institute of nuclear sciences
TRANSCRIPT

INS-R-217
CURRENT APPLICATIONS OF SEMICONDUCTOR X-RAY DETECTORS IN CHEMICAL ANALYSIS
by N. E. Whitehead
November 1976
INSTITUTE OF NUCLEAR SCIENCES Department of Scientific end Industrial Research
LOWER HUTT, NEW ZEALAND
SIR (IN$) *

I V i
^ 4 " * \tf!|jiS#l!«t*"***V-="*v* Sv •^^"3IV.S^#>^W! ;>w?«^^ r f m l M ! I , ?_ w l , J r^M i l ( u > ) r M u | W „ , H-4Aft^,,*i'~ lustj^^WJ^Wj^P^ 1 INS.R-317
CURRENT APPLICATIONS OF SEMICONOCUTOR X-KAY DETECTORS IN CHEMICAL ANALYSIS
N.E. Whitehead Institute of Nuclear Sciences,
D.S.I.R., Lover Hutfe, New Zealand.
J«*v ™??
I.N.S, Contribution no. 735

C O N T K N T S
Summary Scope Intrtluction Historical background Development of solid-state detectors Electronic developments Vindowless detectors Calibration of detector Future detector development Nomenclature Excitation methods Matrix effects Data reduction Thin sample techniques Chemical pre-treatment Analysis without standards Accuracy and precision Applications in the field Applications A coi parison of different excitation systems A comparison with other multi-element analysi
techniques The literature of X-ray spectrometry Conclusions Appendix References Figures

Summary In the last few years, semiconductor detectors have been
used as X-ray detectors with great success, and the routine rapid accumulation of X-ray spectra is now possiLle. This review surveys the historical development of the detectors, the utilisation, and relative merits of various means of exciting the X-radiation from the elements in the sample* and compares the technique with other methods claiming to offer the capability of simultaneous multi-element analysis. It is concluded that it it of average sensitivity, but offers some advantages from its non-destructive nature, and in some cases its ability to offer information about tho spatial distTibution of elements in a sample. Other types of analysis may also be possible simultaneously. Sample preparation tecluiit|ticu nro reviewed, especially tecluiiques of manufacturing thin samples. An Appendix contains details of the very wide variety of samples which have been analysed. More than 350 references are included.

CUHRKXT APPI TCATTONS OK SKMI.CON!>"C I (>l* X-RAV
DETECTORS IN CHEMICAL ANALWSJS
N.E. Whitehead Institute of Nuclear Sciences,
D.S.I.R., Lower Hutt, Now Zealand.
1. Scop'e This review is concerned with the uses and limitations
of semiconductor X-ray detectors in chemical analysis. It includes material on the improvements in electronic equipment which have led to the successful use of these devices and the different means of exciting the X-rays which they detect. The recent use of these detectors for assaying radioactive substances (both natural and artificial) is included.
Attempts to improve the sensitivity of analysis using semiconductor detectors by such means as chemical pre-treatment of samples, and manufacture of thin samples, are considered. The overall method of analysis is compared with other techniques which claim to provide simultaneous analysis.
The main period covered by the review is from about i960 to early 1975.
2. Introduction Semiconductor X-ray detectors are devices which are being
^ied on a wide scale for the accumulation of X-ray spectra. Radiation, sucli as the emission from a radioactive isotope, is used to excite X-rays from a sample, and these emitted X-rays are all detected simultaneously using a semiconductor detector and hence may be used for simultaneous, multielement analysis. Many analytical methods are capable of determining a range of properties, but normally only one at a time. For example, conventional spectrophotometry can produce a spuctriuii of a visible light source, but only by scanning - by measuring the intensity at one point of the spectrum at n time.
I.N.S. Contribution no. 735

Traditional X-ray fluorescence tan also pc«»duci- .1 i-'.poc t rum by scanning. The essential difference between that and the method to be reviewed is that semiconductor devices produce a spectrum by electronic means - without scanning. I» theory this loads to immense saving in time. Such i>i|uipninnl is potentially valuable to the analytical chemist.
This type of spectrometry relies heavily on the multichannel analyser, an electronic device which examines voltage pulses and sorts them, creating a kind of histogram in which, however, the points or cliannels are so closely spaced that the final result resembles a continuous curve. This, and the rest of the instrumentation owes a great deal to the development done for and by nuclear physicists.
The fact that sales of these detectors are increasing by kO-^0"/') per year (R. Frankel, pers.comm.) shows the increasing importance of this field.
3, Hist ori c.il background
On the evening of 8 November 1895, Roentgen made the discovery that emission from a cathodo-ray tube causes a barium platinocyanide plate to fluoresce. He called these emissions 'X-rays' and practical applications followed within one year.
In 1897» Sagnac discovered that when these X-rays struck a target, radiation other than light was emitted, and Darkla made a long study of these secondary rays. He found that some were the same wavelength as the original rays, bat some were of a wavelength that depended only on the elements present in the target and was not affected by clianges in chemical bonding, temperature, or density. Darkla also showed that there were two characteristic emission wavelengths for onch element, and named those the K and L scries. From this, the concept of electron shells evolved. Furl her work, by Bragg arid Mosoloy and others showed that each series could l>e further subdivided into a number of components, Further sn'ips, called the M on! N series, were found by Sicgbabn and others. F,ach scries was of a longer wavelength (or loss energy) than the preceding series.

').
The v m k of the liraggs showed thai X-rayy wore d iffrac ted by various crystals to a degree vhich depended on their wavelength. This meant that they were physically dispersed in space. This dispersion is the basis of the modern technique known as 'X-ray fluorescence'. By 192't, Siegbahn had constructed an apparatus which relied on this effect. He detected the dispersed X-rays simultaneously by means of a photographic emulsion and in two hours was able to determine all the elements between sodium and uranium vhich were present in the target. The semiconductor X-ray detectors described in this review, developed in the last decade, also cover this range but there is an immense increase in the speed of analysis.
However, the main detector used for X-rays has been the proportional counter, a gas-filled tube containing an anode and cathode with a voltage between them. When an X-ray enters such a counter it causes ionisation and a voltage pulse is produced, the height of which is proportional to the energy of the X-ray. By sorting the sizes of the voltage pulses into different channels, U'sing a multi-channel analyser, it is possible to record a spectrum of X-ray energies. The first use of proportional counters lor X-ray spectrometry appears to
75 be that of Curran et al. The same electronic principles are used in solid-state detectors.
*». Development of solid-state detectors
In the same year, Hofstadter ~ introduced the thallium-doped sodium iodide crystal for detecting gamma rays. A thin crystal was found useful for detecting X-rays and the first X-ray spectrum was recorded by Miller and Wilkinson" .
This was the f.irst solid-state detector but the resolution of such devices or FWIIM (Full Width at Half Maximum height) was 5°',' of the height of the po;iks they produced, compared with 2Vr/i for the proportional counter - the proportion.'! counter was hence preferred fox- X-ray spectrometry*
P I I McKay " showed that it was possible to use the semi
conducting metal germanium as a solid-state analogue of the proportional counter. In the proportional counter two electrodes have gas between them. In a ger-man i m.i counter the germanium is the analogue of tlw gas, and a vol i,ago is applied

• ' » .
across it. Voltage puiM'» ai>? produc-d which are pJ"'>|n»rt" ioual to the gnru;ia-ray or X-iay/penetrating the volume br-tweii the electrodes. Hovcvcr, the fii-st use «>f sucli material was for alpha particle detection. A similar u»e of silicon instead of germanium was demonstrated next (McKay and McAfee ). However, the germanium and silicon available to these workers verc not pure enough to make a successful carina-ray detector. Many spurious pulses (noise) were observed due to the impurities.
2'»8 Pell described a way of neutralising the effects of these impurities. He diffused lithium into either Germanium or silicon at a high temperature. This increases the electrical resistance of the element, lowers the noise, and improves the collection of charge at the electrodes. This
171 technique was rapidly applied (Ko)iler, ) to both gamma-ray spectrometry (Daily et al. ; Freck and Wakefield ) and also to X-ray spectrometry (van der Does de Bye ). The resolution for X-rays was worse than a proportional counter and about equivalent to a sodium iodide crystal. The devices were small (so did not detect X-rays efficiently) and still very noisy. Much of the noise was due to poor electronic amplification of the voltage pulses.
Improvements were made in two directions: the electronics and the quality of the detector material. Improvements in the quality of the detectors themselves wore chiefly a matter of optimising the diffusion conditions so that the best possible final material resulted. This difficult task has been achieved to a good degree. Developments in electronics proceeded at the same time and these two effects resulted in a dramatic improvement in resolution.
In 1968, Aitken was able to report in a review that realistic resolution amounted to 10i» (e.g., Fitzgerald et al .
in other words, significantly bet Lei- than proportional counters. The best figure quoted at that date writ y/o (e.g.,
2TH Palms et al. ). This is equivalent to 330 electron volts (oV) for an X-ray of energy 5»9 keV. From that date tTKivlutin figures were usually expressed in terms of the FW1IM at that X-ray energy and ve filial 1 do likewise in the remainder of I hi." review.

Wli-Ti Friant "*, and Israel ofc a.l. . ' reviewed the field, the resolution vu* still improving and Friant e I a I.
quoted a figure u r 165 eV. This still represents the best resolution for routine u^e in 1975* However, for a few small Uovircs the resolution may be as low as WO eV (Ooulding
127 \ and Jaklevic ) , but these ore not generally commercially available. This resolution is about l.5/«' «*" fJ•- peak height.
5. Electronic developments
Most readers of this review will be chemists, but a knowledge of the electronics involved in instrumentation seems more and more necessary, so this section reviews (in an elementary way) this facet of the development of semiconductor X-ray detectors.
Voltage pulses from a detector are amplified in a charge-sensitive preamplifier, then a main amplifier. A typical circuit for a charge-sen.5it.ive preamplifier is shown in fig. 1. One major improvement was the use of n field-effect transistor- (FKT) in the preamplifier stage. These were mvestiga ted by Hadoka and apparently first used in
0 0 X-ray work by Elad in 196} (cited by Donovan ). It had been found that the electronic noise in semiconductor detectors could be reduced by cooling and it was not surprising in view of the similarity of the basic material that the same was true of FET's. It thus became? usual to cool both the detector and FET to the temporal tire of liquid nitrogen. The detector was therefore mounted on the cud of a' /'rod and the FET nearby. A de-war was filled with liquid nitrogen and the rod immersed in it. Sometimes the rod was deliberately bent so that the detector pointed horizontally or in some other direction but Ihe principle remains the same. In spite of cooling, FKT's wore found to be rather unpredictable in characteristics and it was nrc'S u n v to se'leel Ihe best of a batch. In fact, one nl the major co.t:- of a del eel or/ preamplifier was the immense effort needed to select a j-.ood FET. Se.l (•(• tioti was also iior;o>,-a ry for the (VcifhacU resistor in fig. 1. Anolhej improvement was the use of pulse shaping in the ainp.l i f i. '•• r (Ail I;' 11 ). I' roamp I i I i e r,. produce pulses of varying shape.; |,ul, the pulse;, from the rlet.ef lor »: r. 13. \;o | |

be quite distinctive and it is possible to design circuitry which rejects shapes outside pre-set variable limits. This is now a standard feature of many amplifiers. Germanium and silicon detectors need different time constants; usually about 1 and *i microseconds, respectively.
Other features incorporated which are now leutine were direct current coupling, pole-zero cancellation and active baseline restoration. The first is always desirable in an amplifier but requires the stability of modern amplifiers. The second is designed to eliminate baseline over-shoot, and the third is designed to compensate for drifts in the basic voltage line on which the pulses sit. These drifts arise, among other causes, igh pulse rates and from vibrations in the surroundings of the detector. These vibrations produce pulses known as 'microphonics' which are hundreds of microseconds long, and must be corrected for. One cause of them can be the boiling of liquid nitrogen in the reservoir and they are not completely eliminated by pulse shaping; baseline? restoration is necessary. Hebert and Street replaced the copper rod in their system with a braided copper cable and reported that its flexibility prevented the transmission of vibrations to th' detector, while still being a good conductor of heat. This is a standard fetiture in many detectors now.
A further feature (not available in all systems on the market) is pulse pile-up rejection circuitry. When the count rate becomes very high, two or more pulses may overlap and be treated by the amplifier as a single pulse of much greater energy than ei ther. This may cause Die nppc:inmce of spurious peaks in the- spectrum, of double the energy of the most prominent peaks. These peaks are called 'pile-up' peaks and may be partially, though not entirely, eliminated by suitable circuitry such as was introduced b> Kandiah , and Heed*
The feedback resistor in fig. 1 is necessary because the FET becomes depleted of charge niter a lime and so a resistor trickles some back. The resistor contributes some noise t.i> the system though and decreases the resolution. Goulding et a J .
therefore replaced it with an optoelectronic feedback
130

7.
system. lLT'o are light sensitive and charge is ilnvi-lnpoil in them in proportion to the light input. So a light-emi t ting diode (i.ED) is made to emit light tovards the FET in proportion to the charge passing to the main amplifier. This system was further improved l»y pulsing the J ight-omi tt in.--; diode after charge had accumulated to a pre-sct lcv^l rather than letting it continuously emit light. This ^s an improvement because LEU's do not give a completely linear light output with varying input voltage (Land is et al. ; Landis e I. a 1. ";
Friant ). Another design which replaces th« feedback resistor is called 'dynamic charge restoration' (Elad ) .
This is a special circuit which achieves the same end as the optoelectronic system.
Further electronic sophistication has been used by a fev vorkers, in conjunction vith a modified detector, in an attempt to improve the sensitivity. This circuitry is designed to eliminate peaks with degraded energy.
An examination of fig. 2, which is a typical spectrum of X-rays obtained using a Si(Li) detector and X-rays .('mm an X-ray tube to excite secondary radiation from the? sample, shows the presence of scatter peaks, that is, the radiation from the tube which has been scattered from the sampJ o and reached the detector with little loss in energy. They are the major peaks in any similar X-ray excited X-ray spectrum and are a source of background (which reduce sensitivity) by the mechanism illustrated in fig. 3>'«« There occurs a type of edge effect in which there is inefficient collection by the electrodes of the ionisation products of X-rays which deposit their energy near the edges of the detector. This means that a pulse is produced with less voltage than it should have, and a low energy tail appears in the spectrum (see fig. 2 ) . Those degraded pulses appear to some extent under most of the spectrum. They can be reduced in number by using the detector configuration shown in fig. 3b, This lowers the background by a factor of between ',1 and 10 (Oou I d i.ng and .lakJ.evic '"Moulding
1."'° x e t a I.. ). The ring thus formed is called a 'guard ring*. A double guard ring has also been used as J n fig. 'Jc. J.f a pulse is detected in the centre and .intermedial;" ring fit I he same instant, it is rejected, by special coincidence circuitry,

so that /only pulses occurring in the centre of the detector are allowed to enter the multichannel analyser. This may result in the lower in*; of the background still further - perhaps l»y as much as a factor of ."our. Coincidence circuitry, as already described, is not incorporated in commercial systems. An analyst who buys a complete system will, however, obtain most of the other refinements mentioned previously.
In what, way could improvements in the electronics usefully improve these semiconductor devices? Mainly by further reduction in the noise level, for this is at present the limiting factor for low energies. The contribution to peak width of the detector itself is given by the expression &Epuu>| =
(FEC) 2, where &E is the detector contribution, E is the energy of the photons, £ s the average energy required to produce a hole-electron pair by ionisation in the detector material, mid F is the KUIIO J'actor, which represents the statistics of energy
1 ?S loss due to vibrational processes (Goulding and Jaklevjc " ) . / 97
From recent estimates of this factor (Elad and Gedcke ; Gouldinj: and Jaklevic " ), it seems that, at low energies especially, detector noise is minimal and preamplifier noi.se is the major contributor to peak width. For example, Elad and Gedcke 9 7 state that Tor a peak width of 103 eV at. 1.'l9 kcV, 85 eV was contributed by the preamplifier and associated electronics. These and other contributions to noise are also reviewed by Stroback . Any further major reductions in noise rest in the hands of electronic experts.
6» Windowloss detci:tors
Because of their lithium content the detector." must be kept under vacuum and yet there must he as little absorbing

«>.
material as possible between the sample and Lhe detector. In practice, therefore, a thin window of beryllium 12-25 micron thick is used. This is vacuum tight, and does net contribute measurable X-rays itself, but allows most X - m y s to pass
fi'om elements through. Hivevcr, absorption becomes very high for X-raj «* / ^.-^h 7 less than 11 (sodium), and fluorine and lighter elements cannot be detected unless there is some way of removing the window, which is on'y safe if the analysis is done in a vacuum. Since air itself acts as an absorber for low energy X-rays, there is another reason for working in a vacuum.
1 "52 Jaklevic and Goulding J installed a gate valve in front of their detector, containing in one position a 125
-2 micron beryllium window and in the other a 25 microgram cm window of aluminium ( = 1000 X ) . The stronger window was used to protect the detector until a good vacuum was obtained, when the other window was slid into position. Elements as light as carbon were seen, but further .improvements required the complete removal of the window. Boron X-rays have been
'ihrr observed by SOUK* workers (Musket ) . This moans the total energy range accessible to Si(Li) dotectors is from about 0.2 to 25 koV, and for Ge(Li)»s to 110koV or higher.
Hobert and Street dispensed with a window, but found that good vacuum conditions were needed, and even then carbon dioxide and water tended to condense on the cold surface of the detector. The film formed was thick enough to seriously attenuate the oxygon X-rays. Since they used a Si(Li) detector they wa\"iried it to room temperature undor vacuum which volatilised the film and were able to cool the detector again. Their current experience (pera.comm.) is that no deleterious effects on the detector have been noted after 30-'»0 such cycles. Others to experiment w
w i R d o w l o s f n S j £ L i ) Beezhold 2^; McCoy
206 2?3 139 et al. ; Musket find Bauer 5 Horglotz and Lynch J . Aiiothrr alternative is tlm ut,v of a very high vacuum. A
-8 vacuum of 10 torr is recommended (l<.G. Muskot, pors. conwi. ) . Completely windowlnss detectors fire commercially available
from a number of companies, from one company is also available the compromise solution of 25 |ig oin~" aluminium to protect the

10.
detoctor, together with a series of interlocks to protect the window.
7. CaJihration of detector Energy calibration of semiconductor detectors i.s easily
done by using radioactive sources of known energy, but determining the efficiency is much more difficult. Some workers (Gehrko and Lokken ; Cothern et al. ) have used X-ray-emitting radioisotopes which have been calibrated absolutely by other means but such sources are not as readily available as absolutely calibrated gamma-ray sources so Campbell and McNelles , successfully used gamma-ray sources, and a knowledge of their ratio of gamma-ray to X-ray
281 emission. Another approach (Slvinsky and Ebert ) is to measure the emission from an X-ray tube using a thin sodium iodide detector (which completely absorbs the X-rays) and compare its output with that of a semiconductor detoctor placed in the same position. Unless an analyst is attempting to calibrate his detector so that it may be used without reference to standards during analysis, such procedures should not bo needed.
8. Future detector development Si(l,i) nnd Ge(l,i) detectors both have similar ultimate
resolution, though the Ge(Li) detoctor may be 20-30^ bettor (Muggletoii ). Arc there other possible materials that might be superior? Most materials suggested for use have been developed with gamma-ray spectroscopy in mind: cadmium telluride (uomctimes dopod with cliloride ions) (PaJ.IIIM et al.,
121 j MCI gallium arsenide (Gibbons and Howes ; Fri.ant , ami mercuric iodide (Kri.ant i Swi i-rknwsU i PI a I . . These materials have what is called a larger forbidden
gap than silicon, which means that it takes niox'c X-rays to

11.
product* the same- amount of ionisation, but that the noise level, at a given temperature is less than for silicon. This probably means that the ultimate resolution of these materials will be worse than silicon or germanium, but that they may bo usable even at room fceirperaturo and notice may have* possible use in the field. They have the advantage also of being made of elements with a high Z (atomic nur.iber). This; weans that their efficiency at detecting energetic X-rays wi.l'l bo good. This is already true for germanium when compared with silicon, but these materials should be better still. The possible advantage of using energetic K- X-rays for analysis is that interferences are less in that region. II wevor, tho high Z material of the detector would give rise to prominent escape peaks.
Escape peaks arise from the following medianinm. An X~rny strikes the detector and deposits its energy within it. Ideally, none of this energy is lost, and the resulting ionisation appears at the detector output as a voltage pulse strictly proportional to the energy deposited. However, there is one important possible mechanism of energy loss which may decrease the size of the pulse. The energy of the X-ray does not immediately give ionisation in tho detector nr; its sole effect; secondary X-rays (of silicon, or germanium) are produced within the detector, create their own ionization, and so on. Normally all the secondary X-rays, having a short range, deposit energy only within the detector but in a small fraction of casos the silicon or germanium X-ray may escape from tho detector because it is near tho {surface, nnd that energy is lost to the system. This means the energy deposited by the primary X-ray is less by an amount equal to the energy pf the silicon or germanium X-ray, i.e., 1,7*1 or 9-87 keV, respectively. This happens loss than V/o of tho time for Si(Li) detectors (Frnnzgrote -*; Hcamun and Soloj.ki '* ; Voldsoth
. Actual figures range from 1$ for argon X-rays to 0.3$ for iron X-rays (Brady and Cahill ,33*') It is obvious, however, that if tho X-rays of the detector material are of high energy (such as is found for high Z inn tori a J.) then this phenomenon is more likely to bo soon because there is o

12.
higher probability such X-rays will escape from the detector. So, high Z materials vill be efficient at detecting X-rays but vill in roduce many surplus peaks into the spectrum which are analytically a source of potential interference. For this reason, very f<-v Ge(Li) detectors are used ;i»;tlytically - only a few percent of the number of Si(Li)'s. The situation is still more complicated if the detector is composed of more than one element. It therefore seems unlikely that future materials will exceed the general / . of the Si(Li) detectors available or their resolution; most developments will have to b*» on the electronic side.
One other development in the manufacture of detectors could be said to complete a historical cycle. Before the lithium-drift process was developed, 'pure' germanium was tried as a detector material and found inadequate. However, considerably purer germanium is now available and 'intrinsic' detectors are being made. These are so pure they do not need
13 lithium drifting. As early as 1971 (Baertsch ) a resolution 199 of 900 eV was recorded and Marler and Gelezunas reported
640 eV. Commercially available detectors now have resolutions of about 200 eV. These detectors are nearly an order of magnitude more expensive than conventional Go(Li) detectors of the same volume, but still of course produce the characteristic escape peaks of germanium. However, they can be warmed up to room temperature, a fatal event for a Ge(Li) detector because the lithium 'precipitates'.
Similar work has been done on using intrinsic silicon 26 (Beech and Kbcrhart ) and a resolution of 220 eV was
reported. Doth these intrinsic detectors, however, give their best resolution only at low temperature and in addition, Si(Li) detectors do not suffer from warm-up problems to the same extent aj Ge(Li) detectors.
It would seem that for practical analytical applications none of the above detectors should be considered except, the very popular Si(Li), and in special casos the small volume Ge(Li).

12a.
9. Nomenclature The next section will deal with different methods of
producinc X-ray emission from samples and their various special features. What are analytical methods based on these different methods to be called?
A survey of the literature shovr. that almost every author has his own nomenclature. This is because there is fjf.-neral dissatisfaction with the names proposed so far. The most popular set of initials used is •PIXE*, but one set of authors take these to mean 'particle-induced X-ray emission' while others understand them to stand for 'proton-induced X-ray emission'. The situation is not less cinfused when other modes of excitation are used.
Here a system is proposed which has gained wide acceptance (personal communication with most of the authors involved).

13-
The already understood nomenclature of nuclear physics should be adopted. Thus, when the exciting particle is protons the name should be (p,X) spectrometry, when it is alpha particles (<*,X) spectrometry, when it is X-rays (X,X') spectrometry, when c a m R 1^-rays (Y»X) spectrometry, etc. (it is even possible, though not recommended, to use the standard convention where »x' is any undefined particle, and lo describe the general field of analysis using charged particles; to induce X-ray emission as (x,X) spectrometry.) This has the following advantages:
1. it is the most concise terminology so far; 2. it emphasises the idea of spectrometry, the
distinguishing feature of the use of semiconductor X-ray detectors, and also emphasises thn links with other analytical methods such as alpha spectrometry, gamma-ray spectrometry, etc.;
3. it is capable of indefinite extension to other modes of excitation;
k, any acronym formed is very close in form to the original name;
5. the names, derived as they are from internationally understood terminology are self explanatory, and virtually insensitive to language change.
One other point is that using the term •fluorescence' as some authors have when the mode of excitation is other than X-rays, is an incorrect use of the term.
10. Excitation methods X-rays may be produced from a target by bombarding it with
any particle or radiation which cau«»s electrons to be ejected from their orbits. Some polemics have been indulged in by a number of workers to demonstrate the superiority of various particular methods, but their rolntivo values arc nov becoming clearer.
Different typos of excitation are tabulated in Table 1.

14.
Table 1
Radiation type Source U»«r Protons Deuterons Helium-3 particles Alpha particles
Beta Electron.)
N X-rays
Gamma-rays (X-ray region)
Br emsstrahlung Heavy ions
accelerator M H N
radioisotopes
electron guns " probes
accelerator X-ray tubes radioisotopes
accelerator
Johansson etal. 157 Shiroa et al. Shima et al.
•Z88 288 328 Vatson et al.
Franzgroto • 03 ;
Rhodes 2 2 n Neeb ot at. Jaklevic and Goulding Beaman and Solosky 2 ' Colle 62 Dyer et al. 7 ; ... Jaklrvic et al. ^
Chan and Jones 5 •.-_ La Brecque et al.
Cothern et al. Dittrich and
Cairns et al. 3
152.
7 * ; ' 88 Cothern
(i) Deuterons and helium-3 particles -Deuterons and helium-3 particles must be produced by an
accelerator, and at energies producing a good X-ray yield - the gamma radiation from these is such that a background over the entire spectrum is detected. This is of such a magnitude that these particles have never been seriously used for analysis of the elemental composition. The gamma-ray background arises from reactions like (d,p), (d,a), (d,n) on light nuclei. (ii) Bremsstrahlung -
Bremsstrahlung is radiation which is continuous and does not display sharp peaks such as are seen on an X-ray spectrum. This means that it excites secondary emission relatively regularly over the entire spectrum whereas other sources of fixed energy excite secondary radiation most effectively near the region of maximum emission. It may be produced by allowing beta particles to strike a low atomic number element. Such a
26S source (promethium-1^7 and aluminium) was usod by Rhodes ot al. , for the analysis of sulphur and cobalt in hydrocarbons
88 and by Dittrich and Cothern for the analysis of trace metal particulates in atmospheric samples. It is generally thought that the overall sensitivity obtained with this method of excitation is not competitive with other means of

15-. i/|0 67 v
excitation (IAEA, Cooper, ) by two orders of magnitude. (The reason vhy bremsstrahlung excitation is not so successful as other modes is that in gonei-al the sample scatters a good deal of radiation back into the detector. This creates a high background and reduces the sensitivity markedly.) (iii) Beta sources -
22*1 " Neeb has patented one such application. There ;-ro few applications of beta sources to routine elemental analysis. Zaitsev and Sotskov used a thulium-170 source and found they could analyse elements heavier than rubidium with
ZkU five-percent precision at the 200 ppm level. Pearson describes a rather novel system; he measured the X-ray emission from tritiated erbium films using a Si(Li) detector, the principle being that the beta particles from the tritium excited the erbium X-rays. Pearson was interested in measuring the tritium content rather than the erbium content but possibly for some special applications it might be possible to measure the elemental content of some sample by mixing xt with tritium, though other methods would usually be better. Recently, R.G. Musket (pers.coinm.) has used the tritium within a thin film to excite oxygen X-rays and hence measure the oxygen content.
Other applications of beta particle excitation are reviewed elsewhere (IAEA, J. (iv) Electrons -
Colic ct n.1 . used cl ••<•• Irons from a ?. HoV ncco.lerntor and were able to estimate the amounts of various elements in NBS standards with reasonable accuracy. However, there appeared to be difficulty in estimating elements in concentrations less than about 500 parts-per-million due to the background brem&strahlung produced by the slowing down of electrons in the sample. In spite of this severe limitation, there arc some features of this method which deserve attention. The excitation efficiency is relatively uniform over the range of elements, which means that detection of most of Liu: olcin'Mitr. is possible with one irradiation. Also, the cro.ss-fsection for

1 « > .
ionization >•? elements with ciirrcy is relatively constant for electrons, in contrast to somo t.ther particles, and hence the entire target thickness is sampled with the same X-iay production efficiency. The depth of penetration of electrons at 2 MeV in aluminium is approximately 3.7 mm compared vith about 0.030 mm for protons of the same energy and for molybdenum X-rays about 5 »w (but in tin? latter case the efficiency of excitation decreases faster than for electrons). This means that there is (as for the case of X-ray excitation) relatively little sample preparation required. Both Ge(Li) and Si(Li) detectors were used, but Si(Li) detectors were found generally more useful.
Rather analogous to the above is the use of semiconductor detectors in conjunction with scanning electron microscopes, but there are added problems and the detection limits for most elements are : ,-ound 1000 parts-per-million, or 0. 1$ ^Reed and Ware . Such systems are rarely used for quantitative analysis and we shall mention them only in passing under other headings. Some use has also been made of the electron probe (Nicholson and Hall Desborough and Heidi ).
(v) Radioactive sources -
Radioisotopes are still quite popular as sources and various different ones have been used. Tabic 2 gives a list of sources and authors who have used them in conjunction with Si(Li) or Ge(Li) detectors. The exciting radiation emitted is usually a low energy gamma-ray, or alpha particles.
The most popular sources are Am-2'll and Cd-109. For optimum excitation efficiency, it is necessary to use a number of different sources since the best excitation efficiency is obtained at an energy slightly greater than the X-ray energy of the clement being excited. Strengths of the sources range
3k up to as much as some hundred millicurics. Bonner et al. assert that 30 mCi is optimum for Cd- 109 and 500 mCi for Fe-55. Sources of this strength have the disadvantage that they may require substantial shielding and the puro gamma raya or X-rays from the source tend to be multiply scattered before reaching the sample, which means that there are many X-rays of degraded energy reaching the detector and this increases the background.
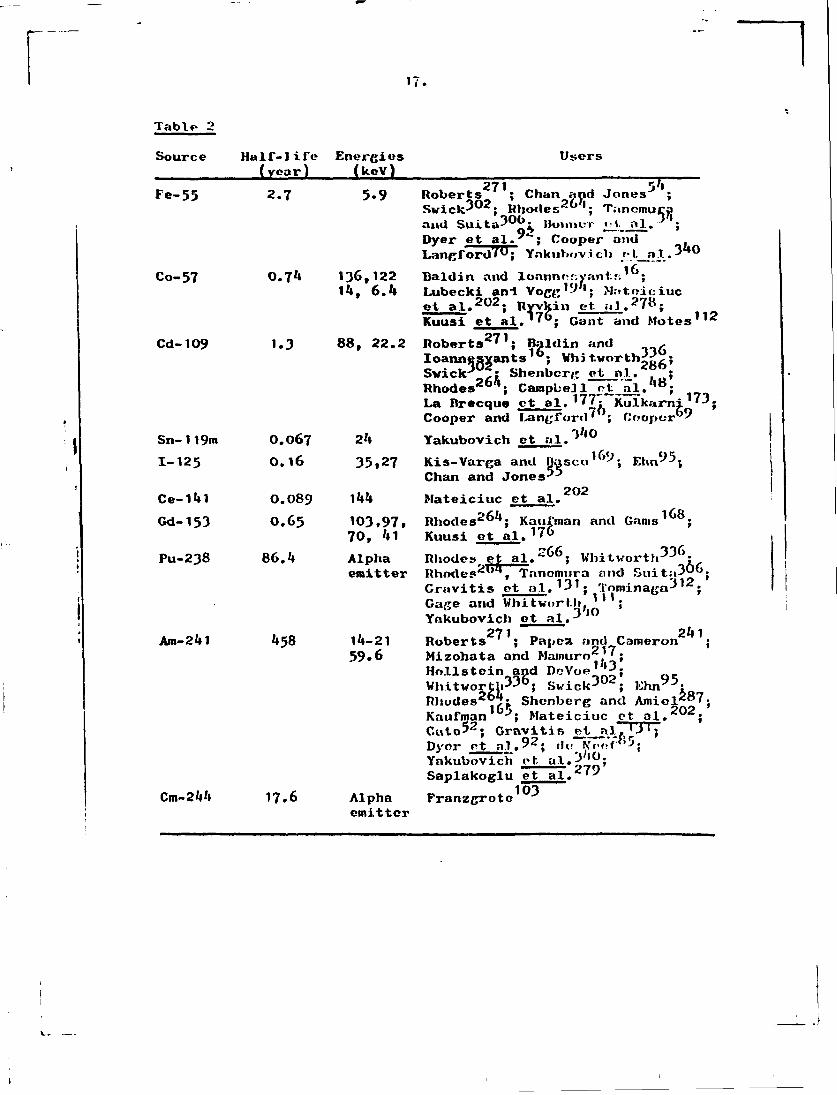
17.
Table 2 Source Half-life Energies Users
(year) (kcV) 271 54
Fe-55 2.7 5.9 Roberts ; Chan and Jones ; Swick 3 0 2; Rhodes 2 6 4; Tanomuca
02 Dyer et al. ; Cooper and Langford?0; Ynkuhovicli /•!._
Co-57 0.74 136,122 Bald in and lonnnor.yant.f;16;
and Suita? 0^ IhnmcT « 1- al. Dyer et al."*"; Cooper and Langford?^; Ynkuhov.i.cli fl_a.1.340
"l6. """ 14, 6.4 Lubecki an-l Vogg1-''1; Matniciuc
et al . 2 0 2 ; Ryvkin et. al. 27 s; Kuusi et al.' 7°; Gant and Motes 1 1 2
Cd-109 1.3 88, 22.2 Roberts 2 7 1; Baldin and , Ioannesvants16; Vhi tvorth^rL; Swick3 ,: Shenbcrg et nl.. ; Rhodes 2 6 1; Campbell otJ^L. , 8; La Brecque ct al. 1 7 7; Kulkarni '3. Cooper and Langford' ; Cooper'"
Sn-119m O.067 24 Yakubovich et al.3**0
1-125 0.16 35,27 Kis-Varga and Basco1^-, Ehn 9 5; Chan and Jones-*-
Ce-141 0.089 144 Mateiciuc et al. Gd-153 0.65 103,97, Rhodes 2 6 4; Kaufman and Gams 1 6 8;
70, 41 Kuusi ot al. 1 7» Pu-238 86.4 Alpha Rhodes et a l . 2 6 6 ; Whitvorth336"
emitter Rhodes et al. " ; Whitworth-'-' ; Rhodes2"', Tnnemura and Suit;»30". Gravitis ot at.*31; Tominaga 3 1 2; Gage and Whitwort.li>(| ' ; Yakubovich et al. 3 , 0
071 24 1 Am-241 458 14-21 Roberts ' ; Papest and Cameron ;
59»6 Mizohata and Marnuro2'7; Hollstein and DeVoe 1» 3; WhitworthVJO; Swick J U*; Ehn^0; Rhodes26:?;. Shenberg and Amiel*87. Kaufman Jx Mateiciuc et al.*"*; Cute*2; Gravitis Si.P.lTT3
Dyor et al.92• d,. NTvt'.Vly5;
Yakubovich et. al.3;»0;
Saplakoglu et al. 7 ^
TJT:
Cm-244 17.6 Alpha Franzgroto emitter
103

IK. The geometry used, and incorporated in most commercial
systems, is illustrated in fig* **(»)• It is a ring-type geometry. An interesting variant on this is described by Kneip and Laurer , and Laurer et ol. in possibly the most efficient geometry described to date. They ndded microspheres of plutonium-238 to the (liquid) sample vhich was enclosed between two layers of flexible piaster, (nee fig. M b ) ) .
95 Another variant used by Ehn who was attempting to measure the amount of silver in photographic products, was to put the sample between t*ie detector and source. The rationale for this was the difficulty of positioning the samples reproducibly. Using the described geometry, and a system which held the film against a firm support by vacuum, an analysis reproducibility of 0.2/6 was attained.
The sensitivity qtioted by various authors varies depending on the basis they use for calculating it (which is rarely
\ 175 69 cited;. Kunzendorf et al. , and Cooper claim detection limits of parts-per-million, but more typical figures 92 are kj ppm for iron (other elements worse), Dyer et ol. ;
37 112 less than 100 ppm, BurkhaIter ; 100 ppm, Gant and Motes
111 278 30 ppm, Gravitis et al. ; hO ppm, Ryvkin et al. In terms of material on air filters this normally
-2 / 269 3*» means a few tons of ngem (Rhodes et al. '', Bonner et al. 177 La Urecque et al. The precision and accuracy as Judged by intnrlaboratory comparison exercises are 5$ and
ifi 10% (Bonner et al. ^ ). The advantages of radioisotope sources are their
compactness, and their independence of power supply nnd power supply fluctuations (and hence their ability to be used in the field). Disadvantages include the lack of sensitivity of analysis compared to other systems of excitation and the presence of a continuous source.

J 8a.
(vi) X-ray tubes -
Accompanying the improvements in detectors there have been improvements in the production of X-rays from vacuum tubes. It was realised that sensitivity was a function of the background and that the normal type of X-ray tube produced a substantial background. In a normal X-ray tube (fig. 5 a ) • the X-rays emerge from the tube at right angles to the incident electron beam. In the configuration favoured by many modern workers (e.g., Jaklevic et al. •* ) , shown in fig. 5b, a transmission anode is used. The radiation which reaches the

l y .
sample has actually pu.s.ifl titrutigh the JUHMIC- itself ami been filtered. This procedure greatly reduces the enertcy spread s>f the beam. Such tubes can be relatively low power. Another modification of the X-ray tube attempts to avoid the occurrence of pulses which overlap and are rejected by the pwlse-pile-up electronics. This particular problem lias been solved by Jaklevic et al. who use a pulsed excitation .-.ourcc. When the electronics are processing a pulse, the X-ray tube is turned off momentarily so that no more pulses are produced. This system does not lead to loss of resolution and increases the maximum count rate possible by a factor of h. The electronics arc able to process pulses at near optimum rate. However, the prominent scatter peaks are the ultimate limit on spectrum accumulation rate.
A further improvemeut is the use of secondary fluorescence (fig. 5c). Merc the X-rays from the rhodium anode strike another target of molybdenum which emits X-rays of sharply defined energy with little background (Daldin and Ioannesyants •
Porter y ; Pate ; Larsen and Karttunen ). However, the efficiency of this process is much less and relatively high power tubes must be used. The actual improvement in analysis sensitivity can be as much as a factor of 10, however. Such systems are commercially available.
348 Perry et al. tried to achieve the same end by mono-chromatisation of the tube X-rays with appropriate filters, as
257 17 did Puumalainen ct al, , and Baldin and loannesyants ,
"""""~~" 296 297 while Sparks et al. ' used monochromatisation by
diffraction. Both these processes require high initial X-ray intensity.
It had been observed by Barkla, quite early, that X-rays emitted from a target were polarised and this phenomenon has been used also in an attempt to reduce the background in X-ray spectrometry, and improve the sensitivity. Dzubay ct al. and Kaufman and Camp ' used a geometry illustrated in fig. 6 in which source, sample and detector are all at right angles to each other. Tho best polarisers appear to t>v boron carbides (D.C. CnmD, pers.comm.), A 3-5-fold improvement in

20.
signal-to-noi.'jo rntio is claimed >»ut the count rate .!.«•• reduced by more than two orders of magnitude. A slightly more efficient system, vhich relies on the selective transmission of polarised radiation through a single, high quality crystal, is described by Howell ct al. 7
Since backgrounds may be lowered by monochromatisation, it is faix- to ask whether any improvement may be obtained by monochromatisation between the sample and detector as well, although this might initially seem to remove the spectral character of X-ray spectrometry. There is much merit in this question. An examination of fig. 2 shows that most of the photons entering the detector are in fact not really useful in the analysis. In fact, they are deleterious. Let us take the situation where the X-ray peaks in fig. 2 ait very small in comparison with the scatter peaks. In theory one nan increase the power on the tube and accumulate a better spectrum, but it proves that the scatter peaks are the limiting factor; the electronics can only handle a limited number of pulses per second (some tens of thousands, but even at that point the resolution starts to deteriorate), so to gain a reasonable spectrum a vast amount of time would be required. If there was some way of preventing the scatter peaks reaching the detector, it would be possible to reduce the overall time of analysis by possibly several orders of magnitude. In effect, this means that the sensitivity is improved.
It may also happen that there may be another poak in the spectrum which is unusually intense compared with the others. Such a peak may also become limiting for tin* rate at which the spectrum can be accumulated and limit the sensitivity of analysis for smaller peaks. Accordingly, a method of eliminating these particular X-rays before they reach the detector would also be most useful.
121 12? Verba ct at. '•* ' suggested a combination of crystal monochromators find Si(Li) detectors could perform this function, but a hotter system liar, been introduced 1»y .'{parks
29o ?01 VOS Pt ol. 7 * -'J .IM,| Sparks and Harris * . . A inrmorhromator and slitn is introduced between the sample and the <l»-l.r«:t.or and nnolJifr is used routinely between the source awl cample to

21.
ensure the pvir.it y of the imltioui, ri.itiiat.ion. The n»-iiin/;c»iin;n t is illustrated ii fig. ?• 1'hr mouocliroinntnrs ar<; mado of doubly-curved crystals of graphite and cost several thousand dollars each, because they must be moulded at several thousand degrees centigrade (Sparks ot M . ). Graphito in used because of its relatively high diffraction efficiency (more than fivo times better than il:; jiuarest competitor). The mono-chromators are adjustable so that any desired spectral range can be made to impinge on the detector and it is therefore possible to eliminate scatter peaks or some other prominent peak in a spectrum. Inevitably a penalty is paid for this in terms of the count rate; many photons are lost in the diffraction process and the initial intensity mtiut bo high. However, the system described by Sparks et al. is claimed to
q be ultimately capable of measuring one part in 10 of many elements. At present they have demonstrated i.heir ability to detect 0.05 ppm of mercury ir. 30 minutes counting time. The limiting factor for sensitivity v!'ll still be tbo intensity of the greatest remaining peak in the spectrum. Potentially such a monochromator system can isolate a spectral line, thus potentially removing the limit on the resolution of Si(Li) detector and Ge(Li) detector, but also it would be no longer necessary to use a Si(Li) dctoctoi at all, any counter would do.
Another possibility of monochromator use (P.S. Ong, pers.comm.) is to use a singly-curved crystal (in this case of lithium fluoride) to focus radiation into a line. This is especially well suited for the analysis of (a) certain types of air filter samples in the form of linear ».troaks, (b) needle biopses, (c) electrophoresis bands.
In spite of limitations, the hybrid system "monochromator-Si(Li)" will be increasingly useful.
As with radioisotope excitation, the excitation of the X-rays of an element is optimum if the exciting radiation is more energetic but not too much - (though this may sometimes be balanced by a greater intensi' - being available from a target of much higher atomic ntotiber), so a number of sources are desirable to cover the entire range. As a result a number of authors have produced l;uben with multiple nnodos

22.
/ 136 51 (Hubert :nid Street, ; Car.r-Brion > or an«me:s that can
pel bo changed relatively simply (Pickles and Cato ). It has 15'* been estimated by Jaklevic ot al. that the efficiency of small X-ray tubes xs equivalent to l6 curies per watt. When it is realised that 25 watts are used in these tubus it will be seen that radioisotope sources cannot easily compete for sensi tiv.' i. ly.
3'iy d i m e mentions that sensitivity criteria can
differ by three orders of magnitude. However, adopting the IUPAC criterion that the sensitivity limit is equal to three times the standard deviation of the background, there appears to be general agreement that the sensitivity for an clement which is not subject to interference is about 20 ngem (Gilfrich et al. ). This is equivalent to a r.onsitivity of 2-9 ppm of sample (Baglan et al. ; Cooper ). If a
-2 guard-ring reject system is adopted this improves to 5 ngem "I Oft
(Goulding and Jaklevic ) , and if secondary fluorescence geometry is adopted the improvement is to 0.3 ppm (1 iigcm ) (Goulding and Jaklevic ). These figures are bettor than the corresponding ones for radioisotope excitation.
It should be noted that these figures are claimed to be achieved in counting times of the order of a fow minuter,. It should also be noted that they are in terms of a thin sample consisting predominantly of carbon, nitrogen and oxygon. A rough estimate is that the figures would be four times worse
117 if the sample was silicaceous (Giauque ot al. ), and considerably better if the sample vns still thinner.
Biological matrices are perhaps artificially rvi.mpln. Most authors, however, have restricted their attention to these, because of the complex matrix corrections which may become necessary otherwise.
If the analyst is prepared to accept a ton-fold or groutoi penalty in sensitivity it is possible to analyse liquids and gases. In this case, the sensitivity is limited (especially at the low energy end) by scattering from the liquid. However, in some casus it is useful to analyse liquid.-., non-destructivoly. Table J gives a comparison of sensitivities obtained analysing the; uamc elements in the iwiinu goomutries

23 .
but in one case on f LI Lei" papT, and in the other, in liquid form in a vial with a very thin Kapton bottom. Table 3
Sensitivity of analysis for aqueous solutions and solids by X-ray spectrometry (Expressed ;•.i 3 x S.I), of background, all figures in ppm)
Mn V 189 &79 6.2 8.3
Element Sr Br As Cu Co 1 Solution 15.7 5.8 22 35 71 1 On filter 3-0 2.5 2.7 k.J '1.3 Taken from Whitehead 3 3 325 , and Wallace
Perhaps the ultimate in X-ray sources may be synchrocyclotron radiation (P. Goulding, pers.comm.). This is available at such high intensity that it may be highly filtered to become monochromatic, but there will still be more than enough intensity for the purposes required. The radiation is also inherently polarised. Lastly, it must be mentioned that any specified energy is available. Such sources will obviously not become generally available, but are excellent research tools.
In summary, X-ray excitation is versatile and will often be the method of choice. (vii) Prot ons -
The possibility of using protons from an accelerator for induction of X-ray emission attracted a good deal of attention
157 with tho publication of the work of Johansson et al. -12 This paper claimed that IO g of many elements might be
do terminable using semiconductor detection. However, (a) the criterion of sensitivity was relatively relaxed, (b) the effects of interferences were not adequately considered, (c) the difficulties of sample preparation wore iindcr-catlmfilfrt, (d) tho radiation resistance of samples and backings to proton bombardment was over-estimated. However, the paper provoked n groat deal of useful work on tho method.
Protons aro produced from accelerators, in tlio main, of the Van do Grnaff typo. They do not penetrate morn than *»0 micron into a silicacoous matrix. Since their penetrating power is much less than X-rays, the sample oi»/,e analysed is very much smaller; and X-ray absorption effects are negligible

except for very l.L{_;lit <:]'.'ineivts. Because of tho t'a:;o with which protons can be stopped, irradiations must be done under vacuum.
Protons have properties which make analysis, using them, quite different from either X-rays or radioisotopes: (i) they have significant mass, (ii) they arc electrically charged, (iii) cross-section data differ markedly from those for X-rays, (iv) their interactions with nuclei can potentially be exploited for analysis.
Because of their mass, protons give up considerable energy to the sample volume stopping them. This means that if the proton energy is 2.5 MeV, and the current is 'lO nA (both typical figures), 0.1 watt is dissipated in 0.05 cm . Neglecting heat transfer by radiation and conduction, it may be calculated that a sample could suffer a rise of temperature of as much as 30 C in one minute. This means thnt there is a high probability of thermal destruction of some samples unless they are deliberately cooled. If any of the elements volatilise on heating (and this will be encouraged by va m ) , the analysis will yield erroneous results. Alexander ot al. and Valkovic
317 et al. found that potassium, copper, and bromine, tended *»0 / to be slightly volatilised, though Cahill (who used
alpha particles) and Campbell did not find statistically significant effects. Aerosol samples seem less subject to these effects than biological samples. A survey by the group at Guelph (B, Orr, pers.comm.) has enabled them to find some conditions in which half a microamp of protons can be used without serious losses. There is some evidence that volatility is minimised if the f.ample vac dried from an acidic solution. (The problem is further documented by Johansson et al.> )
The charge on the protons affords a means of monitoring how many of them are falling on the sample. This is commonly done by electrically isolating the target chamber and recording tho charge collected over the period of the irradiation. Thin charge is usually converted to a series of voltage pulses by a current digitiser and tho pulses collected in a counter. Different runs are usually normalised to u sot amount of charge collected. If the target is very thin, sometimes tho charge

'-:>.
collecting device has been placed well behind tlie tui'gct.
Others have used rotating vanes which intersect the beam well before the target, as a collector and measurer of the target current (•/. Cookson, pers.coim*. ). In other crises, when the pvolon iJLux is very low much more sophisticated techniques must be applied.
It phrmld aJ so be remembered that a certain percentage of a beam after pairing through a collimator may consist of neutral atoms; these will produce X-rays on a target but will not be registered by a current measuring device.
Vhon protons strike a sample, secondary electrons are produced. If these fly off the sample and are lost, the
158 recorded current will also be erroneous. Johansson et al. used 150V bias to suppress these electrons. Others
73 (Coote and Whitehead ) constructed the chamber in such a way that the secondary electrons did not escape and the total overall change in the charge was measured. If the sample is not a conducting one, then it may become quite highly charged, and charge digitisation may be inaccurate. Worse still, the electrical fields involved may produce intense bremsstrahlung from electrons slowing down, which produce a high background under the entire spectrum. There are two ways of avoiding charge build-up: (i) coat the sample with a very thin layer of carbon, or (ii) place a hot filament near the target. The electrons from the filament will neutralise the excess charge (Shabason et al. ) . This latter approach creates difficulties with measurement of the actual charge collected and can deposit a layer >f > ngsten on the target, but the former approach may introduce extra impurities, take extra time, and introduce an error in the analysis because it is portly carbon, not only the sample, which is being analysed. Obviously the answer is to ensure that the carbon film is very pure and very thin.
In the case of excitation by X-rays, the sensitivity decreased towards the regions of lower Z. For protons the reverse is seen, the sensitivity is best for low Z elements and decreases nt higher ?/. The situation is complicated by
(see fig. 3)

26.
bremsstrahlung as seen in the : ;»cctrum of fig. P. Tbi;; bi-omsstrahiung results chiefly irom the slowing down of electrons in nuclear fields and has a cut-off point of *» m e*E^ n c/M , vliere m e is the mass of the electron, M that of the particle and E±nc is the incident energy. This means that tlie most sensitive region for analysis is in the middle of the spectrum because the crfiss-scctions for X-ray production are high at low Z. This sensitivity is reduced in the bremsstrahlung region, whercau at high Z there L no brerosstrahlung but the cross-sections fall rather rapidly. This is true not only for protons but other particles such as alpha particles (Perry and Brady ). .
There are two ways currently baing investigated of reducing the bremsstrahlung background. The first arises from the observation that a proportion of it is produced not in the sample itself, but in the surrounding target holder, and sample chamber. It seems the electrons produced by particle bombardment may be ejected from the sample, in some cases. It may be possible to use high magnetic fields to deflect them to a remote locality, and preliminary results suggest the idea is sufficiently promising to merit more detailed appraisal, which is currently in progress (M. Mantel, pers.comm. ).
The second way of reducing bremsstrahlung arises from the fact that it is not Isotropic. Therefore, suitable choice of beam-samplo-detcctor geometry enables one to reduce the background by as much as a factor of 2 (M. Thomas, pors.comm.).
Although the sensitivity is not optimum, the low 7. peaks in an X-ray spectrum oxer tod 5 • 2.'; MeV proton.1; art; still very prominent because the background under tliein is so high. This means that pile-up peaks are worse in proton excitation than for X-ray excitation. It is possible to avoid l,hi.« problem by putting a low Z filter between the target and tins
, 318 detector. Kilters used include polystyrene (V.-i.l kovi.e ct a.1 .
332 276 Wheeler et .-.l. ), Incite (Rudolph nfc nl. »
Gordon and Krnner. " ), 250 micron Mylar (McCoy el. nl, """ 73 and Whatman's no. 'l 1 filter paper (Coote and Whitehead These filters attenuate the low energy X-rays

dramatically, nn<l also prevoi: I. ycatti'V'l ju-ni.niiii reaching tlio detecto)-. Selective filters have been used to emphasise other parts of the spectrum (l)odurd ft al. ).
Protons are scattered from the target to all parts of tho chamber containing then, T.> p: .-wnii I lie '; t ,>y:: lint:; jiroducfil from reaching the detectors, it is advisable to coat the chamber vith filter paper of lti.{;h purity (Uob.iiison ot, al,,*
o"3 or some other innocuous substance. Colli; ot al. used a Cu-Al-plastic combination. Protons are also scattered directly towards the detector and if the beryllium window is thin they may pass through creating large spurious pulses. These pulses are also prevented from reaching the detector by the filter, which can readily stop 2.5 MeV protons. If specific analysis of the low Z components i..*; desired it is possible to run tho accelerator at a lower voltage, nay, 1 MeV when the brenisstrahlung is lower in energy, the sensitivity for the low Z elements is better and the protons are not sufficiently energetic to penetrate the beryllium window from which the filter lias now been removed. However, operation at two energies is less convenient and an ingenious vay out has
13/1 been suggested by Harrison and Eldred and T.A. Cahill (pers.comm.). They use a filter with a pinhole in it. This procedure greatly attenuates the low energy X-rays but does not eliminate them completely. Good coverage of a wide range of energies is achieved.
Tho other use of scattered protons is for supplementary analysis. The protons are scattered by the sample in a way which depends on the atomic numbers of the* elements involved. A semiconductor detector designed to detect particles, rather than X-rays, is put near the sample and from the particle spectrum (if the target is thin enough) it is possible to determine elements from hydrogen up to about sodium (Larson
185 98% et al. ; Feldman ct al. ).. Use of thick targets demands tho use of much more complex data reduction techniques, though this appears to bo increasingly tractable, Pew workers have installed a dual system such ,' this, however. This typo of nuclear scattering is one example eif nuclear reactions which will be occurring at these encrgio.'*, Rome of these nuclear

28.
reactions can tl!"f>r" t.L<ally produce X-ray;; which potentially may be seen in the X-ray spectrum, but the cross-sections are such that no interference will be noted in practice. Only at about lO MeV do such X-ray interferences become visible (Herman et al. ).
It is alro possible in theory to perform Auger analysis simultaneously Li»u r.aining information about chemical structure
22"! as well as chemical composition (Musket and Uaucr ).
Since liquid samples volatilise in a vacuum, it is generally difficult to analyse these using proton excitation,
327 though Walter et al. "* found that crude oil could be introduced into a vacuum vithout significant volatilisation. Hovever, the n.nin application to analysis of liquids is by Groenwall J . He used a system where the beam of protons passed through a very thin layer of the polyimide Kapton into a cell containing a sample of motor fuel which was being analysed for bromine and lead. The analysis was satisffctory but the protons caused radiation damage to the fuel and decomposition products built up as a layer on the window.
139 Herglotz and Lynch , who used X-i'ay excitation instead, 28 I did not encounter this problem. See also Seaman and Shone
Earliej the technique of pulsing an X-ray tube was mentioned, which gives maximum count rate with minimum distortion of the spectrum. A similar technique is possible with an accelerator. Tn this case, the beam is generally deflected momentarily, using electrostatic plates instead of turning the accelerator beam off (Thibeau ct nl. ;
I3U . Harrison ant' !;.t.d 1 M' ; Nelson, per:;, comin. ) > Thi:, if, a more complicated system than that for X-ray tube control.
One feature true of a charged particle beam, not true of a beam of X-ray.s, is the case with which it can bo .sleeved by electrostatic or magnetic lenses (though ins tinmen Is incorporating beam.? of scanner! X-rays are available (Palmborg, pers.comin. )), This maizes it possible to focus or do-focus a beam on a sample, and more important, to scan a sample to gain information about the spatial distribution of elements. This technique is veil known for tno electron probe anil the scanning

2!>.
electron inicio.if-i);p'-, but relatively few groups have m;>dc charged particle probes with light ions, thoti.'di the ion probe (for example, argon atoms) is being used increasingly.
252 The proton probe h'is been used by Poole and Shaw ,
Cookiion t-1 .'> 1. ' " , Dimlup e>. al. , Peisach o I. n 1. " , 79 ''23 S7
Deconninck , Musket and Jlauor , Cbo et al.^ , specifically for the production of X-rays, though
other groups and those workers have also used other nuclear reactions for the determination of spatial localisation of elements .
There appears to be two approaches to proton probe production, firstly, to rely on collimation to achieve the small bdun size; secondly, to rely on magnetic focussing. Spot sizes realised with the methods are 25 and 3 microns, respectively (Cookson vt al. , Dun .lop cl al. ). With the first approach, the sample itself must be moved, since the collimators which define it are fixed. The other seems rather more flexible.
One group (Doconninck et rtl. . pers. conm. ) has found the use of the standard copper m i e n mesli disk used for electron microscopy completely prevents charge build-up, A small disk is placed in physical contact with the sample, and is grounded. The beam is scanned within a square delimited by the mesh of the disk.
It should finally be noted that the maximum y.'eld of X-rays is obtained with protons at 10 MeV (Herman ct al. ) , but since brornsstrahlung increases faster than yield (Valkovic
317 1'»0 '4 1 '**
et al. 2.2 MeV is stated as tho optimum energy (Herman et al.
(viii) Alpha particles -
Alpha particles have not been used as extensively as protons and most of tho same comments apply to thoin an to protons except that the maximum yield of X-rays is at '18 MeV
126 and the optimum sensitivity at about 15 MeV (Cordon and Kraner ), In general it may be said that excitation with alpha
particles gives better sensitivity for tho higher Z element in comparison with protons, but for the intermediate range of

30.
67 elements the sensitivity is not as good (Cooper ). An entirely viable analysis system can be built on this excitation
39 method, however, and Cahill ~" has performed simultaneous (a,a) spectroscopy and routinely determines a very wide rang* of elements with highly automated instrumentation. Protons as an excitation source seem generally preferable, especially since the alpha particles require much higher energy accelerators. (ix) Heavy ions -
Various types of heavy ions have been used to excite X-rays from samples (Deconninck et al. { - Cairns et al. ;
60 ^ M ._ ^ . 2*6, Close and Bearse ; Peisach et al. ) with
140, energies as high as 120 NeV (Purser * ). . The maximum yield occurs at about 200-300 NeV (Herman at al. ) and the optimum sensitivity is presumably about 80 MeV.
Vith heavy ions there is the possibility of another phenomenon intruding! molecular X-rays. It is observed that when a beam of heavy ions strikes a target, there in a possibility of transient formation of pseudomolecules from which X-rays of a much higher than expected energy may be
2 las observed (Metz ). This does not seem to have been used frequently for analytical purposes. The situation as regards overall sensitivity does not seem clear. A sensitivity of a
2 56 few tens of nanograms per cm is claimed by Chemin et al. , 284 . 10 ppra by Shabaaon et al. This suggests that heavy ions are slightly inferior to other particles commonly
140 285 used and this is confirmed by Herman et al. and Sharma J
who compared protons, alpha particles, and heavy &ons. 90 Duggan et al. claim a sensitivity of about a picogram of an element, but this is for an isolated element on a very thin film and does not take adequate account of the background produced by nuclear reactions and electron bremsstrahlung. There are two possible pitfalls in the use of heavy ions.
Firstly, there is some possibility of the composition of the matrix influencing the X-ray cross-sections; and secondly, there is also the possibility of changes in the shape, and ratio of X-ray peaks from a single element, depending on the molecular structure within the sample (R.L. Watson, pere.comm,),

31.
Vork continues on the use of such ions, however, because the cross-sections for X-ray production are sometimes of the
4 order of 10 barns. Other excitation methods have cross-sections tvo orders of magnitude lover.
Finally, it must be mentioned that whether for protons, alpha particles, or heavier ions, accelerators do not usually operate efficiently at the region of a few nanoamps which is the optimum current range. They are designed to deliver micro-amps, and hence a means must be provided to reduce the intensity of the beam. Many workers have used collimation (Herman et al. l f c lj Walter et al. 3 2 7 ; Bearse et al. 2 5t
Campbell et al. 8j Jundt et al. 3; tout the material chosen must be. such that it is either a long way away from the detector and sample or is made of a material such as graphite (Plocchini et al. ),] which does not contribute to significant X-rays* Another approach is to use a beam sieve. The beam is intercepted by a tantalum plate with very small holes drilled in it. The amount of the beam that passes through is focussed to a point and then scanned over the sample. This can be done in a simple way by using a magnetic double quadrupole lens and to each half, applying alternating current of differing frequency (O'Brien et al. ^ ).
11. Matrix effects Since the X-rays and gamma-rays from some sources referred to
above /penetrate the sample to a relatively great depth it follows that the X-rays emitted may be strongly absorbed before emerging from the sample. This may lead to (a) non-linear calibration curves of emission versus concentration for some elements (Cate 5 ; . Chan and Jones ' " ) , though others are 287 linear over extended ranges (Shenberg and Amiel. $ Gant and Motes 1 1 2j Kaufman l 6 5 j Oravitis et al. 1 3 1f (b) increased problems due to matrix.effects. Primary fluorescent X-.ra/s absorbed within the sample tend to excite secondary fluorescence from other elements in the material, which means that- the apparent composition obtained may differ from the real one. The problems are well known in traditional X-ray fluorescence and six methods of approach ere discussed below.

3?.
1. It is possible to use a variety of : Londardr mid calculate the effects of various component* on the absorption
266 103 of X-rays. Rhodes ot al. ; Franzgrote \ and de 85 ———^
Neef » describe such procedures. 2. Ano!.l«f\r approach ir; an ilerutiv om t via Mi*;
computer. The apparent composition is taken and from tabulated absorption coefficients the actual emission that should bo seen is derived. This is compared with the observed emissions and the concentrations changed, and the emissions re-calculated until the degree of divergence is lower than some arbitrary
l69 minimum. Kis-Varga and Basco have applied t h i s to manganese nodule analysis. So far more use has been made of this technique in traditional X-ray i'.uoresccnce than with semiconductor detectors, perhaps because in traditional X-ray fluorescence corrections are made for one element at a time.
3. A third method of allowing for matrix effects is to use the ratio of the fluorescent to the Compton radiation as
19*> vas done by Lubocki and Vogg . This relics on the principle that the intensity of scattered radiation from a sample could be a measure of its X-ray absorption coefficients.
h. It is possible to reduce the thickness of the sample until the absorption of X-rays is minima).. This has the disadvantage that the sensitivity is decreased but the increased accuracy compensates for this. It will be intuitively apparent that the definition of what constitutes a thin sample must be arbitrary, depending on the degree of absorbanco one
26/1 is prepared to tolerate, but Rhodes has given a definition which has gained acceptance. Me defines a thin sample as one obeying the criterion:
mass ^ 0. 1/n where u in cm /g is the sum of the mass absorption coefficients*
2 17 M-Lzolinta and Namtiro have nugget; |.cd that Wic criterion should be that the sample departs y$> from n condition of non-absorbancc. This criterion is twice as stringent as that of Rhodes;.
5* Matrix corrections are also possible by measuring tho

33.
absorption coefficient of the sample directly (Sparks and Ogle )i that is, by passing X-rays through the sample and
measuring the absorption. 6. Another solution is to dilute samples vith a suitable
substance like high purity cellulose, so that the matrix is effectively the same in all cases (Glauque et al. '''; Swick-*02).
297 Of these five methods, -6. is most precise though with not as good sensitivity.
12. Data reduction The areas of the peaks in an X-ray spectrum are related to
these areas the concentrations of the elements in the sample and/must bo obtained by mathematical methods. There are a number of methods available for processing the accumulated spectrum. The most simple, the height of a peak as the measure of the concentration of the element, does not appear to be used. Some authors, however, perform simple integration of the number of counts in a peak (Cooper ). The other extremes are represented by those users who have modified the gamma-ray code SAMPO for X-ray use (B. Gordon, M. Hillman, M. Peisach, pers.comin. ). Usually, there is a background to be removed and there are a number of ways of doing this.
Perhaps the most sophisticated approach is that marketed commercially in one system, where the spectrum is converted into its Fourier transform. Very, approximately this is a representation of the occurrence of high and low frequency events in the original spectrum. The background is represented in the low frequency area, and is removed. The transformation is reversed and the peaks are then displayed free of background. This approach seems successful, though orror

3«».
calculation becomes complex. Fast Fourier transform algorithms available these days m a n that only a Moderate astount of computer time is used.
The sort traditional approach to background removal is to take two points cither side of a peak, where there is no significant contribution frost the peak itself and interpolate a straight line bctveen thea. This line is assumed to represent the background. The difficulty is that a spectrum may veil be sufficiently complex to prohibit the operation. Peaks may overlap and other approaches are better.
One of these is to record a background in the configuration in which th« analysis will be done and subtract this from the final spectrum, using a computer (Harrison and Eldred ; Cahill ct al. Since the shape of the background is a function of the elements in the sample, the shape of the blank will seldom correspond exactly to the background in the real sample. It is therefore adjusted in shape until it does fit and the result subtracted.
Another method of background subtraction is to perform five-point smoothing on the spectrum, and then to repeat this smoothing one thousand ti.-n«»o. This strange procedure has the result of completely smoothing out the peaks present, so that only a spectrum representing the background remains. This background spectrum is then subtracted from the original spectrum. If large peaks are present, a distorted final background spectrum is obtained, and the procedure must be modified. One must compare the smoothed and unsmoothed spectrum point by point and select the point which is numerically the smaller of each pair. This Masses the whole process towards reducing the size of any peaks present, and gives an adequate measure of the background, when the many smoothings are complete. Contrary to expectation, the amount of computer cime used is relatively small (H. Kaufman, pers.comm.),
The remaining peaks are fitted by a least square* procedure. This involves fitting known peak shapes to the spectrum obtained. Peaks obtained in a typical X-ray spectrum are approximately Gaussian in shape and fitting this function

3 . " J .
to a series of points involves n non-linear least square fitting procedure (Valkovic et al. ; , Jundt ot al. ; Tominaga 3 ' 2 ; Waj U-r ot al. 3 2 7 ; Heath V ^ i Kunzendorf 1 7'
which is usually a little time-consuming on a medium-sized computer. A MI;O!«I.|I • ng procucliu'o i r; usually employer! before this is done, because the randomness of the points in the curve is slightly oisturbetl by the fat: I. tli.it they are recorded in channels a set. distance apart. Simpler programs are used in time sharing systems (Marr UJUI Campbell ). The actual identification of the peaks depends on where they are and most workers locate them using either a cross-
/ 1'3'l ?0'> correlation technique (Harrison and Eldrod ; McCallum ;
Heath • ) , or taking the second derivative of the spectrum. The former authors maintain that the first approach is better.
Using location programs it is possible to locate a peak to within 12 eV (Cahill et al. ; Harrison and Eldred )
which is usually sufficient to differentiate it from other elements omitting X-rays in the vicinity. However, a few interferences cannot easily be treated in this manner such as the interference of the lead L-alpha with the arsenic K-alpha at 10.5 koV. To decide on the concentration of these, the other lines which do not suffer interference must be used I.Walter ), or in other terms, the whole series of lines for a given element must be fitted at once. Some programs
/ 159 have not allowed for interferences at all (Johansson et al.
The limit;, to thin system are determined by the staticl :i c.r,. Thus, even if its position is known very accurately, it is not possible to determine accurately the magnitude of a vory small amount of an element in close proximity to that of a large amount of an adjacent element, a point, well emphasised by
122 Gilfrich .
The width of the Gaussian fitted must vary with onorcy, so allowance must bo made for this. Another complication is introduced by the possibility of the resolution (that is, the peak width) varying with the couit rate. This is vory hard to

3<».
allow for i>v!'ii 11 the cunt i\t'«- if- loiowu. Some workers prefer to record a library of actual spectra, store them in the computer and compare these with the real spectrum {Cooper ) .
Another feature rarely inoi-po' a led in programs is allowance for escape peaks and (especially in (p,X) spectrometry) any pile-up peaks, and provision for M- X-rays. Many programs, however, incorporate ••• <•:• I cul ation of the goodno:;r: of fit (expressed in terms of the chi-squared value).
The most sophisticated program currently available appears to be that of II. Kaufman (pers.comm.). It involves simultaneous non-linear least-squares fitting of 12 parameters and many
2 linear parameter:;, and consistently yields "X values of lens than or near to I. The parameters arc only allowed to vary between p r e s e t l i m i t s or i n c a r i l n ^ i ^ s r e s u l t s may be o b t a i n e d . The average analysis time per spectrum is five minutes on a computer equivalent in speed to a CDC 6.500, and requires 32k to run. A description of the principles involved has been published (Kaufman ).
In contrast, using other criteria, quite simple programs 136 may yield acceptable results. Hebert and Street
describe a system which relies mainly on .simple integration of peaks, but yields overall elemental concentrations for rock which ultimately sum to 100 +_ 0.5$.
This may mean that for many applications great sophistication is not necessary. Some workers maintain they have found little difference with tlie use of complex as opposed to simple programs (if. Hudolph, pers.ooinm. ). (An intorlaboratory comparison exercise suggested the same conclusion (Camp '.;t a.1.. , pers. conim. ) This issue is being discussed at the 1975 Denver conference, and Advances in X-ray Analysis, vol. 19» should be consul ted for details.
13. Thin sample techniques
There is considerable advantage in keeping the sampl e ;i> thin ns possible, and considerable effort lias been devoted to

37.
the manufacture- of thin samples am! l>:ickj •»;•,.*•. This L:; als;o useful in conventional X-ray fluorescence. furity of backing material is essential and for this reason glass fibre filter paper is universally discarded; it has high level of v.Lnc, iron
PA*** and barium, and other elements (Rbod'*.*- otM.1 . ' ) . Normal cellulose filter papers, however, are surprisingly puro if they have been prepared villi ;n m- i.*l wash in 1.1K- manufacture. Those recommended as having specialty low ) i-vc.'l :; «>l' trac:e elements
?67. , 'I5,'i6> include Whatman* s no. k^ (Rhodes ct al.*" ; Camp ) , but Whatman's no. '»2 seems as good and Whatman's no. hk, which is
o thinner than the usual 6 mgem is better still (Whitehead, 197f». unpublished).
/ l'l Other backings used include Mylar (Raglan ct al. ; Hobert
and Street 3 ; Cahill ct al. ' f 1 ; Rudolph ot a l . 2 7 ; Watson opCi O I F I O I
et al. ; Umbarger el --11. y ; FloccMnj pt al. ; Cooper 70 "*
and Langford ) which is usually less beam-resistant than the polymide Kapton (Flocchini et al. ; Whitehead ; Umbarger et al. ; Gordon and Kruiier ; Cahi'Ll c t. nl . "; Barnes et al.'' ), This material has the advantage of being able to endure tiie effects of strong acid. The same cannot be .said of Formvar, which 1ms, however, tin* advantage of being purer and being able to be prepared in the thickness desired. However, Formvar films are easily ruptured (Baglan et. a.1. ; Walter et al. ; Hearse et al. J) and are stronger if alumini ,«>ed (Valkovic ;
Q 117 16l Alexander et al. ; Valkovic et al. ; Jundt ct al. ) .
;*.5 Polycarbonate film has been used bv Camp ; Cate and Pickles
.53 and Jundt et al. 163 Teflon has been vised but compared with other materials gives a higher background for the same thickness (Cahill et al. "; Flocchini et nl. ). Dobtaphan has also been used (Johansson et al. ) but gives four times the spectral background of equivalent carbon film. Carbon films have been extensively used. They can be made very thin, but are fragile (Young ; Christenscn et. n.l. ; Johansson et al. p ; Umbargor ot a 1. '; Gordon and Kraner " ; Herman et al. ; Rudolph rt a.1. ). Their content of trace elements can be low.
t,

38.
Another film which can be prepared from an organic solution in the thickness desired is polystyrene (Johansson
160 227v et al. ; Nelson et al. J. This may be suitably prepared by stretching while hot (P.S. Ong, pers.comm.). Membrane filters are preferred by many, especially when aerosol samples are being examined. Of these, favourites are Millipore (Nelson ot al. '; , Barnes et al. ; Camp ), and
/ 46 343 119 Nucleopore (Camp ; Young et al. ; Giauque nt al. ;
Nelson et al. ' ) . . Colle ot al. ^ soaked photographic lens tissue with the solution to be examined.
The sample may sometimes tend to detach from these type of backings on irradiation (Johansson et al. ; Brady and
36 — — — Cahill ), therefore another foil is put on top of the sample forming a kind of envelope (P.S. Ong, pers.comm.). A
53 similar type of system is advocated by Cate and Pickles and J.V. Nelson (pers.comm.). Here a thin layer of paraffin or an adhesive is sprayed on. Such systems are useful for X-ray excitation but pose dangers foz* charged particle work, in view of the stopping power they present to the beam.
It is even possible to analyse a sample in the form of a / 53
thin film by dusting it onto adhesive tape (Cate and Pickles ; Cothern ct al. ). (The resulting precision is
surprisingly £ 1-2$). The trace element content of these tapes varies (Hebert and Street-* ).
Another technique described by various authors (Akselsson et al. 5{ Herman ot al. 1/'°»11*1. Campbell ct al. j
163 327 Jundt ot al. ; Walter et al. ) is to cut very thin microtome sections of a sample and mount thorn on a suitable backing foil such as carbon. The normal paraffin embedding technique appears to add trace elements to the sample and microtoming of a sample frozen by liquid nitrogen or other means appears superior.
Many samples can be simply driod onto a thin backing (though the thinnest backings tend to rip whon drying occurs), 119% but for other solid samples, Giauque et al. ') have evolved a worthwhile system: the sample dust (finer than 32'j mesh) is scattered in a suitable evacuated chamber out of
,}

y>.
vhic I: i ";idb a tiltei tluuugh uhich the air is being e\<vaeted. A put C" of ;iir is ;tdmi I ted to the chamber via a suitable valve and the dust is then caught up and deposited on the filter.
Another technique of thin film manufacture wa3 introduced 16" by j'j.lly and White . This involves the honnigonj sation
oT the sample in a Waring bleudor Pnd then spraying A* onto a thin backing, tiding a uebuliser. This technique has the disadvantage Tor harder substances that impurities are very likely to be introduced by the blades of the blendor (Jundt J ) t
327 but has an appealing universality. Walter et al. have used a similar technique. Some workers have found the technique gives pool- recoveries (Brady and Cahill J.
A further technique of thin sample preparation exploited by Bowman ct al. " is the specific analysis of hydrogen sulphide in waters. The sulphur is estimated by plating out onto a silver surface, which takes more than an hour, but yields finally nn overall sensitivity of 100 ppb for hydrogen sulphide in water.
It is also possible to homogenise a polar substance in a non-polar substuiicc such as chloroform, and simply filter it (D. Govilon, pcrs.cumin. ). Some elements tend to be more hoino-genef.joly distributed than others under this system.
Another type of system should also be mentioned. Scattering Cciuses background and lowers the sensitivity, so if scattering can be reduced improvements result. A novel way of doing this is to place a thin layer of the sample on an optically flat surface and cause a narrow beam of X-rays to strike the sample at a very small angle, such that reflection takes place in the sample, not the backing. The X-rays are not significantly attenuated when they emerge and sensitivities
— 2 2 3 of 5 ngem can be obtained (Aiginger and Wobrauschek , J
3*» 1 x Yoneda and Horiuchi y. If thin samples are used, thin standards may be used.
These may take the form of paper or tissue soaked with the standard (Colle '; Camp et al. ' ) .

'»0.
For (X,X') spectrometry, cellulose powder has been soaked in the standard solution, dried, and then pressed into pellets (Giauque et al. which would seem more satisfactory.
306 Tanemura and Suita adopted the novel approach of making 74 thin polymer films as standards, while Cothern et al.
used metal deposits of known weight sputtered onto a thin aluminium backing. Other workers have measured the amount of material on a thin backing by optical menus. It should also be mentioned that one commercial firm has produced homogeneous standards of various elements in uniform thin sheets of gelatine (Cothern et al. ). Another rather elaborate but apparently successful technique is intended for electron microprobe work, but could be used elsewhere: Rowse et. al. '
took oxides and silicates of various elements ground to 0.5 micron and made a slurry which was embedded, using a resin. The resulting block was microtomed and thin standards resulted.
In most cases it would seem simpler to use one of the thin plastic backings and evaporate a known amount of a solution of ono or more elements on it, but care must be taken to avoid inhomogeneities.
It is possible, however, if the sample is initially in solution, or at least in a liquid form, to use internal standards. Such an element must be chosen to be easily distinguishable from others in the spectrum and must be present in the sample in negligible quantities. Such elements include palladium (Bearse et al. -*), , nickel and vanadium (Ong et *l»f^?'
It •* T^2 -* • Camp ), caesium (Wheeler et al. . ; Pape
p/if) T^2 et al. ) anrl yttrium (Wheeler ctjih J J ).' However, in the latter case it would appear that the precision of analysis was actually lessened by the introduction of the internal standard.
All techniques of thin film preparation, however, have one major disadvantage: they ultimately do not improve the dotectability of minor components of the sample. The resolution of the detector is limited and the ultimate limit to counting rate and dctectability is the major components in the material. It is for this reason that many workers (e.g., 138 Goulding and Jaklevic mainly consider the type of oauiple

l*\.
which is largely composed of carbon, hydrogen and oxygen, none of which contribute X-rays to the spectrum. However, even for these, the count rate is limited by iron, and also cobalt X-rays (next in atomic number to iron) are usually not seen in X-ray spectra beine obscured by the iron X-rr»ys. Jt is clear that the full potential of X-ray spectrometry cannot be realised unless chemical treatments are resorted to, to remove the bulk elements from the sample. This has th<> difficulty that chemical treatments of various kinds may add elements at trace 'levels, possibly at levels approximately those in the sample. It also raises the possibility of incomplete separation of the element(s) sought, and vastly decreases accuracy of analysis.
If one is prepared to sacrifice some sensitivity, many techniques are available for preparing samples in bulk form. The traditional lithium metaborate fusion used in more conventional X-ray fluorescence work has been used by Hebert and Street J , and Giauque et al. " . I f the sample is a suspension, it is of course possible to filter it through a filter paper and a suitable sample is immediately obtained (Rhodes ; Giauque et al. •• Giauque and Jaklevic .
It is also possible to compress the target material into 119
a self-supporting slab under about 25 tonnes (Giauque et al. This must be done between layers of thin clean plastic.
(Another technique is to eliminate matrix, effects by mixing the sample thoroughly with either graphite (Bodart et al. .
or cellulose powder (Giauque and Jaklovic ), or polyacrimide gel (Kulkarni ). ^k^ Chemical pre-treatment
One of the first procedures for concentrating the elements in the sample is to ash the sample. Core must bo taken with this procedure ns some elements such as zinc and lend,
29 volatilise from some samples on dry ashing. Bergman ct n.l.
.suggest that the addition of sulphur prevents loss of some volatiles but it is not clear how general this is. Vet ashing is much more elegant because the resulting oolution can

U2.
be dried onto a suitable backing. Wet ashing is done using a preliminary oxidation with nitric acid and perchloric acid. There is some risk, and standard references should be consulted for safety precautions.
These procedures have the result of removing any organic matrix which may be present. The usual ash content of a biological sample is of the order of 5?&t so in theory a 20-fold concentration may be obtained, but it will be observed that the elements which are count-rate limiting remain the same as before. This does not improve the detectability of elements whose X-rays are alongside those which are more intense. The
lit same comment applies to freeze-drying (Bonner et al. J ). The overall result of this is that on ashing, as reported by Ong et al. J J
t the background improved by a factor of 3 327 only. Walter et al. also concur in. this. It must be
concluded that the results of ashing and freeze-drying techniques are hardly worth the trouble.
Other chemical treatments have included precipitation, ion-exchange concentration and solvent extraction. These are also applicable to X-ray fluorescence. Precipitation has boon
188 I8'i used by Law and Campbell - , and Larsen and Karttunen , 203 and Mathieson , but it should be noted that to precipitate a trace element it may be necessary to add a significant amount of another element, and this may counterbalance any concentration achieved. The precipitant automatically becomes the count-rate-limiting element. However, effective use can be made of selective organic chelators which can precipitate metals. No other measurable X-rayn are added to the spectrum. The simplest system (Brady and Cahill J ), uses precipitation as dithiocarbamate complexes.
Solvent extraction has been attempted for tungsten 1 lift
(Hubbard and Green ) and probably other elements as well. The degree of selectivity may be high and often the organic materials which are used can be removed fairly easily. On the basis of the above quoted work, and unpublished work of this author, it would seem that determination limits of about 0.1 ppm arc possible.

h).
A /'.'real dc.il <>[' wm I; has been ruuccn tratod oil ion exchange procedures. 'Jhere are two possibilities hc~e: the element of interest, may bo oluted from the column and the cluato dried onto a backing, or the resin itself may be counted in some instances. Another approach is to use a filter paper impregnated with ion-exchange resin.
Liquid ion exchangers can also bo used and are r.ai.d to be faster (Van Niekerk and J)_» Wet " ). Since the application will depend so much on the sample and elements to be analysed, only general comments can be made here. In general, the higher the charge on a cation, the stronger it is attached to a cttion resin, but the elutirtg reagent may alter this considerably. It is relatively simple to separate alkali metals (common major components) from the others present ( K . C . Bearse, pers. coinra, ). It is seldom important to remove anions in the some way because they are usually low Z elements and as such do not give rise to X-rays capable of causing interferences in the spectrum. II .should be noted, however, that some of the rarer metals whi'h have the ability to form oxyanions are strongly retained by anion exchange resin. For example, chromium, molybdenum and tungsten were found to be absorbed (and scparntrd) on anion exchangers by Pakbolkov and Panikarovskikh ** ).
Ion exchange concentration has also been used by Van Niekerk and D P Wet • * (iron, manganese, aluminium, and sulphate - yields averaged 90^); Bergmann et al. (nickel, crbalt, iron, chromium, manganese and titanium! calcium was removed u:>ing citrate) Huhhard and Green . Fuk.Hsawa
(impurities in tantalum; aluminium in zirconium; cadmium in uranium; col).ill. in nickel; metals in water; aluminium in
327 steels and impurities in aluminium), and Walter et al. o Other authors (Allen and Rose ) have commented on the effect of particle size on the apparent concentration of the clement copper (using conventional X-ray fluorescence). The resin had to be fine (56 micron, 2'i0 mesh) for there to be no effect noticeable.
19 I Lienor describes the manufacture of sulphonated

' • ' » .
polystyrene splicrcs lining hot concentrated sulphuric acid. Sinco those are only supox-ficially sulphnnatcd, much Pastor exchange is possible. It is possible to extend the principle to sulphonating the surface of polystyrene she ;ts. Cations ahsorbod on those forr>i M very thin layer .indeed and there should be no X-ray absorption effects, though the ion-exchange capacity would he very low. Thir. has been exploited by DJucrnholm and Loderer , and Holynska cfc al. ° The time taken for absorption of 90$o of cations onto such u ;iiu'i'ui:c is about four hours for some elements, but detection limits of 0.01 -0.1 ppm are claimed.
It is possible to buy or manufacture special ion exchange 189 material which has chelating groups. Leyden describes
the use of the material known as Chelex 100, and gives curves of the pH dependence of extraction. Thir. material was used by Agarwal et al. for concentration and analysis of trace
1 /in metals in urine. Holynska describes the use of Dowex A1 and the extraction of lead, mercury, copper, zinc, and iron, Dingmau et al . describe the extraction of a number of metals on dithiocarbamato resins. Diothyldithiocarbamidate adsorbed on the surface of carbon particles has also been used (jackworth et al. ).
Much work has centred on ion-exchange disks because the end result of absorbing a sample onto them i .* a thin rjboet of material containing the elements of interest. Absorption corrections are minimal or absent. The disks are made commercially by a paper-making process and incorporate both rosin and cellulose. Fti r=omr instance.'-, both rati onir: and anionic material is combined in the one disk. The initial work
292 SO on these was by Spano and Green and Campbell et a.l. J
The usual procedure is to use a kind of filter-typo apparatus and pour the solution through five or more times. This is better than a batch equilibration technique which is much slower. As would be expected from analogy with normal ion-exchange systems, the alkali metals are not held very strongly by a cation disk and certainly not quantitatively. However, the recovery, if 10 disks were used, was about 98$ over 'lO minutes. The difficuJ. ty experienced was that thorn was

' • - > .
a gradient of concentration of the elements through the pile of disks and even on«* disk cove different readings oil different sides, usiii;; conventional X-ray fluorescence. Other work using these disks has been done by Bergmann ot al. " , who determined vanadium «>•> I :• i ckol in sub-ppm amounts in potroloimi,
270 Robert and Yalles vho state that it is theoretically possible to determine 0.01 ppm for must elements (iu;.i.ng radio-
305 isotope excitation) and the work of Tnckctt et__al. The latter authors determined lead in dinner-ware by a leaching process, then isolating the lead via ion-exchange paper. They
-3 claimed a detection limit of 7»2 x 10 ppm. 1HH Law and Campbell used a resin paper which actually
contained Chelox 10O, arm found rather similar absorption conditions were needed fur various metal ions compared with normal ion-exchange piij»>!i-s. They suggest the use of radioisotopic tracers to monitor the yields. This has been done (Whitehead ) ' . . Kadioisotopes corresponding to each element of interest are added and the resultant disk monitored using a high resolution Ge(Li) detector of large volume. The criterion for a radiotracer in this instance is that it must emit gamma radiation. The Ge(Li) detector is used for gamma spectroscopy and the resolution is so high that there is no interference froD the peaks of the various radioisotopes which can therefore be determined simultaneously and independently. This technique could be described as multi-tracer use.
This careful monitoring of yields seems important if high precision is to be obtained. It is quite clear that the presence of large amount:-, of (s-ay) calcium :in a sample may grossly affect the amounts of other metals collected on a resin and some form of yield monitoring is essential. Given this, and some rather more sophisticated means of controlling which ions are collected (for example, by adding chelating agents to the solution), the technique may be useful indeed for some specific ions.
It must be emphasised that selective control of ion collection is necessary or the method may be of little use. It must finally bo said about all these chemical methods that if

he.
they concentrate all elements equally, ve still arrive back at the old problem of the largest peak in the spectrum determining the sensitivity of analysis of all the rest, or if the method is selective, ve may legitimately enquire why a multi-element analysis technique such as X-ray spectrometry was wanted in the first place? It may therefore be the case that there is little point in much of the effort put into chemical pre-concentration methods, unless for some reason no other method* of analysis aro available.
15* Analysis without standards In neutron activation analysis, provided the cross-section
of a reaction and the flux of neutrons is accurately known, and if the sample having been irradiated is counted in conditions in which the response of the detector in known exactly, then in theory the amount of an element is calculable from the radioactivity measured. Similarly, in principle, given the knowledge of the characteristics of a gas chromatography column and the response of a detector, it is theoretically possible to derive from the area of the peak a measure of the amount of substance present. However, for the latter case the theory is so complex that standards are usually used. In the case of neutron activation, the case is a little better and measurement without standards is sometimes attempted, though the accuracy may be as poor as ± 30#. Attempts have ' been made, in the case of X-ray spectrometry, to do away with standards and they have been partially successful.
If an attempt is made to derive absolute concentrations without using standards, it is necessary to know the amount of incident radiation, the cross-section for production of X-rays from the particular element, the exact geometry of the apparatus, the geometry of the detector, the absorption of the X-rays by the sample and the beryllium window of the detector and the absolute efficiency of the detector among other parameters. The list is formidable, but the attempt has been made. Colle et al. , who usod X-ray spectrometry, claimed (using cross-sections measured from standards on tissues) an accuracy of a few percent. Giauquo and Jaklevlc
used X-ray excitation, a single thin film standard and

31? many calculation:*; Tonij i.a;;a has proposed an ii«|.i ovemeiit using two thick samples of elements and interpolation, which also is claimed to give an accuracy of several percent. lie states that the accuracy is chiefly limited, by the nass-absnrpl ion coef t'ieient which is not usually kj.ovn or t:;tlculahlc
17ft to within the stated error. Luine and Tukia have attempted analysis wit.'-.-it standards using ( Y I X ) spectrometry. Various authors have a.ltempted an analogous exorcise for charged particles. Here the limiting factor for accuracy is
*101 said to be the cross-sections (Sunier et al. ) and these have therefore been the subject of investigation by a number
/ 90 2hf*i of authors (Duggan et al, ; Peisach et al, /. At present the agreement of cross-section experimental results with theory and between themselves is good for alpha particles, but for protons there is continuing uncertainty (K.L. Watson, pers. coiiim.). Other d.'tu are tabulated by Spaulding ,
6 190 Aksclsson and Johansson , and Li and Vatson • Other authors vho have attempted standard 1 ess analysis"- include
112 28'. 1'lU Groenwall ; Shaba son et al. ; Herman et al. ; 13-'i ~~""""""* Harrison and Eldrcd . (However, most authors appear to irradiate a standard clement or two in their apparatus to eliminate? some of the variables.)
It seems that such exercises on samples which have been analysed by many other techniques, usually give answers within £ 10% of other analyses.
A system to reduce dependence on standards relies on normalisntion to the scatter peaks which occur in the r.poctrura. The intensity of r.«*nt I.<T peaks an' a function 01 the amount of material from which the X-rays have been scattered, ami all the peaks in the spectrum may hence be normalised to a unit weight of scatterer. This exercise is most appropriate when comparing samples of similar composition.
In tin- case (iT (X,X') spectrometry, therefore, one normalises to the scatter peak corresponding to the element whoso X-rays were used to irradiate the sample; for (p»X) or (a,X) spectrometry one nust normalise to the breiiir.:l rahlung. The former procedure has been used by P.S. Ong (pers.comm.) and

kx.
P. Smythe (peis.cuflH.) and co-workers, and an overall accuracy of 1/4 is claitaefl. The latter procedure lias been used (Whitehead " 5 ) in the study of wool.
l6. Accuracy and precision Whether without standards or v»*n, effort has already
been devoted tr- inter-lal»oratory comparison exercises Tor users of semiconductor X-ray spectrometer* (Camp ; Camp et al.
Two have been carried out - a third is in progress. It was concluded that both particle and photon excitation gave similar results; the accuracy was JK 10%, the precision about 1?t. The samples were specially prepared air filters and presented additional problems in the way of analysis, as mentioned under that heading. Since standards are available (Rhodes ) or can be made, it is preferable to use them.
A second inter-comparison exercise (Camp ct al.. pers. com,) confirmed the conclusions of the previous study, and also suggested that the results obtained using various excitation modes in conjunction with Si'Li) detectors wero as accurate or more accurate than those obtained with the other techniques represented. This conclusion must be taken to apply only to an air filter type of sample.
For various types of samples, precisions quoted at regions where counting statistics are not a limiting factor are: 5# (Watson et al. 3 2 8 ) (a,X); 1-2# (Hubert and Street 1 3 )
(X,X'); rock analysis, 1j£ (Pickles and Cate. ) (y,X); uranium and plutonium r.amples, 10/4 (Kaufman "*) (y,X); in vivo studies, 0.6 - h.'.i'/, (Kis-Varj;a and Masco ) (y,X); manganese nodules, 0.2J* (Ehn 5 ) (y,X); film, 2-*»# (Kunzendorf et al. •}) (y,X); manganese nodules, yfa (Dcaman and Solosky 2 l ) (e,X). It is claimed (Wheeler et at. J 3 2 ) . that X-ray excitation generally yields better precision than charged particle excitation. Thir. statement :lr. true for thick, non-homogeneous samples. Since the penetration of charged particles in a thick sample is much less than that of X-rays, the volume of sai.-iplc irradiated i; much smaller and Iionce sampling errors tend to reduce the precision of analysis. However, for a thin s.impl.c such at. an air filter the results

v.>.
appear to give tompnrabl e provision (Camp ot a.) . ),
17. Applications in the field
Some industrial uses of semiconductor X-ray spectrometers 1 'i 1 1 3 'l
are given by I'r1 ';•. ruul l -.meron ; GHirko and fl'iic ; and Camp . The latter mentions the use of the equipment to differentiate between variolic types of seals, their nr.o in cement manufacture and in the measurement of sulphur -in petroleum. Gehrke and Cole describe possible ana l.ys ir> of steel from a distance (while still very hot) for its manganese content. Valkovic mentions thickness determinations for tin and zinc.
As early as 1967» the use of semiconductor devices -was considered for use in the field (Senftle and Tanner ). The apparatus was mounted in a vehicle but the por.'i .';teirl. problem of cooling hindered its use. Use of this technique in outer space research from rockets was briefly considered (MacGregor et at. ), and the Crab Nebula was observed by a rocket borne Si(Li) (it. Frankel, pers.comm,). There are also plans to orbit an intrinsic germanium detector and a Si(Li) in the High Energy Astrophysical Observatory, but as far as X-ray detection is concerned there seem at present to be two main approaches: firstly, to keep equipment in the field not longer than a week (when the normal liquid nitrogen container will still be adequately full) (Aylrncr et nl. ); secondly, to design a cryogenic refrigerator for field use. In the latter case (Mailer and Gelezunas ), a Solvay cycle ;iyr.tr.111 war-; shown to bo acb-ouate tr> reduce the temperature of a detractor to liquid nitrogen temperature in 1.5 hours. Some problems with microphonics were noted, and in general it is considered existing systems are either expensive or unreliable (R, Frankel, pers.conim. ).
17'» 19') Kun/ftirlorf ; LocvtxTg and Kurr/.nndorf ; and
Niewodniczanski , have used radioisotope sources and modern detectors for geochemical exploration. Other aspects of the problem are reviewed by Langhoxnrich et a3.
It should bo noted that proportional counters wore used for the lunar expeditionr, rather than HOIIIL conch jr tor dotoctorr.

r ) 0 .
because of t h e \: i yutS* ui '• • i •••,u irenienL. 'J111 X-i . iys {iriiiiucciJ by s o l a r p a r t i c l e s s t r i k i n g lunar mater ia l were measured ( jagodu et al, ). Field emission tubes have been suggested for ~ ~""" 207 vise outside laboratories (McCrary and Van „Vorous ) t but radioisotope r.imrcs tin' • • referred fot t.hoir simplicity.
' * Applientjous The Appendix should l.o consulloil for ;i detail'!'* 1 i •; I.. It
is clear that an enormous variety of samples have been analysed.
(i) Air filters It is clear that considerable effort ban been devoted
to the analysis of air filters. The inter-laboratory exercise mentioned earlier centred around this type of sample and a number of specific problems arose. If the filter surface is duR t too smooth the incoming/paiticles tend to bounce off the surface. If it is too porous, they penetrate a considerable distance into the filter and it becomes nearly impossible to prepare adequate standards. There is the added problem that absorption effects become significant for the X-ray.-; of the lighter elements (jaklevic. ot al. ; Cooper ) f though these problems were thought to be not significant by the former workers, who obtained rcr.iil.ts within 10$ of those obtained by neutron activation analysis* However, they suggested irradiating the sample at two X-ray energies to obtain a measure of the surface composition ana bulk composition. Payne
123 and Gilfrich et al. mention that tho ur.ual competitive tccliniquo, atomic absorption, has some difficulty with this sort of mnrpj r . fWnce many of tin? partirl er. in the filter are refractory, complete ionisation and hence a true signal, may not be obtained in a flame. X-ray spectrometry was more sensitive than atomic absorption by a factor of 3 for most elements surveyed.
* To overcome the offer If of particle size and ol.bor 27 inhomogeneities, Hodart nt al. , who used protons for
their analysis, actually pulverised their filters and mixed them with graphite before irradiation, ft is doubtful whether the increased homogeneity warrants the effort involved in Ibis. It would seem that X-i ;y .• pectrometry i« n good method of

3«.
analysis i or this typo of samp.lt>f but that the problems caused by absorption of low-energy X-rays have not yet been solved.
Preliminary results (Criss, pers.comm.) suggest (rather surprisingly) that particle geometry effects aro at their worst when secondary fluort»:=» rp-" generic try j .«? used.
G--*) Tn vivo work i? .-» uw-Ai). possibility vitli excitation using X-rays or radioisotopes and will often result, in a lownr dose of radiation to tb-.* animal or human than using injected radio-tracers. In this mode of operation, a flexible geometry is essential. The technique has been used for measurement of the rate of clearance of heavy metals from Ihe lungs (Kaufman •
Kaufman and Gams ) t measurement of blood flows* and «£e 1*17
using xenon as a tracer (Kaufmun ;. Hellouin )• In the latter case it was found that the yield of X-rays was so low that a sodium iodide detector was required. Attempts have 311 also been made to scan the thyroid for iodine (Tknchenko el. nl.
and to detect iodinated contrast agents (Kaufman ). A commercial thyroid scunner based on a Si(Li) is now available for purchase.
A unique application of (oc,X) spectrometry to in vivo work, though the analysis need not necessarily bo done on a live specimen, is the tagging of mosquitoes with small spots of differing multi-element standards. A retrieved mosquito can thus be identified uniquely (T.A. Cahill, pers.coinm. - see also Umbargcr and Malanify 31\
(lii) Forensic work is a potentially fruitful use of semiconductor X-ray detectors because in many cases thn prior composition of the sample is not known. Some use also has been made of those instruments for the detection of gunshot residues (Mathioson -*; Barnes et al. ). The hands are rinsed with dilute nitric acid, and the rinsings concentrated and evaporated on Kapton. Elements such as antimony, barium and lend are seen.'/'another application Whitehead ct al., (1975-submitted to "Forensic Science") wore able to ehow thut a hole in clothing was made by a bullet. Lead was found around the hole, even if blood soaked. It will bo obvious that X-ray excitation is much simpler than conventional tocJuiiquos in this case.

5'.
(iv ) Naturally radioactive samples - It is possible to conveniently measure the amounts of some elements by the natural emission of gamma rays of low energy, or X-rays that they emit. This has been applied to members of the uranium and thorium decay serir'.. Ger— •itium detector;: nr« used exclusively for this and the enrichment of uranium-2.35 has been measured (Mironov ot al. '" ) l»y comparing the X-rays emitted by all the uranium present with the specific gamma-ray emission Tor uranium-235. Other members of the series can also be analysed for in this way (Gurvich et al. ) but for bulky samples there tend to be absorption problems which reduce the accuracy of such an analysis. Plutonium, an element which demands careful chemistry, has been the subject of assay by this technique (Pickles and Cage ; Camp ;
ig/i Larsen and Karttunen ), but the electronics should be modified to take into account the high gamma-ray overload which results from trying to assay highly radioactive solutions.
The X-rays from fission fragments have also been detected using Si(Li)'s; potentially on»? can rapidly determine a very wide range of fission yields, but in practice the statistics are poor (T. Zabel, pers.comm.).
(v) Artificially radioactive species - Attention has been given to the possibility of estimating the products of neutron activation analysis by X-ray spectrometry rather than conventional high resolution gamma-ray counting. A useful list
198 of practical possibilities is given by Mantel and Amicl . In some cases the X-ray incr.r.Mrcmcnts are superior and this will in the future be an important supplement to gamma-ray counting techniques. However, the efficiency of counting may be low for somo X-rays (e.g., bromino), and it may be better to sacrifice resolution for sensitivity and use a thin sodium iodide detector (Poisacli _ot a.t, ), -The technique in also
~ 211 reviewed by Mcnkc et al. J
The assay c.in bo used in somo places whore previously radiochemical separations were needed (Lavl ), and has the advantage that the ubiquitous Ha-2k does not interfere (Weaver ). In the region of the rare earths, tho gamma

5!K
ray spectra which art? obtained «n neutron irradiation ore vex-y complex. The X-ray spectra obtained are much less complex (Mantel and Ami el ) though the situation is still complicated by some interferences and the presence of a few gamma rays of low energy vhicl> overlap Hie X-ray region. Interferences can potentially be resolved by exploiting the different decay rates of the various species.
The products of irradiation with particle's other than neutrons have been investigated by La Drecque et al. )»' who bombarded samples with heavy lens. In these cases it may be necessary to differentiate between X-rays resulting from radioactive decay and X-rays produced in the sample by the gamma ray flux which will often be present. This can easily be done (Urabarger and Malanify ) by irradiating the sample with more gamma rays. If the X-ray emission increased it was due to secondary effects, if not, it was radioactive in origin.
If, instead of neutrons, high energy charged particles of low mass arc used other analytical possibilities emerge.
80 Decotuiinck et a.l, found, when they attempted to use *»9 MeV H„ (i.e., singly charged hydrogen molecules) to excite K- X-rays of heavy elements, that nuclear reactions produced nuclei which were themselves X-ray emitters and interferred
11 in some cases. Bodart and Deconninck J exploited this at much lower energy (less than 3 MeV) in determining the thickness of very thin coatings. Tils was possible because the coatings consisted of very light elements a.-.d nuclear reactions wore occurring. Resonances, i.e., a vastly enhanced production of the daughter nuclei at certain critical energies, were observed. Incoming particles are steadily reduced in energy as they traverse a thin layer of material. If under the layer is a thick layer of the element of interest, and the energy of the incident beam is changed, there will come a point when the energy of the beam roaching the thick layer is just correct to cause a sudden increase in the amount of the X-rays emitted. From the known incident enercy nnd * n c known energy required for the resonance, the amount of energy lost in the thin layer can be calculated and this is a measure of the

5>*.
thickness of th layer. Bodart ami IKrcoiinj nek also irradiated the surface at different angles vhich amounts to the same thing *• varying the energy of the incident Wum. The technique, which will be fmiliar to those who use nuclear reaction product*- for the some end, 1ms th«* I:»••*•' lotions of destroying the surface through slow abrasion.
The most .•*; golf i cant work on the use of* rliorgert particles for the production of X-ray emitting nucJiin-r .in clxwiicnl
2 08 analysis is the work of NcGinley ', and MnGinley and 209 Schweikert • They used protons and deuterons of 20 and
*l0 MeV and produced short-lived nuclides from light elements which were then transported in a pneumatic rabbit to a Si (Li) detector and counted* Other emissions from the samples were also counted. Interferences (from more than on*: element giving nuclides emitting the same X-ray) were '.om;i.dered and tabulated. The method appears to be usable to the region of 1 ppm for many elements, based on analysis of NHS standard glasses. The sensitivity of course varies erratically from element to element, depending on the cross-section for a particular reaction. The absolute detection limits were demonstrated to be often superior to atomic absorption analysis and sometimes to neutron activation anrr.lyr.ir>, but this is still calculated on the basis of a single element being present and there being no interferences.
In theory, information would also be obtainable by measuring X-rays in coincidence with particle.'; emitted, but apart from the work of Swinth , ' and T.JI. Pierce (pers. comm.), I Mis cipproanh hns not I--OM; favoured,
fri) Sundry applications - Several authors mention the possibility of using energy dispersive X-rny analysis together with diffrnctometry. This gives information about the elements
/ 20/| responsible for the different phases (Sparks and Gedcke J Martin and Klein 2 0 1 , ',
Semiconductor detectors hove been used for mna&itring the chemical shift of on X-ray, i.e., the change in energy when the element emitting the X-ray is bonded in rf S I ferenl ways. Now the cltoiificn.1 shift of an X-ray peak i« of the order of a few mV at most, compared with the resolution of u Si(Li) detector of perhaps 160 eV. It is clear therefore (since peak-

55-
fittittg programs cannot fit to closer than a!>i>ut lO oV) that the resolution of the crystal monochromator is essential. One may therefore ask what is the point of using a Si(Li)
77 detector at all. Deconninck " _ points out that there is a \-fv y »i;:«ifleant r»-tlucti«n in background using smch a device. He therefore used a Si(Li) to examine X-rays after passing through a crystal monuclironator. lie also maintains that the ime ol' protons gives much wore intense excitation and better Geometry, since they can be focussed to a snail spot. However, this is of doubtful value since considerable damage to the chemical structure results by this bombardment, and complications arise in the form of satellite peaks also.
Another advantage of the use of protons for solid state work on crystals is the possibility of exploiting channelling. This phenomenon occurs- «rln*n a charged particle enters a crystal at a certain critical angle and is able to penetrate a considerable distance, because the alignment of the atoms in the lattice forms a channel which the charged particle can travel along ~nd furthermore, the electrostatic forces tend to keep it in the channel. If there is an impurity in the lattice, which blocks a channel, and it may be possible to detect this fairly selectively from the X-rays it emits. This approach has been used by Chemin et al. < who studied germanium crystals and used a beam of heavy ions detecting emergent X-rays with a Si(Li) detector. It requires very careful control of the angle of incidence of the beam* This technique is one requiring specialised equipment and will only find its place in laboratories needing information on solid r.tate conditions,
Another possible application which has been used increasingly is a technique called "beam-foil spectroscopy", in which a beam of ions is passed through n thin foil nnd emerges on the other side highly ionised, due to its collision with other atoms on the way through. The resultant ions emit light which gives information about the ionised state. In principle it is possible to use Si(Ll) detection to detect the X-rays which will also ho given off (Oono and Bickel " ) , but thu counting times ure many hours.

56.
19. A compares'*;* of &1 ffcrent*»xcita*ion .Hy»tea»3 It Is now possible to buy complete X-ray-tube-excited
systems, complete with mini-computers. In general, these are tube-excited, and some exploit secondary fluorescence gsomstry to maximise the sensitivity, Kmy chemists w.iS.1 prei>r a system like this, but those with access to workshop facilities will possibly prefer to make a tube- themselves*, following a design such as described by Jaklevic et al. J Access to modular electronics is definitely required if such devices as pulse-pile-up rejectors and pulsed excitation or guard-ring coincidence electronics are to be used; many of those useful features are not readily available in all complete commercial systems.
If a system is used which is partly commercial and partly built to design, it may be possible to obtain a much greater versatility. It may be possible to design the geometry in such a way that largo samples can be inserted, which is not always the case with commercial systems. Radioisotope excitation often permits similar usage but the sensitivity is somewhat more limited.
The use of accelerators and charged particle beams is much less simple. Since the irradiation is done in a vacuum the sample size is strictly limited; no workers appear to have used a beam which has been allowed to pass through i\ foil into air. Setting-up time for a run may take hours, unless the system is in continuous use, because of the overall complexity of it, fleam alignment, vacuum problems, beam instability, are all difficult.!>:n '/hich must be fnced. However* the increased versatility available from beam scanning and surface examination may compensate for this.
Some workers have tried to examine the comparative costs of analysis by various systems, and unfortunately Howm polemics have resulted, but it is cleor tbwl wbatuvor syntem is used, a high throughput is possible. As a 'real-life' example, RliodcB » using radioisotopic oxcitation, quotes an analysis total of 7000 samples in threo yenrs.

57.
The University of California at Davis is processing about 15,000 samples of air filters per year using one day per month of accelerator time and not much mere for the analysis by computer (T.A. Cahill, pers.corns,..). The Lawrence, Berkeley, laboratory is processing about 25,O0O per year using (X,X* ) spectrometry (J.M. Jaklevic, pers.comm.). The cost per sample works out somewhat cheaper for the accelerator based Hystem, even including allowance for depreciation on the accelerator. The reason for this apparently paradoxical, result is that (a,X) spectrometry is inherently better suited to air filter analysis, because the samples are so thin. Analysis time is about 70 seconds compared with half-an-hour for the (X,X*) spectrometry system. The difference would become even more pronounced if the accelerator system was used full time, as the (X,X*) -spectrometry system is.
The advantage of accelerator based systems in this . . instance would disappear rapidly if a thicker sample, more suited to the (X,X») spectrometry system was employed. One can make the generalisation that for optimum samples for each method, the costs are comparable, and of tve order of $5 per sample (T.A. Cahill, pers.comm.; ORTEC Ltd, pers.comm.). Considering the amount of information obtained, thin is vastly cheaper than a technique like atomic absorption spectroscopy.
123 It should also be mentioned that Gilfrich et al. , *
comparing such spectrometric systems with a conventional X-ray system, employing many crystals in fixed positions and hence able to analyse simultaneously as many elements as the machine contains crystals, comment that given a throughput of 5»000 air filter samples per year the cost of analysis per sample would work out at |1.%7 in spite of the fact that such an instrument costs over a hundred-thousand dollars. If the elements to be analysed are known beforehand, as is often the case, such a system may be preferable. Overall, a multicrystal X-ray fluorescence system is cheaper, per sample, even if allowance is made for the faet that ths accelerator-based system is operating only one day per Month.
Zt would appear then, for applications where a rmry large number of routine analyses are performed, a multi-crystal

58.
system may bo comparabln or better than systems* based on semiconductor X-ray detectors. However, for research it is extremely important to gather as much data as possible even if some of it proves irrelevant, and low-cost, low-volume, high-information systems aro superior. Thus a semiconductor-based system is better. It is also better when the composition of the sample is completely unknown. It will usually bo a matter of taste or style or availability whether a biam of charged particles, or a flux of X-rays from a tube is used for excitation. An (x,X* ) spectrometric system has the advantage of being able to give more information - especially about very light elements, and about spatial localisation of elements.
20. A comparison with other multi-element analysis techniques Simultaneous multi-element analysis is always deniroble,
if possible, because it leads to is—suss savings in time, and money. However, there are a number of criterion that any method must be ablo to meet.
1. The analysis conditions must be able to be the same for each element. If conditions must be optimised for different elements, or groups of elements, simultaneity is lost.
2. Since many clomenta in nature are present in very small amounts, the sensitivity must be high.
3. Interferences Must be known and able to be allowed for* k. Overall analysis tine should be as short as practicable. 5* The system must be able to analyse simultaneously
elements which differ in concentration by many orders of magnitude.
The last point is seldom given enough emphasis. An examination of fig. 9 shows that elemental abundances differ by as much as nine orders of magnitude or decades* To ask any method to measure such different concentration is obviously demanding a great deal. The crucial difficulty is thn detection device* A photomultiplier tube can give a useful signal over six orders of magnitude, which is unusually large, but can only determine one light level at a time. Photographic film, which is a medium capable of use for measuring many events simultaneously, has a dynnmic range of only four ordors of magnitude. The other main doteetion systems used are those

59.
for detecting gamma rays, and X-rays. Both of these are techniques to which Poisson statistics apply, and Many of the same comments apply to each. In both cases, a spectrum is obtained vith the use of a multi-channel analyser, and these devices have limited storage capacity* A typicnl fi&trB for the maximum counts that can be stored in any one channel is 6 10 . This means that the largest analytically useful range of such an instrument is about four orders of magnitude, since at least 10 to 10 counts are presumably needed for adequate quantification of a peak. However, if a computer is used programmed as a multi-channel analyser, there is the possibility of each channel containing 10 counts, if sufficient precision is used. This indicates that a sufficiently wide range is theoretically available to match the Ymry vide range of concentrations found in nature.
It is next important to ask whether the electronics available can cope with count rates which differ by nine orders of magnitude. It is obvious that if the electronics are run at the maximum possible rate which avoids spectrum distortion, the counts due to the very smallest concentrations in the sample will only be 10**™ of the total. If, for a Ge(Li) a realistic maximum count rate is 10 counts per second, and a
k 2 maximum realistic counting time per sample is 10 seconds, 10 counts will be accumulated only for species whose concentration is 10" of the most common component in the spectrum. This means there is a fundamental limitation on the decades any radioactive counting method can handle| about six at best. For a Si(Li) detector, and the analytical times usually quoted of about 10^ seconds, it would seem that the absolute maximum number of decades possible is five. This means that if the sample constituent contributing the most counts is the major one, not less than 10 ppm may be estimated, and if a multichannel analyser is used rather than a computer for accumulating the spectrum, not less than 100 ppm.
In practice, this is pessimistic because the major components of most samples are not contributors to the spectra, but the principles must be remembered.

60.
The final limitation in u spec trim of X-rays or gsmma-raya, etc., is that of resolution.
Considering; sort especially the cases ef those methods which rely on radioactive counting techniques (e.g., X-ray spectrometry) it is possible to determine how the resolution of the system affects the problem of interference. It is clear intuitively that the better the resolution the less this problem will be. The usual method' of resolving these interferences is by least-squares fitting, using a computer and tho efficiency of the procedure will also depend on the algorithm.
To determine how the separation of two peaks affects the error on the determination of the smaller peak, a program was constructed, details of which will be presented elsewhere (Manning and Whitehead, 1975, to be published), which constructed synthetic spectra and then used a peak-fitting algoritiim to determine the areas of both peaks and the errors on those areas. The results of this, for various ratios of peak areas, are presented in fig:* 11. These show that if the separation is less than about 0.2 of the FVHM then the error is very large, even if the peaks are equal in area. Also demonstrated is the fact that if the ratio of the smaller peak to the larger peak is 0.0O2, then the separation must be more than a FVHM to attain a peak with an area three times its standard deviation.
Further examination of the results gives actual limits for some real situations encountered in X-ray spectrometry (assuming 10 counts in the large peak, and 165 eV resolution). Cobalt is an element of interest; it is situated in the spectra adjacent to iron* What ratio of cobalt to iron is necessary before the cobalt can be estimated with an error which is one-third of the peak area? The answer is that this occurs when the ratio is 1 to 100. A similar question could bet what is the ratio of tungsten to copper that is determinable in a sample, given that the L- X-ray of tungsten lies between the prominent X- X-rays of copper and zinc? Calculation shows that tho ratio may be (since tungsten is nearly two FVHM units away from either the copper or sine peaks) 1 to 500, or even less.

01.
It is cli-.T! tii.it i lie typo of simultaneous. multi-elf--w*nt instrumentation al»lo tu handle the greatest dynamic i-;»n|je is that where the radiation emitted by the sample is physically dispersed in space, and a separate detector is used for each element (e.g., the multi-crystal spectrometer). Since the system can be designed so that the degree of separation is very hi.;;li indei n tt\r- limitations of a mul li-efiann«J s»naJyt*er do not apply, nor arc electronic limitations so severe.
Commercial systems exist which exploit this idea for X-rays. They consist of as many as 20 crystals adjusted to specific angles, each isolating the X-rays of a specific element. This type of instrumentation does not exist for gamma rays. Systems like this do exist for arc spectroscopy, and are known as direct reading spectrographs. Tim principle is the same; the different optical wave-lengths are physically dispersed in space, and individual detectors detect the photons corresponding to each element.
Recently (ilutler et al. -* ) t . a system previously confined to single, sequential analysis has been extended to simultaneous multi-element analysis. The technique is known as atomic fluorescence and requires the sample be in a liquid form. The liquid is aspirated into a plasma, which very efficiently atomises most molecules present, and an intense light source excites them electronically. The light emitted passes through a grating monochromator, and is physically dispersed in space. Fixed slits intercept radiation of various wave-lengths, and each has its own photomultiplier tube. With this instrumentation analysis time is about. 90 seconds per sample, and the number of elements which can be estimated simultaneously without significant interference is as many as 48, Sensitivities are of the order of 1 ppm for most elements in the parent material, but much better for samples which do not require dissolving in some solvent. Non-metallic elements can usually not be analysed by this technique. However, such a system appears more versatile than cither the multi-crystal spectrometer just described, or any instrument bnsetl on semi conductor X-ray detectors. The cost of such a multi-clement atomic fluorescence spectrometer in slightly Irr.s thou for a

62.
multi-crystal X-ray spectrometer, being about t75t°°°i "n** cost of analysis per sample (owing to the necessity or dissolving most samples) will remain, however, ssich higher thajK^or other systems.
There are six analysis techniques which claim simultaneous analysis of a wide rang* of elements at one timet X-ray spectrometry, neutron capture, photon activation, arc spectroscopy, spark source mass spectrometry, neutron activation analysis and multi-element atomic fluorescence. It is difficult to compare these directly, but an attempt has boon made in fig. 10, except for multi-element atomic fluorescence. The concentrations ousted are in terms of a logarithmic scale which means that errors will affect the visual appearance to a small extent only. All limits of _ detection quoted are in terms of tho amount of a single element, with no interferences being: present.
(i) X-rav spectrometry - The limits here are derived from (p,X) spectrometry (Robinson et al. ) ' and the criterion adopted is that the minimum determinable amount is that which gives a signal equal to three times the standard deviation of the background. One micro-Coulomb of 2,5 HeV protons is the dose and the irradiation time is about 10 minutes.
(ii) Neutron capture - This Method relies on detecting the gamma rays emitted when a nucleus captures a neutron and falls from an excited state to the ground state before undergoing radioactive decay. These gamma rays are emitted in
-1'» about 10 sec and hence their detection must take place at the same time as irradiation with neutrons. Thin method should be distinguished from neutron activation analysis, which measurs the gamma rays emitted by the nucleus as it decays after it has fallen to the ground state. This means that the two nethods will in general detect different gamma rays for tho same element but that the snnsitivities will tend to be related because in both cases there is reliance on initial neutron capture. It is clearly not possible to put a gamma ray detector in a reactor, so neutron beams are extracted from tho reactor, or a californium-252 source is used. Even so, the detector must be well shielded from the beam irradiating tho sample.

i'»3.
This method has boon mainly considered for analysis of ore bodies and the data on which tho figure is based aro modified from Heukclman (and based on his experimental conditions) with other data from Tiwari and Senftlo ot al. Tin.- i-.'-»n.»i t ivity cril .-r.L'.m i:; that tho signal must be twine the standard deviation of the background. The neutron dose is
R *> 2 x 10 n/em'/spc for '»(> minuti-s. The figures shown are bettor detection limits than those quoted by many workorr..
(iii) Photon activation - This method is similar to neutron activation in that the decay of radionuclides is meas -red some time after the irradiation. Gamma rays of energy greater than 10 MeV are used and usually one or more neutrons are ejected from the inn leus. The procedure therefore gives as products different isotopes compared with noutron activation, but they may also be detected by their gamma rays. Sensitivity
290 figures have been taken from Slunecko and Kosta and are for one hour ,s irradiation at 20 MoV.
(iv) Arc spectroscopy - The sample is subjected to an electrical discharge and the visible and ultra-violet spectrum is recorded (usually on a photographic plate). The data are
218 drawn from Morrison . (v) Spark source mass spectrometry - The sample is
introduced into a mass spectrometer and the individual masses separated physically in space to strike a photographic plate.
218 The data are from Morrison . (vi) Neutron activation analysis - Tho sample is irradiated
in a reactor for as long an a week, or half saturation. A 12 2 flux of 10 n/cin /sec is assumed and *»0 cps are assumed
detectable for the product radioactive nucleus. The data are 218 again from Morrison , slightly modified.
Even given the different conditions described, it appears from the figure that noutron capture is u.'unJly much Icar. senstive than the other methods, and spark-source mass spectrometry much more sensitive. Neutron activation is a consistently sensitive method. X-ray spectrometry is slightly worse than average, for most elements, but this does not take account of interference,. Interferences exist f'oi' all those

6'i.
methods. They are especially severe for analysis of light elements by photon activation because the nuclei produced are usually positron emitters and any gamma ray spectra are dominated to such nn extent by the 5 1 1 koV annihilation peak that analysis for eleitii.itIs simultaneously har. limited applicability.
Interferences c-an ilso exist for neutron activation, but these are more generally duo to such things as a large amount of Ka-2h being produced and producing substantial con I. ri.hnt.Lnn to the spectrun, or complexity where, for example, a mixture of rare earths may give a spectrum so complex that analysis is very hard.
It is difficult to compare the interferences in different methods but X-ray spectrometry appears about average, though the range of elements that can be analysed simultaneously will be considerably less than for either neutron activation or spark-source mass spectrometry. Mass spectrometry suf'forn from interferences in the low Z region. X-rav spectrometry has one of the cheapest capital costs and neutron activation one of the greatest. Analysis by the latter method is also very much the slowest of the ones tabulated. Irradiation of a dozen samples may take a week in which Lime 500 or more might be analysed by X-ruy spectrometry.
The maximum sample size is largest for neutron capture and smallest for («,X) spectrometry; (X,X') spectrometry deals with samples nhout the same size as arc spectroscopy. Sample preparation is minimal, for X-ray spectrometry, as it it)r.o ;i« for most others ol the techniques named, except arc spectroscopy.
21. The literature of X-rnv spectrometry OOP
Useful reviews are given by Woldseth (on the general 11 f
principles and properties of the detectors), by Valkovic (on applications), Ooulding nml Jaklovic '" (mainly photon excitation). The u.';e of radioisotope excitation souree;-. in this type of work is r.-viewed regularly by Idiocies (e.g., "' ), A good review of the possibilities of proton excitation in given by Folknic'mu . Other reviews ore by Oericko -*, Further

65. 22 reviews in languages other than English are by Bauciin ,
Friant (French), Coiante , Fiorani and Gilson (Italian). 179
Other reviews relevant to the topic are by Lai and Kupoor , 195 2kl 255
Lukas , Peisach and Pretorius , Porter and Woldsoth . Sources of further information may be found in tho literature of various commercial manufacturers. As far as journals are concerned 'X-ray spectrometry' contains useful material but most other articles are in various standard analytical Journals such as 'Journal of Radioanalytical Chemistry*, 'Radiochemical and Radioanalytical Letters', and 'Analytical Chemistry'. The journal 'Nuclear Instruments and Methods' should be watched for new developments on the instrumental side, as should the 'Journal of Applied Physics'.
Each year, a conference on X-ray fluorescence is held in Denver in the United States, in August. The proceedings of these conferences are published as 'Advances in X-ray Analysis' and are another standard reference source.
The situation in the field of X-ray spectrometry is also reviewed every two years amongst the comprehensive reviews published by the journal 'Analytical Chemistry'.
22. Conclusions The last few years have seen a very rapid growth in the
use of semiconductor X-ray detectors which parallels the rapid application of X-rays themselves soon after they were discovered. This has depended heavily on the rapid improvement of detector materials, and electronic processing of the signals from the detectors. Dramatic improvements in either the materials or the electronic processing seem very unlikely in the future, nor will very different materials supersede those at present in use except for specialised applications.
The Si(Li) detectors available at present can routinely detect X-rays from elements as heavy as, or heavier than, sodium and special windowless detectors can detect elements as light as carbon with lesser sensitivity, Ge(Li) detectors do not find such extensive application as Si(Li)'s.

66.
There are numerous modes of excitation of X-rays from samples. This reviewer recommends the following methods of analysis for various samples and applications: - for laboratory use (low throughput , y non-routine applications) -
(X tX») spectrometry; for laboratory use where spatial information, or very small samples are involved - (p,X) or (et,X) spectrometry; for field use - (YIX) spectrometry;
- for minimum cost with very large numbers of similar samples - X-ray spectrometry using a multi-crystal instrument. In general, the sensitivity for analysis of an isolated
amount of a single element is of the order of a few nanograms. The overall accuracy is about 10JJ and the precision 1j£.
In analysis using (y,X) and (X,X') spectrometry, matrix effects must be taken into account whereas there is no necessity for this using (p,x) and (a,X) spectrometry. Computer calculations may be required for adequate matrix effect correction or alternatively some of the techniques available for thin target preparation may be used. These eliminate matrix effects, but seldom increase the number of elements seen.
It must also be said that such preparation techniques will often be quite impractical for a large survey, in view of the time, and hence cost involved. This comment also must apply to the various chemical protreatment methods available. If these are genuinely multi-elemental, not just concentrating one specific element, then there are added problems with accuracy of analysis, because some of the concentration procedures are not necessarily quantitative.
Computer calculation is required fov extracting peak areas from a spectrum. There are a wide range of programs available to do this which differ greatly in sophistication. There is some doubt as to the optimum level, and further work is needed in this aroa.

<>7.
In coifipti ri >-<>n with other multi-clement analysis techniques X-ray spectrometry is of aver»(;o sensitivity, and exhibits this sensitivity mainly in the Z region 19-39» for most samples. Lsss sample preparation is required thun for many other techniques, mid the method if. «;ften to be considered non-destructive. Ultimately, it is inferior to a multicrystal spectrometer with fixed slits, but considerably cheaper.
The method is finding particular use in the fields of air filter analysis, iii vivo analysis, forensic science, analysis of naturally and artificially radioactive species, and the very vide range of applications will undoubtedly increase in the next few years. Chemists should find increasing uses for these semiconductor X-ray detectors, nnd so will physicists, biologists, ecologists, and even those from some fields of the arts. It is unusual to find ana analytical method with such wide applicability. It can only be hoped that automation of the analysis will increasingly free the user to think intelligently about the meaning of the results his multi-element analysis system is producing at such relatively great speed.

68.
APPENDIX Applications - In this section in table 3 current applications are tabulated with comments on some of the more interesting aspects.
Tnb.le 3 Sample Blood
Serum
Leaves
Hair
Wool Cell membranes Algae
Reference 127 Goul.ding and Jakl evic 1 _,
Jaklevic and Goulding j? Giauque and Jaklevic Campbell et al. 1 2 f-Gordon and Kraner Young et al.3^3 Young3^ 6
Malanify and Umbarger Bearse et al.25
Johansson ot al. Johansson et Peisach
158
7*5 159
et al.' Goulding et al. 1 29 Rudolph et a.l.277 Wheeler et al.331 Camp'15 Ong et al. Walter et Gouldin. Oberlin
23k
al. and
31
327 Jeklevic 127
Sparks et al. 296 7^ Cothern ct al _„ Deconninck^0, Doconninck McGxnloy 2 0 8
Whitehead333 Gordon and Kroner
et al. 317 et
ot al.
ot al.
al. aT7331 y»3
318' Valkovic Valkovic Wheeler Y o u ; i(7 (••<.• i n . - - , „ / ' Gordon and Kraner Menke et al.2!? Barnes
Demortier „.,, 316 Valkovxc
82
20 Cooper and Langford Smythe (pers.comm.)
70
335 Whitehead""- 7
D e m o r t i e r o t a l . 3') 2
8k
Yourift Young c t n l . D e c o n n i n c k ' "
3'»3

69.
Sample
Plankton
Perch
Swordfish
Enzymes
Carp
Tissues
Bone
Neoplastic tissues
Muscle
Milk
Wine
Finger and toe nails
Urine
Mussel
Tissues
Fish
Renal calculus
Flour
He Terence 343 loung ot at.
342 Young h
Young et al. 3'»3 Young ot, al.
Lochmueller Pt al Walter et .-11.3*7 Young et al.3 *3
192
Walter et al. 327
er
317 Valkovic et al. Walter et al.327 Campbell et al.^ 8
Rudolph et al. 277 Malanify and Unbar.5 Jundt et al. l 63 Knudson (pers.comm.)
7 Alberi et al. Gordon (pers, coinni. ) Knudson (pers.conun. )
196
Walter et al. 327 Campbell et Va1knvir3">'
al. 48 Valkovic- ../-_ Jundt et al. Gordon (pers.comm.)
Dacaner et al. 11
Valkpvic" Camp^5
Valkoyic Orr
ltoyd 236 316
Valkovic 316 Cooper and Langford Uh-i t-«V.f.i.r»33 *
70 Whitehead
Malanify and Umbarger 196
153 Jaklcvic and Goulding-:,-Goulding and J a k l e v i c . 2 Q Goulding and Jaklevic
Herman et al. Wheeler ot nl.
I40,l4l 331
Akselsson et al.
Alexander et al.. 8
Cooper and Lanfjford
Seaman e t a l .
70

70. Sainpl ** Reference Paper 127 Walter et nl.-^ 1
Butterfly ving 327 Walter et al. ' Teeth 79 Deconninck Cloth Walter et nl.3*"7
Organic compounds Uucfnrt and T>«»c»s:iiiiiick Saliva C.»rflon (pcrr . coi"m. )
Synovial membrane Rudolph (pvrs.coM*.)
Viruses Rudolph (pers.comm.)
Tubulin Dearse (per:?. c o m . )
Flower organs Whitehead 3 3 5
Resin Whitehead 3 3
Nematode's Whitehead^"
Vorm casts Whitehead 3 3 5
Water Paue et al. „„,„
Sediments
Coal
Silicates and oxides
Rocks and minerals
Walter et »!.. Cordon and Krai
327 126
uortlon and Jvrapgr Cookson c t n l . * , . « •Tohtmsson c t a l . Johansson r t n l .
3*»3 Young c t a l . Ginuque' *'( Catfr and p i c k l e s M a t l . i o g g n ^ 3
Rhodes
159
53
Sparks c t a l 297 298 Sparks and Ofj.T.e-^.
Shab.isoit rl- n l .
Rowse e t a l . Praii2gTote^°3
27/1
136 llebi.Tt and S t r e e t Dnldin and Ionimraynnta G r a v i t i s ot nl . 1 3 -
18
Mati'iciuc nt a l . Tnnn«r and l»rinkerl ioff VhJ M-hoad3'* '
307

7 1 .
Sample BtfTorcncp
Ores and ore-processing effluent
Petroleum products
Manganese nodules
Ores
Water
Air filters
•SI Cnrr-Brion' Holynaka1** Loevborg and Kunzendorf Grofnwall -* ^, Cookson et al., J
Rhodes et n l . 2 6 9
264 Rhodes Johansson et al.
193
Til:
Kis-Varga ainl Banco " Kunzendorf et al. WJ
Lubecki and Vogc jhi Papez and Cameron Aylmer e t a l , 1 0
Kunzendorf•'* Loevborc and Kunzendorf Pierce3I»0
193
Kraushaar ot Whitehead^T* Perry and Brady'
172 al. 249
Tancmura c™!:«5,tt
and Suita a l . ? 1
306
Camp Johansson Johansson
et ol. et al.
159 153 Jaklevic and G o u l d i n g 1 2 _
Couldinc. and Ja.klcvic 2Qi * Rhodes 2 6' 1
Mizoliata and Mainuro Cate52 G i l f r i c h e t a* '*J
C a h i l l 3 9 C a h i l l e t a l .
217
e t a l . 'II 267 ,268
a l .
160
Rhodes e t Swick302~ Payne*7^ Cooper ' D i t t r i c h and Coibcrn Robinson e t a l . '^ Giauque e t a l . * ' 9 Camp e t a l . * * * ^ ^ Rhodes e t a l . Joliansson ot a l . Nelson e t alm^f R o b e r t s * ' ' » 3 0
Wedberg c t n l . • ; , , J a k l e v i c c t a l . J J
Harrison find Kldrcd Giauque ^ 7 2
Tom i nana Goulding and Johansson ££ Giaiiquu c;t f 1.1. Manuiro rt n l . '^' Wood and Mritli tenon
83
\V\
J a k l g v i c 128
339

72.
Sample
Air filters (ctd)
Metals
Chalcogenide films
Germanium
Photographic film
Forensic samples
Archaeological
Wear studies
Metals
Reference
Bodart ct «1. Deconninck Akselsson e t a l . Young et al.'J^J Walter ot al.3^7 Qng (pers.comm.) ., _. , 208 McGmley Zellor and Forestier Ilollstein and De Voe Musket and B a u e r 2 2 0 ' 2 2 1
Peisach ot. al. IQK Shabason ot al. Clayton et al.59 Voskamp3«iJ
3'«5 IV3
Rhodes et al. 266 Laine and Tukia 178 Peisacli et al. 2l»6 Close and Bearse 60
Thomas et al. 309
56 Chemin et al. -Peisach o t a l . 4 b
Ehn 95
Barnee _ Quinn et
et TT 258 19,20,21
Gage and Wtiitworth Whitworth33o Mathi.eson P a t e 2 ^
111
231 Oberlxn J
Gordon and Perry and
Kraner ,2&9
126 Brady
Whitehead-0-' McCallumJ 0^ Wallacf»32'* Deconninck and Stroobants Bennett and D'Auria
81
Gehrke et nl. 116
Mateiciuc et al. Whitworth31 Pemorticr -' Gehrke and Col
202
1I'» 2'n
Papez and Cameron •\oc\ Giauque and Jaklevic Kuusi ot n l . 1 ? 6
Cookson and Pilling Whitehoad J J J
66
Flame retardant Nelson and Kelly

Sample Glass
Oxide films
Charcoal Clay Insulator content Soil Coins In vivo studies
Actinides, uranium and trailsuranics
71».
•
Boforonc<»
Dziutiikuyski and Hykior Duvison7 _„ - 0
Deconninck',''^ Whitehead^* McGiiiley208
Port f?-53 2"J Utuipr and Musikot ' ,
Nuskut and Duuer *fc•' " J
Needham ot nl. 2 2 6* 2 2- 1* Hovel, ct al.^(»2
333
rt 9 3
Whitehead Deconninck and Fontaine (pers.comm. )
,333 Whitehead' Whitehead 33*
335 Whitehead Hollouin'J* Kaufman1?? Woldseth J J O
TkacUcnko et al. Kaufman and Gams
31» 168
Pickles and Cate 2 5!^ 2, 5 1
Shenberg and Amicl' Cant and Notes ^ Bacharov et nl. .gj. Larson nnd Karttunen Winters337 Mironov et al. i n-Gurvich et al, J J
Camp^5

y».
REFERENCES 1 M. Agarwal, R.B. Ui'mittt, l.G. Stump and J.M. D'Auria,
Anal. Chew. ^7, 92'» (1975). 2 H. Aiging;er and P. Vobrauschek, Hucl. titat. Moth. 11&,
157 (197*0. i ~ " 3 II. Aigincor and P. Wobrauschek, Anal*.Chew. *7, 852 (1975)..
k D.V. Aitkcn, Trans. l.E.K.K. Nucl. Sci. KS-15, 1<> (t9«8)« i i _ _ _ _ _ _ ~ — . _ ^ v
5 R. Akstlsson, T.B. .1ohHn*;."<m and f-.A.E. Johansson, Nord. Hvtr. Tidskr. 53. 11 (l9?2).
***** 6 R. Akselson and T.B. Johansson, Z. Phys. 266, 2*5 (l97'»). 7 J.L. Alberi, H.V. Kramer, P. Bradley-Moore and II.L. Atkins,
Trans. I.E.E.E. Nucl. Sci. NS-2J. 635 (197*). 8 M.E. Alexander, E.K. Biec-rt, J.K. J onus, R.S. Thurston,
V. Valkovic, R.N. WheelCJ , C.A. Winftato ami T. '/.abel, Int. J. APPI. Radiat. Isotopes 25, 229 (197*).
9 A.L. Allen and V.C. Rose, Adv. X-ray Analysis 15, 53;» (1972). 10 J.A. Aylmor, G.J. Clarke, P.L. Kislor, R,.T. Ho J mo;;, p.
Huppurt, P.J. Mathvw, G.H. Nevman and A.U. Vylie, CS1MQ Minerals Research Laboratories Annual Report (197&) p. 2*1.
11 M. Dacaner, J. Broadhurst, T. Hutchison and J. Lilley, Proc. Natn. Acad. Sci. U.S.A. 70, 3**23 (1973).
^ — _ - — ^ — — 1 ^ _ — _ ^ ^
12 N. Dacharov , T . Dragncv . S . Geor / j iev , J . K.-iramaiiova, T . Ruskov and T . Tomov, ftudiochein. R a r t i o a n a l v t . L e t t e r s 1*., 1 ( 1 9 7 3 ) .
13 R.D. B a e r t s c h , T r a n s . I . E . E . E . Kwcl . S c i . NS-18, 166 ( 1 9 7 1 ) .
^k R .J . B a n i a n , A.B. B r i l l and J . A . P a t t o n , T r a n s . I . E . E . E . N u c l . S c i . NS-20 . 379 ( 1 9 7 3 ) .
15 N.A. B n i l y , R . J . G r a i n i e r ;.nd J . V . Mayer s IteVj^Jir.itmt. I n s t r u m . 3J£, 865 ( 1 9 6 ' ) .
16 S.A. B a l d i n and L.M. I o a n n e s y a n t s , I n s t r u m s . Exp. T e c h . i j , hZh (1971 ) .
17 S.A. ttaldin and L.M. loatiiicsynritf*, Yiub-in I 'r i lm ••».'* frr. I ' l , 1'»3 (»97» ) .
18 S.A. B a l d i n and L.M. l o a t i n c s y a n t s , Yadcrn 1 ' r i bo r»Ht r . 15 , hk (1971 ' ) .
19 B.K. B a r n e s , L .E . B o s n i a n , G.lf.R. K e g e l , f ' .C. M;il.!iur and P.V. q u i n n , H u l l . Am. Vhvs. Son, r-,2 18 , 630 ( 1 9 7 3 . ) .
20 B.K. B a r n e s , L .E . Brvthinn, G.11.H. Krr:<'1 , f»,0. Mnlbiir find P. q u i n n , J . l ? a d i o i ' n n h t , n u m . Vj, 13 ( 1 9 7 3 ' ) .

7!».
21 P..K. Barrios, L.K. K-\;tiian, O.II.R. K-.-el, K.C. Nathur ;..H! P. Quiun, J. Kr>.,U<...t>alvt. Chaw. l6, *53 (*973).
22 G. Baudin, CEA-COXK-2250 (1973) Saclay, Franc*. 23 ¥. Bauer am! R.G. Musket, Aool. »hvs. %&. 26o6 (1973) . 24 D.H. Ueawan and L.F. Solosky, Analvt. Chew. 4£, '39B ( 1972). 25 tt.C. Bearse, II.A. Close, J.J. Malanify ami C.J. l*»li;«r|»«r,
Analvt. Ch.—. J&, 1*99 ( 197*). 26 . „M. Beech and J.E. Ebcrhardt, AAEC/K-297 (t973)t l*icas
Heights, New South Vales, Australia. 27 V. Beeshold, SLA-73-5303 (»973)» Alberouerque, New Mexico, United States. 28 R.B. Bennett and J.N. D'Auria, Int. J. Apnl. Radiat.
Isotopes 25, 361 (197*). 29 J.G. Ber&nann, C.H. Krhardt, L. Granatelli and J.l,. Janik,
Analyt. Chcei. 39, 1258 (l9»7). 30 J.O. Iterftiwinn, C.ll. Erhartlt, L. Grauatelli and J.L. Janik,
Analyt. Chew. 3J.» 1331 (19©7). 31 F. Hodart and G. Deconninck, LARN-7J2 (ca. 1973 - undated)
Nawur, Belgiuw. 32 F. Bodart and G. ftecoiminck. Int. J. Kow-dostruct. Test.
4. 171 (1972). 33 P. Bodart, G. Deconninck, J. Hon toy ami S. Vilk, Radiochew.
Radioanalvr. Lf»tt«?rs 13., l6l (1973). 3*» N.A. Bonner, F. Bazan and B.C. Camp, UCRL-5088 (1973)*
Livermore, California, United States. 35 H. Bovwan, A. Hebcrt, II. Vollenberg and F. Asaro, LBL-2966
(197M, Berkeley, California, United States. 36 F.P. Brady and T.A. Cahill, UCD-CNL-l66 (1973), Davie,
California, United States. 37 P.G. BurkhaIter, Aoolied Low Energy X- and Gawwa-rava 1&7,
(1971). Gordon and Breach, New York. 38 C.C. Butler, R.N. Kniseley and V.A. Fassel, Analvt. Chew.
kj, 825 (1975). 39 T.A. Cahill, Bull. Aw. Phvs. Hoe. 1J , 505 (1972). kO T.A. Cahill, UCD-CNL 162 (1972), Davis, California, United
States. *»1 T.A. Cnhill, R.G. Flocchini ani P.J. Fecney, Trans. Aw.
Nucl. Soc. 17, 102 (1973). 42 T.A. Cnhill, II, Sowmcrvillc, R. Flocchini. F.P. Brady and
N.F. Peck, Bull. Aw. l'hvs. Soc. s.2 16, 5'»5 0971).

76. **3 J.A. Cairns, D.F. Molloway and R.S. Nelson, Adv. X-ray-
Anal, iji, 173 (1970). *»*» J.A. Cairns and R.S. Nelson, Radiat. Effects 7. 163 (1970). *»5 D.C. Camp, Trans. Aw. Nucl. Soc. 17, 107 (1973). h(t D.C. Camp, Trans. Am. Wucl. Soc. 1£, 106 ( 1973). *»7 D.C. Camp, J.A. Cooper and J.R. Rhodes, X-ray spectrometry
3, *»7 (I97'i). 48 J.L. Campbell, A.V. Herman, L.A. McNelles, B.ll. Orr and
R.A. Villoughby, Adv. X-ray Anal. IJ, ky[ (l97'«). h9 J.L. Campbell and L.A. McNelles, Nucl. Instrum. Meth. 98,
1*33 (1972). ' 50 V.J. Campbell. E.F. Spano and T.E. Green, A.ialyt. Chem.
38, 987 (1966). 51 K.G. Carr-Brion, X-ray spectrometry 2, 63 (1973). 52 J.L. Cate, UCRL-51038 (l97l), Berkeley, California, United
States. 53 J.L. Cate and W.L. Hckles, UCRL-5^22 (l973)f Berkeley,
California, United States. 5^ P-L. Chan and W.B. Jones, Adv. X-ray Anal, ijl, 102 (1970). 55 P.L. Chan and W.B. Jones, Adv. X-ray Anal. 15, 209 (1972). 56 J.F. Chemin, l.V. Mitchell and F.W. Saris, J. Appl. Phys.
fj5, 532 (197'0-57 2.H. Cho, M. Singh and A. Mohabbatizadch, Trans. I.E.E.E.
Nucl. Sci. NS-21. 622 (197^). 58 C.J. Christensen, J.M. Khan and W.F. Drunnor, Rev. Scient.
Instrmn. 38, 20 (1967). 59 C.G. Clayton, T.W. Packer and J.C, Fisher, Proccdiiifcs of
the syrnpor. i.un on the uc-f of nuclear technique;; in the basic metal industries, ifelsinki, 31 July-** August, 1972, IAEA, p. 319.
60 D.A. Close and R.C. Bearse, Nucl. Instrum. Meth. 97. 607 (1971). "~
61 D. Coiante, RT/F,C-(72)6 (1972), Rome, Italy. 62 R.P. C0II6, High energy electron impact ionization,
Rensselaer Polytechnic, New York (1973) (Thesis). 63 R.P. Colle, I.J. Preiss, A. Li-Scholz and W. Scholz,
J. Radioannlvt. Chem. ijl, 87 0973). 6U J.A. Cooknnn, A.T.O. Fcrgunon find F.J). I'll l.i/ir;, Hod.ioanfilyt.
Chcm. 13,, 39 (1972).

f t
65 J.A. Cookson, M. Ivanovich and F.I). Pilling, AERK-PR/NP/18, (•97'-)t Harwell, Nirrlc. hiro, United Kingdom.
66 J.A. Cookson and F.D. Pilling, Thin solid films 19, 331 (1973). ~
67 J.A. Cooper, BNWL-SA-^O (1973), Richland, Wnsliinpton, I'nitod States.
68 J.A. Cooper, Nucl. fnstrum. Meth. H>6, 525 (1973). 69 J.A. Cooper, Trans. Am. Nucl . Soc. V£, 103 (1973). 70 J.A. Cooper and J.w. Langfoi'd, Tr:tce uuustance& in
environmental health, 6 (1973)» Columbia, Missouri, United States. "*
71 J.A. Cooper, L.A. Rancitelli and T.N. Tanner, BNWL-1751, 112 (1973), Richland, Washington, United States.
72 G.E. Coote, IA'S-K-129 (I973)t lover Hutt, New Zealand. 73 G.K. Coote and N.E. Whitehead, INS-R-lM (197;0» J'ower
llutt, New Zealand. Jk C.R. Cothern, 11.L. Manuel and R.J. Millette, X-ray
Spectrometry 3, 53 (l97z»). 75 S.C. Currau, J. Angus and A.L. Cockcroft, Nature. London
163, 302 (iy'18). 76 C D . Davison, LBL-12'»0 (1972), Berkeley, California,
United States.
77 G. Deconninck, LARN-7^1 (ca. 1973 - undu od), Namur, Belgium. 78 G. Doconninck, CONF 721069-26 (1972), OIK Ridge, Tennessee,
United Status. 79 G. Deconniurk, J. Radioanalvt. Chom. JJ, 29 (l973). 80 G. Deconniurk, J.C. Gondrand, P, M'jrchez and N. Longequeuo,
J. Barlionna.lvt. Chem. J&, 183 0973). 81 G. Doconninck and J. Stroobants, LARN-73'* (ca. 1973 -
undated), Namur, Belgium. 82 G.F.A. Demortier, Radiochem. Kadioanalyt. Letters 20, 197 (WO-83 G.F.A. Domorticr, J. Rad i oannlvt. Chem. ?J\. 'l7 (1975). 8'+ G.F.A. Demortier, Proceedings of the symposium on mid.oar
activation techniques in thoi life .sciences. Bled, 10-1'» April, 1972, IAKA, p. 23.
85 J. de Noef, Knergy dispersive X-ray fJuoroscencc analysis with radioisotope excitation, Ghent State University, Belgium (1973) (flu sis).

78. 86 G.A. Desborough and R.H. Heidi, APPI. Spcctrosc. gjr, *56
(1973). 87 J.F. Dingraan, K.M. Glai,s, E.A. Milano and S. Siggia,
Analvt. Chw. (16, 77* (197*). 88 T.R. Dittrich and C.R. Cothern, J. Air Pollut. Control
Ass. 21, 716 (1971). 89 P.P. Donovan, Trans. I.E.E.B. Nucl. Sci. NS-13. 21 (I966). 90 J.L. Duggan, V.L. Beck, L. Albrecht, L. Munz and J.D.
Spaulding, Adv. X-ray anal. 15. *07 (1972). 91 V.H. Dunlop, J.V. Verba, J.W. Sunier, G.V. Brock and A.B.
Holman, Bull. Aw. Pnva. Soc. a.2 17. 5*8 (1972). 92 G.R. Dyer, D.A. Gedcke and T.R. Harria, Adv. X-ray anal.
£ , 228 (1972). 93 B. Dziunikowski and £. Rykiert, Sgklo Ceram. 2*, 166 (1973). 9* T.G. Dzubay, B.V. Jarrett and J.N. Jaklevic, Kucl. Instrum.
Meth. nj, 297 (197*). 95 E. Ehn, X-ray Spectrometry 2. 27 (1973). 96 E. Elad, Trans. I.E.E.B. Nucl. Sci. NS-19. *03 (1972). 97 E. Elad and D.A. Gedcke, Proceedings of 6th International
Conference on X-ray Optics and Microanalysis. Osaka, 5-10 September, VJ1, University of Tokyo, p. 263.
98 L.C. Feldman, J.M. Poate, P. Ermanis and F. Schwartz, Thin solid films j9. 81 (1973).
99 P. Fiorani and R. Gilson, Ingogn. Nucl. 13,, 19 (1972). 100 R. Fitzgerald, K. Keil and K.F.J. Heinrich, Science 159,
528 (1968). **-
101 R.G. Flocchini, P.R. Penney, R.J. Sommerville and T.A. Cahill, Nucl. Instrum. M«th. 100, 397 (1972).
102 F. Folkmann, J. Phvs. E. Scicnt. Instrum. 8, (1975)» 103 E.J. Franzgrote, Adv. X-rav anal. 15., 388 (1972). 10* D.V. Freck and J. Wakefield, Nature I J, 669 (1962). 105 A. Friant, CEA-CONF-15*1 ( 19.70), Saclay, France. 106 A. Friant, CEA-C0NF-18*0 (1971), Saclay, France. 107 A. Friant, CEA-CONF-225? (1973), Saclay, France. 10H A. Friant, CEA-C0NF-2308 (1973), Saclay, France. 109 A. Friant, R. Gra», A. Liivin and C. Saliou, CEA-N-1*01
(1970), Fontenay-Aux-Rosos, 110 T. Fukasawa, Bunsoki Knenlm 22. *53 (1973).

79. 111 S.J. Gage and J.II. Whitvorth, J. Radxoanulyt. Chew. 15»
337 (1973). *"~ 112 P.L. Gant and B.C. Motes, Trans. Aw. Nucl. Soc. 12, 518
(1969). "~ * O D.A. Gedcke, X-ray Spectrometry J, 129 (1972). 11* R.J. Gehrke and M.S. Cole, Adv. X-rav anal. 15, 276 (1972). 115 R.J. Gehrke and R.A. Lokken, Nucl. Instruct. Moth. 97,
219 (1971). ~ 116 R.J. Gehrko, L.L. Packor, B.A. Woody and V.A. Ux-uton,
Applied Low Energy X- and Gamma-rays. Gordon and Breach, New York (1971) P. 33.
117 R.D. Giauque, Trans. Aw. Kucl. Soc. 17. 104 (1973). 118 R.D. Giauque, L.Y. Goda and M.S. Brown, Envir. Scl.
Technol. 8. 436 (1974). 119 R.l). Giauque, F.S. Goulding. J.M. Jaklevic and R.H. Pchl,
Anuiyt. Chem. 4J, 671 (1973). 120 R.D. Giauque and J.M. Jaklevic, Adv. X-rav anal. 15,
164 (1972). "~ 121 P.E. Gibbons and J.H. Howes, Trans. I.E.E.E. Nucl. Sci.
NS-19., 353 (1972). 122 J.V. Gilfrich, Adv. \-rav anal. 1j>, 1 (1973). 123 J.V. Gilfrich, P.G. Burkhalter and L.S. Birka, Analvt.
Chem. 4jy, 2002 (1973). 124 V.M. Goldschmidt, Geochemistry. Clarendon, Oxford (l95*»). 125 B.M. Gordon and H.W. Kraner, Trace Substances in
Environmental Health 5, University of Missouri, Columbia (1971), p. 419.
126 B.M. Gordon and H.W. Kraner, J. Radioanalvt. Chem. 12, 181 (1972). ""
127 F.S. Goulding and J.M. Jaklevic, UCRL-20625 (1971), Livermore, California, United States.
128 F.S. Goulding and J.M. Jaklevic, Ann. Rev. Nucl. Sci. 23, *»5 (1973).
129 F.S. Goulding, J.M. Jaklevic, B.V. Jarrett and D.A. Landis, Adv. X-rav anal. 1£, 470 (1972).
130 F.S, Goulding, J. Walton and D.F, Malone, Nucl. Instrtun. M«th. 7J, 273 (1969).
131 V.L. Gravitis, R.A. Greig and J.S. Watt, Proc. Australas. Inst. Min. Metall. 2kQ. 1 (1974)
132 J. Oroenwall, LUNP-7308 (1973)» Lund, Sweden.

8 0 .
133 M. Yu. G u r v i c h , A.N. Z l u i k o v s k i i , N .V . M e l ' n ic l tc i iko and R . I . Plut.M i'.<<. , 'it I- iViinl. Melody Genkhlui iya Is:«.l e d o v a n i y n M a t e r i a l y t;«*okli.iwiya K o n f e r o n t s i y a 1970» ( 1 9 7 2 ) , p . 115*
13*» J.F. Harrison and R.A. Eldred, Adv. X-ray anal. 17 t
560 (19~*»). "~ 135 R.L. Heath. A»lv. >-ray anal. 15, 1 (197:*).
136 A.J. Heberl and K. -Street, Analyt. Chew. 46, 203 (197*0. 137 Y. Helloui», CIA-!'-'! "326, Sacl»y, Frnncn. 138 R. Henkelmaiin, !?adi ochim. Acta IJi, I69 (l97l). 139 H.K. Herglotz and D.R. Lynch, Adv. X-ray anal. YJ,
509 (197*»). 140 A.W. Herman, I...A. -'cNelles and J.L. Campbell, Int. J.
Appl. Radiat. Isotopes 24, 677 (1973). 141 A.W. Herman, L.A. McNellcs and J.L. Campbell, Nunl.
Instrum. M»>tlu Hr>, 429 (1973). 142 R. llofstadter, Phys. Rev. 7j*. 10O (1948). 143 M. Hollstniu and J.R. De Voe, J. Radioanalyt. Chem. 6,
139 (1970). ~~ 144 R. Holynska, Spe«-( rochitn. Acta B27, 287 (1972). l4p B. Holynska, Radiochom. Radioanalyt. Letters 17. 313 (1974). 146 B. Ilolyitskii, M. LesKko and E. Nahlik, J. Radioanalyt.
Cliom. JO, '«>1 ( «9V"J). 147 R.M. HowelJ, W.L. Pickles and J.L. Cato, Adv. X-ray anal.
18, 265 (1975). 148 G.L. Hubbard and I.E. Green, Analyt. Chem. 38, '»28 (1966). 149 IAEA, Radiuiuotopt. X-ray Tluorescence spectrometry.
Technical Report 115 (1970), Vienna. 150 II. T.. Israel, J*.W. Lier and E, Storm, Nunl. Inst, rum. Moth.
9J, 1«H (1971). 151 E. Jackwcrlh. J. Lohmar and G, Wittier, Z. Analyt. Chem.
2JJ0, 6 (197'»). 152 J.M. Jaklov.ic and F.S. Colliding, Trans. l.E.E.E. Nticl. Sc i .
NS-1£, 187 (1971). 153 J.M. Jnk.Lev.M-. and I'.S. Gouldiii£, Trans. f.F,,E.K. Niicl. Sci.
NS-19, 3Hh (197:.'). 15'> J.M. Jaklcvi.c, Tt,:•-. Giauqtio, D.F. Mulone and W.l,. Sonrlrs,
Adv. X-ray anal . 1J>, 266 (1972). 155 J.M. Jakluvic, J',:., Gouldintf, 11.V. Jarrelt arid J.I), Meii£,
LRL-17'»3 (1973), I'crkeloy, California, United Slates.

81. 156 J.M. Jaklevic, F.S. Goulding and D.A. Landis, Tvann.
I.K.K.E. Xucl. Sci. NS-19. 392 (1972). 157 T.B. Johansson, R. Akselsson and S.A.E. Johansson, Kucl.
Instrmn. Neth. 8U. 1*1 (1970) . 158 T.B. Johansson, R. Akselsson and S.A.E. Johansson,
LUNP-7109 (1971). Lund, Sweden. 159 T.B. Johansson, R. Akselsson and S.A.E. Johansson,
Adv. X-ray Anal. 1?. 373 097*). 160 T.B. Johansson, J.V. Nelson, R.E. Van Griekcn and J.V.
Winchester, Trans. Am. Nucl. Soc. ]J, 103 (1973). 161 T.B. Johansson, R.E. Van Grieken. J.V. Nelson and J.V.
Winchester, Ana lvt. Chew, kj. 855 (1975). 162 R.K. Jolly, and H.B. White Jr, Nucl. Instrum. Meth. 97.
103 (1971). 163 F.C. Jundt, K.H. Purser, H. Kubo and E.A. Schenk,
J. Histochem. Cytochem. 22, 1 (197*). A6k K. Kandiah, Nucl. Instrum. Meth. 95. 289 (1971). 165 L. Kaufman, Trans. Am. Nucl. Soc. 17t 108 (1973). 166 L. Kaufman, Adv. X-rav Anal. 18, (1975). 167 L. Kaufman and D.C. Camp, Adv. X-ray Anal. J8,, 2*7 (1975). 168 L. Kaufman and G. Gams, Trans. I.E.E.E. Nucl. Sci. NS-21,
( mmmmmia«wmmimmimmaa«aHHmMHeBawimmmHmmm* ^Ww
197*). 169 M. Kis Verge and J. Basco, Izotoptechnika 1£, 83 (1971*). 170 T.J. Kneip and G.R. Laurer, AnalytChem. kk, 57A (1972). 171 T.R. Kohlor, NAS-NRC-Pub. 871, 193 (l96l) Washington, United States. **" 172 T.J. Kraushaar, R.A. Ristinen, H. Ruaolph and W.R. Smythe,
Bull. Am. Phvs, Soc. 3.2 Jj6, 5*5 (1971). 173 P.V. Kulkarni, Spectrochemical Analysis Utilizing energy
Dispersive X-ray Fluorescence Induced by a Radioisotope Source, Rensselaer Polytechnic, New York (1973) (Thesis).
17* H. Kunzendorf,RIS0-M-l6l0 (1973), Ri»o, Denmark, 175 H. Kunzendorf, G. Priedrich, and W. Plueger, Erzmetall.
2J, 85 (197*). 176 J. Kuusi, M. Virtanen and P. Jauho, Trans. Am. Nucl. Soc.
j£r 130 (1971). 177 J.J. La Brecque, P.V. Kulkarni and I.L. Preiss, J.
RadioanalvtChem. U^ 455 (1973). 178 E. Laine and L. Tukia, X-rav Spectrometry 2. 115 0$>73).

82.
179 M. Lai and S.S. Kopoor, Proceedings of the Nuclear Physics and Solid State Physics Symposium. Bombay. 1-** February, 1972, Institute of Atomic Energy (1972), p.503.
180 D.A. Landis, P.S. Goulding and J.M. Jaklevic, Nucl. Instrum. Meth. 8J. 211 (1970).
181 D.A. Landis, F.S. Coulding and B.V. Jarrett, LBL-320 (l97l)i Berkeley, California, united States.
182 D.A. Landis, P.S. Goulding, R.H. Pehl and J.T. Walton, Trans. I.E.E.B. Nucl. Sci. NS-18. 115 (l97l).
183 A.P. Langheinrich, J.V. Porster and T.A. Linn, Proceedings of the symposium on the use of nuclear techniques in the basic metal industries. Helsinki. 31 July-fr August. 1972. IAEA, p.*29.
18*» R.P. Lars en and J.O. Karttunen, Adv. X-ray Fluorescence 17, 571 (197*).
185 H.T. Larson, R.B. Liebert, D. Miljanic, V. Valkovic, T. Zabel and G.C. Phillips, Bull. Am. Phvs. Soc. 17» 5*8 (1972). *"*
186 G.P. Laurer, T.J. Kneip, M.E. Vrenn and M. Eisenbud, COO-30'»0-Xl972), Chicago, United States.
187 N. Lavi, Radiochem. Radioanalvt Letters ££, 177 (197*). 188 S.L. Law and V.J. Campbell, Adv. X-rav Anal. 17, 279 (197*).
Aft'
189 D.E. Ley den, Adv. X-ray Anal. 1J, 293 (197*). 190 T.K. Li and R.L. Vatson, Phvs. Rev. A9. 157* (197*). 191 K.H. Lieser, Radiochem. Radioanalyt Letters 18L. 323 (197*). 192 C.H. Lochmueller, J. Galbraith, R.L. Walter and R.E.
Willis, Analvt.Biochem. 57, 618 (197*). 193 L. Loevborg and H. Kunzendorf, RISO-256 (1972), Riso, Donmar):. 19* A. Lubecki and H. Vogg, KFK-1728 (l973)f Karlsruhe, Germany, 195 J.R. Lukas, IFA-CRD-55-1973 (I973)f Bucharest, Rumania. • 196 J»J. Malanify and C.J. Umbarger, Bull. Am. Phvs. Soc. s.2
)Xf 5*8 (1972). 197 T. Mamuro, Y. Matsuda, A. Mizohata and T. Matsumani,
Radiation Centre Osaka Prefect 13th Annual Report (1973). 198 M. Mantel and S. Ami el, AnalytShem. **, 5*8 (1972). 199 J.M. Marler and V.L. Gelezunas, Trans. I.K.K.E. Nucl. Sci.
KS-20, 522 (1973).

8'}.
200
201
202
203
20'»
205
206
207
208
209
210
211
212
213
2)U
215
216
217
218
219
220
11.E. ifcij-r JJIIU U . J . Campbe l l , Adv. X-rny Anal . Ifi, 20(> ( 1 9 7 3 ) .
G.W. Martin and A.S. Klein, Adv. X-ray Ami. 15, 25;» (1972). V. Mateiciuc, L. Aldea, M. Constantincscu and T. Stadnizov, nt'in. A-nalfl.. ^. 12K (l97'.'). J.M. Mathicson, My r^^ra^AfKtl^ 17, 318 (197'). J.M. Ma thiouiHi, V.L. Da Gi-n^nano and J.P. Hurley, PJ»\C-135 (1969), Puerto Rico, p. '»51. G.J. McCallum, INS-R-157 (197*») Lover Mutt, New Zealand, p.21, J.D. McCoy, .1.1.. Duggan, E.L. Robinson and S.J. Cipolla, Proceedings of the symposium on the use of nuclear techniques in the basic metal industries. Helsinki, 31 July-'» August, 1972, IAEA, p. 591. J.H. McCrary and T. Van Vorous, Adv. X-ray Anal. 15i 285 (1972). "" J.R. McGinley, Novel Techniques in Charged Particle Activation Analysis, Texas A. and M. University (l97*») (Thesis).
J.R. McGinley and E.A. Schweikcrt, J. Radioanaftt.Chcm. 16, 385 (1973). G.A. McGregor, I. Turiel, R. Bcttcnliauseu and A. Iantuono, Rev. Scient. Jnstrtim. h2, 35 (197 0* K.G. McKay, Phys. Rev. 76, 1537 09*»9). K.G. McKay and K.ll, McAfee, Phvs. Rev. 91, 1079 (1953). H. Mcnke, Ch. Leszczynski and M. Weber, Radiochcm. Radloanal>t.Lottors 1J 217 (1973). V.D. Metz, Science 177, 156 (1972). N.1I. Miller and II.G. Wilkinson, Phvs. Rev. 82, 981 (l95l). I.I. Mironov, O.N. Suetnov and S.A. Baldin, Yadcrn. Priborostr. 12, 101 (1970).
— — - — — — r*s A. Mizohata and T. Mamura, Radiation Centre Osaka Prefect, Annual Report 13th (1972) pTTcT! G.H. Morrison, Trace Analysis} Physical Methods, Interscirnce, London (1965). A.M.P. Mufigloton, Nucl. .Tnstrum, Moth. 101. 113 (1972). R.G. Musket and W. Hauer, Acpl. Phvs. Letters 20. '«11 (1972 ). /"

8-'..
221 H.G. Musket anil V. Uauer, Appl. PUvs. Letters 2U, h$h (1972b).
222 H.G. Muskot and V. Bauer, .1. Appl. Phvs. &3, b7»6 (1972 ).
223 R.G. Mu»k«t and V. Bauer, Thin Solid Films 19, <"'9 (1973). — — _ — ——. y v f
22*1 K.H. N c e b , If. N e i . l l and I I . J . V e i s s e , German P a t e n t 2 l ' l l>79* ( 1 9 7 3 ) .
2 2 5 P . B . Necdhara, T . J . D r i s c o l l and N.«;. I«;n», A p p l . I'liy;;. L e t t e r s 2 1 . 502 ( 1 9 7 2 ) .
2 2 6 P . B . Ncedham, II .V. L e a v e n w o r t h and T . J . D r i s c o l l , J . E l e c t r o c h e m . S o c . 1 2 0 , 7 7 8 ( 1 9 7 3 ) .
2 2 7 J .W. N e l s o n , I . W i l l i a m s , T . B . J o h a n s s o n , R . E . Van G r i e k e n , K.R. Chapman and J . ¥ . W i n c h e s t e r , T r a n s . I . K . . E . E . N u c l . S c i . N S - 2 1 . 6 1 8 ( 197'»)«
2 2 8 K. l l . N e l s o n and II. T . K e l l y , T e x t . Cliero. C o l o r , 5 . 2 9 ( 1 9 7 3 ) .
2 2 9 J . N i e w o d n i c z a n s k i , Z c s z . Nauk. Akod. C o r n . - H u t n . Krakov Mat . F i z . Chem. 8 5 ( 1 9 7 1 ) .
2 3 0 W.A. N i c h o l s o n and T . A . H a l l , J . P h y s . K. S c i e i i l . - . I n s t r u m . _ Jj>, 781 ( 1 9 7 3 ) .
2 3 1 I . C . O b e r l i n , CEA-R-'l 5hh ( I 9 7 ; » ) , S a c l a y , F r a n c o .
2 3 ? I.'.J. O'Drxen , G. O o o t c and K . P . P o h l , l N S - n - 1 1 9 ( 1 9 7 2 ) , Lower H u t t , New Z e a l a n d .
2 3 3 P . S . Onct F . L . Cheng and G. S r o k a , Adv. X - r a y A n a l . 1 7 , 2 6 9 ( 1 9 7 ' » ) . " ~ ~
2 3 4 P . S . One, P.K. Lund, C . E . L i t t o n and D .A . M i t c h e l l , Adv. X - r a v A n a l . 1&, 12'» ( 1 9 7 3 ) .
2 3 5 H. O o t i a a ' / ^ . S . D i c k c l , N u c l . I n s t r u m . Moth . I I O , 'l97 ( 1 9 7 3 ) .
2 3 6 n.M. Orj" r Tk-vo.l',-,'•. -:\*. v.'' p r o t o n - 'Mtlncrtl .X-ray ' >>t r,r i 011 s p e c t r o s c o p y , U n i v c i ' s i t y o f G u c l p h , OnLar.in ( l9Vi>) ( T h e s i s ) .
2 3 7 V . S . P a k h o l k o v , V.E.Pai i ikftn>vskikh, Jzv. V y s s . U c h e b . - Z a r e d . Kliim. Kliiin. T c k l m o l . 1 0 , 168 ( 1 9 ^ ' 7 ) .
2 3 8 J .M. Pnlm.3, P, Vermgopnla Rao and U.K. Wood, Nut: I., lnr.trui i i . Moth. 6(i . 3 1 0 ( 1 9 6 8 ) .
2 3 9 J . M . Palms;, 11.E. Wood, O.M. P u c k o t t ami K. Z n n i o , Nucl . Inst i - i im. Moth. 9 8 , 597 ( » 9 7 2 ) .
2*10 A. P a p o , J . C . S o n s , P. VSnty.f A. G;i I J maim, M,E. G o v e , 0 . GuLllaiiiiif ami 1),M. f i l n p i n , NncJ., I n M r n m . Mtl l i . 10 ' j , 161 ( 1 9 7 2 ) . """ " "~ ""' "

2' l l K. P;«pw.£ an<l J . F . lu imcron, l'rococtUn;:.s «»f t h e symposium on t h e n>" o f n i i c ' ' a v t e c l u t i g u P 3 i n thr? l i a s i i ; niet:>l I i i ' l u s t r i o s , H e l s i n k i , 3 * Ju ly - f r A u g u s t , 1 9 7 2 , lAKA, p . 2 1 .
2*12 B.D. Pate, Trans. Am. Mm:I. Soc. 1J, 107 (»973)« 2'l3 J.S. Payne, Texas. Wept, of Health Report (197?). 2hk J.K. Pearson, Anpt. Spectrosc. 27, k$0 (1973). 2'l5 M. Pqisach, B. Maziere, C. Loc'h, D. Coraar and C.
Kollershohn, SUNT-23 ( 1973 ), Faure, South Africa. 2U0 M. Peisach, D.A. iN.-w-tou, I'.F. Puck and T.R. Piciru,
J . Hadioantlyt.Chrm. 1f>, Ml 5 (1973 )• 2^7 M. Peisach and R. Pretorius, Proc. Natl. Conf. Technological
Appl. Kucl. Techniques. Pelindaba, 1972, Atomic Energy Board, p.1.
2&8 E.M. Pell, J. Appl. Phvs. 3), 291 (i960). 2^9 S.K. Perry and F.P. Brady, Kucl. lustrum. Meth. IO&,
389 (1973). ~*~ 250 W.L. Pickles and J.L. Cato, URCL-7'»7 17 (1973), Livcrmoro,
California, United States.
251 W.L. Pickles and J.L. Cote, Adv. X-ray Anal. ]£, 337 (l97'l)« 252 D.M. Poole and J.L. Shaw, Proceedings of the 5th Inter-
nntional Conference on X-ray Optics and Microanalysis. 19o8, Springer-Verlac, Berlin (19°9) p.319.
253 L. Porte, LYCEN-7236 (1972), Lyon, France. 25*1 D.E. Porter, X-ray Spectrometry 2. 85 (1973). 255 D.E. Porter and R. Woldseth, AnaJvtChom. I>5, 60*1A (1973). 256 K.H. Purser, Dull. Am. Phvs. Soc. s.2 17, 50*» (1972). 257 P. Finmmlninen, P. Suominon, J. Hattula und H, Kaln,
JU-BU-73/9 (1973), Jyvaskyla, Finland. 258 P.W. Quiiui, U.K. Marnoa, L.E. Beghiun and G.H.R. Kegel,
Dull. Am. Phvs. Soc. s.2 1JJ, 636 (1973). 259 V. Radeka, BNL-6953 (1962), Upton, New York, United Stater,, 260 S.J.n. Reed, J. Phys. E. Sclent. Ins brum. 5, 99*» ( 1972). 261 S.J.B. Reed and N.G. Ware, X-ray Spectrometry^ 69 (1973). 262 C. Revel, M. !)a Cunha RcJo, J.P. Moriaux, C. Schneider,
N. llillert and R. Dourguillot, J. Racllofiralyt.Clicm. 17, 101 (1973).
263 J.R. Rhodes, CONF-720'iO'l ( 1972) f Oak Ridge t Tennossoe, United States, p. 2218.
U

26'l J.H. Rhodes, 'I'mib. A.K.K.K. Kuc.I. Sci. r!ii»£1,, <»*>» (l9'l'l). 265 J.H. Rhode*, T. Florkowski and J.F. Cameron, AERE-R-3925
(1962), Harwell, United Kingdom. 266 J.R. Rhodes, C.B. Hunter, D.L. Kellog,':, R.T>. 5iebor{r and
T. Knruta, Adv. X- ray Anal. 1^, 127 (lT»'»). 267 J.R. Rhodes, A.II. Prndzynski, C.B. Hunt or, J.S. Payne,
and J.L. Liiidgrcn, Knvir. Sci. Tcchnol. <>. *J2? ( 1972 ). 268 J.R. Rhodes, A.II. 1'rndzynski and R.D. S:» oliorc, Analyst.
Instrum. 10, 1 3 ( »972 ). 269 J.R. Rhodes, A. Pradzynski, R.D. Sieber<; and T. Furuta,
Applied Low Enerfry X- and Gamma-rays. Gordon and Breach, New York, (1971), p.317.
270 A. Robert and R. Valles, Radiochem. Hadi«>nnn1yt.Lott«»rs 15, 279 (1973).
271 N.J. Roberts, UCRL-57*»99 ( 197*0, Livormore, California, United States. ' 272 D.C. Robinson, N.K. Whitehead and G.E. Cooto, INS-R-131
(1973), Lower Hutt, New Zealand. 273 D.C. Robinson, N.K. Whitehead and G.E. Coote, Chem. N.Z.
39, 81 (l97'»). 27^ J.O. Rowse, V.B. Jepson, A.T. Bailey, N.A. ClJnipson and
P.M. Sopor, J. Phy.s. K. Sri.ont. Instrum. J, 512 (197Z«). 275 H. Rudolph, J.K. Klivor, T.J. Kraushnar, n.ll. H.istJnen and
W.R. Smytho, Analyt Inst rum. IjO, 151 (1972 )• 276 H. Rudolph, J.J. Kraushaar, R.A. Ristinen and W.R. Smythe,
Bull. Am. Phys. Soc. s.2 16., 5^5 ( '971). 277 H. Kudolp.i, R.A. Ristinen, W.R. Smytlie, A. Alfrey and
S.R. Cantiguglia, Bull. Am. Phys. Soc. 1_7, 5f»8 ( 1972 ). 278 S.M. Ryvkin, D.A. Gopanov, A.N. Zhukovr.ky, N.T. Komynk,
R.J. 1'lotnikov, N.H. SLrckan and A.K. Khuc a:iiiov, Nuc.t. Inst.rtun. Moth. 95. 177 ( 1971 ) .
279 A. Snp.lakog.lu, I. Ozogln, M. Tan and M. Erdogan, Technical J. (Turkish AKC) 1, hi (197'0-
280 C O . Seaman, H. Martin ami A. Ward, Iki.11 . Am. Phys. Soc. s.2 J&» W (1973).
281 G.O. Seaman and K.C, Shane, Nuc.l. Instrutn, Me_th. 126, '»73 (1975).
282 F.E. SunftJe, II.I). Moore, D.N. Leep, A. EJ-Kacly and D. DufToy, Nucl. lustrum. Moth. 93, '»25 ( 1 S>71) -
1 1 J . .L ^m^tf

87.
263 F.L. Si'tiftl.' .IIU! A.H. TUMIH r, OUNL-l.lc-10 (Vol. l) (l9<>7), Oakrid;£e, Tt-nnosKpf, United States, p. fiO'J.
28'l L. Shabason, U.L. Cohen, (.5.11, Wedbertf and K.C. Chan, J. Appl. Pit vs. {•£., »»7'»9 (1973).
285 D.D. Sliarm.i, Mea.sitr'Mierit <>!' 1 •> 1 ;,i i it.1 >>tal PoJariy.sitJon oi" Beta Rays using a Lens Spectrometer and Inner Shell Ionization by Protons, Univerrity of Nevada (l97l) (Tbosis).
286 C. Shonberg, A. Aladjem and S. Ainio.1 , J. Radioanalvt»Chem. 20. 77 ( 197*0-
287 C. Shenberg and S. Amiel, Analyt Cliem. **3. 1025 (1971).
288 K. Shima, M. Sakisaka, M. Kokuda, T. Yamamoto and I. Makino, Jop. J. Appl. Phvs. 9. 1297 (1970).
289 V.W. Slivinsky and P.J. Kbert. Nucl. Instrum. M>;th. 71. 346 (1969).
290 J. Slunecko and L. Kosta, Chcm. Listy 65,, 1009 (1971). 291 W.D. Metz, Science 177, 15^ (1972).
292 E.F. Spano ami T.K, Green, Annlyt.Chcm. 38, 13*»1 (1966). ~ " " " " ' • " • • * « * * •
293 C.J. Sparks, O.H. Cavin, L.A. Harris and J.C. Ogle, CONF-7506l3-'t ( J974), Oak Uidgo, Tennessee, United States.
29*» C J . Sparks and D.A. Godcko, Adv. X-ray Anal. 1j5,t 2'»0 (1972).
295 C.J. Sparks and L.A. Harris, CONF-721 13*»-1 (1972), Oak Ridge, Tennessee, United tstates.
296 C.J. Sparks, L.A. Harris and O.D. Cavin, ORNL-NSF-EATC-1 (1973), Oak "idge, Tennessee, United States, p. 2*»7.
297 C.J. Sparks, J.C. Ogle, H.W. Dunn and L.D. Hulett, 0RNI.-NSF-EATC-6 (1973 ) , Oak Ridge, Tennessee, United States,
298 C.J. Sparka and J.C. Ogle, OITNL-NSF- JCATC-11 (1975), Oak Ridge, Tennessee, United States, p. 112,
299 J.D. Spaulding, CONF-700322 (1970), Oak Ridge, Tennessee, United States, p. 113,
300 H. Stroback, LYCEN-7282 (1972), Lyon, Franco.
301 J.W. Sunicr, G.W. Brock and-J.W. Verba, Bull. Atn. Phys. Soc. s.2 H, 5^8 (1972).
302 W.A. Svick, NTIS AD report 763728 (1973)» Springfield, Virginia, United States

t*s.
303 S.l*. Svierkou-ski, G.A. Ariu.mtruut and I*. K.lehiior, Trans. I.E.K.E. Nucl. Sci. NS-2I. 302 (197M-
30«i K.L. Swintli, Trans. I.E.K.K. Nucl. Sci. NS-18, 125 (1971). 305 S.L. Tackett, C M . Bender, T.R. Brunncr, D.J. Duncan,
II. r»dak, R.K. TNmtilo, J.F. Miller, K.A. Mot.kov and A. McAuley, Analyt.Lettera 6. 355 (1973).
306 T. Tanemura and M. Suita, Radio-isotopoR 21^ 6'l I (1972). 307 A.11. Tanner and J.M. Brinkorhoff, Appl J od Low Kner^ X-
and Gamma-rays. Gordon and Breach, New i'ork, 097l)'"p»''73» 308 H. Thibeau, J. Stadel, W. Cline and T.A. Cahill, Nucl.
lustrum. Meth. rM, 615 (1973). 309 J.P. Thomas, L. Porte, J. Engerran, J.C. Viajn and
J. Tousset, Kucl. Instrum. Meth. UJ, 579 (l97'»)' 310 P.N. Tivari, Bodiochem. Radioanalyt.Lettors G, 3^3 (l97l). 311 V.I. Tkachenko, L.D. Soshin and K.I. Schekin, Med, Radiol.
l^, 69 (197**). 312 11. Tominaen, Nucl. Instrum. Meth. Y\h,, 65 (197;»)» 313 C.J. Umbarccr and J.J. Malanify, Nucl. Instrurn. Moth.
lOO, 201 (1972). 31*» C.J. Uini>ar£jor and J.J. Malanify, Int. J. Applied Radiat.
Isotopes 2'j. 381 (1972). 315 C.J. Umbareer, R.C. Hoarse, D.A. Close and J.J. Malanify,
Adv. X-ray Anal. )6j 102 (1973). 316 V. Valkovic, Con temp. Phys. 1^, *»15 (1973). 317 V. Valkovic, 11.11. Leibert, T. Zabel, II.T. Liirson, 1).
Miljnnic, R.M. Wheeler and G.C. Phillips, Nucl. lustrum. MfUi. IJ/j, 573 (197,»).
318 V. Valkovic, 1). Miljiiiiic, H.M. Wheeler, K. U. Loibort, T. Zabel and O.C. Phillips, Nntiiro/2>q. 5'»3 (1973).
London, 319 J.A.W. Von d e r Does do Bye, P h i l i p s R e s . Mop, U>, 85 ( l 9 6 l ) .
320 J . N . Van N i e k e r k and J . F , De Wet , NnlurJyflff i ' " 3P0 ( i 9 6 0 ) .
321 J.W. Verba , J . V . S u n i e r , G.W. Brock and A.H, Ho.lman, B u l l . Am. Pl iys . Soc . s . 2 17. 5'»8 (1972 ) .
322 J.W. Verba , J .W. S u n i e r , I!.T. Writfht, J . S.1.fiu», A.H. Holnian and J . G . K u l l o c k , J . Hndioannlyt Client. \2, 171 (1972 ) .
323 A .P . Voskamp, Adv. X-rnv Ana l . 17, 12'» (l97'>)«

8i>.
32't G. Wallace, JNS-l; IT7 ( I97'«) Lower Ilutt, .\c-.. /.<• I.md, p . 2 1 .
323 G. Wallace, INS-.*:-'i2 ( 197'l ) , Lover Ilutt, New /.calami.
326 F..I. waiter, Trans. I.F.r.K. Kwcl. Sci. KS-17* 196 ( 1970). 327 R.L. W.»l.t or, P.D. "mi.;, W.F. Gittknecht am! J.-» Joyce,
Analyt£hem. f»l>, bhj (197 V).
3?8 K.L. Watson, J.K. .»jurseUi and R.V. Howard. N»»- I. Iimtrmn. Moth. 93/t 69 ( 197 » ).
329 J.N. Weaver, Aw. L>b. S, 36 (1973). 330 G.H. Wedberg, K.C. Chan, B.L. Cohen and J.O. Frohliger,
Bull. Am. Phys. Soc. s.2 18, 580 (1973). 331 R.M. Wheeler, R.B. Liebert, D. Miljanic, G.C. Pliillip.t,
V. Valkovic and T. Zabel, Bull. Am. Phvs. Soc. s.2 18, 63^ (1973).
332 R.M. Wheeler, H.B. Liebert, T. Zabel, R.P. CliaLi.rvedi, V. Valkovic, G.C. Phillips, P.S. Ong, E.L. Cl»en<; •*»«« M. Hrgovcic, Hod. Pliys. 1, 68 (197*0.
333 N.K. Vhitchend, INS-R-I36 (1973), Lower Ilutt, Nov 'Zealand. 33;» N.K. Vliitelio.id, INS-K-1*l*i (197*1 )t Lower Ilutt, New Zealand.
p.9. 335 N.K. Whitehead, G. Wallace and C.J. McCallum, IN5;-R-157
( 197*» )» Lower Ilutt, New Zealand. 336 J.R. tfhitworth, Development and Application oi a .Systematic
Approach to Elemental Analysis in Forensic Investigation, University of Texas, Austin, Texas ( 1971) (Thesir.).
1337 W.I. Winters, AUH-2913 (1973), Richland, Washington, United States. 338 R. Woldsoth, All You^Ever Wanted to Know about X-ray
Eneigy Spectrometry Burlinghame, Kevex Corporation (1973). 339 W.G. Wood and J.M. MaU«ici,on, Arlv. X-ray Anal. 16. 13'l
(1973). 3^0 A.L. Yakubovicli, S.M. Przhiyalcovskii and G.N. Tsameryan,
Atomn. F.ncr<;. 3£, 2'»1 (1972). 3*»1 Y. Yonedn and T. Horiuchi, Hnv.Sricnt.Instruni. h2, IO69 (1971). 3^2 F.C. Young, J. W»hh. Acad. Sci. 62. 153 (1972). 3'»3 K.C. Yotnif:, M.L. Kousli and P,G. Herman, 1M_1_;._J. Appl.
RruUat. tM.1 i.pow ?'», 153 (1973). 3'»'» E,I. Znitsev and Y.)'. Solskov, J. H.idioiin 11 ?MMi»-m. 18,
31 (1973). "'

9».
3*l5 C. i&cllor mid R. *"orvst i i»v, P> r>cu»M.tin;:.-s «»f t h e . t n t r r eu t t iwwt l Confe rence on X- ray A n a l y t i c a l Methods* T o u l o u s e . 6 September 197 *» P* H7»
3^6 T . B . P i e r c e , J , B a d i o a n a l y t . Chewt. 1 J , 535 ( 1 9 7 3 ) .
3^7 n . ( J . Musket , Xi ic l . Tn ?=trr:.i. Moth. M J , M"i ( 1 9 7 M *
348 S.K. P e r r y , P . P . Ih-ndy, T.A. C a h i l l , R. F1.occh.inJ, N . S . K i n g , R. V y r l c k ami J . C . doling, B u l l . Aw. P h y s . S f r t y . ? y , t2o (1972).
3^9 L .A. C a r r i e , A n a l v t . Chew. 4 0 . 586 (196H) .
350 N. J a g o d u , K. K u M o r s c h k y , R. F r a n k , P . D j o r k h o l a and P . G r a y , T r a n s . X . E . E » g f X u c l . Sei» KS»21 . »9% ( t 9 7 4 K
35* S» B4oernhol.iB,and l,.M. L e d e r e r , X u c l . I n a t r u r o . Metti. 15* 233 1*962).

f
J ' l"1 T
r'T u
1J Figure 1 Preamplifier circuit diagram (adapted from Valkovic.
w - • —

X-ray spectrum Bowen's kale
Mo X-rays Elastics I ly
scattered 9) I >~
a - "i
0 3
91 •C o o
M 0 - f. ". 3 J f i
3
?
tv
1 U~ 8 12
Energy (fceV) 16 20

X-ray ?
-ve
>
1 \
<1 Guard ring
"sP 0*A
l l i l l l > M U'^Ht IMI
Coincidence circuitry
Double guard ring
Figure 3 Detector configurations (adapted from ValkovicJ

Sample
Source
Si (Li)
Figure 4
Geometries used for radioisotope excitation
Sample Pu-238
^^microsphere
Si (Li)

Traditional system
Transmission anode
Secondary fluorescence
Figuro 5 X-ray tubo configurations

« u i
«. i o C o
•H • •» -rt IS • JS
• 11 t. a »» r» »• t to
• t i u c

Graphite monochromators
X-ray tube Si(Li) detector
Fieur* 7 Schematic eiaffrar: of goom»try ua*d fo: doubl* monochroRjatitation.

syunoo
j

• ft
Crusta! abundances
Figure 9 Cruetal abundances of element*.
-JL 90

LIN r Xa t- Mf
Minimum detectable a m o u n t
JLJ[&A
Amount of elcmenx present in the largest size rock sample these .-r.othods handle (typical crustal rock)
X-ray spectrometry Neutron capture Photon activation Arc spectroscopy Spark source mass spectrometry Neutron activation
B
Hbl Auk
r, nni LM „ Rb
i Sr
in
Sc
mm D
-. Cs r Ba
Z r
Mbiiua it
i. Hf
n .
kill Jl
. Al i. S i L. P ytuy L Ge
m " • II
AS
ffi r. si
1- C l
y r Cd r I n
ii L. Sn j . Sb * . _ T e
LI tiiEal Ml lit M14 fc§ Pb
EMIJ
mil . IT i L B i Po At.
j . La L. Ce
AyEilLi Ac t- Th hi Pa U
Pm i. Sm
I Eu ^ Gd . Tb Dy . Ho . E r . Tm Yb Lu
|jul U id i i l i u U F i g u r e 10 " ' —
Sensitivities for six methods of multi-clement analysis. Heights of columns are sensitivities for amounts of a single isolated element neglecting interferences.

• <
•
8 i 10 « O
0> m ! 1
*S- S ! o a
»5S o »
>ka3d ^OUILU /o,' ^O/.?G SA^B; jy ...... J