hmm sampling and applications to gene finding and alignment european conference on computational...
Post on 19-Dec-2015
219 views
TRANSCRIPT

HMM Sampling and Applications toGene Finding and Alignment
European Conference on Computational Biology 2003
Simon Cawley* and Lior Pachter+
and thanks to Eli Rusman
* Affymetrix+ UC Berkeley Mathematics Dept

Conservation of alternative splicing between human and
mouse• Modrek and Lee: 40-60% of human genes
have alternative splice forms. Nature Genetics 2002.
• Nurtdinov et al. 75% of human alternative splice forms are conserved in mouse.
Human Molecular Genetics 2003.
Can we develop ab-initio methods for detecting conserved alternative splice sites?
A
A
C
A
T
T
A
G
AA G A T T A C C A C A
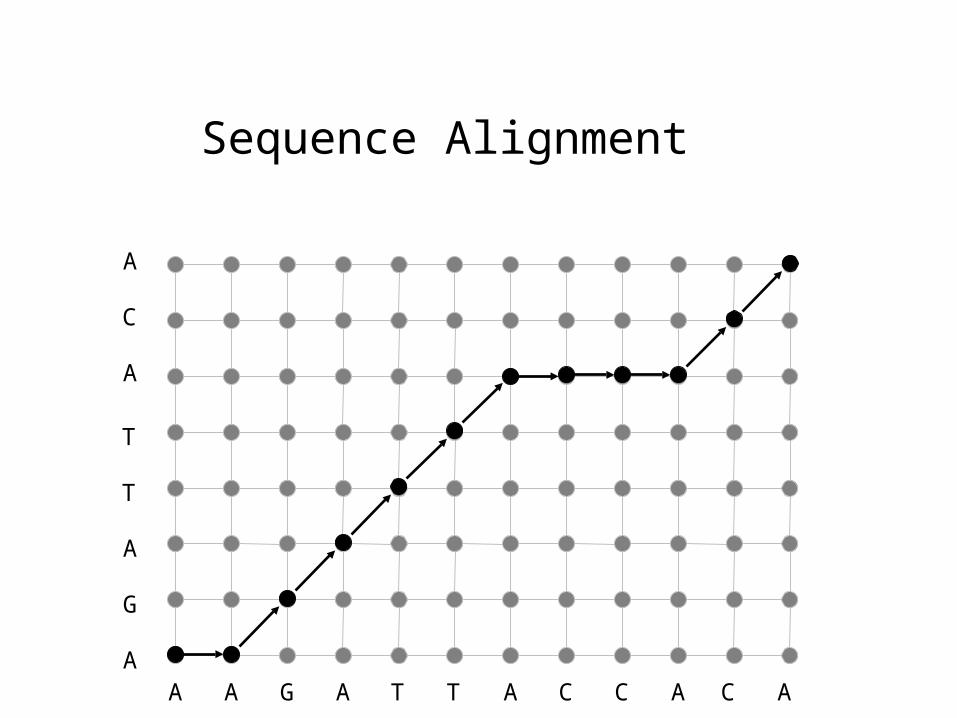
Sequence Alignment

A
A
C
A
T
T
A
G
AA G A T T A C C A C A
max
Finding the optimal alignment

ai,j = w ai-1,j + w ai,j-1 + si,j ai-1,j-1
A
A
C
A
T
T
A
G
AA G A T T A C C A C A
Alignment forward variables for positions [1,i] and [1,j]
in each sequence
Match/mismatch probabilities forpositions i,j in each sequence
gap probabilities
Sampling to find alternative alignments

Linear Space Sampling
Sequences length T,U
To obtain k samples
Time complexity: O(TU+k(T+U))
Memory requirements: O(T+U)
Hirschberg’s divide and conquer algorithm
Time complexity: O(TU)
Memory requirements: O(T+U)
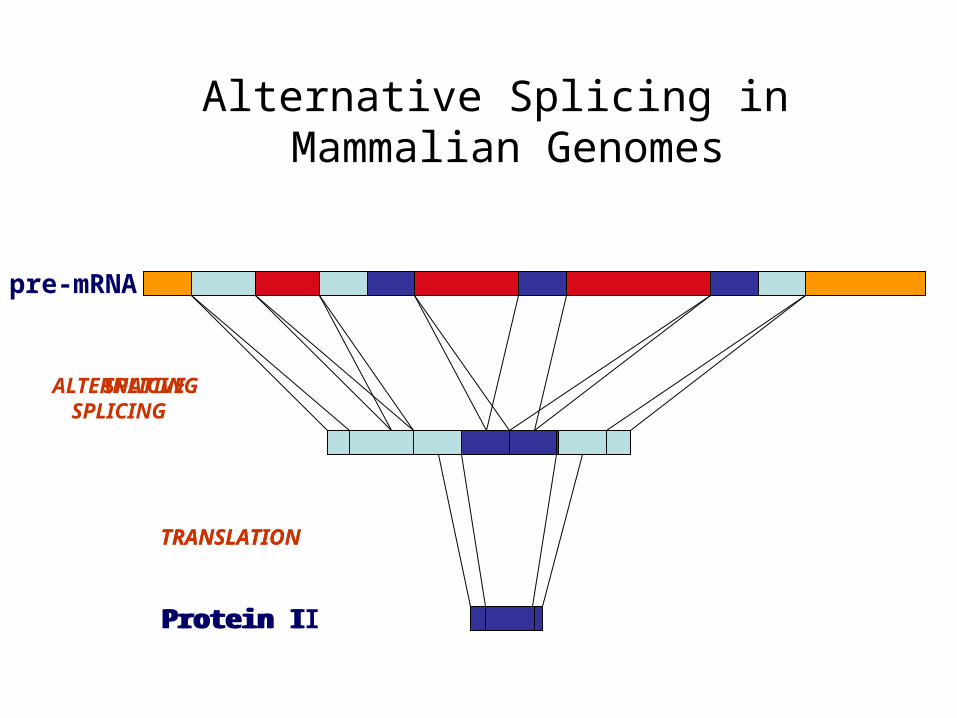
Alternative Splicing in Mammalian Genomes
pre-mRNA
TRANSLATION
SPLICING
Protein I
ALTERNATIVE SPLICING
Protein II
TRANSLATION

M. Alexandersson, S. Cawley, L. Pachter, SLAM- Cross-species gene finding and alignment with a
generalized pair hidden Markov model, Genome Research, 13 (2003) p 496-502
Cross-species simultaneous gene finding
and alignment

Modeling gene features
5’ 3’
Exon 1 Exon 2 Exon 3Intron 1 Intron 2
CNS CNS CNS
[human]
[mouse]

The SLAM hidden Markov model

SLAM components• Splice site detector
– VLMM
• Intron and intergenic regions– 2nd order Markov chain
– independent geometric lengths
• Coding sequence– PHMM on protein level
– generalized length distribution
• Conserved non-coding sequence– PHMM on DNA level

SLAM input and output
• Input:– Pair of homologous sequences.
• Output:– CDS and CNS predictions in both sequences.– Protein predictions.– Protein and CNS alignment.

http://bio.math.berkeley.edu/slam/

Input:

Output:



Methodology for identifying alternative splice sites
• Compiled SLAM gene predictions for the human, mouse and rat genomes.
• Identified a set of 3400 human/mouse/rat gene triples with consistent predictions from hs/mm and hs/rn analyses.
• For each triple, sampled sub-optimal parses from hs/mm and hs/rn runs
• Collected alternative exons (non-Viterbi exons) that appeared in both the hs/mm and hs/rn runs
• Examined overlap with RefSeq genes, mRNAs and ESTs

SLAM whole genome predictions
• Built a whole genome homology map (Colin Dewey)http://baboon.math.berkeley.edu/~cdewey/homologyMaps/
• Pre-aligned the homologous blocks to reduce the SLAM search space (Nicolas Bray using AVID)
http://baboon.math.berkeley.edu/mavid/http://hanuman.math.berkeley.edu/kbrowser/
• Ran SLAM on the resulting blockshttp://bio.math.berkeley.edu/slam/mouse/http://bio.math.berkeley.edu/slam/rat/
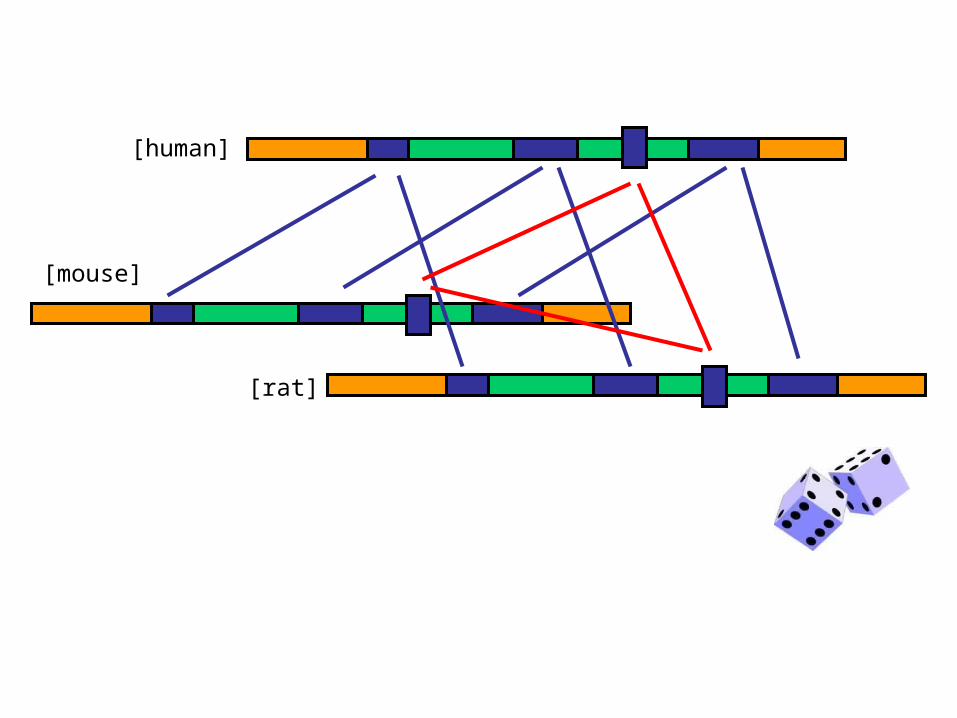
[human]
[mouse]
[rat]




Comparing predicted alternative exons to ESTs and
mRNAshuman/mouse/rat alternative
exonshuman/mouse alternative
exons
EST/mRNANo
EST/mRNA EST/mRNANo
EST/mRNA
Gene count 29 344 461 3296
Alt. Exon count 29 441 557 7240
Shifties 28 209 262 2227
Newbies 1 232 295 5013

Conclusions
• Sampling is memory efficient, fast, and should be used routinely for alignment applications.
• Conserved alternative splice forms can be detected ab-initio.
• The extent of alternative splicing conservation is currently unclear. Sampling provides an alternative approach for investigating this problem- one that is not sensitive to biases in EST data.
• Problem: design effective and scalable validation strategies for alternative splice sites.