introduction to next generation sequencing (ngs) data analysis jenny wu genomics high throughput...
TRANSCRIPT

Introduction To Next Generation Sequencing (NGS) Data Analysis
Jenny WuGenomics High Throughput Facility
UCI

Outline• Goals : Practical guide to NGS data processing• Bioinformatics in NGS data analysis
– Basics: terminology, data formats, general workflow etc.– Data Analysis Pipeline
• Sequence QC and preprocessing• Downloading reference sequences• Sequence mapping• Downstream analysis workflow and software
• RNA-Seq data analysis• Concepts: spliced alignment, normalization, coverage, differential expression.• Tuxedo suite: Popular RNA-Seq pipeline • Data visualization with Genome Browsers.• RNA-Seq pipeline software: Galaxy vs. shell scripting• Downstream Pathway analysis
• ChIP-Seq data analysis workflow and software• NGS bioinformatics resources• Summary

Why Next Generation SequencingOne can generate hundreds of millions of short sequences (up to 250bp) in a single run in a short period of time with low per base cost.
• Illumina/Solexa GA II, HiSeq 2500, 3000,X• Roche/454 FLX, Titanium• Life Technologies/Applied Biosystems SOLiD
Reviews: Michael Metzker (2010) Nature Reviews Genetics 11:31Quail et al (2012) BMC Genomics Jul 24;13:341.

Why Bioinformatics
(wall.hms.harvard.edu)
Informatics

Bioinformatics Challenges in NGS Data Analysis
• “Big Data” (thousands of millions of lines long)– Can’t do ‘business as usual’ with familiar tools– Impossible memory usage and execution time – Manage, analyze, store, transfer and archive huge files
• Need for powerful computers and expertise– Informatics groups must manage compute clusters– New algorithms and software are required and often time
they are open source Unix/Linux based.– Collaboration of IT experts, bioinformaticians and biologists

Basic NGS Workflow
Olson et al.

NGS Data Analysis Overview
Olson et al.

Outline• Goals : Practical guide to NGS data processing• Bioinformatics in NGS data analysis
– Basics: terminology, data formats, general workflow etc.– Data Analysis Pipeline
• Sequence QC and preprocessing• Downloading reference sequences: query NCBI, UCSC databases.• Sequence mapping• Downstream analysis workflow and software
• RNA-Seq data analysis• Concepts: spliced alignment, normalization, coverage, differential expression.• Tuxedo suite: Tophat, Cufflinks and cummeRbund • Data visualization with Genome Browsers.• RNA-Seq pipeline software: Galaxy vs. shell scripting
• ChIP-Seq data analysis workflow and software• NGS bioinformatics resources• Summary

TerminologyExperimental Design:• Coverage (sequencing depth): The number of nucleotides from reads
that are mapped to a given position.• Paired-End Sequencing: Both end of the DNA fragment is sequenced,
allowing highly precise alignment. • Multiplexed Sequencing: "barcode" sequences are added to each
sample so they can be distinguished in order to sequence large number of samples on one lane.
Data analysis:• Quality Score: Each called base comes with a quality score which measures
the probability of base call error.• Mapping: Align reads to reference to identify their origin.• Assembly: Merging of fragments of DNA in order to reconstruct the original
sequence.• Duplicate reads: Reads that are identical. Can be identified after mapping.• Multi-reads: Reads that can be mapped to multiple locations equally well.

What does the data look like?Common NGS Data Formats
For a full list, go to http://genome.ucsc.edu/FAQ/FAQformat.html

File Formats• Reference sequences, reads:– FASTA– FASTQ (FASTA with quality scores)
• Alignments:– SAM (Sequence Alignment Mapping)– BAM (Binary version of SAM)
• Features, annotation, scores:– GFF3/GTF(General Feature Format)– BED/BigBed– WIG/BigWig
http://genome.ucsc.edu/FAQ/FAQformat.html

FASTA Format (Reference Seq)

FASTQ Format (Illumina Example)
@DJG84KN1:272:D17DBACXX:2:1101:12432:5554 1:N:0:AGTCAACAGGAGTCTTCGTACTGCTTCTCGGCCTCAGCCTGATCAGTCACACCGTT+BCCFFFDFHHHHHIJJIJJJJJJJIJJJJJJJJJJIJJJJJJJJJIJJJJ@DJG84KN1:272:D17DBACXX:2:1101:12454:5610 1:N:0:AGAAAACTCTTACTACATCAGTATGGCTTTTAAAACCTCTGTTTGGAGCCAG+@@@DD?DDHFDFHEHIIIHIIIIIBBGEBHIEDH=EEHI>FDABHHFGH2@DJG84KN1:272:D17DBACXX:2:1101:12438:5704 1:N:0:AGCCTCCTGCTTAAAACCCAAAAGGTCAGAAGGATCGTGAGGCCCCGCTTTC+CCCFFFFFHHGHHJIJJJJJJJI@HGIJJJJIIIJGIGIHIJJJIIIIJJ@DJG84KN1:272:D17DBACXX:2:1101:12340:5711 1:N:0:AGGAAGATTTATAGGTAGAGGCGACAAACCTACCGAGCCTGGTGATAGCTGG+CCCFFFFFHHHHHGGIJJJIJJJJJJIJJIJJJJJGIJJJHIIJJJIJJJ
Read RecordHeader
Read BasesSeparator
(with optional repeated header)
Read Quality Scores
Flow Cell ID
Lane TileTile
Coordinates
Barcode
NOTE: for paired-end runs, there is a second file with one-to-one corresponding headers and reads.
(Passarelli, 2012)

Outline• Goals : Practical guide to NGS data processing• Bioinformatics in NGS data analysis
– Basics: terminology, data formats, general workflow etc.– Data Analysis Pipeline
• Sequence QC and preprocessing• Downloading reference sequences: query NCBI, UCSC databases.• Sequence mapping• Downstream analysis workflow and software
• RNA-Seq data analysis• Concepts: spliced alignment, normalization, coverage, differential expression.• Tuxedo suite: Tophat, Cufflinks and cummeRbund • Data visualization with Genome Browsers.• RNA-Seq pipeline software: Galaxy vs. shell scripting
• ChIP-Seq data analysis workflow and software• Scripting Languages and bioinformatics resources• Summary

General Data Pipeline

Why QC?Sequencing runs cost money • Consequences of not assessing the Data • Sequencing a poor library on multiple runs –
throwing money away!
Data analysis costs money and time• Cost of analyzing data, CPU time $$• Cost of storing raw sequence data $$$• Hours of analysis could be wasted $$$$• Downstream analysis can be incorrect.

How to QC?
http://www.bioinformatics.babraham.ac.uk/projects/fastqc/, available on HPCTutorial : http://www.youtube.com/watch?v=bz93ReOv87Y
$ module load fastqc$ fastqc s_1_1.fastq;

FastQC: Example

Outline• Goals : Practical guide to NGS data processing• Bioinformatics in NGS data analysis
– Basics: terminology, data formats, general workflow etc.– Data Analysis Pipeline
• Sequence QC and preprocessing• Downloading reference sequences• Sequence mapping• Downstream analysis workflow and software
• RNA-Seq data analysis• Concepts: spliced alignment, normalization, coverage, differential expression.• Tuxedo suite: Tophat, Cufflinks and cummeRbund • Data visualization with Genome Browsers.• RNA-Seq pipeline software: Galaxy vs. shell scripting
• ChIP-Seq data analysis workflow and software• Scripting Languages and bioinformatics resources• Summary

Premade Genome SequenceIndexes and Annotation
http://ccb.jhu.edu/software/tophat/igenomes.shtml

The UCSC Genome Browser Homepage
Get genome annotation here!
General information
Specific information—new features, current status, etc.
Get reference sequences here!

Downloading Reference Sequences
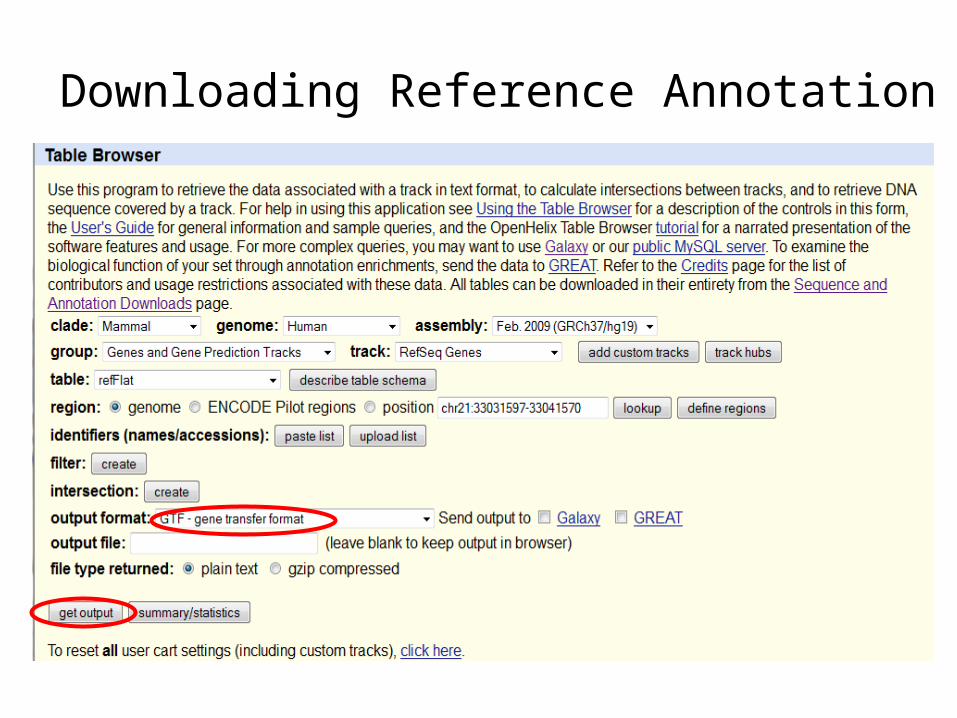
Downloading Reference Annotation

Outline• Goals : Practical guide to NGS data processing• Bioinformatics in NGS data analysis
– Basics: terminology, data formats, general workflow etc.– Data Analysis Pipeline
• Sequence QC and preprocessing• Downloading reference sequences: query NCBI, UCSC databases.• Sequence mapping• Downstream analysis workflow and software
• RNA-Seq data analysis• Concepts: spliced alignment, normalization, coverage, differential expression.• Tuxedo suite: Tophat, Cufflinks and cummeRbund • Data visualization with Genome Browsers.• RNA-Seq pipeline software: Galaxy vs. shell scripting
• ChIP-Seq data analysis workflow and software• Scripting Languages and bioinformatics resources• Summary

Sequence Mapping Challenges• Alignment (Mapping) is often the first step once
analysis-read reads are obtained.• The task: to align sequencing reads against a known
reference.• Difficulties: high volume of data, size of reference
genome, computation time, read length constraints, ambiguity caused by repeats and sequencing errors.

How to choose an aligner?• There are many short read aligners and they
vary a lot in performance(accuracy, memory usage, speed and flexibility etc).
• Factors to consider : application, platform, read length, downstream analysis, etc.
• Constant trade off between speed and sensitivity (e.g. MAQ vs. Bowtie).
• Guaranteed high accuracy will take longer.• Popular choices: Bowtie, BWA, Tophat, STAR.

Outline• Goals : Practical guide to NGS data processing• Bioinformatics in NGS data analysis
– Basics: terminology, data formats, general workflow etc.– Data Analysis Pipeline
• Sequence QC and preprocessing• Downloading reference sequences: query NCBI, UCSC databases.• Sequence mapping• Downstream analysis workflow and software
• RNA-Seq data analysis• Concepts: spliced alignment, normalization, coverage, differential expression.• Tuxedo suite: Tophat, Cufflinks and cummeRbund • Data visualization with Genome Browsers.• RNA-Seq pipeline software: Galaxy vs. shell scripting
• ChIP-Seq data analysis workflow and software• Scripting Languages and bioinformatics resources• Summary

Application Specific Software

Outline• Goals : Practical guide to NGS data processing• Bioinformatics in NGS data analysis
– Basics: terminology, data formats, general workflow etc.– Data Analysis Pipeline
• Sequence QC and preprocessing• Downloading reference sequences: query NCBI, UCSC databases.• Sequence mapping• Downstream analysis workflow and software
• RNA-Seq data analysis• Concepts: spliced alignment, normalization, coverage, differential expression.• Tuxedo suite: Tophat, Cufflinks and cummeRbund • Data visualization with Genome Browsers.• RNA-Seq pipeline software: Galaxy vs. shell scripting
• ChIP-Seq data analysis workflow and software• Scripting Languages and bioinformatics resources• Summary


Two Major Approaches 1. Gene or Exon level differential expression (DE):
DESeq2, EdgeR, DEXSeq…2. Transcripts assembly :
Trinity, Velvet-Oasis, TransABySS, Cufflinks, Scripture…

RNA-Seq Pipeline for DE

RNA-Seq: Spliced Alignment
http://en.wikipedia.org/wiki/File:RNA-Seq-alignment.png“Systematic evaluation of spliced alignment programs for RNA-seq data”Nature Methods, 2013
• Some reads will span two different exons
• Need long enough reads to be able to reliably map both sides
• Use a splice aware aligner!

How much sequence do I need?
• Oversimplified answer:20-50M PE/sample (Human/Mouse)
Depends on: – Size and complexity of transcriptome. – Goal of experiment: DE, transcript discovery.– Tissue type, library type, RNA quality, read length,
single-end…

RNA-Seq: Coverage
• Coverage in RNA-Seq is highly non-uniform• Within a single exon, there are regions with high
coverage and regions with zero coverage.• They change when the library preparation protocol
is changed.• The binding preferences of random hexamer primers
explain them only partially.
We simply hope that this averages out over the whole transcript !

RNA-Seq: NormalizationGene-length bias• Differential expression of longer genes is more significant because long genes yield more reads
RNA-Seq normalization methods:• Scaling factor based: Total count, upper quartile,
median, DESeq, TMM in edgeR• Quantile, RPKM (cufflinks)• ERCCNormalize by gene length and by number of reads mapped, e.g. RPKM/FPKM (reads/fragments per kilo bases per million mapped reads)

RNA-Seq: Differential Expression
Discrete vs. Continuous data: Microarray florescence intensity data: continuous
Modeled using normal distribution
RNA-Seq read count data: discrete
Modeled using negative binomial distribution
Microarray software can NOT be directly used to analyze RNA-Seq data!

Outline• Goals : Practical guide to NGS data processing• Bioinformatics in NGS data analysis
– Basics: terminology, data formats, general workflow etc.– Data Analysis Pipeline
• Sequence QC and preprocessing• Downloading reference sequences: query NCBI, UCSC databases.• Sequence mapping• Downstream analysis workflow and software
• RNA-Seq data analysis• Concepts: spliced alignment, normalization, coverage, differential expression.• Popular RNA-Seq pipeline: Tuxedo suite, HTSeq-DESeq• Data visualization with Genome Browsers.• RNA-Seq pipeline software: Galaxy vs. shell scripting
• ChIP-Seq data analysis workflow and software• Scripting Languages and bioinformatics resources• Summary

Popular RNA-Seq DE Pipeline
(The Tuxedo Protocol)
Pipeline 1 Pipeline 2
(The Alternative Protocol)

http://www.nature.com/nprot/journal/v7/n3/full/nprot.2012.016.html
Classic RNA-Seq (Tuxedo Protocol)
2. Transcript assembly and quantification
1. Spliced Read mapping
3. Merge assembled transcripts from multiple
samples
4. Differential Expression analysis
SAM/BAM
GTF/GFF

Classic vs. Advanced RNA-Seq workflow

1. Spliced Alignment: Tophat
http://www.nature.com/nprot/journal/v7/n3/full/nprot.2012.016.html
$ tophat -p 8 -G genes.gtf -o C1_R1_thout ptgenome C1_R1_1.fq C1_R1_2.fq
$ tophat -p 8 -G genes.gtf -o C1_R2_thout ptgenome C1_R2_1.fq C1_R2_2.fq
$ tophat -p 8 -G genes.gtf -o C2_R1_thout ptgenome C2_R1_1.fq C2_R1_2.fq
$ tophat -p 8 -G genes.gtf -o C2_R2_thout ptgenome C2_R2_1.fq C2_R2_2.fq

2.Transcript assembly and abundance quantification: Cufflinks
Cufflinks: a program that assembles aligned RNA-Seq reads into transcripts, estimates their abundances, and tests for differential expression and regulation transcriptome-wide.
http://www.nature.com/nprot/journal/v7/n3/full/nprot.2012.016.html
$ cufflinks -p 8 -o C1_R1_clout C1_R1_thout/ accepted_hits.bam
$ cufflinks -p 8 -o C1_R2_clout C1_R2_thout/ accepted_hits.bam
$ cufflinks -p 8 -o C2_R1_clout C2_R1_thout/ accepted_hits.bam
$ cufflinks -p 8 -o C2_R2_clout C2_R2_thout/ accepted_hits.bam

3. Final Transcriptome assembly: Cuffmerge$ cuffmerge -g genes.gtf -s genome.fa -p 8 assemblies.txt
$ more assembies.txt
./C1_R1_clout/transcripts.gtf
./C1_R2_clout/transcripts.gtf
./C2_R1_clout/transcripts.gtf
./C2_R2_clout/transcripts.gtf
http://www.nature.com/nprot/journal/v7/n3/full/nprot.2012.016.html

4.Differential Expression: Cuffdiff
CuffDiff: a program that compares transcript abundance between samples.
http://www.nature.com/nprot/journal/v7/n3/full/nprot.2012.016.html
$ cuffdiff -o diff_out -b genome.fa -p 8 –L C1,C2 -u merged_asm/merged.gtf ./C1_R1_thout/accepted_hits.bam, ./C1_R2_thout/accepted_hits.bam ./C2_R1_thout/accepted_hits.bam, ./C2_R2_thout/accepted_hits.bam

Cufflinks and related resources• Pachter, L. Models for transcriptquantification from RNA-Seq.arXiv preprint arXiv:1104.3889 (2011).• Trapnell C, Williams BA, PerteaG, Mortazavi AM, Kwan G, vanBaren MJ, Salzberg SL, Wold B,Pachter L.Transcript assembly andquantification by RNA-Seqreveals unannotated transcriptsand isoform switching duringcell differentiationNature Biotechnology doi:10.1038/nbt.1621
• Roberts A, Trapnell C, DonagheyJ, Rinn JL, Pachter L.Improving RNA-Seq expressionestimates by correcting forfragment biasGenome Biology doi:10.1186/gb-2011-12-3-r22• Roberts A, Pimentel H, TrapnellC, Pachter L.Identification of noveltranscripts in annotatedgenomes using RNA-SeqBioinformatics doi:10.1093/bioinformatics/btr355

Alternative Pipeline with HTSeq
HTSeq
DESeq2/edgeR
Tophat2,
$ htseq-count -f bam C1_R1_thout/sorted.bam -s no –o hsc/C1_R1.counts
$ htseq-count -f bam C1_R1_thout/sorted.bam -s no –o hsc/C1_R1.counts
$ htseq-count -f bam C1_R1_thout/sorted.bam -s no –o hsc/C1_R1.counts
$ htseq-count -f bam C1_R1_thout/sorted.bam -s no –o hsc/C1_R1.counts

HTSeq Output: Gene Count Table
…
…

DESeq2
http://www.bioconductor.org/packages/2.12/bioc/html/DESeq2.html

Downstream Analysis
Pathway and functional analysis:• Gene Ontology over representation• Gene Set Enrichment Analysis (GSEA)• Signaling Pathway Impact Analysis• Software
DAVID, GSEA, WGCNA, Blast2go, topGO..IPA, GeneGO MetaCore

Outline• Goals : Practical guide to NGS data processing• Bioinformatics in NGS data analysis
– Basics: terminology, data file formats, general workflow – Data Analysis Pipeline• Sequence QC and preprocessing• Obtaining and preparing reference • Sequence mapping• Downstream analysis workflow and software
• RNA-Seq data analysis• spliced alignment, normalization, coverage, differential expression.• Tuxedo suite: Tophat/Cufflinks parameters setting, cummeRbund • Data Visualization• RNA-seq pipeline software: RobiNA, Galaxy
• ChIP-Seq data analysis workflow and software• Open source pipeline software with Graphical User Interface• Summary

Integrative Genomics Viewer (IGV)http://www.broadinstitute.org/igv
Available on HPC. Use ‘module load igv’ and ‘igv’

Visualizing RNA-Seq mapping with IGV
http://www.broadinstitute.org/igv/UserGuideIntegrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration.Thorvaldsdóttir H et al. Brief Bioinform. 2013

Outline• Goals : Practical guide to NGS data processing• Bioinformatics in NGS data analysis
– Basics: terminology, data formats, general workflow etc.– Data Analysis Pipeline
• Sequence QC and preprocessing• Downloading reference sequences: query NCBI, UCSC databases.• Sequence mapping• Downstream analysis workflow and software
• RNA-Seq data analysis• Concepts: spliced alignment, normalization, coverage, differential expression.• Tuxedo suite: Tophat, Cufflinks and cummeRbund • Data visualization with Genome Browsers.• RNA-Seq pipeline software: Galaxy
• ChIP-Seq data analysis workflow and software• Scripting Languages and bioinformatics resources• Summary

Galaxy: Web based platform for analysis of large datasets
http://hpc-galaxy.oit.uci.edu/roothttps://main.g2.bx.psu.edu/
Galaxy: A platform for interactive large-scale genome analysis: Genome Res. 2005. 15: 1451-1455

Outline• Goals : Practical guide to NGS data processing• Bioinformatics in NGS data analysis
– Basics: terminology, data formats, general workflow etc.– Data Analysis Pipeline
• Sequence QC and preprocessing• Downloading reference sequences: query NCBI, UCSC databases.• Sequence mapping• Downstream analysis workflow and software
• RNA-Seq data analysis• Concepts: spliced alignment, normalization, coverage, differential expression.• Tuxedo suite: Tophat, Cufflinks and cummeRbund • Data visualization with Genome Browsers.• RNA-Seq pipeline software: Galaxy vs. shell scripting
• ChIP-Seq data analysis workflow and software• Scripting Languages and bioinformatics resources• Summary

What is ChIP-Seq?• Chromatin-Immunoprecipitation (ChIP)- Sequencing
• ChIP - A technique of precipitating a protein antigen out of solution using an antibody that specifically binds to the protein.
• Sequencing – A technique to determine the order of nucleotide bases in a molecule of DNA.
• Used in combination to study the interactions between protein and DNA.

ChIP-Seq Applications
Enables the accurate profiling of
• Transcription factor binding sites• Polymerases• Histone modification sites• DNA methylation

A View of ChIP-Seq Data• Typically reads (35-55bp) are quite sparsely
distributed over the genome. • Controls (i.e. no pull-down by antibody)
often show smaller peaks at the same locations
Rozowsky et al Nature Biotech, 2009

ChIP-Seq Analysis Pipeline
Sequencing
Short readSequences
Base Calling Read QC Short read
Alignment
Enriched Regions
Peak Calling
Combine with gene expression
Motif Discovery
Visualization with genome
browser
Differential peaks

ChIP-Seq: Identification of Peaks• Several methods to identify peaks but they mainly fall into 2
categories:– Tag Density– Directional scoring
• In the tag density method, the program searches for large clusters of overlapping sequence tags within a fixed width sliding window across the genome.
• In directional scoring methods, the bimodal pattern in the strand-specific tag densities are used to identify protein binding sites.
• Determining the exact binding sites from short reads generated from ChIP-Seq experiments
– SISSRs (Site Identification from Short Sequence Reads) (Jothi 2008)
– MACS (Model-based Analysis of ChIP-Seq) (Zhang et al, 2008)

ChIP-Seq: Output• A list of enriched locations
• Can be used:– In combination with RNA-Seq, to determine the
biological function of transcription factors– Identify genes co-regulated by a common
transcription factor– Identify common transcription factor binding
motifs

Resources in NGS data analysis
• Stackoverflow.com

SummarySummary
Thank you!
• NGS technologies are transforming molecular biology.
• Bioinformatics analysis is a crucial part in NGS applications – Data formats, terminology, general workflow– Analysis pipeline– Software for various NGS applications
• RNA-Seq and ChIP-Seq data analysis• Pathway Analysis• Data visualization• Bioinformatics resources