islet xenotransplantationuu.diva-portal.org/smash/get/diva2:163939/fulltext01.pdf · comprehensive...
TRANSCRIPT

Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Medicine 1317
Islet XenotransplantationAn Experimental Study of Barriers to Clinical
Transplantation
BY
PETER SCHMIDT
ACTA UNIVERSITATIS UPSALIENSISUPPSALA 2004


This thesis is based on the four papers below. These will be referred to in the text by their roman numerals.
I Adenovirus-mediated expression of human CD55 or CD59 protects adult porcine islets from complement-mediated cell lysis by human serum.Schmidt P, Goto M, Le Mauff B, Anegon I and Korsgren O. Transplantation. Vol. 75, 697-702, No. 5, March 20031.
II A new murine model of islet xenograft rejection: Graft destruction is dependent on a major histocompatibility-specific interaction between T-cells and macrophages.Schmidt P, Krook H, Maeda A, Korsgren O and Benda B. Diabetes. Vol. 52, 1111-18, May 20032.
III Acute cellular islet xenograft rejection in MyD88-deficient mice.Schmidt P, Krook H, Goto M and Korsgren O. Manuscript.
IV Dynamics of porcine endogenous retrovirus expression after fetal islet xenotransplantation to athymic and normal rats. Schmidt P, Forsman A, Andersson G, Blomberg J and Korsgren O. Manuscript.
Reprints of were made with permission from the publishers
1Copyright 2003 Lippincott Williams & Wilkins 2Copyright 2003 American Diabetes Association

CONTENTS
INTRODUCTION ..........................................................................................9 Diabetes mellitus ........................................................................................9
Transplantation as a cure for Insulin-dependent diabetes....................10 Xenotransplantation .................................................................................10
Solid organ xenotransplantation ..........................................................11 Islet xenotransplantation......................................................................12
Adult versus fetal islets ...................................................................12 The choice of location for implantation of islets ............................13
Immunological barriers to clinical xenotransplantation ...........................14 Cells and proteins of the blood ............................................................14
Blood cells ......................................................................................14 Cytokines ........................................................................................15 The complement system..................................................................15 The coagulation system...................................................................17
Rejection of vascularized xenografts...................................................19 Hyperacute rejection .......................................................................19 Acute vascular rejection..................................................................19 Cellular rejection of solid organs ....................................................20
Islet xenograft rejection .......................................................................21 IBMIR .............................................................................................21 Cellular rejection.............................................................................22
Accommodation and general tolerance................................................23 The risk of zoonosis in xenotransplantation.............................................24
Porcine endogenous retroviruses .........................................................25 PERV release and in vitro transmission..........................................25 Infection in humans?.......................................................................26 PERV pathogenicity........................................................................27
Animal models of xenotransplantation.....................................................27 Models of xenograft rejection .........................................................27 PERV in animal models ..................................................................28

Genetically modified donors ....................................................................29 PERV and genetically modified animals ........................................29
AIM OF THE STUDIES ..............................................................................31 General aims.............................................................................................31 Specific aims ............................................................................................31
Paper I: ............................................................................................31 Paper II:...........................................................................................31 Paper III: .........................................................................................32 Paper IV: .........................................................................................32
RESEARCH DESIGN AND METHODS ....................................................33 Ethics........................................................................................................33 Preparation and culture of islets ...............................................................33
Human islets (Paper I).....................................................................33 Adult porcine islets (API; Paper I and IV)......................................33 Fetal porcine islet-like cell-clusters (ICC; Paper II-IV)..................33 Rodent islets (Paper II) ...................................................................34
Adenoviral vectors and transduction procedures (Paper I) ......................34 Adenoviral vectors ..........................................................................34 Transduction procedures .................................................................34
Flow cytometry analysis of protein expression (Paper I) .........................35 Preparation of single cell suspensions.............................................35 Expression analysis .........................................................................35
Human serum cytotoxicity assay (Paper I)...............................................35 Animals and transplantation procedures ..................................................36
Paper II............................................................................................36 Animals ......................................................................................36 Transplantation procedures ........................................................36
Paper III ..........................................................................................37 Paper IV ..........................................................................................37
Immunohistochemistry.............................................................................37 Paper I .............................................................................................37 Paper II and III ................................................................................37 Paper IV ..........................................................................................38
Real-time quantitative RT-PCR ...............................................................38 Paper II and III ................................................................................38 Paper IV ..........................................................................................39
RT activity in islet culture supernatants (Paper IV) .................................40

RESULTS AND DISCUSSION ...................................................................41 Paper I ......................................................................................................41
Expression of human RCAs in API after adenoviral transduction..41 Cytotoxicity of human sera against transduced and normal API ....42 Alternatives to adenoviral transduction ..........................................44
Paper II .....................................................................................................45 The transfer model ..........................................................................45 Demonstration of T cell dependence...............................................46 Evaluation of intragraft cytokine mRNA expression......................47 Demonstration of xenospecificity ...................................................48 Demonstration of MHC-restriction .................................................49
Paper III....................................................................................................50 Toll-like receptors ...........................................................................50 Immune cell infiltration and cytokine mRNA kinetics. ..................51 ICC xenograft rejection persists in MyD88-deficient mice ............53
Considerations with the quantification of cytokine and chemokine expression using real-time RT-PCR (Paper II and III) ........................54
Paper IV ...................................................................................................55 PERV expression in ICC transplanted to rats .................................55 PERV content in islet cultures ........................................................57 Risk estimates .................................................................................58
CONCLUSIONS ..........................................................................................60
FUTURE ASPECTS.....................................................................................61
ACKNOWLEDGEMENTS..........................................................................63
REFERENCES .............................................................................................65

ABBREVIATIONS
ADCC Antibody-dependent cell-mediated cytotoxicity ANOVA Analysis of variance APC Antigen presenting cell API Adult porcine islets AVR Acute vascular rejection CTL Cytolytic T lymphocyte CVF Cobra venom factor DAF Decay-accelerating factor DTH Delayed-type hypersensitivity DXR Delayed xenograft rejection EC Endothelial cell ERV Endogenous retrovirus HAR Hyperacute rejection HERV Human endogenous retrovirus HLA Human leukocyte antigen Hsp 70 Heat shock protein 70 IBMIR Instant blood-mediated inflammatory reaction IDDM Insulin dependent diabetes mellitus ICC (Fetal) islet-like cell cluster Ig ImmunoglobulinIFN InterferonIL InterleukinLPS Lipopolysaccharides MAC/C5b-9 Membrane attack complex MBL Mannan binding lectin MCP Membrane co-factor protein MCP-1 Monocyte chemoattractant protein 1 MHC Major histocompatibility complex MIP-1 Macrophage inflammatory protein 1MMuLV Moloney murine leukemia virus mRNA Messenger ribonucleic acid NK cell Natural killer cell PAMP Products of microbial metabolism PBMC Peripher blood mononuclear cells PERV Porcine endogenous retrovirus

RANTES Regulated upon activation in normal T-cells, expressed, probably secreted RCA Regulator of complement activation RT-PCR Reverse transcriptase polymerase chain reaction sCR1/TP10 Soluble complement receptor 1 SLA Swine leukocyte antigen Th T helper TNF Tumor necrosis factor Tx Transplantation WT wildtype XNAs Xenoreactive natural antibodies

9
INTRODUCTION
This thesis discusses the appealing possibility of using isolated islets of Langerhans from pigs as a cure for insulin-dependent diabetes mellitus.
The success of allotransplantation during the past decades together with the current deficit of human organs, has lead to an increasing interest in xenotransplantation. However, several obstacles before entering into the clinic have been identified, and many of them remain unsolved. These can roughly be subdivided into four categories: ethical, physiological, immunological and infectious barriers. From an islet perspective, the main focus of this thesis has been on the last two barriers. Since the field of xenotransplantation has traditionally been associated with whole organs e.g. kidneys and hearts, similarities and differences between solid organ and islet xenograft rejection will also be discussed.
Diabetes mellitus Diabetes mellitus includes a number of metabolic disorders leading to uncontrolled high blood glucose levels. The prevalence of diabetes varies considerably between ethnical and regional populations but is estimated to affect 2-4 % in the global population or more than 150 million adults worldwide (1, 2). The majority of these patients are diagnosed with type 2 diabetes resulting from insufficient insulin production, impaired cellular glucose uptake or peripheral insulin resistance. The remaining 5-10 % suffer from insulin-dependent diabetes mellitus (IDDM) or type 1 diabetes, in most cases caused by an autoimmune destruction of the pancreatic islets of Langerhans, most often initiated at an early time in life (1, 2).
While tremendous improvements have been made in monitoring IDDM over the past decades, the disease is still associated with severe secondary complications related to the gradual development of vasculopathy and neuropathy. Although well maintained blood glucose control through daily insulin injections has been shown to markedly delay the onset of such complications, coronary disease, end-stage renal failure, amputations and blindness remain highly over-represented among IDDM patients. A minority also suffers from more acute problems related to a great difficulty in controlling blood glucose levels despite scrupulous monitoring. IDDM

10
patients with this disabling form of the disease have a high risk of entering into the life-threatening state of hypoglycaemia.
Transplantation as a cure for Insulin-dependent diabetes Since insulin injections can merely be regarded as a treatment, not sufficient to restore the glucose metabolism, the only current cure for IDDM is transplantation of a whole pancreas organ or isolated islets of Langerhans. The former procedure is well established, but comprises major surgery with a relative high risk of complications due to the fragility of the pancreas. Therefore, this option is offered mainly to patients who have already developed many of the secondary complications, including end-stage renal failure, but are still physically fit to undergo simultaneous pancreas-kidney transplantation. Transplantation of isolated islets is a fairly new approach that has become an appealing alternative following several breakthroughs during the recent years (3). The major advantage is that the procedure of intraportal infusion is far safer for the patient and allows for repeated transplantations of new cells if necessary. Moreover, an irreversible rejection episode will not have any clinical symptoms and hence does not require removal of the necrotic graft. Islet transplantation can therefore be motivated at an earlier stage of the disease, as a mean to avoid secondary complications, and can also be offered to patients that would otherwise not sustain whole pancreas transplantation.
However, the apparent advantages of restoring physiological blood glucose metabolism have to be balanced against a life-long treatment with immunosuppressive drugs, something that increases the risk for infections and certain forms of tumours and is associated with organ toxicity. For this reason, transplant candidates are currently selected among patients with unstable IDDM or those who have already received a donated kidney (and are on an immunosuppressive regimen anyway).
At present, an acute shortage of islets greatly limits the application of islet transplantation. This is, at least in part, due to the general deficit of donated organs available for transplantation. For this reason, much effort has been made in finding an alternative source of islets. In the future, stem cells may offer a solution to this problem, but in a more short-term perspective animal tissue have the highest potential to become a clinical reality.
Xenotransplantation Successful xenotransplantation of not only islets, but also other cells, tissues or solid organs would offer a much needed alternative for the increasing number of patients waiting for an allograft.

11
The modern era of human-to-human transplantation was initiated in the early 1960´s. At the same time there were also several clinical attempts with vascularized xenografts from primates. However, the poor outcome of these efforts together with the acceptance of brain death, which yielded more donors, soon decreased the interest in xenotransplantation. During the 1980´s, the arrival of more sophisticated immune suppressive drugs (i.e. cyclosporine A) markedly increased the success and broadened the application of allotransplantation. The increasing number of potential recipients lead to an organ deficit and there was renewed interest in xenotransplantation (4).
From an immunological point of view, the most suitable donors are found among the to man phylogenetically closest species, the old world primates (e.g. apes and baboons). However, since the early clinical trials, a variety of considerations have changed the general opinion in favour of the pig as the donor of choice. The pig is of appropriate size, is by far more adapted to large scale farming and it will be more acceptable for the general public that a domesticated species otherwise used for meat production is also a source for organ harvest. In addition, pigs have a short generation time, are available in controlled and homogenous breeds and during the past decade elaborate methods for genetic manipulation of pigs have been developed.
Providing that the barriers to clinical xenotransplantation are overcome, using the pig as the source of tissue would actually be superior to human transplants in some important respects. The transplantation could be planned in advance, allowing for minimal ischemia times and proper preparation of both the pig donor and the human recipient. In addition, it would be possible to create genetically homogenous pig strains, i.e. the properties of the donor would be well characterized and identical in every transplantation. In such a strain, one or more genes could be inserted and/or knocked out if necessary.
Solid organ xenotransplantation Because of very limited clinical experience, it is uncertain whether porcine organs (providing immunological acceptance) will function adequately in the human body. However, it has been demonstrated that both kidneys and hearts can function well in non-human primates (5-7). Also, the requirement of pig organs to function equally well as their human counterparts may prove not to be necessary. A porcine liver is less likely to offer a life-long substitute, because of the inability of taking care of the extensive biochemical metabolism normally present here. Still, such a transplant may be useful as a short-term life saving measure while waiting for a suitable human liver (8). As opposed to life-threatening cardiac, hepatic or pulmonary failure, patients with end-stage renal failure have an alternative to transplantation, i.e. blood dialysis. In the long run, in terms of costs, quality of life and patient survival, transplantation will still be the better option.

12
Although there are certain metabolic incompatibilities in a porcine kidney, these defects do not appear to be crucial for the overall health of the human recipient. For instance, porcine erythropoietin (with limited function in humans) can be replaced by the exogenously administered human equivalent (9).
Islet xenotransplantation In islet allotransplantation, the general shortage of human organs is even more accentuated by the fact that many suitable organs are selected for whole pancreas transplantation. Moreover, isolation of islets from a pancreas is a procedure that only has a 20-50% success rate and most IDDM patients also need islets from multiple donors in order to achieve insulin-independence. It is evident that an additional source of tissue would be highly appreciated.
As far as physiological compatibility of pig islets is concerned, it is known from early clinical experience that porcine insulin can be successfully used for treating IDDM in humans. Since the glucose metabolism is similar between these two omnivorous species, pig islets are also likely to respond adequately to changes in blood glucose levels in a human recipient. It is not clear to what extent there is a risk of reoccurrence of the autoimmune disease (that initially caused IDDM in the patient) after an islet transplantation. However, there are some indications that using pig islets may actually be an advantage to human islets in avoiding this (10, 11).
There is some limited clinical experience with pig islet xenotransplantation. Between 1990 and 1993, 10 IDDM patients with a kidney allograft were transplanted with fetal islets either intraportally or under the kidney capsule (8 and 2 of the recipients, respectively). No clinical benefits were observed, but porcine C-peptide was recorded up to 400 days after the procedure in four of the patients and surviving porcine endocrine cells were found in a biopsy taken from one of the patients three weeks after transplantation (12, 13).
Adult versus fetal islets Both adult porcine islets (API) and fetal islet-like cell clusters (ICC) are candidates in future clinical islet xenotransplantation. ICC have the advantage of being considerably easier to generate (no purification necessary). Isolation of API, which are prepared in much the same way as human islets, is more complicated and the resulting islet yield and quality has proven to be more difficult to reproduce, especially from young pigs (14, 15). However, ICC require approximately 4 weeks to differentiate into insulin producing cells (16) while API have the potential to immediately cure IDDM after transplantation, which makes monitoring of islet function early after transplantation possible. In addition, there are certain phenotypic

13
differences that may influence the choice between the two cell types in a future clinical programme. For example, ICC will increase in cell mass after transplantation unlike API (16), and may also have a greater resistance to ischemia than adult tissue. Another difference is the expression of the Gal (1,3)Gal epitopes (the important role of this antigen in xenotransplantation is discussed later). While ICC express high levels of Gal (1,3)Gal, it appears that the endocrine cells that constitute pure API preparations exhibit no or very low amounts, also after culture (17-19). Notably, with the recent generation of (1,3)galactosyltransferase knockout pigs this obvious advantage in favour of API may no longer be present (20,21).
The choice of location for implantation of islets It is still not established which would be the best site of implantation for optimal islet graft survival. In addition to the clinically used intraportal route of infusion and the transplantation beneath the kidney capsule that is frequently used in small animal models, other sites of implantation e.g. the spleen and the skin have also been tested, often in syngeneic models. In rodents, the renal subcapsular site is superior to other sites. The reason for this may be the relative absence of phagocytic/antigen presenting immune cells and less lymphatic drainage in the kidney compared to e.g. the liver or the spleen (22-25). In addition to the below described instant blood-mediated inflammatory reaction (IBMIR) observed after intraportal transplantation, there are other features that possibly makes the liver a suboptimal site. The glucose levels in portal blood are comparably higher since the portal vein is derived from small intestine capillaries. This could cause a greater stress to islets implanted into the liver before they are revascularized and lead to decreased graft survival (26). In favour of intraportal transplantation, there are some indications that angiogenesis after transplantation of dispersed islets is more efficient compared to islets transplanted in clusters (as in renal subcapsular grafts) and this is likely to be even more important in larger animals or humans when a much greater islet mass is required (27).
Despite the drawbacks with IBMIR and possibly other issues regarding the liver, the convenient and comparably safe way of intraportal infusion has made this the preferred clinical strategy. Unless new data or safer methods of using other sites (e.g. by laparoscopic techniques) are developed, it is likely that this is the route by which xenogeneic islets will be transplanted in the future.

14
Immunological barriers to clinical xenotransplantation
Cells and proteins of the blood Although xenograft rejection is viewed as an immunological process, it is tightly linked with certain physiological incompatibilities such as coagulation. To prevent damage to the transplant, it is necessary to understand the complex interactions between the different cells and proteins in the blood that are present to maintain both physiological and immunological homeostasis.
A detailed description of the blood and the immune system is beyond the scope of this book but a brief introduction to the different cellular and molecular components is given here.
Blood cells The cells in the blood are derived from bone marrow stem cells and include erythrocytes, platelets and leukocytes. In this context, the O2/CO2transporting erythrocytes are of minor importance, although they have been shown to play a role in the process of coagulation (28).
Platelets are pinched-off cytoplasmic fragments of megakaryocytes that are important for immediate prevention of blood loss after damage of a vessel. Platelets have receptors that allow them to attach to collagen and other molecules found in damaged blood vessels in a process enabled by interaction with von Willebrand factor (VWF). Once activated, platelets can release a variety of factors to attract other platelets or trigger the coagulation cascade. A platelet plug is fairly loose and is the first defence before coagulation is triggered and enforces the plug by infiltration of fibrin threads (29).
The leukocytes are the typical immune cells and are to a large extent also found outside the blood stream in various tissues. They include granulocytes, monocytes and lymphocytes. The majority of these cells are granulocytes, or the polymorphonuclear cells, which are further divided into neutrophils, basophils and eosinophils. They have the capacity of releasing a variety of inflammatory mediators and also act by phagocytosis. The monocytes are large cells that circulate the blood stream for some time before they migrate into tissue where they mature into phagocytic macrophages or antigen presenting dendritic cells. Granulocytes and monocyte derived cells are part of the innate (unspecific) immune system. As key cells in adaptive immunity, the lymphocytes are the most sophisticated immune cells with the ability to mediate specific immune mechanisms and to induce immunological memory. T lymphocytes can either be directly cytolytic (through MHC class I recognition) or act as helper cells in indirect activation

15
of effector cells (through MHC class II recognition), while B lymphocytes are responsible for production of antibodies. In addition, there exist subtypes of lymphocytes with more unspecific actions, such as the NK cells.
CytokinesCytokines are small secretory peptides with high turnover used by the immune system in cell-to-cell communication over distances. They are important both in the activation phase of an immune response, stimulating growth and differentiation of immune cells, and in the later phases as activators and directors of different effector cells. In addition, cytokines are essential for normal haematopoiesis in the bone marrow. Chemokines are a subdivision of cytokines that primarily mediate chemotaxis. The action of cytokines is rather complex since they not only have effects on multiple subtypes of immune cells but also display many overlapping properties and often function as regulators of other cytokines. Because of this redundancy, the use of single cytokine antagonists or gene knockouts often have marginal functional outcome.
Although being a somewhat simplified view, the general opinion is that there exist two major subsets of CD4+ T helper cells depending on the cytokines which they secrete. Th1 cells, which produce IL-2, IL-12 and INF-, are primarily associated with the defence against intracellular microbes
and are needed for activation of cytolytic CD8+ T cells. Th2 cells, which secrete IL-4, IL-5 and IL-10, trigger IgE and eosinophil-mediated immune reactions in response to allergens or parasites and mediate activation of the antibody producing B cells (30).
The complement system The complement system is a fast acting part of the innate immune system and constitutes a group of serum proteins that functions through cascade activation. This is an old defence mechanism, found down to very primitive life forms, with a primary function of eliminating invading pathogens. The effects of complement activation are manifold and include opsonization, release of anaphylatoxins, chemotaxis and direct cell lysis.
The central protein in the complement system is C3 that can be activated by three different pathways: the classical, mannan-binding lectin (MBL) or alternative pathways. The classical pathway is triggered following binding of C1 to the Fc domains of IgM or IgG bound to an antigen and is the most rapid way by which antibodies mediate their effect. The MBL pathway activates the classical pathway at C4 and C2 level via a complex between MBL and the MBL-associated serine proteases MASP-1 and MASP-2, homologous to C1. It is an antibody-independent reaction triggered by carbohydrates on microbial surfaces (31, 32). The alternative pathway is elicited either directly as a result of spontaneous C3 tick-over formation of soluble C3b (iC3) or by deposited C3b as a secondary event to the other two

Classical pathwayC1q binding to Ab-Ag complexes
(C1qr2s2)
C9
MBL pathway Alternative pathway
Terminal pathway
Spontaneous breakdownof C3 (tick-over)
MBL binding to oligosaccharideson virus or cell surfaces
C3b
C3a
Factor BC3bB
Alternative C3convertase
Classical C3convertase
MBLcomplex
MASP-1 & 2
MBLC1 C1active
C2
C2b
C4b
C4b2a
C4
Mg2+
C3bBb Factor D
C3a
C3 convertases
convertases(C4b2a3b & C3bBb3b)
C3
C5 C5a
C6C7C8
C5b678C5b C5b-9(MAC complex)
CELL LYSIS
C5
Ba
Schmidt, 2004
Figure 1. A schematic view of the complement activation cascade.
pathways. The alternative pathway may be considered as a complementarydefence in the absence of antibodies, since it is activated by cell walls of gram negative (through LPS) and gram-positive bacteria, certain viruses andalso other types of foreign cell surfaces such as tumour cells (33). Uponactivation of C3, the cascade will continue by the terminal pathway whichultimately leads to formation of a membrane attack complex (MAC) withability to penetrate cell membranes and cause cell lysis.
Inappropriate complement activation on autologous surfaces would bedetrimental, particularly to host cells exposed to blood such as the erythrocytes and endothelial cells lining vessel walls. A family of plasma and membrane bound proteins, the regulators of complement (RCA), has therefore evolved to control the amplification of this cascade system. The C1 inhibitor (C1INH) and C4 binding protein (C4bp) control the classicalpathway, factor H inhibits the alternative pathway and decay-acceleratingfactor (DAF, CD55), membrane co-factor protein (MCP, CD46) and complement receptor 1 (CR1, CD35) interfere with both pathways. At theend of the cascade, CD59 prevents the insertion of the MAC complex into the cell membrane. With the exception of C1INH and CD59, the RCAs either act as co-factors in the factor I-mediated inactivation of surface-boundC3b and C4b and/or possess decay accelerating properties to dissociate C3and C5 convertases.
A variety of complement inhibitory molecules, peptides or antibodies have been discovered or developed. However, none of them have to date reached beyond limited clinical testing since most of them have severe side
16

effects or are causing disruption of systemic complement function whichmakes them unsuitable for clinical use (34). The most widely used complement inhibitory agents in experimental xenotransplantation are cobravenom factor (CVF) and during the past decade soluble complementreceptor 1 (sCR1, TP10)(35). In recent studies, heparin, dextran and other polyionic molecules have also been found to have complement inhibitoryproperties useful in xenotransplantation. The advantage with these agents isthat there is considerable clinical experience since they are used also forother purposes (34, 36, 37).
The coagulation systemSimilarly to the complement system, coagulation involves cascade activation of plasma proteins. Three central events can be identified in blood clotting. First, a complex of factors called prothrombinase is formed throughtriggering of either the extrinsic or the intrinsic pathways. Secondly, theprothrombinase complex catalyzes the conversion of prothrombin into thrombin. In the third and final step, enzymatic properties of thrombintransform fibrinogen into fibrin which in turn polymerize and build a mesharound the platelets to form the blood clot. The extrinsic arm is elicited in the presence of exposed tissue factor, a molecule mainly expressed insubendothelial layers of vessels and on activated endothelium and
Kallikrein Prekallikrein
Foreign surfacesHMW
kininogen
Intrinsic pathway
Polymerization
Damaged tissueActivated monocytesActivated endotheliumFXIIaFXII
Prothrombinase
BLOOD CLOT
FX
FIXaFIX
FXIaFXI
FVIIFVIIa
FXa
Tissue factor (FIII, Thromboplastin)
Extrinsic pathway
ThrombinProthrombin
Fibrinogen FibrinFibrin
FibrinFibrin
FVaPhospholipids
Ca2+Ca2+ Ca2+
Ca2+
Ca2+
FV
Plasmin
Fibrinolysis
(FIV)
(FI)
(FII)
Schmidt, 2004
(FIIa)
FIIa
FVIII FVIIIaFIIa
Kallikrein Prekallikrein
Foreign surfacesHMW
kininogen
Intrinsic pathway
Polymerization
Damaged tissueActivated monocytesActivated endotheliumFXIIaFXII
Prothrombinase
BLOOD CLOT
FX
FIXaFIX FIXaFIX
FXIaFXI FXIaFXI
FVIIFVIIa
FXa
Tissue factor (FIII, Thromboplastin)
Extrinsic pathway
ThrombinProthrombin
Fibrinogen FibrinFibrin
FibrinFibrin
FibrinFibrin
FibrinFibrin
FibrinFibrin
FVaPhospholipids
Ca2+Ca2+ Ca2+
Ca2+
Ca2+
FV
Plasmin
Fibrinolysis
(FIV)
(FI)
(FII)
Schmidt, 2004
(FIIa)
FIIaFIIa
FVIII FVIIIaFIIa
FVIII FVIIIaFIIaFIIa
Figure 2. A simplified schematic description of the coagulation cascade.
17

monocytes. The intrinsic arm is a comparably slower mechanism and is initiated on foreign surfaces. During the repairing of a damaged vessel,fibrinolysis will ultimately dissolve the clot by degradation of fibrin. This is achieved by entrapment in the blood clot of plasminogen which is later converted to active plasmin following secretion of tissue plasminogenactivator (tPA) from the vascular endothelium (38, 39).
To counteract unwanted blood clotting, a variety of parallel anticoagulantstrategies exists. In addition to the smoothness of the endothelium, which in it self prevents contact activation, the inner vascular surface is covered withan endothelial glycocalyx which is a platelet and coagulation factor repelling layer composed of e.g. glycoproteins, glycolipids and proteoglycans. Some of these molecules also have direct anticoagulant properties such asthrombomodulin and tissue factor pathway inhibitor (TFPI) (40, 41).Excessive thrombin is efficiently removed from the blood by adsorption tothe fibrin threads as they develop during clot formation as well as bycombination with circulating or glycocalyx bound antithrombin III. Also, the fibrinolysis forms an important mean as to dissolve harmful clots, especiallyin the microcirculation (38).
Rejection of xenografts
Allotransplantation Xenotransplantation
Cellular rejection
IBMIRHAR
DXR/AVR
IsletsAllo + Xeno
Allotransplantation Xenotransplantation
Cellular rejection
IBMIRHAR
DXR/AVR
IsletsAllo + Xeno
Figure 3. An overview of the different immunological responses triggered aftertransplantation depending on the type of graft. Allografts are normally destroyedthrough cellular rejection while this process in solid organ xenografts is preceded byhyperacute rejection (HAR) and acute vascular rejection (AVR). Pig islets are notvascularized at the time of transplantation and therefore escape HAR and AVR. However, both allogeneic and xenogeneic islets appear to elicit a harmful instantblood-mediated inflammatory response (IBMIR) after intraportal infusion.
18

19
Rejection of vascularized xenografts
Hyperacute rejection Typically, the first immunological hurdle to overcome in pig-to-human solid organ xenotransplantation (and in old world monkey animal models) is the presence of preformed xenoreactive natural antibodies (XNAs). Most of these antibodies are directed against Gal (1,3)Gal sugar residues on the pig cells. These epitopes are formed through enzymatic action of
(1,3)galactosyltransferase which is expressed in all mammals except for humans, apes and old world monkeys. These primates instead use
(1,2)fucosyltransferase to form substance H from which the A and B blood group antigens are constructed (42). Thus, Gal (1,3)Gal can be considered as a blood group antigen foreign to all humans. After reperfusion of a vascularized pig organ these XNAs, that can constitute as much as 4 % of the total circulating IgM, are deposited on the endothelial cells (ECs) of the vessel walls (43). Within minutes to hours this leads to classical pathway activation of the complement cascade and the following damage of the endothelium leads to disruption of the microcirculation with interstitial haemorrhage and edema. Platelet consumption and formation of microthrombi produce ischemia and the graft is lost in what is called a hyperacute rejection (HAR) (44, 45). Importantly, expression of RCAs on the porcine endothelium have only minor protecting effects against complement since the action of these molecules are largely species restricted (46-48).
Traditionally, HAR have been prevented by plasmapheresis to remove circulating antibodies and/or by systemic inhibition of complement using e.g. sCR1 or CVF (49). During the past decade, similar (but more graft specific) effects have been obtained using pigs with transgenic expression of human RCAs e.g. DAF, CD59 and MCP (50-52). More importantly, preliminary results from experiments with the (1,3)galactosyltransferase knockout pigs, that lack expression of the Gal (1,3)Gal epitopes, has given hope as to finally eliminate HAR as an obstacle in xenotransplantation (20, 21).
Acute vascular rejectionRemoving the preformed XNAs or inhibiting complement activation prior to transplantation will only prolong graft survival for a couple of days to weeks until another type of immunological response is triggered. This reaction, described as an acute vascular rejection (AVR) or in some models delayed xenograft rejection (DXR), is somewhat more complex and in many aspects qualitatively different from HAR (53, 54). For example, AVR is sometimes observed in concordant animal models where HAR normally does not occur (45). Common features of AVR pathology include T cell independent

20
leukocyte infiltration, dominated by macrophages and to a lesser extent NK cells and neutrophils (55), edema and ischemia induced by microvascular thrombi following fibrin formation rather than platelet consumption (45, 56).
Activation of graft endothelium appears to be central in AVR. Unlike the rapid EC activation that is observed in HAR, which mainly involves cell separation and extravasation of fluids and blood cells, in AVR it is characterized by gradual loss of protective molecules (e.g. heparan sulphate, thrombomodulin, TFPI and CD39) and upregulation of proinflammatory adhesion molecules (e.g. E- and P-selectin, ICAM-1 and VCAM-1), cytokines/chemokines (e.g. IL-1, IL-6, IL-8, TNF- and MCP-1) and prothrombotic molecules (e.g. tissue factor) (40, 49). The mechanisms leading to this proinflammatory and procoagulant state of the endothelium have been subject to much speculation and are likely to involve several more or less interacting processes (45, 56). Although it has been shown that AVR can proceed in the absence of antibodies (57), deposition of reappearing XNAs is frequently seen in grafts lost from AVR possibly causing indirect (through complement) or direct EC activation (58). FcR-mediated leukocyte activation and antibody-dependent cell-mediated cytotoxicity (ADCC) involving NK cells have also been implicated in AVR (59-61). Complement activation is generally regarded as to be of substantially less importance than in HAR, but some studies have suggested that low grade systemic complement activation in response to antibody deposition may still be sufficient to induce EC activation and trigger AVR (62).
Another way to look at AVR is the mere fact that many proteins, that in the human body or after allotransplantation interact according to given rules, only partially or not at all are able to interact properly in the xenotransplantation setting. Examples of such porcine proteins that function poorly in humans are TFPI and the RCAs (46, 47, 49, 63). This concept of molecular incompatibility may prove to be the greatest challenge in avoiding AVR since this would require targeting of a great number of protein interactions (57).
Cellular rejection of solid organs The mechanisms of cell-mediated pig xenograft rejection have been difficult to study in both non-human primate and rodent models, since the strength and immediacy of HAR and AVR in these discordant combinations have rarely allowed prolonged xenograft survival.
It was initially predicted that the molecular incompatibilities in xenotransplantation would render the cellular mechanisms less functional and thus weaker compared to the allosituation. Disappointingly, the cellular response was instead found to be even stronger and much more difficult to inhibit using conventional immunosuppression. First, there exists a direct response by human T cells against porcine cells (both CD4+/SLA class II and CD8+/SLA class I) implying that many of the molecular interactions

21
involved in T cell function remain intact in this species combination. In addition, it has been shown that there is an indirect T cell mediated response of a much greater magnitude than in allotransplantation, probably reflecting a significant amount of circulating CD4+ T cells able to respond to the large number of peptide xenoantigens processed by human APC and presented on HLA class II (4, 64, 65). Also NK cells may be of greater importance in xenotransplantation, since it appears that certain human inhibitory receptors on these cells are unable to interact with SLA class I (65).
It is likely that the acute cellular response is essentially the same irrespective of what tissue is selected for transplantation. Rejection models involving non-vascularized transplants such as islets (which for reasons discussed below escape HAR and AVR) will therefore be useful also for understanding acute cellular rejection of solid organs.
Islet xenograft rejection The rejection of islets differs, at least in the acute phases, from that of vascularized transplants. In contrast to organs, islets do not have any blood supply at the time of transplantation. Instead, the islet graft induces revascularization of recipient origin in a process that is morphologically completed within 1-2 weeks (27). As a consequence, islet grafts have the advantage of escaping typical HAR and AVR, even after revascularization (66). However, since the preferred way of transplanting islets is by means of intraportal infusion, an islet xenograft will still be in direct contact with the recipient blood during the immediate period following transplantation which may elicit other harmful reactions.
IBMIRIn contrast to allotransplantation of a vascular pancreas the use of isolated islets normally requires multiple donors to achieve insulin independence. The reason for this does not appear to depend on low islet yield after isolation but rather on the fact that a large portion of the islets are destroyed during the immediate time period following transplantation. This observation will most likely apply also to intraportally transplanted porcine islets.
In 1999, a possible mechanism for this was proposed by Bennet et al. who demonstrated that both allogeneic and xenogeneic pig islets trigger coagulation and complement activation when exposed to fresh human blood in vitro in what was described as an instant blood-mediated inflammatory reaction (IBMIR) (67). It was recently discovered that human islets express tissue factor, which may explain why coagulation is triggered in the allosetting (68). To what extent this latter finding is true also for porcine islets is unclear, but it appears that complement activation is of relatively greater importance for porcine islets. In a study where cynomolgus monkeys received API intraportally, it was shown that the islets were acutely damaged

22
by an inflammatory reaction and that pre-treatment of the recipient with sCR1 had a protective effect (69). As mentioned previously, an important difference between fetal and adult porcine islets is that while ICC express Gal (1,3)Gal sugar residues, API are largely devoid of this epitope. In accordance with this finding, single cell preparations of fetal islets, but not adult islets, were shown to bind human Ig after incubation in fresh human serum (18). Despite this difference, single cell preparations from both types of islets are still equally susceptible to lysis in fresh human serum and removal of anti-Gal (1,3)Gal antibodies only reduces lysis of fetal islet cells (18). Therefore, it appears that while cell lysis of ICC is mediated by complement activation via the classical pathway, API are susceptible to complement-mediated cell destruction mainly through antibody-independent pathways. Both CD59 and MCP are expressed on pig islet cells but since the function of these RCAs to a high degree appears to be species restricted, this may provide an explanation why API trigger antibody independent complement activation in contact with human blood (46-48, 70).
Cellular rejection Still, if the islet xenograft escapes this acute damage due to IBMIR it will be subject to acute cellular xenograft rejection. This process has been widely investigated using pig-to-rodent models where API or ICC are implanted under the kidney capsule of mice or rats. Typically, the rejection of the islets takes place during the first 10 days after transplantation and is characterized by a massive infiltration of macrophages with T cells seen accumulating in the periphery of the graft. In contrast to rodent islet allograft rejection, which requires both CD4+ and CD8+ T cells, pig-to-rodent ICC xenograft rejection appears to be predominantly a CD4+ T cell dependent process (71, 72).Furthermore, experiments with Ig-deficient mice has shown that antibodies are not vital for inducing fetal porcine ICC rejection (although this process may be facilitated by XNAs) (73) and eosinophils and NK cells also seem to be of minor importance (74, 75).
The morphological pattern of the infiltrating cells resembles that of a delayed type hypersensitivity reaction (DTH), which is the prototype of a Th1-associated response, and recent studies has proposed a major role for Th1-associated cells at the initial stages of rejection followed by a Th2-associated response in the later phases (76). Although some drug combinations have been found to prolong xenograft survival, many agents that are used in the clinic to prevent allograft rejection have been found to be insufficient for protecting islet xenografts in pig-to-rodent models. This indicates that xenograft rejection may not only be a stronger reaction, but also differ in the underlying mechanisms (77, 78).
In a study, where ICC were transplanted under the kidney capsule of cynomolgus monkeys, a clinically more relevant model, it was demonstrated that the cellular infiltrate differs from that observed in rodents (79). Instead

of being dominated by macrophages (as observed in the rodent models) the infiltrate in the ICC grafts of the monkeys constituted mainly of CD8+ T cells. This suggests that a T cell mediated cytotoxicity of a similar or ahigher degree compared to that of an allogeneic response would be presentalso in humans. Interestingly, when monkeys in the same study were on an immunosuppressive protocol (Cyclosporine A and deoxyspergualin), the CD8+ T cell infiltration was markedly reduced whereas the infiltration of macrophages persisted. The immunohistochemical evaluations of these grafts better correlated with the observations made in the pig-to-rodent models. It may be speculated that the cellular rejection of an ICC xenograft in both rodents and primates involve a T cell dependent infiltration of activated macrophages reflecting indirect recognition, but that in immunocompetent primates this is overshadowed by a qualitatively differentcellular mechanism, constituting of xenoreactive CTLs directly recognizingSLA class I molecules.
Xeno(Primate)
Xeno(Rodent)
DirectCTL
IndirectDTH-like
Allo Xeno(Primate)
Xeno(Rodent)
DirectCTL
IndirectDTH-like
Allo
Figure 4. While allograft rejection is mediated primarily by cytolytic T cellsthrough direct MHC-recognition, pig-to-rodent xenograft rejection has a DTH-likeimmunopathological pattern reflecting indirect MHC-recognition. Pig-to-primatexenograft rejection appears to be a mix between these two mechanisms.
Accommodation and general toleranceInducing tolerance to foreign tissue as a mean to avoid the medical complications of immunosuppressive protocols has long been the ultimate goal in the field of transplantation. This is particularly evident in the case of xenotransplantation considering the strong immunological response that would need to be repressed. Acute heart or liver failure and possibly someother disabling disorders including kidney failure and highly unstable
23

24
IDDM, may motivate heavy immunosuppression. However, for diseases where there is an alternative therapy it will not be acceptable. The vast majority of IDDM patients normally do not have acute problems in controlling blood glucose levels by exogenous administration of insulin. For these people, the risks with a life-long immunosuppressive treatment exceed the benefit of an islet xenotransplant. Therefore, this therapy can not be considered unless a tolerogenic, or at least a considerably milder, immunosuppressive protocol can be introduced.
Under rare conditions, a discordant vascularized xenograft can escape HAR and even AVR. This phenomenon, termed accommodation, was first observed in kidney allografts transplanted across the AB0 barrier, and is typically observed when antidonor antibodies have been removed prior to transplantation. If a second xenograft is transplanted after the reappearance of the depleted antibodies it will be rejected while the accommodated graft remains resistant to the humoral response. This has been shown to be due to a change in antigen presentation and acquired resistance of the graft endothelium (80-82).
However, such an accommodated graft will still be promptly destroyed by means of cellular rejection and in order to avoid this immune response, additional measures will have to be taken. In fact, the pig-to-man xenograft rejection is so much stronger than rejection in the allogeneic situation that it may prove essential to induce immunological unresponsiveness to at least some of the most important antigenic molecules. One such approach, which has also been evaluated in allotransplantation, is to induce mixed chimerism. Here, the aim is to transfer donor bone marrow stem cells to the recipient to maintain tolerance in the reconstituted patient. The level of chimerism does not necessarily have to be high, but the outcome appears to be dependent on the presence of donor dendritic cells in the recipient thymus. Another strategy, with similar intention of eliminating anti-pig reactive T cells, has been to co-transplant part of the thymus from the pig donor (4, 83). One advantage in using animal donors is that it will be possible to use genetically identical tissue for tolerance induction therapies before an intended graft is transplanted.
The risk of zoonosis in xenotransplantation As is the case also in allotransplantation, grafts from animals may include various infectious agents. Although this risk is considered to be smaller with pigs than with primates (84), bacterial and viral contamination, such as the porcine cytomegalovirus can pose a threat to the transplanted patient (85).Although most known infectious agents found in pigs can probably be eliminated, porcine endogenous retroviruses (PERV) represent a unique concern.

25
Porcine endogenous retroviruses Like in all animals, the porcine genome contains many loci coding for endogenous retroviruses (ERV). These viruses are by definition inherited and may for that reason be particularly difficult to remove. In humans none of these ERV (=HERV) have been shown to be replication-competent but in the case of pigs it has been known for several decades that PERV particles are released from a variety of pig cell-lines (86, 87). Ever since it was recognized by Patience and co-workers in 1997 that PERV could also be transmitted to human cells in vitro there has been a vivid debate regarding the safety of clinical xenotransplantation (88). It is recognized that some retroviruses, which cause harmless infections in their natural host, can lead to severe disease when transmitted to other species (84). The main concern has been that with extensive use of pig tissue in transplantation, uncontrollable viral infections may be created with a risk of jeopardizing the health of not only the patients but in the worst case the whole non-transplanted population. As a result, clinical trials have been strictly regulated in most countries and health authorities have favoured a precautionary approach awaiting further research.
PERV release and in vitro transmission Three classes of replication-competent PERV, gammaretroviruses PERV A, B and C, have been identified in the porcine genome (89, 90). Of these mainly PERV A and B seem to have tropism for human cells (90).Replication-competent PERV were first identified from immortalized pig cell lines. Since then, functional PERV have been isolated from a variety of primary cell cultures including endothelial cells and PBMC and found to be able to infect different human cells (88, 91-94). The majority of these in vitro PERV transmission studies were conducted on immortalized human cell lines and, although such reports exist (94, 95), it appears that infecting primary cell cultures are more difficult to achieve.
An analysis of PBMC taken from a set of pigs from different breeds indicates that the release of PERV particles varies, not only between breeds, but also between individuals within the same breed (96). In addition, PERV production may depend on the tissue selected for transplantation (97).Interestingly, there is a recent report of a strain of miniature swine that consistently does not transmit PERV to human cells in vitro (98). Depending on the type of pig cells to be engrafted, different properties of the released PERV may also be expected. In humans, an important way of inactivating retroviruses from non-primate mammals are through preformed antibodies directed against Gal (1,3)Gal sugars on the virus envelope (Fig. 5) (99). As mentioned previously, the endocrine cells of API do not express this epitope and as a consequence any PERV released from such cells would presumably escape this defence mechanism (19).

Figure 5. An important way to eliminateretroviruses is through antibody-dependent complement neutralization.The Gal (1,3)Gal antigen is normallyexpressed on PERV produced in a pig butwill be absent on virus particles buddingfrom human cells and possibly also frompig islets.
While it is recognized that the titre of PERV produced in pig cells is generally rather low compared to many other retroviruses (100), it is well known that the expression of
many retroviruses can be induced by different chemical and biologicalagents such as cytokines and steroid hormones (101, 102). It is thereforelikely that in a transplantation situation the PERV production is influencedby the immunological response in the patient and possibly also directly bythe immunosuppressive agents.
Gal(1,3)Gal
PERVGal(1,3)Gal
PERV
Infection in humans?The pig is a domesticated species that has been living close to humans forseveral thousands of years as a source of food. It might therefore be argued that if PERV transmission to humans is more than a theoretical possibility, it would already have taken place. Although this proves that transmission does not readily occur, clinical xenotransplantation represent a new setting where several of the natural immunological defences against retroviruses areovercome. In a transplantation situation there are no mechanical barriers to infection by microbes, i.e. the skin and mucosal layers in the gastrointestinaltract and the lungs. Further, the various protocols needed to suppress theimmune system to protect the xenograft, will also hamper the cellular and humoral immune defence against PERV. Similarly, as discussed later,genetic manipulation of the donor tissue in order to moderate the rejection process may further increase the risk of PERV particles escaping the immune system.
If ultimately PERV is transmitted to adjacent human cells, the production of virus particles will possibly be altered. Upon serial passage in human celllines, significant increases in viral titre and also production of PERV with higher tropism for human cells in vitro have been demonstrated (93, 94). Inaddition, such viruses are adapted to escape some of the natural immunological barriers against retroviruses (88). In the worst case scenario aprimary infection could lead to an increased titre of virolysis resistant PERV
26

with high tropism for human cells, resulting in an escalating systemicinfection in the patient.
Notably, before PERV was ever considered a risk factor, many patientshad already been exposed to pig tissue, mainly in trials evaluating the effectof different cell therapies. Since then, much effort have been made in developing reliable diagnostic tools able to detect the known human tropic PERV subtypes, but also to discriminate between an actual PERV infection and merely remaining pig cells (microchimerism) in the recipient. Using such techniques, several retrospective studies have been undertakeninvestigating the possible virus transmission to such patients, but so far none of them have provided evidence for any PERV infection (103-108). This isindeed reassuring data, but one has to bear in mind that in most cases the pig cells in these patients survived only for a short period of time,immunosuppression was relatively mild and did not include any systemiccomplement inhibition, and in no case were the patients treated with graftsderived from genetically modified pigs.
PERV pathogenicity The ultimate question concerning the potential pathogenicity of PERV is whether transmission to human cells would pose a real threat to the health of the graft recipient or even the general public. Although PERV has not been shown to be pathogenic in pigs it is at this stage very difficult to estimate their potential effects in humans. The only qualified prediction would be that their mere ability to infect human cells could lead to oncogenicity, especiallyin heavily immunosuppressed patients.
However, the risk-benefit estimate will be in favour of xenotransplantation from the point of view of a patient with an end-stageorgan disease or for that matter a disabling form of diabetes. Highly sensitive methods for detection of replication-competent PERV are available thatcould be used to carefully monitor xenotransplant recipients. The main issueto be addressed is whether PERV poses a potential threat to the non-immunosuppressed population, and in that perspective the risk of PERV transmission will be considerably lower.
Animal models of xenotransplantation
Models of xenograft rejection Depending on the species combination, xenotransplantation is said to be either concordant or discordant reflecting the grade of immunologicalincompatibility (109). This terminology mainly applies to vascularizedxenografts. Generally, the more phylogenetically distant the species are, the more likely the combination is discordant. Discordant models of
27

28
xenotransplantation (e.g. pig-human/rat/mouse) are characterized by rapid rejection through typical HAR. In some discordant models (e.g. guinea pig-rat) HAR is elicited through alternative pathway complement activation, thus in the absence of preformed antibodies. In concordant models (e.g. old world monkey-human and mouse-rat), rejection is slower and primarily involves cellular mechanisms.
Since rodent transplantation models offer a cheap and convenient mean of performing experiments at a large scale, the majority of the data regarding porcine islet xenograft rejection comes from studies using this species combination. Moreover, many basic immunological experiments can be performed in rodents due to the vast availability of inbreed MHC-compatible (syngeneic) or genetically modified strains of these species. It is however important to bear in mind that these results can not without caution be related to the clinical situation. For instance, the Gal (1,3)Gal barrier does not exist between pigs and rodents and the cellular rejection appears to be independent of direct antigen recognition due to great differences in MHC recognition and cytokine specificity between the species. A clinically more relevant recipient species is an ape or an old world monkey since they are closely related to humans. Such experiments, however, raise substantially more ethical concerns, are costly and from a practical point of view considerably more difficult to perform.
PERV in animal models One matter complicating the study of in vivo transmission of PERV is that the virus is likely to have different tropisms depending on the species, making results from animal experiments difficult to translate to the clinical situation. PERV transmission into non-human primates, arguably the most relevant species, has only been reported from in vitro studies (110) but the invivo data from the studies published to date suffer from many of the same limitations as the retrospective studies involving human subjects (111-113).
With respect to the potential large-scale rodent and other small animal models, their relevance to clinical xenotransplantation remains controversial. Unlike humans and other old world primates, rodents and other mammals express Gal (1,3)Gal sugars and therefore lack natural antibodies directed against PERV expressing this epitope. PERV receptors have been demonstrated in both rat and mouse cell lines (90). Recent papers reported invivo PERV transmission to mouse and also implanted human cells following transplantation of porcine islets to athymic (nu/nu) or SCID mice (114-116).The possibility of the murine endogenous retrovirus influencing PERV infectivity in these animals as well as the fact that these mice are incapable of mounting any cellular or humoral response make these data difficult to relate to the pig-to-human situation. In addition, several of the commonly used mouse strains are known to have defective complement systems. Taken

29
together, these circumstances propose that mice, immune deficient or not, may be more susceptible than humans to PERV infection.
All attempts so far to establish productive PERV infection to rats in vivohave failed and it appears that this species is not suitable for viral transmission studies (94, 117). However, rat models may still be of importance when investigating the in vivo induction of intragraft PERV expression during inflammation, rejection, under the influence of different immunosuppressive agents or as a model to screen for antiviral drugs. For these purposes it is in fact an advantage that PERV transmission to infiltrating and adjacent rodent cells does not occur, since this would confound the experimental data obtained from analysis of the transplanted xenograft.
Genetically modified donors Since HAR has been the primary immunological hurdle to successful pig-to-human xenotransplantation, substantial effort has been made in developing genetically modified pigs that will not trigger complement activation. The primary target during the past decade has been to develop transgenic pigs expressing on their endothelium high levels of human RCAs, including DAF, MCP and CD59 (50-52). Recently, the reported cloning of
(1,3)galactosyltransferase-knockout pigs lacking the expression of Gal (1,3)Gal sugar residues has given new hope as to finally eliminating HAR completely (21). Although the need for preventing HAR will be of less importance in the case of islets, for reasons that were discussed previously, complement regulatory properties and a lack of Gal (1,3)Gal expression will most likely be an advantage also for non-vascularized xenografts.
PERV and genetically modified animals As a mean to prevent graft rejection, both transgenic and gene knockout pigs have been made available during the past decade. Unfortunately, such measures taken will most probably eliminate some of the natural immunological barriers against retroviruses (99). When budding from host cell plasma membranes, the PERV particles incorporate part of the cell membrane including membrane-associated proteins and Gal (1,3)Gal-positive glycoproteins. As a result PERV particles deriving from transgenic pigs expressing RCAs will have an innate defence against complement-mediated lysis. In parallel, viruses deriving from pigs lacking the Gal (1,3)Gal epitope will not be targets for the preformed Gal (1,3)Gal reactive natural antibodies present in the human blood. To what extent such modifications are enough to create more infectious PERV is uncertain. In a recent study where human CD59 was incorporated into PERV it was demonstrated that while complement-mediated lysis of these particles was

30
indeed reduced, the same viruses where incapable of infecting human cells after incubation with human serum (118). Similar experiments on PERV isolated from the (1,3)galactosyltransferase knockout pigs have not yet been published.
In other words the PERV produced in such genetically modified pigs would share many of the features with those that are produced in a human cell, and will theoretically have a much higher viability in a human recipient. It will be necessary that every created genetically modified pig strain be evaluated independently, since the outcome of combined genetic modifications with regard to PERV infectivity will be very difficult to predict.

31
AIM OF THE STUDIES
General aims To achieve the ultimate goal of bringing islet xenotransplantation into the clinic, the underlying mechanisms of IBMIR induced by porcine tissue and the subsequent cellular xenograft rejection need to be further characterized. As a tool to gain this knowledge, immunosuppressive agents and protective islet manipulations can be applied in established in vitro and in vivo models and will also be necessary in order to develop effective and safe protocols to prevent the different components of porcine islet xenograft rejection. To ensure that PERV does not represent an unacceptable safety concern in clinical xenotransplantation, further studies of this endogenous retrovirus are essential.
Specific aims
Paper I: As a mean to protect API from complement-mediated damage in human blood the aim of this study was to examine the possibility of inducing a transgene expression of human DAF or CD59 using adenoviral vectors and to study their functional effects after exposure of transduced islet cells to fresh human sera.
Paper II: The aim of this experiment was to study pig-to-mouse ICC xenograft rejection in the absence of the unspecific inflammatory response typically seen after the transplantation due to surgical trauma. The model design was used to clarify issues regarding the recruitment of immune cells to the site of the graft and the mechanisms behind the subsequent specific rejection.

32
Paper III: By using a strain of MyD88-/- knockout mice, the aim was to evaluate whether profound defects in Th1 immunity and disruption of an important way of communication between cells in innate and adaptive immunity would influence the outcome of pig-to-mouse xenotransplantation.
Paper IV: The aim of this study was to examine the dynamics of PERV expression in ICC after transplantation to normal or athymic (nu/nu) rats and to further investigate and compare the expression of PERV in fetal (ICC) and adult (API) islets in culture.

33
RESEARCH DESIGN AND METHODS
A brief description on material and methods used in the different papers is presented here. Detailed information is given in paper I-IV, respectively.
EthicsAll animal experiments were approved by the Research Ethics Committee of Uppsala University and performed in accordance with local institutional and Swedish national rules and regulations. Human islets were isolated after appropriate consent for multiorgan donation.
Preparation and culture of islets
Human islets (Paper I) Human islets were isolated according to a modified Ricordi method, followed by purification on a continuous density gradient (119, 120). Cold ischemia time was 8-10 hours. The islet preparations were of good quality but were available for experimental use since the total islet yield was to low for clinical transplantation. The islet preparations were placed in untreated culture flasks and kept at 37°C for 6 days.
Adult porcine islets (API; Paper I and IV)Islets were isolated from the pancreata of adult Landrace pigs according to a modified Ricordi protocol (119). Cold ischemia time was approximately 2 hours. The API were cultured in flasks at 37°C for 2 days until used.
Fetal porcine islet-like cell-clusters (ICC; Paper II-IV) Pregnant sows from a local stock were killed at 70 5 days of gestation. The foetuses were kept on ice during transport to the laboratory. After aseptic removal, the pancreatic glands were minced into 1-2 mm
3fragments in cold
Hanks' solution and then treated with collagenase during vigorous shaking according to a protocol established in the department (121, 122). The digested tissue was washed and explanted into culture dishes kept at 37 C.On day 4 of culture, all free-floating fragments (diameter <0.7 mm) were

34
considered to be ICC, and were harvested without any further purification step.
Rodent islets (Paper II)Male inbred C57BL/6J and C57BL/KsJ mice served as donors for syngeneic and allogeneic implantation, respectively. Male Sprague-Dawley rats were used to obtain islets for concordant xenogeneic transplantation. Pancreatic islets from rodents were prepared by a collagenase digestion method. Groups of approximately 150 mouse or rat islets were cultured at 37 C free-floating for 1-2 days or 4-5 days, respectively.
Adenoviral vectors and transduction procedures (Paper I)
Adenoviral vectors The three different virus vectors used were replication-defective E1 and E3 deleted adenoviral (Ad) serotype 5 vectors. The hDAF cDNA was under the transcriptional control of the human elongation factor 1- promoter (123).The adenoviral vector Ad.hCD59 coding for human CD59 contains the hCD59 cDNA under the control of the Rous sarcoma virus promoter (124).The adenoviral vector Ad.mB7.1 contains murine B7.1 cDNA under the control of the murine cytomegalovirus promoter (125). All Ad vectors were produced in 293 cells providing the E1 and E3 gene. Vector particles were purified on CsCl gradients and titres were established by a standard plaque forming unit assay using 293 cells in agar culture.
Transduction procedures At the time of transduction the API were sedimented and then washed in serum-free culture medium. The islets from each isolation were divided into four groups and were subsequently transduced with either Ad.hDAF, Ad.CD59 or control m.B7.1 or left untreated. The islets were then cultured for an additional three days before experiments were continued. Along the experiment, control islets not exposed to adenoviral vector were treated in the same way as transduced islets.

35
Flow cytometry analysis of protein expression (Paper I)
Preparation of single cell suspensions Single-cell suspensions from the API were prepared on day 3 after transduction using a modification of the method described by Kohnert and Hehmke (126). The islets were suspended in trypsin and then incubated and agitated for 2-3 min at 35-37°C. The suspension was gently aspirated and flushed using a pipette to enhance single-cell dispersion. Cell aggregates were then allowed to settle and the supernatant containing suspended single cells was collected. Remaining cell aggregates were treated with trypsin once or twice again as described above. The number and viability of the cells were measured by trypan blue staining. Islet cell viability after trypsin digestion was >90% for API single-cell suspensions. Human islet cells were prepared in the same way six days after isolation.
Expression analysis Porcine islet cell surface expression of hDAF, hCD59 and mB7.1 was detected by flow cytometry analysis. 105 single cells were incubated with antibody and subsequently washed and suspended in PBS with 0.5% BSA. Human CD59 was detected using a mouse anti-human CD59 monoclonal antibody, and after washing, the cells were then incubated with secondary phycoerythrin labelled rabbit anti-mouse antibody before analysis. Human DAF was detected using a fluorescein isothiocyanate conjugated mouse anti-human DAF monoclonal antibody and the control protein mB7.1 was detected by a fluorescein isothiocyanate conjugated hamster anti-mouse CD80 monoclonal antibody. The hDAF and hCD59 antibodies were tested negative for cross-reactivity with porcine DAF and CD59. Non-transduced islet cells were used as a reference to estimate the number of positive cells in each of the three different transduction experiments.
Human serum cytotoxicity assay (Paper I)105 dissociated islet cells suspended in PBS with 0.5% BSA were incubated with newly thawed human complement active AB-serum to a dilution of 1/3 for 30 minutes at 37 C. Control cells were incubated in the same way with the corresponding heat-inactivated serum (preincubated at 56 C for 30 minutes). The proportion of viable cells was detected by flow cytometry analysis using propidium iodide (1 µg/105 cells) added immediately prior to analysis to define lytic cells. The percentage of cytotoxicity was calculated as (1-[% living cells after incubation with complement active serum/ %

36
living cells after incubation with the corresponding heat inactivated serum])x100.
All results from the human serum cytotoxicity assay are expressed as mean SEM. Mean values were compared using Friedman´s one way analysis of variance (ANOVA) with significance set at =0.05 (Fig. 8).
Animals and transplantation procedures
Paper II
AnimalsTo generate transfer donors, ICC transplantation was performed in: 1) Male or female inbred B6 mice (H-2b; Tables 1-3); 2) Male homozygous mutant B6 mice with a targeted disruption of the membrane exon of the Ig µ-chain gene (H-2b; Table 1); 3) Male inbred BALB/c mice (H-2d; Table 3) and 4) Male outbred NMRI mice (H-2q; Table 3). Transfer experiments were performed in: 1) Male inbred athymic B6 (nu/nu) mice (H-2b; Tables 1-3); 2) Male inbred athymic BALB/c (nu/nu) mice (H-2d; Table 3) and 3) Male outbred athymic NMRI (nu/nu) mice (H-2q; Table 3).
Transplantation procedures A summary of the different transfer experiments is given in figure 9 and tables 1-3. Fetal porcine ICC were implanted, using a braking pipette, through an incision in the left renal capsule of avertin-anaesthetized animals. One week to more than one year prior to transfer, recipient athymic (nu/nu)mice were either implanted with 3 µl fetal porcine ICC alone (Tables 1 and 3), or together with an additional graft composed of 150 B6 islets, BKs islets or rat islets (Table 2). Animals used for intragraft mRNA analysis received two 3 µl ICC grafts (Table 1).
Immunocompetent donor mice, used for generating grafts to be transferred, received two 3 µl ICC grafts and were killed after six days. The grafts, at this stage infiltrated with immune cells, were excised and then either left untreated, subjected to two cycles of freeze-thawing, or irradiated (15-Gy) before transfer into the peritoneal cavity of the recipient mice.
In some transfer experiments, the graft-bearing kidney of the recipient athymic (nu/nu) mice was removed and the graft prepared for immunohistological evaluation. Five weeks later, the same mice were re-transplanted with 3 µl fetal porcine ICC under the capsule of the remaining kidney. After another six days the animals were killed, and the grafts removed and prepared for evaluation (Table 1).
Some of the recipient athymic B6 (nu/nu) mice implanted with ICC grafts from MHC-mismatched donors (Table 3) were NK1.1+ cell depleted by

37
means of repeated intraperitoneal injections of an anti-NK1.1 monoclonal antibody prior to transfer until the end of experiments (75, 127, 128). To generate immune sera, male inbred B6 mice were injected intraperitoneally with ICC on repeated occasions. Intraperitoneal injections of the serum to the recipient athymic (nu/nu) mice (Table 1) were either given in daily portions or as a single injection at the time of transplantation.
Paper III Male homozygous mutant B6 mice with a targeted disruption of the MyD88 gene were studied. Normal male inbred B6 mice were used as controls. All mice were avertin-anaesthetized and transplanted with one 2 µl ICC graft under the kidney capsule. 3, 6 or 12 days after transplantation the animals were killed and the graft prepared for either immunohistochemistry or mRNA analysis.
Paper IV Normal or athymic (nu/nu) male Lewis rats, anaesthetized with pentobarbital were transplanted with two grafts composed of 2 µl ICC. 1, 3, 5 or 12 days after transplantation the animals were killed and the grafts prepared for immunohistochemistry or mRNA analysis.
Immunohistochemistry
Paper I Intact porcine islets were pelleted by centrifugation, snap-frozen in pre-cooled isopentane and subsequently stored at 70 C. Serial sections, 6 µm thick, were incubated with mouse monoclonal antibodies to human CD59 or human DAF. This was followed by incubation with rabbit anti-mouse Ig antibody. After a final incubation with a monoclonal mouse PAP reagent the alkaline phosphatase reaction was developed using AEC Substrate Chromogen. For detection of murine B7.1 sections were incubated with a hamster monoclonal antibody to murine B7.1 followed by incubation with a goat anti-armenian hamster Ig antibody and subsequently a peroxidase conjugated donkey anti-goat Ig.
Paper II and III Animals were killed at different time points after transplantation/transfer. The grafts were excised with a margin of approximately 3 mm and stored at -70 C. Incubations of 6 µm thick serial sections were made with rat anti-mouse monoclonal antibodies to F4/80, Mac-1, MHC class II, CD3, CD4 or CD8, followed by rabbit anti-rat IgG antibody. After a final incubation with a monoclonal rat APAAP reagent the alkaline phosphatase reaction was

38
developed using BCIP/NBT/INT. The frequency of the different cell phenotypes infiltrating the xenograft and ICC survival was assessed semi-quantitatively.
Paper IV At various time points after transplantation, the animals were killed and the graft excised and stored at -70 C. Serial 6 µm sections were incubated with monoclonal antibodies to rat ED1, ED2, TCR, CD4 or CD8 followed by incubation with rabbit anti-mouse IgG antibody. After incubation with monoclonal mouse peroxidase-anti-peroxidase (PAP) reagent, the peroxidase reaction was developed by incubations in a carbazole-containing buffer. Surviving ICC and the frequency of the different cell phenotypes infiltrating the xenograft was assessed semi-quantitatively.
Real-time quantitative RT-PCR The grafts used for mRNA analysis were peeled off the kidneys with minimal amounts of underlying kidney tissue, and were immediately snap-frozen in liquid nitrogen and stored at –70 C until use.
Paper II and III mRNA from the grafts was extracted, immobilized onto oligo(dT)-coated manifold supports (129) and reversely transcribed to cDNA. 5´ nuclease assays for quantitative analysis of the obtained cDNA was performed using the iCycler iQ RealTime PCR Detection System (Bio-Rad Laboratories).
The cDNA sequences for murine -actin, TNF- , IL-1 , IL-4 and IL-10 were obtained from GenBank and the primer and probe sequences were designed using PrimerExpress software. To avoid amplification of genomic DNA, the primer-probe sets were designed to span exon-exon boundaries. The probes were labelled with 6-carboxyfluorescein (FAM) at the 5’ end and 6-carboxytetramethylrhodamine (TAMRA) at the 3’ end. For analysis of murine IL-2, IL-12p40, IFN- , MCP-1, MIP-1 and RANTES, PCR amplifications were performed using Pre-developed TaqMan Assay Reagents designed not to amplify genomic DNA. The results were represented as threshold cycle values, or Ct values, which are estimates of the amplification cycle number when the fluorescence exceeds a specified threshold value (130, 131). All of the tissue samples were run in triplicates for cDNA synthesis and in the PCR amplifications. Known amounts of amplicons, generated by the different primer pairs, were diluted and run in all PCR amplifications. Standard curves, created by plotting Ct values versus the log of the amount of cDNA template in the respective dilution, were then used to calculate the initial quantity of cDNA template in the tissue samples. No template controls, i.e. cDNA substituted with water, and RT- controls,

39
where reverse transcriptase had been left out in the earlier cDNA synthesis, were run together with the samples in all PCR amplifications, to screen for possible contamination and genomic amplification.
Paper IV RNA was isolated using an RNeasy mini-kit, followed by treatment with RNase-free DNase I to avoid DNA contamination and reversely transcribed to cDNA. The primers used for PCR amplification of PERV cDNA sequences were designed to detect PERV A, B and C. A relatively conserved portion of the integrase portion of the pol gene was used. The PERV specificity was confirmed by analysis of pig, human and mouse DNA. Since the product is detected with SYBR Green, the primer design was focused on avoiding secondary structures such as hairpin-loops and primer-dimers. Histone 3.3 is a conserved protein among mammals that is evenly expressed in different tissues, regardless of the cell cycle stage (132, 133), and was therefore used as a reference gene product. Histone 3.3 primers were based on those described in (133) and modified to meet the new real-time PCR conditions. The probe was labelled with 6-FAM at the 5´ end and an internal Dark Quencher attached to a thymidine 16 base pairs from the 5´end.
All QRT-PCR analyses were carried out in duplicates using a Rotor-Gene real time PCR cycler. For the SYBR Green analysis of PERV, a melt-curve analysis was performed immediately after each PCR run. Known amounts of amplicons, generated by the PERV primers or by the histone 3.3 probe and primers, were diluted and run as standards in all PCR amplifications. Standard curves, created by plotting Ct values versus the log of the amount of cDNA template in the each dilution, were then used to calculate the initial quantity of cDNA template in the tissue samples. No template controls, i.e. cDNA substituted with water, and RT- controls, where reverse transcriptase had been left out in the cDNA synthesis, were run for each sample in all PCR amplifications, to screen for possible contamination and efficiency of the DNase I treatment.
The sensitivity of the real-time quantitative PCR protocol for PERV was evaluated using genomic DNA obtained from PK15 cells and diluted in the range of 30 ng to 300 fg. This range would correspond to approximately 104-0.1 porcine genome equivalents, assuming that 6 pg corresponds to one diploid genome. The PK15 DNA was run together with a dilution series of PERV amplicons generated from the primer pair. Results indicated that the PK15 genome contains 18 copies of PERV A, B and C, which is in accordance with previous estimates (88). No signal was obtained for 0.1 copies of the PERV amplicon, indicating a sensitivity of the protocol down to a few copies of PERV nucleic acid. To confirm species-specificity, the PERV primers were tested using cDNA extracted from rat kidney tissue and human and murine DNA. All controls were negative.

40
RT activity in islet culture supernatants (Paper IV) RT activity in the collected culture supernatants was analysed using the Cavidi HS-kit Mn2+ RT method. Samples were run in duplicates according to the manufacturer´s protocol. To calculate RT activity, a dilution series of Moloney murine leukaemia virus (MMuLV) standard with known amounts of RT activity were run simultaneously. Culture supernatants of PK15 cells and islet culture medium alone were included as positive and negative controls, respectively.

RESULTS AND DISCUSSION
Paper I: Adenovirus-mediated expression of human CD55 or CD59 protects adult porcine islets from complement-mediated cell lysis by human serum
Expression of human RCAs in API after adenoviral transduction In paper I it was demonstrated that expression of the human complementinhibitory proteins DAF or CD59 can be induced on adult porcine islets using adenoviral vectors.
To obtain maximal expression of hDAF or hCD59 a total of three days ofculture was required after transduction, which would be a clinically suitable timeframe. The number of positively stained cells and the level of expression were markedly higher in islet cells transduced with the control gene mB7.1compared to the islet cells transduced with hDAF or hCD59. In the latter cases, positive cells were only detected on a sub-population of the isletsingle cells (Fig. 6). In the immunohistochemical evaluation, isletstransduced with hDAF or hCD59 had a similar degree of expression while islets transduced with mB7.1 had only a slightly higher number of positive cells. Replication-defective adenoviral vectors are generally thought not tobe able to penetrate and infect more than a few cell-layers in a tissue (134,135). In accordance with this, mainly smaller islets not exceeding 20 cells in
A B
41

E
DC
Figure 6. Flow cytometryevaluation of cells prepared fromintact adult porcine islets. Dotplots of cells prepared fromtrypsin-digested intact islets (A) and such cells after exposure tohuman complement active serumdiluted 1/3 (B). Gatedpopulations indicate viable cellsnot stained by propidium iodide(X-axis: forward scatter, Y-axis: FL2 fluorescence for propidium iodidedetection). Histograms showing the expression of hDAF (C), hCD59 (D) andmB7.1 (E) on cells prepared from transduced intact islets (unfilled histograms)as compared to cells derived from untreated control islets (filled histograms).
diameter had a homogenous expression of the three different transgenes. Inlarger islets, expression was restricted to the 5-15 outmost cell-layers of the islet surface (Fig. 7).
Since adenoviral transduction will not lead to insertion of DNA into thegenome of the infected cell, gene expression will be only transient (136).Hence, in many of the islets mainly cells in the periphery will be expressingthe transduced gene and only for a limited time. However, since only thesurface of the islets will be directly exposed to blood after intraportal infusion, an expression of human RCA on these cells would most likely besufficient to inhibit complement activation. Moreover, within 1-2 weeks after intraportal transplantation, the islets induce revascularization ofrecipient origin (27). At this stage the islets will no longer be in directcontact with circulating blood and protection against complement activationwill be of less importance.
Cytotoxicity of human sera against transduced and normal API
42
Cell viability after incubation in heat-inactivated human sera was similarbetween the four different groups of porcine cells while being slightly higherfor human cells. Experiments on human islet cells exposed to fresh humansera showed that these cells were not susceptible to lysis. Incubation of

Figure 7. Immunohistochemical stainings of cryosectioned (6 µm) adultporcine islets. In both pictures the islets were transduced to express humanCD59. The islets were prepared for immunohistochemistry on day three aftertransduction with adenoviral vector (105 pfu/islet equivalent). All cells in thesmaller islets were expressing the transgene. In larger islets, however,expression was restricted to cells located near the islet surface (Left,magnification x 20). A gradient of transduction was found with intensivelyexpressing hCD59 cells in the periphery of the islets and with weakly positivecells bordering the core of unstained cells (Right, magnification x 40).
porcine islet cells transduced with the control vector mB7.1 and non-transduced controls in complement active human sera resulted in an equallyextensive cell-lysis, while cells obtained from islets transduced with either of the two RCAs were partially protected from lysis. Cells expressing hCD59 were in turn slightly more resistant to lysis than cells expressing hDAF.
The endogenous expression of CD59 in both human and porcine islets is found on all islet cells (70). The transduced pig islets in this experimentappeared to generate a similar level of hCD59 on the cells located in the periphery of the islets. Still, the induced RCA expression was not sufficient to fully prevent cell lysis in the serum cytotoxicity assay. A plausibleexplanation for this would be that all islet cells were included in the assay.Because transduction with hDAF and hCD59 was conducted on intactporcine islets, there was no protective gene transfer to the cells in the core ofthe islets, as evidenced by immunohistochemical stainings and flowcytometry analysis.
43
Despite a similar degree of protein expression on the islet cells, hCD59 was more effective than hDAF to prevent direct cell-lysis in this model.CD59 blocks the final assembly of the MAC complex resulting in cell lysis,while DAF accelerates the decay of C3 convertases earlier in thecomplement cascade. It is therefore conceivable that CD59 is more effectivethan DAF in preventing lysis in this particular model. In whole blood, DAF activity will potentially have additional beneficial effects related to the reduced formation of the pro-inflammatory complement cleavage productsC3a and C5a.

Figure 8. Cytotoxicity of complement active human sera in a 1/3 dilutionagainst human and porcine islet cells. Five different types of islet cells were analyzed. Untreated human islet cells (2 isolations, 9 serum-islet isolationcombinations), untreated control porcine islet cells (7 isolations, 16combinations) and porcine islet cells with induced expression of the controlprotein mB7.1 (7 isolations, 16 combinations), hCD59 (5 isolations, 13combinations) or hDAF (7 isolations, 16 combinations). Data are presentedas mean cytotoxicity percentage + SEM of the different serum-islet isolationcombinations. ( ) indicates a difference in cytotoxicity compared tountreated porcine islet cells (p 0.05). (†) indicates difference incytotoxicity compared to porcine cells transduced with Ad.hDAF (p 0.05).
The data from paper I demonstrate that an induced expression of hDAF and hCD59 on porcine islet cells has a protective effect against complement-mediated lysis in human serum. There are however other factors to beconsidered if this strategy is to be tested in vivo. Previous studies on APIexposed to fresh human blood have shown that in addition to complementactivation, also the coagulation system and leukocyte infiltration are part of the triggered IBMIR (69). As have already been concluded from experimentswith human islets, in whole blood exposed to porcine islets it may prove necessary to prevent also the platelet aggregation and coagulation activation in order to prevent secondary complement activation and islet ischemia due to microvessel thrombosis (67, 68, 137).
Alternatives to adenoviral transduction
44
A concern regarding the in vivo use of adenoviral vectors, is that the infection itself trigger inflammatory and adaptive immune responses (138).

In clinical islet xenotransplantation this would counteract the beneficialeffects of a transgenic protein expression induced to limit IBMIR. The use ofother types of vectors, e.g. lentiviruses, or recently developed adenoviral vectors with less intrinsic immunogenicity may circumvent this problem.Alternatively, transgenic pigs could be used that on the islet cells express high levels of protective proteins. In the different transgenic pigs created to date, the main goal has been to avoid HAR and hence to induce hightransgene human RCA expression on the vascular endothelium.Unfortunately, the expression in other tissues may not be equally high and when a strain of transgenic pigs expressing hDAF was examined, no or only minimal amounts of hDAF were detected in isolated islets (70). Thisindicates that it may be necessary to create new gene constructs where thetransgenes are regulated by promoters active in islets, e.g. the insulin promoter.
Paper II: A new murine model of islet xenograft rejection
The transfer model
The transfer model uses the fact that athymic (nu/nu) nude mice do not have functional T lymphocytes. Consequently, the B lymphocytes cannot switch from IgM to IgG. These animals permanently accept a porcine ICC grafttransplanted beneath the kidney capsule. The innate immune system,however, largely remains intact and e.g. HAR can be triggered in the same
Figure 9. Experimental design in paper II.
45

way as in normal mice. Immunocompetent animals reject an ICC xenograft within a week and at this time a large number of infiltrating immune cellsare found within the graft. In these experiments, athymic (nu/nu) mice,bearing a tolerated ICC xenograft for one week up to more than a year,received intraperitoneally ICC xenografts undergoing rejection in immunocompetent animals. This new model makes it possible to studyrejection in the absence of the unspecific inflammatory reaction typicallyseen in conventional models, caused by the surgery during thetransplantation procedure. A schematic description of the experimentaldesign in paper II is outlined in figure 9 and the outcome in the different transfer experiments described below is summarized in Tables 1-3.
Demonstration of T cell dependence Six days after transfer, ICC xenografts in athymic (nu/nu) B6 mice wererejected with no or only a few remaining porcine cells. Rejection was notinduced if the transferred graft had been repeatedly freeze-thawed or irradiated, pointing at the requirement of viable cells. In rejecting animals,the pattern of cellular infiltration resembled that seen in normal B6 mice
after ICC transplantation. The process was somewhat faster after transferwhen compared to ICC xenograft rejection in normal mice and involvedfewer T cells in the process.
Table 1. Outline of the experimental transfer model. Transfer of immunocompetent cells or immune serumto athymic B6 (nu/nu) mice previously transplanted with ICC xenografts.
Donor of transferredmaterial
Transferred material Observation timeafter transfer (days)
Number ofanimals
Rejectionafter transfer
Evaluation a
B6 ICC xenograft 2 5 No IH, PCRB6 ICC xenograft 3 9 No IH, PCRB6 ICC xenograft 4 8 No IH, PCRB6 ICC xenograft 6 12 Yes IH, PCRB6 Freeze-thawed ICC xenograft 6 2 No IHB6 15 Gy-irradiated ICC xenograft 6 3 No IHB6 Ig -/- ICC xenograft 6 4 Yes IHB6 Immune serum 7 2+2b No IHB6 ICC xenograft 6+42c 6 Yes/Yes IH
aIH = Immunohistochemistry, PCR = Real time quantitative RT-PCRbMice were injected with either 0.5 ml serum at the time of transplantation or 0.1 ml serum days 1-5 aftertransplantation.cMice on the alternative protocol, i.e. ICC xenografts taken from the removed left kidney on day 6 aftertransfer and the grafts taken from the right kidney of the same re-transplanted mice 6 weeks after transfer.
Six weeks after transfer, the athymic (nu/nu) mice were capable ofrejecting a newly implanted porcine ICC xenograft demonstrating that a state of memory was induced at the time of transfer. The only cells capable ofmediating this type of immunological memory are sensitized lymphocytes.Since intraperitoneal injections of immune serum did not produce any signs of ICC xenograft destruction and transfer from Ig-deficient mice readilyinduced rejection, it could be concluded that the process is dependent on sensitized T cells. Because the molecular incompatibilities in MHC
46

47
recognition are too great, xenogeneic pig islets largely provoke the indirect pathway of antigen presentation in rodents (71). Activated CD4+ T cells recruit massive numbers of macrophages to the site of the graft, and these cells function as effector cells in the rejection (139, 140). Since the inflammatory reaction triggered by the transplantation procedure has already worn off at the time of rejection, we conclude that the macrophages were recruited to the ICC graft as a crucial effector component in a specific immune response.
In transplantation, the main trafficking of activated T-cells is thought to occur from lymph nodes to the blood and then to the graft. T-cells will be retained within the graft until it is totally rejected, at which point they will die or remain as memory cells. The onset of rejection in the transfer model was observed to be more rapid than in native immunocompetent mice, clearly demonstrating that there must be a significant number of highly activated T-cells escaping apoptosis and wandering from the transferred xenograft into the circulation.
Evaluation of intragraft cytokine mRNA expression We observed a marked increase in mRNA expression of the three analysed chemokines in the grafts, MCP-1 and MIP-1 by day three and four and RANTES by day four, in parallel with the first appearance of infiltrating T cells. These chemokines are potent chemoattractants for monocytes/macrophages and have been shown to upregulate -integrins on monocytes and enhance the transendothelial migration of these cells (141).Initially the source of these chemokines is most probably the transferred sensitized xenospecific CD4+ T-cells within the graft, but in the later phases of the rejection process activated ECs and effector macrophages may also be contributing.
As mentioned in the introduction, previous findings have implicated Th1 cells as important contributors at the initial stages of ICC xenograft rejection followed by Th2-associated responses in the later phases (76). In the transfer model, there was a concurrent up-regulation of all transcripts analysed, including both Th1-associated (IL-12p40 and IFN- ) and Th2-associated (IL-4 and IL-10) cytokines, by day four after transfer. This discrepancy may be explained by the possibility that the T cells mediating rejection in the athymic (nu/nu) mice are phenotypically and functionally similar to T cells present in a primary xenograft 10-12 days after transplantation. Notably, the transferred T cells were harvested from the donor animals on day 6 after transplantation and rejection in the athymic (nu/nu) mice was completed after an additional 4-6 days. At this stage both Th1- and Th2-associated cytokines are expressed in the xenograft.
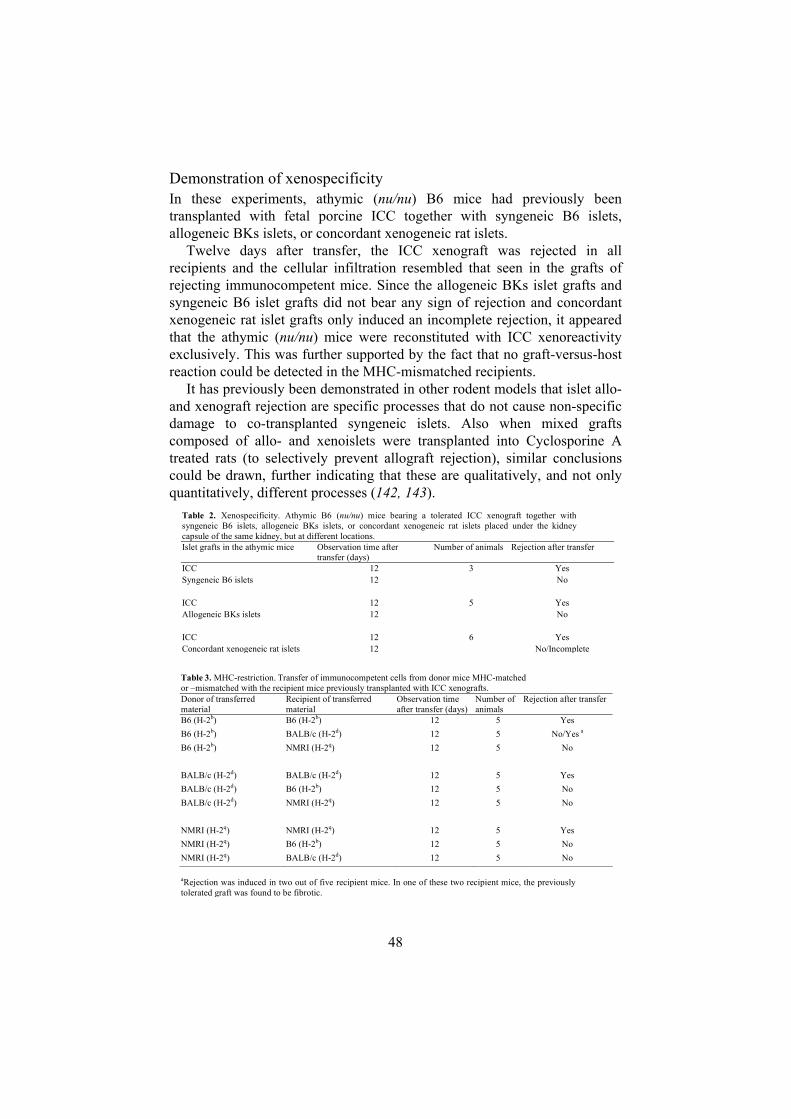
Demonstration of xenospecificity In these experiments, athymic (nu/nu) B6 mice had previously been transplanted with fetal porcine ICC together with syngeneic B6 islets, allogeneic BKs islets, or concordant xenogeneic rat islets.
Twelve days after transfer, the ICC xenograft was rejected in allrecipients and the cellular infiltration resembled that seen in the grafts of rejecting immunocompetent mice. Since the allogeneic BKs islet grafts andsyngeneic B6 islet grafts did not bear any sign of rejection and concordantxenogeneic rat islet grafts only induced an incomplete rejection, it appearedthat the athymic (nu/nu) mice were reconstituted with ICC xenoreactivityexclusively. This was further supported by the fact that no graft-versus-host reaction could be detected in the MHC-mismatched recipients.
It has previously been demonstrated in other rodent models that islet allo- and xenograft rejection are specific processes that do not cause non-specific damage to co-transplanted syngeneic islets. Also when mixed grafts composed of allo- and xenoislets were transplanted into Cyclosporine A treated rats (to selectively prevent allograft rejection), similar conclusionscould be drawn, further indicating that these are qualitatively, and not onlyquantitatively, different processes (142, 143).
Table 2. Xenospecificity. Athymic B6 (nu/nu) mice bearing a tolerated ICC xenograft together withsyngeneic B6 islets, allogeneic BKs islets, or concordant xenogeneic rat islets placed under the kidneycapsule of the same kidney, but at different locations.Islet grafts in the athymic mice Observation time after
transfer (days)Number of animals Rejection after transfer
ICC 12 3 YesSyngeneic B6 islets 12 No
ICC 12 5 YeAllogeneic BKs islets 12 No
ICC 12 6 YeConcordant xeno
s
sgeneic rat islets 12 No/Incomplete
Table 3. MHC-restriction. Transfer of immunocompetent cells from donor mice MHC-matchedor –mismatched with the recipient mice previously transplanted with ICC xenografts.Donor of transferredmaterial
Recipient of transferredmaterial
Observation timeafter transfer (days)
Number ofanimals
Rejection after transfer
B6 (H-2b) B6 (H-2b) 12 5 YesB6 (H-2b) BALB/c (H-2d) 12 5 No/Yes a
B6 (H-2b) NMRI (H-2q) 12 5 No
BALB/c (H-2d) BALB/c (H-2d) 12 5 YesBALB/c (H-2d) B6 (H-2b) 12 5 NoBALB/c (H-2d) NMRI (H-2q) 12 5 No
NMRI (H-2q) NMRI (H-2q) 12 5 YesNMRI (H-2q) B6 (H-2b) 12 5 NoNMRI (H-2q) BALB/c (H-2d) 12 5 No
aRejection was induced in two out of five recipient mice. In one of these two recipient mice, the previouslytolerated graft was found to be fibrotic.
48

Demonstration of MHC-restrictionDemonstration of MHC-restrictionIn the third subset of experiments, athymic (nu/nu) mice from three differentstrains previously transplanted with fetal porcine ICC received a transferred graft from either MHC-matched or -mismatched donors. To rule out thepossibility that NK cells in the athymic (nu/nu) mice were capable of destroying the MHC-mismatched T cells, which would influence theexperimental outcome, some recipient animals in the MHC-mismatchedcombinations were NK cell depleted by means of monoclonal NK1.1-antibodies prior to transfer.
In the third subset of experiments, athymic (nu/nu) mice from three differentstrains previously transplanted with fetal porcine ICC received a transferred graft from either MHC-matched or -mismatched donors. To rule out thepossibility that NK cells in the athymic (nu/nu) mice were capable of destroying the MHC-mismatched T cells, which would influence theexperimental outcome, some recipient animals in the MHC-mismatchedcombinations were NK cell depleted by means of monoclonal NK1.1-antibodies prior to transfer.
12 days after transfer of grafts from MHC-matched donors, ICC xenograft rejection was evident in all three strains of mice. Even though xenografts were heavily infiltrated with immune cells, some intact ICC were still seenin athymic (nu/nu) BALB/c and NMRI recipient mice. Transfer of graftsfrom MHC-mismatched donors did not produce any signs of ICC xenograftdestruction in the athymic (nu/nu) recipients, including mice depleted ofNK1.1+ cells, apart from two out of five mice in one of the groups (B6 toathymic (nu/nu) BALB/c).
12 days after transfer of grafts from MHC-matched donors, ICC xenograft rejection was evident in all three strains of mice. Even though xenografts were heavily infiltrated with immune cells, some intact ICC were still seenin athymic (nu/nu) BALB/c and NMRI recipient mice. Transfer of graftsfrom MHC-mismatched donors did not produce any signs of ICC xenograftdestruction in the athymic (nu/nu) recipients, including mice depleted ofNK1.1+ cells, apart from two out of five mice in one of the groups (B6 toathymic (nu/nu) BALB/c).
Figure 10. The observed requirement of MHC-compatibility may have beenpresent on two levels. Some animals displayed grafts completely devoid of T-cells indicating a failure of the CD4+ T-cells to find the ICC xenograft linedwith MHC-mismatched host ECs, while grafts in other animals displayedCD4+ T-cells within the xenograft area apparently unable to activate MHC-mismatched macrophages.
I.I.
II.
Endothelium
MHC-mismatch MHC-match
=
= CD4+ T cell
Macrophage
ICC XENOGRAFT
KIDNEY
= TCR
= MHC II + pig peptide
I.I.
II.
Endothelium
MHC-mismatch MHC-match
=
= CD4+ T cell
Macrophage
ICC XENOGRAFT
KIDNEY
= TCR
= MHC II + pig peptide
I.I.
II.
Endothelium
MHC-mismatch MHC-match
=
= CD4+ T cell
Macrophage
ICC XENOGRAFT
KIDNEY
= TCR
= MHC II + pig peptide
49

50
These different outcomes not only indicated that the effector macrophages during the rejection were of host athymic (nu/nu) mouse origin but also showed that the infiltration of macrophages is a secondary event to the activation and infiltration of T cells. Together with the previously demonstrated dependence of CD4+ T cells in islet xenograft rejection (71),these observations suggest that a direct interaction between the T cell receptor (TCR) on transferred xenospecific CD4+ cells and MHC class II on host cells is required to induce ICC xenograft rejection. Morphological studies on ICC xenografts removed from athymic (nu/nu) mice in the various MHC-mismatched groups indicate that the requirement of MHC-compatibility might be present on two levels. Some groups displayed grafts completely devoid of T-cells indicating a failure of the CD4+ T-cells to find the ICC xenograft lined with MHC-mismatched host ECs, while grafts in other groups displayed CD4+ T-cells within the xenograft area apparently unable to activate MHC-mismatched monocytes/macrophages (Fig. 10). All mouse strains in the experiment were equally mismatched with regard to MHC class I (H-2K, -D and -L) as well as to MHC class II (I-A and I-E) molecules, thus differences in MHC-mismatch do not provide an explanation for these observations.
Paper III: Acute cellular islet xenograft rejection in MyD88-deficient mice The findings in paper II illustrate that a key issue in preventing acute cellular xenograft rejection is to understand the interaction between the activated CD4+ T cells and the effector macrophages. The Toll-like receptors (TLRs) are important for the communication between innate and adaptive immunity and the role of this signalling system was examined in paper III.
Toll-like receptors TLRs play an important role in the innate immune response to microbial pathogens through recognition of products of microbial metabolism (PAMPs) such as LPS, peptidoglycans and other molecules that are not produced by the host cells. TLRs are expressed primarily on APCs and are involved in the regulation of costimulatory molecules and cytokine expression (144). In addition to recognition of the non-self peptide/MHC complex, this is a requirement for T cell activation, and the function of the TLRs therefore constitutes a link between the innate recognition of non-self antigens with that of adaptive immunity. All TLRs signal through an adapter protein, MyD88, although recent finding indicate that some TLRs may also utilize MyD88-independent pathways (145-147). MyD88-/- knockout mice therefore have an impairment in signalling through all types of TLRs. An

51
issue of immunological debate has been how the immunological defence is able to discriminate between Th1 (intracellular pathogens and different bacteria) and Th2 (multicellular eukaryotic parasites and allergens) adaptive immune responses. Increased knowledge about TLRs has lead to the hypothesis that TLR signalling is used by the innate immune system as a mean to initiate Th1, but not Th2, adaptive immunity. Schnare et al. demonstrated that MyD88-deficient mice, in response to complete Freund´s adjuvant mixed with ovalbumine, were incapable of generating Th1-dependent IgG2a isotype antibodies while IgE isotype switching, which is dependent on Th2-derived cytokines, were unaffected. When exposed to mycobacteria, MyD88-deficient dendritic cells did not upregulate the expression of the co-stimulatory molecules B7.1 or B7.2 or MHC class II and produced no IL-12 (148). In addition to mediating signal transduction by TLRs, MyD88 is required for IL-1 and IL-18 signalling. IL-18 (as well as IL-12) is known to induce IFN- production of T cells. MyD88-deficient mice have no apparent intrinsic defect in T or B cell function (148).
Most previous studies of TLR signalling have involved different infectious models and only one previous report have studied this mechanism in the context of transplantation (149). Here it was demonstrated that TLR signalling is activated in response to a vascularized allograft and that MyD88-/- animals are unable to reject a minor antigen mismatched allograft. However, acute allograft rejection in mice with a deficiency in MyD88 can be restored by an adoptive transfer of primed spleen cells from normal donors or the presence of normal APCs from either the donor or the recipient, indicating that this mechanism is acting at the initiation phase of the immune response. Whether rejection of fully MHC-mismatched allografts is equally impaired in MyD88-/- recipients remains to be investigated.
Several previous studies have investigated the mechanism of islet xenograft rejection in animal models using different strains of knockout mice and various specific cytokine antagonists (74, 150-152). However, the outcomes of these studies have shown that the immune response essentially does not depend solely on any one of the targeted cytokines, illustrating the great redundancy exhibited by these molecules. Only when IL-2, INF- and TNF- was inhibited simultaneously, islet xenograft rejection could be prevented, and this only in a minority of the animals (152). These observations founded the rationale for paper III, where the aim was to study islet xenograft rejection in a strain of mice deficient in MyD88, as a mean to observe the process in animals with a more generalized deficiency in Th1-immunity (148, 153).
Immune cell infiltration and cytokine mRNA kinetics.On day 3 after transplantation, a majority of the ICC remained intact in both the MyD88-/- and the control WT animals. A moderate infiltration of F4/80+

0
25
50
75
100
Day 3 Day 6
IL-12p40
0
250
500
750
1000
Day 3 Day 6
IL-4
0
500
1000
1500
2000
2500
Day 3 Day 6
IL-10
0
3 e5
6 e5
9 e5
12 e5
Day 3 Day 6
Wild type
MyD88-/-
-actin
0
250
500
750
1000
1250
Day 3 Day 6
IFN-Figure 11. Intragraft mRNAexpression of the cytokines IFN-, IL12-p40, IL-4, IL10 and the
housekeeping gene -actin atdays 3 and 6 after transplantationof ICC xenografts under thekidney capsule of MyD88-/-(white bars) and wildtype control(black bars) mice. Data are givenas the mean divided by 103
SEM (n=4).
and Mac-1+ macrophages was observed in xenografts of the WT animals andto a slightly lesser extent in the MyD88-/-. Few CD3+ cells were seen in the two groups and occasional CD4+ and CD8+ T cells were detected in the WT animals. On day 6 after transplantation the rejection was almost completedand no or only occasional intact ICC persisted in the MyD88-/- and WT animals. At this stage the xenografts were heavily infiltrated by F4/80+ and Mac-1+ macrophages. CD3+, CD4+, CD8+ and MHC class II+ immune cells were found in few to moderate numbers primarily in the periphery of thegrafts of animals in both groups. On day 12 the xenografts were completelyrejected in all animals. The pattern and the massive number of infiltrating immune cells persisted in the xenografts.
In the intragraft mRNA analysis of grafts taken day 3 aftertransplantation, gene expression of murine -actin, indicating infiltration of
Table 3. Morphological evaluation of fetal porcine ICC xenografts after transplantation underthe kidney capsule of mice with a targeted disruption of the MyD88 gene (MyD88 -/-) andwildtype controls.
Recipient animal n ICCa F4/80b MAC-1 b MHC II b CD3 b CD4 b CD8 b
Wildtype, day3 4 ++(+) ++ ++ 0 + (+) (+)MyD88-/-, day 3 4 ++(+) + +(+) 0 + 0 0Wildtype, day6 4 + +++ +++ +(+) ++ ++(+) +(+)MyD88-/-, day 6 4 + ++(+) +++ (+) ++ ++ +(+)Wildtype, day 12 4 0 +++ +++ + ++ ++ +(+)MyD88-/-, day 12 4 0 +++ +++ +(+) +++ ++ ++aRemaining ICC was graded semiquantitatively into four categories: 0, totally rejectedxenograft; +, occasional ICC; ++, several intact ICC; and +++, intact xenograft.bInfiltrating cells with various phenotypes were divided semiquantitatively into fourcategories: 0, no or occasional cells; +, few cells; ++, moderate to many; and +++, massiveinfiltration.
52

53
immune cells, and IL-10 was found at comparable levels in both groups while IL-4 mRNA expression was found to be low or absent. In contrast, the number of IL12p40 and IFN- mRNA copies was 2-3 times as high in the WT animals. By day 6, at a time when rejection was essentially completed, the mean gene expression of IL-4 and IL-10 was found to be 3-4 times as high in the MyD88-/- animals. It should however be noted that mainly 2 of the mice in this group accounted for this observation. IFN- in particular but also IL12p40 mRNA was expressed at a lower level compared to day 3 and at this stage intragraft levels were similar between the two groups of mice.
ICC xenograft rejection persists in MyD88-deficient mice The MyD88-deficient mice were shown to readily reject an islet xenograft and both the time course and the immunopathological pattern of the rejection did not deviate from that observed in normal animals. Evaluation of intragraft cytokine mRNA expression revealed that mice deficient in MyD88 produced lower levels of the Th1-associated cytokines IFN- and IL12p40 compared to normal animals on day 3 after transplantation, at a time when rejection is still in full progress. At the same time point, there was no difference between the two groups with regard to the Th2-associated cytokines IL-4 and IL-10. This is in accordance with previous findings that TLR signalling is mainly affecting Th1-immunity and that Th2-responses are initiated through TLR-independent pathways (148). Although acute cellular xenograft rejection should not be simplified as to be regarded as an exclusively Th1-driven immune response, one would anticipate that a deficiency in TLR signalling would lead to a delayed response to the xenograft. However with the results in hand, the production of IFN- and IL-12p40 in the MyD88-/- mice, despite being lower compared to normal recipients, appeared to be sufficient to induce rejection in a similar tempo as in the WT animals.
Based on the findings in paper III we can conclude that porcine islet xenograft rejection persists in mice lacking the TLR adaptor protein MyD88. Despite a comparably lower intragraft mRNA expression of INF- and IL-12 the immunopathological pattern and the tempo of the process was similar to that found in normal animals. Since the low levels of these Th1-associated cytokines still induced full-blown islet xenograft rejection, this may partly explain why conventional immunosuppression sufficient to repress allograft rejection is not able to protect an islet xenograft.

Considerations with the quantification of cytokine and chemokine expression using real-time RT-PCR (Paper II and III) In paper II and III, the real-time quantitative RT-PCR technique was appliedto study the ongoing immune response as reflected by cytokine andchemokine mRNA expression. Since this is an indirect analysis of geneproducts, it could be argued that all detected transcripts will not be translatedto a functional protein. However, as for all biosynthesis pathways, the initialstep is the most efficient for regulation and an increase in mRNA expression of a certain gene most likely reflects an immediate requirement of the geneproduct. This is particularly true for cytokines which are highly inducibleproteins with very limited constitutive expression. For small tissue samples,such as the xenografts used in the present studies, it is also difficult usingcurrent techniques to detect secreted proteins without in vitro stimulation.Another drawback with cytokine and chemokine studies at protein level maybe that immune cells carry cytokines in intracellular vessels which could
cause false positive signals in protein expression analyses. In a study of thereliability of RT-PCR versus intracellular cytokine staining for assessment ofintragraft cytokine profiles, the conclusion was that the mRNA-based RT-PCR technique, and not the protein-based technique, reflected the mostlikely cytokine milieu in the grafts (154).
Q
Q
Digestion of the probeduring the DNA synthesisseparates the flourescentdye and the quencher
fwd primer probe
rev primer
Q
Number of cycles
Log
fluor
esce
nce
Treshold
Ct-value
Figure 12. There are several very sensitive PCR-based approaches to quantifynucleotide sequences. TaqMan utilizes a fluorescent probe that emits light onlywhen it is degraded during a PCR amplification cycle. The cycle after which the sample fluorescence attains a threshold value is registered and can be comparedto that of standards with known amounts of amplimer.
54

55
Also to be considered at data interpretation, is that the variation in expression of the different gene transcripts does not necessarily reflect their relative importance. Differences in mRNA degradation, translation efficiency and receptor properties for the cytokines and chemokines remain to be fully characterized. Moreover, both the cDNA synthesis method and the real-time PCR protocol applied in these experiments could differ in efficiency from one target molecule to another. As a consequence, rather than comparing absolute levels of different gene transcripts, the interpretation should be restricted to relating variations of each cytokine and chemokine transcript level at different time points.
Another observation made in paper II and III was that there was a decrease in murine -actin mRNA expression by the time the rejection was completed. -actin is widely used as a reference gene product in mRNA quantification and should be expressed at relatively stable levels (155-157).The reason for this finding remains unclear, but it may be that transcription of this gene is higher in very active cells such as infiltrating immune cells during the peak of a rejection episode.
Paper IV: Dynamics of porcine endogenous retrovirus expression after fetal islet xenotransplantation to athymic and normal rats
PERV expression in ICC transplanted to rats As expected, ICC xenografts from the athymic (nu/nu) Lewis rats remained intact throughout the experiment. However, a moderate infiltration of ED1+
macrophages was noted in all animals. On day 3, two of the animals had a 10-fold increase in expression of PERV RNA within the graft as compared to 2 µl of ICC from the same preparation retrieved just prior to transplantation (referred to as control tissue). No such increase, however, was detected in the third animal. By day 5, all grafts had 2-4 times as much PERV RNA compared to control tissue and by day 12 no PERV RNA could be detected in any of the animals.
One day after transplantation to normal otherwise untreated Lewis rats, the grafts were still intact and largely devoid of infiltrating cells. The amount of PERV RNA detected in the grafts of two of the animals was 10-20 times as high as in control tissue, while the graft of the third animal expressed approximately 4-5 times as much. By day 3, many intact ICC still remained but the first infiltrating immune cells, mainly ED1+ macrophages, were observed. PERV RNA expression was now 3-5 times as high as in control tissue. On day 5 after transplantation, graft destruction was evident although some ICC still remained. At this stage the graft was heavily infiltrated by

ED1+, ED2+ and CD8+ immune cells. TCR+ and CD4+ cells were foundmainly in the periphery of the graft. PERV expression had now decreased tocontrol levels and was absent in one animal. By day 12, the ICC grafts were completely rejected but a large number of immune cells still persisted within the transplant area. At this stage no PERV RNA could be detected. Takentogether, the grafts of all animals as well as the 2 µl of ICC used as control tissue expressed small amounts of PERV RNA. In no case did the expressionexceed 0.4 % of that of histone 3.3, which is generally expressed at around 103 copies per cell (unpublished observation).
ICC
NORMAL OR ATHYMIC RAT
ICC transplantation
1-12 DAYS
ANIMAL EXPERIMENTS
CULTURE EXPERIMENTS
GRAFT EVALUATION
MORPHOLOGY
QRT-PCR
CONTROLCULTURE
DEXAMETHASONE(1 M)
AzaC(2 mg/ml) +IDU (30 mg/ml)
RETROVIRALINDUCERS
IMMUNESUPPRESSANT
NORMALCONDITIONS
API
EVALUATION
SUPERNATANTRT ACTIVITY
QRT-PCR
Figure 13. Outline of the experimental design used in paper IV. Both adult andfetal islets were cultured and analyzed for PERV content, while ICC were transplanted to normal or athymic (nu/nu) nude rats for evaluation of PERVdynamics in vivo.
Athymic (nu/nu) rats lack functional lymphocytes and are therefore incapable of rejecting ICC grafts. Still, a transient inflammatory response characterized by a moderate infiltration of macrophages is found in these animals. Although remaining low throughout the experiment, PERV production was found to be elevated in the grafts during the first days after transplantation in both immunocompetent and athymic (nu/nu) rats,suggesting that an unspecific inflammation, independent of a rejectionprocess, is sufficient to induce a transient elevation of PERV titres immediately after transplantation. In a previous study it was demonstrated
56

that the expression of heat shock protein 70 (hsp70) in ICC is markedlyreduced three days after subcapsular implantation into nude athymic (nu/nu)mice. Interestingly, the same study demonstrated that also ICC in culture have an elevation in hsp70 expression (158).
Thus, an alternative explanation would be that the increase in virus titre is due to ischemia in the islets before they are revascularized in the host. This is supported by the fact that PERV RNA expression was higher in islets inculture (relative ischemia) than in islets after revascularization in the athymic (nu/nu) rats on day 12 after transplantation.
0
200
400
600
800
Pre Tx Day 1 Day 3 Day 5 Day 12
Normal ratsAthymic rats
2 µl ICC
Figure 14. Expression of PERV RNA in ICC xenografts at different days aftertransplantation to normal Lewis rats (triangles) or athymic nude Lewis rats(squares). Also shown is control tissue composed of ICC under normal cultureconditions (circle). The values represent the number of PERV RNA copiesdetected in each graft. Data are expressed as the mean SEM (n=3 at each timepoint).
PERV content in islet culturesRT activity in the supernatants from two of the five ICC groups showedtraces of RT activity. The remaining ICC cultures as well as all six APIisolations had undetectable levels. Supernatants of islets cultured in thepresence of AzaC and IDU or DEX did not contain elevated RT activity compared to the corresponding non-exposed islet preparations. The numberof PERV RNA copies in ICC and API was low and not notably induced inthe presence of AzaC and IDU or DEX in the culture medium atconcentration established to induce endogenous retroviral particle production(102). While PERV RNA copies could be detected in all API and ICC isolations analysed, RT activity in the culture supernatants was onlyconfirmed in ICC from two groups of piglets. This could merely reflect alower sensitivity of the RT assay but could also indicate that despite some
57

58
PERV RNA expression, the remaining islet preparations did not produce functional virions.
While a pure fraction of API constitutes mainly of endocrine cells, ICC prepared from fetal pancreatic tissue have a much more heterogeneous cell content. In addition to immature endocrine and exocrine cells there is also a high proportion of fibroblasts and ductal cells (16). It is therefore possible that the two types of islets differ with respect to PERV expression. The present findings, however, implicate that a similar level of PERV particles is produced in API and ICC in culture. Both in terms of absolute PERV RNA cell content and culture supernatant RT activity as well as when compared to the PERV producing cell line PK15 the expression could be characterized as low. Notably, the animals used to generate porcine islets in this study were taken from a stock of Swedish Landrace pigs. The results therefore do not necessarily reflect the situation in other breeds of pigs that may be used for clinical xenotransplantation.
It might be argued that because of similar levels of PERV production in fetal and adult islets, the choice between API and ICC will not depend on the relative risk of transmitting PERV to xenotransplant recipients. However, the most important way of inactivating retroviruses from non-primate mammals are through preformed XNAs directed against Gal (1,3)Gal sugars on the virus envelope (99). API, as opposed to ICC, express only few epitopes of these sugars and as a consequence any PERV released from such cells would presumably escape this defence mechanism (18). Attempts in our laboratory to evaluate the resistance to complement-mediated virolysis by PERV produced in API and ICC have so far been unsuccessful because of too low virus loads obtained from the islet tissue (unpublished observations). It may be speculated that ICC in this respect have an inherent advantage over API in xenotransplantation, because the fetal tissue will mature to Gal (1,3)Gal-negative endocrine cells only after revascularization, during a period when PERV expression has declined.
After quantifying islet PERV RNA and RT activity of culture supernatants, we conclude that both fetal and adult porcine islets produce low titres of PERV. Although other factors related to the islet cell phenotype may influence retroviral transmission to the recipient, the low level of expression in islet tissue should decrease this risk.
Risk estimates Based on the data obtained in paper IV, we conclude that the greatest risk of PERV transmission will be during a period immediately following the transplantation, when ischemia, inflammation and other non-specific immunological processes could increase retroviral transcription. At the same time, induction therapies including agents suppressing complement activation and the humoral defence may contribute to increase the risk in the early post-transplantation period. In this perspective, antiviral therapies

59
during an initial stage in clinical xenotransplantation could be a valuable strategy to reduce the risk of PERV infection. Although there appears to be a limited number of antiviral drugs used in the clinic that are effective against PERV, possible candidates have been identified, such as azidothymidine (AZT) and a few other related nucleoside analogues (159, 160). Another possibility is to create a vaccine against PERV that could be administered prior to transplantation.
Ultimately, the best option for eliminating the risk of PERV transmission would be to create pig breeds devoid of replication-competent viruses. Initially, assessments argued for this being impossible. With increasing knowledge of the complexity of PERV loci in the porcine genome it has become clear that the majority of loci include deletions and mutations rendering them incapable of encoding replication-competent human-tropic viruses. Although the porcine genome remains to be fully characterized and unknown functional PERV loci may still be discovered, today only a handful of PERV loci have been identified that are able to produce replication-competent viruses (89, 161, 162). It may in fact prove possible by means of selective breeding and knockout techniques to eliminate these loci and this would no doubt dramatically decrease the risk of PERV transmission. It is however still too early to determine whether this can be achieved.
An additional scenario that has been proposed is that two defective PERV RNA particles can be packaged together and that subsequent recombination may lead to the creation of a functional retrovirus. Thus, even when known functional PERV loci have been removed, new infectious PERV could be created by complementation between two defective genomes. Fortunately, the likelihood that this is actually more than a theoretical possibility is estimated to be extremely low (163-165). As an interesting comparison, a similar risk is taken daily by researchers and animal caretakers who work with immune deficient mice implanted with human or porcine tissue. In these animals such recombination between defective endogenous retroviruses from the different species could also, theoretically, create an infectious new type of virus.

60
CONCLUSIONS
Expression of the human complement regulatory proteins DAF or CD59 can be induced on API using adenoviral vectors. Effective gene transfer was restricted to cells in the periphery of the islets.
Cells derived from API transduced to express these RCAs are partially protected from complement-mediated lysis in human serum.
A new experimental model of ICC xenograft rejection in mice was established. Transfer of immunocompetent cells, removed from the site of ongoing fetal porcine ICC xenograft rejection in normal mice, enabled athymic (nu/nu) mice to reject a previously tolerated ICC xenograft.
The model design demonstrated that the infiltrating effector macrophages are part of a specific immune response independent of the inflammatory response caused by the surgical trauma otherwise seen in conventional transplantation models.
It was demonstrated that ICC xenograft rejection is dependent on the transfer of viable T cells, and that these cells induce rejection of the ICC xenograft exclusively. For the rejection to be initiated, MHC-compatibility between the donor and the recipient was required.
MyD88-dependent TLR signalling is not a requirement in acute cellular ICC xenograft rejection. MyD88-deficient mice reject an ICC xenograft in a similar fashion and tempo as normal mice, despite comparably lower expression of the Th1-associated cytokines IL12 and IFN- .
After ICC transplantation to rats, the overall low intragraft PERV expression correlates to and declines with the unspecific inflammatory response inflicted by the surgical trauma and/or the level of ischemia in the islets.
The levels of PERV expression do not differ between API and ICC in culture. The amount of released virus particles is low, both in terms of absolute RNA levels and compared to the PERV producing porcine cell line PK15.

61
FUTURE ASPECTS
In paper I we have shown that adult porcine islets can be effectively transduced to express the complement regulatory proteins DAF and CD59 and that these proteins can protect islet cells from direct complement mediated lysis in human serum. The aim is to take this experiment further to compare the islet protective effect after transduction with that achieved applying systemic blockade of complement activation using blood loop in vitro models or in vivo intraportal transplantation. Since also platelet aggregation and coagulation play an important role in IBMIR this experiment may also include coagulation inhibitors such as melagatran or inactivated factor VII.
We will also use other adenoviral vectors as a mean to study and prevent IBMIR in porcine islets. At present we have access to vectors that will induce transgene expression of hemeoxygenase-1 (HO-1) and soluble or membrane-bound hirudin. HO-1 is expressed as part of a cytoprotective mechanism during cellular stress and has been shown to reduce inflammatory immune-mediated responses and has anti-platelet aggregation properties. Hirudin, a protein derived from leeches, has thrombin binding properties and can be used to selectively inhibit the coagulation-mediated effects of IBMIR.
To further investigate the mechanisms of acute cellular islet xenograft rejection, we will continue to use the transfer model introduced in paper II and conventional pig-to-rodent islet xenograft transplantation under the kidney capsule. Since intraportal infusion is likely to be the preferred way of transplanting also xenogeneic islets, these established rodent models where the graft is transplanted under the kidney capsule will have to be modified to be able to evaluate graft survival also after intraportal islet transplantation to rodents.
With respect to safety, physiology and immunological acceptance, islet xenotransplantation have important advantages compared to vascular organs and are likely to lead the way to a clinical application. In perspective of the encouraging results of clinical allogeneic islet transplantation that have been reported during the past years, the coming research will focus on making porcine islet xenotransplantation a clinical reality. In order to do this, knowledge from previous in vitro and in vivo rodent models will be used in clinically more relevant non-human primate settings.

62
The discovery of transmission of PERV to human cell lines in 1997 suggested that PERV might pose an unacceptable risk in xenotransplantation. Since then, however, much new information about this virus has become available indicating that PERV may not pose as big a threat as was initially feared. Although PERV will remain a safety concern, it will probably not be the ultimate barrier to clinical pig-to-human xenotransplantation.

63
ACKNOWLEDGEMENTS
The work presented in this thesis was carried out at the division of Clinical Immunology, Rudbeck Laboratory, Department of Oncology, Radiology and Immunology, Faculty of Medicine, Uppsala University.
Financial support was granted from the Swedish Medical Research Council (16P-13568 and 16X-12219), Åke Wiberg Foundation, Nordic Insulin Fund, Torsten and Ragnar Söderbergs Foundation, Family Ernfors Fund, Barn Diabetes Fonden, Göran Gustafsson Foundation, Swedish Diabetes Association, Juvenile Diabetes Foundation International, the Knut and Alice Wallenberg Foundation, Förenade Liv Mutual Group Life Insurance Company (Stockholm, Sweden), Clas Groschinsky Fund and Swedish Society for Medical Research.

64

65
REFERENCES
1. King H, Aubert RE, Herman WH. Global burden of diabetes, 1995-2025: prevalence, numerical estimates, and projections. Diabetes Care 1998; 21 (9): 1414.
2. Amos AF, McCarty DJ, Zimmet P. The rising global burden of diabetes and its complications: estimates and projections to the year 2010. Diabet Med 1997; 14 (Suppl 5): S1.
3. Shapiro AM, Lakey JR, Ryan EA, et al. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med 2000; 343 (4): 230.
4. Auchincloss H, Sachs DH. Xenogeneic transplantation. Annu Rev Immunol 1998; 16: 433.
5. Brenner P, Reichenspurner H, Schmoeckel M, et al. Prevention of hyperacute xenograft rejection in orthotopic xenotransplantation of pig hearts into baboons using immunoadsorption of antibodies and complement factors. Transpl Int 2000; 13 Suppl 1: S508.
6. Brenner P, Reichenspurner H, Schmoeckel M, et al. IG-therasorb immunoapheresis in orthotopic xenotransplantation of baboons with landrace pig hearts. Transplantation 2000; 69 (2): 208.
7. Cozzi E, Bhatti F, Schmoeckel M, et al. Long-term survival of nonhuman primates receiving life-supporting transgenic porcine kidney xenografts. Transplantation 2000; 70 (1): 15.
8. Hammer C, Thein E. Physiological aspects of xenotransplantation, 2001. Xenotransplantation 2002; 9 (5): 303.
9. Platt JL. Physiologic barriers to xenotransplantation. Transplant Proc 2000; 32 (7): 1547.
10. Koulmanda M, Qipo A, Smith RN, Auchincloss H, Jr. Pig islet xenografts are resistant to autoimmune destruction by non-obese diabetic recipients after anti-CD4 treatment. Xenotransplantation 2003; 10 (2): 178.
11. Thomas FT, Pittman K, Brezina P, et al. Pancreas islet xenografts but not allografts are resistant to autoimmune disease recurrence following islet transplantation. Transplant Proc 1997; 29 (1-2): 760.
12. Groth CG, Korsgren O, Tibell A, et al. Transplantation of porcine fetal pancreas to diabetic patients. Lancet 1994; 344 (8934): 1402.
13. Reinholt FP, Hultenby K, Tibell A, Korsgren O, Groth CG. Survival of fetal porcine pancreatic islet tissue transplanted to a diabetic patient: findings by ultrastructural immunocytochemistry. Xenotransplantation 1998; 5 (3): 222.

66
14. Krickhahn M, Buhler C, Meyer T, Thiede A, Ulrichs K. The morphology of islets within the porcine donor pancreas determines the isolation result: successful isolation of pancreatic islets can now be achieved from young market pigs. Cell Transplant 2002; 11 (8): 827.
15. Krickhahn M, Meyer T, Buhler C, Thiede A, Ulrichs K. Highly efficient isolation of porcine islets of Langerhans for xenotransplantation: numbers, purity, yield and in vitro function. Ann Transplant 2001; 6 (3): 48.
16. Korsgren O, Jansson L, Eizirik D, Andersson A. Functional and morphological differentiation of fetal porcine islet- like cell clusters after transplantation into nude mice. Diabetologia 1991; 34 (6): 379.
17. Rayat GR, Rajotte RV, Elliott JF, Korbutt GS. Expression of Gal alpha(1,3)gal on neonatal porcine islet beta-cells and susceptibility to human antibody/complement lysis. Diabetes 1998; 47 (9): 1406.
18. Bennet W, Bjorkland A, Sundberg B, et al. A comparison of fetal and adult porcine islets with regard to Gal alpha (1,3)Gal expression and the role of human immunoglobulins and complement in islet cell cytotoxicity. Transplantation 2000; 69 (8): 1711.
19. Strokan V, Bennet W, Molne J, Korsgren O, Breimer ME. Distribution of the Galalpha1-3Gal antigen in cultured adult and fetal porcine pancreatic islet cells: an immunoelectron microscopic study. Transplantation 2000; 70 (5): 846.
20. Phelps CJ, Koike C, Vaught TD, et al. Production of alpha 1,3-galactosyltransferase-deficient pigs. Science 2003; 299 (5605): 411.
21. Lai L, Kolber-Simonds D, Park KW, et al. Production of alpha-1,3-galactosyltransferase knockout pigs by nuclear transfer cloning. Science 2002; 295 (5557): 1089.
22. Reece-Smith H, Du Toit DF, McShane P, Morris PJ. Prolonged survival of pancreatic islet allografts transplanted beneath the renal capsule. Transplantation 1981; 31 (4): 305.
23. Head JR, Billingham RE. Immunologically privileged sites in transplantation immunology and oncology. Perspect Biol Med 1985; 29 (1): 115.
24. Jaeger C, Wohrle M, Federlin K, Bretzel RG. Pancreatic islet xenografts at two different transplantation sites (renal subcapsular versus intraportal): comparison of graft survival and morphology. Exp Clin Endocrinol Diabetes 1995; 103 (Suppl 2): 123.
25. Korsgren O, Jansson L. Porcine islet-like cell clusters cure diabetic nude rats when transplanted under the kidney capsule, but not when implanted into the liver or spleen. Cell Transplant 1994; 3 (1): 49.
26. Alejandro R, Cutfield RG, Shienvold FL, et al. Natural history of intrahepatic canine islet cell autografts. J Clin Invest 1986; 78 (5): 1339.
27. Menger MD, Yamauchi J, Vollmar B. Revascularization and microcirculation of freely grafted islets of Langerhans. World J Surg 2001; 25 (4): 509.

67
28. Hong J, Nilsson Ekdahl K, Reynolds H, Larsson R, Nilsson B. A new in vitro model to study interaction between whole blood and biomaterials. Studies of platelet and coagulation activation and the effect of aspirin. Biomaterials 1999; 20 (7): 603.
29. Schmugge M, Rand ML, Freedman J. Platelets and von Willebrand factor. Transfus Apheresis Sci 2003; 28 (3): 269.
30. Mosmann TR, Schumacher JH, Street NF, et al. Diversity of cytokine synthesis and function of mouse CD4+ T cells. Immunol Rev 1991; 123: 209.
31. Suankratay C, Mold C, Zhang Y, Lint TF, Gewurz H. Mechanism of complement-dependent haemolysis via the lectin pathway: role of the complement regulatory proteins. Clin Exp Immunol 1999; 117 (3): 442.
32. Zhang Y, Suankratay C, Zhang XH, Lint TF, Gewurz H. Lysis via the lectin pathway of complement activation: minireview and lectin pathway enhancement of endotoxin-initiated hemolysis. Immunopharmacology 1999; 42 (1-3): 81.
33. Zhao Z, Termignon JL, Cardoso J, et al. Hyperacute xenograft rejection in the swine-to-human donor-recipient combination. In vitro analysis of complement activation. Transplantation 1994; 57 (2): 245.
34. Morgan BP, Harris CL. Complement therapeutics; history and current progress. Mol Immunol 2003; 40 (2-4): 159.
35. Krych-Goldberg M, Hauhart RE, Subramanian VB, et al. Decay accelerating activity of complement receptor type 1 (CD35). Two active sites are required for dissociating C5 convertases. J Biol Chem 1999; 274 (44): 31160.
36. Laumonier T, Walpen AJ, Maurus CF, et al. Dextran sulfate acts as an endothelial cell protectant and inhibits human complement and natural killer cell-mediated cytotoxicity against porcine cells. Transplantation 2003; 76 (5): 838.
37. Goto M, Johansson H, Maeda A, Elgue G, Korsgren O, Nilsson B. Low molecular weight dextran sulfate prevents the instant blood-mediated inflammatory reaction induced by adult porcine islets. Transplantation 2003; In press.
38. Stump DC, Mann KG. Mechanisms of thrombus formation and lysis. Ann Emerg Med 1988; 17 (11): 1138.
39. Morrissey JH. Tissue factor: an enzyme cofactor and a true receptor. Thromb Haemost 2001; 86 (1): 66.
40. Bach FH, Soares M, Lin Y, Ferran C. Barriers to xenotransplantation. Transplant Proc 1999; 31 (4): 1819.
41. Pries AR, Secomb TW, Gaehtgens P. The endothelial surface layer. Pflugers Arch 2000; 440 (5): 653.
42. Galili U, Shohet SB, Kobrin E, Stults CL, Macher BA. Man, apes, and Old World monkeys differ from other mammals in the expression of alpha-galactosyl epitopes on nucleated cells. J Biol Chem 1988; 263 (33): 17755.

68
43. Parker W, Bruno D, Holzknecht ZE, Platt JL. Characterization and affinity isolation of xenoreactive human natural antibodies. J Immunol 1994; 153 (8): 3791.
44. Rose AG, Cooper DK, Human PA, Reichenspurner H, Reichart B. Histopathology of hyperacute rejection of the heart: experimental and clinical observations in allografts and xenografts. J Heart Lung Transplant 1991; 10 (2): 223.
45. Lin SS, Weidner BC, Byrne GW, et al. The role of antibodies in acute vascular rejection of pig-to-baboon cardiac transplants. J Clin Invest 1998; 101 (8): 1745.
46. Shin ML, Hansch G, Hu VW, Nicholson-Weller A. Membrane factors responsible for homologous species restriction of complement-mediated lysis: evidence for a factor other than DAF operating at the stage of C8 and C9. J Immunol 1986; 136 (5): 1777.
47. Dalmasso AP, Vercellotti GM, Platt JL, Bach FH. Inhibition of complement-mediated endothelial cell cytotoxicity by decay-accelerating factor. Potential for prevention of xenograft hyperacute rejection. Transplantation 1991; 52 (3): 530.
48. Schaapherder AF, Wolvekamp MC, te Bulte MT, Bouwman E, Gooszen HG, Daha MR. Porcine islet cells of Langerhans are destroyed by human complement and not by antibody-dependent cell-mediated mechanisms. Transplantation 1996; 62 (1): 29.
49. Robson SC, Cooper DK, d'Apice AJ. Disordered regulation of coagulation and platelet activation in xenotransplantation. Xenotransplantation 2000; 7 (3): 166.
50. Byrne GW, McCurry KR, Martin MJ, McClellan SM, Platt JL, Logan JS. Transgenic pigs expressing human CD59 and decay-accelerating factor produce an intrinsic barrier to complement-mediated damage. Transplantation 1997; 63 (1): 149.
51. Cozzi E, Tucker AW, Langford GA, et al. Characterization of pigs transgenic for human decay-accelerating factor. Transplantation 1997; 64 (10): 1383.
52. Adams DH, Kadner A, Chen RH, Farivar RS. Human membrane cofactor protein (MCP, CD 46) protects transgenic pig hearts from hyperacute rejection in primates. Xenotransplantation 2001; 8 (1): 36.
53. Bach FH, Winkler H, Ferran C, Hancock WW, Robson SC. Delayed xenograft rejection. Immunol Today 1996; 17 (8): 379.
54. Parker W, Saadi S, Lin SS, Holzknecht ZE, Bustos M, Platt JL. Transplantation of discordant xenografts: a challenge revisited. Immunol Today 1996; 17 (8): 373.
55. Candinas D, Belliveau S, Koyamada N, et al. T cell independence of macrophage and natural killer cell infiltration, cytokine production, and endothelial activation during delayed xenograft rejection. Transplantation 1996; 62 (12): 1920.
56. Blakely ML, Van der Werf WJ, Berndt MC, Dalmasso AP, Bach FH, Hancock WW. Activation of intragraft endothelial and

69
mononuclear cells during discordant xenograft rejection. Transplantation 1994; 58 (10): 1059.
57. Hancock WW. Delayed xenograft rejection. World J Surg 1997; 21 (9): 917.
58. Platt JL, Lindman BJ, Geller RL, et al. The role of natural antibodies in the activation of xenogenic endothelial cells. Transplantation 1991; 52 (6): 1037.
59. Inverardi L, Samaja M, Motterlini R, Mangili F, Bender JR, Pardi R. Early recognition of a discordant xenogeneic organ by human circulating lymphocytes. J Immunol 1992; 149 (4): 1416.
60. Watier H, Guillaumin JM, Vallee I, et al. Human NK cell-mediated direct and IgG-dependent cytotoxicity against xenogeneic porcine endothelial cells. Transpl Immunol 1996; 4 (4): 293.
61. Schaapherder AF, Daha MR, te Bulte MT, van der Woude FJ, Gooszen HG. Antibody-dependent cell-mediated cytotoxicity against porcine endothelium induced by a majority of human sera. Transplantation 1994; 57 (9): 1376.
62. Loss M, Vangerow B, Schmidtko J, et al. Acute vascular rejection is associated with systemic complement activation in a pig-to-primate kidney xenograft model. Xenotransplantation 2000; 7 (3): 186.
63. Kopp CW, Siegel JB, Hancock WW, et al. Effect of porcine endothelial tissue factor pathway inhibitor on human coagulation factors. Transplantation 1997; 63 (5): 749.
64. Dorling A, Lechler RI. T cell-mediated xenograft rejection: specific tolerance is probably required for long term xenograft survival. Xenotransplantation 1998; 5 (4): 234.
65. Samstein B, Platt JL. Xenotransplantation and tolerance. Philos Trans R Soc Lond B Biol Sci 2001; 356 (1409): 749.
66. Korsgren O, Jansson L. Discordant cellular xenografts revascularized in intermediate athymic hosts fail to induce a hyperacute rejection when transplanted to immunocompetent rats. Transplantation 1994; 57 (9): 1408.
67. Bennet W, Sundberg B, Groth CG, et al. Incompatibility between human blood and isolated islets of Langerhans: a finding with implications for clinical intraportal islet transplantation? Diabetes 1999; 48 (10): 1907.
68. Moberg L, Johansson H, Lukinius A, et al. Production of tissue factor by pancreatic islet cells as a trigger of detrimental thrombotic reactions in clinical islet transplantation. Lancet 2002; 360 (9350): 2039.
69. Bennet W, Sundberg B, Lundgren T, et al. Damage to porcine islets of Langerhans after exposure to human blood in vitro, or after intraportal transplantation to cynomologus monkeys: protective effects of sCR1 and heparin. Transplantation 2000; 69 (5): 711.
70. Bennet W, Bjorkland A, Sundberg B, et al. Expression of complement regulatory proteins on islets of Langerhans: a

70
comparison between human islets and islets isolated from normal and hDAF transgenic pigs. Transplantation 2001; 72 (2): 312.
71. Wolf LA, Coulombe M, Gill RG. Donor antigen-presenting cell-independent rejection of islet xenografts. Transplantation 1995; 60 (10): 1164.
72. Gill RG, Wolf L, Daniel D, Coulombe M. CD4+ T cells are both necessary and sufficient for islet xenograft rejection. Transplant Proc 1994; 26 (3): 1203.
73. Benda B, Korsgren O. Xenograft rejection of fetal porcine islet-like cell clusters in normal, T-cell receptor beta-, delta-, beta x delta-; perforin-; or granzyme B-deficient mice. Transplant Proc 1998; 30 (2): 586.
74. Simeonovic CJ, Townsend MJ, Wilson JD, et al. Eosinophils are not required for the rejection of neovascularized fetal pig proislet xenografts in mice. J Immunol 1997; 158 (5): 2490.
75. Karlsson-Parra A, Ridderstad A, Wallgren AC, Moller E, Ljunggren HG, Korsgren O. Xenograft rejection of porcine islet-like cell clusters in normal and natural killer cell-depleted mice. Transplantation 1996; 61 (9): 1313.
76. Krook H, Hagberg A, Song Z, Landegren U, Wennberg L, Korsgren O. A distinct Th1 immune response precedes the described th2 response in islet xenograft rejection. Diabetes 2002; 51 (1): 79.
77. Wennberg L, Karlsson-Parra A, Sundberg B, et al. Efficacy of immunosuppressive drugs in islet xenotransplantation: leflunomide in combination with cyclosporine and mycophenolate mofetil prevents islet xenograft rejection in the pig-to-rat model. Transplantation 1997; 63 (9): 1234.
78. Wennberg L, Song Z, Bennet W, et al. Diabetic rats transplanted with adult porcine islets and immunosuppressed with cyclosporine A, mycophenolate mofetil, and leflunomide remain normoglycemic for up to 100 days. Transplantation 2001; 71 (8): 1024.
79. Soderlund J, Wennberg L, Castanos-Velez E, et al. Fetal porcine islet-like cell clusters transplanted to cynomolgus monkeys: an immunohistochemical study. Transplantation 1999; 67 (6): 784.
80. Platt JL, Vercellotti GM, Dalmasso AP, et al. Transplantation of discordant xenografts: a review of progress. Immunol Today 1990; 11 (12): 450.
81. Platt JL, Fischel RJ, Matas AJ, Reif SA, Bolman RM, Bach FH. Immunopathology of hyperacute xenograft rejection in a swine-to-primate model. Transplantation 1991; 52 (2): 214.
82. Bach FH, Ferran C, Hechenleitner P, et al. Accommodation of vascularized xenografts: expression of "protective genes" by donor endothelial cells in a host Th2 cytokine environment. Nat Med 1997; 3 (2): 196.
83. Sykes M, Zhao Y, Yang YG. Tolerance induction for xenotransplantation. World J Surg 1997; 21 (9): 932.
84. Weiss RA. Retroviral zoonoses. Nat Med 1998; 4 (4): 391.

71
85. Mueller NJ, Barth RN, Yamamoto S, et al. Activation of cytomegalovirus in pig-to-primate organ xenotransplantation. J Virol 2002; 76 (10): 4734.
86. Armstrong JA, Porterfield JS, De Madrid AT. C-type virus particles in pig kidney cell lines. J Gen Virol 1971; 10 (2): 195.
87. Lieber MM, Sherr CJ, Benveniste RE, Todaro GJ. Biologic and immunologic properties of porcine type C viruses. Virology 1975; 66 (2): 616.
88. Patience C, Takeuchi Y, Weiss RA. Infection of human cells by an endogenous retrovirus of pigs. Nat Med 1997; 3 (3): 282.
89. Le Tissier P, Stoye JP, Takeuchi Y, Patience C, Weiss RA. Two sets of human-tropic pig retrovirus. Nature 1997; 389 (6652): 681.
90. Takeuchi Y, Patience C, Magre S, et al. Host range and interference studies of three classes of pig endogenous retrovirus. J Virol 1998; 72 (12): 9986.
91. Martin U, Kiessig V, Blusch JH, et al. Expression of pig endogenous retrovirus by primary porcine endothelial cells and infection of human cells. Lancet 1998; 352 (9129): 692.
92. Wilson CA, Wong S, Muller J, Davidson CE, Rose TM, Burd P. Type C retrovirus released from porcine primary peripheral blood mononuclear cells infects human cells. J Virol 1998; 72 (4): 3082.
93. Wilson CA, Wong S, VanBrocklin M, Federspiel MJ. Extended analysis of the in vitro tropism of porcine endogenous retrovirus. J Virol 2000; 74 (1): 49.
94. Specke V, Tacke SJ, Boller K, Schwendemann J, Denner J. Porcine endogenous retroviruses: in vitro host range and attempts to establish small animal models. J Gen Virol 2001; 82 (Pt 4): 837.
95. Martin U, Winkler ME, Id M, et al. Productive infection of primary human endothelial cells by pig endogenous retrovirus (PERV). Xenotransplantation 2000; 7 (2): 138.
96. Tacke SJ, Kurth R, Denner J. Porcine endogenous retroviruses inhibit human immune cell function: risk for xenotransplantation? Virology 2000; 268 (1): 87.
97. Clemenceau B, Lalain S, Martignat L, Sai P. Porcine endogenous retroviral mRNAs in pancreas and a panel of tissues from specific pathogen-free pigs. Diabetes Metab 1999; 25 (6): 518.
98. Oldmixon BA, Wood JC, Ericsson TA, et al. Porcine endogenous retrovirus transmission characteristics of an inbred herd of miniature Swine. J Virol 2002; 76 (6): 3045.
99. Takeuchi Y, Porter CD, Strahan KM, et al. Sensitization of cells and retroviruses to human serum by (alpha 1-3) galactosyltransferase. Nature 1996; 379 (6560): 85.
100. Blusch JH, Patience C, Martin U. Pig endogenous retroviruses and xenotransplantation. Xenotransplantation 2002; 9 (4): 242.
101. Larsson E, Venables PJ, Andersson AC, et al. Expression of the endogenous retrovirus ERV3 (HERV-R) during induced monocytic differentiation in the U-937 cell line. Int J Cancer 1996; 67 (3): 451.

72
102. Khan AS, Muller J, Sears JF. Early detection of endogenous retroviruses in chemically induced mouse cells. Virus Res 2001; 79 (1-2): 39.
103. Heneine W, Tibell A, Switzer WM, et al. No evidence of infection with porcine endogenous retrovirus in recipients of porcine islet-cell xenografts. Lancet 1998; 352 (9129): 695.
104. Patience C, Patton GS, Takeuchi Y, et al. No evidence of pig DNA or retroviral infection in patients with short- term extracorporeal connection to pig kidneys. Lancet 1998; 352 (9129): 699.
105. Paradis K, Langford G, Long Z, et al. Search for cross-species transmission of porcine endogenous retrovirus in patients treated with living pig tissue. Science 1999; 285 (5431): 1236.
106. Pitkin Z, Mullon C. Evidence of absence of porcine endogenous retrovirus (PERV) infection in patients treated with a bioartificial liver support system. Artif Organs 1999; 23 (9): 829.
107. Elliott RB, Escobar L, Garkavenko O, et al. No evidence of infection with porcine endogenous retrovirus in recipients of encapsulated porcine islet xenografts. Cell Transplant 2000; 9 (6): 895.
108. Dinsmore JH, Manhart C, Raineri R, Jacoby DB, Moore A. No evidence for infection of human cells with porcine endogenous retrovirus (PERV) after exposure to porcine fetal neuronal cells. Transplantation 2000; 70 (9): 1382.
109. Calne RY. Organ transplantation between widely disparate species. Transplant Proc 1970; 2 (4): 550.
110. Blusch JH, Patience C, Takeuchi Y, et al. Infection of nonhuman primate cells by pig endogenous retrovirus. J Virol 2000; 74 (16): 7687.
111. Martin U, Steinhoff G, Kiessig V, et al. Porcine endogenous retrovirus (PERV) was not transmitted from transplanted porcine endothelial cells to baboons in vivo. Transpl Int 1998; 11 (4): 247.
112. Loss M, Arends H, Winkler M, et al. Analysis of potential porcine endogenous retrovirus (PERV) transmission in a whole-organ xenotransplantation model without interfering microchimerism. Transpl Int 2001; 14 (1): 31.
113. Switzer WM, Michler RE, Shanmugam V, et al. Lack of cross-species transmission of porcine endogenous retrovirus infection to nonhuman primate recipients of porcine cells, tissues, or organs. Transplantation 2001; 71 (7): 959.
114. Deng YM, Tuch BE, Rawlinson WD. Transmission of porcine endogenous retroviruses in severe combined immunodeficient mice xenotransplanted with fetal porcine pancreatic cells. Transplantation 2000; 70 (7): 1010.
115. van der Laan LJ, Lockey C, Griffeth BC, et al. Infection by porcine endogenous retrovirus after islet xenotransplantation in SCID mice. Nature 2000; 407 (6800): 90.

73
116. Clemenceau B, Jegou D, Martignat L, Sai P. B. Clemenceau et al.: PERV infection of mouse and human cells by SPF pig islets in nude mice. Diabetologia 2002; 45 (6): 914.
117. Specke V, Schuurman HJ, Plesker R, et al. Virus safety in xenotransplantation: first exploratory in vivo studies in small laboratory animals and non-human primates. Transpl Immunol 2002; 9 (2-4): 281.
118. Takefman DM, Spear GT, Saifuddin M, Wilson CA. Human CD59 Incorporation into Porcine Endogenous Retrovirus Particles: Implications for the Use of Transgenic Pigs for Xenotransplantation. J Virol 2002; 76 (4): 1999.
119. Ricordi C, Finke EH, Lacy PE. A method for the mass isolation of islets from the adult pig pancreas. Diabetes 1986; 35 (6): 649.
120. Brandhorst H, Brandhorst D, Brendel MD, Hering BJ, Bretzel RG. Assessment of intracellular insulin content during all steps of human islet isolation procedure. Cell Transplant 1998; 7 (5): 489.
121. Korsgren O, Jansson L, Sandler S, Andersson A. Hyperglycemia-induced B cell toxicity. The fate of pancreatic islets transplanted into diabetic mice is dependent on their genetic background. J Clin Invest 1990; 86 (6): 2161.
122. Korsgren O, Andersson A, Sandler S. Pretreatment of fetal porcine pancreas in culture with nicotinamide accelerates reversal of diabetes after transplantation to nude mice. Surgery 1993; 113 (2): 205.
123. Mizushima S, Nagata S. pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Res 1990; 18 (17): 5322.
124. Charreau B, Tesson L, David A, et al. Adenovirus-mediated expression of human CD59 on xenogeneic endothelial cells: Protection against human complement-mediated lysis and induction of cellular activation by adenoviral transduction. Xenotransplantation 1997; 4: 212.
125. Putzer BM, Hitt M, Muller WJ, Emtage P, Gauldie J, Graham FL. Interleukin 12 and B7-1 costimulatory molecule expressed by an adenovirus vector act synergistically to facilitate tumor regression. Proc Natl Acad Sci U S A 1997; 94 (20): 10889.
126. Kohnert KD, Hehmke B. Preparation of suspensions of pancreatic islet cells: a comparison of methods. J Biochem Biophys Methods 1986; 12 (1-2): 81.
127. Koo GC, Peppard JR. Establishment of monoclonal anti-Nk-1.1 antibody. Hybridoma 1984; 3 (3): 301.
128. Korsgren M, Persson CG, Sundler F, et al. Natural killer cells determine development of allergen-induced eosinophilic airway inflammation in mice. J Exp Med 1999; 189 (3): 553.
129. Hagberg A, Barbany G, Krook H, Samiotaki M, Landegren U. Expression profiling across many samples via manifold-assisted mRNA processing. Nucleic Acids Res 2000; 28 (11): E54.

74
130. Gibson UE, Heid CA, Williams PM. A novel method for real time quantitative RT-PCR. Genome Res 1996; 6 (10): 995.
131. Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res 1996; 6 (10): 986.
132. Yin H, Medstrand P, Kristofferson A, Dietrich U, Aman P, Blomberg J. Characterization of human MMTV-like (HML) elements similar to a sequence that was highly expressed in a human breast cancer: further definition of the HML-6 group. Virology 1999; 256 (1): 22.
133. Medstrand P, Lindeskog M, Blomberg J. Expression of human endogenous retroviral sequences in peripheral blood mononuclear cells of healthy individuals. J Gen Virol 1992; 73 (Pt 9): 2463.
134. Grace MJ, Xie L, Musco ML, et al. The use of laser scanning cytometry to assess depth of penetration of adenovirus p53 gene therapy in human xenograft biopsies. Am J Pathol 1999; 155 (6): 1869.
135. Bauerschmitz GJ, Barker SD, Hemminki A. Adenoviral gene therapy for cancer: from vectors to targeted and replication competent agents (review). Int J Oncol 2002; 21 (6): 1161.
136. Chengalvala MV, Lubeck MD, Selling BJ, et al. Adenovirus vectors for gene expression. Curr Opin Biotechnol 1991; 2 (5): 718.
137. Ozmen L, Ekdahl KN, Elgue G, Larsson R, Korsgren O, Nilsson B. Inhibition of thrombin abrogates the instant blood-mediated inflammatory reaction triggered by isolated human islets: possible application of the thrombin inhibitor melagatran in clinical islet transplantation. Diabetes 2002; 51 (6): 1779.
138. McCarter SD, Scott JR, Lee PJ, et al. Cotransfection of heme oxygenase-1 prevents the acute inflammation elicited by a second adenovirus. Gene Ther 2003; 10 (19): 1629.
139. Wu G, Korsgren O, Zhang J, Song Z, van Rooijen N, Tibell A. Pig islet xenograft rejection is markedly delayed in macrophage- depleted mice: a study in streptozotocin diabetic animals. Xenotransplantation 2000; 7 (3): 214.
140. Wallgren AC, Karlsson-Parra A, Korsgren O. The main infiltrating cell in xenograft rejection is a CD4+ macrophage and not a T lymphocyte. Transplantation 1995; 60 (6): 594.
141. Vaddi K, Newton RC. Regulation of monocyte integrin expression by beta-family chemokines. J Immunol 1994; 153 (10): 4721.
142. Korsgren O, Jansson L. Characterization of mixed syngeneic-allogeneic and syngeneic-xenogeneic islet-graft rejections in mice. Evidence of functional impairment of the remaining syngeneic islets in xenograft rejections. J Clin Invest 1994; 93 (3): 1113.
143. Wennberg L, Rafael E, Liu J, Sundberg B, Wernersson A, Korsgren O. Allogeneic and xenogeneic islets are rejected by different and specific mechanisms: A study in rodents using a mixed allogeneic-xenogeneic islet transplantation model. Xenotransplantation 1997; 4: 228.

75
144. Barton GM, Medzhitov R. Control of adaptive immune responses by Toll-like receptors. Curr Opin Immunol 2002; 14 (3): 380.
145. Akira S, Hoshino K. Myeloid differentiation factor 88-dependent and -independent pathways in toll-like receptor signaling. J Infect Dis 2003; 187 Suppl 2: S356.
146. Horng T, Barton GM, Flavell RA, Medzhitov R. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature 2002; 420 (6913): 329.
147. Kopp E, Medzhitov R. Recognition of microbial infection by Toll-like receptors. Curr Opin Immunol 2003; 15 (4): 396.
148. Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R. Toll-like receptors control activation of adaptive immune responses. Nat Immunol 2001; 2 (10): 947.
149. Goldstein DR, Tesar BM, Akira S, Lakkis FG. Critical role of the Toll-like receptor signal adaptor protein MyD88 in acute allograft rejection. J Clin Invest 2003; 111 (10): 1571.
150. Benda B, Karlsson-Parra A, Ridderstad A, Korsgren O. Xenograft rejection of porcine islet-like cell clusters in immunoglobulin- or Fc-receptor gamma-deficient mice. Transplantation 1996; 62 (9): 1207.
151. Benda B, Korsgren O. Interleukin-6 in islet xenograft rejection. Transpl Int 2001; 14 (2): 63.
152. Benda B, Lycke N, Holstad M, Korsgren O. Delayed type hypersensitivity-associated cytokines in islet xenotransplantation: limited efficacy of interleukin-2- and tumor necrosis factor-alpha-blockade in interferon-gamma receptor-deficient mice. Xenotransplantation 2000; 7 (3): 206.
153. Adachi O, Kawai T, Takeda K, et al. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 1998; 9 (1): 143.
154. Spriewald BM, Hara M, Bushell A, Jenkins S, Morris PJ, Wood KJ. Differential role for competitive reverse transcriptase-polymerase chain reaction and intracellular cytokine staining as diagnostic tools for the assessment of intragraft cytokine profiles in rejecting and nonrejecting heart allografts. Am J Pathol 2000; 157 (5): 1453.
155. Lion T. Control genes in reverse transcriptase-polymerase chain reaction assays. Leukemia 1996; 10 (9): 1527.
156. Schmittgen TD, Zakrajsek BA. Effect of experimental treatment on housekeeping gene expression: validation by real-time, quantitative RT-PCR. J Biochem Biophys Methods 2000; 46 (1-2): 69.
157. Zhong H, Simons JW. Direct comparison of GAPDH, beta-actin, cyclophilin, and 28S rRNA as internal standards for quantifying RNA levels under hypoxia. Biochem Biophys Res Commun 1999; 259 (3): 523.
158. Sandberg JO, Margulis B, Jansson L, Karlsten R, Korsgren O. Transplantation of fetal porcine pancreas to diabetic or normoglycemic nude mice. Evidence of a rapid engraftment process

76
demonstrated by blood flow and heat shock protein 70 measurements. Transplantation 1995; 59 (12): 1665.
159. Qari SH, Magre S, Garcia-Lerma JG, et al. Susceptibility of the porcine endogenous retrovirus to reverse transcriptase and protease inhibitors. J Virol 2001; 75 (2): 1048.
160. Wilhelm M, Fishman JA, Pontikis R, Aubertin AM, Wilhelm FX. Susceptibility of recombinant porcine endogenous retrovirus reverse transcriptase to nucleoside and non-nucleoside inhibitors. Cell Mol Life Sci 2002; 59 (12): 2184.
161. Niebert M, Rogel-Gaillard C, Chardon P, Tonjes RR. Characterization of chromosomally assigned replication-competent gamma porcine endogenous retroviruses derived from a large white pig and expression in human cells. J Virol 2002; 76 (6): 2714.
162. Czauderna F, Fischer N, Boller K, Kurth R, Tonjes RR. Establishment and characterization of molecular clones of porcine endogenous retroviruses replicating on human cells. J Virol 2000; 74 (9): 4028.
163. Rowe WP. Deformed whiskers in mice infected with certain exogenous murine leukemia viruses. Science 1983; 221 (4610): 562.
164. Stewart MA, Warnock M, Wheeler A, et al. Nucleotide sequences of a feline leukemia virus subgroup A envelope gene and long terminal repeat and evidence for the recombinational origin of subgroup B viruses. J Virol 1986; 58 (3): 825.
165. Overbaugh J, Riedel N, Hoover EA, Mullins JI. Transduction of endogenous envelope genes by feline leukaemia virus in vitro. Nature 1988; 332 (6166): 731.


Acta Universitatis UpsaliensisComprehensive Summaries of Uppsala Dissertations
from the Faculty of MedicineEditor: The Dean of the Faculty of Medicine
Distribution:Uppsala University Library
Box 510, SE-751 20 Uppsala, Swedenwww.uu.se, [email protected]
ISSN 0282-7476ISBN 91-554-5850-5
A doctoral dissertation from the Faculty of Medicine, Uppsala University,is usually a summary of a number of papers. A few copies of the completedissertation are kept at major Swedish research libraries, while the sum-mary alone is distributed internationally through the series Comprehen-sive Summaries of Uppsala Dissertations from the Faculty of Medicine.(Prior to October, 1985, the series was published under the title “Abstracts ofUppsala Dissertations from the Faculty of Medicine”.)