is 548-1 (1964): methods of sampling and test for oils and
TRANSCRIPT

Disclosure to Promote the Right To Information
Whereas the Parliament of India has set out to provide a practical regime of right to information for citizens to secure access to information under the control of public authorities, in order to promote transparency and accountability in the working of every public authority, and whereas the attached publication of the Bureau of Indian Standards is of particular interest to the public, particularly disadvantaged communities and those engaged in the pursuit of education and knowledge, the attached public safety standard is made available to promote the timely dissemination of this information in an accurate manner to the public.
इंटरनेट मानक
“!ान $ एक न' भारत का +नम-ण”Satyanarayan Gangaram Pitroda
“Invent a New India Using Knowledge”
“प0रा1 को छोड न' 5 तरफ”Jawaharlal Nehru
“Step Out From the Old to the New”
“जान1 का अ+धकार, जी1 का अ+धकार”Mazdoor Kisan Shakti Sangathan
“The Right to Information, The Right to Live”
“!ान एक ऐसा खजाना > जो कभी च0राया नहB जा सकता है”Bhartṛhari—Nītiśatakam
“Knowledge is such a treasure which cannot be stolen”
“Invent a New India Using Knowledge”
है”ह”ह
IS 548-1 (1964): Methods of sampling and test for oils andfats, Part I: Methods of sampling, physical and chemicaltests [FAD 13: Oils and Oilseeds]



IS : 548 (Part 1) -1964
Indian StandardMETHODS OF SAMPLING AND TEST
FOR OILS AND FATS
PART 1 SAMPLING, PHYSICAL AND CHEMICAL TESTS
(Revised)Fourteenth Reprint JULY 2007
(Incorporating AmendmentNos. 1 to 4 and Including AmendmentNo.5)
UDC 665.3 : 543
© Copyright 1976
BUREAU OF INDIAN STANDARDSMANAK BHAVAN, 9 BAHADUR SHAH ZAFAR MARG
NEW DELHI 110002
Gr 11 October 1964
(Reaffirmed 20====2010

IS : 548 ( Part I ).1164
CONTENTS
PAOE
o. Foa~woRD 5
1. SCOPE 6
2. TalUllNOLOOY 7
3. SAlfPUNO 9
4. QuAUI'Y OP Ru.OI.NTI 18
5. DaftUIINATION OP MODTtJRE CONTENT 18
6. DanQONATlON 0. INSOLUBLE IIiPURmES 28'
7. I)nnIllNATIOM or ACID VALUE AND FlUtE FATlY ACIDS ••• 29
P. DaftlUllNATlON 0. UNIAPONmABLE MATl'ER ••• 3.
9. DaTaUIJNAnON OP Ma.TlNG POINT "
10. DaTUlllN4T10N 0. bl'llACTIVE INDEX 35
11. DBn.RIIINATION 0. SPECIPIC GRAVITY 39
12. TITU TaT 42
13. DaTZUONAnON OF CoLOuR 45
14. DaT&aMINAnON 0' IODINE VALUE (WIJI) 47
'5. DZTEIUONAnoN or SAPONlnCAnON VALUE 50
, 6. DETERMlNAnON 0" AaTYL V ALUI AND HYDROXYl.VALva 52
17. DaftllMlNAnON o. ALLYL isttnuOCYANATE
s
56

IS I Sl8 (Pa..t I )-1964
18. DETBIlMlJlATlON OF REICHEIlT-MEISSL VALUE
19. DSTEIUlINAnON OF POLENSU VALva
21. DETERMINATION or PEROXIDE VALUE
~GE
57
62
63

18 I 548 ( Part I )-196t
Indian StandardMETHODS OF SAMPLING AND TEST FOR
OILS AND FATSPART I SAMPLING, PHYSICAL AND CHEMICAL TESTS
( Revised)Oils and Fats Sectional Committee. CAFDC 5
(c.u.~ .,.,." 2)
Rt/w.SM/UtIThe Swaatik Oil Milia Ltd, Bombay
Government Tnt House, Calcutta
The Vanupati Manufacturen' Auociation oC India,Bombay
The T.tA Oil Mills Co Ltd, Bombay
Smll.v,SHI.S.SuaaAHMAHYAN
Alailtant Dircctor ( Chem ), lSIS...I R. C. Mila"
Extra AIIistanl Director ( Chcm ), 151
CA.i,...a"Da J. S. BADAMI
M"'*'JSR.' V. A. 'A.ntH ( AI",,..,, to
Dr J. S. Badami)SKa.S. K. Bosl.
SKa' V. V. DAN' ( Allmllllt )SHU C. R. OM
SHa. N. DUlltAcHAaDa B. G. GUNDZ ( Alln"al, )
n. K. C. Guun Indian Agricultural Research Institute, New Delhi511&. K. P.JAIN Directorate of SUlar " Vanupati ( Ministry or Food
&; Agriculture )SHa. F.·O. T. MENEZES ( Al""ud, )
, ... R. K. MALIK Directorate or Marketinl at Inspection (l\iiniltry orFood It Asricuhure)
StUuK. L. CHATT£RJE£ ( Alit,,,,,,)Da 8. D. NARANG Central Committee for Food Standards (Ministry of
Htalth)na M. S. PATEL Indian Central Oilsrtdl Commiucr, HyderabadSMa. H. P. R. RAJA Bombay OilseedJ and Oil, Exchanle Ltd, Bomb• .,
SHK. CHAIANDAS V. MAR1WALA
(Alt"",." )Da H. G. R. REDDY Di~ctor.teGm~ral ofTtchnicallkvelopment5H.' P. V. SHIUKANTA RAO Khadi and VilialC Industria Commission. BombaySMa. L. R. SUD "tiniltry of Defence (R at D)
SHa. A. P. (".MAIt.AVE.TV ( A/tm." )SMal A. WOOPWAaK Hipdustan Levu Ltd, Bombay
Da G. S. HATnANODI (AI'n".',)Oa S4DooPAI., Director, lSI ( &-e,/itie M",.6Ir)
Deputy Director ( Chem )
BUREAU OF INDIAN STANDARDSMANAK BHAVAN, 9 BAHADUR SJ-IAH ZAFARMARG
NEWDELHI 110002
I

IIIM8 ( Part I )-1'"
( CMIIIaMlJt-,." I )
Oils and Fats Subcommittee, CAFDC 5 : I
RI/Jr,ulllinlHiDdUitan Lever Ltd, Bombay
Indian IllItitute of Srience, BanlaloreThe Tata Oil Mills~ Ltd, Bombay
Indian Soap and Toiletries Muen' A80ciation,Calt:utta ,
Directorate or Marketing & Inspection (Miniatry ofFood & Alriculture)
S....I K. L. CHATl'DJu ( AI",,,.,, )SJnuB. S. MODI The Vanupati Manuracturen' A.uociation or India,
BombayDR H. G. R. RBODY Directorate General of Technical DevelopmentSHu L. R. SUD MiDiatryoC Defence ( R It D)
S.... A. P. CHAuAvuTY ( AI"",.,,)
SHU R. K. MAUlt
e-r..rDa G. 8. HAnIANGDI
11..-,IDI V. U. MAaaALLI (AI""'" to
Dr G. S. HattiaDpU)Da K. T. ACIIAYA Rqional Research Laboratory. Hyderabad
Da S. RAGHAv&MDAa 'R.Ao ( AlImuJII)SHu GoItULCHAND J. AOAllWAL Bombay Oilseed. and Oil. Exchange Ltd. Bombay
SHaI CJwwmAi V. MAJUWALA( AI"".)
DaH.R.CAIIASal N. DarKACIIAIl
Da B. G. OUId). (AlInuII)SIIU S. C. GROU
Hinduatan Lever Ltd, Bombay
The Tala Oil oLdilb Co Ltd, Bombay
In personal capacity ( Dt/NI"m",' ofC,.",.1Il lim..loU. Ultiwrsil.1 ofBttmbll.1, BomIJtI,1 )
Directorate of Marketing &. Inlpection (Miniltryof Food at Agriculture)
8DJ X. L. CHATrUJI.& ( AllmJlJII ).... I. D. SINOR Ministryor Defence- ( R" D)
.... R.. P. VUIIA( .4"",.",. )Da A. R. SUIQ)AJlA&AJAM Central Committee Cor Food Standards (Miniltry c6
Health)
Panel for Studying Indian Standards on Oils and Fats, CAFDC 5: 1: 1
c...rDa s. M. PATaL
MtmMrJSDI N. DIIIXACliAil
Da B. G. GUND& ( AI,.",.16)DaJ.G.KANa
.... 1l.K.M.wx
2

(Page 10. clause 11.0) - Add the following new matter at the endof the clause:
'Method A shall be used as a referee method whereas Method B may beused for routine analysis.'
( Page 24, clause ]8.4 ) - Add the following new clauses after 18.4:
119. TEST FOR THE PRESENCE OF OIL SOLUBLE COLOURS INOILS AND FATS
]9.0 Geaeral - Oil soluble colours are colours (both natural includingnature identical and synthetic) soluble in oils and fats.
19.1 OutliDe of the Method - Oils, fats and other interfering substancesare removed from the colours by solvent partition technique using dimethylforrnamide and hexane (3: 1 ), followed by alumina adsorption. Thecolours are detected by reversed phase paper chromatography.
19.2 Apparatu8
19.2.1 For Extraction - a mechanical shaker.
19.2.2 For Column Chromatography - a chromatographic tube made ofglass (2·5 em in diameter and 30 cm in length ).
19.2.3 Separating Funnel - 250 ml capacity.
19.3 Reagents
19.3.1 Hexane
19.3.2 Dimethyl Formamide
19.3.3 Sodium Chloride Solution - saturated.
19.3.4 Alumina Neutral, Brockmann, Activity1,for Example, Made byNationalChemical Laboratory, Punt or of Equioalm: Quality - activated at 100°C for4 hours.
19.3.5 Diethyl Ether
19.3.6 Ethyl Alcohol
19.3.7 Ammonium Hydroxide Solution
19.3.8 Methanol
19.3.9 Petroleum Ether - B. P. 40 to 60°C.
19.3.10 Liquid Paraffin19.3.11 Whatman No. 1 Filter Paper (Rel1ersed Phase) - Prepared by
soaking the paper in 5 percent liquid paraffin in petroleum ether and subsequent drying in air.
19.3.12 Oil Soluble Colours - Butter yellow, Cres orange GN, Sudan IV,Gress Yellow GRN type, etc.
2

19.3.13 Sodium Hydroxide Solution - 5 percent ( ml» ).
19.3.14 Sulphuric Acid Solution - 13 N.
19.3.15 Stannous Chloride Solution in Hydrochloric Acid - Dissolve 112·8 gof stannous chloride in 1iO ml of concentrated hydrochloric acid using heatif necessary. Dilute with water to 1 litre and add a few pieces of tinmetal.
19.3.16 Boric Acid Solution - Dissolve 4 g of boric acid in 100 ml ofconcentrated hydrochloric acid (relative density 1-19).
19.3.17 Anhydrous Sodium Sulphat«
19.3.18 Saturated Solution of Antimony Trichloride in Chloroform
19.3.19 Solvent Mixture - DiethyJ Ether: Alcohol: water: : 5 : 3 : 2.
19.3.20 Soluent Mixture - 80 percent ethyl alcohol in water.
19.4 Procedure
19.4.1 Extraction - Take about 2 to 3 g of the sample in a conical flask,add approximately 50 ml of hexane and then shake for about 10 minutesin a mechanical shaker. Filter it and concentrate the filtrate to about10 mI.
19.4.1.1 Qualitative test fOT presence oj synthetic colours - Divide aportion of the hexane layer into 4 parts and treat with either 13 N sulphuric acid or concentrated hydrochloric acid and water mixtures 4 : 1,2 : 1 or 1 : 1 respectively. If the acid layer or the whole extract changesshade or more particularly develops pink to reddish violet colour, thesynthetic oil soluble colour may be suspected.
19.4.2 Separation of Colours from Interfering Materials - Transfer theconcentrated extract to a separating funnel and add to it dimethylformamide (DMF) about three times the volume of the concentratedextract and shake vigorously. Keep it for a while for separation of thelayers. The DMF layer contains the natural colour as well as the syntheticcolours (if present), leaving oils and fats in the hexane layer. Now takethe DMF layer and again shake it with hexane after addition of about20 ml of water. Retransfer colours ( both natural and synthetic) this timeto the hexane layer. If emulsion is formed add saturated sodium chloridesolution. Concentrate the hexane layer and preferably repeat the abovementioned procedure. If oils and fats still linger in the sample, these willfloat above the coloured extract when the hexane layer is concentrated to0-5 ml. If oil is present repeat the above procedure till free from oil.Make the hexane solution to 10 ml, If curcumin or turmeric is suspected,shake the residual DMF layer after extraction with and separation ofhexane with diethyl ether and an extra amount of water, if necessary andcollect the ether layer. Add the ether layer to the previous hexane. By
3

this treatment any minute curcumin and turmeric will come to the etherlayer.
19.4.3 Separation of Natural Colours from Synthetic Oil Soluble Colours Clamp the chromatographic tube and pour 40 ml of hexane with the nongreasy stopcock in closed position. Place a cotton plug ( if no fritted glassis fixed in the tube) firmly at the bottom of the tube. Pour 40 ml ofhexane followed by anhydrous sodium sulphate to form a 2 to 3 ern layer.Now open the stopcock to give a trickle of solvent and tap the tube. Pouralumina sufficient to give a column of 10 em length into the solvent withconstant tapping. This will give a channel-free column. It is importantthat the top of the alumina is always under the solvent. When the solventlayer reaches about 1 em above the alumina, pour the hexane extract ofcolours-carefully into the tube. Collect the eluent in a beaker. Continuethe elution with four batches of 10 ml hexane. Concentrate the eluatecontaining oil soluble synthetic colours. After elution with hexane pass40 ml of another eluting solvent, namely, petroleum ether: acetone ( 1 : 1 )for complete elution.
19.4.4 Detection of Synth'ti~ Oil Soluble Colours - Cut a reversed phasechromatography paper to a size of 20 X 20 em, Then draw a straight lineby a lead pencil on the paper leaving away 3 ern from the bottom. Spotthe concentrated extract (say 10 ""lor more according to the colourpresent on the solution) and also some known oil soluble colours on theline drawn on the reversed phase chromatography paper leaving 1 emdistance from each other. Then make the paper to a cylindrical shape,staple it and dip in anyone of the solvents ( see 19.3.19 and 19.3.20 ) forascending chromatography. Run for 10 em, The development in the solventgiven under 19.3.19 takes 45 minutes and that in solvent prescribedunder 19.3.20 takes one hour. Take out the paper, dry in air and detectthe colour by comparing with the known colour in respect of shade andRfvalues.
19.4.5 Detection ofNatural Colou,s - Elute the natural colours absorbed onthe column by the solvent methanol : ammonium hydroxide (9: 1 ) andthen examine for natural colours. If colour of the column changes to red.violet after passing the above mentioned solvent, it confirms the presenceof curcumin or turmeric.
19.4.5.1 Detection of annatto, turm,ric and curcumin - Evaporate theextracted layer (see 19.4.5) to dryness, add 10 ml petroleum ether andshake a 17 ml portion of the solution with sodium hydroxide solution.Take out the alkali layer and divide it into two portions to check for thepresence of annatto, turmeric and curcumin. Dilute the first portion withequal volume of water, add a piece of clean white adsorbent cotton toit and keep it overnight. If after washing gently in water the cotton piececontinues to retain straw colour which turns pink by a drop of stannouschloride solution, it confirms the presence of annatto. To the second
4

portion add boric acid solution to make the solution acidic. If red colourappears, turmeric is present and if pink colour appears it indicates thepresence of curcumin, Sometimes turmeric and curcumin are not elutedif present in small amount. Notice the change of colour of the columnfor their detection.
19.4.5.2 Delee/ion ofcarotene - To the non-alkali treated portion addsaturated solution of antimony trichloride in chloroform. If blue colourappears, carotene is present.'
(CAFDC 5)
Printed at : Prabhat OffsetPress. NewDelhi·2

IS I 548 ( Part I )-1_
Indian StandardMETHODS OF SAMPLING AND TEST FOR
OILS AND FATSPART I SAMPLING, PHYSICAL AND CHEMICAL TESTS
( Revised)o. FORE WORD
0.1 This revisedIndian Standard was adopted by the Indian StandardsInstitution on 22 April 1964, after the draft finalized by the Oils and FatsSectional Committee had been approved by the Chemical Division Counciland the Agricultural and FQOd Products Division Council.
0.2 This standard was first published in 1954. With the rapid progress inthe industry of fatty oils in this country and with the new analyticaltechniques devised to control the quality of the oils and check the trendstowards adulteration, the Sectional Committee responsible for drafting thisstandard decided to iSSUfJ a revised standard in order to keep abreast of thelatest developments in the field.
O~2.1 In view of the periodical review and revision of methods for 'detection of adulteration in vegetable oils and fats, the concerned Committee decided to issue this standard in three parts, namely, Part I Methodsof sampling, physical and chemical tests; Part II Methods for purity tests;and Part III Methods of analysis of vegetable oils and fats by gas liquidchromatography (GLe) technique. This change has been effectedthrough Amendment No.3 to this standard.
03 Taking into consideration the views of producers and consumers of oilsand fats .as also the v~ews of the t~ting authorities and of the technologists,the Sectional Committee responsible for the preparation of this standardfelt that th~ methods of t~t should be re~ated .to the current practices andthe need to Improve them In the country In this field. Due considerationhas also been given to the need for alignment with the methods of testprescribed by the. Central Committee for Food Standards, Ministry ofHealth, ~nd the Directorate of Marke.ting and Inspection, Ministry of Foodand Agriculture, Government of India, as also for international co-ordination among standards prevailing in different countries of the worldThese considerations led the Sectional Committee to draw freely from th~following standards and publications:
B.S. 627: 1953 SAMPUNO or FATI AND FATTY Ons, BritishStandards Institution.
5

IS I Sf. ( Part 1)-1964
B.S. 684: 1958 M.TRODI O~ ANALYm o. OIU AND FAn. ilritishStandards Institution,
B.S. 756: 1952 DBAN AND STARK. ApPAaATUI. British StandardsInstitution.
INDIA. MDQlTRY OF FOOD AND AORICULTURE. Methods of S&lY\flingand Testing Vegetable Oils and Fats under Agmark ( MatketingSeries No. 78). New Delhi. Manager of Publications, 1952.
INDIA. MINISTRY OP FOOD AND AORICULTURB. Methods of Samplingand Testing Butter-Fat (Ghee) and Butter under Agmark( Marketing Seria No. 81). New Delhi. Manager or Publications, 1953.
MITItA, S. N. AND So GUPTA" P. N. Detection of Hydrocyanic Acidin Edible Oils ( Mustard Oil). Science and Culture. Vol. 28,No.5, P. 241 (1962).
MaHLBNBAcR.", V. C. AND HOPPER, T. H. Official and TentativeMethods of the American Oil Chemists' Society. Second Editionincluding Additions and Revisiona- 1947 to 1963 inclusive.'Chicago. American Oil Chemists' Society.
O.t This standard is intended for introduction in India ofunifonn methodsof sampling and test for oils and fats. It does not deal with the I~cifications of the materials, but prescribes only the methods of determiningwhether the material conforms to the requirements 9£the individual standard and thus forms a necessary adjunct to the series of Indian Standardspecifications for individual oils and fats.
0.5 Wherever a reference to any Indian Standard appears in this standard,it shall be taken as a reference to the latest version of the standard.
G.I In reportin~ the result of a test or analysis made in accordance withthis standard, if the final value, observed or calculated, is to be roundedoft it shall be done in accordance with IS: 2-1960 Rules for Roundina OftNumerical Values ( Rlvis,d).
1. SCOPE
1.1 This standard lays down the methods of sampling and test for individual oils) rats and fatty materials. It contains definitions of terms \liedin trade arid industry and prescribes the methods for determining moisture,.insoluble impurities, acid value and free fatty acids, unsaponifiable matter,melting point, refractive index.sspecific ~avity, titre of mixed fatty acids,colour, iodine value (Wijs), saponification value, acetyl value andhydroxyl value, allyl isothiocyanate content, Reichert-Meissl value and
6

IS : 5f8 ( Part I )-1114
Pelenske value
1.2 Test methods, neither prescribed in this standard nor specified instandards for individual materials, shall be subject to agreement betweenthe purchaser and the supplier.
1~:J Should any inconsistency be found to exist between the methodsprescribed in this standard and those prescribed in the standard for anindividual material» the latter shall prevail.
2.TEIIMlNOLOGY2.0 Fer the purpose uf this standard, the following definitions shan apply.
2.t Acetyl Val•• - The number of milligrams of potassium hydroxiderequired to neutralize the acetic acid liberated by the hydrolysis of onegram of the acetylated oil or Cat (s" 2.8 ).
2.1.1 Acetyl value of an oil or Cat is a measure of the hydroxyl contentof the material.
2.2 Add V.I.e ••d Free Fatty Add. - The number of milli~.of
potassium hydroxide required to neutralize the free acid present in ODepvn of the oil or fat under the prescribed conditions.
2.2.1 The acidity of the oil or fat indicated by its acid value is frequentlyexpressed as free fatty acids present in the sample.
2.3 Hydrosyl Val.e - The number of milligrams ofJX?tassium hydroxiderequired to neutralize the acetic acid capable of combining by acetyl.tiODwith one gram of the oil or fat ( s" also 201 ).
2.3.1 Hydroxyl value is equivalent to the hydroxyl content of thematerial based on the weight of the unacetylated fat.
2.~ "_able Impuritiee - Dirt, meal and other foreign substancawhich are insoluble in kerosine and petroleum ether under the conditionsof the prescribed test.
2.5 ICMIIae V.I•• (Wij.) - The number of grams of iodine, absorbedper 100 grams of the oil or fat, when determined by using Wijs solution.
2.5.1 The iodine value of the oil or rat gives an indication of the dqreeof unsaturation of the constituent fatty ,iycerides. It is customary to pvethe method employed for its determination, Wijs method is apphcable toall normal oils and fats not containing conjugated systems.
2.6 Meldal Polat - The temperature at which the oil or rat s~ftena orbecomes suffici~nt1y fluid to slip or run u determined by the open..tubecapillary-slip method. In the case or the closed-tube complete-fusion
7

II: 5_ (Part I )-196t
method, it is the temperature at which the oil or fat becomes perfectlyclear and liquid.
2.7 MoI.tare Coateat - The moisture and any other material containedin the oil or rat which is volatile under the conditions of the prescribedtest. .
2.8 Pole•••• V.lae - The number of millilitres of 0·1 N aqueoussodium hydroxide solution required to neutralize the steam volatile,water insoluble fatty acids distilled from 5 g of an oil or fat under theprecise conditions specified in the method.
2.8.1 The Polenske value is the measure of the steam volatile and waterinsoluble fatty acids, chiefty caprylic, capric and lauric acids, present in theoil or fat.
2.9 Refractive lades - The ratio of the velocity of light in vacuum tothe velocity of light in the oil or fat; more generally, it expressed the ratiobetween the sine of the angle of incidence to the sine of the angle of refraction when a ray of light of a known wave-length ( usually 589·3 ml!, themean of the D lines of sodium) passel from air into the oil or fat.
2.18 Relc:hert-Melll.l Valae - The number of millilitres of 0·1 N sodiumhydroxide solution required to neutralize the steam volatile, water solublefatty acids distilled from 5 g of an oil or ral under the precise conditionslpecified in the method.
2.10.1 Reichert-Meissl value is a measure of water 'soluble steamvolatile fatty acids, chiefly butyric and caproic acids, present in oil or fat.
2.11 SapoaJ&cadoa V.I.e - The number of milligrams of ~taaiumhydroxide required to saponify completely one gram of the oil or rat.
2.12 SpecUlc Gravity
2.12.1 specific Gravity ofan Oil- The ratio of the weight in air of agiven volume of the oil at 30°C to the weight in air of an equal volume ofwater at 30°0.
2.12.2 Specific Gravity of a Fat - The ratio of the weight in air of~ givenvolume of the fat at 95°C to the weight in air of an equal volume ofwater at 30°C.
2.13 Titre - The hi,hest temperature attained under standard conditions,during the solidification of the mixed fatty acids obtained from the oil orfat. .
2.1f UuaroDl8able Matter - The fraction' of substances in oils andCau which IS not saponified by caustic alkali, but is soluble in ordinary fatIOlvents.
8

IS I 5i8 ( Part I )-196t
2.14.1 It includes the higher aliphatic alcohols, sterols, pigmentl,hydrocarbons and resinous matter.
s. SAMPLING
3.0 Geaem- It is very difficult to lay down detailed directions forsampling oils and rats that will encompass all conditions and circumstanceswhich may confront the individual charged with the responsibility oftaking the sample. There are many instances in which the experience andjudgement of the individual should prevail. There are, however, certaingeneral rules relating to the drawing. preparation, storage and handling ofsamples which should always govern if the sample is to be representative.These are described under 3.1 to 3.8.
3.1 Ge.era1 Precaatlo.8 la SampUaC
3.1.1 All sampling instruments should preferably be made of stainlesssteel; if made of copper, brass or bronze, these should be nickel-plated, I
3.1.2 All sampling apparatus shall be clean and dry when 'used. Sampling instruments may be cleaned with hot soapy water or other detergent,care being taken to wash away the last traces with scalding hot water. Ifa source of steam is available, the instruments may receive a final cleansingin a jet of steam and it is recommended that this procedure be carried outwhen edible" oils are sampled and where the taste is likely to be of primaryimportance.
3.13 Samples shall not be taken in an exposed place. The samples, thematerial being sampled, the sampling instruments and containers forsamples shall be protected from adventitious contamination. The tcstsamples shall be placed in suitable clean and dry containers.
3.1.4 Samples shall be stored in such a manner that they are protectedfrom light, temperature fluctuations and other abnormal conditions.
3.1.5 Sample containers shall be so filled that the air space above theliquid level shall be 5, to 10 percent of the capacJty of the samplecontainers.
3.2 SampU•• 1a8trameat.
3.2.1 Sampling B,ltu Dr CDII - This instrument is suitable for samplinglarge vessels arid tanks of liquid oil. It consists of a weighted bottleor metal container with removable stopper or cork, to which is attacheda suitable chain, pole or cord. This device is lowered to the variousdesired depths at which the stopper is removed and the container isallowed to fi11 <SI' Fig. I ).
3.2.2 Sdlft/lli", rub,s - The recommended forms of sampling cubaare <a> closed-type sampling tubes, undivided or divided. tor samp1ina
9

IS I 548 ( Part I )-1964
liquid! and semi-liquids that may not be homogeneous; and (b) open typesampling tube for sampling homogeneous liquids.
Fro. 1 SAMPLING BOTTLE OR CAN
3.2.2.1 CIDsld-type stJmpling tub" undividtd - It consists of two concentric metallic tubes closely fitted into each other throughout theirentire length, so that one tube may be rotated within the other. Longitudinal openings of about one-third the circumference are cut in bothtubes. In one position the openings in the two tubes coincide; the sampling tube is open when in this position and adJ!lits the material. Byturning the inner tube through an angle of 180°, it becomes a sealed container. The inner tube may have a diameter of 2Q to 40 mm and isundivided along its length to serve as a single container ( s" Fig. 2).
The two concentric tubes are provided with ports at their bottomends, 10 placed that it becomes possible to drain the material contained inthe instrument through them, when the longitudinal openings coincide.
The len~h of the instrument should be such as to enable it to reachthe bottom ef the container being sampled.
The instrument is inserted closed. the material is admitted by openingit, and finally, it is closed and withdrawn.
10

IS I 548 ( Part I ~196f
CLOSED
•OPEN
SE~X:*'
x
PORTS OPEN
~
x
CLOSEDt:J::.\o
10TTOM VIE WS
Flo. 2 CLOIED-TyPJ: S.uaPUNO TUBa - UNDIVIDED
3.2.2.2 CloSttJ-1yfH I ••pli", tub" dilliMd- It is also of metal andhas n-shaped croq-section. It is provided with compartments alongita length and is opened and closed by means of a closely-fittingshutter which moves up and down throughout the entire length. It maybe from 25 to 50 mm wide (/U Fi,. 3 ).
II

IS. 5f8 ( Part I )-lfHK
:0ENLARGED
SECTION XXENLARGED
SECTION VY
x~
,,~
I II I1 II I
'4I III I
II'I II I
•II"..II...I IU\
y y
1 1
TUBE St-IJTTER

IS. 5. (Part • )-1'"
The instrument is inserted closed. the shutter is pulled out to admitthe material. and the tube h then closed and withdrawn.
3.2.2.3 Opm-Iype sampling I t"b, for Aomogeneoru lifJUitls - I t is madeof metal or thick glass. and may I be of 20 to 40 mm diameter and400 to 750 mm length. The upper and lower ends are conical andnarrow down to 6 to 12 mm diameter. Handling is facilitated by tworings at the upper end. For taking a sample, the instrument is first closedat the top with the thumb or a stopper and lowered until the desired depthis reached. It is then opened for a short time to admit the material andainally closed and withdrawn (III Fig. 4 ).
3.2.3 Sampling SeoO/Js - These instruments are of the open type andare suitable for taking samples of naturally occurring vegetable fats or
6TO 12mmDIA
EE
~o...oo..,
20 TO 40 mm OIA
, TO 40mm t'lt'
FlO. of OPEN-Tvp.. SAMPLING TuBlt FOR HOllooltNltoUi LI~tJlDI

IS I M8 (Part I )-196t
hydrogenated vegetable oils. They are of metal, of leItli-circular 01'C-shaped cross-section, and will bore out a core of the sample throuahthe material being sampled (s•• Fig. 5A and 5B).
o oI'----I'"l---'I
FlO. 5A SAMPLINO ScooP FlO. 5B SAMPLINO ScooP
S.3 ....pIe Coaal.....
S.3.1 The IUIlpIea aha11 be packed in clean dry containen, preferably of8- or tiD-plate, the aamplecontainersahould be almoIt but not completely
14

More than 100
IS : 5t8 ( Part I )-1_
filled. Glass bottles of 500 ml capacity arc recommended for liquid oilsand glass jars for solid vegetable oils or fats.
3.3.2 All sample containers shan be fitted with suitable tight stoppers.Rubber stoppers should not be used to close the containers. In the case of
. glass containers, glass stoppers or new good quality velvet corks should beused and, in the case of tin containers. tin caps should be suitably solderedon the top, avoiding contamination of fat with acid flux. Use of resin asa flux is recommended. Tinfoil or grease-proof paper may be wrappedround the corks to prevent contact of the sample with them, and it isrecommended that this be done in the case of refined or deodorized edibleoils. In the case of oils with high acid value, neither metal containersnor tinfoil is recommended.
3.3.3 The sample containers shan be sealed with sealing wax in such amanner that it is not possible to remove the contents and the label withoutbreaking the imprint of the seal.
3.4 Scale or SampHD,
'.4.1 LDt - All the rontainers in a single consignment of one type andgrade of material drawn from a single batch ofmanufacture shall constitutethe lot. If a consignment is declared to consist of different batches ofmanufacture, the batches shall be marked separately and the groups ofcontainen in each batch shall constitute separate lots. Should the CDnsipment not be uniform in quality, the parts of the consignment whichappear to be similar may be collected together and each quality treatedas a separate lot.
3.4.2 Gross Sample - The general procedure for taking a gross sample isto draw a number of portions from the bulk quantity (see 3.:1.2.1 ) or anumber of portions form all or serveral packages (see 3.4.2.2 ), and mixthem thoroughly. Representative portions of the gross sample shall betransferred to air-tight containers of suitable size for the test samples asdescribed under 3.6.
3.4.2.1 Gross smnple from bulk fuantitits - shall be drawn in quantitiesof not less than 2 kg per 2 000 kg or less.
3.4.2.2 Gross sample from smallP«/cflgtS - When sampling fro~ drums.barrels. etc, the packages from which' the samples are drawn shall beselected at random from the lot. The following schedule is recommendedfer the number of packages to be sampled:
N"",6n tJj PlIlluJgu ill tlu lA, Humbn ofPtdagts 1o be Sampled1 to 4 Each package5 to 100 At least 20 percent with a minimum
of4 packagesAt least 10 percent with a minimum
or 20 packages
15

IS I M8 ( Part I)-19M
By agreement between the purchaser and the supplier, each packagemay be sampled. A minimum of 0-1 percent of the total quantity in thelot or 2 kg, whichever is greater, shall be drawn as the gross sample.
3.5 Procedue
3.5.1 Oils in Bulk in Storage Tanks "lid Tank Wagons
a) Liquid oils
1) SIaliontlTJ - Lower the closed sampling bottle or can (s"Fig. 1) slowly to the required depth, open and fill it atthat depth. Three samples shall be obtained At levels ofone-tenth of the depth of the liquid from the top surface( top sample), one-half of the depth ( middle sample) andnine-tenths of the depth of the liquid from the top surface( lower sample). If foots or water or both are present, abottom sample shall also be taken at the lowest point of thecontainer (bottom sample). All the samples shall bemixed together in a clean dry container, and shall bereduced as described under 3.6.
2) nuring loading or unloildin,f - If the product is completelyliquid and free flowing, the pet-cock method of samplingmay be used. A bleeder line with a pet-cock ( 10 mmminimum inside diameter) is located in a vertical section ..of the pumping line through which the product is continuously flowing. Adjust the pet-cock so that a continuousstream of sample 80ws freely without dripping during theentire pumping period. Collect the sample in a cleandry container and protect it from dirt, water or othercontamination, Mix the entire sample thoroughly andreduce it as described under 3.6.
In the event of a pet-cock not being available. use a convenient container and withdraw approximately 0-5 )1 portions fromthe dis~harge end of the pipe at regular intervals as the productis entering or leaving the tanl' wagon.
b) Solidoils or fau -- It is not possible to sample solid materials intank wagons correctly. If possible, the material should be Hquefied and then sampled as described under 3.5.1(_). Whennecessary to sample solid materials, use the sampling scoop(su Fig. 5A and 58) and withdraw several portions from thetank wagon taken vertically and obliquely towards the ends ofthe wagon. The scoop should pass through the stock until ittouches the sides of the wagon so that a complete core is taken.Soften (6,,' do nol ·",.11) and mix all portions thorqhly, andreduce the sample u described under 3.6.
16

IS I 5f8 ( Part I ).1964
3.5.2 Oils ill 811m". CuU, Drums _ T"""sa) LiquUl or sImi-solid oils- Roll the container to mix the contents
and insert the sampling tube (Stl Fig. 4) slowly through thebung hole or any other convenient opening. If possible, thesample should be drawn from end to end. As soon as the tubeis fully inserted, close the upper constriction with the thumb ora stopper, withdraw the tube and transfer the sample into aclean dry container. Take several portions in this manner fromthis and other packages recommended under 3.f.2.2. Mixthoroughly and reduce the sample as described under 3.6.
b) Solid oils or fiUJ_. Remove the bung and insert the samplingscoop ( se« Fig. 5A and 58) through the opening, pu~h it throughto the opposite end or side, turn it in a complete CIrcle and withdraw with the sample. If possible, the sample should be drawnfrom end to end. Collect several such portions from this and otherpackages recommended under 3.4.2.2. Soften (but do not melt)and mix thoroughly, and finally reduce the sample as describedunder 3.6.
3.5.3 VIrY liard Fats ira Bags, BtUrtls or Casks - If the material is in theform offtakes or loose lumps or pieces, take grab samples of uniform antiproportional size from packages as directed under 3.4.2.2. If the materialIS in the form of large pieces, it should be broken up before taken grabsamples. Mix thoroughly, quarter, and finally reduce the sample asdescribed under 3.6.
3.6 Test aDd Referee Samples
3.6.1 Si(.t of Test Sampu - The minimum ~ize for roach test sample shallbe 0·5 kg.
3.6.2 PrtjJtI1dlion of Test Sampl, - Normally, all the samples drawn asdescribed under 3.5 shall be put into a clean dry receptacle, such as d
bucket or tub, and the contents of this receptacle shall be thoroughly mixedand at least four uniform samples (test sarnples ) shall be drawn therefrom. One test sample shan be sent to the purchaser and one to thesupplier.
3.6.2.1 The supplier shall have the right to be represented at thetime of sampling.
3.6.2.2 The materialleCt over after the preparation of the test samplesshall be at the disposal of the supplier.
3.&.3 Rifn" Sdm/lu - Two of the test samples bearing the seals of thepurchaser and the supplier shall constitut, the referee samples, to be usedIn cue of dispute between the purchaser and the supplier, and shall bekept at a place a,t'eed to between the purchaser and the supplier.
17

IS I 548 ( Part J )-1964
3.7 Tnt lor Acceptaace
3.7.1 &.m;utiDn tmtl Tests - The purchaser may separately examine testsamples of each of the separate qualities ( st, 3.4.2.2) for compliance withthe requirements of the individual specification, or he may prepare, forthe purpose of such examination and at any stage of the progress of theexamination, a composite sample representing the whole of the consignment, by mixing the test samples.
3.7.2 Critni01lI'"Judltmlftl - The lot shall be considered to conform tothe requirements of this standard if the test sample'satisfies all the requirements and passes all the tests. If two or more qualities are examinedfrom a consignment and if one or more of them do not comply with therequirements oftbe specification for that particular material, the purchasershall have the" right to accept only that portion which complies with therequirements, or accept or reject the whole of the consignment.
3.8 Marlda. ofSample eo.talaen
3.8.1 Each sample container after filling shall be sealed and markedwith full details of sampling, the number of packages sampled, the date ofsampling, and other particulars of the consignment.
3.8.2 A label bearing the particulars given under 3.8.1 shall be attached and sealed to every sample container. A recommended Conn of labelis g'ven below:
a) Name and grade of material, .b) Consigner.•................................................................•.••..•c) Size and particulars of consignment .d) Identification of lot•........•...................................................c) Number of packages sampled .f) Place of sampling......•.....••.•••........•....................................g) Date of sampling.................•...•••.•.....•.......•••...•..•.•...•••..••••
4. Q,VALITY OF REAGENTS
4.1 Unless otherwise specified, pure chemicals and distilled water [,.IS: 1070-1960Specification for Water, Distilled QJ.1ality ( RAllisM)] I&allbeemployed in tests.
r.on - ' Pure chemical. • mall mean cbemicala that do not contain impuriti. whichafl't.ct the resul tI or .n~l)'Iia.
5. DE'r:ERMINATION OF MOISTURE CONTENT
5.0 Gea....I- Three methods, namely, (a) air-oven method, (b) hotplate method, and (e) distillation method are employed. The fint two
18

IS : 548 (Part I )-I96f
methods give the moisture and volatile matter content together while thedistillation method gives only the water content. The hot-plate method isuseful for a rapid preliminary screening.
5.0.1 ApplictJbili!y - The air-oven method is applicable to all theordinary oils and fats which have a relatively low moisture content (belowone percent), but not to drying or semi-drying oils or oils of the coconutoil group. The hot-plate method is applicable to all the ordinary oils andrats including coconut oil. Neither of these two methods is applicable tosolvent extracted oils and fats which may contain residues from solventswith fairly high boiling points. The distillation method is applicable to allnormal oils and fats, including emulsions, for the determination of moistureonly at differentiated from moisture and volatile matter. This method i.not applicable to samples of oib or fats containing volatile substancesmiscible with water.
5.0.2 P"ctlUt;on - Since water tends to settle in samples of oils or fats I
which have softened or melted, care shall be taken to mix the samplesthoroughly so as to distribute the water uniformly. Soften the sample withrnt1e heat ( but do nol 111611 ), and mix thoroughly. This general precaution• applicable to all the three methods.
5.03 Rtf"', M,lADd - In cases or dispute" and unless otherwise agreed tobetween the purchaser and the supplier, the moisture· content shall bedetermined by the distillation method.
5.1 Alr-OY•• MetIa_
5.1.1 .A/JIHIralus
5.1.1.1 Moil"", dill. - made of aluminium sheet about 0·45 to0·56 nun thickness, 70 to 80 mm in diameter and 20 mm deep; providedwith tight-fitting slip-over cover.
5.1.1.2 Duieedlor - containing an efficient desiccant. such u phocphorul pentoxide.
5.1.13 Air."Wft - preferably · electrically heated, with te:nperatu.n:control device.
5.1.2 ProutJ.r, - Weigh accurately about 10 g or the oil or rat inte a!noilture dish which has been dried previously. cooled in the desiccatorwnd then weighed. Place the dish in the air-oven for arproximately onehour at 105 ± I·C. Remove the dish (rom the oven. coo In the desiccatorto room temperature and weigh. Repeat this procedure but keep the dishin the oven only for half an hour each time until the difference betweenthe two successive weilhinp does not exceed one milligram, Preservethe heated oil or rat for the detennination of insoluble impurities (," 6 ).
19

IS, 5f8 (Part I )-19&4
5.1.3 C"bltllion
Moisture and volatile m~tter 100 IIIcontent, percent by weight &IB --w
wherew == loss In weight in g of the material upon drying, andW s= weight in g of the material taken for the test.
5.2 Hot-Plate Method
5.2.1 Apparalru5.2.1.1 Glass b,dk" - 100 to 150 ml capacity.
5.2.1.2 Small gltUs rod
5.2.1.3 Desiccator - containing an efficien! desiccant, such as phosphorus pentoxide.
5.2.1.4 Electric hot-plate - with variable heat control.
5.2.2 Procedure - Weigh accurately about 10 g of the oil or fat into theglass beaker which has been previously dried along with the small glassrod, cooled in the desiccator, and weighed. Heat the sample on theelectric hot-plate, stirring continuously with the glass rod. Avoid spattering of the oil or fat which may result from too rapid an ebullition ofmoisture. The apparent end point is judged by the cessation of the risingbubbles of steam as well as by the absence of foam. Allernativt(y, judge theend point bi' placing a clean, dry watch-glass on top of the beaker andobserving when no further condensation takes place on the watch-glass.When the apparent end point has been reached, heat momentarily to thepoint of incipient smoking taking care not to overheat. Cool to roomtemperature in the desiccator and weigh. Preserve the heated oil or fatfor the determination of insoluble impurities (SIt 6 ).
5.2.3 Calculation .Moisture and volatile m~tt~r 100 w
content, percent by weight == -w-where
w .. loss in weight in .. g of the material upon drying, andW -= weight in g of the material taken for the test.
5.3 DI.tillatloa Method
53.1 Ap/JartJtus - The apparatus consists of a glass flask heated bysuitable means and provided with a reflux eondenser discharging into atrap and connected to ~e flask. The connections between the trap and
20

IS : 5f8 ( Part I )-196f
the condenser and the flask should be interchangeable ground glass joints.The trap serves to collect and measure the condensed water, and to returnthe solvent to the flask. The assembly of the apparatus is shown in Fig. 6,and the various components are described below:
a) Flask- a 500- to I OOO-mi ftask of the shape shown in Fig. 6, madeof hard resistance glass, well annealed and as free as possible fromstriae and similar defects.
b) Condenser - a glass water-cooled reflux type condenser, of thedesign and dimensions shown in Fig. 6A. The only mandatorydimensions for the condenser are the external diameters of theinner tube and of the jacket, which shall be 16 to 17 mm and 23 to25 mm respectively. The joints A and B should be neatly finishedas shown In Fig. 6A, particularly the bore at B shall have the 'minimum disturbance. The shoulder above the cone of jointD shall be elongated as shown in Fig. 6A to avoid a sharp reentrant shape which may restrict the free flow of liquid down theinner wall. The cone shall be extended beyond the length appropriate to the joint D, and the lower end ground at an angle ofapproximately 60° to th~ axis. The drainage tip shall be at thefront of the condenser when the lower water connection is to the .left, and the finish shall be either smooth or fire-polished. Wheninserted into the trap, the tip of the condenser shall be 6 to 7 mmabove the surface of the liquid in the trap after distillation conditions have been established. The nominal dimensions of thejoint D are given below:
Nominal Diaof lArge End
ofGround Zone
mm
18-8
Naminal DiaofSmall End of
Ground Zone
mrn
Nominal l,tngth ofGround Zone ~IeQSU1ed
Axially
mm
26
c) Rtceivtrs - otherwise called the trap, made of hard resistanceglass, well annealed and as free as possible from striae and similardefects, provided with ground glass joints, with the shape.dimensions and tolerances given in FiR- 68 and 6C; consistingessentially of the upper chamber, together with the tube andground joint leading t<; the flask, and the graduated tube.
The receivers shall be of two sizes, namely, 2 ml capacityand 10 ml capacity (sel Fig. 6B and 6C): The mandatorydimensions and tolerances for the receivers shall be as given inTable I.
21

11. Me ( Part I ).196~
CONDENSER
OtSTIl\,A'TtONFLASK
FlO. 6 TYPICAL AaS..BLY OP DIWI AJlD STAaK APPAIlATVI
22

IS: Sfa ( Part 1)-1964
25
23 TO 25 00WAll 1 TO 1·5THICK
250
16 TO 17 00WAll 0'7 TO 1 .../THICK ---
2S
1B LENGTH
AllclimeaaioD. ill miUimetra.Flo. 6A CoNDSNI.a
The fthoulder of the upper chamber of the receiver immediately below the conical joint shan be finished square, as shown inFig. 6B and 60. The graduated portion of the receiver shall becylindrical throughout its length. The bottom of the graduatedtube of the 2-ml receiver shall be sealed, the end of the tubebeing approximately hemispherical in shape. The graduatedscales on the receivers shall be numbered and subdivided as shownin Fig. 6B and 60. The graduation marks shall be fine cleanlyetched permanent lines of uniform thickness Iyin~ in planes atright angles to the axis of the tube. The graduation marks shallbe confined to the cylindrical portion of the tube and there shall be
23

IS I M8 ( Part I )-1964
TAILB I MANDATORY DDIENIIONI AND TOLIDlANCE8 POR RECBIVEIl
(Cl•." 5.S.I(c)]
i) Volume, equivaknt to .mallett .ubdiviaion, ml
St. No.
(I) (2)
IQ-ml
(4)
0'1
il) Scale lenllh. mm
iii) Length or cylindrical tube above upperI"aduation mark, mm
iv) Tolerance on capacity, ml
v) Maximum permiaible lealr.a.e rate 01.topcock,ml/min
no evident irregularity in their spacing. In these receivers thenumbered graduation marks shall be carried completely roInd thetube, the shortest graduation marks shall be earned halfwayround the tube, and the graduation marks of intermediate lengthshall be carried approximately two-thirds of the way round thetube and shall project equally at each end beyond the shortestgraduation marks..
The capacity corresponding to any graduation mark is definedas the volume of water at 27°0, expressed in millilitres, requiredto fill the graduated portion to that mark at 27°0, the axis of thegraduated portion being vertical and the lowest point of the watermeniscus being set on the graduation mark. In the case of IO-mlreceiver, the volume of the bore of the stopcock key and thevolume of the jet below the stopcock shall not be included as partof the measured volume.
The error at any point on the receiver scale, and also thedifference between the errors at any two points on the scale, shallnot exceed the figures given for the receivers in Table I.
For the IO-ml receiver, the stopcock shall be of the 2 mmoblique bore having the general design mown in Fig. 68 and 60.The rate of leakage, tested with the stopcock free from grease, thebarrel and key wetted with water, the receiver filled initially withwater to the top of the scale, and the Ir.ey in either fully ahut-oft'position, shalt not exceed tl,le figures given in Table 1.
24

IS: 548 (Part I)-19M
9S! 10100...--
70 - - -1104
90
AU dimCDlioaa ia miWmetra.
Fla. 68 2·ml RaeaIVa.

18. 548 (Part I )-196f
All dimension. in millimctrea.
Flo. GC 10·ml RacBIVIUl
26

IS I 5.8 ( Part I )-1_
(};lantily of MiJU,iGI(Ap/Woxi~tcl.J )
Less than 1 percent 200 gJ to 5 percent 100gMoisture in excess of Proportionally smaller
5 percent quantityPlace the specified quantity o~tnateria1;accurately weighed, in the dis
tillation flask, add an equal volume Qfxylene or toluene, as desired. or atleast 100 ml if less than 100 f of dle material is used, and swirl to mix.Assemble the apparatus and fil the receiver with the solvent by pouring itthrough the condenser until it begins to overftow into the distillation flask.Insert a loose cotton plug in the top of the condenser to prevent condensation of atmOlphtric moisture within the tube. In order that the refluxingmay be under control, wrap the flask and the tube leading to the receiverwith asbestos cloth. Heat the flask 10 that the distillation rate is about100 drops per minute. \Vhen the greater part of the water has distilled
Each receiver shall have permanently ar-d legibly markedon it:I) the abbreviation C ml "2) the inscription '27°0 t to indicate that the receiver is gra
duated for content at 21°C, and3) an identification number shall also appear on the key.
d) H,,,t so.re,- the source of heat may be either an oil-bath or anelectric heater provided with a sliding rheostat or other meansof heat control. The temperature of the oil in the bath should notbe very much higher than the boiling point of xylene or toluene,whichever solvent is used.
e) Copper wi" -long enough to extent through the condenser, withone end twisted into a spiral. The diameter of the spiral shouldbe such that it fits snugly within the graduated portion of the 'receiver and yet may be moved up and down.
5.3.2 ReQgents
5.3.2.1 PuttJSJ~'um dich,omtJt,-sulphuric iJ&id cleaning solution
5.3.2.2 Xyl,ne or loluine - Saturate the xylene or toluene by shakingwith a small quantity of watert and distil. Use the distillate for the determination of moisture.
5.3.3 Procell"rl - Clean the entire apparatus with potassium dichromate..sulphuric acid cleaning solution to minimize the adherence of waterdroplets to the sides of the condenser and the receiver. Rinse thoroughlywith water and dry completely before using. The quantity of materialtaken for the test is determined by the amount of moisture present (wJw),as indicated below:
Moisture Range
27

IS , 548 ( Part I )-1964
over, increase the distillation rate to about 200 drops per minute andcontinue until no more water is collected. Purge the reflux condenseroccasionally during the distillation with 5 ml portions of xylene or tolueneto wash down any moisture adhering to the walls of the condenser. Thewater in the receiver may be made to separate from the xylene or tolueneby moving the spiral copper wire up and down in the condenser andreceiver occasionally, thus causing the water to settle at the bottom of thereceive-r. Reftux until the water-level in the receiver remains unchangedfor 30 minutes and then shut off' the source of heat. Flush the condenserwith either xylene or toluene, as required, making use of the spiral copperwire to discharge any moisture droplets. Immerse the receiver in water atabout 27°C for at least 15 minutes or until the xylene or toluene layer isclear, and then read the volume of water.
5.3.4 Calculatioll
M ' b · h 100 VD"J.olsture eontent, percent y weig t -= -W-where
V ac volume in ml of water,D =- specific gravity of water at. the temperature at which the
volume of water is read, andW -= weight in g of the material taken for the test.
6. DETERMINATION OF INSOLUBLE IMPUIUTIES
6.0 Geaeral- The material is dissolved in hot kerosine and filtered. Theresidue is washed thoroughly with petroleum ether dried at 100 ± 200and cooled in a desiccator till constant in weight,
6.1 Apparata.6.1.1 Gooch Cruei6lt -. prepared with a pad of acid-washed asbestos,
aboul2 mm thick. \Vash the pad with water alcohol and ether. Dry toconstant weight at 100 ± 2°C, ("001 in a desiccator to room temperatureand weigh.
6.1.2 Filltr Flu" - of convenient size.'.13 Gooela CrueilJl, Adtl/Jltr
1.2 aeacea ts
6.2.1 KtrDSinl - conforming to Grade 1 of'5: 1459-1959 Specificationfor Kerosines, Filter the kerosine through a Gooch crucible prior to use.
'.2.2 P,trot,um Ether - conforming to solvent Grade 60/80 of ·15 : 17451961 Specification for.Petroleum Hydrocarbon Solvents•
• Since rcviacd.
28

IS r 5t8 ( Part I )-196-1
1.3 Proc....- Use the whole of the heated oil or rat lell over after thedetermidation of moisture and volatile matter described under 5.1 or 5.2.AllmIlJli",l:1, use a sample prepared in the same manner. Add 50 ml orkerosine to this quantity of the oil or fat and heat on a water-bath todissolve it. Filter through the prepared Gooch crucible with the aid ofvacuum. Wash the container and the crucible with five IO-ml portions ofhot kerosine allowing each portion tn drain before adding the next. \Vashthe crucible thoroughly with petroleum ether to remove all the kerosine.Dry the crucible and contents to constant weight at 100 ± 2°O, cool toroom temperature in a desiccator, and weigh.
6.4 CalcaladoD
Insolubl~ impurities, percent 100 wby weight =- -w-
wherew = gain in weight in g of Gooch crucible, andIV == weight in g of the original material taken for the test
( see 5.1.S and 5.2.3 ).
7. DETERMINATION OF ACm VALUE AND FREE FATI'Y ACIDI
7.0 Geaeral- The acid value is determined by directly titrating thematerial in an alcoholic medium with aqueous sodium or potassium hydroxide solution. Free fatty acid is calculated as oleic, lauric, ricinoleic orpalmitic acids.
7.1 Rea.eat.
7.1.1 Ethyl Alcolaol- ninety-five f!ercent ( by volume ), or rectified spirit[conforming to IS: 323.. 1959 Specification for Rectified Spirit ( Revised) ]neutral to phenolphthalein indicator.
7.1.2 Phtnol/Jlat1ull,in Indicator Solution - Dissolve one gram of phenolphthalein in 100 ml of ethyl alcohol.
Non - \Vhrn tratinl oill or fatl which give dark coloured loap solution, theoblervation of the end point of the titration may be f.cilitated rither <a> by Ulincthymolphthai~in or alkali blue 68 in place of phenolphthalein, or (b) by addin.one millilitre of a 0'1 percent ( wlu ) solution of I:lcthylene blue in water to each 100mJof phenolphthalein indicator solution before the titration.
7.1.3 Stdnda,d AtJ"'OfU PDlassium Hydrox;u 0' Sodium Ifydro%ide Solulions -0'1 N or 0'5 N.
7.2 Procedare - Mix the oil or melted fat thoroughly before weighing.\\'eigh accurately a suitable quantity of the cooled oil or fat in I
29

IS 1548 (Part I )-1964
200-ml conical flask. The weight of the oil or fat taken for the test and thestrength of the alkali used for the titration shall be such that the volume ofalkali requhed for the titration does not exceed 10 ml, Add 50 to100 ml of freshly neutralized hot ethyl alcohol. and about one millilitreof phenolphthalein indicator solution. Boil the mixture for about, '/tveminutes and titrate while as hot as possible with standard aqueous alkalisolution, shaking vigorously during titration.
7.3 Calculatioll
. 56'1 VNACId value == W
where
28'2 VNW
b) Free fatty acids, in terms of lauric acid, 20-0 VNpercent by weight - W
c) Free fatty acids, in terms of ricinoleic 29'& VNacid, percent by weight a:: -y-
d) Free fatty acids, in terms of palmitic 25'6 VNacid, percent by weight - W
v == volume in ml of standard potassium hydroxide or sodiumhydroxide solution used,
N =: normality of standard potassium hydroxide or sodiumhydroxide solution, and
W = weight in g of the material taken for the test,
7.3.1 Free Fatty Acids - The acidity is frequently expressed as the percen- 'tage of free fatty acids present in the sample. The percentage of free fattyacids in most of the oils and fats is calculated on the basis of oleic acid;although in coconut oil and palm kernel oil it is often calculated in termsof lauric acid, in castor oil in terms of ricinoleic acid, and in palm oil interms of palmitic acid. The calculations in terms of different fatty acidsare as follows:
a) Free fat ty acids, in terms of oleic acid,percent by weight
where
v == volume in ml of standard potusiurn hydroxide solutionused,
N - normality of standard potassium hydroxide solution, andW - weight in B of the material taken f~r the telt.

IS: 548 ( Part 1\-1964
I. DETER.MlNATION OF UNSAPONIFIAR.B MATTER
8.0 GeDeral- The material is completely saponified with alcoholic potassium hydroxide solution and extracted with petroleum ether, Thepetroleum ether extract is washed with aqueous alcohol and then againwith water. The washed ether extract is evaporated and the residueweighed. Unsaponifiable matter is this residue minus the fatty acid presentin it, which is determined by titration with sodium hydroxide solution inalcoholic medium.
8.1 Apparata.
8.1.1 F1Qt-Bottomed or Coni,,,' Fla.sk - 250 to 300 ml capacity. An ordinary round, flat-bottomed flask, fitted with a long glass tube which acts asa condenser, may also be used.
8.1.2 Separating Funnels - 500-ml capacity.
8.2 ReageDt.
8.2.1 A/roholic Pol4Ssium Hyd,ox;u Solution - Dissolve 70 to 80 g of potassium hydroxide in an equal quantity of distilled water, and add sufficientaldehyde-free ethyl alcohol (95 percent by volume) or aldehyde-freerectified spirit [conforming to IS: 323-1959 Specification for RectifiedSpirit ( Revised) ], prepared as described under G-3.4 of IS : 323.1959. tomake up to 1 000 ml, Allow to stand overnight, decant the clear liquidand keep in a bottle closed tightly with a cork or rubber stopper,
8.2.2 Ethyl Alcohol- ninety-five percent ( by volume). or rectified spirit[ conforming to IS : 323·1959 Specification for Rectified Spirit ( Revised) ].
8.2.3 Phenolphthalei" Indicator Solrltion -- Dissolve one gram of phenolphthalein In 100 ml of ethyl alcohol.
8.2.4 Petroleum Etla" - conforming to solvent Grade 60/80 of *IS : 17451961 Specification for Petroleum Hydrocarbon Solvents.
8.2.5 Aqutous Alcohol- containing 10 percent (by volume) or ethylalcohol [conforming to -IS: 321-1952 Specification for Ethyl Alcohol( Absolute Alcohol) ].
8.2.6 St"ndard Sodium HJflroM S,I",ioJl- approximately 0·02 N.
8.2.7 Aceton, - free from evaporation residue (su ·15: J70·1950 Specification for Acetone ).
8.3 Proceclare
8.3.1 \\'eigh accurately about 5. of the well-mixed sample into the fluk.Add 50 ml of alcoholic potassium hydroxide solution. Boil ~ntly butsteadily under a reftux condenser for one hour or until the saponification is
• Since revised.
31

11.548 ( Pan I )-l96t
complete. Wash the condenser with about 10 ml of etltyl alcohol. (';00)the mixture and transfer it to a separating funnel. Complete the transferby washing th( ftuk lint with some ethyl alcohol and then with cold water.Altogether. add 50 ml of water to the separating funnel followed by anaddition of 50 ml of petroleum ether. Insert the sto~per and shakevigorously for at least one minute and allow to settle antil both the layersare clear. Transfer the lower layer containing the soap solution to anotherseparating funnel, and repeat the ether extraction at least six times moreusing 50ml of petroleum ether for each extraction. If any emulsion isformed, add a small quantity of ethyl alcohol or alcoholic potassiumhydroxide solution.
1.3.2 Collect all the ether extracts in a separating funnel. Wash thecombined extracts in the funnel three times with 25-ml portions of aqueousalcohol shaking vigorously and drawing off the alcohol-water layer aftereach washing. Again wash the ether layer successively with 20-ml portionsof water until the wash-water no longer turns pink on addition of a fewdrops of phenolphthalein indicator solution. Do not remove any of theether layers. Transfer the ether layer to a tared flask containing a fewpieces of pumice stone, and evaporate to dryness on a water-bath under agentle stream of clean dry air. To remove the last traces of ether, placethe flask in an air-oven at 80 to 90°0 for about one hour. To remove thelut traces of moisture, add a few millilitres of acetone and pass a gentleItream of clean dry air over the surface of the material or evacuate using a'¥att'r vacuum pump at about 500e for about 15 minutes. Cool in adesiccator and weigh. Repeat the evacuating, cooling and weighing until• constant weight is-obtained,
8.33 After ~eighing, take up the residue in 50 ml of warm neutralethyl alcohol, containing a few drops of phenolphthalein indicator solutionand titrate with standard sodium hydroxide solution.
8.4 Calcaladoa
8.f.l Weight in g or the fatty acids inthe extract ( as oleic acid) -= B ~ 0·282 VN
whereY - volume ill ml of standard sodium hydroxide solution, andN r normality of standard sodium hydroxide solution.
1.4.2 Unsaponlflable matter, percent by weight _ 100 (1,- B)
whereA == \\·eight in g of the residue ( '" 1.3.2),B - weight in g of the fatty acids in the extract (,. I.t.l ).
andW .. weight in g of the material taken for the tat.
32

IS I 5f8 ( Part I )-1964
,. DETERMINATION OF MELTING POINT
9.0 CeDenl - Oils and rats are chiefty mixtures of glycerides. They donot exhibit either a definite or a sharp melting point. Therefore, the termc melting point t does not imply the same characteristics that it does withpure crystalline substances. Fats pass through a stage of gradual softeningbefore they become completely liquid. The melting point is, therefore.defined by the specific conditions of the method by which it is determined(s', 2.& ).
9.0.1 The melting point is determined by taking the solid fat inside asmall capillary tube and two methods are prescribed for the purpose. Bothof these are applicable to all types of normal animal and vegetable fats andthe method used shall be specified while stating the results.
9.1 Opea..Tabe CapiUary-5Up Metllod
9.1.1 Apparatus
9.1.1.1 Mtlling point tubes - thin walled, uniformly bored capillaryglass tubes open at both ends and with the following dimensions:
a) Length, 50 to 60 mm;b) Inside diarneter O'B to I') mm; andc) Outside diameter, 1·2 to 1·5 mm,
9.1.1.2 Thermometer -- with 0-20 subdivisions and a suitable range.The thermometer should be checked against a standard thermometer whichhas been calibrated and certified by the National Physical J...aboratory,New Delhi, or any other laboratory recognized for such work.
9.1.1.3 Beaker - with a side-tube heating arrangement. Thielemelting point tube may be used.
9.1.1.4 Heal source - - gas burner or a spirit lamp.
9.1.2 Proeedur, - Melt the sample and filter it through a filter paper toremove any impurities and the last traces of moisture. Make sure that thesample is absolutely dry. Mix the sample thoroughly. Insert a cleanmelting point tube into the molten sample product so that a column of thematerial, about 10 mm long, is forced inJo the tube. Chill the sample inthe tube at once by placing the end of the tube containing the sampleagainst a piece of the ice until the fat has solidified. Place the meltingpoint tube in a test-tube and hold it for one hour either in a refrigerator orIn water maintained at 4 to lOQC. Remove the melting point tube andattach with a rubber band or any other suitable means to the thermometer10 that the lower end of the melting point tube is even with the bottom ofthe bulb of the thermometer. Pour water at about IOoe into the beakeror the Thiele tube, and suspend the thermometer in the centre of theapparatus, so that the lower end or the sample column is about 30 mm-
33

IS a5f8 (Part I )-196f
below the surface of water. Heat the side tube of the apparatus gently, sothat the temperature of the water rises slowly at the rate of 2°C per minutetill the temperature reaches 25°0, and thereafter at the rate of 0-SOC perminute. Note the temperature of the water when the sample columncommences to rise in the melting point tube. Report the average of twosuch separate determinations as the melting point. provided that the readings do not differ by more than 0-5°C.
9.2 CIo.ecI·Tube CompJ-.FuloD Metlaocl
9.2.1 App",atllJ9.2.1.1 Melting /Join' tu6«s - thin-walled, uniformly bored capillary
glass tubes open at both ends and with the following dimensions:a) Length, 50 to 60 mm;b) Inside diameter, 0-8 to 1·1 mm; andc) Outside diameter, 1·2 to 1-5 mm.
9.2.1.2 TAn""""t,, - with 0-20 subdivisions and a suitable range.The thermometer should be checked against a standard thermometerwhich has been calibrated and certified by the National Physical Laboratory,New Delhi, oe any other laboratory recognized for such work.
9.2.13 LilTg' test-tubt
9.2.1." Glass b,der- 600 ml capacity.
1.2.1.5 BlGt s""" - gas burner or electric hot-plate with rheostalcontrol.
1.2.2 Proe,tlur, - Melt the sample and filter it through a filter paperto remove any impurities and the lut traces of moisture. Make sure thatthe sample is absolutely dry. Mix the sample thoroughly. Insert a cleanmelting point tube into the molten product so that a column of the material about 10 mm long is forced into the tube. Cautiously fuse one end ofthe tube (where the II'mple is located ) in a small flame, taking care notto bum the fat_ Place the tube in • beaker and while the fat is still in theliqui41 state. transfer to • reCriserator and hold at 4 'to 10°C overnight(about 16 houri) _ Remove the tube from the refrigerator, and attachwith a rubber band or by any other luitable means to the thermometer 10that the lower end of the melting point tube is even with the bottom of thebulb of the thermometer. Suspend the thermometer in a large test-tubecontaining water and Immerse it in the 6OO-ml beaker which is about halffull of water. The bottom of the thermometer is immersed in the waterabout 30 mm below the surface. Adjust the starting bath temperaturefrom 8 to lO·C below the melting point of the sample at the belinning ofthe test. Agitate the water in the large test-tube as well as in the beakerwith a small stream of air or by other means. and apply heat so u toincrease the bath temperature at the rate of about O-S·C per minute. As

IS I 548l P...t 1)-1964
fats usually pass through an opalescent stage before melting completely. theheating is continued until the liquid in the tube is completely clearthroughout. Observe the temperature at which the liquid becomes clear.Report the average of two such separate determinations as the meltingpoint. provided that the readings do not differ by more than 0·5°C.
10. DETERMINATION OF REFRACTI\"'E INDEX10.1 Apparata.
10.1.1 &jractomele, - Abbe or Butyro refractometer. The temperatureof the refractometer should be controlled to within ±O' I°0 and for thispurpose it should be provided with a thermostatically controlled water-bathand a motor-driven pump to circulate water through the instrument. Theinstrument should be standardized, following the manufacturer's instructions. with a liquid of known purity and refractive index or with a glassprism of known refractive index. Distilled water, which has a refractiveIndex of 1'333 0 at 20'O°C, is a satisfactory liquid for standardization.
10.1.2 Light Source - If the refractometer is equipped with a compensator, a tungsten lamp or a daylight bulb may be used. Otherwise, amonochromatic light, such as an electric sodium vapour lamp, should beused.10.2 Proeeclare - Melt the sample, jf it is not already liquid, and filterthr .ugh a filter paper to remove any impurities and the last traces of rnois-
. lure. Make sure that the sample is completely dry. Adjust the temperatureof the refractometer to 40'0 ± O'l°C or t~ any other desired temperature.Ensure that the prisms are clean and completely dry, and then place a fewdrops of the sample on the lower prism. Close the prisms, tighten firmlywith the screw-head, and allow to stand for one or two minutes. Adjustthe instrument and light to obtain the most distinct reading possible, anddetermine the refractive index.
10.3 Temper.tare Correctio•• - Unless the correction factors arespecified in the detailed specification, approximate corrections shall bemade using the following equation:
R =- R' + K ( T' - T)where
R == the reading of the refractometer reduced to the specifiedtemperature, TOCj
R' == the reading at T'oC;K == constant, 0'000 365 for fats. and 0·000 385 for oils (if
.Abb, ,t/r_tDmttn is w4d ), or- O'55 for fats and 0'58 for oils (if Bulyro rtjraetomttl' is
used); andT' ar the temperature at which the reading R' is taken;T - the specified temperature (generally 40'0°0).. .
35

IS : 548 ( Part I )-t96t
IO.f eo.VenlOD 01 a.tyrO Rerractometer ReatUaJ. to RelraedyeIacUce. - When Butyro refractometer is used, its readings shall be con-verted into refractive indices (1JD ) using Table 2.
TABLE 2 CONVERSION OF BVTYRO REFRACTOMETER READINGS( B) TO REFRACTIVE INDICES ("I)D )
B 'WID B 'JD B "lJD B 'JD B 1ID
0-0 1-4220 4-7 8 9-5 6 14-4 4- 19-5 20-1 1 .'8 9 9'6 7 14-5 5 19-5 :J0-2 2 5'0 1-4260 9-8 8 14-6 6 19-6 4-0-4 3 5-1 I g'g 9 14-7 7 19'7 S0-5 4 5'2 2 10-0 1-4300 If-9 8 19-8 60-6 5 5-.j. 3 lOti 1 15-0 9 20'0 70'" 6 5'5 .. 103 2 15'1 "i34 0 20-1 8o-s 7 S"6 5 10'4 3 IS"! I 20'3 91·0 8 5'7 6 10'5 4 15"4 2 20-. 1tlf380I-I 9 5-9 7 (0-6 5 15-5 3 20'5 I1-2 1'4230 6-0 8 10-1 6 15'6 4 20'6 21-4 1 6-1 9 10'9 7 15'S 5 20'8 31-5 2 6'2 1'4-27 0 11-0 8 15'9 6 20'9 41-6 3 6'4- I 11-1 9 16-0 1 21-1 51-7 4 6'5 2 11'3 1'1310 16'2 8 21'2 6i-s 5 6-6 3 11-4 1 16-3 9 21-3 72'0 6' 6-8 4- I)-S 2 16'4 )'''350 21'. 82'1 7 6'9 5 11-6 3 16-6 I 21'6 92-2 8 7-0 6 11-8 4 16.7 2 21-7 1'43902-4 9 7'1 7 11'9 5 16'8 3 21'8 I2-5 1-424 0 7-2 8 12'0 6 17-0 .. 22'0 22-6 I 7--1 9 12-2 7 17'1 5 22-1 :42-7 2 7'5 1-4280 12'3 8 17-2 6 22·2 4-2·8 :t 7'6 I 12-4 9 17'4 7 22'40 53-0 4 1-7 2 12'5 1-4320 17-5 8 22-5 63.1 5 7'9 3 12-7 I 17-6 9 22'6 73-2 6 8'0 + 12-8 2 17·8 1·4360 22-7 83-3 7 8-1 5 12'9 3 17-9 I 22-9 9
5'5 8 8'2 6 13'0 4 18'0 2 23-0 1'44003'1) 9 8·... 7 1"2 5 18'2 ! 23'2 IS-7 1'4250 8-5 8 13'3 6 IS-S 4 23-5 2S-8 r 8-6 9 13'5 7 18'4 .5 23'• ,4'0 2 8'7 1'4290 13-6 8 18-5 6 23-5 4
4'1 3 8'9 I 13-7 9 18'7 7 23-7 54-2 4- g-O 2 15-8 1'4!50 IS-8 8 23'8 64'3 5 9'1 3 14-0 I 18-9 9 2S-9 74'5 6 9'2 4 14-1 2 19-1 1-4370 24-1 84-6 7 9-4 5 14-2 S 19-2 I 24-2 9
( CMIirnIM)

IS I 548 ( Part I )-1964
TAIU.& 2 CONVBBIION OP aurrao RlDACTOaa:n& aBADINGI( B ) TO aBl'L\CTIVB INDica ( 1)D ) - GMI4
B 1ID B 1JD B 'liD B 1JD B 1ID
24'S 1·441,0 so·8 8 37·5 6 44·' .. 51·' 224·~ 1 30"9 9 37-7 7 44". 5 .51-4 S24·6 2 31'0 J"4-f6 0 37-8 8 44-6 6 "J-6 f24-7 S 31·: 1 37'9 9 ...·7 7 51-7 524'8 4 31'4 2 38·1 t-flj! 0 44-9 8 51"9 625·0 5 31'5 S 38'2 1 4S-0 9 52-0 725-1 6 51-6 .. 38'3 2 45'2 1'4560 52'2 825·2 7 31'8 5 38·5 3 45'3 I 52'3 925-4 8 31-9 6 38·6 .. 4S-S 2 52·5 "461025'5 9 32'1 7 38·7 5 45-6 3 ~-7 125·6 1·442 0 32'2 8 38'9 6 45'7 4 521 225'8 1 :42'3 9 39·0 7 45'9 5 53- S25·9 2 32·5 1-4470 39'2 8 46"0 6 53-I 426-1 3 32'6 I sg.! 9 46·2 7 53'3 526·2 4- 32·8 2 3g., 1·4520 40-3 8 53·.. 626·5 5 32'9 3 39"6 I 46·. 9 53-6 726-5 6 55'0 4- 39·7 2 46-6 1'4570 S3'7 826'6 7 33'2 5 39'9 3 46'7 1 55·9 926-7 8 33·3 6 40-0 .. 46·9 2 54-0 1'462026-9 9 39'S 7 40-1 5 47'0 3 54-2 127·0 1·4430 33'6 8 40'3 6 .'"2 4 54·S 227"1 1 33" 9 40'4 7 47'3 5 54-5 S27·5 2 33'9 1·4480 40'6 8 47'5 6 54·6 427·4 :4 34'0 I 40" 9 47-6 7 M·8 527'S 4- 34'2 2 ~'9 1-4530 47·7 8 55-0 627·7 5 34'S 3 41'0 I 47"9 9 55-I 727-8 6 3i'. f fJ') 2 fB·O 1'.580 55'S 827-9 7 34'6 5 41-S S 48·2 I 55-4 928'1 8 Sf·, 6 41-" .. 48-3 2 55-6 1'46' 028'2 9 54'9 7 41'S 5 48-5 3 55·7 I28·3 t·4+t 0 5S'0 8 41'7 6 48'6 .- 55-9 228"5 1 35-1 9 41·8 7 48'8 5 56-0 328·6 2 35'3 1-4490 42'0 8 48'9 6 .56·2 f28·7 5 35·5 I 42-1 9 49'1 7 56'S 528·9 .. 35'6 2 42'5 1-4540 49'2 8 56"5 629·0 5 35'7 3 42·4 I 49'4 9 56·6 729'2 6 35-8 4 42·5 2 49'5 1"4590 56·8 829·5 7 !6'O 5 42'7 3 49'7 I 56-9 929-4 8 36'1 6 42'S 4 49'S 2 57·1 1"464029·6 9 36'3 7 43'0 5 SO-O 3 57" 129-7 1·4450 36'" 8 43·1 6 50'1 .. 57-4 229·9 I 36'5 9 43'3 7 50-2 5 57·6 ,90"0 2 36·7 1-4500 43-4 I) SO'. 6 57-7 4SO-I S 36'8 I 43'6 9 SO'S 7 57"9 5SO-S 4- 37'0 2 43·7 l'45S 0 50'7 8 58'0 6SO,t 5 97·1 3 4:t·9 1 SO'8 9 58-2 730·6 6 37'2 4 44'0 2 51-0 1·4600 58'3 8so·7 7 37-4 5 44'2 3 51'1 I 58'S 9
(CMIiuItl )
37

IS I 548 ( Part I )-1964
TABLE 2 CONVERSION OF 81JTYRO REFRACTOMETER aEADlNOI( B) TO IUtFBACTIVE INDICES ( "liD ) - co,.~
B "D B 'In B 1JD B "lJD B 'ID
58"6 1'4650 66"2 9 74'0 8 82"4 7 91"1 658'8 1 66-4 1'4700 '.-1 9 82-5 8 91'2 758-9 2 66-5 I 7'-' 1-4750 82-7 9 91"4- ,59-I 3 66"7 2 74'5 1 82-9 1'4800 91'6 959'2 4 66-8 3 14'6 2 83'1 1 91-& 1-485159'4 5 67'0 .. 7.'8 3 83'2 2 92-0 i59-S 6 6;'2 5 75'0 .. 83'. 3 92'159-7 7 67'3 6 7S'1 5 83-6 .. 92'S59-& 8 67-5 7 75'3 6 83-S 5 92'5 4-6&0 9 67'7 8 7S-S 7 83'9 6 92'7 560'2 1'466 0 67'8 9 75°6 8 84') 7 92°9 660°3 I 68'0 1-471 0 75-8 9 84-3 8 .93°0 760-5 2 68'1 J 76°0 1-.760 84-5 9 93-2 860-6 3 68'3 2 76'1 1 84-6 I-fBI 0 93'4 960-8 .. 68-4 3 16-3 2 (H°a I 93-6 1-48606&9 5 68-6 .. 76-3 3 85'0 2 93'8 161-1 6 68-7 .5 76'7 4 85-2 a 94'0 261-2 7 68'9 6 76'8 5 85" 4 M-I 361-4 8 69'1 7 77-0 6 8SoS 5 94'3 ..61-5 9 69'2 8 77-2 7 85-' 6 94-5 s61-7 1'4670 69'" 9 77'3 8 85'9 7 94-7 661'S I 690S )....720 77'5 9 86-0 8 94-8 762-0 2 69'7 1 77-7 1'4-770 86-2 9 95'0 862-2 3 69-9 2. 77'9 1 86'.. 1'4820 95°2 9&2-4 .. 70-0 3 78°1 2 86'6 1 95°f 104CJ:-O62-5 5 70'2 4 78°2 3 86-7 2 95-6 I62-6 6 70'3 5 78°. 4 86°9 3 95-8 262-8 7 70'S 6 78°6 5 87-1 .- 96°0 362-9 8 '0-7 7 78-7 G 87-3 5 96'1 ..63-1 9 10-8 8 78'9 7 87-5 6 96°3 563-2 1'468 0 71"0 9 79-1 8 87°6 7 960S 663-4 I 71'1 1-4730 79'2 9 87-8 8 96°7 763-S 2 71'3 J 79-4 1-.780 88-0 9 96°9 B63-7 3 71-. 2 79-6 I 88°2 1-483 0 97°0 963-8 -to 71°6 3 790S 2 88-' 1 97-2 I'~" a64'0 s 71'S .. SO-O S 88-.5 2 97-4 164-2 6 7J09 5 80-1 f 88-' 3 97'6. 164-3 7 72-1 6 8&3 5 88°9 .. 97-8 364-5 8 72°2 7 8&5 6 89-1 5 98'0 4-64" 9 72·. 8· 8&6 7 89-2 6 98-1 oS64-8 1-469 0 72-5 9 8&8 8 89'4
~98-3 6
6.5-0 I 72-7 1-47.0 81'0 9 &6 98'.56.5-1 2 72-9 J 81'2 1'4790 89-8 9 98-7 86S-3 3 7S·0 2 81-3 I 90-0 1-'" G 98°9 965-4 .. 73°2 S 810S 2 90-2 J 99-1 1-489065'6 5 7S'! .. 81-7 3 90-3 2 99'2 I65-7 6 730S .5 81-9 .. 90-5 :I 99'f 265-9 7 73-7 6 82-0 .5 go-7 .. 99-6 I66-1 8 73'S 7 82-2 6 90-9 5 99-8 ..
100-0 1-489 5
38

IS I 548 ( Part I )-1964
11. DETERMINATION OF SPECMC GRAVITY
11.0 Geaeral-- The specific gravity may be determined with a Westphalhydrostatic balance, or with a specific gravity bottle or pyknometer. Thelatter method shall be adopted as the referee method in cases of dispute.The temperatures at which the specific gravity is determined shall bereported, namely, sp gr 30°C/30°C or sp gr 95°C/30°C.
tl.1 Prep.ratloll of the Materlal- Melt the sample, if necessary, andfilter through a filter paper to remove any impurities and the last traces ofmoisture, make sure that the sample is completely dry. Cool the sampleto 30°C or warm to the desired test temperature.
11.2 We.tplaal Hydro.tatJc BaJa... MetJaocl- Suspend the plummetin the cylinder filled with recently boiled distilled water ~l the test temperature, and place the largest rider on the hook. Adjust the screw onthe base until the pointer is exactly opposite the fixed indicator point.Wipe the plummet and the cylinder to remove the water. Fill the cylinderwith the material at the lame temperature and dip the plummet into thematerial, removing air bubbles, if" any, formed in the eyehole of theplummet, by lifting it from the material. Re-immerse the plummet in thematerial and adjust the height of the balance to ensure that the plummetwill be nearly in the middle of the material when the beam is counterpoised. Place riders on the beam, till the pointer and the fixed indicatorare exactly opposite each other Read the specific gravity from the position of the riders on the beam, beginning with the largest and ending withthe smallest,
11.2.1 Corrections - Sometimes, especially when using the hydrostaticbalance, it is not convenient to make the determination at the specifiedtemperature. The determination may be made at a convenient temperature(T') as near to the specified temeerature (T) as possible and the result corrected a, shown below to the specified temperature. This correction is basedon the average value for the coefficient of expansion of oils and rats(0·000 64 per 1°C) and for water (0·000 23 per 10e). Approximatetemperature corrections may, therefore, be made as follows:
) S ifi · TOC/ToC D' + 0·000 64 ( T' - T)a pee c gravity at - W, + 0.000 23 ( T' _ T)where
D' = density of oil fat at T'OCj
T" - the temperature at which the densities D' and W' weredetermined;
T == the standard temperature. 3Q°C; andW' =- density of water at T'oC.
39

IS : 548 ( Part I )-l96f
b) Specific gravity at TOC/ToC = S' + 0·000 41 ( T' - T)where
S' .: SpeCJhC gravity at T' /T'oC;T' = temperature at which the specific gravity was deter
mined; andT =- standard temperature, 30oe.
11.3 Sped8c Gravity Bottle or Pykaometer Medaocl
11.3.1 Appartllus11.3.1.1 Specifie ,ravil.J hollu or pyknometer - with well-fitting ground
glass joints, To calibrate, clean and dry the bottle or pyknometerthoroughly, weigh and then fill with recently boiled and cooled water atabout 25°0 after removing the cap of the side arm. Fill to overflowingby holding the bottle or pyknometer on its side in such a manner as toprevent the entrapment of air bubbles. Insert the stopper and immerseIn a water-bath at the desired test temperature ±O·2°C_ Keep theentire bulb completely covered with water and hold at that temperaturefor 30 minutes. Carefully remove any water which has exuded from thecapillary opening. Remove from the bath, wipe completely dry, replacethe cap, cool to room temperature and weigh, Calculate the weight ofwater. This is a constant for the bottle or pyknometer, but should bechecked periodically, A specific gravity bottle of about 50 ml capaeityand of either of the two shapes as shown in Fig. 7 is recommended.
11.3.1.2 Waler·balh - maintained at 30·0 ± 0-2°C, or 95·0 ± 0·2°0.as required.
113.1.3 Th,'mDtnlUr - any convenient thermometer of a suitablerange. with 0-1 or 0-2°0 subdivisions. The thermometer should bechecked against a standard thermometer, which has been calibrated andcertified by the National Physical Laboratory, New Delhi, or any otherlaboratory recognized for such work.
113.2 Proeldurl - Fill the bottle with the oil previously cooled to about25uC or the melted rat to overflowing, holding the' bottle on its side insuch a manner as to prevent the entrapment of air bubbles after' removingthe cap of the side arm. Insert the stopper, immerse in the water-bathat 30·0 ± 0·2°C, or at 95·0 ± 0·2°0, as required, aud hold for30 minutes, Carefully wipe off B.ny oil which has come through the capillary opening. Remove the bottle from the bath, clean and dry itthoroughly. Replace the cap of tile side arm, cool to room temperatureand weigh.
11.33 Cal,ulalionA-·B
a) Specific gravity at 30°C/3000 == C B
40
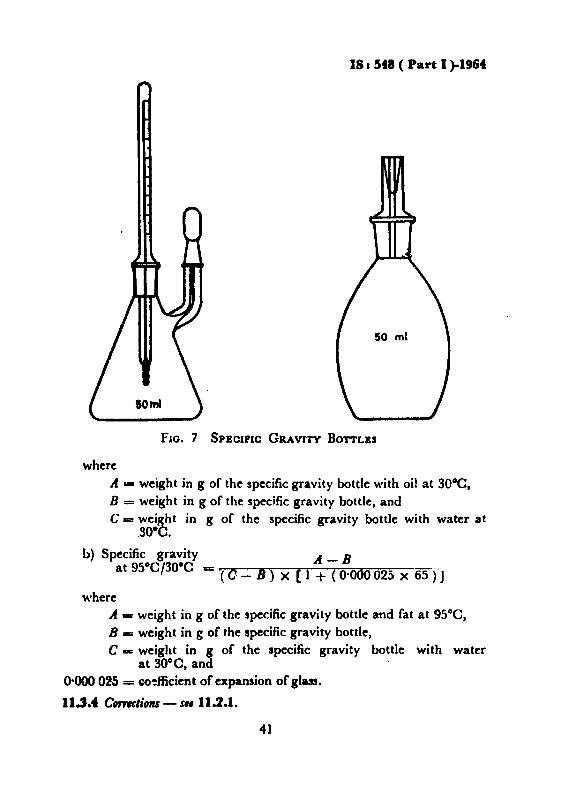
IS I 5t8 ( Part I )-1964
FlO. 7 SPECIFIC GkAVI'rY BOTTLES
where
A .. weight in g of the specific gravity bottle with oil at 3000,B = weight in g of the specific gravity bottle, andC =: weight in g of the specific gravity bottle with water at
.30·0.
b) Specific gravity A _ Bat 95°C/30·C == (C _ B) X [1 + (0·000025 X 65)]
whereA .. weight in g of the specific gravity bottle and fat at 9SoC,
B = weight in g of the specific gravity bottle,C sa weight in g of the specific gravity bottle with water
at 30°0, and0·000 025 == coefficlent of expansion of glass.
11.3.4 C,me';tJIU - tl' 11.2.1.
41

IS I 5f8 ( Part I )-1964
11. TITRE TEST
12.0 General - The fatty oil is saponified with glycerol-caustic potashsolution, and the soap is acidified with dilute sulphuric acid to give fattyacids. The fatty acids obtained are washed and dried. The highesttemperature recorded during the solidification under standard conditionsis the titre.
12.1 Apparatu. - The assembly of the apparatus is shown in Fig. 8, andthe various components are described under 12.1.2 to 12.1 ..8.
12.1.] Saponification Flask - 2.50 to 300 ml capacity.
12.1.2 Low-Form Beak" - of 2 litre capacity, to serve as a water-bath.
12.1.3 JYidt-i\[outh Bettie-- of 450 ml capacity, height 190 mm andinside diameter of neck 38 nun.
12.1.4 Titre Test- Tube - about 100 mm in length, 25 mm in diameterand one millimetre wall thickness, with an etched mark extending aroundthe tube at a distance of 57 mm from the bottom to show the height towhich the tube is to be filled,
12.1.5 Stirrer -- made of glass, stainless steel or monel metal, withone end bent in the form of a loop of 19 mm outside diameter. The upperend may be: formed to suit stirring with hand or attached to a mechanicalstirrer.
12.1.6 lAboralory Thermometer - range 0 to 150°C.
12.1.7 Corks - as shown in Fig. 8.
12.1.8 Titre Thnmomeur - with the following characteristics:
a) Typt- etched stem glass.
b) Liquid- mercury.
c) Filling above liquid- evacuated or nitrogen gas.
d) Tempera/ur, rangl-J1linus 2 to 68°0.e) Subdivisions - O·2°C.f) TOlallength - 385 to 390 mm,
g) Stem dimneter - 6 to 7 mm.
h) SlIm eonst;uetiDn - plain or lens front. The cross-section ofthe lens front type shall be such that it will pus through an8-mm ring gauge but will not enter a 5-mm slot gauge.
j) Bulb dillmtttr - from 5-5 mm to not greater than the diameter ofthe stem.
k) Bulb lIng'"- 15 to 25 mm •
42

f5i
IS s548 (Part I )-196.
2-lITRE lOW-FORMBEAt<ER
CORK EQUAlLV SPACEDTO HOLD THE BOTTLEINTACT
SAMPLE
T~TRE TEST- TUBE
4S0-ml WIDE-MOUTHBOTTLELEAD SHOT CR WEIGHl
Flo. 8 ASUMBLY OF ApPARATUS POR TITRE TEST
m) DisllJ1IUjrom, 6oUo". of1lulb to minIU rc mtl1k - 50 to 60 mm.n) DisltUlUjrom 6rC mtUk 10 ,,,, tif'hirmotMur - 20 to 35 mm.
p) Un"" qf"nelum"d&tl/Jillar.1 b,'wel,. 'III highul grtldutJlion mtl1k and ,h,"JlfJlISiDn eltemabn - 10 mm.
q) E~" ,1uImIJ"- t,o permit heating to at least 85°C.r) TD;jinisIJ -gJass ring.•) Lo",,,"_at;,,, li",s - at each 1°0 mark.t) GrtUlulllions "."b",d - at zero and at each multiple of 2°C.
43

IS : 548 ( Part I )-196f
u) [""""n," - 45 mm, A line shall be etched around the stem)45 mm from the bottom of the bulb. ,
w) Mtuimum Sed"error pnmitt,d al anyJIoinl- 0·2°0.y) S"'"da,di1:;ation - The thermometer shan be standardized at the
melting point of ice and at intervals of approximately 20°0, forthe condition of 45 mm immersion and for an average stem temperature of the emergent mercury column of 25°0.
12.2 ReageDu12.2.1 G~erol.ClJusti& PDtash Solu,ion - Dissolve with the aid of heat,
250 g of solid potassium hydroxide in I 250 g of glycerine. Do nOI: heatabove 140°C.
12.2.2 Dilute Sulplwric Acid - 30 percent by weight obtained bycautiously adding 16 ml of concentrated sulphuric acid, .p gr 1·84 [conforming to IS: 266-1961 Specification for Sulphuric Acid (Rnis,d)] to70 mm of water.
12.23 Methyl Orang, Indicator Solution - Dissolve 0·1 g of methyl orangein 100 ml of water.
12.2.4 Acetone - conforming to *IS: 170-1950 Specification for Acetone.
12.3 p ..ocecIare123.1 Pr'!Jardtioft of Fatty Acids - Weigh about 70 g of the glycerol
caustic potash solution into the saponification flask. Heat to 150°C whilestirring. Add about 30 g of the sample of oil or melted fat and re-heat toabout 150°C. If necessary.. add a little more glvcerol-caustic potash solution to ensure complete saponification. ( Complete saponification is usuallyindicated by an initial change in the appearance of the mass oftenaccompanied by an increase in the viscosity or thickening. The soluticnthen thins out after the reaction is complete 'and assumes a homogeneousappearance. The most common characteristic is that of soap bubbles forming and rising from the surface. Considerable care should be exercised atall tiJ?1e5 to ensure complete saponi~cation.) CooI.lightly and d~lve thesoap 10 300 lot of wacer contained 10 a I OOO-ml beaker. Add dalute sulphuric acid until the solution is distinctly acidic to methyl orange indicator,and place the beaker in a boiling water-bath until the fatty acids coUectas a clear layer at the top. Siphon off the lower aqueous acid layer, add300 ml of hot water, place in the boiling water-bath for a few minutestandagain siphon off the aqueous acid layer. Wash the fatty, acids thrice in thismanner or until the last traces of soap and acid are removed. Theacidification and washing should be done in u short a period as possible,keeping the beaker covered to prevent the oxidation of the fatty acids.After the last wash, allow the fatty acids to settle fOr a few minutes andthen decant them carcfu!ly. Filter through one or two thicknesies of filt~r
·Sinc. reviled.
44

IS 1 548 ( P..rt I )-1964
paper introduced into a conical Aask, and add about 10 ml of acetone.Close the flask with an air-tight cork, carrying a glass tube. Immerse theftask in boiling water and apply suction from a water pump until all6ubbling ceases. Remove the cork and dry the contents of the flask atabout 105°C for at least half an hour.
12.3.2 Dltermination oj Titre - Fill the low-form beaker with water upto two-thirds of its capacity. Adjust the temperature of water between 15°Cand 20°C below the expected titre point when it is not above 35°C, and at20 ± 1°C when it is 35°C or higher. Fill the test-tube up to the mark withthe fatty acid preparation described in 12.3.1, at a temperature 10 to 12°Chigher than the expected titre point. Insert the titre thermometer in thecentre of the sample and adjust its height so that its immersion mark coincides with the top surface of the fatty acid layer. When the temperature ofthe fatty acid comes down to about 10°C higher than the titre point, setthe stirrer moving in a vertical direction at a rate of about 60 complete upand down motions per minute, The temperature of the fatty acid gradually ,comes down and stirring is continued until the temperature remains cons ..tant for 30 seconds. The stirring is stopped when the temperature beginsto rise and the stirrer is raised out of the sample. The highest temperaturerecorded by the thermometer during this rise is the titre point. Duplicatedeterminations should agree within O·2°C.
13. DETERMINATION OF COLOUR
13.0 General- This method determines the colour of oils by comparisonwith Lovibond glasses of known colour characteristics. The colour isexpressed as the sum total of the yellow and red slides us-d to match thecolour of the oil in a cell of the specified size in the Lovibond tintorncter.
13.1 Apparatus
13.1.1 Looibond Tintometer or Colorimeter
13.1.2 Glass Cells - Presently, the Lovibond cell designations generallyemployed in the laboratories in the country, namely, i ill, j in, I in and51 in are recommended, However, with the corresponding metrir desig..nations of the cells, such as 1, 2, 5 and 10 em, also becoming available,their use wiU involve reconsideration of the requirements for colour of oilsand fats.
13.1.3 Filter Paper
13.2 Procedure - Melt the sample, if it is not already liquid, and filterthrough a filter paper to remove any impurities and the last traces ofmoisture. Make sure that the sample is absolutely clear and free fromturbidity. Clean the glass cell of the desired size ( see 13.3.2) with carbontetrachloride and allow it to dry. Fill it with rhe clear filtered sample and
45

IS : 548 (Part 1 )-l96t
place the cell in position in the tintometer, Place along' side of it such red,yell 0\\', blue or neutral Lovibond glass slides or any, combinations of theseas are necessary to match the colour shade of the oil, observing the coloursof the oil and of the combination of the glass slides through an eyepiece.
13.3 Report - Report the colour of the oil in terms of Lovibond units asfollows:
Colour readingin ( • ) cell == (G Y + 5 b R ) or ( a Y + lOb R )
where
a = the sum total of the various yellow ( Y ) slides used, and
b = the sum total of the various red ( R ) slides used.
13.3.1 Although the yellow and red slides required to match the colourshade of an oil in a Lovihond tintometer are assessed separately, it is foundthat to a certain extent these slides arc mutually compensatory. Consequently, different workers may report different values {;)I' the yellow and redunits for the same oil, and the same workers may report different values forthe yellow and red units for the oil examined at different times. To obviatesuch personal errors, a composite factor is to be used for checking thecolour, comprising the sum total of the yellow ( Y ) units and 5 or 10 timesthe total of the red ( R) units as specified for the oil or fat; ( Y + 10 R ) isthe mode of expressing the colour of moderately dark-coloured oils, suchas washed cottonseed oil, whereas (Y + 5 R) is the mode of expressinlthe colour of other vegetable oils.
Y+5RY+5RY+ SR
Y +5RY+5RY+5RY+ lOR
Modi ofExpress;",Colour Reddi,,&
MAHUACottonse~d ( washed
and refined )
Castor
Groundnut
Coconut
Sesame
Mustard
13.3.2 The dimensions of the cell used and the mode of expressing thecolour reading for different oils shall he as follows:
outr« Si{,e-DesignationofCell( in )
1
1
1
I!Ii
.Sizt desililation of the cell used.
46

IS : 548 ( Part I )-196f
14. DETERMINATION OF IODINE VALUE ( WIJS )
If.O Geaeral- The material is treated, in carbon tetrachloride medium,with a known excess of iodine monochloride solution in glacial acetic acid( Wijs solution). The excess 'of iodine monochloride is treated with potassium iodide and. the liberated iodine estimated by titration with sodiumthiosulphate solution.
14.1 Realeat.
14.1.1 Potassism Dichron,at' - conforming to • IS : 250-1953 Specificationfor Potassium Bichromate, Technical and Analytical Reagent,
14.1.2 Concentrated Hydrochloric Acid - conforming to IS: 265-1962Specification for Hydrochloric Acid ( Revised).
14.1.3 Potassium Iodide Solution - Prepare a fresh solution by dissolving10 g of potassiurn iodide free from potassium iodate, in 90 ml of water,
14.1.4 Starch Solutiofl- Triturate 5 g of starch and 0-01 g of mercuriciodide with 30 ml of cold water and slowly pour it with stirring into onelitre of boiling water. Boil for three minutes. Allow to cool and decant offthe supernatant clear liquid.
14.1.5 Standard Sodium ThiosulpluUe Solution - approximately 0·1 N. Dis101ve approximately 24·8 g ofsodium thiosulphate crystals ( NaISs03, 51-1.0 )in water which has been well boiled to free it from carbon dioxide andmake up to I 000 ml, Store the solution in a cool place in a dark.coloured stock bottle with a guard-tube filled with soda lime, After storingthe solution for about two weeks, filter, if necessary, and standardize it asprescribed under 14.1.5.1.
14:.1.5.1 Weigh accurately about s-o g of finely ground potassiumdichromate which has been previously dried to a constant weight at105 ± 2°C into a clean one-litre volumetric flask. Dissolve in water, makeup to the mark; shake thoroughly and keep the solution in a cool darkplace. For standardization of sodium thiosulrhate, pipette 25 ml of thissolution into a clean glass stoppered 250-m conical flask or bottle. Add5 ml of concentrated hydrochloric acid and 15 ml of a 10.percent potassiumiodide solution. Allow to stand in the dark for 5 minutes and titrate themixture with the solution of sodium thiosulphate, using starch solution asan internal indicator towards the end. The end point is taken when theblue colour changes to green, Calculate the normality ( ~V) of the sodiumthiosulphate solution as follows:
25 JY49·03 J"
• Since revised.
47

IS I Stl (P.rt I )-196t
where
W -= weight in g of the potassium dichromate, and
V a: volume in ml of sodium thiosulphate solution requiredfor the titration.
14.].6 Iodine Crystals - re-sublimed,
14.1.7 Acetic Acid - glacial, 99 percent, having a melting point of14·aoC and free from reducing impurities. Determine the melting point ofthe acetic acid and test it for reducing impurities as follows:
a) Determination of melting point - Take a IS-em long test-tube andfill it to about two-thirds with the acetic acid. Insert intothe acid a thermometer satisfying the req uirernents specifiedunder 12.1.8 through a cork stopper fitting the test-tube. Theamount of acid should be at least double the quantity requiredto cover the bulb of the thermometer when the hot tom of thelatter is 12 mm from the bottom of the test-tube. Suspend thistube within a larger test-tube through a cork. Cool the acid byimmersing the assembly in ice water until the temperature is10°C, then withdraw the assembly from the ice water andstir the acid rather vigorously for a few moments, thus causingthe super-cooled liquid to crystallize partially. Take thermometerreadings every 15 seconds and consider as the true meltingpoint. that temperature at which the reading remains constantfor at least 2 minutes.
h) Test for reducing impuritirs (potassium pt,·man.~anatt trst) -- Dilute2 011 of the acetic acid with 10 ml of water and add 2 drops of0·1l': potassium permanganate solution and main tain at 27 ± 2°C.The test shall be taken as having been satisfied if the pink colouris not discharged at the end of two hours.
14.1.8 Chlorine Gas- dry.
14.1.9 Iodine Trichloride ( ICl 3 )
14.1.10 Iodine Monochloride ( lei) - 98 percent chemically pure.
14.1.11 J/li}s Iodine il(Ollochlo,idt Solution - Prepare this solution hy oneof the following three methods, and store in a glass-stoppered bottle ina cool place, protected from light:
a) Dissolve 13 g of iodine in one litre of acetic acid, using gentleheat, if necessary, and determine the strength by titration withstandard sodium thiosulphate solution. Set aside 50 to 100 mlof the solution and introduce chlorine gas into the remainder untilthe characteristic colour change occurs and the halogen contentis nearly' doubled as ascertained again by titration. If the
48

IS I 548 ( Part I )-196t
halogen content has been more than doubled, reduce it by addingthe requisite quantity of the iodine-acetic acid solution. A .lightexcess of iodine does no harm, but avoid an excess of chlorine.
E~ampll:
If the titration of 20 ml of original iodine-acetic acid solutionrequires 22 ml of standard sodium thiosulphate, 20 ml ofthe finished Wijs solution should require between 43 and44 ml (and not more than 44 ml) of the same sodiumthiosulphate solution.
b) Dissolve 8 g of iodine trichloride in approximately 450 ml ofacetic acid. Dissolve separately 9 g of iodine in 450 ml of aceticacid, usin~ heat, ifnecessary. Add gradually the iodine solutionto the iodine trichloride until the colour has changed to reddishbrown. Add 50 ml more of iodine solution and dilute the mixturewith acetic acid till 10 ml of the mixture is equivalent to 20 mlof standard thiosulphate solution when the halogen content isestimated by titration in the presence of an eXCeM of potassiumiodide and water. Heat the solution to 100°0 for 20 minutes.and cool. Prevent access of water vapour in preparing the solution.
c) Dissolve 10 ml of iodine monochloride in about 1 800 ml of alacia!acetic acid ( chemically pure) and shake vigorously. Pipette 5 mlof this, add 10 ml of potassium iodide solution and titrate with0·1 N standard sodium thiosulphate solution, using starch solutionas indicator. Adju'lt the volume of the solution till it is approximately 0-2 N.
14.1.12 Carbon Telrachlorid, or Chloroform - inert to Wijs solution.
1~.2 Procedure -- Melt the sample if it is not already completely liquid,and filter through a filter paper to remove any impurities and the lasttraces of moisture. Make sure that the sample as well as the glassapparatus used is absolutely clean and dry. Weigh accurately, bydifference, an appropriate quantity of the oil or rat between the limitsindicated in col 2 and 3 of Table S, into a clean dry 500-ml iodine flask orwell ground glass-stoppered bottle to which 25 ml of carbon tetrachloridehave been added, and agitate to dissolve the contents. The weight of thesample shall be such that there is an excess of 50 to 60 percent of Wijs solution over that actually needed. Add 25 ml of the Wijs solution and replacethe glass stopper after wetting with potassium iodide solution; swirl forintimate mixing, and allow to stand in the dark for 30 minutes in the caseof non-drying and semi-drying oils and one hour in the case of drying oils.Carry out a blank test simultaneously under similar experimental conditions. After standing, add 15 ml of potassium iodide solution and 100 mJof water, rinsing in the stopper also, and titrate the liberated iodine withltandard sodium thiosulphate solution, swirling the contents of the bottle
49

IS I 5f8 ( Part I )-1964
continuously to avoid any local excess until the colour of the solution isstraw yellow. Add one millilitre of the starch solution and continue thetitration until the blue colour formed disappears after thorough shakingwith the stopper on.
TABLE 3 WEIGHT or OIL OR FAT FOR DETERMINATION OFIODINE VALUE
( CldUSI 14.2 )
IODDIa VALva WZIOHT IN I 0' SAMPLBEu&CT&D -.A..
Maximum Minimum
(I) (2) (3)
U. than 3 10 10
5 6-346 0 5-0770
10 3·1730 2-5384
50 0-661 2 0-5288
100 0·317 3 0-2538
150 0-212 5 0'1700
200 0'158 6 0-1269
WE.IGHING AccuaACY
(4)
.0·001
.0·000 5
.0-000 2
.0·000 2
.0·000 1
.0-000 1
ooס-ס. 1
143 Calcal.doD
. 12'69 ( B - S) NIodine value == ---w -.-----
whereB == Volume in ml of standard sodium thiosulphate solution
required for the blank,S == volume in ml of standard sodium thiosulphate solution
required for the sample,N = normality of the standard sodium thiosulphate solution,
andW == weight in g of the material taken for the test.
15. DETERMINAnON OF SAPONIFICATION VALUE
15.0 Geaeral- The material is saponified by refluxing with a knownexcess of alcoholic potassium hydroxide solution. The alkali consumed forsaponification is determined by titrating the excess alkali with standardhydrochloric acid.

IS I 548 ( Part I )-1964
15.1 Apparata.
15.1.1 Conical Flasks - 250 to 300 ml capacity.
15.1.2 RtJl~ Condens" - any efficient reflux condenser, at least 65 emlong.
15.1.3 Water-Bath or Electric Hot-Plate with Rheostdt Control
15.2 Reale.t.
15.2.1 Alcoholic Potassium Hydroxide Solution - Dissolve 35 to 40 g ofpotassium hydroxide in 20 rot of distilled water, and add sufficientaldehyde-free rectified spirit [conforming to IS: 323-1959 Specificationfor Rectified Spirit ( Revised) ] to make up to 1 000 ml. Allow to stand
. overnight, decant the clear liquid and keep in a bottle closed tight with acork or rubber stopper.
15.2.2 Aldehyde-Free R«tified Spirit - may be prepared according to either,of the two methods given in 15.2.2.1 and 15.'l.2.2.
15.2.2.1 klethod 1- Redistil rectified spirit (conforming to IS: 3231959) over solid caustic soda or caustic potash, add 2 to 3 g of mttaphenylenediamine hydrochloride per litre of rectified spirit, digest at ordinarytemoerature for several days or under a reflux condenser on a steam-bathfor several hours and distil slowly) rejecting the first 100 ml and the last200 ml of the distillate.
15.2.2.2 Method /1 - Reflux 1-2 litres of rectified spirit (confonningto IS: 323-1959) for 30 minutes in a round-bottom flask with 10 g ofcaustic potash and 6 g of granulated aluminium (or aluminium foil).Distil and collect one litre after discarding the first 50 ml.
15.2.3 Phenolphthalein Indicator Solution - Dissolve 1-0 g of phenolphthalein in 100 rnl of rectified spirit ( conforming to IS: 323-1959 ).
NOTE- When testing oils or fats which give dark-eoloured soap lolutlOns, theobservation of the end point of the titration may be facilitated either <a> by ulin,thymolphthalein, or alkali blue 68 in place oi phenolphthalein or (b) by adding onemillilitre bf a 0-1 percent ( rvlv) solution of methylene blue in water to each 100 m1 ofphenolphthalein indicator solution before the titratiol.
15.2.4 Standard Hydrochloric Acid- approximately 0·5 N.
15.3 Procedure - Melt the sample, if it is not already liquid, and filterthrough a filter paper to remove any impurities and the lent traces of rnoisture. Make sure that the sample is completely dry, Mix the samplethoroughly, and weigh accurately by difference about l·S to 2·0 g of thesample in a conical flask. Add 25 rot of the alcoholic potassium hydroxidesolution and connect the reflux air condenser to the flask. Heat the flaskon a water-bath or an electric hot-plate for not more than one hour, Boilgently but steadily until the sample is completely saponified as indicated
51

IS I 548 ( Part I )-1964
by absence of any oily matter and appearance of clear solution. After theflask and condenser have cooled somewhat, wash down the inside of thecondenser with about 10 ml of hot ethyl alcohol neutral to phenolphthalei-.•Add about one millilitre of phenolphthalein indicator solution, and titracewith standard hydrochloric acid. Prepare and conduct a blank determinetion at the same time.
15.4 CalealadoD
.. 56·1 (B - 9 \ NSaponification value - W L:.._
whereB == volume in ml of standard hydrochloric acid required for
the blank,S as volume in ml of standard hydrochloric acid required for
the sample,N c: normality of the standard hydrochloric acid, and
W c::: weight in g of the material taken for the test.
16. DETERMINATION OF ACETYL VALUE AND HYDROXYLVALUE.
16.0 Geaeral - Two methods have been prescribed, namely, Method Aand Method It Being simpler, Method B is preferred.
16.0.1 Method A - A sample of oil or .at is acetylated by reftuxing .withacetic anhydride and the excess anhydride is decomposed with water andsodium bicarbonate solution. The 3aponification value of the washed anddried acetylated oil is determined as in 15.
16.0.2 lvletlrod B - The process consists in acetylating the oil or rat witha measured quantity of acetic anhydride in pyridine decomposing theexcess anhydride by boiling with water and then, after the addition ofsufficient butyl alcohol to give a homogeneous solutien, titrating withalcoholic alkali. A control test with the acetic anhydride and pyridinewithout the oil or rat provides a measure of the acetic anhydride availablefor acetylation; a similar test with the oil or fat and the pyridine withoutthe acetic anhydride provides a measure of the free fatty acid present.From the figures obtained" the acetyl value or the hydroxyl value of the ratis calculated.
16.1 Method A
16.1.1 A.'JHlrlJ,,"
16.1.1.1 B,." -- 800 ml capacity.
52

IS : 5fB ( Pa.-t I )-1964
16.1.1.2 Separ"tingjunnel- 500 ml capacity.
16.1.1,3 Conicaljlasks - 250 to 300 ml capacity.
16.1.1.4 RtJlux condemn - any efficient reflux condenser, at least 65 emlong.
16.1.1.5 Sand-hath or electric hot-plate with rheostat control
16.1.2 RtagentJ
16.1.2.1 Acetic anhydride - containing 95 (0 100 percent ( by weight)or the actual acetic anhydride. Determine the acetic anhydride content asfollows:
a) Weigh accurately about 2 g of the acetic anhydride into a 200-mlglass-stoppered conical flask, cool in ice and add 5 ml of freshlydistilled aniline. Insert the glass stopper immediately, shakevigorously and allow to stand at room temperature for 30 minutes.Wash down the sides of the flask with 50 ml of ice-cold water.Mix well and titrate with previously standardized I N sodiumhydroxide solution, using phenolphthalein as indicator, until thepink colour persists for 10 minutes,
A == yolume in fit of I N sodium hydroxide solution require~
Weight in g ofacetic anhydride taken for the testb) Weigh accurately about 2 g of the acetic anhydride into another
flask, add 50 ml of water, allow to stand for 30 minutes andtitrate with the standard sodium hydroxide solution to the sameend point as above using phenolphthalein as indicator.
B _ Volume in rol of I N sodium hydroxide _solution required- Weight in g of acetic anhydride taken for the test
Acetic anhydride,percent by weight = 10·209 (B-A )
16.1.2.2 Sodium bi(Q,bonate solution - freshly prepared 0·5 percent( wlv ) and neutral to litmus.
16.1.2.3 Anhydrous sodium sulphate
16.1.2.4 AlcoAoli' potassium laydroxide solution - 0·5 N.
16.1.2.5 Phenolphthal,in indicator solution - Dissolve 0'1 g in 100 ml of60 percent rectified spirit.
16.1.2.6 Standard ".1droelllorie tl&id - approximately 0'5 N.
16.1.3 Procedur, - Weigh accurately about 10 g of the material in aconical ftask t add 20 ml of acetic anhydride and boil the mixture under areflux air condenser for about 2 hours. Pour the mixture into a beakercontaining 500 ml of water and boil for 15 minutes. Bubble a stream ofcarbon dioxide or nitrogen through the mixture during boiling to prevent
53

IS I 5f8 (Part I )-1964
bumping. Discontinue boiling, cool slightly, and remove the water with asiphon. Add another 500 ml of water and boil again. Discontinueboiling, cool and transfer the contents of the beaker to a separating funneland reject the lower layer. Wash the acetylated sample successively(a) three times with 50 ml of water, (b) twire with 50 ml of sodium bicarbonate solution, and (c) twice with 50 ml of warm water (60 to 70·C).Drain and remove as much of tlte water as possible» and then transfer theacetylated sample to a beaker and add approximately 5 g of anhydroussodium sulphate. Allow to stand for about one hour with occasionalstirring, Filter through a dry filter paper, preferably in an oven at 100to 110°C, remove the filter paper and) eep the sample in the oven until itis thoroughly dry. Determine the saponification values of the originalmaterial and the acetylated product by the procedure describedunder 15.3.
16.1.4 Calculation
S'-sa) Acetyl value == }.OOO-O-OOO 15 S
S'-Sb) Hydroxyl value == 1.000-0.000 75 S'
whereS' = saponification value after acetylation, andS = saponification value before acetylation ( Stt 15 ).
16.2 MethOd B16.2.1 Reagents
16.2.1.1 Pyridine - Reflux with powdered barium oxide and distil.Use the fraction distilling above 114°C.
16.2.1.2 Acetic anhydride16.2.1.3 Acetylatin, agtnt- Mix one volume of acetic anhydride a.d
seven volumes of pyridine,16.2.1.4 Alcoholic sodium laydroxitl, solut;,,. - Prepare by dissolving
sufficient aqueous caustic soda (60 percent wI") in 95 percent alcohol tomake a 0·30 to 0·35 N solution. Remove the precipitated carbonate byfiltering.
The solution should be standardized against standard acid in thepresence of phenolphthalein before use. The solution remains colourlessfor a long time if kept below 25°C.
16.2.1.5 Normal butyl alcohol
16.2.1.6 Phtnolp1JI"alti" solution - Dissolve 0·1 g in 100 ml of 60 percent rectified spirit.
Non - In the determination upon lubitaDces Bivinl dark-eoloured IOAp IOIutiou,observation of the end point of the titration may be facilitated either (a) by the
54

18 : 548 (Part I )-196f
aubetitution or thymolphthalein or alkali blue 68 for phenolphthalein or (b) by theaddition of one milHlitre of 0'1 percent solution of methylene blue to each 100 mlof the phenolphthalein solution before the titration.
16.2.2 Apparatus - A round-bottomed acetylation flask made of resistanceglass of capacity 150 to 200 ml with a l00-cm ground-in air condensertube.
16.2.3 Procedur«16.2.3.1 Weigh accurately 0'5 to 3'0 g of fat in the acetylation flask.
(The following weights serve as a rough guide for different types ofmaterials: fatty alcohols 0'5 to 0'7 g, castor oil about one gram and ordinaryoils 2 to 3 g.) Measure, or preferably weigh, from a 10-ml burette intothe flask, 5 ml of the pyridine-acetic anhydride mixture. Add the mixturedropwise, and allow no time for drainage. Before attaching the condenser,moisten the neck of the ffask with pyridine to act as a seal, and make surethat the seal is maintained during the acetylation.
16.2.3.2 Mix the fat and the acetylating agent by shaking well, addone or two small pieces of pumice, and boil the contents of the flask I
gently during 60 minutes, maintaining the boiling so that the vapour risesno higher than the bottom end of the condenser. In this way, thepyridine seal needs renewal less often, and there is less tendency for theground-in joint to sieze.
16.2.3.3 Cool the flask to about 50°0 and with a rotary motion toassist in washing the condenser tube, add 5 ml of distilled water, from the ,top of the condenser. Shake the mixture well, and then boil it gently for5 to 10 minutes, shaking the flask two or three times during the boiling.
16.2.3.4 After cooling the flask and the contents to room temperatureand before detaching the condenser, wash the condenser with 30 ml ofbutyl alcohol. Detach the condenser and wash the neck and mouth ofthe flask and the tip of the condenser with a further 20 ml and then, if thecontents of the flask are not homogeneous, add butyl alcohol until theybecome homogeneous. Titrate the free acetic acid with carbonate-free0'35 N sodium hydroxide solution, in the presence of a few drops ofphenolphthalein as indicator.
16.2.3.5 Carry out the same series of operations with 5 ml of pyridineacetic anhydride mixture alone, 'also with a corresponding weight of theoil or fat plus 5 ml of pyridine.
NOTE - Owing to the presence of vapour. of pyridine and butyl alcohol, it i.preferable to carry out the tesll in a fuming chamber.
16.2.~ CalculatiDn56'1 NY
a) Hydroxyl value, H = W
Hb) Acetyl value, A =a J + 0'000 75 fj
55

IS. St8 (Part I )-1964
whereN - normality of sodium hydroxide solution;r - volume ofsodium hydroxide solution in ml corresponding
to-the amount of acetylated rat formed-a+6-,
wherea, band f are respectively the volumes in ml ofsodium hydroxide required by blank with pyridine-acetic anhydr ide mixture, rat p'lus pyridine,and fat plus pyridine plus acetic anhydride; and
W - weight in g of rat or oil taken for test.NOTa- It is to be noted tballl alilht increase in the acetyl value baa been found to
occur with increaaina free fatty acid r.ontent of the sample.
17. DETERMINATION OF ALLYL ISonoOCYANATE
17.0 GeD.al - The oil obtained from black mustard seeds containssinigrin and myrosin which, after maceration with water, yields a volatileoil, the major constituent of which is allyl isothiocyanate. The oilobtained from white mustard seeds contains acrinyl isothiocyanate whichis much less volatile than allyl isothivcyanate.
17.0.1 Method- The allyl isothiocyanate in the oil is steam distilled intoa known excess of silver nitrate solution, and the excess of silver nitrateIOlution .is determined by titration with standard ammonium thiocyanatesolution.
17.1 Apparata.17.1.1 DistiUation FI4Sk - SOO-ml round-bottomed flask.
17.1.2 Any Ejfiaen' Rtftux Cotlturuer-approximately 90 em long.17.1.3 MetJSllri", Flask-200 ml capacity.17.1.4 Waln-Batla
17.2 RealeDt.17.2.1 E'k)l Alcohol- 95 percent (by volume) or rectified spirit (con
forming to IS: 323·1959 Specification for Rectified Spirit (JUlJised)]neutral to pheno phthalein.
17.2.2 Sillier Nit,a', Solutio1l - approximately 0'1 N.17.2.3 Aml'Mnium HyJroxidl Solu,i01l - 10 percent ( will ).17.2.4 Nitrie Acid- conforming to -IS: 264-1950 Specification for Nitric
Acid•
•Sinc:c reviatd.
5&

IS : 5t8 (Part I )-196t
17.2.5 Fmie Ammoni. SulpluJU lrulielJtor - 0·1 percent solution in water.
17.2.6 Standard Ammonium Thiocyanate Solution - approximately 0·1 N.
17.3 Procedare- Weigh accurately about 5 g of the material into a500-ml distillation flask and add to it 25 ml of ethyl alcohol, 250 ml ofwater and a few pieces of pumice stone. Distil the mixture in steam andcollect the distillate in a 200-ml measuring flask containing exactly 25 mlof silver nitrate solution and 10 ml of ammonium hydroxide solution.Collect as rapidly as possible about 150 ml of the distillate. Attach thereflux air condenser to the measuring flask and heat the mixture-for aboutone hour on a boiling water-bath. Cool to room temperature. add waterto make up to 200 ml and filter the contents after shaking. Take 100 01)
of the filtrate, add 6 ml of nitric acid and a few drops of ferric ammoniumsulphate indicator, and titrate with standard ammonium thiocyanatesolution until a permanent red colour is obtained. Carry out a blank testat the same time.
17.4 CalealatioD
Allyl iso~hiocyanate) percent 9·915 ( B - S) Nby weight == . W
where
B -= volume in ml ofstandard ammonium thiocyanate solutionrequired for the blank determination,
S = volume in ml of standard ammonium thiocyanate solutionrequired for the sample,
N == normality of standard ammonium thiocyanate solution,and
W -= weight in g of the sample taken for the test.
II. DETERMINATION OF REICHERT·MEISSL VALUE
18.0 GeBerat - The material is S sponlfied by heating with glycerolsodium hydroxide solution and then split by treatment with dilute sulphuric acid. The volatile acids are immediately steam distilled. The solublevolatile acids in the distillate are filtered out and estimated by titrationwith standard sodium hydroxide solution.
18.1 Apparata. - The assembly of the apparatus for distillation is shownin Fig. 9, and details of the constituent parts are given below:
a) Flal·Bottom Boiling Flask - The flask (A) shall be made of resistanceglass and shall conform to the dimensions given in Fig. 9.

IS. Sfl (Part I )-196f
S20!S
,10 Inl
o 100 rnl
All dimcnsionaln millimetrr••
FIC. 9 REICH£Jt.T-MEI8SL ))ISTILLATIO.. ApPARATUS
59

IS ,5f8 (Part I )-196f
b) Slill-H,atl- The stiJI-head (B) shall be made of glass tubing ofwall thickness 1'25 ± 0'25 mm and shall conform to the shapeand dimensions shown in Fig. 9 and 10.
.....----107·5 t 2'5 ----..t
.---e>o~,----------_.-----+--
J7·5!2·500
All dimen.ion. in mjIJimelret.
Fro. 10 STILL-HEAD FOR RBICHERT-MsISSLDISTILLATION ApPARATUS
A rubber stopper, fitted below the bulb of the longer arm ofthe still-head, and used for connecting it to the flask, shall haveits lower surface 10 mm above the centre of the side-hole of thestill-head.
c) Condenser - The condenser ( C) shall be made of glass and shahconform to the dimensions indicated in FiR' 9.
d) Rtteioer - The receiver ( D) shall be a flask, with two graduationmarks on the neck, one at 100011 and the other at 110 ml.
59

IS I 5f8 ( PArt 1)-19&t
e) Asbtslos Board - An asbestos board ( E), 120 mm diameter, 6 mmin thickness, with a circular hole about 65 mm in diameter, shallbe used to support the flask over the burner. During distillation,the flask shall fit snugly into the hole in the board to prevent theflame from impinging on the surface of the flask above the hole.A new asbestos beard may conveniently be prepared by bevellingthe edge of the hole, soaking in water, moulding the edge with aflask, and drying.
f) Bunsen Barner - The burner (F) should be sufficiently largeto allow the distillation to be completed in the time specifiedin 18.3.2.
18.2 ~"leDt.
18.2.1 Glycerine - conforming to AR Grade of IS: 1796-1960 Speci..fication for Crude Glycerine and Refined Glycerine.
18.2.2 Concentrated Sodium Hydroxide Solution - 50 percent (wlw).Dissolve sodium hydroxide in an equal weight of water and store the solution in a bottle protected from carbon dioxide. Use the clear portion freefrom deposit.
18.2.3 Pumice Stone Grains - approximately 1·4 to 2'0 mm in diameter.
18.2.4 Dilut« Sulphuric A.cid Solution - approximately 1·0 N.
18.2.5 SodIum Hydro%itlJ Solution - 0·1 N solution in water, accuratelystandardized:
18.2.6 Phenolphthalti" Indicator - Dissolve 0·1 g of phenolphthalein intOO ml of 60 percent rectified spirit.
18.3 Procedure
18.3.1 Weigh accurately 5-00 ± 0·01 g of the filtered oil or fat into theboiling flask. Add 20 g of glycerine and 2 ml of the concentrated sodiumhydroxide solution from a burette to which access of carbon dioxide isprevented and whose orifice is wetted before running in the liquid, thefirst few drops from the burette being rejected. Heat the flask and itscontents with continuous shaking on a gauze over the naked flame untilthe fat, including any drops adhering to the upper parts of the flask. hac;been saponified and the liquid becomes perfectly clear. Avoid overheatingduring this saponification. Cover the flask with a watch..glass. and a110\\
the flask to cool a little. Add 90 ml of boiling distilled water which hasbeen vigorously boiled for about 15 minutes. After thorough mixing, thesolution should remain clear. If the solution is not clear ( indicating incomplete saponification) or is darker than light yellqw (indicating overheating ),repeat the saponification with a fresh sample of the oil or fat. If the samp(~
is old. the solution may sometimes be dark and not clear. ·
60

IS : 546 (Part I )-l96t
11".2 Add 0·6 to 0·7 g of pumice stone grains and 50 ml of dilutesulphuric acid, and immediately connect the flask with the distillingapparatus. Place the flask on the asbestos board so that it fits snugly intothe aperture. This will prevent the flame from impinging on the surfaceof the flask above the level of the liquid and avoid superheating of thesystem. After the fatty acids have melted and separated Into a clear liquidlayer on gentle warmlnJ, heat the flask without altering the flame sothat 110 ml of li'Juid dIstils over in the course of 19 to 21 minutes. Thedistillation is considered to begin when the first drop forms in the still-head.Keep the water Bowing in the condenser at a sufficient speed to maintainthe temperature of the out~oing water from the condenser between 15°0and 20·0. Collect the distdlate in a graduated flask.
18.3.3 As soon as 110 ml have distilled over, stop heating the boilingflask and replace the graduated flask by a measuring cylinder of about25 ml capacity to catch drainings. Close the graduated flask with thestopper, and, without ma1cing the contents, place it in a water-bath at 15°Cfor 10 minutes, making sure that the IIO-ml graduation mark is below thelevel of the water. Swirl round the contents of the flask from time to time.Dry the outside of the flask and then mix the distillate by closing the flaskand inverting it four or five times, but do not shake. Filter through a dry9-cm Whatman No.4 filter paper. Reject the first 2 or 3 ml of the filtrateand collect the rest in a dry flask.
Non - The filtrate should 1-.,. free from insoluble Catty acids. When operatins oncoconut oil, it hu been noticed that this is not easily achieved. In cases where liquidinlOluble Cattyacids pass through the filter, transfer the filtrate to a separating (unneland after the separation of the lower aqueous layer, add the insoluble acids to the maiDbulk of insoluble acicb.
11.3.t Pipette 100 ml of the filtrate in a titration flask, add 0·1 ml ofphenolphthalein indicator solution and titrate with standard (0·1 N)sodium hydroxide solution until the liquid becomes slightly pink.
183.5 Run a blank test without the fat but using the same ctuantiticsof reagents and following the same procedure. During the heating withcaustic soda. avoid overheating which will be indicated by the darkening ofthe solution.
18.4 CalcalatiOD - Reichert-Meiss) value :::: ( A - B) X N X 11
where
A - volume in ml of standard sodium hydroxide solutionrequired for the test.
B - volume in ml of standard sodium hydroxide solutionrequired for the blank, and
}/- nonnality of standard sodium hydroxide solution.NOTa- Kirkham, v. H. Au(,s'. Vol 45, P 293 (1920·) and Fine, S. D.]t1WUl"
AuocY'ioft " 0","" A"itul,.,., C1IIJfIiJ". Vol 38,-P 319 ( 1955) have shown that at
61

IS I 5~8 ( Part I )-1964
high altitudes. that ii, low barome:tric presluft:l. the Reichert-Meill. value determination. are lower than those determined at atmospheric presaure. The correction formulaesuggested by Kirkham were verified by Fine.
Since the Reichert-Meiss) value varies with atmospheric prellUre, correction. (or thevalue arc necessary when the determinations are made at places above lea-level or whenbarometric pressure it ftuctuating widely from 760 mm. The corrected Rcichert--Meiulvalue sball be calculated .. follows;
Corre~tcd Reichert- ( RM-10 ) 101160Mellll value == -10"1,---+10
where
RM 1ft observed Reichert-Meisll value, andP == barometric pressure in rom of mercury at the place aDd time of
determination.
19. DETERMINATION or POLENSKE VALUE
19.0 General - The condenser, the 25-ml cylinder and the receiver usedin the Reichert-Meissl value determination are washed into the filter paperthrough which the distillate was filtered for that determination, Afterrinsing, the residue on the filter paper is taken up with ethyl alcohol andtitrated with standard sodium hydroxide solution.11.1 Reagents - All the reagents required for determining the ReichertMeissl value ( see 18.1 ) as well as the following are required.
19.1.1 E(h;'l Alcohol- 90 percent by volume, and neutral tophenolphthalein.
19.2 Apparatus - the same apparatus as that required for the determination of Reichert-Meiss] value ( see 18.2 ).
193 Procedure - After determining the Reichert-Meissl value, detach thestill-head and wash the condenser with three successive l.5-ml portions ofcold distilled water, passing each washing separately through the measuringcylinder, the 1] O-ml flask with stopper, and the filter paper nearly lillin!the paper each time to the brim and draining each washing before filteringthe next. The last 10 ml of wash-water should require not more than onedrop of 0·1 N sodiurn hydroxide solution for neutralization. Discard all thewashings.
Dissolve the insoluble acids by· three similar washings of thecondenser, the measuring cylinder, the 1JO-ml flask with stopper, and thefilter paper with 15-ml portions of ethyl alcohol. Combine the! alcoholicwashings in a clean flask ( the tolal volume thus amounting to 45 ml }, add0·25 rnl of phenolphthalein indicator solution, and titrate with standard(0'1 N) sodium hydroxide solution until the solution turns slightly pink.
19.4 Calca1atioDPolenske value lID 10 VN
62

IS : 5f. {Part I )-IHf
where
V -= volume in ml of standard sodium hydroxide solutionrequired for the test, and
N - normality of standard sodium hydroxide solution•
. NOTJI- Like Reichert-Meisal value. the Polenske value .1.0 varia when determinatlODI are done at atmOlpheric pressure differing widely from 760 mm (I" II•• ). Thecorrected Poleoake value dial! be calculated AI folloWl:
Correc&ed Polenlke value "" PV Y~54S )where
py - observed Polenlke value, and
P - barODletric pressure in mm of mercury at die place and time ordetermination.
20. DETERMINAnON or PEROXIDE VALUE
20.0 Geaeral-- The peroxide value is a measure of the peroxidescontained in a sample of fat, expressed as milli-equivalents of peroxideper I 000 grams of the material.
20.1 o.tUa. or the Methocl _. The rnarerial in an acetic acid-chloroformmedium, is treated with an aqueous solution of potassium iodide. Theliberated iodine is titrated with standard sodium thiosulphate solution.
W.2 Apparata.
20.2.1 Pi",t,. - Graduated. 1 ml capacity.
20.1.2 CtmiealjtdSk - Glass-stoppered, 250 ml capacity.
21.' R....u20. S.t Ac,'i, .A.eitJ-ChIDrojorm Solution -- Mix three parts by volume
of glacial acetic acid, with 2 parts by volume of chloroform.
20.'.2 PoJ4Ssillm Iodide Solution - Saturated. Prepare saturatedsolution of potassium iodide in recently boiled distilled water. Storein the dark.
20.3.3 Sodium Tlaiosulphate Solution - 0-1 N J accurately standardized.
20.3.4 Sodium Thiosulphat» Solution - 0·01 N. This solution isprepared by diluting 100 ml of accurately standardized solution of 0-1 Nsodium thiosulphate to 1 000 ml with freshly boiled and cooled distilledwater.
20.,-5 StG,,/a Sol"tion - 1 percent.
63

IS I 5f8 (Pan I )-196f
20.4_
a.t.l Wei~h 5·00 ± 0·05 g of sample of rat in a 250-ml II...stoppered conical flask and then add 30 ml of the acetic acid-chloroformIOlution. Swirl the flask until the sample is dissolved. Add 0·5 ml ofsaturated potassium iodide solution. Allow the solution to standexactly one minute with occasional shakin(J and then add 30 ml ordistilled water. Titrate with 0·1 N sodium thiosulphate solution with can.tant and vigorous shaking. Continue titration until the yellow colouralmOit disappean. Add 0-5 ml of starch solution and continue titrationtill the blue colour just disappcan.
2O.t.2 If the titre value is leu than 0'5 ml, repeat the determinationusing 0-01 N sodium thiosulphate lolution.
20.43 Conduct a blank determination of the reagents in the same way.The titration in blank determination should not exceed 0-1 m1 of the0-1 N sodium thiosulphate solution.20.5 Calcaladoa - Peroxide value as milli-equivalents per I 000 gramJsam I (S-B)xNxIOOO
pe - ,
where
.S - volume in ml of sodium thiOlulphate solution used up bythe sample.
B a= volume in ml of the sodium thiosulphate solution uIed upin the blank determination.
N - normality of the sodium thiOlulphate solution. and, =- weight in g of the sample.

BUREAU OFINDIAN STANDARDSHeadquarters:Manak Shavan, 9 Bahadur Shah Zafar Marg. NEW DELHI 110002Telephones: 23230131, 23233375, 23239402 Fax: 91+011 23239399.23239382E• Mall: [email protected] website: http://www.bis.org.in
CentralLaboratory:PlotNo. 20/9, SiteIV. Sahibabad Industrial Area. SAHIBABAD 201010
Regional Offices:Central : Manak Shavan, 9Bahadur Shah ZafarMarg. NEWDELHI 11 0002*Eastern : 1/14CITSchemeVIIM,V.I.~ Road, Kankurgachi, KOLKATA 700054Northern : SCQ 335-336, Sector 34-A, CHANDIGARH 160022Southern: C.I.T. Campus, IVCross Road, CHENNAI600113tWestern: Manakalaya, E9.MIDC, Behind MaroiTelephorl~ Exchange,
Andheri (East), MUMBAI 400093
BranchOffices:'Pushpak', Nurmohamed Shaikh Marg, Khanpur, AHMEDABAD 380001Psenya Industrial Area, 1" Stage, Bangalore-Tumkur Road, BANGALORECommercial-cum-Office Complex, Opp. Dushera Maidan, Arera Colony,
Bittan Market, BHOPAL 46201662-63, Ganga Nagar, UnitVI,BHUBANESHWAR 7510015th Floor, Kovai Towers, 44BalaSundaram Road, COIMBATORE 641018SeQ 21,Sector 12,Faridabad 121007Savitri Complex, 116G.T. Road, GHAZIABAD 20100153/5Ward No. 29,R.G. Barua Road, 5thBy-lane, Apurba Sinha Path,
GUWAHATI7810035-8-56C, L.N. Gupta Marg, Nampally Station Road, HYDERABAD 500001Prithavi RajRoad, Opposite Sharat Overseas Bank, C-Scheme, JAIPUR 30200111/418 B,Sarvodaya Nagar, KANPUR 208005Sethi Bhawan, 2M Floor, Behind Leela Cinema, Naval Kishore Road,
LUCKNOW 226001H.No. 15,Sector-3, PARWANOO, Distt. Solan (H.~) 173220PlotNoA-20-21, Institutional Area, Sector 62,Goutam Budh Nagar, NaiDA201307Patliputra Industrial Estate, PATNA 800013PlotNos. 657-660, MarketYard, Gultkdi, PUNE 411037·Sahajanand House· 3rd Floor, Bhaktinagar Circle, 80FeetRoad,
RAJKOT 360002T.C. No. 21275 (1 &2),NearFood Corporation of India, Kesavadasapuram-Ulloor Road,
Kesavadasapuram, THIRUVANANTHAPURAM 6950041" Floor, Udyog Shavan, VUDA, Siripuram Junction, VISHAKHAPATNAM-03
-satesOfficeisat5Chowringhee Approach, ~O. Princep Street, KOLKATA 700072tSaJes OffIce (WAO) PlotNo.E-9, MiCe, AdNo.8,Behind Telephone Exchange,
Andheri (East), Mumbai-400 0093
Printed by theManager, Government of India PressFarldabad, 2007
Telephone2770002
23237617233786622609285
2254198428329295
560134883949552423452
2403139221 0141229217528614982456508
23201084222328222330122618923
23543624022062262808
242748042378251
25579142712833
23553243
28329295