human rhabdomyosarcoma cells retain insulin-regulated glucose transport activity through glucose...
TRANSCRIPT

m1p
Archives of Biochemistry and BiophysicsVol. 373, No. 1, January 1, pp. 72–82, 2000Article ID abbi.1999.1535, available online at http://www.idealibrary.com on
Human Rhabdomyosarcoma Cells Retain Insulin-RegulatedGlucose Transport Activity through Glucose Transporter 11
Satoshi Ito,*,† Takahiro Nemoto,* Shinobu Satoh,† Hisahiko Sekihara,†Yousuke Seyama,* and Shunichiro Kubota*,2
*Department of Physiological Chemistry and Metabolism, Graduate School of Medicine, University of Tokyo, 7-3-1 Hongo,Bunkyo-ku, Tokyo 113-0033, Japan; and †Third Department of Internal Medicine, Yokohama City UniversitySchool of Medicine, 3-1 Fukuura, Kanazawa-ku, Yokohama 236-0004, Japan
Received August 20, 1999, and in revised form September 23, 1999
We evaluated the expression of glucose transporter(glut) isoforms and its function in RD cells, humanrhabdomyosarcoma, which retain the potential to dif-ferentiate into muscle. Gluts 1, 3, and 4 were expressedin RD cells, as detected by reverse-transcription poly-merase chain reaction and immunocytochemistry. Su-praphysiological concentration (1 mM) of insulin treat-
ent increased 2-deoxy glucose transport by up to.68-fold together with concomitant tyrosine phos-horylation of the insulin receptor b subunit and of
insulin receptor substrate 1. Suppression of glut 1mRNA by 38% by antisense oligonucleotide transfec-tion led to a reduction of basal and insulin-stimulated2-deoxy glucose transport by 38 and 55%, respectively.Suppression of gluts 3 and 4 by antisense oligonucle-otide transfection did not affect both basal and insu-lin-stimulated 2-deoxy glucose transport. Thus, glut 1accounts for the major part of basal and insulin-stim-ulated glucose transport in RD cells. Next, we trans-fected expression vectors carrying human gluts 1 and4 cDNAs into RD cells to add further support for therole of glut 1 in glucose transport. Overexpression ofglut 1 stimulated basal and insulin-stimulated 2-deoxyglucose transport by 1.66- and 1.43-fold, respectively.Glut 4 overexpression did not affect basal and insulin-stimulated 2-deoxy glucose transport. Western blotanalysis using glut 1 antibody showed that glut 1 wasredistributed from intracellular membrane to plasmamembrane. These observations support the notionthat RD cells, with the potential to differentiate intomuscle, retain insulin responsiveness. As human mus-cle cell lines are not available at this point, RD cells
1 This work was supported by a grant under the Ministry of Edu-cation, Science, Sports, and Culture, Japan.
2
To whom correspondence should be addressed. Fax: 81-3-5689-2704. E-mail: [email protected].72
can serve as a useful alternative to human muscle forstudies related to insulin signal transduction and glu-cose transport. © 2000 Academic Press
Key Words: RD cells; glucose transporter; insulin.
Glucose is transported across the cell membrane by afacilitative diffusion process mediated by glucosetransporters, gluts.3 Seven glut isoforms have beencloned to date. Glut isoforms are different in terms oftissue distribution, glucose transport efficiency, andinsulin responsiveness (1, 2). Glut 1, the erythrocyte-type glucose transporter, is a widely expressed isoform,especially in erythrocytes, and is important for basalglucose transport; glut 2 is a high Km isoform expressedin hepatocytes and pancreatic b cells; glut 3 is a low Km
isoform expressed in neurons; and glut 4, the insulin-regulatable glucose transporter, is expressed exclu-sively in fat and muscle, where glucose uptake isacutely stimulated by insulin. With insulin treatmentor muscle contraction, glut 4 translocates from theintracellular pool to the plasma membrane and T tu-bules in rat skeletal muscle (3). In 3T3-L1 adipocytes,the increase in glucose transport with short-term ex-posure to insulin results from translocation of mainlyglut 4 from an intracellular pool to the plasma mem-brane (4, 5). As well as glut 4, gluts 1 and 3 were alsofound to translocate to the plasma membrane, withinsulin stimulation, and probably contribute to insulin-stimulated glucose transport (4–7). In L6 cells, an in-
3 Abbreviations used: glut, glucose transporter; DMEM, Dulbecco’smodified Eagle’s medium; IRS 1, insulin receptor substrate 1; RT-PCR, reverse-transcription polymerase chain reaction; PSS, physio-
logic saline solution; PBS, phosphate-buffered saline; TBS, Tris-buffered saline; FCS, fetal calf serum.0003-9861/00 $35.00Copyright © 2000 by Academic Press
All rights of reproduction in any form reserved.

mskttddmiaeipgw
[
caDieb8a(mRwcC62
73INSULIN-STIMULATED GLUCOSE TRANSPORT IN RD CELLS
sulin-sensitive cell line expressing a smaller amount ofglut 4, compared to adipocytes, glut 1 is the predomi-nant transporter (8, 9). Glut 5, a fructose transporter,is expressed in the small intestine and spermatozoa.Glut 6 is a pseudgene (10), and glut 7 seems to be acloning artifact (11).
The increase of glucose uptake is a characteristic ofcancer cells (12). The acceleration of glucose uptake ismediated by gluts (13), many cancers overexpress glutisoforms, and wide expression of glut 1 in various hu-man cancers has been reported (14). Glut 2 is ex-pressed in hepatic tumors (15, 16). Glut 3 is expressedin gastrointestinal tumors (16), lung carcinomas (17),testicular and ovarian carcinomas (18), and gliomas(19). Glut 4 is rarely expressed in cancer cells, but wasfound to be expressed in a breast cancer cell line,MDA-MB-231 (20).
Insulin-stimulated upregulation of glucose transportin human cancer cells is poorly understood, especiallyin human cells which have a potential to differentiateinto muscle. Binder et al. reported that in MDA-MB-231 cells, which express glut 4, short-time stimulationwith 1 mM insulin increased 2-deoxy glucose transportup to 1.73-fold (20). However, they did not characterizethe isoform of gluts responsible for glucose transport.MDA-MB-231 cells do not have a potential to differen-tiate into muscle.
Experimental in vitro models of animal skeletaluscle cell lines and rat hind limb muscle are used to
tudy insulin-stimulated glucose transport (21–24). Wenow of no human cell line that has myoblastic fea-ures and preserves insulin responsiveness of glucoseransport. RD is a cell line, a human embryonal rhab-omyosarcoma, having variable degrees of myoblasticifferentiation and the potential to differentiate intoyocytes (25, 26). The expression of glut isoforms and
nsulin-regulated glucose transport in RD cells haspparently not been documented. We investigated thexpression of gluts 1 to 4 in RD cells and characterizednsulin-regulated glucose transport in RD cells by sup-ression or overexpression of glut isoforms. To assesslucose transport activity, we used 2-deoxy glucose,hich is taken up via the D-glucose transport system
and phosphorylated by the hexokinase system. Phos-phorylated 2-deoxy glucose is not a substrate of thehexose carrier, so it cannot exit the cell, a factor whichmakes feasible measurement of an unidirectional fluxof 2-deoxy glucose (27). We also used 3-O-methyl glu-cose to determine basal and maximally stimulated glu-cose transport rates. To further characterize 2-deoxyglucose transport in RD cells, we used cytochalasin Band herbimycin A, as indicated. Cytochalasin B is afungal metabolite which binds to glucose transportersand inhibits glucose transport (28), but does not inhibitglucose diffusion across the cell membrane. In the pres-
ence of cytochalasin B, one can measure glucose trans-si
port by a diffusion process. Herbimycin A is a relativelyspecific tyrosine kinase inhibitor. On insulin binding,the b subunit of the insulin receptor undergoes tyrosylautophosphorylation and phosphorylates tyrosyl resi-dues of IRS 1 (29). Herbimycin A blocks this processand inhibits insulin-stimulated glut 4 translocation incultured 3T3-L1 adipocytes (30). In the presence ofherbimycin A, one can evaluate glucose transport un-der conditions where the insulin receptor and IRS 1 arenot activated.
MATERIALS AND METHODS
Materials. Modified Dulbecco’s Eagle’s medium (DMEM), bovineserum albumin (fraction V), and a bicinchoninic acid kit for proteindetermination were obtained from Sigma Aldrich Japan (Tokyo,Japan). Fetal calf serum (FCS) was from Nichirei Corp. (Tokyo,Japan). An RNA-PCR kit was purchased from TaKaRa Ltd. (Otsu,Japan). Specific antibody for gluts 1 (31), 3 (31), and 4 (32) andplasmids carrying the human glut 1 (33) and 4 (34) cDNAs weregenerous gifts from Dr. S. W. Cushman of the National Institute ofDiabetes and Digestive and Kidney Diseases (Bethesda, MD). Fluo-rescent anti-rabbit IgG, horseradish peroxidase-labeled anti-rabbitand -mouse IgG antibodies were from Vector Laboratories, Inc. (Bur-lingame, CA). Human recombinant insulin was from Novo NordiskA/S (Bagsvaerd, Denmark). 2-Deoxy-D-[3H]glucose, 3-O-methyl-
14C]glucose, and L-[3H]glucose were from NEN Life Science Products(Boston, MA). Prestained protein marker broad range was from BioLabs, Inc. (Beverly, MA). Nitrocellulose filters and a protein assaykit were from Bio-Rad Laboratories (Hercules, CA). Anti-phosphoty-rosine antibody was from Transduction Laboratories (Lexington,KY). Anti-insulin receptor b subunit and anti-IRS 1 antibody werefrom Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Skim milk,cytochalasin B, herbimycin A, suramin, and other chemicals werefrom Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Ten-week-old male Wistar rats were from S.L.C. (Shizuoka, Japan). Hu-man brain RNA was from Clontech Laboratories Inc. (Palo Alto, CA).Oligonucleotide primers and thioated oligonucleotides were synthe-sized by Espec Oligoservice Co. (Tsukuba, Japan). pTargeT mamma-lian expression vector was from Promega Co. (Madison, WI). Peni-cillin G, a Lipofectamine Plus reagent, and the DNA molecular sizemarker (100-bp ladder) were from Gibco BRL (Rockville, MD).
Cell culture. RD cells, purchased from Human Science ResearchResources Bank (Osaka, Japan), were grown in DMEM, supple-mented with 10% FCS, and 100 units/ml penicillin G. Cell cultureswere maintained at 37°C and 5% CO2.
Reverse-transcription polymerase chain reaction (RT-PCR) of glu-ose transporters. Total RNA was isolated from the cells using thecid guanidinium–phenol–chloroform method (35). RNase-freeNase was added to the samples and the preparation was then
ncubated at 37°C for 1 h. After phenol–chloroform extraction andthanol precipitation, RNA concentration was measured by absor-ance at 260 nm. First-strand cDNA synthesis was performed from00 ng of total RNA, using specific antisense primers for gluts 1–4nd b actin (Table I), according to the manufacturer’s instructionTaKaRa RNA PCR kit, ver. 2.1). For positive control, rat hind limbuscle RNA (gluts 1 and 4), rat liver RNA (glut 2), and human brainNA (glut 3) were used. For negative control, reverse transcriptionithout reverse transcriptase was performed, as indicated. PCR was
arried out in a Gene Amp PCR Thermal Cycler 9600 (Perkin–Elmer,A), under conditions of denaturation at 94°C for 30 s, annealing at0°C for 30 s, and extension at 72°C for 30 s for 23 cycles (glut 1) and6 cycles (gluts 2–4). After amplification, the PCR products were
ubjected to 1.5% agarose gel electrophoresis and visualized follow-ng ethidium bromide staining. The sequence of the amplified PCR
w(
wwp
dm
sb
ab
acoi
Cb
mS
G
G
G
74 ITO ET AL.
products was verified by sequencing on an ABI PRISM 310 auto-mated sequencer (Perkin–Elmer Co., Norwalk, CT). The intensitiesof the bands were measured using NIH Image software, and theintensities of the bands of glut isoforms were normalized to theintensity of the b actin band, which was amplified simultaneously.
Immunocytochemistry. Cells cultured in glass-bottom dishesere fixed with 4% paraformaldehyde in phosphate-buffered saline
PBS: 137 mM NaCl, 8.1 mM Na2HPO4, 2.68 mM KCl, and 1.47 mMKH2PO4) for 16 h. After three washes with PBS and blocking withPBS containing 5% skim milk for 1 h, the cells were reacted withrabbit polyclonal antibodies specific for gluts 1, 3, and 4 at 1:100dilution for 1 h at room temperature. As a negative control, preim-mune rabbit IgG was used. After three washes with PBS the cellswere incubated with fluorescent anti-rabbit IgG for 1 h. After an-other three washes with PBS, the fluorescent-labeled cells wereviewed under a microscope (Olympus, Tokyo, Japan) under excita-tion at 400–440 nm and emission at greater than 500 nm. Imageswere exposed to ASA 800 film (Fuji Film Co., Tokyo, Japan) for 60 s.
2-Deoxy glucose transport. 2-Deoxy glucose transport was deter-mined, as described (36). Briefly, cells were seeded onto 12-wellcluster dishes (2 3 105 cells/well) and cultured for 12 h. Prior toexperimental manipulations, cells were preincubated with physio-logic salt solution (PSS: 145 mM NaCl, 5 mM KCl, 10 mM Hepes, 1mM MgSO4, 0.5 mM Na2HPO4, and 1.5 mM CaCl2) for 2 h. The buffer
as then replaced with PSS containing insulin and the preparationas incubated for another 20 min. To initiate 2-deoxy glucose trans-ort, the medium was replaced with PSS containing 100 mM 2-deoxy-
D-[3H]glucose (0.25 mCi/well). At the time indicated, dishes wereplaced on ice, and the buffer was aspirated, followed by three washeswith ice-cold PSS. Then, 500 ml of 0.4 N NaOH was added to eachwell to dissolve the cells. After neutralization with 50 ml of 4 N HCl,radioactivity was measured in a scintillation counter (Beckman In-struments Inc., Fullerton, CA). All experiments done in triplicatewere performed four times. For experiments with cytochalasin B, 10mM cytochalasin B dissolved in 0.1% ethanol or 0.1% ethanol wasadded to PSS containing insulin. For experiments with herbimycinA, 1 mg/ml herbimycin A dissolved in 0.1% dimethyl sulfoxide or 0.1%
imethyl sulfoxide was added to the preincubation medium after 90in of preincubation.Immunoprecipitation and Western blotting of phosphotyrosine, in-
ulin receptor, and insulin receptor substrate 1. Cells were incu-ated in serum-free DMEM for 3 h prior to the addition of 0.5 mM
insulin. After 5 min at 37°C, the cells were washed twice withice-cold PBS and scraped into 500 ml of homogenization buffer [40mM Hepes, 150 mM NaCl, pH 7.4, with phosphatase inhibitors (10mM sodium pyrophosphate, 10 mM NaF, and 1 mM sodium vana-
TA
Oligonucleotide Prim
Gene Oligonucleotide primers
Glut 1 Sense 59-cgggccaagagtgtgctaaa-39Antisense 59-tgacgataccggagccaatg-39
lut 2 Sense 59-gtgctgggttccttccagtt-39Antisense 59-aaggtatctggggctttctg-39
lut 3 Sense 59-ccaacttcctagtcggattg-39Antisense 59-aggaggcacgacttagacat-39
lut 4 Sense 59-ggcatgtgtggctgtgccatc-39Antisense 59-gggtttcacctcctgctctaa-39
b-Actin Sense 59-cgtgggccgccctaggcacca-39Antisense 59-ttggccttagggttcaggggg-39
date) and protease inhibitors (0.5 mg/ml aprotinin, 1 mM phenyl-methanesulfonyl fluoride, and 1 mg/ml leupeptin)]. Then, suspended
ws
cells were sonicated using an ultrasonic processor (Dr. HielscherGmbh, Stahnsdorf, Germany). The homogenates were centrifuged at10,000 rpm for 5 min at 4°C and protein concentration of the super-natant was determined by the method of Bradford, using the Bio-Radreagent. The supernatant (500 mg of total protein) was incubated at4°C for 2 h with protein A–Sepharose which was incubated withanti-phosphotyrosine antibody for 1 h before immunoprecipitation.The immune complex was precipitated by centrifugation at 10,000rpm for 1 min, washed three times with staph A buffer (1.8 mMNaH2PO4, 8.4 mM Na2HPO4, 5% Triton X-100, 0.5% deoxycholiccid, and 100 mM NaCl), and boiled in 30 ml of Laemmli sampleuffer (37). The whole-cell lysate (100 mg) and immunoprecipitated
proteins were resolved on 7.5% SDS–PAGE and transferred to nitro-cellulose membrane. After blocking with Tris-buffered saline (TBS:10 mM Tris–HCl, pH 7.6, 150 mM NaCl) containing 3% bovine serumalbumin for 1 h, the membrane was incubated with the appropriateantibodies (anti-phosphotyrosine, anti-insulin receptor b subunit,nd anti-IRS 1 antibodies). The proteins were visualized using theolorimetric method and horseradish peroxidase-labeled anti-rabbitr anti-mouse IgG antibodies. To analyze autophosphorylation of thensulin receptor b subunit and IRS 1 in adipocytes, rat adipocytes
were prepared from epididymal fat pads of 10-week-old male Wistarrats (170–200 g), as described (38, 39). Isolated adipocytes werepreincubated in Krebs–Ringer bicarbonate Hepes buffer (30 mMHepes, pH 7.4, 120 mM NaCl, 4 mM KH2PO4, 1 mM MgSO4, 1 mM
aCl2, and 10 mM NaHCO3) containing 200 nM adenosine and 5%ovine serum albumin and further incubated with 0.5 mM insulin.
After 5 min of incubation the cells were washed with ice-cold TESbuffer (20 mM Tris–HCl, pH 7.4, 1 mM EDTA, and 255 mM sucrose),solubilized in sample buffer (5% SDS, 50 mM dithiothreitol, and 50mM Tris–HCl, pH 6.8) containing phosphatase inhibitors (10 mMsodium pyrophosphate, 10 mM NaF, and 1 mM sodium vanadate),and boiled for 3 min. The infranatant was collected as the sample.After determining protein concentration using a bicinchoninic acidkit for protein determination, 50 mM dithiothreitol was added, andthe samples (100 mg of protein) were boiled for an additional 3 minand subjected to SDS–PAGE and Western blotting.
Antisense oligonucleotides. Thioated oligonucleotides are listedin Table II. Complementary sense oligonucleotides were also used fortransfection. The specificity of each oligonucleotide sequence wasconfirmed by CBI BLAST Sequence Similarity Searching (NationalCenter of Biotechnology Information).
Construction of glut 1 and 4 expression vectors. The SalI frag-ent containing full-length glut 1 and 4 cDNAs was ligated into thealI site of the pTargeT expression vector, and the orientation
I
s Used for RT-PCR
Nucleotide No. Product size GenBank No.
846–865 283 K031951109–1128
90–109 686 J03810756–775
1477–1496 250 M206811707–17261220–1240 414 M207471613–1633
182–202 243 X03672404–424
BLE
er
as confirmed by sequencing on an ABI PRISM 310 automatedequencer.

gr
sficT
Kswwmb
2
dBs
(
d
75INSULIN-STIMULATED GLUCOSE TRANSPORT IN RD CELLS
Transfection of antisense oligonucleotides and glut 1 or glut 4expression vector. Oligonucleotides and expression vector carryingglut 1 or glut 4 cDNA were transiently transfected into RD cellsusing a Lipofectamine Plus reagent, according to manufacturer’sinstructions. Cells (1 3 106) were dispensed into 100-mm dishes andallowed to adhere overnight, followed by exposure to 6.5 ml of amixture of oligonucleotides (20 mM) and expression vectors carryinglut 1 or glut 4 cDNA (2 mg), Lipofectamine reagent, and Pluseagent in DMEM for 3 h at 37°C in a 5% CO2 incubator. Then, the
mixture was removed and replaced with DMEM containing 10%FCS. The cells were grown for 36 h before isolation of total RNA asdescribed above for RT-PCR. Forty-eight hours after transfection,cells were seeded onto a 12-well cluster dish and incubated foranother 12 h for the 2-deoxy glucose transport assay. To evaluatenonspecific effects of transfection, transfection without oligonucleo-tides or vector was performed. Forty-eight hours after transfection,RD cells with mock transfection and glut 1 transfection were furthercultured in DMEM containing 500 mg/ml geneticin in 250-ml flasksfor subcellular fractionation, Western blotting of glut 1, and 3-O-methyl glucose transport assay. Transfection efficiency was evalu-ated using pCMV b vector carrying the b-galactosidase gene and bGal staining kit (both were purchased from Clontech, Carlsbad, CA)according to manufacturer’s instructions. One microgram of pCMV bplasmid was transfected into RD cells using Lipofectamine reagentsas described above. After 48 h RD cells were stained using the b Galtaining kit. Total cell number and positively stained cell number pereld were counted under the microscope. Transfection efficiency wasalculated based on the mean cell number from a total of 10 fields.ransfection efficiencies using 1 and 5 mg of pCMV b plasmid were
3.5 6 1.9 and 6.0 6 2.7%, respectively.Subcellular fractionation and Western blotting of glut 1. To study
the translocation of glut 1, 4 3 107 RD cells with mock transfectionand glut 1 transfection were collected after preincubation for 6 h in
rebs––Ringer bicarbonate Hepes buffer containing 200 nM adeno-ine and 5% bovine serum albumin and further incubated for 1 hith 1 mM insulin. Plasma membranes and microsomal membranesere prepared as described previously (40). Thirty micrograms of theembrane samples was subjected to 10% SDS–PAGE and Western
lot analysis using rabbit polyclonal antibody specific for glut 1 and125I-labeled protein A. Immunoreactivity was quantified with a BAS-000 image analyzer (Fuji Film).3-O-Methyl glucose transport. 3-O-Methyl glucose transport was
etermined as described previously (41), with a little modification.riefly, RD cells with mock transfection and glut 1 transfection wereeeded onto 12-well cluster dishes (1 3 106 cells/well) and cultured
for 12 h. Prior to experimental manipulations, cells were preincu-bated with Krebs–Ringer bicarbonate Hepes buffer containing 200nM adenosine and 5% bovine serum albumin and further incubatedfor 1 h with or without 1 mM insulin. 3-O-Methyl glucose uptake wasdetermined by 30-s incubations in 100 mM 3-O-methyl [14C]glucose
TAB
Thioated Oligonucleotid
Gene Thioated oligonucleotides
Glut 1 Sense 59-atggagcccagcagcaagaa-3Antisense 59-ttcttgctgctgggctccat-39
Glut 3 Sense 59-atggggacacagaaggtcac-3Antisense 59-gtgaccttctgtgtccccat-39
Glut 4 Sense 59-atgccgtcgggcttccaaca-39Antisense 59-tgttggaagcccgacggcat-39
0.25 mCi/well). At the time indicated, dishes were placed on ice, andthe buffer was aspirated, followed by three washes with ice-cold PBS
containing 0.5 mM phroletin to inhibit the efflux of 3-O-methylglucose. Then, 500 ml of 0.4 N NaOH was added to each well to
issolve the cells. After neutralization with 50 ml of 4 N HCl, radio-activity was measured in a scintillation counter. All experimentsdone in triplicate were performed four times.
Statistical analysis. Statistical significance was tested usinganalysis of variance, following Student’s t test. When the P value wasbelow 0.01, the differences were accepted as significant.
RESULTS
RT-PCR of glut isoforms. RT-PCR using specificprimers for gluts 1, 2, 3, and 4 was performed. Asshown in Fig. 1, RD cells expressed gluts 1, 3, and 4.The mRNA expression of glut 2 was undetectable. RT-PCR products from muscle, liver, and brain yieldedpositive results.
Immunocytochemistry. To confirm the expressionof gluts 1, 3, and 4 at protein level, immunocytochem-istry for gluts 1, 3, and 4 in RD cells was performedusing specific antibodies. Figure 2 shows immunostain-ing of glut 1 (A), glut 3 (B), and glut 4 (C) in RD cells.Staining for all transporters is membranous and cyto-plasmic. The nuclei did not stain.
2-Deoxy glucose transport and Western blotting. Todetermine the function of gluts in RD cells, we assessed2-deoxy glucose transport activity in RD cells after ashort-term treatment with insulin. 2-Deoxy glucosetransport was almost linear between 5 and 30 min. Thelowest insulin concentration which made a significantincrease (1.32-fold of increase) in 2-deoxy glucosetransport was 1028 M. Maximum stimulation of insulin(1.68-fold) was observed at a concentration greaterthan 1 mM. Table III shows the effects of cytochalasinB and herbimycin A on 1 mM insulin-stimulated glu-cose transport in RD cells. The pulsation time was 5min. Preincubation with cytochalasin B reduced 2-de-oxy glucose transport to 9.2% (under basal condition)and 5.1% (under insulin-stimulated condition) of con-trol cells. This means that glucose is transportedmostly by gluts and not by a diffusion process. Pre-treatment with herbimycin A decreased insulin-stim-ulated glucose uptake to an unstimulated level. Be-
II
Used for Transfection
Nucleotide No. GenBank No.
180–199 K03195199–180243–262 M20681262–243146–165 M20747165–146
LE
es
9
9
cause insulin-stimulated glucose transport was inhib-

utnPtehe2tP
rrtgnteau
f
, R
76 ITO ET AL.
ited by herbimycin A, we next asked if proteins withtyrosyl residues would be phosphorylated during insu-lin stimulation in RD cells. Serum-starved RD cellswere stimulated with 0.5 mM insulin, and whole-celllysate was subjected to Western blotting with anti-phosphotyrosine antibody (Fig. 3A). Adipocytes fromrat epididymal fat pads were used as a positive control,because insulin caused phosphorylation of 95- and 160-kDa proteins in adipocytes, which represents phos-phorylation of the b subunit of the insulin receptor andIRS 1 (39). Insulin caused phosphorylation of 95- and160-kDa proteins in RD cells (Fig. 3A). Immunoprecipi-tation with anti-phosphotyrosine antibody and blottingwith the anti-insulin receptor b subunit and anti-IRS 1antibodies confirmed that insulin caused phosphoryla-tion of the insulin receptor b subunit and IRS 1 in RDcells (Fig. 3B).
Transfection of antisense oligonucleotides of gluts 1,3, and 4. RD cells express gluts 1, 3, and 4. To eval-
ate which transporter plays a major role in glucoseransport in RD cells, we transfected antisense oligo-ucleotides of gluts 1, 3, and 4 using a Lipofectaminelus reagent kit. Thirty-six hours after transfection,
otal RNA was extracted and the level of expression ofach glut isoform was evaluated by RT-PCR. Sixtyours after transfection, the effect of the suppressedxpression of glut isoforms on 1 mM insulin-stimulated-deoxy glucose transport was evaluated. The pulsa-ion time was 5 min. Figure 4 shows the specific RT-
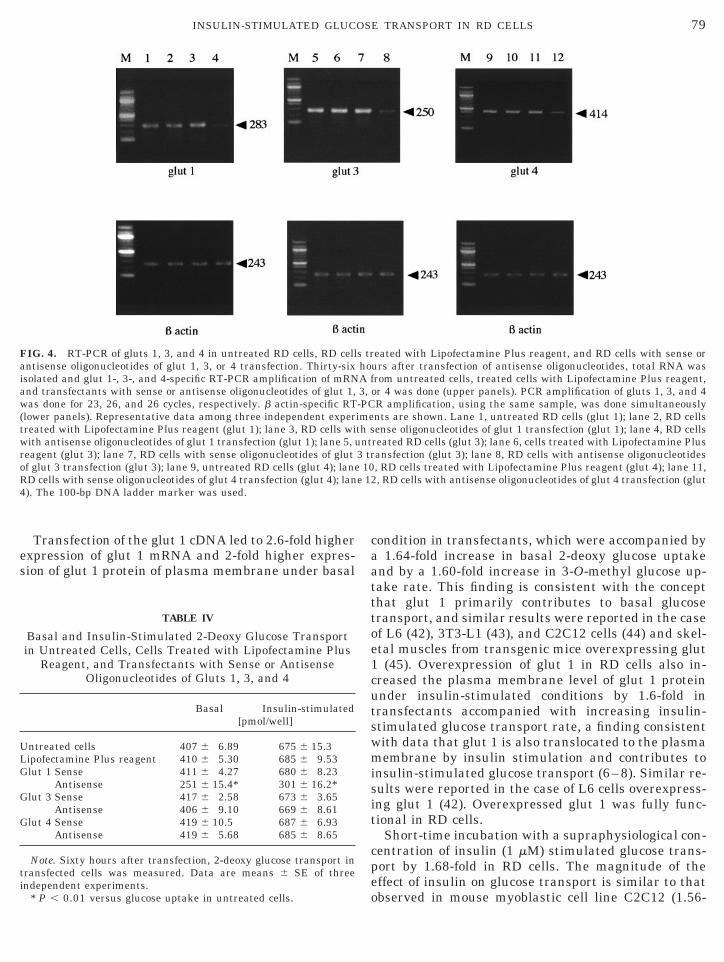
FIG. 1. RT-PCR of glut isoforms. Specific RT-PCR amplification ofor 23, 26, and 26 cycles, respectively (upper panels). b actin-specific
(lower panels). Representative data among three independent experi3, liver (glut 2); lane 4, RD cells (glut 2); lane 5, brain (glut 3); lane 6100-bp DNA ladder marker was used.
CR amplification products of gluts 1, 3, and 4 derived t
from untreated cells, cells treated with LipofectaminePlus reagent, and transfectants with sense or anti-sense oligonucleotides of gluts 1, 3, and 4. The intensityof the bands of glut isoforms was standardized accord-ing to that of b actin. There was no significant differ-ence in the expression of each glut isoform betweencells treated with Lipofectamine Plus reagent andsense oligonucleotide transfectants. The level of mRNAexpression of glut 1 in glut 1 antisense oligonucleotidetransfectant compared with that of untreated cells was62%. Table IV shows glucose transport activity of un-treated cells, cells treated with Lipofectamine Plus re-agent, and transfectants with sense or antisense oligo-nucleotides of gluts 1, 3, and 4. The basal and insulin-stimulated glucose transport activities in the glut 1antisense oligonucleotide transfectant were 61.7 and44.6%, respectively, of that observed in untreated cells.The fold stimulations of glucose transport by insulin inuntreated cells and in antisense oligonucleotide trans-fectant were 1.66 (P , 0.01) and 1.20 (P 5 0.11),espectively. The reduction in glut 1 mRNA effectivelyeduced basal and insulin-stimulated 2-deoxy glucoseransport activities. On the other hand, transfection oflut 3 and 4 antisense oligonucleotides caused no sig-ificant effect on basal and insulin-stimulated glucoseransport activities (Table IV), though the levels ofxpression of the mRNA in transfectants with glut 3nd 4 antisense oligonucleotides were 53 and 57% ofntreated cells, respectively. These results suggest
NA of gluts 1, 3, and 4 from RD cells and control tissues was done-PCR amplification using the same sample was done simultaneouslynts are shown. Lane 1, muscle (glut 1); lane 2, RD cells (glut 1); laneD cells (glut 3); lane 7, muscle (glut 4); lane 8, RD cells (glut 4). The
mRRTme
hat glut 1 is the major transporter and plays a prom-

wd5atamsg
77INSULIN-STIMULATED GLUCOSE TRANSPORT IN RD CELLS
inent role in basal and insulin-stimulated glucosetransport in RD cells.
Overexpression of glut 1 and glut 4. To further in-vestigate the roles of gluts 1 and 4 in glucose transport
FIG. 2. Immunocytochemistry of glut isoforms in RD cells. RD cellsere cultured in glass-bottom dishes and fixed with 4% paraformal-ehyde in PBS for 16 h. The cells were blocked with PBS containing% skim milk for 1 h and then incubated with rabbit polyclonalntibodies specific for gluts 1, 3, and 4 at 1:100 dilution for 1 h. Afterhree washes with PBS, the cells were incubated with fluorescentnti-rabbit IgG for 1 h and the fluorescent-labeled cells were viewedicroscopically under excitation at 400–440 nm (left). The same
amples viewed under visual light are on the right. (A) Glut 1, (B)lut 3, (C) glut 4.
in RD cells, we overexpressed glut 1 and glut 4 sepa-
rately by transfection of the expression vector carryingeither glut 1 or glut 4 cDNA. Thirty-six hours aftertransfection, total RNA was extracted and expressionof each glut isoform was evaluated by RT-PCR. Sixtyhours after transfection, the effect of the overexpressedglut isoforms on 1 mM insulin-stimulated 2-deoxy glu-cose transport was evaluated. The pulsation time was5 min. Figure 5 shows the specific RT-PCR amplifica-tion products of gluts 1 and 4 derived from untreatedcells, cells treated with Lipofectamine Plus reagent,mock transfectants, glut 1 and 4 transfectants, andnormal muscle (positive control). The intensity of thebands of glut isoforms was standardized according tothat of b actin. RT-PCR amplification of glut 1 and 4transfectants without reverse transcriptase producedno PCR product, showing that glut 1 and 4 expressionvectors had been completely destroyed. There was nodifference in the expression of each glut isoform amonguntreated cells, cells treated with Lipofectamine Plusreagent, and mock transfectants. Transfection of gluts1 and 4 led to 2.6-and 2.1-fold higher expressions ofglut 1 and glut 4, respectively, than did untreated cells.Table V shows glucose transport activity of untreatedcells, cells treated with Lipofectamine Plus reagent,mock transfectants, and glut 1 or glut 4 transfectants.The basal and insulin-stimulated glucose transport ac-tivities in glut 1 transfectant were 1.64- and 1.44-foldhigher than that observed in untreated cells, respec-tively. On the contrary, transfection of glut 4 did notaffect basal and insulin-stimulated glucose transportactivities in RD cells, though the expression of glut 4 inthe glut 4 transfectant was 2.1-fold higher than thatobserved in untreated cells (Table V). The magnitudeof increase caused by insulin stimulation was 1.46-foldin the glut 1 transfectant, a value lower than thatobserved in untreated cells (Table V). Figure 6 showsthe dose response of insulin-stimulated 2-deoxy glu-cose transport in the glut 1 transfectant and in un-treated cells. The lowest concentration of insulin to
TABLE III
Effects of Cytochalasin B and Herbimycin Aon Glucose Transport in RD Cells
Control Cytochalasin B Control Herbimycin A[pmol/well]
asal 403 6 15.1 37.1 6 2.65† 419 6 17.3 423 6 4.71nsulin
(1 mM)691 6 15.9* 34.9 6 1.72† 700 6 10.9* 446 6 8.90†
Note. RD cells were pretreated in the presence or absence ofytochalasin B (10 mM) or herbimycin A (1 mg/ml) as described underaterials and Methods. 2-Deoxy glucose uptake was measured for 5in. Data are means 6 SE of three independent experiments.
BI
cMm
* P , 0.01 versus glucose uptake without insulin.† P , 0.01 versus glucose uptake without inhibitors.

ins
78 ITO ET AL.
elicit insulin-stimulated glucose transport was 1028 Min glut 1-transfected cells, which was the same as thatin untreated cells. This suggests that the increased2-deoxy glucose transport in glut 1-transfected cellswas caused by the overexpression of glut and not by anincreased affinity to insulin receptors.
Subcellular fractionation and Western blotting ofglut 1. Figure 7 illustrates the glut 1 protein contentof plasma membrane and intracellular membrane frac-tion in mock transfectants and glut 1 transfectants. Inmock transfectants preincubation with 1 mM insulinresulted in an increase of approximately 1.7-fold inplasma membrane levels of glut 1 protein concomitantwith a 40% decrease in intracellular membrane levelsof glut 1 protein. Insulin caused redistribution of glut 1from intracellular membranes to plasma membranes.Glut 1 transfection increased plasma membrane levelsof glut 1 protein under both basal and insulin-stimu-lated conditions by 2- and 1.6-fold, respectively. In glut1 transfectants preincubation with 1027 M insulin in-creased the plasma membrane level of glut 1 protein by1.3-fold, which was a smaller value than that observedin mock transfectant.
3-O-Methyl glucose transport. Table VI shows basal
FIG. 3. Effect of insulin on phosphorylation of proteins in rat adipwithout 0.5 mM insulin, and the whole-cell lysate was subjected tadipocytes (basal condition); lane 2, adipocytes (treated with insulin);(B) Whole-cell lysate from RD cells treated with or without 0.5 mM inand blotted with an anti-IRS 1 antibody or anti-insulin antibody. Lanwith anti-IRS 1 antibody (treated with insulin); lane 3, blotting wblotting with anti-insulin receptor b subunit antibody (treated with
and maximally insulin-stimulated rates of 3-O-methyl
glucose transport in RD cells with mock transfectionand glut 1 sense transfection. Maximum stimulation ofinsulin caused 1.43-fold higher glucose transport in RDcells with mock transfection. The basal and insulin-stimulated glucose transport activities in glut 1 trans-fectants were 1.60- and 1.56-fold higher than thoseobserved in RD cells with mock transfection, respec-tively.
DISCUSSION
We obtained clear evidence that human rhabdomyo-sarcoma expresses glut 1 at both mRNA and proteinlevels and that glut 1 plays a critical role in glucosetransport. Insulin-induced phosphorylation of insulinreceptor and IRS 1 is an event essential for insulin-stimulated upregulation of glucose transport in RDcells because inhibition of tyrosine phosphorylation byherbimycin A blocked upregulation of glucose uptake.The insulin-mediated signal transduction pathway ofglucose uptake in RD cells is through the insulin re-ceptor and IRS 1, as reported (29). To our knowledgethis is the first documentation that human cells whichhave the potential to differentiate into myoblast retain
tes and RD cells. (A) Adipocytes and RD cells were treated with orestern blotting using an anti-phosphotyrosine antibody. Lane 1,
e 3, RD cells (basal condition); lane 4, RD cells (treated with insulin).in was immunoprecipitated using an anti-phosphotyrosine antibody, blotting with anti-IRS 1 antibody (basal condition); lane 2, blottinganti-insulin receptor b subunit antibody (basal condition); lane 4,ulin).
ocyo Wlansule 1
ith
insulin responsiveness.
e 12
79INSULIN-STIMULATED GLUCOSE TRANSPORT IN RD CELLS
Transfection of the glut 1 cDNA led to 2.6-fold higherexpression of glut 1 mRNA and 2-fold higher expres-sion of glut 1 protein of plasma membrane under basal
FIG. 4. RT-PCR of gluts 1, 3, and 4 in untreated RD cells, RD cellantisense oligonucleotides of glut 1, 3, or 4 transfection. Thirty-sixisolated and glut 1-, 3-, and 4-specific RT-PCR amplification of mRNand transfectants with sense or antisense oligonucleotides of glut 1,was done for 23, 26, and 26 cycles, respectively. b actin-specific RT(lower panels). Representative data among three independent expertreated with Lipofectamine Plus reagent (glut 1); lane 3, RD cells wiwith antisense oligonucleotides of glut 1 transfection (glut 1); lane 5, ureagent (glut 3); lane 7, RD cells with sense oligonucleotides of glutof glut 3 transfection (glut 3); lane 9, untreated RD cells (glut 4); laneRD cells with sense oligonucleotides of glut 4 transfection (glut 4); lan4). The 100-bp DNA ladder marker was used.
TABLE IV
Basal and Insulin-Stimulated 2-Deoxy Glucose Transportin Untreated Cells, Cells Treated with Lipofectamine Plus
Reagent, and Transfectants with Sense or AntisenseOligonucleotides of Gluts 1, 3, and 4
Basal Insulin-stimulated[pmol/well]
Untreated cells 407 6 6.89 675 6 15.3Lipofectamine Plus reagent 410 6 5.30 685 6 9.53Glut 1 Sense 411 6 4.27 680 6 8.23
Antisense 251 6 15.4* 301 6 16.2*Glut 3 Sense 417 6 2.58 673 6 3.65
Antisense 406 6 9.10 669 6 8.61Glut 4 Sense 419 6 10.5 687 6 6.93
Antisense 419 6 5.68 685 6 8.65
Note. Sixty hours after transfection, 2-deoxy glucose transport intransfected cells was measured. Data are means 6 SE of three
independent experiments.* P , 0.01 versus glucose uptake in untreated cells.
condition in transfectants, which were accompanied bya 1.64-fold increase in basal 2-deoxy glucose uptakeand by a 1.60-fold increase in 3-O-methyl glucose up-take rate. This finding is consistent with the conceptthat glut 1 primarily contributes to basal glucosetransport, and similar results were reported in the caseof L6 (42), 3T3-L1 (43), and C2C12 cells (44) and skel-etal muscles from transgenic mice overexpressing glut1 (45). Overexpression of glut 1 in RD cells also in-creased the plasma membrane level of glut 1 proteinunder insulin-stimulated conditions by 1.6-fold intransfectants accompanied with increasing insulin-stimulated glucose transport rate, a finding consistentwith data that glut 1 is also translocated to the plasmamembrane by insulin stimulation and contributes toinsulin-stimulated glucose transport (6–8). Similar re-sults were reported in the case of L6 cells overexpress-ing glut 1 (42). Overexpressed glut 1 was fully func-tional in RD cells.
Short-time incubation with a supraphysiological con-centration of insulin (1 mM) stimulated glucose trans-port by 1.68-fold in RD cells. The magnitude of theeffect of insulin on glucose transport is similar to that
eated with Lipofectamine Plus reagent, and RD cells with sense orurs after transfection of antisense oligonucleotides, total RNA wasrom untreated cells, treated cells with Lipofectamine Plus reagent,or 4 was done (upper panels). PCR amplification of gluts 1, 3, and 4R amplification, using the same sample, was done simultaneouslynts are shown. Lane 1, untreated RD cells (glut 1); lane 2, RD cellsense oligonucleotides of glut 1 transfection (glut 1); lane 4, RD cells
reated RD cells (glut 3); lane 6, cells treated with Lipofectamine Plusansfection (glut 3); lane 8, RD cells with antisense oligonucleotides, RD cells treated with Lipofectamine Plus reagent (glut 4); lane 11,, RD cells with antisense oligonucleotides of glut 4 transfection (glut
s trhoA f3,-PCimeth snt
3 tr10
observed in mouse myoblastic cell line C2C12 (1.56-

1aimtcdgmip
wtwmdccpatwfguft1(m
ULMGG
ti
80 ITO ET AL.
fold increase) (21) and rat vascular smooth muscle cellline A7r5 (1.45-fold increase) (36). An even more potentinsulin-stimulated glucose transport was noted inother cells: 1 mM insulin increases glucose transport by4-fold in 3T3-L1 adipocytes (43). In the case of 3T3-L1dipocytes, the major glucose uptake elicited by insulins mediated by glut 4 (11). In RD cells, glut 1 plays a
ajor role in basal and insulin-stimulated glucoseransport, a result consistent with that observed in L6ells (10, 11). In RD cells suppression of glut 4 mRNAid not affect glucose transport. Transfection of thelut 4 cDNA led to 2.1-fold higher expression of glut 4RNA. However, glut 4 transfectants did not show any
ncrease in basal and insulin-stimulated glucose trans-
FIG. 5. RT-PCR of gluts 1 and 4 in untreated RD cells, RD cellsith mock transfection, and RD cells with glut 1 or glut 4 transfec-
ion. Thirty-six hours after transfection of glut 1 or glut 4, total RNAas isolated and glut 1- and glut 4-specific RT-PCR amplification ofRNA from mock transfectant and glut 1 or glut 4 transfectant was
one. PCR amplification of gluts 1 and 4 was done for 23 and 26ycles, respectively (upper panels). b actin-specific RT-PCR amplifi-ation, using the same sample, was done simultaneously (loweranels). Representative data among three independent experimentsre shown. Lane 1, untreated RD cells (glut 1); lane 2, RD cellsreated with Lipofectamine Plus reagent (glut 1); lane 3, RD cellsith mock transfection (glut 1); lane 4, RD cells with glut 1 trans-
ection (glut 1); lane 5, control muscle (glut 1); lane 6, RD cells withlut 1 transfection (glut 1, without reverse transcriptase); lane 7,ntreated RD cells (glut 4); lane 8, RD cells treated with Lipo-ectamine Plus reagent (glut 4); lane 9, RD cells with mock transfec-ion (glut 4); lane 10, RD cells with glut 4 transfection (glut 4); lane1, control muscle (glut 4); lane 12, RD cells with glut 4 transfectionglut 4, without reverse transcriptase). The 100-bp DNA ladderarker was used.
ort compared with control transfectants. These find- c
ings are consistent with reports that the overexpres-sion of glut 4 did not increase insulin-stimulated glu-cose transport in fibroblasts (46, 47), Chinese hamsterovary cells (48), and C2C12 cells (44). It seems fairlyclear that glut 4 does not play a major role in glucosetransport in RD cells. In vivo, both basal and insulin-stimulated glucose transport are reported to be en-hanced in skeletal muscle and adipose tissue fromtransgenic mice overexpressing glut 4 (49, 50). Wecannot rule out the possibility that another additivegene expression in RD cells would make the expressedglut 4 functionally active. In contrast to gluts 1, 3, and
TABLE V
Basal and Insulin-Stimulated 2-Deoxy Glucose Transportin Untreated Cells, Cells Treated with Lipofectamine
Plus Reagent, Mock Transfectants, and Glut 1and Glut 4 Transfectants
Basal Insulin-stimulated[pmol/well]
ntreated cells 411 6 4.01 696 6 7.17ipofectamine Plus reagent 417 6 8.22 684 6 10.4ock transfectants 417 6 6.16 692 6 11.2lut 1 transfectants 684 6 25.2* 999 6 12.4*lut 4 transfectants 412 6 8.22 680 6 15.4
Note. Sixty hours after transfection, 2-deoxy glucose transport inransfected cells was measured. Data are means 6 SE of threendependent experiments.
* P , 0.01 versus glucose uptake in untreated cells.
FIG. 6. Dose response of insulin-stimulated 2-deoxy glucose trans-port in untreated RD cells and RD cells with glut 1 transfection.Sixty hours after transfection of glut 1, cells were subjected to 2-de-oxy glucose transport assay, as described in the legend to Fig .4. Theresults represent percentage increases over basal uptake in un-treated cells or in glut 1 transfectants elicited by insulin and areexpressed as mean values 6 SE from three independent experi-
ments. *P , 0.01 versus basal glucose transport in untreated RDells or in glut 1 transfectants.
mot
d1g
1
111
1
1
1
1
s
81INSULIN-STIMULATED GLUCOSE TRANSPORT IN RD CELLS
4, glut 2 was not detected in RD cells, a finding noted ingastrointestinal carcinoma (16), pancreas islet tumor(51), and breast carcinoma (52).
At physiological concentrations (1029–10212 M), insu-lin did not significantly increase glucose transport inRD cells. Therefore, RD cells seem to be more insulinresistant than other insulin-sensitive cell lines. Thiscan be explained by the finding that RD cells expressedsmaller amounts of phosphorylated IRS 1 as inducedby insulin, compared to adipocytes.
Other rat and mouse myoblastic cells (L6 andC2C12) were found to undergo myotube formationspontaneously (53, 54). During differentiation, L6 in-creases insulin responsiveness and decreases basalglucose transport, which is accompanied by increasedglut 4 expression and decreased glut 1 expression (22,41). RD cells express the myogenic determination geneMyo D and have the potential to differentiate intomuscle (26). There are reports on the induction of dif-ferentiation in RD cells (55–57). RD cells secrete insu-lin-like growth factor II (58), epidermal growth factor(59), and basic fibroblast growth factor (59), all ofwhich function as autocrine growth factors. Suramin, apolysulfonated naphthylurea with antineoplastic activ-ity (60), blocks this autocrine loop (61–65), inhibits thegrowth of human rhabdomyosarcoma, and induces dif-ferentiation in some human rhabdomyosarcoma celllines, CCA and RMZ-RC2 (59, 66). We attempted toinduce differentiation of RD cells using suramin. Aftertreatment with 100 mg/ml suramin in DMEM supple-
ented with 2% horse serum for 2 weeks, RD cells tookn a spindle shape, but there was no myotube forma-ion (unpublished observation).
In summary, we found that RD cells, a human rhab-omyosarcoma of embryonal histotype, expressed gluts, 3, and 4 and maintains insulin responsiveness, whilelut 1 plays a major role in insulin responsiveness. RD
FIG. 7. Effect of insulin on glut 1 content in the plasma membranesin RD cells with mock transfection and glut 1 transfection. Cells (4 3107) were treated with or without 1 mM insulin and plasma andintracellular membranes from RD cells with mock transfection andglut 1 transfection were subjected to Western blotting using anti-glut1 antibody. Lane 1, plasma membranes from RD cells with mocktransfection (untreated); lane 2, plasma membranes from RD cellswith mock transfection (treated with insulin); lane 3, plasma mem-branes from RD cells with glut 1 transfection (untreated); lane 4,plasma membranes from RD cells with glut 1 transfection (treatedwith insulin); lane 5, intracellular membranes from RD cells with
1mock transfection (untreated); lane 6, intracellular membranes fromRD cells with mock transfection (treated with insulin).
cells can be used to study insulin signal transductionand glucose transport in human muscle.
ACKNOWLEDGMENT
We thank Dr. S. W. Cushman for providing human glut 1 and 4cDNAs and specific antibody for human gluts 1, 3, and 4.
REFERENCES
1. Mueckler, M. (1994) Eur. J. Biochem. 219, 713–725.2. Gould, G. W., and Holman, G. D. (1993) Biochem. J. 295, 329–
351.3. Ploug, T., van Deurs, B., Ai, H., Cushman, S. W., and Ralston, E.
(1998) J. Cell Biol. 142, 1429–1446.4. Piper, R. C., Hess, L. J., and James, D. E. (1991) Am. J. Physiol.
260, C570–C580.5. Calderhead, D. M., Kitagawa, K., Tanner, L. I., Holman, G. D.,
and Lienhard, G. E. (1990) J. Biol. Chem. 265, 13800–13808.6. Zorzano, A., Wilkinson, W., Kotliar, N., Thoidis, G., Wadzinkski,
B. E., Ruoho, A. E., and Pilch, P. F. (1989) J. Biol. Chem. 264,12358–12363.
7. Bilan, P. J., Mitsumoto, Y., Maher, F., Simpson, I. A., and Klip,A. (1992) Biochem. Biophys. Res. Commun. 186, 1129–1137.
8. Koivisto, U. M., Martinez-Valdez, H., Bilan, P. J., Burdett, E.,Ramlat, T., and Klip, A. (1991) J. Biol. Chem. 266, 2615–2621.
9. Evans, J. L., Honor, C. M., Womelsdorf, B. E., Kaplan, E. L., andBell, P. A. (1995) Cell. Signal. 7, 365–376.
0. Kayano, T., Burant, C. F., Fukumoto, H., Gould, G. W., Fan,Y. S., Eddy, R. L., Byers, M. G., Shows, T. B., Seino, S., and Bell,G. I. (1990) J. Biol. Chem. 265, 13276–13282.
1. Burchell, A. (1998) Biochem. J. 331, 973.2. Isselbacher, K. J. (1972) New Engl. J. Med. 286, 929–933.3. Merrall, N. W., Plevin, R., and Gould, G. W. (1993) Cell. Signal.
5, 667–675.4. Younes, M., Lechago, L. V., Somoano, J. R., Mosharaf, M., and
Lechago, J. (1996) Cancer Res. 56, 1164–1167.5. Grobholz, R., Hacker, H. J., Thorens, B., and Bannasch, P. (1993)
Cancer Res. 53, 4204–4211.6. Yamamoto, T., Seino, Y., Fukumoto, H., Koh, G., Yano, H.,
Inagaki, N., Yamada, Y., Inoue, K., Manabe, T., and Imura, H.(1990) Biochem. Biophys. Res. Commun. 170, 223–230.
7. Younes, M., Brown, R. W., Stephenson, M., Gondo, M., andCagle, P. T. (1997) Cancer 80, 1046–1051.
TABLE VI
Basal and Maximally Stimulated 3-O-Methyl GlucoseTransport Rates in RD Cells with Mock Transfection
and Glut 1 Sense Transfection
Basal Insulin-stimulated[pmol/cell/min]
ock transfectants 0.215 6 0.0145 0.310 6 0.0220lut 1 transfectants 0.343 6 0.0210* 0.483 6 0.0171*
Note. 3-O-Methyl glucose transport in transfected cells was mea-ured. Data are means 6 SE of three independent experiments.* P , 0.01 versus glucose uptake in mock transfection.
MG
8. Younes, M., Lechago, L. V., Somoano, J. R., Mosharaf, M., andLechago, J. (1997) Anticancer Res. 17, 2747–2750.

2
2
2
2
23
3
3
3
3
3
3
33
34
4
4
4
44
4
4
4
4
5
5
5555
5
5
5
5
6666
6
6
82 ITO ET AL.
19. Boado, R. J., Black, K. L., and Pardridge, W. M. (1994) BrainRes. Mol. Brain Res. 27, 51–57.
0. Binder, C., Binder, L., Marx, D., Shauer, A., and Hiddemann, W.(1997) Anticancer Res. 17, 4299–4304.
1. Palmer, R. M., Thompson, M. G., Knott, R. M., Champbell, G. P.,Thom, A., and Morrison, K. S. (1997) Biochim. Biophys. Acta1355, 167–176.
2. Mitsumoto, Y., Burdett, E., Grant, A., and Klip, A. (1991) Bio-chem. Biophys. Res. Commun. 175, 652–659.
23. Wojtaszewski, J. F. P., Jakobsen, A. B., Ploug, T., and Richter,E. A. (1998) Am. J. Physiol. 274, E184–E191.
24. Ploug, T., Galbo, H., Vinten, J., Jorgensen, M., and Richter, E. A.(1987) Am. J. Physiol. 253, E12–E20.
25. McAllister, R. M., Melnyk, J., Finkelstein, J. Z., Adams, E. C. J.,and Gardner, M. B. (1969) Cancer 24, 520–526.
26. Tapscott, S. J., Thayer, M. J., and Weintraub, H. (1993) Science259, 1450–1453.
27. Hansen, P. A., Gulve, E. A., and Holloszy, J. O. (1994) J. Appl.Physiol. 76, 979–985.
8. Kahn, B. B., Horton, E. S., and Cushman, S. W. (1987) J. Clin.Invest. 79, 853–858.
9. Morris, F. W., and Kahn, C. R. (1994) J. Biol. Chem. 269, 1–4.0. Elmendorf, J. S., Chen, D., and Pessin, J. E. (1998) J. Biol.
Chem. 273, 13289–13296.1. Maher, F., Vannucci, S., Takeda, J., and Simpson, I. A. (1992)
Biochem. Biophys. Res. Commun. 182, 703–711.2. Vannucci, S. J., Nishimura, H., Satoh, S., Cushman, S. W.,
Holman, G. D., and Simpson, I. A. (1992) Biochem. J. 288,325–330.
3. Mueckler, M., Caruso, C., Baldwin, S. A., Panico, M., Blench, I.,Morris, H. R., Allard, W. J., Lienhard, G. E., and Lodish, H. F.(1985) Science 229, 941–945.
4. James, D. E., Strube, M., and Mueckler, M. (1989) Nature 338,83–87.
5. Chomczynski, P., and Sacchi, N. (1987) Anal. Biochem. 162,156–159.
6. Standley, P. R., and Rose, K. A. (1994) Am. J. Hypertension 7,357–362.
7. Laemmli, U. K. (1970) Nature 227, 680–685.8. Webber, T. M., Joost, J. G., Simpson, I. A., and Cushman, S. W.
(1988) in The Insulin Receptor (Kahn, C. R., and Harrison, L. C.,Eds.), Vol. 2, pp. 171–187, A. R. Liss, New York.
9. Nishimura, H., and Simpson, I. A. (1994) Biochem. J. 302, 271–277.0. Cushman, S. W., and Wardzala, L. J. (1980) J. Biol. Chem. 255,
4758–4762.1. Klip, A., Li, G., and Logan, W. J. (1984) Am. J. Physiol. 247,
E291–E296.
2. Robinson, R., Robinson, L. J., James, D. E., and Lawrence,J. C. J. (1993) J. Biol. Chem. 268, 22119–22126.6
3. Harrison, S. A., Buxton, J. M., Clancy, B. M., and Czech, M. P.(1990) J. Biol. Chem. 265, 20106–20116.
4. Kotliar, N., and Pilch, P. F. (1992) Mol. Endocrinol. 6, 337–345.5. Gulve, E. A., Ren, J. M., Marshall, B. A., Gao, J., Hansen, P. A.,
Holloszy, J. O., and Mueckler, M. (1994) J. Biol. Chem. 269,18366–18370.
6. Haney, P. M., Slot, J. W., Piper, R. C., James, D. E., and Mueck-ler, M. (1991) J. Cell Biol. 114, 689–699.
7. Hudson, A. W., Ruiz, M., and Birnbaun, M. J. (1992) J. Cell Biol.116, 785–797.
8. Asano, T., Takata, K., Katagiri, H., Tsukada, K., Lin, J., Ishi-hara, H., Inukai, K., Hirano, H., Yazaki, Y., and Oka, Y. (1992)J. Biol. Chem. 267, 19636–19641.
9. Hansen, P. A., Gulve, E. A., Marshall, B. A., Gao, J., Pessin,J. E., Holloszy, J. O., and Mueckler, M. (1995) J. Biol. Chem. 270,1679–1684.
0. Shepherd, P. R., Gnudi, L., Tozzo, E., Yang, H., Leach, F., andKahn, B. B. (1993) J. Biol. Chem. 268, 22243–22246.
1. Boden, G., Murer, E., and Mozzoli, M. (1994) Ann. Int. Med. 121,109–112.
2. Brown, R. S., and Wahl, R. L. (1993) Cancer 72, 2979–2985.3. Yaffe, D. (1968) Proc. Natl. Acad. Sci. USA 61, 477–483.4. Yaffe, D., and Saxel, O. (1977) Nature 270, 725–727.5. Melguizo, C., Prados, J., Aneiros, J., Fernandez, J. E., Velez, C.,
and Aranega, A. (1995) J. Pathol. 175, 23–29.6. Crouch, G. D., Kalebic, T., Tsokos, M., and Helman, L. J. (1993)
Exp. Cell Res. 204, 210–216.7. Giovanni, C. D., Landuzzi, L., Frabetti, F., Nicoletti, G., Griffoni,
C., Rossi, I., Mazzotti, M., Scotto, L., Nanni, P., and Lollini, P-L.(1996) Cancer Res. 56, 3898–3901.
8. El-Badry, O. M., Minniti, C., Kohn, E. C., Houghton, P. J.,Daughady, W. H., and Helman, L. J. (1990) Cell Growth Differ.1, 325–331.
9. Giovanni, C. D., Melani, C., Nanni, P., Landuzzi, L., Nicoletti,G., Frabetti, F., Griffoni, C., Colombo, M. P., and Lollini, P. L.(1995) Br. J. Cancer 72, 1224–1229.
0. Hawking, F. (1978) Adv. Pharmacol. Chemother. 15, 289–322.1. Hosang, M. (1985) J. Cell Biochem. 29, 265–273.2. Sato, Y., and Rifkin, D. B. (1988) J. Cell Biol. 107, 1199–1205.3. Betsholtz, C., Johnson, A., Heldin, C., and Westermark, B.
(1986) Proc. Natl. Acad. Sci. USA 83, 6440–6444.4. Coffey, R. J., Leof, E. B., Shipley, G., and Moses, H. L. (1987)
J. Cell. Physiol. 132, 143–148.5. Pollack, M., and Richard, M. (1990) J. Natl. Cancer Inst. 82,
1349–1352.
6. Minniti, C. P., Maggi, M., and Helman, L. J. (1992) Cancer Res.52, 1830–1835.