introduction to sequence analysis - göteborgs...
TRANSCRIPT

Introduction to sequence analysis(Lesk chapter 4)
Problem of sequence alignment - interaction betweenmolecular biology / computer science / statistics
* What are the biological problems ?* Algorithm (dynamic programming)* ‘Simple’ implementation / source code / compiling* Statistics and probability theory of alignments* Common implementations in molecular biology software packages* Results of biological significance

Applications of sequence analysis
Sequencing projects, assembly of sequence dataIdentification of functional elements in sequences, gene predictionSequence comparisonClassification of proteins Comparative genomicsRNA structure prediction Protein structure prediction Evolutionary history

Regulatory elements
PromoterTranslation start
Transcription stop
polyA signal
Transcription start
Translation stop
Exons
Introns
Expression from a eukaryotic gene
Transcription
Translation
DNA
RNA (primarytranscript)
RNA (spliced)
Protein

Applications of sequence analysis
Sequencing projects, assembly of sequence dataIdentification of functional elements in sequences, gene predictionSequence comparisonClassification of proteins Comparative genomicsRNA structure prediction Protein structure prediction Evolutionary history


Overview of methods
1. Comparison
a) IdentityExamples:• finding restriction sites (GAATTC)• pattern matches ((A,G)x4GK[S,T])(Wisconsin package: FindPatterns)
b) Comparing non-identical sequencesAlignmentsPairwise alignment
global alignment (WP: Gap) local alignment (smith-waterman) (WP:Bestfit)

Fasta (WP:Fasta, Tfasta)
Blast (WP / NCBI )blastn compares a DNA sequence to a DNA databaseblastp compares a protein sequence to a protein databasetblastn compares a protein sequences to all possible translation products of a DNA database
Multiple sequence alignmentClustalw (WP: Pileup)

2. Analyzing for property other than a simple linear sequence of letters.Examples: • statistical composition of letters• profile analysis , Position Specific Iterated BLAST (PSI-BLAST)(www.ncbi.nlm.nih.gov/BLAST)• HMMs• Prediction of higher order structure Protein secondary structure (alpha, beta) RNA (folding by base pairing within the molecule)
3. Simple transformation / extraction• Translation, DNA -> protein (W2H: Translate, Map)• Reverse translation• Splicing

Identity. Pattern matching
Pattern matching is used for finding short sequence patterns in asingle sequence, in a group of sequences or in the databases.
Examples of patterns (regular expressions):
GAATTCRecognition site for the restrictionenzyme EcoRI
GDSGGP Typical of serine proteases.
[AG]-x(4)-G-K-[ST] motif A of the ATP/GTP-binding site
C-x(2,4)-C-x(3)-[LIVMFYWC]-x(8)-H-x(3,5)-H zinc finger proteins

Pattern matching with the unix utility ‘grep’
% grep"C.\{2,4\}C...[LIVMFYWC].\{8\}H.\{3,5\}H”
sequence_file
Pattern matching with perl
$seq = ‘TRRCKTTCREQLYSGATGGHHASSHGAQR’;if ($seq =~/C.{2,4}C...[LIVMFYWC].{8}H.{3,5}H/){ print ”Found zinc finger motif”;}
WP: The program Findpatterns uses patterns to search a sequence(s). Theprogram Motifs specifically search a protein sequence or setof sequences for the motifs present in the PROSITE database.

Comparing non-identical sequencesProtein sequence comparison - basic concepts
When two protein sequences are being compared and the similarity isconsidered statistically significant, it is highly likely that the two proteins are evolutionary related.
GWFTREKLREEDHIKKGWFTKEKIREEDHIKK
Two kinds of biological relationships:
Orthologs Proteins that carry out the same function in different species
Paralogs Proteins that perform different but related functions within one organism
Proteins are homologous if they are related by divergence from a common ancestor.

X
X
X1
X
X2
Speciation
Ancestral organism
Organism A
Organism A
Organism B
Organism B
Orthologs
Orthologs

X
X
Xa
X
Xb
Gene duplication
Paralogs
Paralogs

Mouse trypsin -- orthologs -- Human trypsin | | paralogs paralogs | | Mouse chymotrypsin -- orthologs -- Human chymotrypsin

Comparing non-identical sequencesProtein sequence comparison - basic concepts
When two protein sequences are being compared and the similarity isconsidered statistically significant, it is highly likely that the two proteins are evolutionary related.
GWFTREKLREEDHIKKGWFTKEKIREEDHIKK
Two kinds of biological relationships:
Orthologs Proteins that carry out the same function in different species
Paralogs Proteins that perform different but related functions within one organism
Proteins are homologous if they are related by divergence from a common ancestor.

M A K L Q G A L G K R Y
M *A * *K * *I
Q *G * *A * *L * *A * * K * *R *Y
M A K L Q G A L G K R Y
* * * * * * * * * *M A K I Q G A L A K R Y
Comparing 2 sequences - Dotplot analysis
Sequence alignment

M A K L Q L G K R Y
M *A *K * *L * *Q *G *A *L * *G *K * *R *Y *
M A K L Q L G K R Y
* * * * * * * * * *M A K L Q G A L G K R Y
Gap
Sequence alignment
Comparing 2 sequences - Gaps

Gaps are results of mutations (changes in DNA) that occur during evolution
For instance consider this deletion mutation:
AACTTGACGTTGAACTGC
GACTGGGCGTATCTGACCCGCATA
CGGGCACCGGCCCGTGGC
N L T D W A Y R A P
N L T R A P
AACTTGACGTTGAACTGC
CGGGCACCGGCCCGTGGC
DNAprotein
Comparing / aligning two sequences. Gaps

In pairwise comparison gaps cannot be inserted in anunrestricted manner. For these reasons a gap penalty isassigned to gaps. Two parameters frequently used insequence comparison :
-Gap creation penalty-Gap extension penalty
There are two parameters because it is more ’difficult’to create a gap than to extend an existing gap.

M A K L Q G A L G K R Y
M *A * *K * *I
Q *G * *A * *L * *A * * K * *R *Y
M A K L Q G A L G K R Y
* * * * * * * * * *M A K I Q G A L A K R Y
Comparing 2 sequences - Dotplot analysis
Sequence alignment

Cytochrome C
Neurospora_crassa GSVDGYAYTD ANKQKGITWD ENTLFEYLEN PKKYIPGTKM AFGGLKKDKD Stellaria_longipes GSVEGFSYTD ANKAKGIEWN KDTLFEYLEN PKKYIPGTKM AFGGLKKDKD Thermomyces_lanuginosus GSVEGYSYTD ANKQAGITWN EDTLFEYLEN PKKFIPGTKM AFGGLKKNKD Arabidopsis_thaliana GSVAGYSYTD ANKQKGIEWK DDTLFEYLEN PKKYIPGTKM AFGGLKKPKD Aspergillus_niger GQSEGYAYTD ANKQAGVTWD ENTLFSYLEN PKKFIPGTKM AFGGLKKGKE Debaryomyces_occidentalis GQAAGYSYTD ANKKKGVEWT EQTMSDYLEN PKKYIPGTKM AFGGLKKPKD Schizosaccharomyces_pombe GQAEGFSYTE ANRDKGITWD EETLFAYLEN PKKYIPGTKM AFAGFKKPAD Fagopyrum_esculentum GTTAGYSYSA ANKNKAVTWG EDTLYEYLLN PKKYIPGTKM VFPGLKKPQE Sesamum_indicum GTTPGYSYSA ANKNMAVIWG ENTLYDYLLN PKKYIPGTKM VFPGLKKPQE Haematobia_irritans GQAAGFAYTN ANKAKGITWQ DDTLFEYLEN PKKYIPGTKM IFAGLKKPNE Lucilia_cuprina GQAPGFAYTN ANKAKGITWQ DDTLFEYLEN PKKYIPGTKM IFAGLKKPNE Ceratitis_capitata GQAAGFAYTD ANKAKGITWN EDTLFEYLEN PKKYIPGTKM IFAGLKKPNE Sarcophaga_peregrina GQAPGFAYTD ANKAKGITWN EDTLFEYLEN PKKYIPGTKM IFAGLKKPNE Manduca_sexta GQAPGFSYSD ANKAKGITWN EDTLFEYLEN PKKYIPGTKM VFAGLKKANE Samia_cynthia GQAPGFSYSN ANKAKGITWG DDTLFEYLEN PKKYIPGTKM VFAGLKKANE Schistocerca_gregaria GQAPGFSYTD ANKSKGITWD ENTLFIYLEN PKKYIPGTKM VFAGLKKPEE Apis_mellifera GQAPGYSYTD ANKGKGITWN KETLFEYLEN PKKYIPGTKM VFAGLKKPQE Macaca_mulatta GQAPGYSYTA ANKNKGITWG EDTLMEYLEN PKKYIPGTKM IFVGIKKKEE Pan_troglodytes GQAPGYSYTA ANKNKGIIWG EDTLMEYLEN PKKYIPGTKM IFVGIKKKEE Anas_platyrhynchos GQAEGFSYTD ANKNKGITWG EDTLMEYLEN PKKYIPGTKM IFAGIKKKSE Aptenodytes_patagonicus GQAEGFSYTD ANKNKGITWG EDTLMEYLEN PKKYIPGTKM IFAGIKKKSE

Substitution matricesEach amino acid change has a characteristic probability

Global alignmentConsiders similarity across the full extent of the sequences
xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx | | ||||||| | |xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
Local alignment Considers regions of similarity in parts of the sequences only.
xxxxxxx ||||||| xxxxxxx region of similarity


M A K L Q G A L G K R Y
M *A * *K * *I
Q *G * *A * *L * *A * * K * *R *Y
M A K L Q G A L G K R Y
* * * * * * * * * *M A K I Q G A L A K R Y
Comparing 2 sequences - Dotplot analysis
Sequence alignment

Searching databases with FASTA / BLAST
Improvement of speed as compared to local alignment algorithm:
Initial search is for short words.Word hits are then extended in either direction.

Output from Fasta
Fasta searches a protein or DNA sequence data bank version 3.3t04 January 25, 2000Please cite: W.R. Pearson & D.J. Lipman PNAS (1988) 85:2444-2448
../seq/ramp4.seq: 75 aa >ramp4.seq vs /vol1/gcgdata/ncbi_nr/nr.dat librarysearching /vol1/gcgdata/ncbi_nr/nr.dat library
173831120 residues in 553635 sequences statistics extrapolated from 60000 to 552908 sequences Expectation_n fit: rho(ln(x))= 4.8232+/-0.0004; mu= 0.7959+/- 0.022; mean_var=53.2306+/- 9.966, 0's: 686 Z-trim: 26 B-trim: 2227 in 1/63 Kolmogorov-Smirnov statistic: 0.0519 (N=29) at 46
FASTA (3.34 January 2000) function [optimized, BL50 matrix (15:-5)] ktup: 2 join: 36, opt: 24, gap-pen: -12/ -2, width: 16 Scan time: 102.010The best scores are: opt bits E(552908)gi|4585827|emb|CAB40910.1| (AJ238236) ribosome as ( 75) 483 130 1.9e-30gi|7657552|ref|NP_055260.1| stress-associated end ( 66) 426 116 3.7e-26gi|7504801|pir||T23009 hypothetical protein F59F4 ( 65) 251 71 8.5e-13gi|9802529|gb|AAF99731.1|AC004557_10 (AC004557) F ( 77) 145 45 0.00012gi|2498673|sp|Q47415|NRDI_ECOLI NRDI PROTEIN gi|2 ( 136) 105 35 0.22gi|1800061|dbj|BAA16538.1| (D90891) similar to [S ( 217) 105 35 0.33gi|6319639|ref|NP_009721.1| involved in the secre ( 65) 92 31 1.2gi|2498674|sp|Q56109|NRDI_SALTY NRDI PROTEIN gi|1 ( 136) 93 32 1.8

>>gi|4585827|emb|CAB40910.1| (AJ238236) ribosome associa (75 aa) initn: 483 init1: 483 opt: 483 Z-score: 682.4 bits: 130.3 E(): 1.9e-30Smith-Waterman score: 483; 100.000% identity in 75 aa overlap (1-75:1-75)
10 20 30 40 50 60ramp4. MVGAGGAAKMVAKQRIRMANEKHSKNITQRGNVAKTSRNAPEEKASVGPWLLALFIFVVC ::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::gi|458 MVGAGGAAKMVAKQRIRMANEKHSKNITQRGNVAKTSRNAPEEKASVGPWLLALFIFVVC 10 20 30 40 50 60
70ramp4. GSAIFQIIQSIRMGM :::::::::::::::gi|458 GSAIFQIIQSIRMGM 70

>>gi|7504801|pir||T23009 hypothetical protein F59F4.2 - (65 aa) initn: 227 init1: 143 opt: 251 Z-score: 365.3 bits: 71.4 E(): 8.5e-13Smith-Waterman score: 251; 53.846% identity in 65 aa overlap (10-74:1-64)
10 20 30 40 50 60ramp4. MVGAGGAAKMVAKQRIRMANEKHSKNITQRGNVAKTSRNAPEEKASVGPWLLALFIFVVC :. :::. .::.. :::...::::::. . : :.: ..:::..::.::::gi|750 MAPKQRMTLANKQFSKNVNNRGNVAKSLKPA-EDKYPAAPWLIGLFVFVVC 10 20 30 40 50
70 ramp4. GSAIFQIIQSIRMGM :::.:.::. ..:: gi|750 GSAVFEIIRYVKMGW 60

>>gi|2498673|sp|Q47415|NRDI_ECOLI NRDI PROTEIN gi|212121 (136 aa) initn: 66 init1: 41 opt: 105 Z-score: 160.3 bits: 34.6 E(): 0.22Smith-Waterman score: 105; 30.488% identity in 82 aa overlap (3-75:50-125)
10 20 30 ramp4. MVGAGGAAKMVAKQRIRMANEKHSKNITQRGN :.::.: : .: ::. :..:.. . :: gi|249 RLGLPAVRIPLNERERIQVDEPYILIVPSYGGGGTAGAVPRQVIRFLNDEHNRALL-RGV 20 30 40 50 60 70
40 50 60 70 ramp4. VAKTSRNAPEEKASVG---------PWLLALFIFVVCGSAIFQIIQSIRMGM .:. .:: : . .: ::: . : . :. . :...: :. gi|249 IASGNRNFGEAYGRAGDVIARKCGVPWL---YRFELMGTQ--SDIENVRKGVTEFWQRQP 80 90 100 110 120 130

Query Database
blastp Protein Proteinblastn DNA DNAtblastn Protein DNAblastx DNA Proteintblastx DNA DNA

Databases at NCBI
Protein sequence databases
nr All non-redundant GenBank CDS translations+PDB+SwissProt+PIR+PRF
swissprot the last major release of SWISS-PROT
DNA sequence Databases
nr All Non-redundant GenBank+EMBL+DDBJ+PDB sequences(but no EST, STS, GSS, or phase 0, 1 or 2 HTGS sequences)
dbest Non-redundant Database of GenBank+EMBL+DDBJ EST Divisions
dbsts Non-redundant Database of GenBank+EMBL+DDBJ STS Divisions
htgs htgs unfinished High Throughput Genomic Sequences

Output from Blast
BLASTP 2.0.11 [Jan-20-2000]
Reference: Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997), "Gapped BLAST and PSI-BLAST: a new generation of protein database searchprograms", Nucleic Acids Res. 25:3389-3402.
Query= ramp4.seq (75 letters)
Database: nr 457,798 sequences; 140,871,481 total letters
Searching..................................................done
Score ESequences producing significant alignments: (bits) Value
gi|4585827|emb|CAB40910.1| (AJ238236) ribosome associated membr... 126 2e-29gi|3851666 (AF100470) ribosome attached membrane protein 4 [Rat... 126 2e-29gi|3877972|emb|CAB03157.1| (Z81095) predicted using Genefinder;... 74 1e-13gi|3935169 (AC004557) F17L21.12 [Arabidopsis thaliana] 46 3e-05gi|3935171 (AC004557) F17L21.14 [Arabidopsis thaliana] 36 0.048gi|5921764|sp|O13394|CHS5_USTMA CHITIN SYNTHASE 5 (CHITIN-UDP A... 29 3.6
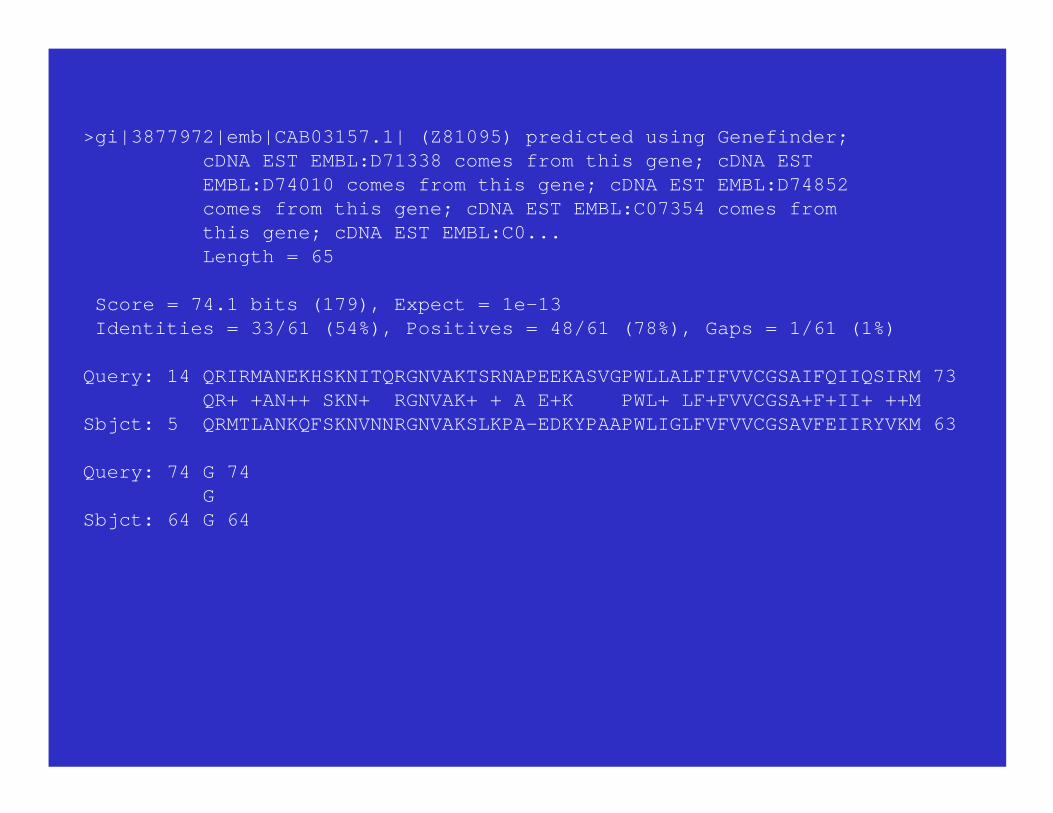
>gi|3877972|emb|CAB03157.1| (Z81095) predicted using Genefinder; cDNA EST EMBL:D71338 comes from this gene; cDNA EST EMBL:D74010 comes from this gene; cDNA EST EMBL:D74852 comes from this gene; cDNA EST EMBL:C07354 comes from this gene; cDNA EST EMBL:C0... Length = 65 Score = 74.1 bits (179), Expect = 1e-13 Identities = 33/61 (54%), Positives = 48/61 (78%), Gaps = 1/61 (1%)
Query: 14 QRIRMANEKHSKNITQRGNVAKTSRNAPEEKASVGPWLLALFIFVVCGSAIFQIIQSIRM 73 QR+ +AN++ SKN+ RGNVAK+ + A E+K PWL+ LF+FVVCGSA+F+II+ ++MSbjct: 5 QRMTLANKQFSKNVNNRGNVAKSLKPA-EDKYPAAPWLIGLFVFVVCGSAVFEIIRYVKM 63
Query: 74 G 74 GSbjct: 64 G 64

In a BLAST search low complexity regions in the query sequence arefiltered out by default
Regions with low-complexity sequence have an unusual composition andthis can create problems in sequence similarity searching. Low-complexity sequence can often be recognized by visual inspection. Forexample, the protein sequence PPCDPPPPPKDKKKKDDGPP has lowcomplexity and so does the nucleotide sequenceAAATAAAAAAAATAAAAAAT. Filters are used to remove low-complexitysequence because it can cause artifactual hits. In BLAST searchesperformed without a filter, often certain hits will be reported with highscores only because of the presence of a low-complexity region. Mostoften, this type of match cannot be thought of as the result of homologyshared by the sequences. Rather, it is as if the low-complexity region is"sticky" and is pulling out many sequences that are not truly related.
Another reason why hits to low-complexity regions in proteins should befiltered out is that such regions often have a disordered 3D structure andare not associated with well-defined biological functions.
BLAST and filtering of low-complexity sequence

Query:295 DDIFGELSSGKNAPKTGGGAKGNNASPAGSGNTKNNGASGADINNYAGQIKSAIESKFYD DDIFGELSSGKNAPKTGGGAKGNNASPAGSGNTKNNGASGADINNYAGQIKSAIESKFYD Sbjct:87 DDIFGELSSGKNAPKTGGGAKGNNASPAGSGNTKNNGASGADINNYAGQIKSAIESKFYD
Query:355 ASSYAGKTCTLRIKLAPDGMLLDIKPEGGDXXXXXXXXXXXXXXXXXXXXSQAVYEVFKN ASSYAGKTCTLRIKLAPDGMLLDIKPEGGD SQAVYEVFKNSbjct:147 ASSYAGKTCTLRIKLAPDGMLLDIKPEGGDPALCQAALAAAKLAKIPKPPSQAVYEVFKN
Query:415 APLDFKP 421 APLDFKPSbjct:207 APLDFKP 213



Multiple sequence alignment
