in copyright - non-commercial use permitted rights ...32672/... · das trockenspinnverfahren 74...
TRANSCRIPT

Research Collection
Doctoral Thesis
Studien auf dem Gebiet der hydrophilen Spinnfasern
Author(s): Jagrović, Petar
Publication Date: 1962
Permanent Link: https://doi.org/10.3929/ethz-a-000090174
Rights / License: In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For moreinformation please consult the Terms of use.
ETH Library

Prom. Nr. 3328
Studien auf dem Gebiet der
hydrophilen Spinnfasern
von der
Eidgenössischen Technischen Hochschule in Zürich
zur Erlangung der Würde eines Doktors
der technischen Wissenschaften
genehmigte
PROMOTIONSARBEIT
vorgelegt von
PETAR JAGROVIC
dipl. Ing.-Chem.
jugoslavischer Staatsangehöriger
Referent: Herr Prof. Dr. H. Hopff
Korreferent: Herr Prof. Dr. H. Zollinger
Verlag Mikrokopie, Mönchen
1962

Fotodrudi: Mikrokopw G.m.b.H. MOndwn 2, Wmtfr. 4

Meinen Eltern und meiner Frau
In Dankbarkeit gewidmet

Leer - Vide - Empty

Meinem sehr verehrten Lehrer
Herrn Prof. Dr. H. Hopff
danke Ich bestens für die wertvollen Ratschläge
und das stete Wohlwollen und Interesse, das er
mir und meiner Arbeit entgegenbrachte.

Leer - Vide - Empty

-7-
INHALTSVERZEICHNIS
Seite
EINLEITUNG 11
THEORETISCHER TEIL 14
1. FASERSTRUKTOB UND WASSEREMPFINDLICHKEIT 14
2. DIE IM TECHNISCHEN MASSTAB HERGESTELLTEN
HYDROPHILEN CHEMIEFASERN 19
2.1. Die Polyvinylalkohoi- und Polyvinyl-acetalfasern 19
2.2. Die Alginatfaser 21
2.3. Die regenerierten Elwelasfasern 22
3. DIE NEUEN HYDROPHILEN CHEMIEFASERN 25
3.1. Polyvinylpyrrolldon 25
3.2. Polyoxymethylen 28
3.3. Polyäthylenoxyd 31
3.4. Die regenerierten Kollagenfasern 35
3.4.1. Chaule des Hautnaterials 36
3.4.2. Struktuinodell und physlkalisoh-ohenisohe Eigenschaften des
Kollagen» 39
3.4.3. Die geschichtliche Entwicklungder Kollagenfaserheratellungund Patentliteratur 43
EXPERIMENTELLER TEIL 47
1. SPINNVEBSUCHE MIT POLYVINYL-
PYRROLIDON 47
1.1. Auagangsaaterial 47
1.2. Spinnversuohe nach de« Nasspinnver-fahren 47
1.2.1. Herstellung der SplnnlBsung 47
1.2.2. Viskosität dar Spinnlösung 47
1.2.3. Fällbader 49
1.2.4. Neutralisation dar aus alka¬
lis ohe» Fällbad ausgesponne¬nen Polyvinylpyrrolidon-Faden 55
1.2.5. Naohhärtung 56
1.2.6. Spinnapparatur 56
1.3. Das Trookenspinnverfahren 58
1.3.1. Spinnlösung 58
1.3.2. Spinnapparatur 58

-8-
Seite
1.4. Physikalische Eigenschaften der
hergestellten Fasern 59
1.4.1. Mechanische Eigenschaften 59
1.4.2. Röntgenographisohe Untersuchung 60
SPINNVERSUCHE MIT POLYOXYMETHY-
L E N 63
2.1. Ausgangsmaterial 63
2.2. Spinnversuche nach den Trockenspinn¬verfahren 63
2.3. Spinnversuche naoh dem Schmelzspinn¬verfahren 64
2.4. Physikalische Eigenschaften der her¬
gestellten Fäden 64
2.4.1. Mechanische Eigenschaften 65
2.4.2. Röntgenographische Untersuchung 65
SPINNVERSUCHE MIT POLYAETHYLEN-
0 X Y D Tl
3.1. Ausgangsmaterial 71
3.2. Spinnversuohe nach dem Schmelzspinn¬verfahren 72
3.2.1. Die Spinnapparatur 72
3.3. Das Trockenspinnverfahren 74
3.3.1. Die Versuche nach den Heiss-
trockenspinnverfahren 74
3.3.2. Das Kalttrookenspinnverfahren 76
3.3.3. Stabilisierungsversuche der
Polyäthylenoxyd-Produkte durch
Acetylierung 80
3.3.4. Hydrophobierungsversuche mit
Hilfe von Komplexbildnern 81
3.4. Das Nasspinnverfahren 86
3.5. Physikalische Eigenschaften der her¬
gestellten Fasern 86
3.5.1. Mechanische Eigenschaften 86
3.5.2. Röntgenographische Untersuchung 91
3.6. Molekulargewichts-Bestimmungen an
Polyäthylenoxydfasern 94
3.6.1. Wahl der Methode 94
3.6.2. Durchführung der Messungen 94

-9-
Seite
4. SPINNVERSUCHE MIT KOLLAGEN-
SUBSTANZEN 101
4.1. Vorversuoba nach dem alkallsohsn
Aufsohlussverfahren 101
4.1.1. Die Spinnapparatur 101
4.1.2. Das Verspinnen 104
4.2. Saures Aufsohlussverfahren 105
4.2.1. Versuche unter fermentâtivem
Abbau 106
4.2.2. Lösungsversuche 107
4.3. Die Spinnversuche Bit Kollagen¬
lösungen 111
4.3.1. Versuche alt Rinderdarmen als
Ausgangsstoff 111
4.3.2. Versuche mit Kalbsblösse-
Abfällen als Ausgangsstoff 114
4.4. Gerbungsversuohe alt Formaldehyd 119
4.5. Weiohnaohung 119
4.6. Physikalische Eigenschaften der her¬
gestellten Fasern 120
4.6.1. Mechanische Eigenschaften 120
4.6.2. Röntgenographisohe Untersuchung 130
ZUSAMMENFASSUNG 134
LITERATURVERZEICHNIS 136

Leer - Vide - Empty

u
EINLEITUNG
Seit H. de Chardonnet1'!« Jahre 1884 die
Nitratkunstseide als erste Kunstfaser im grossteohnisohen
Hasstab herstellte, hat die Produktion der Chemiefasern
eine enorme Entwicklung erlebt. So betrug bereits i« Jahre
1957 in den U.S.A. und Westeuropa der Anteil der künst¬
lichen Fasern für Bekleidungszwecke 27,3 % '. Es sind
verschiedene Kunstfasern neu geschaffen worden, wobei
als Ausgangsmaterial Naturprodukte verwendet (Cellulose,
Eiweiss, Silikate) oder die Fasern vollkommen synthe¬
tisch aufgebaut wurden (vollsynthetische Fasern).
Die Kunstfasern wurden am Anfang als Ersatzprodukte der Na¬
turfasern betrachtet; aber duroh die fortschreitende Ent¬
wicklung und Vervollkommnung wurden auoh solohe Fasern
hergestellt, die in vielen Eigenschaften den Naturfa¬
sern überlegen sind und wertvolle neue Produkte mit
völlig neuen Eigenschaften darstellen (Polyamide, Poly¬
urethane, Polyacrylnitrllfasern usw.). Ihre Anwendungs-
mögliohkeiten beschranken sioh nicht nur auf das Beklei¬
dungsgebiet, sie finden auch für mannigfaltige teohnisohe
Zwecke Verwendung.
Die Synthesefasern weisen andererseits Eigenschaften auf,
die nicht immer von Vorteil sind. So nehmen die voll¬
synthetischen Fasern (mit Ausnahme des. Polyvinylalkoholes)
weniger Feuchtigkeit auf und quellen geringer als die
natürlichen Fasern (vgl. Tab. l).

-12-
TABELLE I
Feuohtlgkeltsaufnahme und Querschnlttsquellung der Natur-,
Régénérât- und synthetischen Fasern,,bei 65 °k relativer
FeuchtigkeitJ'
Faserart Feuchtigkeitsaufnahme(in *)
A/Eiweissfasern:
Wolle
Seide
B/Cellulosefasern:
Baumwolle
Viscoserayon
C/Celluloseesterfasern:
Acetatrayon
D/Synthesefasern:
Polyamide(Nylon)
Polyacrylnitril(Orion)
Polyester(Dacron)
ca. 16,0ca. 11,0
8,513,5-15,5
6,0
ca. 4,0
1-2
ca. 0,5
Die geringe Wasseraufnahme der synthetischen
Querschnittsquellung(in *)
22-26
19-20
20-26
60
6
3,2
5
1
Fasern beruht
auf der kleineren Anzahl hydrophiler Gruppen (Hydroxyl-,
Carboxyl- und Aminogruppen) und auf dem hohen Kristalli¬
sationsgrad und der hohen Packungsdichte, wie auch auf der
besonderen Molekularstruktur.
Die Querschnittsquellung der synthetischen Fasern in Wasser
ist im Vergleich zu den Naturfasern und den Celluloseregene-
ratfasern sehr gering. Dies ist auoh der Grund, weshalb
Materialien aus synthetischen Fasern nach einer Nassbehand¬
lung (unterhalb des Fixlerpunktes) nicht einlaufen.
Rasohes Trocknen, Stabilität gegenüber Dimensionsänderungen
und geringe Knitterneigung der synthetischen Produkte stehen
der schlechten Schweissaugfähigkeit, der schwierigeren Farb-
barkeit und der teilweisen physiologischen Unverträglichkeit
gegenüber.
Die vorliegende Arbelt soll einen Beitrag zur Herstellung
neuer hydrophiler Fasern leisten.

-13-
Als Ausgangsstoffe für die Spinnversuche wurden einerseits
hochmolekulare vollsynthetlaohe Kunststoffe wie Polyvinyl-
pyrrolidon, Polyoxymethylen und Polyäthylenoxyd und anderer¬
seits Naturproteinkörper wie kollagene Substanzen verschie¬
denen Ursprungs verwendet, da alle diese Stoffe physiolo¬
gisch gut verträglich und mit Ausnahme des Polyoxymethylens
stark hydrophil sind und die daraus hergestellten Fasern
in der Medizin Verwendung finden könnten.

-14-
THEORETISCHER TEIL
i. FASERSTRUKTUR UND WASSEREMPFINDLICHKEIT
Die Wasserempfindliohkeit der Fasern ist einerseits von der
Anzahl hydrophiler Gruppen und anderseits von der Uber-
molekularen Struktur, d.h. von der Makromolekulkettenan-
ordnung und deren Verknüpfung abhangig.
Die chemische Zusammensetzung und der makromolekulare Bau
der Cellulose sind durch die Arbeiten von W. N. H a w o r t h'
und von H. Staudinger' einwandfrei bewiesen worden.
Durch eine fortlaufende ß -1,4-Verknupfung von Glucoseresten
bildet sich so durch Polykondensation das Riesenmolekul der
Cellulose. Es lasst sich sofort erkennen, daß pro Glucose-
einheit drei freie reaktionsfähige Hydroxylgruppen vorhanden
sind, die verestert und verathert werden können. Die Faser
musste infolge der freien Hydroxylgruppen wasserlöslich sein,
was aber nicht der Fall ist. Dies deutet darauf hin, dass die
Hydroxylgruppen eines Cellulosemolekuls fur die vom Lösungs¬
mittel ausgehenden Solvatationskrafte nicht verfügbar sind,
sondern dass sie in einer anderen Welse in Anspruch genommen
werden. Bei optischen Untersuchungen stellte C. Nagel i6'
Doppelbrechung fest und zog daraus den Schluss, dass die
Cellulosemolekule in einer regelmassigen kristallähnlichen
Ordnung zueinander stehen. Die Unloslichkeit der Cellulose
in Wasser liegt also im kristallähnlichen Bau begründet; denn
zwischen den Molekülketten sind Nebenvalenzkräfte wirksam,
die durch Wasserstoffbrücken die Hydroxylgruppen benachbarter
Celluloseketten miteinander verbinden. Bei der Baumwolle sind
die Wasserstoffbrücken besonders häufig, und die Faser hat in
der Querriohtung eine höhere Festigkeit als parallel zur
Faserachse. Auch ist die Volumenzunahme bei der Quellung in
beiden Richtungen unterschiedlich.

-15-
Dle Fasermasse 1st aber nicht vollständig kristallin ge¬
ordnet. Neben dem kristallinen Anteil existieren noch
amorphe Faserbereiche, in denen die Fadenmolekule wahllos
durcheinanderliegen. Nach neueren Anschauungen hangt das
Quellvermogen (Aufnahmefähigkeit fur das Wasser) der Faser
vom Anteil der amorphen Bereiche ab. In der Baumwollfaser
7)8)
betragt der amorphe Anteil nach P. Hermans'1'
30-40 %, bei der Viscosefaser 60 <%. Dadurch erklart sich
die fast doppelt so grosse Wasseraufnahme der Viscosefaser
im Vergleich zur Baumwolle.
Die Hydroxylgruppen der Cellulose, die in den amorphen Be¬
reichen nicht durch Van der Waals'sche Kräfte abgesattigt
sind, ziehen Wasser an und umgeben sich mit einer Wasserhulle.
Aufnahme von Wasser und Volumenzunahme, bzw. Quellung, ver¬
laufen gleichzeitig.
Durch Modifizierung der Molekularstruktur von naturlichen
und besonders regenerierten Cellulosefasern kann man hydro-
phobierende Effekte erzielen und sogar quellfeste Pasern
herstellen. Die Vernetzungsreaktionen der Cellulosemole-
kulketten werden meistens durch Methylolverbindungen her-
9 )vorgerufen. Auch durch Behandlung mit Chlormethylathern ,
Diisocyanaten ' und Aethyleniminderivaten ' erhalt man
Cellulosefasern mit völlig veränderten Eigenschaften.
Im Vergleich zu den naturlichen und regenerierten Cellulose¬
fasern weisen die Celluloseesterfasern eine geringere Wasser¬
empfindlichkeit auf. So wird z.B. bei der Celluloseacetat-
faser ein gewisser Anteil der Hydroxylgruppen durch den
Acetylrest "blockiert". Dadurch vermindern sich die Wasser¬
stoffbrücken zwischen den Molekulketten; sie treten nur
dort auf, wo nooh freie, nicht veresterte Hydroxylgruppen
vorhanden sind. Daher zeigt die Acetatfaser eine verminder¬
te Wasseraufnahmefähigkeit und gleichzeitig eine deutliche
Thermoplast! zitat.

-16-
Die veratherte Cellulose weist dagegen eine besonders hohe
Hydrophilie auf. Mit zunehmendem Verätherungsgrad werden
wasserlöslichere Produkte erhalten. So ist z.B. die Oxy-
athylcellulose mit einem Substitutionsgrad von 0,2 loslich
in Alkalien. Die wasserlösliche Oxyäthylcellulose besitzt
12 )einen Substitutionsgrad von mindestens 1,4 . Es wurden
noch von L. Lilienfeld versuchsweise alkalische Cellulose-
atherlosungen in einem Fallbad aus Schwefelsäure (10 %),
Essigsaure (25 %), Ammonchlorid (30 %) und Tannin (20 %)
zu Faden versponnen, doch haben diese bis jetzt keine
technische Bedeutung erlangt. Sie werden aber für Schlich¬
ten und Appreturen verwendet. Nach einem Verfahren der
13 )Firma Johnson & Johnson kann man die hydrophilen und
resorbierbaren chirurgischen Nähfaden aus Carboxymethy1-
cellulose herstellen, indem man als Ausgangsstoff die
Carboxymethylcellulose in mehr oder weniger neutralisierter
Salzform verwendet. Durch Einstellung des Neutralisations¬
grades (DN) der freien Säureresten und des Substitutionsgra¬
des bei der Veratherung ist es möglich, die gewünschte Los-
lichkeit zu steuern. Ein optimaler Neutralisationsgrad (DN)
liegt zwischen 15 und 60 <%. Der optimale Substitutions¬
grad (DS) betragt etwa 2, und die untere Grenze liegt bei
0,7.
Die Struktur der Eiwelssfasern wurde durch Arbeiten von
S p e a k m a n14), A s t b u r yt5\ Z a h n16^,17)
Pauling' und anderen Forschern eingehend unter¬
sucht. Dabei erwies sich, dass alle Eiweissfasern aus
etwa 20 verschiedenen Aminosäuren aufgebaut sind. Diese
sind in parallelliegenden Polypeptidketten amidartlg mit¬
einander verknüpft. Es sind jedoch nicht alle Aoino- und
Carboxylgruppen miteinander verknüpft, sondern es gibt
daneben freie und somit reaktionsfähige Amino- und Carboxyl¬
gruppen im Proteinmolekül. Die meisten hiervon liegen in
Salzform vor, indem Protonen von den Carboxylgruppen auf
die basischen Aminogruppen übertragen werden (Salzbrücken

-1T-
in der Wolle). Neben Salzbrücken existieren bei der Wolle
auoh DisulfldbrUoken, wodurch ebenfalls zwei benachbarte
Polypeptidketten UberbrUokt werden. Es handelt sich dabei
um Querbindungen, die duroh Hauptvalenzkräfte gebildet
werden. Dazu kommen häufig Querbindungen In Form von Wasaer-
stoffbriloken zwischen polaren Imlnogruppen der einen und
Carbonylgruppen der andern parallelliegenden Polypeptidkette.
Diese werden durch Nebenvalenzkräfte gebildet. Im Vergleich
zur Wolle ist die Naturseide einfacher gebaut, da sie nur
aus wenigen Aminosäuren besteht - rund 75 % der Säuren ent¬
fallen auf Glykokoll, Alanin und Serin. Die Querbindungen
bestehen praktisch nur aus Wasserstoffbrtioken. Bei Wasser¬
einwirkung nimmt Wolle mehr Feuchtigkeit auf, da sie gröss-
tentells amorphe Struktur aufweist, Im Gegensatz zu Natur¬
seide, die man als kristallin ansprechen kann. Eine grosse
Rolle spielt die Temperatur des einwirkenden Wassers. Nach
J.B. Speaknan' werden Im Bereioh von 55 bis 80° C
vor allem die Cystinbrücken der Wolle angegriffen; bei
höheren Temperaturen werden auoh die Peptldbindungen hydro-
lislert. Die kunstlichen Eiweissfasern sind infolge ungenü¬
gender Querbindungen noch viel hydrophiler. Sie müssen des¬
halb alle gehärtet, bzw. stabilisiert werden.
Zwischen der kristallinen Struktur der Naturseide und der-
Polyamidfaser besteht eine deutJLlohe Verwandtschaft. Die
Nebenvalenzwirkung erfolgt hier, ähnlich wie bei der Natur¬
seide, zwischen dem Sauerstoffatom der Carbonylgruppe und
der Imidgruppe, wobei die erste als Elektron- und die zweite
19)als Protondonator fungiert . Die Polyamidfasern haben
jedoch im Vergleich zur Naturseide nur wenig Wasserstoff¬
brtioken zwisohen den Molekülketten. Sie werden daher bei
Einwirkung von Wärme erweichen und schmelzen. Beim Nylon 66
kommen auf 100 Kettenatome 14 Imidgruppen und somit 14 Wasser-
stoffbrücken. Wird diese Zahl hither, d.h. der Abstand der
Imlnogruppen kleiner, so resultiert ein höherer Soheelzpunkt ',
Bei der Polyacrylnitrllfaser werden auoh Wasserstoffbin-

-18-
dungan zwisohen den Nitrilgruppen und dan H-Atomen an d«n
benachbarten Kohlenstoffatomen, die ein Wasserstoffatom be-
21 )sitzen, vermutet . Dia Nltrilgruppe, die an solohen Kohlen¬
stoffatomen liegt, lockert die C - H Bindung, wobei auoh die
Nltrilgruppe der parallelliegenden Kette durch dag freie
Elektronenpaar des Stickstoffes die Wasserstoffblndung
begünstigt. Bein dritten grossteohnlsoh hergestellten
Syntheseprodukt - der Polyesterfaser - erklärt sich die
Querbindung durch eine polarisierende Wirkung der Estergruppe22 )
auf die benachbarte Methylengruppe , woduroh das Wasser¬
stoffatob zur Brüokenblldung befähigt wird.
Die hohe Paokungsdlohte, bzw. der hohe Kristallisations¬
grad der erwähnten Synthesefasern, wie auoh die geringere
Anzahl der hydrophilen Gruppen bedingen minimale Wasser¬
aufnahme, was wiederum Ursaohe mehrerer nachteiliger Eigen¬
schaften ist.
Die vollständig hydrophoben Fasern, die keine reaktions¬
fähigen Gruppen enthalten, wie Polyäthylen und Polypropylen-
fasern. zeigen in Wasser und wässrlgen Färbeflotten weder eine
Quellung nooh eine Lockerung, so dass die Wasser-, bzw.
Farbstoffmoleküle, fast nicht einzudringen vermögen.
Die Eigenschaften der Polyvinylalkohol- und Polyvinylaoetal¬
fasern weiohen mit Bezug auf die Hydrophllie wesentlich von
denen der anderen oben erwähnten Fasern ab. Diese werden als
im technischen Hasstab hergestellte hydrophile Fasern im
nächsten Kapitel erwähnt.

-19-
2. DIE IM TECHNISCHEN HASSTAB HERGESTELLTEN HYDROPHILEN
CHEMIEFASERN
2.1. Die Polyvinylalkohol- und Polyvinylacetalfasern
Schon die Entdeoker de» Kunststoffes Polyvinylalkohol,
V.O. H e r m a n n und V. H a e h n e 1 ', haben
1931 versuoht, diesen linearen Polyalkohol zu Fäden zu ver¬
spinnen. Da der Polyvinylalkohol wasserlöslich ist, konnte
man wasserlösliche Fasern herstellen, die spater als chirur¬
gisches Nähmaterial von der Firma B. Braun, Melsungen, unter
der Bezeichnung "Synthofll" In den Handel gebracht wurden.
Den Polyvinylalkohol erhält man duroh Verseifung aus de« ent¬
sprechenden Polyvinylacetat. Das Polyvinylaoetat wird in Metha¬
nol gelost (20-40$lge Losung) und in Gegenwart von wenig Al¬
kall erhitzt.
Um das Produkt wasserlöslich zu erhalten, «uss der Verselfungs-
grad oa. 75 % betragen. Bei der grossteohnlsohen Herstellung
von Polyvinylalkohol führt nan aber die Verseifung bis zu
99 % und mehr. Es 1st bekannt, dass vollständig verseifte
Polyvlnylalkohole sloh in kalten Wasser nicht auflosen, sondern
lediglich quellen. Andererseits verliert der Polyvinylalkohol
seine Wasserlüsliohkeit beim trockenen Erhitzen. I. S a k u -
r a d a schrieb dieses Phänomen der Kristallisation zu *'.
Tatsächlich bestehen zwischen dem Quellungsgrad, dem Kristalli¬
sationsgrad und dem spez. Gewicht enge Beziehuligen: mit stei¬
gendem Kristallisationsgrad, bzw. spez. Qewioht, nimmt das
Quellvermögen von Polyvinylalkohol ab. Versuohe, den Grad
der Kristallisation zu erhöhen, sind laufend unternommen wor¬
den, um Fasern mit grOsserer Festigkeit und geringerer Hydro-
phllle zu erhalten. Diese Arbeiten zeigten unter andere«,
dass der Kristallisationsgrad und damit die Wasserbeständig¬
keit vom Gehalt an 1,2-Glykolstrukturen abhängig sind. Je
mehr 1,2 Glykol-Strukturen vorhanden sind, umso niedriger wird
der Kristallisationsgrad und umsomehr nimmt der Quellungsgrad
zu.

auf.Quellwasser%30etwaFaser
dienimmtC20°vonWasserinEingetauchtg/den.2,3
vonNassfestigkeiteineundg/den.3,2vonreissfestigkeit
Trooken-eineSakuradanachzeigtVinylonfaserDie
anzusehen.PolyacetalfasernalssindFasern
gehärtetenDie%.40-35beträgtFormallsierungsgradDer
0-CH„-0OHOHOH
CHg-CH---CH2-CH-CHg-OH..."^...CH2-CH-CH2-CH-CH2-CH-
(Vernetzung).
KettenverschiedenerHydroxylgruppenzwischenauchoder
KettegleichenderinHydroxylgruppenbenachbartenzwischen
AcetalbildungPolyvlnyalkoholbeimerfolgtKatalysatoren
saurenvonGegenwartinAldehydenmitHartungsprozessBeim
aufweist.SchafwollewieWasseraufnahmeahnlicheeineund
istkochbeständigdieherzustellen,"Vinylon"Faserhartete
ge¬Formaldehydmiteinegelungen,Japanernden1stesund
machen,zuwasserfesternochNachbehandlungeinedurchfaser
Polyvinylalkohol-dieZiel,dashattenForschungsarbeitenDie
sponnen.
ver¬C,100°überTemperaturenbeimeistheiss,Lösungdie
wirdherabzusetzen,ViskositätdieUmSpinnen.beimstung
Wasserverdun¬diebeschleunigenKetoneoderEsterflüchtige
leichtandereoderAlkoholDerAlkohol.%15undWasser
%40Polyvinylalkohol,%45z.B.wieverwendetLösungen
hochkonzentriertemöglichstwerdenTrockenspinnendasFur
stellung.
Viskoseher¬derbeiwieähnlichsindFällbaderDiespricht.
ent¬Viskosespinnlösung-Viskositätüblichenderwaswendet,
ver¬%16-14vonPolyvinylalkoholgehalteinemmitLosungen
wässrlgewerdenNasspinnverfahrendemnachHerstellungZur
möglich.TrockenspinnverfahrendemnachauchalsNass-dem
naohsowohl1stPolyvinylalkoholfasernvonHerstellungDie
-20-

-21-
2.2. Die Alginatfaser
Die Alginatfaser gehört zu den halbsynthetischen Produkten,
da sie aus der aus Seealgen leicht zu extrahierenden Algin-
säure gewonnen wird. Die Molekularstruktur der Alginsäure
wurde von L u n d e H e e n und 0 e y25)
vorgeschlagen
und von einer Reihe anderer Forsoher bestätigt (Hirst^Speakman "«** nhant^^-r-i o-i «
'*
28),
Jones undund
und As t b u r y*"3')« Di* Alginsäure ist eine polymère
d-Mannuronsäure mit Pyranosestruktur.
lain
!00H
OOOH
Wegen der anwesenden Carboxylgruppen ist die Faser schon in
schwach-alkalischer Seifenlösung vollständig löslich. Im
Vergleich mit der Cellulose ist die Alginsäure weniger regel-
mässig gebaut. Die Hydroxyl- und Carboxylgruppen sind in allen
Richtungen verteilt, so dass keine Möglichkeit zur Ausbil¬
dung starker Wasserstoffbrücken besteht. Alginatfasern sind
sehr hygroskopisch (Wasseraufnahme bis 44 %). Bei 65 % re¬
lativer Feuchtigkeit liegt der Wassergehalt zwischen 20 und
35 %. Er steigt mit dem Calciumgehalt an.
Die Trookenfestigkeit beträgt 2,1 g/den.
Die Herstellung der Alginatfaser beruht auf der Extrak¬
tion von Tang mit Sodalösung, wodurch eine dicke, gelatinöse
Masse erhalten wird. Diese Masse wird nach der Filtrierung
gebleicht (NaOCl) und mit Salzsäure gefällt (Alginsäure).
Nach dem Waschen löst man die gewonnene reine Alginsäure
mit Soda zu Natriumalginat. Versponnen wird eine 8-9 #ige
Lösung in einem Fällbad ähnlicher Zusammensetzung, wie es
zum Viskosespinnen verwendet wird, oder neuerdings in einem
salzsauren Calciumchloridfällbad, wodurch lagerbeständigere

-22-
Calciumalginatfasern gewonnen werden.
Die Alginatfasern finden als Trenn- und Stutzfaden in der
Textilindustrie Verwendung.
2.3. Die regenerierten Eiweissfasern
Das Bestrehen, pflanzliche und tierische Proteine aus ihrer
Losung zu Fasern zu verspinnen, ist alt. Die naturlichen
Eiweiisfasern, die sich durch einen angenehmen, warmen Griff
auszeichnen, sind teuer in ihrer Gewinnung. Der groaste Teil
des von den Schafen and Seidenraupen aufgenommenen Eiweisses
dient als Energiequelle. Die Haare, bzw. Faden, sind ein Teil
ihres Lebensprozesses, wobei nur ein kleiner Teil des Eiweisses
in Faserform verbleibt. Zudem ist die Naturseide bezuglich
ihrer Dimensionen und die Schafwolle sowohl hinsiohtlich ihrer
Feinheit als auch ihrer Stapellange beschrankt. Die kunstlichen
Fasern aus Eiweisskorpern sind vor allem aus wirtschaftlichen
Gründen entwickelt worden.
Im Jahre 1894 erhielt A. M i 1 1 a r das erste Patent fur
30 )das Trockenspinnen von Faden aus Gelatine . 10 Jahre spater
fand F. Todtenhaupt ein Verfahren zur Erzeu-
31 )gung von Faden aus dem Casein der Miloh '. Im Jahre 1924
begann A. Ferretti' seine erfolgreichen Arbeiten
über Caseinfasern und brachte diese 1936 zu technischer
Reife. Von wesentlicher Bedeutung für die Produktion seines
"Lanitals" durch die Snia Viscosa S.A. war die Erkenntnis,
dass Salz die Quellung der Fasern zu unterdrücken vermag.
("Lanital" wurde nach dem Zweiten Weltkrieg durch eine ver¬
besserte Caseinfaser "Merinova" ersetzt.) Seither ist die
Entwicklung der regenerierten Eiweissfasern weitergeführt
worden und zwar vor allem in England, Italien, den U.S.A.

-23-
und Japan. Es sind neben den Caseinfasern auch noch andere
Proteinfasern entwickelt und im technischen Masstab herge¬
stellt worden. In England zogen W. T. Astbury und
33 )
Mitarbeiter Erdnusseiweiss als Ausgangsprodukt heran '
und entwickelten eine Faser, die durch ICI unter den Namen
"Ardil" produziert wurde; in den U.S.A. wird heute eine
hochwertige Eiweissfaser "Vioara" aus Zein, einem Eiweiss-
34)körper des Maiskornes, hergestellt . Sojabohneneiweiss
wurde von Japanern zur Erzeugung von "Silkool"-Fasern heran¬
gezogen.
Es ist charakteristisch für alle diese Fasern, dass sie
nicht aus linearen, sondern aus sphärokolloiden Proteinkör¬
pern aufgebaut sind. Bei allen erwähnten Fasern fehlt das
langgestreckte faserbildende Molekül, und aus diesen Grunde
liefern sie kein deutliches Röntgendiagramm. Naoh einer
35 )neueren Theorie ' wird angenommen, dass die Proteinniole-
küle unmittelbar nach dem Verspinnen als "molekulare KUgel-
ohen" vorliegen. Werden diese zu Ketten ausgezogen, unter¬
einander mit Formaldehyd querverknüpft, gestreckt und noch¬
mals mit Formaldehyd in ihrer Lage fixiert, so nehmen die
Molekülketten Fasereigensohaften an.
Da alle heutigen technischen Regeneratproteinfasern aus
sphärokolloiden Proteinen hergestellt sind, ist ihre Er¬
zeugung ähnlich und der Arbeitsgang im allgemeinen folgender:
a) Extraktion der Proteine aus dem Naturstoff und De¬
naturierung durch Säuren, verdünnte Alkalien oder
Harnstoff;
b) Dispergierung, bzw. Lösung der Proteine durch Harn¬
stoff, kalte Alkalilösungen oder Eisessig;
o) Ausspinnen der Proteindispersion von 8 - 35 % Protein¬
gehalt in sauren Salzlösung-Fällbädern;
d) Streokung oder Orientierung der Faden;
e) Härtungsprozess mit neutraler oder saurer Form¬
aldehydlösung ;

können.werden
erzeugtFasernhydrophilereissfesteredarausdasswarten,
er¬zuwarEszeigt.FaserstrukturlinearedeutlicheAufbau
derenKollagensubstanzen,sondernProteine,sphärokolloiden
keinejedochverwendet,Spinnversuchefürgangsprodukt
Aus¬alsNaturproteinkörperauchKunststoffenhydrophilen
vollsynthetischennebenwurdenArbeltvorliegendenderIn
Dehnung.grossezumeistfestigkeit,
Reiss¬geringeNachteile:anfärbbar.leichtsehrundarm
knitter¬mottenecht,filzend,nichtWärmeverhaitungsvermögen,
gutesGriff,wollähnlicherweicher,Eigenschaften:Allgemeine
3030-501,1-1,31,4-1,6Schafwolle
Vergleich:Im
70-8010-110,13-0,170,5-0,6(Régénérât)Wolle
ausKeratinfaser
0,65-0,751,4-1,5acetyliert
(Vicara),Zeinfaser
50420,551,25-1,30normal
(Vicara),Zeinfaser
0,2-0,30,7-0,8Sojabohnenfaser
20-2210-110,2-0,30,7-0,9Erdnussfaser
50400,51,09Caseinfaser
nasstrockennasstrockenrs
%)(inDehnungg/den.)(inReissfestigkeitteraaF
EIWEISSFASERN36'KUENSTLICHENVONDEHNUNGUNDREISSFESTIGKEIT
IITABELLE
Chromsalzen.mitBehandlungoderDesaminierung
Acetylierung,durchFormaldehyd,mitBehandlungmalige
noch¬durchStabilisierung,bzw.Unlöslichmachen,g)
StreckungNochmaligef)
-24-

-25-
3. DIE NEUEN HYDROPHILEN CHEMIEFASERN
3.1. Polyvlnylpyrrolidqn
Polyvinylpyrrolidon gehört zu den zahlreichen Folgeprodukten
der modernen Acetylen-Chemie, die W. R e p p e und Mit-
37 )
arbeiter in der BASF in Ludwigshafen entwickelt haben '.
In Kombination mit Kochsalzlösung und noch anderen Zusätzen
fand Polyvinylpyrrolidon als Blutplasmaersatz während des
Zweiten Weltkrieges umfangreiche Anwendung.
Ausgangsmaterial für das Polyvinylpyrrolidon ist das mono-
mere Vinylpyrrolidon, das sich aus einfachsten Bausteinen wie
Acetylen, Formaldehyd und Ammoniak unter Heranziehung der
38 )
Aethynylierung und Vinylierung gewinnen lässt '.
Polymerisation: Schon 1939 wurde beobachtet, dass sich Vinyl-og\
pyrrolidon unter Einwirkung von Sulfit ' oder Sauerstoffab¬
gebenden Mitteln 'polymerisleren lässt. Das technisch
brauchbare Verfahren entwickelten jedoch erst H. Fikent-
scher und K, Herrle .
Wasserstoffperoxyd hat sich als geeigneter Polymerisations¬
katalysator erwiesen. Die ursprünglich angewandte Substanz-
polymerisation ergab aber infolge der schlechten Wärmeab¬
führung nur gelbe oder braune, uneinheitliche Produkte. Diese
enthielten noch etwa 10 % monomères Vinylpyrrolidon, das
wegen seiner toxischen Eigenschaften durch Extraktion des
Polymerisats mittels Aether entfernt werden muss te.
Es wurde deshalb zur Losungspolymerisation übergegangen.
Auch dafUr ist Wassers toffperoxyd der geeignetste Katalysa¬
tor. Da Vinylpyrrolidon sich in saurer Lösung unter Abspal¬
tung von Acetaldehyd leicht zersetzt, muss die Lösung ge¬
puffert werden. Als Puffersubstanzen bewährten sich vor allem
Ammoniak und allphatische Amine, die gleichzeitig stark akti¬
vierend auf die Polymerisation wirken, so dass man bei re¬
lativ niedrigen Temperaturen arbeiten kann. Damit waren die
bei der Substanzpolymerisation vorhandenen Schwierigkeiten
Überwunden.

-26-
Ein weiterer Vorteil der Losungspolymerisation besteht darin,
dass nach diesem Verfahren Polymerisate von beliebigem Poly-
cerisationsgrad (K-Werte zwischen 10 und 120) erhalten wer¬
den können.
Bei einer Reaktionstemperatur von 70-80° C erhalt man z.B.
unter Zusatz von kleinen Salzmengen mit 2 % 30%iger HgOg-Losung und 40 % Wasser (berechnet auf Vinylpyrrolidon) ein
Polymer mit dem K-Wert 30 (gunstig fur künstliches Blut¬
plasma) und mit einem Zusatz von nur 0,1 % HgO^-Losung ein
Polymer mit dem K-Wert 80.
Die nach dem Losungsverfahren hergestellten Polymerisate
sind völlig klar und farblos und enthalten höchstens 2 %
monomère Anteile.
Der K-Wert der Polymerisate ist unabhängig von 1er Konzen¬
tration des Monomers. Diese beeinflusst nur die Polymeri¬
sationsgeschwindigkeit. Letztere steigt auch mit der Konzen¬
tration des Wasserstoffperoxyds stark an und ist proportional
der Wurzel aus der Ammoniak-Konzentration.
Eigenschaften: Die Wasserloslichkeit des Polyvinylpyrrolidons
ist durch die Laotamgruppierung bedingt. In wassriger Losung
umhüllen die Wassermolekule die Folymerkette besonders in
Nachbarschaft der polaren Stickstoff- und Sauerstoffatome der
Carbonylgruppe, die das Wasser anzuziehen und festzuhalten
vermögen . Physikalische Untersuchungen von Polyvmylpyrro-
lidon-Hydraten (Reaktionswarme, Infrarot-Absorption) weisen
darauf hin, dass sich an das geloste Polyvinylpyrrolidon-
*Die K-Werte stellen eine Funktion des mittleren Molekularge¬wichts dar. Der K-Wert ergibt sich aus der Formel von
H. Fikentscher42):
l06t|rel_
75
o~
1 + 1,5 k . oT *
c = Konzentration in g/100 ml der Losung
,Viskosität der Lösung
"
Viskosität des Lösungsmittels
In der Praxis wird der k-Wert mit 1000 multipliziert, um
Dezimalziffern zu vermeiden.

-27-
MolekUl ungefähr 0,5 Mol Wasser/Monomer-Einheit anlagert4 '.
Dies entspricht der Grössenordnung, wie sie In der Literatur
für die Hydratisierung verschiedener Proteine angegeben wird.
Ausser in Wasser löst sich Polyvinylpyrrolldon unbeschränkt
in verdünnten und konzentrierten wässrigen Lösungen der Mi¬
neralsalze. Trotz seiner hohen Wasserlöslichkeit ist es auch
in zahlreichen organischen Lösungsmitteln löslich, so z.B.
in niedrigen Alkoholen, Glycerin, chlorierten Kohlenwasser¬
stoffen, Dimethylformamid usw. Teilweise löslich ist es in
Dioxan, Aceton und Methylathylketon. Interessant ist, dass
Polyvinylpyrrolidon in Methylenchlorid in Anwesenheit von
Wasser schwer löslich ist. In Aether, Benzol, aliphatischen
und cycloaliphatlschen Kohlenwasserstoffen ist es unlöslich.
In Gegenwart von Wasser Überwiegen jedoch stets die hydrophy-
len Eigenschaften. Ueberschlchtet man die Losung in Chloro¬
form mit Wasser, so geht es in die wässrige Phase über. Aus
wässrigen Läsungen lässt es sich nicht mehr mit organischen
Lösungsmitteln extrahieren.
Polyvinylpyrrolidon ist durch starke Basen und Polyhydro-
phenole, wie Resorcin und Tannin, aus wässrigen Lösungen
ausfällbar.
Die Fasern aus Polyvinylpyrrolidon: Es wurden eingehende
Untersuchungen durchgeführt, um die wässrige Lösung des
Polyvinylpyrrolidons nach dem Nasspinnverfahren zu Fäden
zu verspinnen; es wurde aber auch versucht, die Polyvlnyl-
pyrrolidon-Lösungen in verschiedenen Lösungsmitteln nach dem
Trockenspinnverfahren zu verspinnen. Wie im experimentellen
Teil gezeigt wird, können die Fäden sowohl nach dem einen
als auch nach dem andern Verfahren hergestellt werden.

-28-
Abb. 1
Die Polyvinylpyrrolidon-Faser nach
dem Trookenspinnverfahren hergestellt(Vergrösserung 70 x)
Bedingt durch die minimale Kristallini tat und den kleinen
Orientierungsgrad weisen die Fäden nur eine geringe Reiss¬
festigkeit auf. Die Fasern finden daher für die Textilindus¬
trie keine Verwendung, eignen sich aber in Form von Watte
für medizinische Zwecke, da sie blutstillend wirken und vom
iLenschlichen Organismus gut resorbierbar sind.
Nach den Versuchen mit Polyvinylpyrrolidon, das leicht
wasserlösliche Fasern lieferte, wurde versucht, den polymeren
Formaldehyd (Polyoxymethylen) zu Faden zu verspinnen und somit
auch resorbierbare, aber nicht leicht wasserlösliche Fäden
herzustellen.
3.2. Polyoxymethylen
Die erste vollsynthetische, technisch jedoch nicht brauch¬
bare Faser wurde in den Zwanzigerjähren von H. Stau-
d i n g e r und Mitarbeitern in den Laboratorien der ETH

-29-
în Zurich aus PoXyoxymethylen hergestellt . Es handelte
sich dabei um eine Sublimation des ß-Polyoxymethylens im
Vakuum, wodurch kleine Faserchen entstanden, die nach den
rontgenometrischen Untersuchungen celluloseahnliche Struk¬
tur zeigten. Die Polymere von Formaldehyd können kristallin
sein, wenn sie hohes Molekulargewicht haben. Erst seit Dupont
durch seine "Delrin"-Marken das ausserordentlich hochmole¬
kulare und stabilisierte Polyoxyir.ethylen auf den Markt brach¬
te, besteht die Möglichkeit, auch technisch brauchbare Fasern
herzustellen.
Polymerisation von Fornialdehyd :
Niedere Polyoxymethylene oder, richtiger formuliert, Poly-
oxymethylenglykole sind schon in allen wassrigen Losungen
von Formallehyd enthalten.
HCHO .,H?,°
ynn-Pff -fiH
P-, Bfi-(m n) -CH„-OH
& a 11 £
Sie stehen mit dem Methylenglykol und den höheren Oligo-
reren im Gleichgewicht, das von der Konzentration, dem pH
und der Temperatur abhangt. Sie sind als Polykondensate
des Methylenglykols anzusprechen.
Paraformaldenyd als nachststehendes höheres Oligomer
wird meistens durch Eindampfen der Formaldehydlosungen
unter vermindertem Druck erhalten. Paraformaldehyd ist ein
lineares Polymères und hat die Formel HO(CH„O)nH, wobei
n einen Durchschnittswert von rund 30 hat. Ist n kleiner
als 12, so ist das Produkt löslich in Wasser, Aceton oder
Aether, die höheren Polymere sind jedoch unlöslich. Eine
langsame Auflosung der höheren Polymere in Wasser ist von
einer Hydrolyse begleitet, die zu Bruchstucken von geringe¬
rem Molekulargewicht fuhrt.
Polymere mit hohem Molekulargewicht (über 150.000), von
H. Staudinger Eu-Polyoxymethylene genannt, sind
aus flussigem Formaldehyd bei tiefer Temperatur erhalten

-30-
worden. Die Verflüssigung des Formaldehydes ist leicht
durchzuführen (Kp: -21° C). Formaldehyd bildet auch feste
Polymere, wenn das Gas bei Temperaturen unter 137° C mit
festen Oberflachen in Berührung kommt.
Neben den linearen Polymeren kann man auch cyoliache Polymere
herstellen. Wird eine 60 - 65%ige wassrige Formaldehydlosung
mit 2 # H2S04 destilliert> so lasst sich aus dem Destillat
das cyolische Trimer Trioxymethylen mit Methylenchlorid
extrahieren. Diese farblose Verbindung schmilzt bei 62° C
und siedet bei 115° C ohne Zersetzung oder Depolymerisie-
rung; sie ist in Wasser und organischen Losungsmitteln los¬
lich. Starke Sauren bewirken Depolymerisation. Trioxymethy-
len verspricht als Quelle fur Formaldehyd bei Reaktionen in
nichtwassrigen Losungen in Frage zu kommen. Dieses ringför¬
mige Produkt kann auch in lineare, fasernbildende Polyoxyme-
thylene umgewandelt werden .
Die klassische Arbeit von H. Staudinger und
Mitarbeitern ' über Polyoxymethylene und ihre Derivate
(Diacetate und Dimethylather) hat wertvolle Erkenntnisse
über den Polymerisationsvorgang und die Makromoleküle ge¬
liefert. Diese Polymere hatten Jedoch, selbst wenn sie hohes
Molekulargewicht aufwiesen, keine Bedeutung fur die Erzeu¬
gung von Fasern. Ein Hauptnachteil war die Leichtigkeit, mit
der Depolymerisation - hauptsachlich unter Einwirkung von
Sauren oder Warme - eintritt.
Eine wesentlich grossere Stabilität im Vergleich mit den
ursprunglichen Produkten weisen die Acetylderivate des
Polyoxymethylens auf. Die Acetylierung der endstandigen
Hydroxyl-Gruppen wird mittels Acetanhydrid, eventuell unter
Zusatz von Pyridin als Katalysator, durchgeführt.
Die hochwertigen "Delrin"-Produkte der Firma Dupont stellen
ein so stabilisiertes, hochmolekulares Polyoxymethylen dar,
dass dessen Eigenschaften an die der Polyamide heranreichen.
Die eigenen Versuche, aus solchem hochmolekularem und stabi¬
lisiertem Polyoxymethylen Faden zu verspinnen, ergaben, dass
nach dem Schmelzspinnverfahren, das im experimentellen Teil
beschrieben wird, brauchbare Faden herstellbar sind.

-31-
Abb. 2
Die Polyoxymethylenfaser aus "Delrin"
nach dem Schmelzspinnverfahren gesponnen
(Vergrösserung 70 x)
Neben dem Polyoxymethylen wurde fur die Spinnversuchs
auch das homologe Polyäthylenoxyd verwendet.
3.3. Polyäthylenoxyd
Anregung für diese Versuche war unter anderem das Er¬
scheinen von neuen extremhochmolekularen Polyäthylenoxyd-
Produkten, die die Firma Union Carbide unter der Bezeich¬
nung "Folyox" vor kurzem auf den Markt brachte.
Die ersten Versuche, das hochmolekulare Polyathylenoxyd mit
einem Molekulargewicht von 2500 zu Faden zu verspinnen,
hat E. Saute r48' im Jahre 1933 durchgeführt. Dabei
handelte es sich um Laboratoriumsversuche, das Polyathylen¬
oxyd nach dem Schmelzspinnverfahren zu verspinnen. Die er¬
haltene Faser hat wegen ungenügendem Molekulargewicht und
der damit verbundenen schlechten Eigenschaften keine tech¬
nische Bedeutung erlangt.

-32-
49)
Polymerisation des Aethylenoxyds: Bereits 1859 ' und
1878'wurde über die Polymerisationsfahigkeit von
Aethylenoxyd berichtet. Fingehender befasste sich zu¬
nächst H. Staudinger' damit.
Das monomère Aethylenoxyd ist eine bei 10,7 C siedende
Flüssigkeit, die in Stahlflaschen komprimiert geliefert
wird. Da es leicht spontan unter grosserer Wärmeent¬
wicklung polynierisiert, muss man es durch intensive Trock¬
nung stabilisieren. Da zur Polymerisation eine Spur Was>ser
notwendig ist, findet man an den Kettenenden Hydroxylgrup¬
pen, weswegen das Polyathylenoxyd mit Recht auch Poly-
athylenglykol genannt wird. Es ist auch möglich, direkt
aus Aethylenglykol durch Wasserabspaltung Polymere her¬
zustellen, die allerdings niedermolekular sind.
Aethylenoxyd wird technisch polynierisiert in Anwesenheit
von verschiedenen Katalysatoren wie Kaliumhydroxyd, Zinn
(Iv)-Chlorid und Trimethylamin. Je nach der Art des
Katalysators und der Polynierisationsbedingungen erhalt
uan entweder Produkte mit niederer Kettengliederzahl
(viskose Flüssigkeiten) oder lochcolekulare, wachsartige
Harze, die wasserlöslich sind. Bei letzterem hat sich CaO
als Katalysator fur extremhochmolekulare Verbindungen er¬
wiesen ( "Polyoxn-Markeu der Firma linion Carbide).
Eigenschaften: Die fur die Faserherstellung in Frage kommen¬
den hochmolekularen Produkte sind weisse Pulver mit thermo¬
plastischen Eigenschaften. Die Schmelzpunkte liegen bei
67° C. Sie sind hygroskopisch; der normale Feuchtigkeitsge¬
halt betragt 4 %.
Polyox-Harze sind bei Zimmertemperatur loslich in Wasser,
Eisessig, Acetonitril, lO^igem Ammoniak, Chloroform, Me¬
thylen- und Aethylenchlorid, Formaldehyd (40$ig), Isopropanol
(91%ig) und Trichlorathylen.
Die Harze sind in folgenden heissen Losungsmitteln loslich
(in kalten aber unlöslich):

-33-
Tetraohlorkohlenstoff, Diniethylformamid, Dioxan,
Aethanol, Aethylenoarbonat, Methanol, Methylathylketon,
Propionaldehyd.
Auch in heissem Benzol, Toluol und Xylol sind die Harze lös¬
lich; kalt hingegen quellen sie nur in diesen Lösungsmitteln.
In wasserfreiem Aceton, Aethylàther und Glycerin sind die
Polyox-Harze weder in kaltem noch in heissem Zustand loslich.
Es ist aber charakteristisch fur die Polyox-Harze, dass sie
unter Zusatz von kleinen Mengen gewisser Losungsmittel (sog.
Coupler) auch in Solventien loslich sind, die allein diese
Harze nicht zu losen vermögen. Unter Zusatz von 2-5 % Methanol
kann man z.B. die Polyox-Harze in Benzol bei Zimmertemperatur
gut losen. Methanol ist ausserdem Coupler fur Toluol, Tetrachlor¬
kohlenstoff und Aethylencarbonat.
Es wurde auch festgestellt, dass man zu den in kaltem Aethy-
lenchlorid gelosten Polyox-Harzen mindestens 80 % sonst nicht-
losender Solventien (Aceton, Tetrachlorkohlenstoff, Dimethyl¬
formamid, Dioxan) zugeben kann, ohne dass eine Ausfallung ein¬
tritt. Die so erhaltenen Losungen sind dick und zah.
Werden Polyox-Harze in Wasser gegeben, so quellen sie zuerst,
um dann durch Desintegration von Makromolekulketten und ihrer
Hydratisierung langsam in Losung überzugehen. Die Solvatations-
geschwmdigkeit kann durch Erhitzen und Ruhren beschleunigt
werden. Es ist ratsam, zuerst bis zur Siedetemperatur zu er¬
hitzen, ein paar Minuten sieden zu lassen und dann unter
Ruhren abzukühlen. Eine grossere Ruhrgeschwindigkeit soll man
wegen möglicher Kettenverkurzung von Makromolekülen vermeiden.
500 Touren/min, genügen.
Die Losungen von Polyox-Harzen sind mehr oder weniger trüb,
was durch kolloidal dispergierte Polymerisationskatalysator¬
mengen, die nicht mehr entfernbar sind, verursacht wird.
Die wassrigen Losungen sind gleichzeitig leicht alkalisch,
was den Resten des alkalischen Katalysators (CaO) zuzuschrei¬
ben ist. Die Klarung der Lösung ist durch Ansäuern unter
pH 5,5 leicht möglich.

-34-
Wegen der sehr grossen Viskosität der Läsungen von höher
molekularen Harzen 1st die Konzentration der Lösungen sehr
begrenzt, was natürlich %uch von der Temperatur und der
Natur des Lösungsmittels abhängig ist. Konzentrierte wäss-
rige Lösungen der Polyox-Marke WSR 301 bilden schon ab 15 %
gummielastische Gele, die jedooh für Spinnzwecke ungeeignet
sind.
Die Toxizität der Polyäthylenoxyde Im menschlichen Organismus
wurde oft untersucht. Dabei wurde festgestellt, dass Polymere,
deren Polymerisationsgrad grosser ist als 200, praktlsoh keine
52 )toxischen Erscheinungen zeigen . Glykol allein wirkt toxisch,
weil es den Zellen Wasser entzieht und durch Oxydation in
toxische Oxalsäure übergeht.
Wie aus dem experimentellen Teil zu ersehen ist, lassen sich
die hochmolekularen Polyox-Harze gut verspinnen, wobei die
besten Resultate mit dem Trockenspinnverfahren erzielt wer¬
den. Man erhält starke, hochkristalline und hochorientierte
Fasern, welche gut wasserlöslich sind.
m
Abb. 3
Polyäthylenoxyd-Faser nach dem Trockenspinn-verfahren hergestellt
(Vergrösserung 70 x)

-35-
3.4. Die regenerierten Kollagenfasern
Es wurde auoh versucht, kollagenhaltiges Material, wie
Lederabfälle, Unterleder (Leimleder) und Rinderdärme,
zu lösen, bzw. zu quellen und durch entsprechende Appara¬
turen zu Fäden zu verspinnen.
Die Rohstoffe: Die tierische Haut besteht aus drei in Ent¬
wicklung, Aufbau und chemischer Zusammensetzung grundsätz¬
lich unterschiedlichen' Teilen: der Oberhaut (Epidermis),
der Lederhaut (Corium oder Cutis) und dem Unterhautgewebe
(Subcutis).
Die OBERHAUT mit ihren Bildungsprodukten - Haaren, Schuppen
usw. - kommt nicht als Rohstoff für die Fadenherstellung in
Frage.
Dagegen sind Lederhaut und Unterhautbindegewebe, die haupt¬
sächlich aus Kollagen bestehen, geeignet; insbesondere das
billige Unterhautbindegewebe, das grösstenteils ein Neben¬
produkt, bzw. einen Abfall der Lederfabrikatiou darstellt.
Die LEDERHAUT (Corcium oder Cutis) ist der stärkste Teil
der tierischen Rohhaut und macht durchschnittlich 80 %
ihrer Gesamtdicke aus. Im Gegensatz zu der aus Zellepithel
aufgebauten Oberhaut besteht die Lederhaut aus einem dichten
Flechtwerk von kollagenen Bindegewebefasern. Die Kollagenen
Fasern sind kreuz und quer nach allen Richtungen und Dimen¬
sionen so miteinander verflochten und ineinander verwachsen,
dass niemals ein Anfang oder Ende von Fasern festgestellt
werden kann. Die kollagenen Fasern stellen nämlich Faser¬
bündel und diese Faserbündel wiederum Bündel einer ständig
wechselnden Zahl feinster Kollagenfibrillen dar. Der Raum,
zwischen den Einzelfibrillen ist mit einer Kittsubstanz,
der sogenannten "Interfibrillarsubstanz", gefüllt, wobei die
Einzelfibrillen mit dieser Substanz umhüllt sind. Diese Ei-
weisssubstanz ist alkalilöslich und wird beim Weichen und
Aeschern der Haut in Gerbereien herausgelöst, wodurch eine
Auflockerung des Hautgewebes stattfindet. Die Lederhaut te^lt
sich in zwei Schichten, den oberen Narbenteil, Papillarschicht
genannt, und in die darunterliegende Retikularschicht.

-36-
Erstere enthalt weniger kollagene Bindegewebefasern. Diese
sind viel dünner, und das Fasergeflecht ist oft unterbrochen
und aufgelockert durch die eingelagerten Haarbälge, Talg-
und Schweissdrusen. Dagegen ist die Retikularsohicht charak¬
terisiert durch das dichtverflochtene Fasernetz, ist sehr
stark und gleichzeitig der Haupttrager der mechanischen Ei¬
genschaften der Haut, bzw. des Leders.
Die UNTERHAUT (Subcutis) stellt praktisch eine verbindende
Uebergangsschicht zwischen dem Korper des Tieres und der
eigentlichen Haut dar. Sie besteht aus lockeren, unregel-
massigen Bindegewebsfasern, die stark durchsetzt sind von
Muskelgewebe, Blutgefässen, Nervengewebe und vor allem Fett¬
gewebe. Fur die Lederfabrikation ist die Unterhaut ohne
Bedeutung und wird beim "Entfleischen" der Haut entfernt.
3.4.1. Chemie des Hautmaterials
Die genaue chemische Zusamnensetzung des Hautmaterials
variiert nach Tierart, Gesohlecht, Provenienz, Alter, Er¬
nährung usw. Ebenso zeigen die verschiedenen Teile einer
Haut gewisse Unterschiede in der chemischen Zusammensetzung.
Im Durchschnitt besteht jedoch die von Wasser und Fett be¬
freite tierische Haut fast zu 100 % aus Eiweisstoffen, von
denen das Kollagen allein etwa 98 % ausmacht '. Der Anteil
dus Eiweisstoffes Elastin betragt höchstens 1 %. Dazu kommen
Mineralsalze und geringe Mengen anderer Eiweisstoffe (Globu-
line und Albumine), wie auch Keratin (in der Oberhaut), die
alle bei der Verarbeitung entfernt werden.
Der Wassergehalt von frischer tierischer Haut schwankt in
weiten Grenzen von ca. 50 bis 70 % und ist auch von der Art
und dem Alter der Tiere abhangig.
1. Kollagen: Weitaus der wichtigste und grosste Bestandteil
der Hauptsubstanz ist Kollagen, das ausserdem
auch lie Grundsubstanz der Därme, Knochen und Knorpel dar¬
stellt; teilweise befindet es sich auch in Sehnen, Muskeln
und Fischschuppen.

-37-
Das Kollagen ist kein einheitliches, genau definierbares
Eiweiss; denn es verhalt sich je nach Herkunft verschieden
in Bezug auf Schrumpfung, Quellung, chemische und biologi¬
sche Einwirkungen. Eher stellt es einen Sammelbegriff für
eine Gruppe von Eiweisstoffen ähnlicher Art dar.
Zahlreiche Elementaranalysen des Kollagens weisen schwanken¬
den Stickstoffgehalt auf, der je nach Herkunft des Kollagens
zwischen 17 und 18 % variiert. Besseren Aufschluss über den
chemischen Aufbau des Kollagens als die Elementaranalyse
gibt die Aminosaurenzusammensetzung, ermittelt durch hydro-
lysierenden Abbau mit Mineralsauren . Danach kommen im
Kollagen auf 8 Mol Glykokoll 8 andere Monoaminosauren
(Alanin, Leucin, Phenylalanin, Tyrosm, Serin, Asparagin-
saure, Glutaminsäure), 4 Mol Frolin, 2 Mol Oxyprolin, 1 Mol
Arginin und 1 Mol Lysin. Daraus ergibt sich, dass in einer
Periode von 24 Aminosäuren im Kollagen jede 3. Glykokoll,
jede 6. Prolin, jede 12. Oxyprolin und jede 24. Lysin, bzw.
Arginin, ist. Charakteristisch fur Kollagen ist das Fehlen
der Aminosäuren Cystin und Tryptophan.
Reines, wasserfreies Kollagen 1st ein weisser und spröder
Körper, der in kaltem Wasser und allen organischen Losungs¬
mitteln völlig unlöslich ist. In kalten, verdünnten Säuren
und Alkalien quillt Kollagen, wobei eine beginnende Auf¬
spaltung grosserer Polypeptidketten auftritt. Gleichzeitig
tritt eine Lockerung der Nebenvalenzbindungen zwischen den
Folypeptidketten auf. Die durch diese Quellung hervorgerufene
Strukturveränderung ist nicht völlig reversibel. Bei längerer
Einwirkungsdauer oder höherer Temperatur geht die quellende
und peptisierende Wirkung in eine hydrolysierende über, bei
Alkalien schneller als bei Sauren.
Kollagen schrumpft beim Erwärmen mit Wasser bei 62 - 64° C
auf ca. ein Drittel der ursprunglichen Länge, wird durch¬
sichtig und gummielastisch. Die geschrumpfte Faser weist
kein rontgenographisches Faserdiagramm auf, wohl aber nach

-38-
erfolgter Dehnung. Beim längeren Erhitzen mit Wasser
"verleimt" das Kollagen und geht in wasserlösliche Gelatine
über, wobei die strukturierte Form verloren geht. Die Kri¬
stallstruktur wird dabei zerstört.
Das ungequollene Kollagen iat entgegen zahlreichen Angaben
der Literatur gegenüber den meisten eiweisspaltenden
Enzymen sehr widerstnadsfähig. Dagegen wird gequollenes
Kollagenmaterial (durch verdünnte Sauren, Alkalien oder
Salzlosungen) wie Leim und Gelatine von Trypsin, Pepsin,
Papain und Kathepsin leicht und rasch abgebaut.
2. Elastin: Der zweite Eiweisstoff der tierischen Haut,
Elastin kommt in kleinen Mengen (bis max. 1 %)
in der Papillarschicht der Lederhaut vor und Midet daneben
den Grossteil des elastischen Gewebes in den Sehnen (bis 85 %).
Elastin enthält etwas weniger Stickstoff als Kollagen, durch¬
schnittlich 16,75 io. Die Aminosâurenzusammensetzung ist ähn¬
lich wie beim Kollagen . Elastin zeigt auch das gleiche
Faserdiagramm wie Kollagen. Aus allen diesen Gründen nimmt
man heute an, dass das Elastin eine Umwandlungsform des
Kollagens darstellt. Der Hauptunterschied zwischen Elastin
und Kollagen liegt in der Tatsache, dass Elastin von kochen¬
dem Wasser nicht angegriffen wird, bzw. nicht "verleimt".
im Gegensatz zu Kollagen quillt Elastin in Sauren und Alkalien
ausserordentlich wenig und wird von diesen in der Kälte kaum
angegriffen. Konzentrierte Lauge und heisse Salpetersäure
wirken lösend. Auch gegen Fermente ist Elastin in vorge-
quollender Form weniger empfindlich.
3. Kératine: Kératine sind Bestandteile der verhornten Zellen
der Oberhaut und ihrer Gebilde wie Haare, Hufe,
Klauen usw. Charakteristisch fur Kératine ist der Gehalt an
Schwefel, bzw. der Gehalt an der Aminosäure Cystin, was zu¬
satzliche Disufid-Querbindungen ermöglicht. Dadurch sind

-39-
keratine Polypeptldketten steifer und widerstandsfähiger
gegen Fermente und Wasser. Der kératine Anteil der Tierhaut
wird bei der Verarbeitung entfernt und spielt bei der Her¬
stellung der Kollagenfasern keine Rolle.
An übrigen Proteinen finden sich in der tierischen Haut
Albumine und Globuline, die meist gemeinsam vorkommen.
Albumine sind wasserlöslich, werden aber durch verdünnte
Säuren nicht gefällt und sind schwer aussalzbar. Sie sind
hitzekoagulierbar und chemisch gekennzeichnet durch das
Fehlen der Aminosäure Glykokoll. Sie sind sehr empfindlich
gegen enzymatische Wirkung und gehen entsprechend leicht
in Fäulnis über.
Globuline sind ebenfalls hitzekoagulierbare Eiweisstoffe,
jedoch in Wasser und verdünnten Säuren unlöslich, dagegen
leicht löslich in verdünnten Neutralsalzlösungen und leich¬
ter aussalzbar. Im Gegensatz zu den Albuminen enthalten
Globuline immer Glykokoll.
Diese beiden Eiweisstoffgruppen sind relativ niedrigmolekular
und werden bei der Verarbeitung entfernt.
3.4.2. Strukturmodell und physikalisch-chemische Eigen¬schaften des Kollagens
_
Strukturmodell
Das Kollagen steht gegenwärtig im Mittelpunkt der Eiweiss-
forschung. Die Ergebnisse der morphologischen Strukturfor¬
schung (erhalten mit den Mitteln der Licht-, Elektronen-
und Röntgen-Optik) einerseits und die Resultate der physi¬
kalischen und chemischen Untersuchungsmethoden (Gewinnung
von löslichem Kollagen und Regenerierung zu fibrillären
Strukturen, Bausteinanalyse usw.) andererseits, haben sich
so weit ergänzt, dass ein geschlossenes Bild des Kollagen¬
moleküls entwickelt werden konnte, welches den Erfahrungs¬
tatsachen weitgehend gerecht wird. Besonders das Weitwinkel-
röntgenogramm ist eines der charakteristischsten Merkmale
des Kollagens, und dieses wurde von Rieh und Crick '

-40-
dahln interpretiert, dass über weite Bereiche der Faser¬
struktur hinweg eine dreiadrige Helixkonflguration von Peptid-
ketten vorliegt, in welchen die aus Abbauversuohen wohlbekann¬
te Aminosauresequenz Glycin-Prolin-Hydroxyprolin eingeschlos¬
sen ist, und dass die gesamte Struktur durch Wasserstoff-
brucken stabilisiert wird. Das Modell der dreiadrigen Helix
wird auch gestutzt durch Beobachtungen von D o t y und
Nishihar a '. Das ganze Gebilde ist von hoher Stabi¬
lität, dessen Loslichkeit und chemisches Verhalten gegen
Elektrolyte, komplexbildende Metallkationen usw. auf der
Reaktionsfähigkeit der nach aussen gerichteten Seitenketten
der dieses System aufbauenden Aminosäuren beruht.
Quellung und Loslichkeit
Charakteristisch fur Kollagen ist eine nur langsam Wasser-
aufnahme unter starker Volumenvergrosserung, wobei unter
Quellung erhebliche Wassermengen aufgenommen werden können.
Der Quellungsvorgang, hervorgerufen durch Wasser oder andere
Flüssigkeiten, ist abhangig von der Einwirkungsdauer, der
Temperatur, dem pH-Wert, der Kollagenherkunft und den che¬
mischen und physikalisch-chemischen Eigenschaften des ver¬
wendeten Quellungs-, bzw. Lösungsmittels. In vereinzelten
Fallen tritt auch eine unbegrenzte Quellung mit einem
kontinuierlichen Uebergang aus der festen in die geloste
Form ein. Wegen der starken Verkettung der Polypeptide in
der Längsrichtung der Faserachse und der loseren Vernetzung
in der Querrichtung* ist die Quellung von Kollagenfasern mit
einer Faserverkurzung verbunden.
Die Quellungserscheinung wird im sauren und alkalischen Ge¬
biet gefordert, weil die gleichen elektrischen Ladungen
eine Abstossung der parallelgelagerten Polypeptidketten
hervorrufen und die Quervernetzung der Helixstruktur auf¬
lockern. Bei entgegengesetzten Ladungen werden hingegen die
Zwischenräume verringert und die nebenvalenzartigen Querver¬
bindungen gefestigt. Es ist deshalb verstandlich, dass das
Quellungsminimum bei optimaler Zwitterionen-Ausbildung er¬
reicht wird, also im Gebiet des isoelektrischen Punktes.

-41-
Der isoelektrische Punkt des Kollagens schwankt in weiten
Grenzen,er liegt je nach Herkunft zwischen pH 4,8 und
10,0, bei der Gelatine zwischen pH 4,6 und 8,0. Im Durch¬
schnitt kann die Lage des isoelektrischen Punktes für Kolla¬
gen bei pH 5,2, fUr Gelatine bei pH 5,0 angenommen werden.
Der isoelektrische Punkt der Aminosäuren liegt nämlich zu¬
meist im schwach sauren Gebiet, da der Dissoziationsgrad der
Carboxylgruppen meist grosser ist als der Dissoziationsgrad
der Aminogruppen.
Die Wirkung von Neutralsalzen: Je nach der Konzentration
der Neutralsalzlosungen ist ihre Wirkung verschieden. In
geringen_Konzentrationen rufen die Neutralsalze eine Stei¬
gerung des Quellungseffektes hervor; dem gegenüber entziehen
konzentrierte Losungen das zum Auflosen benotigte Wasser
dem Kollagen, wirken dehydratisierend und fuhren somit eine
Entcjuellung herbei.
Diese Erscheinungen werden folgendermassen erklart:
In geringer Konzentration diffundieren die molekulardis¬
persen Salzmolekule leicht in das Froteingel, und die Kolla-
gemnolekule reagieren mit dem dissozierten Salz unter Bildung
ionisierter Proteinsalze. Diese besitzen eine höhere Loslich-
keit und ein entsprechend stärkeres Quellungsvermogen.
In konzentrierten Neutralsalzlosungen entzieht die sehr
starke Losungstendenz des Salzes dem Proteinkorper Wasser,
setzt den Dissoziationsgrad der Proteinlosung herab und
führt zu einer Koagulation. Die konzentrierten Neutralsalz¬
losungen sind also fähig, die kolloidalen Kollagenlösungen
wieder auszufällen.
Bei der Behandlung von kollagenem Material mit stark verdünn¬
ten Neutralsalzlosungen geht ein gewisser Anteil kollagener
Substanz in Losung. Dieser wurde nach Vorschlag von
J . Gross, J. H. Highberger und F.O.59)
Schmidt'' als "Tropokollagen" bezeichnet.
Durch Erwarmen von sauren Tropokollagenlbsungen Über eine
bestimmte Temperatur (z.B. 35° C bei Kalbshautkollagen

-42-
mit einem pH-Wert von 3,7) tritt eine Denaturierung des
Kollagens ein, die durch Abnahme des negativen Drehwertes
und durch Abnahme der Viskosität der Lösung begleitet wird .
Beide Veränderungen erklären F 1 o r y und W e a w e r
durch die Annahme, dass sich die drei Polypeptidketten aus
ihrer ursprünglichen Helixstruktur "entfalten" (Entspirali-
sierung).
Der Wirkungsgrad der Neutralsalze ist durch die Hofmeister'
sehen Reihen 'gekennzeichnet,
in der Kationen reihe mit der Folge:
Li, Na, K, Rb, Cs, Mg
bei den A n i o n e n mit der Reihenfolge:
Sulfat, Tartrat, Citrat, Acetat,
Fluorid, Chlorid, Bromid, Nitrat,
Jodid, Rhodanid.
Nach der linken Seite der Reihen steigt die dehydratisie-
rende, entquellende und koagulierende Kraft der Salze an;
nach der rechten Seite nimmt der peptisierende, quellungs-
fördernde Effekt zu.
Die vorstehend beschriebene Wirkungsweise hat nur Gültig¬
keit beim isoelektrischen Funkt der Eiweisskörper.
Im sauren oder alkalischen Medium ist die Wirkung ganz ver¬
schieden, in diesem Falle sind einsinnig geladene Proteine
vorhanden, und die Neutralsalzionen, die eine höhere Affini¬
tät zu den Proteinsalzionen haben als das ursprünglich vor¬
handene Gegenion, bewirken einen Ionenaustausch. Dadurch
wird der Dissoziationsgrad des Proteinsalzes herabgedruckt,
und es tritt Entquellung ein. Auf diesem Effekt beruht das
Pickelverfahren, wobei es durch entsprechende Säure-Salz-
Gemische mit genügender Neutralsalzkonzentration möglich
ist, dem Kollagen grössere Säuremengen zuzuführen und seine
chemische Reaktivität zu steigern, ohne dass Quellung ein¬
tritt.

-43-
Die hydrotropisehe Wirkung: Die Förderung
der Quell-, bzw. Lösungsfähigkeit, des Kollagens in Wasser
kann dagegen durch Anwendung sogenannter Hydrotropica, d.h.
nach C. Neuberg solcher Substanzen, welche die
beträchtliche Oberflächen- oder Grenzflächenspannung zwischen
Wasser und Proteinen herabsetzen und die Proteine quellfähiger
bzw. wasserlöslicher, machen, gesteigert werden.
Als hydrotrope Stoffe für Proteine sind organische Aminover-
bindungen wie Harnstoff, Thioharnstoff, Formamid, Acetamid
usw. zu nennen, wobei die NHg-Gruppe der hydrotrop wirksame
Bestandteil ist. Auch aromatische OH-Gruppen, z.B. Phenol,
besitzen stark hydrotropischen Charakter für Proteine.
Bei den Elektrolyten sind vor allem die Sulfonsäuren und
Carbonsäuren der aromatischen Reihe starke Hydrotropica,
wobei die Wirkung ihrer Salze noch ausgeprägter 1st. Die
gleiche hydrotrope Wirkung zeigen auch die vorher erwähn¬
ten anorganischen Neutralsalze in geringer Konzentration,
deren Wirksamkeit durch die Hofmeister1sehen Reihen gegeben
ist.
Eine Erklärung für den Effekt der Hydrotropie wird neben der
rein physikalischen Deutung einer Oberflächenaktivität in
chemischer Hinsicht dadurch gegeben, dass die Mischungslücke
zwischen zwei nicht oder beschränkt mischbaren Substanzen
durch einen dritten, in beiden Substanzen löslichen Stoff
(hydrotropes Reagenz) unter Bildung von aggregierten Additions¬
verbindungen ausgefüllt wird.
3.4.3. Die geschichtliche Entwicklung der Kollagenfaserher-Stellung und Patentliteratur
Wie bereits erwähnt30^, wurde A. Millar schon im
Jahre 1894 das erste Patent für das Trockenspinnen von Fä¬
den aus Gelatine erteilt. Die Millar1sehe "Vanduaraseide"
hatte nur geringe Festigkeit, insbesondere Nassfestigkeit,
und verschwand bald wieder vom Markt. Die weitere Entwick¬
lung ging aber hauptsächlich in Richtung der nichtkollagenen
Eiweissfasern (Casein, Zein usw.), da deren Rohstoffbasis
viel ausreichender war.

-44-
Die technische Produktion von kollagenen Faden war begrenzt
auf die Herstellung von chirurgischen Nahfaden, sogenannten
"Catgut", bzw. Darmsaiten fur Musikinstrumente und Tennis¬
schlager, wobei fur die ersteren Schaf- und fur die letzte¬
ren Rinderdunndarme verwendet wurden. Die zur Verarbeitung
geeigneten Tierdarme sind von beschrankter Lange, so dass
z.B. Catgutfaden praktisch nur bis zu 2,5 m Lange knoten¬
frei hergestellt werden können. Mangelnde Geschmeidigkeit,
nicht immer gleichmassige Zugfestigkeit und schwere und
konplizierte Sterilisation sind weitere Nachteile der Darm¬
saiten.
Die Frkenntnis, das Kollagenhautabfalle, insbesondere iie
billigen Unterhautgewebe, nachdeiu sie einer milden Hydrolyse
unterworfen, mechanisch zerkleinert und homogenisiert wur¬
den, eine verfortbare Masse bilden, führte zur erfolgreichen64 )
Herstellung von künstlichen Wurstdarmen .
Bale» darauf gelang es der Firita C. Freudenberg in Weinheim,
aus. einer ähnlichen Masse gröbere Fasern herzustellen, die
unter dem Namen "Marena" als kunstliches Rosshaar und Pol-
gtermateridl auf dem Markt erschienen .
Die weitere Forschung hatte die Herstellung vun endlosen
Kollagenfaden tun Ziele.
Alle diese Verfahren kann man in zwei Gruppen einteilen:
1. Herstellung von kontinuierlichen Faden durch das
Schneiden von Därmen in schmale Bander, welche dann
an Jen Enden verklebt oder zusammengeknüpft '
werden und weiter durch Zwirnen, Seilen oder Ver¬
flechten zu dickeren Faden vereinigt werden können.
Die hergestellten kontinuierlichen Kollagenfaden
sind gleiehmassig, aber die Herstellung ist kompli¬
ziert und verlangt viel Handarbeit.
2. Herstellung von endlosen Faden aus regeneriertem
Kollagenmaterial: Der Kollagenstoff, der nicht unbe¬
dingt nur von Därmen stammen muss (Hautabfalle,
Spaltleder, Sehnen usw.), wird mittels Alkalien,

-45-
Säuren oder warmem Wasser aufgeschlossen, neutrali¬
siert und durch die entsprechenden Schlitze oder Dü¬
sen zu Bändelten, bzw. Schläuchen, ausgepresst. Diese
können später verzwirnt oder verflochten werden.
In der Literatur wird ein ähnlicher Proteinfasertypus aus
Häuten unter dem Namen "Cuojesco-Faser", jedoch ohne nähere
Angaben, angeführt. Diese Faser soll in Mischung mit ande-
ren Fasern zu Textilerzeugnissen verarbeitet werden .
Erst vor Beginn des 2. Weltkrieges hat man versucht, die
in Kriegszeiten so nötigen und raren Catgutfäden durch re¬
generierte Kollagenfäden aus Sehnen, Abfalleder usw. zu
ersetzen. Die Forschungen sind besonders im Laufe des Krie¬
ges und später fortgesetzt worden und sind noch beute in
vollem Gange.
Alle diese Verfahren zur Herstellung von endlosen regene¬
rierten Kollagenfäden haben sich in der Praxis nicht durch¬
gesetzt wegen des komplizierten Arbeitsvorganges, der
teuren Anlagen und der ungenügenden Festigkeit, bzw. Gleich-
mässigkelt, der Fäden.
Die neuesten Forschungsergebnisse aus den U.S.A. auf die¬
sem Gebiet werden im experimentellen Teil erwähnt werden.
Im Rahmen der vorliegenden Arbeit wurde eine grössere An¬
zahl von Versuchen durchgeführt, um das Kollagenmaterial
in denaturierter Form in organischen Lösungsmitteln zu lö¬
sen und zu Fäden zu verspinnen, was zu positiven Ergebnis¬
sen führte. Die einzelnen Vorgänge werden im nachstehenden
experimentellen Teil beschrieben.

-46-
Abb. 4
Kollagene Multifilfaser,fau Acetonbad ausgefallt und gestreckt
(Vergrosserung lSx)

-47-
XPERIMENTELLER TEIL
1. SPINNVERSUCHE MIT POLYVINYLPYRROLIDON
1.1. Auagangsmaterial
Alle folgenden Versuche wurden mit Polyvlnylpyrrolidon
"LUVISCOL K-90" der Firma B.A.S.F., Ludwigshafen, durchge¬
führt. Die Markenbezeichnung K-90 bezieht sich auf die von
H. Fikentscher eingeführte empirische Molekular¬
gewichtsfunktion K, welche in diesen Falle (K-90) einem
mittleren Molekulargewicht des Polyvinylpyrrolidons von
360.000, bzw. einem Polymerisationsgrad von ca. 3.250 ent¬
spricht.
1.2. Spinnversuche nach dem Nassplnnverfahren
1.2.1. Herstellung der Sginnlösung
Es wurde mit 15, 19 und 21,75#igen wässrigen Lösungen des
Polyvinylpyrrolidons gearbeitet.
Um homogene und blasenfreie Lösungen herzustellen, wurde
dem eingewogenen Polyvinylpyrrolidon-Pulver durch Rühren
siedendes Wasser in Ueberschuss zugegeben, die Masse im
Wasserbad weitergerührt und über Nacht stehengelassen.
Das überschüssige Wasser wurde nachher abgegossen und die
Spinnlösung in die Apparaturreservoire eingefüllt. Die
Spinnlösungen waren durchsichtig, viskos und etwas gelb¬
lich gefärbt.
der_Sp_innlösungen
Die Viskosität der hergestellten Spinnlösungen wurde mit
dem Eppreoht-Strukturviskoslmeter der Firma Contraves,
Zürich, bei 20° C gemessen.
Aus den Rheogrammen (Diagramm l) ist ersichtlich, dass man
nicht eine Gerade, sondern eine Kurve erhält und dass die
Viskosität vom angelegten Geschwindigkeitsgefälle abhängig ist.

-48
MS-C(27,37)
15
75
12
10
To~ lo" srr
WAGRAMM 1
Elnfluss der Konzentration auf die Viskosität
verschiedener wässriger Polyvinylpyrrolidon-Lösungen
D = Geschwindigkeitsgefälle (sec"1)V - Schubspannung (Dyn.oB~ )T = 20° C

-49-
Als günstigste Konzentration hat sioh die 19#ige Lösung
erwiesen, deren Viskosität den normalen Viskosespinnlö-
sungsviskositaten entspricht.
Als Fällbad wurden wässrige Lösungen verschiedener Salze,
Säuren, Basen und organischer Verbindungen bei verschiede¬
nen Temperaturen ausprobiert. In den nachstehenden Tabellen
(III - V) sind die Resultate sämtlicher Versuche zusammen-
gefasst.
Aus den Resultaten ist folgendes zu schliessen:
l) Die mehrwertigen Phenole, Resorcin, Hydroohinon, Pyro-
gallol und Phloroglucin, wirken stark koagulierend, wo¬
bei sich eine 35%ige Resorcinlösung als am günstigsten
erwies. Als optimale Spinnbadtemperatur hat sich die
Temperatur von 40 - 50° C gezeigt. Die weissen, opaken,
aber dicken und elastischen Fäden sind leicht auszieh¬
bar. Im nassen Zustand sind sie genügend fest, um unter
Spannung auf die Trommel aufgespult zu werden. Nachtei¬
lig ist jedoch die Klebrigkeit der Fäden, die ein Zusam¬
menkleben der Fäden zu einem Strang bewirkt, sowie der
Verlust der Elastizität beim Trocknen. Je trockener die
Fäden sind, desto brüchiger und steifer werden sie, und
die auf die Trommel unter Spannung aufgewickelten Fäden
spalten sich in kleine Bruchstücke. Dies deutet daraufhin,
dass die dioken, voluminösen und schwammartigen Fäden,
die sioh im Fällbad formieren, viele mikroskopisch kleine
Fällbadflüssigkeitstropfen okludieren. Diese in der gan¬
zen Fadenmasse fein dispergierten Tröpfchen verursaohen
einerseits den dioken Fadendurchmesser und andererseits
zahlreiche Bruchstellen beim Trocknen.
Um diese Nachteile zu ellminieren, wurde versuoht, den
Fällbädern Elektrolyte, bzw. Salze, Säuren oder Alkalien,

-50-
zuzugeben. Dabei hat sich erwiesen, dass sich nur durch
Zusatz von starken Alkallen bessere Resultate ergeben,
wobei aber eine unerwünschte Färbung der Fäden eintritt.
Gleichzeitig sind die Fäden dann auch stark alkalisoh.
2) Die gerbenden Substanzen, Tannin und Gallussäure, wirken
anders. Die Fäden formieren sich im Fällbad bei erhöhter
Temperatur besonders schnell, erstarren dabei jedoch nicht
in genügendem Masse und sind schleimartig und dehnbar.
Sie zerlliessen leicht beim Ziehen und sind unter Spannung
nicht auf die Trommel aufspulbar.
3) Konzentrierte Lösungen der Salze: Aminonsulfat, Natrium-
nitrit, Natriumsulfit wirken koagulierend, aber nicht in
dem Masse, dass sich Fäden formieren. Man erhält nur eine
voluminöse Ausfällung, die in nassem Zustand gummiartig
ist. Natriumacetat ergibt keine Ausfällung.
Zugabe von Säuren zu den Salzlösungen hat negative Wir¬
kung. In einem Müllerbad konnte man keine Fäden ausspin¬
nen. Variation der Spinnbadtemperatur blieb ohne wesent¬
lichen Einfluss.
4) Die besten Resultate ergeben sich mit stark alkalischen
wässrigen Lösungen. Die Fasern sind glasklar und farblos,
und die Einzelfäden kleben nicht zu einem Strang zusam¬
men. In nassem Zustand sind sie nicht so leicht brüchig,
wohl aber nach der Trocknung.
Die im Kaliunijiydroxydbad ausgefällten Fäden sind stärker
und elastischer als diejenigen aus dem Fällbad mit äqui¬
valenter Menge Natriumhydroxyd.
Aus diesem Grunde wurde noch eine Serie von Kaliumhydroxyd-
badern verschiedener Konzentration hergestellt, um die
optimale Konzentration zu bestimmen. (Tabelle V)
Aus den Versuchsergebnissen geht hervor, dass die opti¬
male Konzentration des Kaliumhydroxyds zwischen 26 und
28 4 liegt.

-51-
TABELLE III
Spinnbäder mit Lösungen der mehrwertigen Fhenple und der
gerbenden Substanzen
Versuchs- F ä 1 1 b a d
Zusammensetzung
1. Resorcln
2. "
3. "
4. "
5. "
6. "
7. "
8. "
9. "
10. Hydrochlnon
11. "
12. "
13. "
14. Pyrogallol
15. "
+ 10 %
16. "
+ 15 %
17. Phloroglucin
10#ige
20%ige
30%ige
40%ige
50#ige
60#ige
35#ige
35#ige
35#ige
155tige
+ 10 *
+ 10 %
+ 20 %(25%ig)
15#ige
It
HC1
it
NaCl
Lsg.
Lsg.
Lsg.
Lsg.
Lsg.
Lsg.
Lsg.
Lsg.
Lsg.
Lsg.
HC1
Na2S04NH.OH
4
Lsg.
n
tt
Temp.(°
42
42
42
42
42
42
20
60
80
50
50
50
50
50
50
50
60
—
F r or a b n 1 9^^^~ Ei X K w w JU X 9
Koagulation der Spinn¬masse, aber ungenügendeFädenformierung
schnellere Koagulation,Fadenstrang aber nicht
stark genug z.Aufspulen
Weisser, dicker u. ela¬
stischer Fadenstrang,
(Einzelfäden kleben zus.)
n n
n n
n n
ungenügend schnelle
Koagulation
schnelle Koagulation
n n
gleich wie Versuohe 3-6
n n n n
n n n n
n n n n
aber gelbe Färbung
gleiche Fadenstränge wie
in den Versuohen 3-6,nuretwas schnellere Koagulat.
gleich wie Versuche 3-6
n n n n
» it n n
18. Gesättigte Tanninlbsung 20
19. ff ff 50
20. " « 80-100
21. Gesättigte Gallussäurelsg. 50
32. " "
+ 10 % Essigsaure 50
Schleimartiger Fadenstrang
Sohwaohe Fadenformierung,Fäden kleben schon im
Spinnbad zusammen
Fäden formleren sich deutl.
sind aber schwach u.klebrig
wie mit Tanninlsg.(Vers,19)

-52-
TABELLE IV
Spinnbäder mit Salzlösungen
Versuchs-
Nr.
F ä 1 1 b a d
Zusammensetzung Temp.(°C)Ergebnis
23. MUllerbad (H2S04 12% 40
245t
ZnCl2
24. Ammonsulfat 60%ige Lag.
25.
26.
27.
28.
29.
30.
31.
32.
33.
35%ige Lsg.
30%ige Lsg.
n n
+ 10% H2S04n n
+ 10% NaOH
27%ige Lsg.
30
30
50
50
50
30
" " 40-90
17,5%ige Lsg. 30
15%ige Lsg.H„S0. 40
+ 12% H„SO.a 4
34. Natriumsulfat 30#ige Lsg.
50-80
30
35. gesätt.Lsg.in n H2S04
36. Natriumsulfit 30%lge Lsg.
30
30
Keine Fädenbildung,lang¬same Ausfällung der
Spinnmasse
Starke Koagulation der
Spinnmasse, aber keine
Fädenbildung. Es fällt
eine gummiartige Masse aus.
wie Versuch 24, nur noch
etwas langsamere Koagulat.
wie Versuch 25
wie Versuch 25
langsame Koagulation der
Spinnmasse, keine Fäden¬
bildung
etwas schnellere Koagula¬tion, keine Fädenbildung
schwache Koagulationder Spinnmasse
sehr schwache Koagula¬tion 4er Spinnmasse
keine Koagulation der
Spinnmasse,die zerfliesst
und sich im Spinnbad auf¬
löst.
Langsame Ausfällung der
Spinnmasse, keine Fäden¬
bildung
Ausfällung der Spinnmassewie mit Ammonsulfat
(Versuch 25)
37. 40-90

-53-
TABELLE IV (Fortsetzung)
Spinnbader mit Salzlosungen
Versuchs- F a 1 1 b a d
Nr. Ergebnis
Zusammensetzung Temp.( C)
38. Natriumbisulfat
30%ige Lsg. 30 Schwache Ausfallungder Spinnmasse, keine
Fadenbildung
39. " " 40-80 " "
40. n CaCl, Losung in 0,02n HC1 30 keine Ausfallung
41. Natriun.acetat gesatt.Lsg. 20 vollständige Lbslichkeit
d. Spinnmasse im Fällbad
42. " " " 50-80 " "

SpinnlösungderKoagulationkeine
51VersuchinalsSpinnlösungderlation
Koagu¬langsamereNoch
zus.klebenEinzelfädenSpinnlsderKoagulation
schnelleUngenügend
bar
dehn¬gutundelastischFäden,formiertegut
44Versuchwie
dehnbar.se
u.teilwei¬steifweniger
aber44,Versuchinwie
Fädenformierterecht
brüchigu.steifjedochEinzelfädenstruktur,mit
FädenglasklareDünne,
bar.
abzieh¬nichtsindFädenschneller,jedocherfolgt
Koagulation25),(VersuchAmmonsulfatmitwiese
Spinnmas¬derAusfällung
20konz.Ammoniumhydroxyd
20Lsg.gesättigteKalkwasser
20Lsg.gesättigteBarytwasser
20Lsg.22%ige"
20Lsg.245&ige"
20Lsg.26f»ige"
20Lsg.28#ige"
20Lsg.30%ige"
20Lsg.32%igeKaliumhydroxyd
20konz.Kalilauge
Lsg.
2032#lgeNatriumhydroxyd
20
50
konz.Natronlauge
Lsg.20#igeNatriumcarbonat
55.
54.
53.
52.
51.
50.
49.
48.
47.
46.
45.
44.
43.
ErgebnisTemp.(°C)Zusammensetzung
dab11äF
Nr.
Versuchs-
Lösungenalkalischenanorganischen,mitSpinnbäder
VTABELLE
-54-

-55-
Die Fäden aus 28%igem Spinnbad behalten Einzelfaserstruk¬
tur, und bei denjenigen aus 26%igem Spinnbad ist schon
ein teilweises Zusammenkleben der Einzelfäden in einem
Strang bemerkbar. Die Spinnbadtemperatur hatte keinen we¬
sentlichen Einfluss auf den Padenformierungsprozess, und
man konnte die Versuche bei Zimmertemperatur (20° C) durch¬
führen.
Konzentriertes Ammoniumhydroxyd und gesättigte Lösungen von
Baryt- und Kalkwasser bleiben ohne Einfluss auf die Koagula¬
tion.
1.2.4. Neutralisation der aus alkalischem Fällbad ausgespon¬
nenen P0iy.viny_lp_y_rrolidon-Fäden_
Um die erzeugten Fäden zu neutralisieren, wurde eine Reihe
von starken und schwachen Säuren verschiedener Konzentrationen
ausprobiert (H^SO^, HC1, Essig- und Oxalsäure). Dabei wurde
festgestellt, dass sich die Fäden in allen sauren wässrigen
Lösungen sofort auflösen.
Aus diesem Grunde wurde die Neutralisation in trockener
HCl-Gas-Atmosphäre versucht. Die Ergebnisse sind positiv
ausgefallen; denn die Fäden wurden durch einfache Durch¬
führung durch HCl-Gas-Atmosphäre vollständig neutralisiert.
1.2^5. Nachhärtung
A) Mit Formaldehyd
Um die ungenügende Faserfestigkeit zu erhöhen, wurde ver¬
sucht, die Fäden mit Formaldehyd nachzuhärten. Die ge¬
wöhnliche, 40#ige wässrige Lösung ergab keine positiven
Resultate, da eine Auflösung der Fäden eintritt. Nach¬
her wurde ein Nachhärten von Fäden in trockener, gas¬
förmiger Formaldehyd-Atmosphäre versucht. Gasförmiger
Formaldehyd wurde durch Erhitzen von dispergiertem Para-
formaldehyd In Paraffinöl erzeugt. Als Resultat bekommt
an dehnbarere Faden, die aber nicht tester geworden sind.
Der Versuch wurde in der gleichen Atmosphäre unter Zusatz

-56-
von trockenem HC1 wiederholt, aber ebenfalls ohne Erfolg.
B) Mit Chromalaunbad
In den Chromalaun-Bädern verschiedener Konzentration
lösen sich die Fäden auf.
C) Thermobehandlungsversuche
Um die leichte Wasserlöslichkeit der Polyvinylpyrrolidon-
Fäden herabzudrücken, wurde versucht, durch thermische
Behandlung eine Vernetzung, wie dies beim Polyvinyl-
alkohol möglich 1st, zu bewirken.
a) Erhitzen in Luft ergab folgende Resultate:
TABELLE VI
Ergebnis
keine merkliche Veränderung
Temperatur
150°
170°
185°
200°
220°
230°
C
C
C
C
C
C
Vergilbung der Fäden,Wasserlöslichkeit unverändert
Rotbräunung der Fäden,Wasserlöslichkeit unverändert
Dunkelviolette Färbung der Fäden
Wasserlöslichkeit unverändert
b) Durch Erhitzen der Fäden auf 100° C in stark alkalischem
Bad wurden diese dauernd unlöslich gemacht.
1.2.6. Sj)innapj>aratur
Für das Nasspinnverfahren wurde eine kleine Viskose¬
nasspinnmaschine verwendet.
Die Spinnlösung wird in den 2-Liter-Behälter aus V4A-Stahl (l)
eingegossen und dieser gut verschlossen. Der Behälter ist
mit einem Manometer (2) und einen EinfUhrungsrohr (3) für
Pressluft (2 Atü) versehen.
Aus dem Behälter wird unter Luftpressdruck die Spinnlösung
zur Zahnradpumpe (4) gedrückt, welche die Lösung wieder in

-57-
JMMPF
Abb. 5
Die Nasspinnmaschine
die Filterkerze (5) presst. Die Lösung wird dort von n.echa-
nischen Verunreinigungen befreit und durch ein biegsau.es
Bleirohr (6) zur Spinndüse (7) weitergeleitet. Die Spinndüse
hat 27 Bohrungen und ist aus Platin gebaut. Die Düse wird
tief in das Fällbad (8) eingetaucht, und die formierten Fa¬
sen werden noch im Bad gestreckt (9) und auf eine perforier¬
te Troiimel (10) über einen hin- und herbeweglichen Leiter
(li) aufgespult.
Die günstige Fällbadtemperatur wird mit indirektem Wasser-
danjpf reguliert (12) und mit einen, Thermometer kontrolliert
(14).
Die nötige Kraft für den Antrieb wird durch einen 0,25 PS
starken Elektromotor (13) zugeführt.
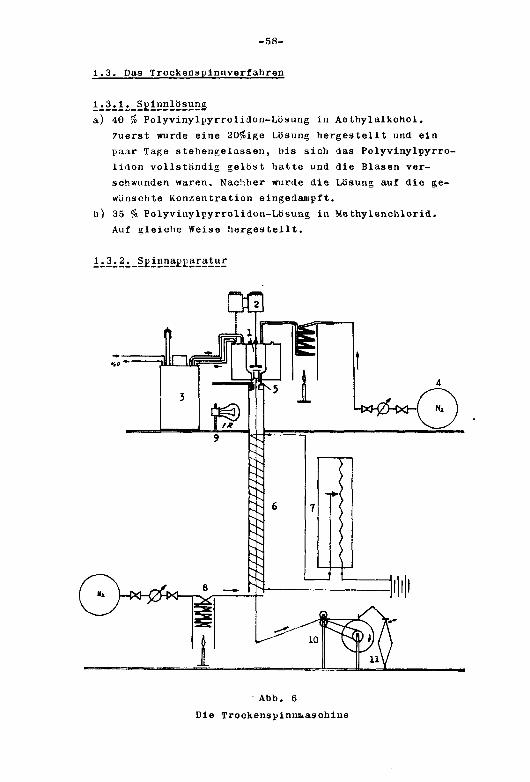
-58-
1.3. Das Trockenspinnverfahren
i •
a) 40 % Polyvinylpyrrolidon-Losung in Aethylalkohol.
Zuerst wurde eine 20$ige Lösung hergestellt und ein
paar Tage stehengelassen, bis sich das Polyvinylpyrro-
lidon vollständig gelost hatte und die Blasen ver¬
schwunden waren. Nachher wurde die Lösung auf die ge¬
wünschte Konzentration eingedampft.
b) 35 io. Polyvinylpyrrolidon-Losung in Methylenchlorid.
Auf gleiche Weise hergestellt.
1.3.2._S£innapjiaratur
Abb. 6
Die Troekenspinniuasohine

-59-
Die Spinnmasse wird in den Behalter (i) eingebracht,
welcher zunächst mit einem Ruhrwerk (2) versehen ist. Die
Temperatur kann mit dem Thermostaten (3) auf die gewünsch¬
te Hohe gebracht werden. Sodann wird nach Erreichen der
gewünschten Temperatur das Ruhrwerk abgestellt und der Be¬
halter luftdicht verschlossen. Hierauf lasst sich mit Hil¬
fe eines Stickstoffdruckes aus der Bombe (4) die Spinnmasse
durch die Düse (5) in das Trocknungsrohr (6) einpressen.
Der erforderliche Druck betragt ca. 1 Atu. Die Trocknung
im Trocknungsrohr, welches mit Hilfe eines Heizbandes und
eines Widerstandes (7) auf die gewünschte Temperatur ge¬
bracht werden kann, erfolgt durch Einblasen eines vorge¬
wärmten StickstoffStroms (8).
Um eine Abkühlung im oberen Teil des Rohres zu verhindern,
wurde dieses zusatzlich mit einer Infrarotlampe (9) bestrahlt.
Die hergestellten Faden werden nach dem Durchlaufen eines
Streckvorganges (10) auf die Spule (ll) aufgewickelt.
1.4. Physikalische Eigenschaften der hergestellten Fasern
1.4.1. Mechanische Eigenschaften
Die Festigkeits- und Dehnbarkeitsmessungen wurden auf dem
6 a e r -Dynamometer ausgeführt in Uebereinstimmung mit
SNV 97 411 und SNV 97 432.
Das Testmaterial wurde bei einer relativen Feuchtigkeit
von 65 % und 20 - 21° C konditioniert.
Die Prufungsresultate sind als Mittelwerte aus je 10 Mes¬
sungen angegeben.
TABELLE VII
Herstellungs- Titer Reiss- Reissfestigkeit Bruch-
Faser verfahren (den) kraft trocken dehnung(g) (g/den) RKM (%)
PolvvinvlDvr-Nasspinnverf.
rolilonfaser (27% KOH, 20°C) 50° 52 °>" *.° 480
Polyvinylpyr- Trockenspinn-rolidonfaser verfahren
Q0(aus Methylen-
1'4UU J5<ä °>25 2.25 °'1
Chlorid)

-60-
Balaatuog
g/dan
0,8
0,6
0,4
0.2
0,0
DIAGRAMM 2
Kraft-Dehnungsdiagramm*von Polyvinylpyrrolidonfasern
N = nach dem Nasspinnverfahren gesponnen
T = nach dem Trockenspinnverfahren gesponnen
*Alle Kraf t-Dehnungsdiagranune sind im gleichen Mass tan an¬
gegeben, damit sie miteinander verglichen werden können.
1.4.2. Rbntgenograp_hische Untersuchung
Das Röntgendiagramm der nach dem Trockenspinnverfahren her¬
gestellten Polyvinylpyrrolidonfasern wurde unter folgenden
Bedingungen auf einem Siemens & Halske Apparat, Typ
"Kristalloflex", aufgenommen:
Dicke des durchstrahlten Präparates: ca. lmm
(Bündel mit genau parallelem Faserverlauf)
Abstand Präparatmitte-Filmmitte:
Blenden: Zylinderblenden 0 75 mm
Strahlung: Cu KoC
Rührenbelastung: 30 kV und 19 mA
Filter: Ni
Belichtungszeit: 8 Stunden
Film: Typon-Röntgenfilm
37,1

-61-
"Qq \
10°
2QO 100
30°
40°
10* 20* JO* 40* 50*
Abb.
50°
7 (a) FaserdiagraniEi einer Polyvinylpyr-
rolidonfaser, nit eingetragenen Radien,
nach welchen aie Photoœeterkurvfin (b)
gewonnen wurden, (c) Darstellung der
relativen Schwärzung der Interferenz
A (Ordinate), willkürlicher Masstab,
als Punktion des Winkelabstandea vom
Aequator (Abszisse).

-62-
Das Röntgendiagramm (a) wird längs verschiedener Radien
photometriscli und jedes Mal die Höhe des Schwarzungsberges
bestirnt t, den die Photouieterkurve von den bestehenden
Interferenzen entwirft (b). Nachher werden die gemessenen
Schwärzungen als Funktion ihres Winkelabstandes vom Aequator
graphisch aufgetragen. So erhält ir.an eine Kurve* (c), welche
den vollständigen Intensitätsabfall längs der Interferenz
aufzeigt.
Die rbntçenograjihische Bestimmung der Polyvinylpyrroiidon-
taser lässt auf geringere Kris tallinitat tier Faser sehlies-
sen, da iie Interferenzen relativ schwach sind. Die ring-
förnige Ausbildung zeiçt, dass praktisch keine Orientierung
vorhanden ist.
*Alle Diagramme mit Photometer- und Sohwärzungskurven sindim gleichen Masstab angegeben, damit sie miteinander ver¬
glichen werden können.

-63-
2. SPINNVERSUCHE MIT POLYOXYMETHYLEN
2.1. Ausgangsmatenal
Die Spinnversuche wurden mit stabilisiertem Polyoxymethylen
"DELRIN" der Firma Dupont durchgeführt. Dabei wurden zwei
"Delrin"-Typen verwendet: "150 x" und "500 x".
Beide Produkte sind in Form eines weissen Granulates zylind¬
rischer Körnung (ca. 3x3 mm) vorhanden.
Das lurohschnittliche Molekulargewicht dieser "Delrin"-
Marken betragt oa. 60.000, bzw. der entsprechende mittlere
Polymerisationsgrad ca. 2.000, wobei sich die erwähnten
Typen nur im ffeichmacherzusatz voneinander unterscheiden.
2.2. Spinnversuohe nach dem Trockenspinnverfahren
Hochmolekulares Polyoxymethylen ist in siedendem Formamid,
Dimethylformaffiid, Nitrobenzol und Diiuethylsulfoxyd loslich.
Die Siedepunkte der Lösungsmittel liegen nahe bei der Zer¬
setzungstemperatur der Polyoxymethylene. Dadurch ist die
Anwendung der genannten Losungsmittel nur beschrankt mög¬
lich, da bei den notwendigen Temperaturen Depolymerisation
der Polyoxymethylene eintreten kann. Dabei muss erwähnt wer¬
den, dass eine hinreichend konzentrierte Losung, wie sie beim
Trockenspinnverfahren notig ist, nicht erreicht werden konn¬
te. Mit Dimethylformamid Hess sich nur eine 5#ige Lösung her¬
stellen.
Noch weniger konzentrierte Losungen sind nach US-Patent
2,775,570 herstellbar. Nach diesem Patent kann man die
2%ige Losung bei 85 - 135° C mit Phenol, p-Chlorphenol,
m-Cresol, <* -Naphthol und Benzylalkohol herstellen. Durch
Zugabe von 2 % A-Pinen kann die Loslichkeit etwas erhöht
werden.
Aus den vorgehend beschriebenen Gründen waren nach dem
Trockenspinnverfahren keine brauchbaren Fäden zu erhalten.

-64-
2.3. Spinnversuche nach dem Sohmelzspinnverfahren
Nach dem Schmelzsplnnverfahren gelang es, die Polyoxymethy-
lene zu Faden zu verspinnen. Als Apparatur wurde die et¬
was umgebaute, früher beschriebene Trockenspinnmaschine
verwendet (Abb. 6). Die Thermostatierung erfolgte hier mit
einet Thermoelement über ein Quecksilberrelais, wobei ein
Elektroruhrer fur gleiohmassige Warmeverteilung des auf
235° C thermostatierten Oelbades sorgte.
Das Auspressen der geschnolzenen Spinnmasse durch die Dü¬
sen von 0,8 und 1,0 mm Durchmesser erfolgte mittels eines
vorgewärmten Stickstoffstromes unter einem Druck von 2 Atu.
Der ausgesponnene Faden wurde zur Verhinderung eines oxy-
dativen Abbaues nach dem Verlassen der Düse im Glasrohr
durch einen kalten Stickstoffström abgekühlt.
Um eine bessere Orientierung der Fas erfasse und dadurch
eine höhere Festigkeit der Faden zu erreichen, wurde der
ausgesponnene Faden noch im warmen, plastischen Zustand
durch einen Streckvorgang bis auf 4-5fache Lange gestreckt.
Wegen des feinen Titers der hergestellten Faden (40 - 185 den)
konnte mit den verfugbaren Apparaturen keine nachträgliche
Kaltstreckung vorgenommen werden.
2.4. Physikalische Eigenschaften der hergestellten Faden
Die Messungen wurden auf gleiche Weise und unter gleichen
Bedingungen wie bei den Polyvinylpyrrolidonfasern ausge¬
führt (Exp. Teil, Kap. 1.4.1.).
Die Ergebnisse sind in der nachstehenden Tabelle VIII zu-
sauiiuengefasst.

-65-
2.4.1. Meohanlsche Eigenschaften
TABELLE VIII
Probe„ .
» SpinnmaasSpinn- Titer Reiss- Reissfestigkeit Bruoh-
duse (den) kraft (g/den) RKM dehnungA In mm (g)
. (*)
1.
2.
3.
4.
"Delrin -
"Delrin -
"Delrin -
"Delrin -
150
150
500
500
X"
X"
X"
x"
1
0
1
0
,0
,8
.0
.8
185
100
175
40
88,3
58,7
105,7
30,2
0
0
0
0
,5
,6
,6
,6
4,5
5,4
5,4
7,2
140
375
220
280
Belastung
g/den
400 500
Dehnung
DIAGRAMM 3
Kraft-Dehnungsdiagranm von PolyoxymethylenfBden1,2,3,4 = ProbemiBJaern
2.4.2, Röntg_enographisohe Untersuchung
Die Röntgenaufnahmen der naoh dem Sohmelzspinnverfahren
hergestellten Polyoxymethylenfäden wurden unter den
gleichen Bedingungen wie bei den Polyvinylpyrrolldonfasern
aufgenommen (Exp. Teil, Kap. 1.4.2.).

-66-
Die röntgenographische Bestimmung der Polyoxymethylenfaser
(Abb. 8 und 9) lässt auf massige Kristallinitat der Fasern
sohliessen, da die Interferenzen nicht besonders stark
sind. Die Orientierung der Faser Nr. 4 (Abb. 9) ist auf
Grund der sichelförmigen Ausbildung der Interferenz wesent¬
lich besser als diejenige der Faser Nr. 1 (Abb. 8).
Diese Resultate stehen in guter Uebereinstimmung mit den
Ergebnissen der mechanischen Untersuchungen. Aus dem Poly-
oxymethylen "Delrin 500 x" lassen sich bessere Fäden her¬
stellen, die noch reissfester werden, wenn eine Düse mit
kleinerem Durchmesser verwendet wird.

-60-
100
au*'
yfi
WBaawawsnxm. <*
Abb. 9
F^serdiagramm (a) des Pölyoxjmethylenfadens Nr. 4
nit seinen Photometarkurren (b) und Dlagraaa dar
relativen Sohwarzung (o)

-68-
or

-«9-
100
Abb. 9
Easerdlagramn (a) des POlyoxymethylenfadens Nr. 4
nlt seinen Photoneterkurren (b) und Diagramm der
relativen Sohwärzung (o)

-70-

-71-
3. SPINNVERSUCHE MIT POLYAETHYLENOXÏD
3.1. Ausgamesmaterial
Die Spinnverauohe wurden mit hochmolekulares Polyathylenoxyd
"POLYOX" der Firma Union Carbide durchgeführt. Dabei wurden
vier "Polyox"-Typen verwendet: Water soluble resins Nr. 35,
205, 301 und Coagulant.
Die in unseren Institut durchgeführten Molekulargewiohts-
bestimmungen nach der Sedimentations- und Diffusionsaethode
haben folgende Werte ergeben (Wasser, 25 C) ':
TABELLE IX
Kunstharz durohschn. Molekulargewicht
Polyox WSR 35 74.000
" " 205 153,000
" " 301 745.000
"
Coagulant 750.000
Dem hohen Molekulargewicht entsprechend ist die Viskosität
der Lösungen sehr hooh.
TABELLE X
Viskosität in Centlpoise von w&ssrlgen Losungen bei 25° C.
Kunstharz
Polyox WSR 35
" " 205
" " 301
" Coagulant
2
5
l*lg<
.000
.500
i Lösung
-
- 4.000
u. sehr
5*lge
225
1500
Lösung
- 375
- 2500
-
-
Die Polyox-Lösungen zeigen wie die Polyvinylpyrrolidonlö-
sungen nicht-Newton'sches Fliessen.
Die Viskosität der wässrlgen Lösungen kann duroh Zugabe von
Salzen wie Harnstoffoxalat oder Caloiumhypochlorlt herabge¬
setzt werden.

-72-
3.2. Spinnversuche nach dem Schmelzapinnverfahren
Ks wurde zuerst versucht, Polyäthylenoxyd mit einer Nylon-
Schaielzspinniuaschine zu verspinnen. Obwohl die Schmelz¬
punkte von Polyox-Harzen schon im Bereich von 65 - 70° C
liegen, konnten sie erst im Bereich von 150° C versponnen
werden, wobei sie im Gegensatz zu den Polyamiden keine dünn¬
flüssige Spinnmasse ergeben. Die Erhöhung der Temperatur der
Spinnmasse auf 240° C (unter Stickstoff) brachte keine we¬
sentlichere Viskositätserniedrigung, so dass wegen mangeln¬
der Dünnflüssigkeit mit der genannten Spinnmaschine keine
brauchbaren Fäden herstellbar sind.
Aus diesem Grunde wurde bei den weiteren Versuchen eine ande¬
re einfache Apparatur verwendet, bei welcher der nötige Aus¬
pressdruck durch einen Kolben statt durch Stickstoff ausge¬
übt wird.
3.2.1. Die Sj>inna££aratur
Abb. 10
Die Schmelzspinnapparatur

-73
Die Apparatur besteht aus einem Druckkolben (l) und einem
dickeren Stahlrohr (2) aus V4A-Stahl, welches mit einer
Bohrung für das Thermoelement (3) versehen ist. Auf den
unteren Teil stellt man die auswechselbare Düse (4) (mit
einem Durchmesser von 0,5 oder 1,0 mm), die mit einer
Mutter (5) am Rohr befestigt wird. Das Rohr hat am oberen
Ende einen Flansch (6), so dass man das Spinnrohr in einer
entsprechenden Klemmvorrichtung befestigen kann.
Das ganze Rohr ist mit einem elektrischen Mantel (7) umgeben,
dessen Heizung thermostatiert wird (8).
Die Polyoxharze werden in Pulverform in das Spinnrohr ein¬
gebracht und mit dem Druokkolben leicht gepresst. Die eigent¬
lichen Spinnversuche werden dann bei verschiedenen Tempera¬
turen durchgeführt.
Die Versuchsergehnisse sind in der nachstehenden Tabelle XI
zusamt enge f as st.
TABELLE XI
Druck
20 Atu
20 Atu
15 Atü
Spinntemperatur ( C
70°
75°
80°
) Resultate
steife Faden, die nicht streck¬
bar sind
steife Faden, die wenig streck¬
bar sind
weichere Faden, die teilweise
streckbar sind
15
15
15
Atu
Atu
Atu
85°
95°
100°
OPTIMALE BEDINGUNGEN FUER
GUT AUSGEBILDETE FAEDEN, DIE
NICHT BRUECHIG SIND UND DIE
MAN KALT STRECKEN KANN
10 Atu
10 Atu 110° weiche Fäden, die nicht mehr so
^__gut streckbar sind
immer weichere Faden, die
im erstarrten Zustand sehr
bruchig und wenig, oder
überhaupt nicht mehr
streckbar sind
5
5
5
2
2
Atü
Atü
Atu
Atu
Atü
120°
130°
140°
150°
160°
2 Atu

-74-
Die nach diesem Verfahren bei optimalen Bedingungen herge¬
stellten Faden lassen sich maximal 5 - 6mal strecken.
Der Fadenquerschnitt ist rund.
3.3. Das Trockenspinnverfahren
Die Eigenschaft der Polyox—Harze, schon bei einem prozent¬
massig kleinen Anteil des Harzes extrem viskose Losungen
zu bilden, und der dementsprechend enorme Anteil des Lösungs¬
mittels in der Spinnlbsung verunmoglichen das Verspinnen nach
dem üblichen Trockenspinnverfahren.
Aus diesem Grunde wurde eine neue Apparatur konstruiert, bei
der der ausgepresste Spinnlosungsstrom direkt mit der beheiz¬
ten Trommeloberflache in Berührung kommt. Die mechanische Be¬
anspruchung des zum Trocknen aufgespulten Fadens ist dadurch
3^3.1. Die_Versuche nach dem Heisstrockensginnverfahren
3.3.1.1. Die Apparatur
Abb. 11
Die Heisstrockenspinnapparatur

-75-
Die Spinnlösung wird aus dem Behälter (l) mit Hilfe eines
Stickstoffdruckes aus der Bombe (2) durch die Düse auf einen
elektrisch beheizten Zylinder (4) ausgepresst. Der Zylinder
ist aus V4A-Stahl gebaut, und seine Drehgeschwindigkeit
wurd durch einen ElektrorUhrer (5) und die Heiztemperatur
mit einem Widerstand (ß) reguliert. Nach dem Verlassen des
Trocknungsweges auf dem Zylinder wird der Faden in der an-
schllessenden Verstreokungsanlage (7) gestreckt und mittels
einer Aufwickelvorrichtung (8) abgezogen.
3.3.1.2. Die Durchführung der Spinnversuche
Als Spinnlösung wurde eine 20#ige Lösung des Polyox VSR 205
verwendet.
Die nötigen Voruntersuchungen wurden auf der Kofler-Heizbank
durchgeführt. Dabei wurde festgestellt, dass bei Temperaturen
bis zu 60 C eine langsame Verdunstung und bei höheren Tem¬
peraturen eine rasche Verdampfung des Lösungsmittels statt¬
findet, was eine entsprechend schnellere Fadentrocknung zur
Folge hat.
Der eigentliche Spinnversuch mit der vorgehend beschriebenen
Apparatur (Abb. 11) wurde auf einer auf 95° C aufgeheizten
Zylinderoberfläohe durchgeführt. Die Stromstärke betrug
ca. 1 A; die Temperatur wurde mittels Testsubstanzen kon¬
trolliert. Zur Erleichterung des Verdampfungsprozesses und
zur Verminderung der Viskosität wurde die Spinnlösung im
Spinnbehälter auf 60° C vorgeheizt. Der erforderliohe Stick¬
stoffdruck bei Anwendung einer Spinndüse von 1 mm Durchmes¬
ser betrug 4 Atü. Unter diesen Bedingungen verursachte der
unvermeidlich grosse Lösungsmittelanteil bei der Verdampfung
im Fadeninnern Gasblasen, so dass viele Hohlräume entstanden,
die zu oinem schlauchartigen Erzeugnis führten.

-76-
Abb. 12
Die Polyathylenoxydfaser nach dem
Heisstrockenspinnverfahren hergestellt(Vergrosserung 10x)
Es wurden irit allen vier Polyox-Typen Versuche durchgeführt,
wobei 20, 15, 10 und 8%ige Losungen in Chloroform angewendet
wurden.
Bei allen Versuchen nach dem Heisstrockenspinnverfahren
wurde im Inneren des Fadens Gasentwicklung hervorgerufen -
bei verdunnteren Losungen in noch stärkeren. Masse.
3^3.2. Das_Kalttrockenspinnverfahren
Die ungenügenden Resultate beim Heisstrockenspinnverfahren
gaben Anlass, ein neues Trockenspinnverfahren zu entwickeln,
welches nicht auf der Verdampfung des Lösungsmittels, son¬
dern auf der Losungsmittelverdunstung beruht.
3.3.2.1. Die Spinnversuche
Zuerst wurde eine Versuchsreihe mit Polyox-Losungen in Chloro¬
form durchgeführt, da Chloroform eine günstige Verdunstungs¬
zahl besitzt (nach H. G n d m m : 2,5). Die Spinnlbsung wurde
auf 60 C vorgewärmt und durch einen Stickstoffdruok durch

-77-
eine Düse von 1 mm Durohmesser ausgepresst. Die Losungen
der niedrigmolekularen Polyox-Typen WSR 35 und WSR 205
(20, bzw. 15%ige Lösungen) konnten mit einem Druck von
1 bis 4 Atü ausgesponnen werden, wobei sich nur die Aus¬
spinngeschwindigkeit änderte und der variierende Druok
ohne Einfluss auf die Formierung des Spinnfandes blieb.
Dagegen durfte beim Verspinnen der Polyox-Typen WSR 301
und Coagulant nicht mit einem grösseren Druck als 1 AtU
vorgegangen werden, da sonst im ausfliessenden Spinnmas¬
senstrahl innere Spannungen entstanden wären und der Faden
einer unerwünschten Kräuselung ausgesetzt worden wäre (Abb.13).
Abb. 13
Die Polyäthylenoxydfaser nach dem Kaltrockenspinnverfahrenbei 0,1 und 1,5 AtU (gekräuselter Teil) gesponnen
(Vergrösserung 2x)
Die Herstellung der Faden erfolgte in der gleiohen Trocken¬
spinnapparatur, wie sie für das Heisstrookenspinnverfahren
angewendet wurde (Abb. ll), mit dem Unterschied, dass dar
Zylinder nicht beheizt, sondern mit einem Luftstrom be¬
lüftet wurde. Während des Drehens wurde der Zylinder seit¬
wärts verschoben, so dass der Faden in diskontinuierlicher
Weise aufgewickelt wurde. Nach erfolgter Trooknung hatten
die Faden einen halbkreisförmigen Querschnitt, was der
glatten Zylinderoberfläche zuzuschreiben ist. Die Fäden
zeigen ein weissliches, etwas trübes Aussehen.

-T8-
Das Naohstreoken: Die hohe Kristallini tat der Polyoxharze
und die niedrige Einfriertemperatur (-50° C) ermöglichten
das kalte Verstreoken des Fadens analog den Polyamidfasern.
Wenn die unverstreokte Faser mit nur nooh sohwaoh orien¬
tierten Makromolekülen rasch vorstreckt wird, so wird sie
nicht allmählich, sondern plötzlich von einem bestimmten
Punkt an dünner. An der Stelle, wo die verstreokte, dünne
Faser an den unverstreokten dickeren Teil angrenzt, bildet
sich eine Einschnürung (Abb. 14). Die Dioke der verstreok-
ten Fäden bleibt weiter konstant.
Abb. 14
StreokungsVorgang der Polyathylenoxydfaser(Vergrösserung 2x)
Die erzeugten vorstreckten Polyäthylenoxydfasern sind
weissglänzend, hoohkrlstallln, sehr weiob, stark und
elastisch. Die Wasserlöslichkeit ist geblieben.

-79-
TABELLE XII
Die Versuche mit verschiedenen Lösungsmitteln
Lösungsmittel
Wasser
Formaldehyd(40*ige Lo¬
sung)
Methanol
Methylen¬chlorid
Methylen-ohlorid/Xylol(1:1)
Temp.(° C)
80
20
65
20
20
% gelöstesPolyox-Harz
10
13,5
4,3
12,1
20
Ergebnis
zu lange Verdunstungszeit
zu lange Verdunstungszeitund Ausscheiden von pul¬verartigem Paraformalde-
hyd. Der Versuch hätte
ein wasserunlösliches
Produkt ergeben sollen,was aber nicht der Fall
war.
Die Löslichkeit der
Polyoxharze ist sogarin siedendem Methanol zu
klein. Beim Verspinnen so
verdünnter Lösungen tritt
unvermeidbar Blasenbil¬
dung auf, die die Reiss¬
festigkeit der Faser
stark herabsetzt.
gute Löslichkeit des
Polyox-Harzes und gün¬stiger Daapfdruok des
Lösungsmittels.
maximale Konzentration
der Spinnlösung ohne Gel¬
bildung. Der Xylolzusatzermöglicht eine gleich-Bässige Verdunstung, wo¬
durch praktisch blasen¬
freie Fäden herstellbar
sind.
Via aus Tabelle XII ersichtlich 1st, ergibt das kombinierte
Lösungsmittel Methylenohlorld/Xylol optimale Resultate.
Zuerst wird das Harz In Methylenchlorid gelöst und dann
die glelohe Menge Xylol beigemischt. Durch Stehenlassen
wird die Lösung auf 30 % der Harzsubstanz aufkonzentriert.
Dabei hat sich gleichzeitig erwiesen, dass die resultieren¬
den Fäden auf das 12faohe gestreokt werden können, wenn noch
etwas Lösungsmittel Im Faden vorhanden ist. Das Verstrecken
erfolgt am vorteilhaftesten 15 Minuten nach dem Verspinnen.

-SO-
In Kapitel 3.5. sind die Resultate der physikalischen
Untersuchungen der hergestellten Fäden zusammengefasst.
3.3.3. Stabilisierungsversuche der Polyäthylenoxyd-Produktedurch Aoet^lierung
44 g des "Polyox"-Harzes "Coagulant" mit einen durchschnitt¬
lichen Molekulargewicht von 750.000 wurden mit 306 cm
Essigsäureanhydrid 4 Stunden am RUckfluss gekooht. Darauf
wurde die Hauptmenge des Anhydrids im Vaouun abdestilliert,
und die Diacetate mit verschiedenem Molekulargewicht erhielt
man in reinem zustand duroh Trocknen im Hoohvaouum.
In Aussehen und Löslichkeit unterscheiden sich die Diaoetate
nicht wesentlich von den Dlhydraten. Ihre Schmelzpunkte lie¬
gen durchwegs um 1 - 2° C tiefer als die der entsprechenden
Hydroxylverbindungen.
Durch die Acetylierung wurde die Wasserunlösliohkeit nicht
beeinflusst. Durch Spinnversuche mit diesen Diacetat-
Polyäthylenoxyden wurden aber nur schwache und brüchige
Fäden erhalten, die nicht brauchbar waren. Da die Viskosi¬
tät dieser Lösungen auch geringer wurde, bedeutet das, dass
beim Acetylieren ein starker Polymerketten-Abbau stattfin¬
det. Dies stimmt mit den Beobachtungen von H. Stau-
d i n g e r'
überein, nach welchen die Polyäthylenoxyd-
Dihydrate bei der Aoetylierung nur bis zum Polymerisations¬
grad 300 beständig sind und eukolloide Polyäthylenoxyde mit
einem Polymerisationsgrad von 2.000 und mehr unter gleichen
Bedingungen weitgehend abgebaut werden.
Die Eigenschaften der Polyäthylenoxyde wurden also, wie er¬
wartet, durch Umsetzungen an den Endgruppen nioht wesentlich
verändert, was verständlich ist, wenn man in Betracht zieht,dass es sich um Stoffe mit sehr hohem Molekulargewicht han¬
delt, bei denen die Endgruppen die physikalischen Eigen¬schaften nur ganz unwesentlich beeinflussen.

-81-
Die Hydrophobierung der Polyox-Harze kann nur auf der Bil¬
dung von Wasserstoff-Briioken zwisohen Sauerstoff aus den
Aether-Gruppen des Makromoleküls und den Wasserstoff-Atomen
aus den Carboxyl-Gruppen irgendeiner Polysäure beruhen '.
0 - CH2-CH2- 9 -CH2-CH2- 0 - CHg-CHjj- 0 CHg-CHg- 0 -
H H H
0 0 0
C=0 C=0 C=0
-R R1 L
Dadurch werden Assoziations-Komplexe gebildet, welche ander«
Eigenschaften aufweisen. Die Wasserlösliohkelt wird stark
herabgesetzt, wie auch die Löslichkeit in den üblichen or¬
ganischen Lösungsmitteln. Wenn man aber an Stelle der Poly-
säure ihr Salz nimmt, gibt es keine Komplexbildung und so¬
mit keinen Hydrophobierungseffekt mehr.
Versuch I: Als Komplex-Partner wurde Polyaorylsäure gewählt
(BASF, K-Wert 25,8)
Reaktionsvorgang : Die beiden Reaktionskomponenten wurden
separat in Wasser gelbst und zwar 20 g Polyox-Harz "Coagulant"
in 500 g Wasser unter Erhitzen und kräftigem Rühren und ander¬
seits 20 g Polyacrylsäure in 100 g Wasser. Dann wurden diese
beiden Komponenten miteinander versetzt. Die Reaktion wurde
bei 80° C und unter kräftigem Rühren durchgeführt. Der pH-
Wert wurde mit Hilfe von Ammoniak im Bereloh von 4 konstant
gehalten« Naoh 10-minütiger Reaktionszeit wurde die Ammoniak¬
zufuhr abgestoppt, und durch weiteres Erhitzen und Rühren
wurde da» vorhandene Ammoniak wieder ausgetrieben, was eine
Herabsetzung des pH-Wertes auf unter 3,5 zur Folge hatte.
Sogleloh bildeten »loh wasserunlösliche Komplexe, die als Nie¬
derschlag ausfielen. Duroh dieses Ausfällen verlor dl« an¬
fänglich hoohviskos« Lösung ihre Eigenschaften, weil unter
diesen Bedingungen beide Beaktlonskomponenten quantitativ
als Komplexe ausfielen und als ober« Schient nur noch fasser
«irttokblieb.

-82-
Die Ammoniakzugabe, die als Inhibitor der Reaktion dient, ist
erforderlich, um die Komplex-Bildung zu verlangsamen, was
eine innigere Mischung beider Komponenten bewirkt.
Als Inhibitoren mit gleicher Wirkung dienen auch Aceton,
Aethanol und Methanol; diese müssen jedoch in beträchtlichen
Mengen angewendet werden (30, 50 bzw. 80 % der gesamten Lö-
sungsmenge).
Bei diesem Versuch wurde der oben erwähnte Niederschlag ab¬
filtriert und bei 110° C getrocknet.
Das günstigste Mischungsverhältnis beider Komponenten ist 1:1.
Wenn aber der Anteil an Polyacrylsäure kleiner und somit der
pH-Wert grosser als 4 wird, tritt keine Komplexbildung ein.
Der optimale pH-Wert für die Waaserstoff-Brückenbildung liegt
somit unter 3,5.
Nach der Trocknung erwies sich das Komplex-Produkt als nicht
mehr thermoplastisch, d.h. es konnte nicht zu Faden verspon¬
nen werden.
Versuch II; Es wurde versucht, bereits fertige Fäden aus
Folyäthylenoxyd nachträglich wasserunlöslich zu machen, in¬
dem man diese in kalte oder warme wässrige Lösungen der
Polyacrylsäure eintauchte.
Dies wurde in drei Versuchsreihen bei Konzentrationen von
10, 20 und 30%igen Polyacrylsäure-Lösungen durchgeführt.
Die Temperaturen variierten von 20 - 100° C.
Die Polyäthylenoxyd-Fäden wurden bei diesen Versuchen durch
die Lösung durchgezogen oder darin stehen gelassen. Dabei
konnte festgestellt werden, dass in allen Fällen eine von
einer starken Quellung begleitete Schrumpfunf der Fäden er¬
folgte. Die Faden wurden klebrig und verloren ihre Faden-
form. Diese Erscheinung tritt desto sohneller ein, je höher
die Behandlungstemperatur und je niedriger die Polyaoryl-
säurekonzentration gewählt wird.

-83-
Daraus folgt, dass eine nachträgliche Behandlung Bit Poly-
acrylsäure In wässrigen Lösungen nicht durohfUhrbar iat.
Versuch III: Da die ersten zwei Versuohe - Herstellung von
fertigen in Wasser schwerlöslichen Polyox-Kompleien, bzw.
die Nachbehandlung der Folyox-Fäden - versagten, wurde ver¬
sucht, die Fäden noch im gequollenen Zustand der Komplex-
Masse aus dem Versuch I vor dem Trocknen zu verspinnen.
Wie bei Versuch I erwähnt, tritt durch Zugabe von wässrlgen
Lösungen der Polyaorylsäuren zu den Polyox-Harz-Lösungen
schon bei Zimmertemperatur Komplex-Bildung ein, bzw. ent¬
steht aus den hochviskosen, kolloidalen Lösungen der Polyox-
Harze, die trübe sind, zuerst eine wasserklare Lösung, die
durch weitere Zugabe von Polyaorylsäure-Lösung beim Erreichen
des pH-Wertes 3,5 plötzlich einen klebrigen, aufgequollenen
Niederschlag liefert.
Bei diesem Versuch wurde ebenfalls das Mischungsverhältnis
der Komponenten 1:1 angewendet. Der gebildete Niederschlag
wurde dekantiert und in einer Zentrifuge vom Überschüssigen
Wasser duroh Schleudern abgetrennt.
Zuerst wurde versucht, die Fäden durch mechanischen Druok
mittels der vorher beschriebenen Kolbendruok-Apparatur
(Abb. 10), Jedoch ohne Heizung, zu verspinnen. Da die hoch¬
viskose, elastisohe Komplex-Masse zu hohe radiale Spannun¬
gen ausübte und sioh eine unregelmässige und stark gekräusel¬
te versponnene Masse bildete, die sioh verklebte und zu ei¬
nem Knäuel zusammenballte, konnten mit dieser Apparatur kei¬
ne gleichmässigen Fäden hergestellt werden.
Danach wurde die Spezlalapparatur fUr hoohvlskose Massen an¬
gewendet, die aus konlsohen Rohren und langen Düsen besteht
und die ebenfalls mit mechanisches Druok betätigt wird.
(Diese Apparatur wurde für das Verspinnen von Kollagen-Fä¬
den entwickelt und wird in Kapitel 4.1.1. beschrieben.)

-84-
Dle Fäden bildeten sich ganz regelmassig und wurden auf eine
Aluminiumspule aufgezogen. Der notwendige Druck betrug
15 AtU, der Durohmesser der Düse 0,5 mm. Die aufgespulten
Fäden wurden naohher im Trockenschrank bei verschiedenen
Temperaturen getrooknet. Die Trocknung erfolgte während 24
Stunden bei 65, 85, 105 und 125° C.
Die Fäden, die bei 65° C getrooknet worden waren, waren glas¬
klar, jedoch noch klebrig, die bei 105° C getrockneten schon
leicht gelb gefärbt und auoh nicht viel verstreokbar und die
bei 125° C getrockneten noch stärker gelb gefärbt. Die be¬
sten Resultate wurden bei 85° C erreicht, wobei die Fäden
glasklar, nicht mehr klebrig und leicht kalt bis zu 500 %
der ursprünglichen Länge verstreckbar waren. Beim Verstreoken
findet eine Orientierung der Molekül-Ketten statt. Die Fäden
sind ausserordentlich welch und knotenfest, aber nicht mehr
kristallin. Die Wasserlöslichkeit der Fäden ist herabgesetzt,
und die Temperaturbeständigkeit nimmt gleichzeitig zu. Die
Fäden zeigen grosse, gummiähnliche Elastizität.
Abb. 15
Kombiniert« Polyäthylenoxyd/Polyaorylsäure-Fassr(Vergrösserung lOx)

-85-
Dadurch konnte nicht nur die Wasserlöslichkeit herabgesetzt,
sondern auf der Basis der Polyox-Harze eine ganz besondere
Art von Fäden hergestellt werden, die grundsätzlich andere
Eigenschaften besitzen als die eigentlichen Polymer-Kompo¬
nenten. Der Faden besteht aus zwei verschiedenen Polymer-
Ketten, die nur durch die van der Waals'sehen Kräfte Über
Wasserstoff-Brücken miteinander verbunden sind.
Infrarot-Spektren:
Es wurden die IK-Absorptlonsspektren einer Folie aus reinem
Polyäthylenoxyd (Polyox-Coagulant) und einer andern hydro¬
phoben Folie aus Polyäthylenoxyd und Polyacrylsäure aufge¬
nommen und verglichen.
Bei der ersten Verbindung (Polyäthylenoxyd) fand man unter
anderem im 3u -Gebiet eine typische OH-Bande, welche ent¬
weder duroh endständige OH-Gruppen des Polyäthylenglykols
oder durch den Wassergehalt hervorgerufen wurde, da das nicht-
hydrophobierte Produkt stark hydrophil ist (normaler Feuch¬
tigkeitsgehalt ca. i %). Im 6m -Gebiet wurden keine Banden
gefunden, im 9p. -Gebiet jedoch typische C-O-C-Bande.
Beim hydrophobierten Produkt hingegen fand man keine OH-Bande
im 3*| -Gebiet, sondern erst bei ca. 3,2jn zwei Absorptions¬
banden, welche vermutlich durch die Bildung von Wasserstoff-
briieken hervorgerufen wurden .
Ferner fand man eine eindeutige Bande bei 5,9JM und eine
andere bei 6,45^ im Gegensatz zum nichthydrophobierten Pro¬
dukt aus reinem Polyäthylenoxyd. Diese 6,45^ -Bande stammt
vermutlich von ionisierten Carboxyl-Gruppen. Solche Fälle
sind in der Literatur ' beschrieben worden, und dies gab
Anlass, anzunehmen, dass bei unserem Produkt ebenfalls sol¬
che Gruppierungen vorhanden sind. Schliesslich fand man
wiederum im Bereich von 9 yi die charakteristische Aether-
bande.

-86-
3.4. Das Nassyinnverfahren
Zu Vergleichszweclten vrarde auch ein Nasspinnversuch durch¬
geführt. Es wurden eine 10$ige wässrige Lösung des Polyox-
Harzes WSIi 205 als Spinnlosung und eine 27%ige Kaliumhydro¬
xyd-Lösung als Fällbad gewählt und die früher beschriebene
Viskosespinniuaschine verwendet (Abb. 5). Die Temperatur des
Spinnbades betrug 20° C. Die Fäden forcierten sich rasch,
und da sie ic. nassen Zustand fest genug «raren, konnte man
sie unter Spannung leicht abziehen uad aufspulen. Die
Faden inlcludieren aber gleichzeitig Spinnoadflüssigkeit, so
<iass ihre Festigkeit nach deu. Trocknen stark abnimmt.
Die Resultate ier Festigkeits- und Dehnbarkeitsmessungen '
sind in nachstehenden Kapitel 3.5. zusanic engefasst.
3.5. Physikalische Eigenschaften der hergestellten Fasern
3.5^1^_Mechanische_Eigenschaften
Die Messungen wurden auf gleiche Weise und unter gleichen
Bedingungen wie bei den Polyvinylpyrrolidonfasern ausge¬
führt (Experimenteller Teil, Kap. 1.4.1.).
TABELLE XIII
Mechanische Werte 4er nach verschiedenen Verfahren herge¬stellten Polytithylenoxydf asern.
Ausgangsuiaterial: Polyox WSR 205 der Firua Union Carbide.
Probe- SpimiL.asse Titer Reiss- Reissfestigkeit Bruch-
Nr. bzw. Verfahren (den) kraft (g/den) (RKM) dehnungSpinnlösung (g) (%)
1. Polyox Schmelz- 560 480 0,85 7,7 9T
WSR 205 spinnverf.2. 15$ige Lsg. Kalttrocken- 660 561 0,9 8,0 78,1
in Chloro- spinnverf.form
3. 10%ige Lsg. Nasspinn- 890 77 0,1 0,9 58,3in Wasser verf.(KOH)

-87-
jBelastung
g/den
20 40 60 80 100
% Dehnung
DIAGRAMM 4
Kraft-Dehnungsdiagramm von Polyäthylenoxydfasern(Probenummern 1-3)
TABELLE XIV
Mechanische Werte der aus verschiedenen Polyox—Harzen nach
dem Kalttrockenspinnverfahren hergestellten Polyäthylenoxyd¬fasern.
Lösungsmittel: Chloroform
Probe- Polyox-Harz Spinnlösung Titer Reiss- Reissfestigkeit Bruch-
Nr. (den) kraft (g/den) (RKM) dehnung(g) (%)
4.
5.
6.
7.
WSR 35
WSR 205
WSR 301
Coagulant
20
15
10
8
des niedrigen Molekulargewichteswegen konnte man keine Fäden ver¬
spinnen.
660 561
410 555
610 971
0,9
1,4
1,6
8,1
12,6
14,4
78,1
58,1
72,7

-88-
100
DIAGRAMM 5
Kraft-Dehnungsdiagramm von Polyäthylenoxydfasern (Probe-nummern 5-7)

-89-
TABELLE XV
Mechanische Werte der bel verschiedenen Bedingungennach dem Kalttrockenspinnverfahren gesponnenen Poly¬
äthylenoxydfasern.
Probe- Polyox- Spinn- Titer Reiss- Reissfestigkeit Bruch-
Nr. Harz Spinnlösung düse (den) kraft (g/den) (RKM) gehnung(fi mm) (g) (*)
8.
9.
10.
11.
12.
13.
14.
WSR 205
WSR 205
WSR 301
WSR 301
Coagu¬lant
Coagu¬lant
Coagu¬lant
12* in
Methylen¬chlorid
12* in
Methylen¬chlorid
10* in
Methylen¬chlorid
20* in
Methylen¬chlorid/
8 * in
Methylen¬chlorid
20* in
Methylen¬
chlorid/
20% in
Methylen¬chlorid/
1,0
0,8
0,8
0,8
0,8
1,0
0,8
800
130
130
150
220
810
330
1260
162
214
280
406
1600
899
1,6 14,4 45,5
1,2 10,8 59,1
1,6 14,4 53,9
1,9 17,1 62,5
1,8 16,2 35,4
2,0 18,0 34,7
2,7 24,3 25,9

-90-
20 40 60 80 100
DIAGRAiMM 6
Kraft-Dehnungsdiagramm von Polyäthylenoxydfasern (Probe-numiuern 8-14)

-91-
Die Kraft-Dehnungsmessungen der hydrophoben Polyäthylen-
pxyd/Polyaorylsäure-Faser ergab folgende Werte:
Titer:
Reisskraft
Reissfestigkeit:
Bruchdehnung
1.600 den
220 g
0,14 g/den
bzw. 1,25 RKM
575 %
Belastung
0 100 200 300 400 500 600
% Dehnung
DIAGRAMM 7
Kraft-Dehnungsdiagraniiu der hydrophoben Polyäthylenoxyd/Poly-aorylsäure-Faser
3.5^2^ Rontgenograjjhi s che_Untersuchung
Die Röntgenaufnahme des nach dem Kalttrockenspinnverfahren
hergestellten Polyäthylenoxydfadens (Probe Nr. 14) wurde
unter den gleichen Bedingungen wie bei der Polyvinylpyrro-
lidonfaser aufgenommen (Experimenteller Teil, Kap. 1.4.2.),
abgesehen vom Abstand Präparatinitte-Filmmitte, der in die¬
sem Falle auf 3,82 cm eingestellt wurde.

-92-
Die Röntgenaufnahme des Polyäthylenoxydfadens (Abb. 16)
stellt ein ausgesprochen schönes Faserliagramn dar. Die
Fasersubstanz weist hohe Kristallinitat und Orientierung
auf.
(Leichter Storreflex links aussen).
100
Abb. 16
Faserdiagran.ni (a) des
Polyathylenoxydfadensmit seinen Photometer¬
kurven (b) und Diagrammder relativen Schwär¬
zung (c).

-93-
iffi
Abb. 16 b

-94-
3.6. Molekulargewiohts-Bestimmunften an Polyäthylenoxydfasern
3.6.1. Wahl der_Methode
Zur Bestimmung der Molekulargewichte kam nur die Ultrazentri¬
fugen-Methode in Betracht. Viskositäts-Messungen in verdünn¬
ter Lösung sind nicht brauchbar, da die fraglichen Produkte
("Polyoxn-Harze der Firma Union Carbide) eine sehr breite
Molekulargewichtsverteilung in der Grössenordnung von
M/M =10-20 aufweisen. Ausserdem sind die Substanzen
strukturviskos. Aenderungen im Molekulargewicht und in der
Molekulargewichtsverteilung können daher zu wenig übersicht¬
lichen Werten führen, da die Viskosität von Molekulargewicht,
Molekulargewichts-Verteilung und Schubspannung abhängt.
Osmotische Messungen sind ebenfalls wegen der breiten Ver¬
teilung nicht durchführbar, da nach den Messungen die Zahlen¬
mittel des Molekulargewichtes in der Grössenordnung von 5000
liegen, was aber bei der breiten Verteilung schon starke Feh¬
ler durch Permeation und Staverman-Effekt hervorrufen kann,72)
wie neulich gefunden wurde . Lichtstreuungsiessungen kamen
ebenfalls nicht in Betracht, da der in den Proben noch vor¬
handene Katalysator CaCOg die Messungen stark stört. Die Pro¬
dukte durften jedoch möglichst nicht weiter gereinigt werden,
da dadurch eventuell nieder- oder hochmolekulare Anteile ent¬
fernt werden.
Als Methode wurde daher das Svedberg-Verfahren über Sedlmen-
tationsgeschwindigkeit und Diffusion gewählt. Es bietet ge¬
genüber dem Archibald-Verfahren den Vorteil, eine mögliche
Paucimolekularität erkennen zu lassen.
3.6^2_. Durchführung der Messungen
Alle Ultrazentrifugen-Messungen wurden in einer Spinco-
Ultrazentrifuge, Modell E-HT mit Temperaturkontrolle und
-regelung bei 25,0 - 0,1° C ausgeführt. Die Geschwindigkeit

-95-
betrug bei den Sedimentationsmessungen stets 59 780 U/min,
ein Druck- oder Konzentrationseinfluss auf die Zeitabhängig¬
keit der Sedimentations-Koeffizienten konnte nicht gefunden
werden. Die Sedimentations-Koeffizienten wurden nach der
Differenzen-Methode aus mindestens je 6 Aufnahmen pro Kon¬
zentration berechnet. Die bei den einzelnen Konzentrationen
errechneten Sedimentations-Koeffizienten s wurden sodann nach
(1) (l/s) = (l/s0) (1 + ksc)
auf die Konzentration 0 extrapoliert.
Die Diffusions-Messungen wurden ebenfalls in der Ultrazentri¬
fuge, aber mit einer Ueberschichtungszelle vom Ventiltyp aus¬
geführt. Die Umdrehungsgeschwindigkeiten wechselten zwischen
ca. 10.000 und 20.000 U/min. Ein Einfluss der Drehzahlen
konnte nicht festgestellt werden. Die Diffusionskoeffizien¬
ten wurden aus den Philpot-Svensson-Aufnahmen nach der Fla¬
chenmethode berechnet. Die so bei den einzelnen Konzentra¬
tionen erhaltenen Diffusions-Koeffizienten D wurden nach
(2) D = Do (1 + kDc)
auf die Konzentration 0 extrapoliert.
Das partielle spezifische Volumen *V beträgt für Wasser und
25° C *V = 0,820 ml/g65*.Aus Sedimentationskoeffizient s
,Diffusionskoeffizient D
o' o
und partiellem spezifischem Volumen *V wurde sodann das
Molekulargewicht MsD Über
RT s
(3) H_
= 2 (d _ Dichte des Lösungs-sD
Do (1 - *Vim)LM
mittels)
bereohnet.
Substanzen: Es wurden drei verschieden behandelte Polyäthylen¬
oxyde gleicher Marke "Polyox WSR 205" und unbehandeltes glei¬
ches Harz (zum Vergleich) untersucht. Die Substanzen wurden
wie folgt bezeichnet:

-96-
0. Polyox WSR 205 unbehandelt
I. Polyox WSR 205 Schmelzspinnverfahren
II. Polyox WSR 205 Kalttrookenspinnverfahren, Losung In
Chloroform
III. Polyox WSR 205 Nasspinnverfahren, kalt, KOH-Fàllbad
Ergebnisse: Die Messungen sind in Tabelle XVI zusammenge-
0
I
II
III
stellt.
TABELLE XVI
HSedimentations-
Koeffizienten
so
(Svedberg)
2,56
1,61
2,25
5,40
Diffusions-
Koeffizienten
Do
(cm /see.)
2,30
2,85
6,3
11,3
Durchschnittliche
Molekulargewichte
MSD
153.000
78.000
49.000
66.000
Aus den Messdaten geht hervor, dass alle Produkte bei den
entsprechenden Behandlungen abgebaut werden. Auffallig ist
jedoch las Verhalten der Sedimentations-Koeffizienten.
Wahren! naL-lich die Diffusions-Koeffizienten erwartungsge-
mass inner hoher liefen ils der Diffusions-Koeffizient des
Ausgangs^roduktes, ist der Sedin.entations-Koef f izient des
Produktes III nicht niedriger, sondern sogar hoher als der
der Ausgangssubstanz.
Un. zu prüfen, ob Jieses Verhalten auf Messfe'ilern beruht,
wurden aus Jen berechneten Molekulargewichten rückwärts
uber bereits vorher aufgestellte Fichbeziehungen69' zwischen
Molekulargewicht und SeJicentations-Koeffizienten und
Diffusions-Koeifizienten die zugehörigen s . und D
ermittelt.

-97-
Sodann wurden die Quotienten s„/s bund Do/D„ ber
g«-
bildet. Die Werte sind in der nachstehenden Tabelle zusam¬
mengestellt.
TABELLE XVII
Experimentell gefundene und berechnete Sedimeutations-
ünd Diffusions-Koeffizienten
Substanz
0
I
II
III
2
1
2
5
8o
,56
,61
,25
,40
D
2
2
6
11
0
,30
,85
,30
,30
8o,
1
1
1
ber.
-
,55
,23
,45
V
2
3
2
ber.
-
,70
,35
,90
"</
1,
1,
3,
o,ber.
-
04
83
72
Do/D
1
1
3
o,ber.
-
,05
,88
,90
Die gefundene, gute Uebereinstimmung zwischen den beiden
Quotienten zeigt, dass der Effekt keinesfalls auf Versuchs¬
fehlern beruhen kann. Eine genaue Betrachtung der Diagramme
der Diffusions-Versuche zeigt aber, dass bei den Versuchen II
und III starke Schultern an der Einmündung der Gradienten¬
kurven in die Basislinien vorhanden sind. Das bedeutet aber,
dass bei der Behandlung der Proben durch die verschiedenen
Verfahren verschieden starke Anteile an niedermolekularer
Substanz (vermutlich Glykolen) entstanden sein müssen.
Die Effekte bei den Sedimentations-Koeffizienten lassen sich
damit wie folgt erklären: bei der Behandlung werden nieder¬
molekulare Anteile in unterschiedlichem Ausnass gebildet.
Bei den Diffusionsmessungen werden diese Anteile nur als Sub¬
stanz mitgemessen, da Ja die gesamte Kurve ausgewertet wird.
Die Diffusions-Koeffizienten werden daher immer höher sein
als der Diffusionskoeffizient der Ausgangssubstanz. Bei den
Sedinientations-Messungen "schwimmt" dagegen die Übriggeblie¬
bene hochpolyuere Substanz sozusagen in der niedermolekularen,
die als zusätzliches Lösungsmittel wirkt. Wenn nun nur wenig

-98-
Niedermolekulares entstanden ist, dann ist sein viskositats-
erhohender Einfluss nur gering. Da aber gleichzeitig ver¬
mutlich relativ viele hochmolekulare Anteile abgebaut wurden,
bekoiiut man eine niedrige Sedimentationskonstante. Wenn nun
aber viel abgebaut wurde, sollte ebenfalls die Sedimentations¬
konstante niedrig sein. Da dies bei der Probe III nicht der
Fall ist und eine höhere Sedin<entationskonstante s gefunden
wurde, muss also neben den. Abbau noch eine Verknüpfung von
Molekülen eingetreten sein.
Die Beobachtung zeigt, dass die verschiedenen berechneten
Molekulargewichte nicht in ihrer Hohe miteinander verglichen
werden dürfen, da in die Sedimentations-Koeffizienten noch
der Viskositatseinfluss mit hineingeht. Aus den Messwerten
darf daher lediglich der Schluss gezogen werden, dass die
Substanzen abgebaut werden, nicht aber eine Reihenfolge des
Abbaus aufgestellt werden kann.

-99-
TABELLE XVIII
Messwerte bei den einzelnen Konzentrationen
Substanz Konzentration
(g/-Dio2
I 1,129
0,819
0,630
0,593
0,404
0,402
0,203
0,200
II 0,766
0,400
0,357
0,197
III 1,098
0,817
0,607
0,397
0,208
Zu Abbildung 17:1 (Versuch S
II (" S
III ( " S
s.10*"
sec"
_
0,718
0,800
-
0,985
-
-
1,252
0,74
1,14
1,15
1,44
_
1,30
1,44
2,20
2,79
120) c = 0,819 ',
133) c = 0,766 i
127) c = 0,817 ?
D.10-
cm /sec
4,07
4,13
3,49
3,36
-
3,60
2,69
-
2,44
3,17
-
5,61
5,13
8,23
7,46
10,0
-
i 12590 U/min
i 19160 U/min
i 13410 U/min
Der Aufnahme-Abstand betrug bel I und II je 16 min, bel
III 8 min. Die gezeigten Bilder entsprechen den Aufnahmen
2-5 der jeweiligen Versuche.

-100-
CdiH
ta

-101-
4. SPINNVERSUCHE MIT KOLLAGENSUBSTANZEN
4.1. Vorversuche nach dem alkalischen Aufschlussverfahren
Zuerst wurde versucht, das bekannte Kunstdarmverfahren zu
modifizieren und die Kollagenmasse durch eine neukonstruierte
Spinnapparatur zu monofilen Fäden zu verspinnen. Den Anlass
dazu gaben Fr. P. 764 642 und DRP 652 824.
Als Ausgangsstogg diente die Rinderunterhaut. Die Quellung
wurde mit Kalkmilch von einer Konzentration von 1,95 % Ca(0H)2,bzw. 15 g CaO/l Wasser, vorgenommen.
Das spez. Gewicht betrug 1,013. Behandlungsdauer 8 Wochen.
Der zu behandelnden Unterhaut wurde Kalkmilch im Mengenver¬
hältnis 1:1 zugefügt. Jeden achten Tag wurde die Kalkmilch
erneuert und das Material umgerührt, um eine gleichmassige
Durchkalkung zu erreichen. Anschliessend wurde das Material
ausgewaschen und 24 Stunden mit n-Salzsäure neutralisiert,
und wieder mit Wasser so lange gespült, bis endlich ein pH-
Wert der Masse von 3 erreicht wurde. Das Material wurde da¬
bei durchsichtig, stark aufgequollen und enthielt 11 %
Trockensubstanz. Die anschliessende Zerfaserung erfolgte in
einer Fleischhackmaschine und die nötige Homogenisierung in
einer Knetmaschine. Zu beachten ist, dass eine zu weit ge¬
triebene Faserzerkleinerung sich auf die Festigkeit des Fa¬
dens nachteilig auswirkt. Darum soll das tierische Hautmateri¬
al nur schonend zerfasert und in gequollenem, plastischem Zu¬
stand weiter verarbeitet werden. Die Entlüftung erfolgte in
einer Industriezentrifuge bei 3.000 u/min. Durch Pressung
der homogenisierten und blasenfreien Masse unter einem Druck
von 20 Atü durch eine enge konische Düse trat Orientierung
der Makromoleküle ein, was die Festigkeit erhöhte.
4.1.1. Die Sj>innap_£aratur
Da alle bekannten, vorher beschriebenen Verfahren zur Her¬
stellung von chirurgischen Fäden aus Kollagenspinnnsasse auf
der Herstellung von Folien oder Schläuchen beruhen, die spä-

-102-
ter durch Schneiden, Drehen, Flechten usw. zu Faden ver¬
formt werden, haben wir versucht, die Faden direkt aus dem
plastifizierten Kollagenmatenal zu verspinnen. Dabei tra¬
ten Schwierigkeiten auf, da bei dieser hochviskosen Spinn-
masse beim Verspinnen zu grosse Radialkrafte auftraten,
was mit der üblichen Druckspinnapparatur zu gekrauselten
Fäden von ungleichmassigem Querschnitt führte. Um diesen
Nachteil auszuschalten, war es nötig, eine neue Apparatur
zu konstruieren, die grundsätzlich aus konischen Einmun-
dungsstucken und langen Düsen besteht. Die konischen Teile
der Spinnapparatur ermöglichten eine weitgehende Faralle-
lisierung der Fibrillen, und die lange Spritzduse bewirkte
eine Verminderung der Radialkrafte und ermöglichte das Ver¬
spinnen von Faden mit rundem und gleichmassigerem Quer- -
schni11.
Die ganze Apparatur ist aus V4A-Stahl gebaut. Der Druck¬
kolben ist zusatzlich noch hart verchromt, um eine gute
Dichtung zu erreichen. Das obere Druckrohr tragt an seinem
unteren Teil einen Flansch, welcher mit Schrauben mit dem
Flansch des unteren konischen Teiles verbunden ist. Dadurch
ist es möglich, die Apparatur auch bei diesem Teil zu öff¬
nen, was nötig ist, um das beim Pressvorgang entstehende
Vacuum auszuschllessen und den Druckkolben fur eine neue
Charge herauszunehmen.
Die Spritzdiise am untersten Teil der Apparatur ist eine
auswechselbare Injektionsnadel von 150 mm Länge.
Die verwendeten Düsen hatten einen Durchmesser von 0,5, 0,7,
0,85, 1,1 und 1,3 mm.
Die beschriebene Apparatur ist aus Abb. 18 ersichtlich.

-103-
Abb. 18
Spinnapparatur für Kollagen- und Polyathylenoxyd/Poly-acrylsaure-Fäden

-104-
4.1.2. Das_Versj> innen
Das aufgequollene, spinnbereite Kollagenniaterial wurde in
das Druckrohr gebracht und mit Hilfe einer hydraulischen
Presse über dem Kolben unter 20 Atu Druck durch die Appara¬
tur gepresst. Die heraustretenden Faden wurden auf eine
AluD.iniumspule gewickelt und getrocknet. Die Trocknung er¬
folgte mit Heissluft bei 55° C. Die Trocknungszeit fur
dünne Faden (Düse von 0,5 mm Durchmesser) war nicht langer
als 10 Minuten.
In einem anderen Versuch wurden die Faden direkt in ein
kaltes Acetonbad gespritzt, wo sie schnell entwassert wur¬
den, sieh besser formten und einen runden Querschnitt auf¬
wiesen.
Fur die Faden aus dickeren Düsen muss te der Weg im Bad ent¬
sprechend verlängert werden.
Die erzeugten Faden zeigten ungenügende Festigkeit und ge¬
ringe Dehnung. Daneben waren die Faden in dieser untiehan-
delten Form spröde und in Wasser stark quellbar.
As-
Abb. 19
Monofile aus Unterhautgewebe hereestellte Kollagenfaser(Vergrosserung 70 x)

-105-
Alle diese Fadenelgensohaften weisen darauf hin, daas der
gewählte Aufsohlusaprozess für Kunstdärme geeignet 1st,
nicht aber für die Herstellung von starken und glelohmässl-
gen Fäden. Es trat dabei eine sohon ziemlioh weltgehende
Hydrolyse ein, die Bit einen Abbau des Kollagens verbunden
war. Aus diesem Grunde wendeten wir uns den milderen sauren
Aufsohlussverfahren zu, woduroh nur ein minimaler hydroly¬
tischer Abbau erfolgt, jedooh eine Quellung alt Lockerung
der Querbindungen zwischen den Polypeptldketten hervorge¬
rufen wird.
Die Eigenschaften der naoh diesen Verfahren hergestellten
Fäden sind in Tabelle XX angegeben.
4.2. Saures Aufsohlussverfahren
Das saure Aufsohlussverfahren ermöglicht schonende Kollagen-
quellung. In neuester Zelt richtet sich die Forsohung in
den U.S.A. auf die Herstellung von Fäden aus regeneriertem
Kollagen, das auf diese Welse zur Spinnmasse verarbeitet
wird73'' 7*'. Beiden Verfahren gemeinsam ist die saure
Quellung des Kollagenmaterials, starke meohanIsche Behand¬
lung, um die Masse zu zerkleinern und homogenisieren,
schwierige Filtrierung und Herstellung eines Gels unter Zu¬
gabe von viel Wasser. Der Feststoffgehalt beträgt nur 0,5
bis 1,5 %, und dies bedingt eine lange Erstarrungszelt im
Fällbad, so das* der Ausstoss bei kontinuierlicher Arbelt
nur 1,8 bis 3,6 ml/min, beträgt.
In den eigenen Arbeiten wurde versuoht, eine konzentriertere
Spinnmasse herzustellen, das Mahlen der Masse zu vermelden
und ein einfacheres und teohnlsch brauchbares Schnellver¬
fahren zu entwickeln. In diesem Sinne wurden zwei Wege be-
sohritten:

verwendbar.nichtpraktischundbruchigwarenfestigkeit,
Reiss¬ungenügendeeineaberzeigtenFadenerhaltenenDie
werden.beobachtetQuellungstarkeeineStundenzweinach
erstkonnte-Zimmertemperaturbei-VersuchdrittenBeim
war.feststellbarbau
Ab¬einMinuten10nachschonwobeidurchgeführt,C60°bei
jedochBedingungen,gleichenbeiwurdeProbeweitereEine
Luft.heissermitfolgte
er¬TrocknungDieversponnen.FadenzuDüsebeschriebeneher
frü¬diedurchundbefreitWasserüberschüssigemvonpressen
Aus¬durchhomogenisiert,MixereinemmitwurdeGutquollene
ge¬soDasneutralisiert.AmmoniakverdünntemmitWasser
mitSpulungsorgfaltigernachwurdeMaterialDasstellen.
fest¬QuellungunterAbbaumerklicheneinenmankonnteStunde
einerVerlaufeImwurde.gehalten2,5bis1,5vonGrenzenden
inSalzsaureverdünntervonZugabedurchpH-Wertderwobei
behandelt,C47°beiPepsing0,04mitwurdenRinderdarmg10
worden.gespultWassermitreichlichundbefreit
Fett&nlagerungenundFleischrestenMuskeln,anhaftendenvon
mechanischundzerschnittennachLangedersindDarmeDie
Verfugung.zurRinderdarmefrischestandenAusgangsstoffAls
vorzurufen.
her¬MediumsaureminEnzymwirkungdurchKollagenmasseder
PiastizitatbessereundTeilabbaueinenversucht,wurdeEs
unter_fermentativem_AbbauVersuche4.2.1.
bringen.zu
LösungsforminHydrotropioavonAnwendungunter
undaufzuschliessensauerKollagenmaterialdas2.
undabzubauenteilweiseMedium
saureminEnzymbehandlungdurchKollagenmasseDie1.
-106-

-107-
Das Pepsin verursacht die Spaltung von NH-CO-Bindungen, wo¬
bei eine starke Molekiilzerkleinerung stattfindet und die
Mechanischen Eigenschaften der Fäden stark beeinflusst wer¬
den.
A.Z.2. Lösungsversuche
Wie erwähnt, ist das Kollagen in trookener Form vollständig
unlöslich, wohl aber quellbar, 'ohne dass ein beträchtlicher
Abbau stattfindet, wenn milde Quellungsmittel angewendet
werden. Es lag dann der Gedanke nahe, das gequollene Kollagen
(bevorzugt in verdünnten Säuren gequollen) als Ausgangsstoff
zu benutzen und in dieser aufgelockerten Form eventuell zu
läsen.
4.2.2.1. Vorbereitung des Ausgangsstoffes
Das Rinderunterhautgewebe, das zur Verfügung stand, enthielt
68,2 # Feuchtigkeit, bzw. 3i,8 % Trockenstoff.
100 g des genannten Kollagenmaterials wurden mit 1500 om
verdünnter-Salzsäure versetzt und 24 Stunden bei Zimmer¬
temperatur stehengelassen. Die erforderliche Menge an kon¬
zentrierter Salzsäure betrug ca. 2 g, um einen pH-Wert von
2,5 bia 3 zu erhalten. Das gequollene Kollagenmaterial wies
7,6 % Trockengehalt auf.
4.2.2.2. Durchführung der Lösungsversuohe
Die ganze gequollene Masse wurde in einer Fleischhaokmasohine
zerschnitten und in einem Kneter homogenisiert. Es wurden Je¬
weils genau 0,5 g gequollenes und homogenisiertes Kollagen
genommen und in Reagenzgläser gebraoht, die Je 10 g Lösungs¬
mittel enthielten.
Die ganze Versuchsreihe wurde unter gleichen Bedingungen
durchgeführt. Die Reagenzgläser wurden in ein auf 20° C
thermostatiertes Wasserbad gebraoht und 24 Stunden stahen-
gelassen. Man wollte den Lösungsvorgang unter milden Be-

-108-
dingungen durchführen, um einen Abbau des hitzeempfindliohen
Kollagens zu vermeiden.
In der nachstehenden Tabelle sind die Resultate der Unter¬
suchungen angegeben.
TABELLE XIX
Lüsungsversuohe von sauer gequollenem Kollagenmaterial
Nr. LOESUNGSMITTEL BEFUND
1 Aceton
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
Acetonitril
Acetophenon
Aethanol
Aether
Acetessigester
Aethylacetat
Aethylbenzol
Aethyl-n Butyloarbinol
Aethylenohlorid
Aethylenglykol
Aethylcellosolve
Allylalkohol
Allylohlorid
o-Ameisensäureäthylester
Amylaoetat
n-Amylalkohol
Benzol
Benzonltril
Butylaoetat
__
0
-
-
0
0
0
0
0
-
0
0
0
0
0
0
0
0

-109-
Nr. LOESUNGSMITTEL BEFUND
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
Butyläther
n-Butylalkohol
tert-Butylalkohol
Chlorbenzol
OC -Chlornaphthalin
Chloroform
Cyanessigsäureester
Cyolohexan
Cyolohexanol
Cyolohexanon
Dekalin
Diäthylamin
Diäthylenglykol
Dichloräthylen
Dlaethylanllln
Dime thyIfonnamid
Dimethylsulfoxyd
Dioxan
Formamid
Glycerin
n-Hexan
Hexamethylphosphamid
m-Kresol
Ligroin
Methanol
0
-
—
0
0
0
0
0
0
0
0
0
++
0
0
+
+++
0
+++
++
0
+
++
0
—

-110-
Nr. LOESUNGSMITTEL BEFUND
46
47
48
49
50
51
52
5*
54
55
56
57
58
59
60
61
62
63
64
65
66
67
Methylenchlorid
Me thylathyIketon
Methylcellosolve
Nitroathan
o-Nitroanisol
Nitrobenzol
Nltromethan
Petrolather
n-Propanol
iso-Propanol
Propylenglykol
Schwefelkohlenstoff
Tetraohlorathan
Tetrachlorkohlenstoff
Tetrahydrofuran
Tetrahydrofurfurylalkohol
Tetralm
Toluol
Triathylaniin
Trichlorathylen
m-Xylol
o-Xylol
0
-
-
-
0
0
-
0
—
-
++
0
0
0
0
0
0
0
0
0
0
0
68 p-Xylol 0
ZEICHENERKLÄRUNG: +++ kolloidale Losung++ starke Quellung+ schwache Quellung0 kein Effekt
schwache Koagulation— starke Koagulation
sehr starke und schnelle Koagulation

-111-
4.3. Die Spinnversuche mit Kollagenlosungen
Auf Grund der durchgeführten Losungsversuche wurden Kollagen-
splnnlosungen hergestellt und zu Faden versponnen.
Als Kollagenausgangsstoffe standen zuerst Rinderdarme und
spater Kalbsblosse-Abfalle zur Verfügung.
4.3^1. Versuche mit Rinderdarmen als Ausgangsstoff
4.3.1.1. Vorbereitung des Rohstoffes.
Die frischen Rinderdarme wurden in einem Kühlschrank bei
-3 C aufbewahrt. Vor jedem Versuch wurde die notige Darm¬
menge in 2 - 3 Stunden bei Zimmertemperatur aufgetaut.
Die Darme wurden der Lange nach zerschnitten und mechanisch
von anhaftenden Muskeln, Fleisehres ten und Fettanlagerungen
befreit. Um weitere unerwünschte Substanzen (Mucosa, Serosa,
Fettresten usw.) zu entfernen, wurde neben der reichlichen
Spulung mit Wasser folgender endgültiger Reinigungsprozess
in Anlehnung an das Am.P. 2,750.251 durchgeführt:
100 g frischer Rinderdarm wurden in gesättigter NaCl-Losung
eingetaucht und 24 Stunden stehengelassen. Dadurch wurde
eine Dehydratiaierung und gleichzeitige Härtung gegen Warme
erreicht. Dl« Schrumpfte»p«r«tur des Gutes variierte zwischen
45 und 50u C. Nachher wurde das Gut mit kaltem Wasser ausge¬
waschen. Die eigentliche chemische Behandlung wurde mit
0,5$iger wassriger Losung des Na-Salzes der Aethylendiamin-
tetraesslgsaure (Trilon B) im Laufe von 18 Stunden bei 38° C
durchgeführt. Der pH-Wert betrug 10,3. Schliesslich erfolgte
eine reichliche Spulung mit kaltem Wasser. Das so behandelte
Kollagenmaterial wurde weltgehend entfettet und von anderen
nichtkollagenen Substanzen befreit.
4.3.1.2. Quellungs- und Losungsvorgang
In den ersten Versuchen (Proben Nr. 3 und 4) wurden 50 g
gereinigtes Darmmaterial zu 750 ml verdünnter Salzsaure
gegeben und 24 Stunden bei Zimmertemperatur stehengelassen.

-112-
Die erforderliche Menge an konzentrierter Salzsaure betrug
ca. 1 ml, um den gewünschten pH-Wert von 2,5 zu erreichen.
Das stark gequollene Gut wies nach dem Abschleudern 8,1 %
Trockengehalt auf. Die ganze gequollene Masse wurde abge¬
schleudert und in kleinere Stucke von ca. 1,5 z 1,5 cm ge¬
schnitten. Der Masse wurden dann 25 g Dimethylsulfoxyd zu¬
gegeben und alles zusammen wurde in einen Laboratoriums¬
mixer (Turmix-500 ff) eingebracht. Im Verlauf von einer Mi¬
nute wurde das Gut kolloidal gelost und homogenisiert. Die
anschliessende Trennung von nichtgelosten Teilen erfolgte
durch eine mit einem Netzsieb (aus V4A-Stahl und 5200 Mesh/
cm ) umwickelten Filterkerze. Die Filtration erfolgte
leicht und schnell unter einem Luftdruck von 2 Atu.
Schon beim Stehenlassen bei Zimmertemperatur geht inner¬
halb von 15 - 20 Minuten das Kollagenmaterial vom Sol- in
den Gelzustand über. Durch leichtes Erwarmen bis max. 60° C
und Ruhren geht die Spinnmasse wieder in den Solzustand zu¬
rück. Die höheren Temperaturen müssen vermieden werden, um
einen Abbau des Kollagens zu verhindern. Das Material ging
beispielsweise bei 80° C wohl kolloidal in Losung; diese
war aber auch nach dem Erkalten nur niederviskos, was als
Zeichen eines Molekulabbaues angesehen werden kann.
Im nächsten Versuch wurde mit verdünnter Essigsaure gear¬
beitet, wobei 6$ige Essigsaure verwendet und der pH-Wert
auf 2,5-3 gehalten wurde. Die Essigsaure gewirkte eine
sehr rasche Quellung, so dass schon nach 4 Stunden genugende
Kollagenquellung festzustellen war, um den weiteren Kosungs-
prozess mit Dimethylsulfoxyd durchzufuhren (Probenummern
5 und 6).
Anschliessend wurde die Quellung mit 0,3%iger Malonsaure
durchgeführt und unter gleichen Bedingungen die Faden ge¬
sponnen, wobei die Quellung unter einem pH-Wert von 2,2
durchgeführt wurde (ProbenumLiern 7 und 8).

-113-
Zum Schluss wurde eine verkürzte alkalische Quellung durch¬
geführt, wobei die Rinderdarme einem 4-wöchigen Aufschluss-
prozess mit Kalkmilch unterworfen wurden (Probenummern 9-19).
Die Lauge enthielt 15 g CaO/l und wurde im Mengenverhältnis
1:1 dem Gut zugegeben. Nach reichlicher Spülung mit Wasser
wurde die Neutralisation mit verschiedenen verdünnten Säuren
vorgenommen (6%ige Essigsäure, O,3#ige Malonsäure und ver¬
dünnte Salzsäure). Ansohliessend wurde wieder mit Wasser ge¬
spült, bis der pH-Wert der Masse sich auf 3 einstellte. Die
Masse wurde 8 Stunden im Kühlschrank bei +4° C aufbewahrt
und in der halben Gewichtsmenge Dimethylsulfoxyd aufgelöst
und versponnen.
Bei den letzten Versuohen (17 - 19) wurden Spinnköpfe mit
mehreren Bohrungen verwendet. Sie hatten 12 in zwei kon¬
zentrischen Reihen angeordnete Bohrungen mit Durchmessern
von 0,8 mm und je 4 mm Länge, bzw. 18 Bohrungen in 3 kon¬
zentrischen Reihen mit Durchmessern von 0,3 mm. Die Ein¬
mündungen in die einzelnen Düsen waren konisch ausgebohrt.
Durch Anwendung dieser neuen Spinnköpfe war es möglich, ei¬
nen niultifilen endlosen Faden herzustellen.
4.3.1.3. Spinnvorgang
Das filtrierte Kollagensol wurde in den Spinnbehälter ge¬
füllt und durch eine Düse mit einem Durohmesser von 1,5 mm
ins Fällbad gepresst und zu monofllen Fäden versponnen.
In den Versuohen, bei denen der Spinnbehälter nicht sofort
nach der Filtration gefüllt werden konnte und das Sol in
Gelzustand überzugehen begann, wodurch eine Viskositätser¬
höhung eintrat, musste der durchsichtige Spinnbehälter mit
der Kollagenmasse zentrifugiert werden, um das Material
von Blasen zu befreien. Diesem Zwecke diente eine grössere
Laboratoriumszentrifuge, in welcher der erwünschte Effekt
nach 10 - 20 Minuten bei 3000 U/min, erreioht wurde.
Das Fällbad: Wie sohon die Lösungsversuohe von aufgequolle¬
nem Kollagenmaterial zeigten, besitzt Aoeton starke Koagu-

-114-
lationsfahigkeit fur das Kollagenmaterial. Ebenso zeigen
niedrige Alkohole gleiche Effekte, wirken aber nicht so
schnell.
Als Fallbad diente ein Glasbehalter, der mit 3 1 frisch
destilliertem Aceton gefüllt wurde. Die Versuche haben
aber gezeigt, dass das Fallbad unbedingt alkalisch sein
muss, um eine Neutralisation dee sauren Kollagenmasse her¬
vorzurufen. Dabei hat sich Ammoniak am besten bewahrt,
weil es fluchtig ist, so dass der Ueberschuss in den neu¬
tralisierten Faden nicht zurückbleibt.
Als minimale AmiLoniakmenge wurden 0,2 Gew.% gefunden. Das
Bad ist nach der Verspinnung von 2 kg Spinnmasse ersohopft
und nuss regeneriert werden. Das Dimethylsulfoxyd ist we¬
gen seines hohen Siedepunktes leicht zu trennen. Bei 13 mm
Hg destilliert das Dimethylsulfoxyd bei 77° C über.
Die Ausstossgeschwindigkeit des monofilen Fadens betrug
10 cm /min., der formierte Faden musste jedoch noch min¬
destens 30 Minuten im Bad liegen, um eine gute Durchsetzung
des dicken Fasergefuges mit Badflussigkeit zu erreichen
und eine gleichzeitige Härtung des Fadens durch Alkali zu
gewährleisten.
Durch Anwendung von Spinnkopfen oit mehreren Bohrungen
(Probenummern 17 - 19) war es möglich, die Spinngeschwindig-
keit auf das Doppelte zu erhohen (20 cm /min.) und die
Härtung im Bad auf 15 Minuten zu verkurzen.
4.3^2. Versucheji.it Kalbsblosse-Abfallen als Ausgangsstoff
4.3.2.1. Alkalischer Aufschluss
380 g Kalbsblosse-Abfalle wurden im 3 1-Becher mit 1500 ml
Kalkmilch mit 15 g CaO/l behandelt. (Spez.Gew. der Kalk¬
milch betrug 1,014). Der Wassergehalt von frischen Abfallen
betrug am Anfang 60 %.
Die Abfalle wurden genau 4 Wochen in Kalkmilch bei 20° C
eingelegt und einmal wöchentlich gekehrt und durchgerührt.

-115-
Naoh Ablauf der Behandlungsdauer wurden die Abfalle reich¬
lich mit kaltem Wasser gespült und nach dem Abtropfen des
anhaftenden Wassers gewogen. Die Wassergehaltszunahme betrug
danach plus 88 g, und das alkalisoh aufgequollene Material
enthielt 67,5 % Wasser (Entsprechend 32,5 # Trockensubstanz).
Anschliessend erfolgte die Neutralisation mit verdünnter
Salzsäure während einer Stunde; dann wurde das Material mit
kaltem, enthärtetem Wasser gründlich gespült, bis der pH-Wert
des Spülwassers auf 2,5 gestiegen war.
In der 5-fachen Menge dieser Spüllösung wurde das Material
weitere acht Stunden in einem gut verschlossenen Glasgefäss
aufbewahrt. Dabei erfolgte eine weitere Quellung des Mate¬
rials mit steigender Wasserzunähme. Das Material hatte da¬
nach nochmals 132 g zugenommen, so dass der Gehalt an Trocken¬
substanz nur noch 25,4 % betrug.
Für die Neutralisation des Materials hat sich verdünnte
Salzsäure sehr gut bewährt, weil bei der Neutralisation das
gebildete Calciunichlorid in Wasser leicht löslich und damit
schnell ausspülbar ist.
Es wurde versucht, das alkalisch vorgequollene Material in
ca. 1,5 z 1,5 cm grosse Stücke zu schneiden und in dieser
Form weiter zu neutralisieren, bzw. nachquellen zu lassen.
In zwei Versuchen (35 und 36) wurde das alkalisch vorge¬
quollene Material (mit 32,7 % Trockensubstanz) im Mixer zu¬
erst fein zerteilt und in dieser Form mit verdünnter Salz¬
säure neutralisiert und nachgequollen. Das nachgequollene
Material wies hierauf 26 % Trockensubstanz auf. Dies steht
in Uebereinstimœung mit den Ergebnissen von anderen Ver¬
suchen; hier ist das Nachquellen in der Masse jedoch gleich-
massig erreicht. Dieses in Dimethylsulfoxyd gelüste Mate¬
rial lässt sich sehr leicht und rasch zu Fäden verspinnen.

-116-
Dle Spinnversuchei Das mit verdünnter Salzsäure naehgequolle-
ne Kollagenmaterial wurde mittels Dimethylsulfoxyd Im Mixer
aufgelöst. Die Lösungen wurden in verschiedenen Konzentratio¬
nen hergestellt, wobei die Lösungen mit 8,5 % Kollagensub¬
stanz die besten Resultate ergaben.
Charakteristisch für diese Versuchsreihe war das leichte
Verspinnen der verdünnten Spinnlösungen, und bei optimalen
Bedingungen konnte man eine Spinngeschwindigkeit von 30 cm /
min. erreiohen.
Der Streokungsgrad erreichte in einzelnen Fällen bis zu
300 %. Die Nachhärtungsdauer im Fällbad betrug im Durchschnitt
15 min. Als Fällbad diente Aceton, das 3 % NH40H (25%ig) ent¬
hielt. Die Temperatur des Fällbades betrug ca. 20° C.
Die frischen Lösungen in Dimethylsulfoxyd waren klar und
Hessen sich in diesem zustand leicht filtrieren und zen-
trifugieren. Nach kürzerer oder längerer Zeit gingen diese
kolloidalen Lösungen in den Gelzustand über, in welchem sie
sehr schwer oder überhaupt nicht zu filtrieren sind. Dage¬
gen ist das Verspinnen nur Im Gelzustand möglich. Deswegen
mussten die verdünnteren Spinnlösungen nach dem Filtrieren
und Zentrifugieren im Kühlschrank gekühlt und erst an-
schliessend versponnen werden.
Es muss betont werden, dass eine zu lange Behandlung im
Mixer einen stärkeren Mizellenabbau zur Folge hatte, was
natürlich die Reissfestigkeit der gesponnenen Fäden herab¬
setzte. Dies wurde schon in der Versuchs-Serie mit Rinder¬
därmen als Ausgangsstoff (versuche Nr. 18 und 19) festge¬
stellt, wobei die beiden Spinnlösungen unter gleichen Be¬
dingungen hergestellt wurden, nur Probe Nr. 19 wurde längerim Mixer behandelt, so dass man die Lösung mit einem sehr
feinen Sieb filtrieren und durch einen Spinnkopf mit feinen
Bohrungen (18 x 0,3) verspinnen konnte. Daduroh entstand
eine geringere Reissfestigkeit, obwohl eine grössere erwar¬
tet wurde.

-117-
In der gleichen Versuchsreihe wurde auch eine Probe mit
Formamid als Lösungsmittel durchgeführt (Nr. 36).
Die Arbeitsbedingungen waren gleich wie bei der Probe Nr. 26.
Die Verspinnung war schwer und sehr langsam (l cm /min.),
und die hergestellten Fäden waren nicht gleichmässig.
4.3.2.2. Saurer Aufschluss
70 von frischen Kalbsblösse-Abfallen wurden in 600 ml ver¬
dünnter Essigsäure bei Einstellung des pH-Wertes auf 2,5
24 Stunden quellen gelassen. Die Gewichtszunahme der Ab¬
fälle nach dem Quellungsvorgang und Abtropfen betrug 23,5 g,
so dass der ursprungliche Trockensubstanzgehalt von 40 %
auf 30 % sank.
Die Spinnversuche: Es wurden die Losungen verschiedener
Konzentrationen hergestellt, wobei die Lösungen mit 7,5
bis 10 % Trockensubstanz die besten Resultate ergaben.
Im Rahmen dieser Versuche wurden auch Proben mit Dimethyl»
sulfoxyd als Lösungsmittel vorgenommen, mit Ausnahme der
Proben Nr. 39 und 37, fur welche eine Mischung von Di-
methylsulfoxyd und Wasser (1:1), bzw. Wasser allein, ge¬
wählt wurde.
Um multifile Fäden herzustellen, wurden Spinnkopfe mit
mehreren Bohrungen (12 x 0,8 und 30 x 0,5) verwendet.
Je nach Spinnbedingungen konnten die einzelnen dünnen Fäden
vollständig getrennt (Abb. 4, Probenuacer 35) oder teilweise
oder vollständig zusammengeklebt zu einem Fadenstrang ausge¬
sponnen werden, was aus den nachfolgenden Abbildungen 20
und 21 ersichtlich ist.

-118-
Abb. 20
Multifile Kollagenfaser mit teilweise
verklebten Einzelfaden (Probenumnier 34)
(Vergrbsserung 70 x)
Abb. 21
Multifile Kollagenfaser mit vollständigverklebten Einzelfaden (Probenummer 32)
(Vergrosserung 70x)

-119-
4.4. Gerbungsversuohe mit Formaldehyd
Die Gerbungsversuche wurden folgendermassen durchgeführt:
10 g Kollagenfasern wurden zuerst mit 5 ml Wasser von
35° C übergössen und im verschlossenen Glasgefass mit
Hilfe einer Schüttelmaschine 10 Minuten im Wasserbad bei
35° C behandelt. Nachträglich wurde die Gerbbrühe, bestehend
aus 5 ml Wasser, 0,2 - 0,4 ml HCHO (40%ig) und 0,2 g NaCl,
zugegeben. Die Behandlung dauerte 30 Minuten bei gleicher
Temperatur. Anschliessend erfolgte Nachgerbung durch all¬
mähliche Zugabe einer 10%igen Natriumblcarbonat-Lö'sung, die
in kleinen Portionen eingeführt wurde. Insgesamt wurden 2 ml
der Lösung verbraucht. Die Proben wurden über Nacht trocknen
gelassen und am nächsten Tag noch 15 Minuten in einem leicht
ammoniakalischen Spülbad bewegt.
4.5. Weichaaohung
Als Vorversuch wurde eine übliche Ledertettung vorgenommen:
10 g Kollagenfasern wurden mit 2 g einer Fettmisohung,
bestehend aus 40 % Talg und 60 % Tran, in ein Glasgefass
gebracht und verschlossen. Das Gefäss wurde darauf in die
Klemme eines Schüttelapparates eingespannt und im Wasserbad
bei 55° C 2,5 Stunden bewegt. Der Versuoh wurde nochmals
wiederholt, wobei die Behandlung 5 Stunden dauerte und das
Fett nicht auf einmal, sondern in 5 Fortionen zu je 0,4 g
der erwähnten Fettmischung zugegeben wurde.
Das Resultat beider Versuche fiel negativ aus, da die Fasern
trockener und spröder geworden waren, was das Eindringen
des Fettes in das Innere der Faser verunmöglichte.
Auf Grund dieser Erfahrungen wurden sämtliche weiteren
Weichmachungsversuche mit Fettlickern durchge¬
führt .
Es wurden folgende Fettlioker verwendet:

-120-
(auf 100 Teile Kollagenfasern)
1. 3 T. Klauenbl
6 T. Turkischrotol
1 T. Kaliseife
190 T. Wasser
2. Turkischrotol in verschiedenen Mengen (4 - 20 T.)
3. Glycerin in einer Menge von 10 - 20 T.
Durchfuhrung: Je 1 g der neutralisierten Kollagenfaden wurde
in Liokergefasse von 10 ml Inhalt gebracht und zuerst mit
10 nil Wasser von 30° C übergössen, verschlossen und im
Trockenschrank 15 Minuten stehengelassen. Dadurch wurden
die Fasern leicht aufgequollen und fur den elf entlichen
Lickerversuch vorbereitet. Die übriggebliebene Wasserinenge
wurde darauf abgegossen und der auf 32 C erwärmte Licker
zugesetzt. Die Lickergefasse wurden nun rasch verschlossen,
in mit Gummidichtung versehende Klemmen des Schuttelapparates
eingespannt und in ein auf 40° C thennostatiertes Wasser¬
bad eingetaucht. Die Dauer der Bewegung betrug 30 Minuten.
Die besten Resultate ergab Turkischrotol, wobei neben dem
Weiohmachungseffekt eine erhebliche Zunahme der Reissfestig¬
keit festgestellt wurde. Eine weitere Streckung der Fäden
konnte bei genügender Menge des absorbierten Turkischrotols
beobachtet werden.
In grösserer Menge gebrauchtes Glycerin ergab eine gute
IVeiclmachungsWirkung, jedoch nahm dadurch die Reissfestig¬
keit ab.
4.6. Physikalische Eigenschaften der hergestellten Fasern
4.6.1. Mechanische Eigenschaften
Die Festigkeits- und Dehnbarkeitsmessungen wurden auf dem
elektronischen Dynamometer "Zwick" ausgeführt in Ueberein-
stimmung mit SNV 97411 und SNV 97432.
Das Testmaterial wurde bei einer relativen Feuchtigkeit von
65 •% und 20 - 21° C konditioniert.

Monofil-Fäden
Bemerkungen
36,3
16,2
17,0
(°A)
Dehnung
4,5
0,50
5,0
0,56
4,3
0,48
(RKH)
(g/den.)
Festigkeit
--
-
Weichmachung
--
-
Werbung
20°C
Aceton
OH
NH
3#
+
2Öfe
Aceton
20°C
Aceton
Fällbad
Dirnethylsulfoxyd
Lösungsmittel
--
-
Jüachquellung
bzw.
Neutralisation
Stunden
24
pH9lU5?5
Essig-
6#-i
ge..
Stunden
24
2,5
=pH
HCl
verd.
Direktsauerverfahren
Aufschluss
Rinderdarm
Ausgangsstoff
54
31"
Probenummern
en
Kollagenfäd
hergestellten
Rinderdarm
aus
der
Werte
Mechanische
XXI
TABELLE
Monofil-Fäden
Bemerkungen
6,6
19,5
(%)
Dehnung
3,6
0,4
2,7
0,3
(RKH)
(g/den.)
Festigkeit
--
Weichmachung
--
Gerbung
20°Q
Aceton
55°C
Luft
Fällbad
Wasser
Lösungsmittel
Stunden
8
3,0
=pH
HCl
verd.
Naehquellung
bzw.
Neutralisation
Wochen
8
CaO/1
g15
Verfahren
Alkalisches
Ausschluss
Unterhautgewebe
Ausgangsstoff
21
1Probenummern
Kollagenfäden
gestellten
her¬
Unterhautgewebe
aus
der
Werte
Mechanische
XX
TABELLE

Monofil-Fäden
Bemerkungen
21,3
29,7
25,2
17,7
19,4
27,2
20,4
(#)
Dehnung
5,4
0,60
8,3
0,92
11,3
1,14
6,6
0,73
5,4
0,60
4,5
0,50
5,9
0,65
(REH)
(g/den.)
Festigkeit
-
Glycerin
%10
--
--
-
Weichmachung
-
(40tf-ig)
HCHO
4%
--
--
Gerbung
20°C
Aceton
NH40H
3?»
+
20°C
Aceton
20°G
Aceton
NH^OH
3#
+
20°C
Aceton
Fällbad
Dimethylsulfoxyd
Lösungsmittel
Stunden
82.5
=pH
Malonsäure
0,3#-ige
Stunden
8
2,5-3
=pH
Essigsäure
èft-ige
--
-
Nachquellung
bzw.
Neutralisation
Wochen
4
Ca0/1
g15
Stunden
24
2,2
=pH
Malonsäure
ige
0,3?
6-
Stunden
24
2,5
=pHsäure
Essig¬
6?S-ige
Verfahren
Alkalisches
Direktsauerverfahren
Aufschluss
in
ra
dr
ed
Ausgangsstoff
12
»10
r~"
ni
R
I_"ZiT17~I_T.""vr."
Probenummern
(Fortsetzung)
XXI
TABELLE

cgj=g=-=:==j—m-—«gras
0,3
x18
0,8
x12
12x0,8
:Spinndüse
Fäden
-Multifll
Fäden
-Monofil
Bemerkungen
16,2
19,0
21,5
22,5
16,3
17,3
24,3
(£)
Dehnung
8,7
0,96
16,6
1,84
16,0
1,78
7,9
0,88
7,2
0,80
6,3
0,70
10,8
1,20
(RKM)
(g/den.)
Festigkeit
Glycerin
205«
rotöl
Türkisch
455
K-Seife
15«
rotöl
Türkisch-
65*KLauenöl
3%
Weichmachung
--
-
(40*ig)
HCHO
4#
--
(40#ig)
HCHO
45<
Gerbung
NH4OH
5«3
+
20°C
Aceton
Fällbad
Dimethylsulfoxyd
Lösungsmittel
Stunden
8
2,5
=pH
HCl
verd.
Nachquellung
bzw.
Neutralisation
Wochen
4
CaO/1
g15
Verfahren
Alkalisches
Aufschlußs
Rinderdarm
Ausgangsstoff
19
18
17
16
15
14
|13'
Probenummern
(Fortsetzung)
XXI
TABELLE

-124-
Eelastung
g/den
10 30 40 50
% Dehnung
DIAGRAMM 8
Kraft-Dehnungsdiagranim von regenerierten Kollagenfasernaus Rinderunterhaut (Frobenummern 1 und 2) und aus
Rinderdami als Ausgangsstoff (Probenuramern 3 - 19).

Fällbad
im
Streckung
300%-ige
zus.geklebt
teilw.
Einzelfàden
zus.geklebt
Einzelfäden
zus.geklebt
Einzelfäden
Bemerkungen
8,0
23,2
7,8
12,1
34,0
8,0
18,8
(5«)
pehnung
19,5
2,16
:
3,8
0,42
4,9
0,54
2,4
0,26
4,0
0,44
3,4
0,38
(RKM)
(g/den.)
Festigkeit
--
6,3
0,70
--
--
Weichmachung
--
--
--
-
Gerbung
0,5
x30
0,8
x12
Spinndüse
NH40H
5«3
+
20°C
Aceton
r'ällbad
10,2
12,7
)-
i.d.Lsg(£
Gehalt
Kollagensubstanz
Dimethylsulfoxyd
Lösungsmittel
Stunden
8
2,5
=pH
HCl
verd.
Nachquellung
bzw.
Neutralisation,
Wochen
4
CaO/1
g15
Verfahren
alisches
Alk
Aufschluss
albsblösse-Abfälle
KAusgangsstoff
26
25
24
23
22
21
20
Probenummern
Kollagenfäden
hergestellten
Kalbsblösse-Abfällen
aus
der
Werte
Mechanische
XXII
TABELLE

Knotenfest
[zus>Reklebt
Streckungüinzelfäden
.
^r16
Knotenfest,
n,
Knotenfest
Knotenfest
Knotenfest
Bemerkungen
78,0
65,3
7,1
73,0
27,6
39,1
56,2
(%)
Dehnung
7,2
0,8
9,0
1,0
3,0
0,33
12,4
1,38
11,7
1,30
7,1
0,79
5,1
0,57
(RKM)
(g/den.)
Festigkeit
Türkischrotöl
5«10
$>15
-Türkischrotöl
155«
Glycerin
$>20
-
Weichmachung
--
-HCHO
254
--
~Gerbung
0,8
x12
0,5
x30
Spinndüse
OH
NH
35«
+
20°C
Aceton
Fällbad
7,25
8,45
i.d.Lsg(jÉ)
Gehalt
Kollagensubstanz-
Dimethylsulfoxyd
Lösungsmittel
Stunden
8
2,5
=pH
HCl
verd.
Nachquellung
bzw.
Neutralisation,
Wochen
4
CaO/1
g15
iVerfahre]
Alkalisches
1Aufschluss
Kalbsblösse-Abfälle
Ausgangsstoff
33
32
31
30
.29
28
27
Probenummern
(Fortsetzung)
XXII
TABELLE

|Knotenfest
Nachquellung
der
vor
verteilt
Fein
Bemerkungen
14,1
17,8
10,2
1,5
11,0
11,1
18,0
(#)
Dehnung
6,5
0,72
3,3
0,37
5,3
0,58
2,9
0,32
6,3
0,70
11,4
1,27
17,4
1,94
(RKM)
(g/den.)
Festigkeit
--
--
-
-
rotöl
Türkisci
103t
Weichmachung
HCHO
25«
--
--
--
Gerbung
0,5
x30
0,8
x12
0,5
x30
Spinndüse
NH40H
35«
+
20°C
Aceton
Fällbad
11,0
12,8
8,45
10,4
i.d.
Lsg.
(5«)
Gehalt
Kollagensubstanz-
sulfoxyd
Dimethyl¬
(1:1)
H20
+
sulfoxyd
Dimethyl¬
sulfoxyd
Dimethyl¬
Wasser
Formamid
Dimethylsulfoxyd
Lösungsmittel
--
--
Stunden
8
2,5
=pH
HCl
verd.
Nachquellung
bzw.
Neutralisation,
Stunden
24
2,5
=pHEssigsäure
verd.
Wochen
4
CaO/1
g15
Sauerverfahren
Direktes
Verfahren
Alkalisches
Ausschluss
Kalbsblösse-Abfälle
ff
ot
sgangs
Aus
40
39
38
37
36
35
34
Probenummern
(Fortsetzung)
XXII
TABELLE

Fällbad
im
Streckung
350#-ige
Bemerkungen
14,6
11,8
22,3
7,0
12,3
12,0
12,1
(#)
Dehnung
13,4
1,48
16,7
1,85
12,6
1,4
9,7
1,08
21,2
2,34
9,0
1,0
5,9
0,66
(RKM)
(g/den.)
Festigkeit
--
--
--
-Weichmachung
HCHO
5«4
HCHO
5«2
[Gerbung
0,5
x30
Spinndüse
NH40H
35«
+
20O.C
Aceton
Fällbad
8,55
10,0
11,0
i.d.Lsg(50
Gehalt
Kollagensubstanz-
Dimethylsulfoxyd
Lösungsmittel
--
--
--
-
Nachquellung
bzw.
Neutralisation
Stunden
24
2,5
=pH
Essigsäure
verd.
ne
Sauerverfahr
ektes
Dir
Aufschluss
albsblösse-Abfälle
Ki
Ausgangsstoff
47
46
45
44
|43
42
41
Probenummern
(Fortsetzung)
XXII
TABELLE

-129-
Jtlaatung
ao
DIAGRAMM 9
Kraft-Dehnungsdiagramm der aus Kalbablösse-Abfällen
hergestellten Kollagenfasern

-130-
4.6.2. Ronts;enograp_hische_Untersuchung
Die Röntgenaufnahmen der aus Rinderdarmen (Probenuiuiier 17)
und dus Kalbsblosse-Abfallen (Probenunimer 43) hergestell¬
ten regenerierten Kollagenfdsern wurden unter den gleichen
Bedingungen wie bei der Polyvinyli-yrrolidonfaser aufge¬
nommen (Exp. Teil, Kap. 1.4.2.), nur wurde der Abstand Prà-
paratmitte-Filmmitte auf 3,82 cm eingestellt.
Die erste Röntgenaufnahme (Abb. 22) weist schwache Inter¬
ferenzen auf. Der Anteil an kristalliner Substanz erscheint
relativ gering. Ein Teil der Interferenzen bildenden Sub¬
stanz zeigt deutliche Orientierung längs der Faser.
Bei der anderen Aufnahme (Abb. 23) sind die kristallinen
Anteile etwas grosser als bei der ersten Aufnahme (Abb. 22).
(Die Schwärzung links aussen ist ^uf eine Stdrreflexion
zurückzuführen.)
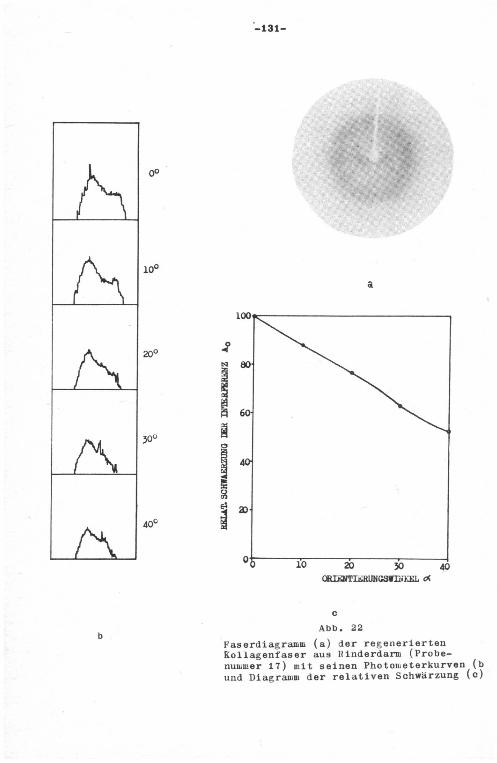
-131-
J\
/\
r\
0°
10°
20°
30°
40°
100
10 20 }0 40
ORnSTIriRUNGSWEiKEL <*
o
Abb. 22
Faserdiagramm (a) der regenerierten
Kollagenlaser aus Rinderdann (Probe-
nun.mer 17) mit seinen Photon,eterkurven (b
und Diagramm der relativen Schwärzung (o)

-132-
8 S S 8
zfcraaraaua vm ounzHTïvraoe "inas
Abb. 23
Faserdiaçranim (a) 1er regenerierten Kolla¬
genfaser aus Kalbsblosse-Abfallen (Probe-
nuc.ii er 43) n.it seinen OhotoUieterkurven (b)
und Diagrau-iD Jer relativen Schwärzung (c)
o
Ä

-133-
Mein verbindlichster und herzlichster Dank gehört
der Firma ETHICON, Inc. Somerville, N.J., fur die
Unterstützung dieser Arbeit,
Herrn PD Dr. H.-G. Elias fur die Molekulargewichts-
bestimniungen,
der Firma Société de la Viscose Suisse, Emmenbrucke,für die mechanischen Messungen,
der Eidgen. Materialprufungs- und Versuchsanstalt,Abt. C, St. Gallen, für die umfangreichen mechani¬
schen Messungen, sowie fur die rontgenographischenund photographischen Aufnahmen
und Herrn H. Muller, Chef der mechanischen Werkstatt
der tech.-ehem. Abt. der ETH, und Herrn F. Aebersold
fur den Bau der notigen Apparate.

-134-
ZÜSAMMENFASSUNG
Es wurde versucht, hydrophile Fasern aus synthetischen und
natürlichen Polymeren nach verschiedenen Verfahren herzustellen.
1. Das hochmolekulare Polyvinylpyrrolidon (Luviscol K-90
der Firma BASF) kann nach dem Nasspinnverfahren aus
wassriger Losung und nach dem Trockenspinnverfahren
aus Losungen organischer Losungsmittel versponnen werden.
i
Bei den Spinnversuchen nach dem Nasspinnverfahren hatten
sich 26 - 28#ige Kaliumhydroxydlosungen als gunstigstes
Fallbad und bei den Versuchen nach dem Trockenspinnver¬
fahren Methylenchlorid als geeignetstes Lösungsmittel er¬
wiesen.
2. Hochmolekulare Polyoxymethylene ("Delrin"-Marken der
Firma Dupont) lassen sich nach dem Schmelzspinnverfahren
bei 235 C zu Faden verarbeiten.
3. Die verschiedenen Polyathylenoxydharze ("Polyox"-Marken
der Fina Union Carbide) können nach dem Schmelz-,
Trocken- und dem Nasspinnverfahren in Fasern überfuhrt
werden. Dabei erwies sich ein neues, modifiziertes
Trockenspinnverfahren am vorteilhaftesten, welches nicht
auf der Verdampfung des Losungsmittels, sondern auf dessen
Verdunstung unterhalb der Siedetemperatur beruht. Als
gunstigstes Losungsmittel bewahrte sich dabei eine Mi¬
schung von Methylenchlorid und Xylol (i:i).
4. Auch Kollagen lasst sich zu Faden verspinnen. Es gelang,
ein Verfahren zu entwickeln, wodurch das alkalisch oder
sauer vorgequollene Kollagenmaterial in Dimethylsulfoxyd
gelost und nach dem Nasspinnverfahren in einem alkalisier-
ten Acetonspinnbad zu Faden versponnen werden konnte.

-135-
5. Es wurde versucht, die Wasserlöslichkeit der hergestell¬
ten hydrophilen Fasern durch verschiedene Hydrophobierungs¬
verfahren zu regulieren.
6. Die physikalischen Eigenschaften der hergestellten Fasern
wurden durch Kraft-Dehnungsmessungen und röntgenographische
Untersuchungen ermittelt.

-136-
LITERATURVERZEICHNIS
1) F.P. 165 349
2) H. Götz, Reyon, Zellwolle 9, 772 (1959)
3) F. Morehead, Text.Res.J. 1J, 96 (1947)
4) W.N. H a w o r t h,J.Cheni.Soo. 1926. 89
J.Chen.Soc. 1932. 2270
Helv. 11, 534 (1928)
5) H. Staudinger, "Die hochmolekularen orga¬
nischen Verbindungen", J. Springer Verlag,Berlin (1932)
6) C. N ä g e 1 i,
"Die Micellartheorie", Ostwald's Klassi¬
ker 227, Akad.V. Leipzig (1928)
7) P.H. Hermans ,Makromol. Chem. (>, 25 (1951)
8) P.H. Hermans,"Contribution to the Physios of
Cellulose Fibres", New York (1946), S.70
9)H. Wenderoth, Melliand Textilber. 30, 205 (1949)
10) H. K r ä s s i g ,Makromol. Chem. .10, 1 (1953)
11) E. E 1 ö d,
P. E t z k o r n,
Z. angew. Ch.51.,45 (1938)
12) E. Ott, H.M. S p u r 1 i n,M.W. G r a f f i n
,
"Cellulose and Cellulose Derivates II", Interscience
Publ. Inc., New York (1954) S.946
13) Brit.Pat. 750,325
14) J. Speakman, Melliand Textilber. 3_3, 823 (1952)
15) W.T. A s t b u r y ,Text. Mercury Argus 125, 1036, 1059
16) H. Zahn, Melliand Textilber. 30, 275 (1949)
17) L. Pauling, R.B. Corey u. H.R. Branson
Proc.Nat.Acad.Sci. USA 3J_, 205 (1951)
18) J.B. Speakman, Melliand Textilber. V]_, 580, 658,736 (1936)
19) J. B r i 1 1,
Z. physikal.Ch. B 5_3, 61 (1943)
20) B. Kramer, Melliand Textilber. 36, 919 (1955)
21) L.G. Ray, Jr., Text. Res. J. 2, 144 (1952)
22) W. Happe, Reyon, Zellwolle 2, 286 (1952)
23) 1.0. Hermann u. W. Haehnel, DRP 685 048
24) T. M o t o y a m a ,Kunststoffe 50, 36 (i960)
25) G. Lunde.E. Heen u. E. Oey, Koll.Z. 83 196
~tl938)
26) E.L. Hirst, J.K.N. Jones u. W.O. J o n e a,
Nature Ijl3, 857 (1939)

34)
35)
36)
Brit
G.B.
R.L.
nach
• Pat
C
W
H.G
r
0
467
0 S
r m
F
812
t 0
I
e 1
r ö
-137-
27) J.B. Speakman u. N.H. Chamberlain,J.Soc.Dyers Col. 60 264 (1944)
28) W.T. A s t b u r y ,Nature 1J>5, 667 (1945)
29) E. Schmidt, Melliand Textilber. £4, 178 (1953)
30) Brit.Pat. 15 522
31) DRP 170 051
32) A. Ferretti, Kunstseide und Zellwolle 2_2, 253 (1940)
33) Brit.Pat. 467 704
n,CD. Evans u. A.K. Smith,
Ind.Eng.Chem. 37, 1194 (1945)
"New Fibres from Proteins", Butter-
worth's Sei. Publ., London 1954
h 1 i c h,
in R. PUMMERER: "Chem.
Textilfasern, Filme und Folien",F. Enke Verlag, Stuttgart 1953, S.566
37) W. R e p p e,
"Neue Entwicklungen auf dem Gebiete der
Chemie des Aoetylens und Kohlenoxyds",Springer Verlag, Berlin, 1949
38) W. R e p p e, "Polyvinylpyrrolidon", Verl. Chemie,Weinheim (1954)
39) DRP 737.663 (A.M. 2,265.450)
40) DRP 757.355 (A.P. 2,335.454)
41) H. Fikentscher u.K. Herrle, Mod.
Plastics (3), 23, 157 (1945)
42) H. Fikentscher, Cellulose Chem. 1J3, 58 (1932)
43) C.E. Schildknecht, "Vinyl and Related
Polymers", J.Wiley & Sons, Inc., N.Y.
(1952), S.669
44) L.E. Miller u. F.A. Hamm, J.Phys.Chem. 57,110 (1953)
45) H. Staudinger, H. Johner,R. S i g n e r
,Z. physikal. Ch. A 126, 425 (1927)
46) H. Staudinger, "Die hochmolekularen organi¬schen Verbindungen Kautschuk und Zellu¬
lose", J. Springer Verlag, Berlin, (1932),S. 224
47) H.W. Kohlschütter u. L. Sprenger,Z. physikal. Ch. B 1(5, 284 (1932)
r,
Z. physikal. Ch. B 21, 162 (1933)
,C. r. 49, 813 (1859)
,Bull. soc. ohim. Fr. 29, 530 (1878)
inger u. 0. Schweizer,B. 62, 2395 (1929)
48)
49)
50)
51)
E.
A.
A.
H.
S
w
w
s
a
u
u
t
u
r
r
a
t
t
t
u
e
z
z
d

-138-
52) W. Beuttner u. K. Steiger-Trippi,Schweiz. Apoth. Zeit. 96, 293 (1958)
53) A. Kuntzel u. K. Buchheimer,
Collegium 1930, 215
54) F. Schneider, Collegium 1940, 97
55) W.H. Stein u. E.G. Miller, J. Biol. Chem. 125,
599, (1938T
56) A. Rich u. F.H. Crick, Nature 17J5, 915 (1955)
A. Rich u.F.H. Crick in "Recent Advances
in Gelatine and Glue Research", Ed.G. Stainsby,
Pergamon Press, New York 1958, S. 20
57) P. Doty u.T. Nishihara in "Recent Advances
xn Gelatine and Glue Research", Ed.G. Stainsby,
Pergamon Press, New York 1958, S. 92
58) W. Grassmann, "Handbuch d. Gerbereichetnie u.
Lederfabrikation" Bd. I, T. l, S. 541
59) J. Gross, J.H. Highberger u.
F.O. Schmidt ,Proc.Nat.Aoad.Sci. U.S.A. 40,
679 (1954"5~
60) K. H a n n i g u. J. Engel, Das Leder 1J2, 213 (1961)
61) P.J. F 1 o r y u. E.S. W e a v e r,
J. Amer. Chet. Soc.
82, 4518 (1960)
62) F. Hofmeister, Naunyn-Schmiedeberg Arch. exp.
Pathol. Pharmakol.
2_4, 247 (1888)
2J_, 395 (l890)
63) C. N e u b e r g ,Bioch. C. J_6, 107 (1916)
64) DRP 650 887
65) DRP 652 874
66) Oe-P. 188 858
67) Oe.P. 189 343
68) G. Coppa-Zuccari, Rayon a. Synth.Textiles 33,44 (I952"r
69) H.-G. Elias,unveröffentlichte Arbeiten
70) H. Staudinger, "Hochmolekulare org. Verbd.",J. Springer Verlag, Berlin, (1932), S.296
71) Union Carbide Chemicals Co, "Advance Technical Infor¬
mation" F-40528/Mai 1959
72) H.-G. Elias u. E. 'Manner, Makromol. Chem. 40,207 (1960)
73) D.A.S. 1101700/61
74) A.A.S. A 1211/59
75) L.J. Bellamy ,"The Infra-red Spectra of
Complex Molecules", Methuen & Co. Ltd., London, 1959

-139-
LEBENSLAUF
Am 19. Juli 1924 wurde ich in Glina (Jugoslavien) geboren.
Die Volksschule und fünf Klassen des Realgymnasiums besuch¬
te ich in Zagreb und die höheren Klassen in Belgrad (Jugos¬
lavien), wo ich im Jahre 1943 auch die Maturitätsprüfung
bestand. Nach einer durch Krieg und Militärdienst beding¬
ten Unterbrechung von drei Jahren konnte ich mich an der Ab¬
teilung fur Chemie der Technischen Hochschule in Zagreb
immatrikulieren und das Studium ein Jahr spater zuerst in
Prag und dann in Belgrad fortsetzen, wo mir an der Chemi¬
schen Fakultät der Technischen Hochschule im November 1952
das Diplom eines Ingenieurs erteilt wurde. Bis Oktober 1958,
als ich unter Leitung von Herrn Prof. Dr. H. Hopff an der
Eidgenossischen Technischen Hochschule in Zurich die vorlie¬
gende Arbeit beginnen konnte, war ich mit industrieller For¬
schung in Jugoslavien beschäftigt. Nachdem ich im S.S. 1960
die Zulassungsprüfung zur Doktorpromotion abgelegt hatte,
konnte ich im W.S. 1961/62 die vorliegende Promotionsarbeit
beendigen.