PJ, Dalgleish AG, McDougal JS, Clapham PR,Weiss RA, Axel R. 1986. The T4 gene encodes the AIDSvirus receptor and is expressed in the immune systemand the brain. Cell 47: 333–348.
Maddon PJ, McDougal JS, Clapham PR, Dalgleish AG,Jamal S, Weiss RA, Axel R. 1988. HIV infection doesnot require endocytosis of its receptor, CD4. Cell 54:865–874.
Margolis L, Shattock R. 2006. Selective transmission ofCCR5-utilizing HIV-1: The “gatekeeper” problem re-solved? Nat Rev Microbiol 4: 312–317.
Marsh M, Helenius A. 2006. Virus entry: Open sesame. Cell124: 729–740.
McClure MO, Marsh M, Weiss RA. 1988. Human immuno-deficiency virus infection of CD4-bearing cells occurs bya pH-independent mechanism. EMBO J 7: 513–518.
McDonald D, Wu L, Bohks SM, KewalRamani VN, Unut-maz D, Hope TJ. 2003. Recruitment of HIVand its recep-tors to dendritic cell-T cell junctions. Science 300: 1295–1297.
McDougal JS, Kennedy MS, Sligh JM, Cort SP, Mawle A,Nicholson JK. 1986. Binding of HTLV-III/LAV to T4þ
T cells by a complex of the 110K viral protein and theT4 molecule. Science 231: 382–385.
Melikyan GB. 2008. Common principles and intermediatesof viral protein-mediated fusion: The HIV-1 paradigm.Retrovirology 5: 111.
Miyauchi K, Kim Y, Latinovic O, Morozov V, Melikyan GB.2009. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 137: 433–444.
O’Brien TR, Winkler C, Dean M, Nelson JA, Carrington M,Michael NL, White GC II. 1997. HIV-1 infection in aman homozygous for CCR5 D32. Lancet 349: 1219.
Orloff GM, Orloff SL, Kennedy MS, Maddon PJ, McDougalJS. 1991. Penetration of CD4 T cells by HIV-1. The CD4receptor does not internalize with HIV, and CD4-relatedsignal transduction events are not required for entry. JImmunol 146: 2578–2587.
Padhan K, Varma R. 2010. Immunological synapse: A multi-protein signalling cellular apparatus for controlling geneexpression. Immunology 129: 322–328.
Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O,Wang N, Lee G, Bartsevich VV, Lee YL, et al. 2008. Estab-lishment of HIV-1 resistance in CD4þ T cells by genomeediting using zinc-finger nucleases. Nat Biotechnol 26:808–816.
Piguet V, Steinman RM. 2007. The interaction of HIV withdendritic cells: Outcomes and pathways. Trends Immunol28: 503–510.
Ping LH, Nelson JA, Hoffman IF, Schock J, Lamers SL,Goodman M, Vernazza P, Kazembe P, Maida M, ZimbaD, et al. 1999. Characterization of V3 sequence heteroge-neity in subtype C human immunodeficiency virus type1 isolates from Malawi: Underrepresentation of X4 var-iants. J Virol 73: 6271–6281.
Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, FarberCM, Saragosti S, Lapoumeroulie C, Cognaux J, ForceilleC, et al. 1996. Resistance to HIV-1 infection in caucasianindividuals bearing mutant alleles of the CCR-5 chemo-kine receptor gene. Nature 382: 722–725.
Saphire AC, Bobardt MD, Zhang Z, David G, Gallay PA.2001. Syndecans serve as attachment receptors for humanimmunodeficiency virus type 1 on macrophages. J Virol75: 9187–9200.
Sattentau QJ, Weiss RA. 1988. The CD4 antigen: Physiolog-ical ligand and HIV receptor. Cell 52: 631–633.
Schuitemaker H, Koot M, Kootstra NA, Dercksen MW, deGoede RE, van Steenwijk RP, Lange JM, SchattenkerkJK, Miedema F, Tersmette M. 1992. Biological phenotypeof human immunodeficiency virus type 1 clones at differ-ent stages of infection: Progression of disease is associated
C.B. Wilen et al.
12 Cite this article as Cold Spring Harb Perspect Med 2012;2:a006866
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
on August 19, 2018 - Published by Cold Spring Harbor Laboratory Press http://perspectivesinmedicine.cshlp.org/Downloaded from

with a shift from monocytotropic to T-cell-tropic viruspopulation. J Virol 66: 1354–1360.
Sherer NM, Jin J, Mothes W. 2010. Directional spread ofsurface-associated retroviruses regulated by differentialvirus-cell interactions. J Virol 84: 3248–3258.
Sohy D, Parmentier M, Springael JY. 2007. Allosterictransinhibition by specific antagonists in CCR2/CXCR4 heterodimers. J Biol Chem 282: 30062–30069.
Sohy D, Yano H, de Nadai P, Urizar E, Guillabert A, JavitchJA, Parmentier M, Springael JY. 2009. Hetero-oligomeri-zation of CCR2, CCR5, and CXCR4 and the proteaneffects of “selective” antagonists. J Biol Chem 284:31270–31279.
Stamatatos L, Levy JA. 1994. CD26 is not involved in infec-tion of peripheral blood mononuclear cells by HIV-1.AIDS 8: 1727–1728.
Stein BS, Gowda SD, Lifson JD, Penhallow RC, Bensch KG,Engleman EG. 1987. pH-independent HIV entry intoCD4-positive T cells via virus envelope fusion to theplasma membrane. Cell 49: 659–668.
Tersmette M, Gruters RA, de Wolf F, de Goede RE, Lange JM,Schellekens PT, Goudsmit J, Huisman HG, Miedema F.1989. Evidence for a role of virulent human immunode-ficiency virus (HIV) variants in the pathogenesis ofacquired immunodeficiency syndrome: Studies onsequential HIV isolates. J Virol 63: 2118–2125.
Theodorou I, Meyer L, Magierowska M, Katlama C, Rou-zioux C. 1997. HIV-1 infection in an individual homozy-gous for CCR5 D32. Seroco Study Group. Lancet 349:1219–1220.
Tilton JC, Doms RW. 2009. Entry inhibitors in the treatmentof HIV-1 infection. Antiviral Res 85: 91–100.
Tremblay M, Meloche S, Gratton S, Wainberg MA, SekalyRP. 1994. Association of p56lck with the cytoplasmicdomain of CD4 modulates HIV-1 expression. EMBO J13: 774–783.
Turville SG, Cameron PU, Handley A, Lin G, Pohlmann S,Doms RW, Cunningham AL. 2002. Diversity of receptorsbinding HIV on dendritic cell subsets. Nat Immunol 3:975–983.
Uchil PD, Mothes W. 2009. HIV entry revisited. Cell 137:402–404.
Ugolini S, Mondor I, Sattentau QJ. 1999. HIV-1 attachment:Another look. Trends Microbiol 7: 144–149.
Vasiliver-Shamis G, Tuen M, Wu TW, Starr T, Cameron TO,Thomson R, Kaur G, Liu J, Visciano ML, Li H, et al. 2008.Human immunodeficiency virus type 1 envelope gp120
induces a stop signal and virological synapse formationin noninfected CD4þ T cells. J Virol 82: 9445–9457.
Vasiliver-Shamis G, Cho MW, Hioe CE, Dustin ML. 2009.Human immunodeficiency virus type 1 envelopegp120-induced partial T-cell receptor signaling createsan F-actin-depleted zone in the virological synapse. JVirol 83: 11341–11355.
Vasiliver-Shamis G, Dustin ML, Hioe CE. 2010. HIV-1 viro-logical synapse is not simply a copycat of the immunolog-ical synapse. Viruses 2: 1239–1260.
Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, WileyDC. 1997. Atomic structure of the ectodomain fromHIV-1 gp41. Nature 387: 426–430.
Welch BD, VanDemark AP, Heroux A, Hill CP, Kay MS.2007. Potent D-peptide inhibitors of HIV-1 entry. ProcNatl Acad Sci 104: 16828–16833.
Welch BD, Francis JN, Redman JS, Paul S, Weinstock MT,Reeves JD, Lie YS, Whitby FG, Eckert DM, Hill CP,et al. 2010. Design of a potent D-peptide HIV-1 entryinhibitor with a strong barrier to resistance. J Virol 84:11235–11244.
Wild C, Oas T, McDanal C, Bolognesi D, Matthews T. 1992.A synthetic peptide inhibitor of human immunodefi-ciency virus replication: Correlation between solutionstructure and viral inhibition. Proc Natl Acad Sci 89:10537–10541.
Wild C, Greenwell T, Matthews T. 1993. A synthetic pep-tide from HIV-1 gp41 is a potent inhibitor of virus-mediated cell-cell fusion. AIDS Res Hum Retroviruses 9:1051–1053.
Wu Y, Yoder A. 2009. Chemokine coreceptor signaling inHIV-1 infection and pathogenesis. PLoS Pathog 5:e1000520. doi: 10.1371/journal.ppat.1000520.
Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V,Abagyan R, Brooun A, Wells P, Bi FC, et al. 2010. Struc-tures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330:1066–1071.
Yang ZY, Huang Y, Ganesh L, Leung K, Kong WP, SchwartzO, Subbarao K, Nabel GJ. 2004. pH-dependent entry ofsevere acute respiratory syndrome coronavirus is medi-ated by the spike glycoprotein and enhanced by dendriticcell transfer through DC-SIGN. J Virol 78: 5642–5650.
Yoder A, Yu D, Dong L, Iyer SR, Xu X, Kelly J, Liu J, Wang W,Vorster PJ, Agulto L, et al. 2008. HIV envelope-CXCR4signaling activates cofilin to overcome cortical actinrestriction in resting CD4 T cells. Cell 134: 782–792.
HIV: Cell Binding and Entry
Cite this article as Cold Spring Harb Perspect Med 2012;2:a006866 13
ww
w.p
ersp
ecti
vesi
nm
edic
ine.
org
on August 19, 2018 - Published by Cold Spring Harbor Laboratory Press http://perspectivesinmedicine.cshlp.org/Downloaded from

10, 20122012; doi: 10.1101/cshperspect.a006866 originally published online AprilCold Spring Harb Perspect Med
Craig B. Wilen, John C. Tilton and Robert W. Doms HIV: Cell Binding and Entry
Subject Collection HIV
Viral Populations and Infected CellsHIV Pathogenesis: Dynamics and Genetics of
John Coffin and Ronald Swanstrom
HIV-1 Pathogenesis: The VirusRonald Swanstrom and John Coffin
Human Immunodeficiency Virus Vaccine Trials
Corey, et al.Robert J. O'Connell, Jerome H. Kim, Lawrence
The T-Cell Response to HIVBruce Walker and Andrew McMichael
HIV TransmissionGeorge M. Shaw and Eric Hunter
HIV-1 Reverse TranscriptionWei-Shau Hu and Stephen H. Hughes
Novel Cell and Gene Therapies for HIVJames A. Hoxie and Carl H. June
HIV Pathogenesis: The Host
RodriguezA.A. Lackner, Michael M. Lederman and Benigno
Strategies for HIV PreventionBehavioral and Biomedical Combination
QuinnLinda-Gail Bekker, Chris Beyrer and Thomas C.
HIV: Cell Binding and EntryCraig B. Wilen, John C. Tilton and Robert W. Doms
HIV-1 Assembly, Budding, and MaturationWesley I. Sundquist and Hans-Georg Kräusslich
Innate Immune Control of HIVMary Carrington and Galit Alter
HIV-1 Assembly, Budding, and MaturationWesley I. Sundquist and Hans-Georg Kräusslich
HIV DNA IntegrationRobert Craigie and Frederic D. Bushman
Vaccine Research: From Minefields to MilestonesLessons in Nonhuman Primate Models for AIDS
Jeffrey D. Lifson and Nancy L. Haigwood TreatmentCurrent Issues in Pathogenesis, Diagnosis, and HIV-1-Related Central Nervous System Disease:
Serena Spudich and Francisco González-Scarano
http://perspectivesinmedicine.cshlp.org/cgi/collection/ For additional articles in this collection, see
Copyright © 2012 Cold Spring Harbor Laboratory Press; all rights reserved
on August 19, 2018 - Published by Cold Spring Harbor Laboratory Press http://perspectivesinmedicine.cshlp.org/Downloaded from