identification of the ureidoglycolate hydrolase gene in the dal gene
TRANSCRIPT

MOLECULAR AND CELLULAR BIOLOGY, Sept. 1985, p. 2279-2288 Vol. 5, No. 90270-7306/85/092279-10$02.00/0Copyright X 1985, American Society for Microbiology
Identification of the Ureidoglycolate Hydrolase Gene in the DALGene Cluster of Saccharomyces cerevisiae
HYANG-SOOK YOO, FRANCIS S. GENBAUFFE, AND TERRANCE G. COOPER*Department of Microbiology and Immunology, University of Tennessee Center for the Health Sciences, Memphis,
Tennessee 38163
Received 26 December 1984/Accepted 30 May 1985
This report describes the isolation of the genes encoding allantoicase (DAL2) and ureidoglycolate hydrolase(DAL3), which are components of the large DAL gene cluster on the right arm of chromosome IX ofSaccharomyces cerevisiae. During this work a new gene (DAL7) was identified and found to be regulated in themanner expected for an allantoin pathway gene. Its expression was (i) induced by allophanate, (ii) sensitive tonitrogen catabolite repression, and (iii) responsive to mutation of the DAL80 and DAL8J loci, which havepreviously been shown to regulate the allantoin degradation system. Hybridization probes generated from thesecloned genes were used to analyze expression of the allantoin pathway genes in wild-type and mutant cellsgrown under a variety of physiological conditions. When comparison was possible, the patterns of mRNA andenzyme levels observed in various strains and physiological conditions were very similar, suggesting that thesystem is predominantly regulated at the level of gene expression. Although all of the genes seem to becontrolled by a common mechanism, their detailed patterns of expression were, at the same time, highlyindividual and diverse.
Saccharomyces cerevisiae can use allantoin as a solenitrogen source by degrading it to urea, which is thenconverted to ammonia and carbon dioxide by themultifunctional protein urea amidolyase (7). The enzymesrequired for metabolizing allantoin to urea are encoded bygenes that are clustered on the right arm of chromosome IXin the order DAL8J-DALJ-DAL4-DAL2 (8). The DAL81gene encodes a positive control element required for expres-sion of the pathway enzymes, while the remaining lociencode the enzymes allantoinase, allantoin permease, andallantoicase, respectively. Conspicuously missing from thecluster is the gene which encodes ureidoglycolate hydrolase.Strains containing mutations of this gene have not beenisolated because of the difficulty in designing a successfulscheme for their selection or identification. If, however, thegene organization followed in the early steps of the pathwayapplies throughout, the ureidoglycolate hydrolase genemight be another component of the DAL gene cluster. Thissituation would conceptually provide a way of isolating theureidoglycolate hydrolase gene and ascertaining its genomiclocation; isolation would be performed via "walking" fromcloned regions of the DAL cluster. To test these hypothesesand begin structural analysis of the large DAL gene cluster,we attempted to clone the DAL2 gene and its flankingregions. This report describes the successful isolation ofDAL2 and the ureidoglycolate hydrolase gene (DAL3) whichwas found nearby. We used probes derived from the clonedgenes to study their expression in wild-type and allantoin-pathway-control mutant strains.
MATERIALS AND METHODSStrains and culture conditions. The strains of S. cerevisiae
and Escherichia coli used in this work are listed in Table 1.All of the culture conditions for growth and transformationhave been described in detail by Sumrada and Cooper (26).
* Corresponding author.
In the present experiments allantoin was provided at a finalconcentration of 0.1% as the sole nitrogen source.Enzyme assays. Allantoicase (DAL2) and ureidoglycolate
hydrolase (DAL3) activities were measured as describedpreviously (5, 14). Transformed and untransformed S. cere-visiae cells were grown in 200 ml of Wickerham minimal-glucose medium with glutamate (0.1%) provided as the sole
TABLE 1. Yeast and bacterial strains used in this work
Organism andstrain Genotype or phenotype
designation
S. ceresisiaeM1285-3C ...... MATa irplhis6 Ivsl daI2M1-2B...... MATa ura3-52 trpiM1062-17D ...... MATa IvsS daI2M96-23A ...... MATTa ade6 leul dal2M1373-6B ...... MATa trpl lysl ura3-52 dal2M1373-24A ...... MATot trpl his6 ura3-52 daI2M1373-7A ...... MATa his6 trpl ura3-52 dal2M1373-12C ...... MATot trpllysl ura3-52 dal2RH218...... MATa trpl CUPI gaI2 SUC2 Mal-VT-20 ...... MATa trpi dal2M970.MA Ta IvsS
MA Tot 1)ys2M1081 . MATa lvsS dal80-1M1081.......~~~~~~~~~~...... . ~-
MATot lYs2 daI80-1M1407 . MATa Ivs2 daI81-2M 47..........
MATa Ivs5 daI81-2W303-la.... MATa leu2-3 trpl-I canl-100 ura3-l ade2
E. coliSF-8...... hsdR hsdM recB recC lopl supE44 ga196
Smr leuiB thil Thr-HB101... lisdR hsdM recA13 supE44 lacZ24 leiuB
proA2 thil Smr
2279

2280 YOO ET AL.
TABLE 2. Allantoicase and ureidoglycolate hydrolase activities in wild-type, mutant, and transformed strains of S. cerevisiae
Activity (nmol/min per mg of protein) with indicated nitrogen source
StrainAllantoicase Ureidoglycolate hydrolase
Ammonia Ureaa Asparagine Glutamate Glutamate mate Glutamateplus urea plus OXLUb Gluta plus OXLU
RH-218 (wild type) 1.07 4.81VT-20 dal2 0.18 0.20 0.4 0.5 3.9 6.3VT-20 transformed with plasmid pTC7 6.45 67.60 4.79 7.3 25.0 67.0 180.0VT-20 transformed with plasmid pTC11 2.2 3.2 5.5 6.6VT-20 transformed and cured 0.71 0.78
a Urea serves as both nitrogen source and inducer.b Glutamate was used as the nitrogen source, and 0.25 mM OXLU was provided as an inducer.
nitrogen source. At a cell density of 50 Klett units, the cellswere harvested and suspended in 2 ml of breakage buffer(0.05 M Tris acetate buffer, pH 7.0, containing 5% glyceroland 100 Rxl of 2-mercaptoethanol per ml). An equal volume ofglass beads was added to the cell suspension, which wasthen mixed intermittently with a Vortex mixer for a total of2 min. Glass beads and cell debris were removed from thesuspension by centrifugation for 15 min at 12,000 x g. Theresulting supernatant solutions were then used as the sourceof protein for the enzyme assays. Protein concentrationswere measured by the method of Lowry et al. (16).
Manipulation of strains and plasmids. All of the methodsemployed in the isolation, analysis, and use of the plasmidsdescribed in this work were those described by Sumrada andCooper (26).
Preparation of chromosomal DNA and Southern blot anal-ysis. Chromosomal DNA was isolated from both His- recip-ient (strain W303-la) and His' transformant strains by themethod of Winston et al. (28). The DNA was then digestedwith restriction endonuclease Sacl, and the resulting frag-ments were resolved in 0.8% agarose gels as described bySumrada and Cooper (26). The resolved fragments weretransferred to nitrocellulose paper as described by Davis etal. (10). The Southern blots were probed with a 0.45-kilobase(kb) AvaI-HindIII fragment that had been radioactivelylabeled via the polynucleotide kinase reaction as describedby Davis et al. (10).
Northern blot analysis. Polyadenylated [poly(A)+] RNAfrom wild-type (strain M970), dal80 (strain M1081), anddal81 (strain M1407) mutant strains was isolated by themethod of Carlson and Botstein (3). Cultures to be used forpoly(A)+ RNA isolation were grown in Wickerham mediumwith proline or asparagine (0.1%) as the sole nitrogen source.Oxalurate (OXLU; 0.25 mM) was added as an inducer whereindicated. Poly(A)j RNA (10 R,g) that had been passaged
, 4,,N,,-1o,> . LC,I
Y1.....KK
twice through an oligodeoxythymidylate column was re-solved on a 1.2% agarose-formaldehyde gel as described bySumrada and Cooper (26). The RNA was transferred tonitrocellulose paper by the method of Fryberg et al. (12). Thefilter was then probed with plasmid pHY9, pHY3, pHY2, orpHY11, which had been radioactively labeled by nick trans-lation (10).
RESULTSIsolation of plasmids able to complement mutations in
DAL2. The first objective of this work was the isolation ofchimeric plasmids containing the DAL2 gene. This wasaccomplished by isolating an allantoicase-negative (dal2)mutant after ethyl methanesulfonate mutagenesis of strainRH-218 (11, 19), which is known from previous work totransform at a high frequency. Allantoicase activity was verylow in the mutant strain (VT-20) compared with that in thewild-type parent (RH-218) (Table 2). The mutant was sepa-rately transformed with two plasmid DNA banks. The firstbank was prepared from yeast strain AB320 by Nasmyth andReed (20). The second bank was constructed with DNA fromstrain X2180-1A by Clarke and Carbon (6). Transformed S.cerevisiae cells were selected on minimal medium withouttryptophan and with allantoin provided as the sole nitrogensource. After 4 days of incubation at 30°C, 30 putativetransformants were isolated. Eight of these transformantswere grown in liquid minimal-allantoin medium. A crudenucleic acid fraction was prepared from each of these eightcultures and used as the source ofDNA to transform E. coliSF-8. Each of the eight yeast DNA preparations yieldedfrom 50 to 125 E. coli transformants. Two of these trans-formants were used as sources of the plasmids pTC7 andpTC11. Plasmid pTC7 was isolated from the bank of
'K
pTC7
q, 3oj%K~ (KT
Isl- 4c9.
KC tCmL,,,//N1 kB
pTC11T
FIG. 1. Restriction maps of plasmids whose inserts complemented dal2 mutations. BglI, BglII, PstI, PvuI, Sall, SmaI, XbaI, and XhoIsites were lacking in the insert pf plasmid pTC7. For the insert of plasmid pTC11, no sites were observed for BglI, BgII, Clal, KpnI, PstIl,PvuI, SacII, Sall, SmaI, XbaI, or XhoI. Diagonal lines indicate flanking vector sequences.
z I'Lle le If, z lr..~ le z Iit I +....
I I I-11I --11-1i P, i p
0$P.
MOL. CELL. BIOL.
i.- -11.1I
:bltl..+9 4c,e le40 V,; +9 .p ..v1( r ( ( 6`1

CONTROL OF ALLANTOIN SYSTEM IN S. CEREVISIAE
TABLE 3. Behavior of an integrated URA3 allelein genetic crosses'"
Assortment of ura3-l4vslCross Integrated Endo- No. ofstrain plasmid nucleaseb asci Parental Non-analyzed type parental Tetratypetye type
M1429 pHY11 Sacl 37 34 0 3M1430 pHY10 BamnHI 37 31 1 5M1431 pHY3 KpnI 39 39 0 0M1432 pHY3 None 23 22 0 1
" Strain M1373-6B was the recipient of the transforming DNA in each case.After transformation, a random transformant was selected and crossed tostrain M1373-24A.
b Nuclease used to digest the plasmid before transformation.
Nasmyth and Reed, and pTC11 came from that of Clark andCarbon.To ascertain whether the two plasmids carried the DAL2
gene, we used them in purified form to transform theallantoicase-negative mutant strain VT-20. Both were capa-ble of transforming the mutant to a wild-type phenotypeat high frequency (greater than 5,000 transformants per pug ofDNA without added carrier DNA). Complementation wasnot seen when plasmid YRp7 was used as the source ofDNAor when DNA was omitted from the transformation reactionmixture.To test whether the presence of plasmid pTC7 resulted in
the appearance of allantoicase activity, we determined theenzyme-specific activity in (i) the transformation recipient(strain VT-20), (ii) a pTC7 transformant of this strain, and(iii) a transformant that had been grown for many genera-tions in rich, nonselective medium. Allantoicase activity wasnearly absent in the dal2 mutant (strain VT-20) (Table 2).After transformation with plasmid pTC7, strain VT-20 ex-hibited more activity in the absence of inducer (minimal-ammonia medium) than did its parent, strain RH-218, grownin its presence (minimal-urea medium). This activity in-creased 10- to 11-fold after induction of the transformant.When a repressive nitrogen source, such as asparagine, was
IS, "Ii(K
5(%pFG8
provided, the level of allantoicase activity decreased 14-foldcompared with that observed in derepressed cells (minimal-urea medium). Allantoicase activity was again found to bevery low when the transformant was grown in rich, nonse-lective medium (strain VT-20, Table 2).
Structure of DAL2-containing plasmids. The structures ofplasmids pTC7 and pTC11 were determined by restrictionmapping (Fig. 1). Plasmids pTC7 and pTC11 contained 6.6-and 3.3-kb inserts, respectively, which possessed a region ofperfect restriction site homology extending for 2.1 kb. Thetwo inserts were in opposite orientation with respect to thetwo vectors, as can be seen by inspecting the flanking vectorsites (Fig. 1).
Integration of pTC7 and pTC11 into the yeast genome.Another way to verify the identity of a plasmid-borne gene isto assess the linkage between a plasmid marker (URA3) andmarkers flanking the chromosomal gene in question afterintegration of the plasmid. The plasmids to be integratedwere constructed by inserting the 7.2- or 3.9-kb EcoRI-SalIfragments from pTC7 and pTC11 (Fig. 1) into vector plasmidYIp5, which had been digested with EcoRI and Sall (Fig. 3).Each of these plasmid preparations, designated pHY5 andpHY6, was linearized at the BamHI site to enhance thefrequency of integration at this site in the genome (21). Bothdigested and undigested versions of the two plasmids wereused as the source of DNA to transform strain M1373-6B.The recipient strain was allantoicase negative and carried theura3-52 mutation. Surprisingly, the undigested plasmidssupported a high frequency of transformation similar to thatseen with pTC7 and pTC11. A significant decrease in trans-formation frequency was observed when the plasmids werelinearized by digestion with BamHI. These characteristicsare expected of a plasmid that contains an ars sequence andis therefore capable of autonomous replication. Since theYIp5 vector does not contain ars sequences, it was likelythat the inserts contained such a sequence. In view of thispossibility, a second plasmid, pHY11, was prepared byinserting the 1.7-kb BamHI-HindIII fragment from plasmidpTC11 into vector YIp5. After digestion with SacI, thisplasmid was used to transform strain M1373-6B. In this case,
pTC7
(rstG
+01 '@
-44-6T
&0
I;e FGI
,0 pFG1
pFG1O
.....1...
FIG. 2. Localization of the DAL2 gene. The designated portions of pTC7 and pTC11 were recloned into the autonomously replicatingvector YRp7. Each of the constructions was then transformed into a daI2 mutant strain and tested for the ability to complement the mutation.Symbols: +, high-frequency transformation with allantoin as the sole nitrogen source; -, no transformation under these conditions.
pTCl1
1. l ,,,,"I ..r/,,, 4,.;.
+ 1 kB
.. ii09,600000*00i 11 11
X, ..................................
-
......F-1..'-",,-'.,-'.'..'.'-.'.."-.-"-.-'.'.'.'.-.'.-..--'-.--'.-..-."-,..'..'.'.'.,"..,-."..'."..-l-,,--.'-.'-""'.-.,
-
".-.- ---.r--------------
VOL. 5, 1985 2281
.- .. -s'4. c, t, §.& , .c
c,lkz (.. o e C.. .F kg Ov .VT-. ( ( (r J

2282 YOO ET AL.
* pTC7
i\\W pHY3 XNN
rY pHY9
pTC11
ec°rONAI IpHY6[~~~ +
reo% PHY15V-CQp
rvol,' pHYlO v
rp
rle pHYI
+
I\l
+ 1 kB.. ........ : ........... ....
Y&pHY12rcoe
pHY2 &U.'+
FIG. 3. Localization of the ARS sequence on the insert of plasmid pTC7. Portions of pTC7 and pTC11 were recloned into theURA3-containing integrative vector YIp5. Each of the constructions was then tested for the ability to support high-frequency transformationof strain M1373-6B, using the URA3 gene as the selected marker. Symbols: +, high-frequency transformation; -, no high-frequencytransformation.
the expected low frequency of transformation was observed.Ura+ transformants were isolated, and after their phenotypewas shown to be stable, these strains were crossed to strainM1373-24A. The Ura+ phenotype segregated in 2+:2- fash-ion in all asci derived from four independent transformants(Table 3). This was the expected result if the sequences
responsible for complementing the defects in ura3 were
carried on the integrated plasmid. The ura3 and lysi gene
pair had a high degree of linkage, i.e., most asci exhibited a
parental ditype array of the markers. Analysis of these datawith the formula of Perkins (22) resulted in an estimateddistance of 5.5 centimorgans between ura3 and lysi. Thesedata are expected if the plasmid is integrated into the dal2gene situated on the right arm of chromosome IX, 4.7centimorgans proximal to the lysi locus.
Location ofDAL2. Sequences complementing mutations inthe daI2 gene were localized on plasmids pTC7 and pTC11by testing the ability of several recloned fragments to trans-form strain VT-20 to the wild-type phenotype. One end ofthe complementing sequences was identified by comparingthe transforming ability of plasmids pFG10 and pHY13 (Fig.2). The insert in plasmid pHY13, which extended from the
left to the BamHI site, was unable to complement themutation in strain VT-20. However, the insert in plasmidpFG10, which extended to the Hindlll site, complementedthe mutation. The other end of the complementing se-quences was identified by using plasmids pFG1 and pFG8.The first plasmid (pFG1), with an insert extending from theright to the BamHI site, was incapable of complementation,but pFG8, with an insert extending to the Sacl site, comple-mented the mutation. In sum, these data suggest that theDAL2 gene is situated between the Hindlll and Sacl sitesthat are common to both pTC7 and pTC11. It may beconcluded that the BamHI site is located within the gene,because segments on both sides of this site were required forcomplementation.
Location of the ars locus. The ars sequence was located onpTC7 and pTC11 by testing the ability of several reclonedfragments derived from these plasmids to support high-frequency transformation when inserted into vector YIp5;uracil prototrophy was used as the selective marker. Oneend of the ars-containing sequence was identified by com-paring the transformation frequencies obtained with plas-mids pHY1 and pHY12 (Fig. 3). Plasmid pHY12, with an
"' """" """" """'...----:--------,. -K-ne-m
:.: ............ I ................11 .il-, ---l I ..L-------L.
MOL. CELL. BIOL.
I0
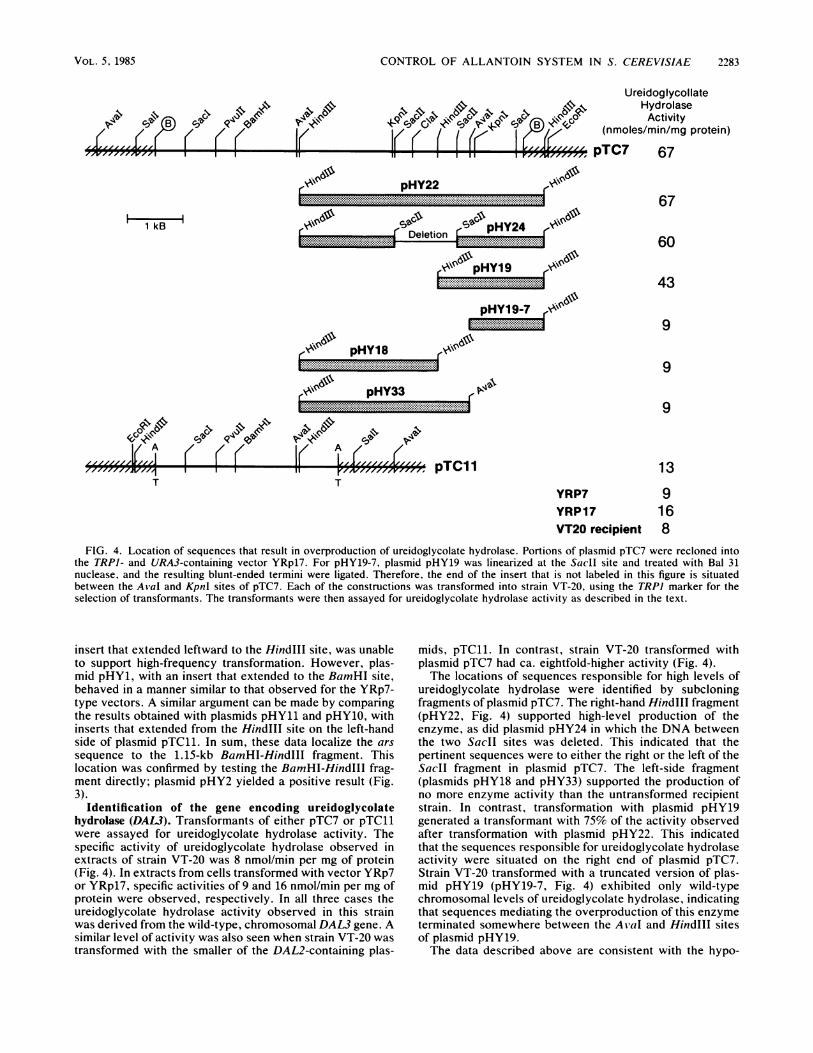
CONTROL OF ALLANTOIN SYSTEM IN S. CEREVISIAE
44 Z C, ,.4C,b O C:90
I I I I Irrr/
r, pHY22I'll.................
................
UreidoglycollateHydrolaseActivity
(nmoles/min/mg protein)
pTC7 67
67St pHY24 \X\<5
.1
pHY19-7 X'Or.
AA,zzpz7zzz,TC1 1T
FIG. 4. Location of sequences that result in overproduction of ureidoglycolate hydrolase. Portions of plasmid pTC7 were recloned intothe TRPI- and URA3-containing vector YRp17. For pHY19-7, plasmid pHY19 was linearized at the SacIl site and treated with Bal 31nuclease, and the resulting blunt-ended termini were ligated. Therefore, the end of the insert that is not labeled in this figure is situatedbetween the AilaI and KpnI sites of pTC7. Each of the constructions was transformed into strain VT-20, using the TRPI marker for theselection of transformants. The transformants were then assayed for ureidoglycolate hydrolase activity as described in the text.
insert that extended leftward to the HindIll site, was unableto support high-frequency transformation. However, plas-mid pHYI, with an insert that extended to the BamHI site,behaved in a manner similar to that observed for the YRp7-type vectors. A similar argument can be made by comparingthe results obtained with plasmids pHY11 and pHY10, withinserts that extended from the HindIII site on the left-handside of plasmid pTC11. In sum, these data localize the ars
sequence to the 1.15-kb BamHI-HindIII fragment. Thislocation was confirmed by testing the BamHI-HindIII frag-ment directly; plasmid pHY2 yielded a positive result (Fig.3).
Identification of the gene encoding ureidoglycolatehydrolase (DAL3). Transformants of either pTC7 or pTC11were assayed for ureidoglycolate hydrolase activity. Thespecific activity of ureidoglycolate hydrolase observed inextracts of strain VT-20 was 8 nmol/min per mg of protein(Fig. 4). In extracts from cells transformed with vector YRp7or YRp17, specific activities of 9 and 16 nmol/min per mg ofprotein were observed, respectively. In all three cases theureidoglycolate hydrolase activity observed in this strainwas derived from the wild-type, chromosomal DAL3 gene. Asimilar level of activity was also seen when strain VT-20 wastransformed with the smaller of the DAL2-containing plas-
mids, pTC11. In contrast, strain VT-20 transformed withplasmid pTC7 had ca. eightfold-higher activity (Fig. 4).The locations of sequences responsible for high levels of
ureidoglycolate hydrolase were identified by subcloningfragments of plasmid pTC7. The right-hand HindIll fragment(pHY22, Fig. 4) supported high-level production of theenzyme, as did plasmid pHY24 in which the DNA betweenthe two SacII sites was deleted. This indicated that thepertinent sequences were to either the right or the left of theSacII fragment in plasmid pTC7. The left-side fragment(plasmids pHY18 and pHY33) supported the production ofno more enzyme activity than the untransformed recipientstrain. In contrast, transformation with plasmid pHY19generated a transformant with 75% of the activity observedafter transformation with plasmid pHY22. This indicatedthat the sequences responsible for ureidoglycolate hydrolaseactivity were situated on the right end of plasmid pTC7.Strain VT-20 transformed with a truncated version of plas-mid pHY19 (pHY19-7, Fig. 4) exhibited only wild-typechromosomal levels of ureidoglycolate hydrolase, indicatingthat sequences mediating the overproduction of this enzymeterminated somewhere between the AvaI and HindlIl sitesof plasmid pHY19.The data described above are consistent with the hypo-
4xL< - i--
.%@4,i
II
I 1 kBDDeletion
pHY18
60
43
9
9
4gt
pHY33 _r
T41$- 4f
9
13
YRP7YRP17VT20 recipient
9168
I3
-------m
VOL. 5, 1985 2283
1% \%4lb C..
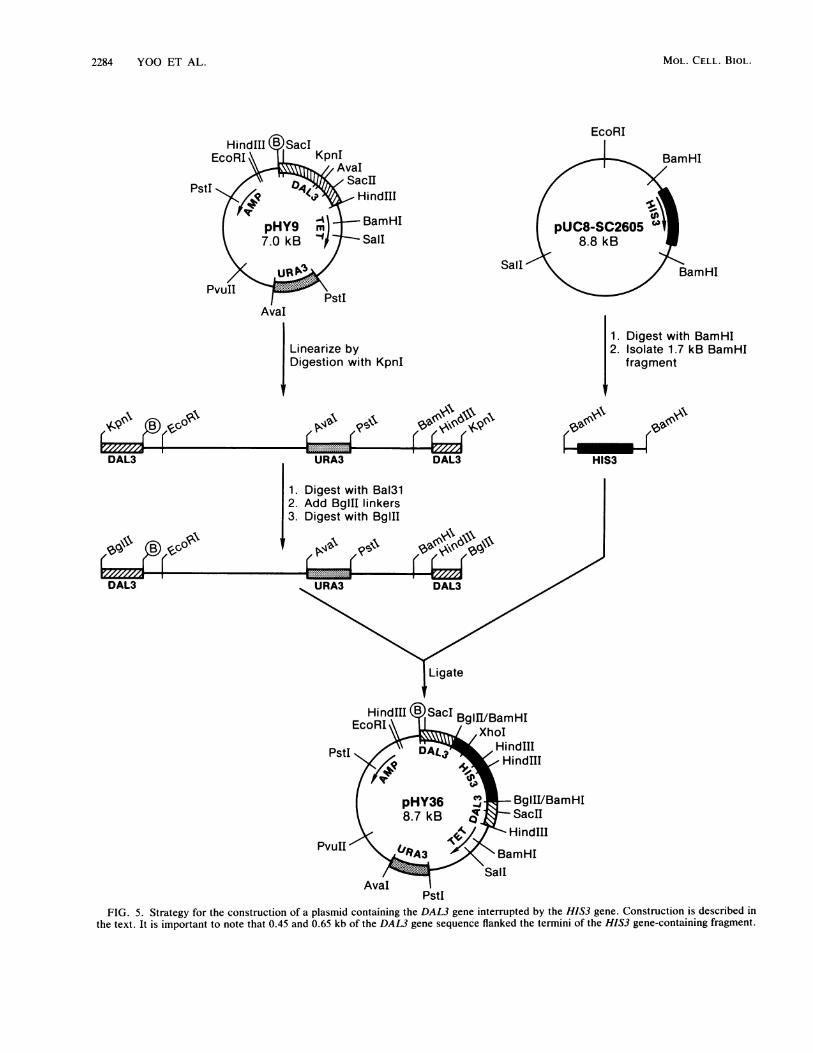
2284 YOO ET AL.
EcoRI
PstI
) <;GOF~P
vz RN
KpnIAvaISacIHindlIl
m A- BamHI¢;_ ~ Sall
BamHI
SaII
I1.2.Linearize by
Digestion with KpnI
DAL3URA3
BamHI
Digest with BamHIIsolate 1.7 kB BamHIfragment
HIS3
1. Digest with Bal3l2. Add BgII[ linkers3. Digest with BglII
Pstl
PvuII '
FIG. 5. Strategy for the construction of a plasmid containing the DAL3 gene interrupted by the HIS3 gene. Construction is described in
the text. It is important to note that 0.45 and 0.65 kb of the DAL3 gene sequence flanked the termini of the HIS3 gene-containing fragment.
IDAL3
r'?s'r'/zzzl03-
,a%, '?C7c P..l
MOL. CELL. BIOL.
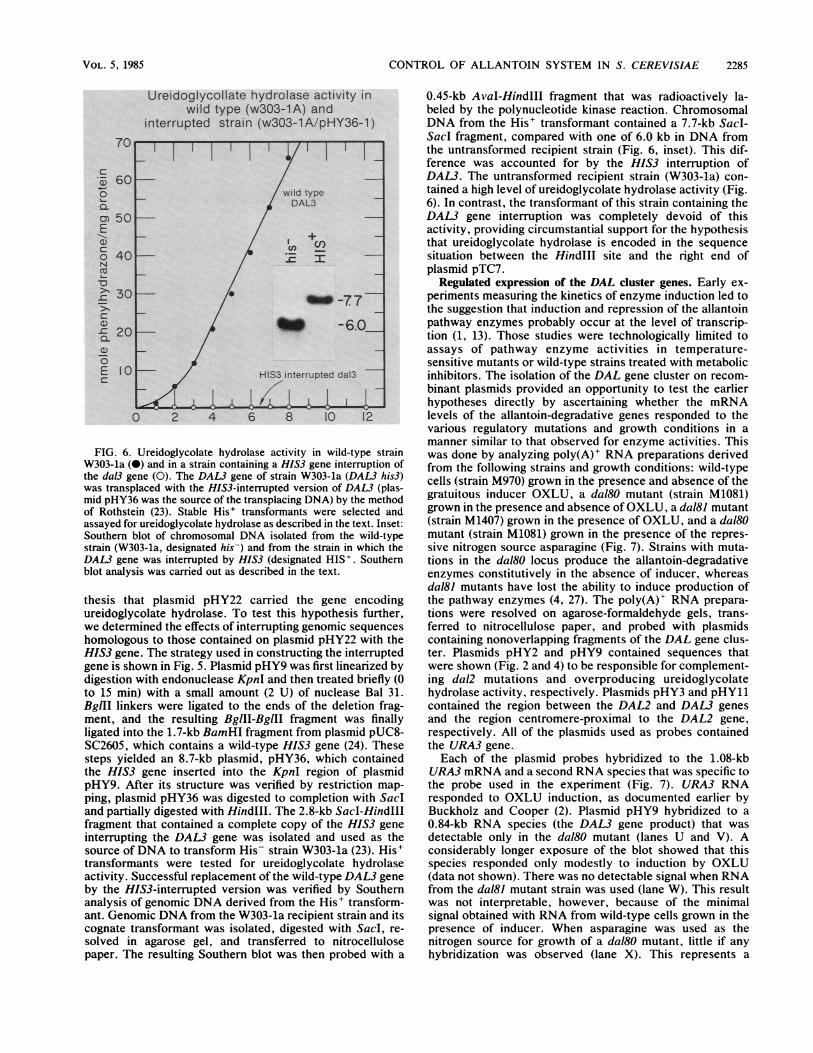
CONTROL OF ALLANTOIN SYSTEM IN S. CEREVISIAE
Ureidoglycollate hydrolase activity inwild type (w303-1A) and
interrupted strain (w303-1A/pHY36-1)70
c
- 60
0~m 50E
CU
a 2a)
E 10c0`3E10
C:
FIG. 6. Ureidoglycolate hydrolase activity in wild-type strainW303-la (0) and in a strain containing a HIS3 gene interruption ofthe dal3 gene (0). The DAL3 gene of strain W303-la (DAL3 his3)was transplaced with the HIS3-interrupted version of DAL3 (plas-mid pHY36 was the source of the transplacing DNA) by the methodof Rothstein (23). Stable His' transformants were selected andassayed for ureidoglycolate hydrolase as described in the text. Inset:Southern blot of chromosomal DNA isolated from the wild-typestrain (W303-la, designated his-) and from the strain in which theDAL3 gene was interrupted by HIS3 (designated HIS'. Southernblot analysis was carried out as described in the text.
thesis that plasmid pHY22 carried the gene encodingureidoglycolate hydrolase. To test this hypothesis further,we determined the effects of interrupting genomic sequenceshomologous to those contained on plasmid pHY22 with theHIS3 gene. The strategy used in constructing the interruptedgene is shown in Fig. 5. Plasmid pHY9 was first linearized bydigestion with endonuclease KpnI and then treated briefly (0to 15 min) with a small amount (2 U) of nuclease Bal 31.BglII linkers were ligated to the ends of the deletion frag-ment, and the resulting BglII-BglII fragment was finallyligated into the 1.7-kb BamHI fragment from plasmid pUC8-SC2605, which contains a wild-type HIS3 gene (24). Thesesteps yielded an 8.7-kb plasmid, pHY36, which containedthe HIS3 gene inserted into the KpnI region of plasmidpHY9. After its structure was verified by restriction map-ping, plasmid pHY36 was digested to completion with SacIand partially digested with HindIlI. The 2.8-kb Sacl-HindIllfragment that contained a complete copy of the HIS3 geneinterrupting the DAL3 gene was isolated and used as thesource ofDNA to transform His- strain W303-la (23). His'transformants were tested for ureidoglycolate hydrolaseactivity. Successful replacement of the wild-type DAL3 geneby the HIS3-interrupted version was verified by Southernanalysis of genomic DNA derived from the His' transform-ant. Genomic DNA from the W303-la recipient strain and itscognate transformant was isolated, digested with Sacl, re-solved in agarose gel, and transferred to nitrocellulosepaper. The resulting Southern blot was then probed with a
0.45-kb AvaI-HindIII fragment that was radioactively la-beled by the polynucleotide kinase reaction. ChromosomalDNA from the His' transformant contained a 7.7-kb Sacl-Sacl fragment, compared with one of 6.0 kb in DNA fromthe untransformed recipient strain (Fig. 6, inset). This dif-ference was accounted for by the HIS3 interruption ofDAL3. The untransformed recipient strain (W303-la) con-tained a high level of ureidoglycolate hydrolase activity (Fig.6). In contrast, the transformant of this strain containing theDAL3 gene interruption was completely devoid of thisactivity, providing circumstantial support for the hypothesisthat ureidoglycolate hydrolase is encoded in the sequencesituation between the HindlIl site and the right end ofplasmid pTC7.
Regulated expression of the DAL cluster genes. Early ex-periments measuring the kinetics of enzyme induction led tothe suggestion that induction and repression of the allantoinpathway enzymes probably occur at the level of transcrip-tion (1, 13). Those studies were technologically limited toassays of pathway enzyme activities in temperature-sensitive mutants or wild-type strains treated with metabolicinhibitors. The isolation of the DAL gene cluster on recom-binant plasmids provided an opportunity to test the earlierhypotheses directly by ascertaining whether the mRNAlevels of the allantoin-degradative genes responded to thevarious regulatory mutations and growth conditions in amanner similar to that observed for enzyme activities. Thiswas done by analyzing poly(A)+ RNA preparations derivedfrom the following strains and growth conditions: wild-typecells (strain M970) grown in the presence and absence of thegratuitous inducer OXLU, a dal80 mutant (strain M1081)grown in the presence and absence ofOXLU, a dal81 mutant(strain M1407) grown in the presence of OXLU, and a daI80mutant (strain M1081) grown in the presence of the repres-sive nitrogen source asparagine (Fig. 7). Strains with muta-tions in the dal80 locus produce the allantoin-degradativeenzymes constitutively in the absence of inducer, whereasdal81 mutants have lost the ability to induce production ofthe pathway enzymes (4, 27). The poly(A)+ RNA prepara-tions were resolved on agarose-formaldehyde gels, trans-ferred to nitrocellulose paper, and probed with plasmidscontaining nonoverlapping fragments of the DAL gene clus-ter. Plasmids pHY2 and pHY9 contained sequences thatwere shown (Fig. 2 and 4) to be responsible for complement-ing dal2 mutations and overproducing ureidoglycolatehydrolase activity, respectively. Plasmids pHY3 and pHY11contained the region between the DAL2 and DAL3 genesand the region centromere-proximal to the DAL2 gene,respectively. All of the plasmids used as probes containedthe URA3 gene.Each of the plasmid probes hybridized to the 1.08-kb
URA3 mRNA and a second RNA species that was specific tothe probe used in the experiment (Fig. 7). URA3 RNAresponded to OXLU induction, as documented earlier byBuckholz and Cooper (2). Plasmid pHY9 hybridized to a0.84-kb RNA species (the DAL3 gene product) that wasdetectable only in the dal80 mutant (lanes U and V). Aconsiderably longer exposure of the blot showed that thisspecies responded only modestly to induction by OXLU(data not shown). There was no detectable signal when RNAfrom the daI81 mutant strain was used (lane W). This resultwas not interpretable, however, because of the minimalsignal obtained with RNA from wild-type cells grown in thepresence of inducer. When asparagine was used as thenitrogen source for growth of a dal80 mutant, little if anyhybridization was observed (lane X). This represents a
VOL. 5, 1985 2285
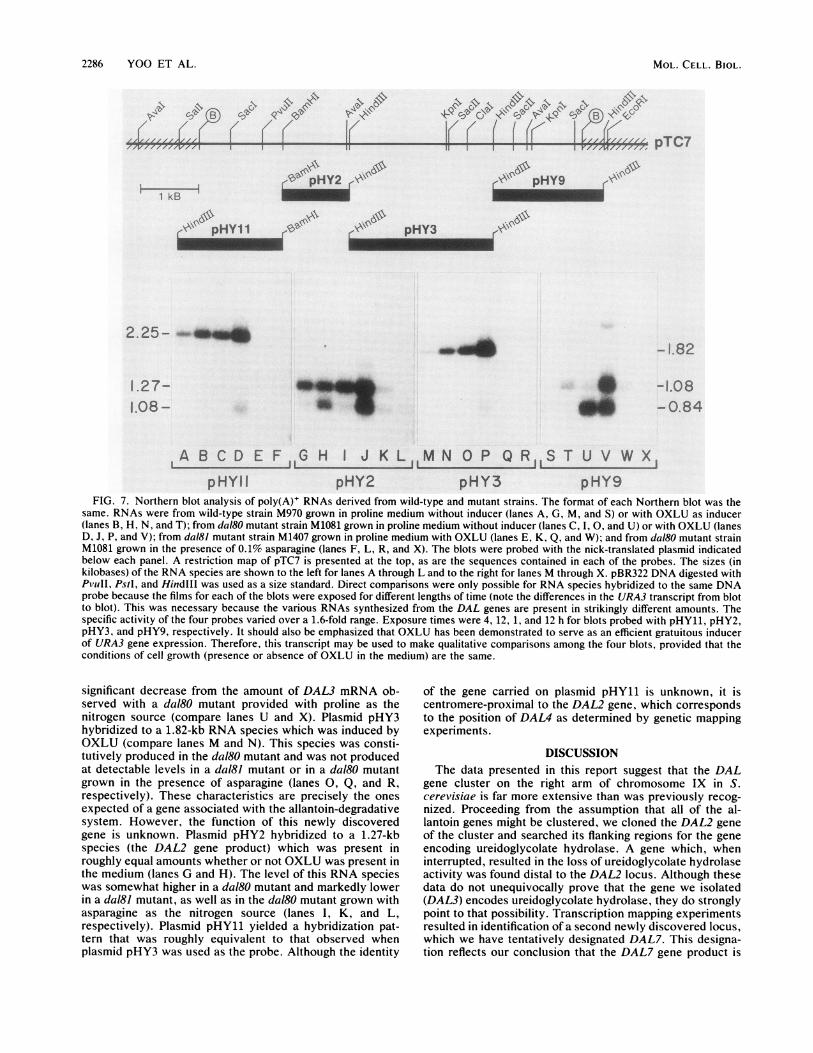
2286 YOO ET AL.
COe 9eA
I; f;- w-=/D,S,,44//2
4A////SfS I
1 kB
H pHY11
I - ,\oA
.".. . ..SpHY9 A\
(---l
-1.82
IA B C D E F,G H
pHYII
I J K LHM N 0 P Q RHS T U V W XipHY2 pHY3 pHY9
FIG. 7. Northern blot analysis of poly(A)+ RNAs derived from wild-type and mutant strains. The format of each Northern blot was thesame. RNAs were from wild-type strain M970 grown in proline medium without inducer (lanes A, G, M, and S) or with OXLU as inducer(lanes B, H, N, and T); from daI80 mutant strain M1081 grown in proline medium without inducer (lanes C, I, 0, and U) or with OXLU (lanesD, J, P, and V); from dal81 mutant strain M1407 grown in proline medium with OXLU (lanes E, K, Q, and W); and from daI80 mutant strainM1081 grown in the presence of 0.1% asparagine (lanes F, L, R, and X). The blots were probed with the nick-translated plasmid indicatedbelow each panel. A restriction map of pTC7 is presented at the top, as are the sequences contained in each of the probes. The sizes (inkilobases) of the RNA species are shown to the left for lanes A through L and to the right for lanes M through X. pBR322 DNA digested withPvuII, PstI, and HindlIl was used as a size standard. Direct comparisons were only possible for RNA species hybridized to the same DNAprobe because the films for each of the blots were exposed for different lengths of time (note the differences in the URA3 transcript from blotto blot). This was necessary because the various RNAs synthesized from the DAL genes are present in strikingly different amounts. Thespecific activity of the four probes varied over a 1.6-fold range. Exposure times were 4, 12, 1, and 12 h for blots probed with pHY11, pHY2,pHY3, and pHY9, respectively. It should also be emphasized that OXLU has been demonstrated to serve as an efficient gratuitous inducerof URA3 gene expression. Therefore, this transcript may be used to make qualitative comparisons among the four blots, provided that theconditions of cell growth (presence or absence of OXLU in the medium) are the same.
significant decrease from the amount of DAL3 mRNA ob-served with a dal80 mutant provided with proline as thenitrogen source (compare lanes U and X). Plasmid pHY3hybridized to a 1.82-kb RNA species which was induced byOXLU (compare lanes M and N). This species was consti-tutively produced in the dal80 mutant and was not producedat detectable levels in a dal8J mutant or in a daI80 mutantgrown in the presence of asparagine (lanes 0, Q, and R,respectively). These characteristics are precisely the onesexpected of a gene associated with the allantoin-degradativesystem. However, the function of this newly discoveredgene is unknown. Plasmid pHY2 hybridized to a 1.27-kbspecies (the DAL2 gene product) which was present inroughly equal amounts whether or not OXLU was present inthe medium (lanes G and H). The level of this RNA specieswas somewhat higher in a dal80 mutant and markedly lowerin a dal8J mutant, as well as in the dal80 mutant grown withasparagine as the nitrogen source (lanes I, K, and L,respectively). Plasmid pHY11 yielded a hybridization pat-tern that was roughly equivalent to that observed whenplasmid pHY3 was used as the probe. Although the identity
of the gene carried on plasmid pHY11 is unknown, it iscentromere-proximal to the DAL2 gene, which correspondsto the position of DAL4 as determined by genetic mappingexperiments.
DISCUSSIONThe data presented in this report suggest that the DAL
gene cluster on the right arm of chromosome IX in S.
cerevisiae is far more extensive than was previously recog-nized. Proceeding from the assumption that all of the al-lantoin genes might be clustered, we cloned the DAL2 geneof the cluster and searched its flanking regions for the geneencoding ureidoglycolate hydrolase. A gene which, wheninterrupted, resulted in the loss of ureidoglycolate hydrolaseactivity was found distal to the DAL2 locus. Although thesedata do not unequivocally prove that the gene we isolated(DAL3) encodes ureidoglycolate hydrolase, they do stronglypoint to that possibility. Transcription mapping experimentsresulted in identification of a second newly discovered locus,which we have tentatively designated DAL7. This designa-tion reflects our conclusion that the DAL7 gene product is
1.27-1.08- J_ -1.08
-0.84
r,\-\\"' pHY3
MOL. CELL. BIOL.
2.25- :----
NIN -IN..Ill \x"\6p

CONTROL OF ALLANTOIN SYSTEM IN S. CEREVISIAE
also associated with the degradation of allantoin. In supportof this conclusion are the observations that expression of thegene was induced by OXLU and subject to nitrogencatabolite repression when the cells were grown in thepresence of a readily used nitrogen source. Expression of thegene is also regulated by the DAL80 and DAL81 geneproducts, as evidenced by the lack of a detectable DAL7transcript in RNA derived from a dal81 mutant and itshigh-level constitutive production in a dal80 mutant. Thesefour characteristics are precisely those observed for DURJ,2gene expression (Genbauffe and Cooper, submitted for pub-lication). The biochemical function carried out by the DAL7gene product remains unknown. Gene interruptions havebeen constructed but have not been particularly illuminatingbecause a null dal7 allele results in slow growth on allantoinrather than a complete loss of the ability to use allantoin asa nitrogeni source. These studies are continuing.The most intriguing aspect of our findings is the unique
genetic organization of the six functionally related genessituated on the right arm of chromosome IX in S. cerevisiae.It is broadly accepted that eucaryotic genes with relatedfunctions are not usually clustered in a manner similar to thatfound in procaryotic organisms. This feature is associatedwith a fundamental difference in their regulation. The pre-dominant regulatory motif in procaryotes is the operon andmulticistronic mRNA. In eucaryotic cells, coordinate orparallel regulation is not accomplished in this way, asreflected by the scattering of functionally related genes. Theallantoin-degradative genes are all individually regulated, asexpected, but yet are situated together. It would be interest-ing to determine whether they were formed through multipleduplications of a single primordial precursor.At present there are contrasting reports about the control
of the allantoin-degrading enzymes. We have repeatedlyreported that allantoin-degradative enzymes are inducible (1,9, 13, 25). Middlehoven, Wiame, and their colleagues, on theother hand, have reported that allantoinase is producedconstitutively (15, 17, 18). The Northern blot experiment(Fig. 7) pointed to a diverse and highly individual behaviorfor each gene. For example, the DAL2 gene appeared to beexpressed constitutively, whereas DAL3 transcripts wereonly observed in dal80 mutant strains. In fact, only theDAL7 gene appeared to show the expected pattern ofexpression. Therefore, it might be concluded either that thegenes are not regulated as was previously suggested on thebasis of enzyme assays or that there is incongruence be-tween the results of the enzyme assays and RNA levels. Thelatter would be expected if the system was regulated in apost-transcriptional manner. The explanation of this paradoxresides both in the strains that were used for the variousexperiments and in the particular genes in question. Lawtherand Cooper presented data which demonstrated the induc-ibility of allantoinase in strain M25 (13). Although theincrease in allantoinase activity was only 7- to 10-fold,compared with a 20- to 50-fold increase for urea amidolyase,inducibility and its kinetic behavior were clearly docu-mented. Lemoine et al. (15) and Middlehoven andHoogkamer-Teniet (17, 18), who reported that allantoinase isproduced constitutively, used strains X1278b and a3962c,respectively. All of the allantoin-degradative enzymes ap-pear to be more inducible in strain M25 than in strain11278b; we have not had an opportunity to conduct exper-iments with strain a3962c. With respect to the question ofcongruence between enzyme and RNA levels, the only datathat may be directly compared are those in Fig. 7 and thoseof Chisholm and Cooper (Table 2 of reference 4).
Ureidoglycolate hydrolase levels were approximately nine-fold higher in a dal80 mutant than in the wild type. DAL3RNA levels behaved similarly (Fig. 7). This relationship mayalso be extended to allantoicase and DAL2 mRNA levels,although in this case less overall difference between thelevels was found between the two strains. In sum, the levelsof enzyme activity and RNA observed are parallel. Thedifferences in the level of induction that were seen in variousstrains remain unexplained. They could possibly derive fromdifferences in (i) the regulatory sequences that flank thestructural genes, (ii) genetic differences in their respectiveregulatory elements, or (iii) differences in the overall geneticbackgrounds that together indirectly generate the observeddifferent responses to the presence of inducer. With properstructural and physiological information, it should be possi-ble for each of these possibilities to be rigorously evaluated.
Finally, we have correlated the drastic decrease in enzymeactivity observed when cells are grown in the presence of arepressive nitrogen source with a similarly dramatic de-crease in the steady-state level of the cognate poly(A)+RNA. In contrast to induction, sensitivity to nitrogencatabolite repression was uniformly observed regardless ofthe strain used, the gene assayed, or the type of assay, i.e.,enzyme or mRNA. This and the dominance of repressionover induction generate the question whether the two typesof regulation are accomplished in the same manner. Thisquestion is currently under investigation.
ACKNOWLEDGMENTSWe thank John Carbon, Louise Clarke, and their colleagues at the
University of California, Santa Barbara, for their generous assistanceduring the initial phases of this work. We thank Roberta Sumrada forthe idea of searching for the ureidoglycolate hydrolase gene in theDAL gene cluster. We also wish to thank Kevin Struhl and RodneyRothstein for providing the plasmid (pUC8-SC2605) and strain(W303-la), respectively, that were used in the gene interruptionstudies. The manuscript was significantly improved by commentsfrom other members of the laboratory.
This work was supported by Public Health Service grantGM-19386 from the National Institutes of Health.
LITERATURE CITED
1. Bossinger, J., and T. G. Cooper. 1976. Execution times ofmacromolecular synthetic processes involved in the inductionof allophanate hydrolase at 15°C. J. Bacteriol. 128:498-501.
2. Buckholz, R. G., and T. G. Cooper. 1983. Oxalurate induction ofmultiple URA3 transcripts in Saccharomyces cerevisiae. Mol.Cell. Biol. 3:1889-1897.
3. Carlson, M., and D. Botstein. 1982. Two differentially regulatedmRNAs with different 5' ends encode secreted and intracellularforms of yeast invertase. Cell 28:145-154.
4. Chisholm, G., and T. G. Cooper. 1982. Isolation and character-ization of mutants that produce the allantoin-degrading enzymesconstitutively in Saccharomyces cerevisiae. Mol. Cell. Biol.2:1088-1095.
5. Choi, K. S., K. W. Lee, and A. H. Roush. 1966. The assay ofyeast ureidoglycolatase. Anal. Biochem. 17:413-422.
6. Clarke, L., and J. Carbon. 1978. Functional expression ofcloned yeast DNA in Escherichia coli: specific complementa-tion of argininosuccinate lyase (argH) mutations. J. Mol. Biol.120:517-532.
7. Cooper, T. G. 1982. Nitrogen metabolism in Saccharomycescerevisiae, p. 39-99. In J. N. Strathern, E. W. Jones, and J.Broach (ed.), The molecular biology of the yeast Saccharomy-ces: metabolism and gene expression. Cold Spring HarborLaboratory, Cold Spring Harbor, N.Y.
8. Cooper, T. G. 1984. Allantoin degradation by Saccharomycescerevisiae-a model system for gene regulation and metabolic
VOL. 5, 1985 2287

2288 YOO ET AL.
integration. Adv. Enzymol. Relat. Areas Mol. Biol. 56:91-139.9. Cooper, T. G., and R. P. Lawther. 1973. Induction of the
allantoin degradative enzymes in Saccharomyces cerevisiae bythe last intermediate of the pathway. Proc. Natl. Acad. Sci.U.S.A. 70:2340-2344.
10. Davis, R. W., D. Botstein, and J. R. Roth. 1980. Advancedbacterial genetics: a manual for genetic engineering, p. 159-176.Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
11. Fink, G. R. 1970. The biochemical genetics of yeast. MethodsEnzymol. 17A:59-78.
12. Fryberg, E. A., K. L. Kindle, N. Davidson, and A. Sodja. 1980.The actin genes of Drosophila: a dispersed multigene family.Cell 19:365-378.
13. Lawther, R. P., and T. G. Cooper. 1975. Kinetics of induced andrepressed enzyme synthesis in Saccharomyces cerev'isiae. J.Bacteriol. 121:1064-1073.
14. Lee, K. W., and A. H. Roush. 1964. Allantoinase assays andtheir application to yeast and soybean allantoinases. Arch.Biochem. Biophys. 108:460-467.
15. Lemoine, Y., E. Dubois, and J.-M. Wiame. 1978. The regulationof urea amidolyase of Saccharomyces cerevisiae. Mating typeinfluence on a constitutivity mutation acting in cis. Mol. Gen.Genet. 166:251-258.
16. Lowry, 0. H., N. J. Rosebrough, A. L. Farr, and R. J. Randall.1951. Protein measurement with the Folin phenol reagent. J.Biol. Chem. 193:265-275.
17. Middlehoven, W. J. 1977. Isolation and characterization ofmethylammonium-resistant mutants of Saccharomyces cereii-siae with relieved nitrogen metabolite repression of al-lantoinase, arginase and ornithine transaminase synthesis. J.Gen. Microbiol. 100:257-269.
18. Middlehoven, W. J., and M. C. Hoogkamer-Teniet. 1981. Re-
pression of catabolic NAD-specific glutamate dehydrogenase ofSaccharomyces cerevisiae by arginine, allantoin and urea.FEMS Microbiol. Lett. 10:307-311.
19. Mortimer, R. K., and D. C. Hawthorne. 1969. Yeast genetics, p.385-460. In A. H. Rose and J. S. Harrison (ed.), The yeasts,vol. 1. Academic Press, Inc., New York.
20. Nasmyth, K. A., and S. I. Reed. 1980. Isolation of genes bycomplementation in yeast: molecular cloning of a cell-cyclegene. Proc. Natl. Acad. Sci. U.S.A. 77:2119-2123.
21. Orr-Weaver, T. L., J. W. Szostak, and R. J. Rothstein. 1981.Yeast transformation: a model system for the study of recom-bination. Proc. Natl. Acad. Sci. U.S.A. 78:6354-6358.
22. Perkins, D. D. 1949. Biochemical mutants in the smut fungusUstilago maydis. Genetics 34:607-626.
23. Rothstein, R. J. 1983. One-step gene disruption in yeast. Meth-ods Enzymol. 101:202-211.
24. Struhl, K., D. T. Stinchcomb, S. Scherer, and R. W. Davis. 1979.High frequency transformation of yeast: autonomous replica-tion of hybrid DNA molecules. Proc. Natl. Acad. Sci. U.S.A.76:1035-1039.
25. Sumrada, R. A., and T. G. Cooper. 1974. Oxaluric acid: anon-metabolizable inducer of the allantoin degradative enzymesin Saccharomyces cerev'isiae. J. Bacteriol. 117:1240-1247.
26. Sumrada, R. A., and T. G. Cooper. 1982. Isolation of the CAR]gene from Saccharomvc es cerevisiae and analysis of its expres-sion. Mol. Cell. Biol. 2:1514-1523.
27. Turoscy, V., and T. G. Cooper. 1982. Pleiotropic control of fiveeucaryotic genes by multiple regulatory elements. J. Bacteriol.151:1237-1246.
28. Winston, F., F. Chumley, and G. R. Fink. 1983. Eviction andtransplacement of mutant genes in yeast. Methods Enzymol.101:211-228.
MOL. CELL. BIOL.