in copyright - non-commercial use permitted rights ...28183/... · design of artificial...
TRANSCRIPT

Research Collection
Doctoral Thesis
Design of artificial microtissues
Author(s): Kelm, Jens Michael
Publication Date: 2005
Permanent Link: https://doi.org/10.3929/ethz-a-005064702
Rights / License: In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For moreinformation please consult the Terms of use.
ETH Library

DISS. ETH No. 16188
Design of artificial microtissues
A dissertation submitted to the
SWISS FEDERAL INSTITUTE OF TECHNOLOGY ZURICH
For the degree of
Doctor of Natural Sciences
Presented by
JENS MICHAEL KELM
Dipl. Biotech. TU Braunschweig
born 24.06.1971
Citizen of
Germany
Accepted on the recommandation of
Prof. Dr. Martin Fussenegger, examiner
Prof. Dr. Sabine Werner, co-examiner
Zurich, 2005

Summary VII
Summary
Design of artificial microtissues (100-400 µm in diameter) by self-assembling of
monodispersed primary or neoplastic/engineered cell lines is gathering momentum in
regenerative medicine, developmental biology and the design of more reliable cell-based
drug-discovery initiatives. In this work, we have refined the hanging drop cultivation
technology, to generate artificial microtissues under controlled conditions and evaluated their
potential to (i) maintain tissue-specific functionality, (ii) produce therapeutic proteins, (iii)
provide complex feeder structures for difficult-to-differentiate cell types, iv) induce
neovascularization and (v) support integration of implants into the host tissue and vascular
network.
Hepatocytes originating from a human hepatocellular carcinoma adopted a polarized
in vivo-like morphology, expressing a number of hepatocyte-specific transcripts
involved in liver metabolism and detoxification (Chapter 2, 8).
We developed the first 3D cell-based high-throughput-compatible in culture ELISA
for VEGF profiling (Chapter 2).
Neonatal rat (NRCs) and mouse cardiomyocytes (NMCs) were assembled to
contracting myocardial microtissues, which retained cardiomyocyte-specific
morphology. For the first time, we were able to grow dissociated adult rat
cardiomyocytes (ARCs) in a 3-dimensional environment (Chapter 3).
Recombinant protein production of lentiviral transduced NRCs and NMCs could be
increased up to 6-fold in microtissue cultures, compared to their monolayer
counterparts (Chapter 3).
Self-patterning to an in vivo-like morphology could be observed in cultures of two and
more cell types according to the differential adhesion hypothesis. In mixed cultures of
human umbilical endothelial cells (HUVECs) and human fibroblasts/hepatocytes,
HUVECs enveloped the microtissues and assembled a barrier between the surrounding
liquid and the tissue (Chapter 2, 5). Sensory neurons originating from embryonic
mouse dorsal root ganglions (DRGs), mixed with mouse-derived fibroblasts and
Schwann cells segregated to the outer surface and formed ganglia-like cap structures.
Outgrowing axons were aligned with myelinating Schwann cells (Chapter 4).

Summary VIII
Microtissues of several cell types were found to express endogenous vascular
endothelial growth factor (VEGF). This was utilized to engineer vascularized
microtissues without addition of any proangiogenic factor (Chapter 5).
Myocardial microtissues placed onto the embryonic chicken chorioallantoic
membrane (CAM) integrated into the CAM and connected to the host vascular
network without supply of any proangiogenic factors (Chapter 6).
Myocardial microtissues composed of neonatal rat cardiomyocytes transplanted into
the pericardial cavity of adult rats entirely integrated within 7 days into the
myocardium (Chapter 6).
Microtissues were successfully used as building blocks to assemble larger-sized tissue
constructs (heart muscle, cartilage, connective tissue, skeletal muscle). Integrating
HUVECs considerably improved connective microtissue assembly, indicating that
endothelial cells might play an important role for tissue integrity (Chapter 6, 7).
Human fibroblast-composed macrotissues placed onto the CAM connected to the
chicken vascular network only when HUVECs had been incorporated, whereas
HUVEC-free macrotissues completely failed to integrate into the CAM (Chapter 7).

Summary IX
Zusammenfassung
Die Entwicklung von artifiziellen Mikrogeweben (100-400 µm im Durchmesser) mittels
zellulärer Reaggregation gewinnt immer mehr an Bedeutung für die regenerative Medizin, in
der Entwicklungsbiologie und der Entdeckung neuer Medikamenten durch verlässlichere Zell
basierender Untersuchungen. In dieser Arbeit haben wir artifizielle Mikrogewebe in
Tropfkulturen unter kontrollierten Bedingungen produziert und deren Potential zur i)
Erhaltung Gewebe-spezifischer Strukturen, ii) Produktion von therapeutischen Proteinen, iii)
Induzierung eines Gefäßsystems in vitro, iv) Induktion von Neovaskularisierung und v)
Integration/Anbindung an ein bestehendes Gefäßsystem untersucht.
Leberzellen, isoliert von einem Leberkarzinoma, behielten die für Hepatozyten
charakteristische polarisierte Zellmorphologie und exprimierten Leber spezifische
Transkripte die für den Leberstoffwechsel wichtig sind wesentlich stärker als
Hepatozyten in klassischen Monolayer Kulturen (Kapitel 2, 8).
Basierend auf die Produktion von Mikrogeweben haben wir einen „in culture“ ELISA
zur Detektion von VEGF entwickelt (Kapitel 2).
Neonatale Ratten und Maus Herzmuskelzellen reaggregierten zu funktionellen
Herzmuskelmikrogewebe in denen die Kardiomyozyten ihre spezifische Morphologie
aufrecht erhielten. Wir konnten zum ersten Mal zeigen, das sich adulte
Kardiomyozyten in einem 3-dimensionalen Verbund kultivieren lassen (Kapitel 3).
Protein Produktion von lentiviral transduzierten, rekombinanten, neonatalen
Kardiomyozyten, konnte in Mikrogeweben im Vergleich zu Monolayer Kulturen um
das 6-fache gesteigert werden (Kapitel 3).
Mischkulturen von zwei oder mehr Zelltypen organisierten sich selbst zu in vivo
ähnlichen Strukturen, so wie es von der differentiellen Adhäsions-Theorie beschrieben
wird. Mischkulturen von humanen Nabelschnur Endothelzellen und humanen
Fibroblasten/Hepatozyten umschlossen jeweils die Endothezellen das Mikrogewebe
und bildeten eine Barriere zwischen dem umgebenden Flüssigkeit und dem Gewebe
(Kapitel 2, 5). Sensorische Nerven isoliert von embryonalen Maus dorsalen
Wurzelganglions vermischt mit Maus Fibroblasten entmischten sich und die Neuronen
bildeten Ganglia ähnliche Strukturen. Die auswachsenden Axone waren assoziiert mit
Schwann’schen Zellen und teilweise myelinisiert (Kapitel 4).

Summary X
Mikrogewebe von verschiedenen Zelltypen produzierten endogenen vaskulären
Endothelzell Wachstumsfaktor (VEGF). Diese Eigenschaft konnte genutzt werden, um
Mikrogewebe zu vaskularisieren ohne Zugabe von proangiogenen Faktoren (Kapitel
5).
Herzmuskelmikrogewebe platziert auf der chorioallantoischen Membran eines
embryonalen Huhns, wanderte ins Membrangewebe ein und induzierte
Revaskularisierung ohne Zugabe zusätzlicher proangiogenen Faktoren (Kapitel 6).
Herzmuskelmikrogewebe welches in den Herzbeutel injiziert wurde, integrierte sich
innerhalb von sieben Tagen ins Herzmuskelgewebe des Empfängers (Kapitel 6).
Mikrogewebe ( m3 Maßstab) konnten erfolgreich als kleinste mögliche
Gewebeeinheiten verwendet werden um größere Gewebe (mm3 Maßstab) zu
generieren. Dabei hat sich herausgestellt, dass Endothelzellen die Reaggregation von
Mikrobindegewebe erheblich verbessern konnten Kapitel (6, 7).
Aus humanen Fibroblasten bestehende Makrogewebe verbanden sich mit dem
Gefäßsystem der chorioallantoischen Membran nur wenn Endothezellen mit ins
Gewebe eingearbeitet wurden. Gewebe ohne Endothezellen wurden abgestoßen und
starben ab (Kapitel 7).

I
Table of Contents
SUMMARY.......................................................................................................................................................VII
ZUSAMMENFASSUNG....................................................................................................................................IX
Chapter 1:
Impact of 3D Cell Culture Technology
2D VS 3D CELL CULTURE................................................................................................................................2
3D CELL CULTURE SYSTEMS ........................................................................................................................4
REFERENCES...................................................................................................................................................... 7
Chapter 2:
Microscale Tissue Engineering Using Gravity-Enforced Cell Assembly
ABSTRACT ......................................................................................................................................................... 11
IMPACT OF MICROTISSUE DESIGN ON REGENERATIVE MEDICINE............................................. 11
SCAFFOLD-FREE MICROTISSUES – HANGING DROP TECHNOLOGY REVISITED...................... 13
DESIGN OF ARTIFICIAL HEPATIC TISSUES............................................................................................ 16
ARTIFICIAL MYOCARDIAL MICROTISSUE ............................................................................................ 18
MICROCARTILAGE......................................................................................................................................... 20
BEYOND TISSUE ENGINEERING – THE FUTURE OF MICROTISSUES IN
BIOPHARMACEUTICAL MANUFACTURING AND HIGH THROUGHPUT DRUGDISCOVERY....22
PERSPECTIVES................................................................................................................................................. 25
ACKNOWLEDGEMENTS................................................................................................................................ 25
REFERENCES.................................................................................................................................................... 25

II
Chapter 3:
Design of Artificial Myocardial Microtissues
ABSTRACT ......................................................................................................................................................... 31
INTRODUCTION............................................................................................................................................... 32
MATERIALS AND METHODS........................................................................................................................ 34
Isolation and three-dimensional cultivation of neonatal rat and mouse cardiomyocytes.................................. 34
Video microscopy............................................................................................................................................. 35
Fluorescence-based characterization of cell morphologies............................................................................... 35
Confocal light microscopy................................................................................................................................ 36
Lentivirus-based transduction technology ........................................................................................................ 36
Quantitative expression analysis of the secreted -amylase (SAMY) ................................................................ 37
RESULTS............................................................................................................................................................. 37
Production of myocardial microtissues............................................................................................................. 37
Immunohistological characterization of myocardial microtissues.................................................................... 41
Characterization of the extracellular matrix of myocardial microtissues.......................................................... 43
Expression of vascular endothelial growth factor (VEGF) by myocardial microtissues .................................. 44
Lentiviral infection of cardiomyocytes............................................................................................................. 46
DISCUSSION ...................................................................................................................................................... 48
ACKNOWLEDGEMENTS................................................................................................................................ 50
REFERENCES.................................................................................................................................................... 51
Chapter 4:
Self-Assembling of Sensory Neurons to Ganglia-like Structures
ABSTRACT ......................................................................................................................................................... 56
INTRODUCTION............................................................................................................................................... 56
MATERIAL AND METHODS.......................................................................................................................... 58
Isolation of mouse embryonic dorsal root ganglia (DRG) and fibroblasts ....................................................... 58
Cell culture and microtissue production ........................................................................................................... 59
Macrotissue assembly....................................................................................................................................... 59
Immunofluorescence-based cell characterization ............................................................................................. 59
Histology .......................................................................................................................................................... 60

III
Confocal light microscopy................................................................................................................................ 60
Transmission Electron Microscopy .................................................................................................................. 61
Gas-inducible ifn- expression ......................................................................................................................... 61
RESULTS............................................................................................................................................................. 62
Cellular re-organization .................................................................................................................................... 62
Development of 3D neuronal structures ........................................................................................................... 63
Long-term cultivation of DRG:MEF microtissue cultures ............................................................................... 66
Assembly of innervated macrotissues............................................................................................................... 67
DISCUSSION ...................................................................................................................................................... 68
ACKNOWLEDGEMENTS................................................................................................................................ 71
REFERENCES.................................................................................................................................................... 71
Chapter 5:
VEGF Profiling and Angiogenesis in Human Microtissues
ABSTRACT ......................................................................................................................................................... 76
INTRODUCTION............................................................................................................................................... 76
MATERIAL AND METHODS.......................................................................................................................... 78
Isolation of primary human aortic fibroblasts................................................................................................... 78
Cell culture ....................................................................................................................................................... 78
Microtissue production ..................................................................................................................................... 79
Fluorescence-based characterization of cell morphologies............................................................................... 79
Toluidine blue staining and immunohistochemistry of paraffin-embedded microtissue sections .................... 80
Confocal light microscopy................................................................................................................................ 80
Transmission Electron Microscopy .................................................................................................................. 81
ELISA-based VEGF quantification .................................................................................................................. 81
RESULTS............................................................................................................................................................. 81
VEGF production profiling of human cell-derived monolayer and microtissue cultures ................................. 81
Self-organization potential of different cell phenotypes in a microtissue format ............................................. 84
VEGF profiling of microtissues assembled from different cell types............................................................... 85
Angiogenesis-based capillary formation in microtissues.................................................................................. 86
Inhibition of angiogenesis in HAF-HUVEC microtissues................................................................................ 92
DISCUSSION ...................................................................................................................................................... 92

IV
ACKNOWLEDGMENTS................................................................................................................................... 95
REFERENCES.................................................................................................................................................... 95
Chapter 6:
Improved Tissue-Transplant Fusion and Vascularization of Myocardial
Micro- and Macrotissues Implanted into Chicken Embryos and Rats
ABSTRACT ....................................................................................................................................................... 101
INTRODUCTION............................................................................................................................................. 101
MATERIAL AND METHODS........................................................................................................................ 103
Preparation of primary cells.............................................................................................................................103
Microtissue Production....................................................................................................................................104
Macrotissue Assembly.....................................................................................................................................104
Immunofluorescence analysis..........................................................................................................................104
Confocal light microscopy...............................................................................................................................105
Transmission electron microscopy ..................................................................................................................105
Microchip-based electrophysiology.................................................................................................................105
Chicken chorioallantoic membrane (CAM) assay ...........................................................................................106
Transplantation of myocardial microtissues into rat hearts .............................................................................106
RESULTS............................................................................................................................................................107
Microtissues assembled from adult cardiomyocytes .......................................................................................107
Microchip-based electrophysiologic analysis of myocardial microtissues ......................................................109
Design and neo-vascularization of higher-order macrotissues assembled from individual myocardial
microtissues .....................................................................................................................................................110
Inter-species vascularization crosstalk enables connection of myocardial microtissues to the chicken embryo
vasculature .......................................................................................................................................................113
Integration of implanted myocardial microtissues into rat hearts ....................................................................115
DISCUSSION .................................................................................................................................................... 118
ACKNOWLEDGMENTS................................................................................................................................. 120
REFERENCES.................................................................................................................................................. 120

V
Chapter 7:
Design of Custom-Shaped Vascularized Tissues Using Microtissue
Spheroids as Minimal Building Units
ABSTRACT ....................................................................................................................................................... 125
INTRODUCTION............................................................................................................................................. 125
MATERIAL AND METHODS........................................................................................................................ 127
Preparation of primary cells.............................................................................................................................127
Cell Culture......................................................................................................................................................127
Microtissue Production....................................................................................................................................127
Macrotissue Assembly.....................................................................................................................................128
Immunohistochemistry ....................................................................................................................................128
Transmission electron microscopy ..................................................................................................................129
Chicken chorioallantoic membrane (CAM) assay ...........................................................................................129
RESULTS........................................................................................................................................................... 130
Microtissue assembly to larger-sized macrotissues .........................................................................................130
Neo-vascularization of scaffold-free macrotissues ..........................................................................................134
Implantation of HMF-HUVEC macrotissues into chicken embryos ...............................................................136
DISCUSSION .................................................................................................................................................... 139
ACKNOWLEDGEMENTS.............................................................................................................................. 141
REFERENCES.................................................................................................................................................. 141
Chapter 8:
Synergies of Microtissue Design, Viral Transduction and Adjustable
Transgene Expression for Regenerative Medicine
ABSTRACT ....................................................................................................................................................... 146
INTRODUCTION............................................................................................................................................. 146
DESIGN OF ARTIFICIAL MICROTISSUES............................................................................................... 147
Directed Cell Differentiation ...........................................................................................................................149
Gene-function analysis ....................................................................................................................................150
Animal-free drug testing and drug discovery ..................................................................................................151
VIRAL TRANSDUCTION............................................................................................................................... 152

VI
Viral vectors for gene therapy .........................................................................................................................153
Gene regulation................................................................................................................................................157
Perspectives .....................................................................................................................................................158
ADJUSTABLE TRANSGENE EXPRESSION .............................................................................................. 158
Key characteristics of an ideal gene regulation system ...................................................................................159
Antibiotic-controlled gene regulation systems ................................................................................................159
Transgene control by chemically induced dimerization ..................................................................................160
Hormone-inducible gene expression................................................................................................................161
Quorum sensing-based transgene modulation .................................................................................................161
Temperature-dependent gene regulation..........................................................................................................162
Gene regulation systems in drug discovery .....................................................................................................162
Use of gene-control systems in biopharmaceutical manufacturing .................................................................163
Outlook ............................................................................................................................................................163
REFERENCES.................................................................................................................................................. 166
ACKNOWLEDGEMENTS.............................................................................................................................. 181
CURRICULUM VITAE ................................................................................................................................... 183

Chapter 1
Impact of 3D Cell Culture Technology

3D Cell Culture Technology 2
2D vs 3D Cell Culture
In vitro cultivation of mammalian cells is predominantly carried out growing cells on
adhesive cell culture surfaces as flat monolayers. However, in their natural environment cells
not only adhere to each other, but are also embedded in an extracellular matrix (ECM)
containing proteins such as collagens, intergrins, laminin, and fibronectin, which affect cell
shape (Goldmann 2002), polarity (Boudreau 2003), tension (Tarone et al. 2000),
differentiation (Bokel et al. 2002) and help to organize communication between the cells
(Schenk et al. 2003) (Figure 1). Local disruption of ECM by pharmacologic or genetic means
results in selective programmed cell death (apoptosis) among adjacent cells (Boudreau et al.
1995). The cell shape of endothelial cells control whether individual cells grow or die,
representing a fundamental mechanism for cell fate regulation within a tissue environment
(Chen et al. 1997). Given this complex mechanical and biochemical interplay of cells in a
tissue, it is no surprise that cells grown in flat monolayers miss biological subtleties (Abbott
2003).
3D structures and interaction between cells and their microenvironment are already
essential in the earliest stages of embryonic development for organization, differentiation and
proliferation (Mathis et al. 2002). For example, the requisite step of cellular condensation
during mesenchymal chondrogenesis is mimicked in vitro in chondrocyte cultures where high
cell densities results in the formation of 3D spheroid structures that are cartilaginous in nature
and associated with the upregulation of ECM components such as type II collagen and
cartilage link protein (Denker et al. 1995). Studies of embryonic chick (calvarial or limb-bud)
cells also confirm the cell density-mediated induction of chondrogenesis (Wong et al. 1995;
Woodward et al. 1999) and demonstrate requirement for cell-cell interactions in this process
(Woodward et al. 1999). Bone development requires the concerted action of several
microenvironmental signals. During osteogenesis cells differentiate into pre-osteoblasts and
then undergo cellular condensation, a process, which precedes osteoblast differentiation and
matrix mineralization (Dunlop et al. 1995). The similarity between these systems of chondro-
and osteogenesis and their concordance with similar processes in the developing embryo
strongly suggest that cell organization into 3D structures is essential for ex vivo tissue
formation (Kale et al. 2000).
Due to the lack of 2-dimensional (2D) cell culture technologies to display tissue-like
phenotypes, biologists are turning more and more to 3-dimensional (3D) cell culture

3D Cell Culture Technology 3
technologies. Radiation biologists have used multicellular tumor spheroids (MCTS) for
around 25 years and their utility is now receiving a wider appreciation. MCTS reproduce the
tumor microenvironment more accurately than 2D cultures (Sutherland 1988; Mueller-Klieser
1997; Hamilton 1998; Kunz-Schughart et al. 1998; Desoize et al. 2000), which has profound
implications for tumor biology, particularly with regard to altered gene expression and
sensitivity to chemotherapeutic agents (multicellular resistance) (Dubessy et al. 2000). Cancer
cells have shown to respond differently to anticancer drugs in 2D and 3D configuration.
Breast cancer cells treated with antibodies against the cell surface receptor 1-integrin
changed their abnormal shape and growth behaviour in 3D culture whereas the effect couldn’t
be observed in monolayer cultures (Weaver et al. 1997). Jacks and Weinberg went as far as to
quote the study of cancer cells in monolayers without including their ECM environment and
neighbouring cells as quaint if not archaic (Jacks et al. 2002) (see also Figure 1).
For tissue engineering initiatives, coaxing cells to form artificial tissues in a reliable
manner is the quintessential engineering design problem that must be accomplished. Tissue
engineering exploits living cells in a variety of ways to restore, maintain or enhance tissues
and organs. It conjures up visions of organs built from scratch in the laboratory with the
potential impact to reduce the need for organ replacement and accelerate the development of
new drugs. Cell-based testing is well established in drug discovery with well-described
models that exist for cancer (Johnson et al. 2001), intestinal absorption (Le Ferrec et al. 2001)
and diabetes (Reed et al. 1999). A cell-based model that is faithful to its in vivo behaviour
offers obvious advantages, such as predictability, savings of time and cost. However, current
models fall short of this ideal (Bhadriraju et al. 2002). Even genetically normal cells, such as
hepatocytes or endothelial cells placed into in cell culture quickly loose their differentiated
gene expression pattern and phenotype (Berthiaume et al. 1996; Kelm et al. 2004). For
example, the hepatitis C virus has infected more than 170 million people worldwide, but
infecting liver cells in vitro is extremely difficult as human hepatocytes quickly loose their
susceptibility to viral infection. In vitro engineered liver tissue may provide a cheaper system
with better control of variables for studying viral infection compared to animal model systems
(Griffith et al. 2002). The more closely in vitro models mimic the morphology and
biochemical processes in the body the more it will allow researchers to reduce the use of
experimental animals even if it will never replace in vivo trials.
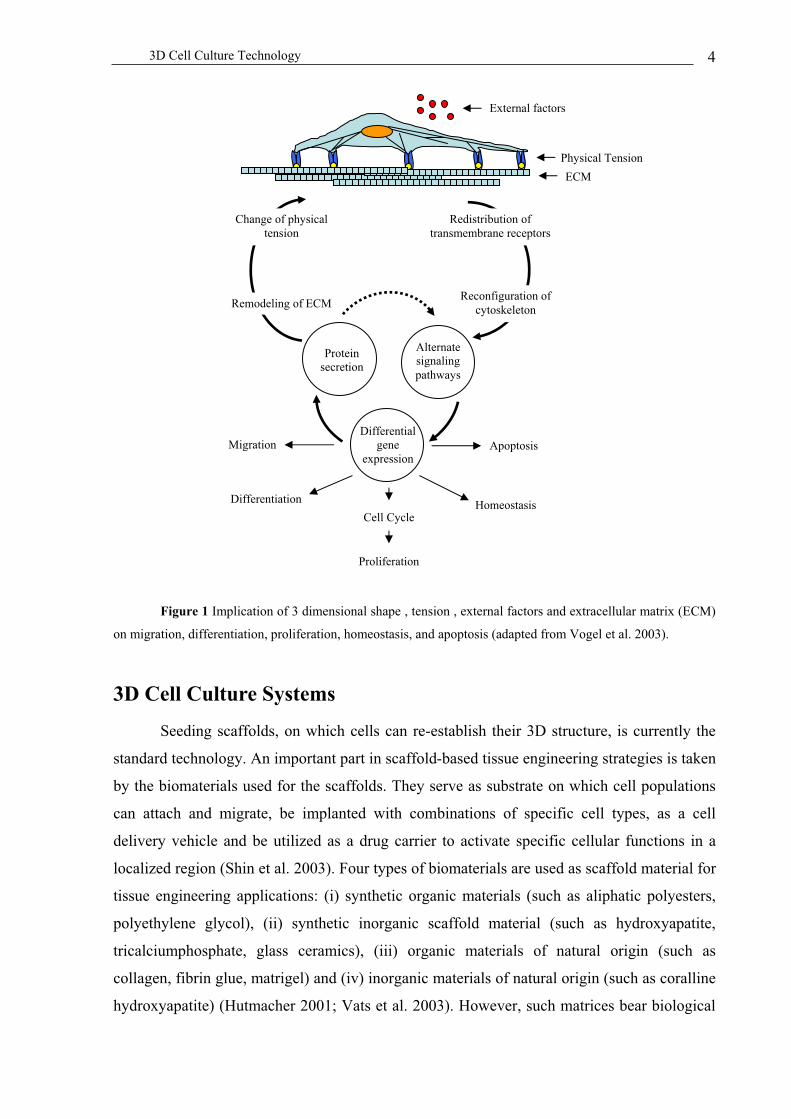
3D Cell Culture Technology 4
Figure 1 Implication of 3 dimensional shape , tension , external factors and extracellular matrix (ECM)
on migration, differentiation, proliferation, homeostasis, and apoptosis (adapted from Vogel et al. 2003).
3D Cell Culture Systems
Seeding scaffolds, on which cells can re-establish their 3D structure, is currently the
standard technology. An important part in scaffold-based tissue engineering strategies is taken
by the biomaterials used for the scaffolds. They serve as substrate on which cell populations
can attach and migrate, be implanted with combinations of specific cell types, as a cell
delivery vehicle and be utilized as a drug carrier to activate specific cellular functions in a
localized region (Shin et al. 2003). Four types of biomaterials are used as scaffold material for
tissue engineering applications: (i) synthetic organic materials (such as aliphatic polyesters,
polyethylene glycol), (ii) synthetic inorganic scaffold material (such as hydroxyapatite,
tricalciumphosphate, glass ceramics), (iii) organic materials of natural origin (such as
collagen, fibrin glue, matrigel) and (iv) inorganic materials of natural origin (such as coralline
hydroxyapatite) (Hutmacher 2001; Vats et al. 2003). However, such matrices bear biological
External factors
ECM
Redistribution of
transmembrane receptors
Reconfiguration of
cytoskeleton
Change of physical
tension
Remodeling of ECM
Differential
gene
expression
Protein
secretion
Alternate
signaling
pathways
ApoptosisMigration
Homeostasis Differentiation
Cell Cycle
Proliferation
Physical Tension

3D Cell Culture Technology 5
information and elicit biological response, which might differ from the response found in the
natural microenvironment (Hunziker 1999). The inability of biomaterial scaffolds to
functionally integrate into surrounding tissue is one of the major roadblocks to developing
new biomaterials and tissue-engineering scaffolds (Vogel et al. 2003). Third generation
biomaterials (biomimetic materials) are capable of eliciting specific cellular response and
directing new tissue formation mediated by specific interactions which can be manipulated by
altering design parameters instead of non-specifically adsorbed ECM proteins (Hench et al.
2002). Despite considerable advances, current approaches to engineering cell-surface
interactions fall short in mimicking the complexity of signals through which surrounding
tissue regulates cell behavior such as induction of angiogenesis (Vogel et al. 2003). However,
one may ask why cells originating from a tissue environment should need a scaffold to rebuild
a tissue-like community.
Figure 2 Illustration of a scaffold without (A) and seeded with cells (B) (photographed by Carnegie
Mellon University, Bone Tissue Engineering Initiative)
An alternative to scaffold-based concepts is the cellular reaggregation, an attempt to
achieve a more or less complete regeneration of tissues from dispersed cells of a particular
origin under controlled conditions (Chapter 2). In contrast to the use of scaffolds, there is only
marginal experience in tissue engineering concerning cellular viability and integration into
host tissue of cellular reaggregates but in general it will reduce post-implantational side
effects to a solely cell-based problem. At least good integration of chondrocytes cultured as
spheroids on human condyle cartilage has been observed so far (Anderer et al. 2002). One of
the principle constraints of the size of tissues engineered in vitro that do not have their own
blood supply is the short distance over which oxygen can diffuse before being consumed
(Griffith et al. 2002). To control the cell number/composition reducing the tissue size to a
minimum, we made use of the hanging drop technology to accumulate cells into a tissue-like
environment (Figure 3, 4). This study exploits the potential of smallest possible tissue units

3D Cell Culture Technology 6
(microtissues) assembled in hanging drops for (i) tissue generation, (ii) cellular organization,
(iii) angiogenic properties and (iv) microtissue graft integration.
Figure 3 Gravity enforced assembly of microtissues in hanging drops. Droplets of a single cell
suspension are placed onto a surface and cultivated upside down. After 1-4 days depending on the cell type, cells
forme a cellular reaggregate
Figure 4 Generation of a multilayer micortissue configuration. After accumulation of a feeder spheroid,
a second cell type of interest is added into the hanging drop. The added cells form an additional cell layer around
the feeder spheroid (Chapter 2, 4).
References
Abbott, A. (2003). "Cell culture: biology's new dimension." Nature 424, 870-2.
Alberts, B., D. Bray, J. Lewis, M. Raff, K. Roberts and J. D. Watson (1994). Molecular
Biology of the cell. New York & London, Garland Publishing, Inc.
Anderer, U. and J. Libera (2002). "In vitro engineering of human autogenous cartilage." J
Bone Miner Res 17, 1420-9.
Surface
Medium
Cells
Initiation of a
Feeder Spheroid
Addition of a
Second Cell TypeDevelopment of a
Cell Layer

3D Cell Culture Technology 7
Berthiaume, F., P. V. Moghe, M. Toner and M. L. Yarmush (1996). "Effect of extracellular
matrix topology on cell structure, function, and physiological responsiveness:
hepatocytes cultured in a sandwich configuration." Faseb J 10, 1471-84.
Bhadriraju, K. and C. S. Chen (2002). "Engineering cellular microenvironments to improve
cell-based drug testing." Drug Discov Today 7, 612-20.
Bokel, C. and N. H. Brown (2002). "Integrins in development: moving on, responding to, and
sticking to the extracellular matrix." Dev Cell 3, 311-21.
Boudreau, N., C. J. Sympson, Z. Werb and M. J. Bissell (1995). "Suppression of ICE and
apoptosis in mammary epithelial cells by extracellular matrix." Science 267, 891-3.
Boudreau, N. J. (2003). "Organized living: from cell surfaces to basement membranes." Sci
STKE 2003, pe34.
Chen, C. S., M. Mrksich, S. Huang, G. M. Whitesides and D. E. Ingber (1997). "Geometric
control of cell life and death." Science 276, 1425-8.
Denker, A. E., S. B. Nicoll and R. S. Tuan (1995). "Formation of cartilage-like spheroids by
micromass cultures of murine C3H10T1/2 cells upon treatment with transforming
growth factor-beta 1." Differentiation 59, 25-34.
Desoize, B. and J. Jardillier (2000). "Multicellular resistance: a paradigm for clinical
resistance?" Crit Rev Oncol Hematol 36, 193-207.
Dubessy, C., J. M. Merlin, C. Marchal and F. Guillemin (2000). "Spheroids in radiobiology
and photodynamic therapy." Crit Rev Oncol Hematol 36, 179-92.
Dunlop, L. L. and B. K. Hall (1995). "Relationships between cellular condensation,
preosteoblast formation and epithelial-mesenchymal interactions in initiation of
osteogenesis." Int J Dev Biol 39, 357-71.
Goldmann, W. H. (2002). "Mechanical aspects of cell shape regulation and signaling." Cell
Biol Int 26, 313-7.
Griffith, L. G. and G. Naughton (2002). "Tissue engineering--current challenges and
expanding opportunities." Science 295, 1009-14.
Hamilton, G. (1998). "Multicellular spheroids as an in vitro tumor model." Cancer Lett 131,
29-34.
Hench, L. L. and J. M. Polak (2002). "Third-generation biomedical materials." Science 295,
1014-7.
Hunziker, E. B. (1999). "Articular cartilage repair: are the intrinsic biological constraints
undermining this process insuperable?" Osteoarthritis Cartilage 7, 15-28.

3D Cell Culture Technology 8
Hutmacher, D. W. (2001). "Scaffold design and fabrication technologies for engineering
tissues--state of the art and future perspectives." J Biomater Sci Polym Ed 12, 107-24.
Jacks, T. and R. A. Weinberg (2002). "Taking the study of cancer cell survival to a new
dimension." Cell 111, 923-5.
Johnson, J. I., S. Decker, D. Zaharevitz, L. V. Rubinstein, J. M. Venditti, S. Schepartz, S.
Kalyandrug, M. Christian, S. Arbuck, M. Hollingshead and E. A. Sausville (2001).
"Relationships between drug activity in NCI preclinical in vitro and in vivo models
and early clinical trials." Br J Cancer 84, 1424-31.
Kale, S., S. Biermann, C. Edwards, C. Tarnowski, M. Morris and M. W. Long (2000).
"Three-dimensional cellular development is essential for ex vivo formation of human
bone." Nat Biotechnol 18, 954-8.
Kelm, J. M., B. P. Kramer, V. Gonzalez-Nicolini, B. Ley and M. Fussenegger (2004).
"Synergies of microtissue design, viral transduction and adjustable transgene
expression for regenerative medicine." Biotechnol Appl Biochem 39, 3-16.
Kunz-Schughart, L. A., M. Kreutz and R. Knuechel (1998). "Multicellular spheroids: a three-
dimensional in vitro culture system to study tumour biology." Int J Exp Pathol 79, 1-
23.
Le Ferrec, E., C. Chesne, P. Artusson, D. Brayden, G. Fabre, P. Gires, F. Guillou, M. Rousset,
W. Rubas and M. L. Scarino (2001). "In vitro models of the intestinal barrier. The
report and recommendations of ECVAM Workshop 46. European Centre for the
Validation of Alternative methods." Altern Lab Anim 29, 649-68.
Mathis, L. and J. F. Nicolas (2002). "Cellular patterning of the vertebrate embryo." Trends
Genet 18, 627-35.
Mueller-Klieser, W. (1997). "Three-dimensional cell cultures: from molecular mechanisms to
clinical applications." Am J Physiol 273, C1109-23.
Reed, M. J. and K. A. Scribner (1999). "In-vivo and in-vitro models of type 2 diabetes in
pharmaceutical drug discovery." Diabetes Obes Metab 1, 75-86.
Schenk, S. and V. Quaranta (2003). "Tales from the crypt[ic] sites of the extracellular matrix."
Trends Cell Biol 13, 366-75.
Shin, H., S. Jo and A. G. Mikos (2003). "Biomimetic materials for tissue engineering."
Biomaterials 24, 4353-64.
Sutherland, R. M. (1988). "Cell and environment interactions in tumor microregions: the
multicell spheroid model." Science 240, 177-84.

3D Cell Culture Technology 9
Tarone, G., E. Hirsch, M. Brancaccio, M. De Acetis, L. Barberis, F. Balzac, S. F. Retta, C.
Botta, F. Altruda, L. Silengo and F. Retta (2000). "Integrin function and regulation in
development." Int J Dev Biol 44, 725-31.
Vats, A., N. S. Tolley, J. M. Polak and J. E. Gough (2003). "Scaffolds and biomaterials for
tissue engineering: a review of clinical applications." Clin Otolaryngol 28, 165-72.
Vogel, V. and G. Baneyx (2003). "The tissue engineeting puzzle: a molecular perspective."
Annu Rev Biomed Eng 5, 441-63.
Weaver, V. M., O. W. Petersen, F. Wang, C. A. Larabell, P. Briand, C. Damsky and M. J.
Bissell (1997). "Reversion of the malignant phenotype of human breast cells in three-
dimensional culture and in vivo by integrin blocking antibodies." J Cell Biol 137, 231-
45.
Wong, M. and R. S. Tuan (1995). "Interactive cellular modulation of chondrogenic
differentiation in vitro by subpopulations of chick embryonic calvarial cells." Dev Biol
167, 130-47.
Woodward, W. A. and R. S. Tuan (1999). "N-Cadherin expression and signaling in limb
mesenchymal chondrogenesis: stimulation by poly-L-lysine." Dev Genet 24, 178-87.

Chapter 2
Microscale Tissue Engineering Using
Gravity-Enforced Cell Assembly
Kelm J.M. and Fussenegger M., (2004) Trends in Biotechnology 22, 201-214

Microscale Tissue Engineering 11
Abstract
Design of artificial microtissues by reaggregation of monodispersed primary cells or
neoplastic/engineered cell lines are gathering momentum in regenerative medicine and
provide insight into the third dimension of cell-cell interactions and underlying regulatory
networks. Recent advances in microtissue production have substantiated the potential of
scaffold-free cell aggregates to (i) maintain tissue-specific functionality, (ii) support seamless
integration of implants into host tissues, (iii) provide complex feeder structures for difficult-
to-differentiate cell types, (iv) be amenable to therapeutic and phenotype-modulating
interventions using latest-generation transduction technologies, (v) produce therapeutic
transgenes at increased levels and (vi) offer tissue-like assay environments to improve drug-
function correlations in current discovery programs. Focusing on liver (liver-specific
detoxification characteristics), heart (interconnection of contractile units) and cartilage
(mechanical properties) we cover the latest on scaffold-free microtissue design.
Impact of microtissue design on regenerative medicine
To some extent, regeneration takes place in the human body throughout life. For
example, blood and skin are continually restored while liver, bone, muscle and blood vessels
have a limited capacity for self-repair (Petit-Zeman 2001). Yet, after traumatic injury or (age-
related) disease including osteoporosis, diabetes, cardiovascular and neurodegenerative
disorders, extensive tissue damage/degeneration exceeds the tissue-encoded repair capacity
resulting in the formation of functionally impaired scar tissue. Irreparably damaged tissue
may either be replaced by medical devices or organ (xeno-) transplantation. However,
medical devices often lack durability and an extensive functional repertoire, donor organs are
in limited supply and ongoing concerns about provirus dissemination and hyperacute rejection
reactions limit the scope of current xenotransplantation protocols (Bouche 2002).
Since most current pharmaceutical interventions may at best retard but not revert
pathologic tissue degeneration, they could only be considered stopgaps as tissue engineering-
based regenerative medicine moves from the realms of science fiction to de novo creation of
artificial organs. As natural organ development is characterized by complex processes
orchestrating assembly of different cell types and integrating a near-infinite number of signals
in space and time design of artificial organs appears like a mission impossible. Until a

Microscale Tissue Engineering 12
system’s view on organs will be available, an optimal balance of harnessing cellular self-
repair programs combined with specific pharmacologic/genetic interventions will dominate
the clinical reality in the immediate future.
Unlimited supply of generic therapeutic cell phenotypes resulting from expansion of
desired primary cells or rational differentiation of multipotent (stem) cells by growth factors
or genetic interventions, managing shape and function, timely coordination of multi-cell type
assembly as well as mastering neoplastic cell expansion are among current challenges of
tissue engineers. Capitalizing on recent pharmacologic advances, proliferation-modulating
factors have become a cornerstone of tissue engineering either to expand cells for implant
production or to functionalize biomaterials for tissue regeneration (Lutolf et al. 2003).
However, the risk of eliciting neoplastic cell characteristics following extended expansion
procedures remains imminent. Positive proliferation control to enable expansion as well as
precise growth arrest to prevent neoplastic outgrowth following implantations are vital issues
which will require increased attention (Fussenegger et al. 1998; Fux in press).
Although expanded (stem) cell populations of desired phenotypes have often been
reported to bear significant therapeutic potential following direct injection into tissue lesions
(heart, cartilage), such therapeutic scenarios lack the scope associated with tissue implants
(Mangi et al. 2003; Rafii et al. 2003). Shaping functional 3D tissue from monodispersed
expanded cell cultures is an ongoing challenge (Abbott 2003). Tissue engineers have thus
relied on material science to provide (functionalized) scaffolds on which tissue cells may
grow and differentiate. Latest-generation scaffolds are branched to enable adequate feeding of
cells in the central layers (Kim et al. 1998; Zandonella 2003), provide biological and
mechanical functions of a native extracellular matrix (Kim et al. 1998), degrade once the
organ or tissue becomes established in the body, and may be designed to release growth
factors or transgene-encoding vectors in response to physiological cues (Lee et al. 2000;
Richardson et al. 2001; Lutolf et al. 2003). Although scaffolds offer unique clinical
opportunities in tissue engineering strategies which require a strict combination of shape and
function (e.g., bladder, bone, cartilage, intestine/stomach, liver, skin) shape-supporting
matrices could be expendable or even less suited for the design of brain and heart structures
(Ochoa et al. 2002) (Table 1). Exemplified by recent advances in the production of artificial
liver, heart and cartilage structures we are covering the latest trends in designing scaffold-free
artificial microtissues.

Microscale Tissue Engineering 13
Table 1 Potential advances of gravity-enforced microtissue design
1) Precise tissue size control owing to a strict correlation between cell number and spheroid
diameter to avoid oxygen and nutrient limitations of the in vitro culture (Kelm et al.
2003).
2) The ease to generate microtissues from different cell phenotypes mimicking natural cell-
type composition (Itskovitz-Eldor et al. 2000).
3) Cell mobility during assembly ensuring intercellular organization including polarization
(Rothermel et al. 2001).
4) Development of an extracellular matrix (Anderer et al. 2002).
5) Compatibility with high-throughput assay systems as well as robotic liquid handling
devices (Layer et al. 1992).
6) Applicability to small volumes and cell numbers (Layer et al. 2002).
7) Mild and natural assembly forces unlikely to interfere with cell regulatory networks.
8) Compatibility with a wide variety of cell types (see Table 2).
Scaffold-free microtissues – hanging drop technology revisited
Strategies harnessing the natural reaggregation potential to assemble monodispersed
cells in a tissue-mimicking manner represent a valuable extension of current scaffold-based
tissue engineering initiatives. Scaffold-free reaggregation of cells to microtissues may occur
following (i) cultivation in shake flasks, gyratory shakers and roller bottles (Furukawa et al.
2001; Kelm et al. 2003; Kelm et al. in press) or on non-adhesive surfaces (Kale et al. 2000),
(ii) centrifugation-based compression (Muraglia et al. 2003), (iii) maintenance in cell culture
inserts (Watzka et al. 2000), or (iv) gravity-enforced assembly of microspheres in hanging
drops (Kelm et al. in press) (Tables 2 and 3).
Originally pioneered for production of embryoid bodies and blastocysts to study the
differentiation potential of stem cells (Wobus et al. 2000), gravity-enforced assembly of
microtissues in hanging drops was found to be compatible with a variety of cell types and
became increasingly popular among tissue engineers (i) to assess tumor-related resistance to
chemotherapeutics in tissue-like cancer models (Bjerkvig et al. 1997; Kunz-Schughart 1999),
(ii) for gene-function analysis of differentiation phenomena and development (Itskovitz-Eldor
et al. 2000) and (iii) the design of functional microlivers, microhearts and microcartilage
(Kelm et al. 2003; Kelm et al. 2003; Kelm et al. in press) (Figure 1; Tables 2 and 3).

Microscale Tissue Engineering 14
Figure 1: Microtissues produced by gravity-enforced assembly of monodispersed cells of a single cell
type. Phase-contrast micrographs of microtissues reaggregated from human aortic fibroblasts (A), human dermal
fibroblasts (B), neonatal rat cardiomyocytes (C), and primary rat hepatocytes (D). (scale bar = 20 µm)
A key benefit of gravity-enforced microtissue design is the mobility of cells during
microtissue formation. Tissues consist of an organized assembly of several cell types a fact,
which should be considered for microtissue design. Although forces orchestrating proper
positioning of different cell types within a tissue remain elusive, cell movements during
development or following implantation of (stem) cells are well established (Brazelton et al.
2000; Clarke et al. 2000). Design of organotypic structures will require detailed understanding
of intercellular crosstalk in cocultures.
In order to get insight into organotypic positioning effects of different cell types, we
have evaluated gravity-enforced microtissue assembly from cocultures of (i) HepG2-HUVEC
(human umbilical vein endothelial cells), (ii) HAF (human aortic fibroblast)-HUVEC as well
as (iii) rat heart-derived cell mixtures. Although completely mixed following seeding in
hanging drops, HUVEC cells always move to the periphery which is reminiscent of the
concentric structures shaped during vascularization (Kelm et al., unpublished; Figure 2A and
B). Also, when cell mixtures reflecting the natural cell-type composition of rat hearts are
cultivated in hanging drops, muscle-specific cell phenotypes were predominantly found at the
periphery of beating microtissues (Kelm et al. in press) (Figure 2C;
http://www.biotech.biol.ethz.ch/martinf/staff/jens.html). These findings exemplify the
C D
A B
CC DD
AA BB

Microscale Tissue Engineering 15
compatibility of gravity-enforced microtissue design with the forces shaping organotypic
structures.
Microtissues could also be used as feeder spheroids, which provide a cell-based 3D
matrix for organotypic microcultivation of difficult-to-study cell types or polarized assembly
of different cell layers (Kelm et al. in press). Primary human keratinocytes (endothelial cells)
assembled tight and seamless cell layers when coated onto fibroblast (hepatocyte)-derived
feeder microtissues in hanging drop cultures (Figure 2C-E).
Owing to the wide variety of different tissues, their in vitro production and
engineering strategies, we prefer to focus on microscale tissue engineering initiatives
currently developed to design artificial (i) hepatic tissue (maintaining liver-specific
detoxification and metabolism), (ii) functional myocardial microtissues beating at human-
compatible frequencies (maintenance of contractile units and specialized cell-cell contacts)
and (iii) cartilage (managing mechanical properties).
Figure 2: Confocal analysis of organotypic microtissue structures assembled from two different cell
types by gravity-enforced reaggregation in hanging drops. Microtissues produced by monodispersed cell
A
D
B
C
E F
AA
DD
BB
CC
EE FF

Microscale Tissue Engineering 16
mixtures (A-C). Hepatocyte (HepG2) – human endothelial cell (HUVEC) (A), human aortic fibroblast (HAF) –
HUVEC (B) mixed cardiomyocyte populations reflecting the natural cell-type composition of the myocard (C)
were cultivated in hanging drops. The entire microtissue was visualized using F-actin-specific staining (red) and
HUVECs were stained for von Willebrand factor (vWF) (green). Cardiomyocytes were monitored by using a
sarcomeric alpha-actinin-specific antibody (green). Premanufactured feeder spheroids were coated with a second
cell type by cultivation in hanging drops (D-F). HepG2 (D) and HAF (E) feeder spheroids were coated with
HUVECs and normal human dermal fibroblasts (NHDF) were coated with keratinocytes (HaCaT) (F). Feeder
tissues were stained for F-Actin (red), HUVECs for vWF (green), HaCaTs for keratin (green). (scale bar =
10 µm)
Design of artificial hepatic tissues
The healthy liver is able to regenerate after injury. However, once damaged by fibrosis
and cirrhosis, resulting from a variety of chronic conditions including alcohol abuse or
infection with hepatitis virus B or C, the liver’s regeneration capacity is compromised. Liver
transplantation is a routine treatment for end-stage liver disease, yet donor organ shortage
continues to be a serious problem (Strain et al. 2002). The liver has a panoply of crucial
functions including production of clotting factors, bile, the regulation of carbohydrate, fat and
protein metabolism, detoxification and breakdown of alcohol and drugs. Most of these
functions are carried out by hepatocytes, which constitute up to 70% of a liver’s cellular
content. Although hepatocytes appear to be the prime candidate for the design of artificial
hepatic tissues, they rapidly lose liver-specific gene expression and become phenotypically
unstable following removal from the complex architecture of the liver (Berthiaume et al.
1996). However, in vitro cultivation of hepatocytes will be key to produce artificial hepatic
tissues for replacement therapies and extracorporal liver-assist devices. Progress in
maintaining the liver-specific phenotype of hepatocytes has been achieved by modulating
tissue culture conditions, providing an extracellular matrix and cocultivating hepatocytes with
other liver cell types (Bhatia et al. 1999; Harimoto et al. 2002).
A variety of different strategies have been pioneered to arrange hepatocytes in 3D
configurations which exhibit epithelial polarization and retain some liver-specific functions
(Hench et al. 2002): (i) Seeding onto preformed matrices (Ambrosino et al. 2003), (ii)
cultivation in soluble matrices (Kamihira et al. 1997), (iii) stacking of monolayers (Harimoto
et al. 2002) and (iv) reaggreagation of monodispersed cells (Kelm et al. 2003) (Tables 2 and
3). Ultrastructural analysis of hepatic HepG2-derived microtissues produced following
gravity-enforced reaggregation in hanging drops revealed seamless integration of single cells
into a compact microliver-like structure (Figure 3A). Individual hepatocytes were embedded
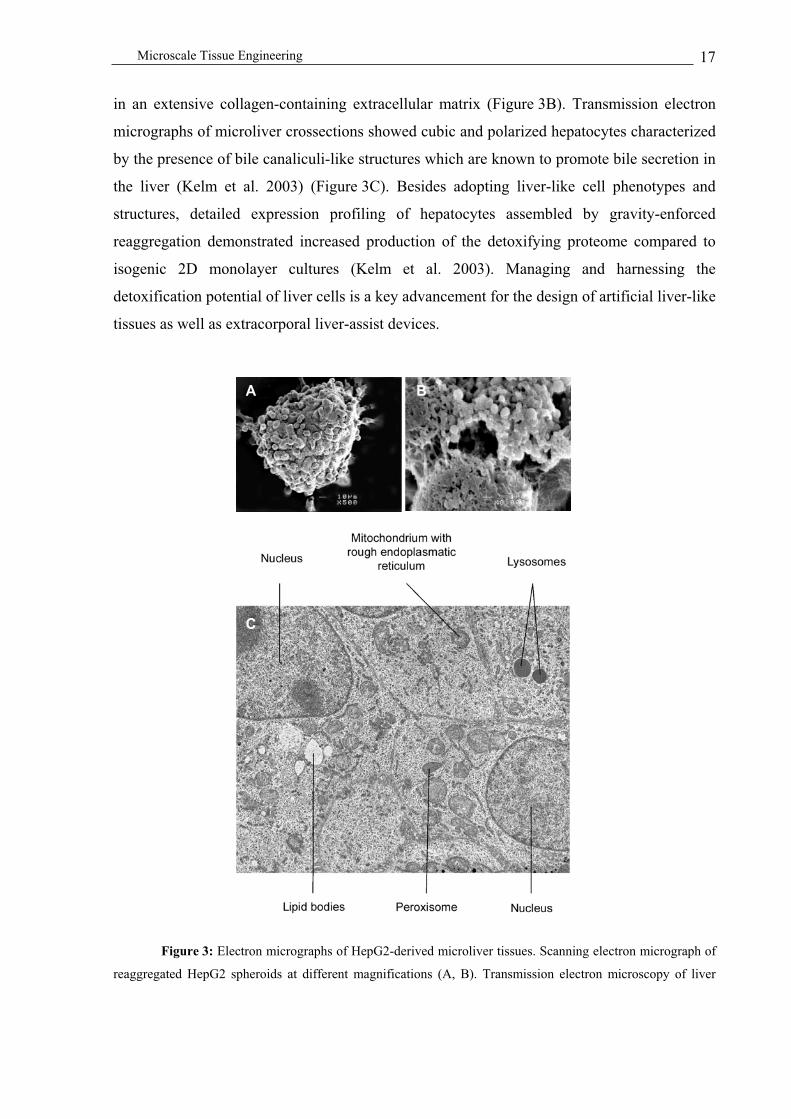
Microscale Tissue Engineering 17
in an extensive collagen-containing extracellular matrix (Figure 3B). Transmission electron
micrographs of microliver crossections showed cubic and polarized hepatocytes characterized
by the presence of bile canaliculi-like structures which are known to promote bile secretion in
the liver (Kelm et al. 2003) (Figure 3C). Besides adopting liver-like cell phenotypes and
structures, detailed expression profiling of hepatocytes assembled by gravity-enforced
reaggregation demonstrated increased production of the detoxifying proteome compared to
isogenic 2D monolayer cultures (Kelm et al. 2003). Managing and harnessing the
detoxification potential of liver cells is a key advancement for the design of artificial liver-like
tissues as well as extracorporal liver-assist devices.
Figure 3: Electron micrographs of HepG2-derived microliver tissues. Scanning electron micrograph of
reaggregated HepG2 spheroids at different magnifications (A, B). Transmission electron microscopy of liver
C
A B
C
A BA B

Microscale Tissue Engineering 18
spheroids revealed intact cubic cells showing hepatocyte-characteristic polarity exemplified by bile canaliculi-
like structures (C).
Artificial myocardial microtissue
Heart diseases including myocardial infarction and heart failures are the most
prevalent pathologies in industrialized countries. Loss of cardiomyocytes accounts for
decreased myocardial function, which may result in total organ failure or trigger
compensatory mechanisms like hypertrophy of the remaining myocardium, activation of
neurohumoral systems and/or autokrine/parakrine stimulation by various growth
factors/cytokines (Zimmermann et al. 2003). The ultimate treatment of end-stage heart failure
remains heart transplantation. However, since available donor organs do not match the
increasing number of patients with heart failures, alternative strategies for restoration of heart
function are a current clinical priority (Miniati et al. 2002). Implantation of functional
cardiomyocytes and other cell types including stem cells has been shown to improve
contractile function in myocardial infarction models. Yet, ongoing clinical trials will have to
confirm the therapeutic impact of these strategies (Barbash et al. 2003; Leobon et al. 2003;
Mangi et al. 2003). An alternative to infusion of single-cell suspensions is the design of
artificial cardiac muscle tissues ex vivo followed by implantation into the diseased heart.
The prevailing 3D cultivation technology in cardiac tissue engineering will have to
unite several key characteristics including (i) long-term maintenance of the contractive
capacity, (ii) multi-cell type cultivation, (iii) potential for self-organization, polarization and
microstructure formation between different cell types, (iv) production of an extracellular
matrix, (v) vascularization, including induction of vascular vessel development and
connection to the host capillary system following implantation, (vi) development of seamless
inter-tissue superstructures and (vii) compatibility with high-efficiency stable gene transfer
technologies to enable cell phenotype-modulating and/or therapeutic interventions. Several
strategies have been developed to produce engineered cardiac tissues including rigid/soluble
matrix-based approaches (Akins et al. 1999; Polonchuk et al. 2000; Teebken et al. 2002)
(particularly successful for shaping heart valves and vessels) and scaffold-free initiatives
(Tables 2 and 3).
We have recently used gravity-enforced reaggregation of pure primary rat and mouse
cardiomyocytes as well as mixed cell populations reflecting the cell type composition of
rodent hearts to design beating heart microstructures ((Kelm et al. in press);

Microscale Tissue Engineering 19
http://www.biotech.biol.ethz.ch/martinf/staff/jens.html). Interestingly, cardiomyocytes
expressed a high degree of organotypic heart tissue phenotypes when arranged in such a
scaffold-free 3D environment. Phenotypic characterization combined with detailed analysis of
muscle-specific cell traits, extracellular matrix components as well as endogenous VEGF
(vascular endothelial growth factor) expression profiles of heart microtissues revealed (i) a
direct cell number - microtissue size correlation (up to 320 m), (ii) inter-microtissue
superstructures, (iii) retention of key cardiomyocyte-specific cell qualities, (iv) a sophisticated
extracellular matrix, (v) a high degree of self-organization exemplified by the tendency of
muscle structures to assemble at the periphery of these myocardial spheroids (Figure 2C) and
(vi) high lentiviral transduction rates for genetic engineering of microhearts. (vii)
Furthermore, myocardial spheroids supported endogenous VEGF expression in a size-
dependent manner, which will likely promote vascularization of heart microtissues produced
from defined cell mixtures, as well as enable connection to the host vascular system following
implantation. Retention of heart-like rod-shaped cardiomyocytes was particularly prominent
when cardiomyocytes were coated onto myofibroblast feeder spheroids. This observation
exemplifies the power of 3D feeder structures for induction and maintenance of specific cell
shapes and phenotypes (Figure 4) (Kelm et al. in press).
Which one of the different myocardial microtissue design concepts will prevail in
future therapies or whether (stem) cell transplantations will succeed remains to be seen in
current clinical trials.
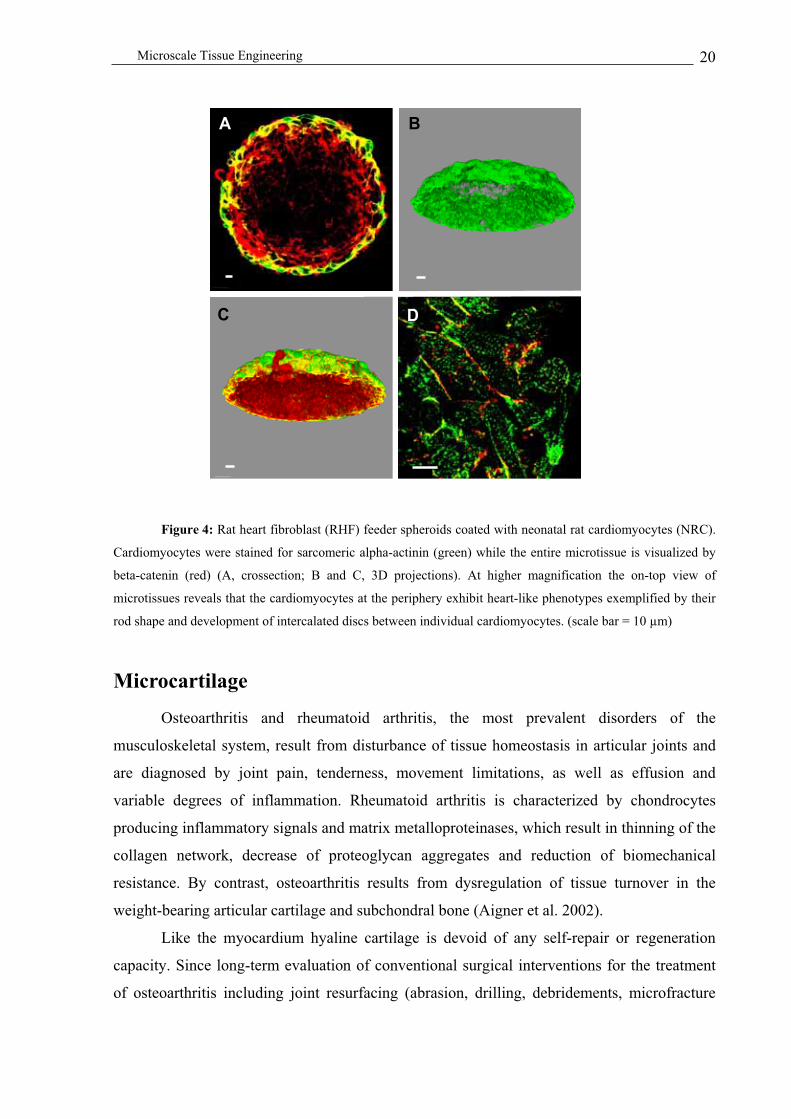
Microscale Tissue Engineering 20
Figure 4: Rat heart fibroblast (RHF) feeder spheroids coated with neonatal rat cardiomyocytes (NRC).
Cardiomyocytes were stained for sarcomeric alpha-actinin (green) while the entire microtissue is visualized by
beta-catenin (red) (A, crossection; B and C, 3D projections). At higher magnification the on-top view of
microtissues reveals that the cardiomyocytes at the periphery exhibit heart-like phenotypes exemplified by their
rod shape and development of intercalated discs between individual cardiomyocytes. (scale bar = 10 µm)
Microcartilage
Osteoarthritis and rheumatoid arthritis, the most prevalent disorders of the
musculoskeletal system, result from disturbance of tissue homeostasis in articular joints and
are diagnosed by joint pain, tenderness, movement limitations, as well as effusion and
variable degrees of inflammation. Rheumatoid arthritis is characterized by chondrocytes
producing inflammatory signals and matrix metalloproteinases, which result in thinning of the
collagen network, decrease of proteoglycan aggregates and reduction of biomechanical
resistance. By contrast, osteoarthritis results from dysregulation of tissue turnover in the
weight-bearing articular cartilage and subchondral bone (Aigner et al. 2002).
Like the myocardium hyaline cartilage is devoid of any self-repair or regeneration
capacity. Since long-term evaluation of conventional surgical interventions for the treatment
of osteoarthritis including joint resurfacing (abrasion, drilling, debridements, microfracture
A B
C D
AA BB
CC DD

Microscale Tissue Engineering 21
techniques or arthroscopic shaving) were of limited success (insufficient repair resulting in
the formation of inadequate resident fibrocartilage), cell-based cartilage regeneration came
into the limelight of current arthritis therapies. As one of the first clinically licensed cell-
based therapies, the Food and Drug Administration (FDA; http://www.fda.gov) approved the
biological production known as autologous chondrocyte implantation (ACI) in 1997 (FDA
Reference No: 96-0372). ACI was based on enzymatic disintegration of cartilage biopsies
followed by selective expansion of chondrocytes and reimplantation into the damaged
cartilage. Unfortunately, chondrocytes lost their differentiated phenotype during in vitro
expansion since the cells were cultured on an inappropriate substrate owing to lack the
requisite characteristic extracellular matrix environment. Although many exercising surgeons
emphasize the good to excellent clinical results (60%-90% of the patients report pain relief
and improved joint functionality), fate and redifferentiation of autologous chondrocyte
implants remain elusive (Hunziker 2002). ACI implants are typically fixed to the defect joint
surface using fibrin glue or resorbable pins but only 8% of the implanted chondrocytes could
be identified within the repaired tissue (Grande et al. 1989). Also, management of a solid
connection between cartilage and bone tissues remains a current clinical focus.
In contrast to ACI-based monolayer cultures, chondrocytes retain their differentiation
status in a 3D environment typically provided by scaffolds supporting initial mechanical
stability and even cell distribution (Risbud et al. 2002). To date, there are only few scaffold-
free approaches available for 3D cultivation/assembly of primary chondrocytes to artificial
cartilage (Tables 2 and 3). Fixation of artificial microcartilage onto native cartilage resulted in
rapid formation of focal adhesion points and seamless integration of the microtissues into the
target cartilage. 3 weeks post fixation, microcartilage-derived cells were located on the native
cartilage surface and showed de novo synthesis of extracellular matrix (Anderer et al. 2002).
Immuno-based confocal analysis of microcartilage (produced by gravity-enforced assembly
of primary human and pig chondrocytes) in hanging drops revealed two distinct cell layers, an
outer one qualified by fibroblast-like cell morphologies and an inner core consisting of a loose
assembly of tubular chondrocytes embedded into an extracellular matrix (Figure 5 C, D; Kelm
et al., unpublished).
Successful therapies for repair and regeneration of cartilage will require assembly of
chondrocytes in a 3D microcartilage configuration compatible with surgical implantation and
fixation, genetic engineering, optimal differentiation and production of an extracellular
matrix. Although gravity-enforced production of scaffold-free microcartilage is expected to
meet with these criteria at a high standard clinical confirmation remains imminent.

Microscale Tissue Engineering 22
Figure 5: Microcartilage produced by gravity-enforced reaggregation of pig and human chrondrocytes
in hanging drops. Phase-contrast micrographs of microcartilage produced from 1200 pig (A) and human (B)
articular chondrocytes. F-Actin-specific staining of pig (C) and human (D) chondrocyte derived microtissues
illustrates the morphological structure of the microcartilage. (scale bar = 20 µm)
Beyond tissue engineering – the future of microtissues in
biopharmaceutical manufacturing and high throughput drug
discovery
Monodispersed cells growing in suspension and protein-free media are currently the
golden standard for large-scale manufacturing of protein therapeutics (Chu et al. 2001).
Although key biopharmaceutical manufacturing parameters such as growth rate, cell density
and specific productivity can be optimized by advanced bioprocess control or specific
molecular interventions in production cell lines, the question remains, whether specific
productivity of suspension cells typically reached in classical bioreactor operation compare
favourably with nature’s well-evolved protein production capacity.
Recent evidence suggested that cells cultivated in microtissues, embedded in a tissue-
like 3D environment or engineered for proliferation-arrested terminal differentiation reach
higher specific productivities compared to proliferation-competent monodispersed suspension
cultures: (i) reprogramming of CHO-K1-, DG44- and NSO-derived cell lines for
A B
DC
AA BB
DDCC

Microscale Tissue Engineering 23
proliferation-controlled terminal differentiation by conditional overexpression of the cyclin-
dependent kinase inhibitors p21Cip1
and p27Kip1
and/or the CAAT/enhancer-binding protein
alpha (C/EBP ) significantly increased specific productivity of (model) product proteins by
up to one order of magnitude (Fussenegger et al. 1998; Meents et al. 2002; Ibarra et al. 2003).
(ii) Encapsulation of mammalian production cell lines in biocompatible hydrogels including
alginate or sodium cellulose sulphate - poly[diallyldimethylammonium chloride]
(PDADMAC) copolymers, results in high-density microspheres showing increased
production compared to isogenic 2D cultures (Weber et al., unpublished). (iii)
Cardiomyocyte-derived microtissues transduced with a secreted reporter gene-encoding
lentivirus produced 6-fold more heterologous protein than control monolayers consisting of
identical cell numbers (Kelm et al. in press). While controlled proliferation technology is
ready for industrial application, microtissue-based biopharmaceutical manufacturing is
currently in an up-scale process. Yet, hardware for industrial manufacturing of
microencapsulated production cell lines is available (Gugerli et al. 2002; Koch et al. 2003)
and gravity-enforced design of microtissues is compatible with standard liquid-handling
robotics.
In addition to provide tissue-mimicking clinical implants or high-performance
aggregates for biopharmaceutical manufacturing, microtissues may reveal a systems’ view on
tissue formation, drug testing and drug discovery. Reaggregated microtissues of tumor cells
have long played a pivotal role in cancer research since multicellular tumor spheroids show
increased proliferative activity and drug resistance similar to solid tumors (Kelm et al. 2003).
Microtumors enable precise analysis of growth constraints (e.g. oxygen and nutrient
consumptions), sensitivities to drugs or radiation, infiltration into non-cancerous tissues as
well as the angiogenic potential. Microtissues are becoming increasingly popular as models
for neurodegenerative disorders toxicology, pharmacology, nutrition and environmental
monitoring (Bhadriraju et al. 2002; Layer et al. 2002).
Drug discovery and diagnostics typically include screening of chemical or metabolic
libraries for therapeutic compounds (antigens, antibodies, nucleotides and peptides) in a
microscale format. Replacing 2D cultures used in classical drug discovery by microtissues
may (i) increase the precision of therapeutic readout, (ii) enable early drug validation at the
tissue level in a cost-efficient animal-free setting and (iii) expand the discovery window into a
yet unknown dimension. Figure 6 exemplifies a prototype high-throughput-compatible ELISA
format which combines target molecule quantification (e.g., vascular endothelial growth
factor; VEGF) with microtissue design (e.g., human aortic fibroblasts, HAF). Multiwell plates

Microscale Tissue Engineering 24
were coated with a VEGF-specific capture antibody and individual wells incubated with
different HAF cell concentrations (500, 2500, 5000, 10000 cells per well). For seven days,
HAF-derived microtissues of different sizes were produced by gravity-enforced reaggreation
in hanging drops. Hypoxia-induced VEGF production correlated directly with the size/cell
number of HAF-derived microtissues and could be quantified using an ELISA-type protocol.
Microtissues are thus compatible with high-throughput profiling of desired proteins.
Figure 6: Microtissue-based high-throughput ELISA-type assay for quantification of desired proteins.
(i) Individual wells of a multiwell plate were coated with a capture antibody specific for the vascular endothelial
growth factor (VEGF) capture antibody. (ii) Monodispersed cell suspensions of human aortic fibroblasts (HAF)
were seeded at various concentrations into each well and the plates were cultivated upside down to enable
formation of hanging drops and gravity-enforced microtissue production. (iii) Microtissues were discarded and
medium removed to quantify microtissue-based VEGF production using an ELISA-type protocol and a
chromogenic readout. (iv) Owing to the direct microtissue size – cell number correlation hypoxia-induced VEGF
production increases with the microtissue diameter.
Coating with the
Capture Antibody
Cultivation of
Microtissues in
Hanging Drops
Developing the
ELISA
500
2500
5000
10000
4000
pg/ml1000
pg/ml
400
pg/ml
VEGF Production
of Different Sized
HAF Microtissues
VEGF Standards
Coating with the
Capture Antibody
Cultivation of
Microtissues in
Hanging Drops
Developing the
ELISA
500
2500
5000
10000
4000
pg/ml1000
pg/ml
400
pg/ml
500
2500
5000
10000
4000
pg/ml1000
pg/ml
400
pg/ml
VEGF Production
of Different Sized
HAF Microtissues
VEGF Standards

Microscale Tissue Engineering 25
Perspectives
“We are a self-assembling organism. That information is there to be captured and
used” says William Haseltine, chairman and chief executive officer (CEO) of Human
Genome Sciences (Rockville, MD) who was among the first to coin the term “regenerative
medicine” to describe new ways of teaching the body to heal itself (Wade 2001). Microtissue
design consisting of reagreggation of biopsy-derived (stem) cell populations or (stem) cell
lines harnesses the organism’s self-assembling programs to provide regenerative medicine
initiatives with clinical tissue implants and new insight into regulatory networks underlying
complex disease phenotypes. “Further in the future”, Haseltine says, “biologists may learn
how to fashion new organs outside the body” (Wade 2000). In that sense, the future has just
begun with the design of artificial microtissues.
Acknowledgements
We are grateful Lars K. Nielsen for providing Figure 3 micrographs. We thank David
Fluri, Beat P. Kramer and Shizuka Hartenbach for critical comments on the manuscript. Work
in the laboratory of M.F. is supported by the Swiss National Science Foundation (grant no.
631-065946).
References
Abbott, A. (2003). "Cell culture: biology's new dimension." Nature 424, 870-872.
Aigner, T. and L. McKenna (2002). "Molecular pathology and pathobiology of osteoarthritic
cartilage." Cell Mol Life Sci 59, 5-18.
Akins, R. E., R. A. Boyce, M. L. Madonna, N. A. Schroedl, S. R. Gonda, T. A. McLaughlin
and C. R. Hartzell (1999). "Cardiac organogenesis in vitro: reestablishment of three-
dimensional tissue architecture by dissociated neonatal rat ventricular cells." Tissue
Eng 5, 103-118.
Ambrosino, G., S. Varotto, S. M. Basso, A. Cecchetto, P. Carraro, A. Naso, G. De Silvestro,
M. Plebani, G. Abatangelo, D. Donato, A. Cestrone, G. Giron and D. F. D'Amico
(2003). "Hepatocyte transplantation in the treatment of acute liver failure:
microencapsulated hepatocytes versus hepatocytes attached to an autologous
biomatrix." Cell Transplant 12, 43-49.

Microscale Tissue Engineering 26
Anderer, U. and J. Libera (2002). "In vitro engineering of human autogenous cartilage." J
Bone Miner Res 17, 1420-1429.
Barbash, I. M., P. Chouraqui, J. Baron, M. S. Feinberg, S. Etzion, A. Tessone, L. Miller, E.
Guetta, D. Zipori, L. H. Kedes, R. A. Kloner and J. Leor (2003). "Systemic delivery of
bone marrow-derived mesenchymal stem cells to the infarcted myocardium:
feasibility, cell migration, and body distribution." Circulation 108, 863-868.
Berthiaume, F., P. V. Moghe, M. Toner and M. L. Yarmush (1996). "Effect of extracellular
matrix topology on cell structure, function, and physiological responsiveness:
hepatocytes cultured in a sandwich configuration." Faseb J 10, 1471-1484.
Bhadriraju, K. and C. S. Chen (2002). "Engineering cellular microenvironments to improve
cell-based drug testing." Drug Discov Today 7, 612-620.
Bhatia, S. N., U. J. Balis, M. L. Yarmush and M. Toner (1999). "Effect of cell-cell
interactions in preservation of cellular phenotype: cocultivation of hepatocytes and
nonparenchymal cells." Faseb J 13, 1883-1900.
Bjerkvig, R., M. Lund-Johansen and K. Edvardsen (1997). "Tumor cell invasion and
angiogenesis in the central nervous system." Curr Opin Oncol 9, 223-229.
Bouche, A. (2002). "Xenotransplantation success for Immerge." Nat Biotechnol 20, 109.
Brazelton, T. R., F. M. Rossi, G. I. Keshet and H. M. Blau (2000). "From marrow to brain:
expression of neuronal phenotypes in adult mice." Science 290, 1775-1779.
Chu, L. and D. K. Robinson (2001). "Industrial choices for protein production by large-scale
cell culture." Curr Opin Biotechnol 12, 180-187.
Clarke, D. L., C. B. Johansson, J. Wilbertz, B. Veress, E. Nilsson, H. Karlstrom, U. Lendahl
and J. Frisen (2000). "Generalized potential of adult neural stem cells." Science 288,
1660-1663.
Furukawa, K. S., T. Ushida, Y. Sakai, M. Suzuki, J. Tanaka and T. Tateishi (2001).
"Formation of human fibroblast aggregates (spheroids) by rotational culture." Cell
Transplant 10, 441-445.
Fussenegger, M., S. Schlatter, D. Datwyler, X. Mazur and J. E. Bailey (1998). "Controlled
proliferation by multigene metabolic engineering enhances the productivity of Chinese
hamster ovary cells." Nat Biotechnol 16, 468-472.
Fux, C. F., M. (in press). "Dual-Regulated Expression of C/EBP-a and BMP-2 Enables
Differential Differentiation of C2C12 Cells into Adipocytes and Osteoblasts." Nucleic
Acids Res.

Microscale Tissue Engineering 27
Grande, D. A., M. I. Pitman, L. Peterson, D. Menche and M. Klein (1989). "The repair of
experimentally produced defects in rabbit articular cartilage by autologous
chondrocyte transplantation." J Orthop Res 7, 208-218.
Gugerli, R., E. Cantana, C. Heinzen, U. von Stockar and I. W. Marison (2002). "Quantitative
study of the production and properties of alginate/poly-L-lysine microcapsules." J
Microencapsul 19, 571-590.
Harimoto, M., M. Yamato, M. Hirose, C. Takahashi, Y. Isoi, A. Kikuchi and T. Okano
(2002). "Novel approach for achieving double-layered cell sheets co-culture:
overlaying endothelial cell sheets onto monolayer hepatocytes utilizing temperature-
responsive culture dishes." J Biomed Mater Res 62, 464-470.
Hench, L. L. and J. M. Polak (2002). "Third-generation biomedical materials." Science 295,
1014-1017.
Hunziker, E. B. (2002). "Articular cartilage repair: basic science and clinical progress. A
review of the current status and prospects." Osteoarthritis Cartilage 10, 432-463.
Ibarra, N., S. Watanabe, J. X. Bi, J. Shuttleworth and M. Al-Rubeai (2003). "Modulation of
Cell Cycle for Enhancement of Antibody Productivity in Perfusion Culture of NS0
Cells." Biotechnol Prog 19, 224-228.
Itskovitz-Eldor, J., M. Schuldiner, D. Karsenti, A. Eden, O. Yanuka, M. Amit, H. Soreq and
N. Benvenisty (2000). "Differentiation of human embryonic stem cells into embryoid
bodies compromising the three embryonic germ layers." Mol Med 6, 88-95.
Kale, S., S. Biermann, C. Edwards, C. Tarnowski, M. Morris and M. W. Long (2000).
"Three-dimensional cellular development is essential for ex vivo formation of human
bone." Nat Biotechnol 18, 954-958.
Kamihira, M., K. Yamada, R. Hamamoto and S. Iijima (1997). "Spheroid formation of
hepatocytes using synthetic polymer." Ann N Y Acad Sci 831, 398-407.
Kelm, J. M., E. Ehler, L. K. Nielsen, S. Schlatter, J. C. Perriard and M. Fussenegger (2004).
"Design of Artificial Myocardial Microtissues." Tissue Eng. 10, 201-214.
Kelm, J. M., B. P. Kramer, V. Gonzalez-Nicolini, B. Ley and M. Fussenegger (2004).
"synergies of microtissue design, viral transduction and adjustable transgene
expression for regenerative medicine." Biotechnol Appl Biochem. 39, 3-16.
Kelm, J. M., N. E. Timmins, C. J. Brown, M. Fussenegger and L. K. Nielsen (2003). "Method
for generation of homogeneous multicellular tumor spheroids applicable to a wide
variety of cell types." Biotechnol Bioeng 83, 173-180.

Microscale Tissue Engineering 28
Kim, S. S., H. Utsunomiya, J. A. Koski, B. M. Wu, M. J. Cima, J. Sohn, K. Mukai, L. G.
Griffith and J. P. Vacanti (1998). "Survival and function of hepatocytes on a novel
three-dimensional synthetic biodegradable polymer scaffold with an intrinsic network
of channels." Ann Surg 228, 8-13.
Koch, S., C. Schwinger, J. Kressler, C. Heinzen and N. G. Rainov (2003). "Alginate
encapsulation of genetically engineered mammalian cells: comparison of production
devices, methods and microcapsule characteristics." J Microencapsul 20, 303-16.
Kunz-Schughart, L. A. (1999). "Multicellular tumor spheroids: intermediates between
monolayer culture and in vivo tumor." Cell Biol Int 23, 157-161.
Layer, P. G., A. Robitzki, A. Rothermel and E. Willbold (2002). "Of layers and spheres: the
reaggregate approach in tissue engineering." Trends Neurosci 25, 131-134.
Layer, P. G., T. Weikert and E. Willbold (1992). "Chicken retinospheroids as developmental
and pharmacological in vitro models: acetylcholinesterase is regulated by its own and
by butyrylcholinesterase activity." Cell Tissue Res 268, 409-418.
Lee, R. J., M. L. Springer, W. E. Blanco-Bose, R. Shaw, P. C. Ursell and H. M. Blau (2000).
"VEGF gene delivery to myocardium: deleterious effects of unregulated expression."
Circulation 102, 898-901.
Leobon, B., I. Garcin, P. Menasche, J. T. Vilquin, E. Audinat and S. Charpak (2003).
"Myoblasts transplanted into rat infarcted myocardium are functionally isolated from
their host." Proc Natl Acad Sci U S A 100, 7808-7811.
Lutolf, M. P., F. E. Weber, H. G. Schmoekel, J. C. Schense, T. Kohler, R. Muller and J. A.
Hubbell (2003). "Repair of bone defects using synthetic mimetics of collagenous
extracellular matrices." Nat Biotechnol 21, 513-518.
Mangi, A. A., N. Noiseux, D. Kong, H. He, M. Rezvani, J. S. Ingwall and V. J. Dzau (2003).
"Mesenchymal stem cells modified with Akt prevent remodeling and restore
performance of infarcted hearts." Nat Med 9, 1195-1201.
Meents, H., B. Enenkel, R. G. Werner and M. Fussenegger (2002). "p27Kip1-mediated
controlled proliferation technology increases constitutive sICAM production in CHO-
DUKX adapted for growth in suspension and serum-free media." Biotechnol Bioeng
79, 619-627.
Miniati, D. N. and R. C. Robbins (2002). "Heart transplantation: a thirty-year perspective."
Annu Rev Med 53, 189-205.

Microscale Tissue Engineering 29
Muraglia, A., A. Corsi, M. Riminucci, M. Mastrogiacomo, R. Cancedda, P. Bianco and R.
Quarto (2003). "Formation of a chondro-osseous rudiment in micromass cultures of
human bone-marrow stromal cells." J Cell Sci 116, 2949-2955.
Ochoa, E. R. and J. P. Vacanti (2002). "An overview of the pathology and approaches to
tissue engineering." Ann N Y Acad Sci 979, 10-26; discussion 35-38.
Petit-Zeman, S. (2001). "Regenerative medicine." Nat Biotechnol 19, 201-206.
Polonchuk, L., J. Elbel, L. Eckert, J. Blum, E. Wintermantel and H. M. Eppenberger (2000).
"Titanium dioxide ceramics control the differentiated phenotype of cardiac muscle
cells in culture." Biomaterials 21, 539-550.
Rafii, S. and D. Lyden (2003). "Therapeutic stem and progenitor cell transplantation for organ
vascularization and regeneration." Nat Med 9, 702-712.
Richardson, T. P., M. C. Peters, A. B. Ennett and D. J. Mooney (2001). "Polymeric system for
dual growth factor delivery." Nat Biotechnol 19, 1029-1034.
Risbud, M. V. and M. Sittinger (2002). "Tissue engineering: advances in in vitro cartilage
generation." Trends Biotechnol 20, 351-356.
Rothermel, A. and P. G. Layer (2001). "Photoreceptor plasticity in reaggregates of embryonic
chick retina: rods depend on proximal cones and on tissue organization." Eur J
Neurosci 13, 949-958.
Strain, A. J. and J. M. Neuberger (2002). "A bioartificial liver--state of the art." Science 295,
1005-1009.
Teebken, O., H. Mertsching and A. Haverich (2002). "Modification of heart valve allografts
and xenografts by means of tissue engineering." Transplant Proc 34, 2333.
Wade, N. (2000). "Teaching the body to heal itself." The New York Times 11/7.
Wade, N. (2001). "Apostle of regenrative meidicine foresees healt of life." The New York
Times 12/18.
Watzka, S. B., J. Lucien, M. Shimada, V. Edwards, H. Yeger, G. Hannigan and J. G. Coles
(2000). "Selection of viable cardiomyocytes for cell transplantation using three-
dimensional tissue culture." Transplantation 70, 1310-1317.
Wobus, A. M., E. Wolf and H. M. Beier (2000). "Embryonic stem cells and nuclear transfer
strategies. Present state and future prospects." Cells Tissues Organs 166, 1-5.
Zandonella, C. (2003). "Tissue engineering: The beat goes on." Nature 421, 884-886.
Zimmermann, W. H. and T. Eschenhagen (2003). "Cardiac tissue engineering for replacement
therapy." Heart Fail Rev 8, 259-269.

Chapter 3
Design of Artificial Myocardial Microtissues
Kelm J.M., Ehler E., Nielsen L.K., Schlatter S., Perriard J.C. and Fussenegger M., (2004)
Tissue Engineering 10, 201-207

Myocardial Microtissue 31
Abstract
Cultivation technologies promoting organization of mammalian cells in three
dimensions are essential for gene-function analyses as well as drug testing and represent the
first step towards the design of tissue replacements and bioartificial organs. Embedded in a
three-dimensional environment, cells are expected to develop tissue-like higher order
intercellular structures (cell-cell contacts, extracellular matrix) which orchestrate cellular
functions including proliferation, differentiation, apoptosis and angiogenesis with unmatched
quality. We have refined the hanging drop cultivation technology to pioneer beating heart
microtissues derived from pure primary rat and mouse cardiomyocyte cultures as well as
mixed populations reflecting the cell type composition of rodent hearts. Phenotypic
characterization combined with detailed analysis of muscle-specific cell traits, extracellular
matrix components as well as endogenous VEGF (vascular endothelial growth factor)
expression profiles of heart microtissues revealed (i) a linear cell number - microtissue size
correlation, (ii) inter-microtissue superstructures, (iii) retention of key cardiomyocyte-specific
cell qualities, (iv) a sophisticated extracellular matrix, and (v) a high degree of self-
organization exemplified by the tendency of muscle structures to assemble at the periphery of
these myocardial spheroids. (vi) Furthermore, myocardial spheroids support endogenous
VEGF expression in a size-dependent manner which will likely promote vascularization of
heart microtissues produced from defined cell mixtures as well as support connection to the
host vascular system following implantation. As cardiomyocytes are known to be refractory to
current transfection technologies we have designed lentivirus-based transduction strategies to
lead the way for genetic engineering of myocardial microtissues in a clinical setting.

Myocardial Microtissue 32
Introduction
Cardiac-related insufficiencies are the major cause of morbidity and associated
mortality in industrialized countries affecting over sixty million patients per year in the USA
alone (Claycomb 1991; Gheorghiade et al. 1998; Hennekens 1998). Despite global initiatives
to foster advances in cardiomyocyte-related tissue engineering this cell type turned out to be
largely refractory to state-of-the-art gene transfer and expansion technologies, particularly in
its terminally differentiated adult phenotype (Datwyler et al. 2001). Yet, cardiac tissue
engineering remains a high priority as transplantations and artificial hearts only cover about
10% of current clinical needs and/or fail to offer any long-term therapeutic perspectives. Even
engineered heart cell suspensions and undersized cell aggregates may be sufficient for the
treatment of smaller heart lesions (Akins 2002).
Although studies on primary cardiomyocyte monolayer cultures produced from
enzymatically dispersed fetal, neonatal, or adult vertebrate hearts have substantiated
intracellular structure-function correlations (Rothen-Rutishauser et al. 1998) and revealed
molecular pathways shaping this complex cell phenotype, two-dimensional cultivation of
cardiomyocytes is likely limited in providing clinically relevant information required for
sophisticated tissue engineering. In contrast to monolayer cultures, three-dimensional
cultivation technologies mimic cardiac tissue-like morphologies and provide a suitable
environment for coordination of cell-cell interaction, self-organization, differentiation and
electrical properties, all of which are essential qualities for the identity and integrity of heart
structures (Eschenhagen et al. 1997; Ross et al. 2001). Also, only three-dimensional
cultivation enables development of an extracellular matrix (ECM) which plays a pivotal role
in differentiation, proliferation and apoptosis regulatory networks (Chen et al. 1997; Aplin et
al. 1999; Giancotti et al. 1999).
The majority of cardiomyocyte-adapted three-dimensional cultivation technologies are
based on artificial scaffolds consisting of (i) titanium dioxide ceramics (Polonchuk et al.
2000), (ii) cell/collagen mixtures (Eschenhagen et al. 1997), (iii) polystyrene microcarrier
beads (Akins et al. 1999), (iv) alginate polymers (Magyar et al. 2001; Dar et al. 2002), and (v)
polyglycolic acid structures maintained in microgravity bioreactors (Freed et al. 1997; Carrier
et al. 1999). Although scaffolds enable artificial organs to grow in a desired shape they may
cause post-transplantation side effects resulting from toxic degradation products, induction of
inflammatory reactions and poor resorption (Yang et al. 2001). Alternative strategies for

Myocardial Microtissue 33
production of scaffold-free artificial tissues are taking advantage of the aggregation of
mammalian cells following (i) cultivation in shake flasks or on non-adhesive surfaces
(Sperelakis 1978), (ii) centrifugation-based compression (Armstrong et al. 2000), (iii)
maintenance in cell culture inserts (Watzka et al. 2000), or (iv) gravity-enforced assembly in
hanging drops (Keller 1995). To date, most attempts to shape artificial cardiac-like cell
aggregates have been based on physical assembly of isolated primary cardiomyocytes since
the emergence of cardiomyocytes in spheroid cultures of human embryonic stem cells
remains stochastic and strategies for rational differentiation of this pluripotent cell type into
cardiac cells are still in its infancy (Schuldiner et al. 2000; Boheler et al. 2002).
A major challenge in the design of (heart) microtissues is timely and well-balanced
induction of vascularization to ensure long-term supply of large artificial tissues with
oxygen/nutrients as well as their connection to the host vascular system following
implantation (Griffith et al. 2002). As a typical cell is connected to the capillary network
within an average perimeter of 60 m in vivo, oversized microtissues may show hypoxia-
/nutrient deprivation-induced cell death in their centers (Intaglietta et al. 1996). Although
localized delivery of heterologous VEGF to cardiac lesions has recently been shown to induce
vascularization and connection of transplanted myoblasts to the coronary artery, overdosing
of this growth factor was associated with the formation of hemangiomas and cell death
(Richardson et al. 2001).
In this study we have refined the hanging drop cultivation technology to design
functional heart microtissues from purified primary neonatal rat (NRC) and mouse (NMC)
cardiomyocytes and from cardiomyocyte-containing cell mixtures reflecting the natural cell
composition of neonatal rodent hearts. These myocardial microtissues could be adjusted in
size and were beating for over 3 weeks. In contrast to monolayer cultures and small-sized
spheroids, myocardial microtissues of 230 11 m in diameter secrete VEGF, which is
expected to facilitate vascularization during production of artificial tissues or following
implantation. In order to enable rational molecular interventions in myocardial microtissues to
refine cell phenotypes or to engineer production of desired protein therapeutics, we have
evaluated lentiviral transduction systems for straightforward transgene delivery into
cardiomyocytes cultivated in two or three dimensions.
We are convinced that the design as well as detailed characterization of myocardial
microtissues bundled with a powerful transduction technology will foster novel opportunities
in cardiac tissue engineering and the treatment of heart diseases.

Myocardial Microtissue 34
Materials and Methods
Isolation and three-dimensional cultivation of neonatal rat and mouse
cardiomyocytes
Newborn rat (Wistar) and mouse (NMRI) hearts were dissected, digested with
collagenase (Worthington Biochemical Corp., Freehold, NJ) and pancreatin (Invitrogen,
Carlsbad, CA) and prepared as described by Auerbach et al. (1999) (Auerbach et al. 1999).
Rat cardiomyocytes were either cultivated as mixed populations reflecting the cell type
composition of rodent hearts (25% cardiomyocytes) or cultured as homogenous populations
following density gradient purification (over 95% cardiomyocytes). The mouse populations
contained up to 50% cardiomyocytes following preplating of isolated cells for 2 h in a tissue
culture dish.
Following isolation, cardiomyocyte-containing cell populations were seeded at
indicated concentrations into 60-well plates (HLA plate, Nunc Inc., Roskilde, Denmark). To
enable gravity-enforced microtissue formation in hanging drops, the 60-well plates were
incubated upside down at 37°C in a humidified atmosphere containing 5% CO2. Pure and
mixed cardiomyocyte cultures were maintained in plating medium (67% Dulbecco`s Modified
Eagle Medium (DMEM; Invitrogen), 17% M199 (Amimed AG, Basel, Switzerland), 10%
horse serum (cat. no. 16050-098, lot no. 3036354D, Invitrogen), 5% fetal bovine serum (FBS;
cat. no. A-15-022, lot no. A01129-242; PAA Laboratories, Linz, Austria), and 1%
penicillin/streptomycin solution (Invitrogen)). After cultivation for 3-4 days in hanging drops
the microtissues were harvested and kept for further analysis in non-adhesive culture dishes
containing maintenance medium (78% DMEM (Invitrogen), 20% M199 (Amimed AG), 1%
horse serum (Invitrogen), 1% penicillin/streptomycin solution (Invitrogen), and 10-4
mM
phenylephrine (Sigma Chemicals, St. Louis, MO). Cardiomyocyte monolayers were
cultivated in fibronectin-coated (10 g/ml human plasma-derived fibronectin (cat. no.
688851, lot no. 14814500, Roche Biochemicals, Basel, Switzerland)) culture dishes using
plating medium. After one day the medium was replaced by maintenance medium.
Cardiomyocyte-coated microtissues cultures were generated by producing a three-
dimensional feeder spheroid using 1’200 myocardial fibroblasts/drop in plating medium.
After 2 days 900 gradient-purified neonatal rat cardiomyocytes were added per feeder
spheroid. After an additional 4-day cultivation in hanging drops the cardiomyocyte-coated
spheroids were harvested and prepared for immunohistochemistry.

Myocardial Microtissue 35
Video microscopy
Phase contrast images of beating myocardial microtissues were recorded using an
inverted microscope (Zeiss, Oberkochen, Germany) equipped with a CCD camera (Kappa
Opto-electronics GmbH, Gleichen, Germany) connected to a video recorder (Panasonic Inc.,
Hamburg, Germany).
Fluorescence-based characterization of cell morphologies
Myocardial microtissues were harvested following 7 days of cultivation in
maintenance medium. After a washing step in 2x phosphate-buffered saline (PBS; 150 mM
NaCl, 6.5 mM Na2HPO4 x 2 H2O, 2.7 mM KCL, 1.5 mM KH2PO4, pH 7.4; Sigma), the
spheroids were fixed for 1 hour in PBS containing 4% paraformaldehyde and subsequently
washed three times for 5 min in phosphate-buffered Triton X-100 (PBT, PBS containing
0.002% Triton X-100; Sigma). The spheroids were then permeated for 30 min in PBS
containing 0.5% Triton X-100. Primary antibodies specific for the indicated proteins as well
as fluorescence-labeled secondary antibodies were diluted in Tris-buffered saline (TBS,
20 mM Tris base, 155 mM NaCl, 2 mM EGTA, 2 mM MgCl2) containing 1% BSA and
incubated for 12 hours (primary antibody) at 4 C and 5 hours (secondary antibody) at room
temperature, respectively. Finally, the myocardial microtissues were washed in PBS and
embedded on glass slides using tris-buffered glycerol (a 3:7 mixture of 0.1 M Tris-HCl
(pH 9.5) and glycerol supplemented with 50 mg/ml n-propyl-gallat). In order to prevent
crunching of the myocardial microtissues between the slide and the cover slip 0.5 mm,
custom-made silicon spacers were used.
For immunofluorescence-based expression profiling the myocardial microtissues were
labeled with monoclonal antibodies specific for (i) sarcomeric- -actinin (Sigma; clone
EA53), (ii) polyclonal titin m8 (kindly provided by Mathias Gautel, King’s College London),
(iii) polyclonal myomesin (Agarkova et al. 2000), (iv) polyclonal -catenin (Sigma; cat no.
C2206), (v) collagen type I (Sigma; clone COL-1), (vi) monoclonal anti-collagen type IV
(Sigma; clone COL-94), (vii) polyclonal laminin (Sigma; cat no. L9393), (viii) polyclonal
fibronectin (Sigma; cat. no. F3648) or (ix) VEGF isoforms 110, 121 and 165 (Santa Cruz
Biotechnology Inc., Santa Cruz, CA; sc-152) and stained with Cy3-coupled secondary anti-
mouse (Jackson Immunochemicals, West Grove, PA; cat. no. 115-165-146) or FITC-coupled
anti-rabbit (ICN Pharmaceuticals, Hyland, CA) antibodies. F-actin was visualized using
A633-coupled phalloidin (Molecular Probes Inc., Eugene, OR). Nuclear staining of

Myocardial Microtissue 36
cardiomyocytes was performed by using the fluorescent dye Draq5 (Biostatus Ltd., Shepshed,
Leicestershire, Great Britain): (i) NRC-derived microtissues were harvested by centrifugation
for 2 min at 50 x g and incubated for 10 min. in 100 �l PBS supplemented with 2 �l of a 5
mM Draq5 stock solution. Prior to immunostaining the microtissues were washed three times
with 1 ml PBS.
Phase contrast as well as fluorescence micrographs of cardiomyocyte monolayers
were recorded using an inverted microscope (HBO 50/AC, Zeiss, Oberkochen, Germany)
equipped with a digital camera (Axiocam HRm, Zeiss).
Confocal light microscopy
The imaging system consisted of an inverted fluorescence microscope (Leica
DMIRB/E, Heerbrugg, Switzerland) equipped with a Leica 20x/10x oil immersion objective,
a confocal scanner (Leica TCS SP1) featuring an argon-helium-neon laser and a Silicon
Graphics workstation (SGI, Schlieren, Switzerland) with Imaris 3D multi-channel image
processing software installed (Bitplane, AG, Zurich, Switzerland (Messerli et al. 1993)).
Lentivirus-based transduction technology
For production of replication-incompetent, self-inactivating lentiviruses a mixture
containing 94 l DMEM, 6 l Fugene 6 (Roche Diagnostics AG, Rotkreuz, Switzerland),
25 mM chloroquine, 1.5 g pLTR-G (encoding the pseudotyping envelope protein VSV-G of
the vesicular stomatitis virus (Reiser et al. 1996)), 1.5 g of the helper construct pCD/NL-
BH* (Mochizuki et al. 1998) and either 1.5 g of the YFP-encoding lentiviral expression
vector pMF351 (5’LTR-+-PhCMV-YFP-3’LTR U3) or the SAMY (secreted -amylase)-
encoding plasmid pMF364 (5’LTR-+-PEF1 -SAMY-3’LTR U3) was transfected into human
embryonic kidney cells (HEK293-T) (Mitta 2002; Schlatter et al. 2002). Following a medium
exchange after 24 h virus production was continued for another 48 h prior to filtration-based
harvesting of lentiviruses from the culture supernatant (0.45 m FP 030/2 filter; Schleicher &
Schuell GmbH, Dassel, Germany) which resulted in typical titers of 2 x 107 virus particles per
ml (Mitta 2002). In a standard transduction setting, 400’000 cardiomyocytes were infected in
a 6-well plate containing 2 ml medium and 200 l lentivirus suspension at a titer of
1x107 CFU/ml. Myocardial microtissues were directly transduced in hanging drops with 2 l
lentivirus suspension (1x107 CFU/ml).

Myocardial Microtissue 37
Quantitative expression analysis of the secreted -amylase (SAMY)
100 l of SAMY-containing cell culture supernatant were centrifuged at 14’000x g for
2 min. 50 l were transferred into an Eppendorf cup also containing 1 ml substrate solution
(45 mg blue starch corresponding to 1 Phadebas®
tablet (cat. no. 10-5380-32, Pharmacia and
Upjohn, Uppsala, Sweden) dissolved in 4 ml H2O) and incubated for 15 min. at 70 C. The
reaction was stopped by addition of 250 l 0.5 M NaOH and the sample was centrifuged for 5
min at 14’000 x g prior to transfer into a 1 ml cuvette and colorimetric quantification at 620
nm (Schlatter et al. 2002).
Results
Production of myocardial microtissues
The cultivation of mammalian cells in hanging drops enables gravity-assisted
assembly of spheroids. Compared to shake-flask cultivation (Sperelakis 1978) or
centrifugation-enforced aggregation strategies (Armstrong et al. 2000), hanging drop-based
microtissues production may be considered as a rather natural three-dimensional cultivation
technology and most suitable for the functional design of complex myocardial spheroids. The
hanging drop cultivation technology was successfully adapted to produce myocardial
microtissues of neonatal rat (NRC) and mouse (NMC) cardiomyocytes. Under optimized
culture conditions 1’200 rat cardiomyocytes form in a single hanging drop a myocardial
spheroid of 130 ± 11 m in diameter with near 100% efficiency within four days (Figures 1
and 2). Microtissues derived from mouse populations of 1’200 cardiomyocytes reached 170 ±
12 µm in diameter at the same timepoint. As myocardium-derived cells are terminally
differentiated and proliferation-inert corresponding myocardial microtissues show an
invariant size/time profile when cultivated for a period of several days. Pure NRC cultures as
well as mixed NRC populations reflecting the cell type composition of rodent heart produce
myocardial microtissues of comparable size (130 ± 11 m; Figure 2A). Since the overall size
of myocardial microtissues is a direct function of the proliferation-incompetent
cardiomyocyte cell number it can be varied over a wide range (NRC: 130 11 µm –
230 11 m; NMC 170 12 µm – 320 19 m) by adjusting the concentration of the cell
suspension between 1’200 to 10’000 cells/hanging drop (Figure 2B). At and above 10’000
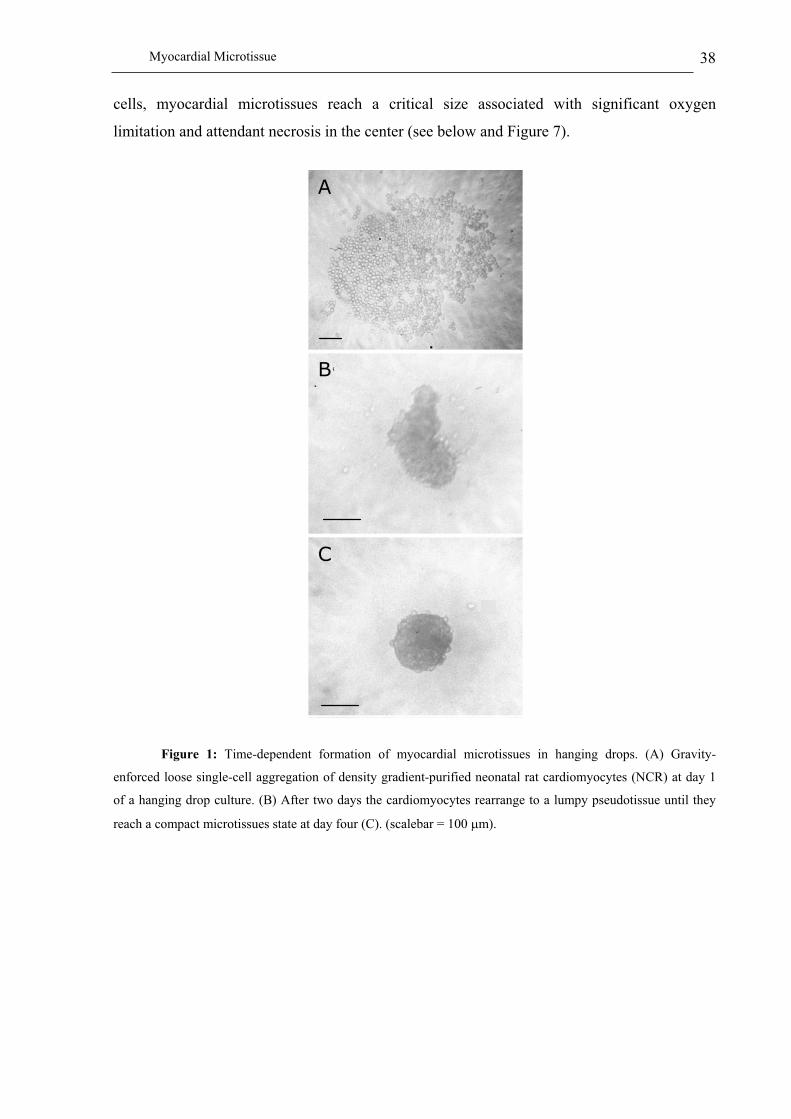
Myocardial Microtissue 38
cells, myocardial microtissues reach a critical size associated with significant oxygen
limitation and attendant necrosis in the center (see below and Figure 7).
Figure 1: Time-dependent formation of myocardial microtissues in hanging drops. (A) Gravity-
enforced loose single-cell aggregation of density gradient-purified neonatal rat cardiomyocytes (NCR) at day 1
of a hanging drop culture. (B) After two days the cardiomyocytes rearrange to a lumpy pseudotissue until they
reach a compact microtissues state at day four (C). (scalebar = 100 m).
A
B
C
AA
BB
CC

Myocardial Microtissue 39
Figure 2: Analysis of tissue growth and cell number/size profiles of neonatal rat (NRC) and
neonatal mouse cardiomyocyte (NMC)-derived myocardial microtissues produced in hanging drop cultures. (A)
Size profiles myocardial microtissues produced from 1’200 density gradient-purified NRCs, mixed NRC
populations (NRC Mix; 1’200 cells reflecting the typical cell type composition of rat hearts) as well as NMC
populations (1’200 cells total) at cultivation day 10. (B) Cell number-size correlations of NRC and NMC-derived
myocardial microtissues cultivated for four days in hanging drops. Since NRCs as well as NMCs are
proliferation-incompetent, the microtissue size is a function of the initial cell concentration inoculated in the
hanging drop.
48 h following cultivation of four-day-old myocardial microtissues in maintenance
medium supplemented with the 1-adrenergic agonist phenylephrine, NRC-derived
4 6 8 10
75
100
125
150
175
200NMC
NRC
NRC MixM
icro
tiss
ue D
iam
ete
r [
m]
Time [D]
100
150
200
250
300
350
400
1200 2500 5000 10000
NRC
NMC
Cell Number
Mic
roti
ssu
e D
iam
ete
r [
m]
4 6 8 10
75
100
125
150
175
200NMC
NRC
NRC MixM
icro
tiss
ue D
iam
ete
r [
m]
Time [D]
4 6 8 10
75
100
125
150
175
200NMC
NRC
NRC MixM
icro
tiss
ue D
iam
ete
r [
m]
Time [D]
100
150
200
250
300
350
400
1200 2500 5000 10000
NRC
NMC
Cell Number
Mic
roti
ssu
e D
iam
ete
r [
m]
100
150
200
250
300
350
400
1200 2500 5000 10000
NRC
NMC
Cell Number
Mic
roti
ssu
e D
iam
ete
r [
m]

Myocardial Microtissue 40
microtissues started rhythmic contraction at beat frequencies of 60 beats per minute which
could be maintained for over three weeks. Figure 3 shows three-week-old beating NRC (Fig.
3A) and NMC (Fig. 3B)-derived myocardial microtissues, the video motion of which can be
accessed at http://www.biotech.biol.ethz.ch/martinf/staff/jens.html. Similar to the situation in
two-dimensional cardiomyocyte cultures, myocardial spheroids rapidly synchronize their
beating frequencies upon inter-microtissue contacts which exemplifies the capacity of
myocardial microtissues to form functional tissue interactions (Figure 4 and data not shown;
(Hertig et al. 1996)). In contrast to NRC-derived microtissues, NMC-based myocardial
spheroids initiate sustained contraction in the absence of any chemical stimuli following
aggregation.
Figure 3: Three-week-old NRC (A) / NMC (B)-derived beating heart microtissue which can be seen in
action at http://www.biotech.biol.ethz.ch/martinf/staff/jens.html. The sound of a human embryonic heart is added
to the motion picture to exemplify the regular contraction of myocardial microtissues at a frequency of 60 beats
per minute. (scalebar = 20 m).
A BA B

Myocardial Microtissue 41
Figure 4: F-actin-based fluorescence micrograph of two interlinked NRC-derived myocardial
microtissues. Four-day-old myocardial microtissues have the potential to form microtissue-microtissue
superstructures and synchronize their contraction frequencies. (scalebar = 10 m).
Immunohistological characterization of myocardial microtissues
As the most physically energetic cell in the body contracting over three billion times
during an average human lifetime, cardiomyocytes are highly differentiated (Severs 2000).
Remodelling three-dimensional heart structures in vitro from isolated cardiomyocytes requires
maintenance of their shape and myofibrils while keeping the extraordinary mechanical
flexibility of this cell type. Also, electrical continuity between the cells has to be maintained
by intercalated disks, specialized cell-cell junctions, which are abundant in heart structures.
We have used a combination of immunofluorescence labelling and confocal microscopy to
characterize whether cardiomyocytes retained in vivo-like striation and cell-cell contacts
when grown as myocardial microtissues. In pure NRC- and NMC-derived myocardial
microtissues striation of myofibrils could be observed after a seven-day cultivation in
maintenance medium (Figures 5A and 5B). Also, -catenin-specific expression throughout
the entire myocardial microtissues indicates tissue-like cell-cell interactions within these
myocardial spheroids (Figures 5D and 5E) while individual cells retain their typical cell
morphologies associated with mature cardiomyocytes (Figure 5H).
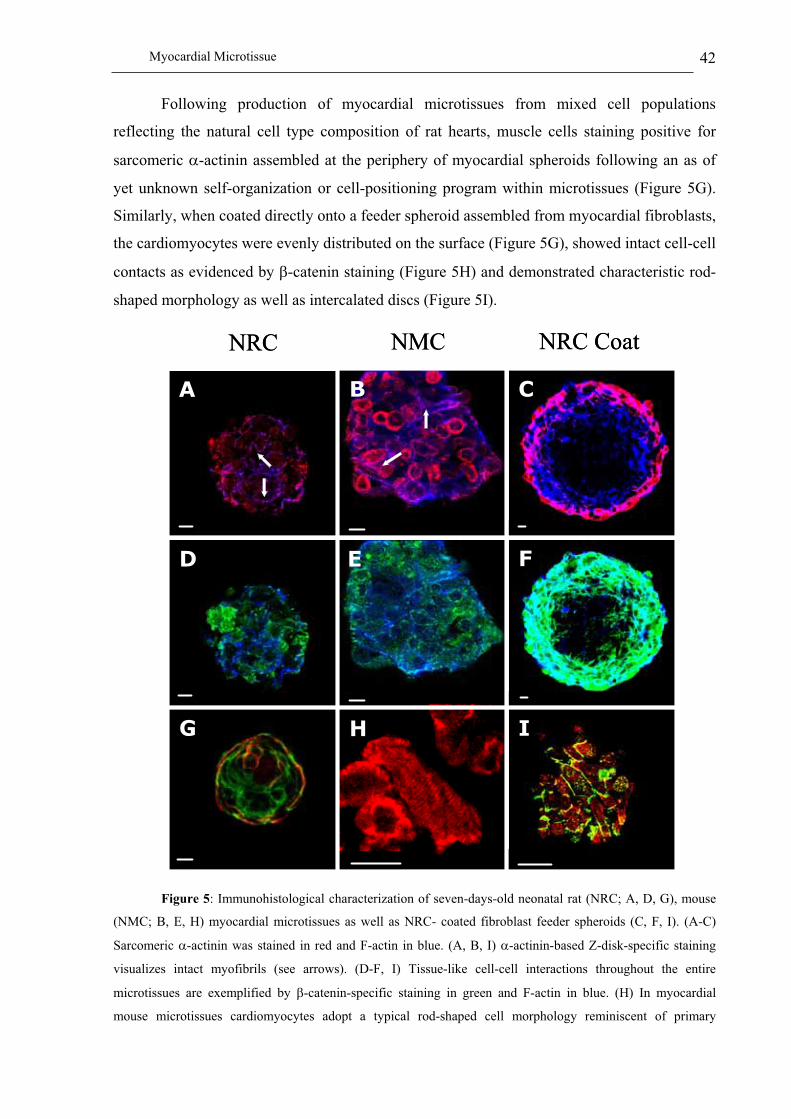
Myocardial Microtissue 42
Following production of myocardial microtissues from mixed cell populations
reflecting the natural cell type composition of rat hearts, muscle cells staining positive for
sarcomeric -actinin assembled at the periphery of myocardial spheroids following an as of
yet unknown self-organization or cell-positioning program within microtissues (Figure 5G).
Similarly, when coated directly onto a feeder spheroid assembled from myocardial fibroblasts,
the cardiomyocytes were evenly distributed on the surface (Figure 5G), showed intact cell-cell
contacts as evidenced by -catenin staining (Figure 5H) and demonstrated characteristic rod-
shaped morphology as well as intercalated discs (Figure 5I).
Figure 5: Immunohistological characterization of seven-days-old neonatal rat (NRC; A, D, G), mouse
(NMC; B, E, H) myocardial microtissues as well as NRC- coated fibroblast feeder spheroids (C, F, I). (A-C)
Sarcomeric -actinin was stained in red and F-actin in blue. (A, B, I) -actinin-based Z-disk-specific staining
visualizes intact myofibrils (see arrows). (D-F, I) Tissue-like cell-cell interactions throughout the entire
microtissues are exemplified by -catenin-specific staining in green and F-actin in blue. (H) In myocardial
mouse microtissues cardiomyocytes adopt a typical rod-shaped cell morphology reminiscent of primary
NMCNRC NRC Coat
D
I
F
C
E
H
BA
G
NMCNRC NRC Coat
D
I
F
C
E
H
BA
G
DD
II
FF
CC
EE
HH
BBAA
GG

Myocardial Microtissue 43
cardiomyocytes freshly isolated from rat hearts. (F) Following cultivation of myocardial microtissues produced
from mixed cell populations reflecting the natural cell type composition of rat hearts for seven days,
cardiomyocytes staining positive for -actinin are preferentially localized at the periphery of spheroids.
(overlapping staining for red ( -actinin) and green (F-actin) results in yellow). (scalebar = 10 m).
Characterization of the extracellular matrix of myocardial microtissues
The ability of artificial microtissues to produce a tissue-specific extracellular matrix
(ECM) typically associated with fundamental processes such as growth, differentiation, cell
migration and invasion will be essential for their use in clinical tissue engineering and gene
therapy (Weber et al. 1994; Giancotti et al. 1999). Development of a collagen I-containing
ECM is key to maintain cardiac mechanics during myocardial remodeling associated with
some heart diseases (Swynghedauw 1999). The natural myocardial ECM consists of collagen
I, fibronectin and proteoglycans forming the pericellular matrix and contains laminin as well
as type IV collagen as major constituents of the basal membrane (Farhadian et al. 1996). We
have performed immunofluorescence-based staining of extracellular matrix components in
order to characterize the ECM of neonatal rat and mouse cardiomyocyte-derived myocardial
microtissues as well as NRCs and NMCs cultivated in a two-dimensional anchorage-
dependent manner (Figure 6).
In cardiomyocyte monolayer cultures neither collagen I nor collagen IV were
produced whereas microtissues derived from pure and mixed NRC populations stained
positive for both collagen types (Figure 6A, C, D, F, H, I). By contrast, NMC-derived
microtissues showed only low-level collagen IV and no collagen I production (Figure 6E, J).
As collagen I is predominantly produced by fibroblasts, microtissues assembled from defined
mixed NRC populations accumulated higher levels of collagen I (Figure 6C). Myocardial
microtissues produced from purified cardiomyocyte cultures showed peripheral surface
expression of collagen I (Figure 6D). On the contrary, collagen IV is produced throughout
purified NRC-derived microtissues whereas myocardial spheroids generated from mixed NRC
populations stain positive for this collagen type predominantly at the spheroid’s surface
(Figure 6H, I). Fibronectin as well as laminin were expressed in two- as well as three-
dimensional NRC cultures (Figure 6K-T).
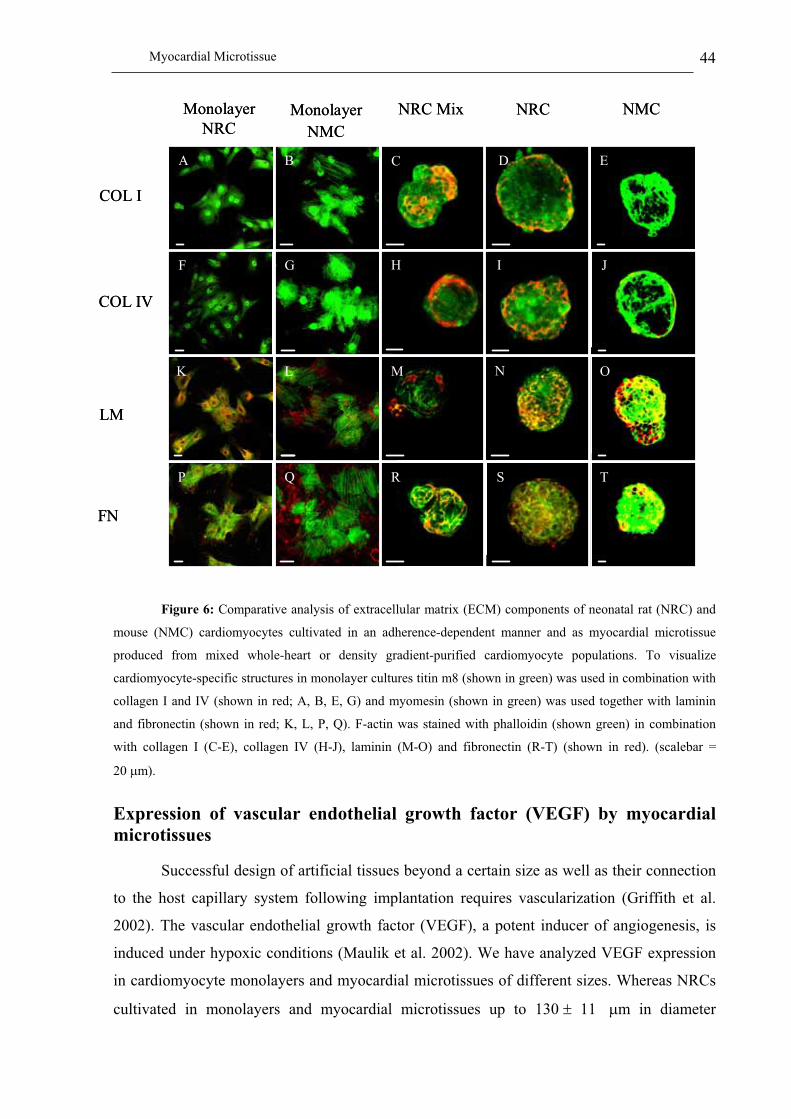
Myocardial Microtissue 44
Figure 6: Comparative analysis of extracellular matrix (ECM) components of neonatal rat (NRC) and
mouse (NMC) cardiomyocytes cultivated in an adherence-dependent manner and as myocardial microtissue
produced from mixed whole-heart or density gradient-purified cardiomyocyte populations. To visualize
cardiomyocyte-specific structures in monolayer cultures titin m8 (shown in green) was used in combination with
collagen I and IV (shown in red; A, B, E, G) and myomesin (shown in green) was used together with laminin
and fibronectin (shown in red; K, L, P, Q). F-actin was stained with phalloidin (shown green) in combination
with collagen I (C-E), collagen IV (H-J), laminin (M-O) and fibronectin (R-T) (shown in red). (scalebar =
20 m).
Expression of vascular endothelial growth factor (VEGF) by myocardial
microtissues
Successful design of artificial tissues beyond a certain size as well as their connection
to the host capillary system following implantation requires vascularization (Griffith et al.
2002). The vascular endothelial growth factor (VEGF), a potent inducer of angiogenesis, is
induced under hypoxic conditions (Maulik et al. 2002). We have analyzed VEGF expression
in cardiomyocyte monolayers and myocardial microtissues of different sizes. Whereas NRCs
cultivated in monolayers and myocardial microtissues up to 130 11 m in diameter
K
A
P
F
COL IV
FN
LM
Monolayer
NRC
COL I
NRC Mix NRCMonolayer
NMC
NMC
B
G
L
Q
C
H
M
R
D
I
N
S
E
J
O
T
KK
AA
PP
FF
COL IV
FN
LM
Monolayer
NRC
COL I
NRC Mix NRCMonolayer
NMC
NMC
B
G
L
Q
C
H
M
R
D
I
N
S
E
J
O
T

Myocardial Microtissue 45
(corresponding to 1’200 NRCs) show no VEGF expression (Figure 7A, C), oversized
(230 11 m in diameter) myocardial microtissues assembled from 10’000 NRCs produce
this growth factor at high levels suggesting hypoxic conditions inside the spheroid (Figure
7E). Similar results were obtained with NMC’s cultivated as monolayers and microtissues
(Figure 7B, D, F).
Figure 7: VEGF (vascular endothelial growth factor)-specific immunostaining of neonatal rat and
mouse cardiomyocytes as monolayers (A, B) and myocardial microtissue cultures (C-F). (A, B) Sarcomeric
alpha-actinin-based striation of cardiomyocytes is shown in red and the presence of VEGF in green. (C, D)
Myocardial microtissue produced from 1’200 NRCs (130 m in diameter) and NMC’s (170 m in diameter),
which was stained with phalloidin (shown in red) and for VEGF expression (shown in green). (E, F) VEGF-
specific immunostaining (green) of oversized myocardial microtissue produced from 10’000 NRC’s and NMC’s
(NRC: 230 m in diameter; NMC: 320 m). (scalebar = 20 m).
Rat Mouse
D
A B
C
E F
Rat Mouse
D
A B
C
E F
DD
AA BB
CC
EE FF

Myocardial Microtissue 46
Lentiviral infection of cardiomyocytes
Therapeutic interventions in mammalian tissues and artificial organs require an
efficient DNA transfer technology. Although adenovirus-based vectors enable remarkable
transduction efficiency in vitro, expression of delivered transgenes remains transient and
immune responses targeted against adenoviral proteins continue to be a major concern
(Schulick et al. 1995). Lentivirus-based transduction technology was shown to be highly
efficient for neonatal as well as adult cardiomyocytes (Rebolledo et al. 1998; Mitta 2002). We
have assessed the potential of third-generation self-inactivating lentiviral particles encoding
either the enhanced yellow fluorescent protein (YFP; pMF351, 5’LTR-+-PhCMV-EYFP-
3’LTR U3; (Mitta 2002)) or the secreted -amylase (SAMY; pMF364, 5’LTR-+-PEF1�-
SAMY-3’LTR U3; (Schlatter et al. 2002)) under control of the human PhCMV or PEF1�
promoters to transduce cardiomyocyte monolayers as well as NRC-derived myocardial
microtissues (Figure 8). pMF351-derived VSV-G (protein G of the vesicular stomatitis virus)-
pseudotyped lentiviral particles transduced NRC monolayer cultures with near 90% efficiency
and infected myocardial microtissues showed continuous bright yellowish fluorescence as a
consequence of viral EYFP delivery to peripheral cells (Figure 8). Interestingly, lentiviral
particles were unable to penetrate myocardial microtissues, as their centers remained
untransduced (Figure 8B).
In order to assess the specific heterologous protein production capacity of mammalian cells
grown in monolayers and microtissues, freshly isolated NRCs were transduced with SAMY-
encoding lentiviral particles (SAMY; pMF364, 5’LTR-+-PEF1 -SAMY-3’LTR U3; Schlatter
et al., 2002). For four days, half of the transgenic population was cultivated as a monolayer
and the other half in hanging drops to form myocardial microtissues. Isogenic control
infections of NRC monolayer cultures revealed no significant transduction-induced
morphological changes compared to mock-infected cultures (data not shown). Whereas the
monolayer culture produced respectable 5 U/cell -amylase NRCs assembled in myocardial
microtissues secreted over 6-fold more SAMY per cell (Figure 9). Owing to their high ectopic
protein production capacity microtissues may eventually become as valuable for in vivo
delivery of protein therapeutics in gene therapy scenarios as for tissue
engineering/replacement therapies.

Myocardial Microtissue 47
Figure 8: Transduction of pMF351 (5’LTR- +-PhCMV-EYFP-3’LTR U3)-derived EYFP (enhanced
yellow fluorescent protein)-encoding lentiviral particles into NRC monolayer (A) and myocardial microtissue
cultures (B). (scalebar for monolayer (A) = 100 m; scalebar for myocardial microtissue (B) = 10 m).
Figure 9: Quantification of SAMY production of 1’200 NRCs transduced with pMF364 5’LTR- +-
PEF1 -SAMY-3’LTR U3)-derived SAMY (secreted -amylase)-encoding lentiviral particles followed by four-day
maintenance in monolayer cultures or hanging drop cultures to form myocardial microtissues.
A BA BS
AM
Y (
U/c
ell)
Microtissue Monolayer
0
10
20
30
40
SA
MY
(U
/cel
l)
Microtissue Monolayer
0
10
20
30
40
0
10
20
30
40

Myocardial Microtissue 48
Discussion
Design and engineering of artificial heart tissues or entire hearts is one of the most
challenging scientific adventures of this millennium. Although advances in cardiac tissue
engineering are overdue since heart diseases are the leading non-infectious cause of death in
industrialized societies (i) production, (ii) rational reprogramming and (iii) in vitro assembly
of cells to form artificial heart structures have not yet become a clinical reality. As the most
energetically active cell in the heart, the cardiomyocyte is considered to be the key cell type
for cardiac tissue engineering (Severs 2000; Noble 2002). However, evolution of
cardiomyocytes which show unique mechanical flexibility, transformation resistance and
energetic activity has resulted in a highly specialized cell phenotype which is inert to current
cell expansion strategies, refractory to most gene transfer technologies and difficult to
assemble in three dimensions due to ongoing contraction (Datwyler et al. 1999; Magyar 1999;
Datwyler et al. 2001; Noble 2002; Pasumarthi et al. 2002).
So far, primary cardiomyocyte cultures from enzymatically dispersed fetal, neonatal or
adult vertebrate hearts are the only reliable source for pioneering cardiac tissue engineering.
Even though cardiomyocytes maintained in monolayer cultures have enabled detailed insight
into major structure- and gene-function correlations (Severs 2000), they are less responsive to
growth and differentiation factors and will therefore reveal fewer secrets relevant for cardiac
tissue engineering compared to cardiomyocytes assembled in three-dimensional cultures
(Armstrong et al. 2000). While three-dimensional cultivation of cardiomyocytes is generally
accepted to be key for cardiac tissue engineering there is ongoing controversy whether design
of artificial heart tissues requires scaffolds. Certainly, latest-generation biocompatible
scaffolds combine shape-supporting capacity with functional and bioactive properties (e.g.
differentiation, wound healing, cell/tissue adhesion, biodegradability) (Hubbell 1999;
Polonchuk et al. 2000). However, some scaffolds have been associated with post-implantation
side effects resulting from toxic degradation products, inflammatory reaction and poor
resorption (Yang et al. 2001). Scaffolds may still be the optimal solution for shaping rigid
tissue implants but they are intuitively less conceivable for the design of mechanically flexible
tissues exemplified by the myocardium (Hoerstrup et al. 2000; Ziegelaar et al. 2002).
The prevailing three-dimensional cultivation technology in cardiac tissue engineering
will have to unite several key characteristics including (i) long-term maintenance of the
beating capacity, (ii) multi-cell type cultivation, (iii) potential for self-organization,

Myocardial Microtissue 49
polarization and microstructure formation between different cell types, (iv) production of an
extracellular matrix, (v) vascularization including induction of vascular vessel development
and connection to the host capillary system following implantation, (vi) development of inter-
tissue superstructures and (vi) compatibility with high-efficiency stable gene transfer
technologies to engineer complementary cell phenotypes or provide therapeutic interventions.
We have evaluated gravity-assisted myocardial microtissue formation following
cultivation of primary neonatal rat and mouse cardiomyocytes in hanging drops. Previously,
the hanging drop technology has generated generic impact on studying differentiation and
lineage control in embryoid bodies and on the production of hepatic microtissues(Magyar et
al. 2001; Boheler et al. 2002; Kelm 2002). Hanging drop-based cultivation of rodent
cardiomyocytes has resulted in myocardial microtissues which form characteristic z-band-
containing myofibrils enabling sustained and regular beating profiles for over three weeks.
Also, these functional myocardial microtissues retain their capacity to form microtissue-
microtissue superstructures which coordinate their beating frequency. Such interactions
between microtissues are of two-fold importance, first, they suggest functional integration
into the host organ following therapeutic implantation, and second, microtissue-microtissue
assembly may represent the building block towards larger-sized artificial tissues. A major
entity for higher order three-dimensional structures is the extracellular matrix, four major
constituents of which are expressed in rat myocardial microtissues: laminin, fibronectin as
well as collagen I and collagen IV. The only difference between rat and mouse cardiomyocyte
microtissues is the lack of collagen I production in mouse-derived spheroids. The
predominant production of collagen type I and IV (rat cardiomyocyte) and collagen type IV
(mouse cardiomyocytes) at the periphery of myocardial spheroids suggests an inside-out
polarization within these microtissues. Such a self-organization force is further exemplified
by the peripheral localization of cardiomyocytes in microtissues produced from mixed heart
cell populations reflecting the typical cell type composition of rodent hearts.
Although myocardial microtissues generated from heart-derived primary
cardiomyocytes cultivated in hanging drops show many of the aforementioned key
characteristics required for production of artificial tissue, their actual impact on cardiac tissue
engineering will depend on the design of artificial tissues beyond a few hundred micrometers
in diameter. As primary cardiomyocytes are terminally differentiated and proliferation-inert,
the size of myocardial microtissues is a direct function of cell number. However, due to
physico-chemical constraints, which limit nutrient and particular oxygen supply in the center
of artificial tissues cardiomyocyte-only-based microtissues cannot go beyond a certain size.

Myocardial Microtissue 50
Interestingly, in contrast to cardiomyocyte monolayer cultures and small myocardial
microtissues (rat: 130 ± 11 m in diameter; mouse: 170 ± 12 m in diameter), oversized
cardiomyocyte-derived spheroids (rat: 230 ± 11 m in diameter; mouse: 320 ± 19 m in
diameter) produce VEGF. Such size-dependent VEGF expression may (i) mediate connection
to the host capillary system following implantation, (ii) enable in vitro vascularization in
multi-cell type-based microtissues, and (iii) eventually foster the design of large, fully
vascularized artificial (mini-) tissues.
Whatever artificial microtissue design will prevail in the future, successful cardiac
tissue engineering as well as heart-targeted gene therapy will require efficient gene transfer
technologies to express desired therapeutic or phenotype-modulating transgenes in
cardiomyocytes and/or myocardial microtissues. Successful transduction of cardiomyocyte
monolayers has been achieved with transgenic adenoviruses as well as Sindbis virus
(Datwyler et al. 1999; Zhou et al. 2000; Datwyler et al. 2001). However, both viruses mediate
only transient gene expression. We have recently designed a new series of lentiviral
expression vectors which transduce in their VSV-G-pseudotyped configuration even the
difficult-to-transfect adult rat cardiomyocytes (Mitta 2002). The same transgenic lentiviral
particles transduce the surface of myocardial microtissues at near 100% efficiency. A
comparative analysis of the production levels between cardiomyocytes cultivated as
monolayer or microtissue revealed that three-dimensional structures produced 6-fold more
reporter protein than two-dimensional cultures.
Complying with most of the aforementioned key tissue characteristics and bundled
with a powerful lentiviral transduction technology, myocardial microtissues are on their way
to establish cardiac tissue engineering as clinical alternatives to heart transplantation and
implantation of artificial non-biological hearts.
Acknowledgements
This work was supported by the Swiss National Science Foundation (grant no. 631-
065946), the Roche Research Foundation (grant no. 118-2001) and the Novartis Foundation
(grant no. 01C41). We thank Evelyne Perriard for isolation of neonatal mouse and rat
cardiomyocytes, Barbara Mitta for production/transduction of lentiviral particles, Stefan
Lange for video microscopy and Valeria Nicolini-Gonzalez as well as Beat P. Kramer for
critical comments on the manuscript.

Myocardial Microtissue 51
References
Agarkova, I., D. Auerbach, E. Ehler and J. C. Perriard (2000). "A novel marker for vertebrate
embryonic heart, the EH-myomesin isoform." J Biol Chem 275, 10256-10264.
Akins, R. E. (2002). "Can tissue engineering mend broken hearts?" Circ Res 90, 120-122.
Akins, R. E., R. A. Boyce, M. L. Madonna, N. A. Schroedl, S. R. Gonda, T. A. McLaughlin
and C. R. Hartzell (1999). "Cardiac organogenesis in vitro: reestablishment of three-
dimensional tissue architecture by dissociated neonatal rat ventricular cells." Tissue
Eng 5, 103-118.
Aplin, A. E., A. K. Howe and R. L. Juliano (1999). "Cell adhesion molecules, signal
transduction and cell growth." Curr Opin Cell Biol 11, 737-744.
Armstrong, M. T., D. Y. Lee and P. B. Armstrong (2000). "Regulation of proliferation of the
fetal myocardium." Dev Dyn 219, 226-236.
Auerbach, D., S. Bantle, S. Keller, V. Hinderling, M. Leu, E. Ehler and J. C. Perriard (1999).
"Different domains of the M-band protein myomesin are involved in myosin binding
and M-band targeting." Mol Biol Cell 10, 1297-1308.
Boheler, K. R., J. Czyz, D. Tweedie, H. T. Yang, S. V. Anisimov and A. M. Wobus (2002).
"Differentiation of pluripotent embryonic stem cells into cardiomyocytes." Circ Res
91, 189-201.
Carrier, R. L., M. Papadaki, M. Rupnick, F. J. Schoen, N. Bursac, R. Langer, L. E. Freed and
G. Vunjak-Novakovic (1999). "Cardiac tissue engineering: cell seeding, cultivation
parameters, and tissue construct characterization." Biotechnol Bioeng 64, 580-589.
Chen, C. S., M. Mrksich, S. Huang, G. M. Whitesides and D. E. Ingber (1997). "Geometric
control of cell life and death." Science 276, 1425-1428.
Claycomb, W. (1991). Proliferative potential of the mammalian ventricular cardiac muscle
cells. The development and regenerative potential of cardiac muscle. J. A. Oberpiller,
M.A. New York, Harwood Academic. 1: 351-362.
Dar, A., M. Shachar, J. Leor and S. Cohen (2002). "Optimization of cardiac cell seeding and
distribution in 3D porous alginate scaffolds." Biotechnol Bioeng 80, 305-312.
Datwyler, D. A., H. M. Eppenberger, D. Koller, J. E. Bailey and J. P. Magyar (1999).
"Efficient gene delivery into adult cardiomyocytes by recombinant Sindbis virus." J
Mol Med 77, 859-864.
Datwyler, D. A., J. P. Magyar, V. Busceti, A. Hirschy, J. C. Perriard, J. E. Bailey and H. M.
Eppenberger (2001). "Recombinant Sindbis virus allows expression and precise

Myocardial Microtissue 52
targeting of proteins of the contractile apparatus in cultured cardiomyocytes." Basic
Res Cardiol 96, 630-635.
Eschenhagen, T., C. Fink, U. Remmers, H. Scholz, J. Wattchow, J. Weil, W. Zimmermann,
H. H. Dohmen, H. Schafer, N. Bishopric, T. Wakatsuki and E. L. Elson (1997).
"Three-dimensional reconstitution of embryonic cardiomyocytes in a collagen matrix:
a new heart muscle model system." Faseb J 11, 683-694.
Farhadian, F., F. Contard, A. Sabri, J. L. Samuel and L. Rappaport (1996). "Fibronectin and
basement membrane in cardiovascular organogenesis and disease pathogenesis."
Cardiovasc Res 32, 433-442.
Freed, L. E. and G. Vunjak-Novakovic (1997). "Microgravity tissue engineering." In Vitro
Cell Dev Biol Anim 33, 381-385.
Gheorghiade, M. and R. O. Bonow (1998). "Chronic heart failure in the United States: a
manifestation of coronary artery disease." Circulation 97, 282-289.
Giancotti, F. G. and E. Ruoslahti (1999). "Integrin signaling." Science 285, 1028-1032.
Griffith, L. G. and G. Naughton (2002). "Tissue engineering--current challenges and
expanding opportunities." Science 295, 1009-1014.
Hennekens, C. H. (1998). "Increasing burden of cardiovascular disease: current knowledge
and future directions for research on risk factors." Circulation 97, 1095-1102.
Hertig, C. M., S. Butz, S. Koch, M. Eppenberger-Eberhardt, R. Kemler and H. M.
Eppenberger (1996). "N-cadherin in adult rat cardiomyocytes in culture. II. Spatio-
temporal appearance of proteins involved in cell-cell contact and communication.
Formation of two distinct N-cadherin/catenin complexes." J Cell Sci 109, 11-20.
Hoerstrup, S. P., R. Sodian, J. S. Sperling, J. P. Vacanti and J. E. Mayer, Jr. (2000). "New
pulsatile bioreactor for in vitro formation of tissue engineered heart valves." Tissue
Eng 6, 75-79.
Hubbell, J. A. (1999). "Bioactive biomaterials." Curr Opin Biotechnol 10, 123-129.
Intaglietta, M., P. C. Johnson and R. M. Winslow (1996). "Microvascular and tissue oxygen
distribution." Cardiovasc Res 32, 632-643.
Keller, G. M. (1995). "In vitro differentiation of embryonic stem cells." Curr Opin Cell Biol
7, 862-869.
Kelm, J., Timmins,N., Brown,C.J., Fussenegger,M., Nielsen,L.K. (2003). "An efficient
cultivation method to generate homogeneouse multicellular tumor spheroids that can
be applied to a wide variety of cell types." Biotechnol Bioeng. 83, 173-180.

Myocardial Microtissue 53
Magyar, J. P., M. Nemir, E. Ehler, N. Suter, J. C. Perriard and H. M. Eppenberger (2001).
"Mass production of embryoid bodies in microbeads." Ann N Y Acad Sci 944, 135-
143.
Magyar, J. P. E., H. M. (1999). Factors involved in the cell cycle arrest of adult rat
cardiomyocytes. Cell Engineering. M. Al-Rubeai. Dordrecht, Kluwer Academic
Publishers. 1: 239-254.
Maulik, N. and D. K. Das (2002). "Potentiation of angiogenic response by ischemic and
hypoxic preconditioning of the heart." J Cell Mol Med 6, 13-24.
Messerli, J. M., H. T. van der Voort, E. Rungger-Brandle and J. C. Perriard (1993). "Three-
dimensional visualization of multi-channel volume data: the amSFP algorithm."
Cytometry 14, 725-735.
Mitta, B., Rimann,M., Ehrbar,M., Djonov,V., Ehrengruber,M.U., Kelm,J., Fussenegger,M.
(2002). "Advanced modular self-inactivating lentiviral expression vectors for
multigene interventions in mammalian cells and in vivo transduction." Nucleic acids
research 30, e113
Mochizuki, H., J. P. Schwartz, K. Tanaka, R. O. Brady and J. Reiser (1998). "High-titer
human immunodeficiency virus type 1-based vector systems for gene delivery into
nondividing cells." J Virol 72, 8873-8883.
Noble, D. (2002). "Modeling the heart--from genes to cells to the whole organ." Science 295,
1678-1682.
Pasumarthi, K. B. and L. J. Field (2002). "Cardiomyocyte cell cycle regulation." Circ Res 90,
1044-1054.
Polonchuk, L., J. Elbel, L. Eckert, J. Blum, E. Wintermantel and H. M. Eppenberger (2000).
"Titanium dioxide ceramics control the differentiated phenotype of cardiac muscle
cells in culture." Biomaterials 21, 539-550.
Rebolledo, M. A., P. Krogstad, F. Chen, K. M. Shannon and T. S. Klitzner (1998). "Infection
of human fetal cardiac myocytes by a human immunodeficiency virus-1-derived
vector." Circ Res 83, 738-742.
Reiser, J., G. Harmison, S. Kluepfel-Stahl, R. O. Brady, S. Karlsson and M. Schubert (1996).
"Transduction of nondividing cells using pseudotyped defective high- titer HIV type 1
particles." Proc Natl Acad Sci U S A 93, 15266-15271.
Richardson, T. P., M. C. Peters, A. B. Ennett and D. J. Mooney (2001). "Polymeric system for
dual growth factor delivery." Nat Biotechnol 19, 1029-1034.
Ross, R. S. and T. K. Borg (2001). "Integrins and the myocardium." Circ Res 88, 1112-1119.

Myocardial Microtissue 54
Rothen-Rutishauser, B. M., E. Ehler, E. Perriard, J. M. Messerli and J. C. Perriard (1998).
"Different behaviour of the non-sarcomeric cytoskeleton in neonatal and adult rat
cardiomyocytes." J Mol Cell Cardiol 30, 19-31.
Schlatter, S., M. Rimann, J. Kelm and M. Fussenegger (2002). "SAMY, a novel mammalian
reporter gene derived from Bacillus stearothermophilus alpha-amylase." Gene 282,
19-31.
Schuldiner, M., O. Yanuka, J. Itskovitz-Eldor, D. A. Melton and N. Benvenisty (2000). "From
the cover: effects of eight growth factors on the differentiation of cells derived from
human embryonic stem cells." Proc Natl Acad Sci U S A 97, 11307-11312.
Schulick, A. H., K. D. Newman, R. Virmani and D. A. Dichek (1995). "In vivo gene transfer
into injured carotid arteries. Optimization and evaluation of acute toxicity."
Circulation 91, 2407-2414.
Severs, N. J. (2000). "The cardiac muscle cell." Bioessays 22, 188-199.
Sperelakis, N. (1978). "Cultured heart cell reaggregate model for studying cardiac
toxicology." Environ Health Perspect 26, 243-267.
Swynghedauw, B. (1999). "Molecular mechanisms of myocardial remodeling." Physiol Rev
79, 215-262.
Watzka, S. B., J. Lucien, M. Shimada, V. Edwards, H. Yeger, G. Hannigan and J. G. Coles
(2000). "Selection of viable cardiomyocytes for cell transplantation using three-
dimensional tissue culture." Transplantation 70, 1310-1317.
Weber, K. T., Y. Sun, S. C. Tyagi and J. P. Cleutjens (1994). "Collagen network of the
myocardium: function, structural remodeling and regulatory mechanisms." J Mol Cell
Cardiol 26, 279-292.
Yang, S., K. F. Leong, Z. Du and C. K. Chua (2001). "The design of scaffolds for use in
tissue engineering. Part I. Traditional factors." Tissue Eng 7, 679-689.
Zhou, Y. Y., S. Q. Wang, W. Z. Zhu, A. Chruscinski, B. K. Kobilka, B. Ziman, S. Wang, E.
G. Lakatta, H. Cheng and R. P. Xiao (2000). "Culture and adenoviral infection of adult
mouse cardiac myocytes: methods for cellular genetic physiology." Am J Physiol
Heart Circ Physiol 279, H429-436.
Ziegelaar, B. W., J. Aigner, R. Staudenmaier, K. Lempart, B. Mack, T. Happ, M. Sittinger, M.
Endres, A. Naumann, E. Kastenbauer and N. Rotter (2002). "The characterisation of
human respiratory epithelial cells cultured on resorbable scaffolds: first steps towards
a tissue engineered tracheal replacement." Biomaterials 23, 1425-1438.

Chapter 4
Self-Assembly of Sensory Neurons into Ganglia-Like
Microtissues
Kelm J.M., Ittner L.M., Born W., Djonov V. and Fussenegger M., (2005) J. Biotech., in press

Artificial Ganglia 56
Abstract
Unraveling intra- and inter-cellular signaling networks managing cell-fate control,
coordinating complex differentiation regulatory circuits and shaping tissues and organs in
living systems remain major challenges in the post-genomic era. Resting on the laurels of
past-century monolayer culture technologies, the cell culture community has only recently
begun to appreciate the potential of three-dimensional mammalian cell culture systems to
reveal the full scope of mechanisms orchestrating the tissue-like cell quorum in space and
time. Capitalizing on gravity-enforced self-assembly of monodispersed primary embryonic
mouse cells in hanging drops, we designed and characterized a three-dimensional cell culture
model for ganglion-like structures. Within 24 hours, a mixture of mouse embryonic
fibroblasts (MEF) and cells, derived from the dorsal root ganglion (DRG) (sensory neurons
and Schwann cells) grown in hanging drops, assembled to coherent spherical microtissues
characterized by a MEF feeder core and a peripheral layer of DRG-derived cells. In a time-
dependent manner, sensory neurons formed a polar ganglion-like cap structure, which
coordinated guided axonal outgrowth and innervation of the distal pole of the MEF feeder
spheroid. Schwann cells, present in embryonic DRG isolates, tended to align along axonal
structures and myelinate them in an in vivo-like manner. Whenever cultivation exceeded ten
days, DRG:MEF-based microtissues disintegrated due to an as yet unknown mechanism.
Using a transgenic MEF feeder spheroid, engineered for gaseous acetaldehyde-inducible
interferon- (ifn- ) production by cotransduction of retro-/lenti-viral particles, a short six-
hour ifn- induction was sufficient to rescue the integrity of DRG:MEF spheroids and enable
long-term cultivation of these microtissues. In hanging drops, such microtissues fused to
higher-order macrotissue-like structures, which may pave the way for sophisticated bottom-up
tissue engineering strategies. DRG:MEF-based artificial micro- and macrotissue design
demonstrated accurate key morphological aspects of ganglions and exemplified the potential
of self-assembled scaffold-free multicellular micro-/macrotissues to provide new insight into
organogenesis.
Introduction
Self-assembly is a basic phenomenon responsible for structural organization of living
systems. Although the differentiation of specific cell phenotypes and the layout map of cell

Artificial Ganglia 57
position in the early development of multicellular organisms are under strict genetic control
and imprinted in a localization-dependent manner, it is the process of cellular self-assembly,
mediated by cell-cell and cell-extracellular matrix (ECM) interactions, which fine-tunes the
organization of a particular cell quorum during histogenesis and organogenesis (Jakab et al.
2004; Wang et al. 2004). Steinberg’s pioneering hypothesis for the self-assembly of
embryonic cells was based on the concept of tissue fluidity and the assumption that
embryonic tissues can, in many respects, be regarded as liquids. In particular, on non-
adhesive surfaces or in suspension, multicellular aggregates reach their lowest free-energy
state by adopting a spherical shape, similar to liquid droplets (Steinberg 1963). The follow-up
differential adhesion hypothesis explains the liquid-like behavior as a function of tissue
surface and interfacial tensions resulting from cohesive (e.g., N-cadherin-mediated) and
adhesive (e.g., integrin-mediated) forces (Beysens et al. 2000; Lauffenburger and Griffith
2001; Steinberg 1963; Tepass et al. 2000). Akin to assembly and structure-shaping forces in a
multicellular embryonic developmental state, monodispersed cells derived from living tissues
retain their imprinted potential to self-assemble into defined in vivo-like structures
(Armstrong 1989; Duguay et al. 2003; Kelm and Fussenegger 2004; Layer et al. 2002;
Steinberg and Foty 1997).
In the vertebrate nervous system, the molecular crosstalk between neurons and glial
Schwann cells is key to maintaining and extending brain structures and innervated tissues
(Chen et al. 2003; Gordon-Weeks 2004). The neurons’ expression of trophic factors trigger
glial cell-fate commitment and glial cells then control survival, differentiation and
interconnection of associated neurons (Lemke, 2001). Although many neuronal differentiation
factors have been identified to date, the details of differentiation control and its coordination
with cell differentiation in neighboring tissues remain largely elusive (Chotard and Salecker
2004; Edlund and Jessell 1999; Griffiths and Hidalgo 2004; Lemke, 2001).
Since in-vivo experiments resolved several of the multifaceted phenomena underlying
neuro-, glia- and ganglia-genesis, future advances in this field will depend on information
gathered from complementary in vitro cell culture systems. The technology for sustained
cultivation of neurons as well as glia cells in a primary cell (co-)culture format will thus play
a pivotal role in deciphering the molecular events orchestrating cell-cell and cell-ECM
interaction in the developing nervous system (Zahir and Weaver 2004). Owing to the
aforementioned occurring in developing tissues, primary cells may lose their tissue-specific
phenotype following isolation and cultivation in a classical two-dimensional cell culture

Artificial Ganglia 58
format (Abbott 2003; Zhang 2004). Furthermore, the culture media, matrices and the absence
of neighboring cell communities may alter morphogen levels/gradients, which could bias the
readout of two-dimensional culture settings (Svenningsen et al. 2003). For these reasons, a
variety of sophisticated cell culture systems have been devised, which include multiple cell
types and consider the requirements for the development and organization of the peripheral
nervous system (Gingras et al. 2003; Suuronen et al. 2004b, Pittier et al. 2004). A priori,
sensory neurons and Schwann cells, derived from embryonic dorsal root ganglions (DRGs)
cocultivated with embryonic fibroblasts, may mimic the complex reciprocal interactions
between neurons and glia cells as well as either of these cell types with the connective tissue.
We have used gravity-enforced self-assembly of mouse DRG-derived cells, cocultured with
mouse embryonic fibroblasts (MEFs) in hanging drops, to create a robust three-dimensional
cell culture system to provide further insight into gangliogenesis.
Material and Methods
Isolation of mouse embryonic dorsal root ganglia (DRG) and fibroblasts
Eight to 12 DRGs, isolated from each E12 (embryonic day 12) embryo of time-mated
NMRI mice ((Taconic M&B A/S, Ry, Denmark) and from each E15 embryo of time-mated
Wistar rats (Janvier Elevage, Le Genest Saint Isle, France), were treated with Hank’s solution
(0.25% trypsin, Invitrogen, Carlsbad, CA) for 30 min. at 37°C, centrifuged for 3 min. at 130g
(800 rpm) and resuspended as monodipersed DRGs in neurobasalTM
medium (Invitrogen)
supplemented with 20% newborn calf serum (cat. no. P30-0400, lot no. P230704, PAN
biotech GmbH, Aidenbach, Germany) and 10 ng/ml nerve growth factor (2.5S-NGF;
Invitrogen). Cell-specific labeling of dissociated embryonic DRG populations was performed
using the PKH26 cell linker kit according to the manufacturer’s instructions (Sigma
Chemicals, Buchs, Switzerland). Mouse embryonic fibroblasts (MEF) were isolated from E14
embryos derived from time-mated mice (ICR-M-TKneo2, (Stewart et al. 1987)). Following
removal of the head and liver, the embryos were transferred to an ice-cold trypsin/EDTA
(0.05%/0.02%) solution (Pan Biotech GmbH) and incubated for 12h at 4°C. Subsequently, the
trypsin/EDTA solution was removed and the cells plated in Dulbecco’s modified Eagle
medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS, cat. no.
3302-P231902, lot no. P231902, Pan Biotech GmbH). Neonatal rat heart fibroblasts (RHF)
were isolated from newborn Wistar rats (Janvier Elevage). The hearts were dissected, digested

Artificial Ganglia 59
with collagenase (Worthington Biochemical Corp., Freehold, NJ) and pancreatin (Invitrogen)
and processed as described by Auerbach and coworkers (Auerbach et al. 1999).
Cell culture and microtissue production
MEFs, RHFs and NIH/3T3 (ATCC No. CRL-1658) were expanded in DMEM
supplemented with 10% FBS, 1% non-essential amino acids (Invitrogen) and 1%
penicillin/streptomycin solution (Sigma Chemicals). Monolayer fibroblast cultures were
trypsinized and mixed with monodispersed DRG populations at a cell ratio of 5x103/6x10
2
(MEF/DRG) prior to seeding into 60-well plates at indicated cell densities (HLA plate,
Greiner-Bio One, Frickenhausen, Germany). In order to enable gravity-enforced microtissue
formation in hanging drops, the 60-well plates were incubated upside down. Multicellular
microtissues were maintained in neurobasalTM
medium supplemented with 20% newborn calf
serum and 10 ng/ml NGF. For mixed MEF-DRG monolayer cultures, 4x105 MEFs were
seeded into 35 mm cell culture dishes and proliferation inactivated by adding 10 µg/ml
mitomycin C (Sigma Chemicals) for 2 h. After washing the monolayers three times in
phosphate-buffered saline (PBS; 150 mM NaCl, 6.5 mM Na2HPO4x2H2O, 2.7 mM KCL,
1.5 mM KH2PO4, pH 7.4), 7.5x104 DRG-derived cells were added.
Macrotissue assembly
Cylindrical agarose moulds (4% agarose in PBS [Sigma Chemicals]) containing a
3x10 mm casting chamber were produced from custom-designed Teflon models. In order to
produce a single macrotissue 900 microtissues were transferred to an agarose mould and
cultivated under static culture conditions in neurobasalTM
medium supplemented with 20%
newborn calf serum and 10 ng/ml NGF for two days at 37°C in a humidified 5% CO2-
containing atmosphere.
Immunofluorescence-based cell characterization
Microtissues were prepared for immunochemistry either as entire microtissues or as
frozen sections. Microtissues were harvested, washed twice in PBS, fixed for 1 h in 4%
paraformaldehyde-containing PBS, washed three times for 5 min. in phosphate-buffered
Triton X-100 (PBT; 0.002% Triton X-100 in PBS) and subsequently permeabilized for
60 min. in 0.5% Triton X-100-containing PBS. Primary antibodies, specific for desired

Artificial Ganglia 60
proteins as well as fluorochrome-conjugated secondary antibodies were diluted in 1% BSA-
containing Tris-buffered saline (TBS; 20 mM Tris base, 155 mM NaCl, 2 mM EGTA, 2 mM
MgCl2) and sequentially incubated with permeabilized microtissues for 12h at 4 C, with three
PBS washings between incubation periods. The microtissues were then washed in PBS and
embedded on glass slides using Tris-buffered glycerol (a 3:7 mixture of 0.1 M Tris-HCl
[pH 9.5] and glycerol supplemented with 50 mg/mL n-propyl-gallat). Tailored 0.5 mm
spacers were used to prevent squashing of the microtissues between the slide and cover slip.
The following primary antibodies were used: beta-catenin (rabbit, polyclonal, Sigma
Chemicals), BrdU (mouse, monoclonal, Sigma Chemicals), Brn-3A (mouse, monoclonal,
Chemicon, Hofheim, Germany), fibronectin (mouse, monoclonal, Sigma Chemicals), Gap-43
(mouse, monoclonal, Oncogene, Cambridge, MA, USA), laminin (rabbit, polyclonal, Sigma
Chemicals), myelin binding protein (MBP ; rat, monoclonal, Serotec, Düsseldorf, Germany),
N-cadherin (rabbit, polyclonal, BD Bioscience AG, Basel, Switzerland), neurofilament
(rabbit, polyclonal, Juro Supply GmbH, Luzern, Switzerland), neurofilament (mouse,
monoclonal, cat. Sigma Chemicals), Sox10 (mouse, monoclonal, kindly provided by Lukas
Sommer), and stained with Cy3-coupled secondary anti-mouse (Jackson Immunochemicals,
West Grove, PA, USA), Cy2-coupled anti-rat (Jackson Immunochemicals) or FITC-coupled
anti-rabbit (ICN Pharmaceuticals, Hyland, CA, USA) antibodies. F-actin was visualized using
A633-coupled phalloidin (Molecular Probes Inc., Eugene, OR, USA).
Histology
Macrotissues were washed once in PBS (Sigma Chemicals) and fixed for 4h at 4°C in
PBS containing 4% paraformaldehyde (Sigma Chemicals). Tissues were embedded in
TissueTec (Merck Eurolab AG, Dietikon, Switzerland) and frozen for 30 min at –25°C.
Sections (10 µm ) were generated using an Ultracut device (Zeiss, Feldbach, Switzerland).
Standard protocols were used for hematoxilin/eosin stainings on frozen sections as described
by Sheehan and Hrapchak (Sheehan and Hrapchak 1980).
Confocal light microscopy
The imaging system consisted of an inverted fluorescence microscope (Leica
DMIRB/E, Glattbrugg, Switzerland) equipped with Leica oil immersion objectives
(40x/20x/10x), a confocal scanner (Leica TCS SP1) featuring an argon and helium-neon laser

Artificial Ganglia 61
and a Silicon Graphics Workstation (SGI, Schlieren, Switzerland) with Imaris 3D multi-
channel image processing software installed (Bitplane, AG, Zurich, Switzerland (Messerli et
al. 1993)).
Transmission Electron Microscopy
For electron microscopic studies microtissues were fixed by total immersion in 0.1M
cacodylate buffer (pH 7.4, 350 mOsm) containing 2.5% glutaraldehyde. Tissue blocks were
postfixed in osmium tetroxide, block-stained using uranyl acetate, dehydrated by increasing
the ethanol concentrations and embedded in Epon 812 according to Djonov et al. (Djonov et
al., 2000) (all chemicals from Merck Eurolab AG, Dietikon, Switzerland). Semithin 1-µm
sections were stained with toluidine blue and visualized using a Leica DMIRB/E light
microscope (Leica Microsystems, Glattbrugg, Switzerland). Ultrathin sections, 80 to 90 nm
thick, were picked up on Formvar-coated (polyvinyl formal; Fluka Chemie AG) copper grids,
double-stained with lead citrate (Merck Eurolab AG) and uranyl acetate and monitored on a
Philips EM 400 electron microscope (FEI AG, Zurich, Switzerland).
Gas-inducible ifn- expression
Retroviral particles encoding 5’LTR-driven expression of Aspergillus nidulans’ AlcR
and the G418 resistance gene (neo), constitutively transcribed by the phosphoglycerate kinase
promoter (PPGK) (pWW506; 5’LTR-+-alcR-PPKG-neo-3’LTR) (Weber et al. 2004), were
produced using the packaging cell line GP-293 (Clontech, Basel, Switzerland; (Burns et al.
1993)) according to manufacturer’s protocol. Lentiviral particles encoding ifn- , controlled by
the AlcR-dependent acetaldehyde-inducible promoter PAIR (pWW430; 5’LTR-+-PAIR-ifn- -
3’LTR U3) (Weber et al. 2004), were produced as described before (Mitta et al. 2002). MEFs
were co-transduced with pWW506-derived retroviral (4 x 104
transducing units (TU/ml)) and
pWW430-derived lentiviral (1.4 x 105 TU/ml) particles (quantified as described by Mitta and
coworkers (Mitta et al. submitted)), mixed with DRG-derived monodispersed cells (see
above) and cultivated in hanging drops for five days. Thereafter, ifn- expression was induced
for 6h by gaseous acetaldehyde, evaporating from a 50 µl acetaldehyde-ethanol (1:4) solution
in the lid of a multi-well hanging-drop cultivation plate. IFN- production was quantified
with a Hu-ifn- ELISA kit according to the manufacturer’s protocol (R&D Systems,
Minneapolis, MN, USA).
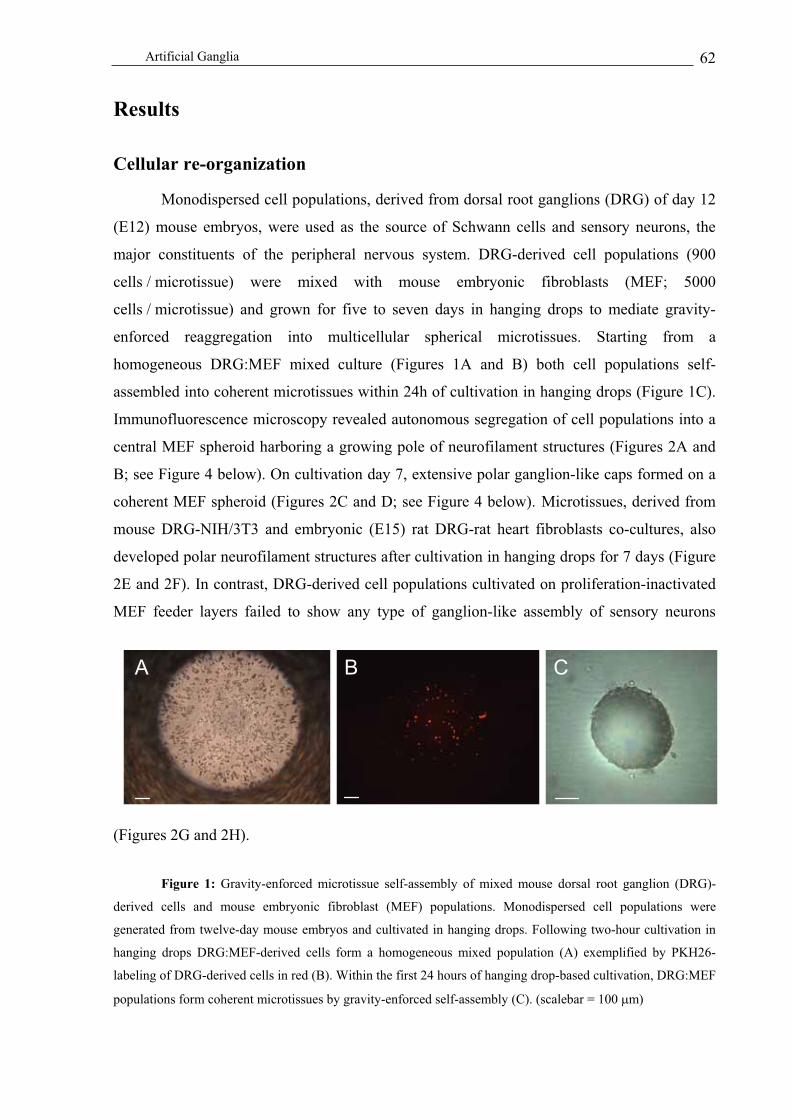
Artificial Ganglia 62
Results
Cellular re-organization
Monodispersed cell populations, derived from dorsal root ganglions (DRG) of day 12
(E12) mouse embryos, were used as the source of Schwann cells and sensory neurons, the
major constituents of the peripheral nervous system. DRG-derived cell populations (900
cells / microtissue) were mixed with mouse embryonic fibroblasts (MEF; 5000
cells / microtissue) and grown for five to seven days in hanging drops to mediate gravity-
enforced reaggregation into multicellular spherical microtissues. Starting from a
homogeneous DRG:MEF mixed culture (Figures 1A and B) both cell populations self-
assembled into coherent microtissues within 24h of cultivation in hanging drops (Figure 1C).
Immunofluorescence microscopy revealed autonomous segregation of cell populations into a
central MEF spheroid harboring a growing pole of neurofilament structures (Figures 2A and
B; see Figure 4 below). On cultivation day 7, extensive polar ganglion-like caps formed on a
coherent MEF spheroid (Figures 2C and D; see Figure 4 below). Microtissues, derived from
mouse DRG-NIH/3T3 and embryonic (E15) rat DRG-rat heart fibroblasts co-cultures, also
developed polar neurofilament structures after cultivation in hanging drops for 7 days (Figure
2E and 2F). In contrast, DRG-derived cell populations cultivated on proliferation-inactivated
MEF feeder layers failed to show any type of ganglion-like assembly of sensory neurons
(Figures 2G and 2H).
Figure 1: Gravity-enforced microtissue self-assembly of mixed mouse dorsal root ganglion (DRG)-
derived cells and mouse embryonic fibroblast (MEF) populations. Monodispersed cell populations were
generated from twelve-day mouse embryos and cultivated in hanging drops. Following two-hour cultivation in
hanging drops DRG:MEF-derived cells form a homogeneous mixed population (A) exemplified by PKH26-
labeling of DRG-derived cells in red (B). Within the first 24 hours of hanging drop-based cultivation, DRG:MEF
populations form coherent microtissues by gravity-enforced self-assembly (C). (scalebar = 100 m)
A B CA B C

Artificial Ganglia 63
Figure 2: Auto-segregation of mouse-derived DRG:MEF microtissues into a polar DRG cells-derived
cap and a MEF-based feeder spheroid following prolonged cultivation in hanging drops. Concomitant with
microtissue formation 24h post seeding of mixed DRG:MEF populations into hanging drops, DRG-derived cells
segregate into a peripheral cell layer on a MEF-based core spheroid (A, B). On cultivation day 7, DRG-derived
cells form polar ganglion-like filament structures on the MEF feeder spheroid (C, D). Polar assembly of neurons
could also be observed after co-cultivation of mouse-derived DRGs with NIH/3T3 (E) and rat-derived DRGs
with rat heart fibroblasts (F) for 5 days in hanging drops. Microtissues are visualized by phase-contrast (A, C)
and immunofluorescence microscopy (B, D), showing neurofilament-specific staining of DRG-derived cells in
green and an F-actin-specific expression by the MEF spheroid in red. (G, H) Fluorescence micrographs of
monolayer cultures derived from 12 day-old mouse embryos grown on a two-dimensional proliferation-
inactivated MEF feeder layer. The MEF feeder layer is visualized by F-actin-specific staining (red) and the
neuronal network stains positive for neurofilament (green) (G). Brn-3A-positive staining specifies neurons as
sensory neurons (Brn-3A shown in red, neurofilament shown in green) (H). (scalebar = 30 m)
Development of 3D neuronal structures
To demonstrate that the ganglion-like cap structures originated from self-organization
of sensory neurons we co-visualized neuron-(Brn-3A) and neurofilament-specific cell staining
of microtissues on cultivation days 1, 3, 5 and 7. 24h post seeding of the DRG:MEF co-
culture in hanging drops, a peripheral assembly of sensory neurons was already prominent
(Figure 3A). In a three-day-old microtissue, neurofilament structures had developed and
continued to grow for four more days (Figures 3B-D). Images of bromodeoxyuridine (BrdU, a
mitotic indicator) did not reveal significant cell division, suggesting that ganglion-like
structures had assembled from sensory neurons or neuronal precursor cells present in the
DRG cell preparations (Figures 3E and 3F). Detailed analysis of ganglion-like structures by
G
H
E
F
A
B
C
D
GG
HH
EE
FF
AA
BB
CC
DD

Artificial Ganglia 64
3D projection of scanning confocal micrographs revealed the full scope of polar ganglion
assembly along with an axon-like outgrowth of sensory neurons covered by Schwann cells
staining positive for Sox10, a Schwann cell-specific transcription factor (Lobsiger et al. 2002)
(Figure 4). Axonal structures covered with Schwann cells were myelinated in 10-day-old
microtissues as shown by myelin binding protein (MBP)–specific immunofluorescence
(Figures 5A and 5B). This observation was confirmed by ultrastructural analysis (Figure 5C).
Neuron-specific growth-associated protein 43 (Gap-43) is typically expressed in
outgrowing axons during development, regeneration, axonal sprouting and synaptic plasticity.
In all of these processes, Gap-43 is a major constituent of axonal growth cones managing
axon motility and pathfinding (Wiese et al. 1992). We have detected Gap-43 in axonal
structures developed from ganglia-like cap structures (Figure 6A). During development and
regeneration, axon guidance is mediated by different cellular factors as well as by
extracellular matrix (ECM) components including laminin and fibronectin. In DRG:MEF-
derived microtissues, ECM-based stimuli are provided by the MEF feeder spheroid
expressing high levels of laminin and fibronectin (Figures 6B and 6C). Furthermore, it has
recently been established that cell type-specific segregation of co-cultured cell populations is
mediated by cadherin-subtype-specific interactions (Duguay et al. 2003). In DRG:MEF-
derived 3D co-cultures, N-cadherin is exclusively expressed in sensory neurons, suggesting a
role of this intercellular adhesion protein in the self-assembly of sensory neurons into
ganglion-like structures (Figure 6D).
Figure 3: Assembly and proliferative activity of neurofilament cap structures in DRG:MEF
microtissues. DRG:MEF microtissues cultivated for 1 (A), 3 (B), 5 (C) and 7 (D) days in hanging drops were
visualized by Brn-3A (red; A-D) and neurofilament (green; A-D) expression as well as bromodeoxyuridine
E
FD
CA
B
E
FD
CA
B

Artificial Ganglia 65
(BrdU) incorporation (red; E, F) by immunofluorescence. On cultivation day 1, sensory neurons assemble at the
periphery of a MEF-based core spheroid and produce polar ganglion-like neurofilament structures, which exhibit
low proliferative activity (G, H). (A-D scalebar = 20 m; E, F scalebar = 5 m)
Figure 4: Three-dimensional confocal micrograph projection of neurofilament structures developed in a
5 day-old DRG:MEF spheroid. (A) Ganglia-like neurofilament structures (green). (B) Ganglia-like
neurofilament structures (green) showing Brn-3A expression specific for sensory neurons (red). (C) Schwann
cells, stained for specific expression of the transcription factor Sox10 (red), accumulate alongside neurites
(green). (scalebar = 30 m)
Figure 5: Immunofluorescence micrographs of axon-covering myelin sheaths visualized by
neurofilament- (red, (A)) and myelin-binding protein- (MBP) specific co-staining (green, (B)). Transmission
electron microscopy (TEM)-based ultrastructural analysis confirmed myelination (arrow) of axonal structures
(asterisk) (C). (A, B scalebar = 10µm; C scalebar = 1 µm)
AA BB CAAAA BBBB CC
CA
B
CAA
BB

Artificial Ganglia 66
Long-term cultivation of DRG:MEF microtissue cultures
DRG:MEF-derived microtissues, cultivated for more than ten days, lost their tissue
integrity and disintegrated. Interferon- (IFN- ), approved in the past decade for the
treatment of relapsing-remitting multiple sclerosis, is a well-known factor in preserving
neuronal structures (Kappos 2004; Njenga et al. 2000). Using the aforementioned DRG:MEF-
derived microtissue design we grew wild-type DRG-derived cells on a transgenic MEF
(MEFtransgenic) feeder spheroid, engineered for inducible IFN- production using the gas-
inducible AIR (acetaldehyde-inducible regulation) technology (Weber et al. 2004). MEFs
were cotransduced with retro- and lenti-viral particles, which mediate constitutive expression
of the Aspergillus nidulans AlR transactivator (pWW506; 5’LTR-+-alcR-PPKG-neo-3’LTR;
retrovector) and enable AlcR-dependent IFN- expression from the acetaldehyde-inducible
promoter PAIR (pWW430; 5’LTR-+-PAIR-ifn- -3’LTR U3; lentivector). IFN- production by
MEFtransgenic feeder spheroids was induced for 6h by placing acetaldehyde into the lid of multi-
well hanging drop cultivation plates containing 5-day-old DRG:MEFtransgenic microtissue
cultures. Acetaldehyde, evaporating from the lid, dissolves into the hanging-drop culture
media at regulation-effective concentrations and induces PAIR-driven IFN- production
(Weber et al. 2004). Such a 6-h induction profile resulted in IFN- levels of 235 9 pg/ml in
the supernatant of the microtissue culture (Figure 7A). Following ifn- induction, all the
spheroids were harvested, washed three times in phosphate-buffered saline (PBS) to remove
the remaining acetaldehyde and cultivated for another nine days in hanging drops. Toluidine
blue-stained sections of DRG:MEFtransgenic microtissue, cultivated in the presence and absence
of acetaldehyde, demonstrated that IFN- production prevented time-dependent microtissue
disintegration. Whereas spheroids cultivated for 14 days in the absence of IFN- production
disintegrated, microtissues subjected to pulsed ifn- expression maintained a homogeneous
tissue format and the aforementioned cellular tissue organization (Figures 7B-E).
Ultrastructural analysis confirmed exclusive disintegration of IFN- -repressed feeder
spheroids highlighted by dissolution of the nulcear membrane and emergence of digestive
vacuoles filled with cell debris (Figures 7C and 7E).

Artificial Ganglia 67
Figure 6: Immunofluorescence micrographs of 7 day-old DRG:MEF spheroids stained for Gap-43 (A;
shown in green), laminin (B; shown in green), fibronectin (C, shown in green) and N-cadherin (D, shown in
green). Neurofilament is shown in red throughout (yellow in overlay with green) (A-D). (A, B, C scalebar =
20 m; D scalebar = 10 m)
Figure 7: Gas-inducible interferon- (ifn- ) expression preserves the integrity of DRG:MEF spheroids.
DRG:MEF spheroids, containing MEFs engineered for gas-inducible ifn- expression, were analyzed for IFN-
production on cultivation day 5 (A), and the integrity of microtissues exposed to IFN- (B, C) or no IFN- (D,
E) was scored by toluidine blue-based staining semi-thin sections as well as transmission electron micrographs
(TEM) on cultivation day 14. (B, D scalebar = 20 m; C, E scalebar = 1 µm)
Assembly of innervated macrotissues
While microtissues represent a powerful model system for the study of multicellular
tissue affairs, tissue engineers will require larger tissue structures for clinical applications. We
have therefore designed an agarose-based casting mould for assembly of scaffold-free
ganglia-like microtissues to higher-order macrotissues. 900 microtissues, each composed of
5’000 MEFs with 600 DRGs, were seeded into the agarose mould. After two days in the
mould culture, spheroids had assembled into a single coherent 3 mm-thick macrotissue
cylinder (Figure 8A). Hematoxylin- and eosin- (H&E) staining confirmed that microtissues
A C DBA C DB
+ acetaldehyde - acetaldehyde
IFN
-[p
g/m
l]
0
50
100
150
200
250
300
A
14d + IFN- 14d - IFN-
B D
C E
+ acetaldehyde - acetaldehyde
IFN
-[p
g/m
l]
0
50
100
150
200
250
300
A
+ acetaldehyde - acetaldehyde
IFN
-[p
g/m
l]
0
50
100
150
200
250
300
0
50
100
150
200
250
300
A
14d + IFN- 14d - IFN-
BB DD
C E

Artificial Ganglia 68
completely fused to the macrotissue although spherical substructures were still be anticipated
at some places within the macrotissues (Figure 8B-D). Cavities resulting from inter-spheroid
fusion were filled by fibroblasts (Figures 8B-D). Neurons assembled preferentially at the
periphery of macrotissues but failed to form specific poles typical of MEF:DRG microtissues
(Figures 8E, F).
Figure 8. Macrotissues produced from 900 5 day-old DRG:MEF-derived microtissues assembled in an
agarose mould for 2 days (A). Hematoxylin and eosin-based staining of frozen sections (B-D) and
immunochemical staining of neurofilament (green) and F-actin (red; E, F). (A, scalebar = 500 µm; B-F scalebar
= 20 µm)
Discussion
Although many of the key cellular and extracellular regulatory components have been
identified over the past decade, including soluble growth factors, insoluble matrix factors and
receptors on the cells themselves, the fundamental principles by which cells organize into
structured tissues remains largely elusive. Yet, only the ability to understand and control
spatial distributions of multiple cell populations will enable rational approaches to tissue
engineering applications where precise multicellular organization in three-dimensional
structures is required. Particularly important tissue engineering objectives include
vascularization and innervation of tissues (McIntire 2002; Nomi et al. 2002; Suuronen et al.
2004a; Suuronen et al. 2004b).
B
A
F
EC
DB
A
F
EC
D

Artificial Ganglia 69
A sensory nerve supply is crucial for optimal tissue function and to enhance wound
healing of its target tissue. We have designed and characterized the first tissue-engineered
three-dimensional culture system that morphologically and physiologically reproduces a
peripheral nerve regeneration process in vitro by co-culturing mouse embryonic fibroblasts
and DRG-derived cells in a scaffold-free format. Short-range cell-cell interactions are
important for controlling cellular organization and cell-fate decisions, as exemplified by
neural crest stem cells (Hagedorn et al. 2000). Since connective tissue is a key regulator of
axonal outgrowth it should be included in a three-dimensional in vitro model of the peripheral
nervous system development (Gingras et al. 2003). In order to alleviate potential interference
of scaffold material and scaffold-breakdown products with short-range cell-cell interactions,
we conceived a completely cell-based 3D co-culture model, embedding embryonic dorsal root
ganglia (DRG) cell populations in a 3D fibroblast feeder environment.
The aim of tissue engineering is not only to create desirable organs, but also to better
understand the fundamental mechanisms and principles of biological organization. Classical
tissue engineering has been based on seeding cells into biodegradable polymer scaffolds or
gels, culturing and expanding the composite cell-scaffold material in sophisticated bioreactors
and implanting the resulting tissue into the recipient organisms where maturation of the new
organ takes place (Griffith and Naughton 2002). During early embryogenesis, sequential well-
orchestrated forces rearrange cells into structures, which are imprinted by follow-up
molecular cell-fate decisions and an extensive extracellular matrix (ECM) coordinating the
interface between differentiating/differentiated cells. Gravity-enforced cell assembly of
primary embryonic cells in hanging drops is a straightforward technology for producing
organized, multi-cell type-based three-dimensional cell-culture models. Owing to their high-
throughput compatibility – both for production and analysis – microtissues derived from
mixtures of a few hundred monodispersed primary cells represent a flexible tissue culture
system for studying classical tissue engineering strategies, generating readout in drug
discovery and testing initiatives with unmatched precision and fostering unprecedented
insight into developmental principles (Lauffenburger and Griffith 2001; Zhang 2004).
As predicted by the differential adhesion hypothesis, which suggests cell type-specific
assembly based on net forces resulting from cadherin-mediated interactions between identical
cell types and cell-ECM interactions (Duguay et al. 2003; Lauffenburger and Griffith 2001;
Steinberg 1962a; Steinberg 1962b), embryonic DRG-derived sensory neurons mixed with
MEFs segregated from the fibroblasts to assemble a polar cap structure on a MEF-derived

Artificial Ganglia 70
feeder spheroid. Sensory neurons exclusively expressed N-cadherin, an intercellular adhesion
molecule shaping and maintaining the pole structure. Following polar cap assembly, the
sensory neurons formed an organized axonal outgrowth in a time-dependent manner.
Formation of axon-like structures likely resulted from re-assembly of existing sensory
neurons rather than from proliferation-based outgrowth, since BrdU-based assessment of cell-
cycle activities of sensory neurons were scored too low to account for the observed massive
organization of axonal fibers. Gap-43, the growth-associated protein known to coordinate and
guide axonal outgrowth, was strongly expressed in three-dimensional neuronal structures.
Furthermore, Schwann cells, specified by expression of the transcription factor Sox10,
showed a high in vivo-like tendency to align along nerve fibers. Thus, DRG:MEF-based
microtissues enabled key structures, which are relevant for detailed studies of peripheral nerve
system development and tissue innervation, including polar N-cadherin-based assembly of
ganglion-mimicking structures, guided Gap-43-mediated outgrowth of axon-like fibers,
sensory neuron-Schwann cell interactions and myelination of axons. Most of these
morphologic and cellular crosstalk characteristics fail to establish to a similar extent in an
isogenic two-dimensional cultivation system, suggesting that maintenance of cells in a tissue-
like format is essential for gaining further insight into aspects of cell-cell/cell-matrix
interactions relevant to tissue engineering (Cukierman et al. 2001).
Micro-scale tissue engineering – the production of microtissues – is a science at the
interface of engineering and organ replacements and exploits three-dimensional multi-cell
type cultures (i) for obtaining new insight into inter-cellular crosstalk (Hynds et al. 2004), (ii)
as building blocks for higher-order tissue design (Jakab et al. 2004) and (iii) for high-
throughput-compatible drug screening and testing (Bhadriraju and Chen 2002). Currently
available two-dimensional cell culture systems are limited and unreliable with respect to their
screening and testing readout as many drug (target) functions are sensitive to a multicellular
microenvironment (Bhadriraju and Chen 2002).
To date, only a few cell culture systems exist, all of which rely on scaffolds or
matrices to assemble sensory neurons, Schwann cells and connective tissue into a three-
dimensional format (Gingras et al. 2003; Suuronen et al. 2004b). Our strategy for producing
microtissues from monodispersed embryonic DRG populations by gravity-enforced self-
assembly represents the first scaffold-free approach to generating ganglia-like structures,
compatible with high-throughput assays and designed to discover/study factors triggering
cell-fate/morphologic control of sensory neurons and Schwann cells. These cell types are

Artificial Ganglia 71
currently in the limelight because of their association with different untreatable
neurodegenerative diseases (England and Asbury 2004).
As shown by a variety of recent examples, scaffold-free micro-scale tissue engineering
will expand our knowledge of developmental phenomena and will almost certainly impact
future drug testing and discovery initiatives.
Acknowledgements
We thank Maurice Kleber and Lukas Sommer for their generous advice and supply of
primary cells as well as Lucilla Nobbio and David Fluri for critical comments on the
manuscript. This work was supported by the Swiss National Science Foundation (grant no.
631-065946), the Swiss State Secretariat for Education and Research within EC Framework 6
and Cistronics Cell Technology GmbH, Einsteinstrasse 1-5, CH-8093 Zurich, Switzerland.
References
Abbott A., (2003). "Cell culture: biology's new dimension." Nature 424, 870-872.
Armstrong P.B., (1989). Cell sorting out: the self-assembly of tissues in vitro. Crit. Rev.
Biochem. Mol. Biol. 24, 119-149.
Beysens D.A., Forgacs G., Glazier J.A., (2000). Cell sorting is analogous to phase ordering in
fluids. Proc. Natl. Acad. Sci. U.S.A 97, 9467-9471.
Bhadriraju K., Chen C.S., (2002). Engineering cellular microenvironments to improve cell-
based drug testing. Drug Discov. Today 7, 612-620.
Burns J.C., Friedmann T., Driever W., Burrascano M., Yee J.K., (1993). Vesicular stomatitis
virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer
and efficient gene transfer into mammalian and nonmammalian cells. Proc. Natl.
Acad. Sci. U.S.A 90, 8033-8037.
Chen S., Rio C., Ji R.R., Dikkes P., Coggeshall R.E., Woolf C.J., Corfas G., (2003).
Disruption of ErbB receptor signaling in adult non-myelinating Schwann cells causes
progressive sensory loss. Nat. Neurosci. 6, 1186-1193.
Chotard C., Salecker I., (2004). Neurons and glia: team players in axon guidance. Trends
Neurosci. 27, 655-661.
Cukierman E., Pankov R., Stevens D.R., Yamada K.M., (2001). Taking cell-matrix adhesions
to the third dimension. Science 294, 1708-1712.

Artificial Ganglia 72
Duguay D., Foty R.A., Steinberg M.S., (2003). Cadherin-mediated cell adhesion and tissue
segregation: qualitative and quantitative determinants. Dev. Biol. 253, 309-323.
Edlund T., Jessell T.M., (1999). Progression from extrinsic to intrinsic signaling in cell fate
specification: a view from the nervous system. Cell 96, 211-224.
England J.D., Asbury A.K., 2004. Peripheral neuropathy. Lancet 364, 1664-1665.
Gingras M., Bergeron J., Dery J., Durham H.D., Berthod F., (2003). In vitro development of a
tissue-engineered model of peripheral nerve regeneration to study neurite growth.
Faseb J. 17:2124-2126.
Gordon-Weeks P.R., (2004). Microtubules and growth cone function. J. Neurobiol. 58, 70-83.
Griffith L.G., Naughton G., (2002). Tissue engineering--current challenges and expanding
opportunities. Science 295, 1009-1014.
Griffiths R.L., Hidalgo A., (2004). Prospero maintains the mitotic potential of glial precursors
enabling them to respond to neurons. Embo J. 23, 2440-2450.
Hagedorn L., Floris J., Suter U., Sommer L., (2000). Autonomic neurogenesis and apoptosis
are alternative fates of progenitor cell communities induced by TGFbeta. Dev Biol
228, 57-72.
Hynds D.L., Rangappa N., Ter Beest J., Snow D.M., Rabchevsky A.G., (2004). Microglia
enhance dorsal root ganglion outgrowth in Schwann cell cultures. Glia 46, 218-223.
Jakab K., Neagu A., Mironov V., Markwald R.R., Forgacs G., (2004). Engineering biological
structures of prescribed shape using self-assembling multicellular systems. Proc. Natl.
Acad. Sci. U.S.A 101, 2864-2869.
Kappos L., (2004). New aspects in the treatment of multiple sclerosis with interferon beta-1b.
J. Neurol. 251 Suppl 4, IV1.
Kelm J.M., Fussenegger M., (2004). Microscale tissue engineering using gravity-enforced cell
assembly. Trends Biotechnol. 22, 195-202.
Lauffenburger D.A., Griffith L.G., (2001). Who's got pull around here? Cell organization in
development and tissue engineering. Proc. Natl. Acad. Sci. U.S.A 98, 4282-4284.
Layer P.G., Robitzki A., Rothermel A., Willbold E., (2002). Of layers and spheres: the
reaggregate approach in tissue engineering. Trends Neurosci. 25, 131-134.
Lemke G., (2001). Glial control of neuronal development. Annu Rev Neurosci 24, 87-105.
Lobsiger C.S., Taylor V., Suter U., (2002). The early life of a Schwann cell. Biol. Chem. 383,
245-253.
McIntire L.V., (2002). Vascular assembly in engineered and natural tissues. Ann. N. Y. Acad.
Sci. 961, 246-248.

Artificial Ganglia 73
Messerli J.M., van der Voort H.T., Rungger-Brandle E., Perriard J.C., (1993). Three-
dimensional visualization of multi-channel volume data: the amSFP algorithm.
Cytometry 14, 725-735.
Mitta B., Rimann M., Ehrengruber M.U., Ehrbar M., Djonov V., Kelm J., Fussenegger M.,
(2002). Advanced modular self-inactivating lentiviral expression vectors for multigene
interventions in mammalian cells and in vivo transduction. Nucleic Acids Res. 30,
e113.
Njenga M.K., Coenen M.J., DeCuir N., Yeh H.Y., Rodriguez M., (2000). Short-term
treatment with interferon-alpha/beta promotes remyelination, whereas long-term
treatment aggravates demyelination in a murine model of multiple sclerosis. J.
Neurosci. Res. 59, 661-670.
Nomi M., Atala A., Coppi P.D., Soker S., (2002). Principals of neovascularization for tissue
engineering. Mol. Aspects Med. 23, 463.
Pittier R., Sauthier F., Hubbell J.A., Hall H., (2004). Neurite extension and in vitro
myelination within three-dimensional modified fibrin matrices. J. Neurobiol. 63, 1-14.
Sheehan D.C., Hrapchak B.B., 1980. Theory and practice of histotechnology. Battelle Press,
Ohio.
Steinberg M.S., (1962a). Mechanism of tissue reconstruction by dissociated cells. II. Time-
course of events. Science 137, 762-763.
Steinberg M.S., (1962b). On the mechanism of tissue reconstruction by dissociated cells. I.
Population kinetics, differential adhesiveness. and the absence of directed migration.
Proc. Natl. Acad. Sci. U.S.A 48, 1577-1582.
Steinberg M.S., (1963). Reconstruction of tissues by dissociated cells. Some morphogenetic
tissue movements and the sorting out of embryonic cells may have a common
explanation. Science 141, 401-408.
Steinberg M.S., Foty R.A., (1997). Intercellular adhesions as determinants of tissue assembly
and malignant invasion. J. Cell Physiol. 173, 135-139.
Stewart C.L., Schuetze S., Vanek M., Wagner E.F., (1987). Expression of retroviral vectors in
transgenic mice obtained by embryo infection. Embo J. 6, 383-388.
Suuronen E.J., McLaughlin C.R., Stys P.K., Nakamura M., Munger R., Griffith M., (2004a).
Functional Innervation in Tissue Engineered Models for In Vitro Study and Testing
Purposes. Toxicol. Sci. 82, 525-533.

Artificial Ganglia 74
Suuronen E.J., Nakamura M., Watsky M.A., Stys P.K., Muller L.J., Munger R., Shinozaki N.,
Griffith M., (2004b). Innervated human corneal equivalents as in vitro models for
nerve-target cell interactions. Faseb J. 18, 170-172.
Svenningsen A.F., Shan W.S., Colman D.R., Pedraza L., (2003). Rapid method for culturing
embryonic neuron-glial cell cocultures. J. Neurosci. Res. 72, 565-573.
Tepass U., Truong K., Godt D., Ikura M., Peifer M., (2000). Cadherins in embryonic and
neural morphogenesis. Nat. Rev. Mol. Cell Biol. 1, 91-100.
Wang F., Dumstrei K., Haag T., Hartenstein V., (2004). The role of DE-cadherin during
cellularization, germ layer formation and early neurogenesis in the Drosophila
embryo. Dev. Biol. 270, 350-363.
Weber W., Rimann M., Spielmann M., Keller B., Baba M.D., Aubel D., Weber C.C.,
Fussenegger M., (2004). Gas-inducible transgene expression in mammalian cells and
mice. Nat. Biotechnol. 22, 1440-1444.
Wiese U.H., Ruth J.L., Emson P.C., (1992). Differential expression of growth-associated
protein (GAP-43) mRNA in rat primary sensory neurons after peripheral nerve lesion:
a non-radioactive in situ hybridisation study. Brain Res. 592, 141-156.
Zahir N., Weaver V.M., (2004). Death in the third dimension: apoptosis regulation and tissue
architecture. Curr. Opin. Genet. Dev. 14, 71-80.
Zhang S., (2004). Beyond the Petri dish. Nat. Biotechnol. 22, 151-152.

Chapter 5
VEGF Profiling and Angiogenesis in Human
Microtissues
Kelm J.M., Diaz Sanchez-Bustamante C., Ehler E., Hoerstrup S.P., Djonov V., Ittner L. and
Fussenegger M., (2005) J. Biotech., in press

Microtissue Vascularization 76
Abstract
Owing to its dual impact on tissue engineering (neovascularization of tissue implants)
and cancer treatment (prevention of tumor-induced vascularization), management and
elucidation of vascularization phenomena remain clinical priorities. Using a variety of
primary human cells and (neoplastic) cell lines assembled in microtissues by gravity-enforced
self-aggregation in hanging drops we (i) studied size and age-dependent VEGF production of
microtissues in comparison to isogenic monolayer cultures, (ii) characterized the self-
organization and VEGF-production potential of mixed-cell spheroids, (iii) analyzed VEGF-
dependent capillary formation of HUVEC (human umbilical vein endothelial cells) cells
coated onto several human primary cell spheroids and (iv) profiled endostatin action on
vascularization in human microtissues. Precise understanding of vascularization in human
microtissues may foster advances in clinical tissue implant engineering, tumor treatment, as
well as drug discovery and drug-function analysis.
Introduction
Transplantation remains the preferred clinical intervention for the treatment of organ
failures. Owing to a global shortage in donor organs alternative strategies providing
bioengineered tissue for replacement of damaged, injured or missing tissues are of high
clinical priority (Griffith and Naughton 2002). An artificial tissue with more than a few cubic
millimeters cannot survive by simple diffusion and requires formation of new blood
capillaries to supply essential nutrients/oxygen and enable connection to the host vascular
system following implantation (Griffith and Naughton 2002; Mooney and Mikos 1999).
Likewise, tumors do not grow beyond a few millimeters unless they become vascularized by
directing an ingrowth of capillaries from adjacent blood vessels (Folkman and Kaipainen
2004). Therefore, precise control of pro- and anti-angiogenic activities is of prime clinical
interest in current cancer therapy and tissue engineering initiatives (Bergers and Benjamin
2003; Nomi et al. 2002). The maturation of nascent vasculature, formed by vasculogenesis or
angiogenesis, requires recruitment of mural cells, generation of an extracellular matrix and
specialization of the vessel wall for structural support and regulation of vessel function. In
addition, the vascular network must be organized to provide parenchymal cells with sufficient
nutrients. All of these processes are orchestrated by physical forces, as well as a panoply of

Microtissue Vascularization 77
ligands and receptors whose spatio-temporal expression patterns are tightly regulated (Jain
2003; Yancopoulos et al. 2000).
Angiogenesis is a morphogenic process of new blood capillaries emerging from
preexisting vessels which consists of six major steps including (i) vasodilatation of the
parental vessel, reducing endothelial cell contact, (ii) degradation of the basement membrane
by a variety of proteolytic enzymes, (iii) migration and proliferation of endothelial cells at the
spearhead of new vessels, (iv) production of the capillary lumen and formation of tube-like
structures, (v) basement membrane synthesis and (vi) recruitment of vascular smooth muscle
cells. The sequence of molecular events resulting in angiogenesis requires precise fine-tuning
of multiple signaling pathways, cell-cell and cell-matrix interactions (Jain 2003).
Vascular endothelial growth factor (VEGF), a major regulator of neovascularization
under physiological and pathological conditions (Gerber et al. 1999), is produced in five
homodimeric isoforms (VEGF121, VEGF145, VEGF165, VEGF189, VEGF206), which differ in
their expression levels and their localization. VEGF family members are involved in (i) the
formation of immature vasculature (VEGF-VEGF receptor 2-mediated signaling in
angioblasts results in formation of the dorsal aorta and the cardinal vein) (Yancopoulos et al.
2000); (ii) induction of migration and proliferation of endothelial cells (Conway et al. 2001);
(iii) vessel dilatation and sprouting in the presence of angiopoietin-2 (Tsigkos et al. 2003),
(iv) stabilization of immature vasculature (VEGF-induced platelet-derived growth factor
secretion of endothelial cells facilitates recruitment of mural cells) (Blau and Banfi 2001); (v)
sequestration of angiopoietin 2 which destabilizes vessels (Tsigkos et al. 2003); (vi)
suppression of apoptosis (Folkman 2003), (vii) branching, remodeling and pruning of
vasculature (protease-mediated release of matrix-sequestrated VEGF) (Peirce and Skalak
2003) and (viii) vessel specialization (arterial growth promoted by VEGF-VEGF receptor 2-
neuropilin 1 signaling). Recent studies indicate that VEGF action goes beyond vascularization
and may be involved in neurogenesis (Jin et al. 2002), as well as growth- and survival-
modulation of chondrocytes (Schipani et al. 2001).
The biological effects of VEGF are extremely dose dependent. Loss of even a single
allele results in fatal vascular defects in the embryo (Ferrara and Alitalo 1999) and
insufficient levels of VEGF lead to post-natal angiogenesis and ischemic heart disease
(Carmeliet et al. 1999). Several control circuits are at work to balance VEGF action. Hypoxia-
mediated induction of hypoxia-inducible factor (HIF-)1 resulting in VEGF production, and
endostatin, a matrix-associated protease-mediated cleavage product of collagen XVIII,
inhibiting VEGF-induced mobilization of endothelial cells are some examples (Schuch et al.

Microtissue Vascularization 78
2003; Semenza 2003). A variety of strategies for therapeutic angiogenesis have been designed
including (i) delivery of recombinant angiogenic molecules through controlled-release
devices (Jain and Carmeliet 2001) and (ii) functionalized matrices (Richardson et al. 2001;
Zisch et al. 2003) or (iii) transfection/transduction of (engineered) angiogenesis-modulating
cDNAs (Elson et al. 2001; Isner 2002).
Although most of today’s knowledge on vascularization regulatory networks has been
derived from in vitro monolayer cultures (Vailhe et al. 2001), three-dimensional (3D)
cultivation technologies may reveal further insight. Current 3D models include formation of
primitive vascular networks in vitro by coculturing endothelial cells with mural cells or their
precursors (Hirschi et al. 1999; Korff et al. 2001). However, several groups have suggested
that blood vessels of artificially pre-vascularized cells/tissues remain essentially self-
contained and do not become connected with the surrounding vasculature (Lee et al. 2000;
Springer et al. 1998).
Based on our previous observations that VEGF production in myocardial microtissue
is strictly correlated to cell number and microtissue size (Kelm et al. 2004a; Kelm et al.
2004b) we have established an entirely human cell-based microtissue format to provide new
insight into VEGF production, angiogenesis and blood vessel formation.
Material and Methods
Isolation of primary human aortic fibroblasts
In order to obtain primary human aortic fibroblasts (HAF), de-endothelialized vessel
segments of the human aorta were minced and cultivated in a 37°C humidified 5% CO2-
containing atmosphere and Dulbecco's modified Eagle's medium (DMEM, Invitrogen,
Carlsbad, CA, USA) supplemented with 10% fetal calf serum (FCS; cat. no. A-15-022, lot no.
A01129-242; PAA Laboratories, Linz, Austria) and 1% penicillin/streptomycin solution
(Invitrogen). Pure HAFs which had migrated out of the tissue pieces after 10 to 14 days were
serially passaged and expanded for 4 to 6 weeks under aforementioned conditions to desired
cell numbers.
Cell culture
Human primary aortic fibroblasts (HAF), the hepatocellular carcinoma cell line
(HepG2, DSMZ: ACC 180), newborn human foreskin fibroblasts (Hs68, ATCC: CRL-1635)

Microtissue Vascularization 79
and human fibrosarcoma cells (HT-1080, ATCC: CCL-121) were expanded as monolayer
cultures in DMEM supplemented with 10% FCS. Human umbilical vein endothelial cells
(HUVEC, No. C-12200; Lot 0111701), normal human dermal fibroblasts (NHDF, No. C-
12300; Lot 1070402) and human umbilical artery smooth muscle cells (HUASMC, No. 252-
05; Lot 1222) were obtained from PromoCell (Heidelberg, Germany). HUVECs (PromoCell,
No. C-22110), HUASMCs (PromoCell, No. C-22162) and NHDFs (PromoCell, No. C-23010)
were expanded in monolayer cultures using PromoCell’s endothelial cell media supplemented
with 10% FCS (HUVECs and HUASMCs only). Human articular chondrocytes (HAC, kindly
provided by Millenium Biologix) were cultured in DMEM/F12 (Invitrogen) supplemented
with 10% FCS. All cell types were cultivated at 37°C in a humidified 5% CO2-containing
atmosphere.
Microtissue production
Monolayer cultures of desired cell types were trypsinized and single-cell suspensions
seeded at indicated cell densities into 60-well plates (HLA plate, Greiner-Bio One,
Frickenhausen, Germany). In order to enable gravity-enforced microtissue formation in
hanging drops, the 60-well plates were incubated upside down. Pure, multicellular and coated
microtissues were produced/maintained in DMEM medium (Invitrogen) supplemented with
10% FCS and 1% penicillin/streptomycin solution. Following cultivation for 2 to 8 days in
hanging drops the microtissues were harvested for further analysis. Onion skin-like
multicellular microtissues were produced in two steps: (i) production of the core feeder
spheroid by 2-day cultivation in hanging drops followed by (ii) cocultivation of feeder
spheroids and monodispersed coating cells (for example HUVECs) in hanging drops.
Fluorescence-based characterization of cell morphologies
Microtissues were harvested, washed twice in phosphate-buffered saline (PBS;
150 mM NaCl, 6.5 mM Na2HPO4x2H2O, 2.7 mM KCL, 1.5 mM KH2PO4, pH 7.4; Sigma
Chemicals, St. Louis, MO), fixed for 1h in PBS containing 4% paraformaldehyde and
subsequently washed three times for 5 min in phosphate-buffered Triton X-100 (PBT, PBS
containing 0.002% Triton X-100; Sigma). The microtissues were then permeabilized for
60 min in PBS containing 0.5% Triton X-100. Primary antibodies specific for desired proteins
as well as fluorescence-labeled secondary antibodies were diluted in 1% BSA-containing
Tris-buffered saline (TBS, 20 mM Tris base, 155 mM NaCl, 2 mM EGTA, 2 mM MgCl2) and

Microtissue Vascularization 80
sequentially incubated with microtissues for 12 hours at 4 C with three PBS washings in
between. Finally, the microtissues were washed in PBS and embedded on glass slides using
Tris-buffered glycerol (a 3:7 mixture of 0.1 M Tris-HCl (pH 9.5) and glycerol supplemented
with 50 mg/ml n-propyl-gallat). Tailored 0.5 mm silicon spacers were used to prevent
crunching of the microtissues between slide and cover slip. Immunofluorescence-based
analysis of microtissues required antibodies specific for human von Willebrand factor (F3520;
Sigma), platelet-endothelial cell adhesion molecule-1 (PECAM-1; P8590, Clone WM-59;
Sigma), alpha-smooth muscle actin (A2547, clone 1A4; SIGMA), V 3 integrin (ab7167,
Abcam, Camebridge, UK) and/or vascular endothelial growth factor (VEGF) isoforms, 121
and 165 and 189 (sc-152; Santa Cruz Biotechnology Inc., Santa Cruz, CA) all of which were
visualized using Cy3-coupled anti mouse (Jackson Immunochemicals, West Grove, PA; cat.
no. 115-165-146) and FITC-coupled anti-rabbit secondary antibodies (ICN Pharmaceuticals,
Hyland, CA). F-actin was stained using A633-coupled phalloidin (Molecular Probes Inc.,
Eugene, OR).
Toluidine blue staining and immunohistochemistry of paraffin-embedded
microtissue sections
Microtissues were harvested, washed once in PBS (Sigma) and fixed for 2h at 4°C in
PBS containing 4% paraformaldehyde (Sigma). Following stepwise dehydration in ethanol,
microtissues were embedded in paraffin (Fisher Scientific, Wohlen, Switzerland). Toluidine
blue (Fluka Chemie AG, Buchs, Switzerland) staininigs of 5µm microtissue sections was
performed as described before (Sheehan and Hrapchak 1980). Immunohistochemical staining
included incubation of rehydrated 5µm microtissue sections with von Willebrand factor-
specific antibodies (F3520; Sigma) for 30 minutes at room temperature. Microtissue sections
were subsequently visualized for von Willebrand factor production using the Vectastain ABC
method (Vector Laboratories, Burlingame, CA) and the Metal Enhanced DAB Substrate
(Pierce Biotechnology, Rockford, IL). For antigen unmasking, microtissue sections were
heated for 5 minutes in 10 mM sodium citrate buffer (pH 5.8). Endogenous peroxidase
activity was blocked with 3% hydrogen peroxide prior to incubation with primary antibodies.
Confocal light microscopy
The imaging system consisted of an inverted fluorescence microscope (Leica
DMIRB/E, Glattbrugg, Switzerland) equipped with a Leica 20x/10x oil immersion objective,

Microtissue Vascularization 81
a confocal scanner (Leica TCS SP1) featuring an argon and helium-neon laser and a Silicon
Graphics Workstation (SGI, Schlieren, Switzerland) with Imaris 3D multi-channel image
processing software installed (Bitplane, AG, Zurich, Switzerland (Messerli et al. 1993)).
Transmission Electron Microscopy
For electron microscopic studies spheroids were fixed by total immersion in 0.1M
cacodylate buffer (pH 7.4, 350 mOsm) containing 2.5% glutaraldehyde. Tissue blocks were
postfixed in osmium tetroxide, block-stained using uranyl acetate, dehydrated by increasing
ethanol concentrations and embedded in Epon 812 according to Djonov et al. (Djonov et al.
2000) (all chemicals from Merck Eurolab AG, Dietikon, Switzerland). Semithin 1 µm
sections were stained with toluidine blue and visualized using an Olympus Vanox BHS light
microscope (Olympus AG, Volketswil, Switzerland). Ultrathin sections of 80-90 nm
thickness were cut, picked up on Formvar-coated (polyvinyl formal; Fluka Chemie AG)
copper grids, double-stained with lead citrate (Merck Eurolab AG) and uranyl acetate, and
monitored on a Philips EM 400 electron microscope (FEI AG, Zurich, Switzerland).
ELISA-based VEGF quantification
VEGF production was quantified in the culture supernatants of confluent monolayers
and/or microtissue cultures using the DuoSET®
enzyme-linked immunosorbent assay
(ELISA) by R&D systems (Minneapolis, MN) according to the manufacturer’s instructions.
Results
VEGF production profiling of human cell-derived monolayer and
microtissue cultures
ELISA-based technology was used to quantify vascular endothelial growth factor
(VEGF) production by human cell lines and primary cells grown as monolayers or assembled
as microtissues. For unbiased VEGF profiling monolayer cultures were grown to confluence
and growth factor production compared to microtissues over a period of 24h. Whereas the
adult-derived human aortic fibroblasts (HAF adult; 0.21 ng/h*cell ± 0.025), the neoplastic cell
line HT-1080 (0.43 ng/h*cell ± 0.032) and the primary human articular chondrocytes (HAC;
1.21 ng/h*cell ± 0.055) produced considerable VEGF amounts, growth factor levels of

Microtissue Vascularization 82
primary human dermal fibroblasts (NHDF), primary children-derived human aortic fibroblasts
(HAF child) and the human newborn foreskin fibroblast cell line Hs68 were undetectable
(Figure 1A). Scaffold-free microtissues derived from the same cell types were generated
using gravity-enforced self-assembly of monodispersed cells. Based on previous observations
suggesting cell type-specific cell number – microtissue size correlations we have used tailored
cell concentrations to obtain microtissues of 350 µm in diameter following a 3-day cultivation
period (Kelm et al. 2003). Whereas Hs68 spheroids failed to produce detectable VEGF levels,
NHDF- (0.09 ng/h*cell ± 0.028), HAF child- (0.27 ng/h*cell ± 0.028), HAF adult-
(0.92 ng/h*cell ± 0.026) and HT-1080- (1.55 ng/h*cell ± 0.231) derived microtissues showed
increased growth factor production compared to isogenic monolayer cultures. Only HAC
spheroids’ specific VEGF production (0.09 ng/h*cell ± 0.011) was inferior to corresponding
monolayer cultures (Figure 1B). For detailed analysis of microtissue size - VEGF production
profiles, gravity-enforced cell assembly of selected cell types was initiated using 500, 2’500,
5’000 and 10’000 cells per spheroid. Plotting of microtissue size vs. VEGF production
revealed cell type-specific correlations (Figure 2).
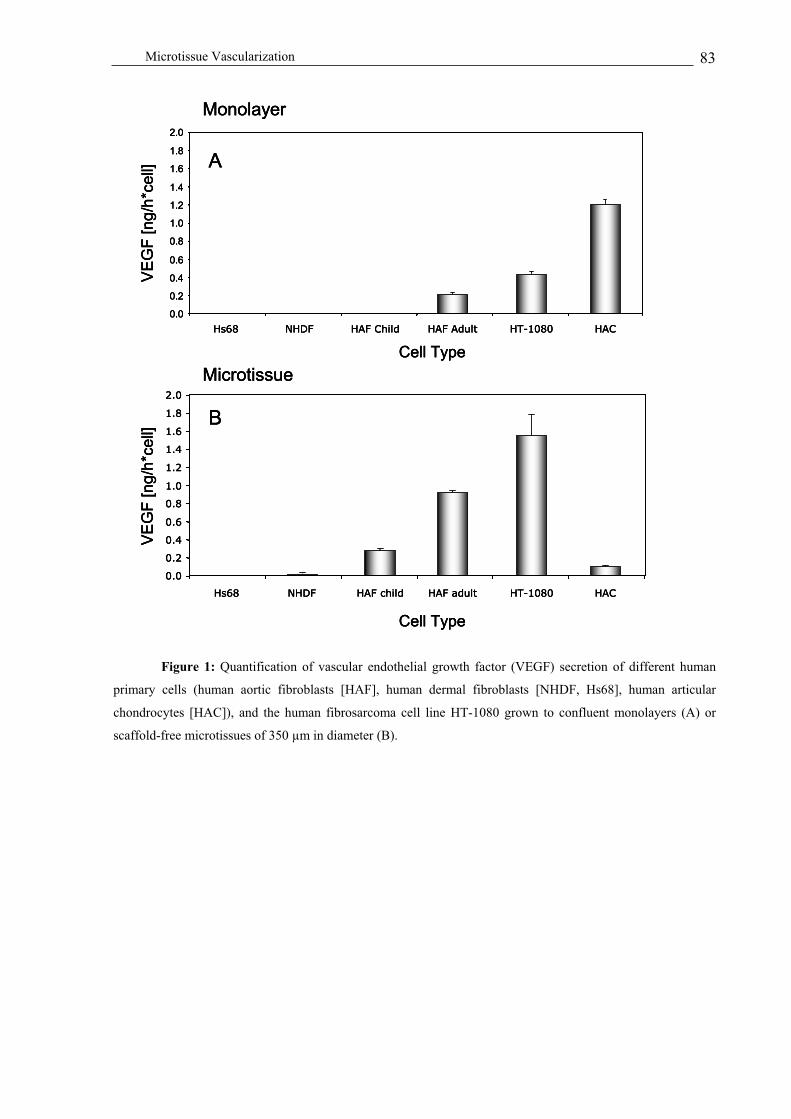
Microtissue Vascularization 83
Figure 1: Quantification of vascular endothelial growth factor (VEGF) secretion of different human
primary cells (human aortic fibroblasts [HAF], human dermal fibroblasts [NHDF, Hs68], human articular
chondrocytes [HAC]), and the human fibrosarcoma cell line HT-1080 grown to confluent monolayers (A) or
scaffold-free microtissues of 350 µm in diameter (B).
Cell Type
VE
GF
[n
g/h
*ce
ll]
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
2.0
NHDF HAF ChildHs68 HAF Adult HT-1080 HAC
Monolayer
A
Hs68 NHDF HACHAF child HAF adult HT-1080
Cell Type
VE
GF
[n
g/h
*ce
ll]
Microtissue
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
2.0
B
Cell Type
VE
GF
[n
g/h
*ce
ll]
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
2.0
NHDF HAF ChildHs68 HAF Adult HT-1080 HAC
Monolayer
A
Cell Type
VE
GF
[n
g/h
*ce
ll]
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
2.0
NHDF HAF ChildHs68 HAF Adult HT-1080 HAC
Monolayer
A
Hs68 NHDF HACHAF child HAF adult HT-1080
Cell Type
VE
GF
[n
g/h
*ce
ll]
Microtissue
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
2.0
B
Hs68 NHDF HACHAF child HAF adult HT-1080
Cell Type
VE
GF
[n
g/h
*ce
ll]
Microtissue
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
2.0
B

Microtissue Vascularization 84
Spheroid Diameter [µm]
VE
GF
[pg
/ml*
h]
0
100
200
300
400
0 200 400 600 800 1000
HT-1080
HAF Adult
HAF Child
HepG2
Spheroid Diameter [µm]
VE
GF
[pg
/ml*
h]
0
100
200
300
400
0 200 400 600 800 1000
HT-1080
HAF Adult
HAF Child
HepG2
HT-1080
HAF Adult
HAF Child
HepG2
HAF Adult
HAF Child
HepG2
Figure 2: Microtissue size – VEGF production profiles of primary human aortic fibroblast- (HAF, adult
and child), human fibrosarcoma cell- (HT-1080) and human hepatocellular carcinoma cell- (HepG2) derived
microtissues produced by 3-day gravity-enforced assembly in hanging drops.
Self-organization potential of different cell phenotypes in a microtissue
format
Blood vessels exhibit a particular architecture, with endothelial cells lining the blood
vessels and forming a cellular interface between the bloodstream and the surrounding tissue.
In order to investigate self-organization forces underlying cell movement during angiogenesis
we aggregated HAF:HUVEC (10’000:1’000 cells) and HepG2:HUVEC (1’200:600)
suspension cocultures to mixed microtissues in hanging drops. After 7 days, gravity-enforced
assembly resulted in HAF:HUVEC- and HepG2:HUVEC-derived microtissues of 800 µm and
350 µm in diameter, respectively. Most importantly, intra-microtissue forces separated
HAF/HepG2:HUVEC populations which resulted in concentric HAF/HepG2-inside:HUVEC-
outside structures reminiscent of blood vessel cross-sections (Figure 3A and B). By contrast,
human umbilical artery smooth muscle cell (HUASMC):HUVEC-derived microtissues
(600:600) were characterized by an inner HUVEC core enveloped by a HUASMC layer
(Figure 3C).
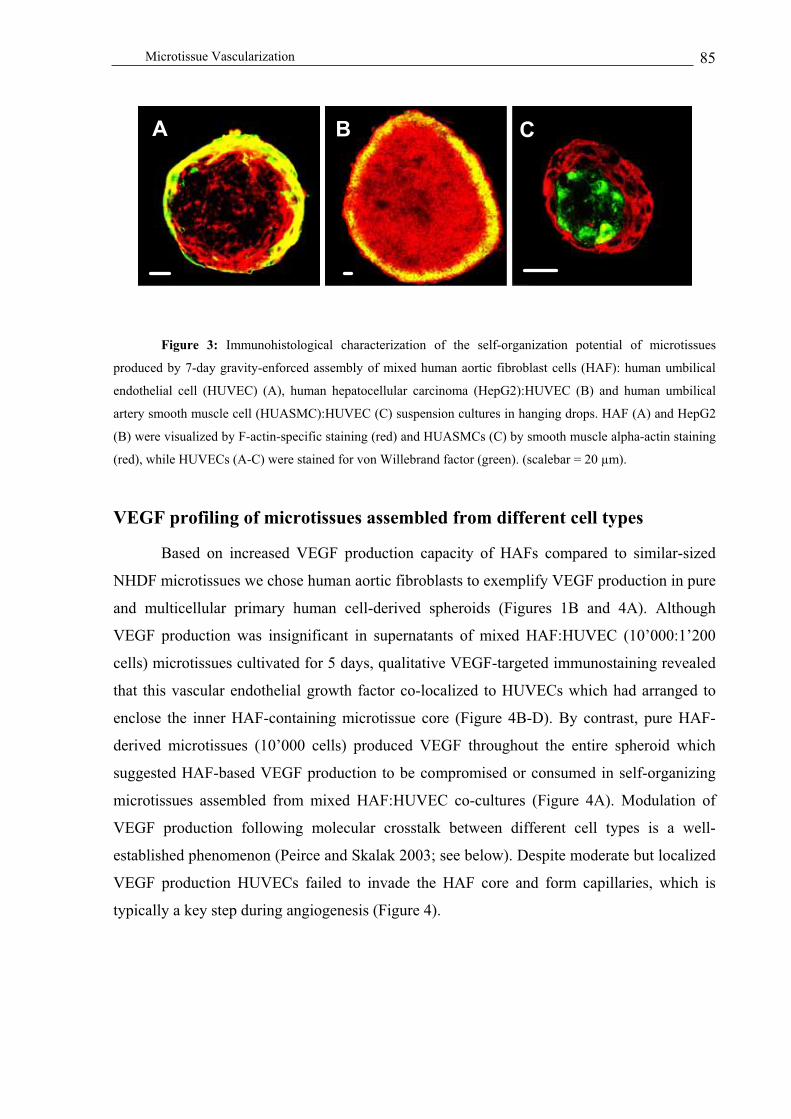
Microtissue Vascularization 85
Figure 3: Immunohistological characterization of the self-organization potential of microtissues
produced by 7-day gravity-enforced assembly of mixed human aortic fibroblast cells (HAF): human umbilical
endothelial cell (HUVEC) (A), human hepatocellular carcinoma (HepG2):HUVEC (B) and human umbilical
artery smooth muscle cell (HUASMC):HUVEC (C) suspension cultures in hanging drops. HAF (A) and HepG2
(B) were visualized by F-actin-specific staining (red) and HUASMCs (C) by smooth muscle alpha-actin staining
(red), while HUVECs (A-C) were stained for von Willebrand factor (green). (scalebar = 20 µm).
VEGF profiling of microtissues assembled from different cell types
Based on increased VEGF production capacity of HAFs compared to similar-sized
NHDF microtissues we chose human aortic fibroblasts to exemplify VEGF production in pure
and multicellular primary human cell-derived spheroids (Figures 1B and 4A). Although
VEGF production was insignificant in supernatants of mixed HAF:HUVEC (10’000:1’200
cells) microtissues cultivated for 5 days, qualitative VEGF-targeted immunostaining revealed
that this vascular endothelial growth factor co-localized to HUVECs which had arranged to
enclose the inner HAF-containing microtissue core (Figure 4B-D). By contrast, pure HAF-
derived microtissues (10’000 cells) produced VEGF throughout the entire spheroid which
suggested HAF-based VEGF production to be compromised or consumed in self-organizing
microtissues assembled from mixed HAF:HUVEC co-cultures (Figure 4A). Modulation of
VEGF production following molecular crosstalk between different cell types is a well-
established phenomenon (Peirce and Skalak 2003; see below). Despite moderate but localized
VEGF production HUVECs failed to invade the HAF core and form capillaries, which is
typically a key step during angiogenesis (Figure 4).
A B CAA BB CC

Microtissue Vascularization 86
Figure 4: Qualitative VEGF expression analysis of 5-days old pure human aortic fibroblast (HAF)-
derived microtissues and microtissues assembled from mixed HAF: human umbilical vein endothelial cell
(HUVEC) cultures. (A) VEGF-specific immunohistological staining (green) of pure HAF microtissues. (B-D)
HAF:HUVEC microtissues were stained for F-actin (blue, entire spheroid) (B), HUVECs were visualized by
Willebrand factor expression (red) and VEGF was monitored in green (C). (scalebar = 30 µm).
Angiogenesis-based capillary formation in microtissues
Initial experiments designed to characterize angiogenesis in hepatic microtissues were
based on HUVEC-coated HepG2 spheroids (HepG2 core – HUVEC shell; HepG2-HUVEC)
produced by cocultivating HepG2 spheroids (initiated with 500 cells), preassembled for 2
days in hanging drops (170 23 µm), with monodispersed HUVECs (900 cells). Since
HepG2 remain proliferation-competent in a microtissue format (Kelm et al. 2003), HepG2-
HUVEC and HepG2-only microtissues reached similar diameters of 400 50 µm after 7 days
post coating (Figure 5). Following invasion of the HepG2 core spheroids, HUVECs formed
multicell-based microvessels characterized by increased V 3 integrin expression typical for
A B
DC
AA BB
DDCC

Microtissue Vascularization 87
neovascular endothelial cells (Brooks et al. 1994; Koh et al. 2004). By contrast, microvessel
formation could not be observed in HepG2-only control spheroids (Figure 5).
Figure 5: Capillary formation in hepatic microtissues. Coated HepG2-HUVEC (A-D) and pure
HepG2 (E, F) spheroids grown for 7 days in hanging drops. Toluidine blue-stained sections of HepG2-HUVEC
(A) and HepG2-only (E) microtissues. Immunohistologic von Willebrand factor-specific staining of endothelial
cells (dark brown) in HepG2-HUVEC (B, C) microtissues reveals HUVEC-based vessel formation (C, arrow;
see also Figure 9). Immunocytologic staining of entire cells for A633-coupled phalloidin (red) and V 3 integrin
(green) shows capillaries with peripheral V 3 integrin expression (arrows) in HepG2-HUVEC (D) but not in
HepG2-only microtissues. (scalebar = 20µm).
Instead of generating HAF:HUVEC microtissues by gravity-enforced assembly of
mixed HAF and HUVEC cultures (see above and Figure 4), we coated pure HAF microtissues
of different size (125 µm – 335 µm; produced by 2-day gravity-enforced assembly) by
cocultivation with 600 – 1’200 monodispersed HUVECs in hanging drops (Figure 6). Coated
HAF-HUVEC microtissues (HAF core - HUVEC shell) of different size were cultivated for 6
days prior to analysis of VEGF-induced HUVEC migration into the inner HAF core (Figure
6A-E). Whereas HUVECs failed to penetrate HAF core microtissues of 125 µm in diameter
(500 fibroblasts) (Figure 6A), these endothelial cells infiltrated HAF microtissues assembled
from 2’500 (225 µm in diameter) and 5’000 (285 µm in diameter) cells likely following the
B
D F
A
E
CBB
DD FF
AA
EE
CC

Microtissue Vascularization 88
HAF-generated hypoxia-induced VEGF gradient (Figure 6B, C). In oversized HAF-HUVEC
microtissues (10’000 cells, 335 µm in diameter) HUVECs assembled into extensive capillary
structures inside the HAF core (Figure 6D). Interestingly, HUVEC-coated NHDF
microtissues (233 µm 12 µm) were able to induce HUVEC invasion and capillary formation
despite NHDF’s low VEGF production (Figure 6F).
Figure 6: Size-dependent induction of angiogenesis-based capillary formation in multicellular
microtissues. Human aortic fibroblast (HAF)-derived spheroids of different size/cell number ([A], 125/500; [B],
225/2’500; [C], 285/5’000; [D], 335/10’000, diameter [µm]/cells) were assembled as microtissues for 2 days,
coated with 300 (A), 600 (B), 1’000 (C) or 1’200 (D) monodispersed human umbilical endothelial cells
(HUVEC) and cultivated for 7 days in hanging drops. (E) Inset of (D) showing a branching capillary at higher
magnification. (F) NHDF-derived microtissue (233µm/10’000 cells) coated with HUVEC (600 cells). (A-F)
HUVEC-specific markers PECAM-1 (platelet-endothelial cell adhesion molecule-1; red) and von Willebrand
factor (green) were immunostained. (scalebar = 20 µm).
Detailed HUVEC migration kinetics of HAF-HUVEC microtissues revealed that
HUVECs (i) assembled on the surface of preformed HAF spheroids in a shell-like manner 2
days post coating (Figure 7A), (ii) HUVECs started infiltrating the HAF core spheroid at day
4 (Figure 7B) and (iii) formed a well-structured capillary system on and beyond day 6 post
CA B
D FE
CCAA BB
DD FFEE

Microtissue Vascularization 89
coating (Figure 7C). Interestingly, VEGF secretion of HAF-HUVEC microtissues was lower
compared to pure HAF spheroids suggesting that either crosstalk between HAFs and
HUVECs is modulating VEGF secretion or VEGF is bound/taken up by HUVECs (Figure 8).
Figure 7: Kinetics of angiogenesis-dependent capillary formation in human umbilical endothelial cell
(HUVEC, 600 cells)-coated human aortic fibroblast (HAF; 335µm/10’000 cells) spheroids. HUVEC-specific
staining of PECAM-1 (platelet-endothelial cell adhesion molecule-1, red) and von Willebrand factor (green) on
days 2 (A), 4 (B) and 6 (C) post coating. (scalebar = 30 µm).
A B CAA BB CC
A
Time [h]
VE
GF
[p
g/m
l]
100
300
500
700
900
48 96 144 192
HAF
HAF-HUVEC
A
Time [h]
VE
GF
[p
g/m
l]
100
300
500
700
900
48 96 144 192
HAF
HAF-HUVEC
HAF
HAF-HUVEC
HAF
HAF-HUVEC

Microtissue Vascularization 90
Figure 8: VEGF production kinetics of human umbilical vein endothelial cells (HUVEC)-coated on
human aortic fibroblast (HAF) spheroids and pure HAF-derived spheroids over 6 days. 125 µm (A, 500 cells) or
335 µm (B, 10’000 cells) HAF-derived spheroids optionally coated with 600 HUVECs.
Ultrastructural characterization of HAF-HUVEC microtissues 7 days post coating
demonstrated advanced development of vascular structures. In early developmental stages
many endothelial cells are characterized by intracellular lumen formation (Figure 9A-C).
Endothelial cells increase their lumen size, loose organelles and progressively attenuate
thereby adopting morphologies of the capillary endothelium (Figure 9C). At an advanced
stage of differentiation HUVECs exhibit a well-developed vesicular system which may
assemble to form typical transendothelial transport channels (Figure 9D). Similar to native
vessels HAFs and HUVECs develop a panoply of cell-cell interactions (Figure 9E).
BV
EG
F [
pg
/ml]
Time [h]
5000
15000
25000
35000
48 96 144 192
HAF
HAF-HUVEC
BV
EG
F [
pg
/ml]
Time [h]
5000
15000
25000
35000
48 96 144 192
HAF
HAF-HUVEC
VE
GF
[p
g/m
l]
Time [h]
5000
15000
25000
35000
48 96 144 192
HAF
HAF-HUVEC
HAF
HAF-HUVEC
HAF
HAF-HUVEC

Microtissue Vascularization 91
Figure 9: Ultrastructure of cocultured HAF-HUVEC spheroids 7 days post coating. (A) Endothelial
cells are typically characterized by intracellular lumen formation (+). Endothelial cells are tightly covered by
human aortic fibroblasts (asterisk). The periendothelial space is indicated by arrowheads. (B) Capillary like-
structures result from fusion of endothelial cell (asterisks) protrusions. (C) Advanced differentiation stages are
characterized by enlarged lumina and organelle-free attenuated endothelial cells. Boxed regions are enlared in D
and E. (D) Vesicles typically open to the luminal (lv) and abluminal (av) sites or form transendothelial channels
(ch). Tight junctions (tj) and zonula adhaerens (za) demonstrate well-established endothelial-periendothelial cell
contacts. (E) The periendothelial space contain fibrils (asterisks) and cellular protrusions which often result from
HAF-HUVEC cell contacts (arrowheads).
A
E
DC
BA
E
DC
B

Microtissue Vascularization 92
Inhibition of angiogenesis in HAF-HUVEC microtissues
The C-terminal cleavage product of collagen XVIII known as endostatin is a key anti-
angiogenic factor (Bloch et al. 2000; O'Reilly et al. 1997). We evaluated endostatin action on
HAF microtissues (10’000 cells) coated with 1’200 HUVECs. Endostatin (1 g/ml) was either
administered during or 2 days post coating. 6 days post coating the HAF-HUVEC
microtissues were immunoprofiled for PECAM-1 and von Willebrand factor expression.
Whereas endostatin was able to prevent angiogenesis when supplied during coating, later
addition of this antiangiogenic factor to mature HAF-HUVEC microtissues failed to inhibit
HUVEC-based capillary formation in the HAF core (Figure 10B and 10C). Similar to addition
of endostatin, HAF spheroids coated with a mixture of HUVECs and human umbilical artery
smooth muscle cells (HUASMC) (HAF-HUVEC/HUASMC; 10’000-600/600 cells) did not
develop any HUVEC-based capillary systems confirming the potential of HUASMCs to
inhibit VEGF-mediated vascularization (data not shown) (Peirce and Skalak 2003).
Figure 10: Endostatin-mediated angiogenesis suppression in human aortic fibroblast (HAF)
microtissues coated with human umbilical vein endothelial cells (HUVEC). Endostatin impact on HUVEC-
driven capillary formation in HAF microtissues was monitored following administration of this anti-angiogenic
factor at coating (A) as well as 2 days post coating (B). Untreated HAF-HUVEC microtissues were used as
control (C). HAF-HUVEC spheroids were cultivated for 6 days and vascularization was analyzed by PECAM-1-
(red) and von Willebrand factor- (green) specific staining. (scalebar = 20µm).
Discussion
Endothelial cells are the central organizational unit of (micro-)vascular structures.
Their lineage commitment, expansion, organization, and assembly into ordered and tissue-
specific interconnecting vascular structures are required for embryonic development,
CBA CCBBAA

Microtissue Vascularization 93
organogenesis, wound healing, reproductive tissue cycles, tumorigenesis and a number of
other pathological conditions involving inflammation (Daniel and Abrahamson 2000). The
elucidation of the molecular basis of tissue-specific angiogenesis remains largely elusive,
owing to the panoply of complex interactions which have to be temporally and spatially
coordinated (Eliceiri and Cheresh 2001). In order to obtain new insight into
neovascularization of human adult tissues, bioengineered tissues or development of tumor
vessels 3D, angiogenesis models are the preferred systems to study complex cell-cell
interactions and signal integration circuits managing the balance of pro- and anti-angiogenic
cellular activities in a clinically relevant manner (Hirschi et al. 1999; Niklason et al. 1999;
Vailhe et al. 2001).
We have refined the hanging drop technology to generate fully size-controlled
multicellular tumor spheroids (MCTS)/microtissues of a wide variety of different cell types
which exhibited size/cell number-dependent expression of the vascular endothelial growth
factor (VEGF) [31, 46]. Microtissue size-dependency of VEGF expression is a result of
hypoxic conditions at the center of oversized spheroids which induce HIF-1 a transcription
factor controlling VEGF production (Semenza 2001). Although VEGF can induce mature
vessels at an appropriate dosage and appears to have a relatively wide therapeutic window,
the balance between clinical benefit and pathologic side effect is likely to differ with genetic
predisposition, age and disease status (Blau and Banfi 2001). The tumor cell used in this study
(HT-1080) produced increased VEGF levels in monolayers compared to isogenic cultures of
primary human mesodermal cells. Moreover, MCTS exclusively assembled from HT-1080
cells exhibited a near linear cell number-VEGF production profile while expression of this
growth factor reached a plateau in microtissues composed of 2’500 to 5’000 non-tumorigenic
cells (HAF, NHDF). VEGF expression under normoxic conditions in monolayer cultures and
the linear correlation between cell number-VEGF secretion indicates increased hypoxia-
independent VEGF production in tumor cells included in this study. Although absolute
VEGF/microtissue size profiles of different human primary cells revealed cell type- and
donor-specific differences their relative levels remained comparable. As an exception, VEGF
expression profiles of HAC microtissues were decreased compared to control monolayer
cultures which supported findings by Schipani and coworkers suggesting HIF-1 -independent
VEGF induction in chondrocytes (Schipani et al. 2001).
An ideal bioengineered tissue will have to be assembled from multiple organ-specific
cell types, while retaining hierarchical cellular architecture and supporting
endovascularization compatible with connection to the native vascular systems of organs.

Microtissue Vascularization 94
Despite induction of angiogenesis by growth factors, 3D self-organization of different cell
types is a prerequisite for engineering of functional and fully vascularized tissue equivalents.
Mixed cultures of HepG2:HUVEC, HAF:HUVEC and HUASMC:HUVEC had shown a high
degree of self-organization mimicking in vivo cellular architecture: HUVECs moved to the
periphery of the microtissue building the barrier to the surroundings. Interestingly, mixed
HUASMC:HUVEC microtissues exhibited the opposite HUVEC-inside/HUASMC-outside
setup which contrasts previous HUVEC-outside/HUASMC-inside organotypic cultures
assembled using liquid methylcellulose. These findings exemplify modulation of self-
organization forces within microtissues by culture additives/matrices (Korff and Augustin
1999; Korff et al. 2001).
The interplay between environmental and genetic impact on tumor
angiogenesis/proliferation is complex and remains largely elusive (Carmeliet and Jain). A
major challenge is to establish a generic cell culture model mimicking tumor development
and angiogenesis of a wide variety of tumorigenic cell types (Kelm and Fussenegger 2004;
Kelm et al. 2003). Although all cancer cell lines used in this study produced VEGF, we have
chosen HepG2 as tumor model since hepatocellular carcinomas are among the most malignant
cancers affecting over 1 million patients per year (Kim et al. 2002). Although HepG2-derived
MCTS produced little VEGF compared to other 3D-assembled cancer cell lines (DU-145,
MCF-7, HT-1080), VEGF production was sufficient to induce vascularization in coated
HepG2-HUVEC microtissue and reduced core necrosis compared to pure HepG2-derived
MCTS.
The development of cell-based therapies to assist/replace diseased tissue is among the
most promising initiatives in regenerative medicine (Petit-Zeman 2001). Yet, a major
challenge on our way towards the design of artificial organs is vascularization management to
provide oxygen/nutrient supplies and removal of waste products (Griffith and Naughton 2002;
Zandonella 2003). Precise knowledge of the impact of tissue microenvironments on
metabolism, proliferation and growth-factor production will be essential for elucidation of
microvessel formation (McIntire 2002). Although often declared an engineering target, VEGF
production can be fine-tuned by multiple cell types arranged in 3D structures in a self-
sufficient manner (Elson et al. 2001; Isner 2002; Jain and Carmeliet 2001; Richardson et al.
2001; Zisch et al. 2003). HAF-HUVEC and NHDF-HUVEC microtissues have exemplified
that a high angiogenic potential can be induced by controlled cell assembly without
administration of any growth factor. 6 days post coating, cellular crosstalk combined with
hypoxia-induced VEGF production established a dense microvessel network, as a

Microtissue Vascularization 95
consequence of which VEGF production decreased significantly. Interference of
HUASMC:HUVEC coated onto HAF spheroids abolished capillary formation (data not
shown) which was reminiscent of HUASMC-mediated repression of vascularization in vivo
(Sato and Rifkin 1989). Capillary formation in HAF-HUVEC cultures could also be
prevented by addition of the well-known antiangiogenic factor endostatin, but only when
supplied at the time of coating. Interestingly, 2 days post coating the angiogenic machinery of
HAF-HUVEC microtissues was set for capillary formation and could no longer be impaired
or reversed by endostatin.
Microtissue design by gravity-enforced assembly of monodispersed cells in hanging
drops enables (i) precise size control, (ii) flexible cell-type composition, (iii) self-organization
of multiple cell types, (iv) intra-/inter-cellular crosstalk, (v) in vivo-mimicking of cellular
architecture and (vi) self-sufficient and auto-controlled vascularization of oversized spheroids.
Owing to the low cell number required for production of microtissues, this 3D culture system
is expected to be compatible with high-throughput drug screening and multi-level gene
function analysis. Also, with their auto-angiogenesis potential, microtissues could represent
the optimal format for (autologous) cell therapies, a vision which will have to be substantiated
by further in vivo studies.
Acknowledgments
We thank, Cornelia C. Weber, as well as Beat P. Kramer for critical comments on the
manuscript, Jean-Claude Perriard for his ongoing support, Millenium Biologix for providing
human articular chondrocytes and Heike Hall for supply of anti-integrin antibodies. Work in
the laboratory of M.F. is supported by the Swiss National Science Foundation (grant no. 631-
065946) and a special research grant by the ETH Vice President for Research.
References
Bergers G., L.E. Benjamin (2003). "Tumorigenesis and the angiogenic switch." Nat Rev
Cancer 3, 401-410.
Blau H.M., A. Banfi (2001). "The well-tempered vessel." Nat Med 7, 532-534.
Bloch W., K. Huggel, T. Sasaki, R. Grose, P. Bugnon, K. Addicks, R. Timpl, S. Werner
(2000). "The angiogenesis inhibitor endostatin impairs blood vessel maturation during
wound healing." Faseb J 14, 2373-2376.

Microtissue Vascularization 96
Brooks P.C., R.A. Clark, D.A. Cheresh (1994)." Requirement of vascular integrin alpha v beta
3 for angiogenesis." Science 264, 569-571.
Carmeliet P., R.K. Jain (2000). "Angiogenesis in cancer and other diseases." Nature 407, 249-
257.
Carmeliet P., Y.S. Ng, D. Nuyens, G. Theilmeier, K. Brusselmans, I. Cornelissen, E. Ehler,
V.V. Kakkar, I. Stalmans, V. Mattot and others (1999). "Impaired myocardial
angiogenesis and ischemic cardiomyopathy in mice lacking the vascular endothelial
growth factor isoforms VEGF164 and VEGF188." Nat Med 5, 495-502.
Conway E.M., D. Collen, P. Carmeliet (2001). "Molecular mechanisms of blood vessel
growth." Cardiovasc Res 49, 507-521.
Daniel T.O., D. Abrahamson (2000). "Endothelial signal integration in vascular assembly."
Annu Rev Physiol 62, 649-671.
Djonov V., M. Schmid, S.A. Tschanz, P.H. Burri (2000). "Intussusceptive angiogenesis: its
role in embryonic vascular network formation." Circ Res 86, 286-292.
Eliceiri B.P., D.A. Cheresh (2001). "Adhesion events in angiogenesis." Curr Opin Cell Biol
13, 563-568.
Elson D.A., G. Thurston, L.E. Huang, D.G. Ginzinger, D.M. McDonald, R.S. Johnson, J.M.
Arbeit (2001). "Induction of hypervascularity without leakage or inflammation in
transgenic mice overexpressing hypoxia-inducible factor-1alpha." Genes Dev 15,
2520-2532.
Ferrara N., K. Alitalo (1999). "Clinical applications of angiogenic growth factors and their
inhibitors." Nat Med 5, 1359-1364.
Folkman J. (2003). "Angiogenesis and apoptosis." Semin Cancer Biol 13, 159-167.
Folkman J., A. Kaipainen (2004). "Genes tell lymphatics to sprout or not." Nat Immunol 5,
11-2.
Gerber H.P., K.J. Hillan, A.M. Ryan, J. Kowalski, G.A. Keller, L. Rangell, B.D. Wright, F.
Radtke, M. Aguet, N. Ferrara (1999). "VEGF is required for growth and survival in
neonatal mice." Development 126, 1149-1159.
Griffith L.G., G. Naughton (2002). "Tissue engineering--current challenges and expanding
opportunities." Science 295, 1009-1014.
Hirschi K.K., S.A. Rohovsky, L.H. Beck, S.R. Smith, P.A. D'Amore (1999). "Endothelial
cells modulate the proliferation of mural cell precursors via platelet-derived growth
factor-BB and heterotypic cell contact." Circ Res 84, 298-305.
Isner J.M. (2002). "Myocardial gene therapy." Nature 415, 234-239.

Microtissue Vascularization 97
Jain R.K. (2003). "Molecular regulation of vessel maturation." Nat Med 9, 685-693.
Jain R.K., P.F. Carmeliet (2001). "Vessels of death or life." Sci Am 285, 38-45.
Jin K., Y. Zhu, Y. Sun, X.O. Mao, L. Xie, D.A. Greenberg (2002). "Vascular endothelial
growth factor (VEGF) stimulates neurogenesis in vitro and in vivo." Proc Natl Acad
Sci U S A 99, 11946-11950.
Kelm J.M., E. Ehler, L.K. Nielsen, S. Schlatter, J.C. Perriard, M. Fussenegger (2004a).
"Design of Artificial Myocardial Microtissues." Tissue Eng 10, 201-214.
Kelm J.M., M. Fussenegger (2004). "Microscale tissue engineering using gravity-enforced
cell assembly." Trends Biotechnol 22, 195-202.
Kelm J.M., B.P. Kramer, V. Gonzalez-Nicolini, B. Ley, M. Fussenegger (2004b). "Synergies
of microtissue design, viral transduction and adjustable transgene expression for
regenerative medicine." Biotechnol Appl Biochem 39, 3-16.
Kelm J.M., N.E. Timmins, C.J. Brown, M. Fussenegger, L.K. Nielsen (2003). "Method for
generation of homogeneous multicellular tumor spheroids applicable to a wide variety
of cell types." Biotechnol Bioeng 83, 173-180.
Kim K.R., H.E Moon, K.W. Kim (2002). "Hypoxia-induced angiogenesis in human
hepatocellular carcinoma." J Mol Med 80, 703-714.
Koh J.T., H. Kook, H.J. Kee, Y.W. Seo, B.C. Jeong, J.H. Lee, M.Y. Kim, K.C. Yoon, S. Jung,
K.K. Kim (2004). "Extracellular fragment of brain-specific angiogenesis inhibitor 1
suppresses endothelial cell proliferation by blocking alphavbeta5 integrin." Exp Cell
Res 294, 172-184.
Korff T., H.G. Augustin (1999). "Tensional forces in fibrillar extracellular matrices control
directional capillary sprouting." J Cell Sci 112, 3249-3258.
Korff T., S. Kimmina, G. Martiny-Baron, H.G. Augustin (2001). "Blood vessel maturation in
a 3-dimensional spheroidal coculture model: direct contact with smooth muscle cells
regulates endothelial cell quiescence and abrogates VEGF responsiveness." Faseb J
15, 447-457.
Lee R.J., M.L. Springer, W.E. Blanco-Bose, R. Shaw, P.C. Ursell, H.M. Blau (2000). "VEGF
gene delivery to myocardium: deleterious effects of unregulated expression."
Circulation 102, 898-901.
McIntire L.V. (2002). "Vascular assembly in engineered and natural tissues." Ann N Y Acad
Sci 961, 246-248.

Microtissue Vascularization 98
Messerli J.M., H.T. van der Voort, E. Rungger-Brandle, J.C. Perriard (1993). "Three-
dimensional visualization of multi-channel volume data: the amSFP algorithm."
Cytometry 14, 725-735.
Mooney D.J., A.G. Mikos (1999). "Growing new organs." Sci Am 280, 60-65.
Niklason L.E., J. Gao, W.M. Abbott, K.K. Hirschi, S. Houser, R. Marini, R. Langer (1999).
"Functional arteries grown in vitro." Science 284, 489-493.
Nomi M., A. Atala, P.D. Coppi, S. Soker (2002). "Principals of neovascularization for tissue
engineering." Mol Aspects Med 23, 463.
O'Reilly M.S., T. Boehm, Y. Shing, N. Fukai, G. Vasios, W.S. Lane, E. Flynn, J.R. Birkhead,
B.R. Olsen, J. Folkman (1997). "Endostatin: an endogenous inhibitor of angiogenesis
and tumor growth." Cell 88, 277-285.
Peirce S., T.C. Skalak (2003). "Microvascular remodeling: a complex continuum spanning
angiogenesis to arteriogenesis." Microcirculation 10, 99-111.
Petit-Zeman S. (2001). "Regenerative medicine." Nat Biotechnol 19, 201-206.
Richardson T.P., M.C. Peters, A.B. Ennett, D.J. Mooney (2001). "Polymeric system for dual
growth factor delivery." Nat Biotechnol 19, 1029-1034.
Sato Y., D.B. Rifkin (1989). "Inhibition of endothelial cell movement by pericytes and
smooth muscle cells: activation of a latent transforming growth factor-beta 1-like
molecule by plasmin during co-culture." J Cell Biol 109, 309-315.
Schipani E., H.E. Ryan, S. Didrickson, T. Kobayashi, M. Knight, R.S. Johnson (2001).
"Hypoxia in cartilage: HIF-1alpha is essential for chondrocyte growth arrest and
survival." Genes Dev 15, 2865-2876.
Schuch G., J.V. Heymach, M. Nomi, M. Machluf, J. Force, A. Atala, J.P. Eder, J. Folkman, S.
Soker (2003). "Endostatin inhibits the vascular endothelial growth factor-induced
mobilization of endothelial progenitor cells." Cancer Res 63, 8345-8350.
Semenza G.L. (2001). "HIF-1, O(2), and the 3 PHDs: how animal cells signal hypoxia to the
nucleus." Cell 107, 1-3.
Semenza G.L. (2003). "Angiogenesis in ischemic and neoplastic disorders." Annu Rev Med
54, 17-28.
Sheehan D.C., Hrapchak. 1980. Theory and practice of histotechnology. Ohio: Battelle Press.
Springer M.L., A.S. Chen,P.E. Kraft, M. Bednarski, H.M. Blau (1998). "VEGF gene delivery
to muscle: potential role for vasculogenesis in adults." Mol Cell 2, 549-558.
Tsigkos S., M. Koutsilieris, A. Papapetropoulos (2003). "Angiopoietins in angiogenesis and
beyond." Expert Opin Investig Drugs 12, 933-941.

Microtissue Vascularization 99
Vailhe B., D. Vittet, J.J. Feige (2001). "In vitro models of vasculogenesis and angiogenesis."
Lab Invest 81, 439-452.
Yancopoulos G.D., S. Davis, N.W. Gale, J.S. Rudge, S.J. Wiegand, J. Holash (2000).
"Vascular-specific growth factors and blood vessel formation." Nature 407, 242-248.
Zandonella C. (2003). "Tissue engineering: The beat goes on." Nature 421, 884-886.
Zisch A.H., M.P. Lutolf, M. Ehrbar, G.P. Raeber, S.C. Rizzi, N. Davies, H. Schmokel, D.
Bezuidenhout, V. Djonov, P. Zilla and others (2003). "Cell-demanded release of
VEGF from synthetic, biointeractive cell ingrowth matrices for vascularized tissue
growth." Faseb J 17, 2260-2262.

Chapter 6
Improved Tissue-Transplant Fusion and
Vascularization of Myocardial Micro- and
Macrotissues Implanted into Chicken Embryos and
Rats
Kelm J.M., Djonov V., Hoerstrup S.P., Guenter C. I., Ittner L.M., Greve F., Hierlemann A.,
Perriard P.C., Ehler E. and Fussenegger M. (submitted)

Myocardial Microtissue Implants 101
Abstract
Cell-based therapies and tissue engineering initiatives are gathering clinical momentum
for next-generation treatment of tissue deficiencies. Capitalizing on gravity-enforced self-
assembly of monodispersed primay cells in hanging drops we have produced adult and neonatal
rat cardiomyocyte-based myocardial microtissues which could optionally be vascularized
following coating with human umbilical endothelial cells (HUVEC). Within myocardial
microtissues, individual cardiomyocytes showed native cell shape and morphologies and
established electrogenic coupling via intercalated disks. This resulted in the coordinated
contracting microtissues, which was recorded by means of a novel multi-electrode CMOS
microchip. Myocardial microtissues ( m3 scale), coated with HUVECs and cast in a custom-
shaped agarose mould, assembled to coherent macrotissue (mm3 scale), characterized by an
extensive capillary network with typical vessel ultrastructures. Following implantation into
chicken embryos, myocardial microtissues produced the vascular endothelial growth factor
(VEGF), which recruited the embryo’s capillaries to functionally vascularize the rat-derived
tissue implant. Similarly, transplantation of rat myocardial microtissues into the pericardium of
adult rat resulted in time-dependent resorption of myocardial microtissues and co-alignment of
implanted and host cardiomyocytes within seven days. Myocardial microtissues and custom-
shaped macrotissues, which are either vascularized in vitro or recruit the host vasculature
following in vivo implantation, exemplify the potential of artificial tissue implants for
regenerative medicine.
Introduction
The heart is the first organ to form in the embryo and all subsequent activities in life
depend on its function. The past decade has witnessed decisive advances in understanding cardiac
function and dysfunction, both genetically and molecularly levels. Although such insights into
the mechanisms of heart development and disease have stimulated new therapeutic opportunities
for the prevention and palliation of cardiac pathogenesis, mortality rates associated with heart-
related pathologies remain at the top of disease statistics in industrialized countries (Olson 2004).
Since cardiac myocytes lose their ability to divide after birth, the regenerative capacity of adult

Myocardial Microtissue Implants 102
heart tissue is limited, and substantial cell loss or dysfunction, such as occurs during myocardial
infarction, is largely irreversible and may lead to progressive heart failure (Pasumarthi and Field
2002).
Transplantation of excitable myogenic cells within the dysfunctional zone is a possible
therapy for restoring cardiac function. For example, initial studies with injected skeletal muscle
cells showed promise; this strategy is, however, unlikely to be suitable for long-term therapy
owing to the failure of the injected cells to become electrically coupled to the heart and to
different contractile properties between cardiac and skeletal muscle cells (Murry et al. 2004).
Although several cell phenotypes have been tested, cardiomyocytes emerged as the preferred cell
type because of their inherent structural, electrophysiological and contractile properties (Reinecke
et al. 1999). However, clinical applications are hampered by the paucity of cell sources for
human cardiomyocytes and by the limited evidence of direct functional integration of host and
donor cells (El Oakley et al. 2001; Rubart et al. 2003; Olson 2004). Nevertheless, injection of
bone marrow and stem cells derived from cloned mouse embryos into the border zone of the
infarcted myocardium have been reported to promote cardiac regeneration and improve cardiac
function in animal models (Kehat et al. 2004; Lanza et al. 2004). However, there is disagreement
as to the efficiency with which exogenous stem cells can colonize the heart and adopt a
cardiomyocyte cell fate (Kehat et al. 2004; Murry et al. 2004; Nygren et al. 2004). Recently,
human embryonic stem cell-derived cardiomyocytes successfully paced the ventricle in swine
with complete heart block, showing that transplanted cardiomyocytes can in principle survive,
function and integrate with host cells and operate as a biological alternative to the electronic
pacemaker (Kehat et al. 2004). However, embryonic stem cells can form teratomas in vivo which
presents another set of therapeutic challenges.
To date, cardiomyocyte-based prototype cell therapy initiatives capitalized on the
transplantation of fetal, neonatal (NRCs) and adult (ARCs) rat cardiomyocytes (Reinecke et al.
1999). While ARCs died 24h after transplantation, 50% of grafted NRCs survived the first day
but more than 90% perished within the first week (Zhang et al. 2001). Despite several attemps to
limit the death of grafted cell suspensions by antiapoptosis engineering (Datta et al. 1999),
localized high-dose application of single cells remains technically challenging. Therefore, pre-
transplantation assembly of monodispersed cells to tissue-like structures is considered to be an
alternative. A variety of scaffold-containing (for example, collagen and ceramics) and scaffold-

Myocardial Microtissue Implants 103
free (for example, stacking of monolayers and reaggregation) strategies have been designed to
produce three-dimensional cardiac tissue constructs, some of which have been transplanted
successfully (Shimizu et al. 2002; Zimmermann et al. 2002; Zandonella 2003; Zimmermann and
Eschenhagen 2003; Kelm et al. 2004).
The potential hurdles in cardiac tissue engineering are the requirements that
cardiomyocyte-derived artificial tissues become seamlessly integrated into the damaged
myocardium without becoming a substrate for arrhythmogenesis, and that they be able to induce
neovascularization of the regenerated myocardium. Capitalizing on gravity-enforced self
assembly of primary cardiomyocytes in hanging drops we recently reported the production of
highly adhesive and functionally beating myocardial microtissues. They produced the vascular
endothelial growth factor in a size-dependent manner, suggesting that they may stimulate
angiogenesis after transplantation (Kelm et al. 2004). In this study we (i) pioneered production of
myocardial microtissues from adult rat cardiomyocytes, (ii) designed a novel CMOS microchip
for assessing of electrongenic microtissue performance, (iii) assembled myocardial microtissues
to larger fully vascularized tissue implants using custom-shaped agarose moulds, which (iv)
managed successful vascularization crosstalk following the grafting of myocardial microtissues
into chicken embryos and (v) provided seamless integration after implantation into the
pericardium of adult rats. We are convinced that microtissue-based scale-up, resulting in fully
vascularized, custom-shaped, scaffold-free and transplantation-ready tissue units will improve
cell-based cardiac-related therapies in the not-too-distant future.
Material and Methods
Preparation of primary cells
Neonatal rat cardiomyocytes (NRCs) were isolated from dissected newborn rat (Wistar;
Janvier Elevage, Le Genest Saint Isle, France) hearts by digestion with collagenase (Worthington
Biochemical Corp., Freehold, NJ) and pancreatin (Invitrogen, Carlsbad, CA, USA), according to
protocols by Auerbach and colleagues (Auerbach et al. 1999). Adult rat cardiomyocytes (ARCs)
were prepared as described elsewhere (Eppenberger and Zuppinger 1999). NRCs and ARCs were
kept in plating medium (67% Dulbecco’s Modified Eagle Medium [DMEM; Invitrogen], 17%

Myocardial Microtissue Implants 104
M199 [Amimed AG, Basel, Switzerland], 10% horse serum [cat. no. 16050-098, lot no.
3036354D, Invitrogen], 5% fetal bovine serum [FBS; cat. no. 3302-P231902, lot no. P231902;
PAN Biotech GmbH, Aidenbach, Germany], 1% penicillin/streptomycin solution [Invitrogen]).
Human umbilical vein endothelial cells (HUVECs, PromoCell, Heidelberg, Germany) were
expanded in endothelial cell growth medium (PromoCell, cat. no. C-22010) supplemented with
10% FBS (PAN Biotech GmbH, Aidenbach).
Microtissue production
After isolation, cardiomyocytes were seeded at indicated cell concentrations and cell-type
compositions into 60-well plates (HLA plate, Greiner-Bio One, Frickenhausen, Germany). To
enable gravity-enforced self-assembly of microtissues in hanging drops, the 60-well plates were
incubated upside down at 37°C in a humidified atmosphere containing 5% CO2.
Macrotissue assembly
In order to assemble microtissues to larger-sized artificial macrotissues we designed
cylindrical agarose casting moulds, 3 mm in diameter. Negative Teflon®
casting moulds were
filled with 4% agarose (Sigma Chemicals, Buchs, Switzerland) in phosphate-buffered saline
(PBS; 150 mM NaCl, 6.5 mM Na2HPO4 x 2 H2O, 2.7 mM KCL, 1.5 mM KH2PO4, pH 7.4) to
generate non-adhesive positive casting moulds to imprint the desired shape on the macrotissue.
Microtisses of indicated cell types and cell numbers were transferred to the desired agarose
moulds and cultivated in cell type-specific media under static conditions at 37°C in a humidified
atmosphere containing 5% CO2.
Immunofluorescence analysis
Microtissues were washed once in PBS (Sigma Chemicals) and fixed for 4h at 4°C in
PBS containing 4% paraformaldehyde (Sigma Chemicals). Immunofluorescence-based analysis
of microtissues was performed as described elsewhere (Kelm et al. 2004) using primary
antibodies specific for sarcomeric- -actinin (mouse monoclonal, Sigma Chemicals; clone EA53),
connexin-43 (rabbit polyclonal, Zymed, San Francisco, CA, USA), or myomesin (mouse
monoclonal (Grove et al. 1984)) was used and stained with FITC-coupled secondary anti-mouse

Myocardial Microtissue Implants 105
(Jackson Immunochemicals, West Grove, PA, USA) or Cy3-coupled anti-rabbit (ICN
Pharmaceuticals, Hyland, CA) antibodies. Cell nuclei were stained with 1 g/ml 4',6-diamidino-
2-phenylindole (DAPI, Molecular Probes Inc., Eugene, OR, USA).
Confocal light microscopy
The imaging system consisted of an inverted fluorescence microscope equipped with 20x
or 10x oil immersion objectives and a confocal scanner (Zeiss LSM510; Carl Zeiss AG,
Feldbach, Switzerland) with argon and helium neon lasers installed. Images were processed using
up-to-date Zeiss software (Carl Zeiss AG).
Transmission electron microscopy
Tissue samples were fixed by immersion in 0.1M cacodylate buffer (pH 7.4, 350 mOsm)
containing 2.5% glutaraldehyde. Tissue blocks were postfixed in osmium tetroxide, block-stained
using uranyl acetate, dehydrated by sequential incubation in increasing ethanol concentrations
and embedded in Epon 812 according to Djonov and coworkers (Djonov et al. 2000) (all
chemicals from Merck Eurolab AG, Dietikon, Switzerland). Semi-thin 1 µm sections were
stained with toluidine blue and visualized by an Olympus Vanox BHS light microscope
(Olympus AG, Volketswil, Switzerland). Ultra-thin sections of 80-90 nm were cut with a
diamond knife and picked up on Formvar-coated (polyvinyl formal; Fluka Chemie AG) copper
grids, double-stained with lead citrate (Merck Eurolab AG) and uranyl acetate and monitored on
a Philips EM 400 electron microscope (FEI AG, Zurich, Switzerland).
Microchip-based electrophysiology
The 5 5 mm2 bio-electronic chip has been designed as an array of 4 4 platinum
electrodes (20 20 µm2). An individual circuitry block manages signal conditioning of a single
four-electrode array, which enables simultaneous recording in the absence of multiplexer
switching. Electrode-recorded cell signals are high-pass filtered (100 Hz cut-off) to prevent input
saturation by signal differences between reference and measurement electrodes, intensified (gain
10x, 100x) by a programmable gain amplifier in the circuitry block, low-pass filtered (4 KHz
adjustable cut-off) and digitalized by external circuitries (Greve et al., unpublished). Microtissues

Myocardial Microtissue Implants 106
(see above for assembly) were prepared for electrogenic recording by cultivating them for six
days in non-adhesive cell culture dishes (Greiner-Bio One). Myocardial microtissues (10 days
old) were placed on the electrodes and incubated at 37°C and 5% CO2. Within 2 h, myocardial
microtissues attached to the platinum electrodes; contraction was initiated by adding of 10-4
mM
of the -adrenergic agent phenylephrine (Sigma Chemicals) before recording of microtissue
signals.
Chicken chorioallantoic membrane (CAM) assay
Chicken embryos were cultured using the shell-free method (Ribatti et al. 2001). Artificial
tissues were grafted atop the growing chicken chorioallantoic membrane (CAM) on embryonic
day 9 and cultured for two days.
Transplantation of myocardial microtissues into rat hearts
Prior to transplantation, myocardial microtissues were labeled for 30 min in Hanks’
balanced salt solution (HBSS, Sigma Chemicals) supplemented with 10 mM CellTrackerTM
(CMTMR, Molecular Probes) and then washed three times in HBSS. Adult rats were treated with
the analgesic Temgesic (300 µl prior to surgery; Essex Chemie AG, Luzern, Switzerland) by
intraperitoneal injection and then anesthetized in an isoflurane chamber. Anesthetized rats were
endotracheally intubated and ventilated with positive-pressure (0.6 l/min, 3 ml tidal volume,
oxygen-supplemented (2 ml) air) using a Harvard ventilator (Harvard Apparatus, Hollisten,
Massachusetts, USA). Under general anesthesia a 1 cm long incision was made directly posterior
to the xiphoid. The chestwall was lifted up in order to expose the diaphragm. Myocardial MTs
were injected into the pericard through the membraneous part of the diaphragm. The injection
hole was sealed with fibrin glue (Tissucolduo, Baxter, Unterschleissheim, Germany). The
abdominal incision was closed in layers with 4-0 silk running sutures. After surgery, the rats were
kept warm and in isolation until they were fully awake when they were returned to their filter
cages. Postoperatively, the rats were injected with decreasing intraperitoneal Temgesic every six
hours for two days. Rats were sacrificed after one (n= 4), four (n=4) and seven days (n=4). The
control group was injected with HBSS and was sacrificed after seven days (n=2).

Myocardial Microtissue Implants 107
Results
Microtissues assembled from adult cardiomyocytes
Although several cell types are currently considered for myocardial tissue repair (Hassink
et al. 2003), only cardiomyocytes form intercalated disks, which are essential for
electrophysiological coupling. In contrast to neonatal cells, which will probably raise ethical
questions, allogenic adult cardiomyocytes are routinely isolated from the auricular cordis and
represent an alternative cell source for future cardiac tissue engineering. However, cultivation is a
challenge, which has prevented the 3D assembly of adult rat cardiomyocytes (ARCs), and
monodispersed ARCs implanted into rat hearts failed to survive for more than 48 h (Reinecke et
al. 1999). Using gravity-enforced self-assembly we produced artificial myocardial microtissues
after a one-week cultivation of monodispersed ARCs in hanging drops (Figures 1A and B).
Within the myocardial microtissues, ARCs adopted a native rod-shaped cell phenotype,
established inter-cardiomyocyte contacts via gap junctions (Figure 1C) and produced the vascular
endothelial growth factor (Figure 1D), similar to NRC-derived microtissues (Kelm et al. 2004).
After 14 days in culture (seven days in hanging drops, followed by seven days in standard
culture) ARCs at the microtissues’ periphery had aligned and were functionally interconnected
via intercalated disks (Figure 1G), while VEGF was produced throughout the entire microtissue
(Figure 1H). These results exemplify that adult cardiomyocytes arranged in a scaffold-free
microtissue retain their native cellular reorganization capacity as well as their angiogenic
potential.
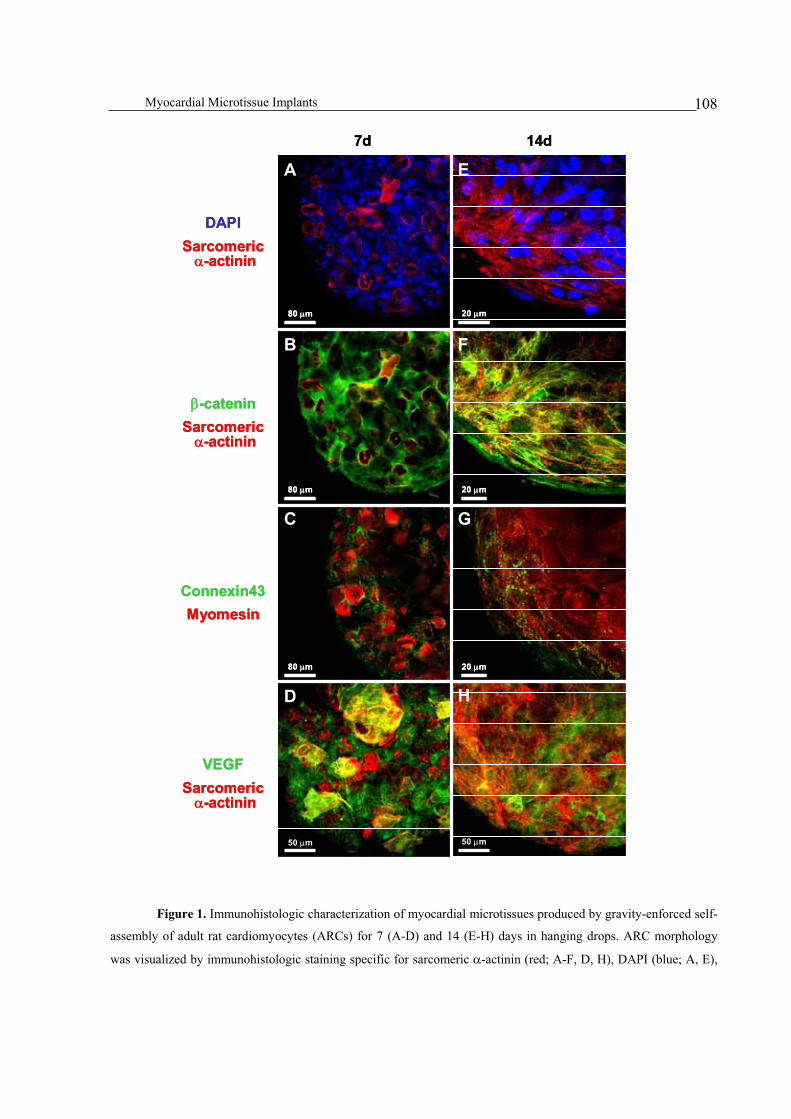
Myocardial Microtissue Implants 108
Figure 1. Immunohistologic characterization of myocardial microtissues produced by gravity-enforced self-
assembly of adult rat cardiomyocytes (ARCs) for 7 (A-D) and 14 (E-H) days in hanging drops. ARC morphology
was visualized by immunohistologic staining specific for sarcomeric -actinin (red; A-F, D, H), DAPI (blue; A, E),
7d 14d
DAPI
Sarcomeric-actinin
-catenin
Sarcomeric-actinin
Connexin43
Myomesin
VEGF
Sarcomeric-actinin
A E
B F
C G
D H
80 m
80 m
80 m
50 m
20 m
50 m
20 m
20 m
7d 14d
DAPI
Sarcomeric-actinin
-catenin
Sarcomeric-actinin
Connexin43
Myomesin
VEGF
Sarcomeric-actinin
A E
B F
C G
D H
80 m80 m
80 m80 m
80 m80 m
50 m
20 m20 m
50 m
20 m20 m
20 m20 m

Myocardial Microtissue Implants 109
-catenin (green; B, F), myomesin (red; C, G), connexin 43 (green; C, G) and vascular endothelial growth factor
(VEGF; green; D, H).
Microchip-based electrophysiologic analysis of myocardial microtissues
To assess inter-cardiomyocyte crosstalk and coordination of electrogenic activity across
NRC-derived microtissues we used metal oxide semiconductor (CMOS) microchip technology
(Greve et al., unpublished). The CMOS chip was designed as a four-block, 20 20 µm2 array,
each containing four platinum electrodes, a circuitry block harboring a high-pass filter and a
programmable amplifier as well as a central external low-pass filter and digitalization units
(Figures 2A and B). Using this CMOS microchip technology we recorded the electrogenic
activities of myocardial microtissues (assembled from 10,000 NRCs for 10 days) for two hours
following phenylephrine-mediated contraction stimulation using the -adrenergic agent
phenylephrine. Although electrogenic signals, recorded by extracellular electrodes represent local
changes in the membrane potential of the individual cardiomycocyte the overall signal is
modulated by the electrically coupled cell quorum (Hescheler et al. 2004). Starting with 0.25 mV
the recorded current increased to 1.75 mV, probably reflecting the initial beating of few
cardiomyocytes which spreads throughout the microtissue and results in coordinated contractions
(Figure 2C). This observation confirms electrophysiologic coupling among cardiomyocytes
embedded in an artificial microtissue structure.

Myocardial Microtissue Implants 110
Figure 2. Microchip-based electrophysiology. The 5 5 mm2 CMOS bio-electronic microchip, mounted on a
ceramic dual-inline (DIL) package (A), was designed as an array of 4 4 platinum electrodes (20 20 µm2),
surrounded by a nitride passivation area and a reference electrode. External circuitries manage signal processing and
chip control. Each four-electrode block harbors a circuitry unit for signal conditioning, which enables the
simultaneous readout of the four electrodes (B). Electrogenic microchip readout of a myocardial microtissue
assembled from 10,000 neonatal rat cardiomyocytes, cultivated for 10 days, placed and maintained for 2 h on an
individual electrode and stimulated by addition of the -adrenergic agent phenylephrine (10-4 mM) (C).
Design and neo-vascularization of higher-order macrotissues assembled from
individual myocardial microtissues
Since inter-microtissue interactions are expected to be similar to graft-host connection,
underlying molecular forces can be exploited to assemble macrotissue supra-structures from
individual microtissues. In designing larger-sized artificial tissue structures for clinical
applications, vascularization, necessary to sustain metabolic activities of the tissue core, will be
A B
C
A B
C

Myocardial Microtissue Implants 111
the prime size-limiting parameter. Previous studies exemplified in-vitro vascularization of human
aortic fibroblast-based core microtissues, whose VEGF production recruited peripheral human
umbilical endothelial cells (HUVEC) to develop a capillary system (Kelm et al. 2005). In order to
produce vascularized myocardial macrotissues from individual microtissues, 300 myocardial
microtissues, assembled from 10,000 NRCs (four-day assembly of core spheroid) coated with
1,200 HUVECs (five days coating in hanging drops), were cast in a custom-designed cylindrical
4% agarose mould, 3 mm in diameter. The agarose mould, produced from a Teflon®
mould,
provided an adhesion-free shape constraint for inter-microtissue crosstalk. Microtissues were
allowed to aggregate in agarose moulds for four days and the resulting macrotissues were kept
for another four days in non-adhesive agarose-coated culture dishes. During maturation, NRC-
HUVEC microtissues assembled into coherent macrotissues characterized by a peripheral layer
of HUVECs exhibiting their typical longitudinal cell morphology (Figure 3A-C). NRC-only
control macrotissues were less coherent and exhibited ruffled tissue structures, probably the
result of apoptosis progression evidenced by nuclear chromatin condensation, apoptotic bodies
and cytoplasm vacuolization (Figures 3E-G). Ultrastructural analysis revealed two types of
developing capillaries but only in HUVEC-containing NRC macrotissues (Figure 3G, H): (i)
tubular structures shaped by two or more endothelial cells connected by intercellular junctions
(Figure 3G) and (ii) lumen formation by a single endothelial cell, which corresponds to a
seamless capillary (Figure 3H).
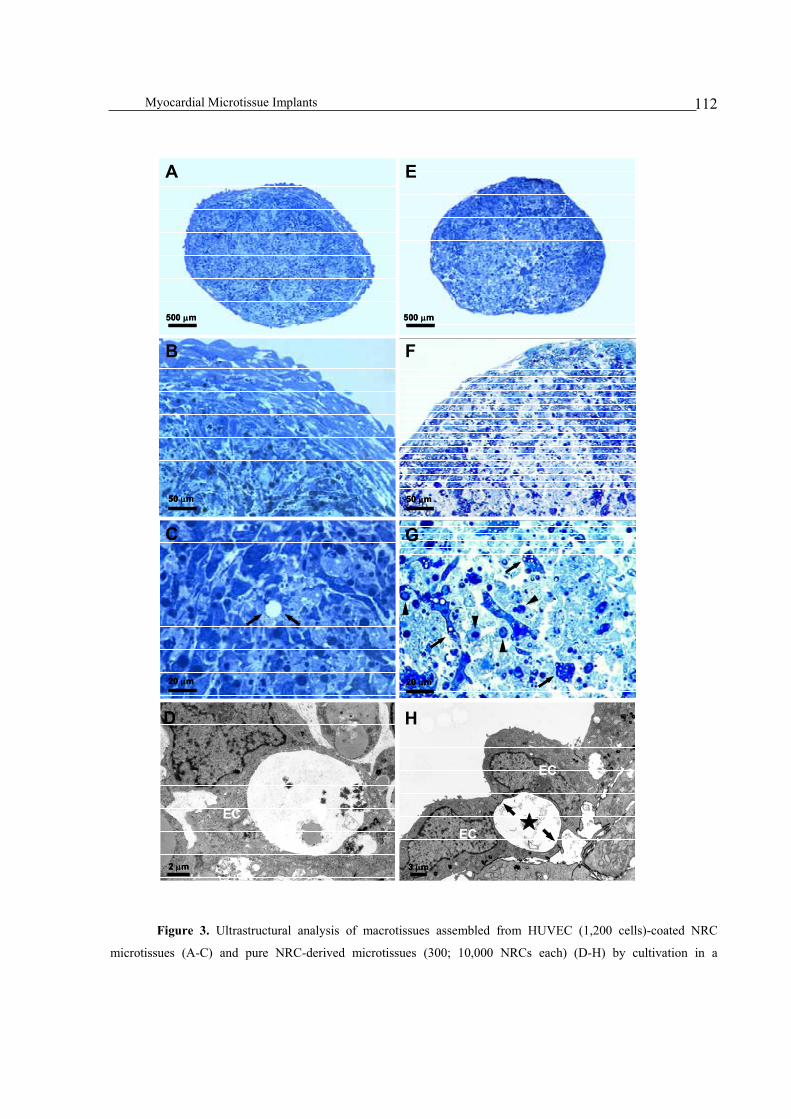
Myocardial Microtissue Implants 112
Figure 3. Ultrastructural analysis of macrotissues assembled from HUVEC (1,200 cells)-coated NRC
microtissues (A-C) and pure NRC-derived microtissues (300; 10,000 NRCs each) (D-H) by cultivation in a
EC
EC
H
3 m
EC
2 m
D
20 m
C
F
50 m
20 m
G
E
500 m
A
500 m
50 m
B
EC
EC
H
3 m
EC
EC
H
3 m3 m
EC
2 m
D
EC
2 m2 m
D
20 m
C
20 m
C
20 m20 m
C
F
50 m
F
50 m50 m
20 m
G
20 m20 m
G
E
500 m
E
500 m500 m
A
500 m
A
500 m500 m
50 m
B
50 m50 m
B

Myocardial Microtissue Implants 113
cylindrical agarose mould (3 mm in diameter) for four days followed by four-days in non-adhesive culture dishes.
Toluidine blue staining of macrotissue sections reveal that pure NRC macrotissues are less coherent and develop
vacuoles (arrows) and apoptotic bodies (arrowheads) (D-H). In contrast, macrotissues produced from HUVEC-
coated NRCs show assembly of HUVECs at the macrotissue’s periphery (A, B) as well as development of capillaries
(C, arrow). Transmission electron micrographs NRC-HUVEC macrotissues reveal developing capillaries
characterized by lumen structures initiated within individual HUVECs (asterisk; arrow indicates lumen) (G) as well
as tubular lumen structures shaped by two or more HUVECs (asterisk; arrows indicate junctions) (H).
Inter-species vascularization crosstalk enables connection of myocardial
microtissues to the chicken embryo vasculature
Connection of artificial tissues to the host vasculature is essential for successful tissue
engineering (Nomi et al. 2002). We have previously reported that rat-derived myocardial
microtissues, exceeding 230 m in diameter (corresponding to 10,000 cells), show hypoxia-
induced VEGF production, which is expected to be one of the key factors for host tissue-
mediated vascularization of artificial tissue implants (Kelm et al. 2004). To study microtissue-
host vascularization crosstalk we implanted NRC-derived myocardial microtissues atop the
chorioallantoic membrane (CAM) of chicken embryos. Morphological analysis at the
microtissue/CAM interface revealed complete integration of the NRC-derived microtissue into
the chicken CAM (Figure 4A). Following a well-evolved vascularization scheme chicken and rat
cells inter-operated seamlessly to establish a fully functional joint vasculature (Figure 4B-C).
Microtissue-based VEGF production was sufficient to recruit the chicken embryo’s vasculature
to invade and fully vascularized the microtissue graft. Ultrastructural analysis of microtissue
implants revealed mature pericyte-covered microvessels (Figure 4E, F). Efficient recruitment of
host vascularization by microtissue-mediated VEGF production suggests that prevascularization
of artificial tissue implants may be dispensable for some transplantation scenarios.
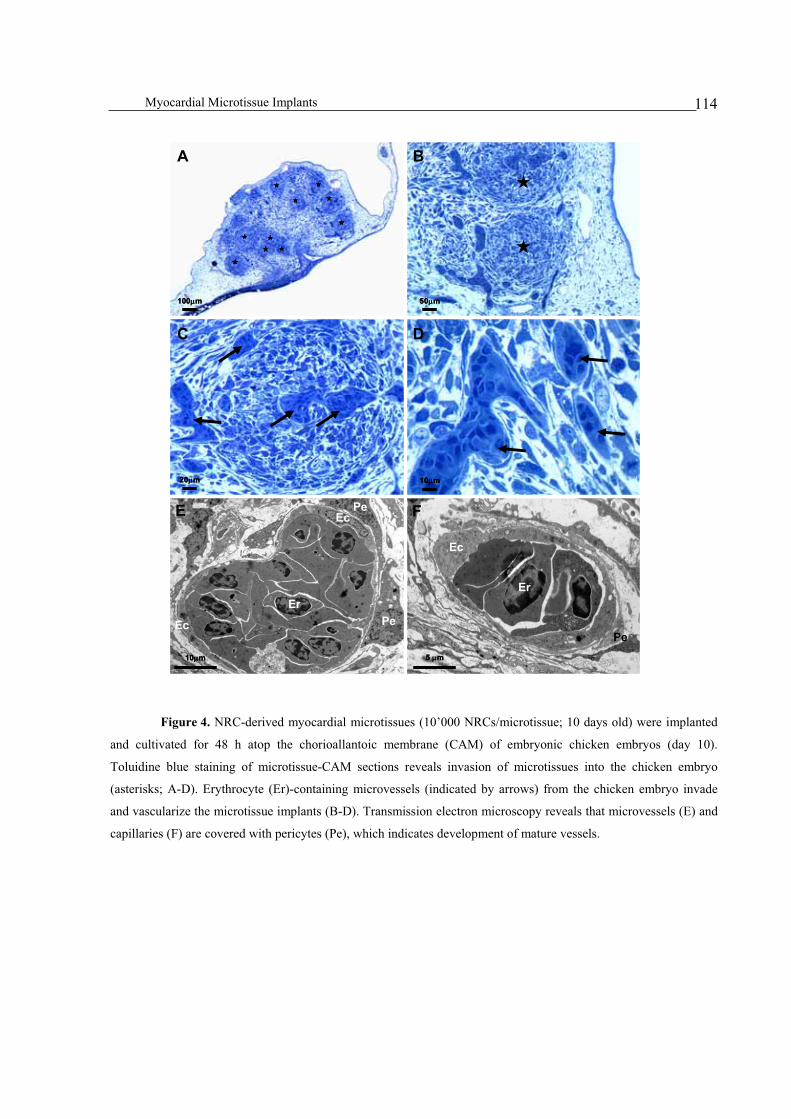
Myocardial Microtissue Implants 114
Figure 4. NRC-derived myocardial microtissues (10’000 NRCs/microtissue; 10 days old) were implanted
and cultivated for 48 h atop the chorioallantoic membrane (CAM) of embryonic chicken embryos (day 10).
Toluidine blue staining of microtissue-CAM sections reveals invasion of microtissues into the chicken embryo
(asterisks; A-D). Erythrocyte (Er)-containing microvessels (indicated by arrows) from the chicken embryo invade
and vascularize the microtissue implants (B-D). Transmission electron microscopy reveals that microvessels (E) and
capillaries (F) are covered with pericytes (Pe), which indicates development of mature vessels.
A
D
E
Er
Er
Pe
PeEc
Ec
Ec
Pe
C
B
F
10 m 5 m
20 m 10 m
100 m 50 m
A
D
E
Er
Er
Pe
PeEc
Ec
Ec
Pe
C
B
F
10 m 5 m
20 m 10 m
100 m 50 m
A
D
E
Er
Er
Pe
PeEc
Ec
Ec
Pe
C
B
F
10 m10 m 5 m5 m
20 m20 m 10 m10 m
100 m100 m 50 m50 m

Myocardial Microtissue Implants 115
Integration of implanted myocardial microtissues into rat hearts
To characterize in-vivo integration of myocardial microtissues into the heart of adult rats
and evaluate their potential as minimal therapeutic tissue engineering units we injected NRC-
derived microtissues into the pericardial cavity. Myocardial microtissue implants were produced
by gravity-enforced self-assembly of 2,500 monodispersed NRCs for four days in hanging drops
and stained with a fluorescent CellTrackerTM
to trace tissue implants in the animal (Zhang et al.
2001). Implantation required a surgery (15 min); a 10 mm incision was made behind the xiphoid
of anesthetized adult rats followed by chest wall lift-up, injection of 300 microtissues (in 200 l
HBSS) into the pericardial cavity, sealing of the puncture with fibrin glue and suturing of the
abdominal incision. The rats were sacrificed one, four or seven day(s) post surgery. Fluorescence
microscopy revealed that CellTrackerTM-labbeled microtissues had penetrated the myocard by
day 4 (Figure 5). Confocal microscopy analysis of sarcomeric -actinin-specific fluorescent
staining of frozen heart explant sections revealed that myocardial microtissues were already
being incorporated into the rat heart 24 h post implantation (Figure 6A, B). Four days after
infection, the myocardial microtissue was completely embedded in the rat heart, but its spherical
shape was retained (Figure 6C, D). However, by day 7, the transplanted myocardial microtissues
had completed seamless resorption in the absence of structural abnormalities and displayed a
longitudinal cell shape, characteristic of the neighboring host cardiomyocytes. (Figure 6E, F).
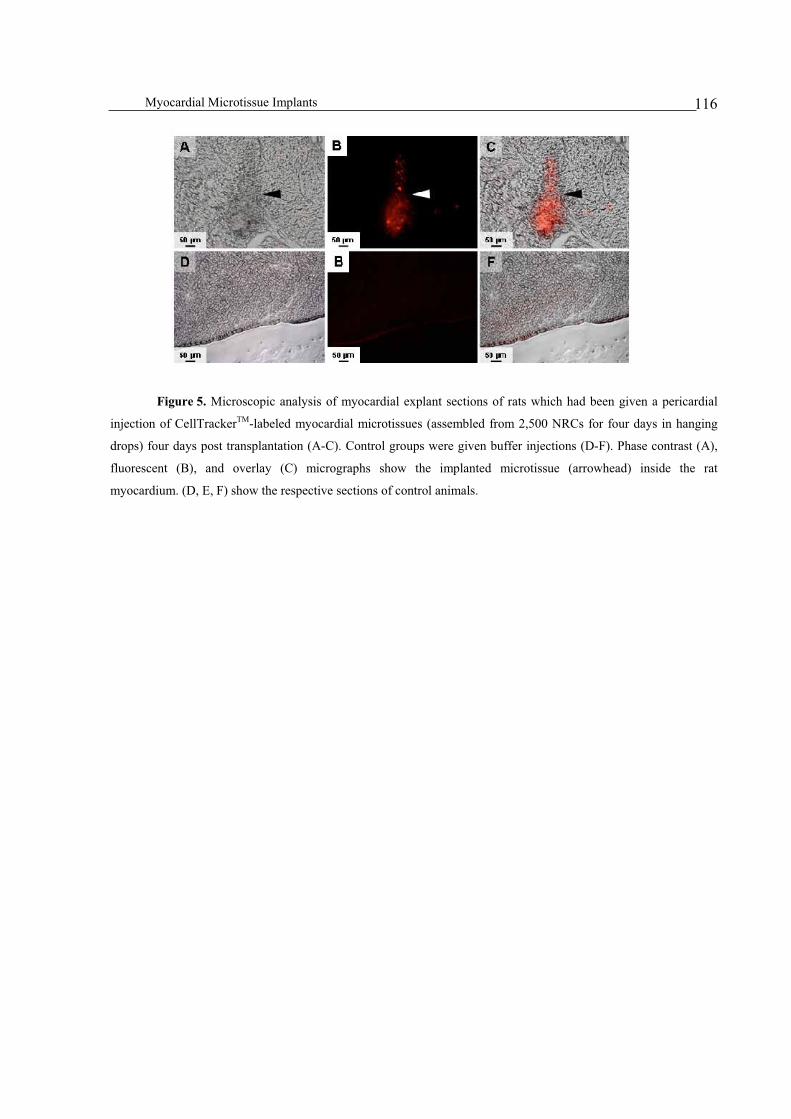
Myocardial Microtissue Implants 116
Figure 5. Microscopic analysis of myocardial explant sections of rats which had been given a pericardial
injection of CellTrackerTM-labeled myocardial microtissues (assembled from 2,500 NRCs for four days in hanging
drops) four days post transplantation (A-C). Control groups were given buffer injections (D-F). Phase contrast (A),
fluorescent (B), and overlay (C) micrographs show the implanted microtissue (arrowhead) inside the rat
myocardium. (D, E, F) show the respective sections of control animals.
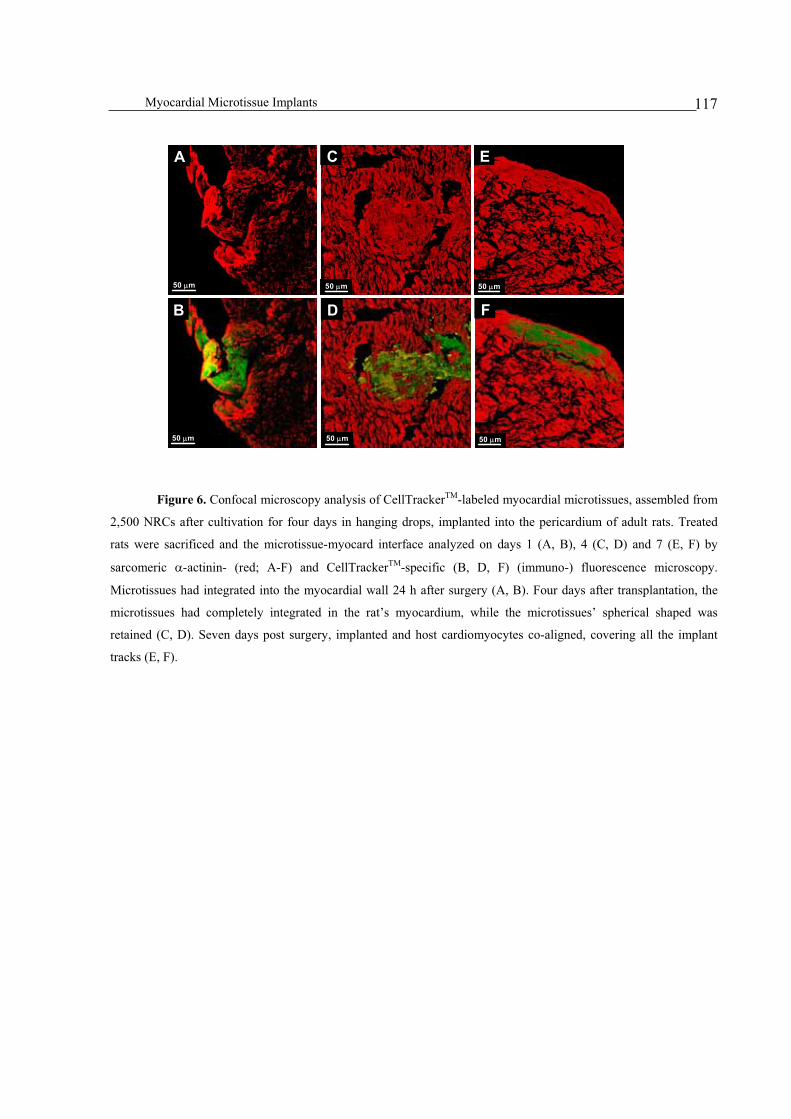
Myocardial Microtissue Implants 117
Figure 6. Confocal microscopy analysis of CellTrackerTM-labeled myocardial microtissues, assembled from
2,500 NRCs after cultivation for four days in hanging drops, implanted into the pericardium of adult rats. Treated
rats were sacrificed and the microtissue-myocard interface analyzed on days 1 (A, B), 4 (C, D) and 7 (E, F) by
sarcomeric -actinin- (red; A-F) and CellTrackerTM-specific (B, D, F) (immuno-) fluorescence microscopy.
Microtissues had integrated into the myocardial wall 24 h after surgery (A, B). Four days after transplantation, the
microtissues had completely integrated in the rat’s myocardium, while the microtissues’ spherical shaped was
retained (C, D). Seven days post surgery, implanted and host cardiomyocytes co-aligned, covering all the implant
tracks (E, F).
50 m
C
50 m
D
50 m
E
50 m
F
50 m
A
50 m
B
50 m
C
50 m50 m
CC
50 m
D
50 m50 m
DD
50 m
E
50 m50 m
EE
50 m
F
50 m50 m
FF
50 m
A
50 m50 m
AA
50 m
B
50 m50 m
BB

Myocardial Microtissue Implants 118
Discussion
Because of the limited regeneration capacity of the adult heart damage to the
muscle usually results in irreversible cardiac dysfunction. The design of myocardial regeneration
strategies is thus important to the patient because of the limitations of standard replacement
therapies. Recent reports suggested that endogenous cardiac stem cells may proliferate in the
myocardium under certain conditions and that stem cells infiltrate from the bone marrow to the
heart where they are expected to contribute to repair (Messina et al. 2004; Schuster et al. 2004).
However, discussions are ongoing as to whether bone marrow-derived cells, which penetrate
target tissues, are reprogrammed by the native tissue environment or fuse with target cells
requiring repair (Nygren et al. 2004; Pomerantz and Blau 2004). In the past decade, cardiac cell
transplantation has emerged as the leading strategy for replacing damaged or diseased
myocardium (Melo et al. 2004). Several transplantation initiatives have shown that direct
injections of cell suspensions of fetal neonatal cardiac myocytes into experimental myocardial
infarcts improved the remodeling and function of the heart (Reffelmann et al. 2003). Others have
replicated those encouraging results by using skeletal myoblasts (Menasche et al. 2001), bone
marrow-derived cells (Orlic et al. 2002) or embryonic stem cells (Gepstein 2002; Kehat et al.
2004). Although post transplantation survival was rather limited, only cardiomyocytes enabled
seamless electrogenic cell-host tissue contacts devoid of arrhythmias (Rubart et al. 2003).
To solve the problems associated with single-cell injections, tissue engineers advanced
scaffold design to (i) provide an extracellular matrix substitute for the infarct area, (ii) supply a
temporary support for self or implanted cells and (iii) control the size, shape, strength and
composition of the graft in vitro (Leor and Cohen 2004; Olson 2004). There are a variety of
techniques for constructing beating cardiac patches for transplantation: (i) Shimizu and
colleagues grew rat cardiomyocyte monolayers on polymer surfaces, which were then detached,
stacked and allowed to fuse before subcutaneous implantation into rats (Shimizu et al. 2002). Six
months post transplantation, the cardiac patch was beating and had been infiltrated by host blood
vessels. (ii) Eschenhagen and coworkers produced rings of engineered cardiac muscle by seeding
neonatal rat cardiomyocytes either into a scaffold or by mixing them with collagen and shaping
the tissue in a ring template (Zimmermann et al. 2002; Zandonella 2003; Zimmermann and
Eschenhagen 2003). Instead of seeding cells into scaffolds, cardiac tissues could also be

Myocardial Microtissue Implants 119
assembled by organ printing (Jakab et al. 2004). This prototype strategy uses a cell printer which
sequentially plots cells onto a thin thermo-reversible gel support following a software blueprint
of the desired organ shape.
No matter how sophisticated and functionalized scaffolds become, they will remain alien
to the native tissue, with the risk for complications. With the advent of myocardial microtissues,
produced by gravity-enforced self-assembly of monodispersed cardiomyocytes in hanging drops,
the question as to whether scaffolds may become dispensable must be posed again (Kelm et al.
2004). Microtissue production is straightforward and highly flexible, enabling the design of pure
or multi-cell type spheroids as well as cell layers coated onto a core feeder spheroid using a
variety of different cell types including embryonic stem cells, chondrocytes, hepatocytes, retinal
cells, tumor cells and cardiomyocytes (Itskovitz-Eldor et al. 2000; Anderer and Libera 2002;
Layer et al. 2002; Kelm and Fussenegger 2004; Timmins et al. 2004). Thus far, microtissues have
provided a welcome system to for studying tissue assembly, inter-cellular crosstalk and drug
function in a tissue-like in vitro context (Kelm et al. 2005). However, little is known about the
potential of microtissues for regenerative medicine. Recent advances in myocardial microtissue
design have (i) established a linear cell number-microtissue size correlation, (ii) suggested inter-
microtissue superstructures, (iii) shown key cardiomyocyte-specific cell qualities and have
resulted in the (iv) development of an extracellular matrix, (v) the coordinated contraction of the
microtissue’s cell quorum and (v) the size-dependent production of VEGF (Kelm et al., 2004).
Using a novel CMOS-based microchip design we assessed the electrogenic characteristics
of myocardial microtissues (Heer et al. 2004). Electric signal readout from NRC-derived
microtissues revealed that the contraction of cardiomyocytes was coordinated, suggesting
functional electrochemical/mechanical coupling by intercalated disks prominent in these tissue
structures. Refinement of the interface between the biological systems and the microchip is
expected to (i) enable quality control of cardiac tissue implants, (ii) allow critical evaluation of
tissue explants of patients suffering from cardiac-related pathologies and optimize related
diagnosis and (iii) provide a platform for sophisticated drug discovery and/or drug-function
analysis.
The future success of cardiac tissue engineering in regenerative medicine will depend on
the design of larger tissues but with increased tissue size control of vascularization to prevent

Myocardial Microtissue Implants 120
ischemia will become more important. Capitalizing on native inter-microtissue interactions we
assembled individual microtissues to functional macrotissues. Control of shape, which is often
thought to be an exclusive asset of scaffolds, was achieved by casting individual microtissues
into custom-designed agarose moulds. Micro- as well as larger cardiac tissues were vascularized
in-vitro by co-assembly of HUVEC and cardiac myocytes, which resulted in a time-dependent
development of a robust capillary network across the myocardical tissues ready to be connected
to native tissues. Indeed, 60 hours after transplantation of myocardial microtissues into chicken
embryos, vascular crosstalk, resulting in the functional connection of the graft tissue to the
embryo, had been established. Furthermore, artificial heart tissues, implanted into the
pericardium of adult rats, integrated seamlessly into the myocardium, confirming in-vivo
compatibility of artificial heart patches.
With several sophisticated scaffold-based/free technologies available and
prototypic organ printing on the rise, electrogenic coupling, under the control and management of
vascularization on track, cardiac tissue engineers are fortunate to have a complementary
technology portfolio at their disposal enabling them to design clinical therapies in the forseeable
future. By combining the aforementioned technologies with high-throughput-compatible tissue
assembly systems and the newest bioprocess engineering tailored for the mass-production of
desired cell phenotypes, tissue engineers may succeed in providing next-generation strategies for
regenerative medicine.
Acknowledgments
We thank Evelyne Perriard for providing neonatal rat cardiomyocytes and Semjidmaa
Dashnyam for isolating of adult rat cardiomyocytes as well as Krystyna Sala-Szymanska for
preparing of the histological samples. This work was supported by the Swiss National Science
Foundation (grant no. 631-065946) and the Swiss State Secretariat for Education and Research
within EC Framework 6.
References
Anderer, U. and J. Libera (2002). "In vitro engineering of human autogenous cartilage." J Bone
Miner Res 17, 1420-1429.

Myocardial Microtissue Implants 121
Auerbach, D., S. Bantle, et al. (1999). "Different domains of the M-band protein myomesin are
involved in myosin binding and M-band targeting." Mol Biol Cell 10, 1297-1308.
Datta, S. R., A. Brunet, et al. (1999). "Cellular survival: a play in three Akts." Genes Dev 13,
2905-2927.
Djonov, V., M. Schmid, et al. (2000). "Intussusceptive angiogenesis: its role in embryonic
vascular network formation." Circ Res 86, 286-292.
El Oakley, R. M., O. C. Ooi, et al. (2001). "Myocyte transplantation for myocardial repair: a few
good cells can mend a broken heart." Ann Thorac Surg 71, 1724-1733.
Eppenberger, H. M. and C. Zuppinger (1999). "In vitro reestablishment of cell-cell contacts in
adult rat cardiomyocytes. Functional role of transmembrane components in the formation
of new intercalated disk-like cell contacts." Faseb J 13 Suppl, S83-89.
Gepstein, L. (2002). "Derivation and potential applications of human embryonic stem cells." Circ
Res 91, 866-876.
Grove, B. K., V. Kurer, et al. (1984). "A new 185,000-dalton skeletal muscle protein detected by
monoclonal antibodies." J Cell Biol 98, 518-524.
Hassink, R. J., A. Brutel de la Riviere, et al. (2003). "Transplantation of cells for cardiac repair."
J Am Coll Cardiol 41, 711-717.
Heer, F., W. Franks, et al. (2004). "CMOS microelectrode array for the monitoring of
electrogenic cells." Biosens Bioelectron 20, 358-366.
Hescheler, J., M. Halbach, et al. (2004). "Determination of electrical properties of ES cell-derived
cardiomyocytes using MEAs." J Electrocardiol 37 Suppl, 110-116.
Itskovitz-Eldor, J., M. Schuldiner, et al. (2000). "Differentiation of human embryonic stem cells
into embryoid bodies compromising the three embryonic germ layers." Mol Med 6, 88-
95.
Jakab, K., A. Neagu, et al. (2004). "Organ printing: fiction or science." Biorheology 41, 371-375.
Kehat, I., L. Khimovich, et al. (2004). "Electromechanical integration of cardiomyocytes derived
from human embryonic stem cells." Nat Biotechnol 22, 1282-1289.
Kelm, J. M., C. Diaz S-Bustamante, et al. (2005). "VEGF Profiling and Angiogenesis in Human
Microtissues." Journal of Biotechnology in press.
Kelm, J. M., E. Ehler, et al. (2004). "Design of Artificial Myocardial Microtissues." Tissue Eng
10, 201-214.

Myocardial Microtissue Implants 122
Kelm, J. M. and M. Fussenegger (2004). "Microscale tissue engineering using gravity-enforced
cell assembly." Trends Biotechnol 22, 195-202.
Lanza, R., M. A. Moore, et al. (2004). "Regeneration of the infarcted heart with stem cells
derived by nuclear transplantation." Circ Res 94, 820-827.
Layer, P. G., A. Robitzki, et al. (2002). "Of layers and spheres: the reaggregate approach in tissue
engineering." Trends Neurosci 25, 131-134.
Leor, J. and S. Cohen (2004). "Myocardial tissue engineering: creating a muscle patch for a
wounded heart." Ann N Y Acad Sci 1015, 312-319.
Melo, L. G., A. S. Pachori, et al. (2004). "Gene and cell-based therapies for heart disease." Faseb
J 18, 648-663.
Menasche, P., A. A. Hagege, et al. (2001). "Myoblast transplantation for heart failure." Lancet
357, 279-280.
Messina, E., L. De Angelis, et al. (2004). "Isolation and expansion of adult cardiac stem cells
from human and murine heart." Circ Res 95, 911-921.
Murry, C. E., M. H. Soonpaa, et al. (2004). "Haematopoietic stem cells do not transdifferentiate
into cardiac myocytes in myocardial infarcts." Nature 428, 664-668.
Nomi, M., A. Atala, et al. (2002). "Principals of neovascularization for tissue engineering." Mol
Aspects Med 23, 463.
Nygren, J. M., S. Jovinge, et al. (2004). "Bone marrow-derived hematopoietic cells generate
cardiomyocytes at a low frequency through cell fusion, but not transdifferentiation." Nat
Med 10, 494-501.
Olson, E. N. (2004). "A decade of discoveries in cardiac biology." Nat Med 10, 467-474.
Orlic, D., J. M. Hill, et al. (2002). "Stem cells for myocardial regeneration." Circ Res 91, 1092-
1102.
Pasumarthi, K. B. and L. J. Field (2002). "Cardiomyocyte cell cycle regulation." Circ Res 90,
1044-1054.
Pomerantz, J. and H. M. Blau (2004). "Nuclear reprogramming: a key to stem cell function in
regenerative medicine." Nat Cell Biol 6, 810-816.
Reffelmann, T., J. Leor, et al. (2003). "Cardiomyocyte transplantation into the failing heart-new
therapeutic approach for heart failure?" Heart Fail Rev 8, 201-211.

Myocardial Microtissue Implants 123
Reinecke, H., M. Zhang, et al. (1999). "Survival, integration, and differentiation of
cardiomyocyte grafts: a study in normal and injured rat hearts." Circulation 100, 193-202.
Ribatti, D., B. Nico, et al. (2001). "Chorioallantoic membrane capillary bed: a useful target for
studying angiogenesis and anti-angiogenesis in vivo." Anat Rec 264, 317-324.
Rubart, M., K. B. Pasumarthi, et al. (2003). "Physiological coupling of donor and host
cardiomyocytes after cellular transplantation." Circ Res 92, 1217-1224.
Schuster, M. D., A. A. Kocher, et al. (2004). "Myocardial neovascularization by bone marrow
angioblasts results in cardiomyocyte regeneration." Am J Physiol Heart Circ Physiol 287,
H525-532.
Shimizu, T., M. Yamato, et al. (2002). "Fabrication of pulsatile cardiac tissue grafts using a novel
3-dimensional cell sheet manipulation technique and temperature-responsive cell culture
surfaces." Circ Res 90, e40.
Timmins, N. E., S. Dietmair, et al. (2004). "Hanging-drop multicellular spheroids as a model of
tumour angiogenesis." Angiogenesis 7, 97-103.
Zandonella, C. (2003). "Tissue engineering: The beat goes on." Nature 421, 884-886.
Zhang, M., D. Methot, et al. (2001). "Cardiomyocyte grafting for cardiac repair: graft cell death
and anti-death strategies." J Mol Cell Cardiol 33, 907-921.
Zimmermann, W. H., M. Didie, et al. (2002). "Cardiac grafting of engineered heart tissue in
syngenic rats." Circulation 106, I151-157.
Zimmermann, W. H. and T. Eschenhagen (2003). "Cardiac tissue engineering for replacement
therapy." Heart Fail Rev 8, 259-269.

Chapter 7
Design of Custom-Shaped Vascularized Tissues
Using Microtissue Spheroids as
Minimal Units
Kelm J.M., Djonov V., Ittner L.M., Born W., Hoerstrup S.P. and Fussenegger M. (submitted)

Macrotissue Generation 125
Abstract
Tissue engineering strategies are gathering clinical momentum in regenerative medicine
and are expected to provide excellent opportunities for the therapy of difficult-to-treat human
pathologies. Being aware of the requirement to produce lager-sized artificial tissue implants for
clinical applications, we used microtissues, produced by gravity-enforced self-assembly of
monodispersed primary cells, as minimal tissue units to generate scaffold-free vascularized
artificial macrotissues in custom-shaped agarose moulds. Mouse myoblast, pig and human
articular chondrocyte (PAC, HAC) as well as human myofibroblast- (HMF) derived microtissues
(µm3 scale) were all amalgamated to coherent macrotissues (mm
3 scale) of the desired shape and
to native tissue ultra-structures. Macrotissues, assembled from the human umbilical vein
endothelial cell (HUVEC)-coated HMF microtissues, developed a HUVEC-managed vascular
system, which functionally connected to the chicken embryo’s vasculature following
implantation. The design of scaffold-free vascularized macrotissues is a first step towards the
scale-up and production of artificial tissue implants for future tissue engineering initiatives.
Introduction
Tissue engineering integrates engineering principles and biological systems in order to
create therapeutic replacement structures (Vacanti and Langer 1999). Tissue development and
maturation is a process of high biological complexity and requires precise orchestration of cell
fate control, metabolic activities, structure-function correlations and crosstalk between and
among different cell populations in space and time. Mastering the custom-shaped assembly of
multiple cell types in an innervated and neovascularized three-dimensional (3D) format remains a
major challenge for current tissue engineering initiatives (Peirce and Skalak 2003; Mol et al.
2004). Pioneering strategies of tissue engineering were based on seeding cells into biodegradable
polymer scaffolds, culturing them in bioreactors and implanting the resulting tissue into the
recipient organism, where maturation of the new organ takes place. Scaffolds were always
thought to be important for tissue engineering as they provide biological, physical and chemical
cues to guide cellular differentiation and assembly in the third dimension (Hubbell 2003; Shin et

Macrotissue Generation 126
al. 2003). Despite advances in biomaterial science, which led to scaffolds with fewer
transplantation side effects resulting from toxic degradation products, the induction of
inflammatory reactions and poor resorption (Yang et al. 2001), most scaffolds fail to promote
vascularization unless they have been specifically functionalized (Vogel and Baneyx 2003).
Since the discovery that monodispersed cells aggregate to spheroids by gravity-enforced self-
assembly in hanging drops and the observation that multicellular spheroids self-organize into
functional tissue-like units according to the differential adhesion hypothesis, scaffold-free
microtissues have been considered as an alternative to artificial scaffold-containing tissues
(Steinberg and Foty 1997; Layer et al. 2002; Kelm and Fussenegger 2004). Self-assembly of
monodispersed cells to microtissues has been successful with a variety of different cell types
including (i) reconstruction of retinal layers (Rothermel and Layer 2001), (ii) design of functional
myocardial microtissues (Kelm et al. 2004), (iii) production of artificial ganglia (Kelm et al.
2005), (iv) construction of cartilage (Anderer and Libera 2002), and tumor cell lines (Kunz-
Schughart et al. 2004). Microtissues assembled in hanging drops attain diameters of only a few
hundred micrometers (100-500 µm in diameter). Beyond this size, diffusion-based nutrient and
oxygen supplies become limiting. In living tissues, cells rely on a capillary network within a
perimeter of 100-150 micrometers for oxygen and nutrient supplies (Tsai et al. 2003).
As well as the innervation and neovascularization of artificial tissues, interconnection to
respective host systems after implantation remain major challenges of current tissue engineering
initiatives (Lauffenburger and Griffith 2001). Neovascularization may be supported by (i)
functionalizing scaffolds with pro-angiogenic factors (Richardson et al. 2001; Zisch et al. 2003;
Zisch et al. 2003; Hall and Hubbell 2004), (ii) ectopic expression of pro-angiogenic factors
(Ajioka et al. 2001) or (iii) by taking advantage of cell-based in-vitro vascularization (Timmins et
al. 2004; Kelm et al. 2005).
For microtissue technology to make an impact on regenerative medicine, microtissues
must be scaled up to larger fully vascularized macrotissues. We have therefore determined
whether microtissues meet those requirements, by assembling microtissues to custom-shaped
neovascularized macrotissues and implanted them into chicken embryos. The observation that
artificial tissues, produced by microtissue scale-up and integrated into host tissues, were
functionally connected to the host vascular system post implantation suggests future
opportunities for applying microtissue-based tissue engineering to regenerative medicine.

Macrotissue Generation 127
Material and Methods
Preparation of primary cells
The preparation of primary human myofibroblasts (HMFs) included mincing arterial
mammary segments followed by cultivation at 37°C in a humidified 5% CO2-containing
atmosphere in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, Carlsbad, CA)
supplemented with 10% fetal bovine serum (FBS; cat. no. 3302-P231902, lot no. P231902; PAN
Biotech GmbH, Aidenbach) and 1% penicillin/streptomycin solution (Invitrogen). Pure HMFs,
which had migrated from the tissue pieces after 10 to 14 days, were serially passaged and
expanded to the desired cell numbers in advanced DMEM (Invitrogen) supplemented with 10%
FBS and GlutaMax (Invitrogen) for four to six weeks under the above aforementioned
conditions.
Cell Culture
Pig (PAC) and human articular chondrocytes (HAC) (kindly provided by Millenium
Biologix, Schlieren, Switzerland) were expanded in DMEM/F12 (Invitrogen) supplemented with
10% FBS, mouse myoblasts C2C12 (ATCC CRL-1772) in DMEM (Invitrogen) supplemented
with 20% FBS (PAN Biotech GmbH, Aidenbach) and human umbilical vein endothelial cells
(HUVECs, PromoCell, Heidelberg, Germany) in endothelial cell growth medium (PromoCell,
cat. no. C-22010) supplemented with 10% FBS. All cell types were cultivated at 37°C in a
humidified 5% CO2-containing atmosphere.
Microtissue Production
After isolation and expansion, the cells were seeded at the indicated cell concentrations
and cell-type compositions into 60-well plates (HLA plate, Greiner-Bio One, Frickenhausen,
Germany). In order to enable gravity-enforced self-assembly of the microtissues in hanging
drops, the 60-well plates were incubated upside down at 37°C in a humidified atmosphere
containing 5% CO2. The specific cultivation conditions for microtissue production are outlined in
Table 1.

Macrotissue Generation 128
Macrotissue Assembly
In order to assemble microtissues to larger artificial tissues we designed two different
agarose casting moulds, (i) a cylindrical mould, 3 mm in diameter, and (ii) a ring-shaped mould,
1 mm thick (Figure 1A). Negative Teflon®
casting moulds were filled with 4% agarose (Sigma
Chemicals, Buchs, Switzerland) in phosphate-buffered saline (PBS; 150 mM NaCl, 6.5 mM
Na2HPO4 x 2 H2O, 2.7 mM KCL, 1.5 mM KH2PO4, pH 7.4) to generate non-adhesive positive
casting moulds to imprint the desired shape on the macrotissue. Microtissues of the indicated
types and numbers of cells were transferred to the respective agarose moulds and cultivated in
cell type-specific media under static conditions at 37°C in a humidified atmosphere containing
5% CO2 (see Table 1 for specific culture conditions).
Immunohistochemistry
The tissues were fixed in 2% paraformaldehyde, rinsed in 15% sucrose solution and
stored at -20°C in 70% ethanol. All the samples were dehydrated by sequential incubation in
increasing ethanol concentrations and embedded in paraffin wax. 3 µm sections, produced using
an Ultracut device (Zeiss, Feldbach, Switzerland), were transferred to gelatinized micro-slides
and air-dried overnight at 37°C. The samples were de-waxed in xylene (three changes),
rehydrated in ethanol and rinsed in Tris-buffered saline (TBS, 20 mM Tris base, 155 mM NaCl, 2
Table 1. Culture media used for cell expansion, microtissue production and macrotissues assembly
Cell Type Cell Expansion
Medium
Microtissue Assembly
Medium
Microtissue Cell
Number
Microtissue
Assembly Time
Microtissues Per
Macrotissue
Tissue Assembly
Time
C2C12 DMEM
+ 20% FBS
DMEM
+ 10% HS 10’000 4 [d] 1’200 10 [d]
PAC/HAC DMEM/F12
+ 10% FBS
DMEM
+ 10% HuS 5’000 5 [d] 600 14 [d]
HMF Adv. DMEM
+ 10% FBS
DMEM
+ 10% HuS 10’000
4 [d] + 4 [d] (HUVEC coating)
300 7 [d]
HUVEC ECGM
+ 10% FBS
___ 1’200 ___ 1’200 ___
Abbreviations: advanced DMEM, cell culture medium (Invitrogen); C2C12, mouse myoblast cell line; DMEM, Dulbecco`s Modified Eagle Medium
(Invitrogen); DMEM/F12, culture medium (Invitrogen); ECGM, endothelial cell growth medium (PromoCell); HAC, human articular chondrocytes;
HMF, human myofibroblasts; HS, horse serum (cat. no. 16050-098, lot no. 3036354D, Invitrogen); HUVEC, human umbilical vein endothelial cells;FBS, fetal bovine serum (PAN Biotech GmbH); HuS: human serum (cat. No. P30-2501, lot no. P520221, PAN Biotech GmbH); PAC, pig articular
chondrocytes.

Macrotissue Generation 129
mM EGTA, 2 mM MgCl2) (two changes). Endogenous peroxidase activity was eliminated by
treatment with 0.3% hydrogen peroxide for 10 min. The sections were subsequently heated for 15
min in a microwave oven (180 W) and blocked by incubation for 10 min in TBS containing 1%
casein (Sigma Chemicals). All sections were then incubated for 15 h at 4°C in TBS containing
primary antibodies specific for VEGF (rabbit polyclonal, Santa Cruz Biotechnology, Santa Cruz,
CA, USA) and CD31 (mouse monoclonal, Dako, Glostrup, Denmark). Then, the sections were
exposed to an affinity-purified biotinylated secondary antibody ([anti-mouse EO 433, anti-rabbit
EO 353, Dako, Glostrup, Denmark] diluted 1:200 in TBS) for 45 min at room temperature,
washed three times in TBS and then treated with the straptavidin-biotin-complex/horseradish
peroxidase (Dako, Glostrup, Denmark) for another 45 min at room temperature. The reaction
product was visualized by exposing the sections to 3-amino-9-ethylcarbazole or 3.3-
diaminobenzidine (Sigma Chemicals), which were then mounted in Aquatex (Merck, Darmstadt,
Germany). The negative controls were stained with non-specific mouse and rabbit sera.
Transmission electron microscopy
Micro- and macro-tissues were fixed by immersion in 0.1 M cacodylate buffer (pH 7.4,
350 mOsm) containing 2.5% glutaraldehyde. The tissue blocks were postfixed in osmium
tetroxide, block-stained using uranyl acetate, dehydrated by sequential incubation in increasing
ethanol concentrations and embedded in Epon 812 according to Djonov and coworkers (all
chemicals from Merck Eurolab AG, Dietikon, Switzerland) (Djonov et al. 2000). Semi-thin 1 µm
sections were stained with toluidine blue and visualized using an Olympus Vanox BHS light
microscope (Olympus AG, Volketswil, Switzerland). Ultra-thin sections of 80-90 nm were cut
using a diamond knife and picked up on Formvar-coated (polyvinyl formal; Fluka Chemie AG)
copper grids, double-stained with lead citrate (Merck Eurolab AG) and uranyl acetate and
monitored on a Philips EM 400 electron microscope (FEI AG, Zurich, Switzerland).
Chicken chorioallantoic membrane (CAM) assay
Chicken embryos were cultured according to the shell-free method (Ribatti et al. 2001).
Artificial tissues were grafted onto the growing chicken chorioallantoic membrane (CAM) at
embryonic day 10 and cultured for 3.5 days.

Macrotissue Generation 130
Results
Microtissue assembly to larger-sized macrotissues
Although microtissues enable new insight into intercellular crosstalk, specific 3D cell
phenotypes and tissue assembly, they are considered too to be small for generating clinical
impact in tissue engineering. Capitalizing on the tissue-like cell morphologies adopted in
microtissues we evaluated their potential to serve as building blocks for larger artificial tissues.
Therefore, 1,200 microtissues, produced from 10,000 mouse myoblasts (C2C12) aggregated in
hanging drops for four days, were assembled to macrotissues in custom-designed ring-shaped
1 mm thick agarose moulds for three days (Figure 1A). The resulting macrotissues were cut once
to produce a tissue string, which was cultivated for another week in agarose-coated culture
dishes. Macroscopic examination revealed that individual microtissues had amalgamated to a
coherent tissue structure (Figure 1B). Toluidine blue staining of 1 µm sections showed highly
organized tissue morphology reminiscent of muscle fibers composed of individual multinucleated
cells (Figures 1C-E). Although ultra-structural analysis revealed a typical longitudinal shape of
muscle cells, myofibrils did not develop, most likely due absence of mechanical forces and/or
electrogenic stimulation (Figure 1F) (Fink et al. 2000; Benjamin and Hillen 2003).

Macrotissue Generation 131
Figure 1. Production of C2C12-derived macrotissues. (a) Agarose mould generated from a negative
Teflon® form to assemble C2C12-derived microtissues to 1 mm thick ring-shaped muscle macrotissues. The ring-
shaped casting chamber is visualized by a blue dye. (b) C2C12-derived macrotissue assembled from 1,200 C2C12
microtissues, each containing 10,000 myoblasts, for three days and cultivated for another seven days in non-adhesive
agarose-coated plates. (d, e) Toluidine blue-stained semi-thin sections of muscle macrotissues reveals a coherent
tissue structure containing regions with a high content of multi-nucleated cells (d, e, arrows). Ultrastructural analysis
shows that longitudinally oriented muscle cells are interconnected by cell-cell contacts (F, circle).
ba
c d
e f
2 mm 0.5 mm
50 m 10 m
10 m 5 m
bbaa
cc dd
ee ff
2 mm 0.5 mm0.5 mm
50 m50 m 10 m10 m10 m
10 m10 m10 m 5 m5 m

Macrotissue Generation 132
Six hundred cartilage microtissues, assembled from 5,000 pig articular chondrocytes
(PAC) or human articular chondrocytes (HAC) for five days in hanging drops, were shaped into a
cylindrical macrotissue (3 mm in diameter) by cultivation in a specific agarose mould for seven
days in the absence of collagen-inducing factors like TGF- or ascorbic acid. Artificial cartilage
tissues were subsequently cultivated for another week in agarose-coated culture dishes. PAC-
derived microtissues had amalgamated into coherent tissues (Figure 2A) characterized by the
ubiquitous production of collagen (Figures 2B-F). Akin to the PAC-composed tissues, HAC-
derived artificial cartilage showed areas of increased collagen production (Figures 3A-F).
Toluidine blue staining revealed bright islet-like structures surrounded by squamous
chondrocytes with a palisade arrangement (Figures 3A-D), which ultrastructural analysis,
revealed to be a robust fibrillary network (Figures 3E and F). Artificial cartilage chondrocytes
exhibited an active euchromatin nucleus with a prominent nucleolus, a cytoplasm with a high,
rough endoplasmic reticulum content and a plasma membrane characterized by numerous
filopodia extending out into the surrounding extracellular matrix (Figures 3E and F).
Figure 2. Pig articular chondrocyte (PAC) microtissues (600, each containing 5,000 PACs) assembled in a
cylindrical agarose mould (3 mm in diameter) to a coherent artificial cartilage (a). Toluidine blue staining of semi-
thin paraffin sections of PAC-derived macrotissues tissues shows a robust fibrillary network embedded in an
extensive extracellular matrix (b, c, asterisks), and van Gieson staining reveals collagen production in light red (d-f,
asterisks).
c 20 ma
c
b 0.5 mm
d 0.5 mm
e
e 50 m
f
f 20 m
0.5 mm c 20 mcc 20 m20 maa
c
b 0.5 mm
c
bb 0.5 mm0.5 mm
d 0.5 mm
e
dd 0.5 mm0.5 mm0.5 mm
e
e 50 m
f
e 50 m50 m
f
f 20 mff 20 m20 m
0.5 mm0.5 mm

Macrotissue Generation 133
Figure 3. Human articular chondrocyte (HAC) microtissues (600, each containing 5,000 HACs) assembled
in a cylindrical agarose mould (3 mm in diameter) to a coherent artificial cartilage. Toluidine blue staining shows
bright islet-like structures (asterisk) surrounded by squamous chondrocytes in palisade arrangement (a-d).
Transmission electron micrographs (e, f) revealed that the islets are composed of robust fibrillary network embedded
in an amorphous matrix (e, f, asterisks). Artificial cartilage chondrocytes exhibited an active euchromatin nucleus
with a prominent nucleolus, a cytoplasm with a high, rough endoplasmic reticulum content (arrow head) and a
plasma membrane characterized by numerous filopodia (arrows) extending out into the surrounding extracellular
matrix (e, f).
a b
c d
e f
0.5 mm 25 m
10 m 10 m
5 m 1 m
aa b
cc dd
ee ff
0.5 mm0.5 mm 25 m25 m
10 m10 m 10 m10 m
5 m5 m 1 m1 m

Macrotissue Generation 134
Neo-vascularization of scaffold-free macrotissues
The aforementioned experiments have established microtissues as functional building
blocks, which can be assembled into scaffold-free tissue structures following self-controlled
assembly in adhesion-free agarose moulds. Along the way to designing larger artificial tissue
structures for clinical applications, will be the prime size-limiting parameter vascularization,
which sustains metabolic activities of the tissue core. Previous studies exemplified the in-vitro
vascularization of human aortic fibroblast (HAF)-based core microtissues, whose VEGF
production recruited peripheral HUVECs to develop a vascular structure (Kelm et al. 2005). In
order to pioneer in-vitro vascularized macrotissues we produced core human myofibroblast
(HMF) spheroids by cultivating 10,000 cells for four days in hanging drops, coating them with
1,200 HUVECs for four days in hanging drops and assembling 300 HMF-HUVEC spheroids in
the aforementioned cylindrical agarose mould. The HMF-HUVEC macrotissue aggregated for
three days and was cultivated for further 4 days in an agarose-coated culture dish. HMF-HUVEC
microtissues assembled into a coherent macrotissue (Figure 4A), characterized by a peripheral
layer of HUVECs (Figure 4C) reminiscent of inter-microtissue forces, which separated mixed
HMF-HUVEC populations into a HUVEC-coated HMF core spheroid during microtissue
assembly in hanging drops. In contrast, pure HMF macrotissues were less coherent and had a
ruffled surface (Figures 4B and D). Moreover, immunohistologic analysis of the endothelial cell-
specific surface marker CD31 revealed a dense network of endothelial cells throughout the HMF-
HUVEC macrotissue (Figures 4E, G, H), a cell network which was not be observed in pure HMF
control tissues (Figure 4F). The equivalence of cell type-specific reorganization (HMF-core,
HUVEC-shell) within multicellular micro- and macrotissues suggests that microtissues can
indeed be considered to be minimal tissue units, which can be up-scaled to larger tissue implants.
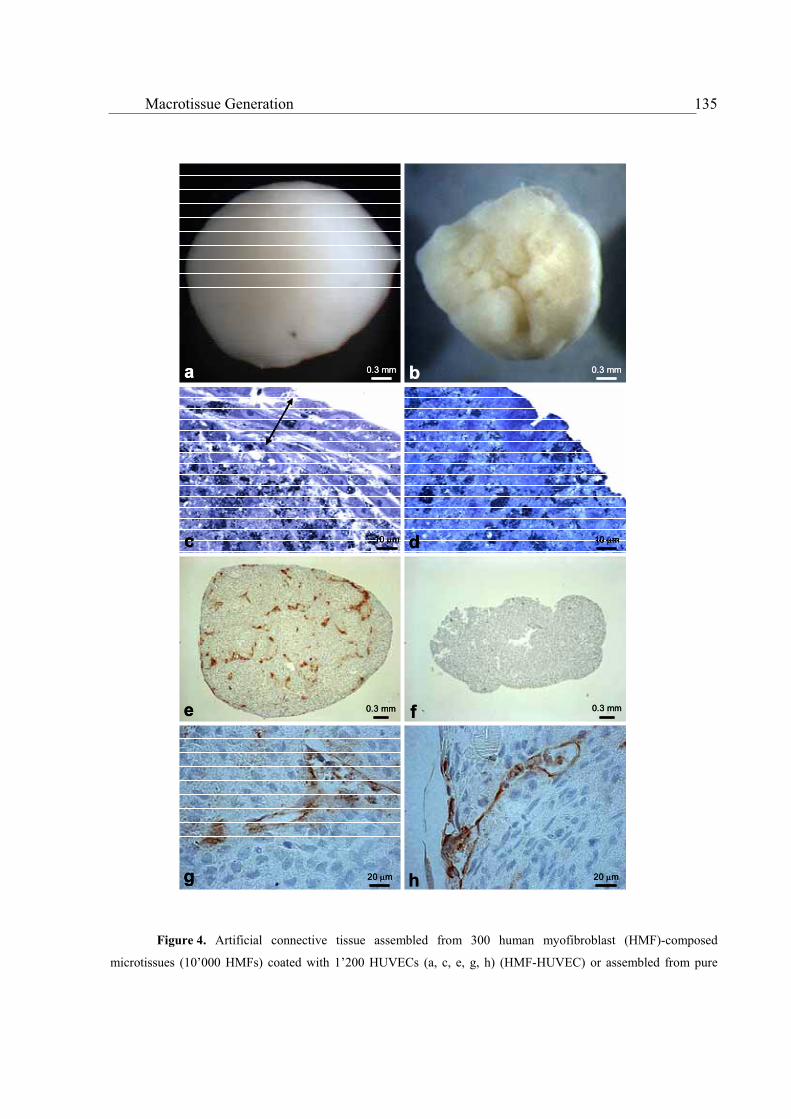
Macrotissue Generation 135
Figure 4. Artificial connective tissue assembled from 300 human myofibroblast (HMF)-composed
microtissues (10’000 HMFs) coated with 1’200 HUVECs (a, c, e, g, h) (HMF-HUVEC) or assembled from pure
10 m
a b
c d
e f
g h
0.3 mm 0.3 mm
10 m
0.3 mm0.3 mm
20 m 20 m
10 m10 m10 m10 m
aa bb
cc dd
ee ff
gg hh
0.3 mm0.3 mm 0.3 mm0.3 mm
10 m10 m
0.3 mm0.3 mm0.3 mm0.3 mm
20 m20 m 20 m20 m
10 m10 m

Macrotissue Generation 136
HMF microtissues (b, d, f). While HMF-HUVEC macrotissues were coherent and showed a smooth tissue surface
(a) tissues composed of pure HMF microtissues displayed a ruffled surface (b), which was confirmed by toluidine
blue stained semi-thin sections (d). Sections of HMF-HUVEC showed exclusive assembly of HUVECs at the
macrotissue’s periphery (c; double-head arrow). Immunohistologic analysis of the endothelial cell-specific surface
marker CD31 (brown staining) revealed a HUVEC-shaped capillary network exclusive to HMF-HUVEC
macrotissues (e, g, h) (f, control macrotissue composed of HMFs only).
Implantation of HMF-HUVEC macrotissues into chicken embryos
In-vivo vascularization crosstalk between artificial and living tissues was assessed by
implanting in-vitro prevascularized HMF-HUVEC macrotissues (see above) atop the
chorioallantoic membrane (CAM) of chicken embryos. Neovascularized HMF-HUVEC
macrotissues integrated into the CAM 84 hours post implantation and were fully connected to the
chicken embryos’s vasculature (Figure 5A). At the macrotissue-CAM interface, a dense network
of erythrocyte-containing microvessels was detected, suggesting inter-species compatibility of
vascular systems (Figures 5B and C). Furthermore, ultra-structural analysis revealed that
individual HUVECs had developed intracellular lumen and that inter-HUVEC crosstalk had
established small microvessels with lumen filled with erythrocytes (Figure 5G). Regular
HUVEC-formed capillaries were characterized by an activated endothelium with intra-luminal
protrusions, a multitude of transport vesicles and an extracellular matrix formed by deposition of
collagen fibers (Figure 5H). In contrast, control macrotissues assembled from HUVEC-free HMF
populations were rejected, scar tissue with no signs of vascular crosstalk developed between the
control tissue and the chicken embryo (Figures 5D-F).
Immunohistological analysis of the human endothelial cell-specific surface marker CD31
confirmed the development of a vascular system across the macrotissue-embryo interface
(Figures 6A and B). Interestingly, HUVECs were found to accumulate preferentially at the
macrotissue-CAM interface managing inter-tissue vascularization. Some HUVECs even migrated
to the chicken embryo’s mesenchym (Figures 6C and D). VEGF-specific staining of macrotissue
implants showed high-level expression of this growth factor in non-vascularized implanted
control macrotissues known to suffer from hypoxia in their core (Kelm et al. 2004). However,
pre-vascularized macrotissue implants, fully connected to the embryo’s vasculature, exhibited

Macrotissue Generation 137
low levels of VEGF expression in the macrotissue, thus confirming a sufficient oxygen supply
inside the implant (Figures 6E and F).
Figure 5. Vascularization crosstalk of human myofibroblast (HMF)-derived macrotissues transplanted onto
chicken embryos. Artificial connective tissue assembled from human umbilical vein endothelial cell (HUVEC)-
Er
Er
a d
b e
c f
g h
1 mm 1 mm
100 m 100 m
50 m 50 m
5 m 5 m
b
c
e
Er
Er
aa dd
bb ee
cc ff
gg hh
1 mm1 mm 1 mm1 mm
100 m100 m 100 m100 m
50 m50 m 50 m50 m
5 m5 m 5 m5 m
b
c
e

Macrotissue Generation 138
coated HMF (HMF-HUVEC) (a-c, g, h) or pure HMF microtissues (d-f) were grafted and cultivated atop the chicken
embryo’s chorioallantoic membrane (CAM) for 3.5 days. Toluidine blue staining of semi-thin sections across the
HMF(-HUVEC)/CAM interface revealed a significant angiogenic response of the chicken embryo exemplified by
large vessels invading the human tissue implant (b, arrows) as well as multitude of capillaries within the artificial
connective tissue (c, arrows). In contrast, pure HMF control macrotissues are not connected to the chicken embryo’s
mesenchyme and vasculature and remain non-vascularized. The graft is rejected as the chicken embryo develops an
enlarged CAM epithelium at the CAM-HMF tissue interface (e, f, asterisks). Transmission electron micrographs
show that (i) microvessels are filled with erythrocytes (Er) (g, h), (ii) sprouts of seamless non-perfused capillaries
formed by single (g, arrow head) or (iii) by adjacent endothelial cells (g, arrow). Most capillaries are activated, and
show a thick endothelium characterized by intra-luminal protrusions of trans-cellular vesicles (h, arrowhead). The
extracellular matrix shows deposition of collagen (h, arrows).
Figure 6. Immunohistologic analysis of human umbilical vein endothelial cell (HUVEC)-coated human
myofibroblast (HMF) (a, c, d, e) and pure HMF (b, f) microtissues assembled to macrotissues and grafted onto
chicken embryos’ chorioallantoic membrane (CAM) for 3.5 days. HUVEC-formed tubular capillary structures,
d
+ HUVEC - HUVEC
+ HUVEC - HUVEC
CD31 CD31
VEGFVEGF
ca
c
e
b
d
f
1 mm 1 mm
100 m 50 m
1 mm1 mm
d
+ HUVEC - HUVEC
+ HUVEC - HUVEC
CD31 CD31
VEGFVEGF
caa
cc
ee
bb
dd
ff
1 mm1 mm 1 mm1 mm
100 m100 m 50 m50 m
1 mm1 mm1 mm1 mm

Macrotissue Generation 139
staining positive for CD31, connected to CAM vessels and invaded the chicken embryo’s mesenchyme (c, d,
arrows). In contrast, HMF-only macrotissues show no vascular connection and remain separated from the host tissue
by a thick epithelium (see also Figure 5). Vascularization of the HMF-HUVEC tissues result in lower production of
the human vascular endothelial growth factor (VEGF) (e), compared to the HMF-pure control tissue (f).
Discussion
So far, classical tissue engineering has been based on seeding expanded cell populations
into synthetic scaffolds or hydrogels to provide a desired shape and implanting the resulting
artificial tissues into the recipient organism, where maturation of the new organ or integration
into host tissues occurred (Hoerstrup et al. 2000; Hench and Polak 2002; Shin et al. 2003; Langer
and Tirrell 2004). Despite decisive advances in the design and functionalization of the scaffold
during the past decade two major challenges remain: (i) Scaffolds fail to mimic essential natural
extracellular matrix (ECM) functions with temporal and spatial intricacy, and (ii) cell-scaffold
interactions lack the signal integration complexity observed for inter-cellular crosstalk in three
dimensions (Richardson et al. 2001; Vogel and Baneyx 2003; Lutolf and Hubbell 2005).
Alternative strategies for the design of scaffold-free tissues took advantage of stacking cell
monolayers (Shimizu et al. 2002) or capitalized on the self-assembly of monodispersed cells to
microtissues (Steinberg and Foty 1997; Kelm and Fussenegger 2004). Unencumbered by scaffold
materials, cells that assemble into microtissues develop their natural microenvironments, control
tissue dynamics and adopt in vivo-like structures. Non-limiting examples include self-assembly
of myocardial microtissues, Kelm et al. 2004), microganglia-like structures (Kelm et al. 2005),
hepatic microtissues (Kelm et al. 2003), artificial cartilage (Anderer and Libera 2002), embryoid
bodies (Itskovitz-Eldor et al. 2000), and retinal structures (Layer et al. 2002). Microtissues have
since been appreciated as 3D tissue culture models and have been used successfully in this study
to assemble larger tissues in vitro.
Mammalian cell aggregates have been shown to display a high cell type-specific affinity,
which may result in the assembly of larger tissues. For example, transgenic Chinese hamster
ovary cells, embedded in hydrogel droplets, fused to ring-like structures (Jakab et al. 2004).
Using cell-specific microtissues to assemble (i) skeletal muscle (C2C12, derived from mouse
myoblasts), (ii) cartilage (derived from pig and human articular chondrocytes) and (iii)
connective (derived from human myofibroblasts) macrotissues, we have substantiated the use of

Macrotissue Generation 140
microtissues as minimal building blocks to generate larger scaffold-free artificial tissue at a mm3
scale. Macroscopic and ultra-structural analyses revealed that all macrotissues displayed tissue-
specific morphologies based on. C2C12 displayed skeletal muscle-specific, longitudinal,
multinucleated cell morphology, and chondrocytes were embedded in a collagen matrix even
without supplements of collagen-inducing substances like ascorbic acid or TGF- .
Increasing the size of artificial tissue beyond a certain threshold diameter will elicit
nutrient and oxygen limitations in the tissue’s core. Nature has evolved vascularization to ensure
metabolic supplies to individual cells in a tissue within a perimeter of 100 µm (Tsai et al. 2003)
Tissue engineers have elaborated two fundamental strategies to mimic vascularization in artificial
tissues: (i) using cells transgenic for the production of pro-angiogenic factors or scaffolds
functionalized with pro-angiogenic factors to recruit tissue-resident endothelial cells for implant
vascularization (Richardson et al. 2001; Hall and Hubbell 2004), (ii) in-vitro pre-vascularization
of implants using purified pro-angiogenic factors or specifically designed scaffold compositions
to enable rapid connection to the host vascular system (Borges et al. 2003; Wu, et al. 2004). The
use and release of pro-angiogenic factors remains problematic due to the inherent instability of
these proteins in vivo (Yancopoulos et al. 2000) and the risk of uncontrolled side effects
including angiomagenesis (Carmeliet 2000). In-vitro implant pre-vascularization using
endothelial cells was difficult as these cells became apoptotic after being embedded in gelatin,
fibrin, collagen or matrigel matrices (Nomi et al. 2002)
We found that coating the desired tissue spheroids with HUVECs results in
perfectly vascularized microtissues as HUVECs migrate to the microtissue’s core following the
established VEGF gradient. Unlike in matrices, HUVECs remain viable in a microtissue
environment (Kelm et al. 2005). As minimal tissue building blocks, HUVEC-vascularized
myofibroblast microtissues perfectly assembled to vascularized macrotissues, which connected to
chicken embryo’s vasculature following implantation. However, non-vascularized HUVEC-free
macrotissue implants were rejected. This observation substantiates the need for the pre-
vascularization of tissue implants beyond a certain size.
An ideal artificial tissue transplant should show three major characteristics: (i) adopt
tissue-typical cell morphologies and tissue integrity, (ii) exhibit seamless and functional
integration into the target tissue without production of significant scar tissue and (iii) connect to

Macrotissue Generation 141
the host vascular and nervous systems. Microtissue-based production of prevascularized tissues
meets all these criteria. With the tissue engineering details in place, bioengineers will have to
take over in order to design mass-production strategies for microtissues and provide clinics with
transplantation-ready artificial tissues.
Acknowledgments
We thank Sirpa Price for isolating of primary human myofibroblasts and Krystyna Sala-
Szymanska as well as Bettina de Breuyn for preparing of the histological specimens. This work
was supported by the Swiss National Science Foundation (grant no. 631-065946) and the Swiss
State Secretariat for Education and Research within EC Framework 6.
References
Ajioka, I., R. Nishio, et al. (2001). "Establishment of heterotropic liver tissue mass with direct
link to the host liver following implantation of hepatocytes transfected with vascular
endothelial growth factor gene in mice." Tissue Eng 7, 335-344.
Anderer, U. and J. Libera (2002). "In vitro engineering of human autogenous cartilage." J Bone
Miner Res 17, 1420-1429.
Benjamin, M. and B. Hillen (2003). "Mechanical influences on cells, tissues and organs -
'Mechanical Morphogenesis'." Eur J Morphol 41, 3-7.
Borges, J., M. C. Mueller, et al. (2003). "Engineered adipose tissue supplied by functional
microvessels." Tissue Eng 9, 1263-1270.
Carmeliet, P. (2000). "VEGF gene therapy: stimulating angiogenesis or angioma-genesis?" Nat
Med 6, 1102-1103.
Djonov, V., M. Schmid, et al. (2000). "Intussusceptive angiogenesis: its role in embryonic
vascular network formation." Circ Res 86, 286-292.
Fink, C., S. Ergun, et al. (2000). "Chronic stretch of engineered heart tissue induces hypertrophy
and functional improvement." Faseb J 14, 669-679.

Macrotissue Generation 142
Hall, H. and J. A. Hubbell (2004). "Matrix-bound sixth Ig-like domain of cell adhesion molecule
L1 acts as an angiogenic factor by ligating alphavbeta3-integrin and activating VEGF-
R2." Microvasc Res 68, 169-178.
Hench, L. L. and J. M. Polak (2002). "Third-generation biomedical materials." Science 295,
1014-1017.
Hoerstrup, S. P., R. Sodian, et al. (2000). "Functional living trileaflet heart valves grown in
vitro." Circulation 102, III44-49.
Hubbell, J. A. (2003). "Materials as morphogenetic guides in tissue engineering." Curr Opin
Biotechnol 14, 551-558.
Itskovitz-Eldor, J., M. Schuldiner, et al. (2000). "Differentiation of human embryonic stem cells
into embryoid bodies compromising the three embryonic germ layers." Mol Med 6, 88-95.
Jakab, K., A. Neagu, et al. (2004). "Engineering biological structures of prescribed shape using
self-assembling multicellular systems." Proc Natl Acad Sci U S A 101, 2864-2869.
Kelm, J. M., C. Diaz S-Bustamante, et al. (2005). "VEGF Profiling and Angiogenesis in Human
Microtissues." J. Biotechnol. in press.
Kelm, J. M., E. Ehler, et al. (2004). "Design of Artificial Myocardial Microtissues." Tissue Eng
10, 201-214.
Kelm, J. M. and M. Fussenegger (2004). "Microscale tissue engineering using gravity-enforced
cell assembly." Trends Biotechnol 22, 195-202.
Kelm, J. M., L. M. Ittner, et al. (2005). "Self-assembly of sensory neurons into ganglia-like
microtissues." J. Biotechnol. in press.
Kelm, J. M., N. E. Timmins, et al. (2003). "Method for generation of homogeneous multicellular
tumor spheroids applicable to a wide variety of cell types." Biotechnol Bioeng 83, 173-
180.
Kunz-Schughart, L. A., J. P. Freyer, et al. (2004). "The use of 3-D cultures for high-throughput
screening: the multicellular spheroid model." J Biomol Screen 9, 273-285.
Langer, R. and D. A. Tirrell (2004). "Designing materials for biology and medicine." Nature 428,
487-492.
Lauffenburger, D. A. and L. G. Griffith (2001). "Who's got pull around here? Cell organization in
development and tissue engineering." Proc Natl Acad Sci U S A 98, 4282-4284.

Macrotissue Generation 143
Layer, P. G., A. Robitzki, et al. (2002). "Of layers and spheres: the reaggregate approach in tissue
engineering." Trends Neurosci 25, 131-134.
Lutolf, M. P. and J. A. Hubbell (2005). "Synthetic biomaterials as instructive extracellular
microenvironments for morphogenesis in tissue engineering." Nat Biotechnol 23, 47-55.
Mol, A., C. V. Bouten, et al. (2004). "Review article: Tissue engineering of semilunar heart
valves: current status and future developments." J Heart Valve Dis 13, 272-280.
Nomi, M., A. Atala, et al. (2002). "Principals of neovascularization for tissue engineering." Mol
Aspects Med 23, 463.
Peirce, S. M. and T. C. Skalak (2003). "Microvascular remodeling: a complex continuum
spanning angiogenesis to arteriogenesis." Microcirculation 10, 99-111.
Ribatti, D., B. Nico, et al. (2001). "Chorioallantoic membrane capillary bed: a useful target for
studying angiogenesis and anti-angiogenesis in vivo." Anat Rec 264, 317-324.
Richardson, T. P., M. C. Peters, et al. (2001). "Polymeric system for dual growth factor delivery."
Nat Biotechnol 19, 1029-1034.
Rothermel, A. and P. G. Layer (2001). "Photoreceptor plasticity in reaggregates of embryonic
chick retina: rods depend on proximal cones and on tissue organization." Eur J Neurosci
13, 949-958.
Shimizu, T., M. Yamato, et al. (2002). "Fabrication of pulsatile cardiac tissue grafts using a novel
3-dimensional cell sheet manipulation technique and temperature-responsive cell culture
surfaces." Circ Res 90, e40.
Shin, H., S. Jo, et al. (2003). "Biomimetic materials for tissue engineering." Biomaterials 24,
4353-4364.
Steinberg, M. S. and R. A. Foty (1997). "Intercellular adhesions as determinants of tissue
assembly and malignant invasion." J Cell Physiol 173, 135-139.
Timmins, N. E., S. Dietmair, et al. (2004). "Hanging-drop multicellular spheroids as a model of
tumour angiogenesis." Angiogenesis 7, 97-103.
Tsai, A. G., P. C. Johnson, et al. (2003). "Oxygen gradients in the microcirculation." Physiol Rev
83, 933-963.
Vacanti, J. P. and R. Langer (1999). "Tissue engineering: the design and fabrication of living
replacement devices for surgical reconstruction and transplantation." Lancet 354 Suppl 1,
SI32-34.

Macrotissue Generation 144
Vogel, V. and G. Baneyx (2003). "The tissue engineering puzzle: a molecular perspective." Annu
Rev Biomed Eng 5, 441-463.
Wu, X., E. Rabkin-Aikawa, et al. (2004). "Tissue-engineered microvessels on three-dimensional
biodegradable scaffolds using human endothelial progenitor cells." Am J Physiol Heart
Circ Physiol 287, H480-487.
Yancopoulos, G. D., S. Davis, et al. (2000). "Vascular-specific growth factors and blood vessel
formation." Nature 407, 242-248.
Yang, S., K. F. Leong, et al. (2001). "The design of scaffolds for use in tissue engineering. Part I.
Traditional factors." Tissue Eng 7, 679-689.
Zisch, A. H., M. P. Lutolf, et al. (2003). "Cell-demanded release of VEGF from synthetic,
biointeractive cell ingrowth matrices for vascularized tissue growth." Faseb J 17, 2260-
2262.
Zisch, A. H., M. P. Lutolf, et al. (2003). "Biopolymeric delivery matrices for angiogenic growth
factors." Cardiovasc Pathol 12, 295-310.

Chapter 8
Synergies of Microtissue Design, Viral
Transduction And Adjustable Transgene
Expression For Regenerative Medicine
Kelm J.M., Kramer B.P., Gonzalez-Nicolini V., Ley B. and Fussenegger M., (2004)
Biotechnology and Applied Biochemistry 39, 3-16
Microtissue
Design
Transduction
Gene Control
Gene Therapy
Gene-Function
Analysis
Drug
Discovery
Tissue engineering
Animal-Free
Drug Testing
Cell-Phenotype Engineering
Biopharmaceutical
Manufacturing

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 146
Abstract
In the past decade, regenerative medicine has evolved as an interdisciplinary field
integrating expertise from the medical, life and material science communities. Recent
advances in tissue engineering, gene therapy, gene-function analysis, animal-free drug testing,
drug discovery, biopharmaceutical manufacturing and cell-phenotype engineering have
capitalized on a core technology portfolio including artificial microtissue design, viral
transduction and precise transcription dosing of therapeutic or phenotype-modulating
transgenes. We provide a detailed overview on recent progress in these core technologies and
comment on their synergistic impact on current and future human therapies.
Introduction
Tissue engineering and gene therapy may be defined as rational therapeutic
interventions to restore diseased gene or tissue functions. In the past two decades advances in
gene therapy and tissue engineering have resulted in a variety of therapeutic advances to
generate and/or reinstate tissue function of skin (Beele 2002), cartilage (Hardingham et al.
2002), bone (Jadlowiec et al. 2003), intestine (Perez et al. 2002), myocardium (Eschenhagen
et al. 2002), pancreas (Prokop 2001) or liver (Samuel 2003). As tissue engineering and gene
therapy are becoming increasingly mature fields they start to merge on their way to foster
novel opportunities in regenerative medicine. Next-generation advances in regenerative
medicine require a specific technology portfolio including (1) systems for cultivation of
(primary) cells in three dimensions (3D) to form artificial (micro-) tissues, (2) efficient
transduction technologies to place preferred transgene configurations onto any target
chromosome (Pfeifer et al. 2002) and (3) human-compatible gene regulation technology for
adjustable molecular interventions as well as rational reprogramming of mammalian cells to
achieve desired cell phenotypes (Carmeliet 2000; Lee et al. 2000). Besides for regenerative
medicine (tissue engineering and gene therapy) these three core technologies also form the
integral basis for other therapeutic areas including (i) gene-function analysis (Greenfield
2000), (ii) drug discovery (Aubel et al. 2001), (iii) animal-free drug testing (Bhadriraju et al.
2002) and biopharmaceutical manufacturing (Fussenegger et al. 1998; Meents et al. 2002).

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 147
Figure 1: Integration of three core technologies is the basis for advanced regenerative medicine (tissue
engineering and gene therapy): (i) Microtissue design, (ii) transduction and (iii) gene control. These three key
technologies merge to provide impact on (i) animal-free drug testing, (ii) biopharmaceutical manufacturing, (iii)
cell phenotype engineering (targeted differentiation), (iv) drug discovery, and (v) gene-function analysis.
Design of artificial microtissues
It has been shown for a wide variety of different cell types such as bone (Kale et al.
2000), liver (Khalil et al. 2001), cartilage (Girotto et al. 2003), and heart muscle cells
(Eschenhagen et al. 1997) that 3D cultivation is an absolute requirement to maintain cell-
specific functionality in vitro (Berthiaume et al. 1996), (Chen et al. 1997). In many cases,
cells cultivated in 2D and 3D differ in their expression profiles for 5 1 and 5 3 integrins,
paxillin and other cytoskeletal components, their phosphorylation status of focal adhesion
kinases and an extracellular matrix (ECM) is exclusively produced by 3D cultures
(Cukierman et al. 2001; Kelm et al. in press).
Beyond providing a physical support for cells the ECM modulates cell differentiation
(Edwards et al. 1998) as well as tissue remodelling (Badylak 2002) and manages inter-tissue
contacts (Giancotti et al. 1999). Also, cell surface receptors interacting with the ECM
integrate growth-modulating external signals and convey them to the cells’ signal
Microtissue
Design
Transduction
Gene Control
Gene Therapy
Gene-Function
Analysis
Drug
Discovery
Tissue engineering
Animal-Free
Drug Testing
Cell-Phenotype Engineering
Biopharmaceutical
Manufacturing
Microtissue
Design
Transduction
Gene Control
Gene TherapyGene Therapy
Gene-Function
Analysis
Gene-Function
Analysis
Drug
Discovery
Drug
Discovery
Tissue engineeringTissue engineering
Animal-Free
Drug Testing
Animal-Free
Drug Testing
Cell-Phenotype EngineeringCell-Phenotype Engineering
Biopharmaceutical
Manufacturing

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 148
transduction cascades (Streuli 1999). For example, neonatal mouse and rat cardiomyocytes
cultivated in 2D lack collagen IV whereas this ECM component is highly expressed in
myocardial microtissues (Kelm et al. in press).
The importance of the ECM, cell morphology and differentiation status as well as cell-
cell contacts for the integrity of any tissue has been well established (Martin et al. 2002).
However, recent advances in polymer science were required to design artificial support for 3D
cultivation of desired cell types. To date, two different strategies for 3D cultivation of
mammalian cells are available: Cultivation in/on artificial scaffolds and reaggregation.
Although collagens (Eschenhagen et al. 1997), foams (Fukuda et al. 2003), and hydrogels
(Khalil et al. 2001) have been shown to enable 3D cell culture, novel biocompatible and
biodegradable polymer scaffolds in particular those functionalized with wound-healing and
growth-promoting factors have become a standard in the production of artificial tissue
constructs (Richardson et al. 2001). Despite their decisive advantages for reshaping 3D
structures, scaffolds have been associated with undesired post-translational effects,
inflammation originating from toxic degradation products, poor resorption and high
production costs (Yang et al. 2001).
The reaggregation approach represents an alternative to the use of artificial scaffolds
for 3D cultivation (Layer et al. 2002). Reaggregation is based on the principle that cells in
suspension self-aggregate under appropriate culture conditions to form spheroids. The use of
spheroids has a long tradition in anticancer research since neoplastic cells arranged in a
multicellular 3D configuration show increased resistance to cytotoxic drugs (multicellular
resistance; MCR) compared to isogenic cells grown as monolayer which enables a more
precise assessment of in vivo drug performance (Desoize et al. 2000). Various cell types
have been cultivated as cellular reaggregates including undifferentiated embryonic bodies
(Itskovitz-Eldor et al. 2000) and neurospheres (Nunes et al. 2003) or terminally differentiated
cardiomyocytes (Kelm et al. in press) and hepatocytes (Khaoustov et al. 1999). Spheroids
produced from undifferentiated progenitor or stem cells display increased proliferation in 3D
compared to 2D monolayer cultures (Chen et al. 1997). By contrast, many cell aggregates
assembled from terminally differentiated cells typically proliferate as monolayers but fail to
do so in a 3D configuration in which they exclusively produce an extracellular matrix (see
above).
Classical cultivation systems for induction of reaggregation include non-adhesive
culture dishes, spinner flasks and roller bottles. Unfortunately, these culture technologies do
not enable control of spheroid size. Only the hanging drop technology which consists of

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 149
cultivating cell suspensions of defined cell number in specific multiwell plates incubated
upside down, provide efficient size management. Gravity-enforced reaggregation of the cell
suspension in the hanging drop produces spheroids of specific size which correlates with the
cell number of the inoculated suspension. Size control was recently established for
cardiomyocytes (Kelm et al. in press) and several neoplastic cell lines including hepatocytes
(Kelm et al. 2003). Control of microtissue size is key to avoid oxygen and nutrient limitations
in the spheroid center. For example, we have recently observed hypoxia-induced expression
of vascular endothelial growth factor (VEGF) in rat myocardial microtissues of 230 µm in
diameter whereas smaller-sized microtissues of 120 µm failed to secrete VEGF (Kelm et al. in
press). Management of size-dependent hypoxia-controlled VEGF expression in artificial
microtissues may prevent the risk for tumor formation resulting from sustained angiogenesis
(Lee et al. 2000).
Directed Cell Differentiation
Cell-based therapies require substantial supplies of specific cell phenotypes. Although
autologous cell material is preferred, the availability of desired cell phenotypes is often
limited. Therefore, different strategies have been designed to produce sufficient supplies of
desired cell phenotypes: (i) precise reprogramming of cell-cycle regulatory networks to
enable cell-cycle reentry of terminally differentiated cells for expansion, followed by
induction of a G1-specific growth arrest post implantation to prevent development of
neoplastic cell phenotypes (Blau et al. 1997; Fux et al. 2001), (ii) expansion of stem cells and
progenitor cells by addition of purified growth factors (Schuldiner et al. 2000), cocultivation
of different cell types (Condorelli et al. 2001) or transduction of growth and transcription
factor-encoding genes (Horb et al. 2003), (iii) rational reprogramming of desired mammalian
cells by conditional overexpression of specific differentiation and dedifferentiation factors
(Fux in press; Fux in press).
Future success of cell and tissue engineering for ex vivo expansion will be
based on technologies managing temporal proliferation control of mammalian cells through
well-balanced expression of growth-promoting and growth-suppressing genetic elements
(Mulligan 1993). Independent control of growth-promoting and -inhibiting genes requires
combination of two compatible regulation systems, such as the PipOFF and TetOFF systems
(see below). A recent example includes dual-regulated expression of p27Kip1 sense and
antisense (Fux et al. 2001).

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 150
Directed differentiation of human embryonic stem cells for tissue engineering is still
in its infancy but holds great promises for clinical applications, in particular for proliferation-
inert cell phenotypes including cardiomyocytes. To date, directed differentiation of a single
cell phenotype is still a scientific dream. Schuldiner and coworkers scored the differentiation
impact of eight different growth factors which resulted in cell populations displaying different
cell phenotypes and exemplified that most growth factors induce various differentiation
programs (Schuldiner et al. 2000). An alternative strategy to obtain single-phenotype
populations from differentiated stem cells is by sorting, although the cell material produced
may fail to reach clinical quantities at reasonable cost (Kolossov et al. 1998).
Understanding interactions of key (trans-) differentiation factors requires sophisticated
in vitro model systems. A well-investigated setting is the transdifferentiation of pancreatic
cells to hepatocytes based on the pancreatic cell line AR42J-B13 (Shen et al. 2000).
Furthermore, Fux and coworkers implemented dual-regulated expression of (i) MyoD and
Msx-1 and (ii) C/EBP- and BMP-2 for precise differentiation control of C2C12 cells into
osteoblasts, adipocytes and myotubes (Fux in press; Fux in press).
Gene-function analysis
A major determinant of cellular function and phenotype is the tissue transcriptome.
Transcriptome analysis enables elucidation of human diseases (Golub et al. 1999), reveals
targets of drug candidates and helps to determine cDNAs for therapeutic interventions
(Greenfield 2000). Also, global transcriptome changes resulting from cultivation of cells in
3D instead of 2D may provide critical information required for regenerative medicine. We
have recently produced hepatic microtissues by cultivating the human hepatocarcinoma cell
line HepG2 in hanging drops. HepG2-derived microtissues display several of the phenotypic
characteristics of normal liver cells when cultivated in 3D (Khalil et al. 2001; Kelm et al.
2003). The transcriptome of HepG2 grown in monolayers and in 3D was analyzed using a
macroarray containing 3528 human genes (Atlas™ Human 3.6 Array; Clontech, Palo Alto,
CA). Several liver-specific, metabolism-associated and detoxifying proteins were upregulated
in microtissues probably as a consequence of induction of HNF-3 , a transcription factor
modulating a variety of liver-specific target genes (Figure 2). Hepatocyte-specific gene
expression is known to be controlled by several transcription factors including HNF-1,
CCAAT/enhancer binding protein (C/EBP), HNF-3, HNF-4 and HNF-6 (Duncan et al. 1998).

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 151
Based on our transcriptome studies HNF-3 may be a key target to reprogram liver-specific
cell phenotypes.
Figure 2: Expression analysis of the human hepatocarcinoma cell line HepG2 cultivated as 3D
microtissues compared to 2D monolayer cultures. In order to cover the functionality of liver cells, expression
profiles of metabolic and detoxifying genes were analyzed using a macroarray containing 3528 genes (Atlas™
Human 3.6 Array; Clontech, Palo Alto, CA). Most of the liver-specific genes were exclusively upregulated in
micortissues suggesting that 3D cultivated is privileged in providing liver-specific functions.
Animal-free drug testing and drug discovery
Screening for novel drugs and design of new drug tests require reproducible cell-based
models which mimic in vivo-like tissue structures. The fact that many established cell-based

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 152
systems designed for drug testing show little correlation between in vitro and in vivo results
exemplifies the need for novel culture systems assessing drug action in a tissue-like
environment (Bhadriraju et al. 2002). 3D multicellular tumor spheroids have long been
suggested as an alternative to animal tests for scoring efficacy of drugs against solid tumors.
Owing to decisive advances in 3D cell culture technology in recent years, protocols for 3D
cultivation of a wide variety of primary cells and cell lines are now available and complete the
cell-based drug screening portfolio (Padron et al. 2000; Braeckmans et al. 2002). For
example, progress in reshaping fully functional myocardial microtissues beating over 3 weeks
may provide a high throughput-compatible assay platform for the detection of myocardium-
stimulating or beating frequency-modulating drugs (Kelm et al. in press). We believe that
synergies resulting from combined use of molecular therapeutic interventions and tissue-
mimicking 3D cultivation systems will foster novel opportunities in regenerative medicine.
Viral Transduction
The ability to manipulate the genetic constitution of any living system has come along
with various technologies. The strategies being pursued in this context cover a wide variety of
different areas from biopharmaceutical manufacturing and gene-function analysis to screening
systems and clinical applications. Clinical applications include some aspects of tissue
engineering, tissue and organ transplantation as well as gene therapy which has already
generated promising impact in clinical trials (Grossman et al. 1994; Blaese et al. 1995;
Cavazzana-Calvo et al. 2000). The science of gene transduction is based on administration of
genetic information in order to equip target cells with new protein-production capacities or
eliminate expression of disease-inducing determinants (see Amor et al. for a review (Amor
2001)). Whereas transduction of functional genes is used to complement single-gene defects
or mendelian diseases gene transfer-based repression of specific gene function may provide
therapeutic opportunities for acquired illnesses including heart disease and cancer where gene
therapy may have its greatest public-health impact (see Scollay et al. for a review (Scollay
2001)). Efficient transfer of vectors encoding therapeutic transgenes is key to the success of
any aforementioned gene-based therapy. Major challenges include precise integration of the
therapeutic transgene on the target chromosome as well as its timely and adjustable
expression. Currently available gene transfer technologies can be divided into two categories:
viral and non-viral vectors. Non-viral vectors also known as synthetic gene delivery systems
include liposomes, DNA-ligand conjugates, naked DNA, ballistic gene delivery and CaPO4

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 153
transfection (Curiel et al. 1992; Felgner et al. 1994; Geissler et al. 1994; Gao et al. 1995;
Dzau et al. 1996). The use of viral vectors for gene delivery holds great promise for basic
research and therapeutic applications. Viral vectors enable researches to monitor functions,
replace, correct, express or block expression of target genes, tag cells for fate determination or
change the physiological state of specific cell populations. At present, there are five major
classes of viral vectors used in clinical trials: oncoretroviruses, lentiviruses, adenoviruses,
adeno-associated viruses (AAVs) and herpes simplex-1 viruses (HSV-1s) (for review see Kay
et al. (Kay et al. 2001) and Koostra et al. (Kootstra et al. 2003)). Emerging viral transduction
systems include those derived from baculoviruses (Sarkis et al. 2000), alpha-viruses (Huang
1996) (for review see Schlesinger et al. (Schlesinger et al. 1999)), and vaccinia viruses which
are currently in clinical trials for cancer gene therapy (Peplinski et al. 1998).
Viral vectors for gene therapy
Most of the oncoretroviral vectors currently designed for gene therapy applications are
based on the moloney murine leukemia virus (MMLV) (Miller et al. 1993), which was used
for the pioneering human gene therapy trials tailored for the treatment of severe combined
immunodeficiency (SCID) (Blaese et al. 1995). Retroviral vectors are typically pseudotyped
with glycoproteins derived from the vesicular stomatitis virus (VSV-G) to expand the tropism
of chimeric viral particles. Following infection of the target cell, the genomic RNA of
oncoretroviruses is reverse transcribed into linear double-stranded DNA by the virion’s
transcriptase (Telenitsky 1997). Since the oncoretroviral virion is devoid of any nucleus-
targeting sequences or protein complexes access to the target chromosome for stable
integration requires cell division. Although oncoretroviral particles can only transduce
mitotically active cells their efficient transgene delivery and random integration of therapeutic
genetic information into target chromosomes which ensures sustained gene expression
rendered them the preferred gene transfer tool for gene therapy trials in the past decade
(Miller et al. 1990; Roe et al. 1993). However, results from recent clinical trial raised
concerns about oncoretrovirus-mediated induction of protooncogenes and dedifferentiation
factors following integration on the patient’s chromosome (Li et al. 2002; Schroder et al.
2002). Indeed, 2 out of 11 patients successfully treated for SCID later developed a leukemia-
like disorder due to integration of the oncoretroviral vector adjacent to the oncogene LMO2
(Hacein-Bey-Abina et al. 2003). Therefore, technologies for site-specific integration of
therapeutic gene cargos at preselected inert chromosomal locations is a current research focus

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 154
of the gene therapy community (Groth et al. 2000; Olivares et al. 2002). The most
encouraging clinical results associated with retroviral gene delivery were two young children
both of which suffered from a type of inherited immune disorder known as SCID. Both
children continue to live a normal life although long-term reconstitution from transduced
progenitor cells remained rather low. Prior to gene therapy the children were treated with a
novel drug to complement missing adenosine deaminase (ADA) function which may have
prevented the selective outgrowth of the transduced progenitor cells (Bordignon et al. 1995).
Lentiviruses which belong to the retroviral family recently came into the limelight of
current gene therapy initiatives since they are able to transduce non-dividing cells (Sadaie et
al. 1998). Most available lentiviral vectors adapted for secure gene transfer have been derived
from human HIV-1. Their inherent tropism for CD4+ T cells and macrophages has prompted
therapeutic approaches to treat HIV infection and AIDS. Pseudotyped with VSV-G
glycoproteins lentiviral particles are able to transduce muscle cells including cardiomyocytes
(Kelm et al. in press), hepatocytes (Naldini et al. 1996), hematopoietic stem cells (Naldini et
al. 1996; Kafri et al. 1997; Miyoshi et al. 1999), lung cells (for the treatment of cystic fibrosis;
(Goldman et al. 1997)), neurons (for gene therapy of Parkinson’s disease (Blomer et al.
1997)) and even chicken and insect cells (Mitta et al. 2002). Because HIV-1 is a human
pathogen, there is ongoing concern about the use of HIV-1-based lentiviral vectors for gene
therapy although third-generation lentivectors are devoid of recombination-competent
nucleotide stretches and fail to deliver any virus protein-encoding cistrons to the target cells.
Nevertheless, chimeric vectors based on non-human lentiviruses continue to be a current
research focus of the gene therapy community although the risk associated with transduction
of such viruses remains to be established (Browning et al. 2001). HIV-1-based vectors are
already in clinical trials for the treatment of HIV infections, yet their use in other gene therapy
settings will have to await clearance of remaining safety issues (Browning et al. 2001; Ikeda
et al. 2003). A DNA-based vaccine containing human immunodeficiency virus type 1 (HIV-1)
was tested for safety and host immune response in 15 asymptomatic HIV-infected patients
raising hopes since vaccine administration failed to induce local or systemic reactions
(MacGregor et al. 2000). Also, a phase I open clinical trial on the safety and tolerability of
single escalating doses of autologous CD4+ T cells transduced with HIV-antisense envelope
(env; VRX496) is currently conducted by the same group.
Adenoviral vectors have become a standard for gene therapy since they efficiently
transduce dividing as well as non-dividing cells, can be concentrated to high titers and
mediate high-level transgene expression from their episomal dsDNA genomes. A distinct

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 155
advantage of adenoviral vectors is their capacity to transduce target cells at low temperatures
which enables gene transfer prior to tissue or organ transplantation (Csete et al. 1994). First-
generation adenoviruses which only harbored deletions in one or two early genes (E1 and E3)
elicited a strong immune response against viral proteins which resulted in rapid elimination of
transgene expression (Kafri et al. 1998). In 1999, a patient treated with transgenic second-
generation adenoviruses against ornithine transcarbamylase deficiency even passed away
during a clinical trial (Raper et al. 2002). The development of the gutless adenoviral vector
which is devoid of most viral sequences prevented elimination of transduced cells by the host
immune system, failed to elicit inflammation in the target organ and showed reduced cellular
infiltration (Morsy et al. 1998). Yet, immune responses specific for the antigenic viral capsid
proteins cannot be alleviated to date. Gutless adenoviral vectors enable prolonged transgene
expression from their episomal non-replicating genomes. Thus, in dividing cells, transgene
expression may be lost over time due to dilution of episomal virions while transgenic
adenoviral episomes are prone to degradation in non-dividing cells. Adenoviral gene delivery
is currently the number-two transduction system in clinical trials. Adenoviral vectors seem to
be best for the treatment of (i) vascular and coronary artery diseases in which transient
transgene expression is preferred (Laitinen et al. 2000; Zhu et al. 2000), (ii) in therapies which
require short-term expression (Baltzer et al. 2000; Lai et al. 2001) and (iii) for cancer gene
therapy in which cellular toxicity and immunogenicity may increase antitumor effects
(Crystal et al. 1997; Brenner et al. 2000). The most prominent example of adenovirus-based
cancer gene therapy (already in phase II clinical trials) is the mutant adenovirus ONYX-015,
which is devoid of E1B gene expression. ONYX-015 can replicate in and lyse p53-deficient
target cells but remains replication-deficient in host cells expressing functional p53.
Mutations in p53 continue to be the major cause of cancer in humans (Bischoff et al. 1996;
Heise et al. 1997; Khuri et al. 2000).
Like adenoviruses, adeno-associated viruses have a broad tissue tropism. However,
transgenic particles derived from adeno-associated viruses integrate stably at specific sites on
the target chromosomes of dividing and non-dividing cells while showing little cytopathy
(Linden et al. 1996). Patients who received intramuscular injections of AAV vectors
transgenic for human factor IX expression in a clinical trial for hemophilia B showed
significant clinical benefits associated with reduced requirements for factor IX infusions. In
addition, these patients tolerated high AAV doses without eliciting vector-associated toxicity
or showing signs indicative for germ line transmission (Kay et al. 2000). Other clinical trials
for liver-based treatment of hemophilia are currently underway as are efforts for AAV-based

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 156
complementation of cystic fibrosis (protocols and abstracts available at
www.wiley.co.uk/genmed/clinical/ & www.dhhs.gov/). Recent advances in AAV vector
design included split units which concatamerize in a head to tail orientation upon transfer into
the nucleus. However, controversial results regarding efficiency of the system suggested that
further refinement of this concept will be required to solve packaging size limitation
associated with AAV vectors (Sun et al. 2000; Nakai et al. 2002). More recently, hybrid viral
vectors have been designed in an effort to combine transduction efficiency of adenoviral
vectors with stable long-term transgene expression associated with AAV and retroviruses
(Linden et al. 1996; Zheng et al. 2000).
Recombinant herpes simplex viral (HSV) vectors display a large transgene cloning
capacity. Currently, the major disadvantage of HSV-based vectors is their cellular and
immunological toxicities. Development of amplicon vectors, which are containing the HSV
origin of replication plus packaging signal and helper-free packaging systems which alleviate
contamination with replicating helper virus have further reduced cytopathic effects and
induction of immune responses of HSV-based transduction systems (Spaete et al. 1982;
Cunningham et al. 1993). The high neurotropism of HSV-derived vectors has initiated
therapies for malignant glioma (Markert et al. 2000). Additional clinical trials for HSV-
derived treatment of malignant melanoma and colon cancer are currently underway (protocols
and abstracts available at www.wiley.co.uk/genmed/clinical/ & www.dhhs.gov/).
Although oncoretroviral, lentiviral, adenoviral, AAV- and HSV-based vectors are all
currently used in clinical trials every application requires careful consideration of particular
characteristics associated with each transduction system (Table 1). Future advances in viral
transduction technologies will likely witness decisive refinements to increase gene-transfer
efficiency and safety. This may be achieved by generating chimeric viruses which harbor the
best genetic traits of several currently available systems (Lieber et al. 1999; Zheng et al.
2000). Also, ideal viral gene-transfer vectors will enable precise dosing of therapeutic
transgene in a gene therapy setting (see below).

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 157
Table 1: Comparison of viral vectors for gene delivery in clinical trials.
Gene regulation
Control of expression levels and duration of transgenes encoded on viral vectors
continues to be a challenge. First-generation retroviral vectors only supported constitutive
proviral transgene expression driven by the 5’ long terminal repeat (5’ LTR) (Gale et al.
2000). Then, heterologous promoters were introduced to achieve and/or restrict transgene
expression. Therefore, transcriptional targeting makes use of gene regulatory elements which
enable exclusive expression of therapeutic transgenes in specific cell types, for example the
promoters of (i) albumin in hepatocytes (Balague et al. 2000), (ii) synapsin and neuron-
Broad range, preferably
neurons
Broad range,
except for
hematopoietic
cells
Broad rangeBroad rangeOnly dividing
cells Target
Efficient transduction
of neural cells, largest
packaging
capacity
Integration into the target
genome, absence of
immune
response
Transduction of
a broad range of
cells, growth to
very high titers, efficient gene
expression
Integration
into the target
genome, Transduction
non-dividing
cells
Integration
into the target
genome,
absence
immune
response
Advantages
Disadvantages
Safety Issues
Pre-primed
patients
Inflamatory
Response
Transgene
Expression
Transgene Size
Genome
Cytotoxicity, transient
transgene
expression
Insertional
mutagenesis, Small
packaging
capacity,
safety
concerns, low
transduction
efficiencies
Episomal
transgene
expression,
Transgene
expression lost
over time,
immunogenic
Insertional
mutagenesis,
safety
concerns
associated
with HIV-derived
lentiviruses
Random
integration may
induce
oncogenesis,
low viral titers,
transduction limited to
dividing cells
Cytotoxicity Insertional
mutagenesis Inflamation
Insertional
mutagenesis
Insertinal
mutagenesis
Yes Yes Yes Only HIV-1
patients No
Strong (small,
amplicon) None
Strong
(small, gutless) NoneNone
transient stabletransientstablestable
40 Kb (150 Kb,
amplicon) 4.5 Kb
5-8 Kb (30-35 Kb,
gutless) 10 Kb8.5 Kb
dsDNA ssDNA dsDNA RNARNA
Herpes simplex
virus
Adeno-associated
virus AdenovirusLentivirusOncoretrovirus

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 158
specific enolase in neurons (Klein et al. 1998; Glover et al. 2003) and (iii) myosin light chain
1 in muscle cells (Shi et al. 1997). Furthermore, some regulatory elements have been reported
to promote transgene expression exclusively in tumors (e.g., carcinoembryonic antigen
(CEA)) (Lan et al. 1997) and hepatocellular carcinomas (e.g., alpha-fetoprotein, AFP) (Ido et
al. 1995; Cao et al. 1999).
More recently, the use of chimeric transcription control systems responsive to
antibiotics and other small molecules have enabled design of viral vectors for adjustable
transgene expression (see below for an overview on gene control systems). For example,
lentiviral vectors harboring tetracycline-, rapamycin- and progesteron analogue-responsive
gene expression components have provided regulated transgene expression following
transduction into a variety of different cell lines and tissues (Oligino et al. 1998; Pollock et al.
2000; Vigna et al. 2002). However, none of the transduction systems engineered for regulated
therapeutic transgene expression have reached clinical trials yet.
Perspectives
To date, viral transduction is the most efficient and flexible technology for transfer of
therapeutic transgenes into most cell types and tissues. Over 500 clinical trials have been or
are being conducted 60% of which focus on cancer treatment. Most of the vectors currently
enrolled in clinical trials are derived from oncoretroviruses and adenoviruses. However, a
great percentage of these vectors are still assessed for their safety following a particular gene-
delivery protocol (phase I). Detailed information on past and ongoing clinical trials is
available on the homepages of the National Institutes of Health (www.dhhs.gov/) or the
journal of gene medicine (www.wiley.co.uk/ genmed/clinical/).
Adjustable Transgene Expression
Control of gene expression in space and time is crucial for many applications in gene
therapy and tissue engineering. Several studies have demonstrated, that different levels of
important transcription factors govern distinct fates in differentiation and metabolism
(Duncan et al. 1998). Niwa and coworkers have expressed the transcription factor Oct-3/4 at
three precise levels, which resulted in three distinct fates of embryonic stem cells (endoderm,
retention of pluripotent phenotype, and trophectoderm) (Niwa et al. 2000). Another example
of therapeutic intervention which requires precise dosing is expression of the vascular
endothelial growth factor (VEGF), which has a high therapeutic potential for the induction of

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 159
angiogenesis/arteriogenesis in ischemic tissues (see above). Yet, sustained VEGF expression
results in the development hemangiomas which may even be fatal in mice (Carmeliet 2000).
Key characteristics of an ideal gene regulation system
Gene therapy and tissue engineering require effective gene regulation systems which
comply with the following criteria at a high standard: (i) high-level expression under induced
conditions, (ii) low leaky expression under repressed conditions, (iii) bioavailability and (iv)
pharmacokinetic profiles in favouring rapid switching of the transgene expression status, (v)
low immunogenicity of the gene control configuration, (vi) lack of interference with host
regulatory networks and (vii) compact genetic design to limit pleiotropic effects associated
with repeated molecular interventions on the host chromosome.
Antibiotic-controlled gene regulation systems
The antibiotic-controlled gene regulation systems are prevalent in basic research and
clinical applications. These systems consist of antibiotic-responsive promoters, transcription
modulators (activators and repressors) and antibiotics, which adjust binding of the modulators
to cognate operator sequences contained in the target promoters (Figure 3). The transcription
modulators consist of prokaryotic DNA-binding proteins fused to transcription activator (for
example, VP16 of Herpes simplex virus, (Triezenberg et al. 1988) (OFF-systems) or the
silencer domains (for example, KRAB of the human kox-1 gene (Moosmann et al. 1997)
(ON systems). Transgene expression controlled by OFF-type systems are repressed by
regulating antibiotics, while induction of ON systems requires administration of regulating
substances. To date, three antibiotic-responsive gene regulation systems are available. The
pioneering Tet system is responsive to tetracycline antibiotics (Gossen et al. 1992). This
system has seen many refinements in the past decade and now consists of an entire family of
different tetracycline-dependent transactivators and promoters (Baron et al. 1997; Kamper et
al. 2002; Krueger et al. 2003). The Tet systems and their derivatives have been extensively
used for basic in vitro and in vivo research (Fux et al. 2001; Malleret et al. 2001; Mallo et
al. 2003) as well as in prototype gene therapy settings tailored for bone regeneration
(Moutsatsos et al. 2001), reversion of -thalassemia (Samakoglu et al. 2002), and anemia in
mice (Sommer et al. 2002).

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 160
In the past years, two novel antibiotic-responsive gene regulation systems have been
developed following the generic design principle of the Tet systems. The PIP and the E.REX
systems adjust desired transgene expression in response to clinically licensed antibiotics of
the streptogramin and macrolide classes (Fussenegger et al. 2000; Weber et al. 2002). These
systems are compatible with each other and enable simultaneous regulation of up to three
independent transgenes within a single cell thus providing unmatched regulation complexity
for future clinical interventions.
Figure 3: Molecular setup of antibiotic-, quorum-sensing- and temperature-responsive gene regulation
systems following the same design concept. A DNA-binding protein (DBP) consisting of a bacterial response
regulator fused to a mammalian transcription-activation domain (TA; typically Herpes simplex-derived VP16)
binds to a specific operator module and induces polymerase (poly)-mediated gene of interest (goi) transcription
from minimal promoters (Pmin) in a molecule-responsive manner. With the exception of rTetR- and TraR-derived
proteins which bind chimeric target promoters in the presence of regulating molecules, transactivator-promoter
interactions of all other systems only form in the absence of modulating substances.
Transgene control by chemically induced dimerization
While antibiotic-responsive gene control systems rely on antibiotic-induced allosteric
changes of the modulator’s DNA-binding affinity chemically induced dimerization (CID)
reconstitutes a chimeric transactivator by heterodimerizing a DNA-binding and a
transactivating domain in an inducer-dependent manner. Latest-generation CID systems use
Operator Pmin goi pA
DBP
TA
DBP
TA
DNA-binding Protein (DBP’s)
•TetR
•rTetR
•PIP
•E
•TraR
•scbR
•RheA (at 37 °C)
Poly
Removed from operator by
•Tetracycline
•Absence of Tetracycline
•Pristinamycin
•Erythromycin
•Absence of 3-oxo-C8-HSL
•SCB1
•41 °C
Operator Pmin goi pAOperator Pmin goi pA
DBPDBP
TATA
DBP
TATA
DNA-binding Protein (DBP’s)
•TetR
•rTetR
•PIP
•E
•TraR
•scbR
•RheA (at 37 °C)
PolyPoly
Removed from operator by
•Tetracycline
•Absence of Tetracycline
•Pristinamycin
•Erythromycin
•Absence of 3-oxo-C8-HSL
•SCB1
•41 °C

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 161
rapamycin and non-immunosuppressive derivatives to heterodimerize an artificial ZFHD1-
binding domain fused to three tandem copies of FKBP with FRAP fused to the p65
transactivation domain of human NF- B (Rivera et al. 1996). In the presence of rapamycin
ZFHD-FKBP3-(FRAP-p65)3 binds and activates chimeric promoters containing twelve
ZFHD1 binding sites (Rivera et al. 1996; Pollock et al. 2002). The rapamycin-induced CID
system has been reported to mediate low leaky expression under non-induced conditions.
However, only sophisticated versions of the system reach the maximum expression levels
typically associated with the Tet and other antibiotic-responsive transcription control systems
(Go et al. 2002). The rapamycin-based CID system has already been successfully used for
gene therapy approaches in animal models (Auricchio et al. 2002; Auricchio et al. 2002).
Hormone-inducible gene expression
Prior to the advent of antibiotic-adjustable and dimerizer-inducible transcription
control systems technology for hormone-responsive transgene modulation had been
developed. Steroid receptors belong to a family of ligand-inducible transcription factors
which contain different DNA- and hormone-binding domains (Beato 1989). Fusion of the
hormone-binding domain to the DNA-binding domain of the yeast Gal4 protein as well as the
VP16 transactivation domain created an artificial transactivator whose binding to promoters
harboring specific Gal4 operator sites was hormone-adjustable. Three hormone-dependent
transcription control systems have been designed which are adjustable by either estradiol
(Braselmann et al. 1993), the progesterone analogue RU486 (Wang et al. 1994) or the insect
molting hormone ecdysone (No et al. 1996). However, due to pleiotropic impacts of these
hormones/hormone analogues on human physiology their use in the clinics may be limited.
Quorum sensing-based transgene modulation
Prokaryotes manage inter- and intrapopulation communication by quorum-sensing
molecules including acylated homoserine lactones which bind to receptors in target cells and
initiate specific regulon switches by modulating the receptor’s affinity to cognate promoters
(Bassler 2002). This bacterial cross-talk system has recently been adapted for controlling
transcription of desired transgenes in mammalian cells (Neddermann et al. 2003; Weber et al.
2003). Homoserine lactone-based mammalian gene regulation systems follow the generic
design principle of aforementioned antibiotic-adjustable control modalities. Despite clinical

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 162
compatibility ongoing use of antibiotics for controlling therapeutic transgene expression
remains controversial. Non-physiologically active regulating agents devoid of any pleiotropic
side effects would be preferred inducers. First in vivo validation of butyrolactones for
regulation of transgenes in mice did not indicate any side effects (Weber et al. 2003). Yet,
studies including long-term administration of this substance remain to be done. The existence
of near infinite bacterial quorum-sensing molecules/receptor pairs provides an inexhaustive
pool for the design of novel mammalian gene control systems in the not-too-distant future.
Temperature-dependent gene regulation
Fine-tuning of heterologous transgene expression in the absence regulating molecules
would represent a decisive advantage for manufacturing of difficult-to-produce protein
therapeutics. Two low-temperature-inducible gene regulation systems have been designed
(Boorsma et al. 2000; Weber et al. 2003): (i) Following a temperature shift from 37°C to
29°C, a mutated viral RNA-dependent RNA replicase initiates an RNA amplification loop
resulting in high expression levels of the transgene (Boorsma et al. 2000). (ii) The second
cold-inducible gene control system uses a Streptomyces albus-derived thermosensor whose
binding and activation of chimeric promoters could be modulated by 1°C steps between 37°C
and 41°C in DT-40 cells (Weber et al. 2003). Application of temperature-controlled gene
regulation systems will likely remain restricted to in vitro use as environmental temperature
changes are incompatible with precise titration of transgene expression in clinical settings.
Gene regulation systems in drug discovery
Applications of gene regulation systems go far beyond the mere regulation of desired
transgenes. Instead of using known antibiotics to regulate a specific transgene, gene
regulation systems configured for expression of a reporter gene can be used to find novel
inducing substances. Since prokaryotic antibiotic resistance regulators used for the desgin of
tTA, PIT or ET recognize almost any commercially available tetracycline, streptogramin or
macrolide antibiotic those proteins are likely able to detect antibiotic core structures. Aubel
and coworkers established a mammalian screening system for the detection of streptogramin
antibiotics. Chinese hamster ovary (CHO) cells transgenic for pristinamycin-responsive SEAP
(human secreted alkaline phosphatase) expression were used to screen for the presence of
streptogramin antibiotics in culture supernatants of Streptomyces isolates. This screening

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 163
system was sensitive compared to traditional antibiogram-based assays and provided a three-
in-one readout: (i) streptogramin core structure, (ii) bioavailability and (iii) non-cytotoxicity
of the detected substance (Aubel et al. 2001). Weber and coworkers refined this approach by
capitalizing on the antibiotic-responsive binding of bacterial resistance-response proteins, the
antibiotic “biosensors”, to their cognate operator sequences. The ELISA-type molecular
biosensor enables the detection of tetracycline, streptogramin and macrolide antibiotics in
spiked liquids including milk and serum with nanogramm precision. Binding of the
biosensors to their cognate DNA chemically linked to a solid surface is converted into an
immuno-based colorimetric readout correlating with specific antibiotic concentrations. Such
technologies will likely have far-reaching implications in the development of novel anti-
infective drugs and the enforcement of antibiotic bans in stock farming.
Use of gene-control systems in biopharmaceutical manufacturing
Regulated gene expression systems are not only of key interest for gene therapy
interventions, but also for reprogramming of mammalian cells lines to increase production of
protein therapeutics. Chinese hamster ovary (CHO-K1)-derived production cell lines
engineered for conditional expression of the cycline-dependent kinase inhibitors p21 or p27
arrested in the G1-phase of the cell cycle and exhibited up to 30-fold higher specific
productivities compared to proliferation-competent cells (Fussenegger et al. 1998; Meents et
al. 2002). Similar studies with hybridoma cells confirmed increased production of
proliferation-controlled cell lines (Watanabe et al. 2002).
Outlook
Although gene therapy originally set out to cure genetic diseases, most of today’s
clinical research and trials focus on the treatment of acquired multigenic disorders including
cancer and heart illnesses. These diseases have a more complex cause and require the
regulated expression of more than one therapeutic transgene. Next-generation gene therapy
scenarios will have to cope with the replacement of entire regulatory networks and signaling
cascades. Major parts of the toolbox required for multiregulated multigene therapeutic
interventions have recently been completed. Multicistronic and bidirectional expression
configurations enable simultaneous regulation of several transgenes (Baron et al. 1995;

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 164
Fussenegger et al. 1998; Fux et al. 2003; Fux et al. 2003). Furthermore, recent studies have
shown that all antibiotic-responsive gene control system are compatible and could be used for
independent control of up to three different transgenes (Weber et al. 2002).
Current approaches have linked different compatible gene regulation systems such that
the output of a first-level adjustable promoter controlled expression of a transactivator or
transrepressor which in turn activates expression of a second-level promoter which then
drives desired transgene transcription (Aubrecht et al. 1996). In some configurations target
promoters were designed to be responsive to different transactivators and could integrate
several internal signal into a one-gene expression readout (Hoshikawa et al. 1998; Freundlieb
et al. 1999). For example, Imhof and coworkers conceived a regulatory network consisting of
a hybrid promoter which was downregulatable by a constitutively expressed TetR-derived
transrepressor and inducible by a Gal4-VP16 transactivator (Imhof et al. 2000). Since Gal4-
VP16 was under control of its own target promoter this configuration enabled autocatalytic
coexpression of the transactivator and the gene of interest.
Recent advances in artificial gene network design capitalized on cascade-like
interconnection of three independent gene control systems. In this configuration the
tetracycline-dependent promoter drives tTA expression in an autoregulatory manner along
with cocistronic expression of the macrolide-dependent transactivator (ET). ET targets its
macrolide-responsive promoter controlling expression of the pristinamycin-dependent
transactivator (PIT) which in turn fine-tunes transcription of the gene of interest. When
transcription modulation is exerted by addition of either tetracycline, erythromycin or
pristinamycin the three-step regulatory network exhibited unprecedented regulation
characteristics: (i) absence of regulating antibiotics resulted in maximum transgene
expression, (ii) addition of tetracycline yielded precise 70% expression, (iii) administration of
erythromycin reduced expression of the gene of interest to 40% of maximal levels and (iv)
was completely repressed following addition of pristinamycin (Figure 3) (Kramer et al. 2003).
The three-step regulatory network enabled precise expression levels following administration
of clinical doses of regulating antibiotics. Adjusting desired transgenes using a single
antibiotic-responsive gene regulation system requires dosing of the specific antibiotic with
nanogram precision which remains challenging in the human body.
Advanced gene network including toggle switches (Figure 4) (Gardner et al. 2000),
oscillators (Elowitz et al. 2000), and logical transcription control (Hasty et al. 2002; Buchler
et al. 2003) which enable new dimensions in artificial transgene control have only been put
into practice in Escherichia coli. Until functional implementation of these latest-generation
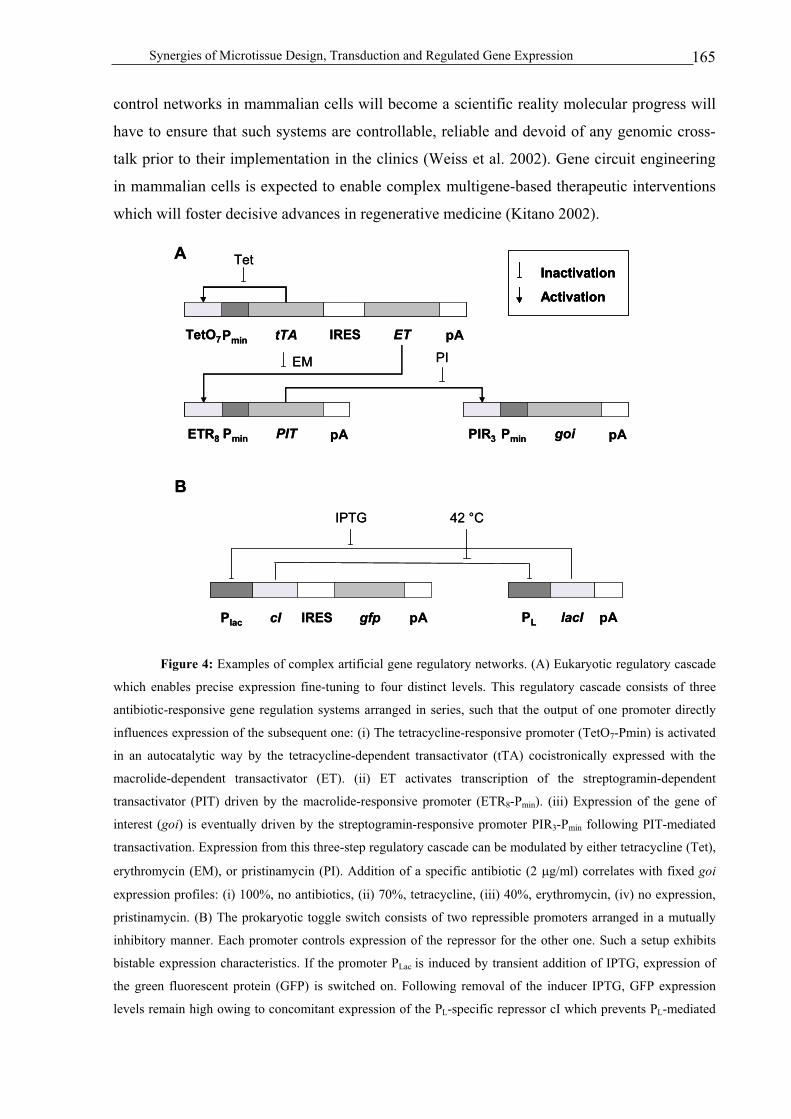
Synergies of Microtissue Design, Transduction and Regulated Gene Expression 165
control networks in mammalian cells will become a scientific reality molecular progress will
have to ensure that such systems are controllable, reliable and devoid of any genomic cross-
talk prior to their implementation in the clinics (Weiss et al. 2002). Gene circuit engineering
in mammalian cells is expected to enable complex multigene-based therapeutic interventions
which will foster decisive advances in regenerative medicine (Kitano 2002).
Figure 4: Examples of complex artificial gene regulatory networks. (A) Eukaryotic regulatory cascade
which enables precise expression fine-tuning to four distinct levels. This regulatory cascade consists of three
antibiotic-responsive gene regulation systems arranged in series, such that the output of one promoter directly
influences expression of the subsequent one: (i) The tetracycline-responsive promoter (TetO7-Pmin) is activated
in an autocatalytic way by the tetracycline-dependent transactivator (tTA) cocistronically expressed with the
macrolide-dependent transactivator (ET). (ii) ET activates transcription of the streptogramin-dependent
transactivator (PIT) driven by the macrolide-responsive promoter (ETR8-Pmin). (iii) Expression of the gene of
interest (goi) is eventually driven by the streptogramin-responsive promoter PIR3-Pmin following PIT-mediated
transactivation. Expression from this three-step regulatory cascade can be modulated by either tetracycline (Tet),
erythromycin (EM), or pristinamycin (PI). Addition of a specific antibiotic (2 g/ml) correlates with fixed goi
expression profiles: (i) 100%, no antibiotics, (ii) 70%, tetracycline, (iii) 40%, erythromycin, (iv) no expression,
pristinamycin. (B) The prokaryotic toggle switch consists of two repressible promoters arranged in a mutually
inhibitory manner. Each promoter controls expression of the repressor for the other one. Such a setup exhibits
bistable expression characteristics. If the promoter PLac is induced by transient addition of IPTG, expression of
the green fluorescent protein (GFP) is switched on. Following removal of the inducer IPTG, GFP expression
levels remain high owing to concomitant expression of the PL-specific repressor cI which prevents PL-mediated
Pmin tTA pAIRES ETTetO7
Pmin PITETR8 pA Pmin goiPIR3 pA
Tet
PIEM
Inactivation
Activation
A
B
Plac cI pAIRES gfp lacIPL pA
IPTG 42 °C
Pmin tTA pAIRES ETTetO7
Pmin PITETR8 pA Pmin goiPIR3 pA
Tet
PIEM
Inactivation
Activation
Inactivation
Activation
A
B
Plac cI pAIRES gfp lacIPL pA
IPTG 42 °C

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 166
expression of the Plac repressor LacI. GFP expression will be repressed following a temperature shift to 42 °C
which induces PL and promotes LacI-mediated shut down of Plac.
References
Amor, D. (2001). "Gene therapy. Principles and potential applications." Aust Fam Physician
30, 953-958.
Aubel, D., R. Morris, B. Lennon, M. Rimann, H. Kaufmann, M. Folcher, J. E. Bailey, C. J.
Thompson and M. Fussenegger (2001). "Design of a novel mammalian screening
system for the detection of bioavailable, non-cytotoxic streptogramin antibiotics." J
Antibiot (Tokyo) 54, 44-55.
Aubrecht, J., P. Manivasakam and R. H. Schiestl (1996). "Controlled gene expression in
mammalian cells via a regulatory cascade involving the tetracycline transactivator and
lac repressor." Gene 172, 227-231.
Auricchio, A., G. P. Gao, Q. C. Yu, S. Raper, V. M. Rivera, T. Clackson and J. M. Wilson
(2002). "Constitutive and regulated expression of processed insulin following in vivo
hepatic gene transfer." Gene Ther 9, 963-971.
Auricchio, A., V. Rivera, T. Clackson, E. O'Connor, A. Maguire, M. Tolentino, J. Bennett and
J. Wilson (2002). "Pharmacological regulation of protein expression from adeno-
associated viral vectors in the eye." Mol Ther 6, 238.
Badylak, S. F. (2002). "The extracellular matrix as a scaffold for tissue reconstruction." Semin
Cell Dev Biol 13, 377-383.
Balague, C., J. Zhou, Y. Dai, R. Alemany, S. F. Josephs, G. Andreason, M. Hariharan, E.
Sethi, E. Prokopenko, H. Y. Jan, Y. C. Lou, D. Hubert-Leslie, L. Ruiz and W. W.
Zhang (2000). "Sustained high-level expression of full-length human factor VIII and
restoration of clotting activity in hemophilic mice using a minimal adenovirus vector."
Blood 95, 820-828.
Baltzer, A. W., C. Lattermann, J. D. Whalen, S. Ghivizzani, P. Wooley, R. Krauspe, P. D.
Robbins and C. H. Evans (2000). "Potential role of direct adenoviral gene transfer in
enhancing fracture repair." Clin Orthop, S120-125.
Baron, U., S. Freundlieb, M. Gossen and H. Bujard (1995). "Co-regulation of two gene
activities by tetracycline via a bidirectional promoter." Nucleic Acids Res 23, 3605-6.

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 167
Baron, U., M. Gossen and H. Bujard (1997). "Tetracycline-controlled transcription in
eukaryotes: novel transactivators with graded transactivation potential." Nucleic Acids
Res 25, 2723-2729.
Bassler, B. L. (2002). "Small talk. Cell-to-cell communication in bacteria." Cell 109, 421-4.
Beato, M. (1989). "Gene regulation by steroid hormones." Cell 56, 335-344.
Beele, H. (2002). "Artificial skin: past, present and future." Int J Artif Organs 25, 163-73.
Berthiaume, F., P. V. Moghe, M. Toner and M. L. Yarmush (1996). "Effect of extracellular
matrix topology on cell structure, function, and physiological responsiveness:
hepatocytes cultured in a sandwich configuration." Faseb J 10, 1471-1484.
Bhadriraju, K. and C. S. Chen (2002). "Engineering cellular microenvironments to improve
cell-based drug testing." Drug Discov Today 7, 612-620.
Bischoff, J. R., D. H. Kirn, A. Williams, C. Heise, S. Horn, M. Muna, L. Ng, J. A. Nye, A.
Sampson-Johannes, A. Fattaey and F. McCormick (1996). "An adenovirus mutant that
replicates selectively in p53-deficient human tumor cells." Science 274, 373-376.
Blaese, R. M., K. W. Culver, A. D. Miller, C. S. Carter, T. Fleisher, M. Clerici, G. Shearer, L.
Chang, Y. Chiang, P. Tolstoshev and et al. (1995). "T lymphocyte-directed gene
therapy for ADA- SCID: initial trial results after 4 years." Science 270, 475-480.
Blau, C. A., K. R. Peterson, J. G. Drachman and D. M. Spencer (1997). "A proliferation
switch for genetically modified cells." Proc Natl Acad Sci U S A 94, 3076-3081.
Blomer, U., L. Naldini, T. Kafri, D. Trono, I. M. Verma and F. H. Gage (1997). "Highly
efficient and sustained gene transfer in adult neurons with a lentivirus vector." J Virol
71, 6641-6649.
Boorsma, M., L. Nieba, D. Koller, M. F. Bachmann, J. E. Bailey and W. A. Renner (2000).
"A temperature-regulated replicon-based DNA expression system." Nat Biotechnol 18,
429-432.
Bordignon, C., L. D. Notarangelo, N. Nobili, G. Ferrari, G. Casorati, P. Panina, E. Mazzolari,
D. Maggioni, C. Rossi, P. Servida and et al. (1995). "Gene therapy in peripheral blood
lymphocytes and bone marrow for ADA- immunodeficient patients." Science 270,
470-475.
Braeckmans, K., S. C. De Smedt, M. Leblans, R. Pauwels and J. Demeester (2002).
"Encoding microcarriers: present and future technologies." Nat Rev Drug Discov 1,
447-456.

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 168
Braselmann, S., P. Graninger and M. Busslinger (1993). "A selective transcriptional induction
system for mammalian cells based on Gal4-estrogen receptor fusion proteins." Proc
Natl Acad Sci U S A 90, 1657-1661.
Brenner, M. K., H. Heslop, R. Krance, M. Horowitz, D. Strother, J. Nuchtern, B. Grilley, E.
Martingano and K. Cooper (2000). "Phase I study of chemokine and cytokine gene-
modified autologous neuroblastoma cells for treatment of relapsed/refractory
neuroblastoma using an adenoviral vector." Hum Gene Ther 11, 1477-1488.
Browning, M. T., R. D. Schmidt, K. A. Lew and T. A. Rizvi (2001). "Primate and feline
lentivirus vector RNA packaging and propagation by heterologous lentivirus virions."
J Virol 75, 5129-5140.
Buchler, N. E., U. Gerland and T. Hwa (2003). "On schemes of combinatorial transcription
logic." Proc Natl Acad Sci U S A 100, 5136-5141.
Cao, Y., U. Karsten, G. Otto and P. Bannasch (1999). "Expression of MUC1, Thomsen-
Friedenreich antigen, Tn, sialosyl-Tn, and alpha2,6-linked sialic acid in hepatocellular
carcinomas and preneoplastic hepatocellular lesions." Virchows Arch 434, 503-509.
Carmeliet, P. (2000). "VEGF gene therapy: stimulating angiogenesis or angioma-genesis?"
Nat Med 6, 1102-1103.
Cavazzana-Calvo, M., S. Hacein-Bey, G. de Saint Basile, F. Gross, E. Yvon, P. Nusbaum, F.
Selz, C. Hue, S. Certain, J. L. Casanova, P. Bousso, F. L. Deist and A. Fischer (2000).
"Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease."
Science 288, 669-672.
Chen, C. S., M. Mrksich, S. Huang, G. M. Whitesides and D. E. Ingber (1997). "Geometric
control of cell life and death." Science 276, 1425-1428.
Condorelli, G., U. Borello, L. De Angelis, M. Latronico, D. Sirabella, M. Coletta, R. Galli, G.
Balconi, A. Follenzi, G. Frati, M. G. Cusella De Angelis, L. Gioglio, S.
Amuchastegui, L. Adorini, L. Naldini, A. Vescovi, E. Dejana and G. Cossu (2001).
"Cardiomyocytes induce endothelial cells to trans-differentiate into cardiac muscle:
implications for myocardium regeneration." Proc Natl Acad Sci U S A 98, 10733-
10738.
Crystal, R. G., E. Hirschowitz, M. Lieberman, J. Daly, E. Kazam, C. Henschke, D.
Yankelevitz, N. Kemeny, R. Silverstein, A. Ohwada, T. Russi, A. Mastrangeli, A.
Sanders, J. Cooke and B. G. Harvey (1997). "Phase I study of direct administration of
a replication deficient adenovirus vector containing the E. coli cytosine deaminase

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 169
gene to metastatic colon carcinoma of the liver in association with the oral
administration of the pro-drug 5-fluorocytosine." Hum Gene Ther 8, 985-1001.
Csete, M. E., K. E. Drazan, M. van Bree, D. F. McIntee, W. H. McBride, A. Bett, F. L.
Graham, R. W. Busuttil, A. J. Berk and A. Shaked (1994). "Adenovirus-mediated
gene transfer in the transplant setting. I. Conditions for expression of transferred genes
in cold-preserved hepatocytes." Transplantation 57, 1502-1507.
Cukierman, E., R. Pankov, D. R. Stevens and K. M. Yamada (2001). "Taking cell-matrix
adhesions to the third dimension." Science 294, 1708-1712.
Cunningham, C. and A. J. Davison (1993). "A cosmid-based system for constructing mutants
of herpes simplex virus type 1." Virology 197, 116-124.
Curiel, D. T., E. Wagner, M. Cotten, M. L. Birnstiel, S. Agarwal, C. M. Li, S. Loechel and P.
C. Hu (1992). "High-efficiency gene transfer mediated by adenovirus coupled to
DNA-polylysine complexes." Hum Gene Ther 3, 147-154.
Desoize, B. and J. Jardillier (2000). "Multicellular resistance: a paradigm for clinical
resistance?" Crit Rev Oncol Hematol 36, 193-207.
Duncan, S. A., M. A. Navas, D. Dufort, J. Rossant and M. Stoffel (1998). "Regulation of a
transcription factor network required for differentiation and metabolism." Science 281,
692-695.
Dzau, V. J., M. J. Mann, R. Morishita and Y. Kaneda (1996). "Fusigenic viral liposome for
gene therapy in cardiovascular diseases." Proc Natl Acad Sci U S A 93, 11421-1425.
Edwards, G. M., F. H. Wilford, X. Liu, L. Hennighausen, J. Djiane and C. H. Streuli (1998).
"Regulation of mammary differentiation by extracellular matrix involves protein-
tyrosine phosphatases." J Biol Chem 273, 9495-9500.
Elowitz, M. B. and S. Leibler (2000). "A synthetic oscillatory network of transcriptional
regulators." Nature 403, 335-338.
Eschenhagen, T., M. Didie, F. Munzel, P. Schubert, K. Schneiderbanger and W. H.
Zimmermann (2002). "3D engineered heart tissue for replacement therapy." Basic Res
Cardiol 97 Suppl 1, I146-152.
Eschenhagen, T., C. Fink, U. Remmers, H. Scholz, J. Wattchow, J. Weil, W. Zimmermann,
H. H. Dohmen, H. Schafer, N. Bishopric, T. Wakatsuki and E. L. Elson (1997).
"Three-dimensional reconstitution of embryonic cardiomyocytes in a collagen matrix:
a new heart muscle model system." Faseb J 11, 683-694.

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 170
Felgner, J. H., R. Kumar, C. N. Sridhar, C. J. Wheeler, Y. J. Tsai, R. Border, P. Ramsey, M.
Martin and P. L. Felgner (1994). "Enhanced gene delivery and mechanism studies
with a novel series of cationic lipid formulations." J Biol Chem 269, 2550-2561.
Freundlieb, S., C. Schirra-Muller and H. Bujard (1999). "A tetracycline controlled
activation/repression system with increased potential for gene transfer into
mammalian cells." J Gene Med 1, 4-12.
Fukuda, J., K. Okamura, K. Nakazawa, H. Ijima, Y. Yamashita, M. Shimada, K. Shirabe, E.
Tsujita, K. Sugimachi and K. Funatsu (2003). "Efficacy of a polyurethane
foam/spheroid artificial liver by using human hepatoblastoma cell line (Hep G2)." Cell
Transplant 12, 51-58.
Fussenegger, M., X. Mazur and J. E. Bailey (1998). "pTRIDENT, a novel vector family for
tricistronic gene expression in mammalian cells." Biotechnol Bioeng 57, 1-10.
Fussenegger, M., R. P. Morris, C. Fux, M. Rimann, B. von Stockar, C. J. Thompson and J. E.
Bailey (2000). "Streptogramin-based gene regulation systems for mammalian cells."
Nat Biotechnol 18, 1203-1208.
Fussenegger, M., S. Schlatter, D. Datwyler, X. Mazur and J. E. Bailey (1998). "Controlled
proliferation by multigene metabolic engineering enhances the productivity of Chinese
hamster ovary cells." Nat Biotechnol 16, 468-472.
Fux, C. and M. Fussenegger (2003). "Bidirectional expresison units enable streptogramin-
adjustable gene expression in mammalian cells." Biotechnol Bioeng 83, 618-625.
Fux, C., S. Moser, S. Schlatter, M. Rimann, J. E. Bailey and M. Fussenegger (2001).
"Streptogramin- and tetracycline-responsive dual regulated expression of p27(Kip1)
sense and antisense enables positive and negative growth control of Chinese hamster
ovary cells." Nucleic Acids Res 29, E19.
Fux, C., W. Weber, M. Daoud-El Baba, C. Heinzen, D. Aubel and M. Fussenegger (2003).
"Novel macrolilde-adjustable bidirectional expression modules for coordinated
expression of two different transgenes in mice." J Gene Med 5, 1067-1079.
Fux, C. F., M. (2004). "Dual-Regulated Expression of C/EBP-a and BMP-2 Enables
Differential Differentiation of C2C12 Cells into Adipocytes and Osteoblasts." Nucleic
Acids Res. 32, e1
Fux, C. L., D. Fussenegger, M. (2004). "Dual-Regulated MyoD- and Msx1-based
interventions in C2C12 Cells Enable Precise Myogenic/Osteogenic/Adipogenic
Lineage Control." J Gene Med. 6, 1159-1169.

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 171
Gale, M., Jr., S. L. Tan and M. G. Katze (2000). "Translational control of viral gene
expression in eukaryotes." Microbiol Mol Biol Rev 64, 239-280.
Gao, X. and L. Huang (1995). "Cationic liposome-mediated gene transfer." Gene Ther 2, 710-
722.
Gardner, T. S., C. R. Cantor and J. J. Collins (2000). "Construction of a genetic toggle switch
in Escherichia coli." Nature 403, 339-342.
Geissler, E. K., J. Wang, J. H. Fechner, Jr., W. J. Burlingham and S. J. Knechtle (1994).
"Immunity to MHC class I antigen after direct DNA transfer into skeletal muscle." J
Immunol 152, 413-421.
Giancotti, F. G. and E. Ruoslahti (1999). "Integrin signaling." Science 285, 1028-1032.
Girotto, D., S. Urbani, P. Brun, D. Renier, R. Barbucci and G. Abatangelo (2003). "Tissue-
specific gene expression in chondrocytes grown on three-dimensional hyaluronic acid
scaffolds." Biomaterials 24, 3265-3275.
Glover, C. P., A. S. Bienemann, M. Hopton, T. C. Harding, J. N. Kew and J. B. Uney (2003).
"Long-term transgene expression can be mediated in the brain by adenoviral vectors
when powerful neuron-specific promoters are used." J Gene Med 5, 554-559.
Go, W. Y. and S. N. Ho (2002). "Optimization and direct comparison of the dimerizer and
reverse tet transcriptional control systems." J Gene Med 4, 258-270.
Goldman, M. J., P. S. Lee, J. S. Yang and J. M. Wilson (1997). "Lentiviral vectors for gene
therapy of cystic fibrosis." Hum Gene Ther 8, 2261-2268.
Golub, T. R., D. K. Slonim, P. Tamayo, C. Huard, M. Gaasenbeek, J. P. Mesirov, H. Coller,
M. L. Loh, J. R. Downing, M. A. Caligiuri, C. D. Bloomfield and E. S. Lander (1999).
"Molecular classification of cancer: class discovery and class prediction by gene
expression monitoring." Science 286, 531-537.
Gossen, M. and H. Bujard (1992). "Tight control of gene expression in mammalian cells by
tetracycline-responsive promoters." Proc Natl Acad Sci U S A 89, 5547-5551.
Greenfield, A. (2000). "Applications of DNA microarrays to the transcriptional analysis of
mammalian genomes." Mamm Genome 11, 609-613.
Grossman, M., S. E. Raper, K. Kozarsky, E. A. Stein, J. F. Engelhardt, D. Muller, P. J. Lupien
and J. M. Wilson (1994). "Successful ex vivo gene therapy directed to liver in a
patient with familial hypercholesterolaemia." Nat Genet 6, 335-341.
Groth, A. C., E. C. Olivares, B. Thyagarajan and M. P. Calos (2000). "A phage integrase
directs efficient site-specific integration in human cells." Proc Natl Acad Sci U S A 97,
5995-6000.

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 172
Hacein-Bey-Abina, S., C. von Kalle, M. Schmidt, F. Le Deist, N. Wulffraat, E. McIntyre, I.
Radford, J. L. Villeval, C. C. Fraser, M. Cavazzana-Calvo and A. Fischer (2003). "A
serious adverse event after successful gene therapy for X-linked severe combined
immunodeficiency." N Engl J Med 348, 255-256.
Hardingham, T., S. Tew and A. Murdoch (2002). "Tissue engineering: chondrocytes and
cartilage." Arthritis Res 4 Suppl 3, S63-68.
Hasty, J., D. McMillen and J. J. Collins (2002). "Engineered gene circuits." Nature 420, 224-
30.
Heise, C., A. Sampson-Johannes, A. Williams, F. McCormick, D. D. Von Hoff and D. H.
Kirn (1997). "ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific
cytolysis and antitumoral efficacy that can be augmented by standard
chemotherapeutic agents." Nat Med 3, 639-645.
Horb, M. E., C. N. Shen, D. Tosh and J. M. Slack (2003). "Experimental conversion of liver
to pancreas." Curr Biol 13, 105-115.
Hoshikawa, Y., K. Amimoto, R. Mizuguchi and M. Hatakeyama (1998). "Highly controlled
heterologous gene expression through combined utilization of the tetracycline-
repressible transactivator and the lac repressor." Anal Biochem 261, 211-218.
Huang, H. V. (1996). "Sindbis virus vectors for expression in animal cells." Curr Opin
Biotechnol 7, 531-535.
Ido, A., K. Nakata, Y. Kato, K. Nakao, K. Murata, M. Fujita, N. Ishii, T. Tamaoki, H. Shiku
and S. Nagataki (1995). "Gene therapy for hepatoma cells using a retrovirus vector
carrying herpes simplex virus thymidine kinase gene under the control of human
alpha-fetoprotein gene promoter." Cancer Res 55, 3105-3109.
Ikeda, Y., Y. Goto, Y. Yonemitsu, M. Miyazaki, T. Sakamoto, T. Ishibashi, T. Tabata, Y.
Ueda, M. Hasegawa, S. Tobimatsu and K. Sueishi (2003). "Simian immunodeficiency
virus-based lentivirus vector for retinal gene transfer: a preclinical safety study in
adult rats." Gene Ther 10, 1161-1169.
Imhof, M. O., P. Chatellard and N. Mermod (2000). "A regulatory network for the efficient
control of transgene expression." J Gene Med 2, 107-116.
Itskovitz-Eldor, J., M. Schuldiner, D. Karsenti, A. Eden, O. Yanuka, M. Amit, H. Soreq and
N. Benvenisty (2000). "Differentiation of human embryonic stem cells into embryoid
bodies compromising the three embryonic germ layers." Mol Med 6, 88-95.
Jadlowiec, J. A., A. B. Celil and J. O. Hollinger (2003). "Bone tissue engineering:recent
advances and promising therapeutic agents." Expert Opin Biol Ther 3, 409-423.

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 173
Kafri, T., U. Blomer, D. A. Peterson, F. H. Gage and I. M. Verma (1997). "Sustained
expression of genes delivered directly into liver and muscle by lentiviral vectors." Nat
Genet 17, 314-317.
Kafri, T., D. Morgan, T. Krahl, N. Sarvetnick, L. Sherman and I. Verma (1998). "Cellular
immune response to adenoviral vector infected cells does not require de novo viral
gene expression: implications for gene therapy." Proc Natl Acad Sci U S A 95, 11377-
11382.
Kale, S., S. Biermann, C. Edwards, C. Tarnowski, M. Morris and M. W. Long (2000).
"Three-dimensional cellular development is essential for ex vivo formation of human
bone." Nat Biotechnol 18, 954-958.
Kamper, M. R., G. Gohla and G. Schluter (2002). "A novel positive tetracycline-dependent
transactivator (rtTA) variant with reduced background activity and enhanced
activation potential." FEBS Lett 517, 115-120.
Kay, M. A., J. C. Glorioso and L. Naldini (2001). "Viral vectors for gene therapy: the art of
turning infectious agents into vehicles of therapeutics." Nat Med 7, 33-40.
Kay, M. A., C. S. Manno, M. V. Ragni, P. J. Larson, L. B. Couto, A. McClelland, B. Glader,
A. J. Chew, S. J. Tai, R. W. Herzog, V. Arruda, F. Johnson, C. Scallan, E. Skarsgard,
A. W. Flake and K. A. High (2000). "Evidence for gene transfer and expression of
factor IX in haemophilia B patients treated with an AAV vector." Nat Genet 24, 257-
261.
Kelm, J. M., E. Ehler, L. K. Nielsen, S. Schlatter, J. C. Perriard and M. Fussenegger (2004).
"Design of Artificial Myocardial Microtissues." Tissue Eng. 10, 201-214
Kelm, J. M., N. E. Timmins, C. J. Brown, M. Fussenegger and L. K. Nielsen (2003). "Method
for generation of homogeneous multicellular tumor spheroids applicable to a wide
variety of cell types." Biotechnol Bioeng 83, 173-180.
Khalil, M., A. Shariat-Panahi, R. Tootle, T. Ryder, P. McCloskey, E. Roberts, H. Hodgson
and C. Selden (2001). "Human hepatocyte cell lines proliferating as cohesive spheroid
colonies in alginate markedly upregulate both synthetic and detoxificatory liver
function." J Hepatol 34, 68-77.
Khaoustov, V. I., G. J. Darlington, H. E. Soriano, B. Krishnan, D. Risin, N. R. Pellis and B.
Yoffe (1999). "Induction of three-dimensional assembly of human liver cells by
simulated microgravity." In Vitro Cell Dev Biol Anim 35, 501-509.
Khuri, F. R., J. Nemunaitis, I. Ganly, J. Arseneau, I. F. Tannock, L. Romel, M. Gore, J.
Ironside, R. H. MacDougall, C. Heise, B. Randlev, A. M. Gillenwater, P. Bruso, S. B.

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 174
Kaye, W. K. Hong and D. H. Kirn (2000). "a controlled trial of intratumoral ONYX-
015, a selectively-replicating adenovirus, in combination with cisplatin and 5-
fluorouracil in patients with recurrent head and neck cancer." Nat Med 6, 879-885.
Kitano, H. (2002). "Computational systems biology." Nature 420, 206-210.
Klein, R. L., E. M. Meyer, A. L. Peel, S. Zolotukhin, C. Meyers, N. Muzyczka and M. A.
King (1998). "Neuron-specific transduction in the rat septohippocampal or
nigrostriatal pathway by recombinant adeno-associated virus vectors." Exp Neurol
150, 183-194.
Kolossov, E., B. K. Fleischmann, Q. Liu, W. Bloch, S. Viatchenko-Karpinski, O. Manzke, G.
J. Ji, H. Bohlen, K. Addicks and J. Hescheler (1998). "Functional characteristics of ES
cell-derived cardiac precursor cells identified by tissue-specific expression of the
green fluorescent protein." J Cell Biol 143, 2045-2056.
Kootstra, N. A. and I. M. Verma (2003). "Gene therapy with viral vectors." Annu Rev
Pharmacol Toxicol 43, 413-439.
Kramer, B. P., W. Weber and M. Fussenegger (2003). "Artificial regulatory networks and
cascades for discrete multi-level transgene control in mammalian cells." Biotechnol
Bioeng 83, 810-820.
Krueger, C., C. Berens, A. Schmidt, D. Schnappinger and W. Hillen (2003). "Single-chain Tet
transregulators." Nucleic Acids Res 31, 3050-3056.
Lai, C. M., M. Brankov, T. Zaknich, Y. K. Lai, W. Y. Shen, I. J. Constable, I. Kovesdi and P.
E. Rakoczy (2001). "Inhibition of angiogenesis by adenovirus-mediated sFlt-1
expression in a rat model of corneal neovascularization." Hum Gene Ther 12, 1299-
1310.
Laitinen, M., J. Hartikainen, M. O. Hiltunen, J. Eranen, M. Kiviniemi, O. Narvanen, K.
Makinen, H. Manninen, M. Syvanne, J. F. Martin, M. Laakso and S. Yla-Herttuala
(2000). "Catheter-mediated vascular endothelial growth factor gene transfer to human
coronary arteries after angioplasty." Hum Gene Ther 11, 263-270.
Lan, K. H., F. Kanai, Y. Shiratori, M. Ohashi, T. Tanaka, T. Okudaira, Y. Yoshida, H.
Hamada and M. Omata (1997). "In vivo selective gene expression and therapy
mediated by adenoviral vectors for human carcinoembryonic antigen-producing
gastric carcinoma." Cancer Res 57, 4279-4284.
Layer, P. G., A. Robitzki, A. Rothermel and E. Willbold (2002). "Of layers and spheres: the
reaggregate approach in tissue engineering." Trends Neurosci 25, 131-134.

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 175
Lee, R. J., M. L. Springer, W. E. Blanco-Bose, R. Shaw, P. C. Ursell and H. M. Blau (2000).
"VEGF gene delivery to myocardium: deleterious effects of unregulated expression."
Circulation 102, 898-901.
Li, Z., J. Dullmann, B. Schiedlmeier, M. Schmidt, C. von Kalle, J. Meyer, M. Forster, C.
Stocking, A. Wahlers, O. Frank, W. Ostertag, K. Kuhlcke, H. G. Eckert, B. Fehse and
C. Baum (2002). "Murine leukemia induced by retroviral gene marking." Science 296,
497.
Lieber, A., D. S. Steinwaerder, C. A. Carlson and M. A. Kay (1999). "Integrating adenovirus-
adeno-associated virus hybrid vectors devoid of all viral genes." J Virol 73, 9314-
9324.
Linden, R. M., P. Ward, C. Giraud, E. Winocour and K. I. Berns (1996). "Site-specific
integration by adeno-associated virus." Proc Natl Acad Sci U S A 93, 11288-11294.
MacGregor, R. R., J. D. Boyer, R. B. Ciccarelli, R. S. Ginsberg and D. B. Weiner (2000).
"Safety and immune responses to a DNA-based human immunodeficiency virus (HIV)
type I env/rev vaccine in HIV-infected recipients: follow-up data." J Infect Dis 181,
406.
Malleret, G., U. Haditsch, D. Genoux, M. W. Jones, T. V. Bliss, A. M. Vanhoose, C.
Weitlauf, E. R. Kandel, D. G. Winder and I. M. Mansuy (2001). "Inducible and
reversible enhancement of learning, memory, and long-term potentiation by genetic
inhibition of calcineurin." Cell 104, 675-686.
Mallo, M., B. Kanzler and S. Ohnemus (2003). "Reversible gene inactivation in the mouse."
Genomics 81, 356-360.
Markert, J. M., M. D. Medlock, S. D. Rabkin, G. Y. Gillespie, T. Todo, W. D. Hunter, C. A.
Palmer, F. Feigenbaum, C. Tornatore, F. Tufaro and R. L. Martuza (2000).
"Conditionally replicating herpes simplex virus mutant, G207 for the treatment of
malignant glioma: results of a phase I trial." Gene Ther 7, 867-874.
Martin, K. H., J. K. Slack, S. A. Boerner, C. C. Martin and J. T. Parsons (2002). "Integrin
connections map: to infinity and beyond." Science 296, 1652-1653.
Meents, H., B. Enenkel, R. G. Werner and M. Fussenegger (2002). "p27Kip1-mediated
controlled proliferation technology increases constitutive sICAM production in CHO-
DUKX adapted for growth in suspension and serum-free media." Biotechnol Bioeng
79, 619-627.
Miller, A. D., D. G. Miller, J. V. Garcia and C. M. Lynch (1993). "Use of retroviral vectors
for gene transfer and expression." Methods Enzymol 217, 581-599.

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 176
Miller, D. G., M. A. Adam and A. D. Miller (1990). "Gene transfer by retrovirus vectors
occurs only in cells that are actively replicating at the time of infection." Mol Cell Biol
10, 4239-4242.
Mitta, B., M. Rimann, M. U. Ehrengruber, M. Ehrbar, V. Djonov, J. Kelm and M.
Fussenegger (2002). "Advanced modular self-inactivating lentiviral expression vectors
for multigene interventions in mammalian cells and in vivo transduction." Nucleic
Acids Res 30, e113.
Miyoshi, H., K. A. Smith, D. E. Mosier, I. M. Verma and B. E. Torbett (1999). "Transduction
of human CD34+ cells that mediate long-term engraftment of NOD/SCID mice by
HIV vectors." Science 283, 682-686.
Moosmann, P., O. Georgiev, H. J. Thiesen, M. Hagmann and W. Schaffner (1997). "Silencing
of RNA polymerases II and III-dependent transcription by the KRAB protein domain
of KOX1, a Kruppel-type zinc finger factor." Biol Chem 378, 669-677.
Morsy, M. A., M. Gu, S. Motzel, J. Zhao, J. Lin, Q. Su, H. Allen, L. Franlin, R. J. Parks, F. L.
Graham, S. Kochanek, A. J. Bett and C. T. Caskey (1998). "An adenoviral vector
deleted for all viral coding sequences results in enhanced safety and extended
expression of a leptin transgene." Proc Natl Acad Sci U S A 95, 7866-7871.
Moutsatsos, I. K., G. Turgeman, S. Zhou, B. G. Kurkalli, G. Pelled, L. Tzur, P. Kelley, N.
Stumm, S. Mi, R. Muller, Y. Zilberman and D. Gazit (2001). "Exogenously regulated
stem cell-mediated gene therapy for bone regeneration." Mol Ther 3, 449-461.
Mulligan, R. C. (1993). "The basic science of gene therapy." Science 260, 926-932.
Nakai, H., C. E. Thomas, T. A. Storm, S. Fuess, S. Powell, J. F. Wright and M. A. Kay
(2002). "A limited number of transducible hepatocytes restricts a wide-range linear
vector dose response in recombinant adeno-associated virus-mediated liver
transduction." J Virol 76, 11343-11349.
Naldini, L., U. Blomer, F. H. Gage, D. Trono and I. M. Verma (1996). "Efficient transfer,
integration, and sustained long-term expression of the transgene in adult rat brains
injected with a lentiviral vector." Proc Natl Acad Sci U S A 93, 11382-11388.
Naldini, L., U. Blomer, P. Gallay, D. Ory, R. Mulligan, F. H. Gage, I. M. Verma and D.
Trono (1996). "In vivo gene delivery and stable transduction of nondividing cells by a
lentiviral vector." Science 272, 263-267.
Neddermann, P., C. Gargioli, E. Muraglia, S. Sambucini, F. Bonelli, R. De Francesco and R.
Cortese (2003). "A novel, inducible, eukaryotic gene expression system based on the
quorum-sensing transcription factor TraR." EMBO Rep 4, 159-165.

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 177
Niwa, H., J. Miyazaki and A. G. Smith (2000). "Quantitative expression of Oct-3/4 defines
differentiation, dedifferentiation or self-renewal of ES cells." Nat Genet 24, 372-376.
No, D., T. P. Yao and R. M. Evans (1996). "Ecdysone-inducible gene expression in
mammalian cells and transgenic mice." Proc Natl Acad Sci U S A 93, 3346-3351.
Nunes, M. C., N. S. Roy, H. M. Keyoung, R. R. Goodman, G. McKhann, 2nd, L. Jiang, J.
Kang, M. Nedergaard and S. A. Goldman (2003). "Identification and isolation of
multipotential neural progenitor cells from the subcortical white matter of the adult
human brain." Nat Med 9, 439-447.
Oligino, T., P. L. Poliani, Y. Wang, S. Y. Tsai, B. W. O'Malley, D. J. Fink and J. C. Glorioso
(1998). "Drug inducible transgene expression in brain using a herpes simplex virus
vector." Gene Ther 5, 491-496.
Olivares, E. C., R. P. Hollis, T. W. Chalberg, L. Meuse, M. A. Kay and M. P. Calos (2002).
"Site-specific genomic integration produces therapeutic Factor IX levels in mice." Nat
Biotechnol 20, 1124-1128.
Padron, J. M., C. L. van der Wilt, K. Smid, E. Smitskamp-Wilms, H. H. Backus, P. E. Pizao,
G. Giaccone and G. J. Peters (2000). "The multilayered postconfluent cell culture as a
model for drug screening." Crit Rev Oncol Hematol 36, 141-157.
Peplinski, G. R., K. Tsung and J. A. Norton (1998). "Vaccinia virus for human gene therapy."
Surg Oncol Clin N Am 7, 575-588.
Perez, A., T. C. Grikscheit, R. S. Blumberg, S. W. Ashley, J. P. Vacanti and E. E. Whang
(2002). "Tissue-engineered small intestine: ontogeny of the immune system."
Transplantation 74, 619-623.
Pfeifer, A., M. Ikawa, Y. Dayn and I. M. Verma (2002). "Transgenesis by lentiviral vectors:
lack of gene silencing in mammalian embryonic stem cells and preimplantation
embryos." Proc Natl Acad Sci U S A 99, 2140-2145.
Pollock, R. and T. Clackson (2002). "Dimerizer-regulated gene expression." Curr Opin
Biotechnol 13, 459-467.
Pollock, R., R. Issner, K. Zoller, S. Natesan, V. M. Rivera and T. Clackson (2000). "Delivery
of a stringent dimerizer-regulated gene expression system in a single retroviral
vector." Proc Natl Acad Sci U S A 97, 13221-13226.
Prokop, A. (2001). "Bioartificial pancreas: materials, devices, function, and limitations."
Diabetes Technol Ther 3, 431-449.
Raper, S. E., M. Yudkoff, N. Chirmule, G. P. Gao, F. Nunes, Z. J. Haskal, E. E. Furth, K. J.
Propert, M. B. Robinson, S. Magosin, H. Simoes, L. Speicher, J. Hughes, J. Tazelaar,

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 178
N. A. Wivel, J. M. Wilson and M. L. Batshaw (2002). "A pilot study of in vivo liver-
directed gene transfer with an adenoviral vector in partial ornithine transcarbamylase
deficiency." Hum Gene Ther 13, 163-175.
Richardson, T. P., M. C. Peters, A. B. Ennett and D. J. Mooney (2001). "Polymeric system for
dual growth factor delivery." Nat Biotechnol 19, 1029-1034.
Rivera, V. M., T. Clackson, S. Natesan, R. Pollock, J. F. Amara, T. Keenan, S. R. Magari, T.
Phillips, N. L. Courage, F. Cerasoli, Jr., D. A. Holt and M. Gilman (1996). "A
humanized system for pharmacologic control of gene expression." Nat Med 2, 1028-
1032.
Roe, T., T. C. Reynolds, G. Yu and P. O. Brown (1993). "Integration of murine leukemia
virus DNA depends on mitosis." Embo J 12, 2099-2108.
Sadaie, M. R., M. Zamani, S. Whang, N. Sistron and S. K. Arya (1998). "Towards developing
HIV-2 lentivirus-based retroviral vectors for gene therapy: dual gene expression in the
context of HIV-2 LTR and Tat." J Med Virol 54, 118-128.
Samakoglu, S., D. Bohl and J. M. Heard (2002). "Mechanisms Leading to Sustained
Reversion of beta-Thalassemia in Mice by Doxycycline-Controlled Epo Delivery from
Muscles." Mol Ther 6, 793-803.
Samuel, D. (2003). "Bioartificial liver: present and future." Transplantation 75, 1769-70.
Sarkis, C., C. Serguera, S. Petres, D. Buchet, J. L. Ridet, L. Edelman and J. Mallet (2000).
"Efficient transduction of neural cells in vitro and in vivo by a baculovirus-derived
vector." Proc Natl Acad Sci U S A 97, 14638-14643.
Schlesinger, S. and T. W. Dubensky (1999). "Alphavirus vectors for gene expression and
vaccines." Curr Opin Biotechnol 10, 434-439.
Schroder, A. R., P. Shinn, H. Chen, C. Berry, J. R. Ecker and F. Bushman (2002). "HIV-1
integration in the human genome favors active genes and local hotspots." Cell 110,
521-529.
Schuldiner, M., O. Yanuka, J. Itskovitz-Eldor, D. A. Melton and N. Benvenisty (2000).
"Effects of eight growth factors on the differentiation of cells derived from human
embryonic stem cells." Proc Natl Acad Sci U S A 97, 11307-11312.
Scollay, R. (2001). "Gene therapy: a brief overview of the past, present, and future." Ann N Y
Acad Sci 953, 26-30.
Shen, C. N., J. M. Slack and D. Tosh (2000). "Molecular basis of transdifferentiation of
pancreas to liver." Nat Cell Biol 2, 879-887.

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 179
Shi, Q., Y. Wang and R. Worton (1997). "Modulation of the specificity and activity of a
cellular promoter in an adenoviral vector." Hum Gene Ther 8, 403-410.
Sommer, B., C. Rinsch, E. Payen, B. Dalle, B. Schneider, N. Deglon, A. Henri, Y. Beuzard
and P. Aebischer (2002). "Long-term doxycycline-regulated secretion of
erythropoietin by encapsulated myoblasts." Mol Ther 6, 155-161.
Spaete, R. R. and N. Frenkel (1982). "The herpes simplex virus amplicon: a new eucaryotic
defective-virus cloning-amplifying vector." Cell 30, 295-304.
Streuli, C. (1999). "Extracellular matrix remodelling and cellular differentiation." Curr Opin
Cell Biol 11, 634-640.
Sun, L., J. Li and X. Xiao (2000). "Overcoming adeno-associated virus vector size limitation
through viral DNA heterodimerization." Nat Med 6, 599-602.
Telenitsky, A. G., S.P. (1997). Retroviruses. Coffin. New York, Cold Spring Harbor Lab:
121-160.
Triezenberg, S. J., R. C. Kingsbury and S. L. McKnight (1988). "Functional dissection of
VP16, the trans-activator of herpes simplex virus immediate early gene expression."
Genes Dev 2, 718-729.
Vigna, E., S. Cavalieri, L. Ailles, M. Geuna, R. Loew, H. Bujard and L. Naldini (2002).
"Robust and efficient regulation of transgene expression in vivo by improved
tetracycline-dependent lentiviral vectors." Mol Ther 5, 252-261.
Wang, Y., B. W. O'Malley, Jr., S. Y. Tsai and B. W. O'Malley (1994). "A regulatory system
for use in gene transfer." Proc Natl Acad Sci U S A 91, 8180-8184.
Watanabe, S., J. Shuttleworth and M. Al-Rubeai (2002). "Regulation of cell cycle and
productivity in NS0 cells by the over-expression of p21CIP1." Biotechnol Bioeng 77,
1-7.
Weber, W., C. Fux, M. Daoud-El Baba, B. Keller, C. C. Weber, B. P. Kramer, C. Heinzen, D.
Aubel, J. E. Bailey and M. Fussenegger (2002). "Macrolide-based transgene control in
mammalian cells and mice." Nat Biotechnol 20, 901-907.
Weber, W., R. R. Marty, N. Link, M. Ehrbar, B. Keller, C. C. Weber, A. H. Zisch, C.
Heinzen, V. Djonov and M. Fussenegger (2003). "Conditional human VEGF-mediated
vascularization in chicken embryos using a novel temperature-inducible gene
regulation (TIGR) system." Nucleic Acids Res 31, E69.
Weber, W., R. Schoenmakers, M. Spielmann, M. Daoud-El Baba, M. Folcher, B. Keller, C.
C. Weber, N. Link, P. van de Wetering, C. Heinzen, B. Jolivet, U. Séquin, D. Aubel,
C. J. Thompson and M. Fussenegger (2003). "Streptomyces-derived quorum-sensing

Synergies of Microtissue Design, Transduction and Regulated Gene Expression 180
systems engineered for adjustable transgene expression in mammalian cells and mice."
Nucleic Acids Res 31, e71.
Weiss, R., S. Basu, S. Hoosangi, A. Kalmbach, D. Karig, R. Mehreja and I. Netravali (2002).
"Genetic Circuit Building Blocks for Cellular Computation, Communications, and
Signal Processing." Natural Computing.
Yang, S., K. F. Leong, Z. Du and C. K. Chua (2001). "The design of scaffolds for use in
tissue engineering. Part I. Traditional factors." Tissue Eng 7, 679-689.
Zheng, C., B. J. Baum, M. J. Iadarola and B. C. O'Connell (2000). "Genomic integration and
gene expression by a modified adenoviral vector." Nat Biotechnol 18, 176-180.
Zhu, H. L., A. S. Stewart, M. D. Taylor, C. Vijayasarathy, T. J. Gardner and H. L. Sweeney
(2000). "Blocking free radical production via adenoviral gene transfer decreases
cardiac ischemia-reperfusion injury." Mol Ther 2, 470-475.

181
Acknowledgements
I would like to take the chance to thank all those people who worked with me, helping to
improve the final outcome of this thesis.
Prof. Dr. M. Fussenegger: for giving me the opportunity to carry out my research in your
research group and to take advantage of your scientific experience. I appreciated very much
the scientific freedom and your scientific advice.
Prof. Dr. S. Werner: for being my coexaminer.
Dr. Elisabeth Ehler: for introducing me into the world of fluorescent antibodies and confocal
microscopy. I welcomed very much that I could ask every question to understand the cell
biology of heart muscle cells.
PD Dr. Valentin Djonov: for getting me into contact with the CAM analysis and interpretation
of histological specimens.
Prof. Dr. Simon Hoerstrup: for your advice.
Prof. Dr. Jean-Claude and Evelyne Perriard: for supplying me with primary cardiomyocytes
and suggestions whenever it was necessary.
Dr. Lars M. Ittner: for your motivating discussions and excellent help during my thesis.
Nadja Guiliani: for your continuous help and contribution to this work during your
apprenticeship.
Carlota Diaz Sanchez-Bustamante and Bettina Ley: for your ambitious support during your
diploma thesis.
David Fluri: for standing by my side in many battles and for creating a wonderful working
atmosphere in the lab.

182
Stefan Schlatter: for introducing me into the secrets of molecular biology and the conventions
of Swiss people. Unfortunately you left already after 1.5 years the lab to go to Australia.
The rest of the Fussi group and Peter Steiner: for their help and beer drinking assistance.

JENS KELM DEPARTMENT OF SURGICAL RASEARCH UNIVERSITY HOSPITAL
CH-8091 Zurich [email protected]
183
CURRICULUM VITAE
PERSONAL DATA
Name: Jens Kelm, Dipl. Biotech.
Date of Birth: 24 June 1971, Muelheim / Ruhr, Germany
Nationality: German
Current Position: Post-doctoral fellow at the University Hospital
Work Address: Jens Kelm
University Hospital Zurich
Tissue Engineering and CellTransplantation
LAB F-41
Raemistrasse 100
CH-8091 Zurich, Switzerland
Private Address: Jens Kelm
Baerenbohlstrasse 35
CH-8046 Zuerich, Switzerland
Tel: +41 1 3718328

JENS KELM DEPARTMENT OF SURGICAL RASEARCH UNIVERSITY HOSPITAL
CH-8091 Zurich [email protected]
184
EDUCATION
April 2005 – present Scientific assistant at the division of Tissue Engineering
and cell transplantation at the department of surgical
research and Clinic for Cardiovascular Surgery,
University Hospital, Zurich, Switzerland.
January 2001 – March 2005 Doctoral fellow at the Institute of Biotechnology, ETH
Zuerich, Switzerland; Ph.D. thesis under supervision of
Prof. Dr. Martin Fussenegger, and Prof. Dr. Sabine
Werner.
Title: “Design of Artificial Microtissues”.
September 2000 Graduation in Biotechnology, at the Technical University
of Braunschweig, Germany
September 1999 – April 2000 Diploma thesis at the Institute of Chemical Engineering,
University of Queensland, Australia, under supervision
of Prof. Dr. Ralf-Rainer Mendel (TU-Braunschweig) and
Prof. Dr. Lars Keld Nielsen.
Title: “Developing an Experimental Cell Cluster Model
to Evaluate Strategies for Tissue Engineering
Applications”.
September 1998 – March 1999 Semester work at the German Research Centre for
Biotechnology (GBF), Braunschweig, Germany, under
supervision of PD Dr. Roland Wagner. Title: Adaptation
of Immortalized Human Hepatocytes to Protein-Free
Culture Conditions”.

JENS KELM DEPARTMENT OF SURGICAL RASEARCH UNIVERSITY HOSPITAL
CH-8091 Zurich [email protected]
185
October 1994 Started as a student in the biotechnology program at the
Technical University of Braunschweig, Germany.
April 1988 – July 1993 High school, Muelheim, Germany, receiving the high-
school diploma.
April 1980 – March 1986 Primary school, Muelheim, Germany.
APPRENTICESHIP
August 1992 – June 1994 Vocational education as Biological Technical Assistant
(BTA) at the full-time vocational school for technical
assistants, Olsberg, Germany.
PUBLICATIONS
Schlatter S., M. Rimann, J. Kelm, and M. Fussenegger (2002). “SAMY, a novel mammalian
reporter gene derived from Bacillus stearothermophilus alpha-amylase”. Gene 282, 19-31.
Mitta B., M. Rimann, M.U. Ehrengruber, M. Ehrbar, V. Djonov, J. Kelm, and M. Fussenegger
(2002). “Advanced modular self-inactivating lentiviral expression vectors for multigene
interventions in mammalian cells and in vivo transduction” Nucleic Acids Res. 1;30(21):e113.
Kelm, J. M., N. E. Timmins, C. J. Brown, M. Fussenegger and L. K. Nielsen (2003). “Method
for generation of homogeneous multicellular tumor spheroids applicable to a wide variety of
cell types.” Biotechnol Bioeng 83, 173-80.
Kelm, J. M., E. Ehler, L. K. Nielsen, S. Schlatter, J. C. Perriard and M. Fussenegger (2004).
"Design of Artificial Myocardial Microtissues." Tissue Eng. 10, 201-214

JENS KELM DEPARTMENT OF SURGICAL RASEARCH UNIVERSITY HOSPITAL
CH-8091 Zurich [email protected]
186
Kelm, J. M., B. P. Kramer, V. Gonzalez-Nicolini, B. Ley and M. Fussenegger (2004).
“Synergies of Microtissue Design, Viral Transduction and Adjustable Transgene Expression
for Regenerative Medicine.” Biotechnol Appl Biochem. 39, 3-16
Kelm J.M. and M. Fussenegger (2004). “Microscale tissue engineering using gravity-enforced
cell assembly.” Trends Biotechnol. 22, 195-202.
Fux C., D. Langer, J.M. Kelm, W. Weber, and M. Fussenegger (2004). “New-generation
multicistronic expression platform: pTRIDENT vectors containing size-optimized IRES
elements enable homing endonuclease-based cistron swapping into lentiviral expression
vectors.” Biotechnol Bioeng. 86, 174-87.
Kelm J.M., C. Diaz Sanchez-Bustamante, E. Ehler, S.P. Hoerstrup, D. Djonov, L.M. Ittner and
M. Fussenegger (2005). “VEGF profiling and angiogenesis in human microtissues.” J.
Biotechnol. (in press).
Diaz Sanchez-Bustamante C., J. M. Kelm, B. Mitta and M. Fussenegger (2005). “Heterologous
Protein Production Capacity of Mammalian Cells in 2D and 3D cultures”. (in press).
Kelm J.M., V. Djonov, L.M. Ittner, W. Born, S.P. Hoerstrup, and M. Fussenegger
(submitted). “Design of Custom-Shaped Vascularized Tissues Using Microtissue Spheroids
as Minimal Building Units ”.
Kelm J.M., V. Djonov, S. P. Hoerstrup, C. I. Guenter, L. M. Ittner, F. Greve, A. Hierlemann,
J. C. Perriard, E. Ehler and M. Fussenegger (submitted). “Tissue-Transplant Fusion and
Vascularization of Myocardial Micro-and Macrotissues Implanted into Chicken Embryos and
Rats.”

JENS KELM DEPARTMENT OF SURGICAL RASEARCH UNIVERSITY HOSPITAL
CH-8091 Zurich [email protected]
187
POSTER PRESENTATIONS
Kelm J.M., P.D. Munro, S. Henning, S.C. Warren, C.J. Brown, and L.K. Nielsen (2001).
Homogenous Cell Clusters for Tissue Engineering Applications; The 17th
meeting of the
European Society of Animal Cell Technology Meeting, Tylösand, Sweden
Kelm J.M., E. Ehler., N. Timmis, L.K. Nielsen, M. Fussenegger (2001). “Exploring the Third
Dimension: Cell Culture Technology to Generate Microtissue” 1st Biennial Meeting of the
European Tissue Engineering Society, Freiburg, Germany
Kelm J.M., E. Ehler, and M. Fussenegger (2002). “Design of Artificial Microtissues”. 4. D-
Biol Symposium, Davos, Switzerland
Kelm J.M., Diaz-Sanchez-Bustamante C., and M. Fussenegger (2005). “VEGF Profiling and
Angiogenesis in Human Microtissues“.The 19th
meeting of the European Society of Animal
Cell Technology Meeting, Harrogate, United Kingdom
ORAL PRESENTATIONS
Design of Artificial Myocardial Microtissues, (2003) The 18th
meeting of the European
Society of Animal Cell Technology Meeting, Granada, Spain
Design of Artificial Microtissues: A Novel Concept for Cell-based Therapies and Tissue
Engineering, (2005) Biochemical Engineering XIV, Harrison Hot Springs, Canada