in copyright - non-commercial use permitted rights ...27450/... · peroxynitrite, but it is also...
TRANSCRIPT

Research Collection
Doctoral Thesis
Peroxynitrous acid and vitamin C
Author(s): Kurz, Christophe Robert
Publication Date: 2004
Permanent Link: https://doi.org/10.3929/ethz-a-004834723
Rights / License: In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For moreinformation please consult the Terms of use.
ETH Library

Diss. ETHNr. 15620
Peroxynitrous acid and vitamin C
Abhandlung zur Erlangung des Titels
DOKTOR DER NATURWISSENSCHAFTEN
der
Eidgenössischen Technischen Hochschule Zürich
Vorgelegt von
Christophe R. Kurz
Dipl. Chem. ETH
Geboren am 26. Mai 1972
von Vechingen (BE)
Angenommen auf Antrag von
Prof. Dr. Willem H. Koppenol, Referent
Prof. Dr. Christian Schöneich, Koreferent
Dr. Reinhard Kissner, Koreferent
2004
1

2

To see what everyone has seen
and think what no one has thought.
By Albert Szent-Györgyi
The investigations reported in this thesis have been supported by the ETH and
the Swiss Nationalfonds.
3

4

CONTENTS
1. Danksagung 9
2. Objectives of the thesis 11
3.A Summary 13
B Zusammenfassung 17
References 21
4. Introduction 25
4.1 Peroxynitrite 25
4.1.1 History 25
4.1.2 Properties of peroxynitrite 26
4.1.3 Synthesis of peroxynitrite 29
4.1.4 Physiological relevance of peroxynitrite 33
4.2 Ascorbate 35
4.2.1 History 35
4.2.2 Properties of vitamin C 36
4.2.3 Synthesis of vitamin C 38
4.2.4 Resorption and excretion 41
4.2.5 Physiological relevance of vitamin C 41
4.2.6 Vitamin C as a scavenger 43
4.3 References 45
5. Material and Methods 53
5.1. Reagents 53
5.2. Stopped-flow spectrophotometrie studies 54
5.2.1 Equipment 54
5.2.2 Electrochemistry 54
5.2.3 Monohydroascorbate 55
5.2.4 Monohydroascorbate and carbon dioxide 55
5

5.3 Cyclic voltammetry 55
5.4 EPR 56
5.5 Ion chromatography 56
5.6 Simulation by Feldberg method 57
5.7 Simulation ofkinetic reactions 57
5.8 References 58
6. On the Chemical and Electrochemical One-Electron Reduction of
Peroxynitrous acid 61
Abstract 61
Results 62
Discussion 64
Appendix 69
References 72
Supplemental information 75
7. Rapid scavenging of Peroxynitrous Acid by Monohydroascorbate 77
(Free Radical Biol. Med. 35:1529-37; 2003)
Abstract 77
Results 78
Discussion 84
Speculations as to the structures of the intermediates 90
References 92
8. Mechanism of the Reaction of Peroxynitrite with L-Ascorbate
studied at physiological pH 95
Abstract 95
Results 95
Discussion 101
References 107
6

9. The Reaction of Peroxynitrite with Carbon Dioxide and Vitamin C..
109
Abstract 109
Results 109
Discussion 113
References 121
10. Curriculum vitae 123
7

8

1. DANKSAGUNG
Der Dank gilt vielen Leuten. Sie alle haben mich während meiner Arbeit
unterstützt, sei dies in Ratschlägen, aufstellenden Worten oder gar
Lösungsvorschlägen.
Herzlich bedanken möchte ich mich bei Herrn Prof. Dr. W. H. Koppenol
welcher mir die Doktorarbeit ermöglichte und mir den Freiraum gab meinen
Forscherdrang auszuleben.
Vielen Dank auch an Herrn Prof. Dr. Christian Schöneich welcher sich für die
Übernahme des Koreferates bereiterklärte.
Herzlichen Dank auch an Herrn Dr. Reinhard Kissner, der mich während der
ganzen Dissertationszeit unterstützt hat und stets geduldig bereit war, meine
vielen Fragen zu Beantworten.
Ebenfalls Bedanken möchte ich mich bei Herrn Dr. Thomas Nauser und Frau
PD Dr. Susanna Herold welche mir mit ihrem Wissen helfend zur Seite standen.
Danke auch an alle Kollegen und Kolleginnen der Gruppe Koppenol und
Herold, welche mich unterstützten, und ich viele lustige Stunden verbringen
konnte: Alina, Anastasia, Changyuan, Francesca, Gabi, Jolanta, Martin, Patrick,
Peter, Renate, Shiva, Xiuqiong.
Zum Schluss möchte ich mich bei den vielen Heinzelmännchen bedanken, die
das Chemiegebäude unterhalten. Sei es das Putzpersonal, die Schalterdamen, der
Hausdienst oder die Mensatruppe. Allen sei hier gedankt.
9

10

2. OBJECTIVES OF THE THESIS
Peroxynitrite is a potent cytotoxic species produced in vivo. It is known to
oxidise and nitrate proteins, lipids and DNA in vitro. One crucial parameter in
this context is the reduction potential of peroxynitrous acid, which appears to be
the more reactive form of peroxynitrite. The reduction potential allows to
estimate the ability of peroxynitrous acid to react with other compounds which,
in turn, permits to predict which compounds would be good scavengers of
peroxynitrite. Therefore we investigated the reduction potential of peroxynitrous
acid.
Another objective was to determine whether monohydroascorbate, a
physiological antioxidant, protects proteins and lipids against peroxynitrite.
Monohydroascorbate is considered to be a very efficient scavenger of free
radicals. It has been that monohydroascorbate protects very well against
peroxynitrite, but it is also known that the rate constant of the reaction between
peroxynitrous acid and ascorbate is very low (Bartlett, D.; Church, D. F. ;
Bounds, P. L.; Koppenol, W. H., The kinetics of oxidation of L-ascorbic acid by
peroxynitrite, Free Radical Biol. Med. 18:85-92; 1995, Squadrito, G. L.; Jin, X.;
Pryor,W. A., Stopped-flow kinetics of the reaction of ascorbic acid with
peroxynitrite, Arch. Biochem. Biophys. 322:53-59; 1995). These observations
are in contradiction to each other, and for this reason we reinvestigated the
reaction of peroxynitrous acid with monohydroascorbate.
11

12

3.A SUMMARY
Micromolar concentrations of both nitrogen monoxide and superoxide are
produced by activated macrophages during the immune response, which very
likely leads to formation of peroxynitrite given the diffusion-controlled rate of
1.6xl010M *s *
[1]. Among the reactive oxygen species, peroxynitrite and its
conjugated acid are of biological importance. Peroxynitrous acid and/or its
deprotonated form oxidizes thiols [2-5], nitrates tyrosines [6-11] and tryptophan
[12,13], and initiates lipid peroxidation [14,15]. Both one-electron oxidations
and oxygen-transfer have been reported [16-18].
The reduction potential of the ONOOH, H+ / N02", H20 couple is a crucial
parameter to predict whether peroxynitrous acid can oxidize a compound.
Values of 2.14 V and 2.0 V were published by Merényi and Lind [19], and
Koppenol and Kissner [20], respectively, and refer to standard conditions. These
values are based on determinations and estimates of the standard Gibbs energy
of formation of peroxynitrous acid. To our knowledge, no direct electrochemical
determination has been reported in the literature. The oxidation of the
peroxynitrite anion has been investigated electrochemically and a value of
0.51±0.02 V has been reported for E°(ONOOVONOO ) [21].
Therefore, we investigated in one part of this thesis the reduction potential of
peroxynitrous acid. Peroxynitrous acid was reduced by cathodic linear sweep
voltammetry at a gold electrode and by iodide at pH 3.2 to 5.6. The cathodic
reduction wave was identified by measuring its decay in time, which was the
same as with observations by optical spectroscopy. The iodide oxidation was
followed by optical measurement of the triiodide formation. Both reductions
show one-electron stochiometry, with the product naa = 0.23 ± 0.04 and an
iodine yield of around 0.5 equivalents per equivalent of peroxynitrous acid. The
13

voltammetric reduction was irreversible up to scan rates of 80 Vs_1. Both
reductions were pH independent in the range studied. The voltammetric
reduction is most likely an irreversible elemental reaction followed by a
chemical decay which cannot be observed directly. Because of the pH
independence, we conclude that both reductions have a common short-lived
intermediate, namely [HOONO]*-. This type of radical ion has been identified in
the reduction of nitrite with hydrated electrons in pulse radiolysis experiments.
The pH independence of the one-electron reductions also suggests that estimates
of the one-electron standard reduction potential of peroxynitrous acid are in
error. See chapter 6.
A lot of efforts have been done to identify a suitable peroxynitrite scavenger in
vivo.
Monohydroascorbate has already been investigated in 1995 by Bartlett et al. [22]
and Squadrito et al. [23]. They published a rate constant for the reaction of
235 M'V1 at 25°C. From the observation of ascorbyl radicals it was concluded
that a one-electron oxidation had taken place.
However many experiments show that the protective ability of
monohydroascorbate against peroxynitrite-mediated damage is much higher
than could be expected from this rate constant [24-27]. The published small rate
constant of the reaction between peroxynitrite and monohydroascorbate is in
contradiction to these observations. Therefore, we reinvestigated in one part of
this thesis the reaction between peroxynitrite and monohydroascorbate.
Data recorded from the reaction between monohydroascorbate and peroxynitrite
suggest a complex mechanism. The peroxynitrite anion does not react with
monohydroascorbate. For the initial reaction of peroxynitrous acid with
monohydroascorbate, a second-order rate constant of kj of 5 x 105±1 M_1s_1 and
Oil 1
an equilibrium constant K of 1 x 10 M were determined. We observed three
intermediates of which the third has an absorption maximum at 345 nm and a
14

pKa of 7.6 ± 0.2. Both the protonated and the deprotonated forms of this third
intermediate can react with a second monohydroascorbate to form nitrite,
monohydroascorbate, and dehydroascorbate. In addition, there is a
monohydroascorbate-independent decay path that yields nitrate and
monohydroascorbate. Therefore, at low monohydroascorbate concentrations, the
third intermediate decays to nitrate and monohydroascorbate, while at
monohydroascorbate concentrations greater than 4 mM, this third intermediate
reacts with a second monohydroascorbate to form nitrite, dehydroascorbate, and
monohydroascorbate.
EPR experiments indicate that the yield of the ascorbyl radical is 0.24 % relative
to the initial peroxynitrous acid concentration, and that this small amount of
ascorbyl radicals is formed concomitantly with the decrease of the absorption at
345 nm. Thus, the ascorbyl radical is not a primary reaction product. Under the
conditions of these experiments, no homolysis of peroxynitrous acid to nitrogen
dioxide and hydroxyl radical is observed. See chapter 7 and 8.
The more reactive peroxynitrous acid is not the most toxic form of peroxynitrite
in vivo. Even more toxic is the peroxynitrite anion when it reacts with carbon
dioxide to form l-carboxylato-2-nitrosodioxidane at a rate constant of
3 x K^lVrV1 at 25°C [28,29]. It would be useful to know whether the rate
constant of the reaction between peroxynitrous acid and monohydroascorbate is
fast enough to prevent the reaction of the peroxynitrite anion with carbon
dioxide or whether it even interacts with the products of the reaction with carbon
dioxide.
In the presence of carbon dioxide, peroxynitrite reacts catalytically with
monohydroascorbate at a very fast rate. Low monohydroascorbate
concentrations prevent the formation of the carbonate radical anion and nitrogen
dioxide, the intermediates produced from the reaction of peroxynitrite with
carbon dioxide. Moreover, at low monohydroascorbate concentrations only
15

small amounts of ascorbyl radicals, relative to the initial monohydroascorbate
concentration, are produced. If the monohydroascorbate concentration is higher
than that of peroxynitrite, the ascorbyl radical concentration increases. Under
these conditions, an alternative pathway, which involves the reaction with a
second monohydroascorbate molecule, is observed. Stopped-flow experiments
indicate that at pH 7, the reaction between peroxynitrous acid and
monohydroascorbate competes with the reaction between peroxynitrite anion,
monohydroascorbate, and carbon dioxide. At pH 6, the main pathway involves
the reaction of monohydroascorbate with peroxynitrous acid. See chapter 8.
A role as peroxynitrite scavenger under physiological conditions seems very
likely.
16

3.B ZUSAMMENFASSUNG
Aktivierte Makrophagen produzieren mikromolare Konzentrationen von
Stickstoffmonoxid und Superoxid, welche mit einer diffusionskontrollierten
Geschwindigkeit von
I.öxIO^mV1 miteinander reagieren [1]. Unter den reaktiven
Sauerstoffmetaboliten ist Peroxynitrit und seine Säure von biologischem
Interesse. Persalpetrige Säure und/oder ihre deprotonierte Form oxidieren Thiole
[2-5], nitrieren Tyrosine [6-11] und tryptophane [12,13], und lösen
Lipidperoxydation aus [14,15]. Es wurden Einelektronen- und Sauerstofftransfer
Beobachtet [16-18].
Das Reduktionspotential ist ein kritischer Parameter für die Voraussage, ob eine
andere Substanz oxidiert werden kann oder nicht. Für persalpetrige Säure
wurden Werte von 2.14 V und 2.0 V durch Merényi und Lind [19], respektive
durch Koppenol und Kissner [20] publiziert. Die Berechnungen beruhen auf
Standardbedingungen. Beide Werte basieren auf gemessenen und geschätzten
Werten für die freie Bildungsenergie von Persalpetriger Säure. Nach unseren
Kenntnissen wurde keine direkte Bestimmung publiziert. Die Oxidation von
Peroxynitritanion wurde elektrochemisch von Amatore et al. untersucht und er
publizierte einen Wert von 0.51 ± 0.02 V für E°(ON0070NOO ) [21].
Deswegen untersuchten wir elektrochemisch das Reduktionspotential der
Persalpetrigen Säure. Persalpetrige Säure wurde durch Iodid, pH 3.2 und 5.6,
und kathodische Linearvorschub-Voltammetrie, an einer Goldelektrode bei
pH 3.2 und 5.6, reduziert. Die kathodische Reduktionswelle wurde durch ihre
Zerfallszeit identifiziert, welche identisch mit der am UV-Vis
Spektrophotometer gemessenen ist. Die Iodidoxidation wurde optisch, durch die
Messung von Triiodid, verfolgt. Beide Reduktionen zeigen eine Ein-Elektron
Stöchiometrie mit dem Produkt naa = 0.23 + 0.04 und einer Iod Ausbeute von
17

etwa 0.5 Equivalenten pro Equivalent Persalpetriger Säure. Die voltammetrische
Reduktion war bis zu einer Vorschubgeschwindigkeit von 80 Vs"1 irreversibel.
Beide Reduktionen waren im gemessenen pH-Bereich pH-unabhängig. Bei der
gemessenen voltammetrischen Reduktion handelt es sich höchstwahrscheinlich
um eine irreversible Elementarreaktion, welche von einem chemischen Zerfall
gefolgt ist. Der Zerfall konnte nicht direkt beobachtet werden. Wegen der pH-
Unabhängigkeit schlussfolgern wir, dass beide Reduktionen das gleiche,
kurzlebige Zwischenprodukt, nämlich [ONOO]"2 /[ONOOH]" ,besitzen. Ein
analoges Radikal wurde in der Pulsradiolyse, durch die Reaktion von Nitrit mit
hydrierten Elektronen, identifiziert. Die gefundene pH-Unabhängigkeit weist
darauf hin, dass die berechneten Einelektronen-Standardreduktionspotentiale
fehlerhaft sind.
Grosse Anstrengungen wurden unternommen um einen passenden Peroxynitrit
Scavenger für die in vivo Anwendung zu finden.
Ascorbat wurde bereits im Jahre 1995 durch Bartlett el al. [22] und Squadritto et
al. [23] untersucht. Beide publizierten eine Geschwindigkeitskonstante von
235 M_1s_1 für 25°C. Die Beobachtung von Ascorbylradikalen wurde als eine
Ein-Elektron Oxidations-Reaktion gedeutet.
Jedoch viele Experimente zeigen dass Ascorbat viel besser vor Peroxynitrit
verursachten Schäden schützt, als durch die Geschwindigkeitskonstante erwartet
werden könnte [24-27]. Die publizierte Geschwindigkeitskonstante für die
Reaktion zwischen Peroxynitrit und Ascorbat von 235 M_1s_1 ist sogar im
Widerspruch zu den experimentellen Beobachtungen. Deswegen untersuchten
wir erneut die Reaktion zwischen Peroxynitrit und Vitamin C.
Die gemessenen Daten der Reaktion zwischen Peroxynitrit und Ascorbat deuten
auf einen komplexen Mechanismus. Peroxynitritanion reagiert nicht mit
Ascorbat. Für die Initialreaktion zwischen Persalpetriger Säure und Ascorbat
18

wurde eine zweite-Ordnungs Geschwindigkeitskonstante kj von
5 x 105±1 M^V1, mit einer Gleichgewichtskonstante K von 1 x 103±1 M^V1
bestimmt. Wir beobachteten drei Zwischenprodukte, wovon das Dritte ein
Absorbtionsmaximum bei 345 nm und einen pKa von 7.6 + 0.2 besitzt. Beide,
die protonierte und deprotonierte Form, können mit einem zweiten
Ascorbatmolekül reagieren und ergeben schlussendlich Nitrit, Ascorbat und
Dehydroascorbat. Zusätzlich existiert ein Ascorbat unabhängiger Zerfall,
welcher Nitrat und Ascorbat produziert. Deswegen zerfällt das dritte
Zwischenprodukt bei tiefen Ascorbat Konzentrationen zu Nitrat und Ascorbat
während bei Ascorbat Konzentrationen grösser als 4 mM, das Zwischenprodukt
mit einem zweiten Ascorbat reagiert und Nitrit, Ascorbat und Dehydroascorbat
ergibt.
EPR Experimente zeigen eine relative Ascorbylradikal Ausbeute von 0.24 %
gegenüber der ursprünglich eingesetzten Peroxynitrit Konzentration. Diese
kleine Menge Ascorbylradikal wird gleichzeitig mit der Abnahme der 345 nm
Absorbtion gebildet. Daher kann das Ascorbylradikal nicht das primäre
Reaktionsprodukt sein. Unter den experimentellen Bedingungen konnte keine
Homolyse von Persalpetriger Säure zu Stickstoffmonoxid und Hydroxylradikal
beobachtet werden. Siehe Kapitel 7 und 8.
Die reaktivere Form, Persalpetrige Säure, ist nicht die gefährlichste Form von
Peroxynitrit in vivo. Die viel gefährlichere Form von Peroxynitrite ist das
Peroxynitritanion wenn es mit Kohlendioxid reagiert und das schädliche 1-
carboxylato-2-nitrosodioxidane mit einer Geschwindigkeitskonstante von
3 x 104 M'V1 bei 25°C bildet [28,29]. Es wäre sehr hilfreich zu wissen, ob die
Geschwindigkeitskonstante der Reaktion zwischen Persalpetriger Säure und
Ascorbat schnell genug ist, um die Reaktion von Peroxynitritanion mit
Kohlendioxid zu verhindern oder ob Ascorbat sogar selber mit den Produkten
der Reaktion mit Kohlendioxid interagiert.
19

In Anwesenheit von Kohlendioxid reagiert Peroxynitrit in einer sehr schnellen
katalytischen Reaktion mit Ascorbat. Tiefe Ascorbat Konzentrationen
vermeiden bereits die Bildung von Carbonatradikalanion und Stickstoffdioxid,
Zerfallsprodukte der Reaktion zwischen Peroxynitritanion und Kohlendioxid.
Ausserdem werden bei tiefen Ascorbat Konzentrationen (kleiner als die
Peroxynitrit Konzentration) nur wenige Ascorbylradikale, relativ zur Ascorbat
Konzentration, gebildet. Ist die Ascorbat Konzentration grösser als diejenige
von Peroxynitrit, steigt die Ascorbylradikal Konzentration. Unter diesen
Bedingungen wird ein zweiter Reaktionsweg sichtbar, welcher die Reaktion mit
einem zweiten Ascorbat Molekül beinhaltet. Stopped-Flow Experimente bei
pH 7 weisen darauf hin, dass die Reaktion zwischen Persalpetriger Säure und
Ascorbat mit der Reaktion zwischen Peroxynitritanion, Ascorbat und
Kohlendioxid konkurriert. Bei pH 6 kann die Hauptreaktion bereits der Reaktion
von Persalpetriger Säure mit Ascorbat zugewiesen werden. Siehe Kapitel 9.
Ascorbat als Peroxynitrit Scavenger unter physiologischen Bedingungen scheint
sehr wahrscheinlich.
20

REFERENCES
[1] Nauser, T.; Koppenol, W. H. The rate constant of the reaction of
superoxide with nitrogen monoxide: Approaching the diffusion limit. J.
Phys. Chem. A 106:4084-4086; 2002.
[2] Koppenol, W. H.; Moreno, J. J.; Pryor, W. A.; Ischiropoulos, H.;
Beckman, J. S. Peroxynitrite, a cloaked oxidant formed by nitric oxide
and superoxide. Chem. Res. Toxicol. 5:834-842; 1992.
[3] Radi, R.; Beckman, J. S.; Bush, K. M.; Freeman, B. A. Peroxynitriteoxidation of sulfhydryls. J. Biol. Chem. 266:4244-4250; 1991.
[4] Gatti, R. M.; Radi, R.; Augusto, O. Peroxynitrite-mediated oxidation of
albumin to the protein- thiyl free radical. FEBSLett. 348:287-290; 1994.
[5] Kalyanaraman, B.; Karoui, H.; Singh, R. J.; Felix, C. C. Detection of thiylradical adducts formed during hydroxyl radical- and peroxynitrite-mediated oxidation of thiols - A high resolution ESR spin-trapping study
atQ-band(35 GRz).Anal. Biochem. 241:75-81; 1996.
[6] Ischiropoulos, H.; Zhu, L.; Chen, J.; Tsai, M.; Martin, J. C; Smith, CD.;
Beckman, J. S. Peroxynitrite-mediated tyrosine nitration catalyzed by
superoxide dismutase. Arch. Biochem. Biophys. 298:431-437; 1992.
[7] Ramezanian, M. S.; Padmaja, S.; Koppenol, W. H. Hydroxylation and
nitration of phenolic compounds by peroxynitrite. Chem. Res. Toxicol.
9:232-240; 1996.
[8] Beckman, J. S. Oxidative damage and tyrosine nitration from
peroxynitrite. Chem. Res. Toxicol. 9:836-844; 1996.
[9] Gow, A.; Duran, D.; Thorn, S. R.; Ischiropoulos, H. Carbon dioxide
enhancement of peroxynitrite-mediated protein tyrosine nitration. Arch.
Biochem. Biophys. 333:42-48; 1996.
[10] Lymar, S. V.; Jiang, Q.; Hurst, J. K. Mechanism of carbon dioxide-
catalyzed oxidation of tyrosine by peroxynitrite. Biochemistry 35:7855-
7861; 1996.
[11] Van der Vliet, A.; Eiserich, J. P.; O'Neill, C. A.; Halliwell, B.; Cross, C.
E. Tyrosine modification by reactive nitrogen species: A closer look.
Arch. Biochem. Biophys. 319:341-349; 1995.
[12] Alvarez, B.; Rubbo, H.; Kirk, M.; Barnes, S.; Freeman, B. A.; Radi, R.
Peroxynitrite-dependent tryptophan nitration. Chem. Res. Toxicol. 9:390-
396; 1996.
21

[13] Padmaja, S.; Ramezanian, M. S.; Bounds, P. L.; Koppenol, W. H.
Reaction of peroxynitrite with L-tryptophan. Redox. Rep. 2:173-177;1996.
[14] Radi, R.; Beckman, J. S.; Bush, K. M.; Freeman, B. A. Peroxynitrite-induced membrane lipid peroxidation: The cytotoxic potential of
superoxide and nitric oxide. Arch. Biochem. Biophys. 288:481-487; 1991.
[15] Darley-Usmar, V. M.; Hogg, N.; O'Leary, V. J.; Wilson, M. T.; Moncada,S. The simultaneous generation of superoxide and nitric oxide can initiate
lipid peroxidation in human low density lipoprotein. Free Radical Res.
Commun. 17:9-20; 1992.
[16] Al-Ajlouni, A.; Gould, E. S. Electron transfer. 133. Copper catalysis in
the sulfite reduction of peroxynitrite. Inorg. Chem. 36:362-365; 1997.
[17] Masumoto, H.; Kissner, R.; Koppenol, W. H.; Sies, H. Kinetic study of
the reaction of ebselen with peroxynitrite. FEBS Lett. 398:179-182; 1996.
[18] Maurer, P.; Thomas, C. F.; Kissner, R.; Rüegger, H.; Greter, O.;
Röthlisberger, U.; Koppenol, W. H. Oxidation of nitrite by peroxynitrousacid. J. Phys. Chem. A 107:1763-1769; 2003.
[19] Merényi, G.; Lind, J. Thermodynamics of peroxynitrite and its C02-adduct. Chem. Res. Toxicol. 10:1216-1220; 1997.
[20] Koppenol, W. H.; Kissner, R. Can ONOOH undergo homolysis. Chem.
Res. Toxicol. 11:87-90; 1998.
[21] Amatore, C; Arbault, S.; Bruce, D.; de Oliveira, P.; Erard, M.;
Vuillaume, M. Characterization of the electrochemical oxidation of
peroxynitrite: Relevance to oxidative stress bursts measured at the singlecell level. Chem. Eur. J. 7:4171; 2001.
[22] Bartlett, D.; Church, D. F.; Bounds, P. L.; Koppenol, W. H. The kinetics
of the oxidation of L-ascorbic acid by peroxynitrite. Free Radical Biol.
Med. 18:85-92; 1995.
[23] Squadrito, G. L.; Jin, X.; Pryor, W. A. Stopped-flow kinetic study of the
reaction of ascorbic acid with peroxynitrite. Arch. Biochem. Biophys.322:53-59; 1995.
[24] Whiteman, M.; Halliwell, B. Protection against peroxynitrite-dependent
tyrosine nitration and al-antiproteinase inactivation by ascorbic acid. A
comparison with other biological antioxidants. Free Radical Res. 25:275-
283; 1996.
22

[25] Lee, J.; Hunt, J. A.; Groves, J. T. Rapid decomposition of peroxynitrite by
manganese porphyrin-antioxidant redox couples. Bioorg. Med. Chem.
Lett. 7:2913-2918; 1997.
[26] Balavoine, G. G. A.; Geletii, Y. V. Peroxynitrite scavenging by different
antioxidants. Part I: Convenient assay. Nitric Oxide: Biol. Chem. 3:40-54;1999.
[27] Kirsch, M.; de Groot, H. Ascorbate is a potent antioxidant against
peroxynitrite-induced oxidation reactions - Evidence that ascorbate acts
by re-reducing substrate radicals produced by peroxynitrite. J. Biol.
Chem. 275:16702-16708; 2000.
[28] Lymar, S. V.; Hurst, J. K. Rapid reaction between peroxonitrite ion and
carbon dioxide: Implications for biological acitivity. J. Am. Chem. Soc.
117:8867-8868; 1995.
[29] Arteel, G. E.; Briviba, K.; Sies, H. Protection against peroxynitrite. FEBS
Letters 445:226-230; 1999.
23

24

4. INTRODUCTION
4,1 Peroxynitrite
4.1.1 HistoryN O Peroxynitrite was mentioned for the first time in 1901 by Bayer
O O et al. [1]. They discovered that a mixture of nitrous acid and
t/ hydrogen peroxide had unusual strong oxidizing properties. In
1907 Raschig proposed for the first time the formula HN04 for "Übersalpetrige
Säure" [2] which was used until Gleu and Roell published in 1929 the correct
formula ONOOH [3]. In 1952 Halfpenny and Robinson suggested, based on
hydroxylated and nitrated products, that peroxynitrous acid undergoes homolytic
fission to give hydroxyl radicals and nitrogen dioxide [4]. In 1954 Anbar and
Taube performed isotopic labelling studies with hydrogen peroxide and nitrous
acid at different pH. At high nitrite concentrations they found a quantitative
transfer of one oxygen to nitrite and nitrate, which is not expected for partial
homolysis [5]. Several years later, in 1968, Hughes and Nicklin claimed that
their results did not confirm the theory of peroxynitrite homolysis as well [6].
The lack of interest in peroxynitrite is reflected in the fact that only ca. 40 papers
on peroxynitrite had appeared until 1990. The history of peroxynitrite, up to
1992, was reviewed by Edwards et al. [7].
In 1990 it was postulated that peroxynitrite could be biologically relevant.
Beckman et al.[8] proposed for the first time that peroxynitrite can be produced
in vivo by the reaction of nitrogen monoxide with superoxide. The rate constant
of this reaction has been recently determined to be 1.6 x lO^M^s-1 [9]. This
rate constant is 7 times higher than the superoxide dismutase-catalysed
dismutation of superoxide (2.3 x 109 M *s *; pH-, buffer- and ionic strength
dependent) [10]. It leads to the conclusion that, in vivo, most superoxide could
react with nitrogen monoxide to form peroxynitrite whenever superoxide
dismutase is not present at concentrations 7 times higher then that of nitrogen
25

monoxide. Nitrogen monoxide concentrations are usually below 1 uM. Higher
concentrations are found at inflammatory and where nitrogen monoxide is used
as neurotransmitter. Indications that peroxynitrite is formed in vivo by the
reaction of nitrogen monoxide with superoxide are that superoxide dismutase
extends the biological lifetime of nitrogen monoxide by removing superoxide,
and that a decreased nitration of tyrosine is observed in the presence of
superoxide dismutase [11]. Nitrotyrosine is considered to be marker for
peroxynitrite in vivo. But peroxynitrite is not the only substrate which nitrates
tyrosine. Nitrogen dioxide can also nitrate proteins.
Beckman et al. concluded in 1990 from some scavenger studies with
peroxynitrite that 'a strong oxidant with reactivity similar to hydroxyl radical'
was formed [8]. Since 1952 it is being debated whether peroxynitrous acid can
homolyse to nitrogen dioxide and the hydroxyl radical [12-15] or not [16-19].
However, this question is more of academic interest than of relevance for
biological studies.
Peroxynitrite formed in vivo is more rapidly consumed by reactions with
carbon dioxide, thiols and metalloproteins than it decays via the isomerisation
pathway [20]. The rate constant for the reaction of peroxynitrite with carbon
dioxide is 5.8 ± 0.2 xlO4 jvrV1 at 37°C [21]. This rate constant, combined with
an in vivo concentration of about 1 mM of carbon dioxide, results in a
peroxynitrite half live of approximately 12 ms. Further information about
l-carboxylato-2-nitrosodioxidane and its physiological relevance is given in
chapter 4.1.4.
4.1.2 Properties of peroxynitrite
The pKa of peroxynitrous acid depends on the kind and concentration of the
buffer. Values in the range of 6.5 (1 mM phosphate) to 8.6 (0.1 M borate) have
been found [22]. The high pKa of peroxynitrite found in borate buffer can be
explained that borate is a Lewis acid. For diluted phosphate buffer a pKa value
26

of 6.8 is mostly used in the literature. Further, the decay of peroxynitrite is
strongly influenced by temperature and the presence of transition metals which
catalyse the isomerisation of peroxynitrite. However, in aqueous alkaline
solutions, peroxynitrite anion is stable for many weeks if it is kept frozen in the
dark [23]. There are a couple of possible decay pathways of peroxynitrite. The
term "isomerisation" is used for the rearrangement of peroxynitrous acid to
nitrate (reaction1.3), "homolysis" is used for reaction (1.4), "dimerisation" for
reaction (1.5) and "decay" for any disappearance.
The following table shows
and decay of peroxynitrite:
NO* + Of
ONOO- + H+
HOONO
HOONO
HOONO + ONOO-
[HOONO/ONOO]-
2 ONOO"
ONOO"
u 4.5xl09M1s1 [24]
k5 2.5xl05M~y1 [17]
t.5 25 s"1 [17]
k6 0.25 s"1 [17]
k7 2xl04M~y1 [17]
hv [22]
ki î.ôxio^ivrV1 [9]
k-i 1.7x10 V1 [25]
k2 lxio^ivrV1 [26]
k-2 1.6x10V1 [22]
k3 1.2 s"1 [22]
k4 1x102 s"1 [16]
Table 1: Some rate constants and properties of peroxynitrite.
27
some reaction pathways of the formation, equilibria
. \ - ONOO (1-1)
k2
k-2
k-4
k-5
HOONO (1-2)
^— N07+H+ (1.3)
k4_*OH + N02 (1-4)
[HOONO/ONOO]- (1.5)
k^» 02 + 2N02 (1-6)
^2— 02 + 2N02 (1-7)
^— N03- (1.8)

From quantum mechanical calculations the bond order between the nitrogen and
the first peroxide oxygen is approximately 1.5 [27,28]. This bond order gives the
possibility of the existence of two different structures of peroxynitrite anion.
Until now only the eis structure of peroxynitrite was found spectroscopicaly
[27,29]. Ab initio quantum chemical calculations showed that the eis isomer is
about 12 kJ/mol more stable than the trans isomer and that a barrier of
88-100 kJ/mol limits the isomerisation between the eis and the trans form
[27,28].
N-O^ -N-0
O
trans-peroxynitrite cis-peroxynitriteThe concentration of peroxynitrite is usually determined by UV/Vis-
spectroscopy. The peroxynitrite anion has a characteristic absorption with a
maximum at 302 nm with an extinction coefficient of 1705 M_1cm_1[30].
Peroxynitrous acid has an absorption maximum at 345 nm with a smaller
extinction coefficient of 80 M^cm"1 and an absorption shoulder at 250 nm with
an extinction coefficient of 600 M^cm"1 [31].
28

1800
1600
1400
1200
§ 1000
§ 800
600
400
200
0
w
1 »
1 <
*- U! »
/
/ \
i i»
1 ^
,: :,
1 v
.—^ \
-^v *
-^-—"^ ^
-^ ^
^: **i— *».
250 300 350
A,/nm
400 450
Fig. 1. Normalized spectra of 500 uM peroxynitrite (dotted line) and peroxynitrous acid (solid
line). The spectra were recorded by R. Kissner (ETH Zürich)
4.1.3 Synthesis of peroxynitrite
Peroxynitrite can be synthesised in a few different ways and its purity depends
on the respective method. Several syntheses are described in Gmelin[23] and in
Methods in Enzymology [32]. The most common ones are briefly described
below.
Biosynthesis
Peroxynitrite is formed if both reactants, superoxide and nitrogen monoxide, are
present simultaneously, for example in endothelial cells, activated macrophages,
neuronal cells, neurophils and smooth muscle cells. To decrease the formation
of peroxynitrite to 50%, superoxide dismutase should be present in the
concentration 7 times higher than that of nitrogen monoxide, (see chapter 4.1.1)
29

Chemical synthesis
Autoxidation ofhydroxylamine in alkaline solution [33]
NH2OH +02 +0H- H202 +NO" + H20
N0-+02 ONOO"
In this simple method oxygen is bubbled through an alkaline solution of
hydroxylamine, which leads to the formation of peroxynitrite. The typical yield
of peroxynitrite is about 25%, based on the starting concentrations of
hydroxylamine. The by-product, hydrogen peroxide, can be removed by
treatment with manganese dioxide. Unfortunately, nitrite, nitrate and
hydroxylamine remain.
Reaction between ozone and sodium azide [34,35]
N3" + 203 ONOO-+ N20 + 02
The ozonisation of azid ions in an alkaline solution yields peroxynitrite at
concentrations of up to 80 mM and the solution is free from hydrogen peroxide.
The disadvantage of this method is that the solution contains unreacted azide.
Reaction between hydrogen peroxide and alkyl nitrites [36,37]
H202 + OH" + RONO ONOO" + ROH + H20
The reaction of alkaline hydrogen peroxide with an alkyl nitrite gives
peroxynitrite with a nearly quantitative yield. The synthesis can be carried out
by two different methods.
Method A [37] employs a water-soluble alkylnitrite such as 2-ethoxyethyl
nitrite. However, the solution contains the alcohol at about the same
concentration as peroxynitrite.
Method B [36] is carried out in a two-phase system, containing alkaline
hydrogen peroxide in water and water-insoluble alkyl nitrite. Method B allows
to obtain concentrations of peroxynitrite up to 1 M, and the solution contains
unreacted hydrogen peroxide, nitrite and alkali.
30

Reaction between HN02 and H202 [1,38]
H202 +HN02 H20 + HOONO
HOONO +OH" ONOO"
The easiest and the most widely used method of peroxynitrite production is
based on the nitrosation of H202 in acid solutions, which generates the
peroxynitrous acid which is deprotonated by immediate addition of excess of
alkali. The disadvantage of this method is that nitrate is produced during the
reaction and therefore only contaminated peroxynitrite samples can be obtained.
For the first time this method was modified by Reed et al. [38]. Peroxynitrite
was synthesised in a quenched flow reactor from reagents pushed by a
compressed gas into a T junction. The yield of contaminations dépende on the
mixing temperature and the reaction time. The second modification was applied
by Beckman et al. [39]. Instead of a compressed gas a vacuum was utilised for
reagents mixing, resulting in a final yield of peroxynitrite of about 90 %.
Solid synthesisfrom K02 and NO'[40]
A more complicated method to obtain peroxynitrite is the solid synthesis. Its
advantage is much lower nitrate and nitrite contamination. In this method solid
potassium superoxide is diluted with quartz sand and after flushing the reaction
vessel with argon, nitrogen monoxide is added. The obtained solid peroxynitrite
is mixed with a small amount of manganese dioxide to eliminate hydrogen
peroxide which is produced by the remnants of superoxide upon dissolving in
cooled sodium hydroxide. The solution contains also low concentrations of
nitrite and nitrate.
Synthesis oftetramethylammoniumperoxynitrite in liquid ammonia [41]
In an inert and dry atmosphere tetramethylammonium hydroxide is mixed with
potassium superoxide and shaken under vacuum for three days. Then dry
31

ammonia is added and the tetramethylammonium superoxide, which is
dissolved, is transfered through a filter into another flask. After purification,
tetramethylammonium superoxide is exposed to nitrogen monoxide. Then
ammonia is slowly removed within 2 days (in vacuo). The obtained solid
tetramethylammonium peroxynitrite is of high purity and can be stored at
-70 °C for months.
Peroxynitrite productionfrom SIN-1 [42]
SIN-1 (3-morpholino-sydnonimine) is used for production of very small
concentrations of peroxynitrite over longer time. Initially SIN-1 is oxidised to
SIN-1"+ and generates superoxide from 02. In the next step SIN-1"+ releases a
nitrogen monoxide molecule and becomes inactive. The produced superoxide
and nitrogen monoxide react with each other forming peroxynitrite. It has to be
taken into account that superoxide and nitrogen monoxide are produced
consecutively and therefore they have also the possibility to react with other
compounds instead forming peroxynitrite.
0^ 0-
N
N
N
xcr^NH
SIN-1
O
NO*
N-+\
N
CH
XCN
Scheme 1: Peroxynitrite production by SIN-1SIN-1'
• +SIN-1C
32

4.1.4 Physiological relevancy of peroxynitrite
Peroxynitrite has been proposed to be produced in several cell types, such as
endothelial cells[43], macrophages[44], neuronal cells[45], neutrophils[46] and
smooth muscle cells[47].
At physiological pH peroxynitrite and its protonated form are present at a
ratio of 4 to 1 and therefore isomerise at 37 °C with a rate constant of 1.1 s"1
[22,48].
The unstable protonated form (ONOOH) is most probably more cytotoxic
than the stable, anionic form. But the peroxynitrite anion reacts with carbon
dioxide (physiological concentration of the latter compound is about 1 mM in
blood plasma) with a rate constant of 3 x 104 M'V1 at 25°C, [49] and produces
l-carboxylato-2-nitrosodioxidane(ONOOC02 ) which is proposed to be mainly
responsible for the toxicity of peroxynitrite. In the absence of other reactants,
ONOOC02 undergoes homolysis to nitrogen dioxide and trioxocarbonate(#l-)
radicals with a yield of 4 - 30 % [12,50-52] and isomerises to nitrate and carbon
dioxide.
The cytotoxic reactions of peroxynitrite, e.g. triggering of DNA strand
breakage, inhibition of lipid peroxidation, direct inhibition of mitochondrial
respiratory chain enzymes and other oxidative protein modifications show how
dangerous peroxynitrite in vivo can be.
The mentioned cytotoxic pathways of peroxynitrite show how important it is
to find a substance to prevent these reactions.
33

34

4.2 Vitamin C
HO L-Ascorbic acid (Vitamin C, 2-oxo-L-threo-hexono-l,4-
4.2.1 History
lactone-2,3-ene-diol) was named after its ability to preventH^^H^OH
q a serious illness called scurvy, one of the deficiency
diseases known longest to humankind. The name stems
from German (Antiscorbut- Vitamin). Between
1536 (Jacques Cartier) and 1601 (Sir James Lancaster) sailors discovered that a
diet including lemons, sauerkraut and vegetables can prevent this illness [53]. In
1747, James Lind carried out probably the first controlled clinical experiment
proving that lack of a compound in the diet was responsible for scurvy and that
oranges and lemons were the most effective against this disease [53]. In 1907,
Hoist discovered that guinea pigs are susceptible to scurvy what gave scientists
an opportunity to perform animal studies [54]. In 1920, vitamin C was isolated
from lemons by Zilva, but he did not identify this compound [55]. In 1928
Szent-Györgyi isolated vitamin C, ascribed the vitamin property to this
compound and named it hexuronic acid [56].
The structure of vitamin C was clarified in 1933 by Haworth and Hirst [57],
who proposed to rename vitamin C as ascorbic acid to reflect its antiscorbutic
properties. One year later Reichstein and Grüssner [58,59] published a method
of synthesis of vitamin C, which is used until today. In 1937 Sir Walter Norman
Haworth received the Nobel price in chemistry for investigations of
carbohydrates and vitamin C. In the same year Albert Szent-Györgyi Von
Nagyrapolt received the Nobel price in medicine for his discoveries in the field
of biological oxidation processes, with special reference to vitamin C. In 1965,
vitamin C was renamed as ascorbic acid or L-ascorbic acid by IUPAC-IUB
Comission on Biochemical Nomenclature [60].
The ability of vitamin C to prevent oxidation is used by the food processing
industry to inhibit food degradation and to obtain functional food of high
35

quality. The world-wide production of ascorbic acid is estimated to be around
60'000tperyear[61].
Ascorbate is known to be an excellent antioxidant, but it can also exhibit the
pro-oxidant properties e.g. the ascorbate-driven Fenton reaction [62].
Furthermore, ascorbate has the ability to re-reduce oxidised compounds.
The reaction between peroxynitrite and ascorbate was studied for the first
time in 1995 by Bartlett et al. [63] and one year later by Squadrito et al. [64].
The rate constant for the reaction is 235 M^s-1 at 25°C. From the observation of
ascorbyl radicals it was concluded that a one-electron oxidation had taken place.
4.2.2 Properties ofvitamin C
Vitamin C has two acidic protons with pKa values of 4.04 and 11.34 [65].
Monohydroascorbate, the single protonated form of vitamin C, is unstable in
water in presence of metal and oxygen. Even lowest traces of metals, nM range,
catalyse the oxidation by air and produce hydrogen peroxide and hydroxyl
radicals [66,67]. It is not possible to inhibit the metal catalysed autoxidation of
vitamin C by employing chelators as edta, dtpe, etc. In some cases the catalytic
effect of metals can be even enhanced [66,68]. Multiple literature reports about
the degradation of DNA and various cells culture, including cancer cells, can
probably be attributed to the formation of partially reduced oxygen species in
presence of traces of transition metal ions in the reaction solution or cell culture
media[69]. Most of the ascorbate is oxidised in a one-electron step process
producing ascorbyl radicals which disproportionate, in a second-order reaction
to ascorbate and dehydroascorbate. The ascorbyl radical, which is neither
strongly oxidising nor strongly reducing (E° = -0.174 V for Dasc/Asc" [65]), is
rather unreactive. The weak reactivity of ascorbyl radicals is mostly responsible
for the antioxidant effects of ascorbate: strongly oxidising radicals react with
ascorbate and the much less reactive ascorbyl radicals are formed [69].
Dehydroascorbate is unstable and decomposes rapidly via complex pathway to
36

oxalic and L-threonic acid (under aerobic conditions) or to carbon dioxide and
furfural (under anaerobic conditions) [61]. The optical isomer of L-Ascorbic
acid, D-(-)-Ascorbic acid has practically no vitamin activity because it is not
transported and can not interact with enzymes.
ability of L-ascorbate are the reduction potentials.
HAsc" + H+ 0-9)
Asc2-+H+ 0-10)
HAsc" 0-11)
Asc'"+H+ 0-12)
DAsc + HAsc" 0-13)
HAsc" 0-14)
pKi 4.04 [70]
pK2 11.7 [70]
pK3 -0.45 [72]
E°'i 0.282 V [71]
E°'2 0.05 V [65]
ki 3.5xl07M^s^
atpH5.8
[73]
Table 2: Some properties of vitamin C.
Ascorbic acid (H2Asc) has an absorption maximum at 243.5 nm with an
extinction coefficient of 10000 M^cm1 [74]. Monohydroascorbate (HAsc)
absorbs at 265 nm with an extinction coefficient in the range of 14500 -
14700 M^cm1 [66,74] In this thesis a value of 14500 M^cm1 was taken.
Ascorbyl radical has the highest absorption at 360 nm with an extinction
Of interest for the antioxidant
HAscT =
H2Asc
pK^
"COI
Asc*"+e +H+ !»
pK3HAsc" =s===
2 Asc*- + H+ ki
E°'DAsc + 2e" + H+ ^
37

coefficient of 5000 M ^m 1at pH 5.8 [75]. Dehydroascorbate itself has no
absorption, but products of its decomposition are yellow.
16000 -i
j
14000 A
12000 / \
-r 10000 / \
| / \
-£ 8000 / \^ 6000 / \
4000 / \ /"'" \
2000 y \ _y'
o 4-^^—'"'-•?"•" *--x-—i 1^=-——
220 270 320 370 420
A,/nm
Fig. 2. Normalized spectra of 800 uM ascorbyl radical (dotted line) and 50 urn
monohydroascorbate (solid line). The ascorbyl radical spectrum was recorded after pulse
radiolysis. Both spectra were obtained at pH 5.8 in 0.05 M phosphate buffer.
4.2.3 Synthesis of vitamin C
Vitamin C can not be synthesised by some organism, which lack the enzyme
1-gulonolactone oxidase. Among those species are primates, fruit-eating bats,
guinea pigs...
38

Biosynthesis
The biosynthesis of ascorbic acid in animal liver [76]:
CHO CHO
H-
HO-
H-
H-
-OH
-H
-OH
-OH
CH2OH
D-glucose
HO OH
Ascorbic acid
H-
HO-
H-
H-
-OH
-H
-OH
-OH
COOH
D-glucuronic acid
-gulonolactone*oxidase
Missing enzyme in
primates, guinea pigand bats
COOH
HO-
HO-
H-
HO-
-H
-H
-OH
-H
CH2OH
L-gulonic acid
HO
\
H^
H^ OH
%..(rf)H hlO>^=0
L-gulonic acid y-lactone
39
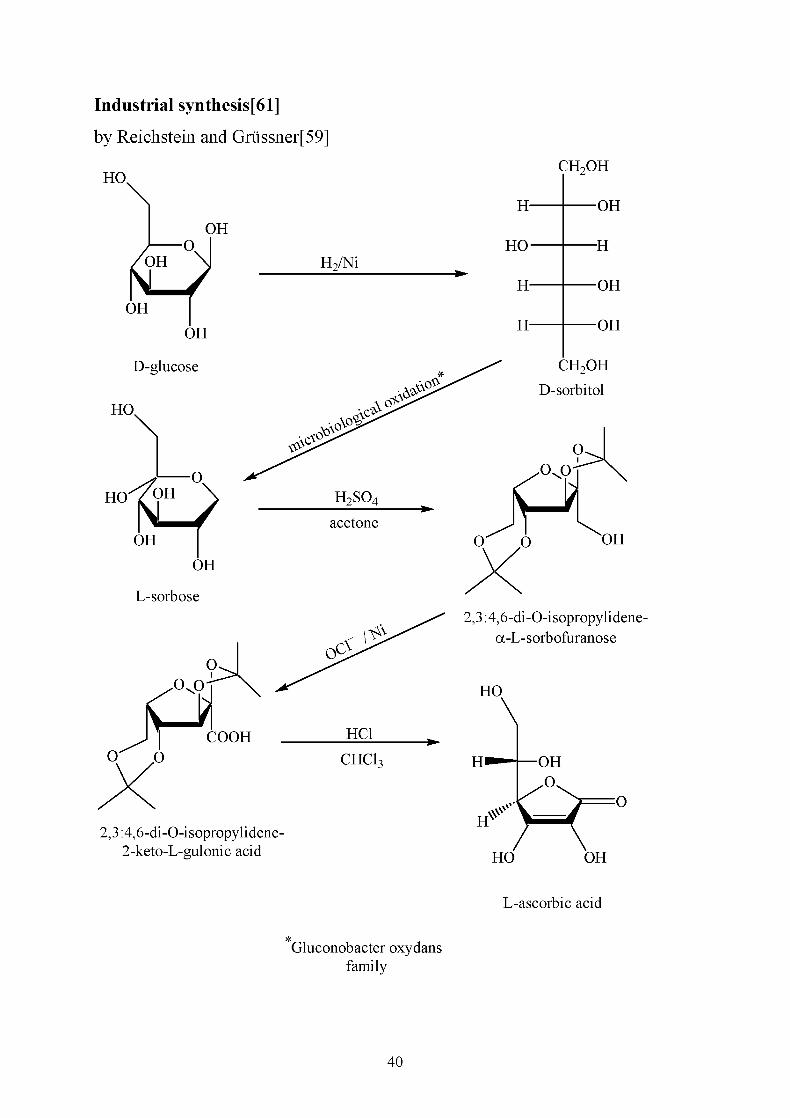
Industrial synthesis[61]
by Reichstein and Griissner[59]
HO.
OH
-o.
où IOH
D-glucose
H2/Ni
H-
HO-
H-
H-
tf»tô
n^°
>\°S*\c£°
0^'^
CH2OH
—OH
—H
—OH
—OH
CH2OH
D-sorbitol
H2S04
acetone
2,3:4,6-di-0-isopropylidene-
2-keto-L-gulonic acid
O
.0^0
b-iO Q X>H
2,3:4,6-di-0-isopropylidene-a-L-sorbofuranose
HO
HO OH
L-ascorbic acid
Gluconobacter oxydans
family
40

Recycling (in human neutrophils) [77]
TT A _
oxidants likeGSSG -*^ ^-*- HAsc ^
GSH — DAsc -*r ^-»* reduced oxidants
It is still unclear if dehydroascorbate is reduced by Dehydroascorbic Acid
Reductase, Protein Disulfide Isomerase or Thiol Transferase.
4.2.4 Resorption and excretion
Ingested ascorbic acid passes unchanged the stomach and is taken up in the
duodenum and transported by an active sodium-dependent mechanism. The rate
of resorption decreases with increasing ascorbate concentration [78].
The largest pools of ascorbate are the liver and the muscles tissue. About
90-95 % of ascorbic acid in the human body is present in the L-ascorbate form,
and the remaining fraction in the oxidised L-dehydroascorbate form (this ratio is
reversed in diabetic persons). Vitamin C is reabsorbed in the kidney, so a
deficiency of vitamin C is prevented. If the ascorbate concentration in plasma
exceeds over 1.2-1.8 mg/100 ml, kidneys stop reabsorption and ascorbic acid
passes to the urine. The formation of oxalic acid by vitamin C is of special
interest. About 40% of the excreted oxalic acid stems from vitamin C. This may
lead to nephrolithiasis. It was found that additional intake of vitamin C increases
the excretion of oxalic acid only insignificantly in non-risk persons of
nephrolithiasis.
4.2.5 Physiologic relevance of vitamin C
Vitamin C is an essential nutrient for the maintenance of human health; it is well
known to act as an intracellular and extracellular scavenger of free radicals and
41

oxidants. Therefore high concentrations of vitamin C are found in eyes and
lungs. In eyes radicals are produced by light-induced homolysis. In lungs
ascorbate protects from oxygen-derived radicals. Physiological concentrations of
ascorbate in various cells and tissues are of about 30 to 150 uM in human
plasma [79],1 mM in human lung and neutrophils [80], 1.3 mM in the liver [81],
7 mM in leukocytes [83] and 10 mM in neurons [82].
L-ascorbate is known to stimulate a number of enzymes, e.g. dioxygenase,
act as a cofactor in hydroxylation, be essential for iron resorption, play an
important role in the proper working of the nervous and endocrine system and
be essential in collagen synthesis, but the detailed mechanisms of numerous
actions of ascorbate are still unclear. Vitamin C takes also part in the redox
cycle of vitamin E, the main antioxidant present in the lipid part of the cells.
Ascorbic acid dissolved in the aqueous medium provokes a fast recycling of Vit.
E through the boundary layer between the aqueous medium and micelles. One-
electron oxidized form of Vit. E, a-tocopherol radical, react with vitamin C with
a second order rate constant of about 106 M-1s-1 [84,85].
The recycling of vitamin E at the expense of vitamin C found in vivo may
explain why vitamin E deficiency occurs very seldom.
Many reports deal with the relationship between vitamin C and the common
cold. A high vitamin C level in the body does not decrease the incidence of the
common cold. However, it consistently decreases the duration of common cold
episodes and the severity of the symptoms.
Therapeutically vitamin C is used for wound healing and enhances also
remarkably the body's immune response in fighting infections. The best known
health effect of ascorbate is the prevention and therapy of scurvy, a disease of
connective tissue, the symptoms of which are gum bleeding, amyotrophia and
tiredness. It has been suggested that L-ascorbate is helpful in preventing cancer
and cardiovascular diseases by protecting the DNA. A daily supplement of
ascorbate to enhance iron-uptake from food is also recommended in the
42

treatment of anemia. As mentioned earlier, the combination of iron, ascorbate
and air lead to radical production [71], therefore an additional intake of
ascorbate with iron should only be applied to persons with anemia. The
proposed daily dose of ascorbate is 120 mg /day (WHO, Aug. 2001)
Some high risk groups, such as smokers, elderly people, diabetics and
alcoholics have a lower vitamin C level than the average of the population.
Sportsmen, sick and stressed people also require a higher daily intake of
vitamin C.
High doses of ascorbic acid over long period of time (years) do not lead to
any side effects [61,78].
4.2.6. Vitamin C as a scavenger
A scavenger was defined by IUPAC in 1997 as follows: A substance that reacts
with (or otherwise removes) a trace component or traps a reactive reaction
intermediate (as in the scavenging of radicals or free electrons in radiation
chemistry). [86]
Ascorbate is known as a very good scavenger for free radicals. Further, it is
often applied in the treatment of the common cold and other diseases. Up to now
there is no substance which is cheaper, better tolerated by the body, and has less
nuisance problems associated with its administration than sodium ascorbate,
NaC6H606H, intravenously and intramuscularly, or ascorbic acid, C6H606H2,
orally.
Is L-ascorbate also a good scavenger for peroxynitrous acid? Peroxynitrous
acid can react with DNA, lipids, thiols and tyrosine residues in proteins. It is of
interest if L-ascorbate is able to scavenge peroxynitrite. A rate constant of
235 M'V1 has been reported in the literature for the reaction between
peroxynitrite and monohydroascorbate. However, many experiments show that
the protective ability of monohydroascorbate against peroxynitrite-mediated
damage is much higher than it can be expected from this rate constant [87].
43

What is necessary for monohydroascorbate to be a good scavenger for
peroxynitrite? The reduction potential of monohydroascorbate should be lower
than that one of peroxynitrous acid and the reaction of the scavenger
(monohydroascorbate) with peroxynitrous acid should be at least 10 times
higher than that of peroxynitrite with any other compound.
At physiological pH peroxynitrite reacts mainly with carbon dioxide, which
is present in a concentration of about 1.3 mM, with a second-order rate constant
of 3 x 104 jyrV1 at 25°C [49,88]. The resulting rate k' is therefore 39 s"1 (k' =
kx[C02]). In order to call monohydroascorbate a scavenger for peroxynitrite, the
physiological first-order rate k' should be higher than 390 s"1. (k' = kx[HAsc ]).
The concentration of monohydroascorbate varies from 100 uM to 10 mM (see
section 4.2.6.). Therefore the second order rate constant should be higher than
3.9 x 104M~1s~1 or even then 3.9 x 106M~1s~1, depending on the ascorbate
concentration.
Thus, in regard to this discrepancy between experimental findigs and kinetic
the reaction of monohydroascorbate with peroxynitrite was reinvestigated.
Results are described in chapter 7 and 8.
44

43 References
[1] Baeyer, A.; Villiger, V. Ueber die salpetrige Säure. Ber. Dtsch. chem.
Ges. 34:755-762; 1901.
[2] Raschig, F. Vorlesungsversuche aus der Chemie der anorganische
Stickstofverbindungen. Ber. Dtsch. chem. Ges. 40:4580-4588; 1907.
[3] Gleu, K.; Roell, E. Die Einwirkung von Ozon auf Alkaliazid.
Persalpetrige Säure I. Zeitschr. anorg. allgem. Chemie 179:233-266;1929.
[4] Halfpenny, E.; Robinson, P. L. Pernitrous acid. The reaction between
hydrogen peroxide and nitrous acid, and the properties of an
intermediate product. J. Chem. Soc. 928-938; 1952.
[5] Anbar, M.; Taube, H. Interaction of nitrous acid with hydrogen peroxideand with water. J. Am. Chem. Soc. 76:6243-6247; 1954.
[6] Hughes, M. N.; Nicklin, H. G. The chemistry of pernitrites. Part I.
Kinetics of decomposition of pernitrous acid. J. Chem. Soc. (A) 450-
452; 1968.
[7] Edwards, J. O.; Plumb, R. C. The chemistry of peroxonitrites. Prog.
Inorg Chem. 41:599-635; 1993.
[8] Beckman, J. S.; Beckman, T. W.; Chen, J.; Marshall, P. A.; Freeman, B.
A. Apparent hydroxyl radical production by peroxynitrite: Implicationsfor endothelial injury from nitric oxide and superoxide. Proc. Natl.
Acad. Sei. USA 87:1620-1624; 1990.
[9] Nauser, T.; Koppenol, W. H. The rate constant of the reaction of
superoxide with nitrogen monoxide: Approaching the diffusion limit. J.
Phys. Chem. A 106:4084-4086; 2002.
[10] Fielden, E. M.; Roberts, P. B.; Bray, R. C; Lowe, D. J.; Mautner, G. N.;
Rotilio, G.; Calabrese, L. The mechanism of action of superoxidedismutase from pulse radiolysis and electron paramagnetic resonance.
Biochem. J. 139:49-60; 1974.
[11] Beckman, J. S.; Ye, Y. Z.; Anderson, P. G.; Chen, J.; Accavitti, M. A.;
Tarpey, M. M.; White, C. R. Extensive nitration of protein tyrosines in
human atherosclerosis detected by immunohistochemistry. Biol. Chem.
Hoppe-Seyler 375:81-88; 1994.
45

[12] Coddington, J. W.; Hurst, J. K.; Lymar, S. V. Hydroxyl radical
formation during peroxynitrous acid decomposition. J. Am. Chem. Soc.
121:2438-2443; 1999.
[13] Gerasimov, O. V.; Lymar, S. V. The yield of hydroxyl radical from the
decomposition of peroxynitrous acid. Inorg. Chem. 38:4317-4321; 1999.
[14] Merényi, G.; Lind, J.; Goldstein, S.; Czapski, G. Peroxynitrous acid
homolyzes into OH and N02 radicals. Chem. Res. Toxicol. 11:712-713;1998.
[15] Richeson, C. E.; Mulder, P.; Bowry, V. W.; Ingold, K. U. The complex
chemistry of peroxynitrite decomposition: New insights. J. Amer. Chem.
Soc. 120:7211-7219; 1998.
[16] Koppenol, W. H.; Kissner, R. Can ONOOH undergo homolysis. Chem.
Res. Toxicol. 11:87-90; 1998.
[17] Kissner, R.; Koppenol, W. H. Product distribution of peroxynitrite decayas a function of pH, temperature, and concentration. J. Am. Chem. Soc.
124:234-239; 2002.
[18] Maurer, P.; Thomas, C. F.; Kissner, R.; Rüegger, H.; Greter, O.;
Röthlisberger, U.; Koppenol, W. H. Oxidation of nitrite by
peroxynitrous acid. J. Phys. Chem. A 107:1763-1769; 2003.
[19] Goldstein, S.; Czapski, G. Indirect oxidation of ferrocyanide - Evidence
against the formation of hydroxyl radicals. Nitric Oxide: Biol. Chem.
1:417-422; 1997.
[20] Koppenol, W. H. 100 Years of peroxynitrite biochemistry and 11 years
of peroxynitrite biochemistry. Redox. Rep. 6:339-341; 2001.
[21] Denicola, A.; Freeman, B. A.; Trujillo, M.; Radi, R. Peroxynitritereaction with carbon dioxide/bicarbonate: Kinetics and influence on
peroxynitrite-mediated oxidations. Arch. Biochem. Biophys. 333:49-58;1996.
[22] Kissner, R.; Nauser, T.; Bugnon, P.; Lye, P. G.; Koppenol, W. H.
Formation and properties of peroxynitrite studied by laser flash
photolysis, high pressure stopped flow and pulse radiolysis. Chem. Res.
Toxicol. 10:1285-1292; 1997.
[23] ONOO" Peroxynitrite. Gmelin Handbook of Inorganic and
Organometallic Chemistry; Nitrogen. Berlin: Springer-Verlag;1996:344-358.
46

[24] Logager, T.; Sehested, K. Formation and decay of peroxynitrous acid: a
pulse radiolysis study. J. Phys. Chem. 97:6664-6669; 1993.
[25] Merényi, G.; Lind, J. Free radical formation in the peroxynitrous acid
(ONOOH)/peroxynitrite (ONOO ) system. Chem. Res. Toxicol. 11:243-
246; 1998.
[26] Eigen, M. Proton Transfer and general acid base catalysis. In Claesson,S. eds. Fast reactions and primary processes in chemical kinetics.
Stockholm: Almqvist&Wiksell; 1967:245-253.
[27] Tsai, J.-H. M.; Harrison, J. G.; Martin, J. C; Hamilton, T. P.; van der
Woerd, M.; Jablonsky, M. J.; Beckman, J. S. Role of conformation of
peroxynitrite anion (ONOO") in its stability and toxicity. J. Am. Chem.
Soc. 116:4115-4116; 1994.
[28] Krauss, M. Electronic structure and spectra of the peroxynitrite anion.
Chem. Phys. Lett. 222:513-516; 1994.
[29] Wörle, M.; Latal, P.; Kissner, R.; Nesper, R.; Koppenol, W. H.
Conformation of peroxynitrite: Determination by crystallographic
analysis. Chem. Res. Toxicol. 12:305-307; 1999.
[30] Bohle, D. S.; Hansert, B.; Paulson, S. C; Smith, B. D. Biomimetic
synthesis of the putative cytotoxin peroxynitrite, ONOO", and its
characterization as a tetramethylammonium salt. J. Am. Chem. Soc.
116:7423-7424; 1994.
[31] Kissner, R. Absorption spectra of peroxynitrite. 2002. Personal
Communication
[32] Uppu, R. M.; Squadrito, G. L.; Cueto, R.; Pryor, W. A. Selecting the
most appropriate synthesis of peroxynitrite. Methods Enzymol. 269:285-
295; 1996.
[33] Hughes, M. N.; Nicklin, H. G. Autoxidation of hydroxylamine in
alkaline solution./. Chem. Soc. (A) 164-168; 1971.
[34] Pryor, W. A.; Cueto, R.; Jin, X.; Koppenol, W. H.; Ngu-Schwemlein,M.; Squadrito, G. L.; Uppu, P. L.; Uppu, R. M. A practical method for
preparing peroxynitrite solutions of low ionic strength and free of
hydrogen peroxide. Free Radical Biol. Med. 18:75-83; 1995.
[35] Uppu, R. M.; Squadrito, G. L.; Cueto, R.; Pryor, W. A. Synthesis of
peroxynitrite by azide-ozone reaction. Methods Enzymol. 269:311-321;1996.
47

[36] Uppu, R. M.; Pryor, W. A. Biphasic synthesis of high concentrations of
peroxynitrite using water-insoluble alkyl nitrite and hydrogen peroxide.Methods Enzymol. 269:322-329; 1996.
[37] Leis, J. R.; Pena, M. E.; Rios, A. A novel route to peroxynitrite anion. J.
Chem. Soc. Chem. Commun. 1993:1298-1299; 1993.
[38] Reed, J. W.; Ho, H. H.; Jolly, W. L. Chemical synthesis with a quenchedflow reactor. Hydroxytrihydroborate and peroxynitrite. J. Am. Chem.
Soc. 96:1248-1249; 1974.
[39] Beckman, J. S.; Chen, J.; Ischiropoulos, H.; Crow, J. P. Oxidative
chemistry of peroxynitrite. Methods Enzymol. 233:229-240; 1994.
[40] Koppenol, W. H.; Kissner, R.; Beckman, J. S. Syntheses of
peroxynitrite: To go with the flow or on solid grounds. Methods
Enzymol. 269:296-302; 1996.
[41] Bohle, D. S.; Glassbrenner, P. A.; Hansert, B. Synthesis of pure
tetramethylammonium peroxynitrite. Methods Enzymol. 269:302-311;1996.
[42] Singh, R. J.; Hogg, N.; Joseph, J.; Konorev, E.; Kalyanaraman, B. The
peroxynitrite generator, SIN-1, becomes a nitric oxide donor in the
presence of electron acceptors. Arch. Biochem. Biophys. 361:331-339;1999.
[43] Palmer, R. M. J.; Ashton, D. S.; Moncada, S. Vascular endothelial cells
synthesize nitric oxide from L-arginine. Nature 333:664-666; 1988.
[44] Ischiropoulos, H.; Zhu, L.; Beckman, J. S. Peroxynitrite formation from
macrophage-derived nitric oxide. Arch. Biochem. Biophys. 298:446-451;1992.
[45] Lafon-Cazal, M.; Culcasi, M.; Gaven, F.; Pietri, S.; Bockaert, J. Nitric
oxide, superoxide and peroxynitrite: Putative mediators of NMDA-
induced cell death in cerebral granule cells. Neuropharmacology32:1259-1266; 1993.
[46] Carreras, M. C; Pargament, G. A.; Catz, S. D.; Poderoso, J. J.; Boveris,A. Kinetics of nitric oxide and hydrogen peroxide production and
formation of peroxynitrite during the respiratory burst of human
neutrophils. FEBS Lett. 341:65-68; 1994.
[47] Schulz, R.; Smith, J. A.; Lewis, M. J.; Moncada, S. Nitric oxide
synthase in cultured endocardial cells of the pig. Br. J. Pharmacol.
104:21-24; 1991.
48

[48] Padmaja, S.; Kissner, R.; Bounds, P. L.; Koppenol, W. H. Hydrogen
isotope effect on the isomerization of peroxynitrous acid. Helv. Chim.
Acta 81:1201-1206; 1998.
[49] Lymar, S.V.; Hurst, J. K. Rapid reaction between peroxonitrite ion and
carbon dioxide: Implications for biological acitivity. J. Am. Chem. Soc.
117:8867-8868; 1995.
[50] Bonini, M. G.; Radi, R.; Ferrer-Sueta, G.; Ferreira, A. M. D.; Augusto,O. Direct EPR detection of the carbonate radical anion produced from
peroxynitrite and carbon dioxide. J. Biol. Chem. 274:10802-10806;1999.
[51] Meli, R.; Nauser, T.; Latal, P.; Koppenol, W. H. Reaction of
peroxynitrite with carbon dioxide: Intermediates and determination of
the yield of C03' and N02\ J. Biol. Inorg Chem. 7:31-36; 2002.
[52] Lymar, S. V.; Hurst, J. K. C02-catalyzed one-electron oxidation by
peroxynitrite: Properties of the reactive intermediate. Inorg. Chem.
37:294-301; 1998.
[53] Moser, U.; Bendich, A. Vitamin C. In Machlin, L. J. eds. Handbook ofVitamins. New York: Marcel Dekker; 1991:195-225.
[54] Hoist, A.; Fröhlich, T. Experimental studies relating to ship beri-beri
and scurvy. II On the etiology of scurvy. J. Hyg. (Cambridge) 7:634-
671; 1907.
[55] Zilva, S. S. The influence of aeration on the stability of the antiscorbutic
factor. The Lancet 197:478; 1921.
[56] Szent-Györgyi, A. Observations on the function of peroxidase systemsand the chemistry of the adrenal cortex. Biochem. J. 22:1387-1409;1928.
[57] Haworth, W. N.; Hirst, E. L. Synthesis of ascorbic acid. J. Soc. Chem.
Ind. London Trans. Commun. 52:645-646; 1933.
[58] Reichstein, T.; Oppenauer, R. Synthese der d- und 1-ascorbinsäure (C-Vitamin). Helv. Chim. Acta 16:1019-1033; 1933.
[59] Reichstein, T.; Grüssner, A. Eine ergibige Synthese der 1-Ascorbinsäure
(C-Vitamin). Helv. Chim. Acta 17:311-328; 1934.
[60] IUPAC Trivial names of miscellaneous compounds of importance in
biochemistry. Biochim. Biophys. Acta. :Gen. Subj. 107:1-4; 1965.
49

[61] Vitamins/Vitamin C. Ullmann's Encyclopedia of industrial chemistry.Weinheim: Wiley-VCH Verlag GMBH; 2003:218-231.
[62] Burkitt, M. J.; Gilbert, B. C. Model studies of the iron-catalysed Haber-
Weiss Cycle and the ascorbate-driven fenton reaction. Free Radical Res.
Commun. 10:265-280; 1990.
[63] Bartlett, D.; Church, D. F.; Bounds, P. L.; Koppenol, W. H. The kinetics
of the oxidation of L-ascorbic acid by peroxynitrite. Free Radical Biol.
Med. 18:85-92; 1995.
[64] Squadrito, G. L.; Jin, X.; Pryor, W. A. Stopped-flow kinetic study of the
reaction of ascorbic acid with peroxynitrite. Arch. Biochem. Biophys.322:53-59; 1995.
[65] Williams, M. H.; Yandell, J. K. Outer-sphere electron transfer reactions
of ascorbate anions. Aust. J. chem. 35:1133-1144; 1982.
[66] Buettner, G. R. In the absence of catalytic metals ascorbate does not
autoxidize at pH 7: ascorbate as a test for catalytic metals. Journal ofBiochemical and Biophysical Methods 16:27-40; 1988.
[67] Miller, D. M.; Buettner, G. R.; Aust, S. D. Transition metals as catalystsof "autoxidation" reactions. Free Radical Biol. Med. 8:95-108; 1990.
[68] Glebska, J.; Grzelak, A.; Pulaski, L.; Bartosz, G. EDTA loses its
antioxidant properties upon storage in buffer. Anal. Biochem. 311:87-89;2002.
[69] Halliwell, B.; Gutteridge, J. M. C. Antioxidant protection by low-
molecular-mass agents: compounds derived from the diet. Free Radicals
in biology andMedicine. New York: Oxford university press; 1999:200-
208.
[70] Handbook of Chemistry and Physics. Internet 3rd electronical edition.
2000. 2000. Electronic Citation
[71] Buettner, G. R.; Jurkiewicz, B. A. Catalytic metals, ascorbate and free
radicals: Combinations to avoid. Radiât. Res. 145:532-541; 1996.
[72] Davis, H. F.; McManus, H. J.; Fessenden, R. W. An ESR study of free-
radical protonation equilibria in strongly acid media. J. Phys. Chem.
90:6400-6404; 1986.
[73] Bielski, B. H. J.; Allen, A. O.; Schwarz, H. A. Mechanism of
disproportionation of ascorbate radicals. J. Am. Chem. Soc. 103:3516-
3518; 1981.
50

Ogata, Y.; Kosugi, Y. Ultraviolet spectra of L-ascorbic acid and cupricascorbate complex. Tetrahedron 26:4711-4716; 1970.
Bielski, B. H. J.; Comstock, D. A.; Bowen, R. A. Ascorbic acid free
radicals. I. Pulse radiolysis study of optical absorption and kinetic
properties. J. Am. Chem. Soc. 93:5624-5629; 1971.
Lewin, S. Vitamin C: London Academic Press; 1976.
Washko, P. W.; Wang, Y.; Levine, M. Ascorbic acid recycling in human
neurophils. J. Biol. Chem. 268:15531-15535; 1993.
Vitamin C. In Eisenbrand, G.; Schreier, P. eds. Römpp-LexikonLebensmittelchemie. Stuttgart: Thieme Verlag; 1995:899-901.
Stocker, R.; Frei, B. Endogenous antioxidant defences in human blood
plasma. In Sies, H. eds. Oxidative Stress: Oxidants and Antioxidants.
London: Academic Press; 1991:213-243.
Washko, P.; Rotrosen, D.; Levine, M. Ascorbic acid in human
neutrophils. Am. J. Clin. Nutr. 54 Suppl.:1221S-1227S; 1991.
Li, X.; Cobb, C. E.; Hill, K. E.; Burk, R. F.; May, J. M. Mitochondrial
uptake and recycling of ascorbic acid. Arch. Biochem. Biophys. 387:143-
153; 2001.
Rice, M. E.; Russo-Menna, I. Differential compartimentalization of
brain ascorbate and glutathione between neurons and glia. Neuroscience
82:1213-1223; 1998.
Griffiths, H. R.; Lunec, J. Ascorbic acid in the 21st century - more than
a simple oxidant. Environ. Toxicol. Pharmacol. 10:173-182; 2001.
Packer, J. E.; Slater, T. F.; Willson, R. L. Direct observation of free
radical interaction between vitamin E and vitamin C. Nature 278:737-
738; 1979.
Scarpa, M.; Rigo, A.; Maiorino, M.; Ursini, F.; Gregolin, C. Formation
of alpha-tocopherol radical and recycling of alpha-tocopherol byascorbate during peroxidation of phosphatidylcholine liposomes.Biochim. Biophys. Acta 801:215-219; 1984.
de Bolster, M. W. G. Glossary of terms used in bioinorganic chemistry.
PureAppl. Chem. 69:1251-1303; 1997.
51

[87] Balavoine, G. G. A.; Geletii, Y. V. Peroxynitrite scavenging by different
antioxidants. Part I: Convenient assay. Nitric Oxide: Biol. Chem. 3:40-
54; 1999.
[88] Arteel, G. E.; Briviba, K.; Sies, H. Protection against peroxynitrite.FEBS Letters 445:226-230; 1999.
52

5. MATERIALAND METHODS
5.7. Reagents
All chemicals used in experiments were of analytical grade or better. Ascorbic
acid, disodium monohydrogen phosphate, sodium dihydrogen phosphate and
sodium nitrite were purchased from Fluka (Buchs, Switzerland). Potassium
superoxide was from Aldrich (St. Louis, MO, USA) or from Aber (Karlsruhe,
Germany), phosphoric acid and sodium hydroxide from Siegfried (Säckingen,
Switzerland), carbon dioxide from Pangs (Luzern, Switzerland), sodium iodide
from Merck (Darmstadt, Germany), and nitrogen monoxide from Linde
(Unterschleissheim, Germany). Peroxynitrite was synthesized according to
Koppenol et al. [1]. The concentration of the peroxynitrite solution was
determined by measuring the absorbance at 302 nm (£302 = 1705 M^cnT1) [2].
Deionised water was purified further by a Millipore Milli-Q unit (Bedford, MA,
USA). Buffers were prepared from salts and acids. All solutions were freshly
prepared and - except the peroxynitrite solutions - were evacuated for five
minutes and then saturated with argon or an argon/carbon dioxide mixture, to
avoid oxidation of monohydroascorbate by dioxygen. The peroxynitrite
solutions were prepared by dilution with 0.010 M sodium hydroxide that was
de-aerated by evacuation for five minutes prior to its addition to the
peroxynitrite stock solution. When peroxynitrite was not mixed with
monohydroascorbate and oxygen had no influence on the results, the solutions
were not evacuated.
53

5.2. Stopped-flow spectrophotometrie studies
5.2.1. Equipment
Kinetic experiments were carried out at 25°C and 37°C and ambient pressure
with an Applied Photophysics SX 17MV, or with Applied Photophysics
SX 18MV stopped-flow spectrophotometer (Leatherhead, Surrey, GB) operated
in the symmetric mixing mode. The pH was measured at the outlet. Were not
different reported, peroxynitrite was synthesized according to Koppenol et al.
[1]. Time-dependent spectra were obtained at 25°C with an OLIS stopped-flow
instrument equipped (Bogart, GA, USA) with an OLIS RSM 1000 rapid
scanning monochromator set to collect up to 1000 spectra per second. When
experiment were made with wavelength higher than 450 nm, filters were used.
5.2.2 One-electron reduction
Kinetics experiments were carried out at 25°C and ambient pressure with an
Applied Photophysics SX 17MV (Leatherhead, Surrey, UK) stopped-flow
spectrophotometer operated in the symmetric mixing mode. The reaction was
initiated by mixing 10, 50 or 100 uM peroxynitrite in 0.01 M sodium hydroxide
with various sodium iodide concentrations (200 uM to 40 mM) in 0.2 M
phosphate buffer at pH 5.8 and at pH3.2. The changes in absorbance were
followed at 350 nm and the pH was determined at the outlet of the stopped-flow
apparatus with a glass electrode. The equilibrium constant of the triiodide
formation
I2 + F + * I3~ was determined to be 930 M"1 in the phosphate buffer
system used, somewhat different from the 710 M"1 found in the literature, [3]
which is a value obtained by extrapolation to zero ionic strength. The extinction
coefficient of I3~ at 350 nm was 28100 M_1cm_1, in good agreement with the
literature. [3]
54

5.2.3. Monohydroascorbate
The reaction was initiated by mixing a peroxynitrite solution in 0.010 M sodium
hydroxide with a monohydroascorbate solution in 0.10 M phosphate buffer
(pH 5.8, 7.2, 8.0 and 8.5). The wavelengths measured were 265 nm
(monohydroascorbate); 320 nm (for peroxynitrite); 345 nm and 360 nm for the
intermediates.
5.2.4. Monohydroascorbate and carbon dioxide
Reactions were initiated by mixing peroxynitrite in a phosphate buffer of pH 12
with monohydroascorbate in phosphate buffer of pH3. The phosphate buffer
concentrations were adjusted to the pH needed. The wavelengths measured were
320 nm (for peroxynitrite); 345 nm (for the intermediates); 360 nm (for the
intermediates; mainly ascorbyl radical); and 640 nm for trioxocarbonate(#-)
radical.
5.3. Cyclic voltammetry
Cyclovoltammograms were measured by a stopped-flow technique. The
apparatus setup is shown in chapter 6.2. (Supplemental information) in Figure 1.
One pump (Dosimat 665 titration unit from Metrohm/Herisau, Switzerland) was
filled with phosphate buffer and the second one with a 2 mM peroxynitrite
solution in 0.01 M NaOH. The pump units were activated synchronously by
computer. The titration devices were both set to a flow of 20 ml/ minute, and the
measuring cell was rinsed with twenty times its volume (Fig. 2S; chapter 6.2.).
The measurements were started by a trigger signal sent by the control unit to the
measuring computer. The control unit consisted of a computer with a 12 bit
analog to digital converter, an AMEL 2049 potentiostat and an AMEL 586
function generator (Milan, Italy). The working electrode was a gold electrode
55

from Metrohm (Herisau, Switzerland) with an active surface of 3.1 mm.The
reference electrode was a silver/silver chloride electrode from Methrom
(Herisau, Switzerland) filled with 0.1 M potassium chloride and 0.1 M sodium
nitrate as bridge electrolyte. The auxiliary electrode was a glassy carbon
electrode from Metrohm (Herisau, Switzerland).
5.4. EPR
Electron paramagnetic resonance spectra were obtained with a Bruker EMX 080
(Karlsruhe, Germany) equipped with a microwave-bridge ER 041 XG and the
dielectric mixing resonator ER4117D-MVT. Data acquisition and analysis
were carried out with ACQUISIT software (Bruker). The solutions (25 uM
peroxynitrite and 250 uM monohydroascorbate) were injected into the cavity
with a KDScientific 220 syringe pump (New Hope, PA, USA) at a flow of
1 ml/min per syringe in a ratio 1:1. The solutions were of the same composition
as those used in the stopped-flow experiments.
The signal intensity of the ascorbyl radical signal was determined by mixing
a freshly prepared cerium(IV) solution (25 uM final concentration) with
monohydroascorbate at pH 5.8 in 50 mM phosphate buffer. Only a few percent
of cerium(IV) react with monohydroascorbate, due to the competing reaction
with phosphate that produces an unidentified, inert cerium-phosphate complex
[4]. The concentration of the small amount of ascorbyl radical produced was
determined by the rate of its bimolecular decay
(3.5 x Kfivry1)^].
5.5. Ion chromatography
Nitrite and nitrate were determined on a Methrom 732 ion separation center
(Herisau, Switzerland) equipped with a Super-Sep IC anion exchanging column
56

(Methrom, Herisau, Switzerland) with a phthalate solution (2.5 mM phthalate,
5% acetonitrile, pH 4.2 (TRIS), A «130 nS/cm) as eluent. [6,7].
5.6. Simulation by Feldberg method
This method is used to simulate cyclovoltammograms to determine the formal
reduction potential. The Feldberg method simulates the concentration gradient
of the substrate to be reduced at the electrode surface. When the onset potential
is reached, the peroxynitrous acid in a small volume next to the electrode is
assumed to be completely reduced, which results in a concentration gradient
between the electrode and the bulk of the solution. To simulate this gradient and
the diffusion of the ions, the solvent around the electrode is divided into small
compartments whereby, for each calculation cycle, the concentrations are only
allowed to equilibrate between adjacent compartments. The depletion rate of
active species at the electrode surface limits and thereby determines the current
[8-10].
5.7. Simulation ofkinetic reactions
Kinetics were simulated based on sets of ordinary differential equations with an
EXCEL macro program that implements a two-step implicit Runge-Kutta
algorithm of 4th order [11]. This method is more stable than explicit integration
techniques, but requires more computation time.
57

References
[1] Koppenol, W. H.; Kissner, R.; Beckman, J. S. Syntheses of peroxynitrite:
To go with the flow or on solid grounds. Methods Enzymol. 269:296-302;
1996.
[2] Bohle, D. S.; Glassbrenner, P. A.; Hansert, B. Synthesis of pure
tetramethylammonium peroxynitrite. Methods Enzymol. 269:302-311;
1996.
[3] Meyerstein, D.; Treinin, A. Charge-transfer complexes of iodine and
inorganic anions in solution. Trans. Faraday Soc. 59:1114-1120; 1963.
[4] Oxidation state +4. In Bailar, J. C; Emeléus, H. J.; Nyholm, R.; Trotman-
Dickenson, A. F. eds. Comprehensive Inorganic Chemistry. Oxford:
Pergamon Press; 1973:97-98.
[5] Bielski, B. H. J.; Allen, A. O.; Schwarz, H. A. Mechanism of
disproportionation of ascorbate radicals. J. Am. Chem. Soc. 103:3516-3518;
1981.
[6] Kissner, R.; Koppenol, W. H. Product distribution of peroxynitrite decay as
a function of pH, temperature, and concentration. J. Am. Chem. Soc.
124:234-239; 2002.
[7] Meli, R.; Nauser, T.; Latal, P.; Koppenol, W. H. Reaction of peroxynitrite
with carbon dioxide: Intermediates and determination of the yield of C03"
andN02'.J. Biol. Inorg. Chem. 7:31-36; 2002.
[8] Feldberg, S. W. Theory of relaxation of the diffusion double layer
following coulostatic charge injection. J. Phys. Chem. 74:87-90; 1970.
58

[9] Feldberg, S. W. Digital simulation: A general method for solving
electrochemical diffusion-kinetic problems. In Bard, A. J. eds.
Electroanalytical chemistry. Marcel Dekker; 1969:199-296.
[10] Mattson, J. S.; Mark, H. B.; MacDonald, H. C. Computers in chemistry and
instrumentation: New York Marcel Dekker; 1972.
[11] Schwarz, H. R. Numerische Mathematik: Stuttgart B.G. Teubner; 1988.
59

60

6. ON THE CHEMICAL AND ELECTROCHEMICAL
ONE-ELECTRON REDUCTION OF
PEROXYNITROUS ACID
CHRISTOPHE KURZT, XIUQIONG ZENGJ, STEFAN HANNEMANNT,REINHARD KISSNERT AND WILLEM H. KOPPENOL*T
^ Laboratorium für Anorganische Chemie, Eidgenössische Technische
Hochschule Zürich (Hönggerberg), Switzerland
^Department of Chemistry at Xixi Campus, ZhejiUniversity, Hangzhou 310028,
China
Abstract— Peroxynitrous acid was reduced by cathodic linear sweep
voltammetry at a gold electrode and by iodide at pH 3.2 to 5.6. The cathodic
reduction wave was identified by measuring its decay in time, which was the
same as with observations by optical spectroscopy. The iodide oxidation was
followed by optical measurement of the triiodide formation. Both reductions
show one-electron stochiometry, with the product naa = 0.23 ± 0.04 and an
iodine yield of around 0.5 equivalents per equivalent of peroxynitrous acid. The
voltammetric reduction was irreversible up to scan rates of 80 Vs"1. Both
reductions were pH independent in the range studied. The voltammetric
reduction is most likely an irreversible elemental reaction followed by a
chemical decay which cannot be observed directly. Because of the pH
independence, we conclude that both reductions have a common short-lived
intermediate, namely [HOONO]*-, which is analogous to [HONO]*~ and
[ONO]*2~. This type of radical ion has been identified in the reduction of nitrite
with hydrated electrons in pulse radiolysis experiments. The pH independence
of the one-electron reductions also suggests that estimates of the one-electron
standard reduction potential of peroxynitrous acid, including our own, are in
error.
61

Results
Voltammetry results.
Electrochemical reduction of
peroxynitrous acid at a
polycrystalline gold electrode with a
linear sweep yielded a cathodic
wave which could be assigned
unambiguously to the substance,
since the peak current at a given
scan rate decreased with the delay
time between the formation of the
acid by mixing peroxynitrite with
dihydrogen phosphate/phosphoric
acid and the onset of the
voltammetric scan. The rate constant
for the first-order peak current
decrease was found to be (1.1
±0.1) s"1 at pH 5.6 and (1.5+0.1) s"1
at pH3.2, quite typical for the
peroxynitrous acid isomerisation at
room temperature [1]. The reduction
wave of peroxynitrous acid was
followed by further reduction waves
at more negative potential. They are
most probably caused by the
reduction of decomposition
products, possibly nitrogen dioxide
and dinitrogen tetroxide. Nitrite as
60
o
oo
o o
o o
o
o
20 H o o
o
o
10- o
©
0 -I 1 1 1
0.0 1.0 2.0 3.0 4.0
(dE/dtf2 / (Vs 1)%
Fig. 1. Peak currents vs. square root of the
scan rate. 1 mM peroxynitrite with 0.1 M
phosphoric acid/phosphate buffers, goldelectrode.
Circles: pH 3.2, diamonds: pH 5.6.
T 1 1 1 1 1 1 1 1 1 1 r
600 400 200 0 -200 -400 -600
El my vs. Ag/AgCI, [CH = 0.1 M
Fig. 2. Reduction waves of peroxynitrousacid at pH 3.2, pH 5.6 and several scan
rates. 1 mM peroxynitrous acid, 0.1 M
phosphoric acid/phosphate buffers, a:
pH 5.6, 1 Vs"1; b: pH 3.2, 1 Vs"1; c: pH 5.6,
4Vs_1; d: pH3.2, 4Vs_1; e: pH5.6,
10Vs_1;f:pH3.2, 10 Vs"1.
40
<
- 30
62

the origin was ruled out, because it yielded no cathodic wave under the same
conditions, in the absence of peroxynitrous acid. Cyclic scans up to 80 Vs-1
showed no re-oxidation wave. The negative peak potential shift for a tenfold
increase in scan rate from 1 Vs-1 and 10 Vs-1 was 190 mV at pH3.2 and
210 mV at pH 5.6. The peak currents were always directly proportional to the
initial peroxynitrous acid concentration and to the square root of the scan rate
for both of the pH values (Fig. 1). The current peaks at pH 3.2 were higher than
at pH 5.6 (Fig. 2) for the same peroxynitrous acid concentrations and scan rates,
but hardly shifted by the different pH. The variation of maximum current
strength with pH was caused by a change in ohmic resistance of the metal-
solution interface depending on the buffer composition. At pH 5.6, there is a
considerable concentration of monohydrogen phosphate present, while at
pH 3.2, dihydrogen phosphate is the only anionic species.
Stopped-flow results.
Peroxynitrous acid oxidises
iodide to iodine like other peroxides
[2,3]. However, it cannot be
determined quantitatively by o 0.e
iodometric titration, since the
relative yield of its reaction with 4
iodide depends on concentrations.
With iodide concentrations below
1 mM, but in at least five-fold excess
peroxynitrous acid, 0.5over
equivalents of iodine were produced
rather precisely. Only with iodide
dilutions below 100 uM, the
isomerisation of the peroxynitrous
oO 0.5
1.0 10.0
[I ] / mM
100.0
Fig. 3. Relative yields of iodine from the
oxidation of iodide by peroxynitrousacid, vs. total iodide concentration. 0.1
M phosphoric acid/phosphate buffers,
circles: [HOONO]imtlai = 25 uM;
squares: [HOONO]imtlai = 5 uM.
63

acid competed with the iodide oxidation, and the iodine yield dropped below 0.5
equivalents. At iodide concentrations above 1 mM, the iodine yield rose above
0.5 equivalents and approached 1 equivalent with high excesses, but never
reached the exact value (Fig. 3). The rate law of the reaction was first order in
both peroxynitrite concentration and peroxynitrous acid concentration, which
means that only these molecules are involved in the rate-determining step. The
second-order rate constant at pH3.2 was found to be (2.8+0.3) x 104M_1s_1
based on the rate of formation of iodine, which was obtained by tranforming the
I3~ absorbance vs. time trace with the law of mass action of the equilibrium
I2 + F« I3~. The reaction rate hardly increased from pH 5.6 to 3.2, the
small change can be attributed to the fact that at pH5.6 about 10% of the
peroxynitrite are in the less reactive ONOO" form.
Discussion
The complete lack of re-oxidation wave and the linear dependence of the
peak current on the square root of the scan rate rule out a quasi-reversible
electrochemical reaction [4]. The remaining conceivable mechanisms are an
irreversible electrochemical reaction El5 case (a), possibly preceded by a
chemical equilibrium, CrEl5 case (b), or a reversible electrochemical reaction
followed by an irreversible chemical one, ErQ, case (c). Process (c) would show
k fRT^ino re-oxidation wave only if the kinetic parameter X = — ~~^\ ,
where kc is the
chemical rate constant based on mole fractions and v the scan rate, were larger
than 0.1 [5]. The negative peak potential shift with a tenfold increase in v should
29.6 mVbe for a truly reversible electrode reaction. Even if the reduction were
nJ
59.1 mVquasi-reversible, the shift should not exceed [6]. The experimental
value of about 200 mV is far too high to account for any concept of type (c). The
64

huge peak potential shift suggests na = 1 for the rate-determining
electrochemical step and a a smaller than 0.5. Processes (a) and (b) remain the
only suitable models. A possible preceding equilibrium (b) could be the
protonation of peroxynitrous acid,
HOONO + H+ < [H2OONO]+ (1)
an idea which is supported by the slightly increased isomerisation rate of
peroxynitrous acid at pH < 3 [7]. An estimated pKa of about 2 for equilibrium
(1) is compatible with this observation. The settling of the protonation
equilibrium would be much faster than the electrochemical reduction, and
therefore the protonation equilibrium should shift the peak potential position by
RT K Xh?oono+AE = ln;7-r with K
anaF K+l XHOono
from the value without equilibrium. K is the conditional equilibrium constant for
a given buffer pH and pKa = 2. With the assumption of a = 0.5, the peak
potentials should differ by about 200 mV for pH 3.2 and 5.6 with na = 1 or by
100 mV with na = 2, given the same scan rates. Experimentally, this was not
found, the peaks appeared at nearly identical potentials for both pH values and
the same scan rates (Fig. 2). The peak potential difference was only 15 mV at
10 Vs-1 and smaller at lower scan rates. The lack of a significant pH dependent
peak shift rules out a protonation equilibrium. Another preceding equilibrium
(b) could be the homolysis of peroxynitrous acid which is claimed to take place
at rate of one third of the isomerisation rate: [8,9]
HOONO +=+ N02* + OH* (2)
This reaction would be almost pH independent below the pKa of
peroxynitrous acid. It is, however, rather unlikely that the cathodic current
measured was caused by the reduction of free OH* radicals, since their steady-
state concentration would be very low due to the fast recombination reactions to
HN03, HOONO and H202. The shape of the cathodic peak and its potential shift
with scan rate also imply a kinetically inhibited reduction, which is not expected
65

for OH* radicals. Since there were no other potential reactants in the
experimental setup, a purely irreversible mechanism of type (a) is consistent
with the data obtained. Linear sweep peaks of reaction type (a) can be analysed
1.857 RTfor ana by means of the relation |EP - Ep/2| =
~
[10]. Averaging of ana
for 11 peaks recorded at pH 3.2 yields ana = 0.26+0.06, and for 9 peaks at
pH 5.6, ana = 0.27+0.03 was found. Plots of ln(ip) vs. Ep have slopes with
ana = 0.21+0.01 for pH 5.6 and ana = 0.22+0.02 for pH 3.2, in good agreement
with the values derived from |EP - Ep/2| .The low values suggest na = 1. The rate-
determining step in the electrochemical reduction of peroxynitrous acid is
obviously the transfer of an electron to the molecule. If the electron transfer
would yield a moderately stable product, the reaction would be at least quasi-
reversible, since a pH independent one-electron transfer is most likely an
elemental reaction. Therefore it must be followed by a very fast and irreversible
chemical decay
HOONO+ e" [HOONO]*" (3)
[HOONO]*" OH" + N02* (4)
[HOONO]*" + H+ H20 + N02* (5)
so that no quasi-reversible signal can be observed with ordinary voltammetry
equipment. The formation of [HOONO]*" as an intermediate is not that far¬
fetched as it looks at first glance: it has been shown by numerous researchers
that nitrite forms N02*2" in pulse radiolysis experiments, when it is treated with
hydrated electrons [11-16].
The iodide oxidation by peroxynitrous acid is hardly pH dependent in the pH
range where peroxynitrite is extensively protonated. The rate law for the iodide
oxidation is ~~vr = k [HOONO] [I"]. This rate law and the constant we
measured are in agreement with the earlier observations by Goldstein and
Czapski [2], but the relative yields are not compatible with the mechanism
66

postulated later [3]. If the iodide oxidation were a two-electron process as
suggested, a yield of one equivalent of iodine per equivalent of peroxynitrous
should be attainable at sufficient iodide excess, which is not the case (Fig. 3).
Since the rate law was determined consistently in all cases, the combined
observations imply that a single electron transfer to HOONO should be rate-
limiting, as it is the case in the electrochemical reduction. Therefore, it is rather
probable that the same intermediate product as in the electrochemical reduction
is formed, [HOONO]*". The radical anion would behave as desribed above. The
scheme explains easily why the first half-equivalent of I2 is formed
stochiometrically even at small iodide concentrations, while the formation of the
second half-equivalent remains incomplete even at high iodide excess, since the
reaction
N02* + I"< N02" + I* (6)
is an equilibrium with a constant of about 10" [17].
Since we found the electrochemical reduction of peroxynitrous acid to be pH
independent, its reduction potential should also not depend on pH.
Thermodynamic calculations of the one-electron reduction potential of
peroxynitrous acid by Merényi and Lind [18] and Koppenol and Kissner [19]
assumed that it is:
ONOOH + H+ + e +=* N02' + H20. (7)
Values of 2.14 V and 2.0 V were published by Merényi and Lind, [18] and
Koppenol and Kissner [19] respectively, and refer to standard conditions. These
values are based on determinations and estimates of the standard Gibbs energy
of formation of peroxynitrous acid, see Appendix. If the proton dependence is
eliminated from equation (7) to
ONOOH + e < » N02' + OH" (8)
the reduction potentials mentioned above can be recalculated. With new values
of Gibbs energy of formation of peroxynitrous acid from Merényi and co¬
workers [20], Lymar and Poskrebyshev [21] and our own value, 1.67 ± 0.03 V,
67

1.65 ± 0.02 V and 1.64 V, respectively, are obtained. For the calculation of the
reduction potential, the following values were used:
A/G°(N02', aq) = 63±1 kJ/mol [22] and AfG°(UO~, aq) = -157.3 kJ/mol [23].
These adjusted values are still insufflent to describe the oxidative properties
of peroxynitrous acid, since they are based on the assumption that N02" and HO
are in equilibrium with HOONO, which is surely not the case according to the
electrochemical results. In order to determine the correct one-electron reduction
potential of peroxynitrous acid by thermodynamic calculation, one would have
to know the Gibbs energy of formation of [HOONO]*".
68

HON02 -*—~-^ HO*, NO2
k
Appendix
Standard Gibbs energy of formation
In 2003 Merényi and coworkers adjusted their former value by studying the
equilibrium ONOOH + H20+ H+ +=+ H202 + HN02 + H+ (9)
and derived a standard Gibbs energy of formation of +(67.4 ± 1.3) kJ mol"1 for
peroxynitrite anion [20].
In the same year Lymar and Poskrebyshev published a value for the standard
Gibbs energy of formation of +(30.8 ± 1) kJ mol"1 [21]. The value was obtained
from a study of the reaction
ONOO « » NO' + 02' (10)
by pulse radiolysis in the presence of methyl viologen.
Three determinations of the
enthalpy of formation of
peroxynitrite have been+162 + 87
published; they originate from
calorimetric measurements of the
heat of isomerisation of H*,O-N00r ^— 0=NOOH
-, -, , -, , TT , f.Fie. 4. Enthalpy cycle according to Mahoney
peroxynitrite to nitrate. Heats 01n„-m 1 ; j
•
+u- uv +- ah^ J(1970), as completed in this publication. All
isomerisation of -38.8 ± 2 kcal values ink! mol1.
mol"1 [24], -37.8 ± 0.4 kcal mol"1 [25] and -163 ±4 kJ mol"1 [26] have been
reported. In relation to the standard enthalpy of formation of nitrate,
-205.0 kJmol"1 [27], the standard enthalpies of formation for peroxynitrite can
be calculated: -43 ±8,-47 + 2 and -42 ± 4 kJmol"1, respectively. We adopt a
value of -43 ± 4 kJ mol"1, where the uncertainty covers all published values.
With this value and the enthalpy of ionisation of peroxynitrous acid, the
thermodynamic cycle of Mahoney [28] can be completed, see Fig. 4. We note
that the enthalpy of homolysis in water, +87 kJ mol1, is quite close to that
computed for the gas phase, +94 kJ mol1 [29], while for the 0-0 bond
69

dissociation energy in the gas phase a value of +82 kJ mol1has been reported
[30].
In 1992, Koppenol et al. estimated that S°(0=NOO") is +188 J K"1 mol"1,
based on S°(N03") = +146 J K"1 mol"1 plus the difference between S°(ONOO*)
and S°(N03'), 42 J K^mol"1 [31]. This resulted in an estimated standard Gibbs
energy of formation of peroxynitrite of +42 kJ mol1, a value that precludes
homolysis. We were criticised [18], because our estimate implies that the
entropies of solution for nitrate and peroxynitrite are the same. Adoption of a
thermodynamic cycle regarding the entropy of peroxynitrite and peroxynitrous
acid [18], with incorporation of the entropy of ionisation, -16 J K"1 mol"1
resulted in an estimate of+58 ± 10 kJ mol"1 for AfG°(ONOO")aq [19]. The large
error is caused for a large part by that in determination of the enthalpy of
ionisation of peroxynitrous acid.
This estimate can be improved. For the entropy of formation of peroxynitrous
acid in various conformations in the gas phase values between +274 and
+281 JK"1 mol"1 have been derived by ab initio methods [29]. These entropies
are significantly lower than our earlier estimate of +301 J K"1 mol"1 [19],
derived from the entropy of oxoperoxonitrogen(»), +293 J K"1 mol"1 [33]. The
+ 293 0=NOO- - 0=NOOHn +278y
8
179
105
smaller entropy is_ ^ 5
probably due to the
ability of
peroxynitrous acid to +285 0=NOO
form an internal
hydrogen bond. We
adopt a value of +106 0=NOO-aq^ — 0=NOOHaq +173
J mo'
^ eFig 5. Entropy cycle [32] with revised values. All values in
average of the two eis-
isomers, from which follows a value of +106 J K"1 mol"1 for ^(ONOO" )aq, see
Fig. 5, and this, in turn, results in a AfG°(ONOO")aq of +65 kJ mol" \ However,
70

this result has still the same uncertainty as the old estimate, and the entropy of
hydration of the peroxynitrite anion is quite negative when compared with other
four-atom monovalent anions [34].
Another estimate can be obtained from a cycle centred on the Gibbs energy of
formation of peroxynitrous acid in the gas phase calculated by ab initio methods
[29]. The Gibbs energy of homolysis for cw,cw-ONOOH in the gas phase is
given as +50.6 kJ mol" \ We note that the entropy change ofthat reaction results
in a TAS term of +45.2 kJ mol"l, which is more than +(38 ± 2) kJ mol"1 as found
for similar reactions. The Gibbs hydration energies of nitrogen dioxide and
hydroxyl are known (together +3 kJ mol"l) and that of peroxynitrous acid can be
estimated as follows. The cost of forming a cavity in water is +17 kJ mol"1,
which is compensated by ca. -8 kJ mol"1 for every hydrogen bond between
solute and solvent [35]. For peroxynitrous acid we assume that it can form half
as many hydrogen bonds as hydrogen peroxide, for which a Gibbs hydration
energy of - 28 kJ mol"1 is calculated from the Henry constant of 7.74 x 104 [36].
These considerations result in a Gibbs energy of hydration of peroxynitrous acid
of - 6 kJ mol"l, and a Gibbs energy of formation of +29 kJ mol"1 (+65 kJ mol"1
for ONOO").
Although these two estimates yield the same value for the standard Gibbs
energy of formation of peroxynitrite, they both involve parameters from an ab
initio study [29] Given the assumption regarding the hydration energy of
peroxynitrous acid, we estimate the error to be smaller than the previous
estimate but still more than 5 kJ mol- 1.
Acknowledgement - This project was supported by the ETH and the Swiss
Nationalfonds.
71

References
[1] Kissner, R.; Nauser, T.; Bugnon, P.; Lye, P. G.; Koppenol, W. H.
Formation and properties of peroxynitrite studied by laser flash
photolysis, high pressure stopped flow and pulse radiolysis. Chem. Res.
Toxicol. 10:1285-1292; 1997.
Goldstein, S.; Czapski, G. Direct and indirect oxidations by peroxynitrite.
Inorg. Chem. 34:4041-4048; 1995.
Goldstein, S.; Meyerstein, D.; van Eldik, R.; Czapski, G. Spontaneousreactions and reduction by iodide of peroxynitrite and peroxynitrate:Mechanistic insight from activation parameters. J. Phys. Chem. A
101:7114-7118; 1997.
Matsuda, H.; Ayabe, Y. Zur Theorie der Randies-Sevcikschen
Kathodenstrahl-Polarographie. ZElektrochem 59:494-503; 1955.
Nicholson, R. S.; Shain, I. Single scan and cyclic methods applied to
reversible, irreversible, and kinetic systems. Anal. Chem. 36:706-723;1964.
Nadjo, L.; Saveant, J. M. Linear sweep voltammetry: kinetic control by
charge transfer and/or secondary cehmical reactions. J. Electroanal. chem.
48:113-145; 1973.
Benton, D. J.; Moore, P. Kinetics and mechanism of the formation and
decay of peroxynitrous acid in perchloric acid solutions. J. Chem. Soc. (A)3179-3182; 1970.
Merényi, G.; Lind, J.; Goldstein, S.; Czapski, G. Peroxynitrous acid
homolyzes into OH and N02 radicals. Chem. Res. Toxicol. 11:712-713;1998.
Lymar, S. V.; Khairutdinov, R. F.; Hurst, J. K. Hydroxyl radical
formation by 0-0 bond homolysis in peroxynitrous acid. Inorg. Chem.
42:5259-5266; 2003.
Bard, A. J.; Faulkner, L. R. Electrochemical Methods. John Wils & sons
Inc.; 1980:222-224.
Elliot, A. J.; McCracken, D. R.; Buxton, G. V.; Wood, N. D. Estimation
of rate constants for nearly-diffusion-controlled reactions in water at high
temperature. J. Chem. Soc. ,Faraday Trans. 86:1539-1547; 1990.
Broszkiewicz, R. K. The pulse radiolysis study ofNaN02 and NaN03
solutions. Bull. Acad. Pol. Sei.,Ser. Sei. Chim. 24:221-229; 1976.
72

[13] Fei, N. S. Determination of the rate constant of a reaction of e-aq with an
acceptor in neutral water by pulsed method. Khimiya Vysokikh Energii
4:178-180; 1970.
[14] Cercek, B. Activation energies for reactions of the hydrated electron.
Nature 223:491-492; 1969.
[15] Baxendale, J. H.; Fielden, E. M.; Keene, J. P. The pulse radiolysis of
aqueous solutions of some inorganic compounds. Proc. Roy. Soc.,Ser. A
286:320-336; 1965.
[16] Thomas, J. K.; Gordon, S.; Hart, E. J. The rates of reaction of the hydratedelectron in aqueous inorganic solutions. J. Phys. Chem. 68:1524-1527;1964.
[17] Barkett, A.; Ottolenghi, M. Laser flash photolysis of aqueous triiodide
solution. Mol. Photochem 6:253-261; 1974.
[18] Merényi, G.; Lind, J. Thermodynamics of peroxynitrite and its C02-
adduct. Chem. Res. Toxicol. 10:1216-1220; 1997.
[19] Koppenol, W. H.; Kissner, R. Can ONOOH undergo homolysis. Chem.
Res. Toxicol. 11:87-90; 1998.
[20] Merényi, G.; Lind, J.; Czapski, G.; Goldstein, S. Direct determination of
the Gibbs' energy of formation of peroxynitrous acid. Inorg. Chem.
42:3796-3800; 2003.
[21] Lymar, S.V.; Poskrebyshev, G. A. Rate of ON-OO- bond homolysis and
the Gibbs energy of formation of peroxynitrite. J. Phys. Chem. A
107:7991-7996; 2003.
[22] Stanbury, D. M. Reduction potentials involving inorganic free radicals in
aqueous solution. Adv. Inorg. Chem. 33:69-138; 1989.
[23] Ross, P. N. Oxygen. In Bard, A. J.; Parsons, R.; Jordan, J. eds. Standard
Potential in Aqueous Solution. New York: Marcel Dekker, INC; 1985:49-
66.
[24] Ray, J. D. Heat of isomerization of peroxynitrite to nitrate and kinetics of
isomerization of peroxynitrous acid to nitric acid. J. Inorg. Nucl. Chem.
24:1159-1162; 1962.
[25] Bohle, D. S.; Hansert, B.; Paulson, S. C; Smith, B. D. Biomimetic
synthesis of the putative cytotoxin peroxynitrite, ONOO", and its
characterization as a tetramethylammonium salt. J. Am. Chem. Soc.
116:7423-7424; 1994.
73

[26] Manuszak, M.; Koppenol, W. H. The enthalpy of isomerization of
peroxynitrite to nitrate. Thermochim. Acta 273:11-15; 1996.
[27] Wagman, D. D.; Evans, W. H.; Parker, V. B.; Schumm, R. H.; Halow, I.;
Bailey, S. M.; Churney, K. L.; Nuttal, R. L. Selected values for inorganicand CI and C2 organic substances in SI units. J. Phys. Chem. Ref. Data
11 (Suppl. 2)37-38; 1982.
[28] Mahoney, L. R. Evidence for the formation of hydroxyl radicals in the
isomerization of pernitrous acid to nitric acid in aqueous solution. J. Am.
Chem. Soc. 92:5262-5263; 1970.
[29] McGrath, M. P.; Rowland, F. S. Determination of the barriers to internal
rotation in ONOOX (X = H, CI) and characterization of the minimum
energy conformers. J. Phys. Chem. 98:1061-1067; 1994.
[30] Olson, L. P.; Bartberger, M. D.; Houk, K. N. Peroxynitrate and
peroxynitrite: A complete basis set investigation of similarities and
differences between these NOx species. J. Am. Chem. Soc. 125:3999-
4006;2003.
[31] Koppenol, W. H.; Moreno, J. J.; Pryor, W. A.; Ischiropoulos, H.;
Beckman, J. S. Peroxynitrite, a cloaked oxidant formed by nitric oxide
and superoxide. Chem. Res. Toxicol. 5:834-842; 1992.
[32] Koppenol, W. H.; Kissner, R. Can 0=NOOH undergo homolysis? Chem.
Res. Toxicol. 11:87-90; 1998.
[33] Benson, S. W. Thermochemical Kinetics: New York John Wiley & Sons;1976.
[34] Marcus, Y. The hydration entropies of ions and their effects on the
structure of water. J. Chem. Soc., Faraday Trans. 1 82:233-242; 1986.
[35] Schwarz, H. A.; Dodson, R. W. Equilibrium between hydroxyl radicals
and Thallium (II) and the oxidation potential of OH(aq). J. Phys. Chem.
88:3643-3647; 1984.
[36] Sander, R. Henry's Law constants. In Linstrom, P. J.; Mallard, W. G. eds.
NIST Chemistry WebBook, NIST Standard Reference Database Number
69. Gaithersburg, MD 20899: National Institute of Standards and
Technology; 2003.
74
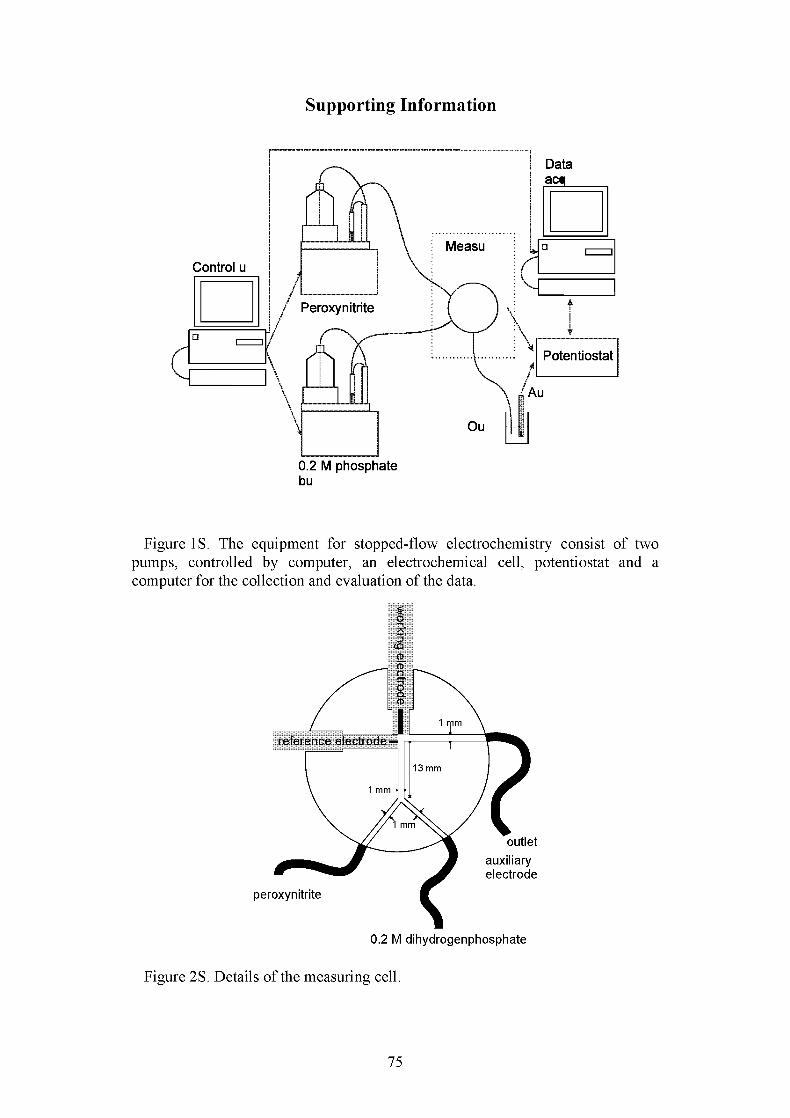
Supporting Information
0.2 M phosphatebu
Figure IS. The equipment for stopped-flow electrochemistry consist of two
pumps, controlled by computer, an electrochemical cell, potentiostat and a
computer for the collection and evaluation of the data.
peroxynitrite
outlet
auxiliaryelectrode
0.2 M dihydrogenphosphate
Figure 2S. Details of the measuring cell.
75

76

7. RAPID SCAVENGING OF PEROXYNITROUS
ACID BY MONOHYDROASCORBATE
(Free Radical Biol. Med. 35:1529-37; 2003)
CHRISTOPHE R. KURZ, REINHARD KISSNER, THOMAS NAUSER,
DANIEL PERRIN and WILLEM H. KOPPENOL*
Laboratorium für Anorganische Chemie, Eidgenössische Technische
Hochschule Zürich (Hönggerberg), Switzerland
Abstract— The reaction of peroxynitrous acid with monohydroascorbate,
over the concentration range of 250 uM to 50 mM of monohydroascorbate at
pH 5.8 and at 25°C, was reinvestigated and the rate constant of the reaction
found to be much higher than reported earlier (Bartlett, D.; Church, D. F. ;
Bounds, P. L.; Koppenol, W. H., The kinetics of oxidation of L-ascorbic acid
by peroxynitrite, Free Radical Biol. Med. 18:85-92; 1995, Squadrito, G. L.;
Jin, X.; Pryor,W. A., Stopped-flow kinetics of the reaction of ascorbic acid
with peroxynitrite, Arch. Biochem. Biophys. 322:53-59; 1995). The new rate
constants at pH 5.8 are kx= 1 x 106M"V and k.x= 500 s"1 for 25°C, and
h= 1.5 x 106 M"V and k.x= 1 x 103 s"1 for 37°C. These values indicate that
even at low monohydroascorbate concentrations most of peroxynitrous acid
forms an adduct with this antioxidant. The mechanism of the reaction
involves formation of an intermediate, which decays to a second intermediate
with an absorption maximum at 345 nm. At low monohydroascorbate
concentrations, the second intermediate decays to nitrate and
monohydroascorbate, while at monohydroascorbate concentrations greater
than 4 mM, this second intermediate reacts with a second
monohydroascorbate to form nitrite, dehydroascorbate, and
monohydroascorbate. EPR Experiments indicate that the yield of the ascorbyl
radical is 0.24 % relative to the initial peroxynitrous acid concentration, and
77

that this small amount of ascorbyl radicals is formed concomitantly with the
decrease of the absorption at 345 nm. Thus, the ascorbyl radical is not a
primary reaction product. Under the conditions of these experiments, no
homolysis of peroxynitrous acid to nitrogen dioxide and hydroxyl radical is
observed. Aside from monohydroascorbate's ability to "repair" oxidatively
modified biomolecules, it may play a role as scavenger of peroxynitrous acid.
Results
Stoppedflow studies
Kinetic traces recorded at 345 nm, 25°C and at pH 5.8 after mixing of
peroxynitrite with different concentrations of monohydroascorbate (0.25 -
50 mM) show an increase in absorption during 20 - 40 ms, followed by
decrease. At monohydroascorbate concentrations higher than 1 mM, the rate
of this previously unobserved increase in absorption depends on
monohydroascorbate (Fig. 1), while a dependence on peroxynitrous acid is
observed over the whole concentration range (not shown).
The decrease in absorption, which begins 50 - 100 ms after mixing, was
measured before at higher monohydroascorbate concentrations [1,2]. The
experiments at these concentrations (Fig. 1, traces C and D) are in agreement
with the cited literature, and show a faster decrease of absorption at higher
monohydroascorbate concentrations. The rate of decrease is not affected by
the peroxynitrous acid concentration.
The insert of Fig. 1 shows that directly after mixing there is a decrease in
absorption. This process clearly had started already during the mixing and
was difficult to quantify. At 345 nm, monohydroascorbate does not absorb,
and the absorption directly after mixing and that after ca. 40 ms are
significantly higher than those expected for peroxynitrous acid alone, see
below. These observations indicate that there may be two intermediates, Im!
and Im2, of which Im2 has an absorption maximum at 345 nm.
78

0.0 0.5 1.0 1.5 2.0 2.5 3.0time /s
Fig. 1. Time course of the absorption at 345 nm after mixing
monohydroascorbate with peroxynitrous acid. 25 uM peroxynitrous acid mixed
with A, 250 uM; B, 1 mM; C, 8 mM, and D, 50 mM monohydroascorbate. All
concentrations indicated are those after mixing. All traces were recorded at
345 nm, 25°C and pH 5.8 (50 mM phosphate buffer). The traces C and D are in
agreement with earlier work [1,2] where higher ONOOH and
monohydroascorbate concentrations were used. At lower monohydroascorbateconcentrations (trace A and B) no dependence on the monohydroascorbateconcentration is observed. Each curve is an average of 12 measurements. Inset:
A fast decrease in absorption that depends on the monohydroascorbateconcentration is observed during the first 6 ms.
The transient absorption with a maximum at 345 nm (Fig. 2, spectra A and
B) has not been observed before. We note that the spectrum of the ascorbyl
radical (Fig. 2, trace C), determined by pulse radiolysis of a dinitrogen
monoxide saturated solution of 0.50 mM monohydroascorbate, shows a
maximum at 360 nm in agreement with the literature [3,4].
79
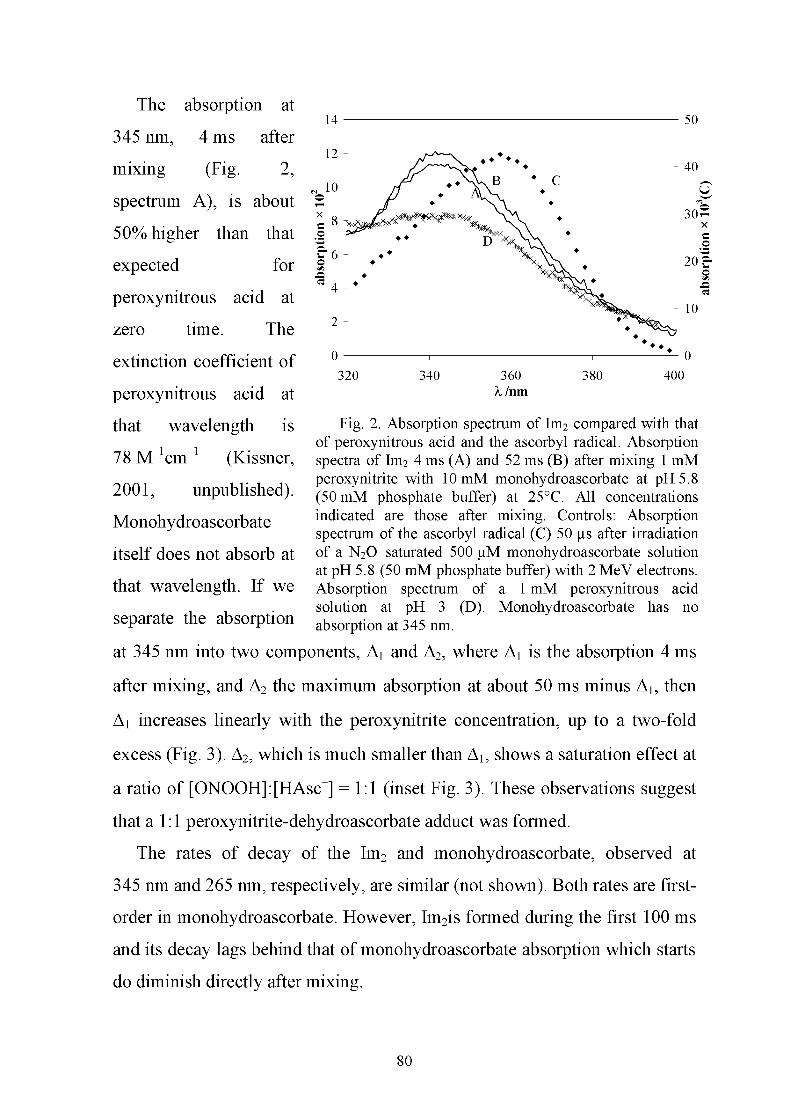
The absorption at
345 nm, 4 ms after
mixing (Fig. 2,
spectrum A), is about
50% higher than that
expected for
peroxynitrous acid at
zero time. The
extinction coefficient of
peroxynitrous acid at
that wavelength is
78 M^cm"1 (Kissner,
2001, unpublished).
Monohydroascorbate
itself does not absorb at
that wavelength. If we
separate the absorption
at 345 nm into two components, Ai and A2, where Ai is the absorption 4 ms
after mixing, and A2 the maximum absorption at about 50 ms minus A1; then
Ai increases linearly with the peroxynitrite concentration, up to a two-fold
excess (Fig. 3). A2, which is much smaller than Ai, shows a saturation effect at
a ratio of [ONOOH] : [HAsc-] = 1:1 (inset Fig. 3). These observations suggest
that a 1:1 peroxynitrite-dehydroascorbate adduct was formed.
The rates of decay of the Im2 and monohydroascorbate, observed at
345 nm and 265 nm, respectively, are similar (not shown). Both rates are first-
order in monohydroascorbate. However, Im2is formed during the first 100 ms
and its decay lags behind that of monohydroascorbate absorption which starts
do diminish directly after mixing.
80
320 340 360
1/nm
380 400
Fig. 2. Absorption spectrum of Im2 compared with that
of peroxynitrous acid and the ascorbyl radical. Absorption
spectra of Im2 4 ms (A) and 52 ms (B) after mixing 1 mM
peroxynitrite with 10 mM monohydroascorbate at pH5.8
(50 mM phosphate buffer) at 25°C. All concentrations
indicated are those after mixing. Controls: Absorption
spectrum of the ascorbyl radical (C) 50 us after irradiation
of a N2Û-saturated 500 uM monohydroascorbate solution
at pH 5.8 (50 mM phosphate buffer) with 2 MeV electrons.
Absorption spectrum of a 1 mM peroxynitrous acid
solution at pH 3 (D). Monohydroascorbate has no
absorption at 345 nm.

o9 -
o8 -
o
s? -
x
§5- o
Pi
-O
a4- O
5" On
2 -
—
1 -
o -
300
UM ONOOH
400 500
0 1
-- 5
n
O
s
J"SB
as
600
time /s
Fig. 3. Kinetics of the formation of Im2 as a function of the peroxynitrous acid
concentration. Ascorbic acid, 250 uM, mixed with peroxynitrous acid A, 25 uM;
B, 50 uM; C, 125 uM; D, 250 uM; E, 375 uM; F, 500 uM. Inset: Absorption after mixing
monohydroascorbate (250 uM final concentration) with variable concentrations of
peroxynitrous acid final concentrations separated into Ai (; absorption 4 ms after mixing)
and A2 (o; absorption difference between Ai and maximal absorption after about 50 ms ).
From measurements over a wide range of monohydroascorbate
concentrations, it is clear that the rates of formation and decay of Im2 are
dependent on the monohydroascorbate concentration. It is implied that the
rate of formation of the first intermediate also depends on the
monohydroascorbate concentration. As shown in Figure 1, Im2 is already
present at very low monohydroascorbate concentrations, while at higher
monohydroascorbate concentrations the rate of formation increases with a
slope of ka = 1.6 x 103 M_1s_1 (Fig. 4).
81
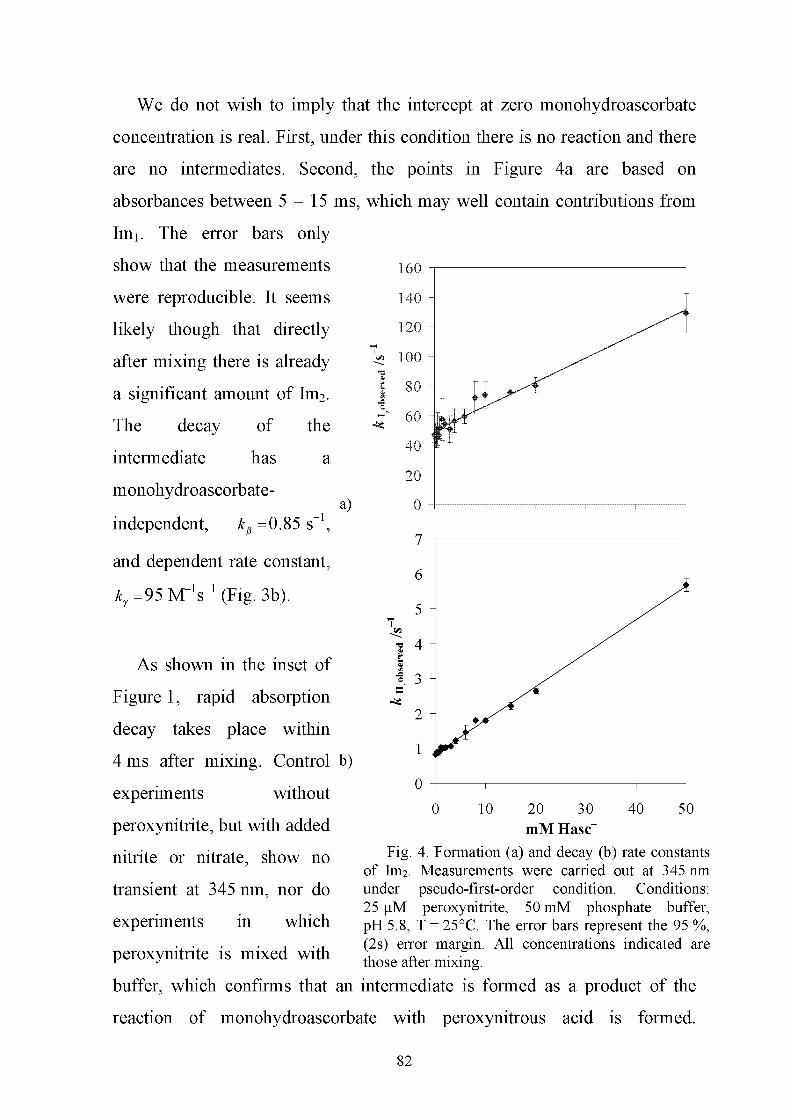
We do not wish to imply that the intercept at zero monohydroascorbate
concentration is real. First, under this condition there is no reaction and there
are no intermediates. Second, the points in Figure 4a are based on
absorbances between 5-15 ms, which may well contain contributions from
Iirii. The error bars only
show that the measurements i60
were reproducible. It seems
likely though that directly
after mixing there is already
a significant amount of Im2.
The decay of the
intermediate has a
monohydroascorbate-
independent, kß =0.85 s-1,
and dependent rate constant,
ytr=95M1s"1(Fig. 3b).
As shown in the inset of
Figure 1, rapid absorption
decay takes place within
4 ms after mixing. Control b)
experiments without
peroxynitrite, but with added
nitrite or nitrate, show no
transient at 345 nm, nor do
experiments in which
peroxynitrite is mixed with
0 10 40 5020 30
mM Hasc"
Fig. 4. Formation (a) and decay (b) rate constants
of Im2. Measurements were carried out at 345 nm
under pseudo-first-order condition. Conditions:
25 uM peroxynitrite, 50 mM phosphate buffer,
pH5.8, T = 25°C. The error bars represent the 95 %,
(2s) error margin. All concentrations indicated are
those after mixing.
buffer, which confirms that an intermediate is formed as a product of the
reaction of monohydroascorbate with peroxynitrous acid is formed.
82

Furthermore, we observed no influence of nitrite, nitrate, or dioxygen on the
kinetics (not shown).
EPR studies
We observed a small
signal with a g-value of
2.005 which is due to
the ascorbyl radical.
This signal was not
detected without
monohydroascorbate or
peroxynitrite. Directly
after mixing
25 uM peroxynitrite and
250 uM monohydro¬
ascorbate, no EPR
signal was seen; instead,
this signal rises for
approximately
2 seconds and
subsequently disappears
by second-order kinetics within 10 seconds (Fig. 5, trace B). Compared with
the optical kinetic trace recorded at 345 nm (Fig. 5, trace A), it is clear that
the concentration of ascorbyl radicals increases while that of Im2 decreases.
The total ascorbyl radical concentration formed during the reaction was
estimated at only 60 nM. Because the ascorbyl radical decays by second-order
kinetics [3,5], its concentration could be estimated to be 20 nM at t = 2 s.
Special care was taken to avoid contamination by dioxygen and transition
metals, which also produce ascorbyl radicals [6].
83
time /s10
Fig. 5. Comparison of EPR and UV-Vis results.
The decay at 345 nm (A) is compared with an EPR
trace, B, that shows formation and disappearance of
ca. 20 nM ascorbyl radical. Trace B increases
concomitant with the decrease of trace A. Curve A is
an average of 12 measurements and curve B an
average of 10 measurements. Both measurements
were made at initial concentrations of 25 uM
peroxynitrite and 250 uM monohydroascorbate in
50 mM phosphate buffer at pH 5.8. All concentrations
indicated are those after mixing.

Product analysis
When 25 uM peroxynitrite and increasing amounts (25 uM - 50 mM) of
monohydroascorbate are mixed, one observes that nitrite increases,
concomitant with a decrease in nitrate. At 2 mM monohydroascorbate equal
amounts of nitrite and nitrate are produced (not shown), and at concentrations
higher than 10 mM saturation is observed. At lower monohydroascorbate
concentrations, most of the peroxynitrous acid is converted to nitrate. Without
monohydroascorbate, peroxynitrous acid yields already 13 % nitrite at pH 5.8,
in agreement with the literature [7].
Discussion
Earlier work
The reaction of peroxynitrous acid with monohydroascorbate has already
been studied by Bartlett et al. [1] and by Squadrito et al. [2]. They concluded
that the reaction was first-order in peroxynitrous acid and in
monohydroascorbate with a rate constant of 235 M" V \ The rapid increase in
absorption during the first 100 ms after mixing (Fig. 1) was not seen by these
workers because vibrations, generated when the flow was stopped, prevented
accurate measurements during 50 ms after mixing. Based on the observation
of the EPR spectrum of the ascorbyl radical, Bartlett et al. [1] concluded that
this radical was a product. We found only a very small EPR signal, and the
corresponding ascorbyl concentration would amount to much less than one
percent of the peroxynitrous acid concentration. The intermediate Im2 which
exhibits an absorbance maximum at 345 nm is thus not identical to the
ascorbyl radical which has an absorption maximum at 360 nm (Fig. 2).
Above pH 5.8 the reaction proceeds more slowly, as was reported earlier
[1]. A manuscript on the reaction of peroxynitrite with monohydroascorbate
near neutral pH is in preparation.
84

ONOO" + H"1
Kr, k-6
ONOOH + HAsc^ ^^
k-i
N(V + H/^
HAsc^ + DAsc + NO?"+H7O9 T Ilj\
Ktz i
ko
[111,^1^ Illl2k_?
kj
HAsc"
345
HAsc"' + NO3" + H
Scheme 1.
Intermediates
The important findings in this paper are that the absorbance at 345 nm ca.
5 ms after mixing is higher than that expected for peroxynitrous acid alone,
that it continues to rise for ca. 0.1 s and then decreases; both processes are
monohydroascorbate-dependent. The increase in absorbance is also dependent
on the peroxynitrous acid concentration. Our attempt to fit these observations
to a reaction scheme that includes a single intermediate was not successful. It
appeared necessary to invoke a second intermediate, appearing prior to Im2,
as shown in Scheme 1. Evidence for the first intermediate is two-fold: at high
concentrations of monohydroascorbate, a decrease in absorption is observed
directly after mixing (inset Fig. 1), and, at that time, there appears to be
already a significant amount of Iirii. We have no data that would provide
information regarding the precise structures of these intermediates. As shown
in inset of Figure 3, the absorption difference A2 shows a saturation effect at
[ONOOH] > [HAsc-]. From this, as suggested by Bartlett et. al. [1], we
conclude that Im2 is a 1:1 adduct of peroxynitrous acid and
monohydroascorbate. We already mentioned that Im2 is not the ascorbyl
radical, as the EPR signal of the ascorbyl radical increases while the
concentration of Im2 decreases (Fig. 5).
Kinetics andMechanism
85

We postulate that Imi is formed very rapidly from monohydroascorbate
and peroxynitrous acid, that Im2 is formed from Imi, and that the former
decays spontaneously and by reaction with a second monohydroascorbate
molecule. Since the concentration of the second intermediate (Im2) increases
with that of peroxynitrous acid (Fig. 3), we conclude that Imi and Im2 are in
equilibrium. We observed the reaction at 345 nm, the absorbance maximum
of Im2; we assume that Imi also absorbs at this wavelength. The rates of
formation of Imi and Im2 increase with the monohydroascorbate
concentration, as shown in Figure 4a. These rates are extracted from the
increase in absorption 5 -15 ms after mixing and are subject to a considerable
error. We assume that, when the monohydroascorbate concentration
approaches zero, kx in Figure 4a is zero. We cannot verify that, because below
500 uM monohydroascorbate no significant rise is observed. The rate
constant for the increase in absorption at 345 nm at monohydroascorbate
concentrations larger than 500 uM (Fig. 4a) is 1.6 x 10 M s.
The decay of Im2, follows the following rate law:
ku,obServed =kß +kr[ascorbate] with kp =0.85 s"1 and ky =95 M_1s_1
The monohydroascorbate-independent pathway yields nitrate and
monohydroascorbate; the decay rate constant kß is smaller than the
isomerization rate constant of peroxynitrous acid at pH 5.8, which is another
indication that at least one intermediate is present. From the faster decay of
Im2 at increased monohydroascorbate concentrations and the product analysis,
we conclude that Im2 interacts with another monohydroascorbate molecule
and produces dehydroascorbate, monohydroascorbate and nitrite.
86

Rate
constantRate value Remark Compound
Absorption Initial
coefficient concentration
ki lxlO^'V a HAsc 0 M'W1250 u.M-
50 mM
k.i 5xl02 s"1 a HOONO 82 M'W1 25 uM
k2 40 s"1 a ONOO" 685 M'W1 —
k.2 5 s"1 a N03 0 M'W1 —
k3 1.2 s"1 b N02 5 M'W1 —
k4 0.85 s"1 c DAsc 0 M'W1 —
k5 lx^M'V c Imi 250 M'W1 —
k6 lxlO^-^s"1 d Im2 310 M'W1 —
k.6 1.58xl03s_1 e
Table 1. Values used for the simulation of the reaction of peroxynitrous acid
with monohydroascorbate at 345 nm and 25°C. a, determined by simulation; b,Kissner et all. [7]; c, rate constant determined by decay of Im2; d, rate constant for
protonation; e, rate constant determined by pKa [13].
Kinetic traces of the reaction between peroxynitrous acid and
monohydroascorbate were simulated for 25°C and 37°C based on Scheme 1.
These simulations were carried out for variable monohydroascorbate
concentrations at fixed concentrations of peroxynitrous acid and reasonable to
good agreement with the experimental results was found. The rate constants
and extinction coefficients used in the simulation are listed in Table 1.
Importantly, the forward rate constants for the reaction of peroxynitrous acid
with monohydroascorbate at pH 5.8 are 1 x 106 M_1s_1 and 1.5 x 106 M_1s_1 at
25°C and 37°C, respectively. The rate constants for the backward reaction are
5 x 102 s"1 and 1 x 103 s"1 at 25°C and 37°C, respectively (not shown). From
these rate constants it can be calculated that at 1 uM peroxynitrous acid and
87

500 uM monohydroascorbate every second peroxynitrous acid molecule is
scavenged by an monohydroascorbate. While the agreement between
simulation and experiment is satisfactory, we do not exclude the possibility of
improvements to this model.
Homolysis or one-electron oxidation?
In the literature contrasting views are expressed with respect to the issue of
peroxynitrous acid to nitrogen dioxide and hydroxyl radicals [7-13]. Both
hydroxyl and nitrogen dioxide radicals react with high rate constants of
7.0 x KflVrV1 [3] and 3.5 x 107 M'V [14] respectively, with
monohydroascorbate and yield the ascorbyl radical. The reaction of
peroxynitrous acid with low concentrations of monohydroascorbate takes
place during 3-4 s (Fig. 1), which is also the time span of the isomerization of
peroxynitrous acid to nitrate at 25°C.
Under these conditions we estimate from our EPR experiments that the
maximal concentration of ascorbyl radical formed during the reaction is about
20 nM. This concentration is too small to be part of the main reaction path. It
is possible that the ascorbyl radicals observed are not produced from the
reaction between monohydroascorbate and peroxynitrous acid at all, but from
the oxidation of monohydroascorbate by dioxygen, a reaction catalyzed by
transition metals present as trace contamination [6,15].
A mechanism consisting of two fast sequential one-electron oxidations,
one by peroxynitrous acid and one by nitrogen dioxide, can be excluded,
because both oxidations would yield ascorbyl radicals.
It is conceivable that the ascorbyl radical was not observed because it
reacted rapidly with another peroxynitrous acid. In that case there would have
been an increase in ascorbyl radicals at higher monohydroascorbate
concentrations. No such increase was observed.
88

Physiological relevance
Can monohydroascorbate compete with carbon dioxide at physiological
pH and temperature? The reaction of peroxynitrite with carbon dioxide
proceeds with a rate constant of 5.8 x 104 M_1s_1 at 37°C [16]. If we assume
that in a very short time 10 uM peroxynitrite were formed in a cell, then the
product distribution of rate constants and concentrations for removal via the
carbon dioxide or the monohydroascorbate pathway can be simulated by
adding the reaction of peroxynitrite with carbon dioxide to Scheme 1. A
simulation for 1 mM carbon dioxide, 100 uM monohydroascorbate (human
serum) and 10 uM peroxynitrite at pH7.3 and 37°C, shows that
monohydroascorbate diverts only ca. 3.5 % of the peroxynitrite (not shown).
At monohydroascorbate concentrations of 10 mM, as found in neurons,
peroxynitrite is trapped to an extent of 67 %. If the reaction between
peroxynitrite and carbon dioxide involves an equilibrium [17], then
monohydroascorbate will, even at low monohydroascorbate concentrations,
curtail the reaction with carbon dioxide. Our studies were carried out at pH
5.8, which may be relevant to infections, where the pH may be significantly
lower than 7.3. Under such conditions monohydroascorbate at physiological
concentrations may provide protection against peroxynitrous acid induced
oxidation and nitration [18-20]. A study of the pH dependence on the reaction
of monohydroascorbate and peroxynitrite is in progress.
89

Speculations as to the structures of the intermediates.
Some efforts to establish the structures of the intermediate were made, but
failed. Therefore we are able to suggest only structures that are based on the
known reaction pathways of peroxynitrite and monohydroascorbate.
A possible reaction scheme between monohydroascorbate and peroxynitrous
acid, including a monohydroascorbate dependent and independent decay
pathway, is drawn in Figure 1.
HO.
H*
HO
"OH h
^°^?—o+
0*)0
\ /u \\J /
/ \ - N-0
HO
HO
?>O
H
HO \ 0\
H V—-SN-0
o-o
A
+ HAsc
HO OIO
H
+ N09
HO O
HO PH ,0
Fig. 1
HAsc
DAsc
H20NO"
HAsc
H20NO"
HO O
H
O" O
90
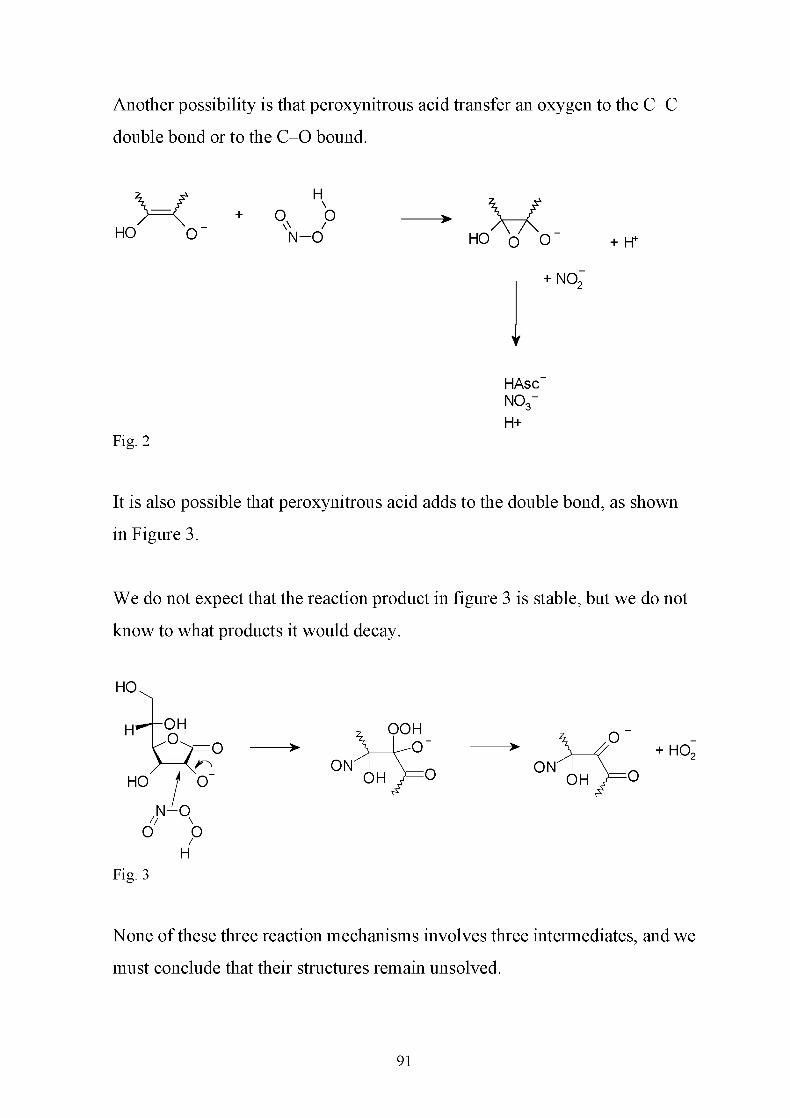
Another possibility is that peroxynitrous acid transfer an oxygen to the C-C
double bond or to the C-0 bound.
HO O
H
o bN-0 HO o O" +HT
+ NO,
Fig. 2
HAsc"
NO3-H+
It is also possible that peroxynitrous acid adds to the double bond, as shown
in Figure 3.
We do not expect that the reaction product in figure 3 is stable, but we do not
know to what products it would decay.
HO.
H'OH
O
HO f
O
O"ON
00H
-O"
OH
,0+ H0,
OON'
OH )=0
N-0// \
o o/
H
Fig. 3
None of these three reaction mechanisms involves three intermediates, and we
must conclude that their structures remain unsolved.
91

Acknowledgement - This project was supported by the ETH and the Swiss
Nationalfonds.
We thank Dr. P.L. Bounds for helpful discussions.
Abbreviation
Imi : First intermediate of the reaction of peroxynitrous acid with
monohydroascorbate.
Im2: Second and last intermediate of the reaction of peroxynitrous
acid with monohydroascorbate.
Peroxynitrite: Means the protonated and the deprotonated form.
References
[1] Bartlett, D.; Church, D. F.; Bounds, P. L.; Koppenol, W. H. The
kinetics of the oxidation of L-ascorbic acid by peroxynitrite. Free
Radical Biol. Med. 18:85-92; 1995.
[2] Squadrito, G. L.; Jin, X.; Pryor, W. A. Stopped-flow kinetic study of
the reaction of ascorbic acid with peroxynitrite. Arch. Biochem.
Biophys. 322:53-59; 1995.
[3] Schöneshöfer, M. Pulsradiolytische Untersuchung zur Oxidation der
Ascorbinsäure durch OH-Radikale und Halogen-Radikal-Komplexe in
wässriger Lösung. Z. Naturforsch. 27b:649-659; 1972.
[4] Bielski, B. H. J.; Comstock, D. A.; Bowen, R. A. Ascorbic acid free
radicals. I. Pulse radiolysis study of optical absorption and kinetic
properties. J. Am. Chem. Soc. 93:5624-5629; 1971.
[5] Bielski, B. H. J.; Allen, A. O.; Schwarz, H. A. Mechanism of
disproportionation of ascorbate radicals. J. Am. Chem. Soc. 103:3516-
3518; 1981.
[6] Buettner, G. R.; Jurkiewicz, B. A. Catalytic metals, ascorbate and free
radicals: Combinations to avoid. Radiât. Res. 145:532-541; 1996.
92

[7] Kissner, R.; Koppenol, W. H. Product distribution of peroxynitrite
decay as a function of pH, temperature, and concentration. J. Am.
Chem. Soc. 124:234-239; 2002.
[8] Maurer, P.; Thomas, C. F.; Kissner, R.; Rüegger, H.; Greter, O.;
Röthlisberger, U.; Koppenol, W. H. Oxidation of nitrite by
peroxynitrous acid. J. Phys. Chem. A 107:1763-1769; 2003.
[9] Coddington, J. W.; Hurst, J. K.; Lymar, S. V. Hydroxyl radical
formation during peroxynitrous acid decomposition. J. Am. Chem. Soc.
121:2438-2443; 1999.
[10] Gerasimov, O. V.; Lymar, S. V. The yield of hydroxyl radical from the
decomposition of peroxynitrous acid. Inorg. Chem. 38:4317-4321;1999.
[11] Merényi, G.; Lind, J.; Goldstein, S.; Czapski, G. Peroxynitrous acid
homolyzes into OH and N02 radicals. Chem. Res. Toxicol. 11:712-
713; 1998.
[12] Richeson, C. E.; Mulder, P.; Bowry, V. W.; Ingold, K. U. The complex
chemistry of peroxynitrite decomposition: New insights. J. Amer.
Chem. Soc. 120:7211-7219; 1998.
[13] Koppenol, W. H.; Kissner, R. Can ONOOH undergo homolysis. Chem.
Res. Toxicol. 11:87-90; 1998.
[14] Alfassi, Z. B.; Huie, R. E.; Neta, P.; Shoute, L. C. T. Temperature
dependence of the rate constants for reaction of inorganic radicals with
organic reductants. J. Phys. Chem. 94:8800-8805; 1990.
[15] Buettner, G. R. In the absence of catalytic metals ascorbate does not
autoxidize at pH 7: ascorbate as a test for catalytic metals. Journal ofBiochemical and Biophysical Methods 16:27-40; 1988.
[16] Denicola, A.; Freeman, B. A.; Trujillo, M.; Radi, R. Peroxynitritereaction with carbon dioxide/bicarbonate: Kinetics and influence on
peroxynitrite-mediated oxidations. Arch. Biochem. Biophys. 333:49-58;1996.
[17] Meli, R.; Nauser, T.; Latal, P.; Koppenol, W. H. Reaction of
peroxynitrite with carbon dioxide: Intermediates and determination of
the yield of C03' and N02'. J. Biol. Inorg. Chem. 7:31-36; 2002.
[18] Kirsch, M.; de Groot, H. Ascorbate is a potent antioxidant against
peroxynitrite-induced oxidation reactions - Evidence that ascorbate acts
93

by re-reducing substrate radicals produced by peroxynitrite. J. Biol.
Chem. 275:16702-16708; 2000.
[19] Balavoine, G. G. A.; Geletii, Y. V. Peroxynitrite scavenging bydifferent antioxidants. Part I: Convenient assay. Nitric Oxide: Biol.
Chem. 3:40-54; 1999.
[20] Whiteman, M.; Halliwell, B. Protection against peroxynitrite-dependent
tyrosine nitration and al-antiproteinase inactivation by ascorbic acid. A
comparison with other biological antioxidants. Free Radical Res.
25:275-283; 1996.
94

8. MECHANISM OF THE REACTION OF
PEROXYNITRITE WITH L-ASCORBATE STUDIED
AT PHYSIOLOGICAL PH
CHRISTOPHE R. KURZ, REINHARD KISSNER, and WILLEM H.
KOPPENOL*
Laboratorium für Anorganische Chemie, Eidgenössische Technische
Hochschule Zürich (Hönggerberg), Switzerland
Abstract— Existing data on the reaction mechanism of monohydroascorbate
with peroxynitrous acid, together with new data on the effect of pH, suggest a
complex mechanism. The peroxynitrite anion does not react with
monohydroascorbate. For the initial reaction of peroxynitrous acid with
monohydroascorbate, a second-order rate constant of kj of 5 x 105±1 M'V1 and
Oil 1
an equilibrium constant K of 1 x 10 M were determined. We observed three
intermediates of which the third has an absorption maximum at 345 nm and a
p£Ta of 7.6 ± 0.2. Both the protonated and the deprotonated forms of this third
intermediate can react with a second monohydroascorbate to form nitrite,
monohydroascorbate, and dehydroascorbate. In addition, there is a
monohydroascorbate-independent decay path that yields nitrate and
monohydroascorbate. EPR Experiments showed that ascorbyl radicals are not
involved in the reaction of peroxynitrite with monohydroascorbate.
Results
Stopped-flow studies
Kinetic traces recorded at 360 nm, 25 °C and at pH 7.2 after mixing 25 uM
peroxynitrite with different concentrations of monohydroascorbate
(0.25 - 50 mM) show an increase in absorption during 0.1 - 0.5 s, followed by
95

decrease (Fig. 1, all concentrations are those after mixing). At
monohydroascorbate concentrations higher than 1 mM, the rate of this increase
in absorption depends, like the magnitude of the absorption, on the
monohydroascorbate concentration (Fig. 1, traces C and D). The rate of increase
and the absorption intensity also depend on the peroxynitrite concentration, but
the decay rate of absorption is independent (data not shown). The absorption
immediately after mixing is significantly higher than that expected for
peroxynitrite alone at the same pH (Fig. 1, trace E). Monohydroascorbate does
not absorb at wavelengths higher than 320 nm. Both the increase and decay rates
of this absorption increase with temperature (inset Fig. 1).
0 2 4 6 8 10 12
time /s
Fig. 1. Time course for the change in absorption at 360 nm after mixing various
concentrations of monohydroascorbate with peroxynitrous acid. Peroxynitrite(25 uM) was mixed with monohydroascorbate at the following concentrations: trace
A, 250 uM; trace B, 1 mM; trace C, 8 mM, trace D, 50 mM and E, without
monohydroascorbate. All concentrations indicated are those after mixing. All traces
were recorded at 360 nm, 25 °C and pH 7.2 (50 mM phosphate buffer). At lower
monohydroascorbate concentrations (trace A and B) no dependence on the
monohydroascorbate concentration is observed. Each curve is an average of
12 measurements. Inset: 25 uM peroxynitrous acid mixed with 250 uM
monohydroascorbate at pH 7.2 at the following temperature: trace A, 25 °C; trace
a, 37 °C. Measurements were made at 360 nm.
96

From pH 5.8 to 8.5 the rates of formation and decay of the absorption at
360 nm decrease with increasing pH (Figs. 2 and 3). The absorption maxima at
3 ms and between 0.1 - 2.3 s, dependent on pH, increased at higher pH (Fig. 2).
Trace a in the inset of Figure 2 was recorded at 360 nm to diminish the
peroxynitrite interference on the absorption of the intermediate. Trace ß was
recorded at 320 nm to maximize the maximum contribution from the
peroxynitrite anion. Trace y represents the peroxynitrite decay without
monohydroascorbate at the same pH and concentration.
time /s
Fig. 2. Kinetics of the formation and decay of an intermediate as a function of
pH. Kinetics traces observed at 360 nm after mixing 250 uM monohydroascorbatewith 25 uM peroxynitrite at the following pH: trace A, pH 8.5; trace B, pH 8.0,trace C, pH 7.2, trace D, pH 5.8. Inset: Time course of the absorption after mixing25 uM peroxynitrite with 250 uM monohydroascorbate at pH8.5. The trace
a (= trace A) was recorded at 360 nm and ß at 320 nm. Trace y is peroxynitritealone measured at pH 8.5 and 320 nm.
97
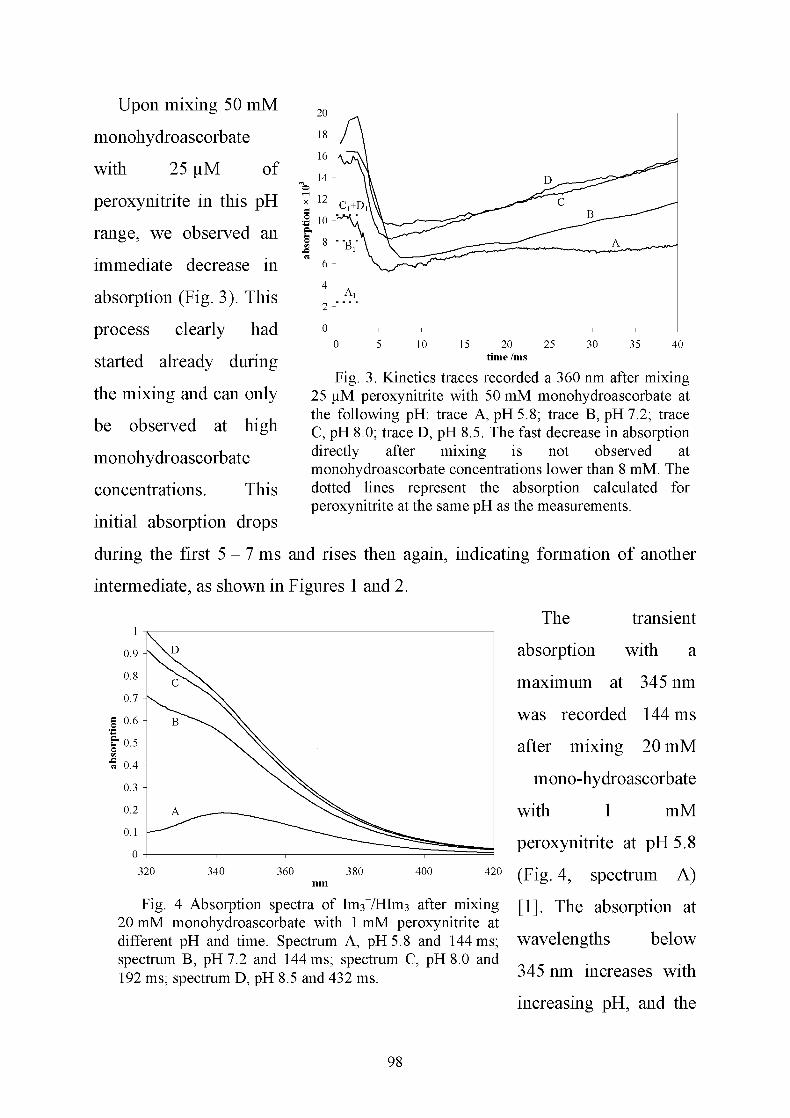
Upon mixing 50 mM
monohydroascorbate
with 25 uM of
peroxynitrite in this pH
range, we observed an
immediate decrease in
absorption (Fig. 3). This
process clearly had
started already during
the mixing and can only
be observed at high
monohydroascorbate
concentrations. This
initial absorption drops
10 15 20
time /ms
25 30 35 40
Fig. 3. Kinetics traces recorded a 360 nm after mixing25 uM peroxynitrite with 50 mM monohydroascorbate at
the following pH: trace A, pH5.8; trace B, pH 7.2; trace
C, pH 8.0; trace D, pH 8.5. The fast decrease in absorption
directly after mixing is not observed at
monohydroascorbate concentrations lower than 8 mM. The
dotted lines represent the absorption calculated for
peroxynitrite at the same pH as the measurements.
during the first 5 - 7 ms and rises then again, indicating formation of another
intermediate, as shown in Figures 1 and 2.
The transient
absorption with a
maximum at 345 nm
was recorded 144 ms
after mixing 20 mM
mono-hydroascorbate
with 1 mM
peroxynitrite at pH 5.8
(Fig. 4, spectrum A)
[1]. The absorption at
wavelengths below
345 nm increases with
increasing pH, and the
320 340 360 380 400 420
Fig. 4 Absorption spectra of Im37HIm3 after mixing20 mM monohydroascorbate with 1 mM peroxynitrite at
different pH and time. Spectrum A, pH5.8 and 144 ms;
spectrum B, pH 7.2 and 144 ms; spectrum C, pH 8.0 and
192 ms; spectrum D, pH 8.5 and 432 ms.
98

transient absorption maximum can no longer be determined (Fig. 4, spectra B,
C, andD).
Control experiments without peroxynitrite, but with nitrite or nitrate added,
show no transient absorption at 345 nm, as in experiments in which
peroxynitrite is mixed with buffer only, which confirms that the intermediate is
formed as a product of the reaction of monohydroascorbate with peroxynitrous
acid. Furthermore, we observed no influence of nitrite, nitrate, or dioxygen on
the absorption kinetics (not shown).
EPR studies
We observe a small signal with a g-value of 2.005 which is due to the
ascorbyl radical. This signal is not detected when monohydroascorbate or
peroxynitrite are absent. Immediately after mixing 25 uM peroxynitrite and
250 uM monohydroascorbate (final concentrations) at pH 7.2, no EPR signal is
observed; instead, the signal intensity rises for approximately 3 seconds and
subsequently disappears within 10 seconds (Fig. 5, trace B). A comparison with
the optical kinetic trace recorded at 360 nm and pH 7.2 (Fig. 5, trace A) shows
that the concentration of ascorbyl radical increases while the absorption
decreases. The total concentration of ascorbyl radical formed during the reaction
at pH5.8 is estimated to be only 60 nM [1]. According to the amount of
ascorbyl radical found at pH 5.8 and given the retarded disproportionation of the
ascorbyl radical at higher pH, we estimate the total ascorbyl radical
concentration formed at pH 7.2 to be below 100 nM.
99

8
time /s
10 12 14
Fig. 5 Comparison of the decay at 360 nm (trace A) with an EPR trace (trace
B) of the ascorbyl radical formation and decay. Trace B, an average of
10 measurements, reaches maximum distinctly later than trace A, after an average
of 12 measurements. Measurements were made at initial concentrations of 25 uM
peroxynitrite and 250 uM monohydroascorbate in 50 mM phosphate buffer at
pH 7.2. All concentrations indicated are those after mixing.
Ion-chromatography
When 25 uM peroxynitrite is mixed with monohydroascorbate at pH 8.1, the
nitrite yield increases with increasing monohydroascorbate concentration and a
corresponding decrease of nitrate. Even at low monohydroascorbate
concentrations, the nitrite yield is already much higher than without
monohydroascorbate (not shown).
100

Discussion
Previous work
From our study of peroxynitrous acid with monohydroascorbate at pH 5.8 we
concluded that; (1) no ascorbyl radical is produced directly, (2) there exist at
least 2 intermediates of which one has an absorption maximum at 345 nm, and
(3) the second-order rate constant of the reaction of peroxynitrous acid with
monohydroascorbate is about 106M1s1 [1]. In this work, we studied the
reaction of peroxynitrite with monohydroascorbate between pH5.8 and 8.5 to
determine the mechanism and the rate constant for the reaction of peroxynitrite
with monohydroascorbate at pH 7.2.
Intermediates
When 25 uM peroxynitrite is mixed with 50 mM of monohydroascorbate, a
fast decrease in absorption during the first 5 ms is observed (Fig. 3). This initial
absorption is much higher than that of peroxynitrite alone and was observed at
all pH values at monohydroascorbate concentrations higher than 8 mM. The
initial high absorption, indicating production of intermediate Im1; decreases
during the first 5 - 7 ms to a value that is still higher than that formed in the
presence of peroxynitrite alone. The decrease in absorption is due to an
intermediate we label Im2. The following increase in absorption with an
absorption maximum at pH 5.8 of 345 nm is assigned to a third intermediate
HIm3 (Figs. 1 and 3). The intermediate Im2 is invoked to explain the absorption
minimum observed after 7-10 ms: otherwise, the absorption would be expected
to increase, decrease, or remain constant, but not show a minimum.
As shown in Figure 2, the difference between the absorption seen at mixing
time and the maximal absorbance at 360 nm increases with pH. When we
assume that only peroxynitrous acid reacts with monohydroascorbate (see
section 'Peroxynitrite anion'), we would expect that less HIm3 is formed at
higher pH (Fig. 2), but the absorption-pH profile contradicts this. To account for
101

the increase in absorption at higher pH, we introduce another species resulting
from deprotonation of HIm3 to form the more strongly absorbing Im3 .We
estimate a p^a of 7.6 ± 0.2 for the HIm3/Im3 acid-base pair by simulation.
Peroxynitrite anion
The decay of the absorption ascribed to HIm3/Im3 proceeds more slowly at
higher pH. We observed that the rate of formation of HIm3/Im3 also decreases
with increasing pH. The spectrum of the third intermediate changes as function
of pH (Fig. 4), with a clear absorption maximum of 345 nm at pH 5.8. At higher
pH, the absorption increases in the same manner as that of peroxynitrite, but
with a maximum below 320 nm. Unfortunately, monohydroascorbate begins to
absorb below 320 nm, and no information could be collected below that
wavelength. The peroxynitrite anion exhibits a maximum at 302 nm with an
absorption coefficient of 1360 M^cm"1 at 320 nm. Since, the signal intensity
after mixing is proportional to the absorption of peroxynitrous acid at each pH
(Fig. 3 and 4), we conclude that at 320 nm and, at t = 0 s, mainly the
peroxynitrite anion absorbs. Of interest is that the decrease in absorption at
320 nm is faster than the decay of peroxynitrite or that of the intermediate
measured at 360 nm (inset Fig. 2).
The observed increase in absorption at 320 nm as function of the pH
immediately after mixing suggests that there is still peroxynitrite present, even
after a few seconds. Furthermore, the relative absorption of HIm3/Im3 decreases
with increasing pH (Fig. 4), implying that only peroxynitrous acid reacts with
monohydroascorbate.
When peroxynitrite is mixed with excess monohydroascorbate at pH 13, the
yellow color of peroxynitrite remains visible for hours.
102

Homolysis or one-electron oxidation?
As we reported earlier, the ascorbyl radical is not a significant intermediate
in the reaction of peroxynitrous acid with monohydroascorbate at pH5.8 [1].
Extension of our earlier EPR experiments to pH range 5.8-8.5 showed that the
ascorbyl radical concentrations remain at about 60 nM and represent a yield of
ca. 0.24 % relative to the initial peroxynitrous acid concentration; this small
amount of ascorbyl radicals forms concomitantly with the decrease of the
absorption at 360 nm. Thus, since the ascorbyl radical is not a primary reaction
product, no one-electron oxidation has taken place, excluding homolysis of
peroxynitrous acid to nitrogen dioxide and hydroxyl radical under the conditions
of these experiments. As mentioned earlier [1], it is possible that the ascorbyl
radicals observed originate from the oxidation of monohydroascorbate by
dioxygen, a reaction that can be catalyzed by trace amounts of contaminating
transition metals[2,3].
Kinetics and mechanism
We postulate from the increased absorption immediately after mixing that
Im! is formed very rapidly from monohydroascorbate and peroxynitrous acid,
indicating that the rate constant kx for the reaction of monohydroascorbate with
peroxynitrous acid to form Imi within 3 ms is high. Imi decays within 10 ms to
form Im2, at which time there is still free peroxynitrite present in the solution, as
indicated by the absorption spectra (Fig 4). The presence of free peroxynitrite
implies that Im2 reverts via Imi to monohydroascorbate and peroxynitrous acid.
Im2 also decays further to HIm3, which is in equilibrium with its deprotonated
form Im3 .The decay rate of this third intermediate strongly depends on the
monohydroascorbate concentration. We conclude, as before [1] that HIm3 can
react with another monohydroascorbate molecule to form nitrite,
monohydroascorbate, dehydroascorbate and water, as can Im3 .Ion-
chromatography of the products obtained from even a small concentration of
103

monohydroascorbate (250 uM) at pH 8.1 shows the presence of nitrite at a
concentration higher than that expected from decomposition [4]; furthermore,
the nitrite yield increases as a function of monohydroascorbate concentration
(data not shown).
ONOO + H+ H++ Im3+
H^sc» HAsc + DAsc + N02 + OH
kl k2 k3^
yc t_ _
,
ONOOH + HAsc ^=?r imi +=*: Im2 ^^ HIm33 4
» HAsc +N03 +H
k.i k-2 k_3k.
9 k\+ HAsc
N03 + H' ^
HAsc + DAsc + N02 +H20
Scheme 1
As shown in the Figure 1, the decay of HIm3 at 250 uM and 1 mM HAsc is
similar at pH7.2, indicating that HIm3 decays via a monohydroascorbate-
independent pathway. Possibly, HIm3 decay results in the formation of
monohydroascorbate and nitrate. A second possibility is that there is a slow back
reaction from HIm3 to Im2. The rates of decay of Im3 (HIm3 and Im3 ) and
monohydroascorbate, observed at 360 nm and 265 nm, respectively, are similar
to those published earlier for reaction at pH5.8, although the
monohydroascorbate decay precedes that of Im3 [1]. Unfortunately, this
simultaneous decay of monohydroascorbate and HIm3 prevents discrimination
of these two pathways, and we are not able to determine whether HIm3
undergoes decay to monohydroascorbate and nitrate or is in equilibrium with
peroxynitrous acid and monohydroascorbate, through Im2 and Imi.
104

Rate_, L , _, , „ , Absorption Initial
A ARate value Remark Compound r~
.
A A A.
constant coetncient concentration
*1 5 x 105±1 M'V a HAsc 0 M'W1250 u.M-
50 mM
£-1 5 x 102±1 s"1 a HOONO 82 M'W1 25 uM
^2 3 ± 1 x 102 s"1 a ONOO" 685 M'W1 —
k-2 3 ± 1 x 101 s"1 a N03 0 M'W1 —
h 2 ± 1 x 102 s"1 a N02 5 M'W1
k-3 3±2x loV1 a DAsc 0 M'W1 —
k4 0.6 ± 0.3 s"1 a, b Imi -400 M'W1 —
h1.2 ±0.3 x 102
M-V a, b Im2 0 M'W1 —
k6 o.uo.imV a HIm3 460 M'W1
h 2.5 x I02s_1 a Im3 1600 M'W1 —
k-7, h lxio^V1 c
k.* 1.6 x I03s_1 d
kg 1.2 s"1 e
Table 1. Values used for the simulation of the reaction of peroxynitrous acid
with monohydroascorbate at 360 nm and 25°C. a = determined by simulation;b = Kurz et al. [1]; c = rate constant for protonation; d = rate constant determined by
pKa [5]; e = Kissner et al. [4].
In contrast to the results obtained at pH 7.2 and lower, the decay traces for
the third intermediate at pH 8.5, when 25 uM peroxynitrite is mixed with
250 uM and 1 mM monohydroascorbate, differ from one another (data not
shown). The monohydroascorbate-dependent and -independent decay rates are
not influenced by pH. We therefore invoke a monohydroascorbate dependent
path that becomes more important at higher pH. We propose that Im3 can react
105

with another monohydroascorbate molecule and form nitrite,
monohydroascorbate, dehydroascorbate and hydroxide.
We simulated the two mechanisms shown in Scheme 1, i.e. with and without
reaction k.3 for the pH range 5.8-8.5 and a monohydroascorbate concentration
range of 250 uM to 50 mM. The simulations suggest a second-order rate
constant kx of 5 x 105±1 M'V1 with a first-order rate constant k_x of 5 x 102±1 s"1
for the back reaction. Further, a pKa value of 7.6 ±0.2 of HIm3 could be
estimated.
Physiological relevance
Can the reaction rate of monohydroascorbate compete for peroxynitrite with
carbon dioxide or other compounds at physiological pH and temperature? The
reaction with carbon dioxide, which is present at around 1 mM in human
plasma, proceeds with a rate constant of 3 x 104M~1s~1 at 25 °C [6] and a
backward rate constant of 1.5 x 106 s_1 [7]. Given that, in both cases, multiple
equilibria are involved, it is not possible to answer this question. However, in
the presence of both carbon dioxide (1 mM) and monohydroascorbate(250 uM),
the ascorbyl radical is observed at pH 8 and higher, but not at lower pH (Kurz,
2004, unpublished). We propose, therefore, that under physiological conditions
(0.1 - 0.2 uM monohydroascorbate) monohydroascorbate competes with carbon
dioxide (1 mM) for peroxynitrite. Monohydroascorbate has been reported to
protect against damage caused by peroxynitrite which has been mostly ascribed
to monohydroascorbate's ability to repair[5,8-10]; here we showed that
monohydroascorbate may prevent damage by acting as scavenger of
peroxynitrous acid.
Acknowledgment - This project was supported by the ETH and the
Schweizerische Nationalfonds.
We thank Dr. P.L. Bounds for helpful discussions.
106

Abbreviation
Imi : First intermediate of the reaction of peroxynitrous acid with
monohydroascorbate. (New intermediate compared with chapter 7.)
Im2: First intermediate of the reaction of peroxynitrous acid with
monohydroascorbate. (Is equal to Imi in chapter 7)
Im3: Third and last intermediate of the reaction of peroxynitrous acid
with monohydroascorbate. (Is equal to Im2 in chapter 7)
Peroxynitrite: Means the protonated and deprotonated form.
References
[1] Kurz, C.; Kissner, R.; Perrin, D.; Nauser, T.; Koppenol, W. H. Rapid
scavenging of peroxynitrous acid by monohydroascorbate. Free Radical
Biol. Med. 35:1529-1537; 2003.
[2] Buettner, G. R. In the absence of catalytic metals ascorbate does not
autoxidize at pH 7: ascorbate as a test for catalytic metals. Journal ofBiochemical and Biophysical Methods 16:27-40; 1988.
[3] Buettner, G. R.; Jurkiewicz, B. A. Catalytic metals, ascorbate and free
radicals: Combinations to avoid. Radiât. Res. 145:532-541; 1996.
[4] Kissner, R.; Koppenol, W. H. Product distribution of peroxynitrite decayas a function of pH, temperature, and concentration. J. Am. Chem. Soc.
124:234-239; 2002.
[5] Koppenol, W. H.; Kissner, R. Can ONOOH undergo homolysis. Chem.
Res. Toxicol. 11:87-90; 1998.
[6] Lymar, S. V.; Hurst, J. K. Rapid reaction between peroxonitrite ion and
carbon dioxide: Implications for biological acitivity. J. Am. Chem. Soc.
117:8867-8868; 1995.
[7] Squadrito, G. L.; Pryor, W. A. Mapping the reaction of peroxynitrite with
C02: Energetics, reactive species, and biological implications. Chem. Res.
Toxicol. 15:885-895; 2002.
[8] Kirsch, M.; de Groot, H. Ascorbate is a potent antioxidant against
peroxynitrite-induced oxidation reactions - Evidence that ascorbate acts
107

by re-reducing substrate radicals produced by peroxynitrite. J. Biol.
Chem. 275:16702-16708; 2000.
[9] Whiteman, M.; Halliwell, B. Protection against peroxynitrite-dependent
tyrosine nitration and al-antiproteinase inactivation by ascorbic acid. A
comparison with other biological antioxidants. Free Radical Res. 25:275-
283; 1996.
[10] Balavoine, G. G. A.; Geletii, Y. V. Peroxynitrite scavenging by different
antioxidants. Part I: Convenient assay. Nitric Oxide: Biol. Chem. 3:40-54;1999.
108

9. THE REACTION OF PEROXYNITRITE WITH
CARBON DIOXIDE AND VITAMIN C.
CHRISTOPHE R. KURZ, REINHARD KISSNER, and WILLEM H.
KOPPENOL*
Laboratorium für Anorganische Chemie, Eidgenössische Technische
Hochschule Zürich (Hönggerberg), Switzerland
Abstract— In the presence of carbon dioxide, peroxynitrite reacts catalytically
with monohydroascorbate at a very fast rate. Low monohydroascorbate
concentrations prevent the formation of trioxocarboanate(#-l) radical and
nitrogen dioxide, the intermediates produced from the reaction of peroxynitrite
with carbon dioxide. Moreover, at low monohydroascorbate concentrations only
small amounts of ascorbyl radicals, relative to the initial monohydroascorbate
concentration, are produced. If the monohydroascorbate concentration is higher
than that of peroxynitrite, the ascorbyl radical concentration increases. Under
these conditions, an alternative pathway, which involves reaction with a second
monohydroascorbate molecule, is observed. Stopped-flow experiments indicate
that at pH 7, the reaction between peroxynitrous acid and monohydroascorbate
competes with the reaction between peroxynitrite anion, monohydroascorbate,
and carbon dioxide. At pH 6, the main pathway involves the reaction of
monohydroascorbate with peroxynitrous acid.
Results
Kinetic traces recorded at pH 7 after mixing 2 mM peroxynitrite with
17 mM carbon dioxide showed a fast decrease in absorption at 640 nm. (Fig. 1,
all concentrations are given after mixing). When 2 mM peroxynitrite was mixed
109

with 17 mM carbon dioxide and 50 uM, 2 mM or 10 mM monohydroascorbate,
a weaker, or no absorption was observed.
Kinetic traces s
recorded at 320 nm at5
4
pH 7 after mixing s*
3a
2 mM peroxynitrite f2
Xi
with 17 mM carbon"
i
dioxide show faster°
-i
decay in the presence of
50 uM, 2 mM, or
10 mM monohydro¬
ascorbate than in the
absence of monohydro¬
ascorbate. Interestingly,
25 50 75
time /ms
100 125 150
Fig 1 Time courses of changes in absorption at
640 nm after mixing peroxynitrite (2 mM) with carbon
dioxide (17 mM) and the following concentrations of
monohydroascorbate: trace A, 0 uM; trace B, 50 uM;
trace C, 2 mM and trace D, 10 mM. All concentrations
are given after mixing. All traces were recorded at 25°C
and pH 7.0 in 50 mM phosphate buffer.
10 20 30
time /ms
40 50 60
Fig. 2 Kinetic traces recorded at 320 nm after mixing 2 mM peroxynitrite with
17 mM carbon dioxide and monohydroascorbate at different concentrations: trace A,0 uM; trace B (x), 50 uM; trace C, 2 mM and trace D (-), 10 mM. All traces were
recorded at 25°C and pH 7.0 in 50 mM phosphate buffer.
110

if peroxynitrite was in excess over monohydroascorbate, the absorption
decreased faster compared to the trace recorded in the absence of
monohydroascorbate. Further, in the presence of monohydroascorbate the decay
rate observed at 320 nm did not change with increasing monohydro-ascorbate
concentration in the range from 50 uM to 10 mM (Fig. 2). The reaction of
peroxynitrite with monohydro-ascorbate, in the absence of carbon dioxide, is too
slow to be responsible for this acceleration in the absorption decay (Fig 3). At
pH 6 the rate of the absorption decay increased with increasing
monohydroascorbate concentration, which is significantly different from results
at pH 7 (cf. Fig 2 and 4). At pH 8, the absorption decay became faster in the
presence of 50 uM of monohydroascorbate and then slowed down with further
increase in monohydroascorbate concentration (Fig. 5). The decrease in
absorption at 320 nm changed also from first-order to second-order kinetics. The
initial absorption was higher with increasing monohydroascorbate
concentrations and the absorption starts to decrease after about 2 ms (Fig. 5).
The rate of the absorption decay, recorded at 320 nm, was maximal at pH 7 and
decreased with changes in pH (cf. Fig. 2, 4 and 5). The absorption spectra at
pH 6, 7 and 8, recorded after mixing 50 uM peroxynitrite with 1 mM carbon
dioxide and 10 mM monohydro-ascorbate have different absorption maxima
(Fig. 6). With decreasing pH the absorption maximum shifts. The spectrum
recorded at pH 8 has a maximum at 360 nm and at pH 6, at 345 nm. The
maximum also decreases with decreasing pH. The high absorption observed at
pH 8 at wavelengths below 340 nm is due to peroxynitrite anion, which absorbs
at 320 nm 15 times more than peroxynitrous acid.
Ill

5
time /s
10
Fig. 3 Kinetic traces recorded at 320 nm after mixing 2 mM peroxynitrite with
different monohydroascorbate concentrations: trace A, 0 uM; trace B, 50 uM; trace C,2 mM and trace D, 10 mM. All traces were recorded at 25°C and pH 7.0 in 50 mM
phosphate buffer.
At pH 8 the kinetic trace recorded at 360 nm after mixing 50 uM
peroxynitrite with 1 mM carbon dioxide disappeard within 0.1 s (Fig. 7). When,
in addition, 100 uM or 1 mM monohydroascorbate is present, the absorption
reaches 0.05and 0.08 in 20 and 30 ms, respectively; this increase is followed by
a second-order decay.
EPR measurements showed an ascorbyl radical spectrum after mixing 2 mM
peroxynitrite with 17 mM carbon dioxide and 10 mM monohydro-ascorbate at
pH8.
Product analysis at pH 8 and low monohydro-ascorbate concentrations
showed that nitrate is nearly quantitatively formed from peroxynitrite.
112

0 50 100 150 200 250 300
time /ms
Fig. 4 Time course of the change in absorption at 320 nm at pH 6 after mixing various
concentrations of monohydro-ascorbate with peroxynitrite and carbon dioxide.
Peroxynitrite (2 mM) was mixed with carbon dioxide (17 mM) and the followingconcentrations of monohydroascorbate: trace A, 0 uM; trace B, 50 uM; trace C, 2 mM
and trace D, 10 mM.
Discussion
l-Carboxylato-2-nitrosodioxidane was proposed to be an intermediate in the
reaction of peroxynitrite with carbon dioxide. This intermediate, the absorption
maximum of which was observed at 600 nm by Papina (unpublished; most
probably trioxocarbonate(#l-) radical), is formed with a rate constant of
3x10^^_1[1].
ONOO" + CQ2<» ONOOCO2 Products
^
As is shown in Figure 1, even equimolar concentrations of vitamin C and
peroxynitrite, in the presence of carbon dioxide, prevent the appearance of the
absorption at 640 nm. This means that either the formation of 1-caboxylato-
2-nitrosodioxidane is completely inhibited or that this intermediate is very
rapidly consumed by vitamin C.
113

2.0
1.8
1.6
1.4 H
J 1.2 H
a.
*0.8
0.6 -
0.4 -
0.2 -
0.0
0 10 15 20 25
time /ms
30 35 40 45 50
Fig.5 Time course for the change in absorption at 320 nm at pH 8 after mixingvarious concentrations of monohydroascorbate with peroxynitrite and carbon dioxide.
Peroxynitrite (2 mM) was mixed with carbon dioxide (17 mM) and the followingconcentrations of monohydroascorbate: trace A, 0 uM; trace B, 50 uM; trace C,2 mM and trace D, 10 mM.
When the reaction between peroxynitrite with carbon dioxide is observed at
320 nm, where mainly peroxynitrite absorbs (Fig. 2), the decrease of the signal
is faster in the presence of vitamin C. As reported in chapter 7, the reaction of
peroxynitrous acid with vitamin C could be fast enough to prevent the reaction
of peroxynitrite with carbon dioxide [2]. Unfortunately, there exist equilibria
between the substrate, peroxynitrous acid, monohydroascorbate, and a third
intermediate (HIm3), which also absorbs at 320 nm and therefore prevent a
straightforward conclusion.
ONOOH + Hasc" Him, - Products (2)
The kinetic traces obtained for the reaction of peroxynitrous acid with
monohydroascorbate decrease much slower in the absence of carbon dioxide and
therefore reaction 2 can not be responsible for the consumption of peroxynitrite
114

within few milliseconds. (Fig. 3) Two possibilities for this fast consumption of
peroxynitrite are proposed:
- peroxynitrous acid reacts with vitamin C whereby the first intermediate Im!
formed within a few milliseconds reacts with carbon dioxide in an
irreversible step (eq. 3),
- l-caboxylato-2-nitrosodioxidane is consumed by monohydroascorbate in a
fast irreversible reaction (eq. 4).
ONOOH + Hasc" «»
Imi+C°2
» Products (3)
ONOO" + CQ2<»
ONOOCO2+ HAsc
» Products (4)
To verify these two possibilities, rates of reaction of peroxynitrite with
carbon dioxide and monohydroascorbate were measured at pH 6. If reaction 3
would occur, the absorption at 320 nm should decrease faster at lower pH. As
shown in Figure 4, at pH 6 the absorption traces disappeared slower compared to
traces recorded at pH 7, which leads us to the conclusion that reaction 3 does not
take place.
Kinetic traces recorded at 320 nm at pH 7 show that the initial absorption is
diminished when monohydroascorbate is present, which indicates that the
reaction has already progressed within the mixing time (Fig. 2). It is of special
interest that, already at very low monohydroascorbate concentrations, 20 times
smaller than the one of peroxynitrite, peroxynitrite is consumed much faster and
the initial absorption decay does not increase further with increasing
monohydroascorbate concentration. In the absence of monohydroascorbate the
decay is much slower. These observations indicate that monohydroascorbate
does not partitcipate in the rate limiting step of this reaction. Further, from
similar kinetic experiments when either peroxynitrite or monohydroascorbate is
in excess, it can be concluded that monohydroascorbate has to react catalytically
with a fast turn over reaction rate. Therefore the following equation can be
proposed:
115

ONOO" + CQ2<ia »ONOOCO2
+ HAsc» HAsc" + Products (5)
k-a kß
Taking into account that the absorption decay at 320 nm at pH 7 is not
accelerated further with increasing monohydroascorbate concentrations and that
at monohydroascorbate concentrations20 times smaller than the one of
peroxynitrite the absorption decay reaches its rate limit, one can conclude that kp
has to be higher than ka.
The maximum of absorption spectra recorded at different pH shifts from
360 nm to 345 nm with decreasing pH (Fig. 6). The absorption spectrum with a
maximum at 360 nm, recorded at pH 8, is similar to the absorption spectrum of
ascorbyl radical (s36o = 5000 M4cm_1 [3]). EPR measurements confirmed the
presence of ascorbyl radicals in the reaction of peroxynitrite with carbon dioxide
and monohydroascorbate (data not shown).
The absorption spectrum with a maximum at 345 nm, recorded at pH 6, is
similar to the spectrum of the intermediate HIm3 recorded after mixing
peroxynitrite with monohydroascorbate in the absence of carbon dioxide (eq. 2)
HIm3 has an extinction coefficient of about 460 M"1cm"1, about 11 times
smaller than the ascorbyl radical[3]. Therefore it has to be present in higher
concentrations than the ascorbyl radical to be registered. At pH 6, a similar
absorption behaviour, independent from carbon dioxide, suggests that mainly
the reaction between peroxynitrous acid and monohydroascorbate is responsible
for the peroxynitrite decay. The high extinction coefficient of ascorbyl radicals
allow to estimate roughly the amount of ascorbyl radicals produced in the
reactions at pH 7 and pH 8. One must realise that both peroxynitrite and HIm3
absorb at 360 nm, but less than the ascorbyl radical. The ascorbyl radicals
concentrations found after mixing 50 uM peroxynitrite with 1 mM carbon
dioxide and 10 mM monohydroascorbate were about 17 uM at pH 8 and 9 uM
at pH 7. As mentioned before, at pH 6 mainly HIm3 is produced and therefore
the reaction with carbon dioxide is only of minor importance. Ascorbyl radicals
116

are observed only when the monohydroascorbate concentration is higher than
the concentration of peroxynitrite.
320 330 340 350 360 370 380 390 400
nm
Fig.6 Absorption spectra of an intermediate after mixing 50 uM peroxynitrite with
1 mM carbon dioxide and 10 mM monohydroascorbate at different pH and times. The
spectrum at pH 8 was recorded 205 ms after mixing; the spectrum at pH 7 at 25 ms
and the spectrum at pH 6 at 25 ms. Additionally, the spectrum of ascorbyl radical
(Asc*-) (11 uM), obtained by pulse radiolysis, and the spectrum of HIm3 (48 uM),obtained at pH 7, by mixing peroxynitrite with monohydroascorbate in the absence of
carbon dioxide are shown.
The ascorbyl radical yield depends, as shown in Figure 7, on the
monohydroascorbate concentration and increases with increasing pH. The pH
dependent increase in the ascorbyl radical concentration correlates with the
amount of protonated and deprotonated peroxynitrite. At higher pH, the
concentration of peroxynitrite anion is higher and therefore more peroxynitrite
can react with carbon dioxide. Many of the reagents and intermediates absorb at
320 nm, among them the ascorbyl radical, which may explain the initial peak
after 2 ms, when 2 mM peroxynitrite is mixed with 17 mM carbon dioxide and
10 mM monohydroascorbate at pH 7 and 8 (Fig. 2 and 5).
One obvious question remains: How are ascorbyl radicals produced? The
production of nitrogen dioxide and trioxocarbonate(#-l) radicals in the reaction
of peroxynitrite with carbon dioxide is well known [4-6]. Rate constants of the
117

reactions between those radicals and monohydroascorbate are sufficiently high
to produce ascorbyl radicals [7,8], but the formation of those radicals is inhibited
because monohydroascorbate scavenges l-carboxylato-2-nitrosodioxidane in a
fast catalytic reaction (Scheme 1).
2 Asc"
+ N02 + C02
+ HAsc
HAsc" + N03 + C02 - X
CCKk «
HT
H2C03hco;
OH"
H+ + ONOO" + C02 =5=^=
+ HAsc
ki
NO, + H+« k'nONOOH + HAsc"
ONOOCO,
-11i t'
Kn
ONOOC02H
h:k24 k_15
2 Asc "+ H
Asc'" +N02 + H+
+ HAsc"
DAsc +HAsc
Asc'"+CO?" +H+
-*-N02 +CO3
+ HAsc
(4% - 30%)
:$: N03" + C02
NO," +2HImj
H++ Im,+ HAsc» HAsc + DAsc + N07 + OH
17—
345 kzo» HAsc +N0, +H+Im2 -^
». HIiri
+ HAsc
HAsc +DAsc + N02 +H20
Scheme 1
Taking these results into account, it can be proposed that the catalytic
reaction between l-carboxylato-2-nitrosodioxidane and monohydroascorbate
proceeds via an intermediate X, which can react with another
monohydroascorbate molecule to form finally 2 ascorbyl radicals, carbon
dioxide and nitrite.
ONOO + CO2-*: ONOOCOo+ HAsc
» X
+ HAsc
HAsc + N03 + C02
2 Asc' +N02 + C02(6)
118

A second possibility of ascorbyl radical formation comes from the catalytic
cycle of ascorbyl radicals, as proposed in Scheme 2. At low
monohydroascorbate concentration no ascorbyl radicals would be observed
because of their fast consumption by peroxynitrite.
Different products are proposed for reactions given by Scheme 2 and
equation 6.
ONOO" + C02 «» ONOOC02"
HAsc» Asc'" + 00^"+ H+ + N02
HAsc" + DAsc
ONOO" + H20 ^H NO9
Asc'
HAsc" + OH" HAsc"
NO +02
H90n2o4 r2
N02 + N03 + 2 H
Scheme 2
For low monohydroascorbate concentrations nitrate is the main product
generated in the reaction given by equation 6, while nitrite is mainly formed in
the reaction presented by Scheme 2. Analysis showed nitrate (quantitatively to
peroxynitrite) to be the major product at pH 8, contradicting the reaction in
Scheme 2.
119
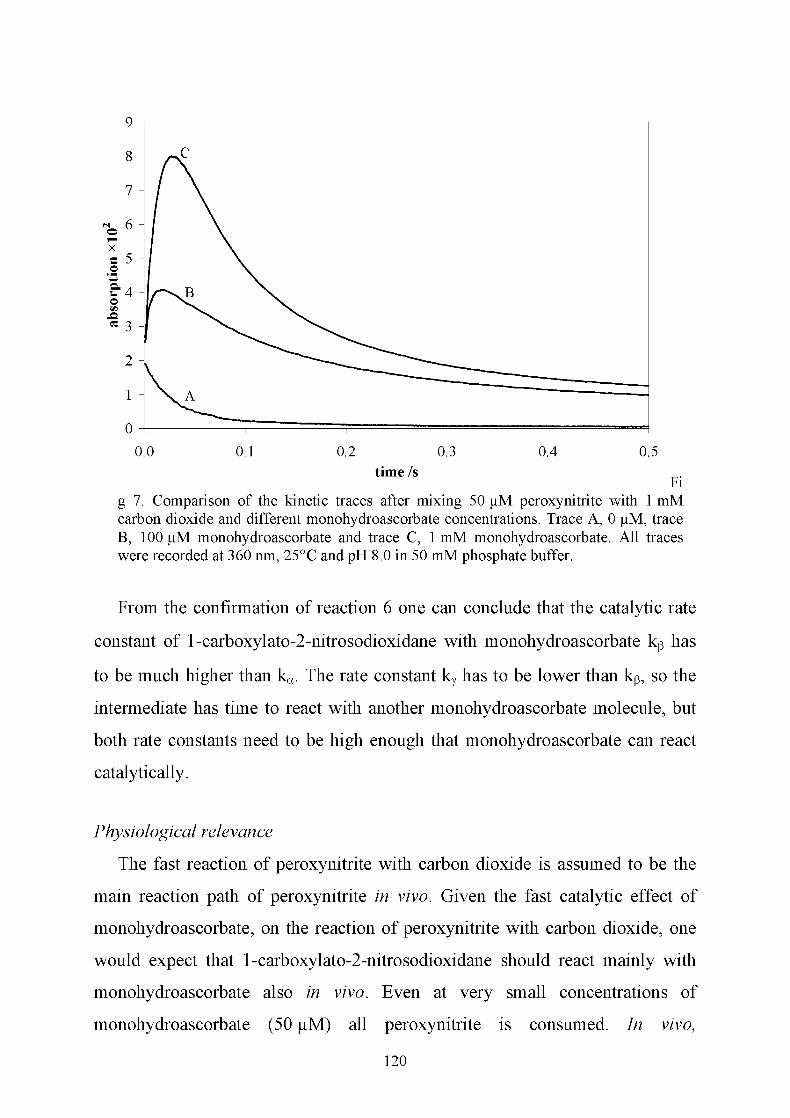
0.0 0.1 0.2 0.3 0.4 0.5
time /sFi
g 7. Comparison of the kinetic traces after mixing 50 uM peroxynitrite with 1 mM
carbon dioxide and different monohydroascorbate concentrations. Trace A, 0 uM; trace
B, 100 uM monohydroascorbate and trace C, 1 mM monohydroascorbate. All traces
were recorded at 360 nm, 25°C and pH 8.0 in 50 mM phosphate buffer.
From the confirmation of reaction 6 one can conclude that the catalytic rate
constant of l-carboxylato-2-nitrosodioxidane with monohydroascorbate kp has
to be much higher than ka. The rate constant ky has to be lower than kp, so the
intermediate has time to react with another monohydroascorbate molecule, but
both rate constants need to be high enough that monohydroascorbate can react
catalytically.
Physiological relevance
The fast reaction of peroxynitrite with carbon dioxide is assumed to be the
main reaction path of peroxynitrite in vivo. Given the fast catalytic effect of
monohydroascorbate, on the reaction of peroxynitrite with carbon dioxide, one
would expect that l-carboxylato-2-nitrosodioxidane should react mainly with
monohydroascorbate also in vivo. Even at very small concentrations of
monohydroascorbate (50 uM) all peroxynitrite is consumed. In vivo,
120

peroxynitrite will be present at smaller concentrations than
monohydroascorbate, which yields ascorbyl radicals as a product. Earlier
investigations with monohydroascorbate and peroxynitrite, without carbon
dioxide, also show a high protection ability of monohydroascorbate against
peroxynitrite induced damage [9-13]. Therefore, it can be concluded that
monohydroascorbate can very efficiently prevent peroxynitrite induced
oxidations and nitrations in vivo.
Acknowledgement - This project was supported by the ETH and the Swiss
Nationalfonds.
References
[1] Lymar, S. V.; Hurst, J. K. Rapid reaction between peroxonitrite ion and
carbon dioxide: Implications for biological acitivity. J. Am. Chem. Soc.
117:8867-8868; 1995.
[2] Kurz, C.; Kissner, R.; Koppenol, W. H. Mechanism of the reaction of
peroxynitrite with L-ascorbate studied at physiological pH. Free Radical
Biol. Med. in progress: 2004.
[3] Bielski, B. H. J.; Comstock, D. A.; Bowen, R. A. Ascorbic acid free
radicals. I. Pulse radiolysis study of optical absorption and kinetic
properties. J. Am. Chem. Soc. 93:5624-5629; 1971.
[4] Bonini, M. G.; Radi, R.; Ferrer-Sueta, G.; Ferreira, A. M. D.; Augusto, O.
Direct EPR detection of the carbonate radical anion produced from
peroxynitrite and carbon dioxide. J. Biol. Chem. 274:10802-10806; 1999.
[5] Goldstein, S.; Czapski, G.; Lind, J.; Merényi, G. Mechanism of
decomposition of peroxynitric ion (02NOO~): Evidence for the formation
of 02" and N02 radicals. Inorg. Chem. 37:3943-3947; 1998.
[6] Meli, R.; Nauser, T.; Latal, P.; Koppenol, W. H. Reaction of peroxynitritewith carbon dioxide: Intermediates and determination of the yield of
C03' andN02'. J Biol. Inorg. Chem. 7:31-36; 2002.
121

[7] Alfassi, Z. B.; Huie, R. E.; Neta, P.; Shoute, L. C. T. Temperature
dependence of the rate constants for reaction of inorganic radicals with
organic reductants. J. Phys. Chem. 94:8800-8805; 1990.
[8] Huie, R. E.; Shoute, L. C. T.; Neta, P. Temperature dependence of the rate
constants for reactions of the carbonate radical with organic and inorganicreductants. Int. J. Chem. Kinet. 23:541-552; 1991.
[9] Kirsch, M.; de Groot, H. Ascorbate is a potent antioxidant against
peroxynitrite-induced oxidation reactions - Evidence that ascorbate acts
by re-reducing substrate radicals produced by peroxynitrite. J. Biol.
Chem. 275:16702-16708; 2000.
[10] Whiteman, M.; Halliwell, B. Protection against peroxynitrite-dependent
tyrosine nitration and al-antiproteinase inactivation by ascorbic acid. A
comparison with other biological antioxidants. Free Radical Res. 25:275-
283; 1996.
[11] Balavoine, G. G. A.; Geletii, Y. V. Peroxynitrite scavenging by different
antioxidants. Part I: Convenient assay. Nitric Oxide: Biol. Chem. 3:40-54;1999.
[12] Lee, J. B.; Hunt, J. A.; Groves, J. T. Manganese porphyrins as redox-
coupled peroxynitrite reductases. J. Am. Chem. Soc. 120:6053-6061;1998.
[13] Lee, J. B.; Hunt, J. A.; Groves, J. T. Rapid decomposition of peroxynitrite
by manganese porphyrin-antioxidant redox couples. Bioorg. Med. Chem.
Lett. 7:2913-2918; 1997.
122

10. CURRICULUM VITAE
Name: Christophe Robert Kurz
Geburtsdatum: 26. Mai 1972
Zivilstand verheiratet
Bürgerort Vechigen (Be)
Ausbildungen
2001 Ausbildung in Menschen und Mitarbeiterführung bei Prof.
R. Steiger, ETH Zürich
2000--2004 Dissertation in der Arbeitsgruppe von Prof. Dr. W. H.
Koppenol am Institut für Anorganische Chemie der ETH
Zürich mit Schwergewicht Peroxynitrit.
1998 Ausbildung zum Strahlenschutz-Sachverständigen am
Paul Scherrer Institut
1993--1999 Chemiestudium an der ETH Zürich mit Spezialisierung
auf Anorganischer Chemie und Analytik.
1988--1992 Matur Typus C am Mathematisch
Naturwissenschaftlichen Gymnasium der Kantonsschule
Zürich
Zusätzliches
2002 Vorlesungen in Vortrags und Diskussionstechnik bei Prof.
R. Steiger, ETH Zürich
2001 Vorlesung in Discovery Management im Zentrum für
Betriebswissenschaften, ETH Zürich
2000-2003 Assistent im Praktikum Allgemeine Chemie I/II an der
ETH Zürich
123

124