inferring selection pressure from positional residue conservation rose hoberman roni rosenfeld...
TRANSCRIPT

Inferring Selection Pressure from Positional Residue Conservation
Rose HobermanRoni Rosenfeld Judith Klein-Seetharaman

Sequence Conservation
Conserved positions in proteins are of functional and structural importance
Many quantitative measures of positional conservation
Applications predict function/structure model protein families/domains evaluate and refine multiple sequence
alignments

Objective
Given a multiple sequence alignment...
1. Which positions are conserved?
2. Identify specific selectional pressures at each position assume amino acids are selected based on
their underlying physical and chemical properties

Related Work
Entropy most common measure
of conservation amino acids as
independent symbols
Property-based smallest set of properties small number (binary)
properties ignores amino acid
frequency
p pii
i
20
ln

Related Work
Mutation-based substitution matrices average over many positions/proteins
Aggregate experimental scales build a similarity matrix “propotypical” properties via dimensionality
reduction average over many properties
By aggregating, loses specificity

250 Properties
1 Hydrophobicity
2 Volume
3 Net charge
4 Transfer free energy
...
248 Average flexibility
249 Alpha-helix propensity
250 Number of surrounding residues

250 Properties
1 Hydrophobicity
2 Volume
3 Net charge
4 Transfer free energy
...
248 Average flexibility
249 Alpha-helix propensity
250 Number of surrounding residues
A C D E F G H I K L 0.66 2.87 0.10 0.87 3.15 1.64 2.17 1.67 0.09 2.77
M N P Q R S T V W Y0.67 0.87 1.52 0.00 0.85 0.07 0.07 1.87 3.77 2.67

Multiple Sequence Alignment
FAMLR...LAMLR...IAMLR...P-EL-...GAELR...PGEIR...L-ELY...L-EVR...I-MLK...WAELR...HAELY...YAILY...WAML-...

Multiple Sequence Alignment
FAMLR...LAMLR...IAMLR...P-EL-...GAELR...PGEIR...L-EVY...L-EVR...I-MLK...WAELR...HAELY...YAILY...WAML-...

Multiple Sequence Alignment
FAMLR...LAMLR...IAMLR...P-EL-...GAELR...PGEIR...L-ELY...L-EVR...I-MLK...WAELR...HAELY...YAILY...WAML-...

Limitations of our approach
Not modelling phylogenetic relationships
Cannot identify property conservation when entropy is low

Variance

Variance
If a property is conserved, the distribution of its values should have lower variance
However, must condition on entropy

Holes

Unimodality
When selection based on a single property, fitness is unimodal
Look for properties for which all amino acids are adjacent

Adjacency Limitations
Definition of a “hole” too strict, fails to model unimodality
Ignores distances between property values
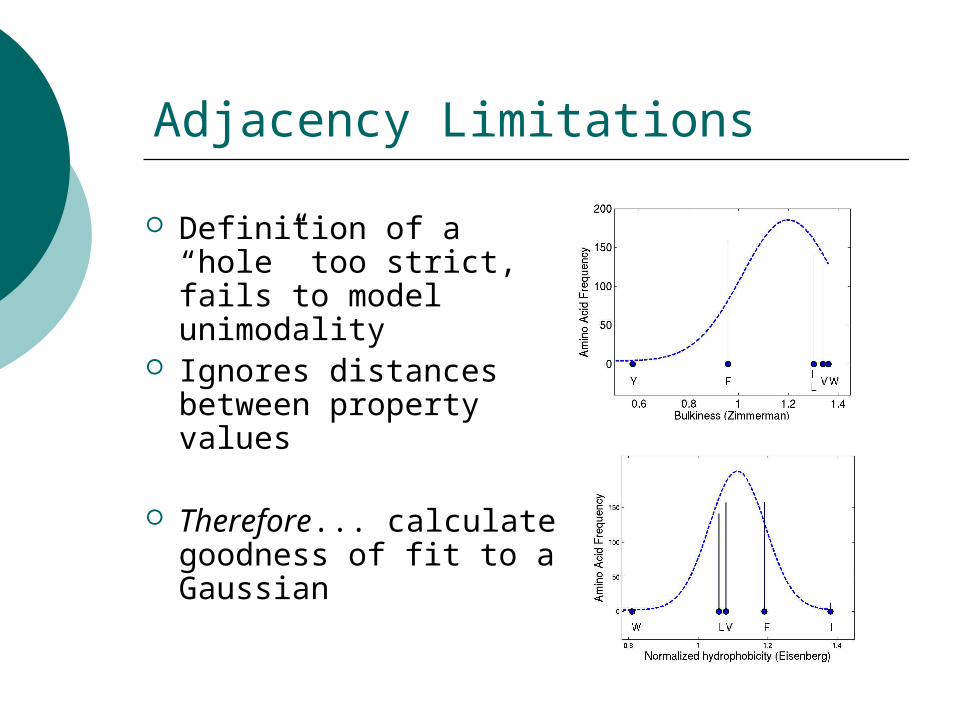
Adjacency Limitations
Definition of a “hole” too strict, fails to model unimodality
Ignores distances between property values
Therefore... calculate goodness of fit to a Gaussian

Gaussian Goodness-of-Fit
Fit a maximum-likelihood Gaussian to amino acid frequencies in property space
Calculate goodness-of-fit to learned Gaussian discretize Gaussian over 20 values calculate statistic 2

Significance
High false discovery rate when low entropy
Calculate significance using shuffled properties
For a position Calculate value for each shuffled property Tells us the likelihood of getting a particular
value by chance Convert to a p-value
2
2

Gaussian Significance I

Gaussian Significance II

Gaussian Significance III

GPCR Family A
7 TM segments Cytoplasmic, TM, and
extracellular domains Responds to a large variety
of ligands Ligand binding allows
binding and activation of a G protein
Diversity in sequences Believed to share similar
structure Only known structure is for
Rhodopsin

False Discovery Rate
properties threshold Expected FD
Positions FDR
5 0.0005 0.63 76 0.8%
5 0.001 1.26 85 1.5%
5 0.005 6.26 136 4.6%
50 0.0005 6.25 103 6.1%
50 0.001 12.34 130 9.5%
240 0.0005 28.61 154 18.6%

Computational Validation
Predict the occurence of amino acids not yet seen in a position of a multiple sequence alignment
Subsample sequences from GPCR alignment of sizes {25, 50, 75, 100, 200}
Baseline: logistic regression with only entropy and variability
New model incorporates the distance of the residue’s value from the mean of the best fitting property Gaussian

Computational Validation

Results for GPCR

Biological Validation
Property conservation in all three domains of GPCR (not just TM) e.g. hydrophobicity conserved in interhelical
loop regions Charge conserved at 134
part of D/E R Y motif of importance to binding and activation of G-protein
Size conserved at 54, 80, 87, 123, 132, 153, 299 helix faces one or two other helices

Biological Validation
Helix-coil equilibrium constant conserved at 278 a helix capping residue at the EC end of helix
VI Dynamic properties conserved
especially in third CP loop in Rhodopsin this is the most flexible
interhelical loop in TM at 215 and 269
close proximity to retinal ligand; the ligand-binding pocket is the most rigid part of crystal structure

Clustered Positions
Positions with high property-conservation similarity are often close in space but distant in primary sequence 129 and 226 and 131 and 219 162 and 297 both face outside of the
bundle at a similar height with respect to the membrane

Novel Hypothesis
175 and 265 highly similar conservation patterns
Both tryptophans in rhodopsin Trp265 in direct contact with retinal ligand,
but when exposed to light, crosslinks to Ala169 instead.
Trp161 has been proposed to contribute to this process
The property conservation patterns suggest Trp175 has a more significant role
This hypothesis can be tested experimentally

Results

Results

More Detailed Results
Helix 1. Proportion of residues 100% buried 3. Hydrophobicity 4. Side chain interaction param 9. Polarity10. Transfer free energy to lipophilic phase20. Membrane buried helix param
Intracellular loop
1. Polarity/Grantham 2. Normalized flexibility/B-values 4. Conformational preference for antiparallel beta 5. Hydrophobicity of physiological L-alpha amino acids 9. Mobilities of amino acids on chromatography paper 10. Relative frequency of extended structure 11. Long range non-bonded energy per atom13. Information value for accessibility
Extracellular loop
1. Normalized flexibility/B-values, average 2. Flexibility param for one rigid neighbor 3. van der Waals param R0 4. Average non-bonded energy per atom 5. Polar requirement 6. Conformational preference for antiparallel beta strands

Future Work
Additional evaluation Consider selection pressure from
more than one property Use multivariate Gaussians Look for adjacency in 2-D property
space Test for unimodality directly Model phylogenetic relationship
between sequences

Conclusions
Method identifies positions with significant evidence that a particular property is conserved
More discriminatory than entropy
Framework is easily extendable to multiple properties

Thanks!

Entropy
Most commonly-used measure of conservation
Based only on frequency of each amino acid Treats amino acids as independent symbols
Amino acids are not uniformly different
p pii
i
20
ln

Property-based Measures
Based on Venn diagram of physical-chemical properties
Find smallest set of properties that explains amino acids
Considers only a small number of property combinations
Only binary properties Ignores amino acid
frequency
Livingstone & Barton, CABIOS, 1993

Hybrid Methods
Stereochemically-sensitive entropy scores Divide amino acids into clusters (show example?) Calculate entropy on reduced alphabet
Clustering is ad hoc Can only consider a limited number properties Still considers each amino acids in a cluster as
uniformly distant from each amino acid in every other cluster
Only models limited number of properties at a very coarse level

More ...
Substitution methods Physical-chemical distance matrices
or aggregate properties DNA, phylogeny, mutation rates

Adjacency Significance
Positions with small number of amino acids will have a high false discovery rate
Add equation and table

Significance
Problem: for positions with low entropy, every property will have low variance very high false positive rate: any combination
of 1 more more properties can explain this! actual explanation may involve several
properties
In this case, multiple property constraints
Cannot determine which one property is conserved
Need to condition on entropy

Significance Testing
What is the probability of a property having low variance in this position purely by chance?
Generate a large set of “random” (shuffled) property scales
show examples of shuffling Calculate variance for each random property The distribution of this statistic can be used to
calculate a threshold for acceptability of false-positives
Show picture here? add error bars?