in vitro assessment of drug-drug interaction potential of...
TRANSCRIPT

1521-009X/41/3/668–681$25.00 http://dx.doi.org/10.1124/dmd.112.049668DRUG METABOLISM AND DISPOSITION Drug Metab Dispos 41:668–681, March 2013Copyright ª 2013 by The American Society for Pharmacology and Experimental Therapeutics
In Vitro Assessment of Drug-Drug Interaction Potentialof Boceprevir Associated with Drug Metabolizing Enzymes
and Transporters s
Xiaoyan Chu, Xiaoxin Cai, Donghui Cui, Cuyue Tang, Anima Ghosal, Grace Chan,Mitchell D Green, Yuhsin Kuo, Yuexia Liang, Cheri M Maciolek, Jairam Palamanda,
Raymond Evers, and Thomayant Prueksaritanont
Merck Sharp & Dohme Corporation, Whitehouse Station, New Jersey
Received October 15, 2012; accepted January 4, 2013
ABSTRACT
The inhibitory effect of boceprevir (BOC), an inhibitor of hepatitis Cvirus nonstructural protein 3 protease was evaluated in vitro againsta panel of drug-metabolizing enzymes and transporters. BOC, aknown substrate for cytochrome P450 (P450) CYP3A and aldo-ketoreductases, was a reversible time-dependent inhibitor (kinact =0.12 minute21, KI = 6.1 mM) of CYP3A4/5 but not an inhibitor of othermajor P450s, nor of UDP-glucuronosyltransferases 1A1 and 2B7. BOCshowed weak to no inhibition of breast cancer resistance protein(BCRP), P-glycoprotein (Pgp), or multidrug resistance protein 2. It wasa moderate inhibitor of organic anion transporting polypeptide (OATP)1B1 and 1B3, with an IC50 of 18 and 4.9 mM, respectively. In humanhepatocytes, BOC inhibited CYP3A-mediated metabolism of midazo-lam, OATP1B-mediated hepatic uptake of pitavastatin, and both the
uptake and metabolism of atorvastatin. The inhibitory potency of BOCwas lower than known inhibitors of CYP3A (ketoconazole), OATP1B(rifampin), or both (telaprevir). BOC was a substrate for Pgp and BCRPbut not for OATP1B1, OATP1B3, OATP2B1, organic cation transporter,or sodium/taurocholate cotransporting peptide. Overall, our datasuggest that BOC has the potential to cause pharmacokineticinteractions via inhibition of CYP3A and CYP3A/OATP1B interplay,with the interaction magnitude lower than those observed with knownpotent inhibitors. Conversely, pharmacokinetic interactions of BOC,either as a perpetrator or victim, via other major P450s and trans-porters tested are less likely to be of clinical significance. The resultsfrom clinical drug-drug interaction studies conducted thus far aregenerally supportive of these conclusions.
Introduction
Boceprevir (BOC), also known as SCH-503034 (Fig. 1), is an antiviralagent for the treatment of hepatitis C virus (HCV) genotype 1 infections.BOC is specifically designed to inhibit HCV nonstructural protein 3protease (Bacon et al., 2011; Poordad et al., 2011), a serine proteaseessential for HCV polyprotein processing and therefore viral replication(Chen and Tan, 2005). BOC binds covalently yet reversibly to thenonstructural protein 3 protease active site through a ketoamide functionalgroup, and elicits potent inhibitory activity in the replicon system alone(Malcolm et al., 2006) or in combination with interferon a-2b andribavirin (Kwo et al., 2010; Foote et al., 2011; Maddur and Kwo, 2011).BOC is a mixture of two diastereomers, SCH-534128 (active isomer)
and SCH-534129 (inactive isomer), that differ in the stereochemicalconfiguration at the third carbon atom (Fig. 1) from the ketoamide end
of the molecule. There is a rapid interconversion between the twodiastereomers, with about 2-fold higher systemic exposure in favor ofthe active isomer in humans. Biotransformation is a major eliminationpathway for BOC across preclinical species and humans. BOC undergoesextensive metabolism involving both cytochrome P450 (P450) CYP3A4/5-mediated oxidation and ketoreduction by cytosolic aldo-keto reductases(AKR)1C2 and AKR1C3 (Ghosal et al., 2011).Drug-drug interactions (DDIs) caused by changes in pharmacoki-
netics and/or pharmacodynamics may lead to drug-induced toxicity oran altered therapeutic effect of a drug. Pharmacokinetic DDIsattributable to alterations of drug absorption, distribution, metabolism,and excretion due to inhibition or induction of drug metabolizingenzymes and/or transporters (Muller and Fromm, 2011) have beencommonly reported in patients receiving polypharmacy. ManagingDDIs is particularly challenging in the treatment of HCV whenconsidering the concomitant medications commonly prescribed forthis patient population who have varying stages of disease, includ-ing cirrhosis, organ transplantation, and coinfection with human
dx.doi.org/10.1124/dmd.112.049668.s This article has supplemental material available at dmd.aspetjournals.org.
ABBREVIATIONS: AKR, aldo-keto reductase; AUC, area under the curve; AZT, 39-azido-39-deoxythimidine; BCRP, breast cancer resistanceprotein; BOC, boceprevir; BSP, bromosulfophthalein; CCK-8, cholecystokinin octapeptide; CHO-K1, Chinese hamster ovary K1 cells; CsA,cyclosporine A; DDI, drug-drug interaction; E217bG, estradiol-17b-D-glucuronide; EA-SG, ethacrynic acid glutathione conjugate; HCV, hepatitis Cvirus; HIV, human immunodeficiency virus; HLM, human liver microsomes; HPLC, high-performance liquid chromatography; KHB, Krebs-Henseleitmodified buffer; Ko143, 3-(6-isobutyl-9-methoxy-1,4-dioxo-1,2,3,4,6,7,12,12a-octahydropyrazino[19,29:1,6]pyrido[3,4-b]indol-3-yl)-propionic acidtert-butyl ester; LC-MS/MS, liquid chromatography–tandem mass spectrometry; LLC-PK1, pig kidney epithelial cell line; MDCKII, Madin-Darbycanine kidney type II cells; MRP, multidrug resistance protein; NTCP, sodium/taurocholate cotransporting peptide; OATP, organic anion-transporting polypeptide; OCT, organic cation transporter; P450, cytochrome P450; PBS, phosphate-buffered saline; Pgp, P-glycoprotein; TCA,taurocholic acid; TDI, time-dependent inhibition; UDPGA, UDP-glucuronic acid; UGT, UDP-glucuronosyltransferases.
668
http://dmd.aspetjournals.org/content/suppl/2013/01/04/dmd.112.049668.DC1Supplemental material to this article can be found at:
at ASPE
T Journals on June 8, 2018
dmd.aspetjournals.org
Dow
nloaded from

immunodeficiency virus (HIV) (Wilby et al., 2012). Most immuno-suppressants and anti-HIV drugs are known to be substrates and/orpotent inhibitors of important drug-metabolizing enzymes (Jimenez-Nacher et al., 2011), such as P450 enzymes (Lin, 2006; Zhou, 2008;Obach, 2009) and UDP-glucuronosyltransferases (UGT) (Kiang et al.,2005; Zhang et al., 2005). Numerous in vitro and in vivo studies havedemonstrated that these compounds are also substrates, inhibitors, orinducers of various drug transporters, including the hepatic uptaketransporters organic anion-transporting polypeptides (OATP1B1)(SLCO1B1), OATP1B3 (SLCO1B3), and OATP2B1 (SLCO2B1),organic cation transporter (OCT1) (SLC22A1), and the sodium/taurocholate cotransporting peptide (NTCP) (SLC10A1), as well as theefflux transporters MDR1 P-glycoprotein (Pgp, ABCB1), multidrugresistance protein (MRP2) (ABCC2), and breast cancer resistanceprotein (BCRP) (ABCG2) (Griffin et al., 2011).It is increasingly recognized that drug transporters have significant
impact on DDIs by modulating the absorption, distribution, metabolism,and excretion of drugs alone or in interplay with drug-metabolizingenzymes (Giacomini et al., 2010). Hepatic elimination of drugs often isa result of the interplay between hepatic uptake/efflux transporters anddrug-metabolizing enzymes. For instance, hepatic elimination of some3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors such asatorvastatin involves OATP-mediated uptake followed by metabolismby CYP3A4 (Lau et al., 2006; Lau et al., 2007). Furthermore, severalefflux transporters such as Pgp, BCRP, and MRP2 are also localized inthe apical membrane of enterocytes. These transporters, together withthe enzymes in the gut, can also modulate the first-pass effect of orallyadministered drugs and thus influence the manifestation of DDIs.We describe the comprehensive in vitro assessment of the potential of
BOC to cause pharmacokinetic interactions at the levels of drug-metabolizing enzymes and transporters. The evaluations include 1)inhibition of major human P450 and UGT enzymes; 2) inhibition ofmajor human drug transporters (OATP1B1, OATP1B3, OATP2B1, Pgp,BCRP, and MRP2); and 3) impact on the enzyme-transporter interplaybetween CYP3A4 or UGTs and OATP1B in human hepatocytes. Wealso evaluated whether BOC is transported by efflux transporters Pgp,BCRP, MRP2, and hepatic uptake transporters OATP1B, OATP1B3,OATP2B1, NTCP, and OCT1. Where applicable, the results arecompared with clinical DDI observations that have been reported, andthe implications for the potential for DDIs with these enzymes andtransporters are discussed.
Materials and Methods
Materials
[14C]BOC (specific activity 56.3 mCi/mmol; purity 98.2% as measured byhigh-performance liquid chromatography [HPLC]) was synthesized by the
Radiochemistry Department of Merck Research Laboratories (Kenilworth, NJ).Unlabeled BOC, SCH-534128, SCH-534129, SCH-629144, and Ko143 (3-(6-isobutyl-9-methoxy-1,4-dioxo-1,2,3,4,6,7,12,12a-octahydropyrazino[19,29:1,6]pyrido[3,4-b]indol-3-yl)-propionic acid tert-butyl ester) were synthesized bythe Chemistry Department, Merck Research Laboratories. [3H]Estradiol-17b-D-glucuronide (E217bG) and [3H]cholecystokinin octapeptide (CCK-8),[3H]prazosin, and [3H]verapamil were purchased from PerkinElmer Life Sciences(Boston, MA). [3H]Pitavastatin, unlabeled pitavastatin, [3H]taurocholic acid(TCA), and [14C]TEA were purchased from American Radiolabeled Chemicals(St. Louis, MO). [3H]Methotrexate was purchased from Moravek (Brea, CA).[14C]Ethacrynic acid glutathione conjugate (EA-SG) was synthesized by theRadiochemistry Department of Merck Research Laboratories. Prazosin,cyclosporine A (CsA), testosterone, 6b-hydroxytestosterone, cortisone, glucose6-phosphate, glucose 6-phosphate dehydrogenase, NADP, UDP-glucuronicacid (UDPGA), alamethicin, estradiol, 39-azido-39-deoxythimidine, nicardi-pine, diclofenac, thymidine, and labetalol were purchased from Sigma-Aldrich(St. Louis, MO). Bromosulfophthalein (BSP) was purchased from MPBiomedicals (Solon, OH). All other reagents were commercially obtained withthe highest analytical purity grade.
Pooled human liver microsomes (HLM) (26 male and 21 female; Lot 37181)were purchased from BD Biosciences Discovery Labware (Woburn, MA).Cryopreserved human hepatocytes (Lot DAC; pooled from five donors) werepurchased from Celsis IVT (Chicago, IL). Membrane vesicles isolated frombaculovirus infected Spodoptera frugiperda (Sf9) cells containing human BCRP(ABCG2) or MRP2 (ABCC2) were purchased from Invitrogen by Life Tech-nologies (Carlsbad, CA).
Cells
Madin-Darby canine kidney type II (MDCKII) cells, MDCKII cells stablytransfected with human BCRP (MDCKII-hBCRP), Chinese hamster ovary K1(CHO-K1) cells, and CHO-K1 cells stably transfected with human OCT1 (CHO-K1-hOCT1) were obtained from Solvo Biotechnology (Budapest, Hungary)under a license agreement and evaluation agreement, respectively. MDCKII, pigkidney epithelial cells (LLC-PK1), and MDCKII or LLC-PK1 cells expressingcomplementary DNA (cDNA) encoding human MDR1 Pgp (MDCKII-MDR1 orLLC-MDR1) were obtained from the Netherlands Cancer Institute (Amsterdam,The Netherlands) under a license agreement.
OATP1B1 (SLCO1B1), OATP1B3 (SLCO1B3), and OATP2B1 (SLCO2B1)stably transfected MDCKII cells (MDCKII-OATP1B1, MDCKII-OATP1B3,and MDCKII-OATP2B1 cells) were generated as described previouslyelsewhere (Monteagudo et al., 2010).
Inhibition Studies with Human P450 and UGT Enzymes in HLM
Evaluation of BOC as a reversible and time-dependent inhibitor of major P450(CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6,CYP2E1, and CYP3A4/5) and a reversible inhibitor of UGT (UGT1A1 andUGT2B7) enzymes was conducted in HLM.
Reversible inhibition to the human P450 enzymes was conducted asdescribed elsewhere (Madan et al., 2002; Walsky and Obach, 2004; Parkinsonet al., 2011). Briefly, incubations were conducted at 37°C in 0.4 ml of
Fig. 1. Chemical structures of boceprevir (BOC) (A) and its diastereomers SCH-534128 (active) (B), and SCH-534129 (inactive) (C).
Drug Interaction Potential of Boceprevir 669
at ASPE
T Journals on June 8, 2018
dmd.aspetjournals.org
Dow
nloaded from

incubation mixtures (pH 7.4) containing potassium phosphate buffer (50 mM),MgCl2 (3 mM), EDTA (1 mM), an NADPH-generating system, and a probesubstrate at concentrations approximately equal to their apparent Km values, asindicated in Table 1. Both midazolam and testosterone were used as probesubstrates for CYP3A4/5. The Ki value for reversible inhibition of CYP3A4/5was determined using midazolam as the probe substrate (1.5–50.0 mM) withseveral BOC concentrations (2.5–100.0 mM). Reactions were initiated with theaddition of an aliquot of an NADPH-generating system and were performed induplicate. Reactions were terminated at 5 minutes by the addition of acetonitrilecontaining the appropriate internal standard. The internal standards were deuteratedmetabolites of the probe substrates: d4-acetaminophen, d5-7-hydroxycoumarin,d6-hydroxybupropion, d5-N-desethylamodiaquine, d4-49-hydroxydiclofenac, d3-49-hydroxymephenytoin, d3-dextrorphan, d2-6-hydroxychlorzoxazone, d3-6b-hydrox-ytestosterone, and d3-19-hydroxymidazolam.
The preincubation-dependent inhibition of BOC was also determined forCYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6,CYP2E1, and CYP3A4/5 by comparison of IC50 values with and without 30minutes of preincubation in the presence of NADPH. The probe substrates andtest concentrations were the same as reversible inhibition conducted withoutpreincubation. To determine kinetic parameters (kinact and KI) for time-dependent inhibition by CYP3A, pooled HLM (1 mg/ml) were preincubated (induplicate) at 37°C with various concentrations of BOC in 100 mM potassiumphosphate buffer (pH 7.4) containing 1 mM EDTA, 6 mM MgCl2, and anNADPH-generating system for 5 to 30 minutes. The incubation mixtures werediluted 10-fold with the same buffer containing 250 mM testosterone (CYP3Aprobe substrate) and an NADPH-generating system. The incubation wascontinued for an additional 10 minutes to monitor the extent of testosterone 6b-hydroxylation.
To determine whether BOC formed a spectrophotometrically detectablemetabolite inhibitory complex with cytochrome P450 (i.e., peaks at approxi-mately 452 nm), an individual HLM sample (H0079, Lot 0010127, XenoTech,LLC) containing high levels of CYP3A4/5 activity (final protein concentration of1 mg/ml, 1.7 nmol P450/mg protein) was added to the sample and referencecuvettes in a buffer mixture consisting of potassium phosphate (50 mM) andMgCl2 (3 mM) for a final volume of 980 ml. Baseline scans from 380 to 520 nmwere recorded on a Varian Cary 100 BIO UV/Vis dual beam spectrophotometer(Varian Analytical Instruments, Palo Alto, CA). BOC was then added to thesample cuvette in 10 ml of methanol for a final incubation concentration of 3 mM.A corresponding volume of the solvent (10 ml of methanol) was added to thereference cuvette. The reactions were initiated with 10 ml of b-NADPH added toboth cuvettes to give a final volume of 1 ml. Continuous scans were conducted everyminute for 15 minutes after the addition of b-NADPH. All scans were conductedat approximately 37°C. Troleandomycin, at a final concentration of 25 mM,
was used as a positive control using the same procedure except that thereference cuvette received a 10-ml aliquot of acetonitrile.
The time-dependent inhibition of CYP3A4/5 (as measured by testosterone6b-hydroxylation and midazolam 19-hydroxylation) was further evaluated bydetermining whether the increased inhibition observed after 30 minutes ofpreincubation with NADPH was reversible after dilution. In this evaluation,BOC (final concentration 3 mM) was preincubated first with HLM (0.05 mg/mlfor midazolam and 0.1 mg/ml for testosterone) for 0, 15, and 30 minutes in thepresence and absence of an NADPH-generating system, without a dilution step.Substrate (100 mM for testosterone and 5 mM for midazolam) was then added,and the incubation was performed for 5 minutes. Second, BOC (0 and 3 mM)were preincubated with HLM (1.25 mg/ml for midazolam and 2.5 mg/ml fortestosterone, which is approximately 25 times the typical incubation con-centration) in the presence of an NADPH-generating system, for 0, 15, and 30minutes. The samples were then diluted 25-fold before being incubated withmarker substrate (200 mM for testosterone and 50 mM for midazolam). Theincubation (at 1/25 the preincubation concentration of BOC and microsomalprotein) was then continued for 5 minutes (to allow formation of any meta-bolites of the marker substrate) and stopped by the addition of the internalstandard and acetonitrile. The residual CYP3A4/5 activity was determined. Inthis study, troleandomycin was used as positive control.
The inhibitory effect of BOC on human UGT1A1-mediated estradiol3-glucuronidation and UGT2B7-mediated 39-azido-39-deoxythimidine (AZT)glucuronidation was evaluated using HLM. Pooled HLM (0.5 mg/ml) wereincubated at 37°C for 20 minutes. For the UGT1A1 assays, the reactionmixtures contained 20 mM estradiol and 0.78 to 100 mM of BOC in 81 mMHEPES buffer (pH 7.0) with 9 mM MgCl2, 5 mM UDPGA, and 25 mg/ml ofalamethicin. Nicardipine was included as positive control inhibitor of UGT1A1(0.78 to 100 mM). For the UGT2B7 assays, the reaction mixtures contained 750mM AZT and 0.78 to 100 mM of BOC in 50 mM potassium phosphate buffer(pH 7.4) with 8 mM MgCl2, 5 mM UDPGA, and 25 mg/ml alamethicin.Diclofenac was included as positive control inhibitor (0.78 to 100 mM). TheUGT reactions were terminated by adding 0.2 ml of ice-cold organic solvent(either methanol or acetonitrile containing 0.3% formic acid for UGT1A1 andUGT2B7 assays, respectively) and thymidine (UGT1A1) or labetalol(UGT2B7) as the internal standards. The samples were spun in a centrifugeat 14,000g for 30 minutes at 4°C. The supernatants were subjected to liquidchromatography–tandem mass spectrometry (LC-MS/MS) analysis.
Covalent Binding of BOC in HLM
Pooled HLM (1 nmol P450/ml) were incubated with [14C]BOC (20 mM) for120 minutes in 0.5 ml of 100 mM potassium phosphate buffer (pH 7.4)
TABLE 1
In vitro evaluation of boceprevir (BOC) as an inhibitor of major cytochrome P450 (P450) and UGT enzymes in human liver microsomes
Enzyme CYP/UGT ReactionSubstrate Concentration
Reversible Inhibition, No PreincubationTime-Dependent Inhibition, 30-Minute
Preincubationa
IC50 (Ki)Inhibition (%) at 100
mMIC50
Inhibition (%) at 100mM
mM mM mM
CYP1A2 Phenacetin O-deethylation 60 .100 22 .100 9.8CYP2A6 Coumarin 7-hydroxylation 0.75 .100 20 .100 7.8CYP2B6 Bupropion hydroxylation 50 .100 2.3 .100 6.9CYP2C8 Amodiaquine N-dealkylation 2.0 .100 25 .100 NACYP2C9 Diclofenac 49-hydroxylation 7.5 .100 3.6 .100 NACYP2C19 S-Mephenytoin 49-hydroxylation 40 .100 25 .100 14CYP2D6 Dextromethorphan O-demethylation 7.5 .100 45 .100 30CYP2E1 Chlorzoxazone 6-hydroxylation 30 .100 NA .100 NACYP3A4/5 Testosterone 6b-hydroxylation 100 .100 41 2.3 6 0.2 95CYP3A4/5 Midazolam 19-hydroxylation 5.0 11 6 1.0 (7.7 6 0.8) 91 1.0 6 0.1 99UGT1A1 Estradiol 3-glucuronidation 20 .100 40 ND NDUGT2B7 39-azido-39-deoxythimidine (AZT)
glucuronidation750 .100 10 ND ND
NA, not applicable (no value was obtained as the rates at the highest concentration of BOC evaluated [100 mM] were higher than the control rates); ND, not determined.a Time-dependent inhibition was determined by comparison of IC50 values with and without preincubation.
670 Chu et al.
at ASPE
T Journals on June 8, 2018
dmd.aspetjournals.org
Dow
nloaded from

containing 3 mM MgCl2 and an NADPH-generating system (0.5 mM NADP,5 mM glucose-6-phosphate, and 1.5 units/ml of glucose-6-phosphate de-hydrogenase). After the preincubation at 37°C for 2 minutes, reactions wereinitiated by addition of BOC. After incubation for 120 minutes, the reactionswere terminated by the addition of 0.5 ml of ice-cold acetonitrile with 1% aceticacid. After vortexing and centrifuging (;10,000g) at 4°C for 10 minutes, thesupernatants were analyzed by HPLC/flow scintillation analyzer. Microsomalpellets were washed 5 times with 0.5 ml of acetonitrile containing 1% aceticacid. After the centrifugation following each washing, the supernatants werecombined and counted for radioactivity. The final pellet was dissolved in 1 mlof BTS-450 (tissue solubilizer; Beckman Coulter, Brea CA), neutralized with6N HCl, and counted for radioactivity.
Uptake and Inhibition Studies in OATP1B1, OATP1B3, OATP2B1, andOCT1 Transfected Cells
Uptake mediated by OATP1B1, OATP1B3, and OATP2B1 was determined inMDCKII cells stably transfected with OATP1B1, OATP1B3, or OATP2B1cDNA, as previously described elsewhere (Monteagudo et al., 2010). OCT1-mediated uptake was measured in CHO-K1 cells stably transfected with OCT1cDNA. For OATP transfected cells, cells were treated with 10 mM sodiumbutyrate (Sigma-Aldrich) for 24 hours to increase the OATP expression beforethe experiment. Cells were dislodged with trypsin EDTA and resuspended inHank’s balanced salt solution plus 10 mM HEPES. Cells were then suspended in96-deep-well plates at a density of 0.6 � 106 cells/well. Uptake was initiated bythe addition of [14C]BOC (1 mM) or the positive control substrates [3H]E217bG(1 mM), [3H]CCK-8 (2.5 nM), [3H]estrone-3-sulfate (0.1 mM), or [14C]TEA (1mM) for OATP1B1, OATP1B3, OATP2B1, and OCT1, respectively. Cells werethen incubated for the indicated time at 37°C, and uptake was stopped by theaddition of ice cold phosphate-buffered saline (PBS), followed by immediatecentrifugation for 1 minute at 3000g at 4°C (Model 5180R; Eppendorf, Hamburg,Germany), and washing of the cell pellets with PBS. Cell pellets wereresuspended in 50% acetonitrile, scintillation fluid (Scintisafe Econo 2; FisherChemicals, Pittsburgh, PA) was added, and radioactivity was determined byliquid scintillation counting in a LS6500 Multipurpose Scintillation Counter(Beckman Coulter). Inhibitory effect of BOC on OATP1B1, OATP1B3, andOATP2B1-mediated uptake was also evaluated in MDCKII cells transfected withOATP1B1, OATP1B3, and OATP2B1. [3H]Pitavastatin (0.1 mM), [3H]BSP (0.1mM), and [3H]estrone-3-sulfate (0.1 mM) were used as probe substrates forOATP1B1, OATP1B3, and OATP2B1, respectively. Uptake of all probesubstrates tested in transfected cells was at least 5-fold higher than in controlcells (unpublished data).
Bidirectional Transport and Inhibition Studies
Bidirectional transport of BOC was assessed in MDCKII, MDCKII-MDR1, andMDCKII-BCRP cells. MDCKII and MDCKII-MDR1 cells were cultured in 96-well transwell culture plates (BD Biosciences, San Jose, CA). [14C]BOC (1 mM)were prepared in Hank’s balanced salt solution with 10 mM HEPES. Substratesolution (150 ml) was added to either the apical (A) or the basolateral (B)compartment of the culture plate, and buffer (150 ml) was added to the compartmentopposite that containing the compound. At 3 hours, 50 ml of sample was removedfrom both sides, and 200 ml of scintillation fluid was added. Radioactivity wasdetermined by liquid scintillation counting in a MicroBeta Wallac Triluxscintillation counter (PerkinElmer). [3H]Verapamil (1 mM) and cyclosporine A(CsA, 10 mM) were used as the positive control substrate and inhibitor for Pgp,respectively. The inhibitory effect of BOC on MDR1 Pgp-mediated [3H]digoxin(0.1 mM) transport was evaluated in LLC-PK1 cells stably transfected with a humanMDR1 Pgp cDNA, as described previously elsewhere (Reitman et al., 2011).
MDCKII and MDCKII-BCRP cells were cultured in 24-well transwellculture plates (BD Biosciences). Twenty-four hours before the experiment, thecells were treated with 10 mM sodium butyrate to increase BCRP expression.[14C]BOC (1 mM) was prepared in Hank’s balanced salt solution with 10 mMHEPES. Substrate solution (500 ml) was added to either the apical (A) or thebasolateral (B) compartment of the culture plate, and buffer (500 ml) was addedto the compartment opposite that containing BOC. The other procedures werethe same as in the Pgp bidirectional transport assay. [3H]Prazosin (5 mM) andKo143 (1 mM) were used as the positive control substrate and inhibitor,respectively.
Vesicular Uptake and Inhibition Studies
Time- and ATP-dependent uptake of BOC was conducted in human MRP2and control vesicles. Membrane vesicles (10 ml) were added to 8-strip tubes(Corning Inc., Corning, NY) at 20 mg/tube. Then 20 ml of [14C]BOC (finalconcentration 2 mM) or [14C]EA-SG (final concentration 2 mM) dissolved intransport buffer (0.25 M sucrose, 10 mM Tris-HCl buffer [pH 7.4], 10 mMMgCl2) were added into the tubes containing vesicles. The mixtures of vesicleand dosing solution were preincubated for 3 minutes at 37°C. Uptake wasinitiated by the addition of 20 ml of ATP-regenerating reagent (5 mM ATP,10 mM creatine phosphate, and 100 mg/ml of creatine phosphokinase intransport buffer) or 20 mL transport buffer, followed by incubation at 37°C forthe indicated time. Uptake was stopped by the addition of 200 ml of ice-coldstop buffer (0.25 M sucrose, 0.1 M NaCl, 10 mM Tris-HCl buffer [pH 7.4])followed by rapid filtration of the reaction mixture onto a prewetted 96-wellglass-fiber type B filter plate (1.0 mm) (Millipore, Billerica, MA). Filterscontaining the membrane vesicles were washed with 200 ml of ice-cold stopbuffer and dried at room temperature, followed by the addition of 25 ml ofscintillation fluid. Radioactivity was determined in a MicroBeta Wallac TriluxScintillation Counter (PerkinElmer). The inhibitory effect of BOC on MRP2- orBCRP-mediated uptake was also evaluated using [14C]EA-SG (2 mM) or[3H] methotrexate (10 mM) as the probe substrate in membrane vesicles containingMRP2 or BCRP, respectively (Chu et al., 2004).
Uptake Studies in Cryopreserved Human Hepatocytes
Uptake of BOC was evaluated as described previously elsewhere (Monteagudoet al., 2010) in cryopreserved human hepatocyte suspension. Briefly, uptake of[14C]BOC into human hepatocytes was determined at 37°C or 4°C, respectively.Cells were resuspended in Krebs-Henseleit modified buffer (KHB) (Sigma-Aldrich) (pH 7.4) in 96-deep well plates (BD Falcon, San Jose, CA) at a densityof 0.2 � 106 cells/well. The cells and dosing solution were preincubated at 37°Cor 4°C for 5 minutes, respectively. Uptake studies were initiated by the additionof 50 mL of [14C]BOC (final concentration 1 mM) or the positive controlsubstrate [3H]E217bG (final concentration 1 mM). The reaction mixtures wereincubated at 37°C or 4°C for the time indicated, and uptake was stopped by theaddition of ice-cold PBS. Other experimental procedures are the same as theuptake assay in transfected cells as described previously. To study sodium-dependent uptake, the uptake of [14C]BOC (1 mM) was determined at 37°C and4°C in KHB and sodium-free KHB at 37°C. [3H]TCA (0.5 mM) was used asa positive control. To measure the kinetic parameters of BOC, the initial uptakerate of [14C]BOC at various concentrations was determined at 0.5 and 3.0 minutesat 37°C. Inhibitory effect of several compounds on initial uptake rate of[14C]BOC (0.5 mM) was measured at 37°C in the presence and absence ofvarious concentrations of the compounds tested.
Inhibition of BOC on Hepatic Uptake and Metabolism in HumanHepatocytes
The inhibitory effect of BOC on the uptake and metabolism of several probesubstrates, including pitavastatin (OATP1B and UGTs), midazolam (CYP3A),and atorvastatin (OATP1B and CYP3A), was evaluated using the same lot ofhepatocytes. For the uptake studies, the impact of BOC on the initial uptake rateof [3H]pitavastatin (1mM) and [3H]atorvastatin (3mM) was studied with humanhepatocytes at 37°C after incubation for 1 and 5 minutes. The substrateconcentrations tested in these studies were well below Km values for pitavastatin(Km = 5 mM; unpublished data) and atorvastatin (Km = 18.9 mM) (Lau et al.,2007) measured in OATP1B1 transfected cells. Other experimental procedureswere as described earlier for the hepatocyte uptake studies.
To assess the effect of BOC on the metabolism of midazolam, pitavastatin,and atorvastatin acid, the formation of their major metabolites, 1-OH-midazolam,pitavastatin-glucuronide, p- and o-OH-atorvastatin acid and atorvastatinlactone, was evaluated in the presence of BOC and positive control inhibitors.Stock solutions of the substrates and inhibitors were prepared in dimethylsulf-oxide followed by dilution with 50% acetonitrile. A 3-ml aliquot of each dilutedstock solution was added to the incubation mixtures to give final substrateand inhibitor concentrations, as indicated later. Human hepatocytes (1.5 or2 million cells/ml) were suspended in William’s E buffer (0.3 ml) containingL-glutamine. Incubations (n = 3 for each inhibitor-substrate pair) were
Drug Interaction Potential of Boceprevir 671
at ASPE
T Journals on June 8, 2018
dmd.aspetjournals.org
Dow
nloaded from

conducted with midazolam (1 mM), pitavastatin (1 mM), or atorvastatin acid (3mM) for 20 minutes at 37°C under a 95% air:5% CO2 atmosphere in theabsence (solvent only) or presence of an inhibitor. The 20-minute reaction timewas chosen to ensure that the inhibitory effects on both OATP1B and CYP3A-mediated metabolite formation could be measured. Ketoconazole (2 mM),rifampin (20 mM), and telaprevir (3 mM) were used as positive controlinhibitors for CYP3A, OATP1B, and both, respectively. In addition, a range ofBOC (0.5–10.0 mM) and telaprevir (0.25–10.0 mM) concentrations was testedto assess the concentration-dependent inhibition on the formation ofmetabolites of atorvastatin. The metabolites formed in the solvent controlincubations were expressed as 100%. Reactions were terminated by theaddition of 0.5 volumes of ice-cold acetonitrile containing the internal standardlabetalol (0.2 or 0.5 mM), followed by vortex-mixing and centrifugation. Theresultant supernatant was analyzed by HPLC-MS.
LC-MS/MS Analysis
Quantification of P450 Enzyme Activity. All P450 probe substratesincluding 19-OH midazolam were analyzed by LC-MS/MS with deuteratedmetabolites as internal standard as described previously elsewhere (Parkinsonet al., 2011).
Quantitation of UGT1A1 Activity. A Sciex API 4000 triple quadrupolemass spectrometer (AB Sciex, Framingham, MA) was used to acquire data.Chromatographic separation was achieved using a Symmetry C18 reversed-phase column (4.6 � 100 mm, 3.5 mm; Waters Corp., Milford, MA) and elutedat a flow rate of 1 ml/min with a mobile phase consisting of A (10% methanolin deionized water containing 0.05% formic acid) and B (10% deionized waterin acetonitrile containing 0.05% formic acid). The gradient conditions were asfollows: 0 to 0.5 minutes at 20% buffer B, 0.5 to 2.0 minutes to 40% buffer B,2.0 to 2.5 minutes to 70% buffer B, 2.5 to 3.5 minutes at 70% buffer B, and 3.5to 3.8 minutes return to 20% buffer B and re-equilibrate until 4.3 minutes with20% buffer B. The instrument was operated in the negative ionization modeusing the electrospray interface, and selected reaction monitoring was used todetermine the specific precursor-ion to product-ion transitions for theglucuronide (447.1/113.0).
Quantitation of UGT2B7 Activity. A Sciex API 4000 triple quadrupolemass spectrometer was used to acquire data. Chromatographic separation wasachieved using a Zorbax SB reversed-phase column (4.6 � 75 mm, 3.5 mm;Waters Corp.) and eluted at a flow rate of 1 ml/min with a mobile phaseconsisting of A (deionized water containing 0.01% formic acid) and B(acetonitrile containing 0.01% formic acid). The gradient conditions were asfollows: 0 to 1.5 minutes at 5% buffer B, 1.5 to 3.0 minutes at 5 to 60% bufferB, 3.0 to 3.5 minutes at 60% buffer B, 3.6 to 4.0 minutes to 95% buffer B, and4.0 to 4.5 minutes return to 5% buffer B. The instrument was operated in thenegative ionization mode using the electrospray interface, and selected reactionmonitoring was used to determine the specific precursor-ion to product-iontransitions for the glucuronide (442.0/125.0).
Quantitation of Metabolites for Midazolam, Pitavastatin, and Atorvas-tatin. LC-MS/MS analysis was performed using a Waters Acquity UltraPerformance LC system coupled to a Waters quadrupole time-of-flight Xevomass spectrometer. HPLC analyses consisted of a Waters Ultra-PerformanceLiquid Chromatography High-Strength Silica T3 column (1.8 mm, 2.1 � 50mm) and a mobile phase with (A) 0.1% formic acid in water and (B) 0.1%formic acid in acetonitrile (constant flow rate of 0.15 ml/min). Hepatocyteincubates were analyzed using the following gradient elution: 0–0.5 minutes,90% A; 7.5 minutes, 5% A; the column was washed at 5% A (1 minute) andequilibrated at 90% A (1 minute) before the next injection. The quadrupoletime-of-flight mass spectrometer was operated under electrospray ionizationpositive-ion mode. The source temperature was set at 100°C while thedesolvation temperature was 600°C. The resolution of the time-of-flightdetection was approximately 8000. Formation of major metabolites ofmidazolam, pitavastatin, and atorvastatin were monitored to assess the effectof inhibitors.
Data Analysis
The Ki value of BOC for the competitive inhibition of CYP3A4/5 wascalculated by eq. 1:
1V¼ Km
Vmax
�1þ ½I�
Ki
�1½S� þ
1Vmax
ð1Þ
where Ki is the inhibition constant; V is the rate of reaction in presence ofinhibitor at a concentration [I] and substrate at a concentration [S]; and Km andVmax are the Michaelis-Menton constants for a given P450 reaction. The datawere fitted in GraFit 4.0 software for Ki determination (Erithacus Software,Horley, United Kingdom). Visual inspection was performed using Eadie-Hofstee plots to confirm the nature of the inhibition.
The first-order rate constants (kobs) of BOC for inactivation of CYP3A4/5 atvarious concentrations were estimated from the initial slopes of a naturallogarithm plot of the percentage of remaining activity versus the preincubationtime. The kinact and KI values were calculated by nonlinear regression analysisof eq. 2 using KaleidaGraph Synergy Software (Reading, PA):
kobs ¼ kinact � ½I�KI þ ½I� ð2Þ
where KI represents the inhibitor concentration that produces a half-maximalrate of inactivation, kinact represents the maximum inactivation rate constant,and [I] is the inhibitor concentration.
The IC50 values for inhibition of metabolism or transporter-mediated uptake/efflux in microsomes and transporter transfected cells were obtained by fittingthe data to eq. 3 by nonlinear regression analysis:
Controlð%Þ ¼ 1001þ I=IC50
ð3Þ
where control (%) represents the metabolism or transporter-mediated uptake/efflux measured in the presence of various concentrations of inhibitor to that inthe absence of inhibitor.
IC50 values for inhibition of uptake in human hepatocytes were obtained byfitting the data to eq. 4:
Controlð%Þ ¼ 1002M
1þ I=IC50þM ð4Þ
where M represents residual control (%) not affected by the inhibitors tested.Prediction of Potential for OATP1B1-Mediated DDIs. The degree of
inhibition of OATP1B1 in humans was estimated by calculating the R value(eq. 5) (Hirano et al., 2006; Giacomini et al., 2010), which represents the ratioof the uptake clearance in the absence of inhibitor to that in its presence:
R ¼ 1þ fu � Iin;max
IC50ð5Þ
where fu represents the blood unbound fraction of the inhibitor, Iin,max representsthe estimated maximum inhibitor concentration at the inlet to the liver, and IC50
was obtained from the in vitro OATP1B1 inhibition study in transfected cell lines.Iin,max was calculated based on eq. 6 (Hirano et al., 2006; Giacomini et al., 2010):
Iin;max ¼ Imax þ Fa � Dose� kaQh
ð6Þ
where Imax is the maximum plasma concentration of the inhibitor, fa is thefraction of the dose of the inhibitor that is absorbed, ka is the absorption rateconstant of the inhibitor, and Qh is the hepatic blood flow rate in humans (1500ml/min). To estimate the Iin,max value, Fa was set at 1, ka was set at 0.03minute–1, and the blood-to-plasma concentration ratio was assumed to be 1.
Kinetic Analysis. Kinetic parameters for uptake of BOC into humanhepatocytes were estimated using nonlinear least-squares data fitting fromeq. 7:
v ¼ Vmax � S
Km þ Sþ Pdif � S ð7Þ
where v is the initial uptake velocity of BOC (pmol/min/106 cells), S is BOCconcentration in the reaction mixture (mM), Km is the Michaelis-Mentonconstant (mM), Vmax is the maximum uptake rate (pmol/min/106 cells), and Pdifis the nonsaturable uptake clearance (ml/min/106 cells).
672 Chu et al.
at ASPE
T Journals on June 8, 2018
dmd.aspetjournals.org
Dow
nloaded from

Results
Evaluation of BOC as an Inhibitor of Major P450 and UGTEnzymes in HLM. Evaluation of BOC reversible inhibition of majorhuman CYPs (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9,CYP2C19, CYP2D6, CYP2E1, and CYP3A4/5) was conducted inHLM with selective probe substrates. We also assessed the reversibleinhibition of BOC to UGT1A1 and UGT2B7, two major UGTisoforms responsible for glucuronidation of most clinically used drugs(Williams et al., 2004).As shown in Table 1, BOC showed minimal inhibition of CYP2B6,
CYP2C9, or CYP2E1. At the highest concentration tested (100 mM),BOC showed only 20–25% inhibition of CYP1A2, CYP2A6, CYP2C8,and CYP2C19, whereas it demonstrated 45% inhibition of CYP2D6.However, its inhibitory effect on CYP2D6, if we assume that IC50 = 100mMis the worst-case scenario, is not likely to be clinically significant, asthe IC50 value was ;33-fold higher than the total plasma Cmax (;3mM) of BOC at the clinically relevant dose. There was also noinhibition of estradiol 3-glucuronidation by UGT1A1 or AZTglucuronidation by UGT2B7 in HLM (IC50 . 100 mM).However, BOC exerted an appreciable inhibition of CYP3A4/5
(measured by midazolam 19-hydroxylation) with an IC50 value of 11 61.0 mM. The inhibition of midazolam hydroxylation was furtherconfirmed to be competitive with a Ki value of 7.7 6 0.8 mM.Interestingly, BOC did not inhibit testosterone 6b-hydroxylation,another functional marker activity for CYP3A4/5 (IC50 . 100 mM).Such substrate-dependent inhibition with CYP3A4/5 has been reportedpreviously for atorvastatin, erythromycin, and quinidine (Obach et al.,2006), and is attributable to the binding of multiple substrates within theactive site of the enzyme (Kenworthy et al., 1999).The CYP3A4/5 inhibition by BOC was also time dependent, as
indicated by the increased inhibitory potency upon 30-minute preincu-bation with NADPH before the addition of CYP3A probe substrates.The IC50 values were 2.3 6 0.2 and 1.0 6 0.1 mM for the inhibitionof testosterone 6b-hydroxylation and midazolam 19-hydroxylation,respectively (Table 1). Time dependency was not observed for any ofthe other P450s tested.Kinetic analysis showed that BOC caused a time-dependent
inhibition (TDI) of CYP3A activity (measured by testosterone 6b-hydroxylation) with kinact and KI values of 0.12 minute21 and 6.1 mM,respectively. Several experiments were conducted to investigatepossible mechanisms for this TDI. BOC did not form a spectropho-tometrically detectable metabolite inhibitory complex with CYP3A4/5, as increases in absorbance between 380 and 520 nm were notobserved in spectral readings of the interactions between BOC and anHLM sample from an individual with high levels of CYP3A4/5activity (Fig. 2A). When HLM were incubated with [14C]BOC (20mM), no radioactivity was detected in the microsomal pellets aftermultiple washings, suggesting that BOC did not show covalentbinding to HLM. Further studies showed that the observed TDImeasured by midazolam 19-hydroxylation was NADPH-dependentand reversible after a 25-fold dilution (Fig. 2, B-D). Similar resultswere also observed when testosterone was used as the probe substrate(unpublished data).
Evaluation of BOC as an Inhibitor for Several Uptake andEfflux Transporters in Transfected Cell Lines or MembraneVesicles. The inhibitory effect of BOC on uptake by humanOATP1B1, OATP1B3, and OATP2B1 was evaluated in MDCKII-OATP1B1, MDCKII-OATP1B3, and MDCKII-OATP2B1 cells. BOCinhibited uptake for OATP1B1, OATP1B3, and OATP2B1 with anestimated IC50 of 18 6 2.4 mM, 4.9 6 1.3 mM, and .50 mM,respectively (Table 2). The inhibitory effects of BOC on several efflux
transporters were also evaluated, including MDR1 Pgp, BCRP, andMRP2. BOC showed no inhibition (,10% decrease in net transport)of MDR1 Pgp-mediated [3H]digoxin (0.1 mM) transport over theconcentration range tested (0.3–300 mM). In membrane vesiclescontaining human BCRP, BOC inhibited ATP-dependent uptake of[3H]methotrexate (10 mM) with an IC50 of 81 6 28 mM (Table 2).Likewise, no inhibition of MRP2-mediated ATP-dependent uptake of[14C]EA-SG (2 mM) was observed (IC50 . 100 mM; Table 2).Evaluation of BOC as a Potential Perpetrator for OATP1B1-
Mediated DDIs. As OATP1B1 is a major contributor to the hepaticuptake of several clinically used drugs such as statins (Shitara andSugiyama, 2006; Muller and Fromm, 2011; Niemi et al., 2011), thepotential for BOC as a perpetrator of inhibition of OATP1B1-mediated hepatic uptake was estimated using R-value analysis (Hiranoet al., 2006; Giacomini et al., 2010). To validate this approach, weassessed OATP1B1 in vitro inhibition by several known OATPinhibitors with various clinical DDI effects as benchmarkingcompounds. These included cyclosporine A (CsA), rifampin, lopinavir,telaprevir, amprenavir, and ritonavir. R values of all tested compoundsat the clinically relevant dose were estimated and compared withclinical DDI data using atorvastatin, pitavastatin, or rosuvastatin asvictim drugs (Table 3). At the clinically relevant dose (800 mg, threetimes daily), the IC50 of BOC for OATP1B1 inhibition (IC50 = 18 62.4 mM) was 18-fold and 2-fold higher than its unbound maximumplasma concentration and unbound maximum concentration at theinlet to the liver, respectively (Table 3). The R value of BOC (R = 1.5at 800 mg, three times daily) was lower than for CsA, rifampin,lopinavir, and telaprevir, which all have been reported to causeclinically significant DDIs with statins (Table 3). On the other hand,the R value of BOC was higher than for amprenavir (600 mg) andritonavir (100 mg), both of which have shown not to cause clinicallysignificant DDIs with rosuvastatin (Table 3).Effects of BOC on Uptake and Metabolism of CYP3A and
OATP1B Substrates in Human Hepatocytes. To further understandthe impact of BOC on hepatic uptake, metabolism, and their potentialinterplay, uptake and metabolism studies were conducted in humanhepatocytes with functional activity of both hepatic uptake trans-porters and drug-metabolizing enzymes. The probe substrates used inthese studies included pitavastatin (OATP1B and UGTs), midazolam(CYP3A), and atorvastatin (OATP1B and CYP3A). For the uptakeexperiments, studies were performed under linear conditions (,5minutes) where metabolism was minimal. As shown in Fig. 3B andTable 4, BOC inhibited hepatic uptake of [3H]pitavastatin (1 mM) withan IC50 of 6.36 2.1 mM. In comparison, ketoconazole, not reported tobe an OATP inhibitor at clinically relevant concentrations, inhibitedpitavastatin uptake with an IC50 of 16.1 6 1.1 mM. In contrast,rifampin, ritonavir, lopinavir, and telaprevir showed more potentinhibition of uptake of pitavastatin with IC50 values of 1.5 6 0.3 mM,1.4 6 0.2 mM, 1.1 6 0.1 mM, and 3.4 6 0.9 mM, respectively(Table 4). Similarly, BOC inhibited uptake of [3H]atorvastatin (3 mM)in human hepatocytes with an IC50 of 7.2 6 1.4 mM, whileketoconazole, rifampin, ritonavir, lopinavir, and telaprevir in-hibited uptake of atorvastatin with IC50 values of 11.9 6 3.1 mM,3.0 6 0.5 mM, 1.0 6 0.1 mM, 1.8 6 0.2 mM, and 4.1 6 0.9 mM,respectively (Fig. 3A; Table 4). Interestingly, estrone sulfate andestropipate, potent prototypical inhibitors for OATP1B1 (Gui et al.,2010), inhibited uptake of pitavastatin (IC50 0.6 6 0.1 mM, 0.8 60.4 mM), but not atorvastatin (IC50 . 50 mM, . 10 mM) (Fig. 3, Aand B; Table 4). This apparent substrate dependency may beattributable to multiple binding sites in OATPs (Noe et al., 2007) orto the contribution of transporters other than OATPs to the hepaticuptake of these drugs.
Drug Interaction Potential of Boceprevir 673
at ASPE
T Journals on June 8, 2018
dmd.aspetjournals.org
Dow
nloaded from
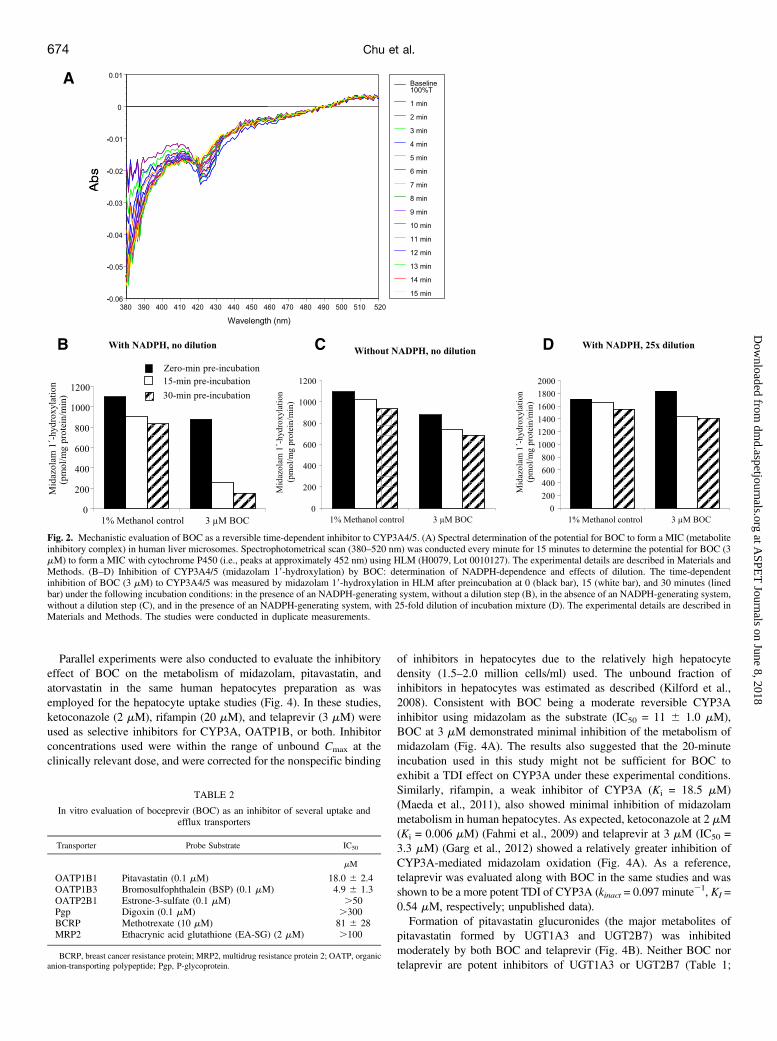
Parallel experiments were also conducted to evaluate the inhibitoryeffect of BOC on the metabolism of midazolam, pitavastatin, andatorvastatin in the same human hepatocytes preparation as wasemployed for the hepatocyte uptake studies (Fig. 4). In these studies,ketoconazole (2 mM), rifampin (20 mM), and telaprevir (3 mM) wereused as selective inhibitors for CYP3A, OATP1B, or both. Inhibitorconcentrations used were within the range of unbound Cmax at theclinically relevant dose, and were corrected for the nonspecific binding
of inhibitors in hepatocytes due to the relatively high hepatocytedensity (1.5–2.0 million cells/ml) used. The unbound fraction ofinhibitors in hepatocytes was estimated as described (Kilford et al.,2008). Consistent with BOC being a moderate reversible CYP3Ainhibitor using midazolam as the substrate (IC50 = 11 6 1.0 mM),BOC at 3 mM demonstrated minimal inhibition of the metabolism ofmidazolam (Fig. 4A). The results also suggested that the 20-minuteincubation used in this study might not be sufficient for BOC toexhibit a TDI effect on CYP3A under these experimental conditions.Similarly, rifampin, a weak inhibitor of CYP3A (Ki = 18.5 mM)(Maeda et al., 2011), also showed minimal inhibition of midazolammetabolism in human hepatocytes. As expected, ketoconazole at 2 mM(Ki = 0.006 mM) (Fahmi et al., 2009) and telaprevir at 3 mM (IC50 =3.3 mM) (Garg et al., 2012) showed a relatively greater inhibition ofCYP3A-mediated midazolam oxidation (Fig. 4A). As a reference,telaprevir was evaluated along with BOC in the same studies and wasshown to be a more potent TDI of CYP3A (kinact = 0.097 minute21, KI =0.54 mM, respectively; unpublished data).Formation of pitavastatin glucuronides (the major metabolites of
pitavastatin formed by UGT1A3 and UGT2B7) was inhibitedmoderately by both BOC and telaprevir (Fig. 4B). Neither BOC nortelaprevir are potent inhibitors of UGT1A3 or UGT2B7 (Table 1;
TABLE 2
In vitro evaluation of boceprevir (BOC) as an inhibitor of several uptake andefflux transporters
Transporter Probe Substrate IC50
mM
OATP1B1 Pitavastatin (0.1 mM) 18.0 6 2.4OATP1B3 Bromosulfophthalein (BSP) (0.1 mM) 4.9 6 1.3OATP2B1 Estrone-3-sulfate (0.1 mM) .50Pgp Digoxin (0.1 mM) .300BCRP Methotrexate (10 mM) 81 6 28MRP2 Ethacrynic acid glutathione (EA-SG) (2 mM) .100
BCRP, breast cancer resistance protein; MRP2, multidrug resistance protein 2; OATP, organicanion-transporting polypeptide; Pgp, P-glycoprotein.
Fig. 2. Mechanistic evaluation of BOC as a reversible time-dependent inhibitor to CYP3A4/5. (A) Spectral determination of the potential for BOC to form a MIC (metaboliteinhibitory complex) in human liver microsomes. Spectrophotometrical scan (380–520 nm) was conducted every minute for 15 minutes to determine the potential for BOC (3mM) to form a MIC with cytochrome P450 (i.e., peaks at approximately 452 nm) using HLM (H0079, Lot 0010127). The experimental details are described in Materials andMethods. (B–D) Inhibition of CYP3A4/5 (midazolam 19-hydroxylation) by BOC: determination of NADPH-dependence and effects of dilution. The time-dependentinhibition of BOC (3 mM) to CYP3A4/5 was measured by midazolam 19-hydroxylation in HLM after preincubation at 0 (black bar), 15 (white bar), and 30 minutes (linedbar) under the following incubation conditions: in the presence of an NADPH-generating system, without a dilution step (B), in the absence of an NADPH-generating system,without a dilution step (C), and in the presence of an NADPH-generating system, with 25-fold dilution of incubation mixture (D). The experimental details are described inMaterials and Methods. The studies were conducted in duplicate measurements.
674 Chu et al.
at ASPE
T Journals on June 8, 2018
dmd.aspetjournals.org
Dow
nloaded from

unpublished data). Rifampin, a known inhibitor for OATP1B, but notUGT1A3 and UGT2B7, displayed the most potent inhibition (.80%),while ketoconazole did not show significant inhibition (Fig. 4B) ofpitavastatin glucuronidation. This is consistent with ketoconazole notbeing a potent inhibitor of OATP1B, UGT1A3, or UGT2B7 at theconcentration tested (2 mM) (Takeda et al., 2006). These resultssuggest that the observed inhibition of pitavastatin glucuronidation byBOC, telaprevir and rifampin (in increasing order) is likely a conse-quence of their inhibition of the OATP1B-mediated hepatic uptakerather than direct inhibition of UGT-mediated glucuronidation.For atorvastatin, a dual substrate for both OATP1B and CYP3A,
BOC showed moderate inhibition of the formation of both oxida-tive (para- and ortho-hydroxylations, both mediated by CYP3A) andtotal metabolites (para- and ortho-hydroxylations and lactone for-mation partly via glucuronidation and other metabolic pathways(Prueksaritanont et al., 2002) to a similar degree (Fig. 4C). Similarly,rifampin equally decreased the oxidative and total metabolism ofatorvastatin, suggesting that under the current experimental conditionsboth BOC and rifampin inhibited the metabolism of atorvastatinmainly by blocking atorvastatin uptake into the hepatocytes. Beinga more potent inhibitor of OATP1B, rifampin inhibited atorvastatin
metabolism (both oxidative and total metabolism) to a greater degreethan BOC. In contrast, ketoconazole and telaprevir exhibited morepotent inhibition toward the atorvastatin oxidative metabolism byCYP3A, as compared with their effect on the total metabolism ofatorvastatin. Additionally, the inhibition of the oxidative metabolismof atorvastatin was strongest with ketoconazole followed by telaprevirand then BOC, in agreement with the rank order of their inhibitorypotency of CYP3A activity as measured by midazolam hydroxylation(Fig. 4A). Interestingly, rifampin, which showed minimal inhibition ofmidazolam metabolism by CYP3A, demonstrated a 20–30% morepotent inhibition of total metabolism of atorvastatin as compared withketoconazole and telaprevir. This further suggested that inhibition byrifampin of the metabolism of atorvastatin was primarily driven by itsinhibition of OATP1B. Across the concentration range tested, BOCshowed less potent and incomplete inhibition of both oxidative andtotal metabolism of atorvastatin as compared with telaprevir (Fig. 4D),consistent with it being a less potent inhibitor of both CYP3A andOATP1B.Transport of BOC by Efflux Transporters Pgp, BCRP, and
MRP2 in Transfected Cell Lines or Membrane Vesicles. InMDCKII control cells, BOC showed low passive permeability (Papp =
TABLE 3
In vitro evaluation of boceprevir (BOC) as a potential perpetrator for OATP1B1-mediated drug-drug interactions
Perpetrator (dose)IC50 (mM)OATP1B1
Cmax, u Iin, max, u R valueVictim (Clinical
DDIs)Clinical DDIs (Fold Increase of AUC) References
mM mM
BOC (800 mg, three times daily) 18 6 2.4 0.79 8.92 1.5 Atorvastatin 2.3 Hulskotte EG et al., 2011CsA (100 mg) 0.3 6 0.1 0.06 0.23 1.8 Pitavastatin 4.5 Shitara and Sugiyama, 2006Rifampin (600 mg) 1.4 6 0.2 0.87 2.47 2.8 Atorvastatin 8 He et al., 2009Lopinavir (400 mg) 0.4 6 0.1 0.3 0.49 2.4 Rosuvastatin 2 Kiser et al., 2008Telaprevir (750 mg) 3.4 6 0.1 1.01 9.84 3.9 Atorvastatin 7.9 Lee et al., 2011Amprenavir (600 mg)a 10.0 6 1.6 1.2 3.57 1.4 Rosuvastatin 1.1 Karlgren et al., 2012Ritonavir (100 mg, twice daily)b 0.8 6 0.2 0.03 0.09 1.1 Rosuvastatin No Busti et al., 2008
AUC, area under the curve; CsA, cyclosporine A; DDIs, drug-drug interactions; OATP, organic anion-transporting polypeptide.a The prodrug fosamprenavir was used in the study.b Coadministered with fosamprenavir.
Fig. 3. Effect of several compounds on the uptake of atorvastatin and pitavastatin in human hepatocytes. Effects of several compounds on initial uptake rate of[3H]atorvastatin (3 mM) (A) and [3H]pitavastatin (1 mM) (B) was evaluated in cryopreserved human hepatocytes. The data are expressed as a percentage of initial uptake ratemeasured at 1 and 5 minutes at 37°C in the presence and absence of inhibitors. The IC50 values for inhibition of atorvastatin and pitavastatin uptake by rifampin (d),telaprevir (s), BOC (j), ketoconazole (u), ritonavir (m), lopinavir (n), estrone sulfate (♦), and estropipate (⋄) are summarized in Table 4. Values shown are mean 6 S.E.for experiments performed in triplicate.
Drug Interaction Potential of Boceprevir 675
at ASPE
T Journals on June 8, 2018
dmd.aspetjournals.org
Dow
nloaded from

4.5 � 1026 cm/s). In both MDCKII-MDR1 (Fig. 5A) and MDCKII-BCRP (Fig. 5B) monolayers, BOC exhibited a greater B-A/A-B ratio(transport from basal to apical [B-A] divided by transport from apicalto basal [A-B]), compared with control cells. This transport wasstrongly inhibited by the Pgp inhibitor CsA (10 mM) and weaklyinhibited by the BCRP inhibitor Ko143 (5 mM), respectively.Verapamil and prazosin, prototypical substrates for Pgp and BCRP,respectively, showed significant higher B-A/A-B ratios in MDCKII-MDR1 and MDCKII-BCRP cells than in control cells, and thistransport was inhibited strongly by CsA and Ko143 (Fig. 5, D and E).
Taken together, these data indicated that despite the endogenoustransport observed in MDCKII cells, BOC was a substrate of MDR1Pgp and BCRP. Similarly, SCH-534128 and SCH-534129, the twodiastereomers of BOC, were also substrates for MDR1 Pgp and BCRP(unpublished data).Similar to control vesicles, uptake of [14C]BOC (2 mM) into MRP2
containing vesicles was not time or ATP dependent (Fig. 5C), suggestingthat BOC was not a substrate of MRP2. [14C]EA-SG (2 mM), a positivecontrol substrate for humanMRP2 (Chu et al., 2004), showed a significantATP-dependent uptake in MRP2 containing vesicles (Fig. 5F), indicatingthe functional activity of MRP2 in these vesicles. SCH-534128 and SCH-534129 were also not substrates for MRP2 (unpublished data).Hepatic Uptake of BOC by Human Hepatocytes and Uptake
Transporters NTCP, OATP1B1, OATP1B3, and OATP2B1, andOCT1. To assess if BOC was a substrate for hepatic uptake transporters,uptake of BOC was determined in cryopreserved human hepatocytes.Uptake of [3H]E217bG (1 mM), [3H]CCK-8 (10 nM), [3H]estrone sulfate(100 nM), [3H]TCA (1 mM), and [14C]TEA (1 mM), prototypicalsubstrates for human OATP1B1, OATP1B3, OATP2B1, NTCP, andOCT1, respectively, showed significant temperature-dependent uptake.This uptake was inhibited by prototypical inhibitors of these transporters(unpublished data). This indicated that the human hepatocytes used in thisstudy had retained the functional activities of these transporters. Asshown in Fig. 6A, the uptake of [14C]BOC (1 mM) into humanhepatocytes was time and temperature dependent. Initial uptake of BOCat 37°C was saturable (Km = 12.4 6 7.4 mM, Vmax = 343 6 150 pmol/
TABLE 4
IC50 values for inhibitory effects of several compounds on uptake of [3H]atorvastatinacid (3 mM) and [3H]pitavastatin (1 mM) in human hepatocytes
CompoundsIC50
Probe Atorvastatin Acid (3 mM) Probe Pitavastatin (1 mM)
mM
Ketoconazole 11.9 6 3.1 16.1 6 1.1Boceprevir 7.2 6 1.4 6.3 6 2.1Rifampin 3.0 6 0.5 1.5 6 0.3Telaprevir 4.1 6 0.9 3.4 6 0.9Ritonavir 1.0 6 0.1 1.4 6 0.2Lopinavir 1.8 6 0.2 1.1 6 0.1Estrone sulfate .50 0.6 6 0.1Estropipate .10 0.8 6 0.4
Fig. 4. Inhibition of the metabolism of midazolam, pitavastatin, and atorvastatin in human hepatocytes. Effects of BOC (3 mM), ketoconazole (2 mM), rifampin (20 mM),and telaprevir (3 mM) on metabolism of (A) midazolam (1 mM), (B) pitavastatin (1 mM), and (C) atorvastatin (3 mM) were evaluated in cryopreserved human hepatocytes.The data were expressed as percentage control of the formation of metabolites. (C) Percentage control of the formation of both oxidative (para- and ortho-hydroxylations)(white bars) and total metabolites (para- and ortho-hydroxylations and lactone formation) (black bars) of atorvastatin were measured. (D) Concentration-dependent inhibitionof BOC (squares) and telaprevir (circles) on both oxidative (j or d) and total (u or s) metabolism of atorvastatin was measured. Values shown are mean 6 S.E. forexperiments performed in triplicate.
676 Chu et al.
at ASPE
T Journals on June 8, 2018
dmd.aspetjournals.org
Dow
nloaded from

min/106 cells, and Pdif = 7.66 1.5 ml/min/106 cells) (Fig. 6B), suggestingthat uptake of BOC into human hepatocytes was a transporter-mediatedprocess. Uptake of SCH-534128, the active form of BOC, also showedtime- and temperature-dependent and saturable uptake (unpublisheddata). To assess whether hepatic uptake of BOC was mediated by NTCP,uptake of 1 mM [14C]BOC into human hepatocytes was also conducted at37°C in sodium-free buffer. As shown in Fig. 6C, uptake of BOC was notsodium dependent, indicating that BOC was not a substrate for NTCP.To further identify potential transporters involved in the hepatic
uptake of BOC, we next examined the uptake of BOC using
MDCKII-OATP1B1, MDCKII-OATP1B3, MDCKII-OATP2B1, andCHO-K1-OCT1 cells. Uptake of [14C]BOC (1 mM) into none of thetransfected cell lines was significantly greater than in control MDCKIIor CHO-K1 cells, respectively (Fig. 7, A–D), suggesting that BOCwas not a substrate of these transporters. Compared with control cells,uptake of positive control substrates [3H]E217bG (1 mM), [3H]CCK-8(2.5 nM), [3H]estrone-3-sulfate (0.1 mM), and [14C]TEA (1 mM) wassignificantly greater in OATP1B1, OATP1B3, OATP2B1, or OCT1transfected cells, indicating the presence of functional transporters inthese cell lines (Fig. 7, E–H, respectively).
Fig. 5. Transport of BOC by MDR1 Pgp, BCRP, and MRP2 in MDCKII-MDR1 and MDCKII-BCRP monolayers and MRP2 containing membrane vesicles. Transcellulartransport of [14C]BOC (1 mM) was evaluated in (A) MDCKII-MDR1 and (B) MDCKII-BCRP monolayers. Papp B-A/A-B ratio in MDR1 or BCRP transfected cells (j) andcontrol MDCKII cells (u) in the absence and presence of cyclosporine A (CsA) (10 mM) or Ko143 (5 mM) was measured at 3 hours. (D–E) Transcellular transport of theprototypical substrates [3H]verapamil (VER) (1 mM) for MDR1 Pgp and [3H]prazosin (PRA) (5 mM) for BCRP measured in transfected cells (j) and control MDCKII cells(u) in the absence and presence of CsA (10 mM) or Ko143 (1 mM), respectively. Time-dependent uptake of [14C]BOC (2 mM) was evaluated in MRP2 (squares) and control(circles) membrane vesicles (C) in the presence (j or d) and absence (u or s) of 5 mM ATP and an ATP-regenerating system. (F) Uptake rate of [14C]EA-SG (2 mM),prototypical substrate of MRP2, measured in MRP2 containing vesicles in the presence (j) and absence (u) of 5 mM ATP and ATP-regenerating system at 5 minutes.Values shown are mean 6 S.E. of experiments performed in triplicate.
Fig. 6. Uptake of BOC in human hepatocytes. (A) Time- and temperature-dependent uptake of [14C]BOC (1 mM) into human hepatocytes at 37°C (j) and 4°C (u),respectively. (B) Kinetic analysis for initial uptake rate of BOC conducted in human hepatocytes at 37°C with obtained Km = 12.4 6 7.4 mM, Vmax = 343 6 150 pmol/min/106 cells, and Pdif = 7.6 6 1.5 ml/min/106 cells. (C) Sodium-dependent uptake of [14C]BOC (1 mM) into human hepatocytes. Time-dependent uptake of [14C]BOC (1 mM)was conducted at 37°C in sodium-free (m) and sodium-containing uptake buffer (j), and at 4°C (u) in sodium-containing buffer. Values shown are mean 6 S.E. ofexperiments performed in triplicate.
Drug Interaction Potential of Boceprevir 677
at ASPE
T Journals on June 8, 2018
dmd.aspetjournals.org
Dow
nloaded from

Consistent with above findings, uptake of [14C]BOC (0.5 mM) wasnot inhibited by BSP, a prototypical inhibitor for anionic transporters.Uptake of BOC was also not inhibited by rifampin, ritonavir, lopinavir,and CsA, but weakly inhibited by rifamycin SV, all known inhibitorsfor OATPs (Fig. 8) (Hirano et al., 2006). These findings confirm thathepatic OATPs do not contribute to uptake of BOC. Quinidine (1–100mM), an inhibitor of organic cation transporters, stimulated rather thaninhibited the uptake of BOC. The mechanism for this unexpectedobservation requires further investigation. As a negative control, ketoco-nazole did not inhibit uptake of BOC (Fig. 8).
Discussion
Our studies indicate that except for CYP3A4/5, BOC did notsignificantly inhibit any of the P450 and UGT enzymes tested.Considering the reversibility and NADPH-dependent nature of theinhibition on CYP3A4/5, the TDI observed in HLM could be causedby a more potent inhibitory effect of oxidative metabolite(s) of BOCgenerated in situ. However, the identity of such inhibitory metabolite(s) is currently unknown because very low levels of these oxidativemetabolites were observed in human plasma after a therapeutic dose ofBOC (unpublished data). Instead, a reductive metabolite formed byAKR, SCH-629144, is the major circulating metabolite in humanplasma. Although it is unlikely that SCH-629144 was responsible forthe increased TDI activity observed in vitro, nevertheless, itscontribution to overall inhibitory effects on CYP3A in vivo couldnot be ruled out. In vitro, SCH-629144 showed reversible inhibition toCYP3A4/5 with similar IC50 values (9.8 mM and 54 mM formidazolam 19-hydroxylation and testosterone 6b-hydroxylation, re-spectively) (Supplemental Table 1) to BOC, and exhibited a time-dependent inhibition to CYP3A4/5 with K I/kinact ratio ;4-fold higherthan that for BOC (Supplemental Fig. 1). The finding that BOC and/orthe reductive metabolite was a TDI of CYP3A4/5, which are present inboth the liver and gut, has been confirmed in clinical DDI studiesdemonstrating that BOC (800 mg, three times daily) increased the
plasma area under the curve (AUC) and Cmax of orally administeredmidazolam (4 mg) by 5.3- and 2.8-fold, respectively (Kiser et al.,2012). Interestingly, this magnitude of inhibition matched reasonablywell with predicted results (;7-fold increase in AUC) using a dynamicmechanistic model-based approach and based simply on the TDIKI /kinact parameters and the clinically observed concentrations ofBOC, without taking into consideration the potential inhibitory effectof SCH-629144 (Prueksaritanont et al., unpublished data). Admit-tedly, the fact that the model provided reasonable prediction does noteliminate the possibility of the involvement of a metabolite in theobserved clinical DDI between midazolam and BOC. Also, this TDIeffect of BOC could conceivably be attributable in part to theincreased AUC of CsA (2.7-fold) and tacrolimus (17-fold) in humanswhen coadministered with BOC, as both are substrates of CYP3A(Hulskotte et al., 2012b).With respect to drug transporters, BOC was not an inhibitor of Pgp
(IC50 . 300 mM) in LLC-MDR1 cells. In contrast, studies in Caco-2cells indicated that BOC was an inhibitor of digoxin (IC50 = 25 mM;unpublished data). The reason for the discrepancy between these twoassay systems is unclear. Caco-2 cells express multiple transporters(Xia et al., 2007), so BOC may affect another uptake or effluxtransporter involved in the transport of digoxin in Caco-2 cells.Therefore, it is reasonable to consider that the inhibition data obtainedwith LLC-MDR1 cells should more accurately predict the effect ofBOC on Pgp-mediated efflux. As such, BOC is unlikely to havea notable inhibitory effect on the Pgp transport at the systemic level(Cmax ;3 mM, 800 mg, three times daily) (Foote et al., 2011).However, local concentrations of BOC in the gut could potentiallyreach a level of 1 mM after the recommended therapeutic dose,a concentration not assessed in vitro. In a clinical DDI study,coadministration of BOC (800 mg, three times daily) with digoxin(0.25 mg, single dose) increased digoxin exposure (AUC and Cmax)slightly (;20%), but not the half-life, presumably due to inhibition ofintestinal not systemic Pgp (Jumes et al., 2012). Likewise, BOC wasalso a weak or non-inhibitor of BCRP (IC50 = 81 mM) and MRP2
Fig. 7. Uptake of BOC into human OATP1B1, OATP1B3, OATP2B1, and OCT1 transfected MDCKII cells. Time-dependent uptake of [14C]BOC (1 mM) was evaluated in(A) MDCKII-OATP1B1, (B) MDCKII-OATP1B3, (C) MDCKII-OATP2B1, and (D) CHO-K1-OCT1 cells. (E–H) Uptake rate of the prototypical substrates [3H]E217bG (1mM), [3H]CCK-8 (2.5 nM), [3H]estrone-3-sulfate (0.1 mM), [14C]TEA (1 mM) for OATP1B1, OATP1B3, OATP2B1, and OCT1 measured at 5 minutes, respectively. (j,uptake in transfected cells; u, uptake by control MDCKII or CHO-K1 cells.) Values shown are mean 6 S.E. of experiments performed in triplicate.
678 Chu et al.
at ASPE
T Journals on June 8, 2018
dmd.aspetjournals.org
Dow
nloaded from

(IC50 . 100 mM), respectively; therefore, significant interactionsmediated via these transporters are not likely.Based on the static R value model and by comparing to known
inhibitors of OATP1B1 that cause clinically significant DDIs withstatins (Table 3), BOC may be classified as a moderate inhibitor ofOATP1B1, and may have the potential to cause DDIs with statins via itsinhibitory effect on OATP1B1. BOC also inhibited OATP1B3 (IC50 =4.9 mM) with an estimated R value of 2.8, assuming that OATP1B3-mediated hepatic uptake is accounting for 100% of the hepatic uptake ofdrugs. Although studied for completeness, the clinical relevance of thisinhibitory effect of BOC on OATP1B3 is currently unclear becauseOATP1B3 is not a major contributor to the hepatic uptake of statinsbased on data from in vitro relative expression/activity factors (Hiranoet al., 2006), and from clinical studies with individuals with geneticpolymorphisms in OATP1B (Niemi et al., 2011). Furthermore, recentclinical studies (Ieiri et al., 2011; Yamada et al., 2011) havedemonstrated that genetic polymorphisms of UGT1A3 but not OATP1B3(SLCO1B3) impact the pharmacokinetics of telmisartan, a selectivesubstrate of OATP1B (Ishiguro et al., 2006).In the case of drugs that are dual substrates of both P450 enzymes
and OATP1B, such as atorvastatin and repaglinide, the fact that BOCis an inhibitor of both OATP1B and CYP3A4/5 could furthercomplicate the scenarios of DDIs. In this study, we used humanhepatocytes, which demonstrate the functional activity of both hepaticuptake transporters and enzymes, to help provide insight into theimpact of BOC on the hepatic elimination of dual OATP1B/CYP3Asubstrates. Together with several probe substrates and appropriatebenchmarking inhibitors, our results suggest that the reduced metabolism
of atorvastatin observed in the presence of rifampin or BOC in hepa-tocytes was largely a consequence of the inhibition of the hepatic uptake,rather than a direct inhibitory effect on CYP3A. These in vitro studies areconsistent with a recent clinical cassette microdose study demonstratingthat hepatic uptake by OATPs is the rate-determining step in the overallhepatic elimination of atorvastatin in humans (Maeda et al., 2011).Importantly, we demonstrated that BOC at clinically relevant concen-trations was a much weaker inhibitor of atorvastatin metabolism inhepatocytes as compared with rifampin, a clinically known OATP1Binhibitor. This finding suggests that BOC should have lesser impact(versus rifampin) on the hepatic elimination of atorvastatin and other dualsubstrates of OATP1B/CYP3A where uptake is the rate-determining step.Indeed, a recent clinical DDI study showed that BOC increased plasmaatorvastatin AUC and Cmax 2.3- and 2.7-fold, respectively (Hulskotteet al., 2011). It is noteworthy that atorvastatin has low intestinalavailability [Fa*fg (intestinal availability) = 0.24] (Shitara, 2011) inhumans, conceivably due to gut CYP3A4 metabolism and Pgp efflux(Hochman et al., 2004). Therefore, the potential of BOC to inhibitatorvastatin gut metabolism might be an additional contributing factor tothe increased systemic exposure of atorvastatin. Also consistent with itsmoderate inhibitory effect on OATP1B1, BOC has been shown toincrease the AUC and Cmax of pravastatin (40 mg) 1.6- and 1.5-fold,respectively (Hulskotte et al., 2011). In humans, pravastatin is eliminatedvia hepatobiliary and renal excretion mediated by hepatic OATP/MRP2and renal OAT3, respectively, with minimal metabolism (Shitara andSugiyama, 2006). As a reference, rifampin caused higher increase inexposure of atorvastatin (.8-fold) and pravastatin (;2.5-fold) in humansafter a single dose of rifampin (Deng et al., 2009; He et al., 2009).
Fig. 8. Effect of various compounds on uptake of BOC into human hepatocytes. Effect of various compounds, including BSP, rifampin, rifamycin SV, ritonavir, lopinavir,cyclosporine A, quinidine, and ketoconazole on initial uptake rate of [14C]BOC (0.5 mM) was investigated in human hepatocytes. The data are expressed as a percentage ofthe initial uptake rate measured at 1 and 5 minutes at 37°C in the presence and absence of inhibitors. Values shown are mean 6 S.E. of experiments performed in triplicate.
Drug Interaction Potential of Boceprevir 679
at ASPE
T Journals on June 8, 2018
dmd.aspetjournals.org
Dow
nloaded from

Furthermore, the greater inhibitory potency of telaprevir relative to BOCtoward both OATP1B and CYP3A, as demonstrated in the present study,also agrees with clinical DDI results that telaprevir (750 mg, three timesdaily) increased plasma AUC and Cmax by 7.88- and 10.6-fold foratorvastatin (Lee et al., 2011), and by 8.96- and 2.86-fold for midazolam(Garg et al., 2012), respectively.To aid in understanding the possible contributions of transporters to the
in vivo disposition of BOC and their implications to DDIs, we assessedthe susceptibility of BOC as a substrate of various drug transporters. BOCas well as its two diastereomers SCH-534128 and SCH-534129 weresubstrates for MDR1 Pgp, and BCRP, but not for MRP2. However, giventhe high therapeutic dose of BOC, Pgp/BCRP activity is likely saturatedand therefore will not significantly impact the intestinal absorption ofBOC. This notion is supported by a recent clinical DDI study (Hulskotteet al., 2012b) that CsA (100 mg), a potent inhibitor of Pgp and BCRP, didnot have a meaningful effect on the pharmacokinetics of BOC.As BOC has low passive permeability and is eliminated primarily
via hepatic metabolism by CYP3A4/5 and aldoketoreductases (Ghosalet al., 2011), uptake transporters may play an important role in thehepatic elimination of BOC. Our studies indicated that uptake of BOCin human hepatocytes was saturable. The active uptake estimated byVmax/Km accounted for ;79% of the total uptake, suggesting thattransporter(s) is/are involved in the hepatic uptake of BOC. In-terestingly, BOC was not a substrate for the major hepatic uptaketransporters tested, and the (novel) transporter(s) contributing tohepatic uptake of BOC remain(s) to be identified. Although possibleinteractions from the unidentified transporter(s) cannot be ruled out,BOC is likely to have low potential as a victim for drug interactionswith the major hepatic uptake transporters tested. This speculation issupported by the finding that BOC exposure is unaffected in healthyvolunteers when codosed with cyclosporine or tacrolimus (Hulskotteet al., 2012b), both broad spectrum inhibitors of drug transporters,including OATPs (Oswald et al., 2011).Recently, drug interactions between BOC and several ritonavir-
boosted HIV protease inhibitors (atazanavir, lopinavir, darunavir)have been reported (Hulskotte et al., 2012a). Unexpectedly, theexposure of all agents (BOC, HIV agents, and ritonavir) wasdecreased. These results cannot be explained simply by inhibition ofdrug metabolizing enzymes or transporters. Ritonavir, atazanavir,lopinavir, and darunavir are inhibitor and/or inducers of multipleenzymes and transporters (Griffin et al., 2011; Jimenez-Nacher et al.,2011), but BOC is not a potent inducer of CYP3A, CYP1A2, andCYP2B6 (unpublished data), and therefore is not expected to be aninducer of Pgp. The underlying mechanism for this complex DDIremains to be investigated, but may involve several interplays at thelevel of metabolic enzymes and transporters, including involvement ofminor or yet to be identified extrahepatic pathways.In summary, our in vitro studies together with clinical DDI
observations suggest that BOC is a relatively potent reversible time-dependent inhibitor of CYP3A and a moderate inhibitor of dualsubstrates of CYP3A and OATP1B for which hepatic uptake is therate-determining step. BOC has low potential to cause pharmacoki-netic interactions by inhibition of other major P450s and transporters,or to be a victim of inhibitors of these known transporters.
Acknowledgments
The authors thank Dr. Lisa A. Shipley for continuous support; Meihong Lin,Tongtong Liu, Jocelyn Yabut, Junying Wang, and Cheng Li for technicalassistance; XenoTech, LLC (Lenexa, KS) for conducting P450 inhibitionstudies; and BD Biosciences Discovery Labware (Woburn, MA) for conductingsome of the Pgp inhibition studies.
Authorship ContributionsParticipated in research design: Chu, Cui, Tang, Ghosal, Palamanda, Evers,
Prueksaritanont.Conducted experiments: Cai, Chan, Green, Kuo, Liang, Maciolek.Performed data analysis: Chu, Cai, Cui, Tang, Ghosal, Chan, Green, Kuo,
Liang, Maciolek, Palamanda.Wrote or contributed to the writing of the manuscript: Chu, Cui, Tang,
Ghosal, Green, Palamanda, Evers, Prueksaritanont.
References
Bacon BR, Gordon SC, Lawitz E, Marcellin P, Vierling JM, Zeuzem S, Poordad F, Goodman ZD,Sings HL, and Boparai N, et al.; HCV RESPOND-2 Investigators (2011) Boceprevir forpreviously treated chronic HCV genotype 1 infection. N Engl J Med 364:1207–1217.
Busti AJ, Bain AM, Hall RG, 2nd, Bedimo RG, Leff RD, Meek C, and Mehvar R (2008) Effectsof atazanavir/ritonavir or fosamprenavir/ritonavir on the pharmacokinetics of rosuvastatin.J Cardiovasc Pharmacol 51:605–610.
Chen SH and Tan SL (2005) Discovery of small-molecule inhibitors of HCV NS3-4A protease aspotential therapeutic agents against HCV infection. Curr Med Chem 12:2317–2342.
Chu XY, Huskey SE, Braun MP, Sarkadi B, Evans DC, and Evers R (2004) Transport ofethinylestradiol glucuronide and ethinylestradiol sulfate by the multidrug resistance proteinsMRP1, MRP2, and MRP3. J Pharmacol Exp Ther 309:156–164.
Deng S, Chen XP, Cao D, Yin T, Dai ZY, Luo J, Tang L, and Li YJ (2009) Effects of a con-comitant single oral dose of rifampicin on the pharmacokinetics of pravastatin in a two-phase,randomized, single-blind, placebo-controlled, crossover study in healthy Chinese male sub-jects. Clin Ther 31:1256–1263.
Fahmi OA, Hurst S, Plowchalk D, Cook J, Guo F, Youdim K, Dickins M, Phipps A, Darekar A,and Hyland R, et al. (2009) Comparison of different algorithms for predicting clinical drug-drug interactions, based on the use of CYP3A4 in vitro data: predictions of compounds asprecipitants of interaction. Drug Metab Dispos 37:1658–1666.
Foote BS, Spooner LM, and Belliveau PP (2011) Boceprevir: a protease inhibitor for the treat-ment of chronic hepatitis C. Ann Pharmacother 45:1085–1093.
Garg V, Chandorkar G, Farmer HF, Smith F, Alves K, and van Heeswijk RP (2012) Effect oftelaprevir on the pharmacokinetics of midazolam and digoxin. J Clin Pharmacol 52:1566–1573.
Ghosal A, Yuan Y, Tong W, Su AD, Gu C, Chowdhury SK, Kishnani NS, and Alton KB (2011)Characterization of human liver enzymes involved in the biotransformation of boceprevir,a hepatitis C virus protease inhibitor. Drug Metab Dispos 39:510–521.
Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R,Fischer V, and Hillgren KM, et al.; International Transporter Consortium (2010) Membranetransporters in drug development. Nat Rev Drug Discov 9:215–236.
Griffin L, Annaert P, and Brouwer KL (2011) Influence of drug transport proteins on the phar-macokinetics and drug interactions of HIV protease inhibitors. J Pharm Sci 100:3636–3654.
Gui C, Obaidat A, Chaguturu R, and Hagenbuch B (2010) Development of a cell-based high-throughput assay to screen for inhibitors of organic anion transporting polypeptides 1B1 and1B3. Curr Chem Genomics 4:1–8.
He YJ, Zhang W, Chen Y, Guo D, Tu JH, Xu LY, Tan ZR, Chen BL, Li Z, and Zhou G, et al.(2009) Rifampicin alters atorvastatin plasma concentration on the basis of SLCO1B1 521T.Cpolymorphism. Clin Chim Acta 405:49–52.
Hirano M, Maeda K, Shitara Y, and Sugiyama Y (2006) Drug-drug interaction between pit-avastatin and various drugs via OATP1B1. Drug Metab Dispos 34:1229–1236.
Hochman JH, Pudvah N, Qiu J, Yamazaki M, Tang C, Lin JH, and Prueksaritanont T (2004)Interactions of human P-glycoprotein with simvastatin, simvastatin acid, and atorvastatin.Pharm Res 21:1686–1691.
Hulskotte EG, Feng HP, Xuan F, van Zutven MG, Treitel MA, Hughes EA, O’Mara E,Youngberg SP, Wagner JA, and Butterton JR (2012a) Pharmacokinetic interactions betweenthe hepatitis C virus protease inhibitor boceprevir and ritonavir-boosted HIV-1 proteaseinhibitors atazanavir, darunavir, and lopinavir. Clin Infect Dis DOI: [published ahead of print].
Hulskotte EG, Gupta S, Xuan F, van Zutven M, O’Mara E, Feng HP, Wagner J, and Butterton J(2012b) Pharmacokinetic interaction between the hepatitis C virus protease inhibitor boceprevirand cyclosporine and tacrolimus in healthy volunteers. Hepatology 56:1622–1630.
Hulskotte EG, Gupta S, Xuan F, van Zutven MG, O’Mara E, Galitz L, Wagner JA, and ButtertonJR (2011) Pharmacokinetic evaluation of the interaction between the HCV protease inhibitorboceprevir and the HMG-CoA reductase inhibitors atorvastatin and pravastatin, in 16th AnnualMeeting of HEP DART; 2001 December 4–8; Kauai, Hawaii.
Ieiri I, Nishimura C, Maeda K, Sasaki T, Kimura M, Chiyoda T, Hirota T, Irie S, Shimizu H,and Noguchi T, et al. (2011) Pharmacokinetic and pharmacogenomic profiles of telmisartanafter the oral microdose and therapeutic dose. Pharmacogenet Genomics 21:495–505.
Ishiguro N, Maeda K, Kishimoto W, Saito A, Harada A, Ebner T, Roth W, Igarashi T,and Sugiyama Y (2006) Predominant contribution of OATP1B3 to the hepatic uptake oftelmisartan, an angiotensin II receptor antagonist, in humans. Drug Metab Dispos 34:1109–1115.
Jiménez-Nácher I, Alvarez E, Morello J, Rodriguez-Nóvoa S, de Andrés S, and Soriano V (2011)Approaches for understanding and predicting drug interactions in human immunodeficiencyvirus-infected patients. Expert Opin Drug Metab Toxicol 7:457–477.
Jumes P, Feng H-P, Xuan F, Youngberg S, Wagner J, and Butterton J (2012) Pharmacokineticinteraction between the HCV protease inhibitor boceprevir and digoxin in healthy adult vol-unteers, in 7th International Workshop on Clinical Pharmacology of Hepatitis Therapy, 2012June 27–28, Cambridge, MA.
Karlgren M, Ahlin G, Bergström CA, Svensson R, Palm J, and Artursson P (2012) In vitro and insilico strategies to identify OATP1B1 inhibitors and predict clinical drug-drug interactions.Pharm Res 29:411–426.
Kenworthy KE, Bloomer JC, Clarke SE, and Houston JB (1999) CYP3A4 drug interactions:correlation of 10 in vitro probe substrates. Br J Clin Pharmacol 48:716–727.
Kiang TK, Ensom MH, and Chang TK (2005) UDP-glucuronosyltransferases and clinical drug-drug interactions. Pharmacol Ther 106:97–132.
680 Chu et al.
at ASPE
T Journals on June 8, 2018
dmd.aspetjournals.org
Dow
nloaded from

Kilford PJ, Gertz M, Houston JB, and Galetin A (2008) Hepatocellular binding of drugs: cor-rection for unbound fraction in hepatocyte incubations using microsomal binding or druglipophilicity data. Drug Metab Dispos 36:1194–1197.
Kiser JJ, Burton JR, Anderson PL, and Everson GT (2012) Review and management of druginteractions with boceprevir and telaprevir. Hepatology 55:1620–1628.
Kiser JJ, Gerber JG, Predhomme JA, Wolfe P, Flynn DM, and Hoody DW (2008) Drug/Druginteraction between lopinavir/ritonavir and rosuvastatin in healthy volunteers. J Acquir ImmuneDefic Syndr 47:570–578.
Kwo PY, Lawitz EJ, McCone J, Schiff ER, Vierling JM, Pound D, Davis MN, Galati JS, GordonSC, and Ravendhran N, et al.; SPRINT-1 investigators (2010) Efficacy of boceprevir, an NS3protease inhibitor, in combination with peginterferon alfa-2b and ribavirin in treatment-naivepatients with genotype 1 hepatitis C infection (SPRINT-1): an open-label, randomised, mul-ticentre phase 2 trial. Lancet 376:705–716.
Lau YY, Huang Y, Frassetto L, and Benet LZ (2007) Effect of OATP1B transporter inhibition onthe pharmacokinetics of atorvastatin in healthy volunteers. Clin Pharmacol Ther 81:194–204.
Lau YY, Okochi H, Huang Y, and Benet LZ (2006) Multiple transporters affect the disposition ofatorvastatin and its two active hydroxy metabolites: application of in vitro and ex situ systems.J Pharmacol Exp Ther 316:762–771.
Lee JE, van Heeswijk R, Alves K, Smith F, and Garg V (2011) Effect of the hepatitis C virusprotease inhibitor telaprevir on the pharmacokinetics of amlodipine and atorvastatin. Anti-microb Agents Chemother 55:4569–4574.
Lin JH (2006) CYP induction-mediated drug interactions: in vitro assessment and clinicalimplications. Pharm Res 23:1089–1116.
Madan A, Usuki E, Burton L, Ogilvie B, and Parkinson A (2002) In vitro approaches for studyingthe inhibition of drug-metabolizing enzymes and identifying the drug metabolizing enzymesresponsible for the metabolism of drugs, in Drug-Drug Interactions (Rodrigues AD, ed), pp217–294, Marcel Dekker, New York.
Maddur H and Kwo PY (2011) Boceprevir. Hepatology 54:2254–2257.Maeda K, Ikeda Y, Fujita T, Yoshida K, Azuma Y, Haruyama Y, Yamane N, Kumagai Y,and Sugiyama Y (2011) Identification of the rate-determining process in the hepatic clearanceof atorvastatin in a clinical cassette microdosing study. Clin Pharmacol Ther 90:575–581.
Malcolm BA, Liu R, Lahser F, Agrawal S, Belanger B, Butkiewicz N, Chase R, Gheyas F, HartA, and Hesk D, et al. (2006) SCH 503034, a mechanism-based inhibitor of hepatitis C virusNS3 protease, suppresses polyprotein maturation and enhances the antiviral activity of alphainterferon in replicon cells. Antimicrob Agents Chemother 50:1013–1020.
Monteagudo E, Fonsi M, Chu X, Bleasby K, Evers R, Pucci V, Orsale MV, Cianetti S, Ferrara M,and Harper S, et al. (2010) The metabolism and disposition of a potent inhibitor of hepatitis Cvirus NS3/4A protease. Xenobiotica 40:826–839.
Müller F and Fromm MF (2011) Transporter-mediated drug-drug interactions. Pharmacoge-nomics 12:1017–1037.
Niemi M, Pasanen MK, and Neuvonen PJ (2011) Organic anion transporting polypeptide 1B1: a geneti-cally polymorphic transporter of major importance for hepatic drug uptake. Pharmacol Rev 63:157–181.
Noé J, Portmann R, Brun ME, and Funk C (2007) Substrate-dependent drug-drug interactionsbetween gemfibrozil, fluvastatin and other organic anion-transporting peptide (OATP) sub-strates on OATP1B1, OATP2B1, and OATP1B3. Drug Metab Dispos 35:1308–1314.
Obach RS (2009) Predicting drug-drug interactions from in vitro drug metabolism data: chal-lenges and recent advances. Curr Opin Drug Discov Devel 12:81–89.
Obach RS, Walsky RL, Venkatakrishnan K, Gaman EA, Houston JB, and Tremaine LM (2006)The utility of in vitro cytochrome P450 inhibition data in the prediction of drug-drug inter-actions. J Pharmacol Exp Ther 316:336–348.
Oswald S, Nassif A, Modess C, Keiser M, Ulrich A, Runge D, Hanke U, Lütjohann D, Engel A,and Weitschies W, et al. (2011) Drug interactions between the immunosuppressant tacrolimus
and the cholesterol absorption inhibitor ezetimibe in healthy volunteers. Clin Pharmacol Ther89:524–528.
Parkinson A, Kazmi F, Buckley DB, Yerino P, Paris BL, Holsapple J, Toren P, Otradovec SM,and Ogilvie BW (2011) An evaluation of the dilution method for identifying metabolism-dependent inhibitors of cytochrome P450 enzymes. Drug Metab Dispos 39:1370–1387.
Poordad F, McCone J, Jr, Bacon BR, Bruno S, Manns MP, Sulkowski MS, Jacobson IM, ReddyKR, Goodman ZD, and Boparai N, et al. SPRINT-2 Investigators (2011) Boceprevir foruntreated chronic HCV genotype 1 infection. N Engl J Med 364:1195–1206.
Prueksaritanont T, Subramanian R, Fang X, Ma B, Qiu Y, Lin JH, Pearson PG, and Baillie TA(2002) Glucuronidation of statins in animals and humans: a novel mechanism of statin lacto-nization. Drug Metab Dispos 30:505–512.
Reitman ML, Chu X, Cai X, Yabut J, Venkatasubramanian R, Zajic S, Stone JA, Ding Y, WitterR, and Gibson C, et al. (2011) Rifampin’s acute inhibitory and chronic inductive drug inter-actions: experimental and model-based approaches to drug-drug interaction trial design. ClinPharmacol Ther 89:234–242.
Shitara Y (2011) Clinical importance of OATP1B1 and OATP1B3 in drug-drug interactions.Drug Metab Pharmacokinet 26:220–227.
Shitara Y and Sugiyama Y (2006) Pharmacokinetic and pharmacodynamic alterations of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors: drug-drug inter-actions and interindividual differences in transporter and metabolic enzyme functions. Phar-macol Ther 112:71–105.
Takeda S, Kitajima Y, Ishii Y, Nishimura Y, Mackenzie PI, Oguri K, and Yamada H (2006)Inhibition of UDP-glucuronosyltransferase 2b7-catalyzed morphine glucuronidation by keto-conazole: dual mechanisms involving a novel noncompetitive mode. Drug Metab Dispos 34:1277–1282.
Walsky RL and Obach RS (2004) Validated assays for human cytochrome P450 activities. DrugMetab Dispos 32:647–660.
Wilby KJ, Greanya ED, Ford JA, Yoshida EM, and Partovi N (2012) A review of drug inter-actions with boceprevir and telaprevir: implications for HIV and transplant patients. AnnHepatol 11:179–185.
Williams JA, Hyland R, Jones BC, Smith DA, Hurst S, Goosen TC, Peterkin V, Koup JR,and Ball SE (2004) Drug-drug interactions for UDP-glucuronosyltransferase substrates:a pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. DrugMetab Dispos 32:1201–1208.
Xia CQ, Milton MN, and Gan LS (2007) Evaluation of drug-transporter interactions using in vitroand in vivo models. Curr Drug Metab 8:341–363.
Yamada A, Maeda K, Ishiguro N, Tsuda Y, Igarashi T, Ebner T, Roth W, Ikushiro S,and Sugiyama Y (2011) The impact of pharmacogenetics of metabolic enzymes and trans-porters on the pharmacokinetics of telmisartan in healthy volunteers. Pharmacogenet Genomics21:523–530.
Zhang D, Chando TJ, Everett DW, Patten CJ, Dehal SS, and Humphreys WG (2005) In vitroinhibition of UDP glucuronosyltransferases by atazanavir and other HIV protease inhibitorsand the relationship of this property to in vivo bilirubin glucuronidation. Drug Metab Dispos33:1729–1739.
Zhou SF (2008) Drugs behave as substrates, inhibitors and inducers of human cytochrome P4503A4. Curr Drug Metab 9:310–322.
Address correspondence to: Dr. Xiaoyan Chu, Merck & Co, RY80-141, 126 EastLincoln Avenue, Rahway, NJ 07065. E-mail [email protected]
Drug Interaction Potential of Boceprevir 681
at ASPE
T Journals on June 8, 2018
dmd.aspetjournals.org
Dow
nloaded from