ibgp 705 biomedical informatics director: prof. kun huang
TRANSCRIPT

IBGP 705 Biomedical Informatics
Director: Prof. Kun Huang

What is Bio(medical)-informatics?
bio·in·for·mat·ics
: the collection, classification, storage, and analysis of biochemical and
biological information using computers especially as applied in molecular
genetics and genomics.
Source: Merriam-Webster's Medical Dictionary, © 2002 Merriam-Webster, Inc.

Myth1 : Bioinformatics is about genomics
• Nucleotide – DNA, RNA, …• Genome – Sequences, chromosomes, expressed data, …• Protein – Sequences, 3-D structure, interaction, …• System – Gene network, protein network, TFs, …• Other – Masspec, microarray, images, lab records, journals, literatures, …
The goal is to understand how the system works.

Myth2 : Data vs. Information
Data
Nucleotide – DNA, RNA, …Genome – Sequences, chromosomes, expressed data, …Protein – Sequences, 3-D structure, interaction, …System – Gene network, protein network, TFs, …Other – Masspec, microarray, images, lab records, journals, literatures, …
Information
GenotypePhenotypeGenotype-Phenotype relationshipSNPs PathwaysDrug targets
Getting data is “easy”, extracting information is hard!

Myth3 : Computer is intelligent
Pros• Repeated work• Accurate storage• Precise computation• Fast communication…
Cons• Cannot generalize• No real intelligence…
The results must be reviewed and validated by biologists. In addition, biologists must have some understanding of how computer processes data (algorithms) – that’s why we need to learn bioinformatics.

Biology – Biomedical informatics – System biology
Biomedical Informatics

BiologyDomain knowledge
• Hypothesis testingExperimental work
• Genetic manipulation• Quantitative measurement• Validation
System SciencesTheoryAnalysisModeling
• Synthesis/prediction• Simulation• Hypothesis generation
InformaticsData management
• DatabaseComputational infrastructure
• Modeling tools• High performance computing
Visualization
System Biology
Understanding! Prediction!

Where does large data come from (who to blame)? High-throughput techniques
Fred Sanger
• Nobel prize in chemistry in 1958"for his work on the structure of proteins, especially that of insulin"
• Nobel prize in chemistry in 1980"for their contributions concerning the determination of base sequences in nucleic acids"

High-throughput techniques
DNA Sequencing
• 1970’s – Nobel prize• 1980’s – Ph.D. thesis• Early 1990’s – Major
research projects• Late 1990’s to now - $20

Human Genome ProjectThe Beginning (1988)
Cold Spring Harbor LaboratoryLong Island, New York

June 26, 2000 at the Whitehouse

Initial Analysis of the Human Genome

http://www.sanger.ac.uk/HGP/draft2000/gfx/fig2.gif

STS – sequence-tagged sites (short segments of unique DNA on every chromosome – defined by a pair of PCR primers that amplified only one segment of the genome)
BAC – Bacterial artificial chromosome, 100-400kb
YAC – Yeast artificial chromosome, 150kb-1.5Mb
Contig – assembled contiguous overlapping segments of DNA from BACs and YACs
ESTs – Expressed Sequence Tags
UniGene Database – a database for ESTs
Genome Mapping

Shotgun Sequencing
• Segments are short ~2kb• Problem with repeated segments or genes
Concepts in Biochemistry, 2nd Ed., R. Boyer

$1000 genome project
SolexaSOLiD454
Re-sequencing using massive parallel sequencer
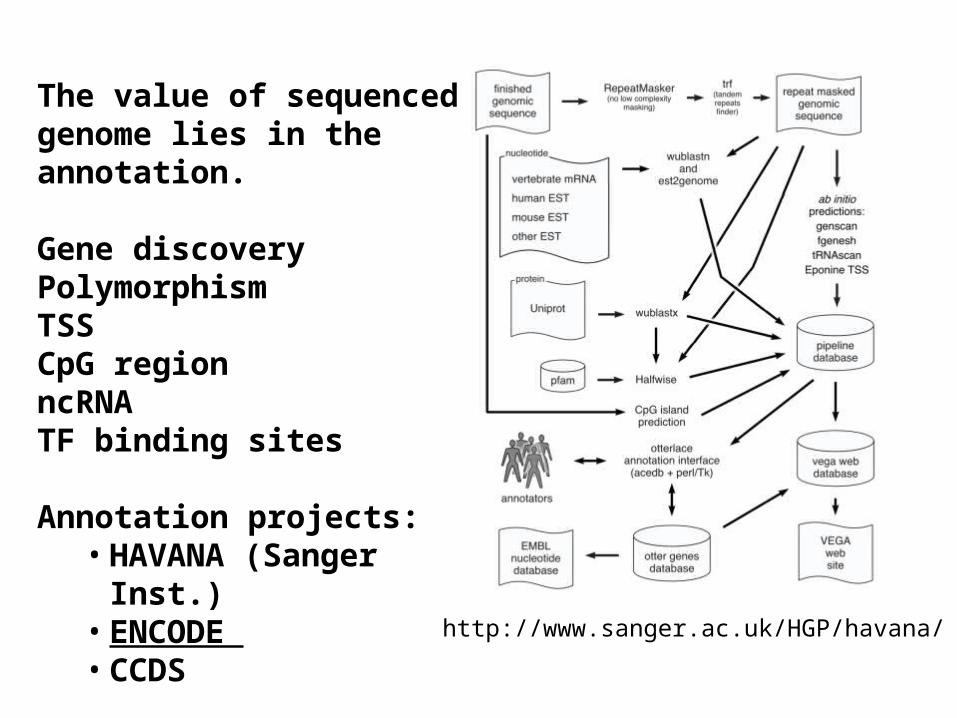
http://www.sanger.ac.uk/HGP/havana/
The value of sequenced genome lies in the annotation.
Gene discoveryPolymorphismTSSCpG regionncRNATF binding sites
Annotation projects:• HAVANA (Sanger Inst.)• ENCODE • CCDS

UCSC Genome Browser


What information do we want to extract?
Science, 9/2/2005 Total genetic difference (# of bases) is 4%35 million single base substitutions plus 5 million insertions or deletions (indels)
The average protein differs by only two amino acids, and 29% of proteins are identical.
Genotype – Phenotype relationship!!!

Phenotype• mRNA level
• Protein expression
• Protein structure
• Cell morphology
• Tissue morphology
• System physiological functions
• Behavior
• …

High-throughput techniquesHigh throughput protein crystalization
Massive parallel sequencing
Mass spectrometry
Microarray
High throughput cell imaging
High throughput in vivo screening
…



“A key element of the GTL program is an integrated computing and technology infrastructure, which is essential for timely and affordable progress in research and in the development of biotechnological solutions. In fact, the new era of biology is as much about computing as it is about biology. Because of this synergism, GTL is a partnership between our two offices within DOE’s Office of Science—the Offices of Biological and Environmental Research and Advanced Scientific Computing Research.
Only with sophisticated computational power and information management can we apply new technologies and the wealth of emerging data to a comprehensive analysis of the intricacies and interactions that underlie biology. Genome sequences furnish the blueprints, technologies can produce the data, and computing can relate enormous data sets to models linking genome sequence to biological processes and function.”

How to extract the information?
Computational tools
• Building the databases
• Perform analysis/extract features
• Data mining
• Classification/statistical learning
• Visualization/representation
Biological information!!!

What we are going to do:
• Search the databases
• Perform analysis
• Present output
Be a salient user!

What we are going to teach:
• Genomics
• Proteomics
• Microarray analysis
• Other aspects
• Ontology
• Imaging informatics
• System biology
• Machine/statistical learning
• Visualization
• Data sources (databases)
• Available tools
• Major issues in using the
databases and tools
• Other resources

Review of Biology
Central dogma

Review of Biology
Operon

Review of Biology
mRNA, cDNA,
exon, intron
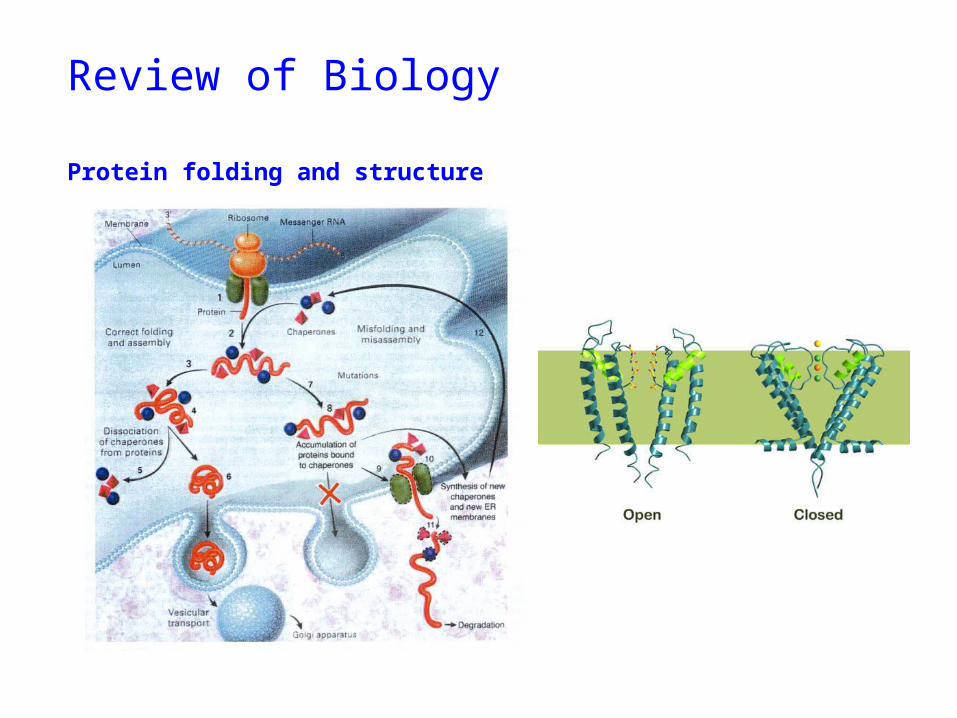
Review of Biology
Protein folding and structure

Databases
GenBank www.ncbi.nlm.nih.gov/GenBank/EMBL www.ebi.ac.uk/embl/DDBJ www.ddbj.nig.ac.jpSynchronized daily.Accession numbers are managed in a consistent way.
AceDBDDJP DNAJJPIDMIPSPHREDPIRPROSITERDPTIGRUNIGENE…

Resources
Local: OSU library
Web: PubMedJSTOR (http://www.jstor.com)http://www.expasy.orghttp://www.genecards.orghttp://www.pathguide.org/
Resources – What’s out there?

PubMed – Entrez
PubMed : http://www.pubmed.gov, http://www.ncbi.nlm.nih.gov/entrez/query.fcgiPubMed training : http://www.nlm.nih.gov/bsd/disted/pubmed.htmlEntrez : http://www.ncbi.nlm.nih.gov/Database/index.html
Entrez is the integrated, text-based search and retrieval system used at NCBI for the major databases, including PubMed, Nucleotide and Protein Sequences, Protein Structures, Complete Genomes, Taxonomy, and others. Click on the graphic below for a more detailed view of Entrez integration.

Entrez Databases

Nucleotide• Gene• Genome• Sequence• mRNA• cDNA• SNP
• Name• Accession number• GI number• Version number• Alias

Accession number, GI number, Version• accession number (GenBank) - The accession number is the unique identifier
assigned to the entire sequence record when the record is submitted to
GenBank. The GenBank accession number is a combination of letters and
numbers that are usually in the format of one letter followed by five digits (e.g.,
M12345) or two letters followed by six digits (e.g., AC123456). • The accession number for a particular record will not change even if the author submits a request to
change some of the information in the record. Take note that an accession number is a unique identifier for
a complete sequence record, while a Sequence Identifier, such as a Version, GI, or ProteinID, is an
identification number assigned just to the sequence data. The NCBI Entrez System is searchable by
accession number using the Accession [ACCN] search field.
• GI (GenBank) - A GI or "GenInfo Identifier" is a sequence identifier that can be
assigned to a nucleotide sequence or protein translation. Each GI is a numeric
value of one or more digits. The protein translation and the nucleotide sequence
contained in the same record will each be assigned different GI numbers. • Every time the sequence data for a particular record is changed, its version number increases and it
receives a new GI. However, while each new version number is based upon the previous version number, a
new GI for an altered sequence may be completely different from the previous GI. For example, in the
GenBank record M12345, the original GI might be 7654321, but after a change in the sequence is
submitted, the new GI for the changed sequence could be 10529376. Individuals can search for nucleotide
sequences and protein translations by GI using the UID search field in the NCBI sequence databases.
• GI number is NOT GeneID.
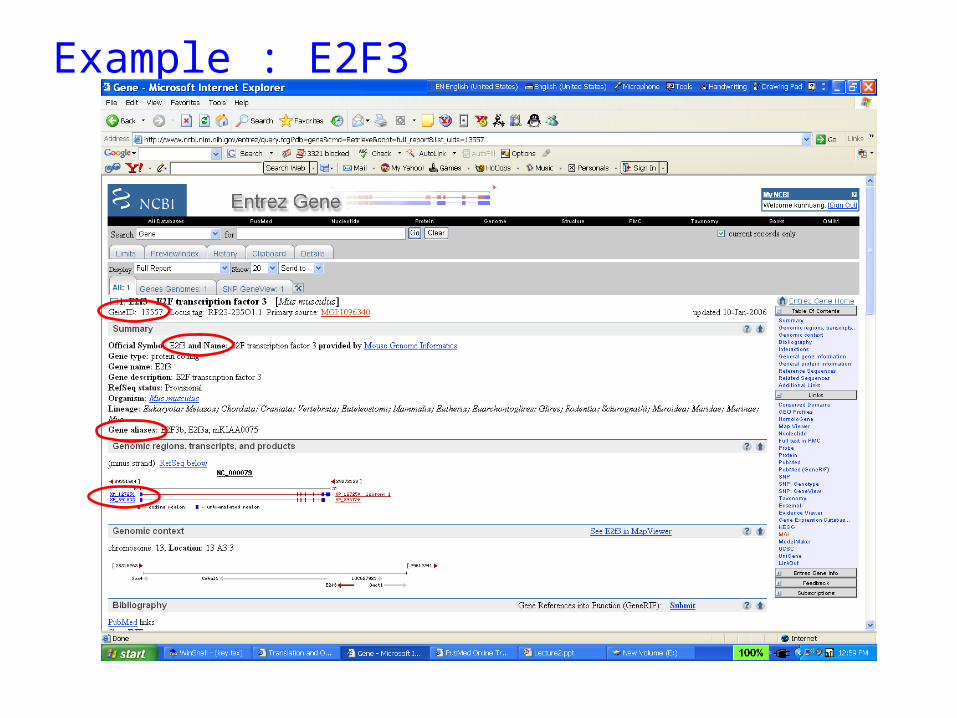
Example : E2F3
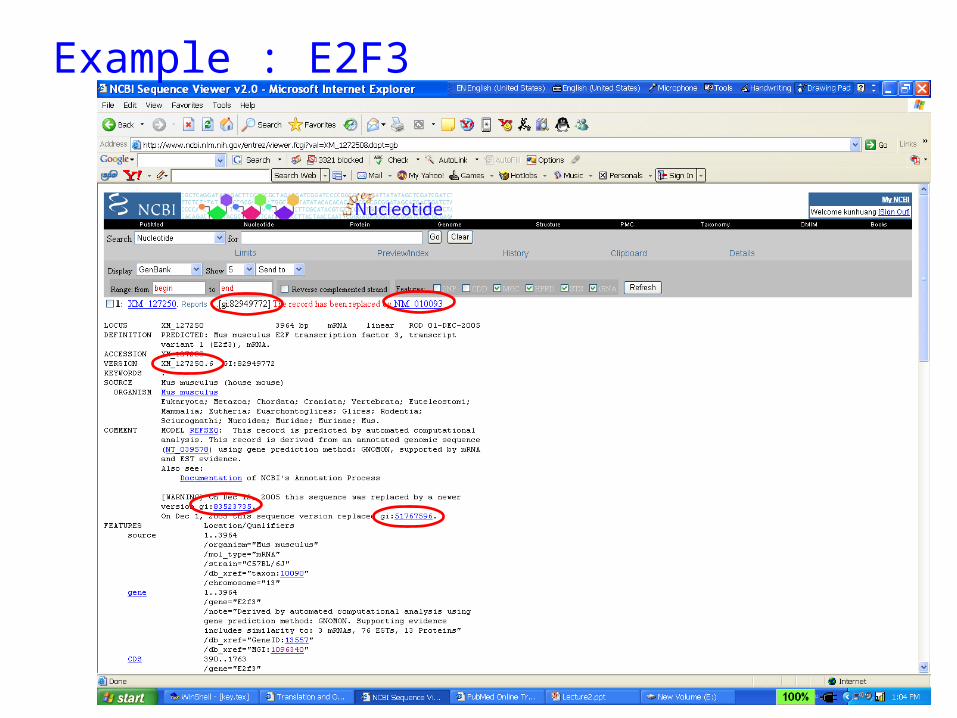
Example : E2F3

Data FormatFASTA (.fasta file) >gi|33469954|ref|NM_000240.2| Homo sapiens monoamine oxidase A (MAOA), nuclear
gene encoding mitochondrial protein, mRNA GGGCGCTCCCGGAGTATCAGCAAAAGGGTTCGCCCCGCCCACAGTGCCCGGCTCCCCCCGGGTATCAAAA GAAGGATCGGCTCCGCCCCCGGGCTCCCCGGGGGAGTTGATAGAAGGGTCCTTCCCACCCTTTGCCGTCC CCACTCCTGTGCCTACGACCCAGGAGCGTGTCAGCCAAAGCATGGAGAATCAAGAGAAGGCGAGTATCGC GGGCCACATGTTCGACGTAGTCGTGATCGGAGGTGGCATTTCAGGACTATCTGCTGCCAAACTCTTGACT GAATATGGCGTTAGTGTTTTGGTTTTAGAAGCTCGGGACAGGGTTGGAGGAAGAACATATACTATAAGGA ATGAGCATGTTGATTACGTAGATGTTGGTGGAGCTTATGTGGGACCAACCCAAAACAGAATCTTACGCTT GTCTAAGGAGCTGGGCATAGAGACTTACAAAGTGAATGTCAGTGAGCGTCTCGTTCAATATGTCAAGGGG AAAACATATCCATTTCGGGGCGCCTTTCCACCAGTATGGAATCCCATTGCATATTTGGATTACAATAATC TGTGGAGGACAATAGATAACATGGGGAAGGAGATTCCAACTGATGCACCCTGGGAGGCTCAACATGCTGA CAAATGGGACAAAATGACCATGAAAGAGCTCATTGACAAAATCTGCTGGACAAAGACTGCTAGGCGGTTT GCTTATCTTTTTGTGAATATCAATGTGACCTCTGAGCCTCACGAAGTGTCTGCCCTGTGGTTCTTGTGGT ATGTGAAGCAGTGCGGGGGCACCACTCGGATATTCTCTGTCACCAATGGTGGCCAGGAACGGAAGTTTGT AGGTGGATCTGGTCAAGTGAGCGAACGGATAATGGACCTCCTCGGAGACCAAGTGAAGCTGAACCATCCT GTCACTCACGTTGACCAGTCAAGTGACAACATCATCATAGAGACGCTGAACCATGAACATTATGAGTGCA AATACGTAATTAATGCGATCCCTCCGACCTTGACTGCCAAGATTCACTTCAGACCAGAGCTTCCAGCAGA GAGAAACCAGTTAATTCAGCGGCTTCCAATGGGAGCTGTCATTAAGTGCATGATGTATTACAAGGAGGCC TTCTGGAAGAAGAAGGATTACTGTGGCTGCATGATCATTGAAGATGAAGATGCTCCAATTTCAATAACCT TGGATGACACCAAGCCAGATGGGTCACTGCCTGCCATCATGGGCTTCATTCTTGCCCGGAAAGCTGATCG ACTTGCTAAGCTACATAAGGAAATAAGGAAGAAGAAAATCTGTGAGCTCTATGCCAAAGTGCTGGGATCC CAAGAAGCTTTACATCCAGTGCATTATGAAGAGAAGAACTGGTGTGAGGAGCAGTACTCTGGGGGCTGCT ACACGGCCTACTTCCCTCCTGGGATCATGACTCAATATGGAAGGGTGATTCGTCAACCCGTGGGCAGGAT TTTCTTTGCGGGCACAGAGACTGCCACAAAGTGGAGCGGCTACATGGAAGGGGCAGTTGAGGCTGGAGAA CGAGCAGCTAGGGAGGTCTTAAATGGTCTCGGGAAGGTGACCGAGAAAGATATCTGGGTACAAGAACCTG
…
>gi|4557735|ref|NP_000231.1| monoamine oxidase A [Homo sapiens] MENQEKASIAGHMFDVVVIGGGISGLSAAKLLTEYGVSVLVLEARDRVGGRTYTIRNEHVDYVDVGGAYV GPTQNRILRLSKELGIETYKVNVSERLVQYVKGKTYPFRGAFPPVWNPIAYLDYNNLWRTIDNMGKEIPT DAPWEAQHADKWDKMTMKELIDKICWTKTARRFAYLFVNINVTSEPHEVSALWFLWYVKQCGGTTRIFSV TNGGQERKFVGGSGQVSERIMDLLGDQVKLNHPVTHVDQSSDNIIIETLNHEHYECKYVINAIPPTLTAK IHFRPELPAERNQLIQRLPMGAVIKCMMYYKEAFWKKKDYCGCMIIEDEDAPISITLDDTKPDGSLPAIM GFILARKADRLAKLHKEIRKKKICELYAKVLGSQEALHPVHYEEKNWCEEQYSGGCYTAYFPPGIMTQYG RVIRQPVGRIFFAGTETATKWSGYMEGAVEAGERAAREVLNGLGKVTEKDIWVQEPESKDVPAVEITHTF WERNLPSVSGLLKIIGFSTSVTALGFVLYKYKLLPRS

Data Format
Other formats
NBRF/PIR (.pir file)Begin with “>P1;” for protein sequence and “>N1;” for nucleotide.
GDE (.gde file) Similar to FASTA file, begin with “%” instead of “>”.

Protein Databases
UniProt is the universal protein database, a central repository of protein data created by combining Swiss-Prot, TrEMBL and PIR. This makes it the world's most comprehensive resource on protein information.The Protein Information Resource (PIR), located at Georgetown University Medical Center (GUMC), is an integrated public bioinformatics resource to support genomic and proteomic research, and scientific studies. Swiss-Prot is a curated biological database of protein sequences from different species created in 1986 by Amos Bairoch during his PhD and developed by the Swiss Institute of Bioinformatics and the European Bioinformatics Institute. Pfam is a large collection of multiple sequence alignments and hidden Markov models covering many common protein domains and families. PDBNCBIhttp://proteome.nih.gov/links.html

ExercisesQuestion 1 - Database search
Find the following genes in GenBank. Write down their accessionnumbers, GI number, chromosome numbers:
Rb1 (human), Rb1 (mouse), Rb1(rat), Rb1(bovine)
Find the protein sequences for the above. Present them in FASTA format.Note: find the most close ones (e.g., if both Rb1 and Rb are present,choose Rb1).
Question 2 – Gene information searchFind the function and alias for the following genes:
TCF3, Col4A1, MMP9 and WASP.
Reading – Entrez tutorialhttp://www.ncbi.nlm.nih.gov/entrez/query/static/help/entrez_tutorial_BIB.pdf