ion mobility measurements and their applications to ... · journal of mass spectrometry, vol. 32...
TRANSCRIPT

JOURNAL OF MASS SPECTROMETRY, VOL. 32, 577È592 (1997)
SPECIAL FEATURE:TUTORIAL
Ion Mobility Measurements and their Applicationsto Clusters and Biomolecules
David E. ClemmerDepartment of Chemistry, University of Indiana, Bloomington, IN 47405, USA
Martin F. JarroldDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA
Ion mobility measurements can be used to obtain structural information for large polyatomic ions in the gas phase.The methods are Ñexible and can be applied to a wide range of chemical systems. This article reviews the develop-ment of these methods and discusses recent applications to complex ions such as atomic clusters and large bio-molecules.
J. Mass Spectrosc. 32, 577È592 (1997)No. of Figs : 15 No. of Tables : 1 No. of Refs : 111
KEYWORDS: ion mobility measurements ; poyatomic ions ; atomic clusters ; large biomolecules
INTRODUCTION
In this article we describe some of the recent work thathas been performed using ion mobility measurements todeduce structural information for polyatomic ions.Determining the structure of a polyatomic ion in thegas phase is a challenging problem, that is usuallyapproached by indirect methods such as dissociation orreactivity studies. Ion mobility measurements haverecently emerged as a technique that can provide moredirect information about the geometries of large polya-tomic ions. The structural information obtained fromthese studies is not as detailed as information fromhigh-resolution spectroscopic studies, but in many casesthe types of spectroscopic studies needed to providestructural information cannot be applied to polyatomicions in the gas phase. While the use of ion mobilitymeasurements to deduce structural information hasgrown in the last Ðve years, both the experimental tech-niques and theoretical methods employed in thesestudies were largely developed over two decades ago.
The mobility of a gas-phase ion is a measure of howrapidly it moves through a bu†er gas under the inÑu-ence of an electric Ðeld. Mobility measurements are per-formed in a drift tube, which contains the bu†er gas andusually has a series of electrodes to provide a uniform
Correspondence to : D. E. Clemmer.Contract grant sponsor : National Science Foundation ; Contract
grant number : CHE-9306900 ; Contract grant number : CHE-9625199 ; Contract grant number : CHE-97XXXXX.
Contract grant sponsor : Petroleum research Fund.
electric Ðeld. The electric Ðeld accelerates the ions, whilecollisions with the bu†er gas decelerate them, leading toa constant drift velocity, The mobility, K, is thevD .ratio of the drift velocity to the electric Ðeld, K \ vD/E,and it contains information about the interactionbetween the ion and the bu†er gas. For atomic ions, themobility depends on the electronic state, as shown byRowe et al.1 in 1980. For a large polyatomic ion, themobility depends on the average collision cross-section.An ion with a large average cross-section undergoesmore collisions with the bu†er gas and travels moreslowly than an ion with a small average collision cross-section. Thus mobility measurements can be used toseparate ions with di†erent geometries. For example,Hagen2 demonstrated in 1979 that structural isomers ofpolycyclic aromatic hydrocarbons could be separatedby their di†erent mobilities. This separation providesthe basis for a sensitive and selective analytical tech-nique, ion mobility spectrometry,3,4 developed byCohen and Karasek in 1970.
Ion mobility spectrometry, or “plasma chromatog-raphyÏ as it was Ðrst called, uses mobility rather thanmass to separate ions. It has been used to detect drugs,chemical warfare agents, explosives and environmentalpollutants.5,6 Several groups have used mobility mea-surements to characterize the size distribution ofaerosol particles and small metal particles.7,8 In addi-tion to information about ion mobilities, drift tubestudies can provide information about ionÈmoleculereaction kinetics and equilibria. The injected-ion drifttube technique, where mass-selected ions are injectedinto the drift tube from an external source, was devel-oped by Hasted and co-workers9 in 1966. This tech-nique has been used by many di†erent groups, including
CCC 1076È5174/97/060577È16 $17.50 Received 10 April 1997( 1997 by John Wiley & Sons, Ltd. Accepted 11 April 1997

578 D. E. CLEMMER AND M. F. JARROLD
those of Warneck, Burke, Lindinger, Johnsen andBiondi and Arnold and co-workers, to measure mobi-lities and study ion chemistry.10h15 The main focus ofthe early work employing this technique was to measuremobilities and study ionÈmolecule reactions importantin understanding the chemistry of the upper atmo-sphere. An advantage of the injected ion drift tube tech-nique is that it can be used with a wide variety ofsources. The early work employed simple electronimpact sources. More recently, Jarrold and co-workers16,17 coupled a pulsed laser vaporization sourceto an injected ion drift tube apparatus and used it tostudy the chemistry of mass-selected cluster ions, focus-ing on silicon clusters. Bowers and co-workers18 subse-quently used a similar approach to study the chemistryof metal cluster ions and to measure the mobilities ofatomic metal ions. They found large di†erences in themobilities of metal ions in di†erent electronic states. In1991, this group reported ion mobility measurementsfor carbon cluster ions showing that di†erent structuralisomers could be resolved.20 This work was timely,because of the enormous interest in fullerenes andcarbon clusters at that time and remarkable because ofthe large number of isomers that were resolved. Obtain-ing structural information for gas phase atomic clustersis a particularly challenging problem and in addition tocarbon clusters,20h23 ion mobility measurements havebeen used to study silicon clusters, germanium clusters,aluminum clusters and a variety of metal-containingcarbon clusters. Bierbaum and co-workers30 haverecently used mobility measurements to study ionÈmolecule clusters.
It is possible that ions that are generated in thesource are not in their lowest energy geometries. Asimple annealing technique makes it possible to use ionmobility measurements to examine the isomerizationprocesses of a polyatomic ion.31 If the ions are injectedinto the drift tube at elevated kinetic energies, collisionswith the bu†er gas lead to a transient heating cycle.While hot, the ions can isomerize and at high injectionenergies they may fragment. Since the transient heatingcycle occurs close to the entrance of the drift tube, therest of the drift tube can be used to probe the geome-tries of the annealed parent ion or the fragments. Thisapproach has been used to provide important informa-tion about isomerization processes in carbon clustersand the mechanism of fullerene formation.32,33
While covalently bound clusters, particularly carbonclusters, have isomers with very di†erent shapes andhence very di†erent mobilities, metal clusters, ionic clus-ters and other polyatomic ions generally have isomerswith similar shapes. The application of ion mobilitymeasurements to these species is hindered by the lowresolution available in injected ion drift tube experi-ments. To overcome this problem, Dugourd et al.34have recently constructed a high-resolution ion mobilityapparatus with a resolving power over a order of mag-nitude better than available from the previous injectedion drift tube experiments. In order to achieve thehigher resolving power, it is necessary to operate withmuch stronger drift Ðelds and higher bu†er gas pres-sures. The resulting experimental conÐguration hasmuch in common with the ion mobility spectrometersdeveloped for analytical applications in the 1970s. With
the increased resolving power, many more structuralisomers have been resolved, even for carbon clusters.
Another exciting development is the recent applica-tion of ion mobility methods to examine the conforma-tions of peptides and proteins in the gas phase. Sincethe development of suitable ionization methods,35h37there has been interest in examining the geometries oflarge biological molecules in the gas phase.38h47 Severalgroups have previously performed ion mobility mea-surements for biological molecules,48h50 although noe†ort was made to deduce structural information in thiswork. Bowers and co-workers51h53 have recentlyreported ion mobility measurements for the peptidebradykinin. The bradykinin ions for this study wereproduced by matrix-assisted laser desorption/ionization(MALDI).37 Only one structure was resolved and thecross-sections deduced from the mobilities were essen-tially independent of temperature. Extensive moleculardynamics studies were performed and suggested that thegas-phase structure of this peptide is dominated by anintramolecular “solvationÏ shell around the charge.53Clemmer, Jarrold and co-workers have performed aseries of ion mobility measurements for protein ionsproduced by electrospray ionization. Electrospray ion-ization generates protein ions in a distribution of chargestates,54 so that ion mobility measurements can be per-formed as a function of the charge. Measurements havenow been performed for cytochrome c,55h58 bovinepancreatic trypsin inhibitor (BPTI),57 apomyoglobin,59lysozyme60 and ubiquitin,61 and many di†erent confor-mations have been resolved. In addition, the “denatur-ationÏ and refolding of these gas-phase proteins hasbeen examined using collisional heating and protonstripping reactions to manipulate the charge state.
EXPERIMENTAL CONSIDERATIONS
Mobilities are measured by determining the amount oftime it takes for a short packet of ions to travel throughthe drift tube. Since the mobility depends on the bu†ergas number density, reduced mobilities, scaled to thenumber density at STP, are usually reported. Thereduced mobility is given by
K0\ L2tD V
]273.2
T]
p760
(1)
where V is the voltage drop across the drift tube, L is itslength, is the drift time, p is the bu†er gas pressure intDTorr (1 Torr \ 133.3 Pa) and T is the temperature ofthe bu†er gas. Since all the parameters in Eqn (1) can bedetermined with an accuracy of better than 1%, it is notdifficult to obtain an absolute accuracy of a few percentin ion mobility measurements and a reproducibility ofbetter than 1%. The parameter that determines an ionÏsenergy in a drift tube is the ratio of the electric Ðeld tothe bu†er gas number density, E/N. At low E/N, wherethe drift velocity is small compared with thermal veloci-ties, the mobility is independent of the Ðeld strength.This is called the low-Ðeld limit. In the high-Ðeld limit,where the drift velocity is much larger than thermalvelocities, the mobility depends on E/N and the ionsmay align to some extent in the drift tube.62 Ion mobil-
( 1997 by John Wiley & Sons, Ltd. JOURNAL OF MASS SPECTROMETRY, VOL. 32, 577È592 (1997)

ION MOBILITY MEASUREMENTS 579
ity measurements that are designed to deduce structuralinformation should usually be performed in the low-Ðeld regime.
If a short packet of ions is injected into the drift tube,the Ñux of ions leaving the drift tube as a function oftime can be calculated from the transport equation.63For a single isomer the drift time distribution is givenby
F(t) \P
dtpP(tp)C[vD ] L /(t [ tp)]
(DT )1@2
]G1 [ exp
C [r024D(t [ tp)
DHexpG[[L [ vD(t [ tp)]2
4D(t [ tp)H
(2)
where is the drift velocity, is the radius of the driftvD r0tube entrance aperture, is the distribution func-dtp P(tp)tion of the packet of ions entering the drift tube, C is aconstant and D is the di†usion constant. Under low-Ðeld conditions D is directly proportional to K, D\
where ze is the charge on the ion and isKkB T /ze,63 kBBoltzmannÏs constant. Comparison of the measured dis-tributions with those calculated with Eqn (2) can revealwhether the measured peak consists of more than oneisomer. The resolution is limited by di†usion of the ionpacket as it travels through the drift tube. If twoisomers have mobilities that di†er by less than the ionpacket expands, they will not be resolved. As describedby Revercomb and Mason64 in 1974, the resolvingpower is given approximately by
tD*t
\A L Eze16kB T ln 2
B1@2(3)
It is apparent from this expression that in order toincrease the resolving power it is necessary to lower thetemperature, increase the drift Ðeld or increase thelength of the drift tube. Lowering the temperature from298 to 77 K increases the resolving power by a factor oftwo. If the drift Ðeld is increased there must be a corre-sponding increase in the bu†er gas pressure in order tokeep the mobilities in the low-Ðeld limit. However, forpressures above around 10 Torr it becomes increasinglydifficult to inject intact polyatomic ions into the drifttube from an external source, since they must overcomethe bu†er gas Ñowing out of the drift tube. As the bu†ergas pressure is increased, higher injection energies areneeded to inject the ions, and then they fragment. Thelength of the drift tube is limited by the expansion of theion packet by di†usion as it travels through the drift
tube. If the drift tube is too long, the ions are lost to thedrift tube walls.
The constraints outlined in the preceding paragraphdeÐne two basic experimental conÐgurations for per-forming ion mobility measurements for polyatomicions :
1. A low-resolution conÐguration, where ions areinjected into the drift tube from an external source.With the bu†er gas pressure limited to less than 10Torr, the drift Ðeld is limited to around 10 V cm~1 andthe drift tube can be up to several tens of centimeterslong. The resolving power of this conÐguration isaround 10È20.
2. A high-resolution conÐguration, where ions aregenerated in a source attached directly to the drift tube.With a bu†er gas pressure of hundreds of Torr, driftÐelds of hundreds of volts per centimeter can beemployed and the drift tube can be up to several meterslong. The resolving power of this conÐguration isaround 200È400.
The injected ion drift tube method and the selectedion Ñow-drift tube developed by Howorka et al.65 fallinto the Ðrst category. An injected ion drift tube appar-atus usually consists of a source to generate the ions, amass spectrometer to select a particular mass to chargeratio, a drift tube, followed by a second mass spectro-meter and ion detector. A wide variety of di†erentsources have recently been employed with this conÐgu-ration, including pulsed laser vaporization,16 pulsedlaser desorption,66 MALDI51 and electrospray ioniza-tion.55 Figure 1 shows a schematic diagram of therecently constructed injected ion drift tube apparatus ofValentine and Clemmer.58 This apparatus is equippedwith an electrospray source. The source has a di†eren-tially pumped desolvation region. A base can be addedto this region to reduce the charge on the electro-sprayed ions through proton stripping reactions. Afterpassing through the desolvation region the ions enterthe vacuum chamber. They are then focused into thedrift tube. There is no initial mass selection in thisapparatus. After passing through the drift tube, the ionsare focused into a quadrupole mass spectrometer andthen detected by an o†-axis collision dynode and dualmicrochannel plates.
Three features of the injected ion drift tube conÐgu-ration deserve to be mentioned.
1. Chemical reactivity/mobility measurements. Areagent can be introduced into the drift tube to examinethe chemical reactivity of the resolved isomers. For
Figure 1. Schematic diagram of injected ion drift tube apparatus at the University of Indiana.
( 1997 by John Wiley & Sons, Ltd. JOURNAL OF MASS SPECTROMETRY VOL. 32, 577È592 (1997)

580 D. E. CLEMMER AND M. F. JARROLD
example, this approach has been used to examine thereactivity of silicon clusters in di†erent geometries.67Recently, Valentine and Clemmer58 have used it toexamine HÈD exchange in cytochrome c.
2. Collisional annealing and dissociation. As the ionsare injected into the drift tube the injection energy isthermalized by collisions with the bu†er gas. Duringthis process some of the injection energy is convertedinto internal energy, heating the injected ion. After theionÏs injection energy is thermalized, and further colli-sions with the bu†er gas remove the ionÏs excess inter-nal energy. While hot the ions may isomerize or, at highinjection energies, fragment. The transient heating andcooling process occurs close to the entrance of the drifttube, so the rest of the drift tube can be used to examinethe geometries of any new isomers or fragment ions thatare formed. Analysis of the injection energy thresholdscan provide information about the activation energiesfor isomerization or dissociation.
3. T hermal annealing. By changing the temperature ofthe drift tube, isomerization processes can be followedas a function of temperature and the results used toderive activation energies. This approach has been usedto follow the isomerization of aluminum clusters26 and,recently, to study the thermal unfolding of gas-phaseproteins.69
This wide variety of di†erent ion mobility experi-ments illustrates the enormous Ñexibility of the injectedion drift tube approach.
The high-resolution conÐguration described aboveemploys much higher drift Ðelds. However, because ofthe high bu†er gas pressure, which is necessary to keepthe drifting ions in the low-Ðeld limit, mass-selectedpolyatomic ions cannot be injected from an external
source. This conÐguration provides the basis for ionmobility spectrometry, the analytical technique basedon the separation of ions by their mobilities. In ionmobility spectrometry, the mobility measurements areperformed at atmospheric pressure and the bu†er gas isgenerally nitrogen or air. Ions are usually produced bya radioactive 63Ni source that produces b-particles. Acomplex series of ionÈmolecule reactions follows theinitial ionization process, ultimately producing analyteproduct ions. The ions are usually detected at the end ofthe drift tube by a collector plate and an ampliÐer. Withthe high bu†er gas pressures employed and because theapparatus is not constructed to high vacuum standards,the formation of large ionÈmolecule clusters, particu-larly those involving water, is avoided by heating thedrift tube. A variety of ionization techniques have beenemployed with ion mobility spectrometry includinglaser desorption70 and electrospray ionization.71
The ion mobility spectrometers that are optimized foranalytical applications are not really suitable for studiesdirected at deriving information about the geometries ofpolyatomic ions. Dugourd et al.34 have recentlydescribed a high-resolution ion mobility apparatusdesigned for this task. A schematic diagram is shown inFig. 2. BrieÑy, the apparatus consists of a source regionwhich is directly coupled to a 63 cm long drift tube. Thesource region and drift tube contain helium bu†er gasat a pressure of D500 Torr. The ions are generated bypulsed laser vaporization or pulsed laser desorption.After formation, the ions are directed by shaped electricÐelds to an aperture in the ion gate that separates thesource region from the drift tube. A concern with thisconÐguration is that neutral species should not beallowed to di†use from the source into the drift tube,
Figure 2. Schematic diagram of the high resolution ion mobility apparatus at Northwestern University.
( 1997 by John Wiley & Sons, Ltd. JOURNAL OF MASS SPECTROMETRY, VOL. 32, 577È592 (1997)

ION MOBILITY MEASUREMENTS 581
because charge transfer may then occur in the drift tubeand this could distort the measured drift time distribu-tions. The ion gate consists of a 0.5 cm diameter by 2.5cm long channel with a uniform electric Ðeld along itslength. A Ñow of helium bu†er gas through the ion gate,from the drift tube into the source region, preventsneutral species from entering the drift tube, while theelectric Ðeld pulls the ions through the ion gate into thedrift tube. The ions then travel along the length ofthe drift tube under the inÑuence of a uniform electricÐeld generated by a stack of isolated rings and a voltagedivider. At the end of the drift tube, some of the ionsexit through a small aperture. They are then focusedinto a quadrupole mass spectrometer and at the end ofthe quadrupole they are detected. Drift time distribu-tions are recorded with a multichannel scaler using thelaser pulse as the start trigger. The drift tube can beheated or cooled so that ion mobility measurements canbe performed as a function of temperature.
At the beginning of this section we mentioned thation mobilities were measured by recording the amountof time it takes for a short packet of ions to travelacross the drift tube. This approach is well suited tosources using pulsed lasers, such as laser vaporization,laser desorption and MALDI sources. If the source gen-erates ions continuously a short pulse of ions can easilybe generated by an electrostatic shutter. However, theduty cycle is generally less than 1% and discarding 99%of the ions is not a concern if there are enough remain-ing to perform the mobility measurements in a reason-able time period. This is often not the case. In 1936,Bradbury and Neilsen72 described another method formeasuring drift times that employs two electrostaticshutters at di†erent positions along the length of thedrift tube. The same sine wave is applied to both shut-ters and the signal is recorded as the frequency of thesine wave is swept. Maxima in the transmitted currentare observed when the drift time is equal to integralmultiples of the half period of the sine wave. The signalrecorded as a function of frequency is an interferogram.Hill and co-workers73 have used Fourier transformtechniques to derive the drift time distributions from theinterferograms. If two gates are employed, 25% of theions are utilized rather than 1%. It is also possible touse only a single entrance gate and record both the inphase and out-of-phase signals using a computer, sothat 50% of the signal is utilized.
MOBILITY CALCULATIONS
Structural information is obtained from ion mobilitymeasurements by calculating the mobilities for trialgeometries and comparing them with the measuredvalues. In the low-Ðeld limit, where the measured mobil-ity is independent of the drift Ðeld, the mobility is givenby63,75
K \ (18n)1@216
C1m
] 1mb
D1@2 ze(kB T )1@2)avg(1, 1)
1N
(4)
where m is the mass of the ion, is the mass of a bu†ermbgas atom and is the average collision integral or)avg(1, 1)collision cross-section. Assuming that there is no align-
ment in the drift tube, which is a reasonable assumptionfor mobilities determined in the low-Ðeld limit, then theaverage collision cross-section can simply be obtainedby averaging over all possible collision geometries.Treating the polyatomic ion as a collection of hardspheres, one for each atom, and assuming hard sphereinteractions between the ion and bu†er gas atom, theaverage cross-section is obtained by averaging the geo-metric cross-section over all possible orientations inspace. We shall refer to this as the hard sphere projec-tion approximation because the geometric cross-sectionis simply the area of the shadow cast by the trialgeometry in collisions with the bu†er gas. This type ofmodel was Ðrst used by Mack76 in 1925. Since com-puters were not available, Mack mounted a beeswaxmodel of the molecule of interest on a two-axis goniom-eter so that it could be orientated in any direction. Thegoniometer was placed between a light source and agraduated screen and the size of the shadow was deter-mined systematically for a variety of orientations. Theprojection approximation, using computers rather thanbeeswax models, has been widely used to calculatemobilities of polyatomic ions in the last fewyears.21,24,26,29,77h79 Recently, Bowers and co-workers52 have described an extension of this modelthat attempts to incorporate long-range interactions inan approximate way so that mobilities can be calculatedas a function of temperature. In this model the hard-sphere contact distance is determined from tabulatedcollision integrals for atomÈatom collisions with a 12È6È4 potential, so the model is e†ectively the projectionapproximation with a temperature-dependent hardsphere contact distance.
While it is obvious that the hard sphere projectionapproximation ignores the long-range interactionsbetween the ion and bu†er gas, this approach alsoignores all the details of the scattering process betweenthe polyatomic ion and bu†er gas atom. in Eqn)avg(1, 1)(1) is really a collision integral that should be calculatedby averaging the momentum transfer cross-section overrelative velocity and collision geometry.63,75 Themomentum transfer cross-section depends on the scat-tering angle, which is the angle between the incomingand outgoing trajectory in a collision between the poly-atomic ion and a bu†er gas atom. The projectionapproximation ignores all these details. Shvartsburgand Jarrold80 have recently described an exact hardspheres scattering model which treats the scattering cor-rectly within the hard sphere limit and they have shownthat the projection approximation signiÐcantly under-estimates the collision integral for some geometries.
The long-range interactions between the bu†er gasand the ion should also not be ignored, but accountingfor them correctly is not trivial. First an e†ective poten-tial, consisting of a sum of interactions between thebu†er gas atom and all the atoms in the polyatomic ion,must be deÐned and then trajectories are run within thispotential to determine the scattering angles. Many tra-jectories must be run to average over the impact param-eter, the relative velocity and the collision geometry.Calculations along these lines have recently beenreported by Mesleh et al.,81 employing a potential con-sisting of a sum of two-body LennardÈJones inter-actions and ion-induced dipole interactions. Table 1
( 1997 by John Wiley & Sons, Ltd. JOURNAL OF MASS SPECTROMETRY VOL. 32, 577È592 (1997)

582 D. E. CLEMMER AND M. F. JARROLD
Table 1. Collision integrals calculated for (C60
)2, (C
60)13
,BPTI and cytochrome c from trajectory calculations,the exact hard spheres scattering model and the pro-jection approximationa
Cross section
(A� 2)Exact hard
Trajectory spheres Projection
Compound calculations scattering approximation
(C60
)2
226 (1.00) 225 (0.99) 216 (0.96)
(C60
)13
813 (1.00) 787 (0.97) 683 (0.84)
BPTI 927 (1.00) 935 (1.01) 767 (0.83)
Cytochrome c 1334 (1.00) 1339 (1.00) 1075 (0.81)
a The quantities in parentheses show the collision integral dividedby the collision integral determined from trajectory calculations.
shows collision integrals calculated for icosahe-(C60)2 ,dral BPTI and cytochrome c using trajectory(C60)13 ,calculations, the exact hard spheres scattering modeland the projection approximation. The values deter-mined using the projection approximation are up to20% smaller than those obtained from trajectory calcu-lations. Clearly, the projection approximation is inade-quate for calculating the collision integrals of largepolyatomic ions. Collision integrals determined fromthe exact hard spheres scattering model are within a fewpercent of those obtained from trajectory calculations.This indicates that the main deÐciency with the projec-tion approximation is that it ignores the details of thescattering process. The di†erence between the valuesdetermined from the exact hard spheres scatteringmodel and from trajectory calculations result frome†ects of the long-range potential between the bu†er gasatom and the polyatomic ion.
It should be obvious from the preceding discussionthat the evaluation of mobilities for comparison withexperimental data is not a solved problem. Also, withthe recent improvements in resolution, more isomers arebeing resolved with more subtle structural di†erences,placing even more stringent demands on the theoreticalmethods. All the methods described above assume arigid geometry and inelastic collisions. Lin et al.82 haveconsidered the e†ects of rotation of the polyatomic ionduring a collision with a bu†er gas atom. For a heliumbu†er gas the e†ect is small. Book et al.78 have reporteda study of the e†ects of vibrational motion on mobilitiescalculated using the projection approximation. This wasdone by averaging over an ensemble of geometries gen-erated by molecular dynamics simulations. Bowers andco-workers52,53 have used a similar approach in theirstudies of ethylene glycol oligomers and bradykinin. Sofar, the e†ects of inelastic collisions have not been con-sidered. A full molecular dynamics treatment is requiredto examine whether inelastic collisions signiÐcantlya†ect the mobilities. The approach to calculating mobi-lities described above is based on the belief that it ispossible to calculate reliable values from the existingtheoretical methods, if the problem is treated with suffi-cient rigor. Wessel and Jurs84 employed a di†erentapproach to predict mobilities. They used multipleregression analysis and neural networks to predict themobilities from structural information and a library ofmeasured mobilities. Here the neural network is trained
to produce the collision integral from structureÈproperty relationships.
APPLICATIONS
Here we brieÑy consider some of the recent applicationsof ion mobility measurements. We will start by describ-ing some work performed on atomic clusters and thenreview some of the recent studies of biomolecules. Thelatter focuses mainly on the conformations, unfoldingand refolding of multiply charged protein ions in thegas phase.
Carbon and metal-containing carbon clusters
Carbon forms directional covalent bonds in a variety ofdi†erent bonding conÐgurations. For these reasons,carbon clusters display many di†erent isomeric forms.Some of these isomers, in particular the chains andsmall monocyclic rings, have been studied for a numberof years85 and much is now known about fullerenes. Ionmobility measurements were Ðrst performed for carbonclusters by Bowers and co-workers.20 Their studies con-Ðrmed the existence of chains and monocyclic rings forthe smaller clusters and revealed the presence of aseveral di†erent ring isomers for the larger ones. Figure3 shows drift time distributions recorded for andC36`produced by laser vaporization of graphite andLaC36`graphite doped with respectively. Figure 3(a)La2O3 ,and (b) show distributions measured for at injectionC36`energies of 50 and 225 eV, respectively. The drift timedistributions that are recorded at 50 eV and below areindependent of injection energy, indicating that the 50eV distribution reÑects the isomer distribution producedby the source. Four distinct peaks are present. Thesepeaks have been assigned to a fullerene at D600 ls, agraphitic sheet at D800 ls, bicyclic ring isomers atD1100 ls and a monocyclic ring at D1200 ls. It isbelieved that the bicylic ring is made by clippingtogether two smaller monocyclic rings. The small shoul-der at D900 ls in Fig. 3(a) is probably due to tricyclic
Figure 3. Drift time distributions recorded for (a) and (b) C36½
and (c) and (d) at 50 eV (top) and 225 eV (bottom).LaC36½
( 1997 by John Wiley & Sons, Ltd. JOURNAL OF MASS SPECTROMETRY, VOL. 32, 577È592 (1997)

ION MOBILITY MEASUREMENTS 583
rings, which become more abundant for larger clusters.When the injection energy is increased the ions are col-lisionally heated as they enter the drift tube and theisomer distribution changes. For the bicylic and tri-C36`cylic rings disappear and the abundance of the mono-cyclic ring increases [compare Fig. 3(a) and (b)].Isomerization of the bicyclic and tricyclic rings to amonocylic ring is driven by strain relief.22 The carbonatoms in the rings are sp1 hybridized and the bicyclicand tricyclic rings are highly strained. It is clear fromthe results shown in Fig. 3 that the amount of ful-C36`lerene present does not increase signiÐcantly as theinjection energy is raised. If the injection energy israised further the monocyclic ring begins to dissociateto smaller ring fragments. Hence small carbon rings donot convert efficiently into fullerenes when collisionallyheated. However, for larger clusters the ring isomers doconvert into fullerenes. The efficiency of this processincreases with increasing cluster size and reaches D80%for These ion mobility and annealing resultsC60` .86suggest that the mechanism of fullerene formationinvolves ring coalescence, followed by isomerization tothe fullerene geometry when the rings are largeenough.87
There has been interest in metallofullerenes since thediscovery of fullerenes.88 Figure 3(c) and (d) show drifttime distributions recorded for with injectionLaC36`energies of 50 and 225 eV, respectively. The same fourisomers are present for as for although theirLaC36` C36` ,relative abundances are di†erent. However, as the injec-tion energy is increased the ring isomers disappear asthey convert into an fullerene and graphite sheet.LaC36`This does not occur for so the metal atom seemsC36` ,to promote the isomerization of the ring isomers intothe fullerene and graphite sheet. Careful comparison ofthe mobilities of the and fullerenes showsC36` LaC36`that they have almost exactly the same mobilities, whichindicates that the metal atom is inside the cage or endo-hedral for The metal atom is endohedral for allLaC36` .
fullerenes with n[ 34, but smaller ful-LaCn` LaCn`lerenes have a non-endohedral metal atom, because themetal atom no longer Ðts inside the cage. For somemetallofullerenes, such as fullerenes with an oddNbCn`number of carbon atoms, the ion mobility measure-ments indicate that the metal atom is networked intothe carbon cage.27
Bowers and co-workers89 have shown that theisomers present for carbon cluster anions are similar tothose present for the cations, except the relative abun-dances are di†erent. For example, the linear chain per-sists to much larger sizes for the anions, a result thathas been attributed to stabilization of the carbene endsof the chain by the extra electrons in the anion. Carboncluster anions have recently been examined using high-resolution ion mobility methods.90 Figure 4 shows ahigh-resolution drift time distribution recorded for C24~ .In this distribution, the feature at 64 ms is due to alinear chain, the three peaks at D57 ms have drift timesclose to those expected from previous low-resolutionmeasurements for a monocyclic ring, while the smallfeatures at D50 ms have drift times expected for bicyclicrings. This distribution can be compared with thatshown in Fig. 3 for where the monocyclic ring atC36` ,D1200 ls and bicyclic ring at D1100 ls are only par-
Figure 4. Drift time distribution recorded for with the high-C24É
resolution apparatus. The distribution was recorded with a heliumbuffer gas pressure of Á500 Torr and a drift voltage of 10 kV.
tially resolved. The existence of several di†erent bicylicring isomers was anticipated because there are anumber of plausible bicyclic ring geometries. However,the presence of several isomers in the monocyclic ringregion is a surprise, since there can only be one mono-cyclic ring. The isomer at D56 ms appears to be the realmonocyclic ring. The geometries of the other isomersare still under investigation.
Sodium chloride nanocrystals
Alkali metal halide clusters are interesting modelsystems because it appears that they adopt the bulkface-centered cubic (fcc) crystal structure at very smallcluster sizes.91h94 The principle evidence for the bulk-like geometries is the presence of magic number clusterscorresponding to complete cuboid geometries. Forexample, for clusters magic numbers at(NaCl)nCl~
and have been(NaCl)13Cl~, (NaCl)22Cl~ (NaCl)37Cl~attributed to completed cuboids with the dimensions3 ] 3 ] 3, 5] 3 ] 3 and 5 ] 5 ] 3. Here, 3] 3 ] 3means a cube with the dimensions 3 atoms ] 3atoms ] 3 atoms, to give a total of 27 atoms, which isthe number of atoms in Figure 5 shows(NaCl)13Cl~.the high-resolution drift time distribution measured forthe 5 ] 5 ] 3 magic number cluster, (NaCl)37Cl~.Three features are present in the drift time distribution.These have been assigned by comparing their mobilitiesto mobilities calculated for geometries optimized usingan ionic potential.95 The middle peak, at 138 ms, hasbeen assigned to the complete 5] 5 ] 3 cuboid, whilethe other peaks have been attributed to geometries withan incomplete face. The peak at 135 ms has been assign-ed to an incomplete 5] 4 ] 4 geometry and that at 145ms to an incomplete 6] 5 ] 3 geometry. The distribu-tion shown in Fig. 5 was recorded with the drift tube at5 ¡C. If the drift tube temperature is increased to slightlyabove room temperature the two features assigned tothe incomplete cuboids convert into the complete5 ] 5 ] 3 geometry, which indicates that this is thelowest energy geometry.
Figure 6 shows drift time distributions recorded forat 7, 33 and 67 ¡C. The three isomers(NaCl)35Cl~
present at low temperature have been assigned to an
( 1997 by John Wiley & Sons, Ltd. JOURNAL OF MASS SPECTROMETRY VOL. 32, 577È592 (1997)

584 D. E. CLEMMER AND M. F. JARROLD
Figure 5. Drift time distribution recorded for with(NaCl)37
ClÉthe high-resolution apparatus. The distribution was recorded witha helium buffer gas pressure of Á500 Torr, a drift voltage of 10 kVand a drift tube temperature of 5 ¡C. The three peaks present havebeen assigned to an incomplete 5 Ã4 Ã4, a complete 5 Ã5 Ã3and an incomplete 6 Ã5 Ã3 cuboid geometries. The structuresshown in the figure were optimized using an ionic potential.
incomplete 5] 5 ] 3, an incomplete 5] 4 ] 4 and an8 ] 3 ] 3 with a single defect. As the temperature israised the 8 ] 3 ] 3 and 5] 3 ] 3 isomers convert intothe 5 ] 4 ] 4 geometry. The drift time distribution inFig. 6 measured at 33 ¡C shows intensity between the
Figure 6. Drift time distributions recorded for at drift(NaCl)35
ClÉtube temperatures of 5, 33 and 67 ¡C. The three peaks presenthave been assigned to an incomplete 5 Ã5 Ã3, an incomplete5 Ã4 Ã4 and an incomplete 8 Ã3 Ã3 cuboid geometries. Thestructures shown in the figure were optimized using an ionicpotential.
peaks assigned to the 8 ] 3 ] 3 and 5] 4 ] 4 geome-tries. This is due to isomerization of the 8 ] 3 ] 3 tothe 5 ] 4 ] 4 at di†erent places along the length of thedrift tube. The amount of time that the ions spend inthe drift tube can be varied by varying the drift voltageand the drift time distributions can be analyzed toobtain the isomerization rates.96 Rate constants havebeen determined in this way as a function of tem-perature for a number of the structural transitions in
clusters. Arrhenius activation energies for(NaCl)nCl~the transitions, determined from the rate constants, are0.3È0.6 eV. Some of these structural transitions, such asthe 8 ] 3 ] 3 to 5 ] 4 ] 4 transition of (NaCl)35Cl~,involve the relocation of close to half the atoms in thecluster, so it is remarkable that the activation energiesare so low. The low activation energies suggest that thestructural transitions occur through a sequence ofsurface di†usion steps, rather than by a single concertedstep.
Protein ions in the gas phase
Intramolecular interactions, such as hydrogen bondsand van der Waals contacts and solvent interactions,both hydrophobic and hydrophilic, contribute to thefree energy of a protein in solution. However, the rela-tive importance of these factors in determining thesolution-phase conformation is still the subject of con-troversy. Studies of unsolvated proteins in the gas phasecan provide direct information about their intramolecu-lar interactions. Ion mobility measurements haverecently been used to examine the gas-phase conforma-tions of both peptides and proteins. Electrospray ioniza-tion produces proteins ions in a distribution of chargestates and the charge state distribution depends on theacidity of the solution. Positive ions result mainly fromprotonation of basic residues while negative ions areproduced by deprotonation of acidic residues. Figure 7shows drift time distributions for the ]7 charge state ofcytochrome c measured as a function of the injectionenergy. The distributions are plotted against a reducedtime-scale obtained by multiplying the true time-scaleby the charge state. This makes it easier to comparedistributions measured for di†erent charge states. Theions for these studies were produced by electrosprayingan unacidiÐed solution where the protein is in its nativeform. The dashed line in Fig. 7 shows the drift timeexpected for the native conformation of cytochromec.97,98 At low injection energies the peak in the drifttime distribution is at slightly shorter times thanexpected for the native conformation, indicating thatthe protein is more compact in the gas phase than insolution. In solution, the solvent provides an e†ectiveforce Ðeld which prevents globular proteins frompacking tightly. Polar side-chains extend out into thesolvent to maximize their interactions and the proteincontains cavities large enough to accommodate watermolecules. In vacuum, intramolecular interactions makethe side-chains collapse on to the protein surface andthe protein packs more tightly. According to moleculardynamics simulations, the radius of gyration of BPTIdecreases by D5% on going from solution tovacuum.99,100 Thus the feature observed at low injec-
( 1997 by John Wiley & Sons, Ltd. JOURNAL OF MASS SPECTROMETRY, VOL. 32, 577È592 (1997)
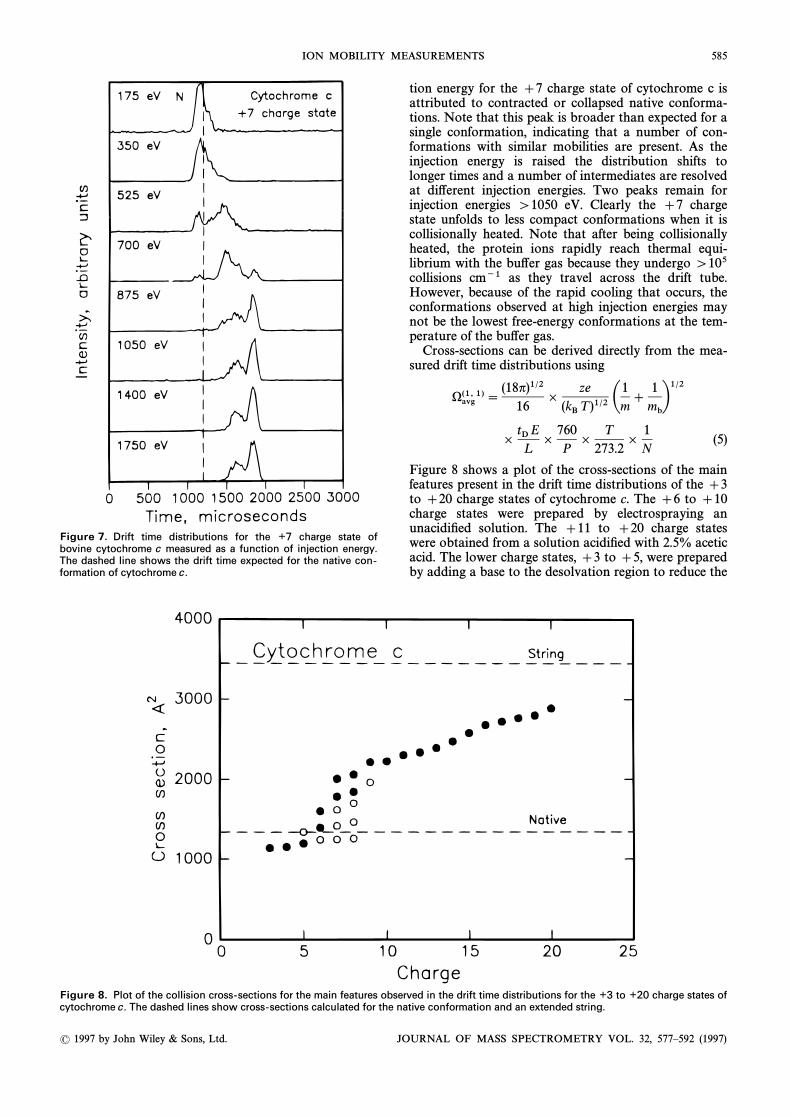
ION MOBILITY MEASUREMENTS 585
Figure 7. Drift time distributions for the ½7 charge state ofbovine cytochrome c measured as a function of injection energy.The dashed line shows the drift time expected for the native con-formation of cytochrome c.
tion energy for the ]7 charge state of cytochrome c isattributed to contracted or collapsed native conforma-tions. Note that this peak is broader than expected for asingle conformation, indicating that a number of con-formations with similar mobilities are present. As theinjection energy is raised the distribution shifts tolonger times and a number of intermediates are resolvedat di†erent injection energies. Two peaks remain forinjection energies [1050 eV. Clearly the ]7 chargestate unfolds to less compact conformations when it iscollisionally heated. Note that after being collisionallyheated, the protein ions rapidly reach thermal equi-librium with the bu†er gas because they undergo [105collisions cm~1 as they travel across the drift tube.However, because of the rapid cooling that occurs, theconformations observed at high injection energies maynot be the lowest free-energy conformations at the tem-perature of the bu†er gas.
Cross-sections can be derived directly from the mea-sured drift time distributions using
)avg(1, 1)\ (18n)1@216
]ze
(kB T )1@2A1m
] 1mb
B1@2
]tD EL
]760P
]T
273.2]
1N
(5)
Figure 8 shows a plot of the cross-sections of the mainfeatures present in the drift time distributions of the ]3to ]20 charge states of cytochrome c. The ]6 to ]10charge states were prepared by electrospraying anunacidiÐed solution. The ]11 to ]20 charge stateswere obtained from a solution acidiÐed with 2.5% aceticacid. The lower charge states, ]3 to ]5, were preparedby adding a base to the desolvation region to reduce the
Figure 8. Plot of the collision cross-sections for the main features observed in the drift time distributions for the ½3 to ½20 charge states ofcytochromec. The dashed lines show cross-sections calculated for the native conformation and an extended string.
( 1997 by John Wiley & Sons, Ltd. JOURNAL OF MASS SPECTROMETRY VOL. 32, 577È592 (1997)

586 D. E. CLEMMER AND M. F. JARROLD
charge by proton stripping reactions. The Ðlled pointsin the Ðgure show cross-sections for features observed athigh injection energies, while the open points show fea-tures observed at lower injection energies. The dashedlines show cross-sections calculated for the native con-formation and an extended string obtained by settingmost ' and ( angles to 180¡. For the low charge statesthe measured cross-sections are slightly smaller than thecross-section calculated for the native conformation.For the ]6 to ]8 charge states a structural transitionoccurs. While conformations, slightly more compactthan the native conformation can be prepared for thesecharge states by electrospraying an unacidiÐed solution,these conformations unfold to less compact ones whencollisionally heated. For higher charge states the cross-sections increase with charge and approach the valuecalculated for the extended string. The unfolding tran-sition that occurs as the charge is increased results fromCoulomb repulsion. Intramolecular interactions, hydro-gen bonds and van der Waals contacts, hold the proteinin a compact, folded conformation. However, as thecharge increases, Coulomb repulsion overcomes theattractive intramolecular interactions and the proteinunfolds. Coulomb repulsion is more important for pro-teins in the gas phase than in water, because water has ahigh dielectric constant.
More information about the stabilities of theobserved conformations can be obtained from measure-ments performed as a function of drift tube tem-perature.101 For the ]5 charge state only a singlefeature is observed in the drift time distributions fromroom temperature up to 300 ¡C and the cross-sectionfor this feature varies only slightly with temperature.Thus temperatures as high as 300 ¡C are not enough tounfold the ]5 charge state of cytochrome c. Cyto-chrome c unfolds at a much lower temperature in solu-tion.102 This demonstrates that the foldedconformations of a gas-phase protein can be much morestable than in solution. However, the results describedabove show that Coulomb repulsion destabilizes foldedconformations in the gas phase. Figure 9 shows drifttime distributions for the ]7 charge state of cyto-chrome c measured as a function of the drift tube tem-perature. The dashed line shows the distributionrecorded at room temperature with a high injectionenergy (2100 eV) while the other distributions wererecorded with a low injection energy (350 eV). As thetemperature is raised the ]7 charge state unfolds, andat 200 ¡C there is a single, relatively narrow peak at thesame position as the smaller peak in the high injectionenergy distribution (shown by the dashed line). Between200 and 300 ¡C a further structural transition occursand the peak moves to the position of the moreunfolded conformation in the high injection energy dis-tribution. These thermal unfolding transitions providean excellent test of the force Ðelds used in moleculardynamics simulations of proteins.
By introducing a base in the desolvation region it ispossible remove protons from highly protonated cyto-chrome c ions and form the low charge states, ]3 to]5. These charge states have compact conformationseven when they are produced from high charge statesthat were generated by electrospraying an acidiÐedsolution where cytochrome c is denatured. The drift
Figure 9. Drift time distributions recorded for the ½7 charge stateof cytochrome c as a function of drift tube temperature. The drifttime-scale has been converted into a cross-section scale so thatdistributions recorded at different temperatures can be easily com-pared. The dashed line shows the distribution measured at highinjection energy (2100 eV). The other distributions were recordedwith a low injection energy (350 eV).
time distributions for the high charge states indicatethat they are unfolded. Thus the folded conformationsobserved for the low charge states must result fromrefolding in the gas phase. The cross-sections for theserefolded conformations are similar to the cross-sectionsfor the compact conformations observed for the ]6 to]8 charge states from an unacidiÐed solution.However, this should not be taken to indicate that therefolded conformations have the native structure. Thenative conformation is expected to be stable in the gasphase because it has a large number of hydrogen bondsand van der Waals contacts. However, it is probablynot the lowest energy gas-phase conformation. Further-more, the native conformation is probably notkinetically accessible without a solvent.
Folding processes similar to those described abovefor cytochrome c have also been observed for the low-charge states of apomyoglobin. The ]6 charge state ofapomyoglobin is particularly noteworthy because itprovides a dramatic example of an activation barrier forgas-phase protein folding.59 Drift time distibutionsmeasured for the ]6 charge state of apomyglobin areshown in Fig. 10. The ions were produced by protonstripping of high charge states generated by electro-spraying an acidiÐed solution. At low injection energiesa partially folded conformation is observed. As theinjection energy is raised the distribution shifts toshorter drift times as the protein refolds. The featureobserved at low injection energies is more compact thanits high charge state precursors and so it is clearly anintermediate in the folding process and there must be an
( 1997 by John Wiley & Sons, Ltd. JOURNAL OF MASS SPECTROMETRY, VOL. 32, 577È592 (1997)
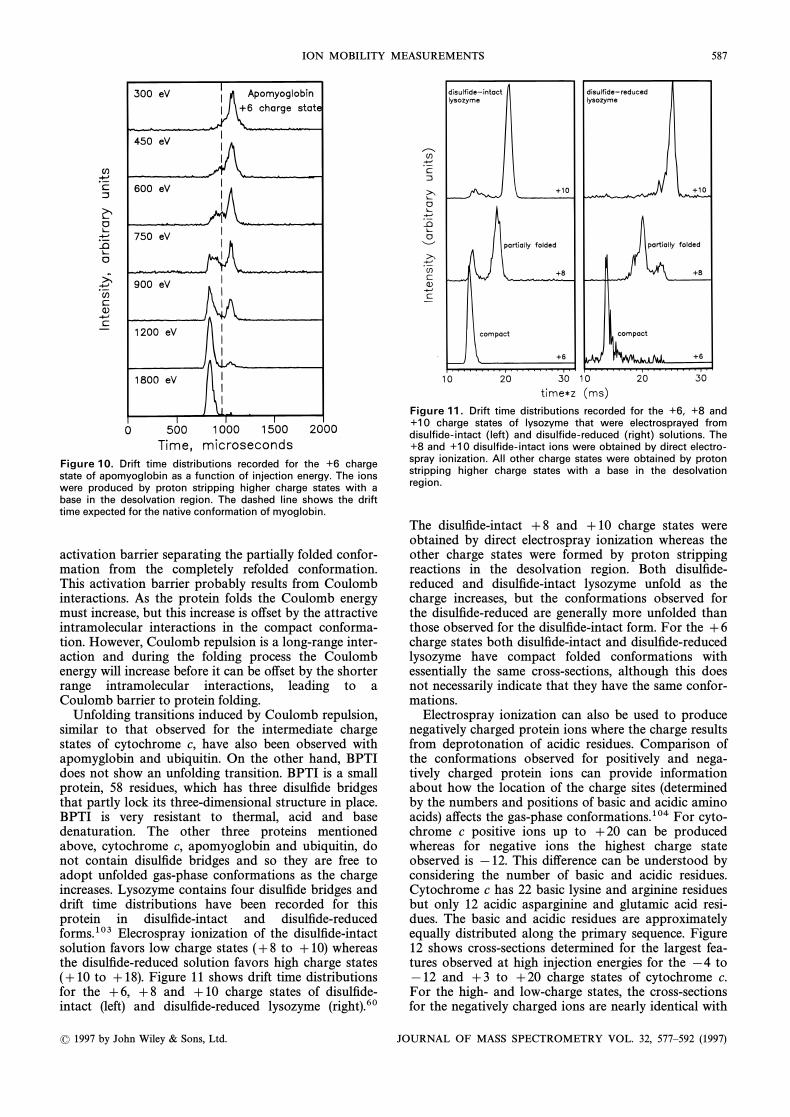
ION MOBILITY MEASUREMENTS 587
Figure 10. Drift time distributions recorded for the ½6 chargestate of apomyoglobin as a function of injection energy. The ionswere produced by proton stripping higher charge states with abase in the desolvation region. The dashed line shows the drifttime expected for the native conformation of myoglobin.
activation barrier separating the partially folded confor-mation from the completely refolded conformation.This activation barrier probably results from Coulombinteractions. As the protein folds the Coulomb energymust increase, but this increase is o†set by the attractiveintramolecular interactions in the compact conforma-tion. However, Coulomb repulsion is a long-range inter-action and during the folding process the Coulombenergy will increase before it can be o†set by the shorterrange intramolecular interactions, leading to aCoulomb barrier to protein folding.
Unfolding transitions induced by Coulomb repulsion,similar to that observed for the intermediate chargestates of cytochrome c, have also been observed withapomyglobin and ubiquitin. On the other hand, BPTIdoes not show an unfolding transition. BPTI is a smallprotein, 58 residues, which has three disulÐde bridgesthat partly lock its three-dimensional structure in place.BPTI is very resistant to thermal, acid and basedenaturation. The other three proteins mentionedabove, cytochrome c, apomyoglobin and ubiquitin, donot contain disulÐde bridges and so they are free toadopt unfolded gas-phase conformations as the chargeincreases. Lysozyme contains four disulÐde bridges anddrift time distributions have been recorded for thisprotein in disulÐde-intact and disulÐde-reducedforms.103 Elecrospray ionization of the disulÐde-intactsolution favors low charge states (]8 to ]10) whereasthe disulÐde-reduced solution favors high charge states(]10 to ]18). Figure 11 shows drift time distributionsfor the ]6, ]8 and ]10 charge states of disulÐde-intact (left) and disulÐde-reduced lysozyme (right).60
Figure 11. Drift time distributions recorded for the ½6, ½8 and½10 charge states of lysozyme that were electrosprayed fromdisulfide-intact (left) and disulfide-reduced (right) solutions. The½8 and ½10 disulfide-intact ions were obtained by direct electro-spray ionization. All other charge states were obtained by protonstripping higher charge states with a base in the desolvationregion.
The disulÐde-intact ]8 and ]10 charge states wereobtained by direct electrospray ionization whereas theother charge states were formed by proton strippingreactions in the desolvation region. Both disulÐde-reduced and disulÐde-intact lysozyme unfold as thecharge increases, but the conformations observed forthe disulÐde-reduced are generally more unfolded thanthose observed for the disulÐde-intact form. For the ]6charge states both disulÐde-intact and disulÐde-reducedlysozyme have compact folded conformations withessentially the same cross-sections, although this doesnot necessarily indicate that they have the same confor-mations.
Electrospray ionization can also be used to producenegatively charged protein ions where the charge resultsfrom deprotonation of acidic residues. Comparison ofthe conformations observed for positively and nega-tively charged protein ions can provide informationabout how the location of the charge sites (determinedby the numbers and positions of basic and acidic aminoacids) a†ects the gas-phase conformations.104 For cyto-chrome c positive ions up to ]20 can be producedwhereas for negative ions the highest charge stateobserved is [12. This di†erence can be understood byconsidering the number of basic and acidic residues.Cytochrome c has 22 basic lysine and arginine residuesbut only 12 acidic asparginine and glutamic acid resi-dues. The basic and acidic residues are approximatelyequally distributed along the primary sequence. Figure12 shows cross-sections determined for the largest fea-tures observed at high injection energies for the [4 to[12 and ]3 to ]20 charge states of cytochrome c.For the high- and low-charge states, the cross-sectionsfor the negatively charged ions are nearly identical with
( 1997 by John Wiley & Sons, Ltd. JOURNAL OF MASS SPECTROMETRY VOL. 32, 577È592 (1997)

588 D. E. CLEMMER AND M. F. JARROLD
Figure 12. Cross-sections measured for negatively charged (deprotonated) and positively charged (protonated) forms of cytochrome c.Only data for the most abundant feature in the drift time distributions recorded at high injection energies are plotted. The dashed lines showthat low and high charge states have similar cross-sections. Intermediate charge states such as the À7 charge states have significantlydifferent cross-sections.
those obtained for their positively charged counterparts.The primary di†erences are observed for the interme-diate charge states where, as discussed above, repulsiveCoulombic interactions are comparable to the attractiveintramolecular interactions that hold the protein in afolded conformation. For these charge states the loca-tion of the charges apparently plays a role in determin-ing which gas-phase conformations dominate the ionmobility distribution. For example, the dominant con-formation observed for the [7 charge state is signiÐ-cantly more folded than the dominant conformationobserved for the ]7 charge state. The unfolding thatoccurs as the charge state increases is somewhat analo-gous to acid denaturation of a protein in solution. Insolution, the compact native state is stable over arestricted pH range near 7 and as the pH is increased ordecreased, it unfolds.105
HÈD exchange has recently been used to examinepeptide106 and protein ions39,40,107,108 in the gasphase. The basic assumption behind these studies is thatthe fraction of exchangeable hydrogens that undergoHÈD exchange can provide a measure of the accessiblesurface area, which in turn can be used to deduce struc-tural information. This information may complementthat obtained from ion mobility measurements. Figure13 shows the results of some HÈD exchange studies ofthe ]8 charge state of horse heart cytochrome c. Horseheart cytochrome c has 198 exchangeable hydrogens, ofwhich 144 are exchanged by the native conformation ina neutral solution. Figure 13 shows drift time distribu-tions and mass spectra measured for the ]8 chargestate with introduced into the drift tube.58 TheD2Odashed line shows a portion of the mass spectrumobtained in the absence of The plots in the upperD2O.half of the Ðgure show results obtained with a low injec-tion energy where the ]8 charge state of cytochrome c
is predominently in compact conformations. Themaximum average mass shift observed under these con-ditions corresponds to the exchange of around 45hydrogens. The lower half of the Ðgure shows resultsobtained at a high injection energy where the protein isin a partially unfolded conformation. The mass spec-trum indicates an average exchange level of D60 hydro-gens. The higher exchange level observed for thepartially folded conformation is in line with expecta-tions. However, the exchange levels are much lowerthan for the native conformation in solution. Further-more, the exchange levels for the extended conforma-tions of the ]8 to ]18 charge states are all
Figure 13. Drift time distributions (left) and mass spectra (right)recorded for the ½8 charge state of horse-heart cytochrome cusing a buffer gas containing 0.3 Torr of The data in theD
2O.
upper half of the figure were recorded using an injection energy of960 eV and those in the lower half with an injection energy of 240eV. The dashed line shows the mass spectrum recorded without
in the drift tube.D2O
( 1997 by John Wiley & Sons, Ltd. JOURNAL OF MASS SPECTROMETRY, VOL. 32, 577È592 (1997)

ION MOBILITY MEASUREMENTS 589
approximately the same, D60, while the ion mobilitymeasurements show that the average cross-sectionincreases substantially over this range. It appears thatthe HÈD exchange levels are insensitive to subtlechanges in conformation for di†erent charge states ofcytochrome c. The observation that the higher chargestates have a constant exchange level is in agreementwith previous HÈD exchange studies for cytochrome cperformed by McLa†erty and co-workers using Fouriertransform ion cyclotron resonance (FT-ICR) spectrom-etry.39,40 However, the exchange levels in drift tubeexperiments were smaller than in the FT-ICR studies.
Studies of gas-phase proteins provide informationabout the intramolecular interactions that are impor-tant in deÐning their solution-phase conformations.However, gas-phase studies also provide newopportunities to study the solvation e†ects that alsoplay an important role in determining the conformationin solution. Starting with an unsolvated protein in thegas phase it is in principle possible to hydrate it sequen-tially and study the hydration process one water mol-ecule at a time. Thermodynamic information can beobtained from equilibrium constant measurements forthe hydration reactions. Kebarle and co-workers109have examined the hydration of a few small polypep-tides. Recently, Woenckhaus and Jarrold101 havereported measurements of the free energy changes forthe adsorption of the Ðrst few water molecules on theunfolded ]7 charge state of cytochrome c and thecompact, refolded ]5 charge state. The equilibriumconstant measurements were performed by addingwater vapor to the helium bu†er gas. With water vaporpressures of around 1 Torr at room temperature, themass spectra showed no evidence for water adsorptionand it was necessary to cool the drift tube to study thehydration reactions. Free energy changes at 271 K weredetermined for adsorption of the Ðrst nine water mol-ecules on the refolded ] 5 charge state and the Ðrst Ðvewater molecules on the unfolded ]7 charge state. Thefree energy changes are shown in Fig. 14, where they arecompared with the free energy changes for the adsorp-tion of the Ðrst few water molecules on GlyH` and
Surprisingly, the unfolded conformationH3O`.109,110(]7 charge state) has smaller initial free energy changesthan the refolded conformation (]5 charge state).However, the free energy changes for initial hydrationof both the unfolded and refolded conformations ofcytochrome c are substantially smaller than the freeenergy changes for initial hydration of andH3O`GlyH`. The free energy changes for the adsorption ofthe Ðrst water molecule on at 293 K are [41,(Gly)nH`[37, [28 and [24 kJ mol~1 for n\ 1È4.109 The sub-stantial di†erences in the free energy changes betweenn \ 2 and 3 has been attributed to cyclization of thelarger polypeptides through hydrogen bonds to carbon-yl oxygens. This intramolecular charge “solvationÏdecreases the free energy change for adsorption of theÐrst water molecule by e†ectively shielding the charge.The small free energy changes for the adsorption of theÐrst few water molecules on to cytochrome c indicatesthat the charge is very e†ectively shielded, presumablyby interactions with a number of carbonyl oxygens.However, it is still not clear why the free energy changesfor the unfolded ]7 charge state are signiÐcantly
Figure 14. Free energy changes for adsorption of the first fewwater molecules on the refolded ½5 charge state and the unfolded½7 charge state of bovine cytochrome c. Free energy changes foradsorption of water on and GlyH½ are shown for compari-H
3O½
son. The dashed line shows the free energy change for adsorptionon bulk water.
smaller than those for the compact refolded ]5 chargestate. This may indicate that the charge is more e†ec-tively shielded in the unfolded ]7 charge state, or theincorporation of some of the adsorbed water moleculesas structural water molecules in the folded ]5 chargestate.
Oligonucleotides
Because of their electronegative phosphate backbone,DNA and RNA can most readily be electrosprayedfrom basic solutions to yield negative ions. Figure 15shows drift time distributions measured for a 10-residueoligonucleotide comprised entirely of thymine bases
Distributions are shown for the [2 to [6 charge(T10).states of where n is the number of dep-[T10[ nH`]n~,rotonated sites. There is a sharp unfolding transitionbetween the [3 and [5 charge states which presum-ably results from Coulomb repulsion. Some typicalstructures obtained for the oligonucleotide usingT10the Insight II molecular modeling software are shown inFig. 15 for the [2, [4 and [6 charge states. Theaverage cross-sections obtained by sampling a largenumber of di†erent structures depend on the location ofthe charges. For the 10-unit homopolymer studied here,which is expected to be deprotonated along the ninephosphodiester linkages, the number of possible com-binations of assigning charges to sites is given by9!/[n !(9[ n) !]. From comparisons of collision cross-sections for modelling results with our experimentaldata it appears that the transition region is best
( 1997 by John Wiley & Sons, Ltd. JOURNAL OF MASS SPECTROMETRY VOL. 32, 577È592 (1997)

590 D. E. CLEMMER AND M. F. JARROLD
Figure 15. Drift time distributions for the É2 to É6 states of T10
.The geometries shown for the É2, É4 and É6 charge states wereobtained from Insight II molecular modeling software.
described when deprotonation sites are dispersed alongthe entire backbone rather than placed on adjacentsites.111
FUTURE PROSPECTS
It is clear from the preceding description of some of therecent applications of ion mobility measurements thatthis area has developed rapidly in the last few years. Inparticular, the studies of biomolecules have only justbegun and ion mobility measurements will be a richsource of information about the conformations, foldingand refolding of these species in the next few years. Thecombination of ion mobility measurements with electro-spray ionization essentially removes the limit on themass of the species that can be studied and so these
methods can be extended to even larger biologicalsystems. An e†ort is currently being made to performmobility measurements for intact viruses. The order ofmagnitude improvement in the resolution availablefrom ion mobility measurements has recently revealedmany more structural isomers for atomic clusters. Highresolution ion mobility measurements for protein ionsare currently under way. A further order of magnitudeimprovement in the resolution is desirable. However,this will be exceedingly difficult to realize simply byextending the current methods, so a new approach mustbe found if the resolution is to be signiÐcantlyimproved. Ion mobility measurements are basically aseparations technique which can be combined withother methods to provide information about the resolv-ed geometries. For example, measurements of theproperties of atomic clusters as a function of thenumber of atoms are not particularly valuable if a dis-tribution of isomers are present. Measurements need tobe performed for the individual isomers. This is alsotrue for biomolecules. The Ðrst studies along these lineshave examined the chemistry of the separated geome-tries. It would be useful to combine the ion mobilitymeasurements with a spectroscopic technique thatwould provide structural information. This is desirablebecause the structural information obtained from amobility measurement is often ambiguous. However, itis difficult to Ðnd a technique that is compatable withlow signal intensities available after separation of thedi†erent geometries. Photoelectron spectroscopy usinga magnetic bottle spectrometer has high sensitivity ande†orts are currently under way to record photoelectronspectra for the isomers resolved in the ion mobilitymeasurements.
Acknowledgements
It is a pleasure to acknowledge the invaluable contributions of ourcolleagues Konstantin Shelimov, Jim Fye, Robert Hudgins, PhilippeDugourd, Alexandre Shvartsburg, Jurgen Woenckhaus, Yi Mao, SteveValentine, Yansheng Liu, Cherokee Hoagland and Ann Countermanto the work described here. We also gratefully acknowledge theNational Science Foundation (CHE-9306900, CHE-9625199 andCHE-97XXXXX) and the Petroleum Research Fund, administered bythe American Chemical Society, for support of this work.
REFERENCES
1. B. R. Rowe, D. W. Fahey, F. C. Fehsenfeld and D. L. Albrit-ton, J.Chem.Phys. 73, 194 (1980).
2. D. F. Hagen,Anal . Chem. 51, 870 (1979).3. M. J. Cohen and F. W. Karasek, J. Chromatogr . Sci . 8, 330
(1970).4. R. H. St. Louis and H. H. Hill, Crit . Rev. Anal . Chem. 21, 321
(1990).5. H. H. Hill, W. F. Siems, R. H. St. Louis and D. G. McMinn,
Anal . Chem. 62, 1201A (1990).6. Y. H. Chen, H. H. Hill and D. P. Wittmer, Int . J . Mass
Spectrom. Ion Processes 154, 1 (1996).7. K. T. Whitby and W. E. Clark, Tellus 18, 573 (1966).8. T. M. Sanders and S. R. Forest, J . Appl . Phys. 66, 3317
(1989).9. Y. Kaneko, M. R. Megill and J. B. Hasted, J. Chem. Phys. 45,
3741 (1966).10. V. Nestler, B. Betz and P. Warneck, Ber . Bunsenges. Phys.
Chem. 81, 13 (1977).
11. R. Thomas, A. Barassin and R. R. Burke, Int . J . MassSpectrom. Ion Phys. 28, 275 (1978).
12. W. Lindinger, E. Alge, H. Stori, R. N. Varney, H. Helm, P.Holzmann and M. Pahl, Int . J . Mass Spectrom. Ion Phys. 30,251 (1979).
13. R. Johnsen, M. A. Biondi and M. J. Hatashi, J . Chem. Phys.77, 2545 (1982).
14. F. Bohringer and F. Arnold, J. Chem.Phys. 77, 5534 (1982).15. H. Bohringer and F. Arnold, Int . J . Mass Spectrom. Ion Pro-
cesses 49, 61 (1983).16. M. F. Jarrold, J. E. Bower and K. Creegan, J. Chem. Phys.
90, 3615 (1989).17. M. F. Jarrold, J. Phys.Chem. 99, 11 (1995).18. P. P. Radi, G. von Helden, M. T. Hsu, P. R. Kemper and M. T.
Bowers, Int . J . Mass Spectrom. Ion Processes 109, 49(1991).
19. P. R. Kemper and M. T. Bowers, J . Phys. Chem. 95, 5134(1991).
( 1997 by John Wiley & Sons, Ltd. JOURNAL OF MASS SPECTROMETRY, VOL. 32, 577È592 (1997)

ION MOBILITY MEASUREMENTS 591
20. G. von Helden, M.-T. Hsu, P. R. Kemper and M. T. Bowers,J . Chem.Phys. 93, 3835 (1991).
21. G. von Helden, M.-T. Hsu, N. Gotts and M. T. Bowers, J .Phys.Chem. 97, 8182 (1993).
22. K. B. Shelimov, J. M. Hunter and M. F. Jarrold, Int . J . MassSpectrom. Ion Processes 138, 17 (1994).
23. J. M. Hunter and M. F. Jarrold, J. Am. Chem. Soc. 117, 103(1995).
24. M. F. Jarrold and V. A. Constant, Phys. Rev. Lett . 67, 2994(1992).
25. J. M. Hunter, J. L. Fye, M. F. Jarrold and J. E. Bower, Phys.Rev. Lett . 73, 2063 (1994).
26. M. F. Jarrold and J. E. Bower, J. Chem. Phys. 98, 2399(1993).
27. D. E. Clemmer, J. M. Hunter, K. B. Shelimov and M. F.Jarrold,Nature (London) 372, 248 (1994).
28. K. B. Shelimov, D. E. Clemmer and M. F. Jarrold, J. Chem.Soc.,Dalton Trans 567 (1996).
29. G. von Helden, N. G. Gotts, P. Maitre and M. T. Bowers,Chem.Phys. Lett . 227, 601 (1994).
30. J. A. de Gouw, L. N. Ding, M. Krishnamurthy, H. S. Lee, E.B. Anthony, V. M. Bierbaum and S. R. Leone, J. Chem. Phys.105, 10398 (1996).
31. M. F. Jarrold and E. C. Honea, J. Am. Chem. Soc. 114, 459(1992).
32. J. M. Hunter, J. L. Fye and M. F. Jarrold, Science 260, 784(1993).
33. G. von Helden, N. G. Gotts and M. T. Bowers, Nature(London) 363, 60 (1993).
34. Ph. Dugourd, R. R. Hudgins, D. E. Clemmer and M. F.Jarrold, Rev. Sci . Instrum. in press.
35. J. J. Monagham, M. Barber, R. Bordolim, E. Sedgewick andA. Taylor,Org.Mass Spectrom. 17, 596 (1982).
36. C. M. Whitehouse, R. N. Dreyer, M. Yamashuta and J. B.Fenn,Anal . Chem. 57, 675 (1985).
37. M. Karas and F. Hillenkamp, Anal . Chem. 60, 2299 (1988).38. B. E. Winger, K. J. Light-Wahl, A. L. Rockwood and R. D.
Smith, J. Am.Chem.Soc. 114, 5897 (1992).39. D. Suckau, Y. Shi, S. C. Beu, M. W. Senko, J. P. Quinn, F.
M. Wampler and F. W. McLafferty, Proc. Natl . Acad. Sci .USA 90, 790 (1993).
40. T. D. Wood, R. A. Chorush, F. M. Wampler, D. P. Little, P. B.O’Connor and F. W. McLafferty, Proc. Natl . Acad. Sci . USA92, 2451 (1995).
41. D. S. Gross, P. D. Schnier, S. E. Rodriguez-Cruz, C. K. Fager-quist and E. R. Williams, Proc. Natl . Acad. Sci . USA 93,3143 (1996).
42. P. D. Schnier, D. S. Gross and E. R. Williams, J. Am. Chem.Soc. 117, 6747 (1995).
43. T. R. Covey and D. J. Douglas, J . Am. Soc. Mass Spectrom.4, 616 (1993).
44. T. R. Covey and D. J. Douglas, J . Am. Soc. Mass Spectrom.4, 616 (1993).
45. B. A. Collings and D. J. Douglas, J . Am. Chem. Soc. 118,4488 (1996).
46. K. A. Cox, R. K. Julian, R. G. Cooks and R. E. Kaiser, J . Am.Soc.Mass Spectrom. 5, 127 (1994).
47. A. P. Quist, J. Ahlbom, C. T. Reimann and B. U. R. Sun-dquist, Nucl . Instrum. Methods Phys. Res. B 88, 164(1994); P. A. Sullivan, J. Axelsson, S. Altmann, A. P. Quist,B. U. R. Sundquist and C. T. Reinmann, J. Am. Soc. MassSpectrom. 7, 329 (1996).
48. R. D. Smith, J. A. Loo, R. R. Loo, M. Busman and H. R.Udseth,Mass Spectrom.Rev. 10, 359 (1991).
49. R. D. Smith, J. A. Loo, R. R. Loo and H. R. Udseth, MassSpectrom.Rev. 11, 434 (1992).
50. D. Wittmer, Y. H. Chen, B. K. Luckenbill and H. H. Hill, Anal .Chem. 66, 2348 (1994).
51. G. von Helden, T. Wyttenbach and M. T. Bowers, Science267, 1483 (1995).
52. G. von Helden, T. Wyttenbach and M. T. Bowers, Int . J .Mass. Spectrom. Ion Processes 146/147, 349 (1995).
53. T. Wyttenbach, G. von Helden and M. T. Bowers, J . Am.Chem.Soc. 118, 8355 (1996).
54. J. B. Fenn, M. Mann, C. K. Meng, S. F. Wong and C. M.Whitehouse, Science 246, 64 (1989).
55. D. E. Clemmer, R. R. Hudgins and M. F. Jarrold, J. Am.Chem.Soc. 117, 10141 (1995).
56. K. B. Shelimov and M. F. Jarrold, J. Am. Chem. Soc. 118,10313 (1996).
57. K. B. Shelimov, D. E. Clemmer, R. R. Hudgins and M. F.Jarrold, J. Am.Chem.Soc. in press.
58. S. J. Valentine and D. E. Clemmer, J. Am. Chem Soc. sub-mitted for publication.
59. K. B. Shelimov and M. F. Jarrold, J. Am. Chem. Soc. inpress.
60. S. J. Valentine, J. G. Anderson, A. D. Ellington and D. E.Clemmer, J. Am.Chem.Soc. submitted for publication.
61. S. J. Valentine and D. E. Clemmer, J. Am. Soc. MassSpectrom. submitted for publication.
62. R. A. Dressler, H. Meyer and S. R. Leone, J. Chem. Phys. 87,6029 (1987); S. R. Leone, in Bimolecular Collisions , editedby M. N. R. Ashford and J. E. Baggott. Royal Society ofChemistry, London, 1989.
63. E. A. Mason and E. W. McDaniel, Transport Properties ofIons in Gases . Wiley, New York (1988).
64. H. W. Revercomb and E. A. Mason, Anal . Chem. 47, 970(1975).
65. F. Howorka, F. C. Fehsenfeld and D. L. Albritton, J. Phys. B12, 4189 (1979).
66. J. M. Hunter, J. L. Fye, N. M. Boivin and M. F. Jarrold, J.Phys.Chem. 98, 7440 (1994).
67. M. F. Jarrold and J. A. Bower, J. Chem. Phys. 96, 9180(1992).
68. M. F. Jarrold and E. C. Honea, J. Phys. Chem. 95, 9181(1991).
69. J. Woenckhaus, Y. Mao and M. F. Jarrold, to be published.70. S. D. Huang, L. Kolaitis and D. M. Lubman, Appl . Spectrosc.
41, 1371 (1987).71. Y. H. Chen, H. H. Hill and D. P. Wittmer, Int . J . Mass
Spectrom. Ion Processes 154, 1 (1996).72. N. E. Bradbury and R. A. Nielsen, Phys.Rev. 49, 388 (1936).73. F. J. Knorr, R. L. Eatherton, W. F. Siems and H. H. Hill, Anal .
Chem. 57, 402 (1985).74. A. A. Shvartsburg, J. Woenckhaus and M. F. Jarrold, to be
published.75. J. O. Hirschfelder, C. F. Curtiss and R. B. Bird, Molecular
Theory of Gases and Liquids . Wiley, New York (1954).76. E. Mack, J.Am.Chem.Soc. 47, 2468 (1925).77. S. Lee, T. Wyttenbach and M. T. Bowers, J . Am. Chem. Soc.
117, 10159 (1995).78. L. D. Book, C. Xu and G. E. Scuseria, Chem. Phys. Lett . 222,
281 (1994).79. G. von Helden, M. T. Hsu, N. G. Gotts, P. R. Kemper and M.
T. Bowers, Chem.Phys. Lett . 204, 15 (1993).80. A. A. Shvartsburg and M. F. Jarrold, Chem. Phys. Lett . in
press.81. M. F. Mesleh, J. M. Hunter, A. A. Shvatsburg, G. C. Schatz
and M. F. Jarrold, J. Phys.Chem. in press.82. S. N. Lin, G. W. Griffin, E. C. Horning and W. E. Wentworth,
J. Chem.Phys. 60, 4994 (1974).83. M. F. Mesleh, G. C. Schatz and M. F. Jarrold, unpublished
work.84. M. D. Wessel and P. C. Jurs, Anal . Chem. 66, 2480 (1994).85. K. S. Pitzer and E. Clementi, J . Am. Chem. Soc. 81, 4477
(1959); R. Hoffmann, Tetrahedron 22, 521 (1966); S. W.McElvaney, B. I. Dunlap and A. O’Keefe, J. Chem. Phys. 86,715 (1987).
86. J. M. Hunter, J. L. Fye, E. J. Roskamp and M. F. Jarrold, J.Phys.Chem. 98, 1810 (1994).
87. N. S. Goroff, Acc.Chem.Res. 29, 77 (1996).88. For a recent review, see D. S. Bethune, R. D. Johnson, J. R.
Salem, M. S. de Vries and C. S. Yannoni, Nature (London)366, 123 (1993).
89. N. G. Gotts, G. von Helden and M. T. Bowers, Int . J . MassSpectrom. Ion Processes 149/150, 217 (1995).
90. Ph. Dugourd, R. R. Hudgins and M. F. Jarrold, to bepublished.
91. J. E. Campana, T. M. Barlak, R. J. Colton, J. J. Decorpo, J.R. Wyatt and B. I. Dunlap, Phys.Rev. Lett . 47, 1046 (1981).
92. R. Pflaum, P. Pfau, K. Sattler and E. Recknagel, Surf . Sci .156, 165 (1985).
93. E. C. Honea, M. L. Homer, P. Labastie and R. L. Whetten,Phys.Rev. Lett . 63, 394 (1989).
94. Y. J. Twu, C. W. S. Conover, Y. A. Yang and L. A. Bloom-field, Phys.Rev.B 42, 5306 (1990).
( 1997 by John Wiley & Sons, Ltd. JOURNAL OF MASS SPECTROMETRY VOL. 32, 577È592 (1997)

592 D. E. CLEMMER AND M. F. JARROLD
95. P. Dugourd, R. R. Hudgins and M. F. Jarrold, Chem. Phys.Lett . in press.
96. P. Dougourd, R. R. Hudgins, J. Tenenbaum and M. F.Jarrold, Phys.Rev. Lett . submitted for publication.
97. G. W. Bushnell, G. V. Louie and G. D. Brayer, J . Mol . Biol .214, 585 (1990).
98. P. X. Qi, D. L. DiStefano and A. J. Wand, Biochemistry 33,6408 (1994).
99. W. F. van Gunsteren and M. Karplus, Biochemistry 21, 2259(1982).
100. M. Levitt and R. Sharon, Proc. Natl . Acad. Sci . USA 85,7557 (1988).
101. J. Woenckhaus and M. F. Jarrold, to be published.102. T. Y. Tsong,Biochemistry 12, 2209 (1973).103. J. A. Loo, C. G. Edmonds, H. R. Udseth and R. D. Smith,
Anal . Chem. 62, 693 (1990).104. Y. Liu, S. J. Valentine and D. E. Clemmer, to be published.105. For recent discussions of the stabilities of proteins as a func-
tion of pH, see A.-S. Yang and B. Honig, J. Mol . Biol . 231,
459 (1993); D. Stigter, D. O. V. Alonso and K. A. Dill, Proc.Natl . Acad. Sci . USA 88, 4176 (1991), and references citedtherein.
106. For a more complete discussion, see S. Campbell, M. T.Rodgers, E. M. Marzluff and J. L. Beauchamp, J. Am. Chem.Soc. 117, 12840 (1995); and references cited therein.
107. B. E. Winger, K. J. Light-Wahl, A. L. Rockwood and R. D.Smith, J. Am.Chem.Soc. 114, 5897 (1992).
108. C. J. Cassady and S. R. Carr, J . Mass Spectrom. 31, 247(1996).
109. J. S. Klassen, A. T. Blades and P. Kebarle, J . Phys. Chem. 99,15509 (1995).
110. Y. K. Lau, S. Ikuta and P. Kebarle, J . Am. Chem. Soc. 104,1462 (1986).
111. C. S. Hoaglund, Y. Liu, A. E. Ellington, M. Pagel and D. E.Clemmer, submitted for publication.
( 1997 by John Wiley & Sons, Ltd. JOURNAL OF MASS SPECTROMETRY, VOL. 32, 577È592 (1997)